Водное базовое покрытие и получение многослойных красочных систем посредством применения базового покрытия - RU2746776C2
Код документа: RU2746776C2
Описание
Настоящее изобретение относится к водному материалу базового покрытия. Настоящее изобретение также относится к способу получения многослойной красочной системы, который включает получение по меньшей мере одной пленки базового покрытия, применяя по меньшей мере один такой водный материал базового покрытия. Кроме того, настоящее изобретение относится к многослойной красочной системе, полученной посредством способа в соответствии с изобретением.
Предшествующий уровень техники
Известны многослойные красочные системы на металлических основах или пластмассовых основах, где примерами при этом являются многослойные красочные системы в секторе автомобильной промышленности. Начиная, в принципе, с металлической основы, многослойные красочные системы указанного вида на металлических основах, как правило, содержат отдельно затвердевшую пленку электроосажденного покрытия, пленку, которая наносится непосредственно на пленку электроосажденного покрытия и затвердевает отдельно, которую обычно называют пленкой грунтовочного покрытия, по меньшей мере одну пленку, которая содержит цветные пигменты и/или эффектные пигменты, и которую, как правило, называют пленкой базового покрытия, а также пленку покровного лака. Пленка базового покрытия и пленка покровного лака, как правило, затвердевают вместе.
Как правило, соответствующие базовое покрытие и пленки покровного лака наносят на пластмассовые основы, которые являются актуальными в секторе компонентов, предназначенных для установки в или на транспортные средства. В некоторых случаях, перед тем, как нанести материал базового покрытия, наносят определенные грунтовки или адгезионные грунтовки.
В частности, в случае металлических основ, существуют подходы, где пытаются обойтись без отдельной стадии отверждения состава покрытия, который был нанесен непосредственно на затвердевшую пленку электроосажденного покрытия (то есть, состав покрытия, называемый грунтовкой в стандартном способе, описанном выше), и в то же время, необязательно, пытаются уменьшить толщину покрывающей пленки, полученной из указанного состава покрытия. В уровне техники, указанную покрывающую пленку, которая, по этой причине, не затвердевает отдельно, в результате, часто называют пленкой базового покрытия (и больше не называют пленкой грунтовочного покрытия) или, для того, чтобы отличить ее от второй пленки базового покрытия, которую наносят поверх нее, ее называют первой пленкой базового покрытия. В некоторых случаях даже пытаются полностью обойтись без указанной покрывающей пленки (в случае чего, затем, непосредственно на пленке электроосажденного покрытия получают только одну так называемую пленку базового покрытия, сверху которой, без отдельной стадии отверждения, наносят материал покровного лака; другими словами, в итоге, отдельная стадия отверждения также не предусматривается). В результате, вместо отдельной стадии отверждения и вместо дополнительной заключительной стадии отверждения, стремятся просто к одной, заключительной стадии отверждения, которая следует за нанесением всех покрывающих пленок, нанесенных на пленку электроосажденного покрытия.
Устранение отдельной стадии отверждения состава покрытия, нанесенного непосредственно на пленку электроосажденного покрытия, является очень преимущественным с экологической и экономической точек зрения. Причина, естественно, в том, что указанное экономит энергию и позволяет производственной операции в целом проходить в соответствии со значительно менее строгими требованиями.
Подобные способы известны и для пластмассовых основ, в случае которых, конечно, пленку электроосажденного покрытия не получают. По этой причине, систему для совместного отверждения, состоящую из первого материала базового покрытия, второго материала базового покрытия, и материала покровного лака, например, наносят непосредственно на пластмассовую основу, которая может получить поверхностно-активирующую предварительную обработку, или помимо прочего на пленку грунтовочного покрытия или на пленку адгезионной грунтовки, которая была нанесена на основу первой.
Также известны способы повторного нанесения при ремонте для восстановления многослойных красочных систем. Следовательно, это способы, в которых необходимо снова получить многослойные красочные системы, которые были получены, как описано выше, но имеющие при этом определенные дефекты. Такие способы повторного нанесения при ремонте осуществляются, например, посредством локального ремонта дефектов (точечный ремонт), или посредством полного повторного покрытия первоначального покрытия, имеющего дефекты (двойное покрытие). В указанном случае, как правило, после локальной зачистки дефектов шлифовальной шкуркой, наносят системы, которые описаны выше, состоящие из грунтовки, базового покрытия, и покровного лака или первого базового покрытия, второго базового покрытия, и покровного лака. Также возможно нанесение только одного базового покрытия и покровного лака, нанесенного на него, за чем следует совместное отверждение. Таким образом, в этом случае, многослойная красочная система с дефектами (первоначальное покрытие) практически служит в качестве основы.
Несмотря на то, что технологические характеристики существующих многослойных красочных систем уже часто являются достаточными, с тем, чтобы соответствовать установленным требованиям производителей автомобилей, продолжает существовать необходимость в их улучшении. Это особенно относится к описанному последним способу получения многослойных красочных систем, в котором, как указано, отдельная стадия отверждения не предусматривается. Однако, даже стандартные способы, описанные ранее выше для получения многослойных красочных систем, все еще нуждаются в оптимизации в указанном отношении.
Особая задача состоит в обеспечении многослойных красочных систем, с помощью которых, во-первых, достигаются очень хорошие оптические свойства (например, устранение следов пузырей или пор), а также, во-вторых, достигается оптимальная механическая прочность, и особенно оптимальные адгезионные свойства. Основная задача состоит в том, чтобы получить хорошие адгезионные свойства в секторе ремонта покрытий.
Проблема
Соответственно, проблема, решаемая с помощью настоящего изобретения, представляла собой обеспечение многослойной красочной системы, и/или составов покрытий, которые подлежат применению при получении таких многослойных красочных систем, которые позволяют устранить указанные выше недостатки. Таким образом, цель состоит в том, чтобы дать возможность получения многослойных красочных систем (первоначальные покрытия), а также повторно наносимых при ремонте многослойных красочных систем, которые наряду с превосходными эстетическими свойствами, также имеют очень хорошие адгезионные свойства.
Техническое решение
Было выявлено, что указанные проблемы были решены посредством водного материала базового покрытия, содержащего по меньшей мере одну водную полиуретан-полимочевинную дисперсию (PD), имеющую полиуретан-полимочевинные частицы, которые присутствуют в дисперсии со средним размером частиц, составляющим 40-2000 нм, и гель-фракцию, составляющую по меньшей мере 50%, где полиуретан-полимочевинные частицы, в каждом случае в прореагировавшем виде, содержат
(Z.1.1) по меньшей мере один полиуретановый преполимер, который включает изоцианатные группы и который включает анионные группы и/или группы, которые могут превращаться в анионные группы, и
(Z.1.2) по меньшей мере один полиамин, который включает две первичные аминогруппы и одну или две вторичные аминогруппы,
а также
по меньшей мере одну водную дисперсию (wD), содержащую полимер, имеющий размер частиц, который составляет 100-500 нм, и полученный при этом посредством последовательной радикальной эмульсионной полимеризации трех разных смесей (А), (Б), и (В), олефинненасыщенных мономеров,
где
полимер, полученный из смеси (А), имеет температуру стеклования, которая составляет 10-65°С,
полимер, полученный из смеси (Б), имеет температуру стеклования, которая составляет -35-15°С, и
полимер, полученный из смеси (В), имеет температуру стеклования, которая составляет -50-15°С.
Водный материал базового покрытия, определенный выше, также будет указан ниже как материал базового покрытия в соответствии с изобретением, и соответственно является объектом настоящего изобретения. Предпочтительные варианты осуществления материала базового покрытия в соответствии с изобретением являются очевидными из описания, приведенного ниже, а также из зависимых пунктов формулы изобретения.
Дополнительным объектом настоящего изобретения является способ получения многослойной красочной системы, где по меньшей мере одну пленку базового покрытия получают посредством применения по меньшей мере одного водного материала базового покрытия в соответствии с изобретением. Кроме того, настоящее изобретение относится к многослойной красочной системе, которая была получена посредством способа в соответствии с изобретением.
В результате применения материала базового покрытия в соответствии с изобретением, получают многослойные красочные системы, которые имеют превосходные эксплуатационные свойства, в частности, превосходные эстетические свойства и, кроме того, очень хорошие адгезионные свойства. Посредством применения материала базового покрытия, также возможно особо высококачественное восстановление многослойных красочных систем, имеющих дефекты. В указанном секторе ремонта покрытий, который является особо проблематичным в отношении адгезии и механической прочности, достигается превосходный профиль свойств.
Подробное описание
Водный материал базового покрытия в соответствии с изобретением содержит по меньшей мере одну, предпочтительно ровно одну, определенную водную полиуретан-полимочевинную дисперсию (PD), причем указанная дисперсия при этом представляет собой дисперсию, в которой присутствующие полимерные частицы основаны на полиуретан-полимочевине. Такие полимеры можно, в принципе, получить посредством традиционного полиприсоединения, например, полиизоцианатов с полиолами, а также полиаминами. Однако, в отношении дисперсии (PD) в соответствии с изобретением, или полимерных частиц, которые она содержит, существуют определенные условия, которые должны соблюдаться, которые изложены ниже.
Полиуретан-полимочевинные частицы, которые присутствуют в водной полиуретан-полимочевинной дисперсии (PD), имеют гель-фракцию, составляющую по меньшей мере 50% (в отношении метода определения смотри раздел Примеров). Кроме того, полиуретан-полимочевинные частицы, которые присутствуют в дисперсии (PD), имеют средний размер частиц (который также называют средней крупностью частиц), который составляет 40-2000 нанометров (нм) (в отношении методов измерений смотри раздел Примеров).
По этой причине, дисперсии (PD), предназначенные для применения в соответствии с изобретением, представляют собой микрогелевые дисперсии. Это связано с тем, что, как уже описано выше, микрогелевая дисперсия представляет собой полимерную дисперсию, в которой, во-первых, полимер присутствует в виде сравнительно небольших частиц, или микрочастиц и, во-вторых, полимерные частицы являются, по меньшей мере частично, внутримолекулярно сшитыми. Последнее означает, что полимерные структуры, которые присутствуют в пределах частицы, приравниваются к типичной макроскопической сетке с трехмерной сетчатой структурой. Однако, если смотреть с макроскопической точки зрения, микрогелевая дисперсия указанного вида все еще является дисперсией полимерных частиц в дисперсионной среде, например, воде. В то время, как частицы также могут частично демонстрировать сшивающие мостики друг с другом (это вряд ли может быть исключено ввиду способа получения как такового), система в любом случае представляет собой дисперсию, имеющую разрозненные частицы, присутствующие в ней, которые имеют измеримый средний размер частиц.
Учитывая, что микрогели представляют собой структуры, которые находятся между разветвленными и макроскопически сшитыми системами и, следовательно, комбинируют характеристики макромолекул с сетчатой структурой, которые являются растворимыми в подходящих органических растворителях, и нерастворимых макроскопических сеток, долю сшитых полимеров могут определять, например, только после выделения твердого полимера, после удаления воды и каких-либо органических растворителей, а также после последующей экстракции. Используемые здесь свойства заключаются в том, что микрогелевые частицы, первоначально растворимые в подходящих органических растворителях, сохраняют свою внутреннюю сетчатую структуру после выделения, и ведут себя подобно макроскопической сетке в твердом материале. В сшивании можно удостовериться с помощью экспериментально доступной гель-фракции. Гель-фракция, в итоге, представляет собой, такую фракцию полимера дисперсии, которая, в виде выделенного твердого вещества, не может подвергаться молекулярно дисперсному растворению в растворителе. В этом случае следует сделать вывод, что, во время выделения полимерного твердого вещества, последующие реакции сшивания дополнительно увеличивают гель-фракцию. Указанная нерастворимая фракция, в свою очередь, соответствует доли полимера, которая присутствует в дисперсии в виде внутримолекулярно сшитых частиц и/или фракций частиц.
В контексте настоящего изобретения выяснилось, что только микрогелевые дисперсии, которые включают полимерные частицы с размерами частиц, находящимися в диапазоне, имеющем значение для изобретения, обладают всеми необходимыми эксплуатационными свойствами. Таким образом, важным моментом, в частности, является комбинация очень маленьких размеров частиц и тем не менее значительной сшитой фракции или гель-фракции. Только таким образом является возможным достичь преимущественных свойств, в частности, комбинации хороших оптических и механических свойств многослойных красочных систем.
Полиуретан-полимочевинные частицы, которые присутствуют в водной полиуретан-полимочевинной дисперсии (PD), предпочтительно имеют гель-фракцию, составляющую по меньшей мере 60%, более предпочтительно, составляющую по меньшей мере 70%, особенно предпочтительно, составляющую по меньшей мере 80%. По этой причине, гель-фракция может составлять до 100% или приблизительно 100%, как например 99% или 98%. По этой причине, в таком случае, весь - или почти весь - полиуретан-полимочевинный полимер находится в виде сшитых частиц.
Полиуретан-полимочевинные частицы, которые присутствуют в дисперсии (PD), предпочтительно имеют средний размер частиц, составляющий 40-1500 нм, более предпочтительно, составляющий 100-1000 нм, дополнительно предпочтительно, составляющий 110-500 нм, и дополнительно предпочтительно, составляющий 120-300 нм. Особенно предпочтительный диапазон составляет от 130 до 250 нм.
Полученная полиуретан-полимочевинная дисперсия (PD) является водной. Выражение "водная" известно специалисту в данной области в указанном контексте. Оно, по своей сути, относится к системе, дисперсионная среда которой исключительно или преимущественно не содержит органических растворителей (которые также называют растворяющими агентами), а вместо этого, напротив, в качестве своей дисперсионной среды содержит значительную долю воды.
Предпочтительные варианты осуществления водной природы, которые определяются посредством применения максимального количества органических растворителей и/или посредством применения количества воды, описаны далее ниже для разных компонентов и систем, например, таких как дисперсии (PD), дисперсии (wD), или материалы базовых покрытий. Полиуретан-полимочевинные частицы, которые присутствуют в дисперсии (PD), содержат, в каждом случае в прореагировавшем виде, (Z.1.1) по меньшей мере один полиуретановый преполимер, который включает изоцианатные группы и который включает анионные группы и/или группы, которые могут превращаться в анионные группы, а также (Z.1.2) по меньшей мере один полиамин, который включает две первичные аминогруппы и одну или две вторичные аминогруппы.
В случае, если в контексте настоящего изобретения указано, что полимеры, такие как полиуретан-полимочевинные частицы дисперсии (PD), например, содержат определенные компоненты в прореагировавшем виде, это означает, что указанные определенные компоненты применяются в качестве исходных соединений во время получения указанных полимеров. В зависимости от природы исходных соединений, определенную реакцию для получения целевого полимера осуществляют в соответствии с разными механизмами. Очевидно, таким образом, во время получения полиуретан-полимочевинных частиц или полиуретан-полимочевинных полимеров, компоненты (Z.1.1) и (Z.1.2) вступают в реакцию друг с другом в результате реакции изоцианатных групп (Z.1.1) с аминогруппами (Z.1.2) с образованием мочевинных связей. В результате, конечно, полимер содержит аминогруппы и изоцианатные группы, которые присутствуют перед этим, в виде мочевинных групп - то есть, в их соответственно прореагировавшем виде. В итоге, при этом, полимер содержит два компонента (Z.1.1) и (Z.1.2), поскольку кроме прореагировавших изоцианатных групп и аминогрупп, компоненты остаются неизменными. Соответственно, для простоты понимания, говорят, что указанный полимер содержит компоненты, в каждом случае в прореагировавшем виде. По этой причине, значение выражения "полимер содержит компонент (X) в прореагировавшем виде" может приравниваться к значению выражения "во время получения полимера, применяли компонент (X)".
Полиуретан-полимочевинные частицы предпочтительно состоят из двух компонентов (Z.1.1) и (Z.1.2) - то есть, их получают из двух указанных компонентов.
Водная дисперсия (PD), например, может быть получена посредством определенного трехстадийного процесса. В качестве части описания указанного процесса, также указаны предпочтительные варианты осуществления компонентов (Z.1.1) и (Z.1.2).
На первой стадии (I) указанного процесса, получают определенный состав (Z).
Состав (Z) содержит по меньшей мере одно, предпочтительно ровно одно, определенное содержащее изоцианатные группы промежуточное соединение (Z.1), имеющее блокированные первичные аминогруппы.
Получение промежуточного соединения (Z.1) содержит реакцию по меньшей мере одного полиуретанового преполимера (Z.1.1), содержащего изоцианатные группы и содержащего анионные группы и/или группы, которые могут превращаться в анионные группы, по меньшей мере с одним полиамином (Z.1.2a), который получают из полиамина (Z.1.2), и который содержит по меньшей мере две блокированные первичные аминогруппы и по меньшей мере одну свободную вторичную аминогруппу.
Полиуретановые полимеры, содержащие изоцианатные группы и содержащие анионные группы и/или группы, которые могут превращаться в анионные группы, в принципе, известны. В контексте настоящего изобретения, для большей простоты понимания, компонент (Z.1.1) называют преполимером. Это связано с тем, что он представляет собой полимер, который следует идентифицировать как предшественник, который при этом применяется в качестве исходного компонента для получения другого компонента, а именно промежуточного соединения (Z.1).
Для получения полиуретановых преполимеров (Z.1.1), содержащих изоцианатные группы и содержащих анионные группы и/или группы, которые могут превращаться в анионные группы, возможно применять алифатические, циклоалифатические, алифатические-циклоалифатические, ароматические, алифатические-ароматические и/или циклоалифатические-ароматические полиизоцианаты, которые известны специалисту в данной области. Предпочтение отдают применению диизоцианатов. Следующие диизоцианаты могут быть указаны в качестве примера: 1,3- или 1,4-фенилендиизоцианат, 2,4- или 2,6-толилендиизоцианат, 4,4'- или 2,4'-дифенилметандиизоцианат, 1,4- или 1,5-нафтилендиизоцианат, простой диизоцианатодифениловый эфир, триметилендиизоцианат, тетраметилендиизоцианат, этилэтилендиизоцианат, 2,3-диметилэтилендиизоцианат, 1 -метилтриметилендиизоцианат, пентаметилендиизоцианат, 1,3-циклопентилендиизоцианат, гексаметилендиизоцианат, циклогексилендиизоцианат, 1,2-циклогексилендиизоцианат, октаметилендиизоцианат, триметилгександиизоцианат, тетраметилгександиизоцианат, декаметилендиизоцианат, додекаметилендиизоцианат, тетрадекаметилендиизоцианат, изофорондиизоцианат (ИФДИ), 2-изоцианато-пропилциклогексилизоцианат, дициклогексилметан 2,4'-диизоцианат, дициклогексилметан 4,4'-диизоцианат, 1,4- или 1,3-бис(изоцианатометил)циклогексан, 1,4- или 1,3- или 1,2-диизоцианатоциклогексан, 2,4- или 2,6-диизоцианато-1-метилциклогексан, 1-изоцианатометил-5-изоцианато-1,3,3-триметилциклогексан, 2,3-бис(8-изоцианатооктил)-4-октил-5-гексилцикло гексен, тетраметилксилилендиизоцианаты (ТМКДИ), такие как м-тетраметилксилилендиизоцианат, или смеси указанных полиизоцианатов. Конечно, также возможно применение разных димеров и тримеров указанных диизоцианатов, таких как уретдионы и изоцианураты. Предпочтение отдают применению алифатических диизоцианатов, таких как гексаметилендиизоцианат, изофорондиизоцианат (ИФДИ), дициклогексилметан 4,4'-диизоцианат, 2,4- или 2,6-диизоцианато-1-метилциклогексан, и/или ж-тетраметилксилилендиизоцианат (м-ТМКДИ). Изоцианат называют алифатическим, когда изоцианатные группы присоединены к алифатическим группам - другими словами, отсутствует ароматический углерод в альфа-положении к изоцианатной группе.
Для получения преполимеров (Z.1.1), полиизоцианаты, как правило, вступают в реакцию с полиолами, в частности, с диолами, с образованием уретанов.
Примеры подходящих полиолов представляют собой насыщенные или олефинненасыщенные сложные полиэфирполиолы и/или простые полиэфирполиолы. В частности, применяемыми в качестве полиолов являются сложные полиэфирполиолы, особенно те, которые имеют среднечисленную молекулярную массу, составляющую 400-5000 г/моль (в отношении метода определения смотри раздел Примеров). Сложные полиэфирполиолы, предпочтительно сложные полиэфирдиолы, указанного вида могут быть получены известным способом посредством реакции соответствующих многоосновных карбоновых кислот, предпочтительно двухосновных карбоновых кислот, и/или их ангидридов, с соответствующими полиолами, предпочтительно диолами, посредством эстерификации. Конечно, необязательно, также дополнительно является возможным, осуществлять пропорциональное применение одноосновных карбоновых кислот и/или одноатомных спиртов для процедуры получения. Сложные полиэфирдиолы предпочтительно являются насыщенными, в частности, насыщенными и неразветвленными.
Примеры подходящих ароматических многоосновных карбоновых кислот для получения таких сложных полиэфирполиолов, предпочтительно сложных полиэфирдиолов, представляют собой фталевую кислоту, изофталевую кислоту, и терефталевую кислоту, из которых изофталевая кислота является преимущественной и, по этой причине, применяется с предпочтением. Примеры подходящих алифатических многоосновных карбоновых кислот представляют собой щавелевую кислоту, малоновую кислоту, янтарную кислоту, глутаровую кислоту, адипиновую кислоту, пимелиновую кислоту, субериновую кислоту, азелаиновую кислоту, себациновую кислоту, ундекандикарбоновую кислоту, и додекандикарбоновую кислоту или, помимо прочего, гексагидрофталевую кислоту, 1,3-цикло-гександикарбоновую кислоту, 1,4-циклогександикарбоновую кислоту, 4-метилгекса-гидрофталевую кислоту, трициклодекандикарбоновую кислоту, и тетра-гидрофталевую кислоту. В качестве двухосновных карбоновых кислот также является возможным применять димерные жирные кислоты, или димеризованные жирные кислоты, которые, как это известно, представляют собой смеси, полученные посредством димеризации ненасыщенных жирных кислот, и которые являются доступными, например, под торговыми наименованиями Radiacid (от компании Oleon) или Pripol (от компании Croda). Применение димерных жирных кислот указанных видов для получения сложных полиэфирдиолов является предпочтительным в контексте настоящего изобретения. Полиолы, применяемые с предпочтением для получения преполимеров (Z.1.1), по этой причине, представляют собой сложные полиэфирдиолы, которые были получены посредством применения димерных жирных кислот. Особенно предпочтительными являются те сложные полиэфирдиолы, при получении которых по меньшей мере 50 мас. %, предпочтительно 55-75 мас. %, применяемых двухосновных карбоновых кислот, представляют собой димерные жирные кислоты.
Примеры соответствующих полиолов для получения сложных полиэфирполиолов, предпочтительно сложных полиэфирдиолов, представляют собой этиленгликоль, 1,2-, или 1,3-пропандиол, 1,2-, 1,3-, или 1,4-бутандиол, 1,2-, 1,3-, 1,4-, или 1,5-пентандиол, 1,2-, 1,3-, 1,4-, 1,5-, или 1,6-гександиол, неопентилгидроксипивалат, неопентилгликоль, диэтиленгликоль, 1,2-, 1,3-, или 1,4-циклогександиол, 1,2-, 1,3-, или 1,4-циклогександиметанол, и триметилпентандиол. Следовательно, предпочтение отдают применению диолов. Такие полиолы или диолы, конечно, также могут применяться непосредственно для получения преполимера (Z.1.1), другими словами вступать в реакцию непосредственно с полиизоцианатами.
Более того, для получения преполимеров (Z.1.1) также возможно применять полиамины, такие как диамины и/или аминоспирты. Примеры диаминов включают гидразин, алкил- или циклоалкилдиамины, такие как пропилендиамин и 1-амино-3-аминометил-3,5,5-триметилциклогексан, и примеры аминоспиртов включают этаноламин или диэтаноламин.
Преполимеры (Z.1.1) содержат анионные группы и/или группы, которые могут превращаться в анионные группы (то есть, группы, которые могут превращаться в анионные группы в результате применения известных нейтрализующих агентов, которые также определены далее ниже, таких как основания). Как известно специалисту в данной области, это, например, группы карбоновой, сульфоновой и/или фосфоновой кислоты, особенно предпочтительно группы карбоновой кислоты (функциональные группы, которые могут превращаться в анионные группы посредством нейтрализующих агентов), а также анионные группы, которые получают из указанных выше функциональных групп, такие как, в частности, карбоксилатные, сульфонатные и/или фосфонатные группы, предпочтительно карбоксилатные группы. Эффектом от введения такой группы, как это известно, является повышение диспергируемости в воде. В зависимости от выбранных условий, определенная пропорция или практически все из указанных групп могут присутствовать в одной форме (например, карбоновая кислота) или в другой форме (карбоксилат). Определенный влияющий фактор заключается, например, в применении упомянутых выше нейтрализующих агентов, которые описаны более подробно далее ниже. Если преполимер (Z.1.1) смешивают с такими нейтрализующими агентами, то, в зависимости от количества нейтрализующего агента, соответствующее количество групп карбоновой кислоты будет превращаться в карбоксилатные группы. Однако, для целей настоящего изобретения, для облегчения понимания, независимо от формы, в которой указанные группы присутствуют, часто выбирают однотипные названия. В случае, если, например, для полимера указано определенное кислотное число, например, для преполимера (Z.1.1), или в случае, если такой полимер называют карбокси-функциональным, то фраза всегда включает не только группы карбоновой кислоты, но также карбоксилатные группы. Если в этом отношении должно быть какое-либо различие, то это делается, например, с использованием степени нейтрализации.
Для того, чтобы ввести указанные группы, во время получения преполимеров (Z.1.1) возможно применять исходные соединения, которые, также, как и группы, которые предназначены для реакции при получении уретановых связей, предпочтительно гидроксильные группы, дополнительно содержат упомянутые выше группы, например, группы карбоновой кислоты. Таким образом, указанные группы вводят в преполимер.
Соответствующие соединения, предусмотренные для введения предпочтительных групп карбоновой кислоты, представляют собой - в той мере, в какой они содержат карбоксильные группы - простые полиэфирполиолы и/или сложные полиэфирполиолы. Предпочтение, однако, отдают применению соединений, которые, в любом случае, имеют небольшую молекулярную массу, и которые имеют по меньшей мере одну группу карбоновой кислоты и по меньшей мере одну функциональную группу, которая является реакционноспособной по отношению к изоцианатным группам, при этом являются предпочтительными гидроксильные группы. Для целей настоящего изобретения, выражение "соединение с низкой молекулярной массой" означает, что в отличие от соединений с относительно высокой молекулярной массой, в частности полимеров, указанные соединения представляют собой соединения, которые могут иметь дискретную молекулярную массу, предпочтительно, как имеют мономерные соединения. Таким образом, соединение с низкой молекулярной массой, является в частности не полимер, поскольку последний всегда представляет собой смесь молекул и должен описываться посредством использования средней молекулярной массы. Термин "соединение с низкой молекулярной массой", предпочтительно, означает то, что указанные соединения имеют молекулярную массу, которая составляет меньше 300 г/моль. Диапазон от 100 до 200 г/моль является предпочтительным.
Соединения предпочтительные в указанном смысле, например, представляют собой одноосновные карбоновые кислоты, содержащий две гидроксильные группы, такие как дигидроксипропионовая кислота, дигидроксиянтарная кислота, и дигидроксибензойная кислота, например. Особенно характерны альфа,альфа-диметилолалконовые кислоты, такие как 2,2-диметилолуксусная кислота, 2,2-диметилолпропионовая кислота, 2,2-диметилолмасляная кислота, и 2,2-диметилолпентановая кислота, в частности, 2,2-диметилолпропионовая кислота.
По этой причине, преполимеры (Z.1.1) предпочтительно имеют карбоксильные функциональные группы. Исходя из содержания твердых веществ, они имеют кислотное число, которое предпочтительно составляет 10-35 мг KOH/г, в частности, 15-23 мг KOH/г (в отношении метода определения смотри раздел Примеров).
Среднечисленная молекулярная масса преполимеров может варьироваться в широком диапазоне, и находится, например, в диапазоне от 2000 до 20000 г/моль, предпочтительно от 3500 до 6000 г/моль (в отношении метода определения смотри раздел Примеров).
Преполимер (Z.1.1) включает изоцианатные группы. Исходя из содержания твердых веществ, он предпочтительно имеет содержание изоцианатных групп, составляющее 0,5-6,0 мас. %, предпочтительно 1,0-5,0 мас. %, особенно предпочтительно 1,5-4,0 мас. % (в отношении метода определения смотри раздел Примеров).
Поскольку преполимер (Z.1.1) включает изоцианатные группы, как правило, гидроксильное число преполимера очевидно будет очень низким. Гидроксильное число преполимера, исходя из содержания твердых веществ, предпочтительно меньше 15 мг KOH/г, в частности, меньше 10 мг KOH/г, и дополнительно предпочтительно, меньше 5 мг KOH/г (в отношении метода определения смотри раздел Примеров).
Преполимеры (Z.1.1) могут быть получены посредством известных и признанных способов, в общей массе или в растворе, особенно предпочтительно посредством реакции исходных соединений в органических растворителях, предпочтительно, таких как метилэтилкетон, при температурах, составляющих, например, 60-120°С, и необязательно с применением катализаторов, обычно применяемых при получении полиуретана. Такие катализаторы известны специалисту в данной области; примером является лаурат дибутилолова. Процедура здесь, конечно, заключается в выборе такого соотношения исходных компонентов, чтобы продукт - то есть, преполимер (Z.1.1) - содержал изоцианатные группы. Также является очевидным, что растворители должны выбираться таким образом, чтобы они не вступали в какие-либо нежелательные реакции с функциональными группами исходных соединений, другими словами, являлись при этом инертными по отношению к указанным группам до такой степени, чтобы они не препятствовали реакции указанных функциональных групп. Получение преполимеров предпочтительно осуществляют уже в органическом растворителе (Z.2), как описано далее ниже, поскольку, в любом случае, присутствие указанного растворителя необходимо в составе (Z), который будет получен на стадии (I) способа.
Как уже указано выше, группы, которые присутствуют в преполимере (Z.1.1) и которые могут превращаться в анионные группы, также могут пропорционально присутствовать в качестве соответствующих анионных групп, например, как результат применения нейтрализующего агента. Таким образом возможно регулировать диспергируемость преполимеров (Z.1.1) и, следовательно, также промежуточного соединения (Z.1), в воде.
Предусмотренные нейтрализующие агенты включают, в частности, известные основные нейтрализующие агенты, такие как, например, карбонаты, гидрокарбонаты, или гидроксиды щелочных металлов и щелочноземельных металлов, например, такие как LiOH, NaOH, KOH, или Са(ОН)2. Также подходящими для нейтрализации и применяемыми с предпочтением в контексте настоящего изобретения являются органические основания, которые содержат азот, такие как амины, такие как аммиак, триметиламин, триэтиламин, трибутиламины, диметиланилин, трифениламин, диметилэтаноламин, метилдиэтаноламин, или триэтаноламин, а также их смеси.
Нейтрализация преполимера (Z.1.1) нейтрализующими агентами, в частности, органическими основаниями, которые содержат азот, может осуществляться после получения преполимера в органической фазе, другими словами, в растворе с органическим растворителем, в частности, с растворителем (Z.2), как описано ниже. Конечно, нейтрализующий агент может также добавляться уже во время или до начала фактической полимеризации, в случае которой, например, исходные соединения, которые содержат группы карбоновой кислоты, затем нейтрализуют.
Если нейтрализация является желательной для групп, которые могут превращаться в анионные группы, в частности, для групп карбоновой кислоты, то нейтрализующий агент может добавляться, например, в таком количестве, чтобы нейтрализовать долю этих групп, составляющую 35%-65% (степень нейтрализации). Предпочтительным является диапазон от 40% до 60% (в отношении метода вычисления смотри раздел Примеров).
Является предпочтительным для преполимера (Z.1.1), когда его нейтрализуют после его получения и до его применения для получения промежуточного соединения (Z.1), как это описано.
Описанное в этой заявке получение промежуточного соединения (Z.1) включает реакцию описанного преполимера (Z.1.1) по меньшей мере с одним, предпочтительно ровно одним, полиамином (Z.1.2a), полученным из полиамина (Z.1.2).
Полиамин (Z.1.2a) содержит две блокированные первичные аминогруппы и одну или две свободные вторичные аминогруппы.
Блокированные аминогруппы, как известно, представляют собой аминогруппы, в которых водородные радикалы на азоте, которые неизбежно присутствуют в свободных аминогруппах, замещаются посредством обратимой реакции блокирующим агентом. По причине блокирования, аминогруппы не могут вступать в реакцию, как это могут свободные аминогруппы, посредством реакции конденсации или присоединения и, по этой причине, в указанном отношении являются нереакционноспособными и, следовательно, отличаются от свободных аминогрупп. Только удаление обратимо приводимого блокирующего агента, снова при этом восстанавливает свободные аминогруппы, таким образом, очевидно, позволяя обычные реакции аминогрупп. Следовательно, этот принцип напоминает принцип скрытых или блокированные изоцианатов, которые также известны в области химии полимеров.
Первичные аминогруппы полиамина (Z.1.2a) могут быть блокированы традиционными блокирующими агентами, например, кетонами и/или альдегидами. В результате, такое блокирование приводит к получению, с высвобождением воды, кетиминов и/или альдиминов, которая больше не содержат каких-либо азот-водородных связей, тем самым предотвращая любые типичные реакции конденсации или реакции присоединения аминогруппы с другой функциональной группой, такой как изоцианатная группа.
Условия реакции для получения блокированного первичного амина указанного вида, такого как, например, кетимин, известны. Таким образом, например, такое блокирование может быть реализовано с подачей тепла к смеси первичного амина с избытком кетона, который одновременно действует как растворитель для амина. Полученную воду реакции предпочтительно удаляют во время реакция, для того чтобы предотвратить другую возможную обратную реакцию (деблокирование) обратимого блокирования.
Условия реакции для деблокирования блокированных первичных аминогрупп также известны как таковые. Так, например, простой перенос блокированного амина в водную фазу является достаточным для смещения равновесия обратно в сторону деблокирования, как результат концентрации давления, которое в итоге оказывает вода, и таким образом приводить к получению свободных первичных аминогрупп, а также свободного кетона, с расходованием воды.
Из указанного выше следует, что в контексте настоящего изобретения приводится четкое различие между блокированными и свободными аминогруппами. Однако, в случае, когда, аминогруппа не указана ни как блокированная, ни как свободная, то ссылаются на свободную аминогруппу.
Предпочтительными блокирующими агентами для блокирования первичных аминогрупп полиамина (Z.1.2a) являются кетоны. Среди кетонов, определенное предпочтение отдают тем кетонам, которые представляют собой органический растворитель (Z.2), как описано далее ниже. Причиной является то, что указанные растворители (Z.2) должны в любом случае присутствовать в составе (Z), который будет получен на стадии (I) способа. Уже было указано выше, что получение таких первичных аминов, блокированных кетоном, в частности, приводит к хорошему эффекту при избытке кетона. По этой причине, в результате применения кетонов (Z.2) для блокирования, возможно соответственно применять предпочтительную процедуру получения блокированных аминов, без какой-либо необходимости в дорогостоящем и затруднительном удалении возможно нежелательного блокирующего агента. Вместо этого, раствор блокированного амина может применяться непосредственно для получения промежуточного соединения (Z.1). Предпочтительные блокирующие агенты представляют собой ацетон, метилэтилкетон, метилизобутилкетон, диизопропилкетон, циклопентанон, или циклогексанон; в частности, предпочтительными являются такие (Z.2) кетоны, как метилэтилкетон и метилизобутилкетон.
Предпочтительное блокирование кетонами и/или альдегидами, в частности, кетонами, и сопутствующее при этом получение кетиминов и/или альдиминов, кроме того, имеет то преимущество, что первичные аминогруппы блокируются избирательно. Присутствующие вторичные аминогруппы, очевидно, могут не блокироваться, и по этой причине остаются свободными. Соответственно, можно без проблем получать полиамин (Z.1.2a), который, также, как и две блокированные первичные аминогруппы, содержит одну или две свободные вторичные аминогруппы, посредством указанных предпочтительных реакций блокирования из соответствующего полиамина (Z.1.2), который содержит свободные вторичные и первичные аминогруппы.
Полиамины (Z.1.2a) могут быть получены посредством блокирования первичных аминогрупп полиаминов (Z.1.2), содержащих две первичные аминогруппы и одну или две вторичные аминогруппы. Подходящими, в итоге, являются все традиционные алифатические, ароматические, или аралифатические (смешанные алифатические-ароматические) полиамины (Z.1.2), имеющие две первичные аминогруппы и одну или две вторичные аминогруппы. Указанное означает, что наряду с указанными аминогруппами, неизбежно могут присутствовать любые алифатические, ароматические, или аралифатические группы. Возможные примеры включают одновалентные группы, расположенные в виде концевых групп на вторичной аминогруппе, или двухвалентные группы, расположенные между двумя аминогруппами.
Органические группы считаются алифатическими в контексте настоящего изобретения, если они не являются ароматическими. Например, группы, которые присутствуют в дополнение к указанным аминогруппам, могут быть алифатическими углеводородными группами, указанные при этом являются группами, которые состоят исключительно из углерода и водорода и не являются ароматическими. Указанные алифатические углеводородные группы могут быть неразветвленными, разветвленными или циклическими, и могут быть насыщенными или ненасыщенными. Указанные группы, конечно, также могут содержать циклические и неразветвленные или разветвленные компоненты. Дополнительная возможность заключается в том, что алифатические группы включают гетероатомы, в частности, в виде мостиковых групп, таких как группы простого эфира, сложного эфира, амида и/или уретана. Возможные ароматические группы являются также известными и не требуют дополнительного разъяснения.
Полиамины (Z.1.2a) предпочтительно имеют две блокированные первичные аминогруппы и одну или две свободные вторичные аминогруппы, и они предпочтительно имеют, в качестве первичных аминогрупп, исключительно блокированные первичные аминогруппы и, в качестве вторичных аминогрупп, исключительно свободные вторичные аминогруппы.
В общем, полиамины (Z.1.2a) предпочтительно имеют три или четыре аминогруппы, указанные группы при этом выбирают из группы блокированных первичных аминогрупп и свободных вторичных аминогрупп.
Особенно предпочтительными полиаминами (Z.1.2a) являются те, которые состоят из двух блокированных первичных аминогрупп, одной или двух свободных вторичных аминогрупп, а также алифатически-насыщенные углеводородные группы.
Аналогичные предпочтительные варианты осуществления действительны для полиаминов (Z.1.2), которые таким образом содержат свободные первичные аминогруппы, а не блокированные первичные аминогруппы.
Примеры предпочтительных полиаминов (Z.1.2), из которых возможно посредством блокирования первичных аминогрупп получать полиамины (Z.1.2a), представляют собой диэтилентриамин, 3-(2-аминоэтил)аминопропиламин, дипропилентриамин, а также N-(2-(4-(2-аминоэтил)пиперазин-1-ил)этил)этан-1,2-диамин (одна вторичная аминогруппа, две первичные аминогруппы для блокирования) и триэтилентетрамин, а также N,N'-бис(3-аминопропил)-этилендиамин (две вторичные аминогруппы, две первичные аминогруппы для блокирования).
Специалисту в данной области понятно, что не в последнюю очередь по причинам, связанным с чисто техническим синтезом, не всегда может происходить теоретически идеальное количественное превращение при блокировании первичных аминогрупп. Например, если блокируется определенное количество полиамина, то пропорция первичных аминогрупп, которые блокируются в процессе блокирования, может, например, составлять 95 мол. % или более (что определяется посредством ИК-спектроскопии; смотри раздел Примеров). В случае, если полиамин в неблокированном состоянии, например, имеет две свободные первичные аминогруппы, и где первичные аминогруппы определенного количества указанного амина затем блокируются, то в контексте настоящего изобретения говорят, что указанный амин имеет две блокированные первичные аминогруппы, если блокируется доля первичных аминогрупп, которые присутствуют в применяемом количестве, составляющая более 95 мол. %. Это связано, с одной стороны, с уже заявленным фактом, что с точки зрения технического синтеза количественное превращение не всегда может быть реализовано. С другой стороны, тот факт, что блокируется более 95 мол. % первичных аминогрупп означает, что основная доля общего количества аминов, применяемых для блокирования, в действительности содержит исключительно блокированные первичные аминогруппы, а именно, ровно две блокированные первичные аминогруппы.
Получение промежуточного соединения (Z.1) включает реакцию преполимера (Z.1.1) с полиамином (Z.1.2) посредством реакции присоединения изоцианатных групп из (Z.1.1) со свободными вторичными аминогруппами из (Z.1.2). В результате, указанная реакция, которая является известной как таковая, приводит к присоединению полиамина (Z.1.2a) к преполимеру (Z.1.1), с образованием мочевинных связей, в результате чего образуется промежуточное соединение (Z.1). Очевидно, что при получении промежуточного соединения (Z.1), предпочтение, таким образом, отдают тому, чтобы не применять какие-либо другие амины, имеющие свободные или блокированные вторичные, или свободные или блокированные первичные аминогруппы.
Промежуточное соединение (Z.1) может быть получено посредством известных и признанных методов, в общей массе или в растворе, особенно предпочтительно посредством реакции (Z.1.1) с (Z.1.2a) в органических растворителях. Является очевидным, что растворители должны выбираться таким образом, чтобы они не вступали в какие-либо нежелательные реакции с функциональными группами исходных соединений, и являлись по этой причине инертными или в основном инертными в своем поведении по отношению к указанным группам. В качестве растворителя, применяемого при получении, предпочтение отдают применению, по меньшей мере пропорционально, органическому растворителю (Z.2), который описан далее ниже, в частности, метилэтилкетону, даже на этой стадии, поскольку в любом случае необходимо, чтобы указанный растворитель присутствовал в составе (Z), который будет получен на стадии (I) способа. Предпочтительно, раствор преполимера (Z.1.1) в растворителе (Z.2) в данном случае смешивают с раствором полиамина (Z.1.2a) в растворителе (Z.2), и описанная реакция может осуществляться.
Конечно, промежуточное соединение (Z.1), полученное таким образом, может нейтрализоваться во время получения или после получения, посредством применения нейтрализующих агентов, которые уже описаны выше, способом, который также описан выше для преполимера (Z.1.1). Тем не менее, является предпочтительным, если преполимер (Z.1.1) будет уже нейтрализован до его применения для получения промежуточного соединения (Z.1), посредством способа, описанного выше, таким образом, что нейтрализация во время или после получения (Z.1) больше не имеет значения. В таком случае, по этой причине, степень нейтрализации преполимера (Z.1.1) может приравниваться к степени нейтрализации промежуточного соединения (Z.1). Таким образом, в случае, если отсутствует дополнительное добавление нейтрализующих агентов вообще в контексте способа, соответственно, то степень нейтрализации полимеров, которые присутствуют в полученных в итоге дисперсиях (PD) в соответствии с изобретением, также может приравниваться к степени нейтрализации преполимера (Z.1.1).
Промежуточное соединение (Z.1) имеет блокированные первичные аминогруппы. Указанное может, очевидно, достигаться тем, что свободные вторичные аминогруппы участвуют в реакции преполимера (Z.1.1) и полиамина (Z.1.2a), но блокированные первичные аминогруппы не вступают в реакцию. Действительно, как уже описано выше, эффектом блокирования является то, что типичные реакции конденсации или реакции присоединения с другими функциональными группами, такими как изоцианатные группы, не могут осуществляться. Указанное, конечно, означает, что условия реакции должны выбираться таким образом, чтобы блокированные аминогруппы также оставались блокированными, для того чтобы тем самым обеспечивать промежуточное соединение (Z.1). Специалист в данной области знает, каким образом установить такие условия, которые возникают, например, в результате реакции в органических растворителях, что является предпочтительным в любом случае.
Промежуточное соединение (Z.1) содержит изоцианатные группы. Соответственно, в реакции (Z.1.1) и (Z.1.2a), конечно, соотношение указанных компонентов необходимо выбирать таким образом, чтобы продукт - то есть, промежуточное соединение (Z.1) - содержал изоцианатные группы.
Поскольку, как описано выше, в реакции (Z.1.1) с (Z.1.2a), свободные вторичные аминогруппы вступают в реакцию с изоцианатными группами, а первичные аминогруппы не вступают в реакцию, благодаря блокированию, таким образом, прежде всего, понятно, что в указанной реакции необходимо, чтобы молярное соотношение изоцианатных групп из (Z.1.1) к свободным вторичным аминогруппам из (Z.1.2a), было больше 1. Этот признак проявляется в неявной форме, тем не менее однозначно и непосредственно из признака, существенного для изобретения, а именно, что промежуточное соединение (Z.1) содержит изоцианатные группы.
Тем не менее, является предпочтительным, когда во время реакции существует избыток изоцианатных групп, как определено ниже. В указанном предпочтительном варианте осуществления, молярные количества (n) изоцианатных групп, свободных вторичных аминогрупп, а также блокированных первичных аминогрупп, удовлетворяют следующему условию: [n (изоцианатных групп из (Z.1.1)) - n (свободных вторичных аминогрупп из (Z.1.2a))] / n (блокированных первичных аминогрупп из (Z.1.2a))=1,2/1-4/1, предпочтительно 1,5/1-3/1, очень предпочтительно 1,8/1-2,2/1, даже более предпочтительно 2/1.
В указанных предпочтительных вариантах осуществления, промежуточное соединение (Z.1), образованное посредством реакции изоцианатных групп из (Z.1.1) со свободными вторичными аминогруппами из (Z.1.2a), имеет избыток изоцианатных групп в отношении блокированных первичных аминогрупп. Указанный избыток, в итоге, достигается посредством такого выбора молярного соотношения изоцианатных групп из (Z.1.1) к общему количеству свободных вторичных аминогрупп и блокированных первичных аминогрупп из (Z.1.2a), чтобы оно было достаточно большим, так, чтобы даже после получения (Z.1) и соответствующего расхода изоцианатных групп в результате реакции со свободными вторичными аминогруппами, оставался соответствующий избыток изоцианатных групп.
В случае, когда, например, полиамин (Z.1.2a) имеет одну свободную вторичную аминогруппу и две блокированные первичные аминогруппы, молярное соотношение между изоцианатными группами из (Z.1.1) и полиамином (Z.1.2a) в очень особенно предпочтительном варианте осуществления установлено как 5/1. Расход одной изоцианатной группы в реакции со свободной вторичной аминогруппой, таким образом, будет означать, что для условия, указанного выше, реализуется соотношение 4/2 (или 2/1).
Доля промежуточного соединения (Z.1) составляет от 15 до 65 мас. %, предпочтительно от 25 до 60 мас. %, более предпочтительно от 30 до 55 мас. %, особенно предпочтительно от 35 до 52,5 мас. %, и, в одном очень особом варианте осуществления, от 40 до 50 мас. %, в каждом случае, из расчета общего количества состава (Z).
Определение доли промежуточного соединения (Z.1) может осуществляться следующим образом: Устанавливают содержание твердых веществ смеси, которая, кроме промежуточного соединения (Z.1), содержит только органические растворители (в отношении метода определения твердых веществ (которое также называют содержанием твердых веществ, смотри раздел Примеров). Таким образом, содержание твердых веществ соответствует количеству промежуточного соединения (Z.1). Таким образом, учитывая содержание твердых веществ смеси, возможно определять или установить долю промежуточного соединения (Z.1) в составе (Z). Учитывая, что промежуточное соединение (Z.1) предпочтительно в любом случае получают в органическом растворителе и, вследствие этого, после получения, в любом случае оно присутствует в смеси, которая кроме промежуточного соединения содержит только органические растворители, этот метод является предпочтительным.
Состав (Z) дополнительно содержит по меньшей мере один определенный органический растворитель (Z.2).
Растворители (Z.2) обладают растворимостью в воде, которая составляет не более 38 мас. % при температуре, составляющей 20°С (в отношении метода определения, смотри раздел Примеров). Растворимость в воде при температуре, составляющей 20°С, составляет предпочтительно менее 30 мас. %. Предпочтительный диапазон составляет от 1 до 30 мас. %.
Растворитель (Z.2) соответственно имеет относительно умеренную растворимость в воде, в частности, не являясь при этом полностью смешиваемым с водой или не обладающий бесконечной растворимостью в воде. При этом растворитель является полностью смешиваемым с водой, когда он может смешиваться с водой в любой пропорции без возникновения разделения, другими словами, без образования двух фаз.
Примеры растворителей (Z.2) представляют собой метилэтилкетон, метилизобутилкетон, диизобутилкетон, простой диэтиловый эфир, простой дибутиловый эфир, простой диметиловый эфир дипропиленгликоля, простой диэтиловый эфир этиленгликоля, толуол, метилацетат, этилацетат, бутилацетат, пропилен карбонат, циклогексанон, или смеси указанных растворителей. Предпочтение отдают метилэтилкетону, который обладает растворимостью в воде, которая составляет 24 мас. % при температуре 20°С.
Следовательно, растворители (Z.2) не являются растворителями, такими как ацетон, N-метил-2-пирролидон, N-этил-2-пирролидон, тетрагидрофуран, диоксан, N-формилморфолин, диметилформамид, или диметилсульфоксид.
Особый эффект от выбора определенных растворителей (Z.2), которые обладают только ограниченной растворимость в воде, состоит в том, что, когда состав (Z) диспергируется в водной фазе, на стадии (II) способа, гомогенный раствор не может быть получен непосредственно. Предполагается, что, вместо этого, присутствующая дисперсия, дает возможность реакциям сшивания, которые происходят как часть стадии (II) (реакции присоединения свободных первичных аминогрупп и изоцианатных групп с образованием мочевинных связей), осуществляться в ограниченном объеме, тем самым, в итоге, позволяя образование микрочастиц, которые определены выше.
Также в качестве имеющих описанную растворимость в воде, предпочтительные растворители (Z.2) обладают точкой кипения, которая составляет не более 120°С, более предпочтительно, составляющий не более 90°С (в условиях атмосферного давления, другими словами 1,013 бар). Указанное имеет преимущества в контексте стадии (III) способа, при этом указанная стадия описана далее ниже, другими словами по меньшей мере частичное удаление по меньшей мере одного органического растворителя (Z.2) из дисперсии, полученной на стадии (II) способа. Причина, очевидно, состоит в том, что в случае применения растворителей (Z.2), которые являются предпочтительными в указанном контексте, указанные растворители могут удаляться посредством дистилляции, например, без одновременного удаления значительного количества воды, которую вводили на стадии (II) способа. По этой причине, нет необходимости, например, в трудоемком повторном добавлении воды, для того чтобы сохранить водный характер дисперсии (PD).
Доля по меньшей мере одного органического растворителя (Z.2) составляет от 35 до 85 мас. %, предпочтительно от 40 до 75 мас. %, более предпочтительно от 45 до 70 мас. %, особенно предпочтительно от 47,5 до 65 мас. % и, в одном очень особом варианте осуществления, от 50 до 60 мас. %, в каждом случае, из расчета общего количества состава (Z).
В контексте настоящего изобретения выяснилось, что в результате определенной комбинации доли, указанной выше для промежуточного соединения (Z.1) в составе (Z), и выбора определенных растворителей (Z.2), возможно, после описанных ниже стадий (II) и (III), получать полиуретан-полимочевинные дисперсии, которые содержат полиуретан-полимочевинные частицы, имеющие необходимый размер частиц, которые дополнительно составляют необходимую гель-фракцию.
Описанные компоненты (Z.1) и (Z.2) предпочтительно в общем составляют по меньшей мере 90 мас. % состава (Z). Предпочтительно, два компонента составляют по меньшей мере 95 мас. %, в частности, по меньшей мере 97.5 мас. %, состава (Z). Особенно предпочтительно, состав (Z) состоит из указанных двух компонентов. В указанном контексте необходимо отметить, что если применяют описанные выше нейтрализующие агенты, то указанные нейтрализующие агенты относят к промежуточному соединению, когда вычисляют количество промежуточного соединения (Z.1). Причина состоит в том, что в указанном случае промежуточное соединение (Z.1) обязательно обладает анионными группами, которые присутствуют в результате применения нейтрализующего агента. Соответственно, катион, который присутствует после того, как указанные анионные группы образовались, также относят к промежуточному соединению.
Если состав (Z), в дополнение к компонентам (Z.1) и (Z.2), включает другие компоненты, то указанные другие компоненты предпочтительно являются именно органическими растворителями. Следовательно, содержание твердых веществ состава (Z) предпочтительно соответствует доле промежуточного соединения (Z.1) в составе (Z). По этой причине, состав (Z) предпочтительно имеет содержание твердых веществ, составляющее 15-65 мас. %, предпочтительно, составляющее 25-60 мас. %, более предпочтительно, составляющее 30-55 мас. %, особенно предпочтительно, составляющее 35-52.5 мас. %, и, в одном особенно предпочтительном варианте осуществления, составляющее 40-50 мас. %.
В частности, по этой причине, предпочтительный состав (Z) содержит в общем по меньшей мере 90 мас. % компонентов (Z.1) и (Z.2), и другие компоненты, отличающиеся от промежуточного соединения (Z.1), включает исключительно органические растворители.
Преимущество состава (Z) состоит в том, что он может быть получены без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и N-этил-2-пирролидон. Соответственно, предпочтительно, состав (Z) содержит менее 10 мас. %, предпочтительно менее 5 мас. %, более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Предпочтительно, состав (Z) совсем не содержит указанных органических растворителей.
На второй стадии (II) способа, описанного в этой заявке, состав (Z) диспергируется в водной фазе.
Является известным, а также следует из уже указанного выше, что на стадии (II), по этой причине, имеет место деблокирование блокированных первичных аминогрупп промежуточного соединения (Z.1). Действительно, как результат переноса блокированного амина в водную фазу, обратимо присоединенный блокирующий агент высвобождается, с расходованием воды, и при этом образуются свободные первичные аминогруппы.
Следовательно, также является очевидным, что полученные свободные первичные аминогруппы затем вступают в реакцию с изоцианатными группами, которые также присутствуют в промежуточном соединении (Z.1), или в деблокированном промежуточном соединении, образованном из промежуточного соединения (Z.1), посредством реакции присоединения, с образованием мочевинных связей.
Также является известным, что перенос в водную фазу означает, в принципе, что изоцианатные группы в промежуточном соединении (Z.1), или в деблокированном промежуточном соединении, образованном из промежуточного соединения (Z.1), могут вступать в реакцию с водой, с удалением диоксида углерода, с образованием свободных первичных аминогрупп, которые могут затем, в свою очередь, вступать в реакцию с изоцианатными группами, которые все еще присутствуют.
Конечно, реакции и превращения, указанные выше, проходят параллельно друг с другом. В итоге, в результате, например, межмолекулярной и внутримолекулярной реакции или сшивания, образуется дисперсия, которая содержит полиуретан полимочевинные частицы с определенным средним размером частиц и с определенной степенью сшивания, или гель-фракция.
На стадии (II) способа, описанного в этой заявке, состав (Z) диспергируется в воде, при этом происходит деблокирование блокированных первичных аминогрупп промежуточного соединения (Z.1) и, посредством реакции присоединения, реакция полученных свободных первичных аминогрупп с изоцианатными группами промежуточного соединения (Z.1), а также с изоцианатными группами деблокированного промежуточного соединения, образованного из промежуточного соединения (Z.1).
Стадия (II) способа в соответствии с изобретением, другими словами диспергирование в водной фазе, может осуществляться любым желательным способом. Указанное означает, что в итоге имеет значение только то, чтобы состав (Z) смешивался с водой или с водной фазой. Предпочтительно, состав (Z), который после получения, например, может находиться при комнатной температуре, другими словами при температуре 20-25°С, или при температуре, повышенной относительно комнатной температуры, которая составляет, например, 30-60°С, может перемешиваться в воде, до получения дисперсии. Вода, которая уже была введена, имеет, например, комнатную температуру. Дисперсия может осуществляться в чистой воде (деионизированной воде), что означает то, что водная фаза состоит исключительно из воды, указанное при этом является предпочтительным. Конечно, кроме воды, водная фаза также может, пропорционально, включать типичные вспомогательные вещества, такие как типичные эмульгирующие вещества и защитные коллоиды. Обобщение данных подходящих эмульгирующих веществ и защитных коллоидов можно найти, например, в Houben Weyl, Methoden der organischen Chemie [Методы органической химии], том XIV/1 Makromolekulare Stoffe [Макромолекулярные соединения], Georg Thieme Verlag, Штутгарт 1961, стр. 411 ff.
Является преимуществом, если на стадии (II) способа, другими словами при диспергировании состава (Z) в водной фазе, соотношение массы органических растворителей и воды выбирают таким образом, что полученная дисперсия имеет соотношение массы воды к массе органических растворителей, составляющее более 1, предпочтительно, составляющее 1,05-2/1, особенно предпочтительно, составляющее 1,1-1,5/1.
На стадии (III) способа, описанного в этой заявке, по меньшей мере один органический растворитель (Z.2) удаляют, по меньшей мере частично, из дисперсии, полученной на стадии (II). Конечно, стадия (III) способа также может включать удаление других растворителей, которые также возможно, например, присутствуют в составе (Z).
Удаление по меньшей мере одного органического растворителя (Z.2) и любых дополнительных органических растворителей может выполняться любым способом, который является известным, например, посредством вакуумной дистилляции при температурах, немного повышенных относительно комнатной температуры, которые составляют, например, 30-60°С.
Полученная полиуретан-полимочевинная дисперсия (PD) является водной (в отношении основного определения "водная", смотри ранее выше).
Особое преимущество дисперсии (PD) в соответствии с изобретением состоит в том, что она может составляться только с очень небольшими долями органических растворителей, но при этом обеспечивает преимущества в соответствии с изобретением, описанные вначале. Дисперсия (PD) в соответствии с изобретением предпочтительно содержит менее 7,5 мас. %, особенно предпочтительно менее 5 мас. %, очень предпочтительно менее 2,5 мас. % органических растворителей (в отношении метода определения, смотри раздел Примеров).
Доля полиуретан-полимочевинного полимера в дисперсии (PD) предпочтительно составляет 25-55 мас. %, предпочтительно 30-50 мас. %, более предпочтительно 35-45 мас. %, в каждом случае, из расчета общего количества дисперсии (определяется, как в описанном выше определении в случае промежуточного соединения (Z.1) посредством содержания твердых веществ).
Доля воды в дисперсии (PD) предпочтительно составляет 45-75 мас. %, предпочтительно 50-70 мас. %, более предпочтительно 55-65 мас. %, в каждом случае, из расчета общего количества дисперсии.
Является предпочтительным, если дисперсия (PD) в соответствии с изобретением состоит по меньшей мере из 90 мас. %, предпочтительно по меньшей мере 92,5 мас. %, очень предпочтительно по меньшей мере 95 мас. %, и даже более предпочтительно по меньшей мере 97,5 мас. % полиуретан-полимочевинных частиц и воды (соответствующее значение получают посредством суммирования количества частиц (то есть, полимера, определенного посредством содержания твердых веществ) и количества воды). Выяснилось, что несмотря на указанную небольшую долю дополнительных компонентов, в частности, таких как органические растворители, дисперсии в соответствии с изобретением в любом случае являются очень стабильными, в частности, стабильными при хранении. Таким образом, объединены два важных преимущества. Во-первых, обеспечиваются дисперсии, которые могут применяться в водных материалах базовых покрытий, где они приводят к эксплуатационным преимуществам, описанным вначале, а также в примерах, приведенных ниже. При этом, во-вторых, достигается соответствующая свобода в составлении водных материалов базовых покрытий. Указанное означает, что в материалах базовых покрытий могут применяться дополнительные доли органических растворителей, которые при этом необходимы, например, для того чтобы обеспечивать соответствующее составление разных компонентов. Но в то же время, водный по своей сути характер материала базового покрытия не затрагивается. Напротив: материалы базовых покрытий, тем не менее, могут составляться с применением сравнительно небольших долей органических растворителей и, следовательно, в частности, имеют хороший экологический профиль.
Даже более предпочтительно, чтобы дисперсия, отличная от полимера, включала только воду и любые органические растворители, например, в виде остаточных фракций, которые не были полностью удалены на стадии (III) способа. По этой причине, содержание твердых веществ дисперсии (PD) составляет предпочтительно 25%-55%, предпочтительно 30%-50%, более предпочтительно 35%-45%, и более предпочтительно все еще находится в соответствии с долей полимера в дисперсии.
Преимущество дисперсии (PD) состоит в том, что она может быть получена без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и N-этил-2-пирролидон. Соответственно, дисперсия (PD) содержит предпочтительно менее 7,5 мас. %, предпочтительно менее 5 мас. %, более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Предпочтительно, дисперсия (PD) является полностью свободной от указанных органических растворителей.
Исходя из содержания твердых веществ, кислотное число полиуретан-полимочевинного полимера, который присутствует в дисперсии, предпочтительно составляет от 10 до 35 мг KOH/г, в частности, от 15 до 23 мг KOH/г (в отношении метода определения смотри раздел Примеров).
Полиуретан-полимочевинный полимер, который присутствует в дисперсии, предпочтительно почти не содержит каких-либо гидроксильных групп, или вовсе не содержит их. Гидроксильное число полимера, исходя из содержания твердых веществ, предпочтительно составляет менее 15 мг KOH/г, в частности, менее 10 мг KOH/г, более предпочтительно даже менее 5 мг KOH/г (в отношении метода определения, смотри раздел Примеров).
Доля одной или большего количества дисперсий (PD), из расчета общей массы водного материала базового покрытия в соответствии с изобретением, предпочтительно составляет 1,0-60 мас. %, более предпочтительно 2,5-50 мас. %, и очень предпочтительно 5-40 мас. %.
Доля полимеров, поступающих из дисперсий (PD), из расчета общей массы водного материала базового покрытия в соответствии с изобретением, предпочтительно составляет от 0,4 до 24,0 мас. %, более предпочтительно 1,0-20,0 мас. %, и очень предпочтительно 2,0-16,0 мас. %.
Определение или установление доли полимеров, поступающих из дисперсий (PD), предназначенных для применения в материале базового покрытия в соответствии с изобретением, может осуществляться посредством определения содержания твердых веществ (которое также называют нелетучей фракцией или фракцией твердых веществ) дисперсии (PD), которая подлежит применению в материал базового покрытия. То же самое касается фракций других компонентов, например, в дисперсии (wD).
Материал базового покрытия в соответствии с изобретением содержит по меньшей мере одну, предпочтительно ровно одну, определенную водную дисперсию (wD), которая содержит определенный полимер.
Получение полимера включает последовательную радикальную эмульсионную полимеризацию трех разных смесей (А), (Б), и (В), олефинненасыщенных мономеров. По этой причине, процесс представляет собой многостадийную радикальную эмульсионную полимеризацию, где I. вначале полимеризуют смесь (А), затем II. полимеризуют смесь (Б) в присутствии полимера, полученного на стадии I., и дополнительно III. полимеризуют смесь (В) в присутствии полимера, полученного на стадии II. По этой причине, все три смеси мономеров полимеризуются посредством радикальной эмульсионной полимеризации, которую в каждом случае проводят отдельно (то есть, на отдельной стадии или, помимо прочего, на отдельной стадии полимеризации), где при этом указанные стадии осуществляются последовательно. С точки зрения времени, стадии могут осуществляться непосредственно одна после другой. В равной степени, является возможным, когда соответствующий реакционный раствор после окончания одной стадии будет храниться на протяжении определенного времени и/или будет переноситься в другой реакционный сосуд, и только затем будет осуществляться следующая стадия. Многостадийное получение определенного полимера предпочтительно не содержит дополнительных стадий полимеризации, дополнительных к полимеризации мономерных смесей (А), (Б), и (В).
Концепция радикальной эмульсионной полимеризации известна специалисту в данной области, и кроме того, опять более подробно разъясняется ниже.
Во время полимеризации указанного вида, олефинненасыщенные мономеры полимеризуют в водной среде, посредством применения по меньшей мере одного растворимого в воде инициатора, а также в присутствии по меньшей мере одного эмульгирующего вещества.
Соответствующие растворимые в воде инициаторы также известны. По меньшей мере один растворимый в воде инициатор предпочтительно выбирают из группы, состоящей из калия, натрия, или пероксодисульфата аммония, пероксида водорода, трет-бутилгидропероксида, 2,2'-азобис(2-амидоизопропан) дигидрохлорида, 2,2'-азобис(N,N-диметиленизобутирамидин) дигидрохлорида, 2,2'-азобис(4-цианопентановой кислоты), и смесей упомянутых выше инициаторов, таких как пероксид водорода и персульфат натрия, например. Также члены указанной предпочтительной группы представляют собой окислительно-восстановительные системы инициатора, которые известны как таковые.
Под окислительно-восстановительными системами инициатора понимают, в частности, те инициаторы, которые содержат по меньшей мере одно содержащее пероксид соединение в комбинации по меньшей мере с одним окислительно-восстановительным соинициатором, примерами при этом являются восстановительные соединения серы, такие как, например, бисульфиты, сульфиты, тиосульфаты, дитиониты или тетратионаты щелочных металлов и соединения аммония, дигидрат гидроксиметансульфинат натрия и/или тиомочевина. Соответственно, возможно применять комбинации пероксодисульфатов со щелочным металлом или водородсульфитами аммония, примерами при этом являются пероксодисульфат аммония и дисульфит аммония. Соотношение массы содержащего пероксид соединения к массе окислительно-восстановительных соинициаторов предпочтительно составляет 50:1-0,05:1.
В комбинации с инициаторами возможно дополнительно применять катализаторы на основе переходных металлов, такие как соли железа, никеля, кобальта, марганца, меди, ванадия, или хрома, например, такие как сульфат железа(II), хлорид кобальта(II), сульфат никеля(II), хлорид меди(I), ацетат марганца(II), ацетат ванадия(III), хлорид марганца(II). Исходя из общей массы олефинненасыщенных мономеров, применяемых в полимеризации, указанные соли переходных металлов обычно применяют в количествах, составляющих 0,1-1000 млн.ч.. Следовательно, возможно применять комбинации пероксида водорода с солями железа(II), такими как, например, 0,5 30 мас. % пероксида водорода и 0,1-500 млн.ч. соли мора, в случае которых диапазоны долей, в каждом случае, основаны на общей массе мономеров, применяемых на соответствующей стадии полимеризации.
Предпочтительно, инициаторы применяют в количестве, составляющем 0,05-20 мас. %, предпочтительно 0,05-10, более предпочтительно от 0,1 до 5 мас. %, из расчета общей массы мономеров, применяемых на соответствующей стадии полимеризации.
Эмульсионную полимеризацию осуществляют в реакционной среде, которая содержит воду в качестве дисперсионной среды и содержит по меньшей мере одно эмульгирующее вещество в виде мицелл. Полимеризация инициируется посредством разложения в воде растворимого в воде инициатора. Растущая полимерная цепь поступает в мицеллы эмульгирующего вещества, и затем в мицеллах происходит дальнейшая полимеризация. В дополнение к мономерам, по меньшей мере к одному растворимому в воде инициатору, и по меньшей мере к одному эмульгирующему веществу, реакционная смесь, следовательно, состоит преимущественно из воды. Указанные компоненты, а именно мономеры, растворимый в воде инициатор, эмульгирующее вещество, и вода, предпочтительно составляют по меньшей мере 95 мас. % реакционной смеси. Предпочтительно, реакционная смесь состоит из указанных компонентов.
По этой причине, очевидно, возможно на каждой отдельной стадии полимеризации добавлять по меньшей мере одно эмульгирующее вещество. Однако, в равной степени является возможным добавлять по меньшей мере одно эмульгирующее вещество только на одной (на первой) или на двух стадиях полимеризации (на первой и на последующей стадии). Количество эмульгирующего вещества в этом случае выбирают таким образом, чтобы существовало достаточное количество эмульгирующего вещества, которое присутствует даже на стадиях, где отдельного добавления не осуществляют.
Эмульгирующие вещества также, в принципе, известны. При этом могут применяться неионные или ионные эмульгирующие вещества, включая цвиттерионные эмульгирующие вещества, а также, необязательно, смеси упомянутых выше эмульгирующих веществ.
Предпочтительные эмульгирующие вещества представляют собой необязательно этоксилированные и/или пропоксилированные алканолы, имеющие 10-40 атомов углерода. Они могут иметь разные степени этоксилирования и/или пропоксилирования (например, аддукты, модифицированные цепями поли(окси)этилена и/или поли(окси)пропилена, состоящими из 5-50 остатков молекул). Также возможно применять сульфатированные, сульфонатированные, или фосфатированные производные указанных продуктов. Такие производные, как правило, применяют в нейтрализованном виде.
В частности, предпочтительными подходящими эмульгирующими веществами являются нейтрализованные сложные эфиры диалкилсульфоянтарной кислоты или алкилдифенилоксид дисульфонаты, которые являются доступными коммерчески, например, такие как EF-800 от компании Cytec.
Эмульсионную полимеризацию преимущественно осуществляют при температуре, составляющей 0-160°С, предпочтительно, составляющий 15-95°С, более предпочтительно, составляющий 60-95°С.
В данном случае является предпочтительным осуществлять полимеризацию в отсутствие кислорода, и предпочтительно в атмосфере инертного газа. Полимеризацию, как правило, осуществляют при атмосферном давлении, при этом применение более низких давлений или более высоких давлений также является возможным. В частности, если в условиях атмосферного давления воды, применяют температуры полимеризации, которые находятся выше точки кипения применяемых мономеров и/или органических растворителей, то обычно выбирают более высокие давления.
Отдельные стадии полимеризации при получении определенного полимера могут, например, осуществляться, как такие, которые называются полимеризациями "с точными дозировками" (также известные как полимеризации "с экономной дозировкой" или "с подачей порциями").
Полимеризация с точными дозировками в смысле настоящего изобретения представляет собой эмульсионную полимеризацию, в которой количество свободных олефинненасыщенных мономеров в реакционном растворе (который также называют реакционной смесью) сводится к минимуму на протяжении всего времени реакции. Указанное означает, что дозированное добавление олефинненасыщенных мономеров осуществляется таким образом, что на протяжении всего времени реакции доля свободных мономеров в реакционном растворе не превышает 6,0 мас. %, предпочтительно 5,0 мас. %, более предпочтительно 4,0 мас. %, в частности, преимущественно 3,5 мас. %, в каждом случае, из расчета общего количества мономеров, которые применяли на соответствующей стадии полимеризации. Дополнительно предпочтительными здесь являются диапазоны концентраций олефинненасыщенных мономеров, составляющие 0,01-6,0 мас. %, предпочтительно 0,02-5,0 мас. %, более предпочтительно 0,03-4,0 мас. %, в частности, 0,05-3,5 мас. %. Например, наивысшая массовая доля, определяемая во время реакции, может составлять 0,5 мас. %, 1,0 мас. %, 1,5 мас. %, 2,0 мас. %, 2,5 мас. %, или 3,0 мас. %, в то время, как все другие значения, которые были определены таким образом, находятся ниже значений, указанных здесь. Общее количество (которое также называют общей массой) мономеров, которые применяли на соответствующей стадии полимеризации, очевидно, для стадии I. соответствует общему количеству мономерной смеси (А), для стадии II. соответствует общему количеству мономерной смеси (Б), и для стадии III. соответствует общему количеству мономерной смеси (В).
Концентрация мономеров в реакционном растворе в данном случае может, например, определяться посредством газовой хроматографии. В этом случае образец реакционного раствора охлаждают жидким азотом сразу же после отбора пробы, и добавляют 4-метоксифенол в качестве ингибитора. На следующей стадии, образец растворяют в тетрагидрофуране и затем добавляют н-пентан, для того чтобы осадить полимер, образованный во время отбора пробы. Жидкую фазу (надосадочную жидкость) затем анализируют посредством газовой хроматографии, применяя полярную колонку и неполярную колонку для определения мономеров, а также плазменно-ионизационный детектор. Обычные параметры газохроматографического определения являются следующим: 25 м кварцевая капиллярная колонка с 5% фенил-, 1% винил-метилполисилоксановой фазы, или 30 м кварцевая капиллярная колонка с 50% фенил-, 50% метил-полисилоксановой фазы, газ-носитель водород, устройство для ввода проб с делением потока 150°С, температура термостата 50-180°С, плазменно-ионизационный детектор, температура детектора 275°С, внутренний эталон изобутилакрилат. Для целей настоящего изобретения, концентрацию мономеров предпочтительно определяют посредством газовой хроматографии, в частности, в соответствии с параметрами, указанными выше.
Доля свободных мономеров может контролироваться различными способами.
Одна из возможностей сохранения доли свободных мономеров низкой представляет собой выбор очень низкого дозированного расхода для смеси олефинненасыщенных мономеров в актуальном реакционном растворе, где мономеры контактируют с инициатором. Если дозированный расход является настолько низким, что все мономеры способны вступать в реакцию практически сразу же, когда они попадают в реакционный раствор, тогда возможно обеспечить, что доля свободных мономеров сводится к минимуму.
В дополнение к дозированному расходу, важным является то, чтобы всегда существует достаточное количество радикалов, присутствующих в реакционном растворе, с тем, чтобы обеспечивать каждому из добавленных мономеров вступать в реакцию чрезвычайно быстро. Таким образом, гарантируется дополнительный рост цепи полимера, и доля свободного мономера сохраняется низкой.
Для указанной цели, условия реакции предпочтительно выбирают таким образом, чтобы подача инициатора начиналась еще до начала дозированного добавления олефинненасыщенных мономеров. Предпочтительно, дозирование начинается по меньшей мере за 5 минут до этого, более предпочтительно по меньшей мере за 10 минут до этого. Предпочтительно, по меньшей мере 10 мас. % инициатора, более предпочтительно по меньшей мере 20 мас. %, очень предпочтительно по меньшей мере 30 мас. % инициатора, в каждом случае, из расчета общего количества инициатора, добавляют до начинала дозированного добавления олефинненасыщенных мономеров.
Предпочтение отдают выбору такой температура, которая позволяет непрерывное разложение инициатора.
Количество инициатора также является важным фактором для достаточного присутствия радикалов в реакционном растворе. Количество инициатора должны выбираться таким образом, чтобы на любой заданный момент времени существовало достаточно доступных радикалов, позволяя добавленным мономерам вступать в реакцию. Если количество инициатора повышается, то в это же время также может вступать в реакцию большее количество мономеров.
Дополнительным фактором определения скорости реакции является реакционная способность мономеров.
По этой причине, контроль доли свободных мономеров может проводиться посредством взаимного влияния количества инициатора, скорости добавления инициатора, скорости добавления мономера, а также как результат выбора мономеров. Не только замедление дозированного добавления, но также повышение исходного количества, но также заблаговременное начало добавления инициатора, служат цели сохранения концентрации свободных мономеров ниже указанных выше пределов.
На любой момент во время прохождения реакции, концентрацию свободных мономеров можно определять посредством газовой хроматографии, как описано выше.
Если указанный анализ выявляет концентрацию свободных мономеров, которая приближается до ограничивающего значения точных дозировок полимеризации, например, в результате небольших долей высокореакционноспособных олефинненасыщенных мономеров, то указанные выше параметры могут применяться для того, чтобы контролировать реакцию. В указанном случае, например, может понижаться дозированный расход мономеров, или может повышаться количество инициатора.
Для целей настоящего изобретения является предпочтительным осуществлять стадии полимеризации II. и III. в условиях точных дозировок. Указанное имеет то преимущество, что образование новых ядер частиц во время указанных двух стадий полимеризации эффективно сводится к минимуму. Вместо этого, частицы, присутствующие после стадии I. (и по этой причине называемые ниже зародышем), могут расти дополнительно на стадии II. в результате полимеризации мономерной смеси Б (по этой причине также называемые ниже ядром). Также является возможным, когда частицы, присутствующие после стадии II. (также называемые ниже полимером, содержащим зародыш и ядро), растут дополнительно на стадии III. в результате полимеризации мономерной смеси В (по этой причине также называемые ниже оболочкой), что приводит в итоге к полимеру, содержащему частицы, которые имеют зародыш, ядро, и оболочку.
Смеси (А), (Б), и (В) представляют собой смеси олефинненасыщенных мономеров. Подходящие олефинненасыщенные мономеры могут быть моно- или полиолефинненасыщенными.
Описанные прежде всего ниже, представляют собой мономеры, которые могут применяться в принципе, и которые являются подходящими для всех смесей (А), (Б), и (В), а также мономеры, которые необязательно являются предпочтительными. Определенные предпочтительные варианты осуществления отдельных смесей будут представлены далее.
Примеры подходящих моноолефинненасыщенных мономеров включают, в частности, моноолефинненасыщенные мономеры на основе (мет)акрилата, моноолефинненасыщенные мономеры, которые содержат аллильные группы, а также другие моноолефинненасыщенные мономеры, которые содержат винильные группы, такие как винилароматические мономеры, например. Термин (мет)акриловый или (мет)акрилат для целей настоящего изобретения включает как метакрилаты, так и акрилаты. Предпочтительными для применения, по меньшей мере, при том, что не обязательно исключительно, являются моноолефинненасыщенные мономеры на основе (мет)акрилата.
Моноолефинненасыщенные мономеры на основе (мет)акрилата могут, например, представлять собой (мет)акриловую кислоту и сложные эфиры, нитрилы, или амиды (мет)акриловой кислоты.
Предпочтение отдают сложным эфирам (мет)акриловой кислоты, имеющим неолефинненасыщенный радикал R.
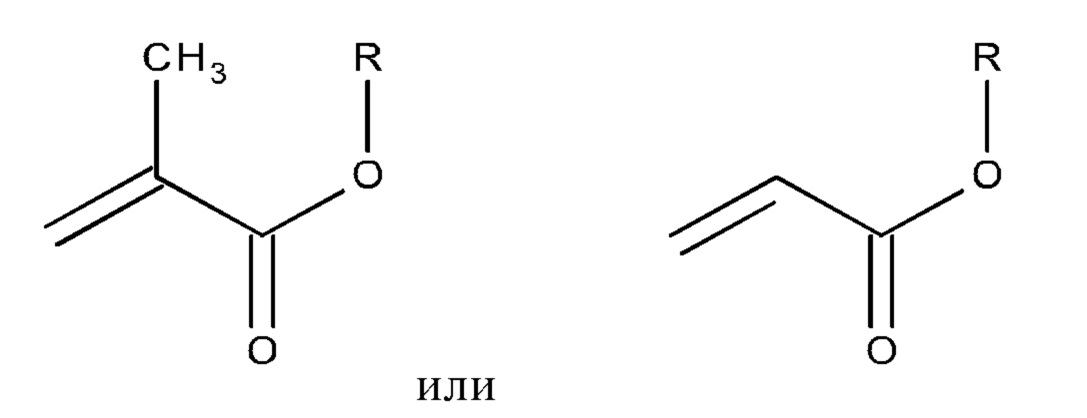
Радикал R может быть насыщенным алифатическим, ароматическим, или смешанным насыщенным алифатическим-ароматическим. Алифатические радикалы для целей настоящего изобретения представляют собой все органические радикалы, которые не являются ароматическими. Предпочтительно радикал R является алифатическим.
Насыщенный алифатический радикал может быть чистым углеводородным радикалом или он может включать гетероатомы из мостиковых групп (например, кислород из групп простого эфира или групп сложного эфира) и/или может замещаться функциональными группами, которые содержат гетероатомы (группы спирта, например). По этой причине, для целей настоящего изобретения, проводится четкое различие между мостиковыми группами, которые содержат гетероатомы, и функциональными группами, которые содержат гетероатомы (то есть, концевые функциональные группы, которые содержат гетероатомы).
По меньшей мере, предпочтение отдают, хотя и не обязательно исключительно, применению мономеров, в которых насыщенный алифатический радикал R представляет собой чистый углеводородный радикал (алкильный радикал), другими словами, тот, который не включает каких-либо гетероатомов из мостиковых групп (например, кислород из групп простого эфира) и также не замещен функциональными группами (группами спирта, например).
Если R представляет собой алкильный радикал, то он, например, может быть неразветвленным, разветвленным, или циклическим алкильным радикалом. Конечно, такой алкильный радикал может также иметь неразветвленные и циклические или разветвленные и циклические структурные компоненты. Алкильный радикал предпочтительно имеет 1-20, более предпочтительно 1-10, атомов углерода.
В частности, предпочтительные мононенасыщенные сложные эфиры (мет)акриловой кислоты с алкильным радикалом представляют собой метил(мет)акрилат, этил(мет)акрилат, пропил(мет)акрилат, изопропил(мет)акрилат, н-бутил(мет)акрилат, изобутил(мет)акрилат, трет-бутил(мет)акрилат, амил(мет)акрилат, гексил(мет)акрилат, этилгексил(мет)акрилат, 3,3,5-триметилгексил(мет)акрилат, стеарил(мет)акрилат, лаурил(мет)акрилат, циклоалкил(мет)акрилаты, такие как циклопентил(мет)акрилат, изоборнил(мет)акрилат, а также циклогексил(мет)акрилат, особенное предпочтение при этом отдают н- и трет-бутил(мет)акрилату и метилметакрилату.
Примерами других подходящих радикалов R являются насыщенные алифатические радикалы, которые содержат функциональные группы, которые включают гетероатомы (например, группы спирта или группы эфира фосфорной кислоты).
Подходящие мононенасыщенные сложные эфиры (мет)акриловой кислоты с насыщенным алифатическим радикалом, замещенным одной или большим количеством гидроксильных групп, представляют собой 2-гидроксиэтил(мет)-акрилат, 2-гидроксипропил(мет)акрилат, 3-гидроксипропил(мет)акрилат, 3-гидроксибутил(мет)акрилат, и 4-гидроксибутил(мет)акрилат, особенное предпочтение при этом отдают 2-гидроксиэтил(мет)акрилату.
Подходящие мононенасыщенные сложные эфиры (мет)акриловой кислоты с группами сложного эфира фосфорной кислоты, например, представляют собой сложные эфиры фосфорной кислоты полипропиленгликольмонометакрилата, такие как коммерчески доступный Sipomer РАМ 200 от компании Rhodia.
Возможные дополнительные моноолефинненасыщенные мономеры, которые содержат виниловые группы, представляют собой мономеры, которые отличаются от описанных выше мономеров на основе акрилата, и которые имеют радикал R' на винильной группе, который не является олефинненасыщенным.

Радикал R' может быть насыщенным алифатическим, ароматическим, или смешанным насыщенным алифатическим-ароматическим, где при этом предпочтение отдают ароматическим и смешанным насыщенным алифатическим-ароматическим радикалам, где алифатические компоненты представляют собой алкильные группы.
В частности, предпочтительными дополнительными моноолефинненасыщенными мономерами, которые содержат винильные группы, в частности, являются винилтолуол, альфа-метилстирол, и в частности стирол.
Также возможными являются мононенасыщенные мономеры, которые содержат винильные группы, где радикал R' имеет следующую структуру:

где радикалы R1 и R2b качестве алкильных радикалов содержат в общем 7 атомов углерода. Мономеры указанного вида являются доступными коммерчески под названием VeoVa 10 от компании Momentive.
Дополнительными, подходящими в принципе мономерами являются олефинненасыщенные мономеры, такие как акрилонитрил, метакрилонитрил, акриламид, метакриламид, N,N-диметилакриламид, винилацетат, винилпропионат, винилхлорид, N-винилпирролидон, N-винилкпролактам, N-винилформамид, N-винилимидазол, N-винил-2-метилимидазолин, и дополнительные ненасыщенные альфа-бета-карбоновые кислоты.
Примеры подходящих полиолефинненасыщенных мономеров включают сложные эфиры (мет)акриловой кислоты с олефинненасыщенным радикалом R''. Радикал R'' может, например, представлять собой аллильный радикал или (мет)акрилоильный радикал.

Предпочтительные полиолефинненасыщенные мономеры включают этиленгликольди(мет)акрилат, 1,2-пропиленгликольди(мет)акрилат, 2,2-пропиленгликольди(мет)акрилат, бутан-1,4-диолди(мет)акрилат, неопентилгликольди(мет)акрилат, 3-метилпентандиолди(мет)акрилат, диэтиленгликольди(мет)акрилат, триэтиленгликольди(мет)акрилат, тетраэтиленгликольди(мет)акрилат, дипропиленгликольди(мет)акрилат, трипропиленгликольди(мет)акрилат, гександиолди(мет)акрилат, и аллил(мет)акрилат.
Более того, предпочтительные полиолефинненасыщенные соединения включают акриловые и метакриловые сложные эфиры спиртов, которые имеют более двух ОН групп, такие как, например, триметилолпропантри(мет)акрилат или глицеролтри(мет)акрилат, а также простой моноаллиловый эфир триметилолпропанди(мет)акрилата, простой диаллиловый эфир триметилолпропан(мет)акрилата, простой моноаллиловый эфир пентаэритритолтри(мет)акрилата, простой диаллиловый эфир пентаэритритолди(мет)акрилата, простой триаллиловый эфир пентаэритритол(мет)акрилата, триаллилсахароза, и пентааллилсахароза.
Также возможными являются простые аллиловые эфиры одно- или многоатомных спиртов, такие как, например, простой моноаллиловый эфир триметилолпропана.
В случае их применения, предпочтительными полиолефинненасыщенными мономерами являются гександиолдиакрилат и/или аллил(мет)акрилат.
Что касается мономерной смеси (А), (Б), и (В), которые применяют на отдельной стадии полимеризации, существуют определенные условия, которые должны наблюдаться, изложенные ниже.
Прежде всего, необходимо указать, что смеси (А), (Б), и (В), в любом случае, отличаются друг от друга. По этой причине, каждая из них содержит разные мономеры и/или разные пропорции по меньшей мере одного определенного мономера.
Смесь (А), предпочтительно но не обязательно, содержит по меньшей мере 50 мас. %, более предпочтительно по меньшей мере 55 мас. %, олефинненасыщенных мономеров, имеющих растворимость в воде, которая составляет менее 0,5 г/л при температуре 25°С. Одним таким предпочтительным мономером является стирол.
Растворимость мономеров в воде могут определять с помощью установления равновесия воздушно-газового пространства над водной фазой (по аналогии со ссылкой на X.-S. Chai, Q.X. Hou, F.J. Schork, Journal of Applied Polymer Science, т. 99, 1296-1301 (2006)).
Для указанной цели, в 20 мл пробирку для отбора проб газа, к определенному объему воды, предпочтительно, составляющему 2 мл, добавляют массу соответствующего мономера, которая имеет такую величину, что указанная масса, в любом случае, не может полностью раствориться в выбранном объеме воды. Дополнительно добавляют эмульгирующее вещество (10 млн. ч., из расчета общей массы отобранной смеси). Для того чтобы получить равновесную концентрацию, смесь непрерывно встряхивают. Надосадочную газовую фазу заменяют инертным газом и, таким образом, равновесие устанавливается снова. В отведенной газовой фазе, устанавливают долю подлежащего определению вещества (предпочтительно посредством газовой хроматографии). Равновесную концентрация в воде могут определять посредством построения графика доли мономера в газовой фазе. Наклон кривой меняется от практически постоянного значения (S1) до существенно отрицательного наклона (S2) по мере того, как избыток доли мономера удаляется из смеси. Равновесная концентрация в данном случае достигается в точке пересечения прямой линии с наклоном S1 и прямой линии с наклоном S2. Описанное определение осуществляют при температуре 25°С.
Мономерная смесь (А) предпочтительно не содержит мономеров с гидроксильными функциональными группами. Также предпочтительно, мономерная смесь (А) не содержит мономеров с кислотными функциональными группами.
Очень предпочтительно, мономерная смесь (А) вообще не содержит мономеров, которые имеют функциональные группы, содержащие гетероатомы. Указанное означает, что гетероатомы, если они присутствуют, присутствуют только в виде мостиковых групп. Указанное верно, например, для моноолефинненасыщенных мономеров, описанных выше, которые основаны на (мет)акрилате и имеют алкильный радикал в качестве радикала R.
Мономерная смесь (А) предпочтительно содержит исключительно моноолефинненасыщенные мономеры.
В одном особенно предпочтительном варианте осуществления, мономерная смесь (А) содержит по меньшей мере один мононенасыщенные сложный эфир (мет)акриловой кислоты с алкильным радикалом и по меньшей мере один моноолефинненасыщенный мономер, который содержит винильные группы, с радикалом, расположенным на винильной группе, который является ароматическим, или который является смешанным насыщенным алифатическим-ароматическим, причем алифатические фракции радикала являются алкильными группами.
Мономеры, которые присутствуют в смеси (А), выбирают таким образом, что полимер, полученный из них, имеет температуру стеклования, которая составляет 10-65°С, предпочтительно, которая составляет 30-50°С.
Для целей в соответствии с изобретением, температуру стеклования Tg определяют экспериментально в соответствии со стандартом DIN 51005 "Термический анализ (ТА) - термины" и стандартом DIN 53765 "Термический анализ - динамическая сканирующая калориметрия (ДСК)". Указанное включает отвешивание 15 мг образца в лодочку для аналитического сжигания и вводили его в аппарат ДСК. После охлаждения до стартовой температуры, осуществляли 1-ю и 2-ю серии измерений с продувкой в атмосфере инертного газа (N2) со скоростью 50 мл/мин со скоростью нагрева, составляющей 10 К/мин, снова охлаждая до стартовой температуры между сериями измерений. Измерения обычно осуществляют в температурном диапазоне от примерно на 50°С ниже ожидаемой температуры стеклования и до примерно на 50°С выше температуры стеклования. Для целей настоящего изобретения, температура стеклования в соответствии со стандартом DIN 53765, раздел 8.1, представляет собой температуру во 2-й серии измерений, при которой достигается половина изменения удельной теплоемкости (0,5 дельта ср). Указанную температуру определяют на основании диаграммы ДСК (зависимость теплопередачи от температуры). Она представляет собой температуру в точке пересечения средней линии между экстраполированными базовыми линиями, до и после стеклования, на графике измерений.
В контексте настоящего изобретения, в случае, когда ссылаются на официальный стандарт, без какого-либо указания официального срока действия, конечно, ссылаются на ту версию стандарта, которая действует на дату подачи заявки, или, если отсутствует действующая версия на указанную дату, на наиболее позднюю действующую версию.
Для эффективной оценки температуры стеклования, ожидаемой при измерениях, может применяться известное уравнение Фокса. Поскольку уравнение Фокса представляет хорошее приближение, основанное на температурах стеклования гомополимеров и их частей по массе, без включения молекулярной массы, оно может применяться в качестве руководства для специалиста в данной области синтеза, позволяя устанавливать желательную температуру стеклования посредством ряда целенаправленных экспериментов.
Полимер, полученный на стадии I. посредством эмульсионной полимеризации мономерной смеси (А), также называют зародышем.
Предпочтительно, зародыш имеет размер частиц, который составляет 20-125 нм (в отношении метода определения смотри раздел Примеров).
Смесь (Б) предпочтительно содержит по меньшей мере один полиолефинненасыщенный мономер, более предпочтительно по меньшей мере один диолефинненасыщенный мономер. Один такой предпочтительный мономер представляет собой гександиолдиакрилат.
Мономерная смесь (Б) предпочтительно не содержит мономеров с гидроксильными функциональными группами. Также предпочтительно, мономерная смесь (Б) не содержит мономеров с кислотными функциональными группами.
Очень предпочтительно, мономерная смесь (Б) вообще не содержит мономеров с функциональными группами, содержащими гетероатомы. Указанное означает, что гетероатомы, если они присутствуют, присутствуют только в виде мостиковых групп. Указанное верно, например, для описанных выше моноолефинненасыщенных мономеров, на основе (мет)акрилата, и которые имеют алкильный радикал в качестве радикала R.
В одном особенно предпочтительном варианте осуществления, мономерная смесь (Б), также в качестве по меньшей мере одного полиолефинненасыщенного мономера, включает по меньшей мере следующие дополнительные мономеры. Во-первых, по меньшей мере один мононенасыщенный сложный эфир (мет)акриловой кислоты с алкильным радикалом, и во-вторых, по меньшей мере один моноолефинненасыщенный мономер, содержащий винильные группы, и имеющий радикал, расположенный на винильной группе, который является ароматическим, или который является смешанным насыщенным алифатическим-ароматическим радикалом, причем алифатическими фракциями радикала являются алкильные группы.
Доля полиненасыщенных мономеров предпочтительно составляет от 0,05 до 3 мол. %, из расчета общего молярного количества мономеров в мономерной смеси (Б).
Мономеры, которые присутствуют в смеси (Б), выбирают таким образом, что полимер, полученный из нее, имеет температуру стеклования, которая составляет -35-15°С, предпочтительно, которая составляет -25-+7°С.
Полимер, полученный в присутствии зародыша на стадии II. посредством эмульсионной полимеризации мономерной смеси (Б), также называют ядром. Таким образом, после стадии II. получают полимер, который содержит зародыш и ядро.
Полимер, который получают в результате стадии II., предпочтительно имеет размер частиц, который составляет 80-280 нм, предпочтительно 120-250 нм.
Мономеры, которые присутствуют в смеси (В), выбирают таким образом, что полимер, полученный из нее, имеет температуру стеклования, которая составляет -50-15°С, предпочтительно, которая составляет -20-+12°С.
Олефинненасыщенные мономеры указанной смеси (В) предпочтительно выбирают таким образом, что полученный полимер, содержащий зародыш, ядро, и оболочку, имеет кислотное число, составляющее 10-25.
Соответственно, смесь (В) предпочтительно содержит по меньшей мере одну альфа-бета ненасыщенную карбоновую кислоту, особенно предпочтительно (мет)акриловую кислоту.
Кроме того, олефинненасыщенные мономеры смеси (В) предпочтительно выбирают таким образом, что полученный полимер, содержащий зародыш, ядро, и оболочку, имеет гидроксильное число, составляющее 0-30, предпочтительно 10-25.
Все упомянутые выше кислотные числа и гидроксильные числа в отношении дисперсии (wD) представляют собой значения, рассчитанные на основе смесей мономеров, которые применяют в общем.
В одном особенно предпочтительном варианте осуществления, мономерная смесь (В) содержит по меньшей мере одну альфа-бета ненасыщенную карбоновую кислоту и по меньшей мере один мононенасыщенные сложный эфир (мет)акриловой кислоты, имеющий алкильный радикал, замещенный гидроксильной группой.
В одном особенно предпочтительном варианте осуществления, мономерная смесь (В) содержит по меньшей мере одну альфа-бета ненасыщенную карбоновую кислоту, по меньшей мере один мононенасыщенный сложный эфир (мет)акриловой кислоты, имеющий алкильный радикал, замещенный гидроксильной группой, и по меньшей мере один мононенасыщенный сложный эфир (мет)акриловой кислоты, имеющий алкильный радикал.
В случае, когда в контексте настоящего изобретения ссылаются на алкильный радикал, без дополнительной конкретизации, то в этом случае всегда понимается чистый алкильный радикал без функциональных групп и гетероатомов.
Полимер, полученный в присутствии зародыша и ядра на стадии III. посредством эмульсионной полимеризации мономерной смеси (В), также называют оболочкой. Таким образом, в результате стадии III. получают полимер, который содержит зародыш, ядро, и оболочку.
После получения, полимер имеет размер частиц, который составляет 100-500 нм, предпочтительно 125-400 нм, очень предпочтительно от 130 до 300 нм.
Доли смесей мономеров предпочтительно гармонизируют друг с другом следующим образом. Доля смеси (А) составляет от 0,1 до 10 мас. %, доля смеси (Б) составляет от 60 до 80 мас. %, и доля смеси (В) составляет от 10 до 30 мас. %, в каждом случае, из расчета суммы отдельных количеств смесей (А), (Б), и (В).
Водная дисперсия (wD) предпочтительно имеет значение рН, которое составляет 5,0-9,0, более предпочтительно 7,0-8,5, очень предпочтительно 7,5-8,5. Значение рН может поддерживаться постоянным во время самого получения как такового, в результате применения оснований, которые определены далее ниже, например, или помимо прочего, может устанавливаться намеренно, после того, как полимер был получен.
В особенно предпочтительных вариантах осуществления, водная дисперсия (wD) имеет значение рН, которое составляет 5,0-9,0, и по меньшей мере один полимер, который присутствует в ней, имеет размер частиц, который составляет 100-500 нм. Даже более предпочтительные комбинации диапазонов являются следующими: значение рН, которое составляет 7,0-8,5, и размер частиц, который составляет 125-400 нм, более предпочтительно, значение рН, которое составляет 7,5-8,5, и размер частиц, который составляет 130-300 нм.
Описанные стадии I.-III. предпочтительно осуществляют без добавления кислот или оснований, известных для регулирования рН. Если при получении полимера, например, применяют мономеры с карбоксильными функциональными группами, что является предпочтительным в контексте стадий III., то рН дисперсии после окончания стадий III может составлять менее 7. Соответственно, добавление основания необходимо для доведения уровня рН до более высокого значения, например, до значения в пределах предпочтительных диапазонов.
Из указанного выше следует, что предпочтительно после стадии III. значение рН, соответственно, регулируется или должно регулироваться, в частности посредством добавления оснований, таких как органические, содержащие азот основания, такие как амин, например, такие как аммиак, триметиламин, триэтиламин, трибутиламины, диметиланилин, трифениламин, N,N-диметилэтаноламин, метилдиэтаноламин, или триэтаноламин, а также посредством добавления гидрокарбоната натрия или боратов, а также смесей указанных выше веществ. Однако, указанное не исключает возможности регулирования рН до, во время, или после эмульсионных полимеризаций или, помимо прочего, между отдельными эмульсионными полимеризациями. Также вполне возможно обойтись без необходимости регулирования уровня рН до желательного значения вообще, вследствие выбора мономеров.
Предпочтительно, измерения значения рН в данном случае осуществляют, применяя прибор для измерения рН (например, прибор для измерения рН Mettler-Toledo S20 SevenEasy), который имеет комбинированный рН-электрод (например, Mettler-Toledo InLab® Routine).
Содержание твердых веществ дисперсии (wD) предпочтительно составляет от 15% до 40%, и более предпочтительно 20%-30%.
Дисперсия (wD) является водной (смотри выше в отношении основного определения). Предпочтительно, чтобы водная дисперсия (wD) содержал долю воды, составляющую 55 - 75 мас. %, особенно предпочтительно 60-70 мас. %, в каждом случае, из расчета общей массы дисперсии.
Дополнительно предпочтительно, если сумма процентов содержания твердых веществ дисперсии (wD) и доли воды в дисперсии (wD) составляет по меньшей мере 80 мас. %, предпочтительно по меньшей мере 90 мас. %. В свою очередь, предпочтительными являются диапазоны от 80 до 99 мас. %, особенно 90 -97,5 мас. %. Здесь, содержание твердых веществ, которое традиционно лишь обозначается единицей "%", подано в "мас. %". Поскольку содержание твердых веществ в конечном итоге также представляет величину процентной массы, то указанный вид представления оправдан. В случае, если, например, дисперсия имеет содержание твердых веществ, составляющее 25%, и содержание воды, составляющее 70 мас. %, то таким образом, определенное выше процентная сумма содержания твердых веществ и доли воды составляет до 95 мас. %.
Соответственно, дисперсия состоит в основном из воды и определенного полимера, и вредные в экологическом отношении компоненты, в частности, такие как органические растворители, присутствуют лишь в незначительной пропорции или вообще не присутствуют.
Доля одной или большего количества дисперсий (wD), из расчета общей массы водного материала базового покрытия в соответствии с изобретением, предпочтительно составляет 1,0-60 мас. %, более предпочтительно 2,5-50 мас. %, и очень предпочтительно 5-40 мас. %.
Доля полимеров, полученных из дисперсии (wD), из расчета общей массы водного материала базового покрытия в соответствии с изобретением, предпочтительно составляет от 0,3 до 17,0 мас. %, более предпочтительно 0,7-14,0 мас. %, очень предпочтительно 1,4-11,0 мас. %.
Для целей настоящего изобретения, принцип, который следует соблюдать для компонентов, предназначенных для применения в материале базового покрытия -например, для компонентов дисперсии (PD), дисперсии (wD), или помимо прочего меламиновой смолы - является следующим (как описано здесь для дисперсии (wD)): в случае возможной конкретизации материалов базовых покрытий, содержащих предпочтительные дисперсии (wD) в определенном пропорциональном диапазоне, применяется следующее. Дисперсии (wD), которые не попадают в предпочтительную группу, конечно, могут все же присутствовать в материале базового покрытия. В этом случае определенный пропорциональный диапазон применяется только к предпочтительной группе дисперсий (wD). Тем не менее, является предпочтительным, когда общая пропорция дисперсий (wD), состоящих из дисперсий из предпочтительной группы и дисперсий, которые не являются частью предпочтительной группы, также подпадала под определенный пропорциональный диапазон.
Таким образом, в случае ограничения пропорциональным диапазоном, составляющим 2,5-50 мас. %, и предпочтительной группой дисперсий (wD), очевидно, указанный пропорциональный диапазон применяется первоначально только к предпочтительной группе дисперсий (wD). Однако, в этом случае, будет предпочтительным, когда диапазон от 10 до 50 мас. % также будет охватывать общее количество всех присутствующих, первоначально включенных дисперсий, состоящих из дисперсий из предпочтительной группы и дисперсий, которые не составляют часть предпочтительной группы. Таким образом, если применяют 35 мас. % дисперсий (wD) из предпочтительной группы, то при этом могут применяться не более 15 мас. % дисперсий из группы, не являющейся предпочтительной.
Материал базового покрытия в соответствии с изобретением предпочтительно содержит по меньшей мере один пигмент. В данном случае ссылаются на традиционные пигменты, придающие цвет и/или оптический эффект.
Такие цветные пигменты и эффектные пигменты известны специалистам в данной области и, например, описаны в Rompp-Lexikon Lacke und Druckfarben, Georg Thieme Verlag, Штутгарт, Нью-Йорк, 1998, страницы 176 и 451. Термины "красящий пигмент" и "цветной пигмент" являются взаимозаменямыми, точно так же, как и термины "оптически эффектный пигмент" и "эффектный пигмент".
Предпочтительные эффектные пигменты, например, представляют собой пластинчатые металлизированные эффектные пигменты, такие как пластинчатые алюминиевые пигменты, золотистые бронзы, оксидированные бронзы и/или пигменты на основе оксида железа и алюминия, перламутровые пигменты, такие как жемчужная эссенция, основные карбонат свинца, оксихлорид висмута и/или пигменты на основе оксида металла и слюды и/или другие эффектные пигменты, такие как пластинчатый графит, пластинчатый оксид железа, многослойные эффектные пигменты, состоящие из ПВД пленок и/или жидкокристаллические полимерные пигменты. Особенно предпочтительными являются пластинчатые металлизированные эффектные пигменты, в частности, пластинчатые алюминиевые пигменты.
Типичные цветные пигменты, в частности, включают неорганические красящие пигменты, такие как белые пигменты, такие как диоксид титана, цинковые белила, сульфид цинка или литопон; черные пигменты, такие как углеродная сажа, железо-марганец черный, или черная шпинель; хроматические пигменты, такие как оксид хрома, оксигидрат хрома зеленый, кобальт зеленый или ультрамарин зеленый, кобальт синий, ультрамарин синий или марганец синий, ультрамарин фиолетовый или кобальт фиолетовый и марганец фиолетовый, оксид железа красный, сульфоселенид кадмия, молибдат красный или ультрамарин красный; коричневый оксид железа, смешанный коричневый, фазы шпинели и фазы корунда или хром оранжевый; или оксид железа желтый, титаноникель желтый, титанохром желтый, сульфид кадмия, сульфид цинка и кадмия, хром желтый или ванадат висмута.
Доля пигментов предпочтительно находится в диапазоне от 1,0 до 40,0 мас. %, предпочтительно в диапазоне 2,0-35,0 мас. %, более предпочтительно в диапазоне 5,0-30,0 мас. %, из расчета общей массы водного материала базового покрытия в каждом случае.
Водный материал базового покрытия предпочтительно дополнительно содержит по меньшей мере один полимер в качестве связующего вещества, который отличается от полимеров, которые присутствуют в дисперсиях (wD) и (PD), в частности, по меньшей мере один полимер, выбранный из группы, состоящей из полиуретанов, сложных полиэфиров, полиакрилатов и/или сополимеров указанных полимеров, в частности, сложных полиэфиров и/или полиуретанполиакрилатов. Предпочтительные сложные полиэфиры описаны, например, в DE 4009858 А1 в колонке 6, строка 53 - колонке 7, строка 61, и колонке 10, строка 24 - колонке 13, строка 3, или в WO 2014/033135 А2, страница 2, строка 24 - страница 7, строка 10, и страница 28, строка 13 - страница 29, строка 13. Предпочтительные полиуретан-полиакрилатные сополимеры (акрилатированные полиуретаны) и их получение описаны, например, в WO 91/15528 А1, страница 3, строка 21 - страница 20, строка 33, и в DE 4437535 А1, страница 2, строка 27 - страница 6, строка 22. Описанные в качестве связующих веществ полимеры предпочтительно имеют гидроксильные функциональные группы, и особенно предпочтительно имеют гидроксильное число в диапазоне от 15 до 200 мг KOH/г, более предпочтительно от 20 до 150 мг KOH/г. Материалы базовых покрытий более предпочтительно содержат по меньшей мере один имеющий гидроксильные функциональные группы полиуретан-полиакрилатный сополимер, более предпочтительно по меньшей мере еще один имеющий гидроксильные функциональные группы полиуретан-полиакрилатный сополимер, а также по меньшей мере один имеющий гидроксильные функциональные группы сложный полиэфир.
Пропорция дополнительных полимеров в качестве связующих веществ может варьироваться в широком диапазоне, и предпочтительно находится в диапазоне от 1,0 до 25,0 мас. %, более предпочтительно в диапазоне 3,0-20,0 мас. %, очень предпочтительно 5,0-15,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Материал базового покрытия в соответствии с изобретением может дополнительно содержать по меньшей мере один типичный сшивающий агент, известный как таковой. Если материал содержит сшивающий агент, то указанный агент предпочтительно содержит по меньшей мере одну аминопластовую смолу и/или по меньшей мере один блокированный полиизоцианат, предпочтительно аминопластовую смолу. Среди аминопластовых смол, меламиновые смолы, в частности, являются предпочтительными.
Если материал базового покрытия содержит сшивающие агенты, то пропорция указанных сшивающих агентов, в частности, аминопластовых смол и/или блокированных полиизоцианатов, очень предпочтительно аминопластовых смол и, среди них, предпочтительно меламиновых смол, предпочтительно находится в диапазоне от 0,5 до 20,0 мас. %, более предпочтительно в диапазоне 1,0-15,0 мас. %, очень предпочтительно в диапазоне 1,5-10,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Материал базового покрытия может дополнительно содержать по меньшей мере один загуститель. Подходящие загустители являются неорганическими загустителями из групп листовых силикатов, такие как литиево-алюминиево-магниевые силикаты. Также, материал базового покрытия может содержать по меньшей мере один органический загуститель, такой, как например, загуститель га основе сополимера (мет)акриловой кислоты и (мет)акрилата или загуститель на основе полиуретана. В данном случае, например, могут применяться традиционные органические ассоциированные загустители, такие, как например, известные ассоциированные загустители на основе полиуретана. Ассоциированные загустители, как известно, являются растворимыми в воде полимерами, которые имеют сильно гидрофобные группы на концах цепи или в боковых цепях, и/или чьи гидрофильные цепи содержат гидрофобные блоки или их концентрации внутри. Как результат, указанные полимеры имеют поверхностно-активную природу, и способны образовывать мицеллы в водной фазе. Подобно поверхностно-активным веществам, гидрофильные участки остаются в водной фазе, в то время как гидрофобные участки поступают в частицы полимерной дисперсии, адсорбируют на поверхности других твердых веществ, таких как пигменты и/или наполнители, и/или образуют мицеллы в водной фазе. По сути, загущающее действие достигается без какого-либо повышения осаждения.
Загустители, как указано, являются доступными коммерчески. Пропорция загустителей предпочтительно находится в диапазоне от 0,1 до 5,0 мас. %, более предпочтительно от 0,2 до 3,0 мас. %, очень предпочтительно от 0,3 до 2,0 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Более того, материал базового покрытия может дополнительно содержать по меньшей мере одну дополнительную активирующую добавку. Примерами таких активирующих добавок являются соли, которые термически разлагаются без остатка или в основном без остатка, полимеры в качестве связующих веществ, которые могут отверждаться физически, термически и/или с использованием актиничного излучения, и которые отличаются от уже указанных в качестве связующих веществ полимеров, дополнительные сшивающие агенты, органические растворители, реакционноспособные разбавители, прозрачные пигменты, наполнители, растворимые в молекулярной дисперсии красящие вещества, наночастицы, стабилизаторы света, вещества против окисления, деаэрирующие вещества, эмульгирующие вещества, добавки скольжения, ингибиторы полимеризации, инициаторы радикальной полимеризации, усилители адгезии, регулирующие текучесть агенты, пленкообразующие вспомогательные вещества контролирующие образование натеков вещества (SCA), замедлители горения, ингибиторы коррозии, воски, сиккативы, биоциды, и матирующие агенты. Такие активирующие добавки применяют в обычных и известных количествах.
Содержание твердых веществ материала базового покрытия может варьироваться в соответствии с требованиями конкретного случая. Содержание твердых веществ зависит в основном от необходимой для нанесения вязкости, в частности, нанесения посредством распыления. Особое преимущество состоит в том, что материал базового покрытия, предназначенный для применения в соответствии с изобретением, при сравнительно высоком содержании твердых веществ способен, тем не менее, иметь вязкость, позволяющую осуществлять соответствующее нанесение.
Содержание твердых веществ материала базового покрытия предпочтительно составляет по меньшей мере 16,5%, более предпочтительно по меньшей мере 18,0%, даже более предпочтительно по меньшей мере 20,0%.
Другими словами, в указанные условиях, при указанном содержании твердых веществ, предпочтительные материалы базовых покрытий имеют вязкость составляющую 40-150 мПа⋅с, в частности, 70-120 мПа⋅с, при температуре 23°С, в условиях усилий сдвига, составляющих 1000 1/с (для дополнительных подробностей в отношении метода определения, смотри раздел Примеров). Для целей настоящего изобретения, вязкость в пределах указанного диапазона в условиях указанных усилий сдвига называют вязкостью распыления (рабочая вязкость). Как известно, покрывающие материалы наносят при вязкости распыления, что означает то, что в указанных имеющихся условиях (большие усилия сдвига) они имеют вязкость, которая, в частности, не слишком высокая, с тем, чтобы получить эффективное нанесение. Указанное означает, что установления вязкости распыления является важным для того, чтобы позволить краске, в принципе, наноситься посредством методов распыления, и обеспечивать возможность получения на основе, которая подлежит покрытию, цельной, однородной покрывающей пленки.
Материал базового покрытия, предназначенный для применения в соответствии с изобретением, является водным (в отношении основного определения "водного", смотри выше).
Доля воды в материале базового покрытия предпочтительно составляет от 35 до 70 мас. %, и более предпочтительно 45-65 мас. %, в каждом случае, из расчета общей массы материала базового покрытия.
Даже более предпочтительным является, когда сумма процентов содержания твердых веществ материала базового покрытия и доли воды в материале базового покрытия составляет по меньшей мере 70 мас. %, предпочтительно по меньшей мере 75 мас. %. Среди указанных количественных параметров, предпочтение отдают диапазонам, составляющим 75-95 мас. %, в частности 80-90 мас. %.
В частности, это означает, что предпочтительные материалы базового покрытия, в принципе, включают только небольшие пропорции вредных в экологическом отношении компонентов, таких как, в частности, органические растворители, относительно содержания твердых веществ материала базового покрытия. Соотношение летучей органической фракции материала базового покрытия (в мас. %) к содержанию твердых веществ материала базового покрытия (по аналогии с указанным выше, в данном случае в мас. %) предпочтительно составляет от 0,05 до 0,7, более предпочтительно от 0,15 до 0,6. Летучую органическую фракцию для целей настоящего изобретения рассматривают как фракцию материала базового покрытия, которая не считается ни частью воды, ни частью содержания твердых веществ.
Другое преимущество материала базового покрытия состоит в том, что он может быть получен без применения неблагоприятных для окружающей среды и вредных для здоровья органических растворителей, таких как N-метил-2-пирролидон, диметилформамид, диоксан, тетрагидрофуран, и N-этил-2-пирролидон. Соответственно, материал базового покрытия предпочтительно содержит менее 10 мас. %, более предпочтительно менее 5 мас. %, еще более предпочтительно менее 2,5 мас. % органических растворителей, выбранных из группы, состоящей из N-метил-2-пирролидона, диметилформамида, диоксана, тетрагидрофурана, и N-этил-2-пирролидона. Материал базового покрытия предпочтительно является полностью свободным от указанных органических растворителей.
Соотношение доли одной или большего количества дисперсий (PD) к доли по меньшей мере одной дисперсии (wD), в каждом случае, из расчета общей массы водного материала базового покрытия в соответствии с изобретением, может корректироваться в соответствии с требованиями отдельного случая, и может варьироваться в пределах широкого диапазона. Следовательно, то же справедливо и в отношении соотношения между долями полимеров, поступающих из дисперсий (PD) и (wD) (которое в каждом случае определяется посредством содержания твердых веществ).
Материалы базовых покрытий могут быть получены посредством применения механизмов для смешивания и методов смешивания, которые являются принятыми и известными для изготовления материалов базовых покрытий.
Дополнительно, настоящее изобретение обеспечивает способ получения многослойной красочной системы, который включает получение по меньшей мере одной пленки базового покрытия, посредством применения по меньшей мере одного водного материала базового покрытия в соответствии с изобретением.
Все утверждения, приведенные выше в отношении материала базового покрытия в соответствии с изобретением, являются также действительными для способа в соответствии с изобретением. Это имеет место, в частности, не в последнюю очередь, для всех предпочтительных, более предпочтительных и очень предпочтительных признаков. Особенно предпочтительно, материал базового покрытия содержит пигмент и, следовательно, является пигментированным.
Соответственно, настоящее изобретение обеспечивает способ, в котором
(1) на основу наносится водный материал базового покрытия,
(2) из покрывающего материала, нанесенного на стадии (1), формируется полимерная пленка,
(3) на полученную пленку базового покрытия наносится материал покровного лака, и затем
(4) пленка базового покрытия затвердевает вместе с пленкой покровного лака,
где водный материал базового покрытия, применяемого на стадии (1), представляет собой материал базового покрытия в соответствии с изобретением.
Указанные способ предпочтительно применяют для получения многослойных цветных красочных систем, эффектных красочных систем, и цветных и эффектных красочных систем.
Водный материал базового покрытия, предназначенный для применения в соответствии с изобретением, обычно наносят на металлические или пластмассовые основы, которые предварительно обрабатывали грунтовкой или грунт-шпатлевкой. Необязательно, указанный материал базового покрытия также может непосредственно наноситься на пластмассовую основу.
В случае, если должна быть покрыта металлическая основа, то предпочтительно, перед нанесением грунтовки или грунт-шпатлевки, ее дополнительно покрывают системой электроосажденного покрытия.
В случае, если при этом покрывают пластмассовую основу, то предпочтительно ее дополнительно подвергают поверхностно-активирующей предварительной обработке перед нанесением грунтовки или грунт-шпатлевки. Способы, наиболее часто применяемые для такой предварительной обработки, представляют собой обработку пламенем, обработку плазмой, обработку коронным электрическим разрядом. При этом обработка пламенем применяется предпочтительном.
Нанесение водного материала базового покрытия в соответствии с изобретением на металлическую основу может осуществляться с толщинами пленок, которые являются обычными в автомобильной промышленности, и находятся, например, в диапазоне от 5 до 100 микрометров, предпочтительно от 5 до 60 микрометров. Указанное осуществляют с помощью использования способов нанесения посредством распыления, таких как, например, распыление с использованием сжатого воздуха, распыление без использования воздуха, нанесение с использованием высокоскоростного вращения, нанесение с помощью электростатического напыления (ESTA), отдельно или в сочетании с распылением с подогревом, например, посредством такого распыления как распыление с применением горячего воздуха.
После того, как водный материал базового покрытия был нанесен, его можно высушить посредством известных способов. Например, (однокомпонентные) материалы базовых покрытий, которые являются предпочтительными, могут подвергаться самоиспарению при комнатной температуре на протяжении 1-60 минут и впоследствии подвергаться сушке, предпочтительно необязательно при немного повышенных температурах, составляющих 30-90°С. В контексте настоящего изобретения, самоиспарение и сушка могут представлять собой испарение органических растворителей и/или воды, в результате чего покрытие становится более сухим, но все еще не затвердевшим или не сформировавшим полностью сшитую покрывающую пленку.
Затем наносится коммерчески доступный материал покровного лака, также посредством обычных способов, при этом толщина пленок, опять-таки, находится в пределах обычных диапазонов, составляющих, например, 5-100 микрометров. При этом двухкомпонентные материалы покровного лака являются предпочтительными.
После того, как материал покровного лака был нанесен, он может подвергаться самоиспарению при комнатной температуре на протяжении, например, 1-60 минут, и необязательно сушиться. Затем покровный лак затвердевает вместе с нанесенным базовым покрытием. В данном случае, например, происходят реакции сшивания, в результате чего на основе получают многослойную цветную и/или эффектную красочную систему в соответствии с изобретением. Предпочтительно, отверждение происходит термически, при температурах, составляющих 60-200°С.
Все толщины пленок, указанные в контексте настоящего изобретения, понимаются как толщины сухих пленок. Таким образом, в каждом случае, толщина пленки представляет собой толщину пленки затвердевшего покрытия. При этом, в случае, если указано, что покрывающий материал наносится с конкретной толщиной пленки, это означает, что покрывающий материал наносится таким образом, что указанная толщина пленки достигается после отверждения.
Пластмассовые основы, как правило, покрывают таким же способом, как и металлические основы. Однако в этом случае, отверждение, как правило, осуществляют при температурах, которые являются намного ниже, и составляют 30-90°С, таким образом, чтобы не вызвать повреждения и/или деформирования основы.
Следовательно, посредством способа в соответствии с изобретением возможно окрашивать металлические и неметаллические основы, в частности пластмассовые основы, предпочтительно кузова автомобилей или их части.
В одном особом варианте осуществления способа в соответствии с изобретением, осуществляют одну небольшую стадию отверждения, по сравнению со стандартной процедурой, как уже описано вначале. Указанное означает, в частности, что покрывающая система для совместного отверждения, содержащая одну или по меньшей мере две пленки базового покрытия, другими словами, в любом случае, первое базовое покрытие и второе базовое покрытие, а также покровный лак, формируется на основе, и затем совместно отверждается. По меньшей мере одно из базовых покрытий, которые применяют в указанной системе, представляет собой материал базового покрытия в соответствии с изобретением. Соответственно, в системе, содержащей по меньшей мере две пленки базового покрытия,, первое базовое покрытие или второе базовое покрытие могут быть материалом базового покрытия в соответствии с изобретением. В равной степени возможным и предпочтительным для целей настоящего изобретения, является то, что оба базовых покрытия могут быть материалами базовых покрытий в соответствии с изобретением.
Система, описанная здесь, формируется, например, на пластмассовой основе, которая, необязательно, была подвергнута поверхностно-активирующей предварительной обработке, или на металлической основе, обеспеченной затвердевшей системой электроосажденного покрытия.
Особенно предпочтительной в указанном случае является конструкция на металлических основах, обеспеченных затвердевшей пленкой электроосажденного покрытия. По этой причине, в указанном варианте осуществления, является важным, чтобы все составы покрытий, нанесенные на затвердевшую систему электроосажденного покрытия, отверждались совместно. Хотя, конечно, возможно отдельное самоиспарение и/или промежуточная сушка, ни одна из пленок не превращается в затвердевшее состояние отдельно.
Для целей настоящего изобретения, отверждение и затвердевшее состояние понимаются в соответствии с их общей интерпретацией специалистом в данной области. Соответственно, отверждение покрывающей пленки означает превращение такой пленки в готовое к применению состояние, другими словами, в состояние, в котором основа, обеспеченная указанной покрывающей пленкой, может перемещаться, храниться, и применяться в соответствии со своим предназначением. Соответственно, затвердевшая покрывающая пленка, в частности, больше не является мягкой или липкой, а вместо этого находится в надлежащем состоянии в виде твердой покрывающей пленки, которая больше не испытывает каких-либо существенных изменений своих свойств, таких как прочность или адгезия к основе, даже в случае, когда ее дополнительно подвергают условиям отверждения, как описано далее ниже.
Настоящее изобретение также обеспечивает способ ремонта многослойных красочных систем, в частности, полученных посредством способа, описанного выше.
Указанный способ, соответственно, представляет собой способ ремонта многослойной красочной системы, где на основе получают одну или, последовательно, по меньшей мере две пленки базового покрытия(-й), а после этого пленку покровного лака, где применяемая при этом основа представляет собой многослойную красочную систему, имеющую дефекты, и при этом все составы покрытий, нанесенные во время способа ремонта, отверждаются совместно. По меньшей мере один из материалов базовых покрытий, которые применяют, при этом представляет собой материал базового покрытия в соответствии с изобретением.
Как известно, при этом является обычным и возможным, в качестве части способа ремонта красочной системы, когда дефекты перед этим подлежат зачистке шлифовальной шкуркой. Также является обычным и возможным, когда способ ремонта применяется только для локального восстановления дефектов (точечный ремонт) или для полного ремонта многослойной красочной системы, имеющей дефекты (двойное покрытие).
Применение материалов базовых покрытий в соответствии с изобретением приводит к получению многослойных красочных систем, которые, кроме превосходных эстетических свойств, также обладают очень хорошими адгезионными свойствами. Это касается как сектора первоначального нанесения покрытий производителем, так и сектора ремонта покрытий.
Примеры
Описание методов
1. Содержание твердых веществ (твердых частиц, нелетучей фракции) Нелетучую фракцию определяют в соответствии со стандартом DIN EN ISO 3251 (дата: июнь 2008 г.). Указанное определение включает отвешивание 1 г образца в алюминиевые лоток, который высушивали перед этим, осуществляя его сушку в сушильном шкафу при температуре 125°С на протяжении 60 минут, охлаждали его в десикаторе, и затем повторно взвешивали. Остаток по отношению к общему применяемому количеству образца соответствует нелетучей фракции. Необязательно может определяться объем нелетучей фракции, если это необходимо, в соответствии со стандартом DIN 53219 (дата: август 2009 г.).
2. Толщина пленок
Толщину пленок определяют в соответствии со стандартом DIN EN ISO 2808 (дата: май 2007 г. ), способ 12А, посредством применения инструмента MiniTest® 3100-4100 от компании ElektroPhysik.
3. Определение стабильности при хранении
Для определения стабильности при хранении составов покрытий, их исследуют как до, так и после хранения при температуре 40°С на протяжении 2 недель, посредством применения ротационного вискозиметра в соответствии со стандартом DIN 53019-1 (дата: сентябрь 2008 г.), и калибруют в соответствии со стандартом DIN 53019-2 (дата: февраль 2001 г.), в условиях контролируемой температуры (23,0°С±0,2°С). Образцы подвергаются воздействию усилий сдвига, вначале на протяжении 5 минут со скоростью сдвига, составляющей 1000 с-1 (фаза нагрузки), и затем на протяжении 8 минут со скоростью сдвига, составляющей 1 с-1 (фаза разгрузки).
На основании данных измерений определяют средний уровень вязкости во время фазы нагрузки (вязкость при высокой скорости сдвига) и уровень после 8 минут фазы разгрузки (вязкость при низкой скорости сдвига), и значения до и после хранения сравнивают друг с другом, посредством вычисления соответствующих процентных изменений. Изменение значения самое большее на 15% считается приемлемым.
4. Оценка частоты встречаемости пузырей и растеканий
Для определения склонности в отношении появления пузырей и растекаемости, в соответствии со стандартом DIN EN ISO 28199-1 (дата: январь 2010 г.) и стандартом DIN EN ISO 28199-3 (дата: январь 2010 г.), многослойные красочные системы получали в соответствии со следующим общим протоколом:
Перфорированную стальную панель, покрытую затвердевшим катодным электроосажденным покрытием (КЭП) (CathoGuard® 800 от компании BASF Coatings GmbH), с размерами 57 см × 20 см (в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.1, версия А), получали по аналогии со стандартом DIN EN ISO 28199-1, раздел 8.2 (версия А). Впоследствии, в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.3, водный материал базового покрытия наносили элетростатически в одно нанесение, в виде клина, с заданной толщиной пленки (толщина пленки высушенного материала) в диапазоне от 0 мкм до 40 мкм. После истечения времени самоиспарения при температуре 18-23°С, составляющего 10 минут (испытание на растекаемость) или без предварительного самоиспарения (испытание на появление пузырей), полученная пленка базового покрытия подвергается промежуточной сушке в камере с принудительной подачей воздуха при температуре 80°С на протяжении 5 минут. В случае испытания в отношении растеканий, панели подвергают самоиспарению и подвергают промежуточной сушке в вертикальном положении.
Определение предела появления пузырей, т.е., толщины пленки базового покрытия, начиная из которой возникают пузыри, осуществляют в соответствии с DIN EN ISO 28199-3, раздел 5.
Определение склонности к растеканиям осуществляют в соответствии с DIN EN ISO 28199-3, раздел 4. В дополнение к толщине пленки, при которой растекание превышает расстояние, составляющее 10 мм, от нижнего края перфорации, также определяют толщину пленки, выше которой начальная склонность к растеканию может наблюдаться визуально на перфорации.
5. Нанесение клиновидных конструкций водного материала базового покрытия
Для оценки частоты встречаемости пор, а также растекания в зависимости от толщины пленки, получают многослойные красочные системы в формате клина в соответствии со следующими общими протоколами:
Вариант А: Первый водный материал базового покрытия в виде клина, второй водный материал базового покрытия в виде непрерывного покрытия
Стальную панель с размерами 30×50 см, покрытую затвердевшим стандартным КЭП (CathoGuard® 800 от компании BASF Coatings), обеспечивают двумя липкими полосками (липкая лента Tesaband, 19 мм) на одном продольном крае, для того чтобы можно было определить разницу в толщине пленки после покрытия.
Первый водный материал базового покрытия наносят электростатически в виде клина с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 0-30 мкм. После самоиспарения при комнатной температуре на протяжении 3 минут, одну из двух липких полосок удаляют, и затем наносят второй водный материал базового покрытия, также электростатически в одно нанесение. Заданная толщина пленки (толщина пленки высушенного материала) составляет 13-16 мкм. После дополнительного времени самоиспарения, составляющего 4 минуты, при комнатной температуре, систему подвергают промежуточной сушке в камере с принудительной подачей воздуха при температуре 60°С на протяжении 10 минут.
После удаления второй липкой полоски, коммерчески доступный двухкомпонентный материал покровного лака (ProGloss® от компании BASF Coatings GmbH) наносят посредством ручного распылителя с подачей самотеком на подвергшуюся промежуточной сушке систему, с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 40-45 мкм. Полученную пленку покровного лака подвергают самоиспарению при комнатной температуре (18-23°С) на протяжении 10 минут; впоследствии, в камере с принудительной подачей воздуха при 140°С на протяжении дополнительных 20 минут происходит отверждение.
Вариант Б: Первый водный материал базового покрытия в виде непрерывного покрытия, второй водный материал базового покрытия в виде клина
Стальную панель с размерами 30×50 см, покрытую затвердевшим стандартным КЭП (CathoGuard® 800 от компании BASF Coatings), обеспечивают двумя липкими полосками (липкая лента Tesaband, 19 мм) на одном продольном крае, для того чтобы можно было определить разницу в толщине пленки после покрытия.
Первый водный материал базового покрытия наносят электростатически с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 18-22 мкм. После самоиспарения при комнатной температуре на протяжении 3 минут, одну из двух липких полосок удаляют, и затем второй водный материал базового покрытия также наносят электростатически в одно нанесение, в виде клина. Заданная толщина пленки (толщина пленки высушенного материала) составляет 0-30 мкм. После дополнительного времени самоиспарения, составляющего 4 минуты при комнатной температуре, систему подвергают промежуточной сушке в камере с принудительной подачей воздуха при температуре 60°С на протяжении 10 минут.
После удаления второй липкой полоски, коммерчески доступный двухкомпонентный материал покровного лака (ProGloss® от компании BASF Coatings GmbH) наносят посредством ручного распылителя с подачей самотеком, на подвергшуюся промежуточной сушке систему, с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 40-45 мкм. Полученную пленку покровного лака подвергают самоиспарению при комнатной температуре (18-23°С) на протяжении 10 минут; впоследствии, в камере с принудительной подачей воздуха при 140°С на протяжении дополнительных 20 минут происходит отверждение.
Вариант В: один водный материал базового покрытия в виде клина
Стальную панель с размерами 30×50 см, покрытую затвердевшим стандартным КЭП (CathoGuard® 800 от компании BASF Coatings), обеспечивают двумя липкими полосками (липкая лента Tesaband, 19 мм) на одном продольном крае, для того чтобы можно было определить разницу в толщине пленки после покрытия.
Водный материал базового покрытия наносят электростатически в виде клина с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 0-30 мкм. После времени самоиспарения, составляющего 4 минуты, при комнатной температуре, систему подвергают промежуточной сушке в камере с принудительной подачей воздуха при температуре 80°С на протяжении 10 минут.
После удаления липкой полоски, коммерчески доступный двухкомпонентный материал покровного лака (ProGloss® от компании BASF Coatings GmbH) наносят посредством ручного распылителя с подачей самотеком, на подвергшуюся промежуточной сушке пленку водного базового покрытия, с заданной толщиной пленки (толщина пленки высушенного материала), составляющей 40-45 мкм. Полученную пленку покровного лака подвергают самоиспарению при комнатной температуре (18-23°С) на протяжении 10 минут; впоследствии, в камере с принудительной подачей воздуха при 140°С на протяжении дополнительных 20 минут происходит отверждение.
6. Оценка частоты встречаемости пор
Для оценки частоты встречаемости пор, многослойные красочные системы получают в соответствии со способами нанесения клиновидных систем водного базового покрытия (вариант А и Б, соответственно), и затем оценивают визуально в соответствии со следующим общим протоколом:
Измеряют толщину сухой пленки всей системы водного материала базового покрытия, состоящей из первого и второго водных материалов базовых покрытий и, для клина толщины пленки базового покрытия, участок 0-20 мкм и участок от 20 мкм до конца клина отмечают на стальной панели.
Поры оценивают визуально на двух отдельных участках клина водного базового покрытия. Количество пор на участок подсчитывают. Все результаты стандартизируют для площади, составляющей 200 см2. Кроме того, необязательно, фиксируют толщину сухой пленки клина водного материала базового покрытия, начиная из которой, поры больше не возникают.
7. Оценка адгезионных свойств после конденсации
Для оценки устойчивости конденсации, исследуемые образцы хранили в камере кондиционирования в условиях проведения испытания СН в соответствии со стандартом DIN EN ISO 6270-2:2005-09 на протяжении периода, составляющего 10 дней. Затем соответствующие металлические панели оценивали визуально, как через один час, так и через 24 часа после удаления из камеры кондиционирования, в отношении образования пузырей, а также в отношении адгезионных свойств.
Частоту встречаемости пузырей оценивали следующим образом, посредством комбинации двух значений:
- Количество пузырей оценивали посредством показателя количества от 1 до 5, где m1 при этом обозначает незначительное количество пузырей, и m5 обозначает очень много пузырей.
- Размер пузырей оценивали посредством показателя размера, опять от 1 до 5, где g1 при этом обозначает очень маленькие пузыри, и g5 обозначает очень большие пузыри.
Обозначение m0g0, соответственно, означает не содержащие пузырей покрытия после подвержения конденсации при хранении, и представляет отличный результат с точки зрения образования пузырей.
Адгезию в отношении ударов мелких камней после подвержения конденсации исследовали в соответствии со стандартом DIN EN ISO 20567-1, способ В. Полученный характер повреждения также оценивали в соответствии со стандартом DIN EN ISO 20567-1.
Дополнительно, в соответствии со стандартом DIN 55662, способ В, проводили испытания струей пара. Царапины (с перекрестной накаткой) выполняли с использованием игл Сиккенса (смотри DIN EN ISO 17872 Приложение А). Оценку результатов испытания струей пара выполняли в соответствии со стандартом DIN 55662, и в частности устанавливали максимальную ширину отслоений в миллиметрах.
Более того, испытания струей пара в соответствии со стандартом DIN 55662, способ В (перекрестную накатку выполняли с использованием игл Сиккенса в соответствии со стандартом DIN EN ISO 17872 Приложение А) осуществляли на основах, которые предварительно подвергались испытанию в отношении ударов мелких камней в соответствии со стандартом DIN EN ISO 20567-1, способ В. Для визуальной оценки характера повреждения, использовали следующую шкалу:
KW0 = отсутствуют изменения на образце
KW1 = небольшая потеря цвета имеющегося повреждения
KW2 = хорошо видимая потеря цвета имеющегося повреждения в покрывающей пленке
KW3 = полное отслоение покрывающей пленки на участке струи пара
KW4 = полное отслоение покрывающей пленки за пределами участке струи пара
KW5 = отслоение всей покрывающей пленки от основы
8. Содержание изоцианатных групп
Содержание изоцианатных групп, также указанное ниже как содержание NCO, определяют посредством добавления избытка 2%-го раствора N,N-дибутиламина в ксилоле к гомогенному раствору образцов в ацетоне/N-этилпирролидоне (1:1 об. %), а также посредством потенциометрического обратного титрования избытка амина 0,1N соляной кислоты в соответствии со стандартом DIN EN ISO 3251, DIN EN ISO 11909, и стандартом DIN EN ISO 14896. На основании доли полимера (твердых веществ) в растворе возможно вычислить обратно содержание NCO полимера, исходя из содержания твердых веществ.
9. Гидроксильное число
Гидроксильное число определяли в соответствии с методом R.-P.

10. Кислотное число
Кислотное число определяли посредством метода в соответствии со стандартом DIN EN ISO 2114 в гомогенном растворе тетрагидрофурана (ТГФ)/воды (9 частей по объему ТГФ и 1 часть по объему дистиллированной воды), посредством применения этанольного раствора гидроксида калия.
11. Эквивалентная масса амина
Эквивалентную массу амина (раствор) применяют для определения содержания амина в растворе, и ее устанавливали следующим образом. Анализируемый образец растворяли в безводной уксусной кислоте при комнатной температуре, и титровали с 0,1N перхлорной кислоты в безводной уксусной кислоте в присутствии кристаллического фиолетового. На основании исходной массы образца и расхода перхлорной кислоты определяют эквивалентную массу амина (раствор), массу раствора основного амина, которая необходима для нейтрализации одного моля хлорной кислоты.
12. Степень маскирования первичных аминогрупп
Степень маскирования первичных аминогрупп определяли посредством ИК спектрометрии, применяя спектрометр Nexus FT-IR (от компании Nicolet) и детектор ИК излучения (d=25 мм, окно из KBr), при максимуме поглощения при 3310 см-1), на основании ряда концентраций применяемых аминов, со станлартизацией до максимума поглощения при 1166 см-1 (внутренний эталон) при температуре 25°С.
13. Содержание растворителя
Количество органического растворителя в смеси, такой как водная дисперсия, например, определяли посредством газовой хроматографии (Agilent 7890А, 50 м кварцевая капиллярная колонка с полиэтиленгликолевой фазой, или 50 м кварцевая капиллярная колонка с полидиметилсилоксановой фазой, газ-носитель гелий, устройство для ввода проб с делением потока 250°С, температура термостата 40-220°С, плазменно-ионизационный детектор, температура детектора 275°С, внутренний эталон н-пропилгликоль).
14. Среднечисленная молекулярная масса
Среднечисленную молярную массу (Mn) определяли, если не указано иное, посредством применения парофазного осмометра 10.00 (от компании Knauer) на основании ряда концентраций в толуоле при 50°С с бензофеноном в качестве калибровочного вещества для определения экспериментальной калибровочной постоянной инструмента, в соответствии с Е.


15. Средний размер частиц в дисперсии (PD)
Средний размер частиц (объемный средний) полиуретан-полимочевинных частиц, которые присутствуют в дисперсии (PD), предназначенной для применения в соответствии с изобретением, для целей настоящего изобретения определяют посредством фотонной корреляционной спектроскопии (ФКС) в соответствии со стандартом DIN ISO 13321.
В частности, для измерений применяли Malvern Nano S90 (от компании Malvern Instruments) при температуре 25±1°С. Инструмент охватывает диапазон размеров от 3 до 3000 нм, и был оборудован 4 мВт He-Ne лазером с длиной волны 633 нм. Дисперсии (PD) разбавляли не содержащей частиц деионизированной водой в качестве дисперсионной среды, до такой степени, чтобы впоследствии их можно было измерить в 1 мл полистирольной кювете с соответствующей интенсивностью рассеянного излучения. Оценку осуществляли, применяя цифровой коррелятор, с помощью программного обеспечения для оценки Zetasizer, версия 7.11 (от компании Malvern Instruments). Измерение проводили пять раз, а потом измерения повторяли на втором, только что полученном образце. Среднее квадратическое отклонение 5-кратных определений составляло ≤4%. Максимальное отклонение арифметического среднего объемного среднего (Об.-среднего) пяти отдельных измерений составляло ±15%. Описанный средний размер частиц (объемный средний) представляет собой арифметическое среднее среднего размера частиц (объемный средний) отдельных составов. Исследование осуществляли посредством применения полистирольных образцов, имеющих подтвержденные размеры частиц, находящиеся в диапазоне между 50-3000 нм.
Очевидно, измерение и метод оценки, описанные здесь, также применяли для определения размера частиц полимера, который присутствует в водной дисперсии (wD).
16. Гель-фракция
Для целей настоящего изобретения, гель-фракцию определяли гравиметрически. В этом случае, прежде всего, полимер, который присутствует в образце, в частности, в водной дисперсии (PD) (начальная масса 1,0 г), выделяли посредством сушки вымораживанием. Вслед за определением температуры отверждения, температура выше которой больше не происходит каких-либо изменений электрического сопротивления образца, когда температура понижается далее, полностью замороженный образец подвергался основной сушке, обычно в диапазоне давлений вакуумной сушки, находящемся между 5 мбар и 0,05 мбар, при температуре сушка, которая на 10°С ниже температуры отверждения. В ходе постепенного повышения температуры нагретых поверхностей размещения до 25°С, полимер быстро сушили вымораживанием, и после времени сушки, обычно составляющего 12 часов, количество выделенного полимера (твердая фракция, которую определяли с помощью сушки вымораживанием) было постоянным, и не испытывало какого-либо дополнительного изменения даже после продолжительной сушки вымораживанием. После сушки при температуре поверхности размещения, составляющей 30°С, в условиях максимально пониженного окружающего давления (обычно в диапазоне между 0,05 и 0,03 мбар) достигается оптимальная сушка полимера.
Выделенный полимер впоследствии спекали в камере с принудительной подачей воздуха при температуре 130°С на протяжении одной минуты, и после этого экстрагировали в избытке тетрагидрофурана (соотношение тетрагидрофурана к твердой фракции = 300:1) при температуре 25°С на протяжении 24 часов. Затем нерастворимую фракцию выделенного полимера (гель-фракцию) отделяли на подходящем фильтре, сушили в камере с принудительной подачей воздуха при температуре 50°С на протяжении 4 часов, и затем взвешивали снова.
Далее убеждались, что при температуре спекания, составляющей 130°С, с изменением времени спекания от одной минуты до 20 минут, гель-фракция, выявленная для частиц микрогеля, является независимой от времени спекания. По этой причине, указанное исключает какое-либо дополнительное увеличение гель-фракции во время реакций сшивания, которые следуют после выделения полимерного твердого вещества.
Гелевую фракцию, определенную таким образом в соответствии с изобретением, также называют гель-фракцией (высушенной вымораживанием), и также могут представлять в мас. %. Причина, очевидно, заключается в том, что указанная фракция представляет собой массовую долю полимерных частиц, которые подверглись сшиванию, как описано вначале в отношении дисперсии (PD), и которая, следовательно, может выделяться в виде геля.
Параллельно, гелевую фракцию, также указанную ниже как гель-фракция (130°С), определяли гравиметрически посредством выделения полимерного образца из водной дисперсии (начальная масса 1,0 г) при температуре 130°С на протяжении 60 минут (содержание твердых веществ). Массу полимера определяли перед этим, в соответствии с процедурой, аналогичной описанной выше, полимер экстрагировали в избытке тетрагидрофурана при температуре 25°С на протяжении 24 часов, и нерастворимую фракцию (гель-фракцию) выделяли, сушили, и повторно взвешивали.
17. Растворимость в воде
Растворимость органического растворителя в воде определяли при температуре 20°С следующим образом. Указанный органический растворитель и воду объединяли в подходящем стеклянном сосуде и перемешивали, и затем смесь выдерживали. Количество воды и растворителя в данном случае выбирали таким образом, чтобы уравновешивание в результате указанного выдерживания приводило к получению двух отдельных фаз. После уравновешивания, для взятия образца водной фазы (то есть, фазы, содержащей скорее воду, чем органический растворитель) применяли шприц, и указанный образец разбавляли в соотношении 1/10 тетрагидрофураном, и посредством газовой хроматографии определяли долю растворителя (в отношении условий смотри раздел 8. Содержание растворителя).
Если независимо от количества воды и растворителя, две фазы не образуются, то растворитель является смешиваемым с водой в любом массовом соотношении. Поэтому указанный бесконечно растворимый в воде растворитель (например, ацетон) в любом случае не является растворителем (Z.2). Изготовление водных дисперсий (wD) и (PD) Дисперсии (wD)
Протокол изготовления, описанный ниже, относится к Таблице А. Мономерная смесь (А), стадия I.
80 мас. % материалов 1 и 2 из Таблицы А вводили в стальной химический реактор (объем 5 л) с обратным холодильником и нагревали до 80°С.Оставшиеся доли компонентов, перечисленные в "Исходной загрузке" в Таблице А, предварительно смешивают в отдельном сосуде. Указанную смесь и, отдельно от нее, раствор инициатора (Таблица А, материалы 5 и 6) капля по капле добавляют одновременно в химический реактор на протяжении 20 минут, при этом доля мономеров в реакционном растворе, из расчета общего количества мономеров, которые применяют на стадии I., не превышает 6,0 мас. % на протяжении всего времени реакции. Впоследствии, осуществляют перемешивание на протяжении 30 минут.
Мономерная смесь (Б), стадия II.
Компоненты, указанные в строке "Моно 1" в Таблице А, предварительно смешивают в отдельном сосуде. Указанную смесь добавляют капля по капле в химический реактор на протяжении 2 часов, при этом доля мономеров в реакционном растворе, из расчета общего количества мономеров, которые применяют на стадии II., не превышает 6,0 мас. % на протяжении всего времени реакции. Впоследствии, осуществляют перемешивание на протяжении 1 часа. Мономерная смесь (В), стадия III.
Компоненты, указанные в строке "Моно 2" в Таблице А, предварительно смешивают в отдельном сосуде. Указанную смесь добавляют капля по капле в химический реактор на протяжении 1 часа, при этом доля мономеров в реакционном растворе, из расчета общего количества мономеров, которые применяют на стадии III,. не превышает 6,0 мас. % на протяжении всего времени реакции. Впоследствии, осуществляют перемешивание на протяжении 2 часов.
После этого реакционную смесь охлаждают до 60°С, и нейтрализующую смесь (Таблица А, материалы 20, 21, и 22) предварительно смешивают в отдельном сосуде. Нейтрализующую смесь добавляют капля по капле в химический реактор на протяжении 40 минут, во время которого значение рН реакционного раствора доводят до значения 7,5-8,5. Продукт реакции впоследствии перемешивали на протяжении дополнительных 30 минут, охлаждали до 25°С, и фильтровали.
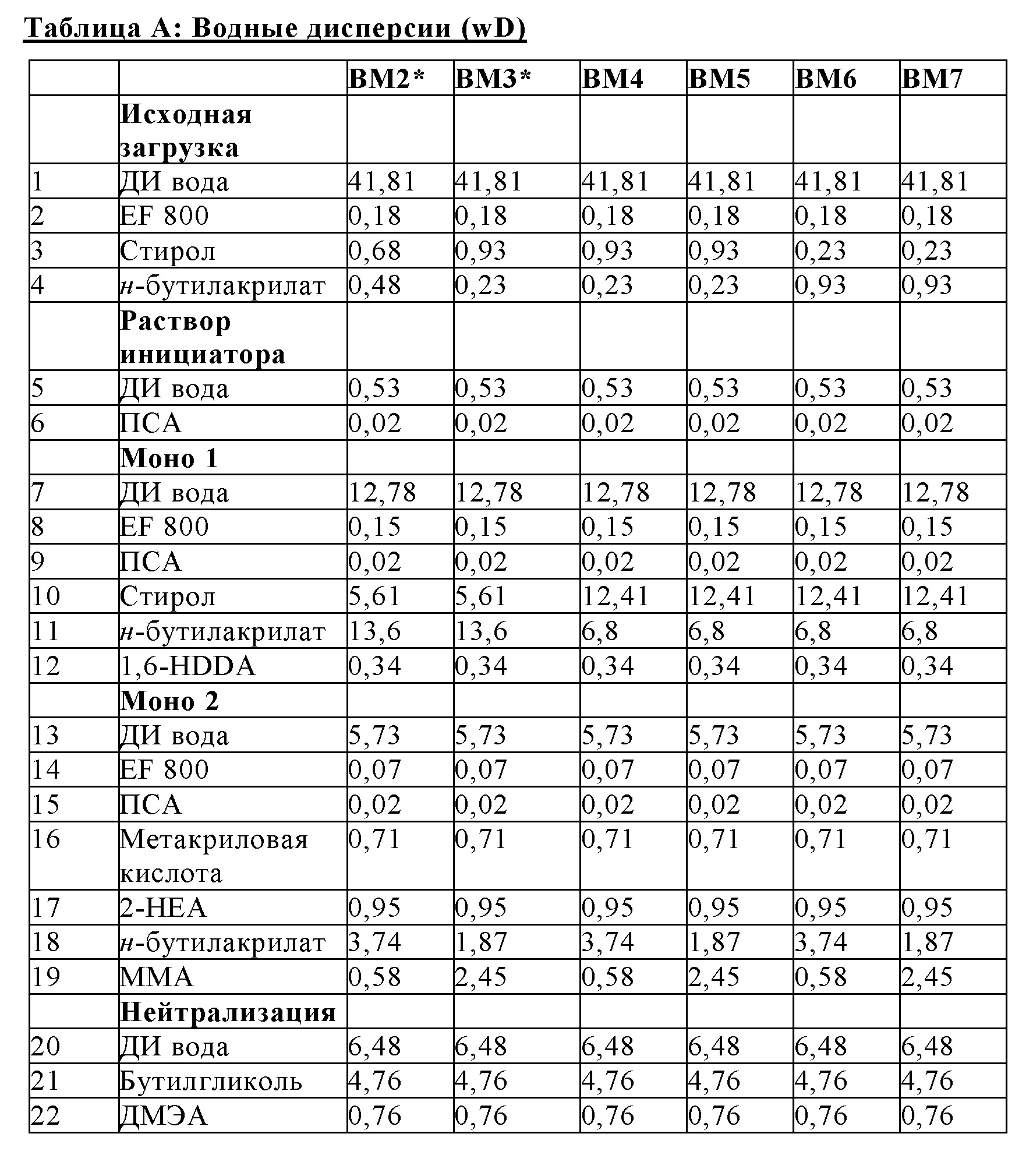

Содержание твердых веществ определяли для того, чтобы отслеживать реакцию. Результаты указаны в Таблице Б:

После каждой стадии и после окончательной нейтрализации, определяли размер частиц. Результаты представлены в Таблице В.

Каждая из указанных мономерных смесей (А), (Б), и (В) (соответствующие "Исходной загрузке", "Моно 1", и "Моно 2") полимеризовали отдельно, и затем определяли соответствующую температуру стеклования полученного полимера. Дополнительно, температуру стеклования определяли для всего полимера после нейтрализации.
Результаты указаны в Таблице Г.
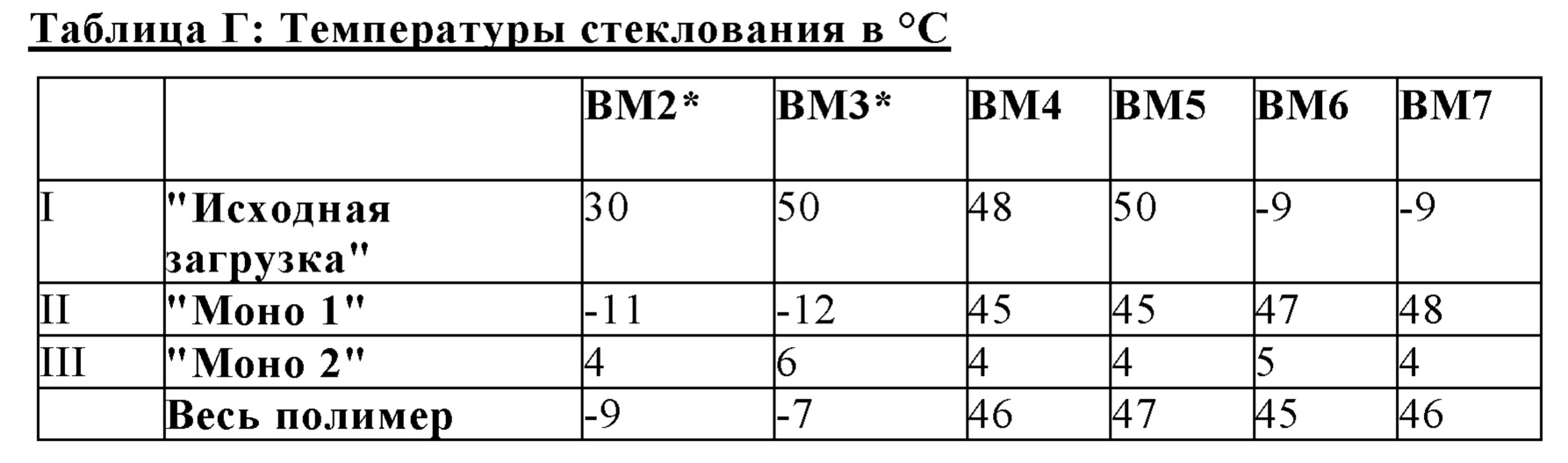

Дисперсия (PD)
Дисперсию (PD1) получали следующим образом. а) Получение частично нейтрализованного раствора преполимера
Для растворения 559,7 частей по массе неразветвленного сложного полиэфирполиола и 27,2 частей по массе диметилолпропионовой кислоты (от компании GEO Speciality Chemicals) в 344,5 частях по массе метилэтилкетона в атмосфере азота применяли реакционный сосуд, обеспеченный мешалкой, внутренним термометром, обратным холодильником, и электрический нагрев. Неразветвленный сложный полиэфирдиол получали перед этим из димеризованной жирной кислоты (Pripol 1012, от компании Croda), изофталевой кислоты (от компании BP Chemicals), и гексан-1,6-диола (от компании BASF SE) (соотношение масс исходных материалов: димерной жирной кислоты к изофталевой кислоте к гексан-1,6-диолу = 54,00:30,02:15,98), и при этом он имел гидроксильное число, которое составляло 73 мг KOH/г, исходя из содержания фракции твердых веществ, кислотное число, которое составляло 3,5 мг KOH/г, исходя из содержания фракции твердых веществ, и вычисленную среднечисленную молярную массу, составляющую 1379 г/моль, и среднечисленную молярную массу, как было определено посредством осмометрии с использованием давления паров, которая составляла 1350 г/моль.
Полученный раствор смешивали при температуре 30°С последовательно с 213,2 частями по массе дициклогексилметан-4,4'-диизоцианата (Desmodur® W, от компании Bayer Material Science), который имел содержание изоцианатных групп, составляющее 32,0 мас. %, и с 3,8 частями по массе дилаурата дибутилолова (от компании Merck). За этим следовал нагрев до 80°С с перемешиванием. Перемешивание продолжали при этой температуре до тех пор, пока содержание изоцианатных групп раствора не стало постоянным, и составляло 1,49 мас. %. После этого 626,2 частей по массе метилэтилкетона добавляли к преполимеру, и реакционную смесь охлаждали до 40°С. Когда достигали температуры 40°С, капля по капле на протяжении двух минут добавляли 11,8 частей по массе триэтиламина (от компании BASF SE), и смесь был перемешивали на протяжении дополнительных 5 минут.
б) Реакция преполимера с диэтилентриаминдикетимином
Впоследствии 30,2 частей по массе 71,9 мас. % раствора диэтилентриаминдикетимина в метилизобутилкетоне (соотношение изоцианатных групп преполимера к диэтилентриаминдикетимину (имеющему одну вторичную аминогруппу) : 5:1 моль/моль, соответствующее двум NCO группам на блокированную первичную аминогруппу) смешивали на протяжении минуты, во время которого температура реакции в течении короткого времени повышалась на 1°С после добавления раствора преполимера. Раствор диэтилентриаминдикетимина в метилизобутилкетон получали перед этим посредством азотропного удаления воды реакции во время реакции диэтилтриамина (от компании BASF SE) с метилизобутилкетоном в метилизобутилкетоне при температуре 110-140°С. Посредством разбавления метилизобутилкетоном, раствор доводили до эквивалентной массы амина, составляющей 124,0 г/экв. ИК-спектроскопия, применяя остаточное поглощение при 3310 см-1, выявила 98,5% блокирования первичных аминогрупп.
Содержание твердых веществ раствора полимера, содержащего изоцианатные группы, было установлено как составляющее 45,3%.
в) Диспергирование и вакуумная дистилляция
После 30 минут перемешивания при температуре 40°С, содержимое химического реактора диспергировали на протяжении 7 минут в 1206 частей по массе деионизированной воды (23°С). Метилэтилкетон дистиллировали из полученной дисперсии в условиях пониженного давление при температуре 45°С, и любые потери растворителя и воды были компенсированы, посредством применения деионизированной воды, в результате чего получали содержание твердых веществ, составляющее 40 мас. %.
В результате получали белую, стабильную дисперсию низкой вязкости, с высоким содержанием твердых веществ, со сшитыми частицами, которые вообще не демонстрировали осаждения даже после истечения 3 месяцев.
Полученная микрогелевая дисперсия (PD1) имела следующие характеристики:

Изготовление материалов водных базовых покрытий
Следующее должно быть принято во внимание в отношении компонентов состава и их количеств, указанных в Таблицах, приведенных ниже. В случае, когда ссылаются на коммерчески доступный продукт или на протокол изготовления, описанный где-либо, то ссылка, независимо от основного обозначения, выбранного для этого составляющего компонента, относится именно к указанному коммерчески доступному продукту или именно к продукту, полученному в соответствии с указанным протоколом.
Соответственно, если компонент состава имеет основное обозначение "меламин-формальдегидная смола", и если для этого составляющего компонента указан коммерчески доступный продукт, то меламин-формальдегидную смолу применяют в виде именно указанного коммерчески доступного продукта. По этой причине, если нужно сделать выводы о количестве активного вещества (меламин-формальдегидной смолы), то необходимо принимать во внимание любые дополнительные составляющие компоненты, присутствующие в коммерчески доступном продукте, например, такие как растворители.
Соответственно, если ссылаются на протокол изготовления компонента состава и, если в результате такого изготовления, например, получают полимерную дисперсию, имеющую определенное содержание твердых веществ, то именно указанную дисперсию применяют. Ключевым фактором является не то, представляет ли собой основное обозначение, которое было выбрано, термин "полимерная дисперсия" или просто активное вещество, например, "полимер", "сложный полиэфир", или "полиуретан-модифицированный полиакрилат". Указанное необходимо принимать во внимание, если нужно сделать выводы в отношении количества активного вещества (полимера).
Все пропорции, указанные в Таблицах, представляют собой части по массе. Пигментные пасты: Изготовление белой пасты Р1
Белую пасту изготавливают из 50 частей по массе титана рутильной формы 2310, 6 частей по массе сложного полиэфира, полученного в соответствии с примером D, колонка 16, строки 37-59 публикации DE 4009858 А1, 24,7 частей по массе дисперсии связующего вещества, полученной в соответствии с заявкой на получение патента ЕР 0228003 В2, страница 8, строки 6-18, 10,5 частей по массе деионизированной воды, 4 частей по массе 2,4,7,9-тетраметил-5-дециндиола, 52% в БГ (доступный от компании BASF SE), 4,1 частей по массе бутилгликоля, 0,4 частей по массе 10%-й концентрации диметилэтаноламина в воде, и 0,3 частей по массе Acrysol RM-8 (доступный от компании The Dow Chemical Company). Изготовление черной пасты Р2
Черную пасту изготавливают из 57 частей по массе полиуретановой дисперсии, полученной в соответствии с WO 92/15405, страница 13, строка 13, - страница 15, строка 13, 10 частей по массе углеродной сажи (углеродная сажа Monarch® 1400 от компании Cabot Corporation), 5 частей по массе сложного полиэфира, полученного в соответствии с примером D, колонка 16, строки 37-59 публикации DE 4009858 А1, 6,5 частей по массе 10%-й концентрации водного раствора диметилэтаноламина, 2,5 частей по массе коммерчески доступного простого полиэфира (Pluriol® Р900, доступного от компании BASF SE), 7 частей по массе бутилдигликоля, и 12 частей по массе деионизированной воды.
Изготовление пасты на основе талька Р3
Пасту на основе талька изготавливают из 49,7 частей по массе водной дисперсии связующего вещества, полученной в соответствии с WO 91/15528, страница 23, строка 26, - страница 25, строка 24, 28,9 частей по массе стеарита (Microtalc IT extra от компании Mondo Minerals B.V.), 0,4 частей по массе Agitan 282 (доступный от компании Miinzing Chemie GmbH), 1,45 частей по массе Disperbyk®-184 (доступный от компании BYK-Chemie GmbH), 3,1 частей по массе коммерчески доступного простого полиэфира (Pluriol® Р900, доступный от компании BASF SE), и 16,45 частей по массе деионизированной воды.
Изготовление пасты на основе сульфата бария Р4
Пасту на основе сульфата бария получали из 39 частей по массе полиуретановой дисперсии, полученной в соответствии с ЕР 0228003 В2, страница 8, строки 6 - 18, 54 частей по массе сульфата бария (Blanc fixe micro от компании Sachtleben Chemie GmbH), 3,7 частей по массе бутилгликоля, и 0,3 частей по массе Agitan 282 (доступный от компании Miinzing Chemie GmbH) и 3 частей по массе деионизированной воды.
Изготовление суспензии алюминиевого пигмента S1
Суспензию алюминиевого пигмента получали посредством использования перемешивающего элемента для смешивания 50 частей по массе бутилгликоля, а также 35 частей по массе коммерчески доступного эффектного пигмента Alu Stapa IL Hydrolan 2192 №5 и 15 частей по массе коммерчески доступного эффектного пигмента Alu Stapa IL Hydrolan 2197 №5 (каждый доступен от компании Altana-Eckart).
Изготовление базовых покрытий в соответствии с изобретением WBM серый А1 и WBM серый А2
Компоненты, перечисленные в Таблице 1.1, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством использования деионизированной воды и диметилэтаноламина доводили до значения рН, составляющего 8, и до вязкости распыления, составляющей 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.

Изготовление сравнительных базовых покрытий WBM серый A3 и WBM серый А4
Компоненты, перечисленные в Таблице 1.2, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством применения деионизированной воды и диметилэтаноламина доводили до значения рН, которое составляло 8, и до вязкости распыления, которая составляла 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.
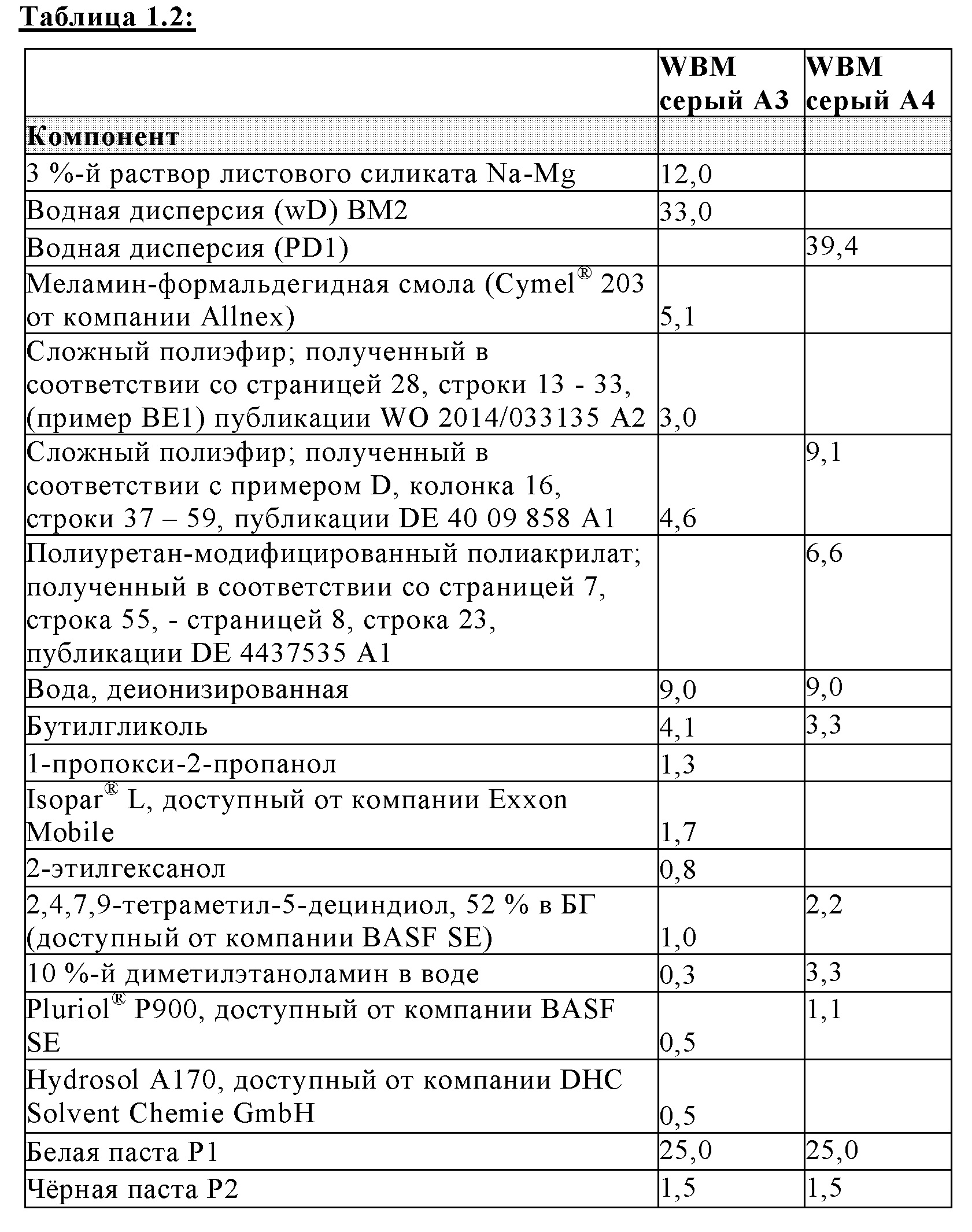
Изготовление базовых покрытий в соответствии с изобретением WBM серебристый В1 и WBM серебристый В2
Компоненты, перечисленные в Таблице 1.3, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством применения деионизированной воды и диметилэтаноламина доводили до значения рН, которое составляло 8, и до вязкости распыления, которая составляла 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.
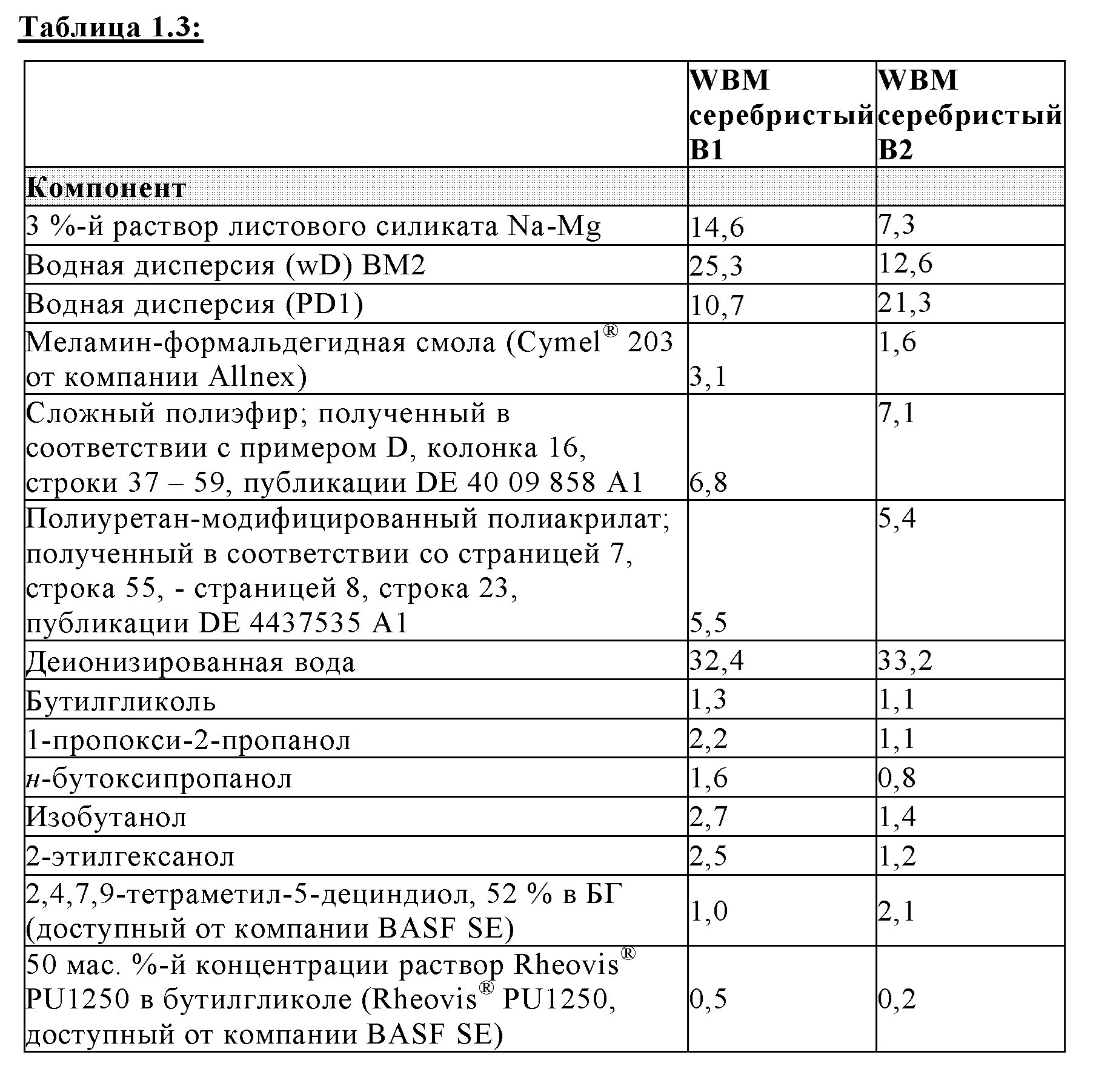

Изготовление сравнительных базовых покрытий WBM серебристый В3 и WBM серебристый В4
Компоненты, перечисленные в Таблице 1.4, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством применения деионизированной воды и диметилэтаноламина доводили до значения рН, которое составляло 8, и до вязкости распыления, которая составляла 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.


Изготовление базовых покрытий в соответствии с изобретением WBM черный В5 - WBM черный В8
Компоненты, перечисленные в Таблице 1.5, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством применения деионизированной воды и диметилэтаноламина доводили до значения рН, которое составляло 8, и до вязкости распыления, которая составляла 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.
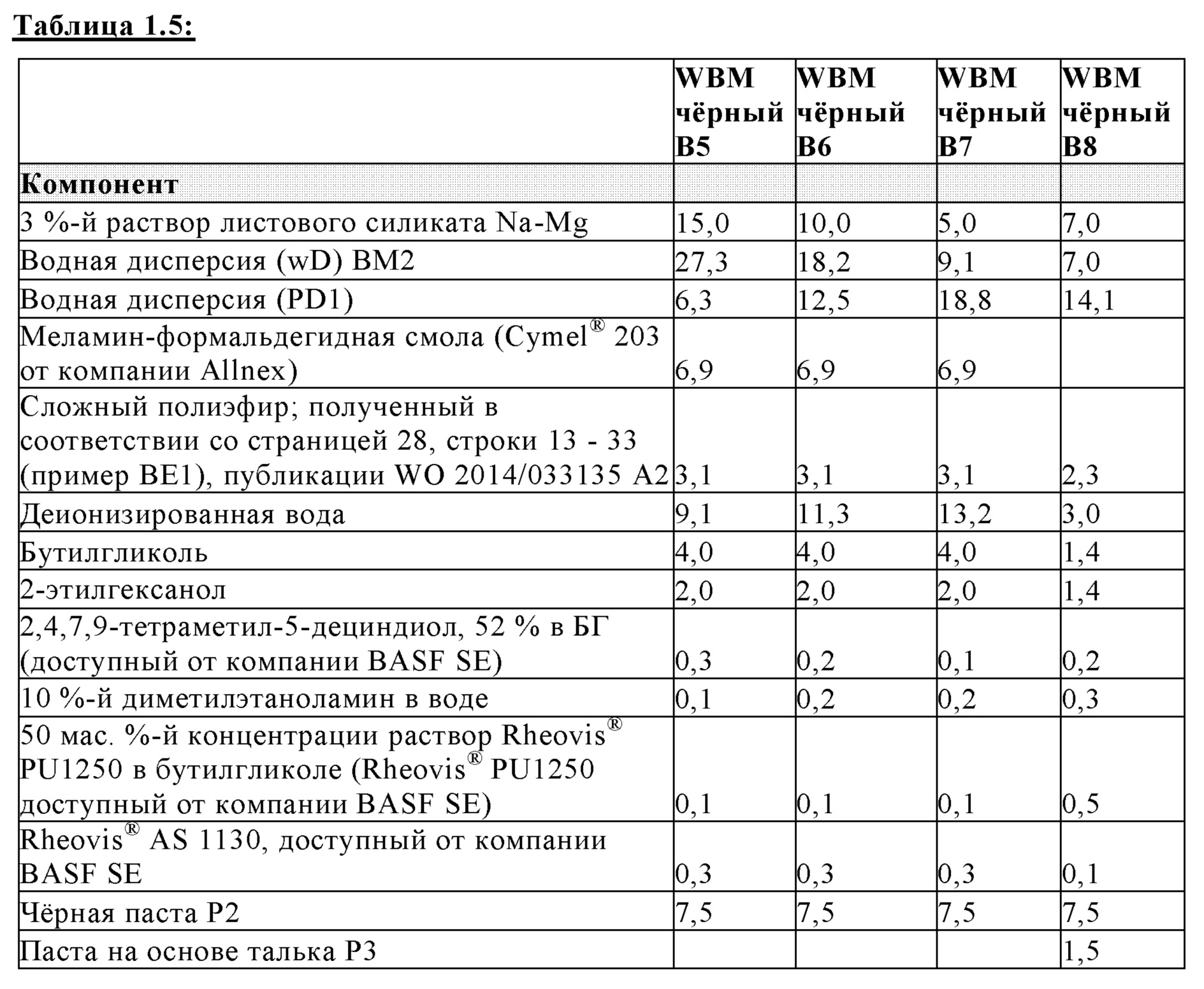
Изготовление базовых покрытий в соответствии с изобретением WBM черный В9 и WBM черный В10
Компоненты, перечисленные в Таблице 1.6, объединяли вместе с перемешиванием в указанном порядке для получения водной смеси. Указанную смесь затем перемешивали на протяжении 10 минут, и посредством применения деионизированной воды и диметилэтаноламина доводили до значения рН, которое составляло 8, и до вязкости распыления, которая составляла 90±10 мПас, в условиях применения усилий сдвига, составляющих 1000 с-1, как было установлено посредством применения ротационного вискозиметра (прибор Rheolab QC с нагревательным устройством C-LTD80/QC от компании Anton Paar) при температуре 23°С.

В то время, как каждое из базовых покрытий в соответствии с изобретением содержат комбинацию двух дисперсии (PD) и (wD) (где представлены разные соотношения масс двух компонентов), сравнительные базовые покрытия содержат только одну из двух дисперсий.
Осуществление исследований базовых покрытий и многослойных красочных систем, полученных посредством применения базовых покрытий Стабильность при хранении, растекания, пузыри:
Посредством применения методов, описанных выше, исследовали все материалы базовых покрытий A1 - А4 иВ1 - В10, в отношении их стабильности при хранении и их растекаемости и появления пузырей. Все материалы оказались стабильными при хранении, при этом ни одно базовое покрытие не продемонстрировало пузырей или растекания. Поры:
В соответствии со способами, описанными выше (варианты А, Б, и В) были получены следующие многослойные системы в виде клина, и затем их исследовали (х = полученная система, - = система отсутствует):

В целом было выявлено, что все базовые покрытия приводят к получению многослойных красочных систем, которые обладают превосходными оптическими и эстетическими свойствами.
Адгезионные свойства:
А) Для моделирования систем повторного нанесения при ремонтных работах следовали следующему общему протоколу:
Перфорированную стальную панель, покрытую затвердевшим катодным электроосажденным покрытием (КЭП) (CathoGuard® 800 от компании BASF Coatings GmbH), с размерами 57 см × 20 см (в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.1, версия А), получали по аналогии со стандартом DIN EN ISO 28199-1, раздел 8.2 (версия А). Впоследствии, в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.3, наносили элетростатически в одно нанесение водный материал базового покрытия WBM серый A1 - WBM серый А4 с заданной толщиной пленки, составляющей 20 мкм. После истечения времени самоиспарения при комнатной температуре, которое составляло 3 минуты, наносили элетростатически второй водный материал базового покрытия (WBM серебристый 1 - WBM серебристый 4) (заданная толщина пленки 15 мкм). После дополнительного периода времени самоиспарения которое, составляло 4 минуты, при комнатной температуре, систему подвергали промежуточной сушке в камере с принудительной подачей воздуха при температуре 60°С на протяжении 5 минут. Впоследствии, посредством применения ручного распылителя с подачей самотеком, на высушенную посредством промежуточной сушки систему наносили коммерчески доступный двухкомпонентный покровный лак (ProGloss® от компании BASF Coatings GmbH), с заданной толщиной пленки, составляющей 40-45 мкм. Полученную пленку покровного лака подвергали самоиспарению при комнатной температуре на протяжении 10 минут, за чем следовало отверждение в камере с принудительной подачей воздуха при температуре 140°С на протяжении дополнительных 20 минут.
На последующей стадии, указанные выше нанесения повторяют, с тем, чтобы смоделировать систему повторного нанесения при ремонтных работах, где адгезия является особенно важной с практической точки зрения. Следовательно, ту же систему получали еще раз, но в указанном случае, очевидно, первый материал базового покрытия наносили не на затвердевшую пленку катодного электроосажденного покрытия, а вместо этого, на предварительно полученную многослойную красочную систему (или на затвердевшую пленку покровного лака). Следовательно, в этом случае, первоначальная многослойная красочная система служила в качестве основы наносимой системы.
Единственное отличие от определенной выше процедуры состоит в температуре заключительной стадии отверждения. В то время, как указанное выше отверждение осуществляли при температуре 140°С, процедура повторного нанесения в случае ремонта включала два разных набора условий, один раз в так называемых условиях недостаточной температуры сушки (125°С вместо 140°С), и один раз в так называемых условиях чрезмерной температуры сушки (155°С вместо 140°С).
Во всех случаях, подвержение конденсации оставляло не содержащую пузырей систему (m0g0) в том же состоянии. Дополнительные результаты представлены в Таблице ниже.

Результаты демонстрируют, что посредством применения базовых покрытий в соответствии с изобретением получают превосходные адгезионные свойства, которые, кроме всего прочего, являются лучшими, чем в случае сравнительных систем.
Б) Далее для моделирования систем повторного нанесения при ремонтных работах соблюдали следующий общий протокол:
Перфорированную стальную панель, покрытую затвердевшим катодным электроосажденным покрытием (КЭП) (CathoGuard® 800 от компании BASF Coatings GmbH), с размерами 57 см × 20 см (в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.1, версия А) получали по аналогии со стандартом DIN EN ISO 28199-1, раздел 8.2 (версия А). Впоследствии, в соответствии со стандартом DIN EN ISO 28199-1, раздел 8.3, в одно нанесение элетростатически наносили водный материал базового покрытия WBM черный В5 - WBM черный В10 с заданной толщиной пленки, составляющей 15 мкм. После истечения времени самоиспарения, составляющего 4 минуты, при комнатной температуре, систему подвергали промежуточной сушке в камере с принудительной подачей воздуха при температуре 60°С на протяжении 5 минут. Впоследствии, посредством применения ручного распылителя с подачей самотеком, на высушенную посредством промежуточной сушки систему наносили коммерчески доступный двухкомпонентный покровный лак (ProGloss® от компании BASF Coatings GmbH), с заданной толщиной пленки, составляющей 40-45 мкм. Полученную пленку покровного лака подвергали самоиспарению при комнатной температуре на протяжении 10 минут, за чем следовало отверждение в камере с принудительной подачей воздуха при температуре 140°С на протяжении дополнительных 20 минут.
По аналогии с процедурой, описанной в А), на последующей стадии, указанные выше нанесения повторяли, с тем, чтобы смоделировать систему повторного нанесения при ремонтных работах. Опять-таки, единственное отличие от определенной выше процедуры состоит в температуре заключительной стадии отверждения.
Во всех случаях, подвержение конденсации оставляло не содержащую пузырей систему (m0g0) в том же состоянии. Дополнительные результаты представлены в Таблице ниже.

Результаты снова демонстрируют, что посредством применения базовых покрытий в соответствии с изобретением, получают превосходные адгезионные свойства, которые, кроме всего прочего, являются лучшими, чем в случае сравнительных систем.
В целом, результаты демонстрируют, что только материалы базовых покрытий в соответствии с изобретением являются подходящими для получения многослойных красочных систем и систем повторного нанесения при ремонтных работах, которые комбинируют превосходные оптические и эстетические свойства с превосходными адгезионными свойствами.
Реферат
Настоящее изобретение относится к водному материалу базового покрытия, способу получения многослойной красочной системы, способу ремонта многослойной красочной системы и к системе повторного нанесения при ремонтных работах. Водный материал базового покрытия содержит по меньшей мере одну водную полиуретан-полимочевинную дисперсию (PD) с полиуретан-полимочевинными частицами, которые присутствуют в дисперсии, и при этом имеют средний размер частиц 40-2000 нм, и гель-фракцию, составляющую по меньшей мере 50%, а также содержит по меньшей мере одну водную дисперсию (wD), которая содержит полимер с размером частиц 100-500 нм, полученный посредством последовательной радикальной эмульсионной полимеризации трех разных мономерных смесей (А), (Б), и (В) олефинненасыщенных мономеров. Указанные полиуретан-полимочевинные частицы в прореагировавшем виде содержат (Z.1.1) по меньшей мере один полиуретановый преполимер, включающий анионные группы и/или группы, которые могут превращаться в анионные группы, и (Z.1.2) по меньшей мере один полиамин, включающий две первичные аминогруппы и одну вторичную аминогруппу. Температура стеклования полимера, полученного из смеси (А), составляет 10-65°С. Температура стеклования полимера, полученного из смеси (Б), составляет -35-+15°С. Температура стеклования полимера, полученного из смеси (В), составляет -50-+15°С. Водный материал базового покрытия позволяет получать многослойные красочные системы, а также повторно наносимые при ремонте многослойные красочные системы, которые наряду с превосходными эстетическими свойствами также обладают хорошими адгезионными свойствами. 5 н. и 11 з.п. ф-лы, 13 табл., 10 пр.
Формула
Документы, цитированные в отчёте о поиске
Водная полиуретановая дисперсия, не содержащая n-метилпирролидона и растворителей, способ ее получения и ее применение
Комментарии