Соединения на основе пиридоксина, обладающие способностью активировать фермент глюкокиназу - RU2644355C1
Код документа: RU2644355C1
Чертежи



Описание
Изобретение относится к физиологически активным веществам на основе бис-пиридоксиновой структуры, в которой два фрагмента пиридоксина соединены алкилсульфидным или алкилдисульфидным линкером, общей формулы I, а также их солям, образованным соляной, бромоводородной, серной, L-винной кислотами и прочими фармацевтически приемлемыми кислотами, разрешенными к применению (Food Additive Status List [Список разрешенных пищевых добавок] [Электронный ресурс]. URL: http://www.fda.gov/Food/IngredientsPackagingLabeling/FoodAdditivesIngredients/ucm091048.htm), которые обладают способностью активировать фермент глюкокиназу, вследствие чего заявленные соединения могут найти применение в медицине для лечения сахарного диабета 2 типа.
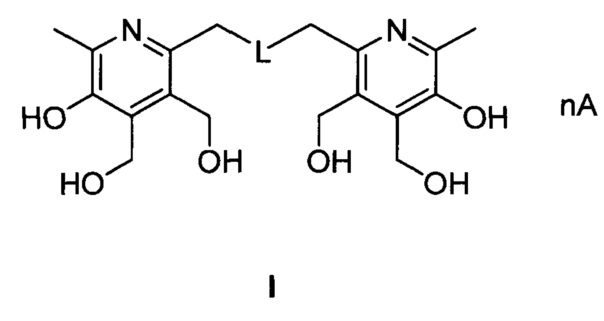
, где:
при L=-S-; n=2, А=HCl (I-1);
при L=-SS-; n=2, А=HCl (I-2);
при L=-S(O)-; n=2, А=HCl (I-3);
при L=-S(O)2-; n=2, А=HCl (I-4);
при L=-S-; n=0 (I-5);
при L=-SS-; n=0 (I-6);
при L=-S(O)-; n=0 (I-7);
при L=-S(O)2-; n=0 (I-8);
при L=-S-; n=2, А=HBr (I-9);
при L=-SS-; n=2, A=HBr (I-10);
при L=-S(O)-; n=2, A=HBr (I-11);
при L=-S(O)2-; n=2, A=HBr (I-12);
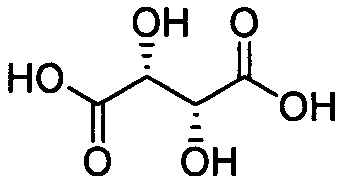
при L=-S-; n=1, A=H2SO4(I-13);
при L=-SS-; n=1, A=H2SO4(I-14);
при L=-S(O)-; n=1, A=H2SO4(I-15);
при L=-S(O)2-; n=1, A=H2SO4(I-16);
при

при

при

при

В современной медицинской практике используются следующие основные группы препаратов, рекомендованные современными алгоритмами и протоколами для лечения пациентов с сахарным диабетом 2 типа: бигуаниды, тиазолидиндионы, препараты сульфонилмочевины, глиниды, ингибиторы ДПП-4, агонисты рецепторов ГПП-1, инсулин [Алгоритмы специализированной медицинской помощи больным сахарным диабетом / под ред. И.И. Дедова, М.В. Шестаковой // Сахарный Диабет - 2015. - Т. 18 - №1S - С. 1-112]. Препараты из вышеперечисленных групп используются как в моно-, так и в комбинированной терапии.
Общими недостатками известных препаратов является: высокий риск гипогликемии, прибавка массы тела, быстрое развитие резистентности. Несмотря на широкий выбор пероральных гипогликемических препаратов, на сегодняшний день велика потребность в создании и внедрении в клиническую практику новых, эффективных и безопасных препаратов для лечения сахарного диабета 2 типа.
Одной из перспективных белковых биомишеней для создания новых противодиабетических средств для лечения диабета 2 типа является фермент глюкокиназа (ГК) [Leighton В. Small molecule glucokinase activators as novel anti-diabetic agents (Низкомолекулярные активаторы глюкокиназы в качестве новых противодиабетических средств) / В. Leighton, A. Atkinson, М.P. Coghlan // Biochem. Soc. Trans. - 2005. - Т. 33 - №2 - 371-374 c.], при этом лечебный эффект зависит от степени активности глюкокиназы. Вещества, обладающие способностью эффективно активировать глюкокиназу - активаторы глюкокиназы - представляют собой новый класс перспективных противодиабетических препаратов, которые обладают надежным гипогликемическим эффектом, связанным с повышением активности глюкокиназы печени и поджелудочной железы, доказанным в доклинических исследованиях.
При этом очень важным является то, что активаторы глюкокиназы вызывают пониженную, по сравнению с другими типами противодиабетических препаратов, клинически значимую гипогликемию в моделях сахарного диабета, не влияют на уровень липидов крови, а также не увеличивают массу тела [Matschinsky F.M. Glucokinase activators (GKAs) promise a new pharmacotherapy for diabetics (Перспектива новой противодибетической терапии с использованием активаторов глюкокиназы (GKAs) / F.М. Matschinsky, D. Porte Jr. // F1000 Med. Rep. - 2010. - Т. 2 - №1.]. Активное исследование и внедрение в клиническую практику этого класса препаратов может способствовать повышению качества и продолжительности жизни у пациентов с сахарным диабетом 2 типа, а также снижению риска возникновения микро- и макрососудистых осложнений [Grimsby J. Allosteric activators of glucokinase: Potential role in diabetes therapy (Аллостерические активаторы глюкокиназы: Потенциальная роль в терапии диабета) // Science (80-.). - 2003. - Т. 301 - №5631 - 370-373 с.].
Глюкокиназа (гексокиназа IV), представляет собой цитоплазматический фермент, катализирующий превращение глюкозы в глюкозо-6-фосфат (G-6-P), тем самым снижая ее концентрацию в клетке. В дополнение к нейрональным/нейроэндокринным клеткам, глюкокиназа (далее - ГК) селективно экспрессируется в панкреатических β-клетках и гепатоцитах. Глюкокиназу часто называют «сенсором глюкозы» в β-клетках. Концентрация глюкозы, при которой ГК демонстрирует полумаксимальную активность, составляет примерно 8.0 ммоль/л.
Три других изотипа гексокиназы (гексокиназа I, гексокиназа II, гексокиназа III) чувствительны к более низким концентрациям глюкозы (<1.0 ммоль/л). Скорость поглощения глюкозы контролируется реакцией ее фосфорилирования, катализируемой ГК и приводящей к образованию G-6-P. В нормальных физиологических условиях и концентрациях ГК может фосфорилировать глюкозу, если ее концентрация выше 1-2 ммоль/л. ГК обладает низким сродством к глюкозе с Km=6-10 ммоль/л (Km - константа Михаэлиса, равная концентрации вещества, при которой скорость реакции составляет половину от максимальной). Кроме того, ГК при физиологических концентрациях не ингибируется глюкозо-6-фосфатом [Reid S. On the developmental properties and tissue interactions of hexokinase (О эволюционных свойствах и взаимодействиях с тканями гексокиназы) / S. Reid, С. Masters // Mech. Ageing Dev. - 1985. - Т. 31 - №2 - 197-212 с.].
Активаторы глюкокиназы активируют ГК печени и поджелудочной железы, то есть имеют двойное действие, что значительно усиливает их гипогликемическую активность.
Анализ выявленного уровня техники на дату подачи заявки показал, что можно выделить основные эффекты, присущие активаторам глюкокиназы:
- увеличение сродства ГК к глюкозе;
- стимулирование фосфорилирования глюкозы в гепатоцитах и в панкреатических островках β-клеток;
- стимулирование гликолиза и синтеза гликогена;
- ингибирование глюконеогенеза и гликогенолиза в гепатоцитах;
- увеличение стимулированной глюкозой секреции инсулина;
- отсутствие влияния на секрецию инсулина при низких концентрациях глюкозы плазмы крови, что снижает риск развития гипогликемии;
- обеспечение имитации индуцированной гипергликемией транслокации ГК из ядра в цитозоль, что позволяет активировать фермент ГК и, как следствие, обеспечивает снижение уровня сахара в крови;
- нормализация показателей гликемии при сахарном диабете 2 типа, то есть приведение показателя содержания глюкозы в крови к норме;
- стимуляция пролиферации и предотвращение апоптоза β-клеток, вызванного глюкозотоксичностью [Oh Y.S. Treatment with glucokinase activator, YH-GKA, increases cell proliferation and decreases glucotoxic apoptosis in INS-1 cells (Лечение с помощью активатора глюкокиназы, YH-GKA, увеличение пролиферации клеток и уменьшение глюкотоксического апоптоза в INS-1 клетках) / Y.S. Oh, Y.-J. Lee, К. Park, Н.Н. Choi, S. Yoo, H.-S. Jun // Eur. J. Pharm. Sci. - 2014. - T. 51 - №1 - 137-145 c.].
Применение соединений, действующих по механизму активации ГК, как в комплексной, так и в монотерапии сахарного диабета 2 типа, позволяет существенно повысить качество и продолжительность жизни пациентов, а также снижает риск возникновения микро- и макрососудистых осложнений, свойственных диабету.
Таким образом, все вышеизложенное обусловливает повышенный интерес в мире к поиску активаторов глюкокиназы как антидиабетических лекарственных средств нового поколения, поскольку препаратов, действующих по этому механизму и разрешенных для клинического применения, в мире на дату представления настоящей заявки не выявлено.
Проведенный заявителем анализ уровня техники - российских и зарубежных патентных баз данных, научной литературы и Интернет-ресурсов показал, что существуют аналоги заявленного технического решения по назначению, способные активировать ГК, которые, однако, обладают существенными недостатками, а именно - недостаточно высокой эффективностью и/или существенными побочными эффектами. Далее заявителем приведена информация об основных препаратах, выявленных заявителем, из числа вышедших на стадию клинических испытаний.
Известно соединение под условным обозначением RO4389620 (Piragliatin) (Hoffman-LaRoche) [Пат. США №2003225283 Substituted-cycloalkyl and oxygenated-cycloalkyl glucokinase activators (Замещенные и окигенированные циклоалкильные активаторы глюкокиназы)], которое было одним из первых активаторов глюкокиназы, интенсивно исследовавшихся в клинике. Препарат изучался в качестве монотерапии, а также в комбинации с метформином и симвастатином, и дошел до II фазы клинического исследования, которое, однако, было прекращено. Причинами прекращения исследований явились:
- недостаточная клиническая эффективность (снижение концентрации глюкозы в крови составило не более 30-35%);
- выявление побочных эффектов у некоторых пациентов (гипогликемия) [Zhi J. Effects of piragliatin, a glucokinase activator, on fasting and postprandial plasma glucose in patients with type 2 diabetes mellitus (Влияние пираглиатина, как активатора глюкокиназы, на уровень глюкозы в плазме крови у пациентов с сахарным диабетом 2 типа натощак и после приема пищи) / J. Zhi, S. Zhai // J. Clin. Pharmacol. - 2016. - Т. 56 - №2 - 231-238 с.].
Известен аналог заявленного технического решения по назначению - соединение под условным обозначением PF-04937319 (Pfizer), дошедшее до II фазы клинических исследований. Однако в 2015 году клинические исследования были прекращены вследствие недостаточной эффективности и нестабильности наблюдаемых эффектов [Amin N.B. Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes (Исследование влияния двух дозировок PF-04937319, системного частичного активатора глюкокиназы, в качестве дополнительной терапии при лечении метформином у взрослых с сахарным диабетом 2 типа) // Diabetes, Obes. Metab. - 2015. - Т. 17-№8 - 751-759 с.].
Известно соединение под условным обозначением ТТР-399 (TransTechPharma) [Пат. WO 2009/022179, Метод-011], которое недавно исследовалось в фазе II клинических испытаний. При этом на дату подачи заявки неизвестно о каких-либо дальнейших исследованиях, очевидно, вследствие недостаточной эффективности и наблюдавшихся побочных эффектов у пациентов, таких как метеоризм и запоры [Valcarce С., Dunn I., Freeman J. ТТР399, a novel, liver selective glucokinase activator: Results from a 10-day pilot study in patients with type 2 diabetes mellitus (T2DM)

Известно соединение под условным обозначением AMG-151 (Array BioPharma) (Пат. ЕР №2573087, Pyridin-2-yl-amino-1,2,4-thiadiazole derivatives as glucokinase activators for the treatment of diabetes mellitus (Пиридин-2-ил-амино-1,2,4-тиадиазолы как активаторы глюкокиназы для лечения сахарного диабета)], которое также вошло в фазу II клинических исследований, которые при этом не завершились успешно вследствие того, что на фоне умеренной эффективности препарат показал побочные эффекты, такие как гипогликемия и гипертриглицеридемия [Katz L. AMG 151 (ARRY-403), a novel glucokinase activator, decreases fasting and postprandial glycaemia in patients with type 2 diabetes (AMG 151 (ARRY-403) - новый активатор глюкокиназы, уменьшающий гликемию у пациентов с сахарным диабетом 2 типа натощак и после приема пищи) // Diabetes, Obes. Metab. - 2016. - Т. 18 - №2 - 191-195 с.].
Известно соединение под условным обозначением GKM-001 (AdvinusTherapeutics) [Matschinsky F.M. Assessing the potential of glucokinase activators in diabetes therapy (Оценка потенциала активаторов глюкокиназы в терапии диабета) / F.М. Matschinsky // Nat. Rev. Drug Discov. - 2009. - Т. 8 - №5 - 399-416 c.], которое также исследовалось в фазе I/II клинических испытаний, которые на момент подачи настоящей заявки продолжаются. При этом опубликованные предварительные результаты этого исследования показали недостаточно высокую терапевтическую эффективность, так как уровень глюкозы снижается не более чем на 20% в наивысшей из применявшихся доз [Ramanathan V., Vachharajani N., Patel R., Barbhaiya R. GKM-001, a liver-directed/pancreas-sparing glucokinase modulator (GKM), lowers fasting and post-prandial glucose without hypoglycemia in type 2 diabetic (T2D) patients (GKM-001 - ориентированный на печень панкреапротекторный модулятор глюкокиназы (ГКМ), понижает глюкозу без гипогликемии у пациентов с сахарным диабетом 2 типа (СД2Т) натощак и после приема пищи) // 72nd Annu Meet Sci Sess Am Diabetes Assoc (ADA). Philadelphia: American Diabetes Association, 2012. C. 293.].
Известен препарат под условным обозначением Sinogliatin (Roche) [Пат. США №20100234285, Benzofuranyl derivatives used as glucokinase inhibitors (Производные бензофуранила, используемые в качестве ингибиторов глюкокиназы)], который также исследуется в фазе II клинических испытаний, при этом публичная информация об их результатах на дату предоставления заявки недоступна.
При этом из описания указанного технического решения известно, что максимальный уровень активации глюкокиназы при помощи известного соединения достигает 60.8%.
Кроме описанных выше препаратов, ряд соединений на дату подачи заявки дошли до фазы I клинических испытаний. Например, речь идет о препаратах RO-4597014, R-1511 (Roche), МК-0599 (Merck), BMS-820132 (Bristol-Myers Squibb), TMG-123 (Teijin Pharma). При этом ни один из них не перешел в фазу II клинических исследований, а имеющиеся на момент подачи настоящей заявки опубликованные данные позволяют высказать предположения либо об ограниченной эффективности, либо о наличии тех или иных побочных эффектов.
Таким образом, основываясь на вышеизложенном, можно сделать вывод о том, что проблема создания эффективных активаторов ГК, разрешенных к клиническому применению, остается на дату подачи настоящей заявки актуальной не только в РФ, но и за рубежом, поскольку максимально выявленный уровень активации глюкокиназы достигает всего 60.8%.
Задачей заявленного технического решения является получение новых соединений, обладающих высокой противодиабетической активностью, которые обеспечивают возможность вывода на рынок новых противодиабетических лекарственных средств, не имеющих аналогов в мире.
Техническим результатом предлагаемого изобретения является создание принципиально новых, не имеющих аналогов в мире по структуре, импортозамещающих, доступных по цене широким слоям населения противодиабетических соединений на основе пиридоксина, обладающих способностью эффективно активировать глюкокиназу, которые потенциально позволят существенно повысить качество и продолжительность жизни пациентов, а также снизить риск возникновения микро- и макрососудистых осложнений.
Задача решается и указанный технический результат достигается получением заявленных новых соединений общей формулы I, обладающих способностью эффективно (на 85-150% относительно базового уровня) активировать глюкокиназу.

Заявленные соединения синтезируют посредством трехстадийного синтеза из исходного 6-гидроксиметильного производного пиридоксина 1 согласно нижеприведенной схеме:

Заявленные соединения I-3 и I-4 синтезируют из соединения I-1 путем окисления последнего пероксидом водорода в уксусной кислоте согласно схеме 2:

Заявленные соединения I-5, I-6, I-7 и I-8 синтезируют из соединений I-1, I-2, I-3 и I-4 соответственно действием основания; заявленные соли, образованные бромоводородной (I-9, I-10, I-11 и I-12), серной (I-13, I-14, I-15 и I-16) и L-винной (I-17, I-18, I-19 и I-20) кислотами получают действием перечисленных кислот на соединения I-5, I-6, I-7 и I-8 согласно схеме 3:

Характеристики новых соединений, полученных в соответствии с вышеприведенной схемой, описаны далее в примерах конкретного выполнения.
Структуры полученных соединений подтверждены методами масс-спектрометрии,1Н и13C ЯМР-спектроскопии. Спектры ЯМР регистрируют на приборе Bruker AVANCE-400. Химический сдвиг определяют относительно сигналов остаточных протонов дейтерированных растворителей (1Н и13С). Температуры плавления определяют с помощью прибора Stanford Research Systems MP А-100 OptiMelt. Контроль за ходом реакций и чистотой соединений проводят методом ТСХ на пластинах Sorbfil Plates. Эксперимент ВЭЖХ/МС сверхвысокого разрешения проведен с использованием масс-спектрометра TripleTOF 5600, АВ Sciex (Германия).
Заявленное техническое решение поясняется Фиг. 1, Фиг. 2, Фиг. 3.
На Фиг. 1 представлена Таблица 1, в которой приведены ГК-активирующие свойства соединений I-1 - I-20, где ЕС50, или полумаксимальная эффективная концентрация, означает концентрацию вещества, которая вызывает эффект, равный половине максимального возможного для данного вещества после истечения некоторого промежутка времени.
На Фиг. 2 представлен график, показывающий влияние I-2 на активность глюкокиназы. По оси X - активирование в %, по оси Y - lg [1-2], М.
На Фиг. 3 представлена Таблица 2 с параметрами острой токсичности соединений I-1, I-2, I-5, I-6, I-9, I-10, I-13, I-14, I-17 и I-18 при внутрижелудочном введении крысам (значения ЛД50).
Примеры конкретного выполнения заявленного технического решения
Пример 1. Синтез дигидрохлорида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-1).
Соединение I-1 получают в 3 стадии из исходного соединения (1).
1 стадия. Навеску 2.00 г (8.4 ммоль) 6-(гидроксиметил)-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-9-ола (1) [Штырлин, Н.В. Новый метод синтеза 6-метил-2,3,4-трис(гидроксиметил)пиридин-5-ола / Н.В. Штырлин, А.Д. Стрельник, Л.П. Сысоева, О.А. Лодочникова, Е.Н Климовицкий, Ю.Г. Штырлин // Журн. орг. химии. - 2009. - Т. 45, Вып. 8. - С. 1274-1275.] суспендируют в 20 мл дихлорметана. К полученной суспензии при перемешивании одной порцией добавляют 2.90 мл (20.9 ммоль) триэтиламина, затем по каплям 1.50 мл (21.0 ммоль) ацетилхлорида. Реакционную смесь перемешивают 2 часа при комнатной температуре. После этого раствор промывают три раза 20 мл воды. Органическую фракцию сушат над сульфатом магния, отфильтровывают и упаривают на роторном испарителе с получением 2.65 г (98%) (9-ацетокси-3,3,8-триметил-1,5-дигидро-[1,3] диоксепино[5,6-с]пиридин-6-ил)метилацетата (2).
2 стадия. Навеску 2.65 г (8.2 ммоль) (9-ацетокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метилацетата (2) растворяют в 32 мл метилового спирта в круглодонной колбе на 100 мл, отдельно приготовляют раствор 2.00 г нонагидрата сульфида натрия (8.3 ммоль) в 16 мл воды. Растворы смешивают при активном перемешивании. Реакционную смесь оставляют на два часа для завершения реакции. По окончании реакции основную часть метилового спирта упаривают на роторном испарителе. В кубовый остаток добавляют 40 мл дистиллированной воды, после чего раствор нейтрализуют до рН=6.5 соляной кислотой. Образовавшийся осадок отфильтровывают и промывают тремя порциями по 15 мл дистиллированной воды с получением 1.50 г (77%) неочищенного бис((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метил)сульфида (3а). Полученный продукт используют на следующей стадии без дополнительной очистки.
3 стадия. Навеску 1.50 г (3.2 ммоль) бис((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метил)сульфида (3а) растворяют в 16 мл 2М соляной кислоты. Полученную смесь нагревают на водяной бане при 40°C в течение 3 часов. По окончании реакции раствор отфильтровывают, фильтрат упаривают на роторном испарителе. Кубовый остаток перекристаллизовывают из метанола с получением 0,92 г (62%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида дигидрохлорида (I-1). Т.пл. 185-187°C. ЯМР1H (400 МГц, D2O), δ, м.д.: 2.56 (с, 6Н, 2СН3); 4.20 (с, 4Н, 2СН2); 4.73 (с, 4Н, 2СН2); 5.08 (с, 4Н, 2СН2). ЯМР13С {1H} (100 МГц, D2O), δ, м.д.: 14.20; 30.08; 55.33; 57.02; 134.01; 140.64; 142.59; 143.59; 152.38. HRMS-ESI: найдено

Пример 2. Синтез дигидрохлорида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида (I-2)
Соединение I-2 получают в 2 стадии из исходного соединения (2).
1 стадия. Навеску 11.90 г (49.5 ммоль) нонагидрата сульфида натрия растворяют в 50 мл воды, к полученному раствору добавляют 1.70 г (52.0 ммоль) серы. Полученную смесь перемешивают при 90°C до полного растворения серы, получая таким образом раствор дисульфида натрия. Навеску 8.00 г (24.7 ммоль) (9-ацетокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метилацетата (2) растворяют в 100 мл метанола. К полученному раствору при активном перемешивании добавляют раствор дисульфида натрия. Реакционную смесь оставляют на два часа для завершения реакции. По окончании реакции основную часть метилового спирта упаривают на роторном испарителе. В кубовый остаток добавляют 40 мл дистиллированной воды, после чего раствор нейтрализуют до рН=6.5 соляной кислотой. Образовавшийся осадок отфильтровывают и промывают 3 порциями по 15 мл дистиллированной воды с получением 5.47 г (87%) неочищенного бис((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метил)дисульфида (3б). Полученный продукт используют на следующей стадии без дополнительной очистки.
2 стадия. Навеску 5.47 г (10.8 ммоль) бис((9-гидрокси-3,3,8-триметил-1,5-дигидро-[1,3]диоксепино[5,6-с]пиридин-6-ил)метил)дисульфида (3б) растворяют в 100 мл 2М соляной кислоты. Полученную смесь нагревают на водяной бане при 40°C в течение 3 часов. По окончании реакции раствор отфильтровывают, фильтрат упаривают на роторном испарителе. Кубовый остаток перекристаллизовывают из смеси вода-изопропанол (1:8) с получением 2.70 г (50%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида дигидрохлорида (1-2). Т.пл. 197-200°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.61 (с, 6Н, 2СН3); 4.19 (с, 4Н, 2СН2); 4.74 (с, 4Н, 2СН2); 5.03 (с, 4Н, 2СН2). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.20; 35.54; 55.61; 57.21; 133.97; 140.69; 142.63; 143.36; 152.52. HRMS-ESI: найдено
Пример 3. Синтез дигидрохлорида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида (I-3)
Соединение I-2 получают из исходного соединения (I-1), получение которого описано в Примере 1.
Навеску 10.00 г (21.3 ммоль) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида дигидрохлорида (I-1) суспендируют в 120 мл уксусной кислоты. К полученной смеси добавляют 3.54 мл (42.6 ммоль) 36% раствора пероксида водорода и 32 мл воды. Реакционную смесь перемешивают при комнатной температуре в течение 4 часов, после чего упаривают на роторном испарителе. Кубовый остаток перекристаллизовывают из изопропилового спирта с получением 8.7 г (84%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида дигидрохлорида (I-3). Т.пл. 183-184°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.62 (с, 6Н, СН3); 4.64 (д,2JHH=14.1 Гц, 2Н, CHaHb-S); 4.79 (с, 4Н, CH2-ОН); 4.89 (д,2JHH=14.1 Гц, 2Н, CHaHb-S); 5.05 (с, 4Н, CH2-ОН). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.52; 51.40; 55.76; 57.09; 133.88; 136.02; 141.91; 144.98; 153.27. HRMS-ESI: найдено
Пример 4. Синтез дигидрохлорида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона (I-4)
Соединение I-2 получают по методике, описанной в Примере 3, с той разницей, что используют 14.30 мл (170 ммоль) 36% раствора пероксида водорода и синтез ведут при температуре 50°C. Получают 5.5 г (51.5%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона дигидрохлорида (I-4). Т.пл. 181-183°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.64 (с, 6Н, 2СН3); 4.82 (с, 4Н, 2CH2OH); 5.08 (с, 4Н, 2СН2ОН). ЯМР13С {1H} (100 МГц, D2O), δ, м.д.: 14.67; 56.05; 57.45; 129.30; 136.67; 141.65; 146.07; 153.87. HRMS-ESI: найдено
Пример 5. Синтез бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-5)
Соединение I-5 получают из исходного соединения (I-1), получение которого описано в Примере 1.
Навеску 2.00 г (4.26 ммоль) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида дигидрохлорида (I-1), растворяют в 50 мл дистиллированной воды. К полученному раствору добавляют 0.72 г (8.57 ммоль) гидрокарбоната натрия. Реакционную смесь перемешивают до прекращения выделения газа. Образовавшийся осадок отфильтровывают и промывают 10 мл воды с получением 1.65 г (98%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-5). Т.пл. 186°C с разложением. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.32 (с, 6Н, 2СН3); 3.89 (с, 4Н, 2CH2S); 4.53 (д,3JHH=4.6 Гц, 4Н, 2CH2OH); 4.77 (с, 4Н, 2CH2OH); 5.02 (т,3JHH=4.6 Гц, 2Н, СН2ОН); 5.73 (уш.с, 2Н, СН2ОН); 9.20 (уш.с, 2Н, ArOH). ЯМР13С {1H} (100 МГц, ДМСО-d6), δ, м.д.: 19.25; 35.48; 56.30; 56.57; 131.04; 133.40; 144.98; 146.42; 148.93. HRMS-ESI: найдено [М+Н]+ 397.1428, C18H24N2O6S, вычислено [М+Н]+ 397.1428.
Пример 6. Синтез бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида (I-6)
Соединение I-6 получают по методике, описанной в Примере 5, из исходного соединения (I-2), получение которого описано в Примере 2. Выход 1.67 г (98%). Т.пл. 182-184°C. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.34 (с, 6Н, 2СН3); 4.11 (с, 4Н, 2CH2S); 4.56 (д,3JHH=4.9 Гц, 4Н, 2CH2OH); 4.78 (с, 4Н, 2CH2OH); 5.05 (т,3JHH=4.9 Гц, 2Н, 2CH2OH); 5.75 (уш.с, 2Н, 2СН2ОН); 9.30 (уш.с, 2Н, ArOH). ЯМР13С {1H} (100 МГц, ДМСО-d6), δ, м.д.: 19.28; 42.81; 56.55; 56.57; 131.58; 133.24; 144.72; 145.56; 149.29. HRMS-ESI: найдено [М+Н]+ 429.1149, C18H24N2O6S2, вычислено [М+Н]+ 429.1149.
Пример 7. Синтез бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида (I-7)
Соединение I-7 получают по методике, описанной в Примере 5, из исходного соединения (I-3), получение которого описано в Примере 3. Выход 1.66 г (98%). Т.пл. 240°C с разложением. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.35 (с, 6Н, 2СН3); 4.39 (д,2JHH=13.4 Гц, 2Н, 2CHaHb); 4.41 (д,2JHH=13.4 Гц, 2Н, 2CHaHb); 4.49 (д,2JHH=12.5 Гц, 2Н, 2CHcHd); 4.54 (д,2JHH=12.5 Гц, 2Н, 2CHcHd); 4.78 (с, 4Н, 2СН2). ЯМР13С {1Н} (100 МГц, ДМСО-d6), δ, м.д.: 19.41; 56.27; 56.30; 56.43; 133.06; 133.57; 140.36; 146.26; 149.58. HRMS-ESI: найдено [М+Н]+ 413.1377, C18H24N2O7S, вычислено [М+Н]+ 413.1377.
Пример 8. Синтез бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона (I-8)
Соединение I-8 получают по методике, описанной в Примере 5, из исходного соединения (I-4), получение которого описано в Примере 4. Выход 1.68 г (98%). Т.пл. 240°C с разложением. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.36 (с, 6Н, 2СН3); 4.52 (д,3JHH=5.1 Гц, 4Н, 2CH2OH); 4.77 (с, 4Н, 2СН2); 4.79 (с, 4Н, 2СН2); 4.94 (т,3JHH=5.1 Гц, 2Н, 2СН2ОН); 5.80 (уш.с, 2Н, 2СН2ОН); 9.47 (уш.с, 2Н, 2ArOH). ЯМР13С {1Н} (100 МГц, ДМСО-d6), δ, м.д.: 19.34; 56.54; 56.56; 57.68; 133.26; 133.99; 137.77; 145.93; 149.91. HRMS-ESI: найдено [М+Н]+ 429.1326, C18H24N2O8S, вычислено [М+Н]+ 429.1326.
Пример 9. Синтез дигидробромида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-9)
Соединение I-9 получают из исходного соединения (I-5), получение которого описано в Примере 5.
Навеску 0.10 г (0.3 ммоль) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида суспендируют в 1 мл дистиллированной воды. К полученной смеси при перемешивании добавляют 0.06 мл (0.5 ммоль) 48% бромоводородной кислоты. Полученный раствор упаривают на роторном испарителе с получением 0.14 г (99%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида дигидробромида (I-9). Т.пл. 185-187°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.59 (с, 6Н, 2СН3); 4.23 (с, 4Н, 2CH2S); 4.75 (с, 4Н, 2СН2ОН); 5.04 (с, 4Н, 2СН2ОН). ЯМР13С {1H} (100 МГц, D2O), δ, м.д.: 14.28; 30.16; 55.41; 57.15; 134.07; 140.64; 142.67; 143.63; 152.43. HRMS-ESI: найдено [М-2Br-Н]+ 397.1428, C18H26Br2N2O6S, вычислено [М-2Br-Н]+ 397.1428.
Пример 10. Синтез дигидробромида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида (I-10)
Соединение I-10 получают по методике, описанной в Примере 9, из исходного соединения (I-6), получение которого описано в Примере 6. Выход 0.14 г (99%). Т.пл. 189-190°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.64 (с, 6Н, 2СН3); 4.21 (с, 4Н, 2CH2S); 4.76 (с, 4Н, 2CH2OH); 5.06 (с, 4Н, 2CH2OH). ЯМР13С {1H} (100 МГц, D2O), δ, м.д.: 14.26; 35.64; 55.66; 57.28; 134.00; 140.71; 142.66; 143.41; 152.55. HRMS-ESI: найдено [М-2Br-Н]+ 429.1149, C18H26Br2N2O6S2, вычислено [М-2Br-Н]+ 429.1149.
Пример 11. Синтез дигидробромида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида (I-11)
Соединение I-11 получают по методике, описанной в Примере 9, из исходного соединения (I-7), получение которого описано в Примере 7. Выход 0.138 г (99%). Т.пл. 171-172°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.64 (с, 6Н, 2СН3); 4.65 (д,2JHH=14.1 Гц, 2Н, 2CHaHb); 4.79 (д,2JHH=13.3 Гц, 2Н, 2CHcHd); 4.83 (д,2JHH=13.3 Гц, 2Н, 2CHcHd); 4.91 (д,2JHH=14.1 Гц, 2Н, 2CHaHb); 5.07 (с, 4Н, 2CH2OH). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.56; 51.45; 55.81; 57.18; 133.88; 136.05; 141.95; 145.00; 153.31. HRMS-ESI: найдено [М-2Br-Н]+ 413.1377, C18H26Br2N2O7S, вычислено [М-2Br-Н]+ 413.1377.
Пример 12. Синтез дигидробромида бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона (I-12)
Соединение I-12 получают по методике, описанной в Примере 9, из исходного соединения (I-8), получение которого описано в Примере 8. Выход 0.14 г (99%). Т.пл. 188-189°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.65 (с, 6Н, 2СН3); 4.79 (с, 4Н, 2CH2OH); 4.84 (с, 4Н, 2СН2ОН). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 12.49; 51.23; 53.88; 55.32; 127.10; 134.49; 139.50; 143.86; 151.70. HRMS-ESI: найдено [М-2Br-Н]+ 429.1326, C18H26Br2N2O8S, вычислено [М-2Br-Н]+ 429.1326.
Пример 13. Синтез сульфата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-13)
Соединение I-13 получают из исходного соединения (I-5), получение которого описано в Примере 5.
Навеску 0.10 г (0.3 ммоль) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида суспендируют в 1 мл дистиллированной воды. К полученной смеси при перемешивании добавляют 0.25 мл (0.3 ммоль) 1М раствора серной кислоты. Полученный раствор упаривают на роторном испарителе с получением 0.12 г (99%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида сульфата (I-13). Т.пл. 240°C с разложением. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.55 (с, 6Н, 2СН3); 4.21 (с, 4Н, 2CH2S); 4.70 (с, 4Н, 2СН2ОН); 4.96 (с, 4Н, 2СН2ОН). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.21; 30.21; 55.33; 56.94; 134.01; 140.76; 142.63; 143.52; 152.33. HRMS-ESI: найдено

Пример 14. Синтез сульфата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида (I-14)
Соединение I-14 получают по методике, описанной в Примере 13, из исходного соединения (I-6), получение которого описано в Примере 6. Выход 0.12 г (99%). Т.пл. 168-170°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.59 (с, 6Н, СН3); 4.22 (с, 4Н, CH2S); 4.70 (с, 4Н, СН2ОН); 4.99 (с, 4Н, CH2OH). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.13; 35.14; 55.51; 57.12; 133.97; 140.66; 142.68; 143.17; 152.44. HRMS-ESI: найдено
Пример 15. Синтез сульфата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида (I-15)
Соединение I-15 получают по методике, описанной в Примере 13, из исходного соединения (I-7), получение которого описано в Примере 7. Выход 0.12 г (99%). Т.пл. 154-156°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.60 (с, 6Н, 2СН3); 4.69 (д,2JHH=14.0 Гц, 2Н, 2CHaHb); 4.75 (д,2JHH=13.6 Гц, 2Н, 2CHcHd); 4.77 (д,2JHH=13.6 Гц, 2Н, 2CHcHd); 4.90 (д,2JHH=14.0 Гц, 2Н, 2CHaHb); 5.02 (с, 4Н, 2CH2OH). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.49; 51.44; 55.68; 57.03; 133.97; 136.03; 141.90; 145.02; 153.23. HRMS-ESI: найдено
Пример 16. Синтез сульфата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона (I-16)
Соединение I-16 получают по методике, описанной в Примере 13, из исходного соединения (I-8), получение которого описано в Примере 8. Выход 0.12 г (99%). Т.пл. 156-160°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.61 (с, 6Н, 2СН3); 4.79 (с, 4Н, 2CH2OH); 5.04 (с, 4Н, 2CH2OH). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.54; 53.31; 56.00; 57.48; 129.13; 136.78; 141.86; 145.97; 153.91. HRMS-ESI: найдено
Пример 17. Синтез L-тартрата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида (I-17)
Соединение I-17 получают из исходного соединения (I-5), получение которого описано в Примере 5.
Навеску 0.10 г (0.3 ммоль) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида суспендируют в 1 мл дистиллированной воды. К полученной смеси при перемешивании добавляют 0.04 г (0.3 ммоль) L-винной кислоты. Полученный раствор упаривают на роторном испарителе с получением 0.14 г (99%) бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфида L-тартрата (I-17). Т.пл. 169-170°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.51 (с, 6Н, 2СН3); 4.14 (с, 4Н, 2CH2S); 4.49 (с, 2Н, 2СНОН); 4.71 (с, 4Н, 2CH2OH); 4.96 (с, 4Н, 2СН2ОН). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.71; 3.48; 55.48; 56.78; 72.64; 133.40; 141.33; 141.50; 143.82; 152.23; 176.07. HRMS-ESI: найдено

Пример 18. Синтез L-тартрата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)дисульфида (I-18)
Соединение I-18 получают по методике, описанной в Примере 17 из исходного соединения (I-6), получение которого описано в Примере 6. Выход 0.13 г (99%). Т.пл. 133-134°C. ЯМР1Н (400 МГц, D2O), δ, м.д.: 2.52 (с, 6Н, СН3); 4.08 (с, 4Н, CH2S); 4.42 (с, 2Н, СНОН); 4.65 (с, 4Н, СН2ОН); 4.94 (с, 4Н, CH2OH). ЯМР13С {1Н} (100 МГц, D2O), δ, м.д.: 14.50; 35.88; 55.64; 56.98; 72.71; 133.54; 140.65; 141.69; 143.66; 152.64; 176.26. HRMS-ESI: найдено
Пример 19. Синтез L-тартрата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфоксида (I-19)
Соединение 1-19 получают по методике, описанной в Примере 17, из исходного соединения (1-7), получение которого описано в Примере 7. Выход 0.14 г (99%). Т.пл. 240°C с разложением. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.35 (с, 6Н, 2СН3); 4.31 (с, 2Н, 2СНОН); 4.38 (д,2JHH=13.2 Гц, 2Н, 2CHaHbS); 4.41 (д,2JHH=13.2 Гц, 2H, 2CHaHbS); 4.48 (дд,2JHH=12.5 Гц,3JHH=3,4 Гц, 2Н, 2CHaHbOH); 4.54 (дд,2JHH=12.5 Гц,3JHH=5.0 Гц, 2Н, 2CHaHbOH); 4.83 (м, 2Н, 2CHaHbOH); 5.73 (уш.с, 2Н, 2СН2ОН); 9-39 (уш.с, 2Н, 22ArOH). ЯМР13С {1Н} (100 МГц, ДМСО-d6), δ, м.д.: 19.38; 56.22; 56.26; 56.36; 72.18; 133.04; 133.58; 140.49; 146.17; 149.37; 173.16. HRMS-ESI: найдено
Пример 20. Синтез L-тартрата бис((5-гидрокси-3,4-бис(гидроксиметил)-6-метилпиридин-2-ил)метил)сульфона (I-20)
Соединение I-20 получают по методике, описанной в Примере 17, из исходного соединения (I-8), получение которого описано в Примере 8. Выход 0.13 г (99%). Т.пл. 240°C с разложением. ЯМР1Н (400 МГц, ДМСО-d6), δ, м.д.: 2.36 (с, 6Н, 2СН3); 4.32 (с, 2Н, 2СНОН); 4.52 (с, 4Н, 2CH2OH); 4.77 (с, 4Н, 2СН2ОН); 4.52 (с, 4Н, 2CH2S); 4.95 (уш.с, 2Н, 2СН2ОН); 5.80 (уш.с, 2Н, 2СН2ОН); 9.49 (уш.с, 2Н, 2ArOH). ЯМР13С {1Н} (100 МГц, ДМСО-d6), δ, м.д.: 19.33; 56.50; 56.55; 57.59; 72.21; 133.40; 134.10;137.70; 145.92; 149.96; 173.20. HRMS-ESI: найдено
Пример 21. Определение активности по отношению к глюкокиназе.
Активность глюкокиназы (ГК человеческой печени рекомбинантная, экспрессированная в Е. coli, Sigma, США) определяют посредством сопряженной реакции образования глюкозо-6-фосфата с генерацией восстановленной формы никотинамидадениндинуклеотида (далее - НАД) с помощью глюкозо-6-фосфатдегидрогиназы (Г6ФДГ L. mesenteroides, 550-1100 ЕД/мг, Sigma, США).
Анализ проводят при 37°C в 96-луночном прозрачном полистирольном планшете с плоским дном (Costar 9018, США) в конечном инкубируемом объеме 210 мкл. Инкубационная смесь содержит: 25 мМ буфера на основе 4-(2-гидроксиэтил)-1-пиперазинэтансульфоновой кислоты (далее - HEPES) (рН 7.2), 25 мМ KCl, 5 мМ D-глюкозы, 1 мМ аденозинтрифосфата (далее - АТФ), 1.8 мМ НАД, 2 мМ MgCl2, 1 мМ дитиотретола, тестируемое соединение или 5% диметилсульфоксид (далее - ДМСО), 1.8 ед/мл Г6ФДГ и 2 мкг/мл ГК. Тестируемые соединения вносят в 5% ДМСО и предварительно инкубируют с ГК в термостатируемом шейкере PST-60HL (Biosan, Латвия) в течение 10 мин до достижения температурного равновесия, а затем инициируют реакцию введением 10 мкл раствора D-глюкозы [Salt D. Sigma quality control test procedure enzymatic assay of glucokinase (EC 2.7.1.2) Principle: P. 7-10] и [Биззаро Т.Ф. et al. Активаторы глюкокиназы. Патент РФ №2242469 по заявке: 2001126559/04, от 20.03.2000 г.].
В соответствии с используемой методикой за меру активности ГК принимают повышение оптической плотности при длине волны 340 нм в течение 20 мин инкубирования после начала реакции. Измерения проводят с помощью микропланшетного ридера Infinite М200 PRO (Тесап, Австрия). В лунки, содержавшие 5% ДМСО без тестируемого соединения, добавляют ГК в количестве, достаточном для того, чтобы за 20 мин инкубирования достичь повышения оптической плотности от 0.08 до 0.1 ед. Предварительными опытами установлено, что реакция ГК в течение этого периода времени была линейной.
При этом активность ГК в контрольных лунках сравнивают с активностью в лунках, содержавших тестируемые активаторы ГК.
Результаты проведенных испытаний по определению активности заявленных соединений по отношению к глюкокиназе представлены на Фиг. 1 и Фиг. 2.
Из данных, приведенных в Таблице 1 (Фиг. 1) и на графике, приведенном на Фиг. 2, видно, что заявленные соединения I-1 - I-20 проявляют высокий уровень активации глюкокиназы. Так, в концентрации 100 мкМ они способны увеличивать активность этого фермента более чем на 85-150% относительно базового уровня (то есть без тестируемого соединения) соответственно.
Наименьшее значение ЕС50 при этом составляет 18.56 мкМ для соединения I-1 и его солевых форм (I-5, I-9, I-13, I-17) соответственно.
Представленный технический результат, проанализированный заявителем по параметру увеличения уровня активности фермента глюкокиназы, превосходит известные заявителю результаты аналогов по назначению.
Так, у препарата Sinogliatin (Roche) [Пат. США №20100234285], дошедшего до фазы II клинических испытаний, максимальный уровень активации глюкокиназы достигает 60.8%. Это существенно ниже, чем у заявленных соединений I-1 - I-20, при использовании которых увеличение уровня активности фермента глюкокиназы достигает 85-150% (см. Фиг. 1, 2).
Таким образом, эффективность заявленного технического решения по уровню активации глюкокиназы (85-150%) выше от 1.4 до 2.5 раз соответственно по сравнению с наиболее эффективным известным препаратом США Sinogliatin (Roche), максимальный уровень активации глюкокиназы которого достигает 60.8%.
Исходя из вышеизложенного и на основании проведенных исследований можно сделать вывод, что соединения I-1 - I-20 являются эффективными активаторами глюкокиназы и, исходя из исследованного заявителем уровня техники, существенно превосходят по активности аналоги заявленного технического решения по назначению. Из уровня техники известно, что величины активирования глюкокиназы, превышающие 50%, могут считаться существенными, что свидетельствует о безусловной перспективности заявленных соединений при использовании по назначению.
Пример 22. Определение острой токсичности на крысах.
Исследование проводили на крысах линии Sprague Dawley (6-8 недель, 180-220 г) обоего пола. Внутрижелудочное введение осуществляли с помощью желудочного зонда животным, лишенным корма не менее чем на 8 часов со свободным доступом к воде. Расчет доз вели на 250 г живой массы крысы. Для исследования токсичности брали, например, наиболее активные соединения I-1, I-2, I-5, I-6, I-9, I-10, I-13, I-14, I-17 и I-18.
Для внутрижелудочного введения крысам в дозе 2000 мг/кг взвешивают навески этих соединений по 4,0 г в полистироловых лодочках на весах Vibra (Shinko Denshi, Япония, кат. № AF 225 DRCE), переносят в мерный цилиндр объемом 40 мл, доводят дистиллированной водой до метки 40 мл в мерной колбе и растворяют. Объем введения 5 мл на 250 г живой массы крысы. Объем введения рассчитывают индивидуально для каждого животного, основываясь на массе тела, зарегистрированной непосредственно перед введением вещества. Доступ к корму возобновляют через час после введения.
Клинический осмотр животных проводят индивидуально после введения на протяжении 30 минут, затем не реже раза в час на протяжении 4-х часов, далее ежедневно 1 раз в день в течение 14 дней. Массу тела регистрируют непосредственно перед введением препарата для расчета объема введения, далее 1 раз в два дня.
Параметры острой токсичности ЛД50 приведены в Таблице 2 (Фиг. 3). На основании проведенных исследований обнаружено, что заявленные соединения являются малотоксичными, поскольку параметр ЛД50 при пероральном применении у крыс превышает 2000 мг/кг массы тела.
Исходя из вышеизложенного, можно сделать вывод, что заявителем решена поставленная задача и достигнут заявленный технический результат - получены новые соединения на основе пиридоксина, обладающие способностью эффективно (на 85-150% относительно базового уровня, т.е. от 1.4 до 2.5 раз) активировать глюкокиназу, что обеспечивает возможность вывода на рынок новых высокоэффективных и безопасных лекарственных средств терапии сахарного диабета 2 типа, не имеющих аналогов в мире по структуре, импортозамещающих, доступных по цене широким слоям населения, которые потенциально позволят существенно повысить качество и продолжительность жизни пациентов, а также снизить риск возникновения микро- и макрососудистых осложнений.
Заявленное техническое решение соответствует критерию «новизна», предъявляемому к изобретениям, так как из исследованного уровня техники не выявлены технические решения, обладающие заявленной совокупностью отличительных признаков, обеспечивающих достижение заявленных результатов.
Заявленное техническое решение соответствует критерию «изобретательский уровень», предъявляемому к изобретениям, так как не является очевидным для специалиста в данной области науки и техники.
Заявленное техническое решение соответствует критерию «промышленная применимость», т.к. может быть реализовано на любом специализированном предприятии с использованием стандартного оборудования, известных отечественных материалов и технологий.
Реферат
Изобретение относится к соединениям на основе пиридоксина общей формулы:где при L=-S-; n=2, А=HCl; при L=-SS-; n=2, А=HCl; при L=-S(O)-; n=2, А=HCl; при L=-S(O)-; n=2, А=HCl; при L=-S-; n=0; при L=-SS-; n=0; при L=-S(O)-; n=0; при L=-S(O)-; n=0; при L=-S-; n=2, А=HBr; при L=-SS-; n=2, А=HBr; при L=-S(O)-; n=2, А=HBr; при L=-S(O)-; n=2, А=HBr; при L=-S-; n=1, А=HSO; при L=-SS-; n=1, А=HSO; при L=-S(O)-; n=1, А=HSO; при L=-S(O)-; n=1, А=HSO;при;при;при;при;которые проявляют высокий уровень активации глюкокиназы. 1 з.п. ф-лы, 3 ил., 23 пр.
Формула




Комментарии