Тетрагидроизохинолиновые соединения или их фармацевтически приемлемые соли, способ их получения, фармацевтическая композиция на их основе и способ связывания допаминовых рецепторов - RU2122999C1
Код документа: RU2122999C1
Чертежи











Описание
Изобретение относится к новым тетрагидроизохинолиновым соединениям и к фармацевтическим составам, способам получения составов и к использованию в анальгезии и в лечении психозов (например, шизофрении), болезни Паркинсона, синдрома Леш-Ньяна, нарушения дефицита внимания или ослабления познавательной способности, или в облегчении лекарственной зависимости или замедленной дискинезии.
Изобретение относится к тетраизохинолиновым соединениям формулы I
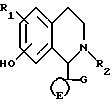
и их фармацевтически приемлемым солям,
где R1 представляет один или более заместителей, выбираемых из H, галогена, гидрокси, алкила с 1 - 3 атомами углерода (необязательно замещенного гидроксилом), алкоксила с 1 - 3 атомами углерода, алкилтио с 1 - 3 атомами углерода, алкилсульфинила с 1 - 3 атомами углерода, алкилсульфонила с 1 - 3 атомами углерода, нитро, циано, полигалоалкила с 1 - 3 атомами углерода, полигалоалкокси с 1 - 3 атомами углерода, фенил (необязательно замещенный одним или более замеcтителями, выбираемыми из галогена, алкила с 1 - 3 атомами углерода, алкокси с 1 - 3 атомами углерода), или R1 является карбамоилом, необязательно алкилированным одной или двумя алкильными группами, каждая из которых независимо с 1 - 3 атомами углерода;
R2 представляет алифатическую группу, содержащую 1 - 3 атома углерода, необязательно замещенных гидрокси или алкокси, содержащими 1 - 3 атома углерода;
E представляет алкиленовую цепь, содержащую 2 - 5 атомов углерода, необязательно замещенных одной или более алкильными группами, содержащими 1 - 3 атома углерода;
G представляет фенил или фенил, замещенный одним или более заместителями, которые могут быть одинаковыми или различными и которые являются независимо алкилом с 1 - 3 атомами углерода, алкокси с 1 - 3 атомами углерода, галогеном, гидрокси, полигалоалкилом с 1 - 3 атомами углерода, полигалоалкокси с 1 - 3 атомами углерода, циано, алкилтио с 1 - 3 атомами углерода, алкилсульфинилом с 1 - 3 атомами углерода, алкилсульфонилом с 1 - 3 атомами углерода, фенилом (необязательно замещенным одним или более заместителями, выбранными из галогена, алкила с 1 - 3 атомами углерода или алкокси с 1 - 3 атомами углерода), карбамоилом, необязательно алкилированным одной или двумя алкильными группами, каждая из которых независимо с 1 - 3 атомами углерода, или G представляет фенильное кольцо, имеющее конденсированное с ним гетероциклическое или ароматическое карбоциклическое кольцо,
и их O-ацилированные производные.
В предпочтительных соединениях формулы I гидроксильная
группа находится в 7 положении. Соответственно, одна из групп предпочтительных соединений данного изобретения представлена формулой II

и их фармацевтически приемлемыми солями,
где R1, R2, E и G определены выше,
и их O-ацилированными производными.
Предпочтительная группа O-ацилированных производных соединения формулы I представлена соединениями формулы III

и их фармацевтически приемлемыми солями,
где R1, R2, E и G определены выше;
R7 представляет ацильную группу, производную от карбоновой кислоты, имеющую 6 - 20 атомов углерода, предпочтительно 7 - 18 атомов углерода.
В более предпочтительных соединениях формулы III R7 представляет гептаноил, деканоил, додеканоил, гексадеканоил или октадеканоил. В наиболее предпочтительных соединениях формулы III группа OR7 находится в положении 7.
В предпочтительных соединениях формулы I, II или III R1 представляет H, галоген, гидрокси, алкил с 1 - 3 атомами углерода, алкокси с 1 - 3 атомами углерода, алкилтио с 1 - 3 атомами углерода, нитро, полифторалкил с 1 - 3 атомами углерода, полифторалкокси с 1 - 3 атомами углерода или фенил, необязательно замещенный фтором, хлором, бромом, метилом или метокси. В более предпочтительных соединениях формулы I, II или III R1 представляет H, фтор, хлор, бром, гидрокси, метил, метокси, фенил или нитро. В особенно предпочтительных соединениях формулы II R1 представляет один из заместителей в положении 6, который является H, фтором, хлором, гидрокси, метилом, метокси или фенилом.
В предпочтительных соединениях формулы I, II или III R2 представляет алкильную группу, содержащую 1 - 3 атома углерода (например, метил или этил), необязательно замещенную гидрокси (например, R2 является 2-гидроксиэтилом) или метокси (например, R2 является 2-метоксиэтилом), или R2 представляет алкенильную группу с 2 - 3 атомами углерода (например, аллил).
В предпочтительных соединениях формулы I, II или III группа E представляет -(CH2)2-, -(CH2)3-, -(CH2)4-, -(CH2)5- или -CH2CMe2 CH2-. В особенно предпочтительных соединениях формулы I или II E представляет -(CH2)2- или -(CH2)3-.
В предпочтительных соединениях формулы I, II или III G представляет фенил или фенил, замещенный одним и более заместителями, которые являются независимо алкилом с 1 - 3 атомами углерода, алкокси с 1 - 3 атомами углерода, галогеном, гидрокси, полифторалкилом с 1 - 3 атомами углерода, полифторалкокси с 1 - 3 атомами углерода или фенилом, необязательно замещенным фтором, хлором, бромом, метилом или метокси, или G представляет нафтил или дигидробензофуран-7-ил.
В более предпочтительных соединениях формулы I, II или III G представляет фенил или фенил, необязательно замещенный метилом, гидрокси, метокси, метилтио, фтором, хлором, бромом, трифторметилом, циано или трифторметокси, или представляет нафтил или группу дигидробензо [b] фуран-7-ил. В особенно предпочтительных соединениях формулы I, II или III G представляет фенил, 2-хлорфенил, 4-хлорфенил, 2,4-дихлорфенил, 3,4-дихлорфенил, 4-фторфенил, 2-бромфенил, 2-метилфенил, 2-метилтиофенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 4-гидроксифенил, 3-трифторметилфенил, 4-трифторметоксифенил, 2-цианофенил, 2-бром-4,5-диметоксифенил, 1-нафтил, 2-нафтил или 2,3-дигидробензо [b]фуран-7-ил.
Конкретными соединениями
формулы I являются:
6,7-дигидрокси-2-метил-1-(1-фенилциклопропил)-1,2,3,4- тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-(3,3-диметил-1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-(1-фенилциклопентил)-1,2,3,4- тетрагидроизохинолин,
6,
7-дигидрокси-2-метил-1-(1-фенилциклогексил)-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклопропил]-6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклопентил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклогексил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил] -2-этил-6,7-дигидрокси-1,2,3,4- тетрагидроизохинолин,
2-аллил-1-[1-(4-хлорфенил)циклобутил] -6,7-дигидрокси-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(3-хлорфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(3,4-дихлорфенил)циклобутил]-6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2,
4-дихлорфенил)циклобутил]-6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-бромфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-бромфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-фторфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-фторфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-[1-(2-метилтиофенил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
6,
7-дигидрокси-2-метил-1-[1-(2-трифторметилфенил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-[1-(3-трифторметилфенил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-[1-(1-(O-толил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-[1-(4-бифенилил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
6,
7-дигидрокси-1-[1-(4-метоксифенил)циклобутил] -2-метил- 1,2,3,4-тетрагидроизохинолин,
6,7-дигидрокси-1-[1-(4-гидроксифенил)циклопентил] -2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[-(2-цианофенил)циклобутил] -6,7-дигидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
6,7-дигидрокси-2-метил-1-[1-(2-нафтил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-2-метил-1-(1-фенилциклобутил] - 1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-2-метил-1-(1-фенилциклопентил] - 1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-2-метил-1-(1-фенилциклогексил] - 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-бромфенил)циклобутил]-7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил]-7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил]-7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)-3,3-диметилциклобутил] -7-гидрокси-6-метокси- 2-метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопептил] -7-гидрокси-6-метокси- 2-метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2,4-дихлорфенил)циклобутил] -7-гидрокси-6-метокси- 2-метил-1,2,3,
4-тетрагидроизохинолин,
7-гидрокси-6-метокси-1-[1-(2-метоксифенил)циклопропил] -2- метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2- метил-(2-гидроксиэтил)-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2- метил-(2-метоксиэтил)-1,2,3,
4-тетрагидроизохинолин,
7-гидрокси-6-метокси-1-[1-(2-метоксифенил)циклобутил] -2- метил-1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-1-[1-(3-метоксифенил)циклобутил]
-2- метил-1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-2-метил-1-[1-(4-трифторметоксифенил)циклобутил] - 1,2,3,4-тетрагидроизохинолин,
1-[1-(2,3-дигидробензо[b]
фуран-7-ил)циклопропил] -7-гидрокси-6- метокси-2-метил-1,2,3,4-тетрагидроизохинолин,
7-гидрокси-6-метокси-2-метил-1-[1-(1-нафтил)циклопропил] - 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-бром-4,5-диметоксифенил)циклобутил] -7-гидрокси-6- метокси-2-метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -7-гидрокси-2-метил-6-фенил- 1,2,3,
4-тетрагидроизохинолин,
6-фтор-7-гидрокси-2-метил-1(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,
4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил- 1,2,3,
4-тетрагидроизохинолин,
1-[1-(2,4-дихлорфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2,4-дихлорфенил)циклопропил] -6-фтор-7-гидрокси-2-метил- 1,2,
3,4-тетрагидроизохинолин,
1-[1-(4-бромфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-бромфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,
4-тетрагидроизохинолин,
6-хлор-7-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
2-аллил-6-хлор-7-гидрокси-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
6-хлор-7-гидрокси-2-метил-1-(3,3-диметил-1-фенил-циклобутил)- 1,2,3,4-тетрагидроизохинолин,
6-хлор-1-[1-(4-хлорфенил)циклобутил] -7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
6-хлор-1-[1-(2-хлорфенил)циклобутил] -7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-бромфенил)циклобутил] -6-хлор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
7-хлор-6-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
5-хлор-8-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
5-хлор-6,
7-дигидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
6,8-дихлор-7-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
7-гидрокси-2-метил-6-нитро-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
6-бром-7-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
7-гидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил]-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил]-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин,
1-[1-(4-хлорфенил)циклобутил]-5-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин,
и их фармацевтически приемлемые соли в виде индивидуальных энантиомеров, рацематов или других смесей
энантиомеров.
Соединения формулы I, II и III могут существовать в виде солей фармацевтически приемлемых кислот. Примеры таких солей включают гидрохлориды, гидробромиды, гидройодиды, сульфаты, нитраты, малеаты, ацетаты, цитраты, фумараты, тартраты, сукцинаты, бензоаты, пальмоаты, метилсульфаты, додеканоаты и соли кислых аминокислот, таких как глютаминовая кислота. Соединения формулы I, II, III и их соли могут существовать в виде сольватов (например, гидратов).
Соединения формулы III имеют высокую липидную растворимость и являются, следовательно, удобными для использования в так называемых депо препаратах, которые предусматривают источник активного соединения, который расположен в теле (например, путем внутримышечной инъекции). Эти соединения могут быть получены в виде фармацевтически приемлемого масла.
Конкретными соединениями формулы III являются:
1-[1-(2-хлорфенил)циклопропил] -7-гептаноилокси-6-метокси-2-метил- 1,2,3,
4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]
-7-додеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гексадеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-октадеканоилокси-6-метокси-2- метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-фтор-2- метил-1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -7-деканоилокси-6-фтор-2- метил-1,2,3,4-тетрагидроизохинолин,
и их фармацевтически приемлемые соли в виде индивидуальных энантиомеров, рацематов или
других смесей энантиомеров.
Специалистам понятно, что соединения формулы I, II и III содержат хиральный центр. Когда соединение формулы I, II и III содержит единственный хиральный центр, оно существует в двух энантиомерных формах. Изобретение включает индивидуальные энантиомеры и смеси этих энантиомеров. Энантиомеры могут быть получены способами, известными специалистам. Такие способы включают обычно разделение посредством образования диастереомерных солей, которые могут быть разделены, например, кристаллизацией; посредством образования диастереомерных производных или комплексов, которые могут быть разделены, например, кристаллизацией, газожидкостной или жидкостной хроматографией; селективным взаимодействием одного из энантиомеров с энантиомер-специфичным реагентом, например ферментативной этерификацией, окислением или восстановлением; или газожидкостной, или жидкостной хроматографией с хиральной фазой, например с хиральным носителем, таким как кварц с присоединенным хиральным лигандом или в присутствии хирального растворителя. Будет отмечено, что там, где желаемый энантиомер превращается в другой химический объект одним из способов разделения, описанных выше, необходима следующая стадия для освобождения желаемой энантиомерной формы. Альтернативно, специфические энантиомеры могут быть синтезированы путем асимметричного синтеза с использованием оптически активных реагентов, субстратов, катализаторов или растворителей или путем превращения одного энантиомера в другой путем асимметрического превращения.
Конкретными энантиомерными формами соединений формулы I являются:
(-)-6-хлор-7-гидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин,
(+)-1-[1-(2-бромфенил)циклобутил]
-6,7-дигидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин гидробромид,
(+)-1-[1-(2-хлорфенил)циклобутил] -6-фтор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин гидробромид,
(+)-1-[1-(2-хлорфенил)циклопропил] -6-фтор-7-гидрокси-2-метил- 1,2,3,4-тетрагидроизохинолин гидробромид,
(+)-1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-6-метокси-2-метил- 1,2,3,
4-тетрагидроизохинолин гидробромид,
(+)-1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-2,6-диметил- 1,2,3,4-тетрагидроизохинолин гидробромид,
(+)-1-[1-(2-хлорфенил)циклопропил]
-7-гептаноилокси- 1,2,3,4-тетрагидроизохинолин,
(+)-1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси- 6-метокси-2- метил-1,2,3,4-тетрагидроизохинолин,
(+)-1-[1-(2-хлорфенил)циклопропил] -7-додеканоилокси- 6-метокси-2- метил-1,2,3,4-тетрагидроизохинолин,
(+)-1-[1-(2-хлорфенил)циклопропил]-7-гексадеканоилокси-6- метокси-2-метил-1,2,3,
4-тетрагидроизохинолин,
(+)-1-[1-(2-хлорфенил)циклопропил]-7-октадеканоилокси-6- метокси-2-метил-1,2,3,4-тетрагидроизохинолин,
(-)-1-[1-(2-хлорфенил)циклобутил]
-7-деканоилокси-6-фтор-2- метил-1,2,3,4-тетрагидроизохинолин,
(+)-1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-фтор-2- метил-1,2,3,4-тетрагидроизохинолин.
Когда соединение формулы I, II или III содержит более одного хирального центра, оно может существовать в диастереомерной форме. Диастереомерные пары могут быть выделены методами, известными специалистам, например, с помощью хроматографии или кристаллизации, а индивидуальные энантиомеры в каждой паре могут быть разделены как описано выше. Данное изобретение включает каждый стереомер соединений формулы I или II и их смеси.
Некоторые соединения формулы I, II или III могут существовать в более чем одной кристаллической форме, и данное изобретение включает каждую кристаллическую форму и их смеси.
Данное изобретение касается также фармацевтических составов, включающих терапевтически эффективное количество соединения формулы I, II или III вместе с фармацевтически приемлемыми разбавителем или носителем. Такие фармацевтические препараты могут быть использованы для анальгезии и при лечении психозов (например, шизофрении), болезни Паркинсона, синдрома Леш-Ньяна, нарушения дефицита внимания, ослабления познавательной способности, облегчения лекарственной зависимости или замедленной дискинезии.
При терапевтическом использовании активное соединение может вводиться орально, ректально, парентерально или наружно, предпочтительно орально. Таким образом, терапевтические составы по данному изобретению могут быть в виде любого известного терапевтического состава для орального, ректального, парентерального или наружного употребления, фармацевтически приемлемые носители, пригодные для использования в таких составах хорошо известны в области фармации. Составы по изобретению могут содержать 0,1 - 90 мас.% активного соединения. Составы изобретения обычно готовят в форме единичной дозы.
Составы для орального применения являются предпочтительными составами изобретения и они представляют собой известные фармацевтические формы для такого употребления, например таблетки, капсулы, гранулы, сиропы, растворы и водные или масляные суспензии. Наполнители, используемые для приготовления этих составов, являются наполнителями, известными в области фармации. Таблетки могут быть получены из смеси активного соединения с наполнителями, например с фосфатом кальция, разрыхляющими агентами, например с кукурузным крахмалом; смазывающими агентами, например стеаратом магния; связывающими веществами, например микрокристаллической целлюлозой или поливинилпирролидоном и другими возможными ингредиентами, известными в этой области, позволяющими таблетировать смесь известными способами. Таблетки могут, если желательно, снабжаться покрытием с использованием известных методов и наполнителей, которые могут включать кишечное покрытие с использованием, например, фталата гидроксипропилметилцеллюлозы. Таблетки могут быть получены способом, известным специалистам, так, чтобы дать замедленный выход соединений данного изобретения. Такие таблетки могут, если желательно, быть получены с покрытием для кишечника с помощью известных способов, например использования ацетатфталата целлюлозы. Подобно этому, капсулы, например твердые или мягкие желатиновые капсулы, содержащие активное соединение с или без добавленных наполнителей, могут быть получены с помощью известных способов и, если желательно, получены с желудочным покрытием известным способом. Содержание капсулы может быть составлено с использованием известных способов так, чтобы дать замедленный выход активного соединения. Таблетки и капсулы могут обычно содержать 1 - 500 мг активного соединения каждая.
Другие составы для орального назначения включают, например, водные суспензии, содержащие активное вещество в водной среде в присутствии нетоксичного суспендирующего агента, такого как натрий карбоксиметилцеллюлоза, и масляные суспензии, содержащие соединение данного изобретения в соответствующем растительном масле, например в арахисовом масле. Активное соединение может быть упаковано в гранулы с или без добавочных наполнителей. Гранулы могут приниматься внутрь пациентом непосредственно или они могут быть добавлены к подходящему жидкому носителю (например, к воде) перед заглатыванием. Гранулы могут содержать разрыхлители, например пару, вызывающую образование пузырьков, состоящую из кислоты и карбонатной или бикарбонатной соли, для облегчения диспергирования в жидкой фазе.
Составы изобретения, пригодные для ректального введения, являются известными фармацевтическими формами для такого введения, например суппозиториями с твердым жиром или полиэтиленгликольными основаниями.
Составы изобретения, пригодные для парентерального введения, являются известными фармацевтическими формами для такого введения, например стерильными суспензиями или стерильными растворами в соответствующем растворителе.
Составы для наружного применения могут содержать матрицу, в которой фармакологически активные соединения данного изобретения диспергированы так, что соединения поддерживаются в контакте с кожей для того, чтобы ввести соединения через кожу. Альтернативно, активное соединение может быть диспергировано в фармацевтически приемлемом креме, геле или в мазевой основе. Количество активного соединения, содержащегося в препарате для наружного применения, должно быть таким, чтобы терапевтически эффективное количество соединения поступило в тот период времени, в течение которого препарат для наружного применения находился в контакте с кожей.
Соединения данного изобретения могут также вводиться путем непрерывного поступления либо из внешнего источника, например путем внутривенного вливания, либо из источника соединения, помещенного внутри тела. Внутренние источники включают имплантированные резервуары, содержащие соединения, предназначенные для подачи, которая непрерывно происходит, например, путем осмоса, и имплантанты, которые могут быть (а) жидкими, такими как суспензия или раствор в фармацевтически приемлемом масле соединения, предназначенного для поступления, например, в виде очень слаборастворимого в воде производного, такого как додеканоатная соль, или соединение формулы III, как описано выше, или (б) твердыми, в форме имплантированного носителя, например синтетической смолы или воскообразного материала, для соединения, предназначенного для подачи. Носитель может быть единственным, содержащим все вещество, или в виде нескольких одинаковых частей, каждая из которых содержит часть соединения, предназначенного для введения. Количество активного вещества, имеющегося во внутреннем источнике, должно быть таким, чтобы терапевтически эффективное количество соединения поступало за длительный период времени.
В некоторых составах может быть выгодным использование соединений данного изобретения в форме частиц очень малого размера, например, таких как полученные путем помола.
В составах данного изобретения активное вещество может быть, если желательно, объединено с другими совместимыми фармакологически активными ингредиентами.
Фармацевтические составы, содержащие терапевтически эффективное количество соединения формулы I, II или III, могут использоваться в анальгезии или для лечения психозов (например, шизофрении), болезни Паркинсона, синдрома Леш-Ньяна, нарушения дефицита внимания или ослабления познавательной способности или в облегчении лекарственной зависимости, или замедленной дискинезии. При таком лечении количество соединения I или II, назначенное орально, ректально или парентерально, в день составляет в пределах 0,1 - 5000 мг, предпочтительно 5 - 500 мг, даваемое в разовой или многократных дозах один раз или более в течение дня.
Далее будут описаны способы получения соединений формулы I. Эти способы являются следующим объектом данного изобретения.
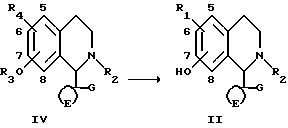
Соединения формулы I могут быть получены расщеплением соединений
формулы IV

где R3 является необязательно замещенной алкильной группой (например, метил или бензил);
R4 является группой R1 или группой, которую можно преобразовать в группу R1.
Диметилирование можно осуществлять путем реакции с бромистоводородной кислотой необязательно в присутствии уксусной кислоты, с трибромидом бора, с пиридин гидрохлоридом, с метантиолатом натрия или с триметилйодсиланом. Дебензилирование можно осуществлять гидролизом, например кислотным гидролизом, или гидрогенолизом, например, с использованием катализатора палладий/древесный уголь. Соединения формулы I, в которых R1 является гидрокси, можно получать путем расщепления соединений формулы IV, в которых группы OR3 или R4 одинаковы (например, метокси или бензилокси). Гидролиз группы R4 будет проходить одновременно с гидролизом группы OR3.
Соединения формулы I можно получать путем алкилирования или алкенилирования соединений формулы V

в условиях, которые не приводят к алкилированию или алкенилированию гидроксигруппы. Например, соединения формулы I, в которых R2 является метилом, можно получать путем метилирования соединений формулы V, например, с использованием формальдегида и муравьиной кислоты или формальдегида и цианборгидрида натрия.
Соединения формулы I, в которых R1 является отличным от H, можно получать путем реакций замещения, которые хорошо известны специалистам. Например, соединения формулы I, в которых R1 является нитро, можно получать путем нитрования соединений формулы I, в которых R1 - H, с использованием азотной кислоты, а соединения формулы I, в которых R1 представляет один или больше атомов хлора, можно получить из соединений формулы I, в которых R1 - H, путем хлорирования, например используя гипохлорит натрия и соляную кислоту.
Соединения формулы II можно получить способами, аналогичными тем, что описаны выше для получения соединений формулы I.
Соединения формулы III можно получить из соединений формулы I реакцией ацилирующим агентом, например хлорангидридом карбоновой кислоты формулы R7Cl или ангидридом карбоновой кислоты формулы (R7)2 O.
Соединения формулы IV можно получить алкилированием соединений формулы VI

например, путем реакции с алкилгалогенидом (например, с метилйодидом) или с алкенилгалогенидом (например, с аллилйодидом или бромидом). Соединения формулы IV можно получать путем восстановительного алкилирования соединений формулы VI, например путем реакции с альдегидом или кетоном в присутствии восстанавливающего агента. Например, соединения формулы IV, в которых R2 является метилом, можно получать путем метилирования соединений формулы VI, например, используя формальдегид и муравьиную кислоту, формальдегид и дигидрофосфит натрия или формальдегид и цианборгидрид натрия.
Соединения формулы IV, в которых R2 является метилом, можно получать путем реакции соединений формулы VII

где R5 является группой R3 в условиях, которые приводят к восстановлению и метилированию соединения формулы VII, например, с формальдегидом и в присутствии восстанавливающего агента, такого как цианборгидрид натрия.
Соединения формулы IV можно получать путем реакции соединений формулы VIII

в которых R6 является группой R2 с соединением формулы IX

в присутствии кислоты, например соляной кислоты.
Соединения формулы IV можно получать
путем восстановления соединений формулы X
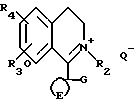
где Q⊖ является подходящим анионом, таким как йодид или метилсульфат с, например, боргидридом натрия, цианоборгидридом натрия, бораном, комплексом боран-диметилсульфида, литийалюминийгидридом или путем каталитического гидрирования. Хиральные восстанавливающие агенты, такие как хиральные триацилоксиборгидриды натрия (например, соответствующие энантиомеры трис-(N-бензилоксикарбонилпролилокси) боргидрид или трис-[N-(2-метилпропилоксикарбонил) пролилокси] боргидрид), хиральные диалкилоксибораны, хиральные оксазаборлидины могут использоваться для получения одного из энантиомеров соединения формулы IV. Один из энантиомеров соединения формулы IV можно получать путем каталитического гидрирования с использованием хирального катализатора. Соответствующим катализатором является комплекс, образующийся путем реакции хирального фосфина (например, 2,3-0-изопропилиден-2,3-дигидрокси-1,4-бис (дифенилфосфино)-бутана) с комплексом переходного металла (например, с хлор(1,5-циклооктадиен)родий (I)димером).
Соединения формулы V можно получать путем гидролиза соединений формулы VI, в которых R4 является группой R1 или группой, которая может быть преобразована в группу R1 способом, подобным тому, что описан выше в отношении соединений формулы I.
Соединения формулы V можно получать путем восстановления соединений формулы VII, в которых R5 является H, например, используя реакции восстановления, подобные тем, что описаны выше для восстановления соединения формулы X. Хиральные восстанавливающие агенты можно использовать для получения одного из энантиомеров соединения формулы V способом, подобным тому, что указан выше для восстановления соединений формулы X.
Соединения формулы VI можно получать путем восстановления соединений формулы VII, в которых R5 является группой R3, способом, подобным тому, что указана выше для получения соединений формулы IV и V.
Соединения формулы VI можно получать путем восстановления соединений формулы XI

например, используя каталитическое гидрирование.
Соединения формулы VI можно получать путем реакции соединений формулы VIII, в которых R6 является H, с соединением формулы IX в присутствии кислоты, например соляной кислоты.
Соединения формулы VII можно получать путем циклирования соединений формулы XII

в котором R5 - H или R3.
Циклизацию можно осуществлять в присутствии конденсирующего агента, такого как оксихлорид фосфора, пентоксид фосфора, пентахлорид фосфора, полифосфорный эфир, полифосфорная кислота, хлорид цинка, соляная кислота, тионилхлорид или серная кислота.
Соединения
формулы VII можно получать путем реакции соединения формулы XIII

с галозамещенной группой формулы X-G, в которой X является гало (например, фтор) в присутствии основания, такого как диизопропиламид лития.
Соединения формулы IX можно получать путем восстановления
арилциклоалканкарбонитрилов формулы XIV

ди-трет-бутилалюминийгидридом или ди-изобутилалюминийгидридом, или восстановления арилциклоалканкарбонилхлоридов формулы XV

три-трет-бутоксиалюминийгидридом.
Соединения формулы X можно получать путем реакции соединения формулы VII, в котором R5 является группой R3, с алкилирующим агентом формулы R2O, например с метилйодидом или с диметилсульфатом.
Соединения формулы XI можно получать путем циклизации соединений формулы XVI

Циклизацию можно осуществлять в присутствии кислоты, такой как серная кислота.
Соединения
формулы XII можно получать путем взаимодействия фенэтиламина формулы XVII

где R5 является H или R3, с арилциклоалканкарбонилхлоридом формулы XV, например, в присутствии органического основания, такого как триэтиламин. Соединения формулы XII можно получать путем конденсации фенэтиламина формулы XVII с арилциклоалканом карбоновой кислоты формулы XVIII

или ее эфира, например, путем связывания или путем действия конденсирующего агента, такого как карбонилдиимидазол. Соединения формулы XIII можно получать путем циклизации соединений формулы XIX

в условиях, подобным тем, что указаны выше для циклизации соединений формулы XII.
Арилциклоалканкарбонитрилы формулы XIV можно получать путем реакции арилацетонитрила формулы XX
G-CH2-CN
с дигалоидным соединением формулы XXI
Z-T-Z'
где
Z и Z', которые могут быть одинаковыми или различными, являются уходящими группами, такими как гало, например хлор или бром, в присутствии основания, такого как гидрид натрия или гидроксид калия.
Арилциклоалканкарбонилхлориды формулы XV можно получать из арилциклоалканкарбоновых кислот формулы XVIII способами, хорошо известными в данной области, например путем реакции с тионилхлоридом.
Соединения формулы XVI можно получать путем реакции соединения формулы XXII

с галоацетальдегиддиметилацеталем, например хлорацетальдегид диметилацеталем.
Арилциклоалканкарбоновые кислоты формулы XVIII можно получать путем гидролиза (например, основного гидролиза) арилциклоалканкарбонитрилов формулы XIV или путем взаимодействия перекиси водорода с арилциклоалканкарбонитрилами формулы XIV в присутствии основания с последующим взаимодействием с азотной кислотой для получения необходимой карбоновой кислоты.
Соединения формулы XIX можно получить путем реакции фенилэтиламина формулы XV с циклоалканкарбонилхлоридом формулы
XXIII

Соединения формулы XXII можно получить путем взаимодействия соединения формулы XXIV

где У является галогеном (например, хлором или бромом) с арилциклоалканкарбонитрилом формулы XIV с последующим восстановлением, например, боргидридом натрия.
Соединения формулы XXIV можно получить путем взаимодействия магния с соединением формулы XXV
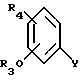
где Y является галогеном (например, хлором или бромом).
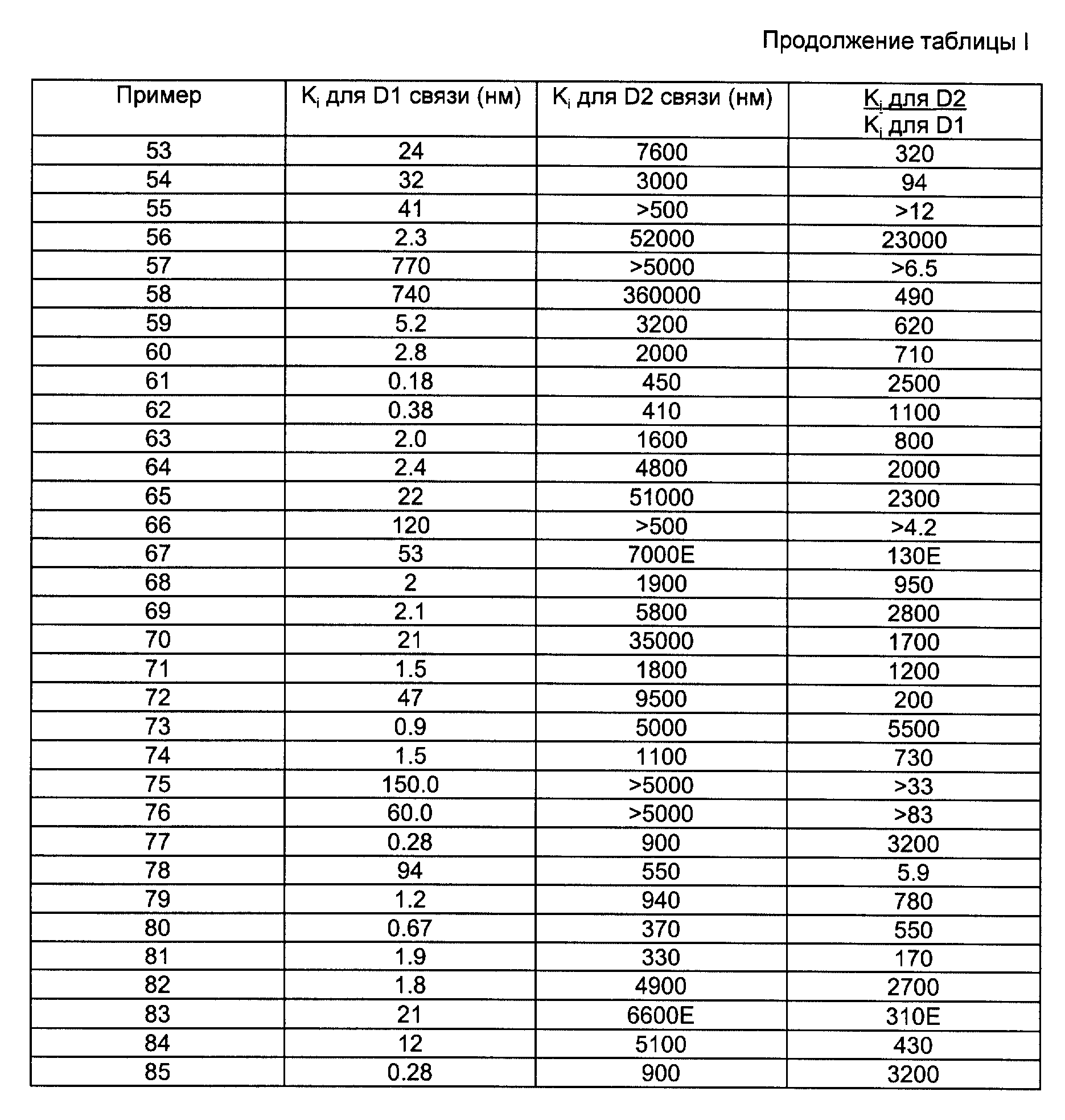
Способность соединений формулы I или II к взаимодействию с допаминовыми рецепторами продемонстрирована в следующих испытаниях, которые определяют способность соединений к ингибированию тритированного лиганда, связывающегося с допаминовыми рецепторами in vitro, в частности с D1 и D2 допаминовыми рецепторами.
Образцы срезов ткани мозга самцов крыс Charles River CD, вес которых составлял 140 - 250 г, гомогенизируют в охлажденном льдом 50 мМ трис-HCl-буфере (pH 7,4, когда измеряют при 25oC для анализов D1 связывания, и pH 7,7, когда измеряют при 25oC для анализов D2 связывания) и центрифугируют в течение 10 мин (при 21,000 g, когда используют для анализов D1 связывания, и 40,000 g, когда используют для анализов D2 связывания). Осадок повторно суспендируют в том же буфере, снова центрифугируют и целевой осадок хранят при -80oC. Перед каждым тестом осадок повторно суспендируют в 50 мМ трис-HCl-буфере, содержащем 120 мМ NaCl, 5 мМ KCl, 2 мМ CaCl2 и 1 мМ MgCl2, при pH 7,4 для анализов D1 связывания и при pH 7,7 с добавлением 6 мМ аскорбиновой кислоты для анализов D2 связывания. Равные части этой суспензии добавляют после этого в пробирки, содержащие лиганд и либо соединение, подлежащее тестированию, либо буфер. Для анализов D1 связывания лиганд тритируют SCH 23390 и смесь инкубируют при 37oC в течение 30 мин перед окончанием инкубации путем быстрого фильтрования. Для анализов D2 связывания лиганд тритируют (S)-сульпиридом и смесь инкубируют при 4oC в течение 40 мин перед окончанием инкубации путем быстрого фильтрования. Неспецифическое связывание определяют экспериментально путем добавления насыщающих концентраций хлорпромазина или спиропиридола для D1 и D2 рецепторов соответственно.
Фильтры промывают охлажденным льдом трис-HCl-буфером и сушат. Фильтры помещают в пузырьки, содержащие сцинциллирующую жидкость и оставляют на период в течение около 20
ч перед тем, как подвергнуть сцинцилляционной спектрофотометрии. Получают кривые замещения для ряда концентраций исследуемого соединения и концентрацию, которая дает 50%-ное ингибирование
специфического связывания. Затем рассчитывают коэффициент ингибирования Ki по формуле

где [лиганд] является концентрацией используемого тритированного лиганда;
KD является равновесной константой диссоциации для лиганда.
Значения Ki, полученные из указанных выше тестов для D1 и D2 связывания, для каждого из целевых продуктов по примерам 1 - 85, приведенных ниже, даны в табл. 1, которая также показывает соотношение между этими двумя значениями и двумя значимыми объектами. В некоторых случаях значения Ki для D2 связывания устанавливают из данных об одной концентрации путем применения уравнения адсорбционной изотермы Ленгмюра. Эти случаи отмечены с помощью "E" в последних двух столбцах табл. 1. В других случаях установить или определить Ki было невозможно и значение Ki дано как большее (>) чем то, которое дало бы применение приведенной выше формулы к наивысшей концентрации, которая вытесняет ≤ 50% лиганда.
Изобретение иллюстрируется с помощью следующих примеров, которые даны всего лишь как примеры. В этих примерах все температуры даны в градусах Цельсия. Целевые продукты каждого из этих примеров характеризуются одними или несколькими из следующих способов анализа: элементный анализ, спектроскопия ядерного магнитного резонанса и инфракрасная спектроскопия.
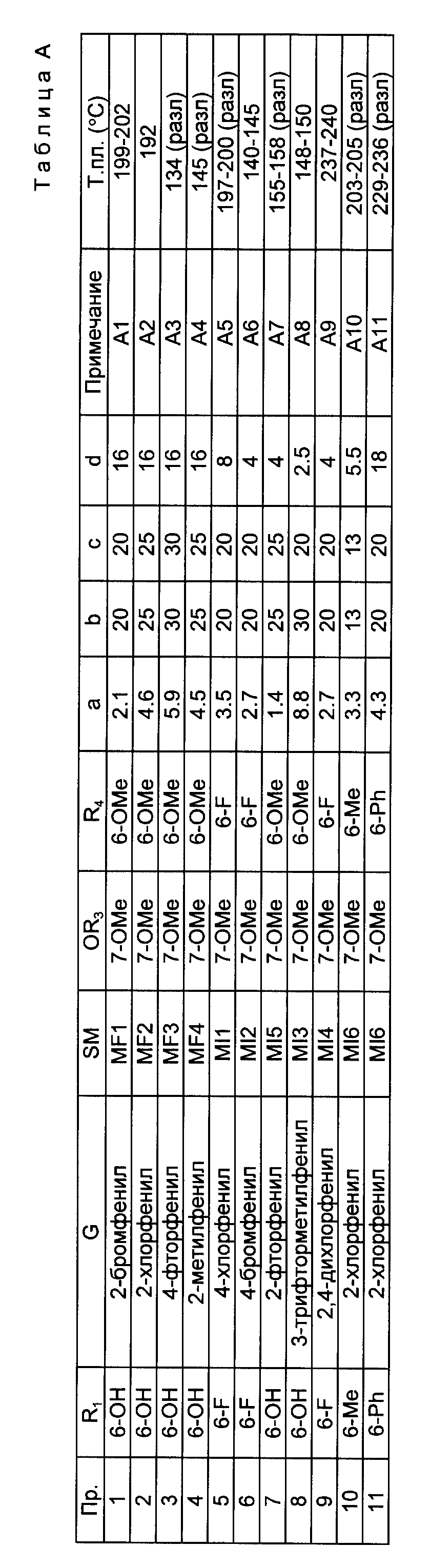
Примеры 1 - 11.
Соединение формулы IV (а, г, полученное как описано в примере, указанном в столбце SM), в котором R2 является метилом, OR3 и R4 определены в табл. A, и E является -(CH2)3-, нагревают с обратным холодильником с 48%-ной водной бромистоводородной кислотой (b, мл) и ледяной уксусной кислотой (с, мл) в течение d ч, чтобы получить соединение формулы II, в котором R1 определено в табл. A, R2 является метилом, а E является -(CH2)3 -. Растворитель удаляют путем выпаривания, а остаток сушат путем повторяющейся азеотропной отгонки с пропан-2-олом. Продукт выделяют в виде его гидробромидной соли (точка плавления в градусах Цельсия дана в столбце, озаглавленном "T.пл."). Способ выделения и любые другие изменения в изложенном выше способе приведены в столбце, озаглавленном "Примеч.".
Примечания к табл. A:
A1 Продукт осаждают из его концентрированного раствора в пропан-2-оле.
A2 Остаток от азеотропной отгонки сушат в вакууме при 100oC в течение 2 ч, затем обеспечивают древесным углем в пропан-2-оле, промывают эфиром, пропан-2-олом (1-2 мл) и эфиром и сушат в вакууме.
A3 Остаток от азеотропной отгонки перекристаллизовывают из пропан-2-ола, получая желаемый продукт.
A4 Остаток от азеотропной отгонки дает желаемый продукт, который используют без дальнейшей очистки.
A5 Реакция проводится в атмосфере азота. Остаток от азеотропной отгонки распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой сушат и обрабатывают щавелевой кислотой. Полученную соль щавелевой кислоты собирают путем фильтрации, промывают эфиром и сушат при 50oC в вакууме. Температура плавления соли щавелевой кислоты дана в последнем столбце табл. A.
A6 Реакцию проводят в атмосфере азота. Остаток после азеотропной отгонки обесцвечивают древесным углем в метаноле и полученный продукт сушат путем азеотропной отгонки с пропан-2-олом. Полученный остаток обесцвечивают древесным углем в этаноле. Раствор дает продукт при выпаривании.
A7 Остаток после азеотропной отгонки обесцвечивают древесным углем в метаноле. Остаток распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой обрабатывают эфирным раствором щавелевой кислоты. Полученный твердый осадок промывают эфиром и сушат в вакууме. Температура плавления соли щавелевой кислоты дана в последнем столбце табл. A.
A8 Реакцию проводят в атмосфере азота. Полученный остаток после удаления растворителя из реакционной смеси растворяют в техническом денатурате и обесцвечивают древесным углем. Растворитель удаляют и остаток промывают эфиром, растворяют в этаноле и обесцвечивают древесным углем. Концентрирование раствора дает желаемый продукт, который промывают эфиром.
A9 Реакцию проводят в атмосфере азота. Растворитель удаляют из реакционной смеси путем отгонки и остаток растворяют в метаноле и обесцвечивают древесным углем. Фильтрование и выпаривание дают остаток, который сушат путем азеотропной отгонки с пропан-2-олом. Продукт кристаллизуют из пропан-2-ола, собирают путем фильтрации, промывают эфиром и сушат в вакууме при 80oC.
A10 Остаток от азеотропной отгонки распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой дает остаток, который нагревают с обратным холодильником с 48%-ной бромистоводородной кислотой (15 мл) и ледяной уксусной кислотой (15 мл) в течение 5 ч. Реакционную смесь нейтрализуют насыщенным водным раствором бикарбоната калия и экстрагируют эфиром. Эфирный экстракт промывают 1 М соляной кислотой. Промывание дает твердый осадок, который сушат в вакууме при 70oC, подщелачивают и экстрагируют эфиром. Экстракт дает остаток, который растворяют в этаноле и обрабатывают эфирным раствором щавелевой кислоты, получая желаемый продукт в виде его соли щавелевой кислоты, температура плавления которого дана в последнем столбце табл. A.
A11 Реакцию проводят в атмосфере азота. Остаток после азеотропной отгонки обесцвечивают древесным углем в метаноле и полученный продукт растирают в порошок в пропан-2-оле. Твердый продукт кристаллизуют из эфира, собирают с помощью фильтрования и сушат в вакууме при 50oC.
Пример 12. 1-[1-(2-Хлорфенил)циклопропил]-7-метокси-2-метил-1,2,3,4- тетрагидроизохинолин (0,25 g, полученный как описано в примере MF7) нагревают на паровой бане при 100oC в растворе 48%-ной бромоводородной кислоты (60 мл) и ледяной уксусной кислоты (60 мл) в течение 16 ч. Смесь нагревают с обратным холодильником в течение 2 ч до тех пор, пока реакция не закончится. Растворитель удаляют в вакууме и остаток сушат путем азеотропной отгонки с пропан-2-олом. Полученную суспензию удаляют путем фильтрации и кристаллизуют из этанола дважды, получая 1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-2- метил-1,2,3,4-тетрагидроизохинолин гидробромид, который характеризуют с помощью элементного анализа.
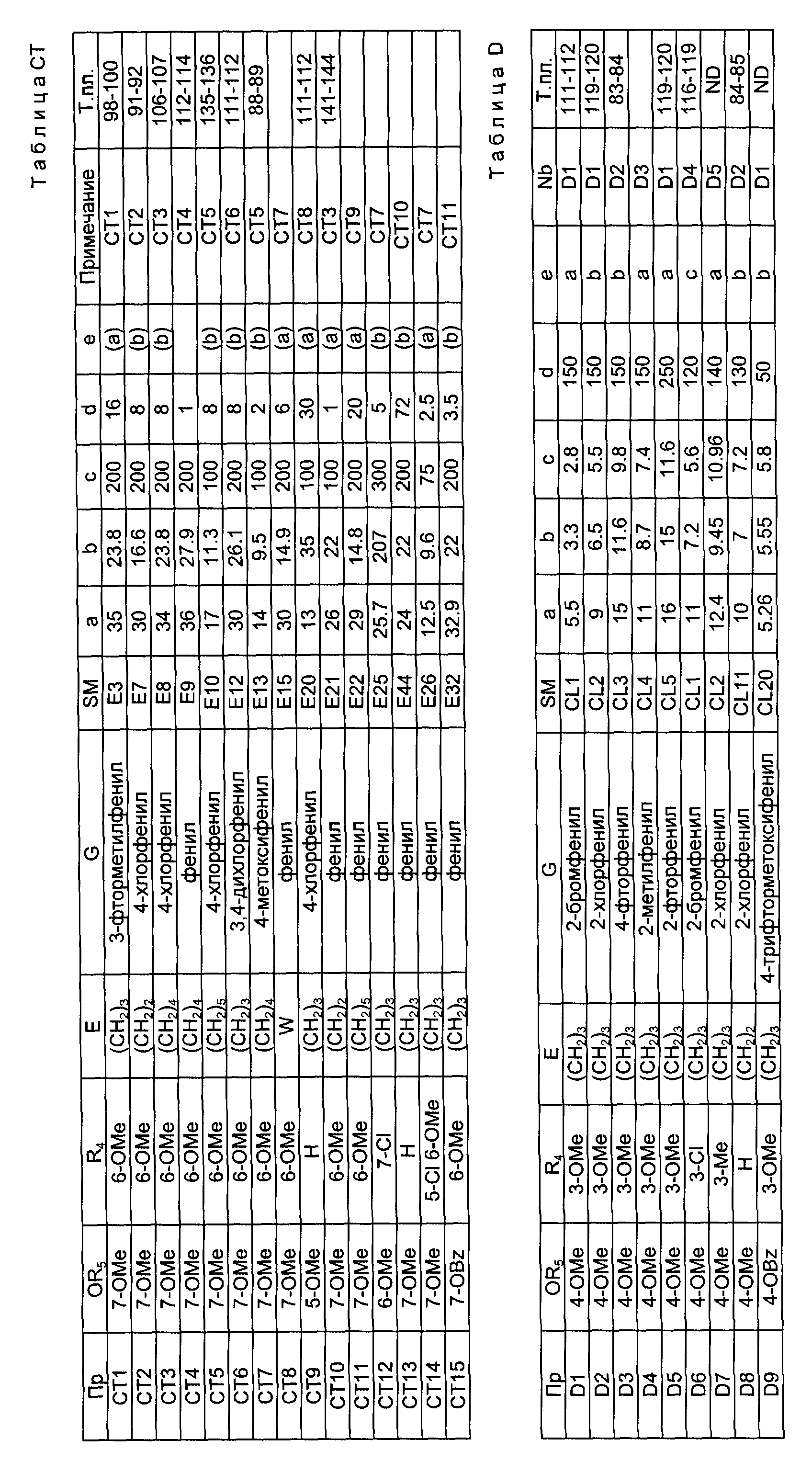
Примеры 13 - 34.

Смесь соединения формулы VI, в которой OR3, R4, E и G определены в табл. AA, часть I (a, г), безводного карбоната калия (b, г), метилйодида (c, г) и ацетона (d, мл) перемешивают при комнатной температуре в течение e ч. Реакционную смесь фильтруют и обрабатывают как представлено в примечаниях к табл. AA, часть I, получая соединение формулы IV, в котором R2 является метилом, OR3, R4, E и G определены в табл. AA, часть I, которое используют не охарактеризовывая в следующей стадии реакции.
Смесь соединения формулы VI, полученного по способу, рассмотренному в предыдущем абзаце (f, г), 48%-ной бромоводородной кислоты (g, мл) и ледяной уксусной кислоты (h, мл) нагревают в азоте с обратным холодильником в течение j ч, получая соединение формулы I, в котором R1 определено в табл. AA, часть II, R2 является метилом, положение гидроксизаместителя показано в столбце, озаглавленном POS в табл. AA, часть II, а E и G определены в табл. AA, часть I. Желаемый продукт выделяют так, как описано в примечаниях к табл. AA, часть II.
Примечания к табл. AA:
В столбце E табл. AA W представляет -CH2CMe2CH2-.
AA1 Следующую порцию метилйодида (0,19) добавляют через 3 ч. Реакционную смесь фильтруют и растворитель удаляют путем выпаривания, получая остаток, который растворяют в дихлорметане. Раствор фильтруют и растворитель удаляют путем выпаривания, получая твердый остаток, который используют в следующей стадии.
AA2 Растворитель удаляют из реакционной смеси и остаток сушат путем азеотропной отгонки с пропан-2-олом. Потом остаток обесцвечивают древесным углем в пропан-2-оле, получая гидробромидную соль желаемого соединения формулы I, которую промывают пропан-2-олом, а затем эфиром и сушат в вакууме. Температура плавления соли дана в последнем столбце табл. AA.
AA3 Желаемый продукт получают путем фильтрования и удаления растворителя из реакционной смеси.
AA4 Растворитель удаляют из реакционной смеси. Раствор обесцвечивают древесным углем в метаноле, фильтруют и удаляют растворитель, получая остаток, который кристаллизуют из пропан-2-ола, получая гидробромидную соль, температура плавления которой дана в последнем столбце табл. AA.
AA5 Растворитель удаляют из реакционной смеси и остаток распределяют между эфиром и водой. Эфирный слой дает желаемый продукт.
AA6 Остаток из концентрированной реакционной смеси распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой дает остаток, который обрабатывают эфирным раствором HCl, получая гидрохлоридную соль, которую перекристаллизовывают из пропан-2-ола. Температура плавления этой соли дана в табл. AA.
AA7 Гидробромидную соль осаждают из реакционной смеси. Температура плавления этой соли дана в табл. AA.
AA8 Остаток от реакционной смеси обесцвечивают древесным углем в метаноле, сушат путем азеотропной отгонки с пропан-2-олом, растирают в порошок в пропан-2-оле и эфире, получая гидрохлоридную соль. Температура плавления дана в табл. AA.
AA9 Остаток после выпаривания реакционной смеси обеспечивают древесным углем в пропан-2-оле. Растворитель удаляют, остаток промывают эфиром и обесцвечивают древесным углем в ацетоне. Растворитель удаляют, получая гидробромидную соль, температура плавления которой не может быть определена, так как продукт разрушается при ~150oC.
AA10 Исходный материал является соединением формулы VI, в которой G является 4-метоксифенилом. Метоксигруппу преобразуют в желаемую гидроксигруппу во время второй стадии реакции. Остаток от реакционной смеси распределяют между водой и дихлорметаном. Органический слой дает желаемый продукт.
AA11 Остаток от концентрирования реакционной смеси распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой дает остаток, который обрабатывают эфирным раствором HCl, получая гидрохлоридную соль, которую перекристаллизовывают из 10:1 смеси этанола и легкого петролейного эфира (температура кипения 60-80oC). Температура плавления этой соли дана в табл. AA.
AA12 Остаток от выпаривания растворителя из реакционной смеси обесцвечивают древесным углем в пропан-2-оле. Остаток промывают петролейным эфиром (температура кипения 60-80oC) и распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой дает масло, которое обрабатывают эфиром, получая осадок, который отделяют путем фильтрования. Фильтрат обрабатывают эфирным раствором щавелевой кислоты, получая соль щавелевой кислоты желаемого продукта, которую перекристаллизовывают из метанола. Температура плавления этой соли дана в табл. AA.
AA13 Остаток от выпаривания растворителя из реакционной смеси обрабатывают водой и экстрагируют этилацетатом. Органический слой дает желаемый продукт, который используют без дальнейшей очистки.
AA14 Остаток от выпаривания растворителя из реакционной смеси обесцвечивают древесным углем в метаноле, сушат путем азеотропной отгонки с пропан-2-олом, получая гидрохлоридную соль, которую перекристаллизовывают из пропан-2-ола. Температура плавления этой соли дана в табл. AA.
AA15 Остаток от выпаривания растворителя из реакционного раствора сушат путем азеотропной отгонки с пропан-2-олом, обесцвечивают древесным углем в метаноле и перекристаллизовывают из пропан-2-ола, получая гидробромидную соль, температура плавления которой дана в табл. AA.
AA16 Остаток от выпаривания растворителя из реакционной смеси распределяют между водой и дихлорметаном, органический слой дает желаемый продукт.
AA17 Остаток от выпаривания растворителя из реакционной смеси распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органический слой дает остаток, который растворяют в смеси 10:1 эфира и пропан-2-ола и обрабатывают эфирным раствором HCl. Желаемое соединение в виде его гидробромидной соли осаждают при охлаждении и перекристаллизовывают из смеси 10:1 пропан-2-ола и метанола. Температура плавления этой соли дана в табл. AA.
AA18 Остаток от выпаривания растворителя из реакционной смеси распределяют между эфиром и водой. Органический слой отделяют, обеспечивают древесным углем, упаривают до половины объема и обрабатывают эфирным раствором HCl, получая желаемый продукт в виде гидрохлоридной соли.
AA19 Остаток от выпаривания растворителя из реакционной смеси распределяют между этилацетатом и насыщенным водным раствором бикарбоната натрия. Органическую фазу обесцвечивают древесным углем и растворитель удаляют, получая остаток, который обрабатывают этанольным раствором HCl, получая желаемый продукт в виде гидрохлоридной соли, которую перекристаллизовывают из ацетона. Температура плавления этой соли дана в табл. AA.
AA20 Остаток от выпаривания растворителя из реакционной смеси распределяют между эфиром и водой, и высушенный эфирный слой обрабатывают этаноловой HCl получая смолу, которую обрабатывают метанолом, получая желаемый продукт в виде его гидрохлоридной соли, которую используют без дальнейшей очистки.
AA21 Остаток от выпаривания растворителя из реакционной смеси сушат путем азеотропной отгонки пропан-2-ола и обеспечивают древесным углем в метаноле. Растворитель удаляют и остаток обрабатывают смесью пропан-2-ола и эфира, получая твердую фазу, которую перекристаллизовывают из смеси 1:1 этанола и эфира, с получением желаемого продукта в виде его гидробромидной соли, температура плавления которой дана в последнем столбце табл. AA.
AA22 Остаток от выпаривания растворителя из реакционной смеси сушат путем азеотропной отгонки пропан-2-ола, получая остаток, который кристаллизуют из пропан-2-ола, получая гидробромидную соль желаемого соединения формулы I, температура плавления которой дана в последнем столбце табл. AA.
AA23 Исходный материал освобождают из его гидрохлоридной соли перед реакцией. Растворитель удаляют из реакционной смеси и остаток распределяют между эфиром и водой. Эфирный слой дает желаемое соединение.
AA24 Остаток из реакционной смеси обесцвечивают древесным углем в метаноле и распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой дает остаток, который обрабатывают эфирным раствором HCl, получая гидрохлоридную соль. Температура плавления этой соли дана в табл. AA.
AA25 Реакционную смесь охлаждают и выливают в воду. Смесь экстрагируют эфиром. Желаемый продукт получают их эфирного экстракта.
AA26 Реакционную смесь выливают в лед, подщелачивают 5н. водным раствором гидробромида натрия и экстрагируют эфиром. Эфирный экстракт дает остаток, который растворяют в эфире. Обработка эфирным раствором щавелевой кислоты дает оксалатную соль желаемого продукта, температура плавления которой дана в последнем столбце табл. AA.
AA27 Растворитель удаляют из реакционной смеси и остаток распределяют между эфирным раствором HCl и пропан-2-олом. Удаление растворителей дает гидробромидную соль желаемого продукта. Температура плавления этой соли дана в последнем столбце табл. AA.
AA28 Остаток из реакционной смеси обрабатывают насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом. Экстракты дают остаток, который обрабатывают смесью 1:10 пропан-2-ола и эфирным раствором HCl, получая гидробромидную соль желаемого продукта. Температура плавления этой соли дана в табл. AA.
AA29 Растворитель удаляют из реакционной смеси и к остатку добавляют воду. Полученную смесь экстрагируют эфиром. Чтобы осадить оксалатную соль продукта добавляют эфирный раствор щавелевой кислоты. Соль растворяют в 1 M водном растворе гидрохлоридного натрия и экстрагируют эфиром. Экстракт дает желаемый продукт (как его свободное основание), который используют без дальнейшей очистки.
AA30 Растворитель удаляют из реакционной смеси и остаток сушат путем азеотропной отгонки пропан-2-ола, обесцвечивают древесным углем в метаноле и опять сушат путем азеотропной отгонки пропан-2-ола, а твердый продукт осаждают путем добавления эфира. Этот цикл растворения/осаждения повторяют, а твердый продукт собирают и промывают смесью 1:5 пропан-2-ола и эфира, получая желаемый продукт в виде его гидробромидной соли, температура плавления которой дана в последнем столбце табл. AA.
AA31 Остаток из реакционной смеси сушат путем азеотропной отгонки пропан-2-ола, чтобы получить остаток, который обесцвечивают древесным углем в метаноле, получая гидробромидную соль. Температура плавления соли дана в табл. AA.
Примеры 35-39.

Смесь соединения формулы I, в которой OR3, R4, E и G определены в табл. AB, часть I (a, г), безводного карбоната калия (b, г), метилйодида (c, г) и ацетона (d, г) перемешивают при температуре окружающей среды в течение e ч. Смесь фильтруют и удаляют, чтобы получить остаток, который распределяют между эфиром и водой. Эфирный слой дает соединение формулы IV, в котором R2 является метилом, а OR3, R4, E и G определены в табл. AB, часть I. После этого соединение формулы IV нагревают с обратным холодильником в растворителе, определенном в столбце f табл. AB, часть II (a - метанол, b - этанол) (g, мл), с концентрированной соляной кислотой (h, мл) в течение j ч, чтобы получить желаемое соединение формулы I, в котором положение гидроксизаместителя определено в столбце, озаглавленном POS в табл. AB, часть II, R1 определено в табл. AB, часть II, R2 является метилом, а E и G определены в табл. AB, часть I. Способ, который используют для получения продукта указан в последующих примечаниях к табл. AB.
Примечания к табл. AB:
Сокращение OBz обозначает бензилокси.
AB1 Растворитель удаляют из реакционной смеси и остаток распределяют между насыщенным водным раствором бикарбоната натрия и этилацетатом. Органический слой дает остаток, который обрабатывают эфирным раствором оксалиновой кислоты, получая желаемый продукт в виде его оксалатной соли, которую перекристаллизовывают из ацетонитрила. Температура плавления соли дана в последнем столбце табл. AB.
AB2 Растворитель удаляют из реакционной смеси, а остаток подщелачивают насыщенным водным раствором бикарбоната натрия и экстрагируют эфиром. Экстракты обрабатывают смесью 4:1 соляной кислоты и пропан-2-ола в эфире. Удаление растворителя дает остаток, который сушат азеотропной отгонкой с пропан-2-олом. Остаток собирают путем фильтрования, промывают эфиром и сушат в вакууме при 60oC, получая желаемый продукт в виде его гидробромидной соли, температура плавления которой дана в последнем столбце табл. AB.
AB3 Растворитель удаляют из реакционной смеси. Остаток подщелачивают насыщенным водным раствором бикарбоната натрия и экстрагируют этилацетатом. Экстракт дает остаток, который растворяют в эфире. Добавляют эфирным раствором оксалиновой кислоты, получая твердый продукт, который кипятят в смеси 9: 1 эфира и ацетата, получая желаемый продукт в виде оксалиновой соли, температура плавления которой дана в последнем столбце табл. AB.
AB4 Растворитель удаляют из реакционной смеси, а остаток обесцвечивают древесным углем в этаноле и сушат путем азеотропной отгонки с пропан-2-олом. Остаток тритируют в этилацетате и растворяют в этаноле. Растворитель удаляют и остаток промывают петролейным эфиром (температура кипения 60-80oC) и сушат при 55oC в вакууме, получая желаемый продукт в виде его гидробромидной соли, температура плавления которой дана в последнем столбце табл. AB.
AB5 Перед нагреванием соединения формулы IV с обратным холодильником соединение растворяют в смеси 1:1 этилацетата и петролейного эфира (10 мл) и элюируют в колонке для флэш (flash) хроматографии, используя ту же смесь растворителей в качестве элюента. Материал, имеющий фактор задержки (Rf) 0,33, собирают и растворитель отгоняют, получая твердую фазу, которую растворяют в этилацетате и пропускают через колонку Florisel®. Материал, имеющий фактор задержки (Rf) 0,33, собирают и растворитель отгоняют, получая смолу. Последнюю растворяют в эфире (60 мл), фильтруют и через фильтрат пробулькивают газообразный хлористый водород то тех пор, пока осаждение не прекращается. Твердую фазу собирают путем фильтрования, промывают эфиром в вакууме 4 ч (т. пл. 173-179oC). После нагревания соединения формулы IV с обратным холодильником растворитель удаляют из реакционной смеси и остаток сушат путем азеотропной отгонки с пропан-2-олом. Остаток тритируют в смеси 1: 3 пропан-2-ола и эфира, а твердый продукт собирают путем фильтрации, промывают смесью 1:3 пропан-2-ола и эфира и сушат в вакууме при 50oC в течение 4 ч, чтобы получить желаемый продукт в виде его гидробромидной соли, температура плавления которой приведена в последнем столбце табл. AB.
Пример 40. Муравьиную кислоту (39 мл) добавляют по каплям при 0oC в атмосфере азота к смеси 1-[1-(2-бромфенил)циклобутил]-6-хлор-7-метокси-3,4-дигидроизохинолина (5,5 г, полученного как описано в примере СА20), боргидрида натрия (3,9 г) и тетрагидрофурана (50 мл). Реакционную смесь перемешивают при температуре окружающей среды в течение 16 ч. Следующую порцию боргидрида натрия (1 г) добавляют к смеси, которую нагревают при 50oC в течение 2 ч. Добавляют воду и подщелачивают смесь путем добавления 50%-ного водного гидроксида натрия. Смесь экстрагируют этилацетатом. Экстракт дает остаток, который очищают путем флэш хроматографии. Смесь очищенного остатка (0,7 г), ледяной уксусной кислоты (10 мл), 48%-ной бромистоводородной кислоты (10 мл) нагревают с обратным холодильником в течение 5 ч. Затем растворитель удаляют и остаток обрабатывают пропан-2-олом. Удаление растворителя дает 1-[1-(2-бромфенил)циклобутил] -6-хлор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид, который промывают эфиром и сушат в вакууме, т.пл. 165-170oC (разл.).
Пример 41. Боргидрид натрия (всего 8 г) добавляют порциями к теплой смеси 7-метокси-1-(1-фенил-циклобутил)-3, 4-дигидроксиизохинолина (15 г полученного как описано в примере СТ13) и технического денатурата (200 мл) в течение 1 ч. Смесь добавляют к воде. Технический денатурат удаляют путем выпаривания, а остаток экстрагируют эфиром. Удаление растворителя дает масло, которое растворяют в ацетоне (250 мл) и перемешивают с метилйодидом (7,57 г) и безводным карбонатом натрия (13,4 г) в течение 1 ч при 50-55oC. Реакционную смесь обрабатывают древесным углем и фильтруют. Удаление растворителя дает остаток, который дигерируют (экстрагируют при повышенной температуре) эфиром. Эфирный раствор фильтруют и удаляют растворитель с получением масла, которое растворяют в ледяной уксусной кислоте (75 мл). 48%-ную бромоводородную кислоту (75 мл) добавляют к смеси и смесь нагревают с обратным холодильником в течение 4 ч. Реакционную смесь добавляют к смеси льда и водного раствора аммиака. Полутвердый продукт осаждают. Верхний жидкий слой удаляют путем декантации, а остаток промывают водой и растворяют в этаноле. Добавляют концентрированную соляную кислоту. Растворитель удаляют путем выпаривания с получением 7-гидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин гидрохлорида (т. пл. 236-240oC), который кристаллизуют из пропан-2-ола.
Пример 42. Смесь 6,7-диметокси-1-[1-(2-нафтил)циклобутил] -1,2,3,4- тетрагидроизохинолина (4,74 г полученного как описано в примере PB20, 37 - 40%-ного водного раствора формальдегида (5,1 мл), ацетонитрила (120 мл) и цианборгидрида натрия (1,3 г) перемешивают при температуре окружающей среды в течение 15 мин. Смесь нейтрализуют добавлением ледяной уксусной кислоты и перемешивают в течение 45 мин. Смесь концентрируют путем выпаривания и подщелачивают 2н. водным раствором гидроксида натрия. Полученную смесь экстрагируют эфиром. Экстракты промывают водным раствором гидроксида натрия и экстрагируют водной соляной кислотой. Кислотный экстракт подщелачивают и экстрагируют эфиром. Эфирный экстракт дает остаток, часть которого (3,86 г) перемешивают с 48%-ной водной бромистоводородной кислотой (40 мл) и с ледяной уксусной кислотой (40 мл) и нагревают при 100oC в течение 2 дней. Растворитель удаляют путем выпаривания и остаток сушат путем азеотропной отгонки с пропан-2-олом. Остаток промывают эфиром и обесцвечивают древесным углем в пропан-2-оле. Смесь фильтруют и получают 6,7-дигидрокси-2-метил-1[1-(2-нафтил)циклобутил] -1,2,3,4-тетрагидроизохинолин 1,1 гидробромид (т. пл. 150-153oC), который сушат в вакууме.
Пример 43. 1 M гидрофосфит натрия (179 мл, получают из фосфорной кислоты (14,7 г), воды (180 мл) и гидрокарбоната натрия (15 г)), затем 37-40%-ный водный раствор формальдегида (94 мл) добавляют в перемешиваемому раствору 7-бензилокси-6-метокси-1-[1-(1-нафтил)циклопропил] -1,2,3,4-тетрагидроизохинолина (9,3 г, полученного как описано в примере RC15) в техническом денатурате (1 л). Смесь перемешивают в течение 16 ч и растворитель удаляют в вакууме. Добавляют к остатку воду (300 мл), а затем водный раствор аммиака в избытке. Продукт экстрагируют эфиром. Экстракты дают остаток (9,1 г), который экстрагируют теплой смесью 5:4:1 петролейного эфира (т.к. 40-60oC), эфира и триэтиламина. Растворители удаляют из экстракта путем выпаривания и остаток очищают путем флэш хроматографии, используя указанную выше смесь растворителей в качестве элюента, с получением 7-бензилокси-6-метокси-2-метил-1-[1-(1-нафтил)циклопропил]-1,2,3,4- тетрагидроизохинолина в виде смолы.
Смолу нагревают с обратным холодильником с этанолом (16 мл) и концентрированной соляной кислотой (16 мл) в течение 30 мин, растворитель удаляют в вакууме и остаток дигерируют холодной водой, получая 7-гидрокси-6-метокси-2-метил-1[1-(1-нафтил)циклопропил]-1,2,3,4- тетрагидроизохинолин гидрохлорид (4,06 г), т.пл. 196-200oC.
Пример 44. Смесь
5-хлор-6-метокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолина (5,5 г, полученного как описано в примере М15), 48%-ной водной бромистоводородной кислоты (50 мл) и ледяной уксусной
кислоты (50 мл) нагревают с обратным холодильником в атмосфере азота в течение 24 ч. Растворитель удаляют путем выпаривания. Остаток обесцвечивают древесным углем в метаноле. Смесь фильтруют и
растворитель удаляют из фильтра. Остаток сушат путем азеотропной отгонки с пропан-2-олом и обесцвечивают древесным углем в метаноле. Смесь фильтруют и растворитель удаляют из фильтра. Остаток сушат
путем азеотропной отгонки с пропан-2-олом и обрабатывают эфиром, получая 5-хлор-8-гидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин гидробромид, т.пл. 197-200oC (разл.)
Пример 45.2 Раствор 1-фенилциклобутанкарбонилхлорида (29 г) в эфире (100 мл) добавляют к смеси 3,4-диметоксифенэтиламина (28 г), триэтиламина (25 мл) и эфира (200 мл). Смесь перемешивают в
течение 1 ч. Реакционную смесь выливают в воду и смесь экстрагируют полифосфатэфиром (200 г). Смесь добавляют к смеси лед/вода и полученную смесь промывают эфиром, подщелачивают избытком водного
раствора аммиака и экстрагируют смесью 1:1 эфира и толуола, а затем этилацетатом. Удаление растворителей из экстрактов дает твердый продукт. Образец этого твердого продукта (40 г) в метаноле (500 мл)
обрабатывают порциями боргидрида натрия (25 г всего). Смесь нагревают с обратным холодильником в течение 16 ч, а затем подкисляют 6н. водной соляной кислотой, подщелачивают водным раствором гидроксида
натрия и экстрагируют этилацетатом. Экстракт дает остаток, часть которого (15 г) обрабатывают по каплям муравьиной кислотой (6,7 г), а затем добавляют 37-40%-ный водный раствор формальдегида (11 мл).
Смесь нагревают с обратным холодильником в течение 5 ч, а затем охлаждают и подщелачивают 5н. водным раствором гидроксида натрия. Полученную смесь экстрагируют эфиром. Высушенный экстракт дает остаток,
который растворяют в эфире, фильтруют и обрабатывают эфирные растворы HCl. Выпаривание дает остаток, который перекристаллизовывают из технического денатурата, получая 6,
7-диметокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин гидрохлорид, т.пл. 130-132oC.
Свободное основание указанного выше продукта (9 г) нагревают с обратным холодильником с 48%-ной бромистоводородной кислотой (200 мл) в течение 16 ч. При охлаждении осаждают твердый продукт, который собирают, промывают водой и обесцвечивают древесным углем в техническом денатурате и фильтруют. Частичное упаривание фильтрата вызывает кристаллизацию 6,7-дигидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин гидробромида, т. пл. 95 - 100oC.
Пример 46. Раствор метилйодида (1,9 г) в ацетоне (20 мл) добавляют по каплям к перемешиваемой суспензии 1-[1-(2,4-дихлорфенил)циклобутил]-6,7-диметокси-1,2,3,4-тетрагидроизохинолина (4 г, полученного как описано в примере RC9) и безводного карбоната натрия (3,1 г) в ацетоне (70 мл). Смесь перемешивают при температуре окружающей среды в течение 90 мин и растворитель удаляют путем выпаривания. Добавляют воду и смесь экстрагируют эфиром. Эфирный слой дает 1-[1-(2,4-дихлорфенилциклобутил]-6,7-диметокси-2-метил-1,2,3,4-тетрагидроизохинолин в виде масла. Часть этого масла характеризуют путем преобразования в его 1,5 оксалатную соль, т.пл. 125-132oC.
Раствор масла (3,2 г, полученного по способу предыдущего абзаца) в дихлорметане (50 мл) охлаждают до -50oC в азоте. Добавляют по каплям 1 М раствор трибромида бора в дихлорметане (24 мл). Смесь перемешивают при температуре окружающей среды в течение 16 ч и охлаждают до -50oC. Медленно добавляют метанол (20 мл). Растворители удаляют выпариванием, а остаток сушат путем азеотропной отгонки с пропан-2-олом. Остаток обесцвечивают древесным углем в метаноле и растворяют в пропан-2-оле. Добавление эфира вызывает осаждение 1-[1-(2,4-дихлорфенил)циклобутил]-6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромида, т. пл. 205 - 210oC.
Пример 47. Раствор метилйодида (0,5 г) в ацетоне (10 мл) добавляют по каплям в перемешиваемую суспензию 1-[1- (2-хлорфенил)циклобутил]-7-метокси-1,2,3,4-тетрагидроизохинолин оксалата (1,42 г, полученного как описано в примере RC11) и безводного карбоната калия (3 г) в ацетоне (80 мл). Смесь перемешивают при температуре окружающей среды в течение 3,5 ч и растворитель удаляют путем выпаривания. Добавляют воду и смесь экстрагируют эфиром. Эфирный слой дает 1-[1-(2-хлорфенил)циклобутил]- 7-метокси-2-метил-1,2,3,4-тетрагидроизохинолин в виде масла.
Раствор масла (1, 1 г, полученного по способу предыдущего абзаца) в дихлорметане (50 мл) охлаждают до -50oC с азоте. Добавляют по каплям 1 М раствор трибромида бора в дихлорметане (10 мл). Смесь перемешивают при температуре окружающей среды в течение 16 ч и охлаждают до -50oC. Медленно добавляют метанол (20 мл). Растворитель удаляют путем выпаривания и остаток сушат путем азеотропной отгонки с пропан-2-олом. Остаток обесцвечивают древесным углем в метаноле, растворяют в пропан-2-оле и нагревают до 35 - 40oC. Добавление эфира дает твердый продукт, который тритируют в теплом пропан-2-оле. Добавляют эфир, получая 1-[1- (2-дихлорфенил)-циклобутил]-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид, т. пл. 216 - 218oC.
Пример 48. Тонко измельченную смесь 4-гидрокси-3-метоксифенэтиламина (9,9 г) и 1-фенилциклопентанкарбоновой кислоты (11,9 г) нагревают при 200oC в атмосфере азота в течение 2 ч. Расплав слегка охлаждают и добавляют к смеси 1 : 1 ледяной уксусной кислоты и воды. Кристаллизуется твердый продукт, который удаляют путем фильтрования и промывают уксусной кислотой. Фильтрат подщелачивают избытком карбоната натрия и экстрагируют эфиром. Экстракт промывают 4н. соляной кислотой. Эфирный экстракт дает остаток, который нагревают с обратным холодильником в атмосфере азота с ацетонитрилом (150 мл) и оксихлоридом фосфора (18,7 мл) в течение 1 ч. Растворитель удаляют и к остатку добавляют воду. Смесь нагревают и добавляют этанол. Полученный раствор нагревают при 90 - 95oC в течение 1 ч и охлаждают. Для растворения твердого продукта добавляют этанол и раствор подщелачивают водным раствором аммиака. Добавляют боргидрид натрия порциями (2,5 г всего) и путем выпаривания удаляют растворители. Остаток распределяют между водой и этилацетатом, получая твердый продукт, который собирают путем фильтрования, промывают водой и сушат на воздухе. Твердый продукт растворяют в смеси 37 - 40%-ного водного раствора формальдегида (27 мл) и муравьиной кислоты (15 мл), а раствор подогревают до 60oC в течение 2 ч. Добавляют лед и смесь подщелачивают водным раствором аммиака. Полученную смесь экстрагируют эфиром. Экстракт сушат, а растворитель удаляют, получая остаток, который растворяют в эфире. Экстракт фильтруют, сушат и добавляют эфирный раствор оксалиновой кислоты.
Твердый продукт осаждают. Эфир удаляют декантированием и добавляют этилацетат, а смесь нагревают с обратным холодильником. Твердый продукт тритируют в этилацетате, собирают путем фильтрования, промывают этилацетатом и сушат на воздухе, получая 7-гидрокси-6- метокси-2-метил-1-(1-фенилциклопентил)-1,2,3,4-тетрагидроизохинолин оксалат, т. пл. 172oC (разл.).
Пример 49. Тонко растертую смесь 4-гидрокси-3-метокси-фенэтиламина (10,45 г) и 1-(4-хлорфенил)циклобутанкарбоновой кислоты (12,17 г) нагревают при 200oC в азоте в течение 2 ч. Расплав охлаждают и добавляют к смеси 1 : 1 ледяной уксусной кислоты и воды. Дальнейшее добавление воды вызывает выпадение осадка, который экстрагируют эфиром. Эфирный слой промывают водным раствором карбоната натрия, а затем 6н. соляной кислотой, и он дает остаток, который нагревают с обратным холодильником в атмосфере азота с ацетонитрилом (227 мл) и оксихлоридом фосфора (27,2 мл) в течение 16 ч. Добавляют воду и технический денатурат, а смесь нагревают в течение 1 ч. Через 16 ч добавляют водный раствор с избытком аммония и лед, а полученный твердый продукт собирают и промывают водой и эфиром, и сушат при 60oC в вакууме. Остаток упаривают с этанолом, а нерастворимый твердый осадок собирают и растворяют в метаноле (250 мл) и воде (50 мл). Боргидрид натрия (всего 2,8 г) добавляют порциями, а затем добавляют воду и избыток разбавленной соляной кислоты. Всплывшую жидкость подщелачивают водным раствором аммиака и полученный осадок экстрагируют эфиром. Эфир удаляют и оставляют остаток нагреваться в течение 1 ч в смеси 37 - 40%-ного водного раствора формальдегида (45 мл) и муравьиной кислоты (27 мл). Смесь оставляют стоять в течение 16 ч, а затем нагревают в течение следующих 30 мин и охлаждают. Добавляют лед и избыток водного раствора аммиака, а полученную смесь экстрагируют эфиром. Удаляют растворитель, а остаток растворяют в эфире. Эфирный раствор щавелевой кислоты добавляют к высушенному раствору, чтобы получить полутвердый продукт, который тритируют в кипящем эфире и дигерируют этилацетатом, получая смолу, которую упаривают с пропан-2-олом, получая 1-[1-(4-хлорфенил)циклобутил]- 7-гидрокси-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин оксалат, т.пл. 202oC (разл.).
Пример 50. Раствор 1-(4-хлорфенил)циклобутанкарбонилхлорида (22,9 г) в эфире (200 мл) добавляют к перемешиваемой смеси 3,4-диметоксифенэтиламина (18,1 г), триэтиламина (13,9 мл) и эфира (300 мл). Смесь перемешивают в течение 1,5 ч, а затем добавляют воду. Эфирный слой дает осадок, который растворяют в дихлорметане (195 г) в атмосфере азота. Смесь выдерживают при 75 - 82oC в течение 16 ч, а затем добавляют к воде (1200 мл). Органическую фазу промывают водой и подщелачивают избытком водного раствора аммиака. Щелочной водный раствор экстрагируют эфиром. Эфирный экстракт дает масло, которое растворяют в метаноле (200 мл). Двенадцать порций боргидрида натрия (всего 12 г) добавляют в течение 20 мин. Смесь перемешивают в течение 16 ч и подкисляют путем осторожного добавления 5н. соляной кислоты. Смеси оставляют стоять в течение 16 ч и полученный твердый продукт отделяют фильтрованием. К фильтрату добавляют воду, затем его подщелачивают и экстрагируют этилацетатом. Экстракт дает осадок, который сушат путем азеотропной отгонки с пропан-2-слоем, получая 1-[1-(4-хлорфенил)циклобутил]-6,7-диметокси-1,2,3,4-тетрагидроизохинолин, образец которого (8,8 г) нагревают при 90 - 95oC с муравьиной кислотой (5 мл) и 38 - 40%-ным водным раствором формальдегида (6,2 мл) в течение 16 ч. Добавляют воду и подщелачивают смесь водным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает смолу, которую нагревают с обратным холодильником с 48%-ной водной бромистоводородной кислотой (100 мл) в течение 3,5 ч. Добавляют древесный угол и технический денатурат (100 мл) и фильтруют смесь. Удаление растворителя из фильтрата дает остаток, который сушат путем азеотропной отгонки с пропан-2-олом. Остаток помещают в смесь 1 : 1 пропан-2-оле и этаноле, а растворитель удаляют путем выпаривания. Остаток нагревают с обратным холодильником в этаноле в течение 30 мин и затем охлаждают. Твердый продукт собирают путем фильтрования и сушат с небольшим количеством холодного этанола, получая 1-[1-(4-хлорфенил)циклобутил] - 6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид, т. пл. 220oC.
Пример 51. Раствор 1-(4-хлорфенил)циклобутанкарбонилхлорида (30,7 г) в эфире (300 мл) добавляют к перемешиваемому раствору 4-метоксифенэтиламина (20,22 г) и триэтиламина (20 мл) в эфире (200 мл). Через 1 ч добавляют воду (200 мл) и эфирный слой удаляют декантацией. Водный слой экстрагируют дихлорметаном. Объединенные органические экстракты дают остаток (46 г), который смешивают с полифосфатэфиром (100 мл) и нагревают на паровой бане в течение 65 ч. Реакционную смесь добавляют к смеси льда и концентрированного водного раствора аммиака и экстрагируют эфиром и этилацетатом. Объединенные органические экстракты дают остаток, который растворяют в этаноле (180 мл). Боргидрид натрия (15,6 г всего) добавляют порциями. Смесь нагревают в течение 1 ч при 90 - 95oC. Добавляют воду, а затем разбавленную соляную кислоту и смесь подщелачивают водным раствором гидроксида натрия, и экстрагируют эфиром. Экстракт охлаждают, фильтруют и добавляют эфирный раствор щавелевой кислоты, получая смолу, которую тритируют в эфире. Остаток подщелачивают водным раствором гидроксида натрия, получая остаток, который очищают с помощью жидкостной хроматографии высокого разрешения, и порцию продукта (1,6 г) нагревают в атмосфере азота в течение 19 ч с ледяной уксусной кислотой (35 мл) и 48%-ной бромистоводородной кислотой (35 мл). Смесь охлаждают и добавляют к смеси льда и 10%-ного водного раствора карбоната натрия, нерастворимый материал, который осаждается, обесцвечивают древесным углем в смеси 6н. соляной кислоты, уксусной кислоты и метанола. Раствор дает остаток, который сушат путем азеотропной отгонки с пропан-2-олом.
Смесь остатка (1 г), формиата натрия (0,2 г), 37 - 40%-ного водного раствора формальдегида (10 мл) и муравьиной кислоты (10 мл) нагревают при 90 - 95oC в течение 20 мин. Смеси позволяют постоять в течение 16 ч и выливают в смесь льда и водного раствора аммиака, который экстрагируют эфиром. Экстракт дает остаток, который очищают с помощью флэш хроматографии и преобразуют в 1-[1-(4-хлорфенил)циклобутил] -7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин 1,35 оксалат, т.пл. 192 - 194oC (разл.).
Пример 52. 1-Фенилциклобутанкарбонилхлорид (20 г) в эфире (100 мл) добавляют к перемешиваемой смеси 3-хлор-4-метоксифенэтиламина (19,1 г), триэтиламина (14 мл) и эфира (100 мл), и полученную смесь перемешивают в течение 1 ч. Добавляют воду и экстрагируют смесь этилацетатом. Экстракт дает твердый продукт (т.пл. 62-64oC), образец которого (14,6 г) нагревают при 90oC с полифосфатэфиром (89 мл) в течение 48 ч. Смесь выливают в лед/воду, подщелачивают водным раствором аммиака и экстрагируют эфиром. Порцию (90% от всего) экстракта добавляют к смеси боргидрата натрия (6 г) и этанола (400 мл). Смесь нагревают с обратным холодильником в течение 90 мин после удаления эфира, а затем удаляют растворитель путем выпаривания, получая остаток, который добавляют к воде. Получившуюся смесь экстрагируют эфиром. Эфирный экстракт добавляют к 1 М водному раствору фосфита натрия (получают из фосфорной кислоты (20,5 г), бикарбоната натрия (21,0 г) и воды (250 мл)), 37 - 40%-ному водному раствору формальдегида (150 мл) и метанола (400 мл). Смесь нагревают с обратным холодильником в течение 17 ч, позволяя эфиру испариться. Затем метанол удаляют путем выпаривания и остаток подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает масло, которое очищают с помощью флэш хроматографии и жидкостной хроматографии высокого разрешения, получая соединение (4,1 г), которое нагревают с обратным холодильником в течение 5 ч с ледяной уксусной кислотой (45 мл) и 48%-ной бромистоводородной кислотой (45 мл). Охлажденную реакционную смесь распределяют между эфиром и 50%-ным водным раствором карбоната натрия. Экстракт дает стеклообразную массу, которую растворяют в эфире и обрабатывают эфирным раствором щавелевой кислоты, получая 6-хлор-7-гидрокси-2-метил-1-(1-фенилциклобутил)- 1,2,3,4-тетрагидроизохинолин оксалат (т.пл. 220 - 223oC).
Пример 53. Раствор 1-(4-хлорфенил)циклобутанкарбонилхлорида (20 г) в эфире (50 мл) добавляют к перемешиваемой смеси 3-хлор-4-метоксифенэтиламина (16,2 г), триэтиламина (13 мл) и эфира (100 мл). Через 1 ч добавляют воду (50 мл) и смесь экстрагируют этилацетатом. Органический слой дает остаток, который перекристаллизовывают дважды из технического денатурата. Порцию продукта (12,6 г) нагревают с полифосфатэфиром (70 мл). Полученный раствор нагревают при 90oC в течение 48 ч, охлаждают и добавляют к смеси льда, концентрированного раствора аммиака и эфира. Эфирный слой промывают, сушат и добавляют к боргидриду натрия (5 г) в этаноле (200 мл), и смесь нагревают с обратным холодильником в течение 90 мин. За это время эфир испаряется, а этанол удаляют. Добавляют воду (200 мл) и полученную смесь экстрагируют эфиром. Экстракт добавляют к смеси 1 М водного раствора фосфата натрия (получают из фосфорной кислоты (16,4 г), бикарбоната натрия (16,8 г) и воды (200 мл)), 37 - 40%-ного водного раствора формальдегида (130 мл) и метанола (300 мл). Эфир удаляют путем выпаривания и добавляют метанол (250 мл). Затем смесь нагревают с обратным холодильником в течение 16 ч и затем метанол удаляют путем выпаривания. Остаток подщелачивают водным раствором аммиака и экстрагируют эфиром. Растворитель удаляют из экстракта и остаток сушат путем азеотропной отгонки с техническим денатуратом, а затем с пропан-2-олом. Высушенный остаток кристаллизуют из пропан-2-ола, получая твердый продукт, который в дальнейшем очищают с помощью жидкостной хроматографии высокого разрешения. Образец этого очищенного продукта (1 г) нагревают при 110 - 115oC в азоте с ледяной уксусной кислотой (10 мл) в течение 6 ч. Затем реакционную смесь охлаждают и распределяют между эфиром и 50%-ным водным раствором карбоната натрия. Эфирный слой дает 6-хлор-1-[1-(4-хлорфенил)циклобутил]- 7-гидрокси-2-метил-1,2,3,4-тетрагидроизохинолин (т.пл. 168 - 171oC).
Пример 54. Раствор 1-(4-метилоксифенил)циклобутанкарбонилхлорида (5,83 г, полученного как описано в примере CL28) в дихлорметане (20 мл) добавляют к раствору 3,4-дибензилоксифенэтиламина гидрохлорида (9,61 г) в дихлорметане (100 мл). Добавляют триэтиламин (20 мл). Через 16 ч подкисляют смесь разбавленной соляной кислотой. Органический слой дает остаток, который нагревают с обратным холодильником в течение 3 ч с оксихлоридом фосфора (20 мл) и ацетонитрилом (200 мл). Удаление растворителя дает остаток, который дигерируют этилацетатом. При охлаждении осаждают твердую дихлорфосфатную соль. Образец (11,6 г) этой соли добавляют порциями к перемешиваемой смеси боргидрида натрия (9,5 г) в техническом денатурате (250 мл). Реакционную смесь нагревают в течение 3 ч при 90 - 95oC. Добавляют смесь боргидрида натрия (3 г) и технического денатурата (150 мл), осторожно кипятят в течение 2 ч. Уменьшают объем реакционной смеси, добавляют воду и полученную смесь экстрагируют эфиром. Эфирный экстракт дает остаток, который растворяют в смеси метанола (190 мл) и 37 - 40%-ном водном растворе формальдегида (65 мл). Полученную смесь смешивают с 1 М водным раствором сульфита натрия (получают из фосфорной кислоты (9,9 г), бикарбоната натрия (10,1 г) и воды (120 мл)) и добавляют метанол (300 мл). Смесь подогревают и дают постоять в течение 16 ч. Супернатант подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает смолу, которую растворяют в смеси метанола (100 мл) и муравьиной кислоты (20 мл) и перемешивают в азоте с 5% палладием на древесном угле (4 г, тип 38 н. ex Johnson Mattey) в течение 3 ч. Смесь фильтруют и добавляют к фильтрату концентрированную соляную кислоту (1,2 мл). Растворитель удаляют путем выпаривания, а остаток сушат путем азеотропной отгонки с пропан-2-олом, смесью пропан-2-ола и толуола, а затем с пропан-2-олом, получая твердый продукт, который тритируют в этилацетате, собирают путем фильтрования, промывают этилацетатом и сушат в вакууме, получая 6, 7-дигидрокси-1-[1-(4- метилоксифенил)циклобутил]-2-метил-1,2,3,4-тетрагидроизохинолин гидрохлорид, т.пл. 85 - 90oC.
Пример 55. Боргидрид натрия (7 г всего) добавляют порциями за 2 ч к раствору продукта по примеру CT15 (25 г) в техническом денатурате (300 мл), который нагревают с обратным холодильником. К смеси, экстрагированной эфиром, добавляют воду. Экстракт дает твердый продукт, который смешивают со смесью метанола (400 мл) и 37 - 40%-ного водного раствора формальдегида (213 мл). Добавляют 1 М раствор фосфита натрия (получают из фосфорной кислоты (33,6 г), бикарбоната натрия (34,4 г) и воды (410 мл)) и смесь нагревают при 90 - 95oC в течение 4 ч. Объем реакционной смеси уменьшают, и смесь подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает масло, которое растворяют в этаноле (270 мл) и нагревают с обратным холодильником в атмосфере азота в течение 30 мин с концентрированной соляной кислотой (270 мл). Смесь охлаждают и добавляют к смеси льда и водного раствора аммиака, которую экстрагируют эфиром. Экстракт дает остаток, который очищают с помощью флэш хроматографии на кварце, используя смесь 4:96 метанола и дихлорметана в качестве элюента. Фракцию, содержащую желаемый продукт, обрабатывают эфирным раствором щавелевой кислоты, получая смолу, которую кристаллизуют из смеси метанола и этилацетата, получая 7-гидрокси-6-метокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин 1,4 оксалат (т.пл. 132 - 133oC).
Пример 56. Раствор 2'-бром-4', 5-диметилоксифенилацетонитрила (136,8 г) и 1,3-дибромпропана (58,7 мл) в диметилсульфоксиде (300 мл) по каплям добавляют за 2 ч к перемешиваемой смеси порошкообразного гемигидрата гидроксида натрия (150 г) и 18-краун-6 (2 г) в диметилсульфоксиде (700 мл) в атмосфере азота при 24-25oC. Перемешивание продолжают в течение следующих двух часов, а затем добавляют смесь воды и льда. Полученную смесь экстрагируют дихлорметаном. Экстракт дает остаток, который кристаллизуют из эфира. Образец этого кристаллизированного материала (67,2 г) нагревают с обратным холодильником в растворе гидроксида натрия (32,5 г) в пропаноле (600 мл) в течение 7 дней. Раствор удаляют, остаток распределяют между водой и этилацетатом. Этилацетатный слой дает 1-(2-бром-4, 5-диметоксифенил)циклобутанкарбоксамид, который нагревают с обратным холодильником с гидроксидом натрия (30 г) и водой (300 мл) в течение 3 дней. Смесь промывают этилацетатом, а водный слой подкисляют и экстрагируют эфиром. Полученную кислоту растворяют в этилацетате (80 мл) и раствор смешивают с раствором 4-бензилокси-3-метоксифенэтиламина (10,8 г) в этилацетате. Осаждается соль, которую нагревают в азоте при 195oC в течение 2 ч и при 205oC в течение 30 мин. Реакционную смесь охлаждают и образовавшуюся стеклообразную массу нагревают с обратным холодильником с оксихлоридом фосфора (22 мл) и ацетонитрилом (200 мл) в течение 4 ч. Реакционную смесь добавляют к смеси льда и водного раствора аммиака. Смесь экстрагируют эфиром. Эфирный экстракт дает остаток, образец которого (5,9 г) дигерируют в эфире. Добавление эфирного раствора щавелевой кислоты дает твердый продукт, который подщелачивают метанольным раствором гидроксида натрия и распределяют между водой и эфиром. Эфирный слой дает остаток, который очищают с помощью флэш хроматографии, получая смолу, которую перемешивают с ледяной уксусной кислотой (20 мл) и метанолом (10 мл) при 0oC в азоте, в то время как порциями добавляют цианоборгидрид (0,8 всего). Затем смесь перемешивают при 20-25oC в течение 16 ч, добавляют к водному раствору гидроксида калия и экстрагируют эфиром. Экстракт дает остаток (3,1 г), который смешивают с метанолом (300 мл), с 37 - 40%-ным водным раствором формальдегида (26 мл) и 1 М водным раствором фосфита натрия (получают из фосфорной кислоты (4,05 г), бикарбоната натрия (4,1 г) и воды (50 мл)) и смеси дают постоять в течение 3 дней. Растворитель удаляют путем выпаривания в вакууме при температуре не менее чем 50oC, а остаток добавляют к смеси льда и водного раствора аммиака и экстрагируют эфиром. Экстракт дает остаток, который растворяют в этаноле (35 мл) и обрабатывают в азоте концентрированной соляной кислотой (35 мл). Смесь кипятят в течение 30 мин, охлаждают, добавляют в смесь льда и водного раствора аммиака и экстрагируют этилацетатом. Экстракт дает остаток, который очищают с помощью флэш хроматографии, получая 1-[1-(2-бром-4,5-диметоксифенил)-циклобутил] -7-гидрокси-6-метокси-2- метил-1,2,3,4-тетрагидроизохинолин, т.пл. 154-157oC.
Пример 57. Продукт по примеру 41 (42) растворяют в воде при 80oC. Раствор подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Экстракт дает осадок, который растворяют в уксусной кислоте (40 мл). Раствор охлаждают до 0oC и добавляют уксусный ангидрид (20 мл), а затем смесь 70%-ной азотной кислоты (1,4 мл), уксусной кислоты (30 мл) и уксусного ангидрида (20 мл). Смесь выдерживают при 5oC в течение 40 мин, а затем добавляют к водному бикарбонату натрия и оставляют на 16 ч, прежде чем экстрагировать этилацетатом. Экстракт дает остаток, который очищают с помощью флэш хроматографии, получая твердый продукт, который растворяют в этилацетате. Добавление эфирного раствора щавелевой кислоты дает твердый продукт, который насыщают тритием в горячем этилацетате, получая 7-гидрокси-2-метил-6-нитро-1-(1-фенил-циклобутил)-1,2,3,4- тетрагидроизохинолин оксалат, т.пл. 172-173oC (разл.).
Пример 58. Раствор гипохлорита натрия (11 мл - 8%
свободного хлора) добавляют при 0oC к смеси продукта по примеру 41 в виде его свободного основания (2 г), ледяной уксусной кислоты (25 мл), воды (20 мл) и концентрированной соляной кислоты
(20 мл). Добавляют ледяную уксусную кислоту (25 мл) и концентрированную соляную кислоту, а затем следующую порцию (8 мл) указанного выше раствора гипохлорита натрия. Смесь перемешивают в течение 20
мин и добавляют твердый метабисульфит натрия в избытке. Смесь подщелачивают водным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает остаток, который растворяют в эфире. Добавление
эфирного раствора щавелевой кислоты дает 6,8-дихлор-7-гидрокси-2-метил-1- (1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин 1,5 оксалат, т.пл.120oC (разл.)
Пример 59. Раствор
1-фенилциклобутанкарбонилхлорида (19,45 г) в дихлорметане (100 мл) добавляют при 10 - 13oC к раствору 3-хлор-4-метоксифенэтиламина (18,55 г) и триэтиламина (30 мл) в дихлорметане (300 мл)
за 40 мин. Смесь перемешивают при 20 - 25oC в течение 3 ч и оставляют стоять в течение 3 дней. Добавляют воду и отделяют органический слой и промывают 2н. соляной кислотой, а затем 1н.
водным раствором гидроксида натрия. Органический слой дает смолу (32,89 г), которую нагревают с обратным холодильником с ксилолом (390 мл) и оксихлоридом фосфора (76,8 мл) в течение 10 ч. Реакционную
смесь добавляют порциями к перемешиваемой смеси водного раствора гидроксида натрия и льда. Температуру оставляют ниже 85oC. Добавляют толуол примерно при 80oC, чтобы растворить
осадок в виде масла. Органический слой отделяют и получают масло, которое кристаллизуют из пропан-2-ола. Кристаллизовавшийся твердый продукт (10 г) растворяют в этаноле (200 мл) и добавляют боргидрид
натрия (2 г) при подогревании. Через 40 мин растворитель удаляют, а остаток обрабатывают водой и экстрагируют эфиром с получением осадка, который нагревают с обратным холодильником с боргидратом
натрия (всего 8 г) и пропан-2-олом (100 мл) всего в течение 7 ч. Смесь охлаждают и добавляют воду, затем водный раствор гидроксида натрия. Смесь экстрагируют эфиром. Эфирный экстракт дает
6-хлор-7-метокси-1-(1-фенилциклобутил)-1,2,3,4-тетрагидроизохинолин, часть которого (8,78 г) растворяют в метаноле (100 мл) и обрабатывают раствором дибензил-L-виноградной кислоты (10,08 г) в метаноле
(50 мл). Растворитель удаляют при 40oC/40 мм Hg, оставляя 70 мл. Эфир добавляют до тех пор, пока не появится тонкий слой осадка, а смесь подогревают, получая прозрачный раствор. При
охлаждении осаждается твердый продукт, который отделяют путем фильтрования. Фильтрат подщелачивают водным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает масло, которое обрабатывают в
метаноле дибензоил-L-виноградной кислотой (7,12 г), а твердый продукт осаждают путем добавления эфира. Образец этого твердого продукта (2,9 г) и бикарбонат натрия (0,35 г) перемешивают в метаноле (50
мл) и добавляют 1 М раствор фосфита натрия, получают из фосфорной кислоты (2,55 г), бикарбоната натрия (2,61 г) и воды (16 мл). Добавляют метанол (50 мл) и смесь оставляют в течение 16 ч,
подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает остаток, который растворяют в ледяной уксусной кислоте (20 мл) и добавляют 48%-ную бромистую кислоту (20 мл) в атмосфере
азота. Смесь нагревают при 100oC в течение 16 ч, а затем с обратным холодильником в течение 6,5 ч, охлаждают, добавляют к смеси льда и водного раствора аммиака и экстрагируют этилацетатом.
Экстракт дает остаток, который очищают с помощью флэш хроматографии, получая (-)-6-хлор-7-гидрокси-2-метил-1-(1-фенилциклобутил)-1,2,3,4- тетрагидрохинолин (т. пл. 73 - 75oC), который имеет
удельное вращение плоскости поляризации [α]D= -139,8°.
Пример 60. 1-[1-(2-Бромфенил)циклобутил]-6,7-диметокси-1,2,3,4- тетрагидроизохинолин (15,7 г,
полученный способом, описанным в примере RB1) растворяют в эфире (1100 мл) и обрабатывают 0,4 М раствором дибензоил-L-виноградной кислоты в эфире (98 мл). Осадок твердого продукта собирают путем
фильтрования и сушат в вакууме. Часть этого твердого продукта (10,5 г) растворяют в кипящем метаноле (350 мл). Раствор оставляют стоять в течение 2 дней, собирают путем фильтрования и
перекристаллизовывают из метанола (-)-1-[1-(2-бромфенил)циклобутил] -6,7-диметокси-1,2,3,4- тетрагидроизохинолин дибензоил-L-тартрат. Дополнительные количества этой соли получают из маточной жидкости,
из которой осаждают твердый продукт с помощью удаления растворителя путем выпаривания, и перекристаллизовывают остаток из метанола. Соль (т.пл. 174-175oC (разл.)) имеет удельное вращение
плоскости поляризации [α]D= (-57,6)-(-61,9)°.
Смесь (-)-1-[1-(2-бромфенил)циклобутил] -6,7-диметокси- 1,2,3,4-тетрагидроизохинолина (1,85 г) (выделенного
из дибензоил-(L)-виноградной соли (3,7 г)), ацетонитрила (60 мл), 37 - 40%-ного водного раствора формальдегида (1,8 мл) и цианборгидрида натрия (0,46 г) перемешивают в течение 15 мин, затем
нейтрализуют ледяной уксусной кислотой и перемешивают еще в течение 45 мин. Смесь концентрируют путем выпаривания и подщелачивают до pH 12 с помощью разбавленного раствора гидроксида натрия. Смесь
экстрагируют этилацетатом, экстракт дает смолу, которую очищают путем флэш хроматографии, используя смесь 1:2 этилацетата и петролейного эфира в качестве элюента и получая
(-)-1-[1-(2-бромфенил)циклобутил] -6,7-диметокси-2-метил-1,2,3,4- тетрагидроизохинолин, который имеет удельное вращение плоскости поляризации [α]D= -33,6°. Выход 1,
6 г. Смесь (-)-1-[1-(2-бромфенил)циклобутил]-6,7-диметокси-2-метил-1,2,3,4 -тетрагидроизохинолина (1,46 г), 48%-ной водной бромистоводородной кислоты (20 мл) нагревают с обратным холодильником в
течение 5 ч. Растворитель удаляют путем выпаривания, а остаток сушат путем многократной азеотропной отгонки с пропан-2-олом. Остаток растворяют в пропан-2-оле и осаждают эфиром. Полученный твердый
продукт сушат в вакууме при 45oC, получая (+)-1-[1-(2-бромфенил)-циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид (т.пл. 207 - 209oC (разл.)),
который имеет удельное вращение плоскости поляризации [α]D= +33,6°.
Пример 61. Раствор 1-(2-хлорфенил)циклопропан карбонилхлорида (25 г) в дихлорметане
(100 мл) добавляют по каплям к энергично перемешиваемой смеси 2-(3-фтор-4-метоксифенил)этиламина гидрохлорида (23,9 г), триэтиламина (70 мл) и дихлорметана (400 мл), а затем смесь перемешивают в
течение 1 ч. Затем добавляют 6н. соляную кислоту в избытке. Полученный раствор промывают водой, сушат над карбонатом калия, а растворитель удаляют в вакууме, получая N-[2-(3-фтор-4-метоксифенил)этил]
-1-(2-хлорфенил)циклопропанкарбоксамид, который добавляют в виде расплава к полифосфатэфиру (365 г) в азоте. Смесь нагревают при 100oC в течение 17 ч, затем добавляют к воде (600 мл) и
промывают эфиром (600 мл). К водной фазе добавляют водный раствор аммиака для достижения pH 8 - 9. Полученный осадок собирают путем фильтрования, промывают водой, сушат на воздухе и
перекристаллизовывают из кипящего ацетонитрила, получая 1-[1-(2-хлорфенил)циклопропил] -6-фтор-7-метокси-3,4 -дигидроизохинолин, т.пл. 172 - 176oC.
Цианборгидрид натрия (10,7 г) добавляют при 0oC к смеси 1-[1-(2-хлорфенил)циклопропил] -6-фтор-7-метокси-3,4-дигидроизо[b] хинолина (26,7 г, получают способом, подобным тому, что описан выше), уксусной кислоты (185 мл) и метанола (95 мл) в атмосфере азота. Смеси позволяют достичь комнатной температуры и перемешивают в течение 22 ч. Смесь выливают в воду и добавляют твердый гидроксид натрия (150 г) в ледяной воде. Продукт экстрагируют эфиром и затем растворитель удаляют в вакууме, получая 1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-метокси-3,4- тетрагидроизохинолин в виде смолы.
Смесь смолы, метанола (2700 мл), 1 M гидрофосфита (690 мл, получают из фосфорной кислоты и гидрокарбоната натрия (57,9 г)) и 37 - 40%-ного водного раствора формальдегида (360 мл) подогревают до 60oC, а затем перемешивают при комнатной температуре в течение 64 ч, метанол удаляют в вакууме и добавляют раствор гидроксида калия (50 г) в воде (500 мл), а затем этилацетат к полученному водному раствору. Органический слой сушат над карбонатом натрия, а растворитель удаляют в вакууме, получая смолу, которую кристаллизуют из ацетонитрила, получая 1-[1-(2-хлорфенил)циклопропил)] -6-фтор-7- метокси-2-метил-1,2,3,4-тетрагидроизохинолин. Выход 18,15 г.
Смесь 1-[1-(2-хлорфенил)циклопропил)-6-фтор-7-мектоси-2-метил -1,2,3,4-тетрагидроизохинолина (18,0 г) в уксусной кислоте (150 мл) и 48%-ной водной бромистоводородной кислоты (150 мл) нагревают с обратным холодильником в атмосфере аргона в течение 220 мин. Растворитель удаляют в вакууме, остаток сушат путем азеотропной отгонки с пропан-2-олом и кристаллизуют из пропан-2-ола, получая 1-[1-(2-хлорфенил)-циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид (18,56 г), т. пл. 240oC (разл.).
Пример 62. 1-[1-(2-Хлорфенил)циклопропил]-6-фтор-7-гидрокси-2- метил-1,2,3,4-тетрагидроизохинолин гидробромид (18 г, получают способом, подобным способу по примеру 61) распределяют между водным раствором аммиака и этилацетатом. Этилацетат удаляют в вакууме и остаток разделяют на две фракции с помощью хиральной препаративной жидкостной хроматографии высокого разрешения на колонке Chiralcel OD, элюируя смесью 97:3 гексана и этанола. Фракцию 1 растворяют в пропан-2-оле и обрабатывают небольшим избытком 48%-ной водной бромистоводородной кислоты. Полученный твердый продукт собирают путем фильтрования и сушат, получая (+)-1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид, который имеет удельное вращение плоскости поляризации [α]D= +2,38°. Выход 7,82 г, т.пл. 250oC (разл.).
Пример 63. Раствор 1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4 -тетрагидроизохинолина гидробромида (1 г, получают способом, подобным способу по примеру 61) в этилацетате (30 мл) смешивают с раствором малеиновой кислоты (0,24 г) в этилацетате (8 мл) и смесь подогревают для образования раствора. Раствор охлаждают, получая 1-[1-(2-хлорфенил)циклопропил] -6-фтор-7-гидрокси-2-метил-1,2,3, 4- тетрагидроихохинолин малеат, т.пл. 183 - 184oC. Выход 0,9 г.
Пример 64. 1-[1-(2-Хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин (8,0 г освобождают из соли, которую получают по примеру 24) разделяют на фракции с помощью хиральной препаративной жидкостной хроматографии высокого разрешения на колонке Chiralcel OD, элюируя смесью 80: 20 гексана и этанола. Остаток, полученный путем удаления растворителя из фракции 1 растворяют в пропан-1-оле и обрабатывают 48%-ной водной бромистоводородной кислотой, получая (+)-1-[1-(2-хлорфенил)-циклобутил]-6-фтор-7-гидрокси- 2-метил-1,2,3,4-тетрагидроизохинолин гидробромид, который имеет удельное вращение плоскости поляризации [α]D= +9,64°, т.пл. 242 - 245oC (разл.). Выход 3,63 г.
Пример 65. 6-Хлор-7-метокси-1-(1-фенилциклобутил)-1,2,3,4- тетрагидрохинолин (1,38 г, получают способом, подобным способу примера 59) растворяют в ацетоне (50 мл) и перемешивают с безводным карбонатом калия (1,16 г) и аллилйодидом (0,78 г) в течение 1 ч. Смесь фильтруют, фильтрат концентрируют и распределяют между водой и эфиром. Эфирный слой дает масло, которое помещают в дихлорметан (30 мл) и охлаждают до -70oC. 1 M раствор трибромида бора в дихлорметане (11 мл) добавляют по каплям и смеси позволяют нагреваться до комнатной температуры. Через 2 ч смесь охлаждают до -60oC и осторожно добавляют метанол (30 мл). Растворители удаляют, а остаток обесцвечивают древесным углем в метаноле. Удаление растворителя дает остаток, который перекристаллизовывают из смеси пропан-2-ола и эфира, получая 2-аллил-6-хлор-7-гидрокси-1-(1-фенилциклобутил)-1,2,3,4- тетрагидроизохинолин гидробромид, т.пл. 194 - 196oC.
Пример 66. Смесь 1-[1-(4-хлорфенил)циклобутил]-6,7-диметокси-1,2,3,4-тетрагидроизохинолина (7,29 г получают по примеру 50), этилйодида (1,76 мл), безводного карбоната калия (5,52 г) и ацетона (100 мл) нагревают с обратным холодильником в течение 16 ч. Реакционную смесь фильтруют, а остаток упаривают со смесью 9:1 петролейного эфира и триэтиламина. Раствор фильтруют и растворитель удаляют получая остаток. Образец этого остатка (4 г) нагревают с обратным холодильником в атмосфере азота с ледяной уксусной кислотой (40 мл) и 48%-ной бромистоводородной кислотой в течение 20 ч. Удаление растворителя дает остаток, который сушат путем азеотропной отгонки с техническим денатуратом, затем с пропан-2-олом и, наконец, со смесью толуола и пропан-2-ола, получая твердый продукт, который промывают пропан-2-олом и сушат в вакууме при 80oC, получая 1-[1-(4-хлорфенил)циклобутил]-2-этил-6,7-дигидрокси-1,2,3,4 -тетрагидроизохинолин гидробромид, т.пл. 213 - 215oC.
Пример 67. Смесь 1-[1-(4-хлорфенил)циклобутил]-6,7-диметокси- 1,2,3,4-тетрагидроизохинолина (7,29 г, получают по примеру 50), аллилбромида (2,66 г), безводного карбоната калия (5,52 г) и ацетона (100 мл) нагревают с обратным холодильником в течение 2 ч. Реакционную смесь фильтруют и растворитель удаляют путем выпаривания, получая остаток, который упаривают со смесью 9:1 петролейного эфира и триэтиламина. Раствор декантируют от остаточного дегтя, фильтруют и растворитель удаляют, получая остаток. Образец этого остатка (3 г) нагревают с обратным холодильником в атмосфере азота с ледяной уксусной кислотой (50 мл) и 48%-ной бромистоводородной кислотой (50 мл) в течение 7 ч. Реакционную смесь добавляют к смеси лед/вода и медленно добавляют водный раствор аммиака в избытке в атмосфере азота. Полученную смесь экстрагируют этилацетатом. Органический слой выпаривают, а остаток растворяют в этилацетате. Добавление раствора щавелевой кислоты в этилацетате дает 2-аллил-1-1-[1-(4-хлорфенил)циклобутил] -6,7-дигидрокси-1,2,3,4- тетрагидроизохинолин оксалат (т.пл. 85oC (разл.)), который сушат на воздухе.
Пример 68. 1-(2-Бромфенил)циклобутанкарбонилхлорид (9,4 г) добавляют в раствору 4-бензилокси-3-метоксифенэтиламина (8 г) и триэтиламина (3,15 г) в этилацетате (50 мл) и тетрагидрофуране (50 мл). Смесь перемешивают в течение 2 дней, добавляют 2 M водный раствор гидроксида калия (50 мл) и перемешивают в течение 20 мин. Водный слой отделяют и экстрагируют этилацетатом. Экстракт промывают 1 M соляной кислотой и рассолом, и получают масло, которое растворяют в ацетонитриле (180 мл). Добавляют оксихлорид фосфора (12 мл) и смесь нагревают с обратным холодильником в течение 2,5 ч. Смесь охлаждают и добавляют к смеси концентрированного водного раствора аммиака (100 мл) и воды (100 мл), смесь перемешивают в течение 10 мин и экстрагируют этилацетатом. Экстракт дает масло, которое насыщают тритием в эфире и полученный твердый продукт перекристаллизовывают из циклогексана, получая 7-бензилокси-1-[1-(2-бромфенил)-циклобутил] -6-метокси-3,4- дигидроизохинолин (т. пл. 115 - 116oC). Образец (3,1 г) этого материала в метаноле (16 мл) и в уксусной кислоте (35 мл) обрабатывают цианборгидридом натрия (1 г). Смесь перемешивают в течение 16 ч, разбавляют водой, подщелачивают 50%-ным водным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает остаток, который растворяется в ацетонитриле (115 мл), а затем добавляют 37 - 40%-ный водный раствор формальдегида (3,2 мл) и цианборгидрид натрия (0,83 г). Смесь перемешивают в течение 15 мин, а затем нейтрализуют уксусной кислотой и перемешивают в течение 45 мин. После концентрирования добавляют 2 М водный раствор гидроксида натрия и смесь экстрагируют этилацетатом. Экстракт дает 7-бензокси-1-[1-(2-бромфенил)циклобутил] -6-метокси-2-метил-1,2,3,4- тетрагидроизохинолин, образец которого (3 г) смешивают с этанолом (70 мл) и концентрированной соляной кислотой (70 мл), а смесь нагревают с обратным холодильником в течение 45 мин. Растворитель удаляют путем выпаривания и остаток сушат путем азеотропной отгонки с пропан-2-олом. Высушенный остаток суспендируют в пропан-2-оле (20 мл) и смесь фильтруют. Фильтрат обесцвечивают древесным углем в метаноле. Путем подщелачивания освобождают свободное основание и растворяют его в эфире. Удаление эфира дает 1-[1-(2-бромфенил)циклобутил]-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолин (т.пл. 48 - 51oC), который сушат в вакууме (около 0,1 мм Hg) в течение 4 ч.
Пример 69. Смесь гидрохлоридной соли 7-бензилокси-1-[1-(2-хлорфенил)циклобутил] -6-метокси-1,2,3,4- тетрагидроизохинолин (2,75 г получают по способу, подобному способу примера RB25), метанола (50 мл) и 37 - 40%-ного водного раствора формальдегида (3 мл) охлаждают до 10oC и добавляют цианборгидрид натрия (1,5 г). Смесь перемешивают в течение 24 ч и растворители удаляют путем выпаривания. Остаток распределяют между этилацетатом и разбавленным водным раствором гидроксида натрия. Органический слой дает смолу, которую растворяют в метаноле (25 мл) и в концентрированной соляной кислоте (25 мл), и нагревают с обратным холодильником в течение одного часа. Растворители удаляют путем выпаривания и затем остаток распределяют между эфиром и насыщенным водным раствором бикарбоната натрия. Эфирный слой дает остаток, который растворяют в пропан-2-оле (100 мл) и 48%-ным водным раствором бромистоводородной кислоты (5 мл). Растворитель удаляют путем выпаривания, а остаток кристаллизуют, затем перекристаллизовывают из пропан-2-ола, получая 1-[1-(2-хлорфенил)циклобутил]-7-гидрокси-6-метокси-2-метил-1,2,3, 4- тетрагидроизохинолин гидробромид, т.пл. 148 - 150oC.
Пример 70. Смесь 7-бензилокси-6-метокси-[1-(2-хлорфенил)-3,3-диметилциклобутил] -1,2,3,4- тетрагидроизохинолина (6,7 г, получают по примеру RC23), метанола (50 мл) и концентрированной соляной кислоты (50 мл) нагревают с обратным холодильником в течение 20 ч. Смесь охлаждают и объем сокращают до 25%. 7-Гидрокси-6-метокси-[1-(2-хлорфенил)-3,3-диметилциклобутил]-1,2,3,4- тетрагидроизохинолин (т. пл. 163- 165oC (разл.)) кристаллизуют и собирают путем фильтрования.
Смесь 7-гидрокси-6-метокси-[1-(2-хлорфенил)-3,3-диметилциклобутил] -1,2,3,4- тетрагидроизохинолина (3,7 г), метанола (50 мл) и 37 - 40% водного раствора формальдегида охлаждают до 5oC. Добавляют цианоборгидрид натрия (1,4 г) и смесь перемешивают в течение 1,5 ч. Растворитель удаляют путем выпаривания, а остаток распределяют между водой и дихлорметаном. Органический слой промывают водным раствором аммиака, а затем рассолом, сушат и удаляют растворитель путем выпаривания, получая смолу, которую растворяют в пропан-2-оле. Добавление 48%-ного водного раствора бромистоводородной кислоты дает 7-гидрокси-6-метокси-2-метил-1-[1-(2-хлорфенил)-3,3- диметилциклобутил] -1,2,3,4-тетрагидроизохинолин гидробромид, т.пл. 202 - 204oC (разл.).
Пример 71. Цианоборгидрид натрия (0,8 г) добавляют к смеси 7-бензилокси-6-метокси-1-[1-(2-метилтиофенил)циклобутил] -3,4- дигидроизохинолина (2,8 г, получают по примеру CA32), уксусной кислоты (20 мл) и метанола (10 мл) при 0oC и перемешивают в течение 60 ч при комнатной температуре. Смесь выливают в воду (300 мл) и экстрагируют дихлорметаном. Органический слой промывают водным раствором аммиака (100 мл), затем рассолом (100 мл) и сушат над сульфатом магния. Растворитель удаляют путем выпаривания, получая смолу. Смолу растворяют в пропан-2-оле и обрабатывают 48%-ным водным раствором бромистоводородной кислоты в избытке. Выпаривание растворителя дает твердый продукт, который насыщают тритием в петролейном эфире (т.к. 60 - 80oC), затем выделяют путем фильтрования, получая 7-бензилокси-6-метокси-1-[1-(2-метилтиофенил)циклобутил]-1,2,3,4- тетрагидроизохинолин.
Смесь 7-бензилокси-6-метокси-1-[1-(2-метилтиофенил)-циклобутил]-1,2,3,4- тетрагидроизохинолина (1,89 г), 37%-ного водного раствора формальдегида (3 мл), метанола (30 мл) и цианборгидрида натрия (0,5 г) перемешивают при комнатной температуре в течение 24 ч. Смесь выливают в воду (100 мл) и экстрагируют дихлорметаном (300 мл). Экстракт промывают разбавленным водным раствором аммиака получая остаток, который растворяют в пропан-2-оле и обрабатывают 48%-ным водным раствором бромистоводородной кислоты. Растворитель удаляют путем выпаривания, получая 7-бензилокси-6-метокси-2-метил-1-[1-(2-метилтиофенил)циклобутил] - 1,2,3,4-тетрагидроизохинолин, который используют без дальнейшей очистки.
7-Бензилокси-6-метокси-2-метил-1-[1-(2-метилтиофенил)- циклобутил] -1,2,3,4-тетрагидроизохинолин (1,7 г) нагревают с обратным холодильником в течение 2 ч с 48%-ной водной бромистоводородной кислотой (15 мл) и ледяной уксусной кислотой (15 мл). Растворитель удаляют путем выпаривания в вакууме, а остаток сушат путем азеотропной отгонки с пропан-2-олом. Затем остаток растворяют в пропан-2-оле, обесцвечивают древесным углем, и растворитель выпаривают, получая 6,7-дигидрокси-2-метил-1-[1-(2-метилтиофенил)циклобутил] -1,2,3,4- тетрагидроизохинолин гидробромид (1,1 г).
Пример 72. Смесь 7-бензилокси-6-метокси-1-[1-(4-трифторметоксифенил)циклобутил] - 1,2,3,4-тетрагидроизохинолина (4,1 г, получают способом, подобным способу по примеру RC18), 37 - 40%-ного водного раствора формальдегида (4,4 мл), ацетонитрила (90 мл) и цианборгидрата натрия (2,32 г) перемешивают при 5oC в течение 15 мин, затем нейтрализуют ледяной уксусной кислотой и перемешивают следующие 16 ч. Смесь выливают в разбавленный водный раствор гидроксида натрия и экстрагируют этилацетатом. Экстракты дают смолу, которую растворяют в эфире и обрабатывают хлористым водородом, получая 7-бензилокси-6-метокси-2-метил-1-[1-(4-трифторметоксифенил) циклобутил] -1,2,3,4-тетрагидроизохинолин в виде гидрохлоридной соли.
Смесь 7-бензилокси-6-метокси-2-метил-1-[1-(4-трифторметоксифенил) циклобутил] -1,2,3,4-тетрагидроизохинолина гидрохлорида (2,94 г), технического денатурата (5 мл) и концентрированной соляной кислоты (65 мл) нагревают с обратным холодильником в течение 45 мин. Смесь концентрируют и сушат путем азеотропной отгонки с пропан-2-олом. Остаток растворяют в эфире и обрабатывают одним эквивалентом щавелевой кислоты, получая 7-гидрокси-6-метокси-2-метил-1-[1-(4-трифторметоксифенил) циклобутил] -1,2,3,4-тетрагидроизохинолин оксалат, который кристаллизуют из ацетонитрила.
Пример 73. Смесь (2-метоксифенол)ацетонитрила (147 г), и 1,2-дибромэтана (168 мл) в диметилсульфоксиде (250 мл) добавляют в течение 1 ч и перемешиваемой суспензии порошкообразного гидроксида калия (250 г) и 18-краун-6 (5 г) в диметилсульфоксиде (1200 мл) при 25oC. Перемешивание продолжают в течение 20 ч. Добавляют воду (1200 мл) и смесь экстрагируют эфиром, получая сырое масло, которое очищают путем отгонки (т.к. 102o/0,25 мбар), получая 1-(2-метоксифенил)циклопропан карбонитрил.
1-(2-Метоксифенил)циклопропан карбонитрил (24 г) нагревают с обратным холодильником в течение 20 ч с 10%-ным водным раствором гидроксида калия (150 мл). После охлаждения раствор промывают толуолом, а затем эфиром. Водный слой подкисляют избытком соляной кислоты, получая 1-(2-метоксифенил)циклопропан карбонил хлорид.
Раствор 1-(2-метоксифенил)циклопропан карбонил хлорида (16 г) в этилацетате (50 мл) добавляют к перемешиваемому раствору 4-бензилокси-3-метокси-фенилэтиламина гидрохлорида (22,3 г) в этилацетате (250 мл) и триэтиламине (30 мл). Смесь перемешивают в течение 3 дней, затем добавляют воду. Органический слой промывают 5 М HCl, затем водой, затем 2 М водным раствором гидроксида натрия и сушат над сульфатом натрия. Выпаривание дает N-[2-(4-бензилокси-3-метоксифенил)этил]-1-(2-метоксифенил) циклопропанкарбоксамид.
Смесь N-[2-(4-бензилокси-3-метоксифенил)этил] -1-(2-метоксифенил) циклопропанкарбоксамида (30,3 г) в ацетонитриле (450 мл) и фосфорилхлориде (50 мл) нагревают с обратным холодильником в течение 80 мин. Растворитель испаряют в вакууме при температуре ниже 50oC и остаток промывают этилацетатом, а затем перемешивают с этилацетатом (300 мл) и 5%-ным водным раствором аммиака при охлаждении льдом (200 мл) в течение 10 мин. Органический слой сушат над карбонатом калия и растворитель удаляют путем выпаривания, получая 7-бензилокси-6-метокси-1-[1-(2-метоксифенил)циклопропил]-3,4- дигидроихолинолин.
Цианборгидрид натрия (7,4 г) добавляют к перемешиваемой смеси 7-бензилокси-6-метокси-1-[1-(2-метоксифенил)циклопропил] -3,4- дигидроизохинолина (23,3 г), уксусной кислоты (125 мл) и метанола (65 мл), охлажденной в смеси лед/вода. Через 16 ч при комнатной температуре смесь добавляют к гидроксиду натрия (110 г) со льдом. Продукт экстрагируют эфиром и органический слой сушат над карбонатом калия, а растворитель удаляют путем выпаривания, получая твердый продукт. Твердый продукт (16,9 г) в техническом денатурате (1800 мл) перемешивают в течение 3 дней с 1 М гидрофосфитом натрия (340 мл, получают из фосфорной кислоты (27,9 г) и гидрокарбоната натрия (28,5 г)) и 37 - 40%-ным водным раствором формальдегида (180 мл). Раствор концентрируют в вакууме до объема 200 мл и подщелачивают карбонатом калия (40 г) в воде (200 мл). Смесь экстрагируют эфиром и растворитель удаляют из экстракта получая смолу, которую очищают путем азеотропной отгонки с техническим денатуратом, а затем с пропан-2-олом, получая 7-бензилокси-6-метокси-1-[1-(2-метоксифенил)циклопропил] -2-метил-1,2,3,4- тетрагидроизохинолин, который нагревают с обратным холодильником в течение 30 мин с этанолом (200 мл) и концентрированной соляной кислотой (200 мл). Растворитель удаляют в вакууме, получая твердый продукт, который сушат путем азеотропной отгонки с пропан-2-олом получая остаток, который растворяют в ацетонитриле (200 мл). Добавляют этилацетат и кипятят смесь. Раствор декантируют и выпаривают в вакууме, получая остаток, который растирают в холодном этилацетате. Фильтрат осаждают, далее кристаллический твердый продукт, который собирают путем фильтрования, промывают этилацетатом и сушат. Полученный продукт является 7-гидрокси-6-метокси-1-[1-(2-метоксифенил)циклопропил] -2-метил- 1,2,3,4-тетрагидроизохинолин гидрохлоридом, т.пл. 118oC.
Пример 74. Смесь 7-бензилокси-1-[1-(2-хлорфенил)-циклопропил]-6-метокси-1,2,3,4- тетрагидроизохинолин гидробромида (5 г, получают по примеру RC14), метанола (100 мл) и 37%-ного водного раствора формальдегида (5 мл) охлаждают до 10oC. Добавляют боргидрид натрия (2,5 г) и смесь перемешивают при комнатной температуре в течение 2 ч. Метанол выпаривают в вакууме, а остаток распределяют между разбавленным водным раствором гидроксида натрия (100 мл) и эфира (2 • 100 мл). Органический слой дает масло, которое растворяют в метаноле (25 мл) и концентрированной соляной кислоте (25 мл) и нагревают с обратным холодильником в течение 2 ч. Растворитель удаляют в вакууме, а остаток растворяют в горячем этаноле, обесцвечивают, фильтруют и выпаривают растворитель. Полученный твердый продукт промывают эфиром, сушат, а затем распределяют между этилацетатом и концентрированным водным раствором аммиака. Органический слой дает масло, которое растворяют в метаноле. Добавляют 48%-ный водный раствор бромистоводородной кислоты и смесь нагревают с обратным холодильником в течение 2 ч. Растворители удаляют путем выпаривания, получая твердый продукт, который перекристаллизовывают из этанола. Твердый остаток промывают эфиром и сушат в вакууме при 50oC, получая 1-[1-(2-хлорфенил)-циклопропил] -7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин гидробромид, т.пл. 150 - 153oC. Выход 2,95 г.
Пример 75. Смесь 7-бензилокси-1-[(2-хлорфенил)-циклопропил]-6-метокси-1,2,3,4- тетрагидроизохинолина (2,1 г, свободное основание выделяют из гидробромидной соли, полученной способом, подобным способу по примеру RC14), ацетона (30 мл), безводного карбоната калия (1,6 г) и 2-метоксиэтилбромида (2,1 г) нагревают с обратным холодильником в течение 6 ч. Через следующие 16 ч при комнатной температуре добавляют карбонат калия (3 г) и 2-метоксиэтилбромид (2,22 г), а нагревание продолжают в течение 6 ч. Смесь фильтруют, остаток промывают ацетоном, а растворитель удаляют из фильтрата в вакууме, получая 7-бензилокси-1-[(2-хлорфенил)-циклопропил]-6-метокси-2-(2- метоксиэтил)-1,2,3,4-тетрагидроизохинолин в виде масла.
Смесь масла, этанола (25 мл) и концентрированной соляной кислоты (25 мл) нагревают с обратным холодильником в течение 30 мин. Растворитель удаляют в вакууме, а остаток сушат путем азеотропной отгонки с этанолом. Полученный остаток насыщают тритием в этилацетате, получая твердый продукт, который собирают путем фильтрации, промывают этилацетатом и сушат при 40oC в вакууме, получая 1-[1-(2-хлорфенил)-циклопропил]-7-гидрокси-6-метокси-2-(2- метоксиэтил)-1,2,3,4-тетрагидроизохинолин (1,5 г), т.пл. 115 - 120oC.
Пример 76. Смесь 7-бензилокси-1-[(2-хлорфенил)-циклопропил]-6-метокси- 1,2,3,4-тетрагидроизохинолина (2,83 г, выделенный из гидробромидной соли, полученной способом, подобным способу по примеру RC14), ацетона (40 мл), безводного карбоната (5,5 г) и 2-бромэтанола (3,6 мл) нагревают с обратным холодильником в течение 18 ч. Смесь фильтруют, твердые продукты промывают ацетоном, а растворитель из фильтрата удаляют в вакууме, получая 7-бензилокси-1-[(2-хлорфенил)-циклопропил] -2-(2-гидроксиэтил)- 7-метокси-1,2,3,4-тетрагидроизохинолин в виде масла.
Смесь масла, этанола (30 мл) и концентрированной соляной кислоты (30 мл) нагревают с обратным холодильником в течение 30 мин. Растворитель удаляют в вакууме, а остаток сушат путем азеотропной отгонки со смесью этанола и толуола. Полученную в результате смолу дигерируют в кипящем этилацетате и остаток сушат при 45oC в вакууме, получая твердый продукт, который собирают путем фильтрации и промывают этилацетатом. Полученный твердый продукт растворяют в теплой воде, а полученный раствор подщелачивают путем добавления небольшого избытка водного раствора аммиака, получая твердый продукт, который растворяют в этилацетате. Полученный раствор сушат над сульфатом магния, а растворитель удаляют в вакууме, получая смолу, которая образует стеклообразную массу при охлаждении. Продукт является 1-[1-(2-хлорфенил)-циклопропил] -7-гидрокси-2-(гидроксиэтил)-6- метокси-1,2,3,4-тетрагидроизохинолином (0,74 г), т. пл. 65 - 70oC, который собирают путем фильтрования и сушат в вакууме, получая (+)-1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин гидробромид, который имеет удельное вращение плоскости поляризации [α]D= +14,6°, т.пл. 154 - 157oC.
Пример 78. Смесь 6,7-диметокси-1-[1-(4-бифенил)циклобутил] -1,2,3,4-тетрагидроизохинолина гидрохлорида (3,5 г, получают по примеру RC20), метанола (50 мл), 37%-ного водного раствора формальдегида (5 мл) и цианборгидрида натрия (2,08 г) перемешивают при комнатной температуре в течение 24 ч. Растворители удаляют в вакууме, остаток распределяют между водным раствором гидроксида натрия и эфиром. Эфирный экстракт сушат над сульфатом магния, раствор фильтруют и растворитель удаляют в вакууме. Остаток растворяют в ледяной уксусной кислоте (30 мл). Добавляют 48%-ную водную бромистоводородную кислоту (30 мл), смесь нагревают с обратным холодильником в азоте в течение 6 ч. Растворитель удаляют в вакууме, а остаток перекристаллизовывают из метанола, получая твердый продукт, который распределяют между концентрированным водным раствором аммиака и эфиром. Эфирные экстракты промывают с рассолом, сушат и фильтруют. Сухой хлористый водород пробулькивают через фильтрат, получая 6,7-диметокси-1-[1-(4-бифенил)циклобутил] -1,2,3,4- тетрагидроизохинолин гидрохлорид (1,1 г), т.пл. 131 - 135oC (разл.).
Пример 79. 1-[1-(2-Хлорфенил)циклопропил] -карбонилхлорид (16,9 г) добавляют по каплям при 0oC в азоте к суспензии 2-(4-метокси-3-метилфенил)этиламина (13 г, получают способом, аналогичным способу примера 10) и триэтиламина (11,8 г) в тетрагидрофуране (200 мл). Реакционную смесь перемешивают при комнатной температуре в течение 16 ч, затем выливают в водный раствор гидроксида натрия и перемешивают в течение 1 ч. Продукт экстрагируют этилацетатом. Экстракты сушат над сульфатом магния, а растворитель концентрируют, получая N-[2-(4-метокси-3-метилфенил)этил]-1-(2-хлорфенил) циклопропанкарбоксамид, который используют без дальнейшей очистки.
Смесь амида и 82 мас.% раствора полифосфатэфира в хлороформе (170 г) осторожно нагревают в течение 16 ч, затем выливают в воду (1200 мл) и смесь промывают эфиром. Водную фазу подщелачивают путем добавления водного раствора аммиака и продукт экстрагируют этилацетатом. Экстракты сушат и концентрируют, получая 1-[1-(2-хлорфенил)циклопропил] -7-метокси-6-метил-3,4- дигидроизохинолин в виде твердого продукта. Выход 14,7 г.
Цианборгидрид натрия (5,3 г) добавляют порциями к раствору 1-[1-(2-хлорфенил)циклопропил] -7-метокси-6-метил-3,4- дигидроизохинолин (14 г) в метаноле (70 мл) и уксусной кислоте (140 мл). Смесь перемешивают в течение 1 ч, концентрируют, а остаток обрабатывают водным раствором гидроксида натрия. Полученную смесь экстрагируют этилацетатом, а экстракты промывают рассолом и сушат над сульфатом магния. 1-[1-(2-Хлорфенил)циклопропил]-7-метокси-6-метил-1,2,3, 4- тетрагидроизохинолин кристаллизуют из экстракта и собирают путем фильтрации (6,5 г).
Смесь 1-[1-(2-хлорфенил)циклопропил] -7-метокси-6-метил-1,2,3,4- тетрагидроизохинолина (6,2 г), метанола (160 мл) и 37 - 40%-ного водного раствора формальдегида перемешивают в течение 15 мин. Добавляют цианборгидрид натрия и перемешивают смесь в течение следующих 10 мин. Затем реакцию нейтрализуют уксусной кислотой и перемешивают 45 мин. Метанол удаляют в вакууме, а остаток обрабатывают водным раствором гидроксида натрия. Продукт экстрагируют этилацетатом. Экстракты промывают водным раствором аммиака, водой и рассолом, а затем сушат над сульфатом магния. Концентрирование дает 1-[1-(2-хлорфенил)циклопропил] -7-метокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин в виде смолы (5,02 г).
Смесь смолы (5,02 г), 48%-ной водной бромистоводородной кислоты (120 мл) и ледяной уксусной кислоты (120 мл) нагревают при 90 - 95oC в течение 16 ч. Реакцию нейтрализуют путем добавления водного раствора гидроксида натрия, продукт экстрагируют этилацетатом. Экстракты концентрируют и остаток перерастворяют в пропан-2-оле (150 мл), содержащем концентрированную водную соляную кислоту (2 мл). Раствор концентрируют и сушат путем азеотропной отгонки с пропан-2-олом, получая твердый продукт. Твердый продукт промывают этилацетатом, получая 1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин гидрохлорид. Выход 4,5 г, т.пл. 159 - 161oC.
Пример 80. 1-[1-(2-Хлорфенил)циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин (2,0 г, выделяют из смеси, которую получают по примеру 79) разделяют путем хиральной препаративной жидкостной хроматографии высокого разрешения на колонке Chiracel OD, элюируя смесью 1:19 пропан-2-ола и гексана. Фракцию 1 преобразуют в ее бромистоводородную соль, используя 48%-ный водный раствор бромистоводородной кислоты в пропан-2-оле и получая (+)-1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин гидробромид. Соль имеет удельное вращение плоскости поляризации [α]D= +5,6°, т.пл. 170 - 175oC (разл.). Выход 0,8 г.
Пример 81. Цианборгидрид натрия (2,8 г) добавляют при 5oC к смеси 1-[2, 4-дихлорфенил)циклопропил]-6-фтор-7-метокси-1,2,3,4- тетрагидроизохинолин гидробромида (10 г, получают по примеру RC4) метанола (100 мл) и 37-40%-ного водного раствора формальдегида (7,2 мл). Смесь подогревают до комнатной температуры и перемешивают в течение 1,5 ч. Растворитель удаляют в вакууме, а остаток распределяют между водой и дихлорметаном. Органическую фазу промывают концентрированным раствором аммиака, затем водой и сушат над сульфатом магния. Растворитель удаляют в вакууме, получая 1-[1-(2,4-дихлорфенил)циклопропил] -6-фтор-7-метоки-2-метил-1,2,3,4- тетрагидроизохинолин в виде смолы, которая твердеет со временем.
Смесь 1-[1-(2,4-дихлорфенил)циклопропил] -6-фтор-7-метокси-2-метил-1,2,3,4- тетрагидроизохинолина (8,5 г), 48%-ной бромистоводородной кислоты (100 мл) и ледяной уксусной кислоты (100 мл) нагревают с обратным холодильником в течение 2 ч. Растворитель удаляют в вакууме, а остаток сушат путем азеотропной отгонки с пропан-2-олом, получая 1-[1-(2, 4-дихлорфенил)циклопропил] -6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолин гидробромид (7,2 г), т.пл. 235 - 237oC.
Пример 82. N, N, N', N'-Тетраметилэтилендиамин (231,4 г) добавляют при комнатной температуре к раствору н-бутиллития (2,5 М, 800 мл) в гексане (2,5 л), а затем раствор 2,3-дигидробензо [b] бутана (102 г) в гексане (25 мл). Смесь перемешивают в атмосфере азота при комнатной температуре 5 ч. Полученную суспензию медленно добавляют к сухому льду (300 г) и гексану (500 мл) в азоте. После перемешивания при комнатной температуре в течение 16 ч смесь разбавляют водой (2 л) и слои разделяют. Водный слой промывают гексаном, подкисляют до pH 1 концентрированной соляной кислотой, охлаждают и осадок собирают путем фильтрования. Последний промывают водой и дихлорэтаном и сушат при 80oC в вакууме, получая 2,3-дигидробензо [b] фуран-7-карбоновую кислоту (39,6 г), т.пл. 164 - 165oC. Гексановый слой фильтруют, сушат над сульфатом натрия и концентрируют. При охлаждении получают последующий продукт (32,7 г), т.пл. 170oC.
Боран-диметилсульфидный комплекс (60 мл) добавляют к перемешиваемому раствору указанной выше кислоты (70,2 г) и тетрагидрофурана (500 мл). Смесь перемешивают в течение 30 минут, затем аккуратно добавляют воду (200 мл). Тетрагидрофуран удаляют в вакууме. Добавляют воду (200 мл), а затем водный раствор гидроксида натрия. Продукт экстрагируют эфиром, а экстракты сушат над карбонатом калия. Растворитель удаляют в вакууме, получая 7-гидроксиметил-2, 3-дигидробензо [b] фуран (50 г) в виде масла.
Раствор указанного выше масла (50 г) в дихлорметане (200 мл) обрабатывают порциями при 20oC тионилхлорида (50 мл) за 10 мин. Раствор подогревают, а растворитель удаляют в вакууме, получая 7-хлорметил-2,3-дигидробензо [b] фуран в виде масла, которое используют без дальнейшей очистки.
Раствор цианида натрия (45 г) в воде (200 мл) добавляют к смеси указанного выше масла в толуоле (200 мл). Добавляют тетрабутиламмонийбромид (2 г). Смесь нагревают с обратным холодильником при энергичном перемешивании в течение 3 ч. После стояния в течение 16 ч смесь обесцвечивают путем добавления древесного угля. Смесь фильтруют, разделяют и органический слой сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая масло (40 г), которое очищают путем отгонки при 144 - 160oC/4 мбар, а затем при 90 - 112oC/0,4 мбар. Дистиллят нагревают с диметилсульфоксидом (50 мл) и цианидом натрия (6 г) при 90 - 95oC в течение 8 ч с удалением влаги. Затем смесь добавляют к воде, а продукт экстрагируют эфиром. Экстракты сушат над карбонатом калия, а раствор удаляют в вакууме, получая масло.
Смесь масла, толуола (60 мл), пиридина (2,2 мл) и фталевого ангидрида (4 г) нагревают при 90 - 95oC в течение 4 ч. Полученный раствор охлаждают, промывают 10%-ным водным раствором карбоната калия, затем разбавленной соляной кислотой. Затем раствор сушат над сульфатом натрия и удаляют растворитель в вакууме, получая масло. Масло отгоняют (т.пл. 120/1 мбар), получая 2,3-дигидробензо [b] фуран-7-илацетонитрил, который быстро отвердевает (16,97 г).
Твердый продукт (16,97 г) расплавляют и растворяют в диметилсульфоксиде (100 мл). Добавляют 1,2-дибромэтан (18 мл) и смесь добавляют со скоростью 1 капля в секунду при 20 - 30oC к перемешиваемой смеси твердого гидроксида калия (50 г), диметилсульфоксида (150 мл) и 18-краун-6 (1,5 г). Смесь перемешивают в течение следующих 16 ч. Добавляют 1,2-дибромэтан (10 мл) и продолжают перемешивать в течение 8 ч. Смеси позволяют стоять при комнатной температуре в течение 16 ч. Добавляют 1,2-дибромэтан (8 мл), смесь перемешивают в течение 6 ч, затем добавляют 1,2-дибромэтан (10 мл) и смесь стоит в течение 3 дней. Смесь добавляют в воду и продукт экстрагируют эфиром. Экстракты сушат над карбонатом калия, растворитель удаляют в вакууме и остаток отгоняют (т. пл. 120oC/0,25 мбар), получая 1-[2,3-дигидробензо[b] фуран-7-ил]циклопропанкарбонитрил в виде твердого продукта (11,2 г).
Смесь указанного выше твердого продукта (11,2 г), гидроксида калия (30 г) и воды (300 мл) перемешивают и нагревают с обратным холодильником в течение 6 ч. Полученный раствор промывают эфиром и водную фазу подкисляют путем добавления избытка соляной кислоты, получая твердый продукт, который собирают путем фильтрования, промывают водой и сушат на воздухе, получая 1-[2,3-дигидробензо [b] фуран-7-ил]-циклопропанкарбоновую кислоту.
Указанную выше карбоновую кислоту (7,72 г) нагревают с тионилхлоридом (20 мл), затем нагревают с обратным холодильником в течение 20 мин. Избыток тионилхлорида выпаривают. Отгонка дает масло (т.пл. 70oC/50 мбар), которое растворяют в этилацетате (50 мл). Раствор добавляют к перемешиваемой смеси 2-(4-бензилокси-3-метоксифенил)этиламина гидрохлорида (15,7 г), этилацетата (200 мл) и триэтиламина (30 мл), и смесь перемешивают в течение 64 ч. Добавляют воду. Этилацетатный слой промывают водой, разбавленным раствором гидроксида натрия, разбавленной соляной кислотой и водой, а затем сушат над сульфатом натрия. Растворитель удаляют в вакууме, получая смолу, которую очищают с помощью флэш хроматографии, используя эфир в качестве элюента и получая N-[2-(4-бензилокси-3-метоксифенил)этил] -1-[2,3-дигидробензо [b] фуран-7-ил] -циклопропанкарбоксамид, который растворяют в ацетонитриле (200 мл). Добавляют фосфорилхлорид (20 мл) и смесь нагревают с обратным холодильником в течение 80 мин. Раствор и избыток фосфорилхлорида удаляют в вакууме, а остаток растворяют в этилацетате. Добавление эфира вызывает выделение масла. Супернатант декантируют и добавляют эфир до тех пор, пока масло не перестает выделяться. Масло насыщают тритием в эфире, затем добавляют к смеси разбавленного водного аммиака и эфира. Эфирный слой сушат над карбонатом калия, а растворитель удаляют в вакууме, получая 7-бензилокси-1-[1-(2,3-дигидробензо [b] фуран-7-ил) циклопропил]-6-метокси-3,4-дигидроизохинолин в виде смолы (11,1 г).
Смесь указанной выше смолы (11,1 г), уксусной кислоты (60 мл) и метанола (30 мл) перемешивают в течение 2 ч, затем охлаждают в смеси лед/вода. Добавляют цианборгидрид натрия в тетрагидрофуране (1 М раствор, 53 мл) и смесь перемешивают в течение 2 ч. Добавляют воду, затем водный раствор аммиака в избытке, и продукт экстрагируют эфиром. Растворитель удаляют в вакууме, получая смолу, которая используется без дальнейшей очистки.
1 М водный гидрофосфат натрия (218 мл, получают из фосфорной кислоты (17,9 г) в воде и бикарбоната натрия (18,3 г)) добавляют к смеси смолы и технического денатурата (1,15 л). Добавляют 37 - 40%-ный водный раствор формальдегида (115 мл) и полученному раствору дают постоять в течение 64 ч. Раствор концентрируют в вакууме до 200 мл, фильтруют и декантируют, удаляя следы смолы. Раствор разбавляют водой, подщелачивают путем добавления водного раствора аммиака в избытке, и продукт экстрагируют эфиром. Экстракты сушат над сульфатом натрия, растворитель удаляют в вакууме, получая масло. Масло очищают с помощью колоночной хроматографии, используя смесь 19:1 эфира и триэтиламина в качестве элюента. Фракции очищают с помощью жидкостной хроматографии высокого давления. Фракции 1 и 2 (9,46 г) объединяют.
Смесь фракций 1 и 2 (9,46 г), 98 - 100%-ной муравьиной кислоты (25 мл) и метанола (125 мл) в атмосфере аргона обрабатывают 10% палладием на древесном угле (3 г), добавляют и пробулькивают аргон в течение 24 ч. Добавляют 48%-ную водную бромистоводородную кислоту (3,6 мл) и смесь фильтруют, а растворитель удаляют из фильтрата в вакууме. Остаток сушат путем азеотропной отгонки с этанолом (100%), получая твердый продукт, который промывают этилацетатом, сушат на воздухе, а затем в вакууме при 45oC.
Твердый продукт растворяют в воде, подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Экстракт сушат над сульфатом натрия, а растворитель удаляют в вакууме, оставляя малый объем. Добавляют эфир и 1-[1-(2,3-дигидробензо[b] фуран-7-ил)циклопропил] - 7-гидрокси-6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин (5,18 г), т.пл. 156oC собирают путем фильтрования и промывают эфиром.
Пример 83. Смесь N-[2-(3,4-диметоксифенил)этил]циклобутанкарбоксамида (44 г, получают способом, подобным способу по примеру E46), оксихлорида фосфора (150 мл) и ацетонитрила (900 мл) нагревают с обратным холодильником в течение 2,5 ч. Охлажденный раствор выливают в разбавленный водный раствор аммиака и продукт экстрагируют этилацетатом. Экстракты промывают рассолом, сушат и растворитель удаляют в вакууме. Полученное масло отвердевает в течение 16 ч. Твердый продукт перекристаллизовывают из петролейного эфира (т. к. 60 - 80oC), получая 1-циклобутил-3,4-дигидро-6,7-диметоксиизохинолин (20 г).
n-Бутиллитий (24,5 мл, 2 М раствор в гексане) добавляют к раствору диизопропиламина (6,85 мл) в тетрагидрофуране (50 мл) при 0oC. Полученный раствор диизопропиламина лития перемешивают при 0oC в течение 20 мин. Добавляют раствор 1-цикло-бутил-3, 4-дигидро- 6,7-диметоксиизохинолина (10 г) в тетрагидрофуране (100 мл). Смесь перемешивают при 0oC в течение 1 ч, затем охлаждают до -70oC и добавляют по каплям 2-фторбензонитрил (4,42 мл). Смесь перемешивают при -70oC в течение 50 мин, затем ей позволяют медленно нагреваться до комнатной температуры. Смесь выливают в соляную кислоту и промывают эфиром. Водную фазу подщелачивают раствором аммиака, а продукт экстрагируют этилацетатом. Полученный твердый продукт (0,6 г) собирают путем фильтрования. Фильтрат промывают рассолом, сушат над сульфатом магния, а растворитель удаляют в вакууме, получая дальнейший твердый продукт. Объединенные твердые продукты перекристаллизовывают из ацетонитрила, получая твердый продукт, который сушат в вакууме, получая 1-[1-(2-цианфенил)циклобутил]-6,7-диметокси-3,4-дигидроизохинолин (5 г).
Смесь дигидроизохинолина (5 г), ацетонитрила (150 мл) и метилйодида (18 мл) нагревают осторожно с обратным холодильником в течение 64 ч. Растворитель удаляют в вакууме, а остаток промывают эфиром. Твердый продукт собирают путем фильтрования и сушат в вакууме, получая 1-[1-(2-цианфенил)-циклобутил] -6, 7-диметокси-2-метил- 3,4-дигидроизохинолин йодид (6,7 г).
Боргидрат натрия (0,465 г) добавляют порциями к смеси 1-[1-(2-цианфенил)циклобутил]-6,7-диметокси-2-метил-3, 4-дигидроизохинолиний йодида (6 г) и метанола (40 мл). Смесь перемешивают в течение 2 ч, а затем выливают в водный раствор гидроксида натрия, а продукт экстрагируют в эфир. Растворитель удаляют из экстрактов, получая смолу, которую растворяют в этилацетате и в небольшом количестве пропан-2-ола. Добавляют 0,4 М раствор (±)-дибензоилвиноградной кислоты в эфире (35 мл), и полученную суспензию концентрируют в вакууме. Оставшийся твердый продукт промывают эфиром, получая 1-[1-(2-цианфенил)циклобутил] -6,7-диметокси-2-метил-1,2,3,4- тетрагидроизохинолин (±)-дибензоилтартрат (7,2 г), т. пл. 109 - 112oC (разл.).
(±)-Дибензоилтартратную соль (2 г) нейтрализуют насыщенным водным раствором гидрокарбоната натрия и экстрагируют этилацетатом. Экстракты сушат, а растворители удаляют, получая 1-[1-(2-цианфенил)циклобутил]-6,7-диметокси-2-метил-1,2,3,4- тетрагидроизохинолинового свободного основания (1 г), которое растворяют в дихлорметане (5 мл). Раствор охлаждают до -70oC и обрабатывают по каплям 1 М раствором трибромида бора в дихлорметане (3 мл). В течение 30 мин смеси позволяют нагреться до комнатной температуры, а затем перемешивают в течение 90 мин. Смесь охлаждают до -70oC и в дальнейшем добавляют дихлорметан (7 мл) и раствор 1 М трибромида бора в дихлорметане (3 мл). Смесь подогревают до комнатной температуры и перемешивают в течение 1 ч, затем охлаждают до -40oC и добавляют метанол в избытке. Смесь нагревают до комнатной температуры и добавляют метанол (100 мл). Растворитель отгоняют в вакууме и триметилборат удаляют путем азеотропной отгонки с метанолом.
Остаток нейтрализуют насыщенным водным раствором гидрокарбоната натрия и экстрагируют этилацетатом. Экстракты обрабатывают эфирной соляной кислотой в избытке. Растворитель удаляют в вакууме, получая 1-[1-(2-цианфенил)циклобутил] -6,7-дигидрокси-2-метил-1,2,3,4- тетрагидроизохинолин (0,4 г), т.пл. 206 - 208oC (разл.).
Пример 84. Добавляют метилйодид (15 мл) к раствору 7-бензилокси-1-[1-(2-хлорфенил)циклопентил] -6-метокси-3,4-дигидроизохинолина (13 г, получают по примеру CA34) в ацетонитриле (100 мл). Смесь нагревают с обратным холодильником в течение 24 ч, охлаждают, а полученный твердый продукт собирают путем фильтрования. Твердый продукт промывают эфиром и сушат на воздухе, получая 7-бензилокси-1-[1-(2- хлорфенил)циклопентил]-6-метокси-2-метил-3,4-дигидроизохинолиний йодид (10 г).
Боргидрид натрия (2,6 г) добавляют порциями к смеси 7-бензилокси- 1-[1-(2-хлорфенил)циклопентил]-6-метокси-3,4-дигидроизохинолиний йодида (10 г) и метанола (250 мл). Смесь перемешивают при комнатной температуре, нагревают с обратным холодильником в течение 1 ч, затем охлаждают до комнатной температуры. Добавляют боргидрид натрия (5 г) и смесь перемешивают при комнатной температуре в течение 30 мин. Полученный твердый продукт собирают путем фильтрования, промывают эфиром и растворяют в ацетоне. Нерастворимые частицы удаляют путем фильтрования и растворитель удаляют из фильтрата в вакууме, получая 7-бензилокси-1-[1-(2-хлорфенил)циклопентан] - 6-метокси-2-метил-1,2,3,4-тетрагидроизохинолин (3,8 г).
Смесь указанного выше тетрагидроизохинолина (3,8 г), метанола (25 мл) и концентрированной соляной кислоты (25 мл) нагревают с обратным холодильником в течение 3,5 ч. Растворитель удаляют в вакууме, а остаток перекристаллизовывают из метанола. Полученный твердый продукт промывают дважды эфиром и сушат, получая 1-[1-(2-хлорфенил)циклопентил] -7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолин гидрохлорид (2,8 г), т.пл. 119 - 121oC (разл.).
Пример 85. Раствор 7-бензилокси-1-[1-(2-хлорфенил)циклопропил]- 6-метокси-3,4-дигидроизохинолина (0,5 г, получают способом, подобным способу по примеру CA28) в дихлорметане (10 мл) добавляют по каплям при -40oC к перемешиваемому раствору натрий трис[N-(2-метилпропилоксикарбонил)пропилокси]боргидрида (2,43 г) в дихлорметане (10 мл). Смеси позволяют достичь комнатной температуры и перемешивают в течение 49 ч. Добавляют разбавленную соляную кислоту (10 мл, 10 мас.%) и перемешивание продолжают в течение 1 ч. Смесь подщелачивают путем добавления насыщенного водного раствора карбоната натрия. Органический слой промывают рассолом, сушат над сульфатом магния и растворитель удаляют в вакууме, получая 7-бензилокси-1-[1-(2- хлорфенил)циклопропил] -6-метокси-1,2,3, 4-тетрагидроизохинолин в виде смолы (0,42 г). Показано, что этот продукт имеет 92%-ный энантиометрический избыток одного из энантиомеров.
Смесь энантиомерно-обогащенного 7-бензилокси-1-[1-(2- хлорфенил)циклопропил] -6-метокси-1,2,3,4-тетрагидроизохинолина (0,41 г), безводного карбоната калия (0,4 г), метилйодида (0,152 г) и ацетона (25 мл) перемешивают при комнатной температуре в течение 2 ч. Растворитель удаляют в вакууме, а остаток растворяют в воде и экстрагируют эфиром. Экстракты концентрируют в вакууме. Смесь остатка, концентрированной соляной кислоты (5 мл) и метанола (5 мл) нагревают с обратным холодильником в течение 2 ч. Растворители удаляют в вакууме и остаток распределяют между насыщенным водным раствором бикарбоната натрия и эфиром. Эфирный слой концентрируют в вакууме, остаток растворяют в пропан-2-оле и подкисляют 48%-ной водной бромистоводородной кислотой. Раствор концентрируют в вакууме, получая (+)-1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолин гидрохлорид (0,24 г).
Пример 86. Добавляют деканоилхлорид (0,45 г) к смеси (+)-1-[1-(2-хлорфенил)циклопропил]
-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолина гидрохлорида (1 г, получают способом, подобным способу примера 77), триэтиламина (0,7 г) и эфира (30 мл). Смесь перемешивают при комнатной
температуре в течение 24 ч, затем промывают водой, 10%-ным водным раствором гидроксида натрия и рассолом. Органический слой сушат над сульфатом магния, фильтруют и концентрируют, получая смолу,
которую очищают с помощью хроматографии на силикагелевой колонке, используя смесь 1:3 эфира и легкой фракции нефти в качестве элюента. Нужные фракции концентрируют в вакууме, получая
(+)-1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6- метокси-2-метил-1,2,3,4-тетрагидроизохинолин (0,95 г) в виде смолы, имеющей удельное вращение плоскости поляризации [α]D= +21,
47°.
Пример 87. Деканоил хлорид (0,66 г) в дихлорметане (5 мл) добавляют к перемешиваемой смеси (+)-1-[1-(2-хлорфенил) циклобутил]-6-фтор-7-гидрокси-2-метил-1,2,3,
4-тетрагидроизохинолина гидробромида (1,5 г, получают способом, подобным способу по примеру 64), триэтиламина (1,06 г) и дихлорметана (25 мл). Смесь перемешивают при комнатной температуре в течение 20
мин, затем добавляют воду (20 мл) и продолжают перемешивать в течение 1 ч. Органический слой промывают насыщенным водным раствором бикарбоната натрия, водой и рассолом, затем сушат над сульфатом
магния, фильтруют и сушат над сульфатом магния, получая (-)-1-[1-(2-хлорфенил)циклобутил] -7-деканоилокси-6-фтор-2-метил-1,2,3,4- тетрагидроизохинолин (1,31 г). Последний имеет удельное вращение
плоскости поляризации [α]D= -5,59°.
Пример 88. Деканоил хлорид (0,61 г) добавляют к перемешиваемой суспензии
(+)-1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолина гидробромида (1 г, получают способом, аналогичным способу по примеру 62) и дихлорметана (25 мл). Добавляют
триэтиламин (1,35 мл) и оставляют, перемешивая 1,5 ч. Затем добавляют воду, органический слой промывают 2н. водным раствором гидроксида натрия, затем водой и сушат над карбонатом калия. Растворитель
удаляют в вакууме и остаточное масло очищают с помощью жидкостной хроматографии с использованием петролейного эфира (т.к. 40 - 60oC) в качестве элюента примесей, а затем 5% эфира в
петролейном эфире (т. к. 40 - 60oC) в качестве элюента продукта. Растворитель удаляют в вакууме, получая (+)-1-[1-(2-хлорфенил)циклопропил]-7-деканоилокси-6-фтор-2-метил-1,2,3,
4- тетрагидроизохинолин (0,85 г), который имеет удельное вращение плоскости поляризации [α]D= +10,6°.
Пример 89. Деканоил хлорид (0,5 г) в дихлорметане и
затем триэтиламин (0,8 г) добавляют в перемешиваемую смесь 1-[1-(2-хлорфенил)циклопропил]-6-фтор-7-гидрокси-2-метил-1,2,3,4- тетрагидроизохинолина гидробромида (0,825 г, получают способом, подобным
способу примера 61) в дихлорметане (20 мл). После перемешивания в течение 1 ч добавляют воду и органическую фазу промывают 2н. водным раствором гидроксида натрия и затем водой. Затем органическую фазу
сушат над сульфатом магния и удаляют растворитель в вакууме, получая 1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-фтор-2-метил-1,2,3,4- тетрагидроизохинолин (0,85 г).
Пример 90.
Гексадеканоил хлорид (0,83 г) в дихлорметане (5 мл) добавляют по каплям к перемешиваемому раствору 1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолина (1,04 г,
получают из (+)-энантиомера гидробромидной соли по способу, подобному способу по примеру 77), триэтиламина (0,91 г) и дихлорметана (20 мл). Смесь перемешивают при комнатной температуре в течение 2 ч,
затем промывают разбавленным раствором гидроксида натрия, водой и рассолом. Органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Полученное масло растворяют в петролейном
эфире (т. к. 60 - 80oC)и промывают многократно водой. Органический слой сушат над сульфатом магния, фильтруют и концентрируют в вакууме, получая (+)-1-[1-(2-хлорфенил)циклопропил]
-7-гексадеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин (1,45 г), который имеет удельное вращение плоскости поляризации [α]D= +21,18°.
Пример
91. Раствор додеканоил хлорида (0,737 г) в дихлорметане (5 мл) добавляют по каплям к перемешиваемому раствору 1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолина
(1,16 г, свободное основание освобождают из (+)-энантиомера гидробромидной соли, которую получают по примеру 77), триэтиламина (1,02 г) и дихлорметана (20 мл). Смесь перемешивают при комнатной
температуре в течение 2 ч. Смесь промывают разбавленным раствором гидроксида натрия, водой и затем рассолом. Органический слой концентрируют в вакууме и остаток распределяют между легкокипящим
петролейным эфиром и водой.
Органический слой промывают водой, сушат над сульфатом магния, фильтруют и растворитель удаляют в вакууме, получая смолу. Смолу растворяют в петролейном
эфире и очищают с помощью флэш хроматографии, используя смесь 1:4 эфира и петролейного эфира в качестве элюента. Удаление растворителя из элюента дает
(+)-1-[1-(2-хлорфенил)циклопропил]-7-додеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолина (0,81 г), который имеет удельное вращение плоскости поляризации [α]D= +25,05°.
Пример 92. Раствор гептаноил хлорида (0,34 г) в дихлорметане (5 мл) добавляют по каплям к перемешиваемому раствору 1-[1-(2-хлорфенил)циклопропил]
-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолина (0,79 г, свободное основание освобождают из (+)-энантиомера гидробромидной соли, которую получают по примеру 77), триэтиламина (0,69 г) и
дихлорметана (20 мл). Смесь перемешивают при комнатной температуре в течение 2 ч. Смесь промывают разбавленным раствором гидроксида натрия, водой и затем рассолом. Органический слой концентрируют в
вакууме и остаток распределяют между легкокипящим петролейным эфиром и водой.
Органический слой промывают водой, сушат над сульфатом магния, фильтруют и удаляют растворитель в вакууме
получая смолу. Смолу растворяют в петролейном эфире и очищают с помощью флэш хроматографии, используя смесь 1:3 эфира и петролейного эфира в качестве элюента. Удаление растворителя из элюента дает
(+)-1-[1-(2-хлорфенил)циклопропил] -7-гептанокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин (0,64 г), который имеет удельное вращение плоскости поляризации [α]D= +23,04°.
Пример 93. Раствор октадеканоил хлорида (0,61 г) в дихлорметане (5 мл) добавляют по каплям к перемешиваемому раствору
1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-6-метокси-2-метил-1,2,3,4- тетрагидроизохинолина (0,69 г, свободное основание освобождают из (+)-энантиомера гидробромидной соли, которую получают по примеру
77), триэтиламина (0,61 г) и дихлорметана (20 мл). Смесь перемешивают при комнатной температуре в течение 2 ч. Смесь промывают разбавленным раствором гидроксида натрия, водой и затем рассолом.
Органический слой концентрируют в вакууме и остаток распределяют между светлым петролейным эфиром и водой.
Органический слой промывают водой, сушат над сульфатом магния, фильтруют и
удаляют растворитель в вакууме получая смолу. Смолу растворяют в петролейном эфире и очищают с помощью флэш хроматографии, используя смесь 1:3 эфира и петролейного эфира в качестве элюента. Последний
дает (+)-1-[1-(2-хлорфенил)циклопропил] -6-метокси-2-метил-7-октадеканоилокси- 1,2,3,4-тетрагидроизохинолин (0,41 г), который имеет удельное вращение плоскости поляризации [α]D= +23,
33°.
Примеры MI.

Смесь соединения формулы VI, в которой OR3, R4 и G являются тем, что определено в табл. MI, и E является -(CH2)3- (a, г), метилйодида (b, г), безводного карбоната калия (c, г) и ацетона (d, мл) перемешивают при комнатной температуре в течение e ч. Смесь фильтруют и растворитель удаляют путем выпаривания. Остаток распределяют между этилацетатом и водой. Этилацетатный слой дает остаток, который обрабатывают как представлено в примечаниях ниже получая желаемое соединение формулы IV, в которой OR3, R4 и G являются тем, что определено в табл. MI, R2 является метилом и E является -(CH2)3-.
Примечания к таблице MI:
ND показывает, что температура плавления не определена.
MI1 Целевой остаток растирают в смеси 5:1 легкокипящего петролейного эфира (т. к. 60 - 80oC) и эфира. Продукт характеризуют с помощью перекристаллизации небольшого количества вещества из легкокипящего петролейного эфира. В табл. МI дана температура плавления перекристаллизованного образца.
MI2 Остаток из начальной реакции распределяют между эфиром и водой. Выпаривание эфирного слоя дает продукт, который характеризуют с помощью перекристаллизации маленького образца из легкокипящего петролейного эфира. В табл. MI дана температура плавления перекристаллизованного образца.
MI3 Продукт характеризуют с помощью преобразования небольшого количества вещества в его гидробромидную соль. Температура плавления этой соли дана в последнем столбце табл. MI.
MI4 Продукт характеризуют с помощью перекристаллизации небольшого количества вещества из петролейного эфира (т.к. 60-80oC). В табл. MI дана температура плавления перекристаллизованного образца.
MI5 Продукт характеризуют с помощью преобразования небольшого количества вещества в его оксалатную соль, температура плавления этой соли дана в последнем столбце табл. MI.
MI6 Остаток из начальной реакции распределяют между эфиром и водным раствором аммиака. Выпаривание эфирного слоя дает твердый продукт, который растворяют в пропан-2-оле (50 мл) и 48%-ном водном растворе бромистоводородной кислоты (20 мл). Выпаривание дает остаток, который сушат путем азеотропной отгонки с пропан-2-олом, затем растворяют в метаноле и полученный твердый продукт тритируют с пропан-2-олом, и кристаллизуют из эфира. Продукт в виде гидробромидной соли используют без дальнейшей очистки.
Примеры MF.

Смесь соединения формулы VI, в которой OR3, R4 и G являются тем, что определено в табл. MI, и E является -(CH2)3 - (a, г), 37 - 40%-ного водного раствора формальдегида (b, мл), цианборгидрида натрия (c, г) и ацетонитрила (d, мл), перемешивают в течение 15 мин. Добавляют ледяную уксусную кислоту для нейтрализации раствора и продолжают перемешивание в течение следующих 45 мин. Смесь концентрируют путем выпаривания и подщелачивают 2н. водным раствором гидроксида калия. Полученную смесь экстрагируют эфиром и эфирные экстракты сушат с водным раствором гидроксида калия. Продукт экстрагируют водным раствором и соляной кислотой. Кислый экстракт подщелачивают и экстрагируют эфиром. Эфирный экстракт дает остаток, который является соединением формулы IV, в которой значения OR3, R4 и G определены в табл. MF, E является - (CH2)3- и R2 является метилом.
Примечания к таблице MF:
MF1 Остаток используют без дальнейшей обработки. Его температура плавления дана в
последнем столбце табл. MF.
MF2 Остаток характеризуют с помощью преобразования небольшого количества вещества в его гидробромидную соль. Температура плавления гидробромидной соли дана в последнем столбце табл. MF.
MF3 Остаток преобразует в его гидробромидную соль, которую перекристаллизуют из пропан-2-ола. Температура плавления этой соли дана в последнем столбце табл. MF.
MF4 Продукт примера RB5 преобразуют в его свободное основание, которое используют в качестве исходного материала. Продукт в форме его свободного основания получают в виде смолы. Спектр ЯМР является совместимым с требуемой структурой.
Пример MF6. Смесь оксалатной соли 1-[1-(2-хлорфенил)циклобутил]-7-метокси-6-метил-1,2,3,4-тетрагидроизохинолина (4,42 г, получают по примеру RC12), метанола (87 мл) и 37-40%-ного водного раствора формальдегида (5,1 мл) охлаждают до 10oC и добавляют цианборгидрид натрия (2,64 г). Через 10 мин смесь оставляют нагреваться до комнатной температуры и перемешивают в течение 24 ч. Реакционную смесь концентрируют и остаток распределяют между этилацетатом и разбавленным водным раствором гидроксида натрия. Органическую фазу промывают водным раствором аммиака, затем сушат и концентрируют, получая 1-[1-(2-хлорфенил)циклобутил] -7-метокси-2,6-диметил-1,2,3,4-тетрагидроизохинолина (3,3 г).
Пример MF7. Смесь 1-[1-(2-хлорфенил)циклопропил] -7-метокси-1,2,3,4-тетрагидроизохинолина (2,5 г, получают по примеру RC10), 37 - 40%-ного водного раствора формальдегида (2,9 мл), цианборгидрид натрия (0,75 г) и ацетонитрила (100 мл) перемешивают в течение 15 мин. Добавляют ледяную уксусную кислоту и продолжают перемешивать в течение следующих 45 мин. Смесь подщелачивают водным раствором гидроксида натрия, затем экстрагируют этилацетатом. Экстракты дают 1-[1-(2-хлорфенил)-циклопропил]-7-метокси-1,2,3,4-тетрагидроизохинолина.
Примеры RB.

Соединение формулы VII, в которой OR5 является группой OR3, как она определена в табл. RB, и R4, E и G является тем, что определено в табл. RB (a, г), в метаноле (b, мл) обрабатывают боргидридом натрия (c, г), который добавляют порциями при перемешивании. Когда тонкослойная хроматография показывает, что восстановление в основном закончено, реакционную смесь концентрируют, добавляют воду и полученную в результате смесь экстрагируют растворителем, указанным в столбце d (a - этилацетат, b - эфир или c - дихлорметан). Экстракты сушат и растворитель удаляют путем выпаривания, получая остаток, который удаляют путем выпаривания, получая остаток, который обрабатывают как указано в примечаниях к табл. RB, получая соединение формулы VI, в которой OR3, R4, E и G являются тем, что определено в табл. RB.
Примечания к
табл. RB:
Сокращение OBz обозначает бензилокси. В столбце E табл. RB W обозначает -CH2CMeCH2-.
RB1 Остаток перекристаллизуют из смеси этилацетат/петролейный эфир, получая желаемый продукт в виде свободного основания, температура плавления которого дана в последнем столбце табл. RB.
RB2 Остаток характеризуют путем преобразования его порции в его оксалатную соль, температура плавления которой дана в последнем столбце табл. RB.
RB3 Остаток очищают путем получения гидрохлоридной соли. Температура плавления этой соли дана в последнем столбце табл. RB.
RB4 Остаток используют в качестве исходного материала для следующей стадии без обработки.
RB5 Остаток характеризуют путем преобразования небольшого количества вещества в гидрохлоридную соль, температура плавления которой дана в последнем столбце табл. RB.
RB6 Реакционную смесь фильтруют и объем ее сокращают наполовину. Желаемый продукт осаждают и собирают путем фильтрования. Температура плавления дана в последнем столбце табл. RB.
RB7 Остаток очищают с помощью флэш хроматографии, получая желаемый продукт, который используют без дальнейшей обработки.
RB8 Продукт перекристаллизуют из петролейного эфира (т.пл. 60-80oC).
RB9 Остаток от реакционной смеси освобождают от триметилбората с помощью азеотропной отгонки с метанолом и распределяют между водой и дихлорметаном. Экстракт дает желаемый продукт, который используют без дальнейшей обработки.
RB10 Желаемый продукт осаждают из реакционной смеси при охлаждении. Продукт промывают метанолом и сушат на воздухе. Его температура плавления дана в последнем столбце табл. RB.
RB11 Добавляют воду к реакционной смеси и желаемый продукт осаждают при охлаждении. Его температура плавления дана в последнем столбце табл. RB.
RB12 Реакционную смесь подкисляют 5н. соляной кислотой и полученный в результате твердый продукт собирают путем фильтрования. Твердый продукт подщелачивают водным раствором гидроксида натрия и полученную смесь экстрагируют эфиром. Эфирный экстракт дает желаемый продукт, который используют без дальнейшей очистки.
Примеры RC.

Раствор соединения формулы VII, в которой OR5 является группой OR3, как она определена в табл. RC, и R4, E и G являются такими, как определено в табл. RC (a, г, получают по примеру, определенному в столбце SM табл. RC) в ледяной кислоте (b, мл) и метаноле (c, мл) при 0oC обрабатывают цианборгидратом (d, г). Смесь перемешивают при комнатной температуре, получая соединения формулы VI, в которой OR3, R4, E и G являются такими, как определено в табл. RC. Реакционную смесь обрабатывают как описано в примечаниях к табл. RC.
Примечания к табл. RC:
RC1 Объем реакционной смеси сокращают путем выпаривания и
остаток распределяют между водой и дихлорметаном. Органический слой промывают с концентрированным водным раствором аммиака и рассолом и затем сушат. Удаление растворителя дает остаток, который
используют без дальнейшей очистки.
RC2 Реакционную смесь выливают в воду и полученную в результате смесь экстрагируют дихлорметаном. Экстракт промывают рассолом и сушат. Удаление растворителя дает твердый продукт, который растирают в петролейном эфире (т. к. 60 - 80oC), перекристаллизуют из этанола и затем растворяют в дихлорметане. Раствор промывают концентрированным водным раствором аммиака и сушат. Удаление растворителя дает желаемый продукт.
RC3 Объем реакционной смеси сокращают путем выпаривания и остаток распределяют между водой и дихлорметаном. Органический слой промывают с концентрированным водным раствором аммиака и рассолом и затем сушат. Удаление растворителя дает остаток, который растирают в смеси легкокипящего петролейного эфира и эфира, получая желаемый продукт, температура плавления которого равна 100 - 102oC.
RC4 Объем реакционной смеси сокращают путем выпаривания и остаток распределяют между водой и дихлорметаном. Органический слой промывают с концентрированным водным раствором аммиака и рассолом, сушат, обесцвечивают и затем удаляют растворитель, получая сироп, который кристаллизуется при стоянии. Продукт промывают эфиром и сушат (т. пл. 109 - 111oC).
RC5 Реакционную смесь разбавляют дихлорметаном и полученный в результате органический слой промывают с концентрированным водным раствором аммиака и сушат. Выпаривание дает желаемый продукт в виде сиропа, который используют в следующей стадии без дальнейшей очистки.
RC6 Реакционную смесь выливают в воду и экстрагируют дихлорметаном. Органический слой промывают концентрированным водным раствором аммиака, а затем сушат. Высушенный органический слой дает желаемый продукт при выпаривании, который используют в следующей стадии без дальнейшей очистки.
RC7 Воду добавляют к реакционной смеси и смесь подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает желаемый продукт, который используют без дальнейшей очистки.
RC8 Реакционную смесь добавляют к смеси лед/вода, подщелачивают водным раствором аммиака и экстрагируют эфиром. Добавляют эфирный раствор щавелевой кислоты, получая желаемый продукт в виде оксалатной соли, которую сушат при 55oC в вакууме.
RC9 Через 24 ч детектируется непрореагировавший исходный материал. Добавляют следующую порцию цианборгидрида натрия (0,22 г) и перемешивают в течение 2 ч. Реакционную смесь распределяют между дихлорметаном и водой. Органический слой промывают концентрированным водным раствором аммиака и получают желаемый продукт, который используют без дальнейшей очистки.
RC10 Через 24 ч детектируется непрореагировавший исходный материал. Добавляют следующую порцию цианборгидрида натрия (0,2 г) и перемешивают в течение 24 ч. Реакционную смесь распределяют между дихлорметаном и концентрированным водным раствором аммиака. Органический слой дает желаемый продукт, который используют без дальнейшей очистки.
RC11 Реакционную смесь сначала перемешивают при 0 - 5oC в течение 2 ч и затем перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь выливают в смесь льда и воды, которую подщелачивают путем добавления концентрированного водного раствора аммиака и экстрагируют дихлорметаном. Экстракт дает желаемый продукт, который используют без дальнейшей очистки.
RC12 Реакционную смесь выливают в воду, подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Экстракт дает смолу, которую растворяют в эфире и обрабатывают эфирным раствором щавелевой кислоты, получая желаемый продукт в виде оксалатной соли, которую сушат в вакууме.
RC13 Реакционную смесь выливают в водный раствор аммиака и экстрагируют этилацетатом. Экстракты промывают водным раствором аммиака и рассолом, затем сушат и концентрируют, получая желаемый продукт, который используют без дальнейшей очистки.
RC14 Объем реакционной смеси сокращают путем выпаривания и остаток распределяют между дихлорметаном и концентрированным водным раствором аммиака. Органический слой сушат, фильтруют и выпаривают в вакууме, получая масло, которое растворяют в пропан-2-оле и обрабатывают 48%-ным водным раствором бромистоводородной кислоты. Выпаривание дает твердую гидробромидную соль, которую перекристаллизуют из эфира.
RC15 Реакционную смесь сначала перемешивают при 0 - 5oC в течение 2 ч, а затем перемешивают при комнатной температуре в течение 24 ч. Реакционную смесь выливают в смесь льда и воды, которую подщелачивают путем добавления концентрированного водного раствора аммиака и экстрагируют дихлорметаном. Экстракты дают остаток, который растворяют в пропан-2-оле (50 мл). Добавляют 48%-ный водный раствор бромистоводородной кислоты (25 мл). Смесь сушат с помощью азеотропной отгонки с пропан-2-олом и кристаллизуют из эфира, получая желаемый продукт в виде гидробромидной соли (т.пл. 227 - 233oC).
RC16 Реакционную смесь обрабатывают способом, подобным способу, описанному в примечании RC6, но продукт растворяют в пропан-2-оле и обрабатывают 48%-ным водным раствором бромистоводородной кислоты. Раствор охлаждают и снимают со стенок полученный твердый продукт, который фильтруют и сушат, получая гидробромидную соль.
RC17 Реакционную смесь выливают в воду и экстрагируют дихлорметаном. Органический слой промывают разбавленным водным раствором аммиака, затем водой, затем сушат над сульфатом мания, фильтруют и концентрируют в вакууме. Остаток растворяют в эфире и пробулькивают хлористый водород. Полученный в результате осадок собирают путем фильтрования, промывают эфиром и сушат в вакууме, получая желаемый продукт в виде гидрохлоридной соли.
Примеры CA.

Смесь соединения формулы VII, в которой OR5, R4, E и G являются такими, как определено в табл. CA (a, г, получают по примеру, указанному в столбце SM табл. CA), оксихлорида фосфора (b, мл) и ацетонитрила (c, мл) нагревают с обратным холодильником. После нагревания в течение d ч смесь подщелачивают водным раствором аммиака и экстрагируют растворителем, определенным в столбце f табл. CA (a - этилацетат, b - дихлорметан, c - эфир). Экстракт дает остаток, который обрабатывают как описано в примечаниях к табл. CA, получая соединения формулы VII, в которых OR3, R4, E и G являются тем, что определено в табл. CA.
Примечания к табл. CA:
Сокращение OBz обозначает бензилокси. В столбце E табл. CA W обозначает -CH2CMeCH2-.
CA1 Остаток используют в качестве исходного материала для следующей стадии без дальнейшей очистки. Температура плавления дана в последнем столбце табл. CA.
CA2 Остаток перекристаллизуют из петролейного эфира (т.пл. 60 - 80oC), получая желаемый продукт, температура плавления которого дана в последнем столбце табл. CA.
CA3 Остаток перекристаллизуют из пропан-2-ола, получая желаемый продукт, температура плавления которого дана в последнем столбце табл. CA.
CA4 Остаток используют без определения физико-химических характеристик.
CA5 Остаток обрабатывают смесью 2:1 пропан-2-ола и эфира, получая продукт в виде бледно-желтого твердого продукта, температура плавления которого дана в последнем столбце табл. CA.
CA6 Реакционную смесь выливают в смесь лед/вода, подщелачивают водным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает остаток, который вводят в горячую смесь эфира и циклогексана. Твердый продукт, который перекристаллизуют из пропан-2-ола, осаждают при охлаждении, получая желаемый продукт, температура плавления которого дана в последнем столбце табл. CA.
CA7 Остаток обрабатывают холодным пропан-2-олом. Желаемый продукт собирают путем фильтрования. Температура плавления которого дана в последнем столбце табл. CA.
CA8 Остаток кристаллизуют из метанола. Температура плавления желаемого продукта дана в последнем столбце табл. CA.
CA9 Остаток обрабатывают эфиром и полученную в результате смесь фильтруют. Желаемый продукт получают из фильтрата и используют без дальнейшей очистки.
CA10 Остаток растирают с петролейным эфиром (т.к. 40 - 60oC), получая желаемый продукт, температура плавления которого приведена в последнем столбце табл. CA.
CA11 Остаток обрабатывают ацетонитрилом. Твердый продукт отделяют путем фильтрования и фильтрат концентрируют, получая смолу, которую очищают с помощью флэш хроматографии. Образец полученного в результате продукта перекристаллизуют из пропан-2-ола, получая желаемый продукт, температура плавления которого дана в табл. CA.
CA12 Остаток тритируют в петролейном эфире (т.к. 60 - 80oC), получая желаемый продукт, температура плавления которого дана в последнем столбце табл. CA.
CA13 Остаток тритируют в петролейном эфире (т.к. 60 - 80oC) и перекристализуют из пропан-2-ола.
CA14 Остаток очищают с помощью флэш хроматографии, получая твердый продукт, температура плавления которого дана в табл. CA.
CA15 Остаток экстрагируют кипящим петролейным эфиром (т.к. 60 - 80oC). Экстракт дает остаток, который перекристаллизуют из пропан-2-ола, получая желаемый продукт, температура плавления которого указана в последнем столбце табл. CA.
CA16 Остаток обрабатывают смесью 1:3 эфира и пропан-2-ола, получая желаемый продукт в виде твердого продукта, температура плавления которого дана в табл. CA.
CA17 Остаток кристаллизуют из этанола. Образец (1 г) перекристаллизуют из этанола, получая твердый продукт, температура плавления которого дана в последнем столбце табл. CA.
CA18 Экстракт промывают разбавленным водным раствором соляной кислоты. Продукт подщелачивают и экстрагируют этилацетатом. Экстракт дает желаемый продукт, который используют без дальнейшей очистки.
CA19 Экстракт промывают рассолом, сушат над сульфатом магния, обесцвечивают древесным углем, фильтруют и растворитель удаляют путем выпаривания, получая твердый продукт. Твердый продукт перекристаллизуют из пропан-2-ола, получая 7-бензилокси-6-метокси-[1-(2-метилтиофенил)циклобутил] - дигидроизохинолин.
CA20 После нагрева в течение 20 ч реакционную смесь выливают в смесь лед/вода и подщелачивают концентрированным водным раствором аммиака. Щелочной раствор экстрагируют этилацетатом и экстракты промывают рассолом, сушат над сульфатом магния и фильтруют. Растворитель удаляют в вакууме, получая смолу, которую используют без дальнейшей очистки.
CA21 После нагрева в течение d ч растворитель удаляют в вакууме и остаток растворяют в этилацетате. Добавляют небольшой избыток водного раствора аммиака. Органический слой дает смолу, которую используют без дальнейшей очистки.
CA22 Экстракт промывают разбавленным водным раствором гидроксида натрия и рассолом. Экстракт дает остаток, который используют без дальнейшей очистки.
Пример CA37. Смесь N-[2-(2-метокси-5-бифенилилэтил] -1-(2- хлорфенил)циклобутанкарбоксамида (12,5 г, получают по примеру E30), оксихлорида фосфора (25 мл) и ацетонитрила (150 мл) нагревают с обратным холодильником в течение 6 ч, растворитель удаляют путем отгонки и остаток добавляют к смеси льда и воды. Смесь экстрагируют дихлорметаном, получая твердый продукт, который перекристаллизуют из пропан-2-ола, получая 1-[1-(2-хлорфенил)циклобутил] -7-метокси-6- фенил-3,4-дигидроизохинолин, т. пл. 149 - 152oC.
Пример CP. Смесь N-[2-(4-метоксифенил)этил]-1-(2-хлорфенил)циклопропанкарбоксамида (2 г, получают способом, подобным способу по примеру D8) и полифосфатэфира (20 мл) осторожно нагревают в течение 12 ч. Смесь выливают в воду и промывают эфиром, затем этилацетатом. Водную фазу подщелачивают водным раствором аммиака и экстрагируют этилацетатом. Экстракт дает твердый продукт, который перекристаллизуют из циклогексана, получая 1-[1-(2-хлорфенил)циклопропил]-7-метокси-3,4-дигидроизохинолин.
Пример CQ. Смесь N-[2-(3-фтор-4-метоксифенил)этил] -1-(2,4- дихлорфенил)циклопропанкарбоксамида (19,7 г, получают способом, подобным способу по примеру E4), 52% полифосфат эфира в хлороформе (200 г) нагревают с обратным холодильником в течение 5 ч, затем охлаждают и выливают на лед. Органический слой отделяют, водный слой экстрагируют дихлорметаном и объединенные органические слои подщелачивают путем добавления водного раствора аммиака, промывают рассолом, сушат над сульфатом магния и фильтруют. Растворитель удаляют в вакууме, получая твердый продукт, который растирают в смеси петролейного эфира (т.к. 40 - 60oC) с пропан-2-олом. Твердый продукт собирают путем фильтрования и сушат, получая 1-[1-(2,4-дихлорфенил)циклопропил]-6-фтор-7-метокси- 3,4-дигидроизохинолин (10,3 г), т. пл. 151 - 154oC.
Примеры CT.

Смесь соединения формулы XII, в которой OR5, R4, E и G являются такими, как указано в табл. CT (a, г, получено по примеру, указанному в столбце SM табл. CT), оксихлорида фосфора (b, мл) и толуола (c, мл) нагревают на паровой бане в течение d ч. Смесь выливают в смесь лед/вода, подщелачивают концентрированным водным раствором аммиака и экстрагируют растворителем, указанным в столбце e табл. CT (a - этилацетат, b - эфир). Экстракт дает остаток, который обрабатывают как описано в примечаниях к табл. CT, получая соединения формулы VII, в которой OR5, R4, E и G являются тем, что указано в табл. CT.
Примечания к табл. CT:
Сокращение
OBz обозначает бензилокси. В столбце E табл. CT W обозначает -CH2CMeCH2-.
CT1 Остаток обрабатывают смесью эфира и петролейного эфира. Полученный в результате твердый продукт собирают путем фильтрования. Его температура плавления дана в последнем столбце табл. CT.
CT2 Остаток кристаллизуют из смеси 4:1:2 эфира, этилацетата и петролейного эфира, получая желаемый продукт, температура плавления которого дана в последнем столбце табл. CT.
CT3 Остаток используют в следующей стадии без дальнейшей очистки. Температура плавления дана в последнем столбце табл. CT.
CT4 Толуол удаляют из реакционной смеси путем выпаривания и остаток выливают в смесь лед/вода. Раствор подщелачивают водным раствором аммиака и экстрагируют эфиром. Экстракт дает желаемый продукт, температура плавления которого дана в последнем столбце табл. CT.
CT5 Остаток кристаллизуют из эфира, получая желаемый продукт. Температура плавления дана в последнем столбце табл. CT.
СТ6 Остаток кристаллизуют из смеси эфира и петролейного эфира. Полученный в результате твердый продукт собирают путем фильтрования. Его температура плавления дана в последнем столбце табл. СТ.
СТ7 Остаток используют в следующей стадии без дальнейшей очистки.
СТ8 Остаток очищают с помощью флэш хроматографии, получая желаемый продукт. Температура плавления дана в последнем столбце табл. СТ.
СТ9 Остаток обрабатывают смесью эфира и петролейного эфира. Полученный в результате твердый продукт собирают путем фильтрования. Продукт используют без дальнейшей очистки.
СТ10 Остаток помещают в эфир и раствор сушат. Добавляют эфирный раствор щавелевой кислоты, получая оксалатную соль, которую промывают эфиром и распределяют между водным раствором аммиака и эфиром. Эфирный слой дает желаемый продукт в виде его свободного основания, которое используют без дальнейшей очистки.
СТ11 Остаток помещают в эфир и добавляют эфирный раствор щавелевой кислоты, получая оксалатную соль, которую промывают эфиром, подщелачивают 30% водным раствором гидроксида калия и экстрагируют эфиром. Экстракт дает масло, которое используют без дальнейшей очистки.
Примеры D.

1-Арилциклоалканкарбонилхлорид формулы XV, в которой E и G являются тем, что указано в табл. D (a, г, получено по примеру, указанному в столбце SM табл. D), добавляют по каплям при 0oC к перемешиваемому раствору фенэтиламина формулы XVII, в которой OR5 и R4 являются тем, что указано в табл. D (b, г) и триэтиламина (c, мл) в эфире (d, мл), образуя соединение формулы XII. Через 16 ч реакционную смесь выливают в воду и экстрагируют растворителем, определенным в столбце e табл. D (a - этилацетат, b - эфир, c - дихлорметан). Экстракт дает остаток, который обрабатывают как указано в примечаниях (Nb) к табл. D.
Примечания (Nb) к табл. D:
D1 Остаток используют без
дальнейшей очистки. Его температура плавления дана в последнем столбце табл. D.
D2 Остаток промывают петролейным эфиром (т.к. 60 - 80oC). Его температура плавления дана в последнем столбце табл. D.
D3 Остаток является стеклообразной массой, температура плавления которой не определена.
D4 Эфир в реакционной смеси заменяют дихлорметаном и реакционную смесь выливают в воду, и подкисляют концентрированной соляной кислотой. Выпаривание дихлорметанового слоя дает остаток, который промывают этанолом и сушат в вакууме. Его используют без дальнейшей очистки. Его температура плавления дана в табл. D.
D5 Реакционную смесь перемешивают в течение 2 дней и затем выливают в воду. Смесь подщелачивают, перемешивают в течение 30 мин, подкисляют, промывают эфиром, подщелачивают и экстрагируют этилацетатом. Экстракт дает желаемый продукт, который используют без дальнейшей очистки.
Примеры E.

Раствор 1-арилциклоалканкарбонилхлорида формулы XV, в которой E и G являются тем, что указано в табл. E (a, г, получают по примеру, указанному в столбце SM табл. E), в эфире (b, мл) добавляют по каплям к перемешиваемому раствору фенэтиламина формулы XVII, в которой OR5 и R4 являются тем, что указано в табл. E (c, г), и триэтиламина (d, мл), в эфире (e, мл), получая соединение формулы XII. Когда реакция завершается, добавляют воду и продукт отделяют как описано в примечаниях к табл. E.
Примечания к табл. E:
Сокращение OBz обозначает бензилокси. В столбце E табл. E W обозначает -CH2CMe2CH2-.
E1 Остаток, который получают путем выпаривания органического слоя реакционной смеси, тритируют в петролейном эфире и затем используют в качестве исходного материала для следующей стадии без дальнейшей очистки.
E2 Остаток, который получают путем выпаривания органического слоя реакционной смеси, используют без дальнейшей очистки.
E3 Остаток, который получают путем выпаривания органического слоя реакционной смеси, перекристаллизуют из пропан-2-ола.
E4 Остаток, который получают путем выпаривания органического слоя реакционной смеси, обрабатывают смесью эфира и петролейного эфира, и собирают путем фильтрования. Температура плавления дана в последнем столбце табл. E.
E5 Раствор фенэтиламина добавляют к раствору карбонилхлорида. Реакционная смесь дает осадок, который собирают путем фильтрации, промывают эфиром и водой, и сушат в вакууме, получая желаемый продукт.
E6 Раствор фенэтиламина добавляют к раствору карбонилхлорида. Продукт выделяют из органической фазы и используют без дальнейшей очистки.
E7 Температура плавления продукта дана в последнем столбце табл. E.
E8 Остаток, который получают путем выпаривания органического слоя реакционной смеси, тритируют в петролейном эфире получая желаемый продукт, температура плавления которого дана в последнем столбце табл. E.
E9 Раствор фенэтиламина добавляют к раствору карбонилхлорида. Продукт выделяют из органической фазы, и его температура плавления дана в последнем столбце табл. E.
E10 Реакцию проводят в атмосфере азота. После добавления воды реакционную смесь экстрагируют этилацетатом и желаемый продукт выделяют из органической фазы, и перекристаллизуют из смеси этилацетата и петролейного эфира. Его температура плавления дана в последнем столбце табл. E.
E11 Продукт, который осаждается после добавления воды, собирают, промывают водой и сушат в вакууме. Его температура плавления дана в последнем столбце табл. E.
E12 Фенэтиламиновый исходный материал получают по примеру P.
E13 После добавления воды реакционную смесь экстрагируют этилацетатом и желаемый продукт выделяют из органического слоя, и используют без дальнейшей очистки.
E14 Фенэтиламиновый исходный материал получают по примеру Q. Водную смесь экстрагируют этилацетатом. Органическую фазу промывают водой, 2н. соляной кислотой, водой и 2н. водным раствором гидроксида натрия. Экстракт дает остаток, который перекристаллизуют дважды из пропан-2-ола, получая желаемый продукт, температура плавления которого дана в последнем столбце табл. E.
E15 После воды добавляют этилацетат и желаемый продукт получают из органической фазы, и обрабатывают смесь эфира и петролейного эфира. Продукт используют без дальнейшей очистки.
E16 После добавления воды реакционную смесь экстрагируют этилацетатом и органическую фазу промывают 1н. соляной кислотой, водой, затем 1н. водным раствором гидроксида натрия, сушат над сульфатом магния и удаляют растворитель, получая желаемый продукт, который используют без дальнейшей очистки.
E17 После добавления воды смесь перемешивают в течение 30 мин, затем органический слой отделяют, промывают разбавленным водным раствором гидроксида натрия, разбавленным водным раствором соляной кислоты и затем рассолом, и фильтруют, фильтрат дает смолу, которую очищают с помощью колоночной хроматографии, используя смесь 1:3 этилацетата и петролейного эфира в качестве элюента, получая желаемый продукт.
E18 После добавления воды смесь перемешивают в течение 1 ч, затем органический слой отделяют и водный слой экстрагируют эфиром. Объединенные органические слои промывают разбавленным водным раствором соляной кислоты, разбавленным водным раствором гидроксида натрия, рассолом и затем сушат над сульфатом магния и фильтруют. Растворитель удаляют из фильтрата в вакууме, получая твердый продукт, который растирают в петролейном эфире (т.к. 60 - 80oC) и собирают путем фильтрования, получая желаемый продукт.
E19 После добавления воды органический слой отделяют, а водный экстрагируют эфиром. Объединенные органические слои промывают разбавленным водным раствором соляной кислоты, разбавленным водным раствором гидроксида натрия и затем сушат над сульфатом магния и фильтруют. Растворитель удаляют из фильтрата в вакууме, получая смолу, которую используют без дальнейшей очистки.
Пример E44. Раствор 1-фенилциклобутанкарбонилхлорида (129 г) в дихлорметане (500 мл) добавляют в азоте за 1,5 ч к перемешиваемой смеси 4-метоксифенэтиламина (99,5 г), дихлорметана (500 мл) и триэтиламина (110 мл). Смесь перемешивают при 20 - 25oC в течение 1 ч и оставляют стоять в течение 16 ч. Реакционную смесь промывают 2н. соляной кислотой, водой и 2н. водным раствором гидроксида натрия, сушат и выпаривают, получая N-[2-(4-метоксифенил)этил]-1-фенилциклобутанкарбоксамид.
Пример E45. Раствор 1-(1-нафтил)циклопропанкарбонилхлорида (7,1 г, получают по примеру CL32) в этилацетате (50 мл) добавляют к перемешиваемой смеси 4-бензилокси-3-метоксифенэтиламина гидрохлорида (9,29 г) в этилацетате (150 мл). Добавляют триэтиламин (25 мл) и перемешивают смесь следующие 64 ч. Органический слой промывают разбавленной соляной кислотой, сушат над сульфатом натрия и растворитель удаляют в вакууме, получая N-[2-(4-бензилокси-3-метоксифенил)-этил]-1-(1-нафтил) циклопропанкарбоксамид (14,1 г).
Пример E46. Циклобутанкарбонилхлорид (19 г) добавляют по каплям при комнатной температуре к перемешиваемому раствору 3,4-диметоксифенэтиламина (1 л). Через 1,5 ч смесь выливают в разбавленную соляную кислоту (1 л) и продукт экстрагируют, получая N-[2-(3, 4-диметоксифенил)-этил]циклобутанкарбоксамид (44 г), который используют без дальнейшей очистки.
Примеры CL.

1-Арилциклоалканкарбонилхлориды формулы XV получают путем нагревания 1-арилциклоалканкарбоновых кислот формулы XVIII (a, г, получают по примеру, указанному в столбце SM табл. CL) и тионил хлорида (b, г) нагревают с обратным холодильником в течение c ч. Требуемый продукт получают путем отгонки при пониженном давлении. Температура кипения дана в последнем столбце табл. CL. Там, где 1-арилциклобутанкарбоновая кислота является коммерчески доступной, это указано с помощью "K" в столбце SM.
В табл. CL W обозначает -CH2CMe2-CH2-.
Пример CL31. 1-(2-Метилтиофенил)циклобутанкарбоновую кислоту (12,1 г, получают по примеру H15) нагревают с обратным холодильником в течение 1 ч с тионилхлоридом (12 мл). Избыток тионилхлорида удаляют путем отгонки и остаток используют без дальнейшей очистки.
Пример CL32. 1-(1-Нафтил)циклопропанкарбоновую кислоту (6,7 г, получают способом, подобным способу по примеру H20) добавляют к тионилхлориду (25 мл) и смесь нагревают при 90 - 95oC в течение 1 ч. Избыток тионилхлорида удаляют в вакууме, получая 1-(1-нафтил)циклопропанкарбонилхлорид (7,1 г), который используют без дальнейшей очистки.
Примеры H.

Смесь 1-арилциклоалканкарбонитрила формулы XIV (a, г, получают по примеру, указанному в столбце SM табл. H), раствор гидроксида калия (b, г) либо в воде (c, мл), либо в этиленгликоле (d, мл) нагревают с обратным холодильником до тех пор, пока реакция не завершится. Подкисление реакционной среды дает требуемую 1-арилциклоалканкарбоновую кислоту, температура плавления которой дана в последнем столбце табл. H.
Примечания к табл. H:
Там, где исходный карбонитрил является известным, это отмечено K в столбце M.
W обозначает -CH2-CMe2CH2-.
ND показывает, что температура плавления не определена.
H1 Продукт перекристаллизуют из смеси этилацетата и петролейного эфира (т.к. 60 - 80oC).
H2 Охлажденный реакционный раствор промывают этилацетатом и фильтруют. Фильтрат подкисляют и экстрагируют этилацетатом. Экстракт дает твердый продукт, который перекристаллизуют из петролейного эфира (т.к. 60-80oC).
H3 Подкисленную реакционную смесь экстрагируют эфиром, экстракты промывают водой и рассолом, затем сушат над сульфатом магния и концентрируют. Остаток перекристаллизуют из петролейного эфира (т.к. 60 - 80oC).
H4 Реакционную смесь выливают в воду, подкисляют и экстрагируют этилацетатом, получая желаемый продукт.
H5 Реакционную смесь выливают в воду, подкисляют и экстрагируют этилацетатом, и получают смолу, которая отвердевает при стоянии. Этот твердый продукт растворяют в 5N водном растворе гидроксида натрия, перемешивают в течение 30 мин, затем промывают этилацетатом. Водную фазу раскисляют и полученный в результате твердый продукт собирают путем фильтрования и сушат на воздухе.
Пример H18. Смесь 1-(4-метоксифенил)циклобутанкарбонитрила (41 г), гидроксида калия (20 г) и диэтиленгликоля (160 г) нагревают с обратным холодильником в течение 3,5 ч. Смесь добавляют в воду, подкисляют разбавленной соляной кислотой и экстрагируют эфиром. Экстракт дает 1-(4-метоксифнил)циклобутанкарбоновую кислоту.
Пример H19. Раствор гидроксида натрия (2,8 г) в воде (4 мл) добавляют к перемешиваемой смеси 1-(3-трифторметилфенил)цикло бутанкарбонитрила (50 г) в техническом денатурате (350 мл). К этому добавляют перекись водорода (100 объемов, 105 мл) при температуре ≈ 40oC. Затем смесь нагревают при 50oC в течение 1 ч, позволяют остыть подкисляют 5%-ной серной кислотой (≈ 40 мл) и концентрируют в вакууме. Остаток распределяют между водой и эфиром. Органические экстракты промывают водой, сушат (MgO4), фильтруют и выпаривают в вакууме, получая бесцветное масло (63 г), которое растворяют в диоксане (350 мл). Добавляют концентрированную соляную кислоту (75 мл) и охлаждают раствор до 5oC. Добавляют раствор нитрита натрия (27 г) в воде (60 мл) по каплям, все еще оставаясь при температуре ≈ 10oC. Затем смесь нагревают при 90 - 95oC в течение 1 суток. Смесь охлаждают и отделяют органический слой. Водный слой экстрагируют эфиром и объединенные органические слои сушат, фильтруют и концентрируют в вакууме, получая 1-(3-трифторметилфенил)циклобутанкарбоновую кислоту.
Пример H20. Смесь 1-(1-нафтил)циклопропанкарбонитрила (60,7 г, получают способом, подобным способу по примеру N9) и 10%-ного водного раствора гидроксида натрия перемешивают и нагревают с обратным холодильником в течение 5 ч. Каждый раз, когда достигается полное растворение, добавляют еще воды до помутнения. Добавляют воду и смесь оставляют стоять в течение 16 ч. Полученный в результате твердый продукт удаляют путем фильтрования. Фильтрат разбавляют водой (800 мл), фильтруют и фильтрат подкисляют путем добавления разбавленной соляной кислоты. Полученный в результате твердый продукт собирают путем фильтрования, промывают водой и сушат на воздухе, получая 1-(1-нафтил)циклопропанкарбоновую кислоту (25,9 г).
Примеры N.

Смесь арилацетонитрила формулы XX (a, г) и 1,3-дибромпропана (b, г) и либо эфира (c, мл), либо диметилсульфоксида (d, мл) добавляют по каплям к перемешиваемой суспензии порошкообразного гидроксида калия (e, г) в диметилсульфоксиде (f, мл). Реакционную смесь обрабатывают способами A, B, C, D или E.
Способ A. Смесь нагревают при 30 - 35oC в течение 3 ч (2 ч для примера N 3) и затем выливают в смесь лед/вода и концентрированной соляной кислоты при температуре ниже 15oC и фильтруют. Экстракт дает масло, которое отгоняют, получая желаемый 1-арилциклобутан ацетонитрил формулы XIV, температура кипения которого дана в табл. N.
Способ B. Смесь перемешивают при комнатной температуре в течение 16 ч и выливают в воду. Смесь экстрагируют этилацетатом. Экстракт дает масло, которое отгоняют, получая желаемый 1-арилциклобутанкарбонитрил формулы XIV, температура кипения которого дана в табл. N.
Способ C. Смесь перемешивают при комнатной температуре в течение 16 ч и выливают в воду. Полученный в результате осадок собирают путем фильтрования и растворяют в эфире. Фильтрат экстрагируют эфиром. Раствор и экстракты объединяют и получают масло, которое отгоняют, получая желаемый 1-арилциклобутанкарбонитрил формулы XIV, температура кипения которого дана в табл. N.
Способ D. Смесь перемешивают при комнатной температуре в течение 3 ч, затем при 30 - 35oC в течение 3 ч, затем выливают в смесь лед/вода и концентрированной соляной кислоты. Смесь промывают эфиром, экстракты промывают водой, сушат, фильтруют, растворитель выпаривают, получая желаемый 1-арилциклобутанкарбонитрил формулы XIV, температура кипения которого дана в табл. N.
Способ E. Исходный нитрил получают по примеру R. Смесь перемешивают при комнатной температуре в течение 1 ч и выливают в воду. Смесь экстрагируют этилацетатом. Экстракт дает масло, которое отгоняют. Фракцию с температурой кипения выше 160oC очищают с помощью флэш хроматографии, используя смесь 1:9 петролейного эфира и этилацетата в качестве элюента. Растворитель выпаривают, получая желаемый 1-арилциклобутанкарбонитрил формулы XIV.
Пример N8. Смесь фенилацетонитрила (47,4 г) и 1,3-дийод-2,2-диметилпропана (131,2 г) добавляют по каплям за 2 ч при 25oC к перемешиваемому раствору порошкообразного гидроксида калия (90,7 г) в сухом диметилсульфоксиде (600 мл) в азотной атмосфере. Смесь перемешивают в течение 1 суток, затем выливают в воду и экстрагируют эфиром (1000 мл). Экстракты обесцвечивают древесным углем, фильтруют и концентрируют в вакууме, получая оранжево-коричневое масло, которое отгоняют в вакууме, получая 2,2-диметил-1-фенилциклобутанкарбонитрил (т. к. 101 - 106oC/0,6 мбар) в виде бледно-желтого масла.
Пример N9. Раствор 1-нафтилацетонитрила (53 г) в 1-бром-2-хлорэтане (35,2 мл) добавляют за 35 мин к энергично перемешиваемой смеси бензилтриэтил хлорида аммония (2 г) и 50% вес/объем, водного раствора гидроксида натрия (190 мл) при примерно 70oC. Смесь нагревают при 75 - 80oC в течение 2 ч, затем добавляют 1-бром-2-хлорэтан (15 мл) и бензилтриэтил аммоний хлорид (1 г) и нагревание продолжают в течение 4,5 ч. После отстаивания в течение 16 ч добавляют 1-бром-1-хлорэтан (10 мл), бензилтриэтил аммоний хлорид (1 г) и твердый гидроксид натрия (20 г). Смесь перемешивают и нагревают при 75 - 80oC в течение 6 ч. Водный слой промывают эфиром и объединенные органические слои дают масло, которое отгоняют (т.к. 135oC/0,07 мбар), получая дистиллят, который отвердевает при охлаждении. Твердый продукт 1-(1-нафтил)- циклопропанкарбонитрил используют без дальнейшей очистки.
Пример N10. Раствор 4-трифторметоксифенилацетонитрила (38,9 г) и 1,3-дибромпропана (39,1 г) в тетрагидрофуране (50 мл) добавляют в аргоне к перемешиваемому раствору 50% гидрида натрия (19,2 г) в тетрагидрофуране (200 мл) и диметилформамиде (25 мл) при 25 - 30oC за 1 ч. Смесь перемешивают при 20oC в течение 1,5 ч, затем при 30 - 40oC в течение 1 ч, затем охлаждают и промывают эфиром. Органический слой промывают водой, сушат над сульфатом магния и растворитель удаляют путем выпаривания. Остаток отгоняют (т. к. 111 - 117oC/10 мбар), получая 1-(4-трифторметоксифенил)циклобутанкарбонитрил (30,98 г).
Пример N11. Смесь 2-хлорфенилацетонитрила (26,5 г), 1,3-дийод- 2,2-диметилпропана (56 г) и диметил сульфоксида (300 мл) добавляют по каплям к перемешиваемой суспензии порошкообразного гидроксида калия (40 г) в сухом диметил сульфоксиде (300 мл). Смесь перемешивают в течение 1 ч, затем выливают в смесь лед/вода и экстрагируют этилацетатом. Экстракты дают масло, которое отгоняют в вакууме, получая 2, 2-диметил-1-(2-хлорфенил)циклобутанкарбонитрил (т. к. 116 - 120oC/1 мбар), который перекристаллизуют из петролейного эфира (т.к. 60 - 80oC).
Пример N12. Смесь 2-хлорфенилацетонитрила (20 г), 1,4-дибромбутана (28,5 г) и диметил сульфоксида (100 мл) добавляют по каплям к перемешиваемой суспензии порошкообразного гидроксида калия (26 г) в сухом диметил сульфоксиде (300 мл). Смесь перемешивают в течение 1 ч, затем выливают в смесь лед/вода и экстрагируют этилацетатом. Экстракты дают масло, которое отгоняют в вакууме (т.к. 112 - 116oC/1 мбар), получая 1-(2-хлорфенил)циклопентанкарбонитрил (16,7 г).
Пример P. Метил йодид (110 г) добавляют по каплям к перемешиваемому раствору 1-хлор-3-метилфенола (100 г) и карбоната калия (194 г) в ацетоне (500 мл). Смесь перемешивают в течение 1,5 ч. Добавляют метилйодид (142 г) и перемешивают смесь в течение следующего часа. Растворитель удаляют путем выпаривания и остаток распределяют между водой и этилацетатом. Органический слой дает масло, которое нагревают с обратным холодильником в течение 2 ч с N-бромсукцинимидом (119 г) и бензоил пероксидом (1 г) в карбонтетрахлориде (250 мл). Смесь охлаждают, фильтруют и фильтрат концентрируют, получая остаток, который отгоняют в высоком вакууме. Фракции собирают между 88 и 94oC/0,4 мбар и перекристаллизуют из петролейного эфира (т.к. 60 - 80oC), получая 2-хлор-5-метоксибензил бромид, т.пл. 51 - 52oC.
Цианид натрия (10,4 г) добавляют порциями к перемешиваемому раствору указанного выше бензилбромида в смеси 2:1 этанола и воды (250 мл). Смесь перемешивают в течение 1 ч при 50oC, выливают в воду и экстрагируют эфиром. Экстракт дает 2-хлор-5-метоксифенилацетонитрил (т.пл. 62 - 65oC). Добавляют 10 М боран-метилсульфидный комплекс (11,3 мл) по каплям в атмосфере азота к нагреваемому с обратным холодильником раствору указанного выше фенилацетонитрила (18,6 г) в тетрагидрофуране (150 мл). Смесь нагревают с обратным холодильником в течение 2 ч и добавляют по каплям разбавленную соляную кислоту, и подкисленную смесь нагревают при 95oC в течение 1 ч. Охлажденную смесь промывают эфиром, подщелачивают разбавленным раствором гидроксида натрия и экстрагируют эфиром. Экстракт дает 2-хлор-5- метоксифенэтиламин в виде желтого масла.
Пример Q. Смесь четвертичного трифенилфосфина палладия (7 г), 3-бром-4-метоксифенэтиламина гидробромида (62,2 г) и толуола (400 мл) перемешивают в азоте с 2 М водным раствором карбоната натрия (200 мл) и затем добавляют раствор фенилборной кислоты (26,8 г) в этаноле (100 мл). Смесь нагревают с обратным холодильником в течение 48 ч, затем охлаждают и обрабатывают 30% водным раствором перекиси водорода (10 мл). Смесь перемешивают при комнатной температуре в течение 1 ч. Водный слой отделяют и экстрагируют эфиром. Эфирные экстракты объединяют с исходной органической фазой. Отгонка дает 4-метокси-3-фенилфенэтиламин, который используют без дальнейшей очистки.
Пример R. Трибромид фосфора (75 г) в толуоле (50 мл) добавляют к перемешиваемой смеси 2-метилтиобензилэтилового спирта (42,5 г) в толуоле (25 мл) за 10 мин при 5oC. Затем смесь перемешивают при 40oC в течение 1 ч, добавляют воду (100 мл) и продолжают перемешивать при комнатной температуре в течение 1 ч. Органический слой отделяют и водный слой экстрагируют этилацетатом. Объединенные органические слои дают масло. Это масло (58,6 г) растворяют в смеси 1:2 воды и этанола, и добавляют цианид натрия (26,5 г) за 15 мин. Смесь перемешивают при 50oC в течение 1,5 ч, затем выливают в воду (500 мл) и экстрагируют этилацетатом. Экстракт дает 2-метилтиофенилацетонитрил.
Пример 94. Использование соединений данного изобретения в производстве фармацевтических составов иллюстрируется с помощью следующего описания. В этом описании термин "активное соединение" означает любое соединение, которое является целевым продуктом одного из предыдущих примеров.
a). Капсулы.
При получении капсул 10 мас. ч. активного соединения и 240 мас. ч. лактозы измельчают и перемешивают. Смесь помещают в твердые желатиновые капсулы, каждая капсула содержит единичную дозу или часть единичной дозы активного соединения.
b). Таблетки.
Таблетки получают из следующих ингредиентов, мас. ч.:
Активное соединение - 10
Лактоза
- 190
Кукурузный крахмал - 22
Поливинилпиролидон - 10
Стеарат магния - 3
Активное соединение, лактозу и немного крахмала измельчают, перемешивают и полученную в
результате смесь гранулируют в растворе поливинил пиролидона в этаноле. Сухой гранулят смешивают со стеаратом магния и остатком крахмала. Затем смесь прессуют в машине для производства таблеток,
получая таблетки, каждая из которых содержит единичную дозу или часть единичной дозы активного соединения.
c). Таблетки с кишечным покрытием.
Таблетки получают по способу (b). Таблетки покрывают обычным способом, используя раствор 20% ацетатфталата целлюлозы и 3% диэтилфталата в смеси этанол:дихлорметан (1:1).
d). Суппозитории.
Для получения суппозиториев 100 мас. ч. активного соединения соединяют с 1300 мас. ч. триглециридной основы суппозиториев и смесь формуют в суппозитории, каждый из которых содержит терапевтически эффективное количество активного ингредиента.
Реферат
Тетрагидроизохинолиновые соединения I, где R1 - водород, галоген, гидрокси, C1-C3-алкил, необязательно замещенный гидрокси, алкокси, алкилтио, алкилсульфинил, алкилсульфонил, нитро, циано, полигалоидалкил, полигалоидалкокси, фенил или карбамоил, необязательно алкированный одной или двумя алкильными группами; R2 - С1-C3-алкил, необязательно замещенный гидрокси или алкокси; Е - С2-С5 -алкиленовая цепь, необязательно замещенная 1 или более алкильными группами; G - фенил или фенил, замещенный алкилом, алкокси, галогеном, гидрокси, полигалоидалкилом; полигалоидалкокси, циано, алкилтио, алкилсульфинил, алкилсульфонил, карбамоил, 1-нафтил, 2-нафтил или 2,3-дигидробензо [в] фуран-7-ил, или их производные O-ацилированные в 5, 6, 7 или 8 положении тетрагидроизохинолинового кольца. Фармацевтические составы на их основе могут использоваться в анальгезии и в лечении психозов (например, шизофрении), болезни Паркинсона, синдрома Леш-Ньяна, нарушения дефицита внимания или ослабления познавательной способности. 4 с. и 13 з.п.ф-лы, 15 табл.

Формула

или их фармацевтически приемлемые соли,
где R1 представляет собой один или более заместителей, которые выбирают из H, галогена, гидрокси, алкила с 1 - 3 атомами углерода, необязательно замещенного гидрокси, алкокси с 1 - 3 атомами углерода, алкилтио с 1 - 3 атомами углерода, алкилсульфинила с 1 - 3 атомами углерода, алкилсульфонила с 1 - 3 атомами углерода, нитро, циано, полигалоидалкила с 1 - 3 атомами углерода, полигалоидалкокси с 1 - 3 атомами углерода, фенила, необязательно замещенного одним или более заместителями, которые выбирают из галогена, алкила с 1 - 3 атомами углерода или алкокси с 1 - 3 атомами углерода, или R1 представляет собой карбамоил, необязательно алкилированный одной или двумя алкильными группами, каждая, независимо, с 1 - 3 атомами углерода,
R2 представляет собой алифатическую группу, содержащую 1 - 3 атомов углерода, необязательно замещенную гидрокси или алкокси группой, содержащей 1 - 3 атомов углерода;
E представляет собой алкиленовую цепь, содержащую 2 - 5 атомов углерода, необязательно замещенную одной или более алкильными группами, содержащими 1 - 3 атомов углерода;
G представляет собой фенил или фенил, замещенный одним или более заместителями, которые могут быть одинаковыми или различными, и которые, независимо, представляют собой алкил с 1 - 3 атомами углерода, алкокси с 1 - 3 атомами углерода, галоген, гидрокси, полигалоидалкил с 1 - 3 атомами углерода, полигалоидалкокси с 1 - 3 атомами углерода, циано, алкилтио с 1 - 3 атомами углерода, алкилсульфинил с 1 - 3 атомами углерода, алкилсульфонил с 1 - 3 атомами углерода, фенил, необязательно замещенный одним или более заместителями, которые выбирают из галогена, алкила с 1 - 3 атомами углерода или алкокси с 1 - 3 атомами углерода; карбамоил, необязательно алкилированный одной или двумя алкильными группами, независимо, каждая с 1 - 3 атомами углерода, или G представляет 1-нафтил, 2-нафтил или 2,3-дигидробензо-[b] фуран-7-ил,
или их производные 0 - ацилированные в 5, 6, 7 или 8 положении тетрагидроизохинолинового кольца.

где R1, R2, E и G принимают значения, указанные в любом из предыдущих пунктов или их производные 0-ацилированные в 5, 6, 7 или 8 положения тетрагидроизохинолинового кольца,
или их фармацевтически приемлемые соли.

где R1, R2, E и G принимают значения, указанные в любом из пп.1 - 9;
R7 представляет собой ацильную группу, производную от карбоновой кислоты, имеющей от 6 до 20 атомов углерода,
или их фармацевтически приемлемые соли.
1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гидрокси-6-фтор-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил]-7-гидрокси-2,6-диметил-1,2,3,4- тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гептоноилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-додеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-гексадеканоилокси-6-метокси-2-метил- 1,2,3, 4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-октадеканоилокси-6-метокси-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклопропил] -7-деканоилокси-6-фтор-2-метил- 1,2,3,4-тетрагидроизохинолин,
1-[1-(2-хлорфенил)циклобутил] -7-деканоилокси-6-фтор-2-метил- 1,2,3,4-тетрагидроизохинолин,
или их фармацевтически приемлемые соли в виде индивидуальных энантиомеров, рацематов или других смесей энантиомеров.
а) деалкилирование или дебензилирование соединения формулы IV
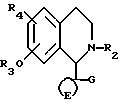
где R3 представляет собой необязательно замещенную алкильную группу;
R4 представляет собой группу R1 или группу, которая может быть превращена в группу R1, причем указанное деалкилирование проводят нагреванием соединения формулы IV, где R3 представляет собой необязательно замещенную алкильную группу, при температуре кипения с бромводородной кислотой, необязательно в присутствии ледяной уксусной кислоты, или с трибромидом бора, а упомянутое дебензилирование проводят кислотным гидролизом или гидрогенолизом соединения формулы IV, где R3 представляет собой бензил, с получением соединения формулы I;
б) необязательное ацилирование продукта стадии а) ацилирующим агентом с получением соединения формулы III, как определено в п.1.
Комментарии