Способ получения 4-амино-3-хлор-5-фтор-6-(замещенных)пиколинатов - RU2539578C1
Код документа: RU2539578C1
Описание
Настоящее изобретение относится к способу получения 4-амино-3-хлор-5-фтор-6-(замещенных)пиколинатов. Более конкретно, настоящее изобретение относится к способу получения 4-амино-3-хлор-5-фтор-6-(замещенных)пиколинатов, в которых 5-фторзаместитель вводят путем галогенидного замещения на ранней стадии в схеме способа.
Патент США № 6297197 B1 описывает, помимо прочего, определенные 4-амино-3-хлор-5-фтор-6-(алкокси или арилокси)пиколинатные соединения и их использование в качестве гербицидов. Патенты США №№ 6784137 B2 и 7314849 B2 описывают, помимо прочего, определенные 4-амино-3-хлор-5-фтор-6-(арил)пиколинатные соединения и их использование в качестве гербицидов. Патент США № 7432227 B2 описывает, помимо прочего, определенные 4-амино-3-хлор-5-фтор-6-(алкил)пиколинатные соединения и их использование в качестве гербицидов. Каждый из этих патентов описывает получение 4-амино-3-хлор-5-фтор-пиколинатных исходных материалов путем фторирования соответствующих 5-(незамещенных)пиколинатов, используя 1-(хлорметил)-4-фтор-1,4-диазониабицикло[2.2.2]октана бис(тетрафторборат). Было бы выгодно получать 4-амино-3-хлор-5-фтор-6-(замещенные)пиколинаты, не применяя непосредственное фторирование пиколината в положении 5 дорогостоящим фторирующим реагентом, таким как 1-(хлорметил)-4-фтор-1,4-диазониабицикло[2.2.2]октана бис(тетрафторборат).
Настоящее изобретение относится к способу получения 4-амино-3-хлор-5-фтор-6-(замещенных)пиколинатов из 3,4,5,6-тетрахлорпиколинонитрила. Более конкретно, настоящее изобретение относится к способу получения 4-амино-3-хлор-5-фтор-6-(замещенного)пиколината формулы I

в которой
R представляет собой (C1-C4)алкил, циклопропил, (C2-C4)алкенил или фенил, содержащий от 1 до 4 заместителей, в качестве которых независимо выбирают галоген, (C1-C4)алкил, (C1-C4)галогеналкил, (C1-C4)алкокси или (C1-C4)галогеналкокси; и
R1 представляет собой (C1-C12)алкил или незамещенный или замещенный (C7-C11)арилалкил;
который включает следующие стадии:
a) фторирование 3,4,5,6-тетрахлорпиколинонитрила (формула A)

источником фторид-иона для получения 3-хлор-4,5,6-трифторпиколинонитрила (формула B)

b) аминирование 3-хлор-4,5,6-трифторпиколинонитрила (формула B) аммиаком для получения 4-амино-3-хлор-5,6-дифторпиколинонитрила (формула C)

c) реакцию фторзаместителя в положении 6 4-амино-3-хлор-5,6-дифторпиколинонитрила (формула C) с гидразином для получения 4-амино-3-хлор-5-фтор-6-гидразинопиколинонитрила (формула D)
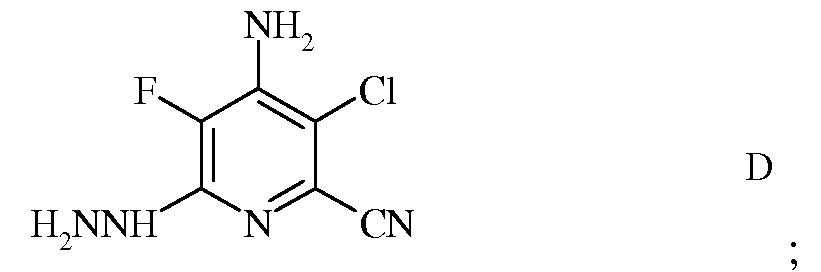
d) галогенирование 4-амино-3-хлор-5-фтор-6-гидразинопиколинонитрила (формула D) источником хлора, брома или йода для получения 4-амино-3-хлор-5-фтор-6-галогенпиколинонитрила формулы E
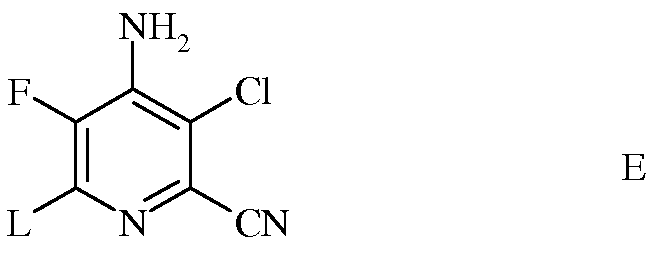
в которой L представляет собой Br, Cl или I;
e) гидролиз и этерификацию 4-амино-3-хлор-5-фтор-6-галогенпиколинонитрила формулы E сильной кислотой и спиртом (R1OH) для получения 4-амино-3-хлор-5-фтор-6-галогенпиколината формулы F

в которой L и R1 являются такими, как определено выше; и
f) сочетание 4-амино-3-хлор-5-фтор-6-галогенпиколината формулы F с арил-, алкил- или алкенилметаллоорганическим соединением формулы G

в которой R является таким, как определено выше, и Met представляет собой Zn-галогенид, Zn-R, три-((C1-C4)алкил)олово, медь или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород или (C1-C4)алкил или совместно образуют этиленовую или пропиленовую группу в присутствии содержащего переходный металл катализатора для получения 4-амино-3-хлор-5-фтор-6-(замещенного)пиколината формулы I.
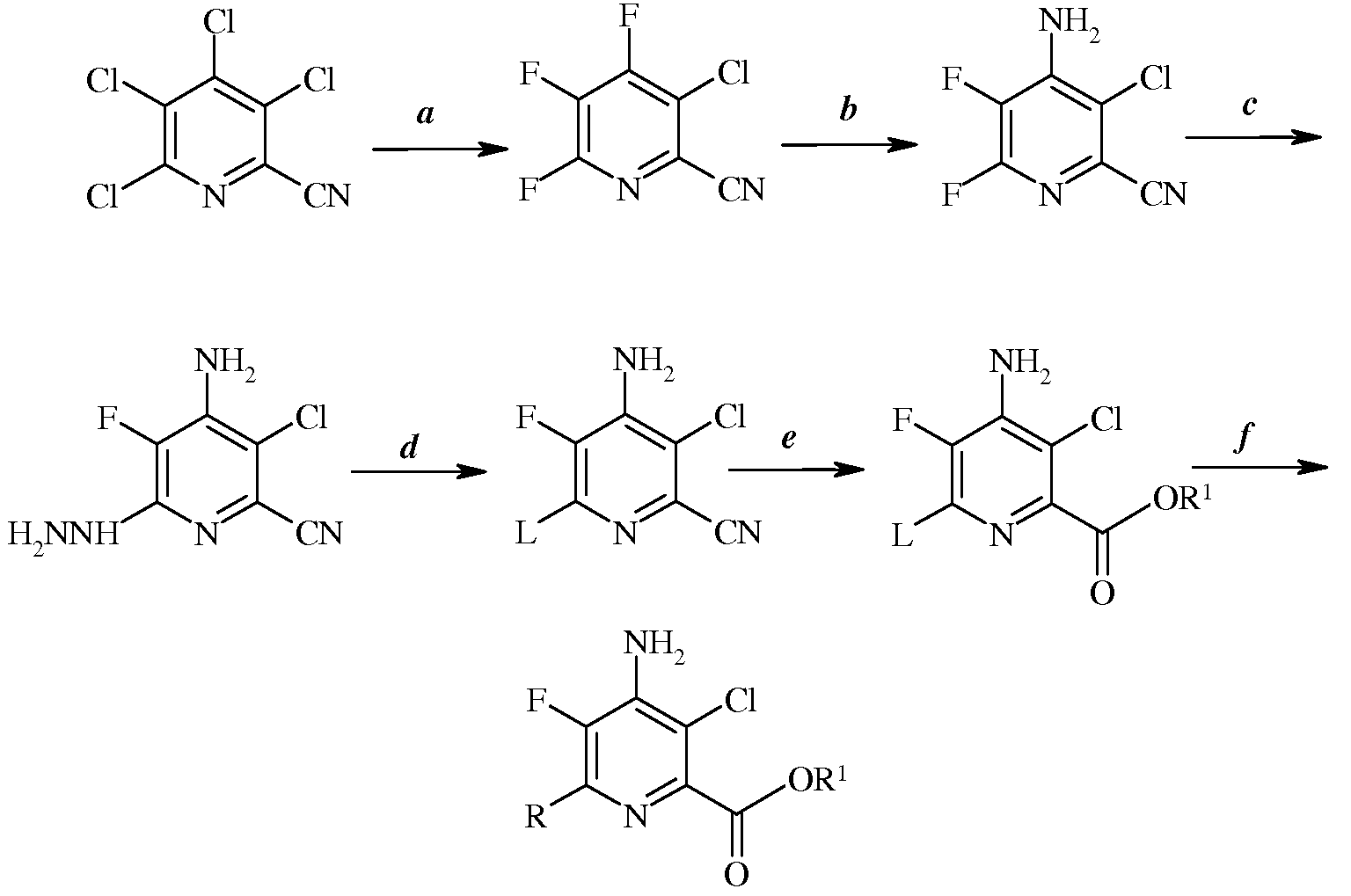
Стадии (a)-(f) можно осуществлять в последовательности, представленной на схеме I, или некоторые из стадий можно менять местами, получая другие последовательности, насколько это допускает совместимость химических реагентов. Например, стадию (f) можно осуществлять перед гидролизом и этерификацией стадии (e).
Схема I
Дополнительный аспект настоящего изобретения относится к усовершенствованному способу увеличения количества выделяемого 3-хлор-4,5,6-трифторпиколинонитрила (формула B)

полученного фторированием 3,4,5,6-тетрахлорпиколинонитрила (формула A)

источником фторид-ионов, где усовершенствование включает следующие стадии:
i) выделение полностью фторированного 3,4,5,6-тетрафторпиколинонитрила (формула H)
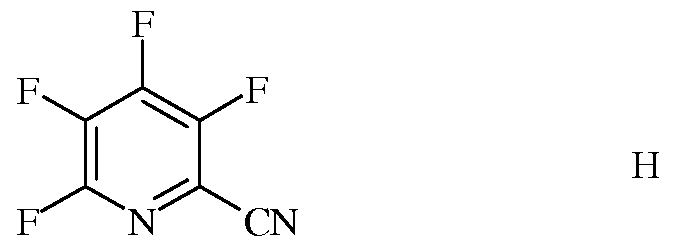
в качестве побочного продукта фторирования 3,4,5,6-тетрахлорпиколинонитрила;
ii) реакция выделенного 3,4,5,6-тетрафторпиколинонитрила (H) (a) с хлоридом лития (LiCl), (b) с 3,4,5,6-тетрахлорпиколинонитрилом (формула A) в присутствии катализатора межфазного переноса или (c) с сочетанием LiCl и 3,4,5,6-тетрахлорпиколинонитрила для получения смеси, которую составляют преимущественно 3,4,5,6-тетрахлорпиколинонитрил, монофтортрихлорпиколинонитрил и дифтордихлорпиколинонитрил; и
iii) возвращение смеси, которую составляют преимущественно 3,4,5,6-тетрахлорпиколинонитрил, монофтортрихлорпиколинонитрил и дифтордихлорпиколинонитрилы в реакцию фторирования для получения 3-хлор-4,5,6-трифторпиколинонитрила (формула B).
Следующий аспект настоящего изобретения представляет собой новый промежуточный продукт, полученный при использовании настоящего способа, а именно соединение:

Подробное описание изобретения
Термины «алкил», «алкенил» и «алкинил», а также производные термины, такие как «алкокси», «ацил», «алкилтио» и «алкилсульфонил», при использовании в настоящем документе включают в пределах своего объема содержащие линейные цепи, разветвленные цепи и циклические радикалы. Если определенно не установлены другие условия, каждый из них может быть незамещенным или содержать один или более заместителей, в качестве которых выбирают, не ограничиваясь этим, галоген, гидрокси, алкокси, алкилтио, (C1-C6)ацил, формил, циано, арилокси или арил при том условии, что заместители являются стерически совместимыми и выполняются правила химической связи и энергии деформации. Термины «алкенил» и «аклкинил» предназначены для включения одной или нескольких ненасыщенных связей.
Термин «арилалкил» при использовании в настоящем документе означает фенил, замещенный алкильной группой, содержащий всего от 7 до 11 атомов углерода, такой как бензильная (-CH2C6H5), 2-метилнафтильная (-CH2C10H7) и 1-или 2-фенетильная (-CH2CH2C6H5 или -CH(CH3)C6H5). Сама фенильная группа может быть незамещенной или содержать один или несколько заместителей, в качестве которых независимо выбирают галоген, нитрогруппу, цианогруппу, (C1-C6)алкил, (C1-C6)алкокси, галогенированный (C1-C6)алкил, галогенированный (C1-C6)алкокси, (C1-C6)алкилтио, C(O)O(C1-C6)алкил, или два соседних заместителя совместно образуют группу -O(CH2)nO-, в которой n составляет 1 или 2, при том условии, что заместители являются стерически совместимыми и выполняются правила химической связи и энергии деформации.
Если определенно не установлены другие ограничения, термин «галоген», а также производные термины, такие как «гало», означают фтор, хлор, бром и йод.
Фенильные группы, содержащие от 1 до 4 заместителей, в качестве которых независимо выбирают галоген, (C1-C4)алкил, (C1-C4)галогеналкил, (C1-C4)алкокси или (C1-C4)галогеналкокси, могут иметь любую ориентацию, но предпочтительными являются 4-замещенный фенильный, 2,4-дизамещенный фенильный, 2,3,4-тризамещенный фенильный 2,4,5-тризамещенный фенильный, и 2,3,4,6-тетразамещенный фенильный изомеры.
4-амино-3-хлор-5-фтор-6-(замещенные)пиколинаты получают из 3,4,5,6-тетрахлорпиколинонитрилов, используя последовательность стадий, включающих фторидное замещение, аминирование, реакцию с гидразином, галогенирование, гидролиз, этерификацию и катализируемую переходными металлами комбинацию. Отдельные стадии можно осуществлять, используя другие последовательности.
3,4,5,6-тетрахлорпиколинонитрил в качестве исходного материала представляет собой известное соединение, которое имеется в продаже.
В реакции фторидного замещения фторированный пиколинонитрил получают, когда соответствующий хлорированный пиколинонитрил реагирует приблизительно с одним эквивалентом источника фторид-ионов на каждого подлежащего обмену хлоридного заместителя в кольце.

Типичные источники фторид-ионов представляют собой фториды щелочных металлов, которые включают фторид натрия (NaF), фторид калия (KF) и фторид цезия (CsF), причем предпочтительными являются KF и CsF.
Фторид четвертичного алкил- или ариламмония или фосфония можно также использовать в качестве источника фторид-ионов или в качестве добавки. Предпочтительно реакцию осуществляют, используя полярный апротонный растворитель или реакционную среду, такую как диметилсульфоксид (DMSO), N-метилпирролидон (NMP), N,N-диметилформамид (DMF), гексаметилфосфорамид (HMPA) или сульфолан. Температура, при которой проводят реакцию, не имеет решающего значения, но обычно она составляет от 60°C до 180°C и предпочтительно от 70°C до 80°C. В зависимости от того какой растворитель используют в конкретной реакции, оптимальная температура будет изменяться. Вообще говоря, чем ниже температура, тем медленнее будет проходить реакция. Данную реакцию проводят, как правило, при перемешивании с достаточно высокой интенсивностью, чтобы поддерживать практически однородно диспергированную смесь реагентов.
При проведении реакции фторирования не имеет решающего значения ни скорость, ни последовательность введения реагентов. Как правило, растворитель и фторид щелочного металла смешивают перед тем, как хлорированный пиколинонитрил добавляют в реакционную смесь. Для типичной реакции требуется, как правило, от 2 до 100 часов, предпочтительно от 3 до 6 часов, и ее обычно проводят при атмосферном давлении окружающей среды.
Хотя точное количество реагентов не имеет решающего значения, оказывается предпочтительным использование количества фторида щелочного металла, которое обеспечивает, по меньшей мере, эквимолярное количество атомов фтора по отношению к числу подлежащих обмену атомов хлора в исходном материале, т. е., по меньшей мере, эквимолярное количество фторида щелочного металла. После завершения реакции желательный продукт выделяют, используя стандартные технологии разделения и очистки веществ, такие как дистилляция, кристаллизация и хроматография.
В типичной реакции фторидного замещения получают смесь продуктов, включающую значительное количество полностью фторированного побочного продукта, а именно 3,4,5,6-тетрафторпиколинонитрила (формула H).

Конечный выход желательного 3-хлор-4,5,6-трифторпиколинонитрила можно повышать, выделяя полностью фторированный побочный продукт 3,4,5,6-тетрафторпиколинонитрил и возвращая его для получения промежуточных продуктов, которые можно вводить в реакцию фторидного замещения. Это можно осуществлять несколькими способами. Реакция 3,4,5,6-тетрафторпиколинонитрила с LiCl или реакция 3,4,5,6-тетрафторпиколинонитрила с избытком 3,4,5,6-тетрахлорпиколинонитрила или сочетание обеих реакций с использованием или без использования растворителей приводит к образованию смесей хлорфторпиколинонитрилов, где 3-хлор изомеры можно использовать в качестве исходного материала для получения желательного продукта. Таким образом, 3,4,5,6-тетрафторпиколинонитрил можно нагревать с избытком LiCl для получения смеси, содержащей преимущественно 3,4,5-трихлор-6-фторпиколинонитрил и тетрахлорпиколинонитрил. Согласно другой методике в реакции выделенного 3,4,5,6-тетрафторпиколинонитрила с избытком 3,4,5,6-тетрахлорпиколинонитрила в присутствии катализатора межфазного переноса получают смесь, которую составляют преимущественно монофтортрихлорпиколинонитрилы и дифтордихлорпиколинонитрилы. Наконец, эквивалентные смеси выделенного 3,4,5,6-тетрафторпиколинонитрила и 3,4,5,6-тетрахлорпиколинонитрила в присутствии катализатора межфазного переноса и от 1 до 3 эквивалентов LiCl образуют смесь, содержащую преимущественно 3,4,5-трихлор-6-фторпиколинонитрил и 3,4,5,6-тетрахлорпиколинонитрил. Эти смеси, которые составляют преимущественно монофтортрихлорпиколинонитрилы и/или дифтордихлорпиколинонитрилы, можно использовать в реакции фторирования, применяя фторид щелочного металла, чтобы получить 3-хлор-4,5,6-трифторпиколинонитрил из 3,4,5,6-тетрафторпиколинонитрила.
В обратной реакции галогенидного замещения нагревают 3,4,5,6-тетрафторпиколинонитрил и от 5 до 10 эквивалентов, предпочтительно 6 эквивалентов LiCl, чтобы получить смесь, содержащую 4,5-дихлор-3,6-дифторпиколинонитрил (3,6-F2-PN), 6-фтор-3,4,5-трихлорпиколинонитрил (6-F-PN) и 3,4,5,6-тетрахлорпиколинонитрил (Cl4-PN). Данную реакцию можно осуществлять без растворителя или в полярном апротонном растворителе или реакционной среде, такой как DMSO, NMP, DMF, HMPA или сульфолан. Часто оказывается удобным проведение реакции в растворителе. Температура, при которой проводят реакцию, не имеет решающего значения, но эта температура составляет обычно от 80°C до 200°C и предпочтительно от 100°C до 150°C.
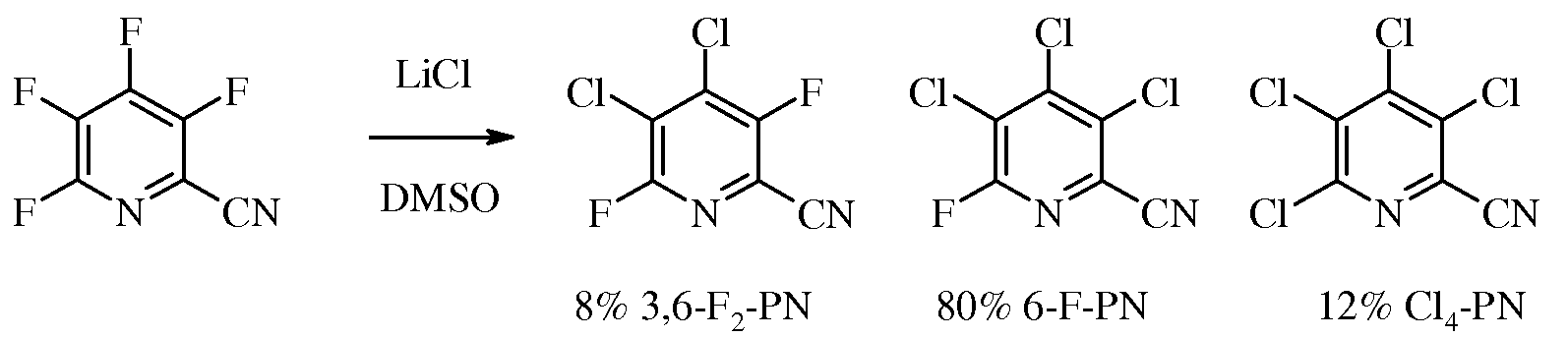
Можно использовать 90% или большую часть смеси для получения 3-хлор-4,5,6-трифторпиколинонитрила путем рециркуляции смеси через реакцию фторидного замещения.
В реакции замещения, в которой замещаются группы фтора и хлора, реагируют 3,4,5,6-тетрафторпиколинонитрил и от 1 до 3 эквивалентов 3,4,5,6-тетрахлорпиколинонитрила, предпочтительно 2 эквивалента 3,4,5,6-тетрахлорпиколинонитрила. Данную реакцию можно осуществлять без растворителя или в полярном апротонном растворителе или реакционной среде, такой как DMSO, NMP, DMF, HMPA или сульфолан. Часто оказывается удобным проведение реакции без растворителя. Реакцию замещения проводят в присутствии добавки. Добавки включают (a) соли четвертичного фосфония, содержащие 10 или более атомов углерода и (b) макроциклические простые полиэфиры, общеизвестные как краун-эфиры. Подходящие в качестве катализаторов краун-эфиры включают, но не ограничиваются этим, 18-краун-6; дициклогексано-18-краун-6; дибензо-18-краун-6; 15-краун-5. Подходящие соли четвертичного фосфония включают соли тетра-н-алкилфосфония, которые являются особенно предпочтительными. Температура, при которой проводят реакцию, не имеет решающего значения, но эта температура обычно составляет от 80°C до 200°C и предпочтительно от 150°C до 180°C.
В типичной реакции замещения, например, в которой реагируют 1 эквивалент 3,4,5,6-тетрафторпиколинонитрила и 2 эквивалента 3,4,5,6-тетрахлорпиколинонитрила, можно получить следующую смесь изомеров: 3,4,5,6-тетра-хлорпиколинонитрил (C14-PN), 3,5-дихлор-4,6-дифторпиколинонитрил (4,6-F2-PN), 3,4-дихлор-5,6-дифторпиколинонитрил (5,6-F2-PN), 4,5-дихлор-3,6-дифторпиколинонитрил (3,6-F2-PN), 6-фтор-3,4,5-трихлорпиколинонитрил (6-F-PN) и 4-фтор-3,5,6-трихлорпиколинонитрил (4-F-PN).
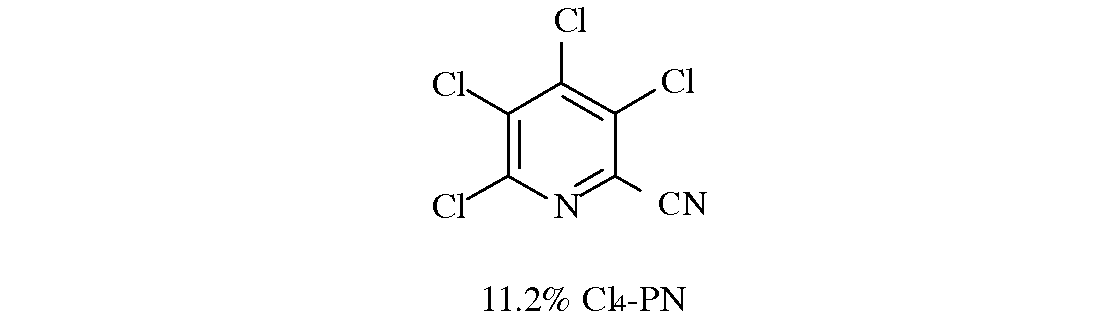

Можно использовать 80% этой смеси для получения 3-хлор-4,5,6-трифторпиколинонитрила путем рециркуляции смеси через реакцию фторидного замещения.
В комбинации обратной реакции галогенидного замещения и реакций замещения реагируют 3,4,5,6-тетрафторпиколинонитрил и от 1 до 3 эквивалентов 3,4,5,6-тетрахлорпиколинонитрила, предпочтительно 1 эквивалент 3,4,5,6-тетрахлорпиколинонитрила и от 1 до 4 эквивалентов, предпочтительно от 1,5 до 2,5 эквивалентов LiCl. Данную реакцию можно осуществлять без растворителя или в полярном апротонном растворителе или реакционной среде, такой как DMSO, NMP, DMF, HMPA или сульфолан. Часто оказывается удобным проведение реакции без растворителя. Реакцию замещения проводят в присутствии добавки. Добавки включают (a) соли четвертичного фосфония, содержащие 10 или более атомов углерода и (b) макроциклические простые полиэфиры, общеизвестные как краун-эфиры. Подходящие в качестве катализаторов краун-эфиры включают, но не ограничиваются этим, 18-краун-6; дициклогексано-18-краун-6; дибензо-18-краун-6; 15-краун-5. Подходящие соли четвертичного фосфония включают соли тетра-н-алкилфосфония, которые являются особенно предпочтительными. Температура, при которой проводят реакцию, не имеет решающего значения, но эта температура обычно составляет от 80°C до 200°C и предпочтительно от 150°C до 180°C.
В типичном сочетании реакций галогенидного замещения и реакций замещения реагируют, например, 1 эквивалент 3,4,5,6-тетрафторпиколинонитрила, 1 эквивалент 3,4,5,6-тетрахлорпиколинонитрила и 1,5 эквивалента LiCl и можно получить следующую смесь изомеров:

Можно использовать 92% этой смеси для получения 3-хлор-4,5,6-трифторпиколинонитрила путем рециркуляции смеси через реакцию фторидного замещения.
В реакции аминирования 4-фторпиколинонитрил реагирует с аммиаком для замещения атома фтора аминогруппой.
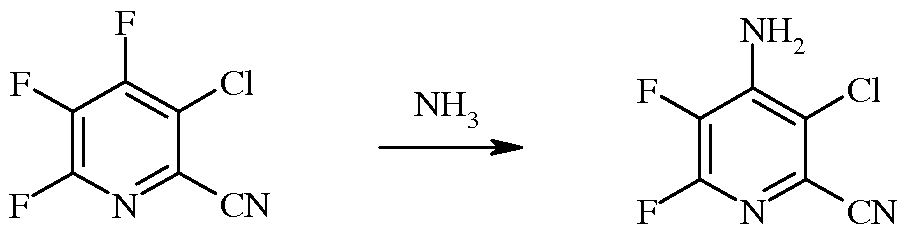
Хотя требуется только стехиометрическое количество аммиака, часто оказывается удобным использование большого избытка аммиака. Часто оказывается удобным использование аммиака, действующего одновременно как реагент и основание, чтобы нейтрализовать фтороводород (HF), образующийся в реакции. В качестве альтернативы, аммиак может присутствовать в форме раствора, такого как водный раствор гидроксида аммония. Реакцию осуществляют без растворителя или в инертном растворителе. Если используют растворитель, то инертные растворители включают, но не ограничиваются этим, спирты, простые эфиры, сложные эфиры, кетоны, DMSO и ароматические растворители. Температура, при которой проводят реакцию, не имеет решающего значения, но эта температура составляет обычно от 0°C до 45°C и предпочтительно от 10°C до 30°C.
Для типичной реакции требуется, как правило, от 0,5 до 5 часов и ее обычно проводят при атмосферном давлении окружающей среды. Желательный продукт выделяют, используя стандартные методики разделения и очистки.
В реакции с гидразином 6-фторпиколинонитрил реагирует с гидразином, замещая атом фтора гидразиновой группой.

Хотя требуется только стехиометрическое количество гидразина, часто оказывается удобным использование избытка гидразина, действующего одновременно в качестве реагента и в качестве основания для нейтрализации фтороводорода, выделяющегося в реакции. Гидразин предпочтительно представляет собой моногидрат. Реакцию осуществляют в инертном полярном растворителе, таком как DMSO, DMF, NMP, ацетонитрил, хлорированный растворитель, простой эфир, тетрагидрофуран (THF) или спирт. Предпочтительной является смесь DMSO и THF. Температура, при которой проводят реакцию, не имеет решающего значения, но она составляет обычно от температуры окружающей среды до 150°C и предпочтительно от 35°C до 70°C.
При проведении реакции с гидразином гидразин растворяют в растворителе и 6-фторпиколинонитрил добавляют в реакционную смесь. Для типичной реакции требуется, как правило, от 0,5 до 5 часов и ее обычно проводят при атмосферном давлении. Желательный продукт выделяют, используя стандартные методики разделения и очистки. Соединения можно легко выделять, разбавляя реакционную смесь ацетонитрилом, после чего следует фильтрование.
В реакции галогенирования 6-галопиколинонитрил получают посредством взаимодействия соответствующего 6-гидразинопиколинонитрила и по меньшей мере одного эквивалента бромирующего, хлорирующего или йодирующего реагента.

Подходящие йодирующие, хлорирующие и бромирующие реагенты включают, но не ограничиваются этим, йод, монохлорид йода, хлор, бром, сульфурилхлорид и сульфурилбромид. Хотя требуется только стехиометрическое количество галогенирующего реагента, часто оказывается удобным использование избытка галогенирующего реагента. Реакцию осуществляют в инертном апротонном растворителе. Можно использовать разнообразные растворители, такие как хлорированные растворители, ацетонитрил, DMSO, диоксан, DMF и вода. Часто оказывается предпочтительным использование хлорированного растворителя. Температура, при которой проводят реакцию, не имеет решающего значения, но она составляет обычно от температуры окружающей среды до 100°C и предпочтительно от температуры окружающей среды до 50°C.
При проведении реакции галогенирования 6-гидразинопиколинонитрил растворяют или суспендируют в растворителе и галогенирующий реагент добавляют в реакционную смесь. Для типичной реакции требуется, как правило, от 0,5 до 24 часов. Желательный продукт выделяют, используя стандартные методики разделения и очистки.
В реакциях гидролиза и этерификации пиколинонитрил реагирует со спиртом (R1OH) в присутствии кислоты Бренстеда (Brønsted) или кислоты Льюиса (Lewis).

Кислоты Бренстеда включают, но не ограничиваются этим, такие кислоты, как хлористоводородная кислота, серная кислота и фосфорная кислота. Кислоты Льюиса включают трифторид бора, тетрагалогениды титана, тетраалкоксиды титана, галогениды цинка, галогенид олова, а также пентафториды фосфора и сурьмы. Кислоты, такие как серная кислота или фосфорная кислота, как правило, используют в стехиометрических количествах. Реакцию осуществляют в (C1-C12)алкиловом спирте или незамещенном или замещенном (C7-C11)арилалкиловом спирте, соответствующем желательному сложному эфиру. Реакцию можно удобно проводить в герметичном реакторе, если температура реакции находится выше температуры кипения спиртового растворителя. При проведении этерификации пиколинонитрил или промежуточный продукт гидролиза пиколинамида добавляют к смеси спирта и кислоты. Хотя температура реакции не имеет решающего значения, часто осуществляют нагревание при температуре от 80°C до 140°C в течение от 2 до 24 часов, предпочтительно при температуре от 100°C до 120°C в течение от 6 до 8 часов. Желательный продукт выделяют, используя стандартные методики разделения и очистки.
В реакции сочетания 6-галопиколинат реагирует с арил-, алкил- или алкенилметаллоорганическим соединением, где металл представляет собой Zn-галогенид, Zn-R, три-(C1-C4алкил)олово, медь, или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород, (C1-C4)алкил или совместно образуют этиленовую или пропиленовую группу в присутствии содержащего переходный металл катализатора.

Термин «катализатор» означает содержащий переходный металл катализатор, в частности палладиевый катализатор, такой как диацетат палладия(II) или дихлорбис(трифенилфосфин)палладий(II), или никелевый катализатор, такой как ацетилацетонат никеля(II) или дихлорбис(трифенилфосфин)никель(II). Кроме того, катализаторы можно получать на месте применения in situ, используя соли металлов и лиганды, такие как ацетат палладия(II) и трифенилфосфин или хлорид никеля(II) и трифенилфосфин. Данные получаемые на месте применения катализаторы можно получать посредством предварительной реакции соли металла и лиганда, после чего следует введение в реакционную смесь, или посредством раздельного введения соли металла и лиганда непосредственно в реакционную смесь.
Как правило, реакции сочетания осуществляют при отсутствии кислорода, используя инертный газ, такой как азот или аргон. Методики, используемые для исключения кислорода из реакционных смесей в реакциях сочетания, такие как вытеснение инертным газом, хорошо известны специалистам в данной области техники. Примеры таких методик описывает книга «Обращение с чувствительными к воздуху соединениями», второе издание под редакцией D. F. Shriver и M.A. Drezdzon, издательство Wiley-Interscience, 1986 г. Катализатор используют в субстехиометрических количествах, составляющих, как правило, от 0,0001 эквивалента до 0,1 эквивалента. Можно необязательно вводить дополнительные количества лиганда для повышения устойчивости и активности катализатора. Кроме того, в реакцию сочетания, как правило, вводят добавки, такие как карбонат натрия, карбонат калия, фторид калия, фторид цезия и фторид натрия. Для реакции сочетания требуется, как правило, от 1 до 5 эквивалентов, предпочтительно от 1 до 2 эквивалентов такой добавки. Воду можно необязательно вводить в реакцию сочетания для повышения растворимости этих добавок. Для реакции сочетания требуется, как правило, от 1 до 3 эквивалентов, предпочтительно от 1 до 1,5 эквивалентов арил-, алкил- или алкенилметаллоорганического соединения. Реакцию осуществляют в инертном растворителе, таком как толуол, THF, диоксан или ацетонитрил. Температура, при которой проводят реакцию, не имеет решающего значения, но эта температура составляет обычно от 25°C до 150°C и предпочтительно от 50°C до 125°C. Для типичной реакции требуется, как правило, от 0,5 до 24 часов. Никакой определенный порядок введения реагентов, как правило, не требуется. Часто оказывается технологически более простым объединение всех реагентов за исключением катализатора и последующее удаление кислорода из реакционного раствора. После удаления кислорода катализатор можно вводить для начала реакции сочетания.
Когда металлоорганическая часть арил-, алкил- или алкенилметаллоорганического соединения представляет собой Zn-галогенид, Zn-R или медь, может оказаться необходимой защита реакционно-способных функциональных групп. Например, если присутствует аминный заместитель (-NHR или-NH2), может потребоваться защита этих реакционно-способных групп. В технике известны разнообразные группы для защиты аминогрупп от реакции с металлоорганическими реагентами. Примеры таких защитных групп описывает книга «Защитные группы в органическом синтезе», третье издание под редакцией T. W. Greene и P.G.M. Wuts, издательство Wiley-Interscience, 1999 г. На выбор металла для использования в металлоорганическом соединении R-Met влияет множество факторов, таких как стоимость, устойчивость, реакционная способность и необходимость защиты реакционно-способных функциональных групп.
Продукты, полученные любым из этих способов, можно выделять традиционными средствами, такими как испарение или экстракция, и их можно очищать стандартными процедурами, такими как перекристаллизация или хроматография.
Следующие примеры представлены, чтобы проиллюстрировать настоящее изобретение.
Примеры
Фторидное замещение
Пример 1a. 3-Хлор-4,5,6-трифторпиколинонитрил
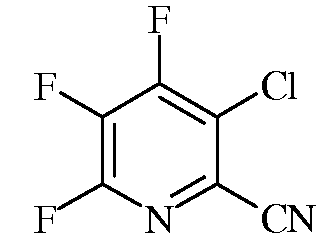
В пятилитровую колбу с механической мешалкой в атмосфере азота загружали DMSO (3820 мл), порошкообразный карбонат калия K2CO3 (42 г) и тонкоизмельченный фторид цезия CsF (1510 г). DMSO (приблизительно 1 л) удаляли путем дистилляции при 75-80°C и 3,5 мм рт. ст. (0,46 кПа). Суспензию охлаждали до 55°C в атмосфере азота перед введением тонкоизмельченного 3,4,5,6-тетрахлорпиколинонитрила (685 г). Введение осуществляли в течение 15-минутного периода при охлаждении для поддержания температуры реакционной смеси ниже 74°C. Температуру выдерживали на уровне от 65 до 70°C при медленном потоке азота в течение 4 часов. Реакционную смесь охлаждали до 40-50°C и выливали в смесь ледяной воды H2O (15 л) и диэтилового эфира Et2O (3 л). После отделения органической фазы водную фазу экстрагировали Et2O (2×2 л). Органические экстракты объединяли, сушили над сульфатом магния (MgSO4), фильтровали и концентрировали путем дистилляции при атмосферном давлении, получая смесь неочищенных продуктов (469 г) в форме светло-коричневого масла. Это масло объединяли с дополнительным материалом, полученным аналогичным образом, получая в сумме 1669 г неочищенного продукта. Это масло дистиллировали в вакууме, используя 30-тарелочную колонку Олдершоу (Oldershaw) в температурном интервале от 80 до 90°C, собирая фракции при 63, 13 и 2 мм рт. ст. (8,4, 1,7 и 0,27 кПа). Из материала, собранного при 13 мм рт. ст. (1,7 кПа), получали 457 г (выход 22%) твердого вещества, которое представляло собой смесь двух хлортрифторпиколинонитрилов в соотношении 93/7. Это твердое вещество перекристаллизовывали при 5°C из смеси гексана (420 г) и Et2O, получая 3-хлор-4,5,6-трифторпиколинонитрил (354 г, чистота 98%) в форме тонких белых игольчатых кристаллов. Небольшой образец перекристаллизовывали второй раз, получая чистоту 99,7% согласно газовой хроматографии (GC); температура плавления 41,5-43°C; спектр ЯМР19F (376 МГц, CDCl3) δ -78,1 (т, JF-F=23,1 Гц, F6), -114,2 (дд, JF-F=18,5, 22,5 Гц, F4), -149,3 (дд, JF-F=18,2, 22,6 Гц, F5); спектр ЯМР13C {1Н} (101 МГц, CDCl3) δ 154,5 (ддд, JF-C=270, 11, 7 Гц, C4), 151,3 (ддд, JF-C=247, 13, 5 Гц, C6), 138,0 (ддд, JF-C=279, 31, 13 Гц, C5), 124,7 (ддд, JF-C=16, 6, 2 Гц, C3), 124,4 (ддд, JF-C=16, 7, 2 Гц, C2), 112,2 (c, CN); электронно-стимулированная масс-спектрометрия (EIMS) m/z 192 ([M]+). Элементный анализ. Вычислено для C6ClF3N2 (%): C, 37,43; N, 14,55. Найдено (%): C, 36,91; N, 14,25.
Из второй фракции при дистилляции (63 мм рт. ст., 8,4 кПа) получали чистый 3,4,5,6-тетрафторпиколинонитрил (525 г, 24%) в форме бесцветного масла; спектр ЯМР19F (376 МГц, CDCl3) δ -77,6 (т, JF-F=23,8 Гц, F6), -133,7 (кв., JF-F=18,8 Гц, F4), -134,2 (ддд, JF-F=24,2, 18,6, 10,1 Гц, F3), -145,3 (ддд, JF-F=24,1, 18,2, 10,2 Гц, F5); спектр ЯМР13C {1Н} (101 МГц, CDCl3) δ 150,4 (дм, JF-C=272 Гц, C3), 148,5 (ддд, JF-C=245, 12, 4 Гц, C6), 147,3 (дм, JF-C=270 Гц, C4), 138,6 (ддд, JF-C=280, 33, 11 Гц, C5), 113,4 (м, C2), 110,20 (c, CN).
Из третьей фракции при дистилляции (2 мм рт. ст., 0,27 кПа) получали 3,5-дихлор-4,6-дифторпиколинонитрил (48 г, чистота 98%) в форме белого твердого вещества; температура плавления 78-79°C; спектр ЯМР19F (376 МГц, CDCl3) δ -63,65 (д, JF-F=18,7 Гц, F6), -92,52 (д, JF-F=18,5 Гц, F4); спектр ЯМР13C {1Н} (101 МГц, CDCl3) δ 162,6 (дд, JF-C=269, 6 Гц, C4), 157,8 (дд, JF-C=245, 5 Гц, C6), 127,6 (дд, JF-C=17, 3 Гц, C3), 123,5 (дд, JF-C=18, 6 Гц, C2), 112,4 (дд, JF-C=36, 21 Гц, C5), 112,3 (CN).
Пример 1b. Обратная реакция галогенидного замещения 3,4,5,6-тетрафторпиколинонитрила с хлоридом лития
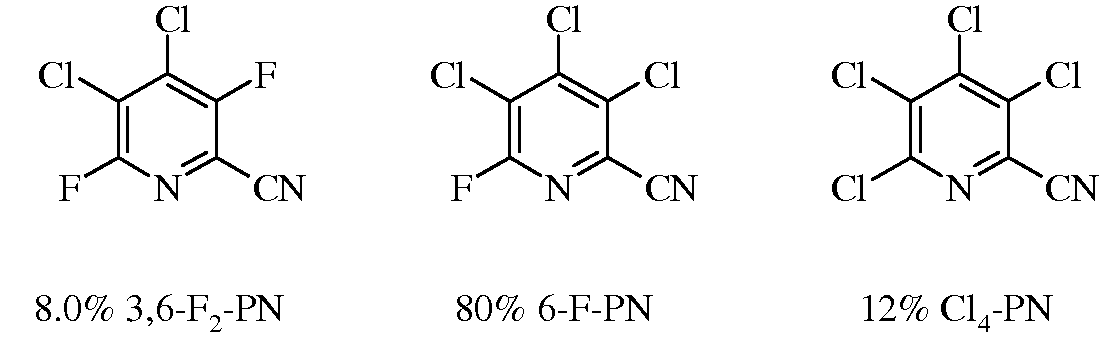
Смесь 3,4,5,6-тетрафторпиколинонитрила (17 г, 0,1 моль) и сухого LiCl (25,4 г, 0,6 моль) нагревали в сухом DMSO (200 мл). За реакцией наблюдали, проводя анализ методом газовой хроматографии аликвот, экстрагированных Et2O из H2O. Сначала реакционную смесь нагревали до 120°C и весь LiCl растворялся. В течение пятиминутного выдерживания при 120°C весь исходный материал и изомеры хлортрифторпиколинонитрила реагировали, образуя смесь 3,6-F2-PN (83%) и 6-F-PN (14%). Температуру реакционной смеси повышали до 135°C и через 75 минут после начала реакции проводили анализ методом газовой хроматографии. Определяли, что смесь представляла собой 3,6-F2-PN/6-F-PN/Cl4-PN смесь в соотношении 8:80:12.
Пример 1ca. Обмен 3,4,5,6-тетрафторпиколинонитрила
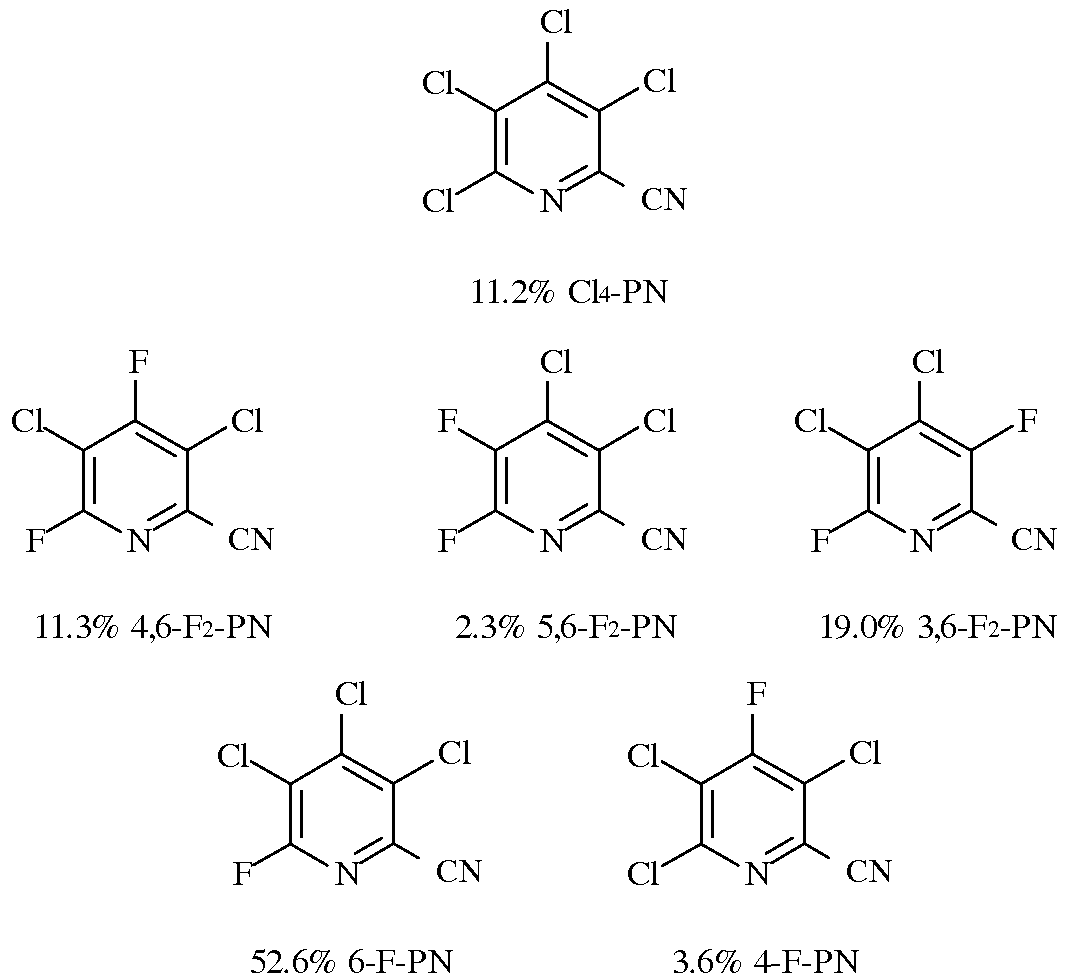
Смесь 3,4,5,6-тетрахлорпиколинонитрила (16,1 г, 66 ммоль) и 3,4,5,6-тетрафторпиколинонитрила (5,9 г, 33 ммоль) нагревали до 160°C в атмосфере азота, получая раствор. В данный раствор при перемешивании вводили хлорид тетрабутилфосфония Bu4PCl (0,36 г, 1,2 ммоль) и раствор выдерживали при 160°C в течение 1 часа. Аликвоту растворяли в метиленхлориде (CH2Cl2) и пропускали через тонкий слой силикагеля перед анализом методом газовой хроматографии. Галогенированные пиколинонитрилы имели следующий состав: 11,2% Cl4-PN; 11,3% 4,6-F2-PN; 2,3% 5,6-F2-PN; 19% 3,6-F2-PN; 52,6% 6-F-PN и 3,6% 4-F-PN. Можно использовать 80% данной смеси в реакции галогенидного замещения, чтобы получать 3-хлор-4,5,6-трифторпиколинонитрил.
Пример 1cb. Выделение после обмена 3,4,5,6-тетрафторпиколинонитрила
В реакционную колбу, снабженную коротким дефлегматором, загружали тонкоизмельченный CsF (35,1 г, 0,23 моль) и сухой DMSO (175 мл). Реакционную смесь перемешивали и нагревали до 70-75°C в вакууме (0,1 мм рт. ст., 13,33 Па) до тех пор, пока DMSO (75 мл) не был удален путем дистилляции. Данную суспензию охлаждали до 50°C в атмосфере азота и вводили полученную ранее расплавленную реакционную смесь (21,7 г). Реакционную смесь нагревали при 70°C в течение 2,5 ч при интенсивном перемешивании. Экстракт диэтилового эфира вводили в воду и анализировали методом газовой хроматографии, обнаруживая, что она содержала: 61% 3,4,5,6-тетрафторпиколинонитрила; 31% 3-хлор-4,5,6-трифторпиколинонитрила; 3,4% 5-хлор-3,5,6-трифторпиколинонитрила и 4,8% 3,5-дихлор-4,6-дифторпиколинонитрила. Этот результат являлся благоприятным по сравнению с типичной чистотой от 38 до 42%, полученной методом газовой хроматографии для неочищенного образца, когда аналогичную реакцию проводили, используя в качестве исходного материала чистый 3,4,5,6-тетрахлорпиколинонитрил.
Пример 1d. Обмен 3,4,5,6-тетрафторпиколинонитрила под действием LiCl
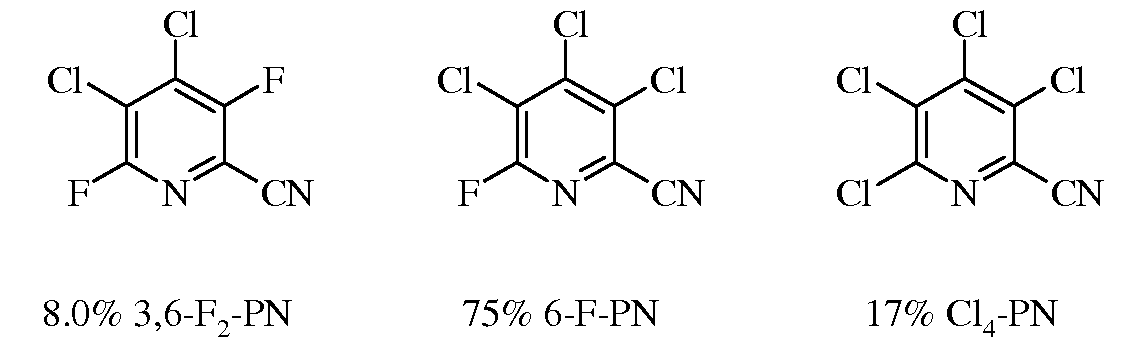
Смесь 3,4,5,6-тетрахлорпиколинонитрила (12,2 г, 50 ммоль) и 3,4,5,6-тетрафторпиколинонитрил (8,8 г, 50 ммоль) нагревали до 160°C в атмосфере азота, получая прозрачный раствор. В него вводили Bu4PCl (0,36 г, 1,2 ммоль). Реакционный раствор выдерживали при 160°C в течение 15 минут перед введением сухого LiCl (4,2 г, 0,1 моль). Через 60 минут вводили дополнительное количество LiCl (2,2 г, 50 ммоль) и реакционную смесь перемешивали в течение 11 часов. Газохроматографический анализ эфирного экстракта из воды определил смесь 3,6-F2-PN/6-F-PN/Cl4-PN в соотношении 8:75:17.
Аминирование
Пример 2. 4-Амино-3-хлор-5,6-дифторпиколинонитрил

Раствор 3-хлор-4,5,6-трифторпиколинонитрила (200 г) в этилацетате (EtOAc) (3 л) охлаждали до 10°C. В этот раствор медленно вводили 14% водный раствор гидроксида аммония (NH4OH) (1296 г), выдерживая температуру в интервале от 18 до 23°C. Водный раствор отделяли от органического раствора. Органическую фазу промывали, используя последовательно разбавленный вдвое (50/50) насыщенный водный раствор NaCl и воду (500 мл) и насыщенный водный раствор NaCl (250 мл). Органическую фазу концентрировали в вакууме при 50°C до объема 500 мл для кристаллизации продукта. К данной суспензии добавляли гептан (1 л) и оставшийся EtOAc удаляли в вакууме, получая конечную суспензию. Твердое вещество отделяли путем фильтрования. Данное твердое вещество промывали пентаном и сушили в вакууме, получая 4-амино-3-хлор-5,6-дифторпиколинонитрил (173,8 г, 90%, чистота 99,6%) в форме белого кристаллического твердого вещества; температура плавления 190-191,5°C; спектр ЯМР13C {lH} (101 МГц, DMSO-d6) δ 150,03 (дд, J=232,4, 12,5 Гц, C6), 144,29 (дд, J=11,4, 6,9 Гц, C4), 133,72 (дд, J=257,9, 30,8 Гц, C5), 122,14 (дд, J=19,6, 4,9 Гц, C2), 119,31 (c, C3), 114,25 (c, CN); спектр ЯМР19F (376 МГц, DMSO-d6) δ -91,24 (д, J=24,2 Гц), -154,97 (д, J=24,2 Гц); электронно-стимулированная масс-спектрометрия (EIMS) m/z 189 ([M]+). Элементный анализ. Вычислено для C6H2ClF2N3 (%): C, 38,02; H, 1,06; N, 22,17. Найдено (%): C, 37,91; H, 1,00; N, 22,02.
Реакция c гидразином
Пример 3. 4-Амино-3-хлор-5-фтор-6-гидразинопиколинонитрил

В раствор гидрата гидразина (3,9 г, 78 ммоль) в THF (15 мл) и DMSO (10 мл) вводили 4-амино-3-хлор-5,6-дифторпиколинонитрил (5 г, 26 ммоль) в форме раствора в DMSO (5 мл). Полученный раствор нагревали при 65°C в течение 45 минут, охлаждали и разбавляли ацетонитрилом (30 мл), осаждая продукт в форме светлого желто-коричневого твердого вещества. Данное твердое вещество сушили в вакууме при 40°C в течение 3 часов, получая 4-амино-3-хлор-5-фтор-6-гидразинопиколинонитрил (5,1 г, 98%); температура плавления 215-220°C (разложение); спектр1H ЯМР (400 МГц, DMSO-d6) 7,9 (широкий сигнал, 1H), 6,5 (широкий сигнал, 2H), 4,0 (широкий сигнал, 2H); спектр ЯМР13C {1H} (101 МГц, DMSO-d6) δ 149,34 (д, J=10,5 Гц, C6), 138,28 (д, J=11,6 Гц, C4), 133,81 (д, J=251,6 Гц, C5), 123,74 (д, J=5,3 Гц, C2), 115,87 (c, C3), 112,57 (c, CN); спектр ЯМР19F (376 МГц, DMSO-d6) δ -154,6; электронно-стимулированная масс-спектрометрия (EIMS) m/z 203 ([M+H]+). Элементный анализ. Вычислено для C6H5ClFN5 (%): C, 35,75; H, 2,50; N, 34,74. Найдено (%): C, 35,97; H, 2,70: N, 35,01.
Галогенирование
Пример 4. 4-Амино-3,6-дихлор-5-фторпиколинонитрил

В суспензию 4-амино-3-хлор-5-фтор-6-гидразинилпиколинонитрила (9,04 г, 44,8 ммоль) в CH2Cl2 (150 мл) вводили сульфурилхлорид (7,20 мл, 89 ммоль). Смесь перемешивали при комнатной температуре в течение 40 часов. Растворитель удаляли при пониженном давлении и остаток разбавляли насыщенным водным раствором бикарбоната натрия (NaHCO3) и EtOAc. Органическую фазу отделяли, сушили над Na2SO4 и фильтровали. Раствор концентрировали и остаток очищали методом хроматографии с силикагелем, получая продукт (7,01 г, 76%) в форме беловатого твердого вещества; спектр ЯМР1H (300 МГц, DMSO-d6) δ 7,55 (c, 2H); спектр ЯМР13C (101 МГц, DMSO-d6) δ 143,26 (д, J=259,2 Гц, C5), 142,69 (д, J=14,0 Гц, C4), 135,53 (д, J=17,6 Гц. C6), 126,07 (д, J=4,4 Гц, C2), 120,14 (д, J=4,3 Гц, C3), 114,36 (c, CN); спектр ЯМР19F (376 МГц, DMSO-d6) δ -132,30 (с); электронно-стимулированная масс-спектрометрия (ESIMS) m/z 203 ([M+H]+), 206.
Гидролиз и этерификация
Пример 5. Метил-4-амино-3,6-дихлор-5-фторпиколинат
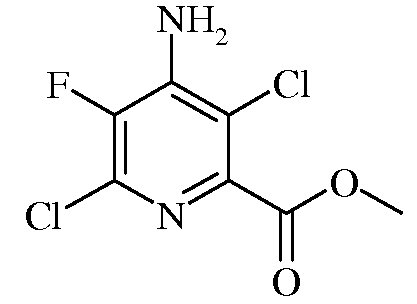
Концентрированную серную кислоту (2,0 мл, 37,5 ммоль) медленно вводили в метиловый спирт (8 мл) при охлаждении. Затем в раствор вводили 4-амино-3,6-дихлор-5-фторпиколинонитрил (0,20 г, 0,97 ммоль) и смесь перемешивали с обратным холодильником в течение 29 часов. Растворитель удаляли в вакууме, остаток выливали на лед и перемешивали в течение 15 минут. Продукт экстрагировали EtOAc (3 раза). Органические экстракты объединяли, промывали концентрированным раствором NaCl, сушили над MgSO4, фильтровали и очищали на колонке с силикагелем, получая метил-4-амино-3,6-дихлор-5-фторпиколинат (0,085 г, 37%) в форме белого твердого вещества: спектр ЯМР1H (400 МГц, CDCl3) δ 5,08 (c, 2H), 3,97 (c, 3H); спектр ЯМР13C (101 МГц, CDCl3) δ 163,57 (c, C=0), 143,29 (д, J=258,1 Гц, C5), 141,73 (д, J=5,1 Гц, C2), 141,05 (д, J=12,7 Гц, C4), 135,32 (д, J=16,8 Гц, C6), 116,26 (c, C3), 53,24 (c, OMe); спектр ЯМР19F (376 МГц, CDCl3) δ -135,63 (с).
Сочетание
Пример 6a. Метил-4-амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метокси-фенил)пиколинат
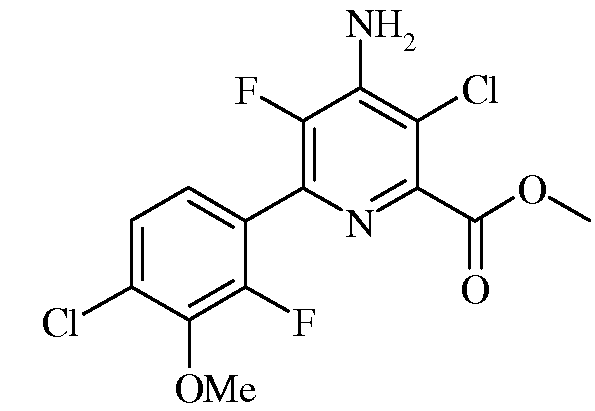
В трехгорлую колбу объемом 250 мл, снабженную обратным холодильником, впуском азота и термопарой, загружали метил-4-амино-3,6-дихлор-5-фторпиколинат (9,965 г, 41,7 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинан (12,74 г, 52,1 ммоль) и KF (4,84 г, 83 ммоль). Добавляли ацетонитрил (78 мл) и воду (26 мл). Реакционную смесь продували азотом. Вводили дихлорбис(трифенилфосфин)палладий(II) Pd(PPh3)2Cl2 (1,477 г, 2,10 ммоль, 5 мол.%) и раствор нагревали при 70°C в атмосфере азота в течение 2 часов. После охлаждения до комнатной температуры образовавшийся осадок отфильтровывали и промывали водой. Осадок растворяли в EtOAc (приблизительно 500 мл) и промывали водой и затем концентрированным раствором NaCl. Органический слой сушили над MgSO4 и растворитель удаляли с помощью роторного испарителя, получая оранжевое твердое вещество, которое сушили в вакуумной печи при 50°C (11,46 г, 76% выход); температура плавления 169-170,5°C; спектр ЯМР1H (400 МГц, DMSO-d6) δ 7,48 (д, J=8,4 Гц, 1H), 7,32 (т, J=7,7 Гц, 1H), 7,15 (c, 2H), 3,96 (c, 3H), 3,90 (c, 3H); спектр ЯМР13C {1Н}(101 МГц, DMSO-d6) δ 164,85 (с), 153,11 (д, J=252,5 Гц), 146,29 (с), 144,52 (д, J=4,3 Гц), 143,74 (с), 142,75 (дд, J=227,1, 14,0 Гц), 136,38 (д, J=13,4 Гц), 128,58 (д, J=3,2 Гц), 125,87 (с), 125,54 (д, J=3,5 Гц), 122,89 (дд, J=13,8, 4,0 Гц), 113,01 (д, J=3,0 Гц), 61,61 (д, J=4,2 Гц), 52,70 (с); электронно-стимулированная масс-спектрометрия (ESIMS) m/z 364 ([M+H]+). Элементный анализ. Вычислено для C14H10Cl2F2N2O3 (%): C, 46,30; H, 2,78; N, 7,71. Найдено (%): C, 46,60; H, 2,68; N, 7,51.
Пример 6b. 4-Амино-3-хлор-5-фтор-6-(4-хлор-2-фтор-3-метоксифенил)пиколинонитрил

Смесь 4-амино-3,6-дихлор-5-фторпиколинонитрила (0,37 г, 1,80 ммоль), 2-(4-хлор-2-фтор-3-метоксифенил)-1,3,2-диоксаборинана (0,549 г, 2,24 ммоль) и KF (0,209 г, 3,59 ммоль) вводили в ацетонитрил (6,75 мл) и воду (2,25 мл). Смесь перемешивали и продували азотом. Вводили Pd(PPh3)2Cl2 (63 мг, 0,1 ммоль) и смесь снова продували азотом. Затем раствор нагревали при 75°C в атмосфере азота в течение 2 часов. После охлаждения образовывался осадок, который выделяли путем фильтрования, промывали водой и сушили в вакууме, получая продукт (0,34 г) в форме беловатого твердого вещества. Водную фазу экстрагировали EtOAc (3 раза) и объединенные органические экстракты промывали концентрированным раствором NaCl, сушили и концентрировали. Методом хроматографической очистки с силикагелем получали дополнительный продукт (0,12 г) в форме белого твердого вещества. Суммарный выход 78%. Спектр ЯМР1H (400 МГц, DMSO-d6) δ 7,50 (дд, J=8,5, 1,4 Гц, 1H), 7,45 (c, 2H), 7,33 (дд, J=8,5, 7,2 Гц, 1H), 3,94 (c, 3H); спектр ЯМР13C {1H} (101 МГц, DMSO-d6) δ 152,97 (д, J=253,2 Гц), 145,73 (д, J=260,8 Гц), 143,82 (д, J=13,7 Гц), 141,83 (д, J=14,7 Гц), 138,45 (д, J=14,8 Гц), 133,93-132,79 (м), 128,93 (д, J=3,3 Гц), 127,74 (с), 126,37-125,10 (м), 122,08 (дд, J=13,6, 3,9 Гц), 119,34 (д, J=4,5 Гц), 114,99 (с), 61,61 (с); спектр ЯМР19F (376 МГц, DMSO-d6) δ -129,00 (дд, J=28,2, 7,0 Гц, 1F), -133,76 (д, J=28,2 Гц, 1F); электронно-стимулированная масс-спектрометрия (ESIMS) m/z 330,1 ([M+H]+).
Реферат
Изобретение относится к способу получения 4-амино-3-хлор-5-фтор-6-(замещенного)пиколината формулы Iгде R представляет собой (C-C)алкил, циклопропил, (C-C)алкенил или фенил, содержащий от 1 до 4 заместителей, в качестве которых независимо выбирают галоген, (C-C)алкил, (C-C)галоалкил, (C-C)алкокси или (C-C)галоалкокси; и Rпредставляет собой (C-C)алкил или незамещенный или замещенный (C-C)арилалкил, включающему фторидное замещение, аминирование, реакцию с гидразином, галогенирование, гидролиз и этерификацию, а также катализируемое переходными металлами сочетание. 3 н. и 1 з.п. ф-лы, 6 пр.
Формула
в которой
R представляет собой (C1-C4)алкил, циклопропил, (C2-C4)алкенил или фенил, содержащий от 1 до 4 заместителей, в качестве которых независимо выбирают галоген, (C1-C4)алкил, (C1-C4)галоалкил, (C1-C4)алкокси или (C1-C4)галоалкокси; и
R1 представляет собой (C1-C12)алкил или незамещенный или замещенный (C7-C11)арилалкил;
который включает следующие стадии:
a) фторирование 3,4,5,6-тетрахлорпиколинонитрила (формула A)
источником фторид-ионов для получения 3-хлор-4,5,6-трифторпиколинонитрила (формула B)

b) аминирование 3-хлор-4,5,6-трифтор-2-пиколинонитрила (формула B) аммиаком для получения 4-амино-3-хлор-5,6-дифторпиколинонитрила (формула C)

c) реакцию фторзаместителя в положении 6 4-амино-3-хлор-5,6-дифторпиколинонитрила (формула C) с гидразином для получения 4-амино-3-хлор-5-фтор-6-гидразинопиколинонитрила (формула D)
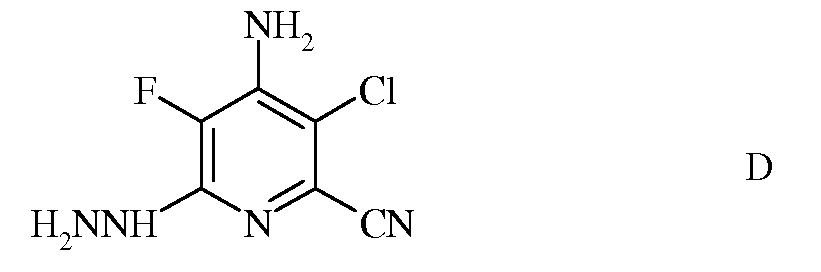
d) галогенирование 4-амино-3-хлор-5-фтор-6-гидразинопиколинонитрила (формула D) источником хлора, брома или йода для получения 4-амино-3-хлор-5-фтор-6-галогенпиколинонитрила формулы E
в которой L представляет собой Br, Cl или I;
e) гидролиз и этерификацию 4-амино-3-хлор-5-фтор-6-галогенпиколинонитрила формулы E сильной кислотой и спиртом (R1OH) для получения 4-амино-3-хлор-5-фтор-6-галогенпиколината формулы F
в которой L и R1 являются такими, как определено выше; и
f) сочетание 4-амино-3-хлор-5-фтор-6-галогенпиколината формулы E с арил-, алкил- или алкенилметаллоорганическим соединением формулы G
в которой R является таким, как определено выше, и Met представляет собой Zn-галогенид, Zn-R, три-((C1-C4)алкил)олово, медь, или B(OR2)(OR3), где R2 и R3 независимо друг от друга представляют собой водород или (C1-C4)алкил, или совместно образуют этиленовую или пропиленовую группу в присутствии содержащего переходный металл катализатора, для получения 4-амино-3-хлор-5-фтор-6-(замещенного)пиколината формулы I.
полученного фторированием 3,4,5,6-тетрахлорпиколинонитрила (формула A)
источником фторид-ионов, где усовершенствование включает следующие стадии:
i) выделение полностью фторированного 3,4,5,6-тетрафторпиколинонитрила (формула H)
в качестве побочного продукта фторирования 3,4,5,6-тетрахлорпиколинонитрила;
ii) реакция выделенного 3,4,5,6-тетрафторпиколинонитрила (H) (a) с хлоридом лития (LiCl), (b) с 3,4,5,6-тетрахлорпиколинонитрилом (формула A) в присутствии катализатора межфазного переноса или (c) с комбинацией LiCl и 3,4,5,6-тетрахлорпиколинонитрила для получения смеси, которую составляют преимущественно 3,4,5,6-тетрахлорпиколинонитрил, монофтортрихлорпиколинонитрил и дифтордихлорпиколинонитрил; и
iii) возвращение смеси, которую составляют преимущественно 3,4,5,6-тетрахлорпиколинонитрил, монофтортрихлорпиколинонитрил и дифтордихлорпиколинонитрилы в реакцию фторирования для получения 3-хлор-4,5,6-трифторпиколинонитрила (формула B).
Комментарии