Алкилсульфонамидхинолины с аффинностью к рецепторам nk-3 - RU2421447C2
Код документа: RU2421447C2
Описание
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Данное изобретение относится к производным хинолина, к содержащим их фармацевтическим композициям и к применению таких соединений в лечении заболеваний или расстройств центральной и периферической нервной системы. Данное изобретение также относится к применению таких соединений в комбинации с одним или более другими агентами ЦНС для потенцирования эффектов других агентов ЦНС. Соединения по данному изобретению также полезны в качестве зондов для локализации рецепторов клеточной поверхности.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рецепторы тахикининов являются мишенями семейства структурно родственных пептидов, которые включают вещество Р (SP), нейрокинин А (NKA) и нейрокинин В (NKB), имеющие общее название "тахикинины". Тахикинины синтезируются в центральной нервной системе (ЦНС) и в периферических тканях, где они проявляют различные виды биологической активности. Известны три рецептора тахикининов, которые называются рецепторами нейрокинина-1 (NK-1), нейрокинина-2 (NK-2) и нейрокинина-3 (NK-3). Рецепторы NK-1 и NK-2 экспрессируются в различных периферических тканях, и рецепторы NK-1 экспрессируются также в ЦНС, тогда как рецепторы NK-3 экспрессируются главным образом в ЦНС.
Рецепторы нейрокининов опосредуют ряд стимулируемых тахикининами биологических эффектов, которые включают: передачу возбуждающих нейронных сигналов в ЦНС и на периферии (например, болевых сигналов), модулирование сократительной активности гладкой мускулатуры, модулирование иммунного и воспалительного ответов, индуцирование гипотензивных эффектов через дилатацию периферической сосудистой сети и стимуляцию секреции эндокринных и экзокринных желез.
Было показано, что в ЦНС активация рецепторов NK-3 модулирует высвобождение дофамина, ацетилхолина и серотонина, что предполагает терапевтическую полезность лигандов NK-3 для лечения ряда расстройств, в том числе тревоги, депрессии, шизофрении и ожирения. Исследования на головном мозге приматов показали присутствие мРНК NK-3 в ряде областей, релевантных для этих расстройств. В исследованиях на крысах было показано, что рецепторы NK-3 локализуются на содержащих МСН (меланинконцентирующий гормон) нейронах в латеральном гипоталамусе и zona incerta, что опять же предполагает терапевтическую полезность лигандов NK-3 для ожирения.
Были разработаны непептидные лиганды для каждого из тахикининовых рецепторов, однако известные непептидные антагонисты рецептора NK-3 имеют целый ряд недостатков, таких как видовая селективность, которая ограничивает возможность оценки этих соединений во многих соответствующих моделях заболеваний. Следовательно, необходимы новые непептидные лиганды рецептора NK-3 для применения в качестве терапевтических агентов и в качестве инструментов для исследования биологических последствий модулирования рецептора NK-3.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Раскрыты соединения, в частности производные алкилсульфонамидхинолина, с аффинностью к рецепторам NK-3 (NK-3r). Эти соединения обладают потенциалом для лечения множества заболеваний, расстройств и состояний, в том числе, но не ограничиваясь ими, депрессию, тревогу, шизофрению, когнитивные расстройства, психозы, ожирение, воспалительные заболевания, в том числе синдром раздраженного кишечника и воспалительное кишечное расстройство, рвоту, преэклампсию, хроническую обструктивную болезнь легких, расстройства, связанные с избыточными гонадотропинами и/или андрогенами, в том числе дисменорею, доброкачественную гиперплазию простаты, рак простаты и рак яичка, при которых полезно модулирование активности рецепторов NK-3.
Раскрытые лиганды к рецепторам NK-3 и их стереоизомеры, энантиомеры, гидролизуемые in vivo предшественники и фармацевтически приемлемые соли представляют собой соединения формулы I,

где:
R1 выбран из Н, С1-4алкила, С3-6циклоалкила и С1-4алкилОС(O)-;
А представляет собой фенил или С3-7циклоалкил;
R2 в каждом случае независимо выбран из Н, -ОН, -NH2, -CN, галогена, С1-6алкила, С3-7циклоалкила, С1-6алкокси и С1-6алкоксиС1-6алкила;
n равно 1, 2 или 3;
R3 в каждом случае независимо выбран из Н, -ОН, -NH2, -NO2, -CN, галогена, С1-6алкила, C1-6алкокси и С1-6алкоксиС1-6алкила;
m равно 1, 2 или 3;
r равно 0, 1, 2 или 3,
R4 выбран из С1-4алкила, С1-6алкоксиС1-6алкила, С3-7циклоалкила и Е-(СН2)Р-, где Е выбран из -NR6R7, -SR6, -SOC1-6алкил, -SO2С1-6алкил, N+(O-)R6R7, -NR6SO2R7, арила и N- или С-связанного 5- или 6-членного ароматического или неароматического гетероциклического кольца, имеющего 1, 2, 3 или 4 атома азота, или его N-оксида, и р равно 0, 1, 2, 3, 4 или 5;
R5 в каждом случае независимо выбран из Н, -ОН, -CN, галогена, -R6, -OR6, -NR6R7, -SR6, -SOR6 и -SO2R6;
q равно 1, 2 или 3;
R8 выбран из Н, C1-5алкильной группы с прямой или разветвленной цепью или С3-5 циклоалкильной группы, причем указанные группы либо не замещены, либо замещены одной или более группировками, выбранными из -ОН,=O, -NH2, -CN, галогена, арила и C1-3алкокси;
где:
R6 и R7 в каждом случае независимо выбраны из Н, C1-6 алкильной группы с прямой или разветвленной цепью, С2-6 алкенильной или алкинильной группы с прямой или разветвленной цепью и С3-7карбоциклической группы, имеющей ноль, одну или две двойные или тройные связи, причем указанные группы либо не замещены, либо замещены одной или более группировками, выбранными из -ОН, =O, -NH2, -CN, галогена, арила и С1-3алкокси; и
когда R4 представляет собой Е-(СН2)Р-, и указанный Е представляет собой N- или С-связанное 5- или 6-членное ароматическое или неароматическое гетероциклическое кольцо или его N-оксид, указанный Е не замещен или имеет 1, 2 или 3 заместителя, независимо выбранных из -ОН, =O, -NH2, -CN, галогена, С1-4алкила, С1-4алкокси, С1-4алкил-СO-, -NR6R7, арила и 5-или 6-членного ароматического или неароматического гетероциклического кольца, имеющего 1, 2, 3 или 4 атома азота; и
когда R1, R2, R3 или R4 представляет собой алкильную, циклоалкильную, алкокси или алкоксиалкильную группировку, указанные группировки не замещены или имеют 1, 2, 3, 4 или 5 заместителей, независимо выбранных в каждом случае из -ОН, -NH2, -CN, фенила и галогена.
Раскрыты также фармацевтические композиции и препараты, содержащие эти соединения, способы их применения для лечения заболеваний и состояний либо отдельно, либо в комбинации с другими терапевтически активными соединениями или веществами, способы и промежуточные соединения, используемые для их получения, их применение в качестве лекарственных средств, их применение в изготовлении лекарственных средств и их применение в диагностических и аналитических целях. В частности, раскрыты соединения, композиции, содержащие их, и способы с их использованием для лечения или предупреждения состояний и расстройств, связанных с множеством заболеваний или расстройств, при которых, как считается, рецепторы NK-3 играют роль.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения по изобретению представляют собой соединения формулы I,

где:
R1 выбран из Н, С1-4алкила, С3-6циклоалкила и С1-4алкилОС(O)-;
А представляет собой фенил или С3-7циклоалкил;
R2 в каждом случае независимо выбран из Н, -ОН, -NH2, -CN, галогена, C1-6алкила, С3-7циклоалкила, С1-6алкокси и С1-6алкоксиС1-6алкила;
n равно 1, 2 или 3;
R3 в каждом случае независимо выбран из Н, -ОН, -NH2, -NO2, -CN, галогена, С1-6алкила, С1-6алкокси и С1-6алкоксиС1-6алкила;
m равно 1, 2 или 3;
r равно 0, 1, 2 или 3,
R4 выбран из С1-4алкила, С1-6алкоксиС1-6алкила, С3-7циклоалкила и Е-(СН2)Р-, где Е выбран из -NR6R7, -SR6, -SОС1-6алкил, -SO2С1-6алкил, N+(O-)R6R7, -NR6SO2R7, арила и N- или С-связанного 5- или 6-членного ароматического или неароматического гетероциклического кольца, имеющего 1, 2, 3 или 4 атома азота, или его N-оксида, и р равно 0, 1, 2, 3, 4 или 5;
R5 в каждом случае независимо выбран из Н, -ОН, -CN, галогена, -R6, -OR6, -NR6R7, -SR6, -SOR6 и -SO2R6;
q равно 1, 2 или 3;
R8 выбран из Н, C1-5 алкильной группы с прямой или разветвленной цепью или С3-5 циклоалкильной группы, причем указанные группы либо не замещены, либо замещены одной или более группировками, выбранными из -ОН, =O, -NH2, -CN, галогена, арила и С1-3алкокси;
где:
R6 и R7 в каждом случае независимо выбраны из Н, C1-6 алкильной группы с прямой или разветвленной цепью, С2-6 алкенильной или алкинильной группы с прямой или разветвленной цепью и С3-7карбоциклической группы, имеющей ноль, одну или две двойные или тройные связи, причем указанные группы либо не замещены, либо замещены одной или более группировками, выбранными из -ОН,=O, -NH2, -CN, галогена, арила и С1-3алкокси; и
когда R4 представляет собой Е-(СН2)Р-, и указанный Е представляет собой N- или С-связанное 5- или 6-членное ароматическое или неароматическое гетероциклическое кольцо или его N-оксид, указанный Е не замещен или имеет 1, 2 или 3 заместителя, независимо выбранных из -ОН,=O, -NH2, -CN, галогена, С1-4алкила, С1-4алкокси, С1-4алкил-СО-, -NR6R7, арила и 5- или 6-членного ароматического или неароматического гетероциклического кольца, имеющего 1, 2, 3 или 4 атома азота; и
когда R1, R2, R3 или R4 представляет собой алкильную, циклоалкильную, алкокси или алкоксиалкильную группировку, указанные группировки не замещены или имеют 1, 2, 3, 4 или 5 заместителей, независимо выбранных в каждом случае из -ОН, -NH2, -CN, фенила и галогена;
их стереоизомеры, энантиомеры, гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
В одном воплощении соединения представляют собой соединения формулы II, то есть соединения формулы I, где r равно 0,
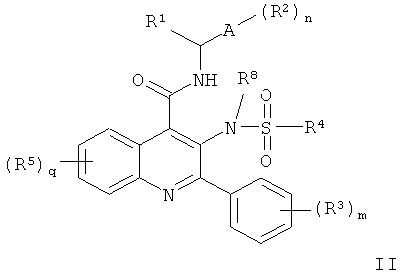
где R1, A, R2, n, R3, m, R5, q, R4 и R8 являются такими, как определено для формулы I, их гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
В другом воплощении соединения представляют собой соединения формулы I, где R1, A, R2, n, R3, m, R5, q, r и R8 являются такими, как определено для формулы I, их гидролизуемые in vivo предшественники и фармацевтически приемлемые соли, и где R4 выбран из С3-7циклоалкила и Е-(СН2)Р-, где Е выбран из -NR6R7, -SR6, -SOC1-6алкил, -SО2С1-6алкил, N+(O-)R6R7, -NR6SO2R7, арила и N- или С-связанного 5- или 6-членного ароматического или неароматического гетероциклического кольца, имеющего 1, 2, 3 или 4 атома азота, или его N-оксида, и р равно 1, 2, 3, 4 или 5.
В другом воплощении соединения представляют собой соединения формулы I, где R1, A, R2, n, R3, m, R5, q, r и R4 являются такими, как определено для формулы I, его гидролизуемые in vivo предшественники и фармацевтически приемлемые соли, и где R8 выбран из Н или С1-5 алкильной группы с прямой или разветвленной цепью или С3-5 циклоалкильной группы, причем эти группы замещены одной или более группировками, выбранными из -ОН, =O, -NH2, -CN, галогена, арила и С1-3алкокси.
В другом воплощении соединения представляют собой соединения формулы I или II, где А представляет собой фенил.
В другом воплощении соединения представляют собой соединения формулы II, где:
А представляет собой фенил;
R1 выбран из С1-4алкила, С3-6циклоалкила и С1-4алкилОС(O)-;
R2 выбран из Н, галогена и незамещенного С1-6алкокси;
R3 представляет собой Н или галоген;
R8 представляет собой Н или метил;
n и m оба равны 1, и
когда R1 или R4 представляет собой алкильную, циклоалкильную или алкоксиалкильную группировку, указанные группировки не замещены или имеют 1, 2, 3, 4 или 5 заместителей, независимо выбранных в каждом случае из -ОН, -NH2, -CN и галогена, их стереоизомеры, энантиомеры, гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
В другом воплощении соединения представляют собой соединения формулы II, где:
А представляет собой фенил;
R1 выбран из С1-4алкила и С1-6циклоалкила;
R2 выбран из Н, галогена и незамещенного С1-6алкокси;
R3 представляет собой Н или галоген;
R8 представляет собой Н или метил;
n и m оба равны 1;
R4 выбран из С1-4алкила и Е-(СН2)Р-, где Е представляет собой замещенное или незамещенное N-связанное 5- или 6-членное ароматическое или неароматическое гетероциклическое кольцо, имеющее 1, 2, 3 или 4 атома азота, и
R5 представляет собой Н, их стереоизомеры, энантиомеры, гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
В еще одном воплощении соединения представляют собой соединения формулы II, где:
А представляет собой фенил;
R1 представляет собой этил или циклопропил;
R2 выбран из Н, F и -ОСН3;
R3 представляет собой Н или F;
R8 представляет собой Н;
каждый из n, m и q равен 1, и
R5 в каждом случае независимо выбран из Н, -ОН и галогена, их стереоизомеры, энантиомеры, гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
В еще одном воплощении соединения представляют собой соединения формулы III или IV

где R1, A, R2, n, R3, m, r, R4, R5 и q являются такими, как определено для формулы I,
их гидролизуемые in vivo предшественники и фармацевтически приемлемые соли.
Конкретные соединения выбраны из:
(1-фенил-пропил)-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 3-этансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 2-фенил-3-трифторметансульфониламино-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 2-фенил-3-(2,2,2-трифторэтансульфониламино)-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 2-фенил-3-(пропан-1-сульфониламино)-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 2-фенил-3-(3,3,3-трифторпропан-1-сульфониламино)-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 3-циклопропансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
(циклопропил-фенил-метил)-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
[1-(3-фтор-фенил)-пропил]-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
[циклопропил-(3-фтор-фенил)-метил]-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты (1-фенилпропил)-амида,
(циклопропил-фенил-метил)-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
[1-(3-фтор-фенил)-пропил]-амида 2-(3-фторфенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
[циклопропил-(3-фтор-фенил)-метил]-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты ((S)-1-фенилпропил)-амида,
((S)-1-фенил-пропил)-амида 3-этансульфониламино-2-фенилхинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 2-фенил-3-трифторметансульфониламино-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 2-фенил-3-(2,2,2-трифторэтансульфониламино)-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 2-фенил-3-(пропан-1-сульфониламино)-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 2-фенил-3-(3,3,3-трифторпропан-1-
сульфониламино)-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 3-циклопропансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
((S)-циклопропил-фенил-метил)-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
[(S)-1-(3-фтор-фенил)-пропил]-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
[(S)-циклопропил-(3-фтор-фенил)-метил]-амида 3-метансульфониламино-2-фенил-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
((S)-циклопропил-фенил-метил)-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
[(S)-1-(3-фтор-фенил)-пропил]-амида 2-(3-фтор-фенил)-3-
метансульфониламино-хинолин-4-карбоновой кислоты,
[(S)-циклопропил-(3-фтор-фенил)-метил]-амида 2-(3-фтор-фенил)-3-метансульфониламино-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 3-(метансульфониламино-метил)-2-фенил-хинолин-4-карбоновой кислоты,
(1-фенил-пропил)-амида 3-(этансульфониламино-метил)-2-фенил-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 3-(метансульфониламино-метил)-2-фенил-хинолин-4-карбоновой кислоты,
((S)-1-фенил-пропил)-амида 3-(этансульфониламино-метил)-2-фенил-хинолин-4-карбоновой кислоты,
N-[(1S)-циклопропил(фенил)метил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамида,
N-[(1S)-циклопропил(3-фторфенил)метил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамида,
N-[(1S)-1-циклогексилэтил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамида,
N-[(1R,2S)-2-гидрокси-1-фенилпропил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамида,
N-[(S)-циклопропил(фенил)метил]-3-[(циклопропилсульфонил)амино]-2-фенилхинолин-4-карбоксамида и
((S)-1-фенил-пропил)-амида 3-(метансульфонилметиламино)-2-фенилхинолин-4-карбоновой кислоты, их стереоизомеров, энантиомеров, гидролизуемых in vivo предшественников и фармацевтически приемлемых солей.
Соединения по настоящему изобретению имеют преимущество в том, что они могут быть более растворимыми, могут легче всасываться и могут быть более эффективными in vivo, могут оказывать меньше побочных эффектов, быть менее токсичными, быть более действенными, более избирательными, могут действовать дольше, меньше метаболизироваться и/или иметь улучшенный фармакокинетический профиль, чем известные соединения, или могут обладать другими полезными фармакологическими или физико-химическими свойствами по сравнению с известными соединениями. С использованием описанных здесь анализов на функциональную активность обнаружено, что соединения по изобретению имеют значения IС50 менее примерно 1 мкМ в отношении рецепторов NK-3, и что многие соединения имеют значения IC50 менее примерно 100 нМ в отношении рецепторов NK-3.
СОКРАЩЕНИЯ И ОПРЕДЕЛЕНИЯ
Если не указано иное, здесь С1-6алкил включает, но не ограничен ими, группировки метил, этил, н-пропил, н-бутил, изопропил, изобутил, трет-бутил, втop-бутил, либо сами по себе, либо в составе другой группы, и алкильные группы могут быть с прямой или разветвленной цепью.
Если не указано иное, здесь С1-6алкокси включает, но не ограничен ими, группировки -О-метил, -О-этил, -О-н-пропил, -О-н-бутил, -О-изопропил, -О-изобутил, -O-тpeт-бутил, -O-втop-бутил, либо сами по себе, либо в составе другой группы, и алкоксигруппы могут быть с прямой или разветвленной цепью.
Если не указано иное, здесь С3-6циклоалкильные группы включают, но не ограничены ими, циклические алкильные группировки циклопропил, циклобутил, циклопентил и циклогексил.
Если не указано иное, здесь С2-6алкенил включает, но не ограничен ими, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил и 3-бутенил.
Если не указано иное, здесь С2-6алкинил включает, но не ограничен ими, этинил, 1-пропинил, 2-пропинил, 1-бутинил, 2-бутинил и 3-бутинил.
Если не указано иное, здесь галогено или галоген относится к фтору, хлору, брому или йоду.
Здесь арил относится к фенилу и нафтилу.
Здесь ароматические или неароматические гетероциклические кольца включают, но не ограничены ими, N- или С-связанный фурил, имидазолил, оксазолил, пирролидинил, тиазолил, тиофенил, пирролил, морфолинил, пиперидинил, пиперазинил, пиразинил, пиридил, пиримидинил, инданил, индолил, хинолинил, изохинолинил, хиназолинил, хиноксалинил, бензо[b]тиофенил, бензоксазолил или бензотиазолил;
DCM относится к дихлорметану;
ЕtOAс относится к этилацетату;
EDC относится к 1-(3-диметиламинопропил)-3-этилкарбодиимиду;
EDTA относится к этилендиаминтетрауксусной кислоте;
HEPES относится к 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты мононатриевой соли; и
TEA относится к триэтиламину.
В описанных здесь способах, если необходимо, гидрокси, амино или другие реакционноспособные группы могут быть защищены защитной группой, как описано в "Protecting groups in Organic Synthesis", 3rd Edition (1999) by Greene and Wuts.
Если не указано иное, реакции проводят в инертной атмосфере, предпочтительно в атмосфере азота, и обычно их проводят при давлении от примерно одной до примерно трех атмосфер (от 101,3 до 303,9 кПа), предпочтительно при давлении окружающей среды (примерно одна атмосфера (примерно 101,3 кПа)).
Соединения по изобретению и промежуточные соединения могут быть выделены из их реакционных смесей стандартными методами.
Соли присоединения кислоты соединений формулы I, которые можно упомянуть, включают соли минеральных кислот, например гидрохлорид и гидробромид; и соли, образованные с органическими кислотами, например формиат, ацетат, малеат, бензоат, тартрат и фумарат.
Соли присоединения кислоты соединений формулы I могут быть образованы в результате взаимодействия свободного основания или его соли, энантиомера или защищенного производного с одним или более эквивалентами соответствующей кислоты. Эта реакция может быть проведена в растворителе или в среде, в которой соль нерастворима, или в растворителе, в котором соль растворима, например в воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или в смеси растворителей, которые могут быть удалены в вакууме или сублимационной сушкой. Эта реакция может представлять собой метатетический процесс, или может быть проведена на ионообменной смоле.
Некоторые соединения формулы I могут существовать в таутомерных или энантиомерных формах, которые все входят в объем изобретения. Различные оптические изомеры могут быть выделены разделением рацемической смеси соединений с использованием общепринятых методов, например фракционной кристаллизацией или хиральной ВЭЖХ. Альтернативно, индивидуальные энантиомеры могут быть получены в результате взаимодействия соответствующих оптически активных исходных веществ в реакционных условиях, не вызывающих рацемизацию.
СИНТЕЗ И СХЕМЫ
Соединения формулы I могут быть получены способами, проиллюстрированными на приведенных ниже схемах.
Схема 1:

Схема 1 иллюстрирует синтез соединений, где r равно 0, и, в частности, синтез соединения Примера 1, где R1 представляет собой этил, А представляет собой фенил и n, q и m равны 0.
Схема 2:

Схема 2 иллюстрирует синтез соединений, где r выше, чем 0, и, в частности, синтез соединения Примера 3.
В общем, соединения по изобретению могут быть получены в результате взаимодействия 2-амино-1-фенилэтанона и алкилсульфонилхлорида в присутствии TEA и DCM с образованием N-(2-оксо-2-фенилэтил)-алкилсульфонамида; обработки алкилсульфонамида NaOH и THF (тетрагидрофуран) в этаноле с образованием 3-алкилсульфониламино-2-фенилхинолин-4-карбоновой кислоты и взаимодействия этой карбоновой кислоты с ариламином в присутствии EDCI (1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид) и НОВТ (1-гидроксибензотриазол) с образованием соединения формулы I.
Другие соединения по изобретению могут быть получены в результате обработки метилового эфира бромалкил-2-фенил-хинолин-4-карбоновой кислоты NaN3 в DMF/THF с образованием его азидопроизводного; обработки этого производного LiOH в смеси этанол/вода с образованием азидокарбоновой кислоты; взаимодействия этой кислоты с ариламином в присутствии EDCI и НОВТ с образованием азидохинолина; обработки этого азидохинолина палладием на углероде в этаноле с образованием амина; и взаимодействия этого амина и алкилсульфонилхлорида в присутствии TEA и DCM с образованием соединения формулы I.
В следующем аспекте изобретение относится к описанным здесь соединениям, где один или более из атомов является радиоактивным изотопом того же элемента. В конкретной форме данного аспекта изобретения соединение мечено тритием. Такие меченые соединения синтезируют либо путем включения меченых исходных веществ, либо, в случае трития, путем обмена водорода на тритий известными способами. Известные способы включают (1) электрофильное галогенирование с последующим восстановлением галогена в присутствии источника трития, например с гидрогенизацией газом тритием в присутствии палладиевого катализатора, или (2) обмен водорода на тритий, который осуществляют в присутствии газа трития и подходящего металлоорганического (например, палладиевого) катализатора.
Соединения по изобретению, меченные тритием, полезны для открытия новых лекарственных соединений, которые связываются с рецептором NK-3 и модулируют его активность посредством агонизма, частичного агонизма или антагонизма. Такие меченные тритием соединения можно использовать в анализах, в которых измеряют вытеснение таких соединений, чтобы оценить связывание лигандов, которые связываются с рецептором NK-3.
В еще одном аспекте изобретение относится к описанным здесь соединениям, дополнительно содержащим один или более атомов радиоактивного изотопа. В конкретном воплощении данного аспекта изобретения соединение содержит радиоактивный галоген. Такие меченные радиоизотопом соединения синтезируют путем включения меченных радиоизотопом исходных веществ известными способами. Конкретными воплощениями данного аспекта изобретения являются воплощения, в которых радиоизотоп выбран из18F,123I,125I,131I,75Вr,76Вr,77Вr или82Вr. Наиболее предпочтительным конкретным воплощением данного аспекта изобретения является воплощение, в котором радиоизотоп представляет собой18F. Такие соединения, содержащие один или более атомов радиоактивного изотопа, полезны в качестве лигандов для позитронно-эмиссионной томографии (ПЭТ) и других применений и методик для определения локализации рецепторов NK-3.
Терапевтическое применение соединений
В другом аспекте изобретение относится к описанным здесь соединениям формулы I и к применению таких соединений в терапии и в композициях, полезных для терапии.
В другом аспекте изобретение охватывает применение описанных здесь соединений для терапии заболеваний, опосредованных действием рецепторов NK-3. Такой аспект охватывает способы лечения или профилактики заболеваний или состояний, при которых полезно модулирование рецептора NK-3, включающие введение терапевтически эффективного количества антагонистического соединения по изобретению субъекту, страдающему указанным заболеванием или состоянием.
В одном воплощении данного аспекта изобретения предложен способ лечения или профилактики расстройств, где расстройство представляет собой депрессию, тревогу, шизофрению, когнитивные расстройства, психозы, ожирение, воспалительные заболевания, включая синдром раздраженного кишечника и воспалительное кишечное расстройство, рвоту, преэклампсию, хроническую обструктивную болезнь легких, расстройства, связанные с избыточными гонадотропинами и/или андрогенами, в том числе дисменорею, доброкачественную гиперплазию простаты, рак простаты или рак яичка, включающий введение фармакологически эффективного количества соединения формулы I пациенту, нуждающемуся в этом.
В еще одном аспекте изобретение относится к применению соединения по изобретению, его энантиомера или его фармацевтически приемлемой соли в лечении или профилактике заболевания или состояния, при котором полезно модулирование рецептора NK-3. Конкретными заболеваниями и состояниями, которые можно лечить, являются депрессия, тревога, шизофрения, когнитивные расстройства, психозы, ожирение, воспалительные заболевания, включая синдром раздраженного кишечника и воспалительное кишечное расстройство, рвота, преэклампсия, хроническая обструктивная болезнь легких, расстройства, связанные с избыточными гонадотропинами и/или андрогенами, в том числе дисменорея, доброкачественная гиперплазия простаты, рак простаты или рак яичка. В более конкретном воплощении предложено применение соединения в лечении или профилактике тревоги, депрессии, шизофрении и ожирения.
В еще одном аспекте изобретение относится к применению соединения по изобретению, его энантиомера или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или профилактики заболеваний или состояний, упомянутых здесь. Конкретным воплощением данного аспекта изобретения является применение соединения по изобретению в изготовлении лекарственного средства для лечения или профилактики депрессии, тревоги, шизофрении, когнитивных расстройств, психозов, ожирения, воспалительных заболеваний, включая синдром раздраженного кишечника и воспалительное кишечное расстройство, рвоты, преэклампсии, хронической обструктивной болезни легких, расстройств, связанных с избыточными гонадотропинами и/или андрогенами, в том числе дисменорею, доброкачественную гиперплазию простаты, рак простаты или рак яичка.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Соединения по изобретению, их энантиомеры и их фармацевтически приемлемые соли можно применять сами по себе или в форме подходящих лекарственных препаратов для энтерального или парентерального введения. В соответствии со следующим аспектом изобретения предложена фармацевтическая композиция, содержащая, предпочтительно, менее 80% и, более предпочтительно, менее 50 масс.% соединения по изобретению в смеси с инертным фармацевтически приемлемым разбавителем, смазывающим агентом или носителем.
Примерами разбавителей, смазывающих агентов и носителей являются:
- для таблеток и драже: лактоза, крахмал, тальк, стеариновая кислота;
- для капсул: винная кислота или лактоза;
- для инъекционных растворов: вода, спирты, глицерин, растительные масла;
- для суппозиториев: натуральные или отвержденные масла или воски.
Предложен также способ изготовления такой фармацевтической композиции, включающий смешивание или компаундирование ингредиентов вместе и формование этих смешанных ингредиентов в таблетки или суппозитории, инкапсулирование ингредиентов в капсулы или растворение ингредиентов с образованием инъекционных растворов.
Фармацевтически приемлемые производные включают сольваты и соли. Например, соединения по изобретению могут образовать соли присоединения кислоты с кислотами, такими как обычные фармацевтически приемлемые кислоты, включая малеиновую, соляную, бромистоводородную, фосфорную, уксусную, фумаровую, салициловую, лимонную, молочную, миндальную, винную и метансульфоновую кислоты.
Соли присоединения кислоты соединений формулы I, которые можно упомянуть, включают соли минеральных кислот, например, гидрохлорид и гидробромид; и соли, образованные с органическими кислотами, такие как формиат, ацетат, малеат, бензоат, тартрат и фумарат. Соли присоединения кислоты соединений формулы I могут быть образованы в результате взаимодействия свободного основания или соли, их энантиомера или защищенного производного с одним или более эквивалентами подходящей кислоты. Эту реакцию можно проводить в растворителе или в среде, в которой соль нерастворима, или в растворителе, в котором соль растворима, например, в воде, диоксане, этаноле, тетрагидрофуране или диэтиловом эфире, или в смеси растворителей, которую можно удалить в вакууме или сублимационной сушкой. Эта реакция может представлять собой метатетический процесс, или ее можно проводить на ионообменной смоле.
Для применений, способов, лекарственных средств и композиций, упомянутых здесь, количество используемого соединения и вводимая доза, разумеется, будет варьировать в зависимости от используемого соединения, способа введения и желательного лечения. Однако, как правило, удовлетворительные результаты получают, когда соединения по изобретению вводят в суточной дозе от примерно 0,1 мг до примерно 20 мг/кг массы тела животного. Такие дозы можно вводить в дробных дозах от 1 до 4 раз в сутки или в форме с пролонгированным высвобождением. Для человека суммарная суточная доза находится в пределах от 5 мг до 1400 мг, более предпочтительно от 10 мг до 100 мг, и стандартные лекарственные формы, пригодные для перорального введения, содержат от 2 мг до 1400 мг соединения, смешанного с твердыми или жидкими фармацевтическими носителями, смазывающими агентами и разбавителями.
Некоторые соединения по изобретению могут существовать в таутомерных, энантиомерных, стереоизомерных формах или геометрических изомерных формах, которые все входят в объем изобретения. Различные оптические изомеры могут быть выделены путем разделения рацемической смеси соединений с использованием общепринятых методов, например фракционной кристаллизацией или хиральной ВЭЖХ. Альтернативно, индивидуальные энантиомеры могут быть получены в результате взаимодействия соответствующих оптически активных исходных веществ в реакционных условиях, не вызывающих рацемизацию.
Иллюстративные соединения по изобретению могут быть получены способами, аналогичными способам, представленым на Схеме 1. Специалистам в данной области понятно, что многие подходящие амины и хлорангидриды кислот и карбоновые кислоты могут быть использованы для образования соединений, входящих в объем объекта, описанного здесь как формула I.
ИЛЛЮСТРАТИВНЫЕ СОЕДИНЕНИЯ
Иллюстративные соединения и способы описывают изобретение путем иллюстрации и примера для ясности понимания. Однако специалистам в данной области после рассмотрения и изучения соединений, способов и методов по данному изобретению будут очевидны модификации и изменения, которые могут быть выполнены без отклонения от сущности или объема изобретения.
Пример 1. 3-[(Метилсульфонил)амино]-2-фенил-N-[(1S)-1-фенилпропил]хинолин-4-карбоксамид(1) (обозначения в Примере 1 относятся к Схеме 1).

К раствору 3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоновой кислоты (1с) (342 мг, 1,0 ммоль), гидрата НОВТ (231 мг, 1,5 ммоль), 4-метилморфолина (276 мкл, 1,5 ммоль) в тетрагидрофуране (50 мл) добавляли EDCI (289 мг, 1,5 ммоль) при комнатной температуре в атмосфере N2. Затем добавляли (S)-1-фенилпропиламин (135 мг, 1,0 ммоль) и эту реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Весь растворитель удаляли в вакууме, и остаток распределяли между этилацетатом и 10% водным раствором бикарбоната натрия, высушивали над сульфатом натрия, а затем концентрировали в вакууме. Остаток очищали хроматографией, элюируя смесью 15-25% этилацетата/гексан, с получением указанного в заголовке соединения (70 мг, 15%) в виде твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.94 (t, 3Н), 1.97 (m, 2Н), 3.44 (s, 3Н), 5.17 (q, 1Н), 5.47 (m, 2Н), 7.32 (d, 2Н), 7.34 (d, 2Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.42 (m, 2Н). МС-ХИАД (масс-спектрометрия с химической ионизацией при атмосферном давлении) m/z=460 (М+1). ЖХМС (жидкостная хроматография/масс-спектрометрия: 2,51 мин.
Исходная кислота, 3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоновая кислота (1 с), была получена следующим образом:
N-(2-оксо-2-фенилэтил)метансульфонамид (1b)
К раствору 2-амино-1-фенилэтанона гидрохлорида (1а) (1715 мг, 10 ммоль) в DMF (20 мл) добавляли TEA (2,8 мл, 20 ммоль). После охлаждения в бане лед - вода медленно добавляли метилсульфонилхлорид (0,77 мл, 10 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Смесь распределяли между дихлорметаном и рассолом, высушивали над сульфатом натрия, а затем концентрировали в вакууме с получением указанного в заголовке соединения (2100 мг, 98%) в виде не совсем белого твердого вещества. МС-ХИАД, m/z=214 (М+1). ЖХМС: 1,19 мин.
3-[(Метилсульфонил)амино1-2-фенилхинолин-4-карбоновая кислота (1 с)
К изатину (441 мг, 3 ммоль) добавляли раствор гидроксида натрия (1,15 г, 29,0 ммоль) в воде (2,5 мл). Полученный коричневый осадок энергично перемешивали при комнатной температуре в течение 20 минут, после чего его нагревали до 85°С. Затем добавляли по каплям раствор N-(2-оксо-2-фенилэтил)метансульфонамида (1b) (639 мг, 3,0 ммоль) в смеси этанол/THF/вода (6,3 мл/1,25 мл/6,3 мл) за 30 минут. Реакционную смесь перемешивали при 85°С еще в течение 4 ч, после чего охлаждали до комнатной температуры. Все органические растворители удаляли в вакууме, и водный остаток концентрировали до объема примерно 6 мл. Этот водный остаток промывали эфиром (3×10 мл), а затем водный остаток подкисляли при охлаждении до рН 4 уксусной кислотой. Образовавшийся осадок собирали, промывали водой и высушивали с получением указанного в заголовке соединения в виде твердого вещества (721 мг, 70,3%).1Н ЯМР (300 МГц, CDCI3) δ 3.11 (s, 3Н), 7.05 (d, 1Н), 7.39 (d, 2Н), 7.64 (m, 2Н), 7.78 (m, 1Н), 8.06 (m, 1Н), 8.19 (m, 1Н), 8.47 (m, 1Н), 10.03 (b, 2Н). МС-ХИАД, m/z=343 (М+1). ЖХМС: 1,07 мин.
Пример 2: 3-(Метилсульфониламинометил)-2-фенилхинолин-4-карбоновой кислоты ((S)-1-фенилпропил)-амид (обозначения в Примерах 2 и 3 относятся к Схеме 2).
К раствору 3-(аминометил)-2-фенил-N-[(1S)-1-фенилпропил]хинолин-4-карбоксамида (2е) (197 мг, 0,5 ммоль) в DCM (30 мл) добавляли триэтиламин (140 мкл, 1,0 ммоль) в атмосфере N2. После охлаждения в бане вода - лед добавляли по каплям метансульфонилхлорид (39 мкл, 0,51 ммоль) и реакционную смесь перемешивали при комнатной температуре дополнительно в течение 2 ч. Смесь промывали рассолом (10 мл), и органическую фазу отделяли и высушивали над сульфатом натрия, а затем концентрировали в вакууме. Остаток очищали хроматографией, элюируя смесью 15-25% этилацетата/гексан, с получением указанного в заголовке соединения (79 мг, 42%) в виде твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.94 (t, 3Н), 1.95 (m, 2Н), 2.99 (s, 3Н), 4.88 (s, 2Н), 5.25 (q, 1Н), 6.66 (b, 2Н), 7.30 (d, 2Н), 7.34 (d, 2Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.20 (m, 2Н). МС-ХИАД, m/z=474 (М+1). ЖХМС: 2,16 мин.
Пример 3. 3-{[(Этилсульфонил)амино]метил}-2-фенил-N-[(1S)-1-фенилпропил]хинолин-4-карбоксамид

По методике, аналогичной описанной в Примере 2, но используя в качестве реагента этансульфонилхлорид (48,6 мкл, 0,51 ммоль), получили указанное в заголовке соединение в виде белого твердого вещества. (90 мг, 38%).1Н ЯМР (300 МГц, CDCI3) δ 0.92 (t, 3Н), 1.32 (t, 3Н), 1.95 (m, 2Н), 3.04 (q, 2Н), 4.87 (s, 2Н), 5.17 (q, 1Н), 5.48 (b, 2Н), 7.25 (d, 2Н), 7.34 (d, 2Н), 7.39 (т, 1Н), 7.78 (т, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.17 (m, 2Н). МС-ХИАД, m/z=488 (М+1). ЖХМС: 2,24 мин.
Исходный амин, 3-(аминометил)-2-фенил-N-[(1S)-1-фенилпропил]-хинолин-4-карбоксамид, для Примеров 2 и 3 был получен следующим образом:
Метил-3-(азидометил)-2-фенилхинолин-4-карбоксилат (2b)
К раствору 3-(бромметил)-2-фенилхинолин-4-карбоксилата (2а) (2000 мг, 5,618 ммоль) в THF/DMF (50 мл/10 мл) добавляли азид натрия (402 мг, 6,18 ммоль), и эту реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 12 ч. Весь растворитель удаляли в вакууме, и остаток распределяли между этилацетатом и рассолом, высушивали над сульфатом натрия, а затем концентрировали в вакууме. Остаток очищали хроматографией, элюируя смесью 10% этилацетата/гексан, с получением указанного в заголовке соединения (1781 мг, 99,7%) в виде беловатого твердого вещества. МС-ХИАД, m/z=319 (М+1). ЖХМС: 2,34 мин.
3-(Азидометил)-2-фенилхинолин-4-карбоновая кислота (2с)
К раствору метил-3-(азидометил)-2-фенилхинолин-4-карбоксилата (2b) (1781 мг, 5,0 ммоль) в метаноле (50 мл) добавляли раствор моногидрата гидроксида лития (674 мг, 28,1 ммоль) в 25 мл воды. Реакционную смесь перемешивали при кипячении с обратным холодильником в течение 4 ч, и остаток подкисляли 1 н. HCl до рН 2. Объем реакционной смеси концентрировали при пониженном давлении. Полученную водную фазу экстрагировали этилацетатом (150 мл). Органическую фазу отделяли и промывали рассолом (20 мл) и высушивали над сульфатом натрия, а затем концентрировали в вакууме. В результате кристаллизации из смеси этилацетат/гексан получили указанное в заголовке соединение (1200 мг, 66%) в виде не совсем белого твердого вещества. МС-ХИАД, m/z=305 (М+1). ЖХМС: 1,09 мин.
3-(Азидометил)-2-фенил-N-[(1S)-1-фенилпропил1хинолин-4-карбоксамид (2d)
К раствору 3-(азидометил)-2-фенилхинолин-4-карбоновой кислоты (2 с) (997 мг, 3,28 ммоль), гидрата НОВТ (760 мг, 4,92 ммоль), 4-метилморфолина (551 мкл, 4,92 ммоль) в тетрагидрофуране (50 мл) добавляли EDC (960 мг, 4,92 ммоль) при комнатной температуре в атмосфере N2. Затем добавляли (S)-1-фенилпропиламин (488,5 мг, 3,62 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 12 ч. Весь растворитель удаляли в вакууме, и остаток распределяли между этилацетатом и 10% водным раствором бикарбоната натрия, высушивали над сульфатом натрия, а затем концентрировали в вакууме. Остаток очищали хроматографией, элюируя смесью 15-25% этилацетата/гексан, с получением указанного в заголовке соединения (1000 мг, 73%) в виде светло-желтого твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.94 (I, 3Н), 2.01 (m, 2Н), 5.17 (q, 1Н), 5.19 (s, 2Н), 5.72 (b, 1Н), 7.21 (d, 2Н), 7.34 (d, 2Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.49 (m, 2Н). МС-ХИАД, m/z=422 (М+1). ЖХМС: 2,47 мин.
3-(Аминометил)-2-фенил-N-[(1s)-1-фенилпропил1хинолин-4-карбоксамид (2е)
К раствору 3-(азидометил)-2-фенил-N-[(1s)-1-фенилпропил]хинолин-4-карбоксамида (2d) (1000 мг, 2,37 ммоль) в этаноле (200 мл) добавляли Pd/C (10%, 1190 мг) и HCl (2 н., 2,5 мл). Реакционную смесь гидрогенизировали при 50 фунт/кв. дюйм (344,738 кПа) Н2 при комнатной температуре в течение 1,5 ч. Катализатор удаляли фильтрованием через толстый слой диатомовой земли, фильтр промывали этанолом, и промывки и этанольный раствор объединяли и концентрировали в вакууме. В результате кристаллизации из дихлорметана и эфира получили указанное в заголовке соединение (940 мг, 92%) в виде соли с соляной кислотой.1Н ЯМР (300 МГц, CDCl3) δ 0.92 (t, 3Н), 2.01 (m, 2Н), 3.18 (b, 3Н), 4.39 (t, 2Н), 5.16 (q, 1Н), 7.30 (d, 2Н), 7.34 (d, 2Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.44 (m, 2Н). МС-ХИАД, m/z=396 (М+1). ЖХМС: 1,70 мин.
Пример 4. 3-[Метил(метилсульфонил)амино]2-фенил-N-[(1S)-1-фенилпропил1хинолин-4-карбоксамид

К раствору 3-[(метилсульфонил)амино]-2-фенил-N-[(1S)-1-фенил пропил]-хинолин-4-карбоксамида (1) (459 мг, 1,0 ммоль) в 5 мл DMF добавляли Cs2CO3(325,8 мг, 1,0 ммоль) и CH3l (62,3 мкл, 1,0 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере N2 в течение 2 ч. Весь растворитель удаляли в вакууме, и остаток распределяли между этилацетатом и 10% водным раствором бикарбоната натрия, высушивали над сульфатом натрия, а затем концентрировали в вакууме. Остаток очищали перекристаллизацией из эфира/СН2Сl2 с получением указанного в заголовке соединения (210 мг, 44,4%) в виде твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.94 (t, 3Н), 1.97 (m, 2Н), 2.71 (s, 3Н), 3.43 (s, 3Н), 5.10 (q, 1Н), 5.14 (b, 1Н), 7.32 (d, 2Н), 7.34 (d, 2Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.42 (m, 2Н). МС-ХИАД, m/z=474 (М+1). ЖХМС: 2,32 мин.
Пример 5. N-[(1S)-циклопропил(фенил)метил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамид (3).

По методике, аналогичной описанной в Примере 1, за исключением того, что использовали (5)-1-циклопропил-1-фенилметанамин в качестве аминного реагента, было получено указанное в заголовке соединение (3) (50%) в виде белого твердого вещества.1Н ЯМР (300 МГц, CDCl3) δ 0.44 (m, 2Н), 0.61 (m, 2Н), 1.57 (m, 1H), 3.06 (s, 3Н), 4.76 (q, 1H), 7.32 (d, 2H), 7.34 (d, 2H), 7.39 (m, 1H), 7.78 (m, 2H), 7.84 (m, 2H), 8.08 (m, 1H), 8.30 (m, 2H), 8.42 (m, 2H). МС-ХИАД, m/z=472 (М+1). ЖХМС: 2,21 мин.
Пример 6. N-[(1S)-циклопропил(3-фторфенил)метил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамид (4)

По методике, аналогичной описанной в Примере 1, за исключением того, что использовали (S)-1-циклопропил-1-(3-фторфенил)метанамин в качестве аминного реагента, было получено указанное в заголовке соединение (4) (35%) в виде белого твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.46 (m, 2Н), 0.66 (m, 2Н), 1.57 (m, 1Н), 3.07 (s, 3Н), 4.99 (q, 1Н), 7.32 (d, 2Н), 7.34 (m, Н), 7.39 (m, 1Н), 7.78 (m, 2Н), 7.84 (m, 2Н), 8.08 (m, 1Н), 8.30 (m, 2Н), 8.42 (m, 2Н). МС-ХИАД, m/z=490 (М+1). ЖХМС: 2,24 мин.
Пример 7. N-[(1S)-1-циклогексилэтил]-3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамид (5)
По методике, аналогичной описанной в Примере 1, за исключением того, что использовали (1S)-1-циклогексилэтанамин в качестве аминного реагента, было получено указанное в заголовке соединение (5) (36%) в виде белого твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 1.12-1.16 (m, 4Н), 1.26 (d, 2Н), 1.45-1.55 (m, 4Н), 1.57 (m, 2Н), 1.92 (m, 1Н), 2.3 (s, 3Н), 4.23 (q, 1Н), 6.62 (d, 1Н), 7.37 (d, 2H), 7.56 (m, 2H), 7.64 (m, 2H), 7.78 (m, 1H), 7.80 (d, 1H), 8.14 (d, 1H). МС-ХИАД, m/z=452 (М+1). ЖХМС: 2,30 мин.
Пример 8. N-[(1R,2S)-2-гидрокси-1-Фенилпропил]3-[(метилсульфонил)амино]-2-фенилхинолин-4-карбоксамид (6)
К раствору 3-метансульфониламино-2-фенил-4-карбоновой кислоты (1 с) (250 мг, 0,73 ммоль), гидрата HOBT (148 мг, 1,1 ммоль), 4-метилморфолина (160 мкл, 1,46 ммоль) в метиленхлориде (15 мл) добавляли EDCI (210 мг, 1,1 ммоль) при комнатной температуре в атмосфере N2. Добавляли смесь (1R,2S)-1-aмино-1-фенилпропан-2-ола гидрохлорида и 4-метилморфолина (193 мкл, 1,75 ммоль) в метиленхлориде (5 мл), и эту реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Раствор распределяли против воды и дважды экстрагировали метиленхлоридом. Объединенные органические экстракты промывали рассолом, высушивали над сульфатом магния, а затем концентрировали в вакууме с добавлением силикагеля. Соединение затем элюировали из силикагеля и подвергали хроматографии, элюируя смесью 15-30% этилацетата/дихлорметан, с получением указанного в заголовке соединения (167 мг, 48%).1Н ЯМР (500,333 МГц, CDCI3) δ 1.10 (d, J=6.6 Гц, 3Н), 2.23 (s, 3Н), 2.69 (d', J=6.7 Гц, 1Н), 4.44 - 4.47 (m, 1Н), 5.35 (dd, J=8.4, 3.3 Гц, 1Н), 7.35 (d, J=7.2 Гц, 1Н), 7.39 (dd, J=6.9, 6.9 Гц, 2Н), 7.44 (d, J=7.5 Гц, 2Н), 7.47-7.55 (m, 4Н), 7.71-7.78 (m, 4Н), 8.16 (d, J=8.4 Гц, 1Н) МС-ХИАД, m/z=476,1 (М+1). ЖХМС: 1,86 мин.
Исходный амин, (1R,2S)-1-амино-1-фенилпропан-2-ола гидрохлорид, был получен из (1S, 2S)-(-)-1-фенилпропиленоксида известными способами.
Пример 9. 3-[(Циклопропилсульфонил)амино1-2-фенил-N-[(1S)-1-фенилпропил]хинолин-4-карбоксамид (7)

По методике, аналогичной описанной в Примере 1, за исключением того, что использовали 3-[(циклопропилсульфонил)амино]-2-фенилхинолин-4-карбоновую кислоту в качестве кислотного реагента, было получено указанное в заголовке соединение (7) (85%) в виде белого твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.79-0.82 (m, 2Н), 0.95 (t, 3Н), 1.09-1.11 (m, 2Н), 1.87-2.07 (m, 2Н), 4.13 (m, 1Н), 5.19 (q, 1Н), 7.06 (d, 1Н), 7.09 (d, 2Н), 7.32 (m, 1Н), 7.37 (d, 2Н), 7.56 (m, 2Н), 7.64 (m, 2Н), 7.78 (m, 1Н), 7.80 (d, 1Н), 7.93 (m, 1Н), 8.18 (d, 1Н). МС-ХИАД, m/z=486 (М+1). ЖХМС: 2,31 мин.
Пример 10. N-[(S)циклопропил(фенил)метил]-3-[(циклопропилсульфонил)амино]-2-фенилхинолин-4-карбоксамид (8)

По методике, аналогичной описанной в Примере 1, за исключением того, что использовали 3-[(циклопропилсульфонил)амино]-2-фенилхинолин-4-карбоновую кислоту в качестве кислотного реагента, было получено указанное в заголовке соединение (8) (45%) в виде белого твердого вещества.1Н ЯМР (300 МГц, CDCI3) δ 0.43 (m, 2Н), 0.63 (m, 2Н), 0.79-1.11 (m, 4Н), 0.95 (t, 3Н), 1.56 (m, 1Н), 3.63 (m, 1Н), 4.75 (q, 1Н), 7.04 (d, 1Н), 7.09 (d, 2Н), 7.32 (m, 1Н), 7.37 (d, 2Н), 7.56 (m, 2Н), 7.64 (m, 2Н), 7.78 (m, 1Н), 7.80 (d, 1Н), 7.93 (m, 1Н), 8.18 (d, 1Н). МС-ХИАД, m/z=498 (М+1). ЖХМС: 2,37 мин.
БИОЛОГИЧЕСКИЕ ТЕСТЫ
Активность связывания рецептора NK-3:
Как правило, активность связывания NK-3r можно оценить, используя анализы, которые проводят, как описано в Krause et al. (Proc. Natl. Acad. Sci. USA 94: 310-315, 1997). Комплементарную ДНК NK-3r клонируют из гипоталамической РНК человека, используя стандартные методики. кДНК рецептора встраивают в подходящий экспрессионный вектор, которым трансфицируют клеточную линию яичника китайского хомячка, и стабильно экспрессирующая клональная клеточная линия может быть выделена, охарактеризована и использована для экспериментов.
Клетки могут быть выращены в среде для тканевых культур по методикам, известным специалистам в данной области, и выделены низкоскоростным центрифугированием. Клеточные осадки могут быть гомогенизированы, суммарные клеточные мембраны могут быть выделены высокоскоростным центрифугированием и суспендированы в забуференном физиологическом растворе. Обычно анализы на связывание рецептора можно проводить путем инкубации подходящих количеств очищенных мембранных препаратов с [125I]-метилРhе7-нейрокинином В в присутствии или в отсутствие тестируемых соединений. Мембранные белки могут быть собраны путем быстрого фильтрования, и радиоактивность может быть считана в сцинтилляционном β-счетчике для планшетов. Неспецифичное связывание можно отличить от специфичного связывания путем использования подходящих контролей, и сродство соединений к экспрессируемому рецептору можно определить путем использования различных концентраций соединений.
Получение мембранных препаратов из клеток СНО, трансфицированных клонированными рецепторами NK-3:
Ген человеческого рецептора NK-3 был клонирован с использованием методов, аналогичных описанным для других человеческих рецепторов NK (Aharony et al., Mol. Pharmacol. 45: 9-19, 1994; Caccese era/., Neuropeptides 33, 239-243, 1999). Последовательность ДНК клонированного рецептора NK-3 отличалась от опубликованной последовательности (Buell et al., FEBS Letts. 299,90-95, 1992; Huang et al., Biochem. Biophys. Res. Commun. 184, 966-972, 1992) наличием молчащей замены одного основания Т>С в нуклеотиде 1320 кодирующей последовательности. Поскольку эта замена является молчащей, клонированный ген обеспечивает первичную аминокислотную последовательность кодируемого рецепторного белка NK-3, идентичную опубликованной последовательности. кДНК рецептора использовали для трансфекции клеток СНО-К1 с использованием стандартных методов, и клон, стабильно экспрессирующий рецептор, был выделен и охарактеризован. Препараты плазматических мембран из этих клеток были получены, как опубликовано (Aharony et al., 1994).
Клетки собирали и центрифугировали для удаления среды. Клеточный осадок гомогенизировали (Brinkman Polytron, три 15-секундных импульса на льду) в буфере, состоящем из 50 мМ Трис-HCl (рН 7,4), 120 мМ NaCl, 5 мМ KCl, 10 мМ EDTA и ингибиторов протеаз (0,1 мг/мл ингибитора трипсина сои и 1 мМ йодоацетамида). Гомогенат центрифугировали при 1000хg в течение 10 мин при 4°С для удаления клеточных обломков. Осадки промывали один раз буфером для гомогенизации. Надосадочные жидкости объединяли и центрифугировали при 40000хg в течение 20 мин при 4°С. Осадок, содержащий мембраны, гомогенизировали на Polytron, как описано выше. Суспензию центрифугировали при 40000хg в течение 20 мин при 4°С, осадок суспендировали в буфере (20 мМ HEPES, рН 7,4, содержащем 3 мМ MgCl2, 30 мМ KCl и 100 мкМ тиорфан) и определяли концентрацию белка. Затем суспензию мембран разводили до 3 мг/мл буфером, содержащим 0,02% BSA (бычий сывороточный альбумин), и быстро замораживали. Образцы хранили при -80°С до использования.
Анализ на активность связывания рецептора NK-3:
Метод анализа на связывание рецептора с [125I]-MePhe7-NKB был модифицирован по сравнению с описанным Aharony et al., J. Pharmacol. Exper. Ther., 274:1216-1221, 1995.
Конкурентные эксперименты проводили в 0,2 мл аналитического буфера (50 мМ Трис-HCl, 4 мМ MnCl2, 10 мкМ тиорфан, рН 7,4), содержащего мембраны (2 мкг белка/реакция), тестируемые конкуренты и [125I]-MePhe7NKB (0,2 нМ). Немеченый гомологичный лиганд (0,5 мкМ) использовали для определения неспецифичного связывания. Инкубации проводили при 25°С в течение 90 мин. Лиганд, связанный с рецептором, выделяли вакуумной фильтрацией в Packard Harvester на планшетах GF/C, предварительно замоченных в 0,5% BSA. Планшеты промывали 0,02М Tris, рН 7,4. Компьютерный анализ констант равновесия связывания (КD и Ki), плотности рецептора (Вmax) и статистический анализ проводили, как опубликовано ранее (Aharony et аl., 1995), используя программное обеспечение GraphPad Prism или IDBS ХLfit.
Функциональная активность NK-3:
Как правило, функциональную активность NK-3 можно оценить в анализах мобилизации кальция в клеточных линиях, стабильно экспрессирующих NK-3r. Мониторинг мобилизации кальция, индуцированной агонистом метилРhе7-нейрокинина В, может быть осуществлен с использованием FLIPR (планшет-ридер с флуорометрической визуализацией) (Molecular Devices), способом, описанным изготовителем. Агонисты могут быть добавлены к клеткам и можно непрерывно считывать флуоресцентные ответы в течение вплоть до 5 мин. Действия антагонистов можно оценить путем предварительной инкубации клеток перед внесением агониста метилРhе7-нейрокинина В. Действие агонистов можно оценить путем наблюдения их собственной активности в такой системе.
Анализ на функциональную активность NK-3:
Клетки СНО, экспрессирующие рецептор NK-3, поддерживали в ростовой среде (среда Ham's F12, 10% ФСТ, 2 мМ L-глутамин и 50 мг/мл гигромицин В). За одни сутки до анализа клетки распределяли в 384-луночные планшеты в среде Ultraculture (Cambrex Bio Science) с 2 мМ L-глутамином до достижения 70-90% конфлюентности. Для количественного определения индуцированной рецептором NK-3 мобилизации кальция клетки сначала промывали аналитическим буфером, состоящим из сбалансированного солевого раствора Хэнкса, 15 мМ HEPES и 2,5 мМ пробенецида, рН 7,4. Затем клетки нагружали красителем Fluo4/AM (4,4 мкМ) в аналитическом буфере. Клетки инкубировали в течение одного часа, а затем промывали аналитическим буфером, подвергали воздействию 0,02-300 нМ сенктида и считывали флуоресцентный ответ, используя FLIPR (Molecular Devices Corporation). Для количественного определения антагонизма агонистического ответа клетки предварительно инкубировали с варьирующими концентрациями тестируемого соединения в течение 2-20 мин, а затем подвергали воздействию 2 нМ сенктида, концентрации, которая сама по себе вызывает 70% максимальный ответ кальция. Полученные данные анализировали, используя программное обеспечение XLfit (изготовитель IDBS) для определения значений ЕС50 и IC50.
Реферат
Настоящее изобретение относится к соединению, которое представляет собой ((S)-1-фенил-пропил)-амид 3-(метансульфониламино)-2-фенил-хинолин-4-карбоновой кислоты, его стереоизомеру, энантиомеру или фармацевтически приемлемой соли. Изобретение также относится к фармацевтической композиции на основе заявленного соединения. Технический результат: получено новое производное метилсульфонамидхинолина, полезное для модулирования рецептора NK-3. 2 н.п. ф-лы.
Формула
Документы, цитированные в отчёте о поиске
Способ получения фенилхинолинкарбоновых кислот или их эфиров,или их фармацевтически совместимой соли
Комментарии