Пиперидин- и пиперазинзамещенные n-гидроксиформамиды в качестве ингибиторов металлопротеиназ - RU2283306C2
Код документа: RU2283306C2
Описание
Настоящее изобретение относится к соединениям, полезным в ингибировании металлопротеиназ, в частности к фармацевтическим композициям, содержащим их, а также их применению.
Соединения по данному изобретению являются ингибиторами одного или более чем одного фермента металлопротеиназы. Металлопротеиназы представляют собой надсемейство протеиназ (ферментов), число которых за последние годы резко увеличилось. Эти ферменты классифицировали на семейства и подсемейства, основываясь на соображениях относительно структуры и функциий, как описано в N.M.Hooper (1994) FEBS Letters 354:1-6. Примеры металлопротеиназ включают в себя матриксные металлопротеиназы (ММП), такие как коллагеназы (ММП-1, ММП-8, ММП-13), желатиназы (ММП-2, ММП-9), стромелизины (ММП-3, ММП-10, ММП-11), матрилизин (ММП-7), металлоэластаза (ММП-12), энамелизин (enamelysin) (ММП-19), МТ-ММП (металлопротеиназы мембранного типа) (ММП-14, ММП-15, ММП-16, ММП-17); репролизин (reprolysin), или адамализин (adamlysin), или семейство MDC, которое включает в себя секретазы и шеддазы (sheddases), такие как ФНО (фактор некроза опухоли)-превращающие ферменты (ADAM10 и ТАСЕ); семейство астацинов, которое включает в себя ферменты, такие как протеиназа для процессинга проколлагена (ППК), и другие металлопротеиназы, такие как аггреканаза, семейство эндотелин-превращающих ферментов и семейство ангиотензин-превращающих ферментов.
Считают, что металлопротеиназы важны при множестве физиологических процессов заболеваний, которые затрагивают ремоделирование ткани, такое как эмбриональное развитие, образование кости и ремоделирование матки во время менструации. Это основано на способности металлопротеиназ расщеплять широкий ряд матриксных субстратов, таких как коллаген, протеогликан и фибронектин. Также считают, что металлопротеиназы важны при процессинге или секреции биологически важных клеточных медиаторов, таких как фактор некроза опухоли (ФНО), и пост-трансляционном протеолизном процессинге или шеддинге биологически важных мембранных белков, таких как низкоаффинный lgE-рецептор CD23 (более полный список см. N.M.Hooper et al., (1997) Biochem. J. 321:265-279).
Металлопротеиназы связаны со многими болезненными состояниями. Ингибирование активности одной или более чем одной металлопротеиназы может быть весьма полезным при таких болезненных состояниях, как например: различные воспалительные и аллергические заболевания, такие как воспаление сустава (в частности, ревматоидный артрит, остеартрит и подагра), воспаление желудочно-кишечного тракта (в частности, воспалительное заболевание кишечника, неспецифический язвенный колит и гастрит), воспаление кожи (в частности, псориаз, экзема, дерматит); при метастазировании опухоли или инвазии; при заболевании, связанном с неконтролируемым расщеплением внеклеточного матрикса, таком как остеоартрит; при заболевании, связанном с резорбцией кости (таком как остеопороз и болезнь Педжета); при заболеваниях, связанных с аберрантным ангиогенезом; усиленном ремоделировании коллагена, связанным с диабетом, заболеванием периодонта (таким как гингивит), изъязвлением роговицы, изъязвлением кожи, послеоперационными состояниями (такими как кишечный анастомоз) и заживлением кожных ран; демиелинизирующих заболеваниях центральной и периферической нервной системы (таких как рассеянный склероз); болезни Альцгеймера, ремоделировании внеклеточного матрикса, наблюдаемом при сердечно-сосудистых заболеваниях, таких как рестеноз и атеросклероз; и хронических обструктивных заболеваниях легких, ХОЗЛ, (например, роль ММП, таких как ММП-12, обсуждается в Anderson & Shinagawa, 1999, Current Opinion in Anti-inflammatory and Immunornodulatory Investigational Drugs, 1(1): 29-38).
Известен ряд ингибиторов металлопротеиназ; различные классы соединений могут иметь различные степени эффективности и селективности в отношении ингибирования различных металлопротеиназ. Авторы изобретения обнаружили новый класс соединений, которые являются ингибиторами металлопротеиназ и представляют особый интерес в ингибировании ММП-13, а также ММП-9. Соединения по настоящему изобретению имеют полезные эффективность и/или фармакокинетические свойства.
ММП-13, или коллагеназу 3, первоначально клонировали из библиотеки кДНК, имеющей происхождение от опухоли груди [J.M.P.Freije et al. (1994) Journal of Biological Chemistry 269(24): 16766-16773]. ПЦР-РНК-анализ (ПЦР-полимеразная цепная реакция) РНК широкого ряда тканей показал, что экспрессия ММП-13 ограничена карциномами груди, поскольку она не найдена в фиброаденомах груди, нормальной молочной железе или молочной железе в состоянии покоя, плаценте, печени, яичнике, матке, простате или околоушной железе или в клеточных линиях рака груди (T47-D, MCF-7 и ZR75-1). После этого наблюдения ММП-13 обнаружили в трансформированных эпидермальных кератиноцитах [N.Johansson et al., (1997) Cell Growth Differ. 8(2): 243-250], сквамозных клеточных карциномах [N.Johansson et al,, (1997) Am. J.Pathol. 151(2):499-508] и эпидермальных опухолях [К.Airola et al., (1997) J.Invest. Dermatol. 109(2):225-231]. Эти результаты позволяют предположить, что ММП-13 секретируется трансформированными эпителиальными клетками и может быть вовлечена в расщепление внеклеточного матрикса и взаимодействие клетка-матрикс, связанными с метастазированием, в частности, как наблюдается, при инвазивных повреждениях при раке груди и при злокачественном росте эпителия в канцерогенезе кожи.
Недавно опубликованные данные означают, что ММП-13 играет определенную роль в обновлении других соединительных тканей. Например, в соответствии с субстратной специфичностью ММП-13 и предпочтением к расщеплению коллагена типа II [P.G.Mitchell et al., (1996) J. Clin. Invest. 97(3):761-768; V.Knauper et al., (1996) The Biochemical Journal 271:1544-1550], предполагали, что ММП-13 играет роль во время первичной оссификации и скелетного ремоделирования [М.Stahle-Backdahl et al., (1997) Lab. Invest. 76(5):717-728; N.Johansson et al., (1997) Dev. Dyn. 208(3):387-397], в деструктивных поражениях суставов, таких как ревматоидный и остеоартрит [D.Wernicke et al., (1996) J.Rheumatol. 23:590-595; P.G.Mitchell et al., (1996) J. Clin. Invest. 97(3): 761-768; O.Lindy et al., (1997) Arthritis Rheum 40(8):1391-1399], и в ходе асептической нестабильности замен тазобедренного сустава [S.Imai et al., (1998) J.Bone Joint Surg. Br. 80(4):701-710]. ММП-13 также вовлечена в хронический периодонтит взрослых, поскольку она локализована в эпителии хронически воспаленной слизистой оболочки ткани десны человека [V.J.Uitto et al., (1998) Am. J.Pathol 152(6):1489-1499], и в ремоделирование коллагенового матрикса в хронических ранах (М.Vaalamo et al., (1997) J.Invest. Dermatol. 109(1):96-101).
ММП-9 (желатиназа Б, 92 кДа, Коллагеназа IV типа, 92 кДа Желатиназа) является секретируемым белком, который впервые очистили, затем клонировали и секвенировали в 1989 г (S.M.Wilhelm et al, (1989) J. Biol. Chem. 264(29): 17213-17221. Published erratum in J. Biol. Chem. (1990) 265(36):22570). Недавний анализ ММП-9 обеспечил превосходный источник подробной информации и ссылок по этой протеазе; Т.Н.Vu & Z.Werb (1998) (In: Matrix Metalloproteinases. 1998. Edited by W.C.Parks & R.P.Mecham. Pp.115-148. Academic Press. ISBN 0-12-545090-7). Следующие пункты взяты из этого обзора Т.Н.Vu & Z.Werb (1998).
Экспрессия ММП-9 ограничена в норме несколькими типами клеток, включая трофобласты, остеокласты, нейтрофилы и макрофаги. Однако ее экспрессия может быть индуцирована в этих же клетках и в других типах клеток некоторыми медиаторами, включая воздействие на эти клетки факторов роста или цитокинов. Это те же медиаторы, которые часто вовлечены в инициирование воспалительного ответа. Как и другие секретируемые ММП, ММП-9 высвобождается в виде неактивного про-фермента, который далее расщепляется с образованием ферментативно активного фермента. Протеазы, необходимые для этой активации in vivo, не известны. Соотношение активной ММП-9 и неактивного фермента далее регулируется in vivo путем взаимодействия с ТИМП-1 (тканевым ингибитором металлопротеиназ-1), естественно встречающимся белком. ТИМП-1 связывается с С-концевым участком ММП-9, что приводит к ингибированию каталитического домена ММП-9. Соотношение между индуцированной экспрессией проММП-9, расщеплением про- до активной ММП-9 и присутствием ТИМП-1 определяет количество каталитически активной ММП-9, которая присутствует в данном месте. Протеолитически активная ММП-9 атакует субстраты, которые включают в себя желатин, эластин и нативные коллагены типа IV и типа V; она не обладает активностью по отношению к нативному коллагену типа I, протеогликанам или ламининам.
Увеличивается количество данных, дающих основание предполагать участие ММП-9 в различных физиологических и патологических процессах. Физиологическая роль включает в себя инвазию эмбриональных трофобластов в эпителий матки на ранних стадиях эмбриональной имплантации; некоторую роль в росте и развитии костей; и миграцию воспалительных клеток из сосудистой сети в ткани. Повышенная экспрессия ММП-9 наблюдается при некоторых патологических состояниях, вовлекая тем самым ММП-9 в болезненные процессы, такие как артрит, метастазирование опухоли, болезнь Альцгеймера, рассеянный склероз и разрыв бляшек при атеросклерозе, ведущий к острым коронарным состояниям, таким как инфаркт миокарда.
В WO-99/38843 заявлены соединения общей формулы
B-X-(CH2)m-(CR1R2)n-W-COY
для применения в производстве лекарственного средства для лечения или предупреждения состояния, связанного с матриксными металлопротеиназами. Конкретно описано соединение N-{1S-[4-(4-хлорфенил)пиперазин-1-сульфонилметил]-2-метилпропил}-N-гидроксиформамид.
Авторы изобретения в настоящее время обнаружили соединения, которые являются сильными ингибиторами ММП-13 и имеют желаемые профили активности.
В первом аспекте изобретения предложены соединения формулы I,

где В представляет собой фенильную группу, монозамещенную по 3- или 4-положению галогеном или трифторметилом или двузамещенную по 3- и 4-положениям галогеном (который может быть одинаковым или разным); или В представляет собой 2-пиридильную или 2-пиридилоксигруппу, монозамещенную по 4-, 5- или 6-положению галогеном, трифторметилом, циано или С1-4алкилом; или В представляет собой 4-пиримидинильную группу, возможно замещенную по 6-положению галогеном или С1-4алкилом;
Х представляет собой атом углерода или азота;
R1 представляет собой триметил-1-гидантоинС2-4алкильную или триметил-3-гидантоинС2-4алкильную группу; фенил или С2-4алкилфенил, монозамещенный по 3- или 4-положению галогеном, трифторметилом, тио, или C1-3алкилом, или C1-3алкокси; фенил-SO2NHC2-4алкил; 2-пиридил или 2-пиридилС2-4алкил; 3-пиридил или 3-пиридилС2-4алкил; 2-пиримидин-SCH2СН2; 2- или 4-пиримидинилС2-4алкил, возможно монозамещенный одним из заместителей: галоген, трифторметил, С1-3алкил, C1-3алкилокси, 2-пиразинил, возможно замещенный галогеном, или 2-пиразинилС2-4алкил, возможно замещенный галогеном.
Любые алкильные группы, упомянутые выше, могут быть неразветвленными или разветвленными.
Предпочтительными соединениями по изобретению являются такие соединения, к которым относится одно или более чем одно из следующего:
В представляет собой 4-хлорфенил, 4-фторфенил, 4-бромфенил или 4-трифторфенил; 2-пиридил или 2-пиридилокси, монозамещенный по 4- или 5-положению, такой как 5-хлор-2-пиридил, 5-бром-2-пиридил, 5-фтор-2-пиридил, 5-трифторметил-2-пиридил, 5-циано-2-пиридил, 5-метил-2-пиридил; в частности, 4-фторфенил, 5-хлор-2-пиридил или 5-трифторметил-2-пиридил;
Х представляет собой атом азота;
R1 представляет собой 3-хлорфенил, 4-хлорфенил, 3-пиридил, 2-пиридилпропил, 2- или 4-пиримидинилэтил (возможно монозамещенный фтором), 2- или 4-пиримидинилпропил, 2-(2-пиримидинил)пропил (возможно монозамещенный фтором); в частности, 2-пиримидинилпропил, 2-(2-пиримидинил)пропил (возможно монозамещенный фтором) или 5-фтор-2-пиримидинилэтил.
Для соединений формулы I, отдельная подгруппа представлена соединениями, где В представляет собой фенильную группу, монозамещенную по 3- или 4-положению галогеном или трифторметилом или двузамещенную по 3- и 4-положениям галогеном (который может быть одинаковым или разным); или В представляет собой 2-пиридильную или 2-пиридилоксигруппу, монозамещенную по 5- или 6-положению галогеном, трифторметилом или циано; или В представляет собой 4-пиримидинильную группу, возможно замещенную по 6-положению галогеном или С1-4алкилом; Х представляет собой атом углерода или азота; R1 представляет собой триметил-1-гидантоинС2-4алкильную или триметил-3-гидантоинС2-4алкильную группу; или R1 представляет собой фенил или С2-4алкилфенил, монозамещенный по 3- или 4-положению галогеном, трифторметилом, тио, или C1-3алкилом, или С1-3алкокси; или R1 представляет собой фенил-SO2NHC2-4алкил; или R1 представляет собой 2-пиридил или 2-пиридилС2-4алкил; или R1 представляет собой 3-пиридил или 3-пиридилС2-4алкил; или R1 представляет собой 2-пиримидин-SCH2CH2; или R1 представляет собой 2- или 4-пиримидинилС2-4алкил, возможно монозамещенный одним из заместителей: галоген, трифторметил, C1-3 алкил, С1-3алкилокси, 2-пиразинил или 2-пиразинилС2-4алкил; любая алкильная группа может быть неразветвленной или разветвленной.
Понятно, что отдельные заместители и количество заместителей на В и/или R1 выбраны так, чтобы избежать стерически нежелательных комбинаций.
Каждое подтвержденное примером соединение представляет собой отдельный и независимый аспект изобретения.
Если в соединениях формулы I существуют оптически активные центры, то авторами изобретения раскрыты все индивидуальные оптически активные формы и их комбинации как индивидуальные специфические воплощения изобретения, а также их соответствующие рацематы. Рацематы могут быть разделены на индивидуальные оптически активные формы с использованием известных методик (см. Advanced Organic Chemistry: 3rd Edition: author J.March, p.104-107), включая, например, образование диастереоизомерных производных, имеющих подходящие оптически активные вспомогательные группы, с последующим разделением, а затем отщеплением вспомогательных групп.
Понятно, что соединения по изобретению могут содержать один или более чем один асимметрически замещенный атом углерода. Присутствие одного или более чем одного таких асимметрических центров (хиральных центров) в соединении формулы I может дать начало стереоизомерам, и в каждом случае нужно понимать, что изобретение охватывает все подобные стереоизомеры, включая энантиомеры и диастереоизомеры и смеси, включающие их рацемические смеси.
В примерах описаны выделение и характеристика некоторых энантиомеров. Энантиомеры могут быть получены путем взаимодействия рацемического вещества с хиральным вспомогательным соединением, разделения образовавшихся диастереоизомеров с помощью хроматографии с последующим дальнейшим отщеплением хирального вспомогательного соединения. Диастереоизомер, элюированный вторым с колонки (с использованием условий, описанных здесь) и затем расщепленный, при исследовании дает более активный энантиомер, В каждом случае авторы изобретения считают, что активный энантиомер имеет S-стереохимическую конфигурацию, но не желают ограничиваться этим начальным определением. Активный энантиомер характеризуется своим производным, элюируемым вторым с разделительной колонки. Использование различных соединений формулы I, альтернативных колонок и/или различных растворителей может влиять на порядок элюирования наиболее активного энантиомера.
В примерах описаны выделение и характеристика некоторых диастереоизомеров. Хроматографическое разделение и дальнейшее исследование выявило то, что более активный диастереоизомер элюируется первым с разделительной колонки (то есть более активный диастереомер характеризуется тем, что он элюируется первым с разделительной колонки). Использование различных соединений формулы I, альтернативных колонок и/или различных растворителей может влиять на порядок элюирования наиболее активного диастереоизомера.
Для соединений формулы I с двумя хиральными центрами авторы изобретения считают, что активный энантиомер имеет S, S-стереохимическую конфигурацию, но не желают ограничиваться этим начальным определением.
В случае существования таутомеров в соединениях формулы I, авторами изобретения раскрыты все индивидуальные таутомерные формы и их комбинации как индивидуальные специфические воплощения изобретения.
Как изложено ранее, соединения по изобретению являются ингибиторами металлопротеиназ, в частности, они являются ингибиторами ММП-13. Каждый из указанных выше признаков для соединений формулы I представляет собой независимое и частное воплощение изобретения. Не желая быть связанными теоретическими соображениями, авторы изобретения считают, что соединения по изобретению проявляют селективное ингибирование относительно любого из вышеуказанных признаков по отношению к любой ММП-1-ингибирующей активности, в качестве неограничивающего примера, они могут проявлять 100-1000-кратную селективность относительно любой ММП-1-ингибирующей активности.
Некоторые соединения по изобретению особенно полезны как ингибиторы аггреканазы, то есть ингибиторы расщепления аггрекана. Некоторые соединения по изобретению особенно полезны как ингибиторы ММП-9 и/или ММП-12.
Соединения по изобретению могут быть предложены в виде фармацевтически приемлемых солей. Такие соли включают в себя соли присоединения кислоты, такие как соли гидрохлорид, гидробромид, цитрат и малеат, и соли, образованные фосфорной и серной кислотой. В другом аспекте подходящими солями являются соли с основаниями, такие как соль щелочного металла, например натрия или калия, соль щелочноземельного металла, например кальция или магния, или соль органического амина, например триэтиламина.
Они могут быть также предложены в виде гидролизуемых in vivo сложных эфиров. Такими являются фармацевтически приемлемые сложные эфиры, которые гидролизуются в организме человека с образованием исходного соединения. Такие эфиры могут быть идентифицированы с помощью введения, например внутривенно, испытуемому животному исследуемого соединения и дальнейшего изучения жидкостей тела исследуемого животного. Подходящие гидролизуемые in vivo сложные эфиры для карбоксигруппы включают в себя метоксиметил и для гидроксигруппы включают в себя формил и ацетил, в частности ацетил.
Для того чтобы использовать соединение формулы I, или его фармацевтически приемлемую соль, или его гидролизуемый in vivo сложный эфир для терапевтического лечения (включая профилактическое лечение) млекопитающих, включая людей, его обычно приготавливают в соответствии со стандартной фармацевтической практикой, в виде фармацевтической композиции.
Таким образом, в другом аспекте настоящего изобретения предложена фармацевтическая композиция, которая содержит соединение формулы I или его фармацевтически приемлемые соль или гидролизуемый in vivo сложный эфир и фармацевтически приемлемый носитель.
Фармацевтические композиции по этому изобретению могут быть введены стандартным образом при болезненном состоянии, подлежащем лечению, например путем перорального, местного, парентерального, трансбуккального, назального, вагинального или ректального введения или путем ингаляции. Для этих целей соединения по настоящему изобретению могут быть приготовлены известными специалистам способами в форме, например, таблеток, капсул, водных или масляных растворов, суспензий, эмульсий, кремов, мазей, гелей, аэрозолей для носа, суппозиториев, тонкоизмельченных порошков или аэрозолей для ингаляции, и для парентерального применения (включая внутривенное, внутримышечное введение или инфузию) стерильных водных или масляных растворов, или суспензий, или стерильных эмульсий.
В дополнение к соединениям по настоящему изобретению фармацевтические композиции по настоящему изобретению могут также содержать или могут совместно вводиться (одновременно или последовательно) с одним или более чем одним агентом, ценным в лечении одного или более чем одного болезненного состояния, упомянутого выше.
Фармацевтические композиции по настоящему изобретению обычно вводят людям таким образом, чтобы, например, получать суточную дозу от 0,5 до 75 мг/кг массы тела (и предпочтительно от 0,5 до 30 мг/кг массы тела). Эта суточная доза может быть дана в разделенных дозах как необходимо, точное полученное количество соединения и путь введения зависят от массы, возраста и пола пациента, подлежащего лечению, и от конкретного болезненного состояния, которое лечат, в соответствии с принципами, известными в данной области техники.
Типично стандартные лекарственные формы содержат приблизительно от 1 до 500 мг соединения по этому изобретению.
Таким образом, в следующем аспекте настоящего изобретения предложено соединение формулы I или его фармацевтически приемлемые соли или гидролизуемый in vivo сложный эфир для применения в способе терапевтического лечения организма человека или животного. В частности, описано применение в лечении заболевания или состояния, опосредованного ММП-13, и/или аггреканазой, и/или ММП-9, и/или ММП-12.
Еще в одном аспекте настоящего изобретения предложен способ лечения болезненного состояния, опосредованного металлопротеиназами, при котором теплокровному животному вводят терапевтически эффективное количество соединения формулы I или его фармацевтически приемлемых соли или гидролизуемого in vivo сложного эфира. Болезненные состояния, опосредованные металлопротеиназами, включают в себя артрит (такой как остеоартрит), атеросклероз, хронические обструктивные заболевания легких (ХОЗЛ).
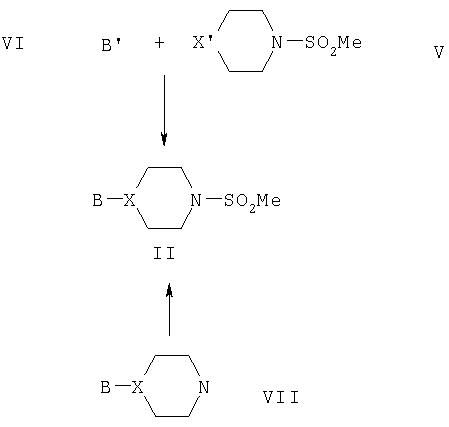
В другом аспекте настоящего изобретения предложен способ получения соединения формулы I или его фармацевтически приемлемых соли или гидролизуемого in vivo сложного эфира, при котором соединение формулы II подвергают взаимодействию с подходящим соединением формулы R1CHO с получением алкена формулы III, который затем превращают в соединение формулы IV, которое является предшественником соединения I и, возможно, после этого осуществляют образование фармацевтически приемлемых соли или гидролизуемого in vivo сложного эфира соединения формулы I, как изложено ниже.

Соединение формулы II можно удобно получить путем взаимодействия соединения формулы V с соединением формулы VI, где В' является предшественником В и X' представляет собой X, или предшественник X, или активированную форму X, подходящую для взаимодействия с В'. Оно также может быть получено из соединения VII так, как показано ниже:
Понятно, что многие из необходимых исходных веществ имеются в продаже. Дополнительно в нижеследующей таблице подробно описаны альдегидные промежуточные соединения и их соответствующие регистрационные номера в Chemical Abstracts.
Альдегиды без регистрационных номеров согласно Chemical Abstracts
3-(2-Пиримидил)пропионовый альдегид. К раствору 2-бромпиримидина (7,95 г, 0,05 М) в ацетонитриле (150 мл) добавляют пропаргиловый спирт (4,2 г, 0,075 М), хлорид бис-(трифенилфосфин)-палладия (II) (750 мг, 1 мМ), иодид меди (100 мг, 0,5 мМ) и триэтиламин (25 мл, 0,25 М) и смесь перемешивают и нагревают при 70°С в течение 2 часов. Затем к реакционной смеси, которую перемешивали и нагревали при 70°С дополнительно в течение 1 часа, добавляют дополнительное количество пропаргилового спирта (2,1 г, 0,038 М), хлорида бис-(трифенилфосфин)-палладия (II) (375 мг, 0,5 мл) и иодида меди (50 мг, 0,25 мл).
Реакционную смесь выпаривают досуха и остаток, который предварительно адсорбируют на диоксиде кремния, подвергают хроматографии. При элюции этилацетатом получали 3-(2-пиримидил)проп-2-ин-3-ол в виде желтого твердого вещества, 4,45 г (66%). ЯМР (ядерный магнитный резонанс) (CDCl3) 2,9 (1Н, t), 4,5 (2Н, d), 7,3 (1H, d), 8,8 (2H, t), МС (масс-спектроскопия) найдено МН+ 135.
3-(2-Пиримидил)проп-2-ин-1-ол (4,45 г, 0, 033 М) растворяют в этилацетате (140 мл), добавляют 10% Pd/C (890 мг) и смесь перемешивают в атмосфере водорода в течение 6 часов. Реакционную смесь фильтруют через целит и фильтрат выпаривают с получением 3-(2-пиримидил)пропан-1-ола в виде желтого масла, 4,15 г (91%). ЯМР (CDCl3) 2,1 (2H, m), 3,2 (2H, t), 3,8 (2H, t), 7,2 (1H, t), 8,7 (2H, d) MC найдено MH+ 139.
3-(2-Пиримидил)пропан-1-ол окисляют с получением 3-(2-пиримидил)пропионового альдегида, используя следующие условия по Сверну (Swern). К оксалилхлориду (14,3 мл), растворенному в дихлорметане (700 мл), добавляют ДМСО (диметилсульфоксид) (21,3 мл), поддерживая температуру ниже -60°С. Через 15 минут медленно добавляют этиловый спирт (20,8 г) растворенный в дихлорметане (20 мл) с последующим добавлением через 30 минут триэтиламина (125 мл). Через 15 минут реакционную смесь оставляют нагреваться до комнатной температуры при добавлении воды (100 мл). Растворители разделяют и органический слой промывают водой (3×150 мл), сушат (MgSO4) и выпаривают с получением масла, которое очищают путем колоночной флэш-хроматографии, элюируя смесью этилацетат/метанол (5%) с получением продукта (8,71 г, 43%) в виде масла. ЯМР (CDCl3) 3,0 (2Н, t), 3,4 (2H, t), 7,1 (1H, t), 8,7 (2H, d), 9,9 (1H, s).
Используя методику, описанную выше, получили следующие альдегиды:
4-(2-пиримидил)масляный альдегид, используя 3-бутин-1-ол вместо пропаргилового спирта. ЯМР (CDCl3) 9,8 (1Н, s), 8,6 (2Н, m), 7,15 (1H, m), 3,0 (2H, m), 2,5 (2H, m), 2,2 (2H, m).
3-(2-пиразинил)пропионовый альдегид, используя 2-бромпиразин вместо 2-бромпиримидина. ЯМР (d6-ДМСО) 9,77 (s, 1H), 8,61 (а, 1H), 8,54 (dd, 1H), 8,46 (d, 1H), 3,10 (t, 2Н), 2,92 (t, 2Н).
4-(2-пиразинил)масляный альдегид, используя 2-бромпиразин вместо 2-бромпиримидина и 3-бутин-1-ол вместо пропаргилового спирта. ЯМР (d6-ДМСО) 9,68 (s, 1H), 8,56 (m, 2Н), 8,49 (m, 1H), 2,80 (t, 2Н), 2,5 (m, 2Н), 1,96 (m, 2Н).
4-(4-трифторметилпиримидин-2-ил)масляный альдегид, используя 2-хлор-4-трифторпиримидин [CAS регистрационный номер 33034-67-2] вместо 2-бромпиримидина и 3-бутин-1-ол вместо пропаргилового спирта.1Н ЯМР (CDCl3): 9,80 (s, 1H), 8,92 (d, 1H, J=5,0 Гц), 7,47 (d, 1H, J=5,0 Гц), 3,11 (dd, 2Н, H=7,5, 7,5 Гц), 2,60 (dd, 2Н, J=6,1, 6,1 Гц), 2,21 (m, 3H).
4-(5-фторпиримидин-2-ил)масляный альдегид, используя 2-хлор-5-фтор-пиримидин [CAS регистрационный номер 62802-42-0] вместо 2-бромпиримидина и 3-бутин-1-ол вместо пропаргилового спирта.1H ЯМР (CDCl3): 9,90 (s, 1H), 8,52 (s, 2Н, J=5,0 Гц), 7,47, 3,47 (m, 2Н), 3,33 (dd, 2H, J=6,8, 6,8 Гц), 3,02 (m, 2H).
4-(4-метоксипиримидин-2-ил)масляный альдегид, используя 2-хлор-4-метокси-пиримидин [CAS регистрационный номер 22536-63-6] вместо 2-бромпиримидина и 3-бутин-1-ол вместо пропаргилового спирта.1Н ЯМР (CDCl3): 9,80 (s, 1H), 8,34 (d, 1H, J=5,0 Гц), 6,55 (d, 1H, J=5,0 Гц), 3,97 (s, 3H), 2,91 (dd, 2H, J=6,8, 6,8 Гц), 2,58 (m, 2H), 2,20 (m, 2H).
4-(5-этилпиримидин-2-ил)масляный альдегид, используя 2-хлор-5-этил-пиримидин [CAS регистрационный номер 111196-81-7] вместо 2-бромпиримидина и 3-бутин-1-ол вместо пропаргилового спирта.1Н ЯМР (CDCl3): 9,79 (s, 1H), 8,51 (s, 2H), 2,99 (dd, 2H, J=7,4, 7,4 Гц), 2,54 (m, 4H), 2,17 (р, 1H, J=7,4 Гц), 1,04 (t, 2H, J=7,2 Гц).
5-(2-пиримидил)пентаналь, используя 2-бромпиримидин и 4-пентин-1-ол вместо пропаргилового спирта:1Н ЯМР (CDCl3): 9,8 (1Н, s), 8,65 (2Н, m), 7,1 (1H, m), 3,0 (2Н, m), 2,5 (2Н, m), 1,9 (2Н, m), 1,7 (2Н, m).
3-(5-бромпиримидин-2-ил)пропионовый альдегид, используя 2-иод-5-бромпиримидин вместо 2-бромпиримидина.1Н ЯМР (CDCl3): 9,90 (s, 1H), 8,70 (s, 2Н), 3,30 (dd, 2Н), 3,0 (dd, 2Н).
4-(4-Пиримидил)-бутан-1-аль. 2,4-Дихлорпиримидин (4,47 г, 0,03 M) растворяют в триэтиламине (250 мл) под аргоном. Добавляют (Ph3P)2PdCl2 (420 мг, 0,006 M), Cul (28 мг, 0,00015 M) и 3-бутин-1-ол (2,36 мл, 0,03 M) и смесь перемешивают при температуре окружающей среды в течение 18 часов. После выпаривания досуха добавляют воду (250 мл) и экстрагируют дихлорметаном. Объединенные органические фазы сушат и выпаривают досуха. Оставшееся масло подвергают хроматографии, элюируя смесью изо-гексан/этилацетат 1:1, с получением 4-(2-хлор-4-пиримидил)-3-бутин-1-ола в виде масла (3,3 г). ЯМР (CDCl3) d 8,5 (d, 1H); 7,3 (d, 1H); 3,9, (t, 2Н); 2,8, (m, 2Н); 1,6, (s, 1H). Macc-спектроскопия показала MH+ 183. Это вещество гидрируют, как описано выше, но в присутствии 1 эквивалента триэтиламина, с получением требуемого насыщенного спирта, который окисляют, используя описанное ранее окисление по Сверну с получением требуемого 4-(4-пиримидил)-бутан-1-аля. ЯМР (CDCl3) d 9,8, (s, 1H); 9,1; (s, 1H); 8,5, (d, 1H); 7,1, (d, 1H); 2,8, (t, 2Н); 2,5, (t, 2Н); 2,1, (m, 2Н). Масс-спектроскопия показала МН- 149.
3-(5-Фторпиримидин-2-ил)пропионовй альдегид. К перемешиваемому раствору (Е)-1-этокси-3-(5-фторпиримидин-2-ил)проп-2-енилэтилового эфира и (Z)-1-этокси-3-(5-фторпиримидин-2-ил)проп-2-енилэтилового эфира (9,7 г, 43 ммоль) в безводном этаноле (100 мл) при комнатной температуре в атмосфере аргона добавляют 10% палладий на активированном угле (1,0 г). Из реакционной колбы затем откачивают воздух и наполняют газообразным водородом. Смесь затем перемешивают в течение 18 часов при комнатной температуре. Реакционную смесь затем фильтруют через слой из целита и выпаривают при пониженном давлении с получением желтого масла (8,7 г, 89%). К раствору этого масла (15 г, 66 ммоль) в ТГФ (тетрагидрофуране) (200 мл) при комнатной температуре добавляют водный раствор соляной кислоты (36 мл 2М раствора, 72 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 3 часов. Реакционную смесь затем разбавляют этилацетатом (100 мл) и рН смеси доводят до рН 9 путем добавления водного раствора гидрокарбоната натрия (насыщенный, 100 мл). Слои затем разделяют и водную фазу экстрагируют этилацетатом (3×100 мл). Объединенные органические экстракты затем сушат (Na2SO4), фильтруют и выпаривают при пониженном давлении с получением 3-(5-фторпиримидин-2-ил)пропионового альдегида (16 г), который использовали без дополнительной очистки.1H ЯМР (CDCl3): 9,90 (s, 1Н), 8,50 (s, 2H), 3,33 (dd, 2H, J=6,9, 6,9 Гц), 3,00 (dd, 2H, J=6,9, 6,9 Гц).
Исходное вещество получили с помощью следующего способа: К раствору 2-хлор-5-фтор-пиримидина [CAS регистрационный номер 62802-42-0] (9,0 г, 6,8 ммоль) и 1-трибутилстаннил-3,3-диэтоксипроп-1-ена (42,8 г, 102 ммоль, смесь E:Z-изомеров 5:1) в сухом ДМФ (диметилформамиде) (140 мл) в атмосфере сухого аргона добавляют последовательно твердый карбонат калия (9,4 г, 68 ммоль), хлорид тетраэтиламмония (11,2 г, 68 ммоль) и хлорид бис(трифенилфосфин)палладия (II) (2,4 г, 3,4 ммоль). Полученную смесь затем нагревают до 120°С в течение 3 часов. Реакционную смесь затем охлаждают до комнатной температуры и разбавляют водой (100 мл) и диэтиловым эфиром (150 мл). Эту смесь затем фильтруют через слой из целита. Слои разделяют и водную фазу экстрагируют диэтиловым эфиром (3×100 мл). Объединенные органические экстракты затем сушат (MgSO4), фильтруют и выпаривают при пониженном давлении. Флэш-хроматография (силикагель, 10% этилацетат в гексанах) затем дала продукт в виде светло-желтого масла и смеси E:Z-изомеров 3:1 (9,7 г, 63%).
Е-изомер:1Н ЯМР (CDCl3): 8,53 (s, 2H), 6,99 (dd, 1Н, J=15,4, 4,1 Гц), 6,86 (d, 1Н, J=15,4 Гц), 5,14 (d, 1Н, J=4,1 Гц), 3,56 (m, 4H), 1,24 (t, 6H, J=7,1 Гц).
Z-изомер:1H ЯМР (CDCl3): 8,57 (s, 2H), 6,65 (d, 1Н, J=12,1 Гц), 6,49 (d, 1Н, J=7,5 Гц), 6,09 (dd, 1Н, J=12,1, 7,5 Гц), 3,70 (m, 4H), 1,21 (t, 6H, J=7,1 Гц).
Аналогичный способ использовали для получения следующих альдегидов с использованием подходящим образом замещенного 2-хлор-пиримидина:
3-(4-метоксипиримидин-2-ил)пропионовый альдегид,1H ЯМР (CDCl3): 9,82 (s, 1Н), 8,34 (d, 1H, J=8,4 Гц), 6,55 (d, 1H, J=7,4 Гц), 3,91 (s, 3H), 3,28 (dd, 2H, J=7,4, 7,4 Гц), 2,99 (dd, 2H, J=7,4, 7,4 Гц).
3-(4-трифторметилпиримидин-2-ил)пропионовый альдегид,1H ЯМР (CDCl3): 9,92 (s, 1H), 8,90 (d, 1H, J=5,0 Гц), 7,47 (d, 1H, J=5,0 Гц), 3,43 (dd, 2H, J=6,8, 6,8 Гц), 3,07 (dd, 2H, J=6,8, 6,8 Гц).
3-(5-этилпиримидин-2-ил)пропионовый альдегид1H ЯМР (CDCl3): 9,91 (s, 1H), 8,49 (s, 2H), 3,31 (dd, 2H, J=6,9, 6,9 Гц), 2,98 (dd, 2H, J=6,9, 6,9 Гц), 2,61 (q, 2H, J=7,6 Гц), 1,26 (t, 3H, J=7,6 Гц).
3,5,5-Триметил-1-пропанальгидантоин

Раствор 3,5,5-триметилгидантоина [CAS (6345-19-3)] (3,5 г, 0,025 моль), 2-(2-бромэтил)-1,3-диоксолана (4,8 мл, 0,041 моль), К2СО3 (8,5 г, 0,062 моль), хлорида бензилтриметиламмония (2,23 г, 0,012 моль) в MeCN (100 мл) нагревают вместе при температуре дефлегмации в течение 24 часов. После охлаждения реакционной смеси до комнатной температуры и фильтрования, фильтрат выпаривают в вакууме. Остаток растворяют ДХМ (дихлорметане), затем промывают водой (×3) перед выпариванием в вакууме. Остаток подвергают азеотропной перегонке с толуолом (×3) с получением желтого масла (5,4 г). Масло затем перемешивают в ТГФ (30 мл) с конц. HCl (4 мл) при комнатной температуре в течение 20 часов. Нейтрализуют водным раствором NaHCO3 и экстрагируют ДХМ (×8). Объединенные органические слои сушат над Na2CO3 и выпаривают в вакууме с получением желтого масла (4,3 г).1H ЯМР (CDCl3): 9,82 (s, 1H), 3,62 (t, 2H), 3,04 (s, 3H), 2,90 (m, 2H), 1,37 (s, 6H).
1,5,5-Триметил-3-пропанальгидантоин

1,5,5-Триметилгидантоин [CAS (6851-81-6)] (5,0 г), 35,0 моль) добавляют к смеси NaOEt (0,02 г, 0,298 ммоль, каталитический) и EtOH (8 мл) и перемешивают под аргоном. Смесь нагревают до 30°С перед медленным добавлением акролеина (2,35 мл) и реакционная смесь экзотермически нагревается до 45°С. Реакционную смесь оставляют охлаждаться до комнатной температуры и перемешивают в течение дополнительных 2 часов. К смеси добавляют АсОН (0,136 мл, 2,4 ммоль) и силикагель (3,5 г) перед выпариванием в вакууме. Продукт на диоксиде кремния подвергают хроматографии на колонке с диоксидом кремния (элюент: смесь 5% ацетон/ДХМ) с получением прозрачного масла (6,2 г). Дополнитнльная очистка остатка на оксиде алюминия (элюент: ДХМ) привела к получению прозрачного масла (2,7 г).1H ЯМР (CDCl3): 9,78 (s,1H), 3,88 (t, 2H), 2,86 (s, 3H), 2,82 (m, 2H), 1,37 (s, 6H).
Аналогичным способом получили 1,5,5-триметил-3-бутанальгидантоин [M+H 213].
3-(3-Хлорфенил)масляный альдегид. Смесь 3-хлориодбензола (2,38 г), ацетата палладия (20 мг), бикарбоната натрия (1,01 г) и кротонового спирта (1,28 г) в N-метилпирролидоне (4 мл) перемешивают и нагревают при 130°С в течение 2 часов. Реакционную смесь оставляют охлаждаться, добавляют воду (50 мл) и смесь экстрагируют диэтиловым эфиром (2×50 мл). Объединенные органические экстракты сушат и остаток, полученный при удалении растворителя, очищают путем хроматографии на диоксиде кремния, элюируя смесью этилацетата и метиленхлорида (1:20), с получением указанного в заголовке соединения в виде масла, выход 519 мг, М-Н=181.
3-(2-Пиридил)масляный альдегид. Получают с помощью окисления по Сверну соответствующего спирта (CAS 90642-86-7).
3-(5-Фторпиримидин-2-ил)масляный альдегид

Концентрированную соляную кислоту (1 мл) добавляют к перемешиваемому раствору 2-[2-(1, 3-диоксолан-2-ил)-1-метилэтил]-5-фторпиримидина (1,1 г) в тетрагидрофуране (10 мл) при температуре окружающей среды, перемешивают в течение 3 часов, затем добавляют твердый гидрокарбонат натрия до нейтрального рН. Смесь выливают на угольный брикет Chemelute (Chemelute cartridge) и промывают этилацетатом (3×20 мл), объединенные органические слои сушат над Na2SO4 и выпаривают в вакууме с получением 3-(5-фторпиримидин-2-ил)масляного альдегида (300 мг, 35%), который используют без дополнительной очистки.
Исходное вещество получали следующим образом:
2-[2-(1,3-диоксолан-2-ил)-1-метилэтил]-5-фторпиримидин

К перемешиваемой суспензии активированного цинка "Rieke" в тетрагидрофуране (21 мл, 1,53 М) добавляют 2-(2-бромпропил)-1,3-диоксолан (6,6 г) в тетрагидрофуране (50 мл), наблюдают повышение температуры от 21 до 40°С, нагревают при 40°С в течение 1 часа, затем оставляют охлаждаться до температуры окружающей среды перед добавлением 2-хлор-5-фторпиримидина (3 г) и хлорида [1, 2-бис(дифенилфосфино)-пропан]дихлорникеля (II) (368 мг). Смесь перемешивают при температуре окружающей среды в течение 4 часов, затем фильтруют через слой из целита и фильтрат выпаривают при пониженном давлении. Затем флэш-хроматография (силикагель, гексан-25% этилацетат в гексанах) дала продукт в виде бледно-желтого масла (1,1 г);1H ЯМР (d6-ДМСО): 8,81 (s, 2H), 4,73 (dd, 1H), 3,66-3,87 (m, 4H), 3,21-3,30 (m, 1H), 2,19 (ddd, 1H), 1,83 (ddd, 1H), 1,27 (d, 3H); m/z 213 (M+1).
2-(2-Бромпропил)-1,3-диоксолан
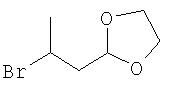
Кротоновый альдегид (9,18 г, 108 ммоль) по каплям добавляют к перемешиваемому раствору бромтриметилсилана (24 г, 156 ммоль) при 0° С, перемешивают в течение 1 часа при 0°С, затем нагревают до комнатной температуры и перемешивают в течение еще 1 часа. Добавляют этиленгликоль (9,5 г, 156 ммоль) и паратолуолсульфоновую кислоту (100 мг) и раствор нагревают до температуры дефлегмации, воду удаляют путем использования аппарата Дина-Старка. После завершения смесь охлаждают до комнатной температуры и промывают водным раствором гидрокарбоната натрия (насыщенный, 2×50 мл). Остаток очищают с помощью вакуумной перегонки с получением 2-(2-бромпропил)-1,3-диоксолана (18,8 г, 40-42°С, 21 мм рт.ст. (2799,8 Па), 89%).
1H ЯМР (CDCl3): 5,05 (dd, 1Н), 4,18-4,33 (m, 1H), 3,84-4,0 (m, 4H), 2,25 (add, 1H), 2,03 (ddd, 1H), 1,75 (d, 3Н).
Аналогичный способ использовали для получения следующих альдегидов, используя подходящим образом замещенные 2-хлор-пиримидин и 1,3-диоксолан:
3-(5-хлорпиримидин-2-ил)пропионовый альдегид

1H ЯМР (CDCl3): 9,90 (s, 1H), 8,60 (s, 2H), 3,32 (dd, 2H), 3,04 (dd, 2H).
3-(5-Хлорпиримидин-2-ил)масляный альдегид

1H ЯМР (CDCl3): 9,85 (s, 1H), 8,60 (s, 2H), 3,65 (m, 1H), 3,14 (dd, 1H), 2,75 (dd, 1H), 1,39 (d, 3H).
3-[2-(6-Хлорпиразинил)]пропионовый альдегид

Диэтилацеталь 3-[2-(6-хлорпиразинил)]пропионового альдегида (200 мг, 0,82 ммоль) обрабатывают 2н. соляной кислотой (450 мкл) в тетрагидрофуране (2,5 мл) при комнатной температуре в течение 18 часов. После доведения рН до 8 с помощью насыщенного водного бикарбоната натрия, реакционную смесь экстрагируют (×3) этилацетатом и органические слои сушат (безводный сульфат натрия), фильтруют и концентрируют в вакууме с получением указанного в заголовке соединения в виде темно-коричневого масла (137 мг, 98%). Это вещество используют без дополнительной очистки.
1H ЯМР (CDCl3): δ 9,85 (1Н, s); 8,4 (2Н, 2 × s); 3,5 (2Н, t); 3,0 (2Н, t).
Исходное вещество получали следующим способом:
диэтилацеталь 3-[2-(6-хлорпиразинил)]пропионового альдегида

Диэтилацеталь 3-[2-(6-хлорпиразинил)]пропиолового альдегида (5,5 г, 22,9 ммоль) в этаноле (55 мл) дегазируют аргоном и добавляют оксид платины (IV) (52 мг, 0,23 ммоль). Из реакционного сосуда откачивают воздух и подают водород при давлении 101325,3 Па. Через 2 суток реакционную смесь концентрируют в вакууме и очищают с помощью флэш-хроматографии, элюируя градиентом 0-50% этилацетата в изогексане, с получением диэтилацеталя 3-[2-(6-хлорпиразинил)]пропионового альдегида в виде светло-желтого масла (1,17 г, 21%).
1H ЯМР (CDCl3): δ 8,4 (1Н, s); 8,35 (1Н, s); 4,5 (1Н, t); 3,75-3,55 (2Н, m); 3,55-3,4 (2Н, m); 2,9 (2Н, dd); 2,1 (2Н, dd); 1,2 (6H, t).
Диэтилацеталь 3-[2-(6-хлорпиразинил)]пропиолового альдегида

К раствору 2, 6-дихлорпиразина (1 г, 6,7 ммоль) и диэтилацеталя пропионового альдегида (1,1 мл, 7,4 ммоль) в ацетонитриле (10 мл) при комнатной температуре в атмосфере аргона добавляют дихлорид бис(трифенилфосфин)палладия (II) (94 мг, 0,13 ммоль) и иодид меди (I) (51 мг, 0,27 ммоль) с последующим добавлением триэтиламина (4,7 мл, 33,6 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и остаток очищают с помощью флэш-хроматографии, элюируя 10-20% этилацетатом в изогексане, с получением диэтилацеталя 3-[2-(6-хлорпиразинил)]пропиолового альдегида в виде желтого масла (660 мг,41%).
1H ЯМР (CDCl3): δ 8,6 (1Н, s); 8,55 (1H, s); 5,5 (1H, s); 3,9-3,75 (2H, m); 3,7-3,4 (2H, m); 1,25 (6H, t)
MC (El+ (ионизация электронным ударом)) 241/243 (МН+).
Альтернативный способ получения соединения формулы I или его фармацевтически приемлемых соли или гидролизуемого in vivo сложного эфира включает в себя взаимодействие соединения формулы II с соединением формулы R1COOR с получением соединения формулы VIII, его превращение в соединение формулы IX, превращение этого соединения формулы IX в алкен формулы III, который затем превращают в соединение формулы IV, которое является предшественником соединения I, и, возможно, затем образование фармацевтически приемлемых соли или гидролизуемого in vivo сложного эфира соединения формулы I, как указано ниже.
Подходящие сложные эфиры формулы R1COOH могут иметься в продаже, или могут быть доступны другим способом, или могут быть получены с использованием, например, методики, аналогичной методике, описанной в примере 10. Понятно, что возможно использование любого сложного эфира формулы R1COOH (где R1 является таким, как определено ранее): -R может быть любой группой, включая, например, алкил, аралкил, гетероарил и так далее.

Соединения по настоящему изобретению могут быть охарактеризованы, например, в следующих исследованиях.
Исследования выделенного фермента
Семейство матриксных металлопротеиназ, включающее в себя, например, ММП-13
Рекомбинантная проММП-13 человека может быть экспрессирована и очищена так, как описано Knauper et al. [V.Knauper et al., (1996) The Biochemical Journal 271:1544-1550 (1996)]. Очищенный фермент может быть использован для контролирования ингибиторов активности следующим образом: очищенную проММП-13 активируют, используя 1 мМ аминофенилртутную кислоту ((АФРК), aminophenylmercuric acid), в течение 20 часов при 21°С, активированную ММП-13 (11,25 нг на образец) инкубируют в течение 4-5 часов при 35°С в буфере для исследования (0,1 М Трис-HCl, рН 7,5, содержащем 0,1 М NaCl, 20 мМ CaCl2, 0,02 мМ ZnCl и 0,05% (мас./об.) Brij 35), используя синтетический субстрат 7-метоксикумарин-4-ил)ацетил-Pro-Leu-Gly-Leu-N-3-(2,4-динитрофенил)-L-2,3-диаминопропионил-Ala-Arg-NH2 в присутствии или в отсутствие ингибиторов. Активность определяют с помощью измерения флуоресценции при λех 328 нм и λem 393 нм. Процент ингибирования вычисляют следующим образом: % ингибирования равен [флуоресценцияплюс ингибитор - флуоресценцияфон], деленные на [флуоресценцияминус ингибитор - флуоресценцияфон].
Подобный протокол может быть использован для других экспрессированных и очищенных проММП с применением условий, касающихся субстратов и буферов, оптимальных для конкретных ММП, например, как описано С. Graham Knight etal., (1992) FEBS Lett. 296(3):263-266.
Семейство адамализина, включающее в себя, например, конвертазу ФНО
Способность заявленных соединений ингибировать фермент конвертазу проФНОα может быть определена с использованием исследования частично очищенного выделенного фермента, при этом фермент получают из мембран ТНР-1, как описано К.М.Mohler et al., (1994) Nature 370:218-220. Активность очищенного фермента и его ингибирование определяют с помощью инкубирования частично очищенного фермента в присутствии или в отсутствие исследуемых соединений, используя субстрат 4',5'-диметокси-флуоресцеинилSer-Pro-Leu-Ala-Gln-Ala-Val-Arg-Ser-Ser-Ser-Arg-Cys(4-(3-сукцинимид-1-ил)-флуоресцеин)-NH2 в буфере для исследования (50 мМ трис-HCl, рН 7,4, содержащем 0,1% (мас./об.) Triton X-100 и 2 мМ CaCl2, при 26°С в течение 18 часов. Величину ингибирования определяют как для ММП-13, но используют λех 490 нм и λem 530 нм. Субстрат синтезируют следующим образом. Пептидную часть субстрата собирали на Fmoc-NH-Rink-MBHA-полистироловой смоле либо вручную, либо на автоматическом пептидном синтезаторе с помощью стандартных способов, включающих в себя применение Fmoc-аминокислот и гексафторфосфата О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония (ГБТУ) в качестве агента сочетания по меньшей мере с 4- или 5-кратным избытком Fmoc-аминокислоты и ГБТУ. Ser1 и Pro2сочетали дважды. Использовали следующую стратегию защиты боковых цепей: Ser1(But), Gln5(тритил), Arg8, 12(Pmc или Pbf), Ser9, 10, 11(тритил), Cys13 (тритил). После сборки, N-концевую Fmoc-защитную группу удаляли с помощью обработки Fmoc-пептидил-смолы в ДМФ. Амино-пептидил-смолу, полученную таким образом, ацилировали путем обработки в течение 1,5-2 часов при 70°С 1,5-2 эквивалентами 4', 5'-диметокси-флуоресцеин-4(5)-карбоксильной кислоты [Khanna & Ullman, (1980) Anal Biochem. 108:156-161), которую предварительно активировали диизопропилкарбодиимидом и 1-гидроксибензотриазолом в ДМФ]. С диметоксифлуоресцеинил-пептида затем одновременно удаляли защиту и отделяли от смолы путем обработки трифторуксусной кислотой, содержащей 5% воды и 5% триэтилсилана. Диметоксифлуоресцеинил-пептид выделяли путем выпаривания, растирания с диэтиловым эфиром и фильтрования. Выделенный пептид подвергали взаимодействию с 4-(N-малеимидо)-флуоресцеином в ДМФ, содержащем диизопропилэтиламин, продукт очищали с помощью ОФ-ВЭЖХ (обращенно-фазовой высокоэффективной жидкостной хроматографии) и окончательно выделяли с помощью сушки сублимацией из водной уксусной кислоты. Продукт характеризовали с помощью времяпролетной масс-спектрометрии с ионизацией посредством лазерной десорбции с использованием матрицы и аминокислотного анализа.
Естественные субстраты
Активность соединений по изобретению как ингибиторов расщепления аггрекана может быть исследована с использованием способов, основанных, например, на описаниях Е.С.Arner et at., (1998) Osteoartritis and Cartilage 6:214-228; (1999) Journal of Biological Chemistry, 274(10), 6594-6601, и антителах, описанных там же. Способность соединений действовать как ингибиторы коллагеназ может быть определена, как описано Т.Cawston and A.Barrett (1979) Anal. Biochem. 99:340-345.
Ингибирование активности металлопротеиназ при активности, основанной на клетке/ткани
Тестирование в качестве агента для ингибирования мембранных шеддаз, таких как конвертаза ФНО
Способность соединений по настоящему изобретения ингибировать клеточный процессинг продукции ФНОα может быть определена на клетках ТНР-1 с использованием ELISA (твердофазного иммуноферментного анализа) для обнаружения высвобожденного ФНО, по существу как описано К.М.Mohler et al., (1994) Nature 370:218-220. Подобным образом процессинг или шеддинг других мембранных молекул, таких как молекулы, описанные в N.M.Hooper et al., (1997) Biochem. J. 321:265-279, может быть тестирован с использованием подходящих клеточных линий и с помощью подходящих антител для обнаружения шед-белка.
Тестирование в качестве агента для ингибирования основанной на клетках инвазии
Способность соединения по настоящему изобретению ингибировать миграцию клеток в исследовании на инвазию может быть определена как описано A.Albini et al., (1987) Cancer Research 47:3239-3245.
Тестирование в качестве агента для ингибирования ФНО-шеддазной активности цельной крови
Способность соединений по настоящему изобретению ингибировать продукцию ФНОα оценивают в исследовании с цельной кровью человека, где для стимулирования высвобождения ФНОα используют ЛПС (липополисахариды). Гепаринизированную (10 Ед/мл) кровь человека, полученную от добровольцев, разбавляют 1:5 средой (RPMI 1640 + бикарбонат, пенициллин, стрептомицин и глутамин) и инкубируют (160 мкл) с 20 мкл исследуемого соединения (трижды) в ДМСО или подходящем носителе, в течение 30 минут при 37°С в увлажняемом (5% CO2/95% воздуха) инкубаторе, перед добавлением 20 мкл ЛПС (E.coli. 0111:В4; конечная концентрация 10 мкг/мл). Каждое исследование включает в себя контроли разведенной крови, инкубируемые только со средой (6 лунок/планшет) или известным ингибитором ФНОα в качестве стандарта. Планшеты затем инкубируют в течение 6 часов при 37°С (увлажняемый инкубатор), центрифугируют (2000 об/мин в течение 10 минут, 4°С), плазму собирают (50-100 мкл) и хранят в 96-луночных планшетах при -70°С перед последующим анализом на концентрацию ФНОα с помощью ELISA.
Тестирование в качестве агента для ингибирования деградации хряща in vitro
Способность соединений по настоящему изобретению ингибировать деградацию аггрекана или коллагеновых компонентов хряща может быть определена, по существу как описано К.М.Bottomley et at., (1997) Biochem. J. 323:483-488.
Фармакодинамический тест
Для оценки характеристик клиренса и биодоступности соединений по настоящему изобретению осуществляют ex vivo фармакодинамический тест с использованием исследования с синтетическим субстратом, описанным выше, или альтернативно ВЭЖХ или масс-спектрометрического анализа. Это общее исследование, которое может быть использовано для определения скорости выведения соединений в пределах класса. Животным (например крысам, мартышкам) дают дозы внутривенно или перорально с растворимым препаратом соединения (таким как 20 мас.%/об. ДМСО, 60 мас.%/об. ПЭГ (полиэтиленгликоль) 400) и в последующие моменты времени (например, 5, 15, 30, 60, 120, 240, 480, 720, 1220 минут) образцы крови отбирают из подходящего сосуда в 10 Ед гепарин. Фракции плазмы получают после центрифугирования и белки плазмы осаждают ацетонитрилом (конечная концентрация 80 мас.%/об.). Через 30 минут при -20°С белки плазмы осаждают с помощью центрифугирования и фракцию супернатанта выпаривают досуха с применением вакуумного испарителя Savant. Осадок растворяют в буфере для исследования и далее анализируют на содержание соединения с использованием исследования с синтетическим субстратом. Кратко, для оцениваемого соединения строят кривую концентрация соединения-ответ. Серийные разведения растворенных экстрактов плазмы оценивают на активность и количество соединения, присутствующее в первоначальном образце плазмы, вычисляют, используя кривую концентрация-ответ, принимая во внимание коэффициент общего разведения плазмы.
Оценка in vivo
Тестирование в качестве анти-ФНО-агента
Способность соединений по настоящему изобретению действовать как ех vivo ингибиторы ФНОα определяют на крысах. Кратко, группам самцов крыс Wistar Alderley Park (АР) (180-210 г) дают дозы соединения (6 крыс) или носителя для лекарства (10 крыс) с помощью подходящего пути введения, например перорального (п.о.), внутрибрюшинного (в.б.), подкожного (п.к.). Через девяносто минут крыс умерщвляют, используя увеличивающуюся концентрацию CO2 и кровь отбирают через полую заднюю вену в 5 Ед натрий-гепарина/мл крови. Образцы крови немедленно помещают на лед и центрифугируют при 2000 об/мин в течение 10 минут при 4°С и собранную плазму замораживают при -20°С для последующего изучения их влияния на продукцию ФНОα с помощью LPS-стимулированной крови человека. Образцы плазмы крови крыс размораживают и 175 мкл каждого образца добавляют ранее определенным образом в 96-ти U-луночный планшет. Затем в каждую лунку добавляют пятьдесят мкл гепаринизированной крови человека, перемешивают и планшет инкубируют в течение 30 минут при 37°С (увлажняемый инкубатор). В лунки добавляют ЛПС (25 мкл, конечная концентрация 10 мкг/мл) и инкубирование продолжают еще 5,5 часов. Контрольные лунки инкубируют только с 25 мкл среды. Планшеты затем центрифугируют в течение 10 минут при 2000 об/мин и 200 мкл супернатантов переносят в 96-луночный планшет и замораживают при -20°С для последующего анализа концентрации ФНО с помощью ELISA.
Анализ данных с помощью специализированного программного обеспечения рассчитывают для каждого соединения/дозы:

Тестирование в качестве агента против артрита
Активность соединения как агентов против артрита тестируют на коллаген-индуцированном артрите (КИА), как определено в D.E.Trentham et al., (1977) J. Exp. Med. 146:857. В этой модели кислый растворимый нативный коллаген типа II вызывает полиартрит у крыс при введении в неполном адъюванте Фрейнда. Подобные условия могут быть использованы для индуцирования артрита у мышей и приматов.
Тестирование в качестве противоракового агента
Активность соединения как противоракового агента может быть оценена, по существу как описано в I.J.Fidler (1978) Methods in Cancer Research 15:399-439, с использованием, например, клеточной линии В16 (описана в B.Hibner et al., Abstract 283, р.75, 10th NCI-EORTC Symposium, Amsterdam, June 16-19, 1998).
Настоящее изобретение проиллюстрировано следующими примерами, но не ограничено ими.
ПРИМЕР 1
N-[1-([4-(4-бромфенил)пиперазино]сульфонилметил)-4-пиримидин-2-илбутил]-N-гидроксиформамид
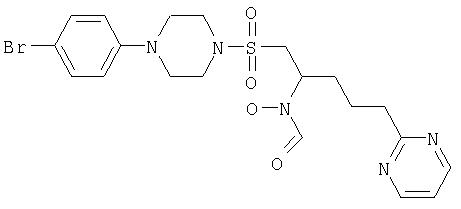
К перемешиваемому раствору N-[1-([4-(4-бромфенил)пиперазино]сульфонилметил)-4-пиримидин-2-илбутил]гидроксиламина (497 мг, 1,0 ммоль) в ТГФ (5,0 мл) и муравьиной кислоты (2,5 мл), охлажденному до 0°С, добавляют предварительно полученную смесь уксусного ангидрида (566 мкл, 6,0 ммоль) и муравьиной кислоты (2,0 мл). Смесь перемешивают при 0°С в течение 1 часа и оставляют достигать комнатной температуры. Растворители удаляют с помощью роторного испарителя и остаток очищают с помощью хроматографии (50 г Silica Bond Elute, элюент 0→15% метанол/дихлорметан), чистые фракции выпаривают, и они кристаллизуются из горячего этилацетата с получением N-[1-([4-(4-бромфенил)пиперазино]сульфонилметил)-4-пиримидин-2-илбутил]-N-гидроксиформамида в виде белого кристаллического порошка (262 мг, 51%).
ЯМР (300 МГц ДМСО-d6) δ/млн-1: 9,87 (s, 1H*), 9,55 (s, 1H*), 8,70 (m, 2H), 8,29 (s, 1H*), 7,98 (s, 1H*), 7,33 (m, 3Н), 6,92 (dd, 2H), 4,68 (m, 1H*), 4,13 (m, 1H*), 3,55-3,31 (m, 5H, частично затемненный), 3,25-3,09 (m, 7H, частично затемненный), 1,80-1,50 (m, 4H).
* - сигналы ротамеров
МС: ES+ (электрораспыление), (M+H)+ = 512, 514 (изотопная модель Br (Br isotope pattern)).
Исходное вещество получают следующим образом:
I) К раствору гидрохлорида 1-(4-бромфенил)пиперазина (5,09 г, 18,3 ммоль) и триэтиламина (7,67 мл) в дихлорметане (100 мл) по каплям добавляют метансульфонилхлорид (2,83 мл, 36,3 ммоль). Смесь перемешивают в течение 1 часа при комнатной температуре, затем добавляют дихлорметан (100 мл). Органические слои промывают водой (2×), рассолом, сушат (Na2SO4), выпаривают в вакууме до желтого твердого вещества, которое кристаллизуется из этанола, промывают его диэтиловым эфиром с получением 1-(4-бромфенил)-4-(метансульфонил)пиперазина (4,74 г, выход 81%) в виде белого пушистого порошка.
ЯМР (300 МГц CDCl3) δ/млн-1: 7,38 (d, 2H), 6,91 (d, 2H), 3,21 (m, 8H), 2,89 (s, 3Н). МС: ES+, (М+H)+ = 318,320 (изотопная модель Br).
II) К 1-(4-бромфенил)-4-(метансульфонил)пиперазину (902 мг, 2,0 ммоль), суспендированному в безводном ТГФ (15 мл), под азотом, охлажденному до температуры между -20 и -30°С, последовательно добавляют бис(триметилсилил)амид лития (1,0 М в ТГФ, 4,0 мл), хлортриметилсилан (217 мг, 2,0 ммоль, 253 мкл) и 4-пиримидин-2-илмасляный альдегид (300 мг, 2,0 ммоль). Смесь перемешивают при -20°С в течение 1 часа, реакцию останавливают насыщенным раствором хлорида аммония и оставляют стоять при температуре окружающей среды в течение ночи. Растворители удаляют под вакуумом и остаток распределяют между дихлорметаном (15 мл) и водой (5 мл), органическую фазу отделяют и подвергают хроматографии (50 г Silica Bond Elute, элюируя градиентом 0→100% этилацетат/гексан) с получением 2-(5-[4-(4-бромфенил)пиперазино]сульфонилпент-4-енил)пиримидина в виде белого кристаллического вещества (759 мг, выход 84%).
МС: ES+, (М+Н)+ = 451,453 (изотопная модель Br).
III) К перемешиваемому раствору 2-((Е)-5-[4-(4-бромфенил)пиперазино]сульфонилпент-2-енил)пиримидина (451 мл, 1,0 ммоль) в ТГФ (10 мл) добавляют гидроксиламин (50%-ный раствор в воде, 500 мкл) и смесь перемешивают в течение ночи. Растворители удаляют в вакууме, осуществляя азеотропную перегонку с толуолом (3×), с получением N-[1-([4-(4-бромфенил)пиперазино]сульфонилметил)-4-пиримидин-2-илбутил]гидроксиламина (497 мг, количественно).
МС: ES+, (М+Н)+ = 484, 486 (изотопная модель Br).
ПРИМЕР 2
N-[1-([4-(5-хлорпиридин-2-ил)пиперазино]сульфонилметил)-3-(5-фторпиримидин-2-ил)пропил]-N-гидроксиформамид
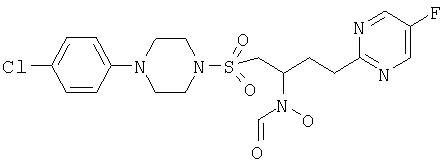
Уксусный ангидрид (0,51 мл) добавляют непосредственно к муравьиной кислоте (2,0 мл), охлажденной до 0°С, и затем добавляют раствор 2-[4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил-3-(гидроксиамино)бутил]-5-фторпиримидина (0,485 г) в тетрагидрофуране (11 мл). Раствор перемешивают при комнатной температуре в течение 3 часов и затем выпаривают в вакууме, полученный остаток подвергают азеотропной перегонке с толуолом и затем его растворяют в метаноле и нагревают до 40°С в течение 30 минут. Раствор выпаривают досуха, а затем добавляют диэтиловый эфир и перемешивают при комнатной температуре в течение 10 минут, твердое вещество отфильтровывают, сушат в вакууме с получением N-[1-([4-(5-хлорпиридин-2-ил)пиперазино]сульфонилметил)-3-(5-фторпиримидин-2-ил)пропил]-N-гидроксиформамида, (0,218 г), т.пл. (температура плавления) 154-155°С. ЯМР (d6-ДМСО 373°К (100°С)): 2,20 (m, 2Н), 2,95 (m, 2Н), 3,23 (dd, 1H), 3,30 (m, 4H), 3,49 (dd, 1H), 3,60 (m, 4H), 4,42 (очень шир. s, 1H), 6,88 (d, 1H), 7,59 (dd, 1H), 8,05 (очень шир. s, 1H), 8,12 (dd, 1H), 8,71 (s, 2H), 9,40 (очень шир. s, 1H); m/z 473 (М+1).
Исходное вещество получали следующим образом:
1) 1-(5-хлорпиридин-2-ил)-4-(метилсульфонил)пиперазин (0, 600 г) перемешивают в безводном тетрагидрофуране (22 мл) под аргоном, затем охлаждают до -10°С перед добавлением бис(триметилсилил)амида лития (4,8 мл 1,0 М раствора в тетрагидрофуране). Смесь перемешивают при -10°С в течение 30 минут и добавляют раствор диэтилхлорфосфата (0,345 мл). Смесь перемешивают при -10°С в течение 15 минут и затем добавляют 3-(5-фторпиримидин-2-ил)пропионовый альдегид (0,334 г), перемешивают при -10°С еще 30 минут. Смесь оставляют нагреваться до комнатной температуры, затем промывают водным хлоридом аммония и экстрагируют этилацетатом. Органические слои сушат над Na2SO4.
Очистка остатка на диоксиде кремния с элюированием смесью 70% этилацетат и 30% гексан привела к образованию смеси 6:4 2-((Е)-4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонилбут-3-енил)-5-фторпиримидина и 2-((Z)-4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонилбут-3-енил)-5-фторпиримидина (0,44 г).
1H ЯМР (CDCl3): 8,55 (d, 1H), 8,48 (s, 1H), 7,46 (dd, 1H), 6,85 (m, 1H), 6,60 (d, 1H),*6,45 (m, 1H), 6,15 (d, 1H),*6,03 (d, 1H), 3,61 (m, 4H), 3,28 (m, 2H), 3,15 (m, 4H),*2,81, (m, 2H); MC (ES+): 412,3 (MH+).
* обозначает неосновной изомер.
2) К раствору 2-((Е)-4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонилбут-3-енил)-5-фторпиримидина и 2-((Z)-4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонилбут-3-енил)-5-фторпиримидина (0,44 г) в тетрагидрофуране (5 мл) добавляют гидроксиламин (1,0 мл, 50%-ный водный раствор). Смесь перемешивают в течение 18 часов, затем разбавляют EtOH (10 мл) и промывают насыщенным раствором хлорида аммония (10 мл). Органический слой сушат над Na2SO4 и выпаривают в вакууме с получением 2-[4-[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил-3-(гидроксиамино)бутил]-5-фторпиримидина (0,483 г).
1H ЯМР (CDCl3): 8,45 (s, 2H), 8,08 (d, 1H), 7,39 (dd, 1H), 6,55 (d, 1H), 5,76 (шир. s, 2H), 3,59 (m, 4H), 3,46 (m, 1H), 3,42 (m, 2H), 3,33 (m, 4H), 3,10 (m, 4H), 2,82 (m, 1H), 2, 15 (m, 1H), 2,01 (m, 1H); MC (ES+): 445,3 (MH+).
ПРИМЕР 3
Получили следующие соединения








Исходные пиперазин- и пиперидин-сульфонамиды, необходимые для синтеза соединений, имелись в продаже или были получены так, как показано ниже:
1-(4-фторфенил)-4-(метансульфонил)пиперазин

К раствору 1-(4-фторфенил)пиперазина (35 г, 194 ммоль) и пиридина (17,5 мл) в безводном дихлорметане (200 мл) при 0°С по каплям добавляют метансульфонилхлорид (20 мл, 258 ммоль). Смесь перемешивают в течение 3 часов при комнатной температуре. Смесь промывают водой и экстрагируют дихлорметаном (2×100 мл). Органические слои сушат с помощью MgSO4 и выпаривают в вакууме. Остаток растирают и промывают метанолом с получением 1-(4-фторфенил)-4-(метансульфонил)пиперазина (39,35 г) в виде белых кристаллов.
1H ЯМР (CDCl3): 7,00 (m, 2Н), 6,90 (m, 2H), 3,40 (m, 4H), 3,20 (m, 4H), 2,83 (s, 3H).
Арил/гетероарилпиперазины и пиперидины, используемые в качестве исходных веществ, имелись в продаже или описаны в научной литературе.
1-(6-Хлорпиримидин-4-ил)-4-мезилпиперазин
Смесь 4,6-дихлорпиримидина (39,4 г), гидрохлорида 1-мезилпиперазина (55,7 г) и триэтиламина (116 мл) в этаноле (500 мл) перемешивают при температуре дефлегмации в течение 4 часов. Смесь затем перемешивают при комнатной температуре в течение 12 часов. Отделившееся твердое вещество собирают фильтрацией, суспензию промывают этанолом (2×80 мл, 160 мл), затем диэтиловым эфиром (150 мл) и сушат с получением 1-(6-хлорпиримидин-4-ил)-4-мезилпиперазина в виде кремового твердого вещества (71,9 г), т.пл. 200-202°С.
ЯМР (d6-ДМСО): 2,88 (s, 3Н), 3,18 (m, 4H), 3,80 (m, 4H), 7,04 (s, 1H), 8, 38 (m, 1H); m/z 277,3 (М+1).
Используя аналогичную методику, гидрохлорид 1-мезилпиперазина, CAS (161357-89-7), подвергают взаимодействию с подходящим хлорпиридином с получением следующих соединений:

2-(4-Пиперидинилокси)-5-хлорпиридин
1) NaH (2,88 г, 66 ммоль, 55% дисперсия в минеральном масле) перемешивают в безводном диметиловом эфире (DME) (200 мл) под аргоном. К суспензии NaH по каплям в течение периода 30 минут добавляют смесь 2,5-дихлорпиридина (8,87 г, 60 ммоль) и 4-гидроксипиперидина (6,67 г, 66 ммоль), растворенную в безводном DME (200 мл). После завершения добавления реакционную смесь нагревают до 82°С в течение 48 часов, поддерживая атмосферу аргона. Реакцию медленно останавливают водой перед удалением большей части ТГФ. Водный слой экстрагируют ДХМ (×3). Органические слои сушат с помощью Na2SO4 и выпаривают в вакууме с получением 2-(4-пиперидинилокси)-5-хлорпиридина в виде желтого масла (12,7 г, количественно).
1H ЯМР,(ДМСО): 8,17 (d, 1H), 7,76 (dd, 1H), 6, 81 (d, 1H), 4,96 (m, 1H), 2,93 (m, 2H), 2,53 (m, 2H), 1,91 (m, 2H), 1,46 (m, 2H); МС (ES+): 213,3 (МН+), 225,3 (MNa+).
Аналогичным способом получили 2-(4-пиперидинилокси)-5-бромпиридин МН + 257,3.
ПРИМЕР 4 - разделение
N-[(1S)-1-({[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил}метил)-4-(пиримидин-2-ил)бутил]-N-гидроксиформамид
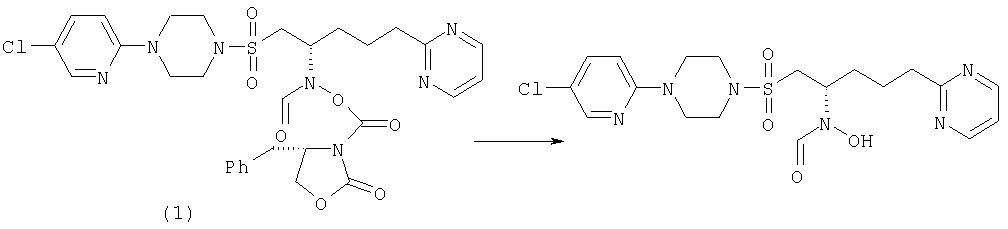
К карбамату 1 (3,8 г, 5,66 ммоль), растворенному в ТГФ (76 мл), добавляют метанол (76 мл) с последующим добавлением воды (38 мл) и к этому раствору добавляют моногидрат гидроксида лития (2,37 г, 56,6 ммоль). После перемешивания в течение 2 часов при комнатной температуре растворители удаляют при пониженном давлении, остаток растворяют в воде (250 мл), промывают этилацетатом (200 мл) и диэтиловым эфиром (2×250 мл). Добавляют насыщенный водный хлорид аммония до тех пор, пока водный слой не будет иметь приблизительно рН 8, и затем экстрагируют дихлорметаном (3×250 мл). Объединенные дихлорметановые экстракты сушат (MgSO4) и выпаривают с получением продукта в виде белого порошка (2,2 г, 83%). Хиральная ВЭЖХ с использованием колонки Chiralpak AS показала, что продукт выделен с 96% энантиомерным избытком (считают, что он имеет S-стереохимическую конфигурацию). Т.пл. (из EtOH) 124,5-126,5°С; [a]D25=-17,2 (МеОН); ЯМР (CDCl3): d 9,9 (шир. s, 1H)*; 8,7 (m, 2H); 8,5 (s, 1H)*; 8,1 (шир. s, 1Н); 8,0 (s, 1H)*; 7,5 (dd, 1H); 7,2 (m, 1H); 6,6 (d, 1H); 4,9 (m, 1H)*; 4,2 (m, 1H)*; 3,7-3,5 (m, 4H); 3,5 (m, 1H)*; 3,4-3,2 (m, 4H); 3,3 (m, 1H)*; 3,1-2,9 (m, 3Н); 2,0-1,6 (m, 4H). МС для C19H25ClN6O4S (M+H) вычислено 469, найдено 469.
* сигналы ротамеров
Стадия А

К обратному гидроксамату 2 (18,76 г, 40 ммоль), растворенному в дихлорметане (300 мл) и охлажденному до 0°С, добавляют триэтиламин (10,4 мл, 75 ммоль) с последующим добавлением (4S)-4-бензил-2-оксазолидинон-3-карбонилхлорида (10,55 г, 44 ммоль) [CAS номер 139149-49-8]. После перемешивания в течение 3 часов при -3-0°С смесь промывают водой (250 мл), сушат (MgSO4) и выпаривают с получением бежевой пены (27,1 г). Диастереоизомеры разделяют с применением препаративной ВЭЖХ, элюируя смесью этилацетат/EtOH (5%). Более полярный диастереоизомер выделили с выходом 35%. МС для C30H34ClN7O7S (M+H) вычислено 672, найдено 672.
Соединение 2 получили, используя способы, раскрытые в примере 2: (M+H 469), т.пл. 131-134°С; ЯМР (ДМСО): 9,8 (1Н, шир.), 8,7 (2Н, m), 8,3 и 7,9 (1Н, s), 8,1 (2Н, s), 7,6 (1Н, m), 6,9 (1Н, m), 4,1 (1Н, шир. m), 3,6 (4Н, m), 3,2 (6Н, m), 2,8 (2Н, m), 1,8(4Н, m).
ПРИМЕР 5
Способом, аналогичным способу, описанному в примере 4, получили следующие соединения:
N-[(1S)-1-({[4-(5-бромпиридин-2-ил)пиперазино]сульфонил}метил)-4-(пиридин-2-ил)бутил]-N-гидроксиформамид
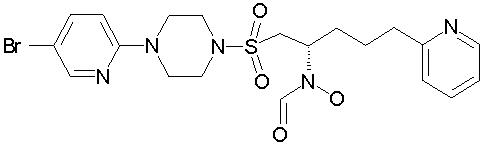
ЯМР (CDCl3): d 11,9 (шир. s, 1Н)*; 8,5 (s, 1Н)*; 8,5-8,4 (m, 1Н); 8,2 (m, 1Н); 8,1 (s, 1Н)*; 7,8-7,7 (m, 1Н); 7,6 (m, 1Н); 7,3-7,2 (m, 2Н); 6, 6 (m, 1Н); 5,0-4,9 (m, 1Н)*; 4,3-4,2 (m, 1Н)*; 3,7-3,6 (m, 4Н); 3,6 (m, 1Н)*; 3,4-3,3 (m, 4Н); 3,3 (m, 1Н)*; 3,1 (dd, 1Н)*; 2,9 (m, 1Н)*, 2,9-2,8 (m, 2Н); 2,1-1,6 (m, 4Н). МС для С20Н26BrN5O4S (M+H) вычислено 514, найдено 514.
* сигналы ротамеров
[а]D25=-14 (c=2,3, МеОН)
Рацемическое исходное вещество получили с использованием способа, описанного в примере 2. M+H=512/514.
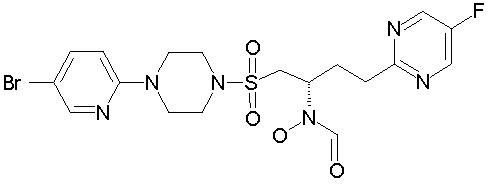
1H ЯМР (ДМСО, 373°К (100°С)); 9,44 (шир. s, 1H), 8,70 (s, 2H), 8,10 (d, 1H, J=2,6 Гц), 8,05 (шир. s, 1H), 7,57 (dd, 1H, J=9,1, 2, 6 Гц), 6,86 (d, 1H, J=9,1 Гц), 4,40 (шир. s, 1H), 3,59 (dd, 4H, J=5,3, 5,0 Гц), 3,47 (dd, 1H, J=14,6, 7,4 Гц), 3,28 (dd, 4H, J=5,3, 5,0 Гц), 3,24 (dd, 1H, J=14,6, 4,3 Гц), 2,93 (m, 2H), 2,16 (m, 2H).
MC (ESI (ионизация электрораспылением)): 473 (МН+)
аd=-11,03(МеОН, с=1,242)
Рацемическое исходное вещество получили в примере 2.
N-[(1S)-1-({[4-(4-фторфенил)пиперазино]сульфонил}метил)-4-(пиримидин-2-ил)бутил]-N-гидроксиформамид
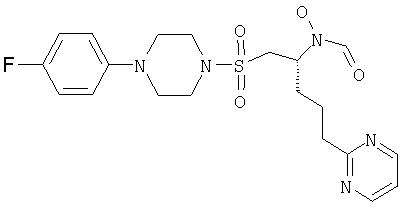
М+Н 452,44; ЯМР (CDCl3) d 9,9 (шир. s, 1H)*; 8,7 (m, 2H); 8,5 (s, 1H)*; 8,05 (s,1H)*; 7,2 (m, 1H); 7,0-6,9 (m, 4H); 4,9 (m, 1H)*; 4,2 (m, 1H)*; 3,5-3,4 (m, 4H); 3,5 (m, 1H)*; 3,2-3,1 (m, 4H); 3,3 (m, 1H)*; 3,1-2,9 (m, 3H); 2,0-1,6 (m, 4H).
* сигналы ротамеров
Рацемическое исходное вещество получали, используя способ, описанный в примере 3. ЯМР (ДМСО) 10,0 (1H, шир. s), 8,6 (2H, m), 8,2 (1H, d), 7,2 (1H, m), 6,9 (4H, m), 4,9 и 4,2 (1H, шир.), 3,4 (6Н, m), 3,0 (6H, m), 1,9 (4H, m).
ПРИМЕР 6 - хроматографическое разделение
N-[(1S)-1-({[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил}метил)-3-(пиримидин-2-ил)пропил]-N-гидроксиформамид и N-[(1R)-1-({[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил}метил)-3-(пиримидин-2-ил)пропил]-N-гидроксиформамид

N-[1-({[4-(5-хлорпиридин-2-ил)пиперазино]сульфонил}метил)-3-(пиримидин-2-ил)пропил]-N-гидроксиформамид, полученный в рацемической форме, разделяют на индивидуальные энантиомерные формы с помощью хроматографического разделения на колонке, набитой Chiralpak AD №AD00CJ-НК002, элюируя этанолом. Биологической активностью обладает соединение, элюированное с колонки вторым; признано, что оно имеет S-стереохимичесмкую конфигурацию.
Энантиомер, элюированный 1-м MH+ 455.
Энантиомер, элюированный 2-м МН+ 455.
Рацемическое исходное вещество получали, используя способ, приведенный в примере 2.
МН+=455. ЯМР (ДМСО) 9,9, 9,6 (1Н, шир. s), 8,6 (2Н, m) 8,3 и 7,9 (1Н, s), 8,1 (1Н, dd), 7,3 (1Н, m), 6,9 (1Н, d), 4,7 и 4,2 (1Н, широкий m), 3,6 (4H, m), 3,4-3,2 (6H, m), 2,8 (2Н, m), 2,1 (2Н, m).
ПРИМЕР 7 - дополнительные примеры хроматографического разделения
Следующие соединения разделяли, используя условия, приведенные в примере 6:
N-[(1S)-1-({[4-(5-трифторметилпиридин-2-ил)пиперазино]сульфонил}метил)-3-(пиримидин-2-ил)пропил]-N-гидроксиформамид и N-[(1R)-1-({[4-(5-трифторметилпиридин-2-ил)пиперазино]сульфонил}метил)-3-(пиримидин-2-ил)пропил]-N-гидроксиформамид
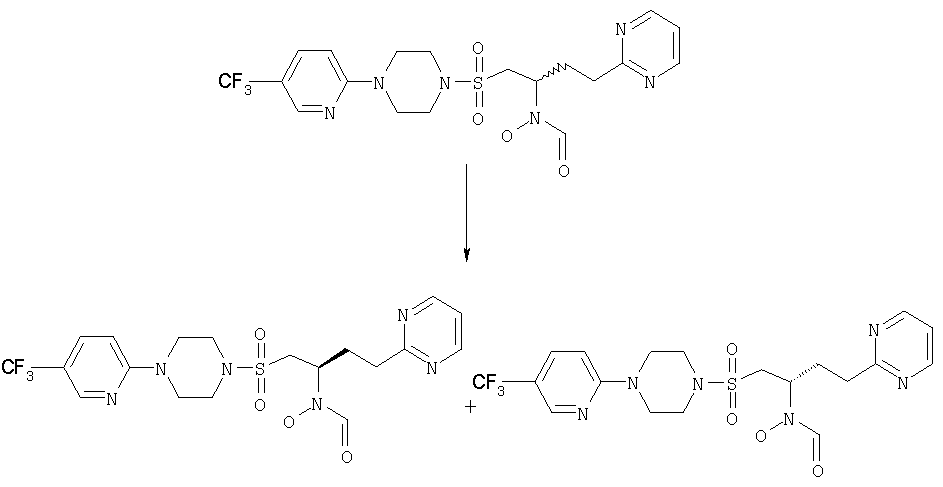
Энантиомер, элюированный 1-м М+Н 489,5.
Энантиомер, элюированный 2-м М+Н 489,5.
Рацемическое исходное вещество получили в примере 3.
N-[(1S)-1-({[4-(5-бромпиридин-2-ил)пиперазино]сульфонил}метил)-4-(пиримидин-2-ил)бутил]-N-гидроксиформамид и N-[(1R)-1-({[4-(5-бромпиридин-2-ил)пиперазино]сульфонил}метил)-4-(пиримидин-2-ил)бутил]-N-гидроксиформамид
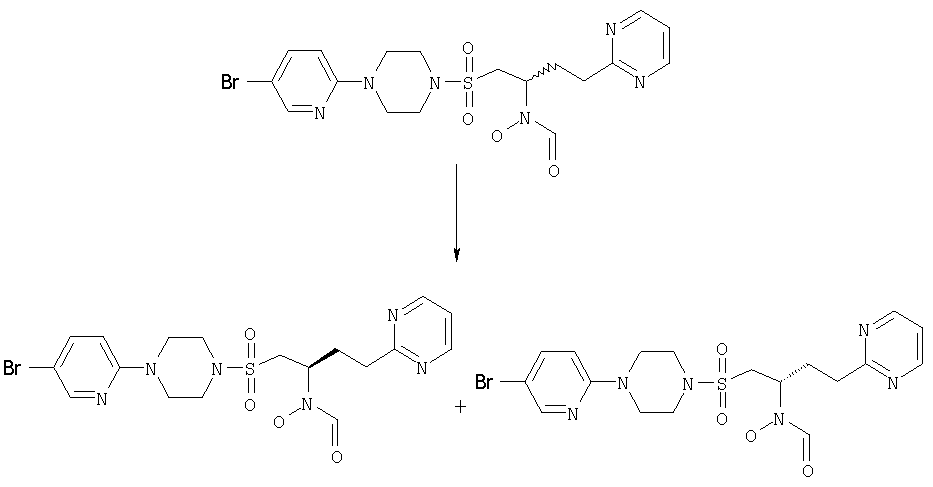
Энантимер, элюированный 1-м М+Н 513/515.
Энантиомер, элюированный 2-м М+Н 513/515.
Рацемическое исходное вещество получали, используя способ, описанный в примере 2: М+Н 513/515.
ПРИМЕР 8
Были получены следующие соединения




В таблице выше:
** показывает соединение (S-энантиомер), полученное способом из примера 6 с использованием колонки Chiralpak AD (250 мм × 4,6 мм) №ADooCE-JJ122 и элюента MeOH/MeCN 15/85;
Диастереоизомеры А и В относятся к порядку элюирования с колонки с диоксидом кремния, элюируемой 3-5% этанолом в дихлорметане (диастереоизомер А является первой элюируемой фракцией, диастереоизомер В - второй).
ПРИМЕР 9
Приводятся данные ЯМР для следующих соединений, перечисленных в примере 8:
N-[(1S)-1-({[4-(5-трифторметилпиридин-2-ил)пиперазино]сульфонил}метил)-4-(пиримидин-2-ил)бутил]-N-гидроксиформамид

ЯМР (CDCl3): δ 10,1 (шир. s, 1H)*; 8,7 (m, 2H); 8,5 (s, 1H)*; 8,4 (шир. s, 1H); 8,1 (s, 1H)*; 7,7 (dd, 1H); 7,2 (m, 1H); 6,7 (d, 1H); 4, 9 (m, 1H)*; 4,2 (m, 1H)*; 3,9-3,7 (m,4H); 3,6 (m, 1H)*; 3,4-3,2 (m, 4H); 3,3 (m, 1H)*; 3,1-2,9 (m, 3Н); 2,0-1,6 (m, 4H).
* сигналы ротамеров
N-({[4-Фторфенилпиперазино]сульфонил}метил)-3-(5-фторпиримидин-2-ил)пропил]-N-гидроксиформамид
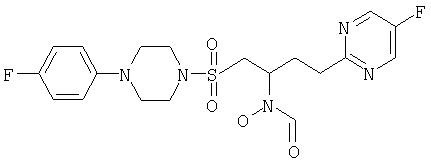
1H ЯМР (ДМСО, 373°К (100°С)): 9,46 (шир. s, 1H), 8,73 (s 2H), 7,08-6,96 (m, 4H), 4,42 (шир. s, 1H, 3,50 (dd, J=14,8, 7,5 Гц, 1H), 3,35 (m, 4H), 3,28 (dd, J=14, 8, 4,4 Гц, 1H), 3,18 (m, 4H), 2,97 (m, 2H), 2,21 (m, 2H).
N-[{1R или 1S)-({[4-Хлорфенилпиперазино]сульфонил}метил)-3-[(3R или 3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид (индивидуальный диастереоизомер А)
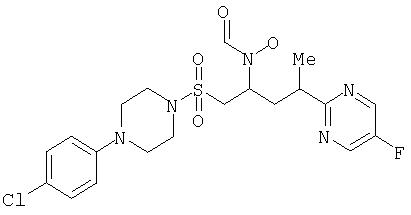
1H ЯМР (CDCl3) (2 ротамера в приблизительно равных количествах): 8,72 (s, 0,5Н),8,57 (d, 2H), 8,25 (s, 0,5H), 7,89 (s, 0,5H), 7,23 (dd, 2H), 6,83 (dd, 2H), 4,94 (секст., 0,5Н), 4,30 (m, 0,5H), 3,57 (dd, 0,5H), 3,44 (m, 2H), 3,37 (m, 2,5H), 3,16 (m, 5,5H), 3,02 (dd, 0,5H), 2,52 (ddd, 0,5H), 2,35 (ddd, 0,5H), 2,02 (dt, 0,5H), 1,89 (ddd, 0,5H), 1,40 (dd, 3H).
N-[(1R или 1S)-({[4-Бромфенилпиперазино]сульфонил}метил)-3-[(3R или 3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид (индивидуальный диастереоизомер А)
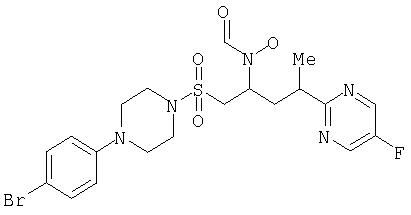
1H ЯМР (CDCl3) (2 ротамера в приблизительно равных количествах): 8,72 (s, 0,5Н), 8,57 (d, 2H), 8,25 (s, 0,5H), 7, 89 (s, 0,5H), 7,38 (dd, 2H), 6,80 (dd,2H), 4,94 (секст, 0,5Н), 4,30 (m, 0,5H), 3,57 (dd, 0,5H), 3,44 (m, 2H), 3,37 (m, 2,5H), 3,16 (m, 5,5H), 3,02 (dd, 0,5H), 2,52 (ddd, 0,5H), 2,35 (ddd, 0,5H), 2,02 (dt, 0,5H), 1,89 (dt, 0,5H), 1,40 (dd, 3H).
N-[(1R или 1S)-({[4-Хлорфенилпиперидино]сульфонил}метил)-3-[(3R или 3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид (индивидуальный диастереоизомер А)

1H ЯМР (CDCl3) (2 ротамера в приблизительно равных количествах): 8,69 (s, 0,5Н), 8,57 (d, 2H), 8,25 (s, 0,5H), 7,89 (s, 0,5H), 7,27 (затемненный), 7,13 (dd, 2H), 4,91 (секст., 0,5Н), 4,30 (m, 0,5H), 3,87 (m, 2H), 3,57 (dd, 0,5H), 3,35 (dd, 0,5H), 3,18 (m, 1,5H), 3,00 (dd, 0,5H), 2,85 (m, 2H), 2,55 (m, 1,5H), 2,35 (ddd, 0,5H), 2,06 (dt, 0,5H) 1,88 (m, 2,5H), 1,7 (затемненный), 1,40 (dd, 3H).
N-[[1R или 1S)-({[3, 4-Дихлорфенилпиперазино]сульфонил}метил)-3-[(3R или 3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид (индивидуальный диастереоизомер А)

1H ЯМР (CDCl3) (2 ротамера в приблизительно равных количествах): 8,62 (s, 0,5Н), 8,55 (d, 2H), 8,22 (s, 0,5H), 7, 86 (s, 0,5H), 7,28 (m, 1H); 6,95 (m, 1H), 6,73 (m, 1H), 4,92 (секст., 0,5Н), 4,30 (m, 0,5H), 3,57 (dd, 0,5H), 3,44 (m, 2H), 3,37 (m, 2,51-1), 3,16 (m, 5,5H), 3,02 (dd, 0,5H), 2,52 (ddd, 0,5H), 2,37 (ddd, 0,5H), 2,04 (dt, 0,5H), 1,89 (dt, 0,5H), 1,40 (dd, 3H).
N-[(1R или 1S)-({[4-(5-Цианопиридин-2-ил)пиперазино]сульфонил}метил)-3-[(3R или 3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид (индивидуальный диастереоизомер А)

1H ЯМР (CDCl3) (2 ротамера в приблизительно равных количествах): 8,72 (s, 0,5Н), 8,55 (s, 2H), 8,41 (s, 1H), 8,22 (s, 0,5H), 7,86 (s, 0,5H), 7,65 (m, 1H), 6,61 (dd, 1H), 4,92 (m, 0,5H), 4,30 (m, 0,5H), 3,78 (m, 4H), 3,57 (dd, 0,5H), 3,38 (m, 2H), 3,30 (m, 2,5H), 3,16 (m, 1,5H), 3,02 (dd, 0,5 H), 2,52 (m, 0,5H), 2,37 (m, 0,5H), 2,04 (dt, 0,5H), 1,84 (dt, 0,5H), 1,40 (dd, 3H).
N-[(1S)-({[4-(4-Фторфенилпиперазино]сульфонил}метил)-3-[(3S)-(5-фторпиримидин-2-ил)бутил]-N-гидроксиформамид

1H ЯМР (ДМСО-d6); 9,9, 9,53 (2s, 1H), 8,78 (s, 2H), 7,98 (d, 1H), 7,12-6,91 (m, 4H), 4,8, 4,17 (2s, 1H), 3,13 (m, 4H), 3,0 (m, 1H), 1,86 (m, 1H), 1,22 (m, 3H).
ПРИМЕР 10
1-({[4-(4-Хлорфенил)пиперазин-1-ил]сульфонил}метил)-3-(5-хлорпиридин-3-ил)пропил](гидрокси)формамид

К муравьиной кислоте (400 мкл, 10,8 ммоль) при 0°С добавляют уксусный ангидрид (102 мкл, 1,1 ммоль) и смесь затем перемешивают при комнатной температуре в течение 15 минут. Смесь затем снова охлаждают до 0°С и по каплям с помощью шприца добавляют раствор 1-(4-хлорфенил)-4-{[4-(5-хлорпиридин-3-ил)-2-(гидроксиамино)бутил]сульфонил}пиперазина (100 мг, 0,22 ммоль) в ТГФ. После перемешивания при комнатной температуре в течение 1,5 часа, летучие вещества удаляют в вакууме и остаток подвергают азеотропной перегонке с толуолом (2 мл). Остаток затем растворяют в метаноле (5 мл) и перемешивают при 40°С в течение 1 часа. После охлаждения до комнатной температуры растворитель выпаривают и остаток растворяют в метаноле (0,5 мл). Затем добавляют диэтиловый эфир (5 мл) и мутную суспензию перемешивают при комнатной температуре в течение 1 часа. Осевшее твердое вещество отфильтровывают, промывают диэтиловым эфиром и сушат в вакууме с получением указанного в заголовке соединения в виде твердого вещества белого цвета с желтоватым оттенком (48 мг, 0,099 ммоль).
1H ЯМР (ДМСО, 373°К (100°С)): 9,55 (шир. s, 1H), 8,43 (d, 1H), 8,41 (d, 1H), 8,17 (шир. s, 1H), 7,76 (dd, 1H), 7,25 (m, 2H), 6,96 (m, 2H), 4,35 (шир. s, 1H), 3,49 (dd, 1H), 3,34 (m, 4H), 3,25 (m, 5H), 2,67 (m, 2H), 2,02 (m, 2H).
MC (ESI): 487,06, 489,04, 490,08 {MH+ 2 × Cl).
Исходное вещество получали следующим образом:
(I) этил-3-(5-хлорпиридин-3-ил)пропаноат

К перемешиваемому раствору этил-(2Е)-3-(5-хлорпиридин-3-ил)проп-2-еноата (338 мг, 1,6 ммоль) [CAS номер 163083-45-2] в безводном этаноле (10 мл) при 0°С в атмосфере аргона добавляют твердый борогидрид натрия (67 мг, 1,75 ммоль). Реакционную смесь оставляют нагреваться до комнатной температуры и перемешивают в течение четырех часов, после чего добавляют дополнительное количество борогидрида натрия (67 мг, 1,75 ммоль). После перемешивания в течение дополнительных 18 часов добавляют насыщенный водный раствор хлорида аммония (5 мл). Летучие вещества удаляют в вакууме и остаток распределяют между водой (10 мл) и этилацетатом (10 мл). Слои разделяют и водную фазу экстрагируют этилацетатом (3×10 мл). Объединенные органические экстракты затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Флэш-хроматография (силикагель, от 20 до 100% этилацетат в гексане) дала указанное в заголовке соединение (132 мг, 0,62 ммоль) и насыщенный спирт (70 мг).
1H ЯМР (CDCl3): 8,43 (m, 1Н), 8,34 (m, 1H), 7,55 (m, 1H), 4,16 (q, 2H), 2,96 (dd, 2H), 2,63 (dd, 2H).
(II) 1-{[4-(4-Хлорфенил)пиперазин-1-ил]сульфонил}-4-(5-хлорпиридин-3-ил)бутан-2-он
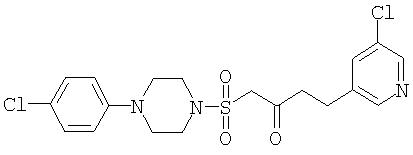
К перемешиваемому раствору 1-(4-хлорфенил)-4-(метилсульфонил)пиперазина (235 мг, 0,85 ммоль) в безводном ТГФ (7,5 мл) при -10°С в атмосфере аргона по каплям в течение 4 минут добавляют раствор LiHMDS (1,71 мл 1,0 M раствора в ТГФ, 1,71 ммоль). Раствор затем перемешивают при этой температуре в течение 40 минут. Затем по каплям через канюлю в течение 5 минут добавляют раствор этил-3-(5-хлорпиридин-3-ил)пропаноата (201 мг, 0,94 ммоль) в ТГФ (1 мл). Реакционную смесь перемешивают при -10°С дополнительно в течение 30 минут перед тем, как остановить реакцию насыщенным водным раствором хлорида аммония (5 мл). Летучие вещества удаляют в вакууме и остаток экстрагируют CH2Cl2 (3×5 мл). Объединенные органические экстракты промывают водой (10 мл) и рассолом (10 мл) перед сушкой (MgSO4), фильтруют и концентрируют в вакууме. Флэш-хроматография (силикагель, 50% этилацетат в гексане) дала указанное в заголовке соединение (228 мг, 0, 52 ммоль) и рекуперированный этил-3-(5-хлорпиридин-3-ил)пропаноат (74 мг, 0,35 ммоль).
1H ЯМР (CDCl3): 8,46 (m,1H), 8,38 (m, 1H), 7,58 (m,11-1), 7,21 (m, 2H), 6, 83 (m, 2H), 3,96 (s, 2H), 3,37 (m, 4H), 3,17 (m, 6H), 2,95 (dd, 2H),
MC (ESI): 442,07, 444,06, 445,1 (MH+ 2 × Cl).
(III) 1-{[4-(4-Хлорфенил)пиперазин-1-ил]сульфонил}-4-(5-хлорпиридин-3-ил)бутан-2-ол

К перемешиваемому раствору 1-{[4-(4-хлорфенил)-пиперазин-1-ил]сульфонил}-4-(5-хлорпиридин-3-ил)бутан-2-она (228 мг, 0,51 ммоль) в системе смеси растворителей CH2Cl2/MeOH (1:1, 5 мл) при комнатной температуре добавляют твердый борогидрид натрия одной порцией. Реакционную смесь перемешивают в течение 40 минут перед тем, как остановить реакцию водной соляной кислотой (1 M, 2 мл). Слои затем разделяют и водную фазу экстрагируют CH2Cl2 (3×5 мл). Объединенные органические экстракты сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенный продукт затем фильтруют через пробку из силикагеля, элюируя 50% этилацетатом в гексане, с получением указанного в заголовке соединения (111 мг, 0,25 ммоль).
1H ЯМР (CDCl3): 8,47 (m, 1H), 8,40 (m, 1H), 7,59 (m, 1H), 7,21 (m, 2H), 6,86 (m, 2H), 4,21 (m, 1H), 3,45 (m, 4H), 3,24 (m, 4H), 3,11 (m, 2H), 2,88 (m, 2H), 1,89 (m, 2H).
(IV) 1-(4-Хлорфенил)-4-{[(1Е)-4-(5-хлорпиридин-3-ил)бут-1-енил]сульфонил}пиперазин
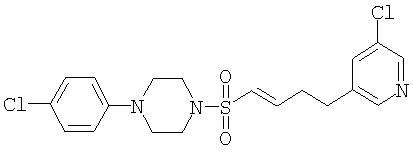
К перемешиваемому раствору 1-{[4-(4-хлорфенил)пиперазин-1-ил]сульфонил}-4-(5-хлорпиридин-3-ил)бутан-2-ола (111 мг, 0,25 ммоль) в безводном CH2Cl2 (2,5 мл) при комнатной температуре в атмосфере аргона добавляют гидрохлорид триметиламина (2 мг, 0,02 ммоль), триэтиламин (52 мкл, 0,25 ммоль), затем метансульфонилхлорид (21 мкл, 0,25 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 30 минут, затем реакцию останавливают путме добавления насыщенного водного раствора бикарбоната натрия (5 мл). Слои разделяют и водную фазу экстрагируют этилацетатом (3×6 мл). Объединенные органические экстракты затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем растворяют в CH2Cl2 (2,5 мл) и обрабатывали триэтиламином (100 мкл, 1,36 ммоль). Через 30 минут реакцию останавливают путем добавления насыщенного водного раствора бикарбоната натрия (5 мл). Слои разделяют и водную фазу экстрагируют этилацетатом (3×6 мл). Объединенные органические экстракты затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Неочищенное вещество использовали на следующей стадии.
МС (ESI): 446,06, 428,06, 430,07 (MH+ 2×Cl).
(V) 1-{4-Хлорфенил}-4-{[4-(5-хлорпиридин-3-ил}-2-(гидроксиамино)бутил]сульфонил}пиперазин

К перемешиваемому раствору 1-(4-хлорфенил)-4-{[(1Е)-4-(5-хлорпиридин-3-ил)бут-1-енил]сульфонил}пиперазина (неочищенного с предыдущей стадии) в ТГФ (10 мл) при комнатной температуре добавляют раствор гидроксиламина (2 мл, 50%-ный водный раствор в воде). Реакционную смесь перемешивают в течение 3 часов при комнатной температуре перед тем, как остановить реакцию насыщенным водным раствором хлорида аммония (5 мл). Слои разделяют и водную фазу экстрагируют этилацетатом (3×10 мл). Объединенные органические экстракты затем сушат (MgSO4), фильтруют и концентрируют в вакууме. Остаток затем очищают с помощью флэш-хроматографии (диоксид кремния, 100% этилацетат) с получением указанного в заголовке соединения (100 мг, 0,22 ммоль).
Данные по биологической активности заявленных соединений
Реферат
Настоящее изобретение относится к пиперидин- и пиперазин-замещенным N-гидроксиформамидам общей формулы I или их фармацевтически приемлемым солям
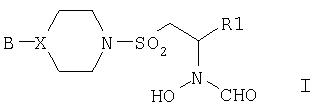
Формула
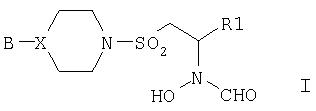


Комментарии