Производные 5-гидразинохинолона, композиция для лечения или профилактики инфекционного заболевания, способ лечения или профилактики - RU2126000C1
Код документа: RU2126000C1
Описание
Это изобретение относится к новым противомикробным соединениям, композициям и способам лечения. В частности, соединения этого изобретения включают хинолон или близкий гетероциклический радикал.
В химической и медицинской литературе описано множество соединений, которые указываются как антибактериальные, то есть обладающие способностью разрушения или подавления роста или репродукции микроорганизмов, таких как бактерии. В частности, противобактериальные вещества включают большое разнообразие соединений естественного происхождения (антибиотики), синтетические или полу-синтетические. Они могут быть классифицированы (например) как аминогликозиды, ансамакролиды, бета-лактамы (включая пенициллины и цефалоспорины), линкозаминиды, макролиды, нитрофураны, олигосахариды, пептиды и полипептиды, феназины, полиены, полиэфиры, хинолоны, тетрациклины и сульфонамиды. Такие антибактериальные и другие противомикробные вещества описаны в Antibiotics, Chemotherapentics, and Antibacterial Agents for Disease Control (M. Grayson, editor, 1982) и E. Gale et al., The Molecular Basis of Antibiotic Action 2-d edition (1981), оба упоминаемых здесь в качестве ссылок.
Механизм действия этих антибактериальных веществ различный. Однако каждый может в основном быть классифицирован как функционирующий по одному или более из четырех путей: ингибирование синтеза или восстановления клеточных стенок; изменение проницаемости клеточных стенок; ингибирование синтеза протеина; или ингибирование синтеза нуклеиновых кислот. Например, бета-лактамовые антибактериальные вещества действуют посредством ингибирования избыточных связывающих пенициллин протеинов (ПСП) в бактериях, которые ответственны за синтез клеточных стенок. С другой стороны, хинолоны действуют путем ингибирования синтеза ДНК бактерии, таким образом препятствуя воспроизводству бактерий.
Не удивительно поэтому, что фармакологические характеристики антибактериальных и других противомикробных веществ и их пригодность для любого данного клинического применения также значительно изменяются. Например, классы противомикробных веществ (и члены внутри классов) могут изменяться по их относительной эффективности в отношении различных типов микроорганизмов и их восприимчивости к развитию устойчивости к микробам. Эти противомикробные вещества могут также отличаться своими фармакологическими характеристиками, таким как их биологическая доступность и биораспределение вещества после его введения в организм (поиск тканей-мишеней). Соответственно, выбор подходящего антибактериального вещества (или другого противимикробного вещества) в любой данной клинической ситуации может быть полным анализом многих факторов, включая тип организма, желательный способ введения и место локализации инфекции, на которую воздействуют.
Фармакологическая литература полна попыток разработок улучшенных противомикробных веществ (то есть, соединений, которые обладают более широким спектром активности, повышенной активностью, улучшенной фармакологией и/или пониженной чувствительностью к развитию невосприимчивости). Например, одна группа противомикробных веществ, которые были разработаны недавно для клинического применения, является хинолонами. Эти соединения включают, например, налидиксиновую кислоту, дифлоксацин, эноксацин, флероксацин, норфлоксацин, ломефлоксацин, офлоксацин, ципрофлоксацин и пефлоксацин. См. C. Marchbanks and M. Dubley, "New Fluoroguinolones", 7 Hospital Therapy 18 (1988); P. Shah, "Quinolones", 31 Prog. Drug Res. 243 (1987); Quinolones - Their Future in Clinical Practice, (A. Percival, editor, Royal Society of Medical Services, 1986); and M. Parry, "Pharmacology and Clinical Uses of Quinolone Antibiotics", 116 Medical Times 39 (1987).
Однако много других попыток по получению улучшенных противомикробных веществ привели к сомнительным результатам. В самом деле. Разработано некоторое число противомикробных веществ, которые являются достоверно клинически приемлемыми в отношении спектра их противомикробной активности, отсутствия склонности к микробной невосприимчивости и фармакологии. Например, хинолоны часто показывают пониженную эффективность в отношении некоторых клинических важных патогенов (например, грамположительных бактерий и/или анаэробных бактерий). Хинолоны также имеют ограниченную водную растворимость, в воде, ограничивающую их биологическую доступность и возможность парентерального дозирования. Они также могут иметь вредные побочные эффекты, такие как желудочно-кишечные расстройства и воздействие на центральную нервную систему (такое как конвульсии). См., M. Neuman and A. Esanu, "Gaps and Perspectives of New Fluoroguinolones", 24 Drugs Exptl. Clin. Res 385 (1988); W. Christ. et al., "Specific Toxicologic Aspects of the Quinolones", 10 Rev. Infections Diseases 5141 (1988); H. Neu, "Clinical Use of the Quinolones", Lancet 1319 (1987); and "Ciprofloxacin; Panacea or Blunder Drug?", J. South Carolina Med. Assoc 131 (March 1989).
Настоящее изобретение касается соединений общей структуры

где (A) (I) (a) R1 обозначает алкил; алкенил; карбоциклическое кольцо, гетероциклическое кольцо, или -N(R6) (R7), где R6 и R7, являются, независимо, водородом, алкилом, алкенилом, карбоциклическим кольцом, гетероциклическим кольцом, или R6 и R7 вместе образуют гетероциклическое кольцо, которое включает азот, к которому они присоединены;
(b) R2 обозначает водород, галоген, низший алкил или низший алкокси, или
(2) R1 и R2 вместе могут образовывать шестичленное гетероциклическое кольцо, которое включает N', и атом углерода, к которому присоединен R2;
(B) R3 обозначает гетероциклическое или карбоциклическое кольцо, и
(C) (I) R4 и R5 обозначают, независимо, водород, низший алкил, циклоалкил, гетероалкил, или - C(=O) - X - R8, где X обозначает ковалентную связь, N, O или S и R8 является низким алкилом, алкенилом, арилалкилом, карбоциклическим кольцом или гетероциклическим кольцом, и
(2) R4 и R5 вместе образуют гетероциклическое кольцо; которое включает азот, к которому они присоединены;
и их фармацевтически приемлемых солей, биогидролизуемых эфиров и сольватов.
Было также обнаружено, что соединения по данному изобретению и композиции, содержащие эти соединения, являются эффективными противомикробными агентами в отношении широкого спектра патогенных микроорганизмов. Авторы также обнаружили, что неожиданно, соединения по данному изобретению обладают значительно повышенной растворимостью в воде при физиологических pH по сравнению с близкими по строению противомикробными веществами, известными в данной области. Это неожиданное свойство может позволить, помимо других вещей, улучшенную фармакологию, включая повышенные уровни сыворотки при введении, простоту приготовления составов и более гибкий режим дозирования.
Настоящее изобретение охватывает некоторые новые хинолоны, способы их получения, дозированные формы и способы введения хинолонов человеку или другим животным, субъектам. Конкретные соединения и композиции для использования по изобретению должны быть, соответственно, фармацевтически приемлемыми. Как здесь используется, таким "фармацевтически приемлемым" компонентом является такой, который подходит для использования для человека и/или животных без нежелательных побочных эффектов (таких как токсичность, раздражение и аллергическая реакция), взятого с приемлемым соотношением польза/риск.
5-(N-Гетерозамещенный амино) хинолоны.
Соединения по этому изобретению, упоминаемые здесь как 5-(N-гетерозамещенный амино) хинолоны", охватывают любые варианты хинолонов (и близких гетероциклических радикалов), имеющих N-гетероаминозамещенный заместитель в 5-ом положении хинолоновой части.
5-(N-Гетерозамещенный амино) хинолоны по этому изобретению включают соединения общей структуры

где (A) (I) (a) R1 обозначает алкил; алкенил; карбоциклическое кольцо; гетероциклическое кольцо; или -N(R6) (R7) (предпочтительно алкил или карбоциклическое кольцо), где R6 и R7 являются, независимо, водородом, алкилом, алкенилом, карбоциклическим кольцом, гетероциклическим кольцом, или R6 и R7 вместе образуют гетероциклическое кольцо, которое включает азот, к которому они присоединены;
(b) R2 обозначает водород, галоген, низший алкил или низший алкокси (предпочтительно галоген); или
(2) (Предпочтительно) R1 и R2 могут вместе образовывать шестичленное гетероциклическое кольцо, которое включает N', и атом углерода, к которому присоединен (R2);
(B) R3 обозначает гетероциклическое или карбоциклическое кольцо (предпочтительно гетероциклическое кольцо); и
(C) (1) (R4 и R5 обозначают, независимо, водород; низший алкил; циклоалкил; гетероалкил; или -C(=O) - X - R8 (предпочтительно, водород или низший алкил), где X обозначает ковалентную связь, N, O или R8 является низшим алкилом, алкенилом, арилалкилом, карбоциклическим кольцом или гетероциклическим кольцом; и
(2) (Предпочтительно) R4 и R5 вместе образуют гетероциклическое кольцо, которое включает азот, к которому они присоединены;
и их фармацевтически приемлемых солей и биогидролизуемых эфиров и сольватов.
Определения и употребления терминов:
Далее перечислены определения для используемых здесь терминов.
"Гетероатом" является атомом азота, серы или кислорода. Группы, содержащие один или более гетероатомов, могут содержать различные гетероатомы.
"Алкилом" является незамещенный или замещенный насыщенный углеводородный цепочечный радикал, имеющий от 1 до 8 атомов углерода, предпочтительно от 1 до 4 атомов углерода. Предпочтительные алкильные группы включают (например) метил, этил, пропил, изопропил и бутил.
"Гетероалкилом" является незамещенный или замещенный насыщенный цепочечный радикал, имеющий от 3 до 8 атомов, содержащий атом углерода и один или два гетероатомов.
"Алкенилом" является незамещенный или замещенный углеводородный цепочечный радикал, имеющий от 2 до 8 атомов углерода, предпочтительно от 2 до 4 атомов углерода, и имеющий, по крайней мере, одну олефиновую двойную связь (включая, например, винил, аллил и бутенил).
"Карбоциклическим кольцом" является незамещенный или замещенный, насыщенный, ненасыщенный или ароматический углеводородный кольцевой радикал. Карбоциклический радикал является моноциклическим или конденсированным, мостиковым или спирополициклической кольцевой системой. Моноциклические кольца содержат от 3 до 9 атомов, предпочтительно от 3 до 6 атомов. Полициклические кольца содержат от 7 до 17 атомов, предпочтительно от 7 до 13 атомов.
"Циклоалкилом" является насыщенный карбоциклический кольцевой радикал. Предпочтительные циклоалкильные группы включают (например) циклопропил, циклобутил и циклогексил.
"Гетероциклическим кольцом" является незамещенный или замещенный, насыщенный, ненасыщенный или ароматический углеводородный кольцевой радикал, содержащий атомы углерода и один или более гетероатомов в кольце. Гетероциклический радикал является моноциклическим или конденсированным, мостиковым или спирополициклической кольцевой системой. Моноциклические кольца содержат от 3 до 9 атомов, предпочтительно от 3 до 6 атомов. Полициклические кольца содержат от 7 до 17 атомов, предпочтительно от 7 до 13 атомов.
"Арилом" является ароматический карбоциклический кольцевой радикал. Предпочтительные арильные группы включают (например) фенил, толил, ксилил, куменил и нафтил.
"Гетероарилом" является ароматический гетероциклический кольцевой радикал. Предпочтительные гетероарильные группы включают (например) тиенил, фурил, пирролил, пиридинил, пиразинил, тиазолил, пиримидин, хинолинил и тетразолил.
"Алкоксилом" является кислородный радикал, имеющий углеводородный цепочечный радикал, где углеводородная цепочка является алкилом или алкенилом (то есть, -O-алкил или -O-алкенил). Предпочтительные группы включают (например) метокси, этокси, пропокси и аллилокси.
"Алкиламином" является аминорадикал, имеющий один или два алкильных заместителя (т.е. -N-алкил).
"Арилалкилом" является алкильный радикал, замещенный арильной группой. Предпочтительные арилалкильны группы включают бензил и фенилэтил.
"Ариламином" является аминорадикал, замещенный арильной группой (т.е. -NH-арил).
"Арилокси" является кислородный радикал, имеющий арильный заместитель (т.е. -O-арил).
"Ацилом" или "карбонилом" является радикал, образованный удалением гидроксигруппы из карбоновой кислоты (т.е. R=C(=O)-). Предпочтительные алкилацильные группы включают (например) ацетил, формил и пропионил.
"Ацилокси" является кислородный радикал, имеющий ацильный заместитель (т.е. -O-ацил); например, -O-C(=O)-алкил.
"Ациламино" является аминорадикал, имеющий ацильный заместитель (т.е. -N-ацил); например, -NH-C (=O)-алкил.
"Гало", "галогеном" или "галогенидом" является атом хлора, брома, фтора или иода. Хлор и фтор являются предпочтительными галогенидами.
Также, как здесь используется, "низшим" углеводородным радикалом (т.е. "низший" алкил) является углеводородная цепочка, содержащая от 1 до 6, предпочтительно от 1 до 4 атомов углерода.
"Фармацевтически приемлемой солью" является катионная соль, образованная по любой кислотной (например, карбоксильной группе или анионной солью, образованной по любой основной (например, амино) группе. Много таких солей известны специалистам, как описано в Международной Патентной Публикации 87/05297, Johnston et al. , опублик. 11 сентября 1987 (включенной здесь в качестве ссылки). Предпочтительные катионные соли включают соли щелочных металлов (такие как натрия и калия) и соли щелочноземельных металлов (такие как магния и кальция). Предпочтительные анионные соли включают галогениды (такие как соли галогенидов).
"Биогидролизуемым эфиром" является эфир 5-(N-гетерозамещенного амино)хинолона, который по существу не препятствует проявлению противомикробной активности соединений или который легко преобразуется in vivo в организме человека или низшего животного с получением противомикробно-активного 5-(N-гетерозамещенного амино)хинолона. Такие эфиры включают такие, которые не препятствуют биологической активности хинолоновых противомикробных веществ. Многие такие эфиры известны специалистам, как описано в Международной Патентной Публикации 87/05297, Johnston et al., опублик. 11 сентября 1987 (включенной здесь в качестве ссылки). Такие эфиры включают низшие алкиловые эфиры, р низшие ацилокси-алкиловые эфиры (такие как ацетоксиметиловые, ацетоксиэтиловые, аминокарбонилоксиметиловые, пивалоилоксиметиловые и пивалоилоксиэтиловые эфиры), лактониловые эфиры (такие как фталидиловые и тиофталидиловые эфиры), низшие алкоксиацилоксиалкиловые эфиры (такие как метоксикарбонилоксиметиловый, этоксикарбонилоксиэтиловые и изопропоксикарбонилоксиэтиловые эфиры), алкоксиалкиловые эфиры, хинолиновые эфиры и алкилациламиноалкиловые эфиры (такие как ацетамидометиловые эфиры).
"Сольватом" является комплекс, образованный сочетанием растворяемого агента (например, 5-(N-гетерозамещенный амино)хинолон) и растворителя (например, вода). См. The Van Nostard Chemist's Dictionary, p. 650 (1953). Фармацевтически приемлемые растворители, используемые согласно настоящему изобретению, включают такие, которые не препятствуют проявлению биологической активности хинолоновых противомикробных веществ (например, вода, этанол, уксусная кислота, N,N-диметилформамид).
Как указано выше и используется здесь, группы заместителей могут быть сами замещены. Такие заместители могут быть замещены одним или более заместителями. Такие заместители включают перечисленные C. Hansch и A. Leo, Subtituentn Constants for Correlation Analysis in Chemistry and Biology (1979), включенному здесь в качестве ссылки. Предпочтительные заместители включают (например) алкил, алкенил, алкокси, гидрокси, оксо, нитро, амино, аминоалкил (например, аминометил и т.д.), циано, галоген, карбокси, алкоксиацетил (например, карбэтокси и т.д.), тиол, арил, циклоалкил, гетероарил, гетероциклоалкил (например, пиперидинил, морфолинил, пирролидинил и т.д.), имино, тиоксо, гидроксиалкил, арилокси, арилалкил и их сочетание.
Группы R1, R2 и R3 образуют любые варианты хинолоновых радикалов, известные специалистам, как обладающие противомикробной активностью. Такими радикалами являются хорошо известные специалистам, описанные в следующих статьях, всех включенных здесь в качестве ссылок: J. Wolfson et al., "The Fluoroguinolones: Structures, Mechanisms of Action and Resistance, and Spectra of Activity in Vitro", 28 Antimicrobial Agents and Chemotherapy 581 (1985); and T. Rosen et al., 31 J. Med. Chem. 1586 (1988); T. Rosen et al., 31 J. Med. Chem. 1598 (1988); G. Klopman et al., 31 Antimicrob. Agents Chemother. 1831 (1987); 31: 1831 - 1840; J. P. Sanchez et al., 31 J. Med. Chem. 983 (1988); J. M. Domagalia et al., 31 J. Med. Chem. 991 (1988); M. P. Wentland et al., in 20 Ann. Rep. Med. Chem. 145 (D.M. Baily, editor, 1986); J. B. Cornett et al., in 21 Ann. Rep. Med. Chem. 139 (D.M. Bailey, editor, 1986); P. B. Fernandes et al., in 22 Ann. Rep. Med. Chem. 117 (D.M. Bailey, editor, 1987); R. Albrecht 21 Prog. Drug. Research 9 (1977); and P. B. Fernandes et al., in 23 Ann. Rep. Med. Chem. 117 (R.C. Allen, editor, 1987).
R1 обозначает предпочтительно алкил, алкенил, арил,
циклоалкил, гетероциклическое кольцо или алкиламино. Более предпочтительно, R1 обозначает этил, 2-фторэтил, 2-гидроксиэтил, трет-бутил, 4-фторфенил, 2,4-дифторфенил, метиламино, 3-оксетанил,
2-фторциклопропил, бицикло[1,1,1]пентан, винил или циклопропил. Циклопропил, 2-фторциклопропил, трет-бутил и 2,4-дифторфенил являются особенно предпочтительными R1 группами.
Предпочтительные 5-(N-гетерозамещенные амино)хинолоны также включают такие, где R1 и R2 вместе образуют 6-членное гетероциклическое кольцо, в соответствии с формулой
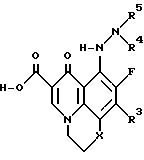
где
X является O, S или CH2;
R2 обозначает предпочтительно хлор, фтор, метокси или метил. Фтор и хлор являются особенно предпочтительными группами R2.
Предпочтительные R3 группы включают азотсодержащие гетероциклические кольца. Особенно предпочтительными являются азотсодержащие гетероциклические кольца, имеющие от 5 до 8 членов. Гетероциклическое кольцо может содержать дополнительные гетероатомы, такие как кислород, сера или азот, предпочтительно азот. Такие гетероциклические группы описаны в патенте США 4599334, Petersen et al., опубл. 8 июля 1986; и в патенте США 4670444, Grohe et al., опубл. 2 июня 1987 (оба включены здесь в качестве ссылки). Предпочтительные R3 группы включают незамещенный или замещенный пиридин, морфолин, диазабицикло[3,1,1] гептан, диазабицикло[2,2,1]гептан, диазабицикло[3,2,1]октан, диазабицикло[2,2,2] октан, имидазолидин и 5-амино-3-азабицикло[4,2,0]гептан, в качестве особенно предпочтительных групп R3, которые включают пиперазин, 3-метилпиперазин, 3-аминопирролидин, 3-аминометилпирролидин, N,N-диметиламинометилпирролидин, N-этиламинометилпирролидин, 3,5-диметилпиридин, N-метилпиперазин и 3,5-диметилпиперазин.
Предпочтительные R4 и R5 группы включают такие, где R4 и R5 вместе образуют гетероциклическое кольцо, содержащее атом азота, к которому они присоединены, такие, где R4 и R5 являются низшим алкилом, такие, где один из R4 или R5 является водородом и другой низшим алкилом, и такие, где оба R4 и R5 являются водородом. Более предпочтительными группами являются такие, где одна из R4 или R5 является водородом и другая алкилом и такие, где оба R4 и R5 являются водородом. Особенно предпочтительными группами являются такие, где оба R4 или R5 является водородом.
Предпочтительные соединения по настоящему изобретению включают такие, которые имеют как R3 группу, которая содержит основной атом азота (включая, например, пиридин, пиперидин, диазабицикло[3,1,1]гептан, диазабицикло[2,2,1] гептан, диазабицикло[3,2,1]октан, диазабицикло[2,2,2]октан и имидазолидин), так и R4 и R5 группы, которые придают атому азота, к которому они присоединены, основной характер (включая, например, когда R4 и R5 вместе образуют гетероциклическое кольцо, содержащее атом азота, к которому они присоединены, оба R4 и R5 являются низшим алкилом, один из R4 и R5 является низшим алкилом и другой водородом или оба R4 и R5 являются водородом). Особенно предпочтительными соединениями являются такие, где R3 группа является одной из пиперазина, 3-метилпиперазина, 3-аминопирролидина, 3-аминометилпирролидина, N,N-диметиламинопирролидина, N-этиламинопирролидина, N-метилпиперазина или 3,5-диметилпиперазина, и оба R4 и R5 являются водородом.
Как здесь используется, "основным атомом азота" является такой, где атом азота обладает свободной парой электронов, которые могут включаться в ионное связывание с любым из множества катионов. Специалисту понятно, что основность атома азота данного радикала будет зависеть от природы ковалентного связывания этого атома азота. См., например, A. Streitwieser and C. Heathcock, Introdution to Organic Chemistry, 2-d edition, pp. 734 - 40 (1981), включенное здесь в качестве ссылки.
Предпочтительные 5-(N-гетерозамещенные
амино)хинолоны включают
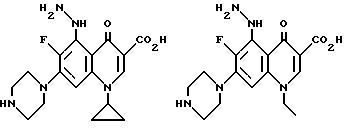
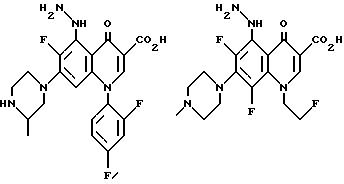
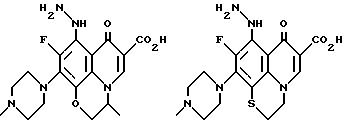

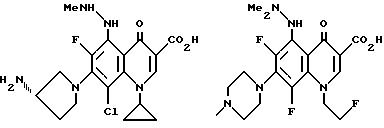
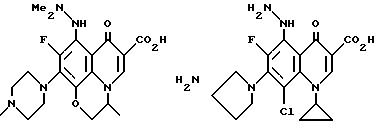

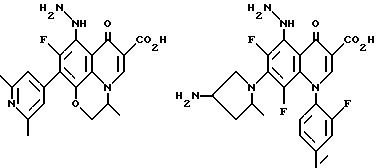

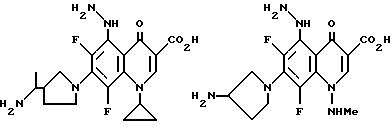
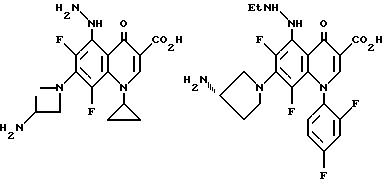
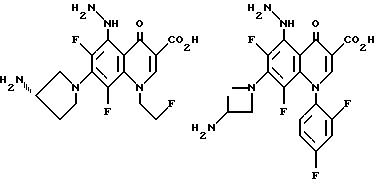

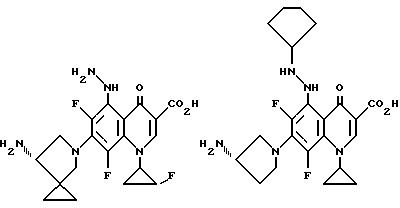

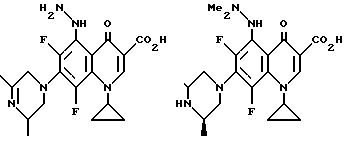

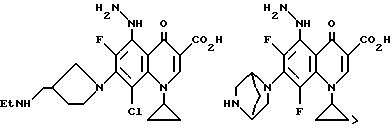
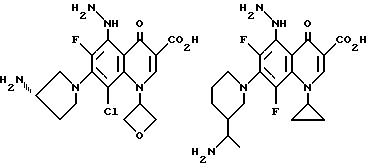
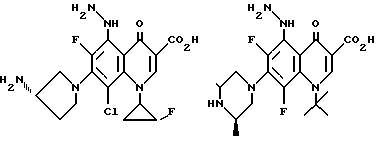

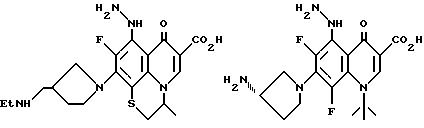

Соединения по данному изобретению используются также в качестве промежуточных продуктов в синтезе новых лактам-хинолонов. Такие соединения описаны в Международной публикации N WO 91/16237, опубликованной 31 октября 1991, включенной здесь в качестве ссылки. Лактам-хинолоны представляют собой любой лактанный радикал, присоединенный, с помощью связывающего радикала, к хинолоновому радикалу в 5-положении хинолона. Лактам-хинолоны включают соединения, имеющие общую структуру
Q - L - B
где Q, L и B определены следующим образом:
(I) Q является структурой, соответствующей формуле (I)

где (A) (1) A1 обозначает N или C (R7),
где (i) R7 является водородом, гидрокси, алкокси, нитро, циано, галогеном, алкилом или N(R8) (R9) (предпочтительно водородом или галогеном), и (ii) R8 и R9 являются, независимо, R8a, где R8a является водородом, алкильным, алкенильным, карбоциклическим кольцевым или гетероциклическим кольцевым заместителями, или R8 и R9 вместе образуют гетероциклическое кольцо, включающее атом азота, к которому они присоединены;
(2) A2 обозначает N или C (R2) (предпочтительно C (R2)); где R2 является водородом или галогеном;
(3) A3 обозначает N или (предпочтительно) C (R5)); где R5 является водородом;
(4) R1 обозначает водород, алкил, карбоциклическое кольцо, гетероциклическое кольцо, алкокси, гидрокси, алкенил, арилалкил или N(R8) (R9) (предпочтительно алкил или карбоциклическое кольцо);
(5) R3 обозначает водород, галоген, алкил, карбоциклическое кольцо или гетероциклическое кольцо (предпочтительно гетероциклическое кольцо);
(6) R4 обозначает гидрокси, и
(7) R7 обозначает R15 или R16X; где R15 является заместителем у L и либо отсутствует, либо обозначает алкил, гетероалкил или алкенил, R16 является заместителем у L и обозначает алкил, алкенил, карбоциклическое кольцо или гетероциклическое кольцо, и X является алкилом, гетероалкилом, алкенилом, кислородом, серой или NH;
(B) за исключением того, что
(1) когда A1 обозначает C(R7), R1 и R7 могут вместе образовывать гетероциклическое кольцо, включающее N', и A1;
(2) когда A2 обозначает C(R2), R2 и R3 могут вместе образовывать -O-(CH2)n-O-, где n является целым числом от 1 до 4;
(3) когда A3 обозначает C(R5), R4 и R5 могут вместе образовывать гетероциклическое кольцо, включающее атомы углерода, к которым присоединены R4 и R5 и атомы углерода формулы (I), к которому присоединены указанные атомы углерода, и
(4) когда A3 обозначает C(R7), R1 и R5 могут вместе образовывать гетероциклическое кольцо, включающее N' и смежный атом углерода, к которому присоединен R5;
(II) B является структурой согласно формуле (II), где L присоединен к R14;
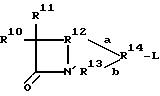
где (A) R10 является водородом, галогеном, гетероалкилом, карбоциклическим кольцом, гетероциклическим кольцом, R8a -O-, R8a CH=N-, (R8)(R9)N-, R17 - C=(CH20)-C(=O)NH - или (предпочтительно) алкилом, алкенилом, R17 - C(= NO - R19)-C(=O)NH - или R18 - (CH2)m -C(=O)NH -;
где (1) m является целым числом от 0 до 9 (предпочтительно от 0 до 3);
(2) R17 обозначает водород, алкил, алкенил, гетероалкил, гетероалкенил, карбоциклическое кольцо или гетероциклическое кольцо (предпочтительно алкил, карбоциклическое кольцо или гетероциклическое кольцо);
(3) R18 обозначает R17, -y1 или CH(y2)(R17);
(4) R19 обозначает R17, арилалкил, гетероарилалкил, -C(R22)(R23)COOH, -C(= O))-R17 или -C(=O)NH - R17, где R22 и R23 являются, независимо, R17 или вместе образуют гетероциклическое кольцо, включающее атом углерода, к которому присоединены R22 и R23 (предпочтительно R17 или C- (R22)((R23)COOH);
(5) R20 обозначает R19, галоген, -y1 или CH(y2) (R17) (предпочтительно R19 или галоген);
(6) y1 обозначает -C-(=O)OR21, -C-(=O)R21, -N(R24)R21 или S(O) p R29 или -OR29; и y2 обозначает y1 или -OH, -SH или -SO3H;
(a) P является целым числом от 0 до 2 (предпочтительно 0);
(c) R25 обозначает R17, NH(R17), N(R17) (R26), O(R26) или S(R26) (предпочтительно R17, NH(R17) или N(R17) (R26); где R26 обозначает алкил; алкенил; карбоциклическое кольцо; гетероциклическое кольцо или (предпочтительно); когда R25 обозначает N(R17) (R26), R26 может содержать радикал, присоединенный к R17 с образованием гетероциклического кольца, и
(7) R21 обозначает R29 или водород; где R29 обозначает алкил; алкенил; арилалкил; гетероалкил, гетероалкенил, гетероарилалкил, карбоциклическое кольцо, гетероциклическое кольцо, или, когда y1 - N(R24) R21 и R21 обозначает R29, R21 и R24 могут вместе образовывать гетероциклическое кольцо, включающее атом азота, к которому присоединен R24 (предпочтительно водород, алкил, карбоциклическое кольцо или гетероциклическое кольцо);
(B) R11 является водородом, галогеном, алкокси или R27C(= O) NH - (предпочтительно водородом или алкокси), где R27 обозначает водород или алкил (предпочтительно водород);
(C) связь "a" обозначает одинарную связь или отсутствие связи, и связь "b" обозначает одинарную связь, двойную связь или отсутствие связи, за исключением того, что обе связи "a" и "b" не могут одновременно отсутствовать;
(D) R12 обозначает -C(R8a)- или -CH2-R28- (предпочтительно -C(R8a)-); где R28 является -C(R8a)-, -O- или -N-, и R28 непосредственно присоединено к N'' в формуле (II) с образованием 5-членного кольца; за исключением того, что если связь "a" отсутствует, тогда R12 обозначает (1) (предпочтительно) -C(R8a) (XI)-, где
(i) X1 обозначает -R21; -OR30; -S(O)гR30, где г обозначает целое число от 0 до 2 (предпочтительно 0); -O(C=O)R30 или N(R30)R31; и
(ii) R30 и R31 являются, независимо, алкильным, алкенильным, карбоциклическим кольцевым или гетероциклическим кольцевым заместителями, и R30 и R31 вместе образуют гетероциклическое кольцо, включающее атом азота, к которому R30 и R31 присоединены, или
(2) -CH2-R32-; где R32 обозначает -C(R8a)(R21), -O- или NR8a, и R32 непосредственно присоединен к N'' в формуле (II) с образованием 5-членного кольца;
(E) (1) если связь "b" обозначает одинарную связь, R13 предпочтительно обозначает -CH(R33)-; или -C(O)NHSO2-, если связь "a" отсутствует; или -C*(R33)-, если R14 содержит радикал R36; где R33 является водородом или COOH (предпочтительно COOH) и C* присоединен к R36 с образованием 3-членного кольца;
(2) если связь "b" обозначает двойную связь, R13 обозначает -C(R33)=; или
(3) если связь "b" отсутствует, R13 обозначает водород, -SO3H, -PO(OR34)OH, -C(O)NHSO2N(R34)(R35), -OSO3H, -CH(R35)COOH или -OCH(R34)COOH
(предпочтительно -SO3H или -C(O)NHSO2N(R34)(R35); где R34 является водородом, алкилом, алкенилом, карбоциклическим кольцом или гетероциклическим кольцом; и R35 является водородом, алкилом, алкенилом или -NHR8a; или (предпочтительно), R13 обозначает -C(O)NHSO2N(R34)(R35), R34 и R35 связаны; и
(F) (1) если связь "a" или связь "b" отсутствует, тогда R14 отсутствует и L непосредственно присоединена к R12 или R13;
(2) если связь "a" или связь "b" являются одинарными связями, R14 обозначает -W-C'''=C(R8a) - R37- или -W-C'''(R36) - R37-; или
(3) (предпочтительно) если связь "a" является одинарной связью и связь "b" обозначает двойную связь, R14 обозначает -C(R8a)(R38)-W-C'''-R37 - или -W-C'''-R37-; R37-;
где (a) W обозначает O; S(O)s, где s обозначает целое число от 0 до 2 (предпочтительно 0); или C (R38),
(b) R36 является водородом, алкилом, алкенилом, -COOH; или, если R13 - C*(R33), R36 может быть присоединен к C* с образованием 3-членного гетероциклического кольца;
(c) R37 отсутствует или является алкилом, алкенилом, карбоциклическим кольцом или гетероциклическим кольцом; и
(d) C''', непосредственно присоединен к R13 с образованием 5- или 6-членного кольца, и
(III) L связывает Q и B; и L является L', -X2t-R39-L' или X3t-R39-L', где L' обозначает Q', -X2-Q'' или X4t-C(=Y3u)-Z-Q'' (предпочтительно -X2-Q'', -X3-Q'', -X4t-C(=Y3u)-Z-Q'');
(1) t и u обозначают, независимо, 0 или 1;
(2) R39 обозначает алкил, алкенил, гетероалкил, гетероалкенил, карбоциклическое кольцо или гетероциклическое кольцо (предпочтительно алкил или алкенил);
(3) X2 является кислородом или S(O)v где v является целым числом от 0 до 2 (предпочтительно 0);
(4) X3 обозначает азот, N(R40), N + (R41)(R42) или R43 - N(R41); и присоединено к R14 одинарной или двойной связью; или, если R14 отсутствует, X3 присоединен к B одинарной или двойной связью (предпочтительно X3 является азотом, N(R40) или N+(R41) (R42);
где (a) R40 обозначает R8a; OR8a или -C(=O)R8a; (предпочтительно R8a);
(b) R41 и R42 являются, независимо, водородом; алкилом, алкенилом, карбоциклическим кольцом, гетероциклическим кольцом или, если R6 обозначает R16 X, R41 и R42 вместе с Q'' могут образовывать гетероциклическое кольцо, как R16;
(c) R43 обозначает N(R41), кислород или серу;
(5) X4 является кислородом, серой, NR40 или R43-NR41 (предпочтительно кислородом, серой или NR40);
(6) Y3 является кислородом, серой, NR40 или N+(R41)(R42);
(7) Y4 является кислородом, или NR41 (предпочтительно кислородом);
(8) L отсутствует или является кислородом, серой, азотом, NR40 или N(R41) - R43 (предпочтительно кислородом, серой, азотом или NR40);
(9) Q' обозначает указанный заместитель R6 для Q; и
(10) Q'' обозначает Q'; или вместе с X2, X3, Z или Z', является указанным заместителем R6 для Q;
и их фармацевтически приемлемые соли и биогидролизуемые эфиры и сольваты.
Предпочтительные лактамсодержащие радикалы включают цефемы, изоцефемы, изо-оксацефемы, оксацефемы, карбацефемы, пенициллины, пенемы, карбапенемы и моноциклические бета-лактамы. Особенно предпочтительными являются цефемы, пенемы, карбапенемы и моноциклические бета-лактамы.
R10 в формуле (II) является любым радикалом, который может быть замещен по активному стереоизомерному положению углерода, смежного с лактамным карбонилом лактама, обладающего противомикробной активностью. (Как используется здесь, термин "лактам, обладающий противомикробной активностью" относится к лактамсодержащему соединению без фрагмента хинолонового заместителя, который обладает противомикробной активностью). Этим "активным" положением является бета (то есть, 7-бета) для цефемов, оксацефемов и карбацефемов (например). Активным положением является альфа для пенемов, карбапенемов, клавемов и клавамов. Подходящие группы R10 будут очевидны любому обычному специалисту.
Способы получения хинолонов и хинолоновых промежуточных соединений, используемых по способам настоящего изобретения, описаны в следующих ссылках, всех включенных здесь в качестве ссылок (включая статью, перечисленные в этих ссылках); 21 Progress in Drug Research, 9 - 104 (1977); 31 J. Med. Chem. , 503 - 506 (1988); 32 J. Med. Chem., 1313 - 1318 (1989); 1987 Liebigs Ann. Chem., 871 - 879 (1987); 14 Drugs Exptl. Clin. Res., 379 - 383 (1988); 31 J. Med. Chem., 983 - 991 (1988); 32 J. Med. Chem., 537 - 542 (1989); 78 J. Pharm. Sci., 585 - 588 (1989); 26 J. Het. Chem., (1989); 24 J. Het. Chem. , 181 - 185 (1987); U.S. Patent 4, 599, 334, 35 Chem. Pharm. Bull. 2281 - 2285 (1987); 29 J. Med. Chem., 2363 - 2369 (1986); 31 J. Med. Chem., 991 - 1001 (1988); 25 J. Het. Chem., 479 - 485 (1988); European Patent Publication 266, 576; European Patent Publication 251, 308, 36 Chem. Pharm. Bull., 1223 - 1228 (1988); European Patent Publication 227,088; European Patent Publication 227,039; European Patent Publication 228,661; 31 J. Med. Chem., 1586 - 1590 (1988); 31 J. Med. Chem., 1598 - 1611 (1988); and 23 J. Med. Chem., 1358 - 1363 (1980).
Обычно, 5-(N-гетерозамещенные амино)хинолоны могут быть получены следующим способом:
[5-галогенхинолон] + H2N - N(R4)(R5) - -> [5-((R4)(R5)N - NH)-хинолон]
где R4 и R5 определены выше и [5-галогенхинолон] является защищенный подходящим
образом 5-галогензамещенный хинолон, где галогеном предпочтительно является хлор или фтор (предпочтительно фтор). Механизм реакции может быть представлен как нуклеофильное ароматическое замещение в
хинолоне 5-галоген заместителя (R4) (R5) NH2 заместителем с образованием 5-(N-гетерозамещенных амино)хинолонов.
Альтернативно, 5-(N-гетерозамещенные
амино)хинолоны могут быть получены следующим образом:
[5,7-дигалогенхинолон] + H2N - N(R4)(R5) - -> [5-((R4)(R5)N
- NH)-7-галогенхинолон]
где
R4 и R5 определены выше и [5,7-дигалогенхинолон] является защищенный подходящим образом 5- и 7-галогензамещенный хинолон, где
галогеном в положениях 5 и 7 независимо является хлор или фтор (предпочтительно фтор). Механизм реакции может быть представлен как избирательное нуклеофильное ароматическое замещение в хинолоне
5-галоген заместителя (R4) (R5)N-NH2 заместителем с образованием 5-(N-гетерозамещенных амино)хинолонов. Реакцию предпочтительно осуществляют в неполярном апротонном растворителе, предпочтительно
бензоле, толуоле, ксилоле и т.д. при повышенной температуре, предпочтительно от 50oC до температуры кипения смеси.
Композиции по данному изобретению включают:
(a)
безопасное и эффективное количество 5-(N-гетерозамещенного амино)хинолона, и
(b) фармацевтически приемлемый носитель.
"Безопасное и эффективное количество" 5-(N-гетерозамещенного амино)хинолона это такое количество, которое является эффективным для ингибирования роста микробов на инфицированном участке, который необходимо обработать, у человека или низшего животного, без нежелательных побочных эффектов (таких как токсичность, раздражение или аллергическая реакция), с соответствующим приемлемым соотношением польза - риск, когда применяют по способу настоящего изобретения. Конкретное "безопасное и эффективное количество" будет, очевидно, зависеть от таких факторов, как конкретное состояние, подвергаемое лечению, физическое состояние пациента, длительность лечения, природа параллельного лечения (если есть), конкретная используемая дозированная форма, используемый носитель, растворимость в нем 5-(N-гетерозамещенного амино)хинолона и дозировочный режим, желательный для композиции.
Композиции по этому изобретению предпочтительно представлены в единичной дозированной форме. Как здесь используется, "единичная дозированная форма" является композицией по настоящему изобретению, содержащей такое количество 5-(N-гетерозамещенного амино)хинолона, которое приемлемо для введения человеку или низшему животному в одноразовой дозе в соответствии с хорошей медицинской практикой. Эти композиции предпочтительно содержат от около 30 мг до около 20000 мг, более предпочтительно от около 50 мг (миллиграмм) до около 7000 мг, более предпочтительно от около 500 мг до около 3500 мг, 5-(N-гетерозамещенного амино)хинолона.
Композиции по данному изобретению могут быть представлены в любой из множества известных форм, подходящих (например) для перорального, ректального, местного или парентерального введения. В зависимости от конкретного желаемого пути введения могут быть использованы разнообразные хорошо известные специалистам фармацевтически приемлемые носители. Они включают твердые или жидкие наполнители, разбавители, гидротропы, поверхностно-активные агенты и образующие оболочки вещества. Могут быть включены необязательные фармацевтически активные вещества, которые практически не мешают проявлению противомикробной активности 5-(N-гетерозамещенного амино)хинолона. Количество носителя, используемого в сочетании с 5-(N-гетерозамещенным амино)хинолоном достаточно для обеспечения практического количества вещества для введения на единичную дозу 5-(N-гетерозамещенных амино)хинолонов. Методы и композиции для приготовления дозированных форм, используемые в настоящем изобретении, описаны в следующих работах, включенных сюда в качестве ссылок: 7 Modern Pharmaceutics, Chapters 9 and 10 (Banker & Khodes, editors, 1979); Lieberman et al., Pharmaceutical Dosage Forms; Tablets (1981); and Ansel, Introduction to Pharmaceutical Dosage Forms; 2-d Edition (1978).
В частности, фармацевтически приемлемые носители для введения в организм включают сахара, крахмалы, целлюлозу и ее производные, солод, желатин, сульфат кальция, растительные масла, синтетические масла, полиолы, альгиновую кислоту, растворы фосфатного буфера, эмульсии, изотонические солевые растворы и свободную от пиргенов воду. Предпочтительные носители для парентерального введения включают пропилен гликоль, этилолеат, пирролидон, этанол и кунжутное масло. Предпочтительно, фармацевтически приемлемый носитель, в композициях для парентерального введения содержится по крайней мере в количестве около 90% по весу от общего веса композиции.
Могут применяться различные пероральные дозированные формы, включая такие как твердые формы в виде таблеток, капсул, гранул и объемные порошки. Эти пероральные формы содержат безопасное и эффективное количество, обычно по крайней мере около 5%, и, предпочтительно, от около 25% до около 50%, 5-(N-гетерозамещенного амино)хинолона. Таблетки могут быть спрессованными, растертыми, покрыты защитным от желудочного сока покрытием, покрытые сахаром, покрытые пленкой, или многослойно спрессованными, содержащими подходящие связующие, смазочные агенты, разбавители, разрыхляющие агенты, окрашивающие агенты, ароматизирующие агенты, облегчающими текучесть агентами и плавящимися агентами. Жидкие пероральные формы включают водные растворы, эмульсии, суспензии, растворы и/или суспензии, получаемые из нешипучих гранул и шипучие препараты, получаемые из шипучих гранул, подслащивающие агенты, плавящиеся агенты, окрашивающие агенты и ароматизирующие агенты. Предпочтительные носители для перорального введения включают желатин, пропиленгликоль, хлопковое масло или кунжутное масло.
Композиции по этому изобретению могут быть также применены субъекту наружно, т. е. прямым нанесением или размазыванием композиции на эпидермальную или эпителиальную ткань субъекта. Такие композиции включают, например, лосьоны, кремы, растворы, гели и твердые вещества. Эти композиции для наружного применения предпочтительно содержат безопасное и эффективное количество, обычно по крайней мере около 0,1% и предпочтительно от около 1% до около 5%. 5-(N-гетерозамещенного амино)хинолона. Подходящие носители для наружного применения предпочтительно остаются на участках кожи в виде длительно действующей пленки и устойчивы к удалению с потом или при погружении в воду. Обычно носитель является органическим по природе и способным к диспергированию или растворению 5-(N-гетерозамещенного амино)хинолона. Носитель может включать фармацевтически приемлемые смягчающие вещества, эмульсификаторы, загустители и растворители.
Данное изобретение включает способы лечения для профилактики инфекционного заболевания человека или другого животного субъекта путем введения безопасного и эффективного количества 5-(N-гетерозамещенного амино)хинолона указанному субъекту. Как здесь указывается "инфекционное заболевание" является любым заболеванием, характеризующимся присутствием микробной инфекции. Предпочтительными методами по данному изобретению являются методы для лечения бактериальной инфекции. Такие инфекционные заболевания включают (например) инфекции центральной нервной системы, инфекции наружного уха, инфекции среднего уха (такие как острый отит), инфекций черепно-мозговых каверн, глазные инфекции, инфекции ротовой полости (такая как инфекция зубов, десен и слизистой оболочки), инфекции верхних дыхательных путей, инфекции низших дыхательных путей, инфекции мочеполовой системы, желудочно-кишечные инфекции, гинекологические инфекции, сепсис, инфекции костной ткани и суставов, инфекции кожи и кожных структур, бактериальный эндокардит, ожоги, антибактериальная профилактика хирургического вмешательства и антибактериальная профилактика пациентов с подавленной иммунной системой (такие как пациенты, получающие противораковую химиотерапию или пациенты с трансплантированными органами).
5-(N-Гетерозамещенные амино)хинолоны и композиции по данному изобретению могут вводиться наружно или внутрь организма. Внутреннее применение включает любой способ введения 5-(N-гетерозамещенного амино)хинолона в ткани тела, например, внутрь оболочек спинного мозга, в спинномозговой канал, внутримышечным, через кожу, внутривенным, внутрибрюшинным, подкожным, подъязычным, ректальным или пероральным введением. Конкретные дозы вводимых противомикробных веществ, а также продолжительность лечения взаимозависимы. Дозировка и режим лечения будут также зависеть от таких факторов, как конкретный используемый 5-(N-гетерозамещенный амино)хинолон, устойчивость инфицированного организма к используемому 5-(N-гетерозамещенному амино)хинолону, способность 5-(N-гетерозамещенного амино)хинолона достигать минимума ингибирующей концентрации на участке инфекции, природа и размер других инфекций (если они есть), личные качества субъекта (такие как вес), совместимость с режимом лечения и присутствие или степень любых побочных эффектов лечения.
Обычно для взрослого человека (весящего приблизительно 70 килограмм) назначается в день от около 75 мг до около 30000 мг, более предпочтительно от около 100 мг до около 20000 мг, более предпочтительно от около 500 мг до около 3500 мг 5-(N-гетерозамещенного амино)хинолона. Режим лечения предпочтительно продолжается от около 1 до около 56 дней, предпочтительно от около 7 до около 28 дней. Профилактический режим (такой как предохранение от вредных инфекций иммуноослабленных пациентов) может продолжаться 6 месяцев или дольше в соответствии с надлежащей медицинской практикой.
Предпочтительным способом парентерального введения является введение путем внутримышечной инъекции. Как известно специалистам, все составы для парентерального введения должны быть стерильны. Для млекопитающих, особенно людей (предполагаемый приблизительный вес тела 70 килограмм) подходящими являются индивидуальные дозы от около 100 мг до около 7000 мг, предпочтительно от около 500 мг до около 3500 мг.
Предпочтительным системным способом введения является пероральный. Предпочитают индивидуальные дозы от около 100 мг до около 2500 мг, предпочтительно от около 250 мг до около 1000 мг.
Наружное применение может использоваться для введения в организм 5-(N-гетерозамещенного амино)хинолона или для обработки локальной инфекции. Количества 5-(N-гетерозамещенного амино)хинолона для наружного применения зависят от таких факторов, как кожная чувствительность, тип и место обрабатываемой ткани, назначаемая композиция и носитель (если есть), конкретный применяемый 5-(N-гетерозамещенный амино)хинолон, а также конкретное заболевание, которое подвергают лечению, и какая желательна продолжительность воздействия на организм (если не локальное воздействие).
Следующие неограничивающие примеры иллюстрируют соединения, композиции, способы и применение настоящего изобретения.
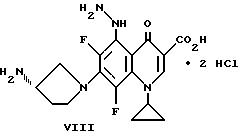
Пример 1. Дигидрохлорид (S)-7-(3-аминопирролидинил)-1- циклопропил-6,8-дифтор-5-гидразино-1,4-дигидро-4-оксо-3- хинолинкарбоновой
кислоты
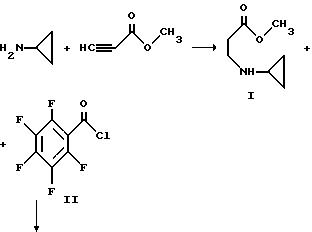


Смесь метилпропиолата (983 г, 11,7 моль) и 500 мл тетрагидрофурана охлаждают до 5oC и в течение приблизительно около часа прибавляют из капельной воронки со скоростью, поддерживающей температуру при 3 - 7oC циклопропиламин (667 г, 11,7 моль), растворенный в 1000 мл тетрагидрофурана. Смесь перемешивают дополнительно в течение часа при 5oC и удаляют ледяную баню. Реакционную смесь перемешивают приблизительно в течение часа при 20 - 25oC, при комнатной температуре в течение 3 часов и оставляют на около 2,5 часа при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток перегоняют в вакууме с получением I.
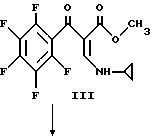
Раствор приблизительно 207 г пентафторбензоилхлорида II (0,90 моль) и 250 мл диоксана охлаждают до 15 - 20oC на бане с ледяной водой и раствор приблизительно 126 г I и 90,9 г триэтиламина (0,90 моль) в 300 мл диоксана добавляют по каплям в течение 5,5 часов. Капельную воронку ополаскивают 50 мл диоксана и реакционную смесь перемешивают при 20oC в течение ночи. Смесь затем подвергают вакуумной фильтрации и остаток дважды промывают порциями 100 мл диоксана. Фильтрат упаривают в вакууме при 25oC и к остатку прибавляют 1000 мл гексана. Большую часть осадка собирают и добавляют к первому. Объединенный продукт затем вновь суспендируют в 1500 мл гексана, быстро перемешивают, фильтруют и сушат в вакууме с получением III.
В 5-литровую трехгорлую колбу, снабженную термометром, трубкой для подачи аргона, механической мешалкой и капельной воронкой помещают приблизительно 14,9 г (0,621 моль) NaH (из промытой гексаном NaH/минеральное масло) и 1000 мл диметилформамида. Эту смесь охлаждают до 15 - 20oC и прибавляют по каплям в течение около 3,5 часов при поддержании температуры при 15 - 20oC приблизительно 181, 5 III (0,542 моль), растворенного в 2 л диметилформамида. Перемешивание продолжают в течение 1,5 часа при этой температуре и затем смесь охлаждают до 10o и прибавляют 500 мл льда и 1 л воды. Смесь нейтрализуют до pH 7 приблизительно 5 мл уксусной кислоты и экстрагируют три раза хлороформом. Высушенные хлороформенные экстракты упаривают с получением взвеси, которую обрабатывают 400 мл кипящего этанола. Полученное твердое вещество фильтруют при комнатной температуре. Дополнительная промывка 100 мл холодным этанолом и последующее вакуумное высушивание дают IV.
Смесь IV (22 г, 0,070 моль) и 2 н H2SO4 (600 мл) перемешивают при 100oC в течение 20 часов и дают остыть до комнатной температуры. Продукт V собирают фильтрацией, промывают водой.
К смеси V (18 г, 0,060 моль) (3S)-трет-бутокси-карбониламинопирролидина (12 г, 0,066 моль) и диметилформамида (130 мл) при 54oC прибавляют по каплям триэтиламин (17 мл, 0,12 моль). Смесь перемешивают при 54oC в течение четырех часов. Прибавляют ацетонитрил (120 мл) и смесь нагревают до 75oC и затем дают ей остыть до комнатной температуры. Смесь охлаждают до 15oC и твердый продукт собирают фильтрацией, промывают ацетонитрилом (2 x 60 мл). Твердый продукт перемешивают в ацетонитриле (180 мл) в течение 10 минут и продукт VI собирают фильтрацией, промывают ацетонитрилом (2 x 60 мл).
Смесь VI (4,0 г 0,0086 моль), ацетонитрила (120 мл) и гидразина моногидрата (4,0 мл, 0,082 моль) кипятят с обратным холодильником в течение 2,5 часов с образованием раствора. Раствор разбавляют ацетонитрилом (100 мл) и перемешивают в течение 2 часов при комнатной температуре. Остаток собирают фильтрацией и нагревают в ацетонитриле (150 мл). Немного нерастворенного вещества удаляют фильтрацией и фильтрат выдерживают при комнатной температуре в течение ночи. Продукт VII собирают фильтрацией и промывают ацетонитрилом.
К смеси VII (4,0 г, 0,0083 моль) и метиленхлорида (85 мл) при комнатной температуре медленно при перемешивании прибавляют насыщенный этанол / HCl (55 мл). Смесь перемешивают при комнатной температуре в течение 4,5 часов и твердый продукт собирают фильтрацией. Это вещество нагревают в CHCl3 (100 мл) и добавляют метанол (10 мл). Смесь охлаждают до комнатной температуры и конечный продукт (VIII) собирают фильтрацией, промывая CHCl3.
Пример 2. Дигидрохлорид (3S)-7-(3-амино-1-пирролидинил)-1- (2,4-дифторфенил)-6,
8-дифтор-5-гидразино-1,4-дигидро-4-оксо-3- хинолинкарбоновой кислоты
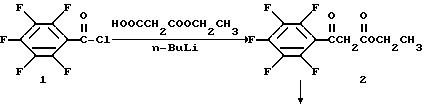
Моноэтил гидромалонат (13,3 г, 0,10 моль) растворяют в тетрагидрофуране (260 мл) и охлаждают до -65oC. Затем по каплям прибавляют 2М н-бутиллития (10 мл, 0,20 моль) при поддерживании температуры ниже -50oC. Реакционную смесь нагревают до -5oC и опять охлаждают до -65oC. Пентафторбензоилхлорид I (7,20 мл, 0,05 моль) растворяют в тетрагидрофуране (32 мл) и прибавляют по каплям при поддержании температуры ниже -50oC. После прибавления реакционную смесь нагревают до -35oC и перемешивают в течение 1 часа. К раствору прибавляют водную HCl (13%, 316 мл) и перемешивают в течение 30 мин. Смесь экстрагируют CH2Cl2 и промывают водной NaHCO3 и затем водой. Органический слой сушат Na2SO4 и концентрируют с получением продукта 2, который существует в виде смеси кетоенольных таутомеров в растворе.
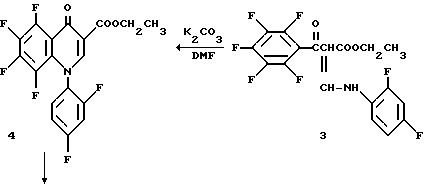
Этиловый эфир пентафторбензоилуксусной кислоты 2 (10 г. 0,035 моль) прибавляют к ангидриду уксусной кислоты (8,5 мл, 0,09 моль) и триэтилортоформиату (10 мл, 0,06 моль). Реакционную смесь нагревают до 110oC в течение 2,25 час. Реакционную смесь концентрируют. Продукт растворяют в этаноле (250 мл) и охлаждают до 0oC. Затем медленно прибавляют 2, 4-дифторанилин (4,7 мл, 0,046 моль) и ледяную баню убирают. Реакционную смесь перемешивают в течение ночи и концентрируют досуха при пониженном давлении. Остаток растирают в петролейном эфире и продукт собирают фильтрацией с получением 3 в виде смеси цис-транс изомеров.
Виниловый амид 3 (9,43 г, 0,022 моль) растворяют в диметилформамиде (57,0 мл) и прибавляют K2 CO3 (9,46 г, 0,068 моль). Реакционную смесь перемешивают в течение ночи и затем концентрируют. Прибавляют метиленхлорид и раствор промывают водой. Органическую фазу сушат над Na2 SO4, концентрируют и сушат в вакууме с получением хинолона 4.
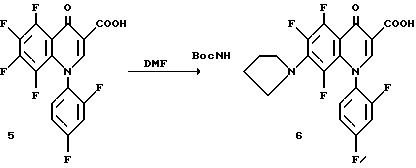
Эфир 4 (9,49 г., 0,021 моль) помещают в раствор 8 : 6 : 1 уксусная кислота/вода/H2SO4 (309 мл) и нагревают до 100oC до завершения реакции. Раствор выливают в ледяную воду и остаток фильтруют. Продукт перекристаллизовывают растворением в CH2 Cl2 и высаживанием гексаном. Твердый осадок собирают с получением кислоты 5. Фильтрат концентрируют и остаток очищают как описано выше из CH2 Cl2 с получением второй партии.
Хинолон 5 (10 г, 0,027 моль) растворяют в диметилформамиде (60 мл) и прибавляют (3S)-трет-бутоксикарбониламинопирролидин (6,0 г, 0,032 моль). Реакционную смесь нагревают до 55oC и в течение более 20 мин прибавляют триэтиламина (7,5 мл, 0,054 моль). Реакция завершается за 45 мин, как определяется ТСХ и нагревание прекращают. Продукт осаждается из раствора и фильтруется. Твердый продукт промывают эфиром. Продукт растворяют в горячем EtOAc и высаживают прибавлением гексана. Твердый продукт фильтруют и сушат в вакууме с получением 6.
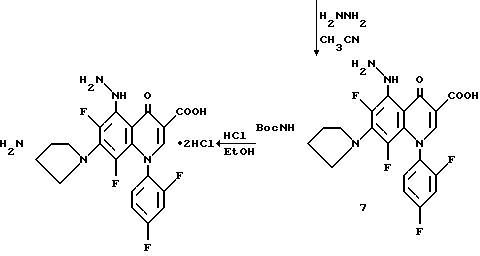
Смесь хинолона 6 (2, 0 г. 0,0037 моль), ацетонитрила (60 мл) и гидразина (0,46 мл, 0,0095 моль) кипятят с обратным холодильником в течение 1,6 час и охлаждают до комнатной температуры. Продукт собирают фильтрацией и перекристаллизовывают из ацетонитрила с получением 7.
Смесь (0,20 г, 0,00036 моль) и насыщенной HCl/EtOH (4 мл) перемешивают при комнатной температуре в течение одного часа и прибавляют еще 4 мл HCl/EtOH. Реакционную смесь перемешивают дополнительно 3 часа и твердый продукт собирают фильтрацией. Твердый продукт растирают в CH2 Cl2 и собирают фильтрацией. Продукт перекристаллизовывают из ацетонитрила/H2O с получением целевого продукта.
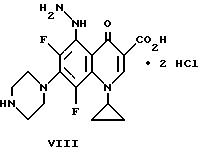
Пример 3. Дигидрохлорид 1-циклопропил-6,8-дифтор-5- гидразино-1,
4-дигидро-4-оксо-7-пиперазинил-3-хинолинкарбоновой кислоты
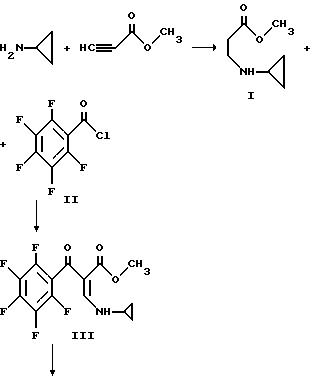

Смесь метилпропиолата (983 г. 11,7 моль) и 500 мл тетрагидрофурана охлаждают до 5oC и в течение приблизительно около часа прибавляют из капельной воронки со скоростью, поддерживающей температуру при 3 - 7oC циклопропиламин (667 г. 11,7 моль), растворенный в 1000 мл тетрагидрофурана. Смесь перемешивают дополнительно в течение часа при 5oC и удаляют ледяную баню. Реакционную смесь перемешивают приблизительно в течение часа при 20 - 25oC, при комнатной температуре в течение 3 часов и оставляют приблизительно на 2,5 часа при комнатной температуре. Растворитель удаляют при пониженном давлении и остаток перегоняют в вакууме с получением I.
Раствор приблизительно 207 г пентафторбензоилхлорида II (0,90 моль) и 250 мл диоксана охлаждают до 15 - 20oC на бане с ледяной водой и раствор приблизительно 126 г I и 90,9 г триэтиламина (0,90 моль) в 300 мл диоксана добавляют по каплям в течение 5,5 часов. Капельную воронку ополаскивают 50 мл диоксана и реакционную смесь перемешивают при 20oC в течение ночи. Смесь затем подвергают вакуумной фильтрации и остаток дважды промывают порциями 100 мл диоксана. Фильтрат упаривают в вакууме при 25oC и к остатку прибавляют 1000 мл гексана. Большую часть осадка собирают и добавляют к первому. Объединенный продукт затем вновь суспендируют в 1500 мл гексана, быстро перемешивают, фильтруют и сушат в вакууме с получением III.
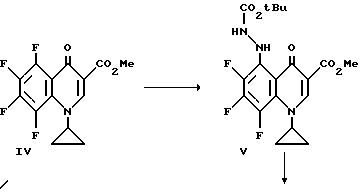
В 5-литровую трехгоролую колбу, снабженную термометром, трубкой для подачи аргона, механической мешалкой и капельной воронкой помещают приблизительно 14,9 г (0,621 моль) NaH (из промытой гексаном NaH/минеральное масло) и 1000 мл диметилформамида. Эту смесь охлаждают до 15 - 20oC и прибавляют по каплям в течением около 3,5 часов при поддержании температуры при 15 - 20oC приблизительно 181, 5 г III (0,542 моль), растворенного в 2 л диметилформамида. Перемешивание продолжают в течение 1,5 часов при этой температуре и затем смесь охлаждают до 10o и прибавляют 500 мл льда и 1 л воды. Смесь нейтрализуют до pH 7 приблизительно 5 мл уксусной кислоты и экстрагируют три раза хлороформом. Высушенные хлороформенные экстракты упаривают с получением взвеси, которую обрабатывают 400 мл кипящего этанола. Полученное твердое вещество фильтруют при комнатной температуре. Дополнительная промывка 100 мл холодным этанолом и последующее вакуумное высушивание дают IV.
Перемешиваемую смесь метилового эфира 1-циклопропил-5,6,7,8-тетрафтор-4-оксо-3-карбоновой кислоты (соединение IV) (8,2 г), триэтиламина (4 мл), трет-бутилкарбазата (3,8 г) и толуол кипятят с обратным холодильником и концентрируют досуха при пониженном давлении. Осадок растворяют в CH2 Cl2 (200 мл) и промывают водой (200 мл) и насыщенным раствором соли (200 мл). Органическую фазу сушат над Na2SO4, фильтруют и фильтрат концентрируют досуха при пониженном давлении. Остаток очищают флэш-хроматографией (силикагель) с 5% Me OH/CH2 Cl2 с получением желаемого C-5 замещенного продукта V.
Смесь приблизительно 3,5 г V. тетрагидрофурана (ТГФ) (30 мл) и 17 мл 1 н NaOH нагревают при 80oC приблизительно в течение 1,5 часов. Реакционную смесь охлаждают на ледяной бане и прибавляют воду (200 мл) с последующим прибавлением ледяной уксусной кислоты (2,3 мл). Осадок фильтруют, промывают водой и эфиром с получением соединения VI.
Смесь приблизительно 2,6 г. VI, 1,3 г. 1-(трет-бутоксикарбонил) пиперазина и пиридина (20 мл) нагревают при 80oC приблизительно в течение 1 часа и реакционную смесь концентрируют досуха при пониженном давлении. Остаток растворяют в CH2 Cl2 (100 мл) и промывают водой, 5% лимонной кислотой и насыщенным раствором соли. Органический слой сушат над Na2SO4, фильтруют и концентрируют досуха. Остаток очищают флэш-хроматографией (силикагель) с 5% MeOH/CH2 Cl2 с получением соединения VII.
К смеси VII (2,3 г.) и CH2 Cl2 (40 мл) при комнатной температуре медленно прибавляют приблизительно 28 мл насыщенного этанола/HCl. Смесь перемешивают при комнатной температуре в течение приблизительно 4,5 часов и продукт собирают фильтрацией. Твердый продукт нагревают в CHCl3 (50 мл) и добавляют метанол (10 мл). Смесь охлаждают до комнатной температуры и продукт собирают фильтрацией с получением соединения VIII.
Пример 4. Противомикробную композицию для парентерального введения в соответствии с настоящим изобретением
готовят с содержанием:
Компонент - Количество
(S)-7-(3-Аминопирролидинил)-1-циклопропил-6,8-дифтор-5- гидразино-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота I - 100 мг/кг
Носитель
Буфер цитрата натрия с (процент от веса носителя)
Лецитин - 0,48
Карбоксиметилцеллюлоза - 0,53
Повидон - 0,50
Метил парабен - 0,11
Пропил
парабен - 0,011
I: 5-(N-гетерозамещенный амино)хинолон, полученный по примеру 1.
Вышеуказанные компоненты смешивают, получая суспензию. Приблизительно 2,0 мл суспензии вводят системно посредством внутримышечной инъекции человеку против инфекции нижних дыхательных путей с наличием Streptococcus pneumonia. Эту дозировку повторяют дважды в день в течение приблизительно 14 дней. Через 4 дня симптомы заболевания ослабевают, свидетельствуя, что патоген практически уничтожен.
Пример 5. Противомикробную композицию с покрытием для защиты от желудочного сока
для перорального введения в соответствии с настоящим изобретением готовят со следующим содержанием ядра таблетки:
Компонент - Количество (мг)
(S)-7-(3-Аминопирролидинил)-1-циклопропил-6,8-дифтор-5- гидразино-1,4-дигидро-4-оксо-3-хинолинкарбоновая кислота I - 350,0
Крахмал - 30,0
Стеарат магния - 5,0
Микрокристаллическая целлюлоза - 100,0
Коллоидный диоксид кремния - 2,5
Повидон - 12,5
I: 5-(N-гетерозамещенный амино)хинолон, полученный по примеру 1.
Компоненты смешивают в объемную смесь. Прессованные таблетки получают с использованием способов табленитрования, известных специалистам. Таблетки затем покрывают суспензией полимера метакриловой кислоты /эфира метакриловой кислоты в изопропаноле/ ацетоне. Человеку с инфекцией мочевого тракта с наличием Escherichia coli перорально вводят две таблетки каждые 8 часов в течение 14 дней. Симптомы заболевания затем ослабевают, свидетельствуя о практическом уничтожении патогена.
Реферат
Изобретение относится к новым противомикробным 5-(N-гетерозамещенные амино) хинолоновым соединениям общей формулы I
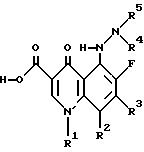
где R1, R2 и R3 образуют любой их множества хинолонов и близких гетероциклических структур подобных тем, которые известны социалистам как обладающие противомикробной активностью, и (2) (а) R4 и R5 являются, независимо, водородом, низшим алкилом, циклоалкилом, гетероалкилом, или -С(=О)-Х-R8, где X является ковалентной связью, N, O или S и R8 является низшим алкилом, низшим алкенилом, арилалкилом, карбоциклическим кольцом, гетероциклическим кольцом, или (b) R4 и R5 вместе образуют гетероциклическое кольцо, которое включает азот, к которому они присоединены, и их фармацевтически приемлемые соли и биогидролизуемые эфиры и сольваты. Кроме того, изобретение относится к композиции, содержащей соединение формулы I, и к способу лечения или профилактики. 4 с. и 14 з.п.ф-лы.
Формула
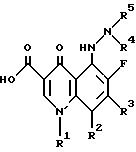
R11 обозначает замещенное или незамещенное 3-6-членное карбоциклическое кольцо;
R22 обозначает галоген;
R33 обозначает замещенное или незамещенное 5-8-членное азотсодержащее гетероциклическое кольцо;
R44 и R55 обозначают, независимо, водород или низший алкил, или их фармацевтически приемлемые соли, или биогидролизуемые эфиры или сольваты.
(3S)-7-(3-амино-1-пирролидинил)-1-(2,4-дифторфенил)-6,8-дифтор-5-гидразино-1, 4-дигидро-4-оксо-3-хинолинкарбоновой кислоты;
(3S)-7-(3-аминопирролидинил)-1-циклопропил-6,8-дифтор-5-гидразино-1,4-дигидро-4-оксо-3-хинолинкарбоновой кислоты, или его фармацевтически приемлемые соли, биогидролизуемые эфиры или сольваты.
Комментарии