Новые промежуточные продукты и способ получения производных камптотецина (срт-11) и родственных соединений - RU2164917C2
Код документа: RU2164917C2
Чертежи


Описание
В данном изобретении описываются и заявляются новые промежуточные продукты и способы синтеза производных камптотецина, например иринотекана, и других соединений, имеющих отношение к синтезу СРТ-11. Описываются также родственные способы и соединения, например новый способ получения маппицина.
Уровень техники
Соединение, которому дано обозначение 14СРТ в этом документе, упоминается в М.
Shamma, D.A. Smithers, V.St. George, Tetrahedron, 1973, 1949-1954.
Асимметрический синтез этого соединения, 14СРТ, описывается в следующих публикациях (сгруппированы автором):
Группа 1.
Н. Terasawa, М. Sugimori, A. Ejima, Y. Tagawa, Chem. Pharm. Bull., 1989, 37, 3382-3385.
A. Ejima, Н. Terasawa, М. Sugimori, Н. Tagawa, J.C.S. Perkin I, 1990, 27-31.
H. Tagawa, Н. Terasawa, A. Ejima, US Patent 4778891 (Oct. 18, 1988).
H. Tagawa, H. Terasawa, A. Ejima, EP 220601 (Oct 14, 1986).
Группа 2.
M.C. Wani, A.W. Nicholas, M.E. Wall, J. Med. Chem. 1987, 2317-2319.
M.C. Wani, A.W. Nicholas, M.E. Wall, US Patent 5053512 (Oct. 1, 1990).
M. E. Wall, M. C. Wani, A.W. Nicholas, G. Manikumar, US Patent 4894456 (Jan. 16, 1990).
M. E. Wall, M.C. Wani, A.W. Nicholas and G. Manikumar, WO 90/03169 (Sep. 28, 1988).
Предпосылки
создания изобретения
Производные камптотецина, например иринотекан, являются эффективными противораковыми лекарственными средствами. В данном изобретении описывается эффективный способ
синтетического получения различных производных капмтотецина, включая иринотекан или СРТ-11, и других полезных соединений, подобных маппицину.
Краткое изложение сущности изобретения
Данное изобретение включает соединения, способы, реакции и реагенты, показываемые здесь в СХЕМАХ, формулах и фигурах. Эти соединения, способы, реакции и реагенты можно использовать для получения
производных камптотецина, например СРТ-11, и других родственных соединений, например маппицина.
Конкретными соединениями, выбранными из соединений, описанных и обозначенных в данном
описании, являются соединения, обозначенные в СХЕМАХ как 2G, 3G, 4G, 5G, 6G, 7GG, 7GA, 8GG, 8GA, 8GB, 9GG, 9GA, 10G, 10G(S), 10G(R), 11G, 11G(S), 11G(R), 12GA-1, 12GA-1(S), 12GA-1(R), 12GA-2,
12GA-2(S), 12GA-2(R), 12GB-1, 12GB-1(S), 12GB-1(R), 12GB-2, 12GB-2(S), 12GB-2(R), 12G, 12G(S), 12G(R), 13G, 13G(S) или 13G(R),
где R1 представляет любой необязательно замещенный
C1-8 алкил, включая низший алкил, C3-10 циклоалкил, низший алкил- C3-10 циклоалкил, алкенил, арил, замещенный арил, алкиларил или замещенный алкиларил, включая бензил
или замещенный бензил;
где R2 представляет H,
а) любой необязательно замещенный алкил, включая C1-8 алкил, алкиларил, включая C1-6 алкиларил, C1-8 алкил-C6 арил, замещенный бензил и незамещенный бензил;
б) -C(O)-R3 или
в) -C(R7)2-O-R3, где каждый R7
независим от другого;
где R3 представляет H, необязательно замещенный C1-8 алкил, включая низший алкил, циклоалкил, алкенил, арил, замещенный арил и алкиларил, или
замещенный алкиларил, включая бензил и замещенный бензил;
где R4 представляет H, необязательно замещенный C1-8 алкил, включая низший алкил, C3-10 циклоалкил,
низший алкил-C3-10 циклоалкил, алкенил, арил, замещенный арил, алкиларил или замещенный алкиларил, включая бензил и замещенный бензил;
где R5 представляет H,
необязательно замещенный C1-8 алкил, включая низший алкил, арил, замещенный арил, или две группы R5 могут быть соединены с образованием циклопентана, или циклогексана, или их
замещенных производных;
где R6 представляет необязательно замещенный C1-8 алкил, низший алкил, включая этил, арил, замещенный арил, алкиларил, замещенный алкиларил,
включая бензил и замещенный бензил, C3-10 циклоалкил, низший алкил-C3-10 циклоалкил, гетероарил или замещенный гетероарил,
где R7 независимо представляет H,
необязательно замещенный C1-8 алкил, включая низший алкил, арил, замещенный арил, алкиларил, замещенный алкиларил, или два R7 могут быть соединены с образованием циклопентана,
или циклогексана, или их замещенных производных;
где R8 представляет необязательно замещенный C1-6 алкил, включая низший алкил, включая трет-бутил, C3-10
циклоалкил, низший алкил-C3-10 циклоалкил, алкенил, арил, замещенный арил, алкиларил или замещенный алкиларил, включая бензил и замещенный бензил.
Другие конкретные соединения по изобретению выбраны из соединений, описанных и обозначенных в описании как 2СРТ, 3СРТ, 4CPT, 5CPT, 6CPT, 7CPT, 7CPTA, 8CPTG, 8CPTA, 8CPTAB, 9CPTG, 9CPTA, 9CPTB, 10CPT, 10CPT(S), 10CPT(R), 11CPT, 11CPT(S), 11CPT(R), 12CPTA-1, 12CPTA-1(S), 12CPTA-1(R), 12CPTA-2, 12CPTA-2(S), 12CPTA-2(R), 12CPTB-1, 12CPTB-1(S), 12CPTB-1(R), 12CPTB-2, 12CPTB-2(S), 12CPTB-2(R), 12CPT, 12CPT(S), 12CPT(R), 13CPT, 13CPT(S) и 13CPT(R), где R1-R9 определяются выше.
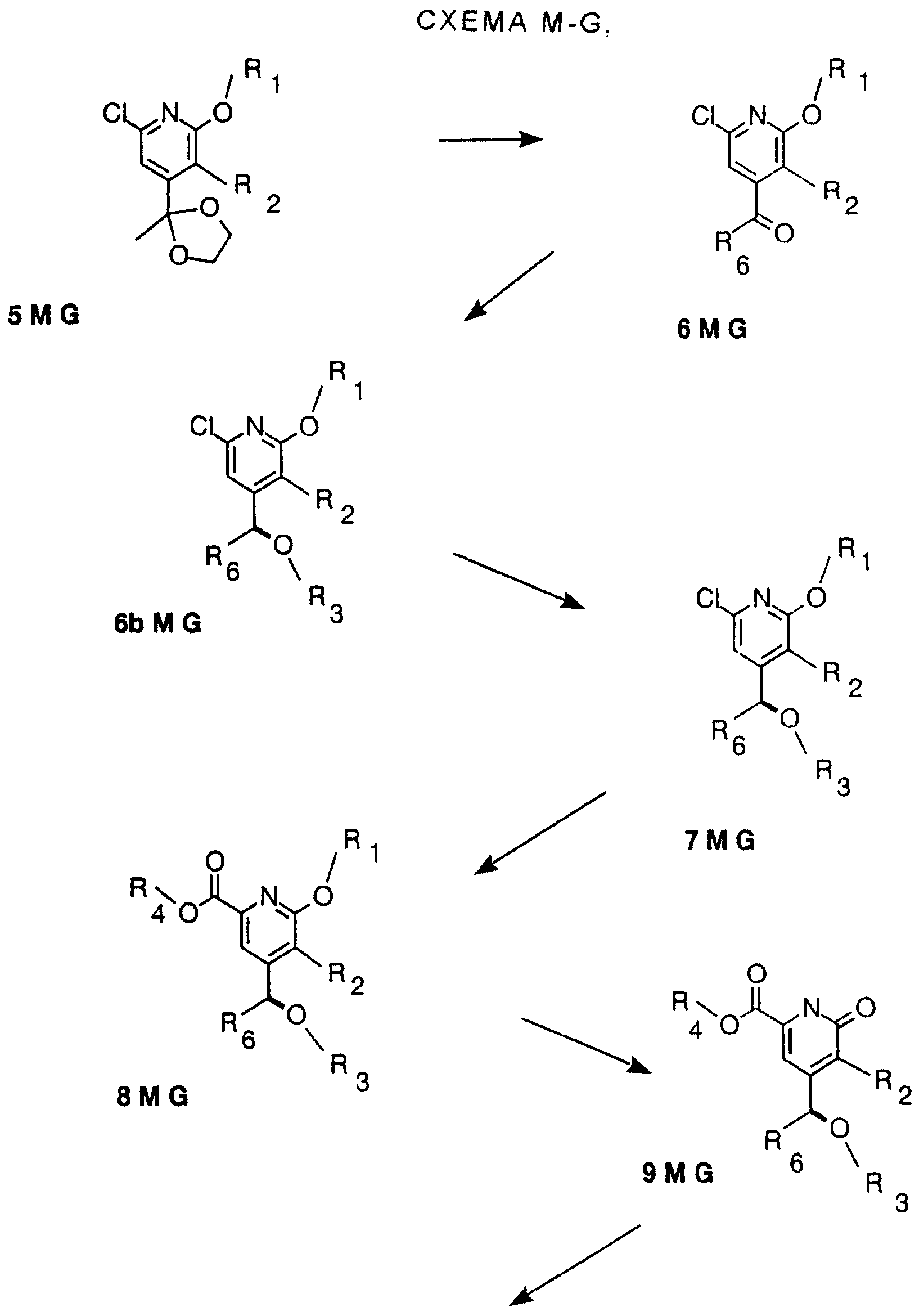
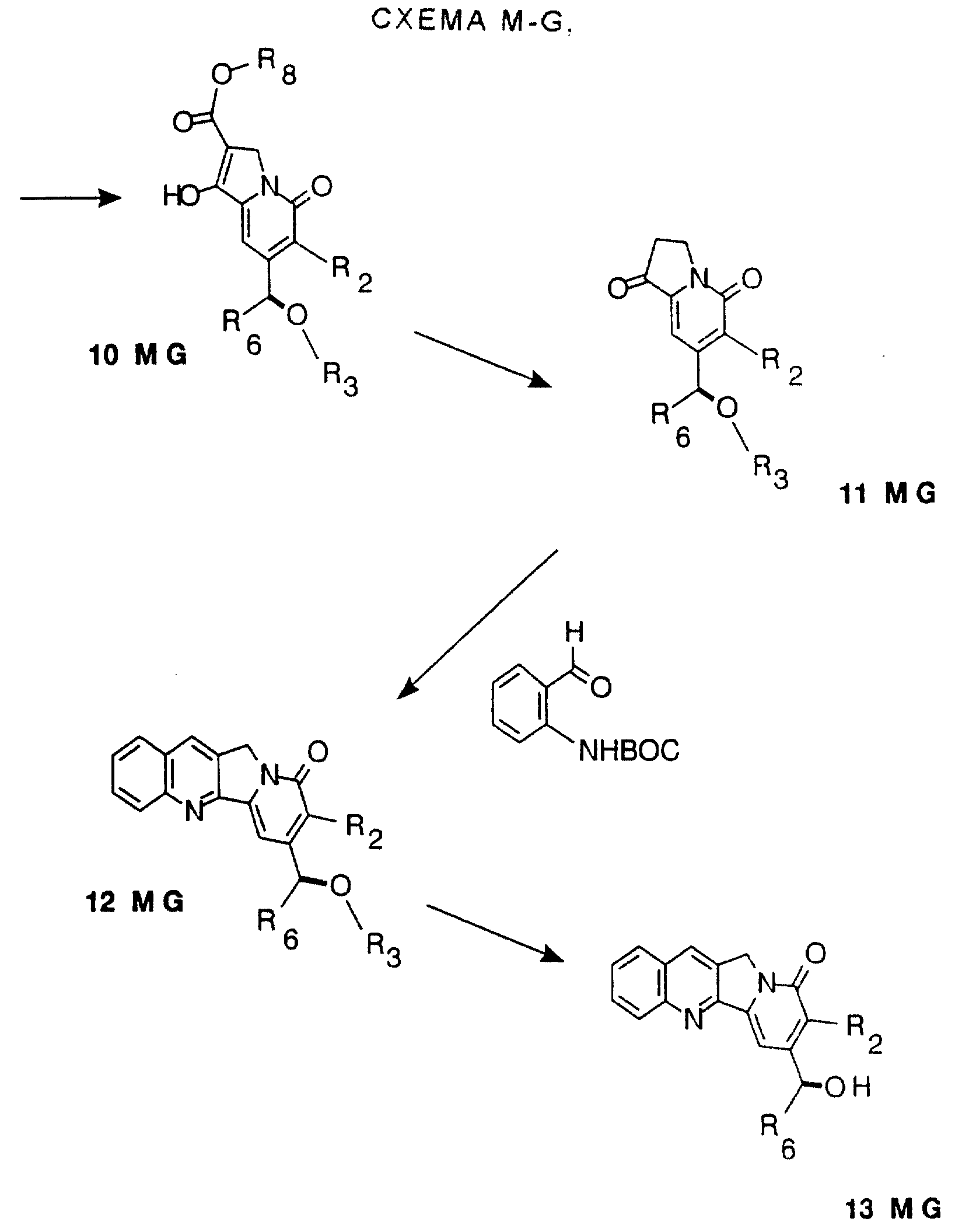
Другие конкретные соединения по изобретению выбраны из соединений, описанных и обозначенных в описании как 6MG, 7MG, 8MG, 9MG, 10MG, 11MG, 12MG, 13MG за исключением 13MG, у которого R6 представляет C1-2 алкил, где радикалы имеют такие же значения, как указанные выше радикалы.
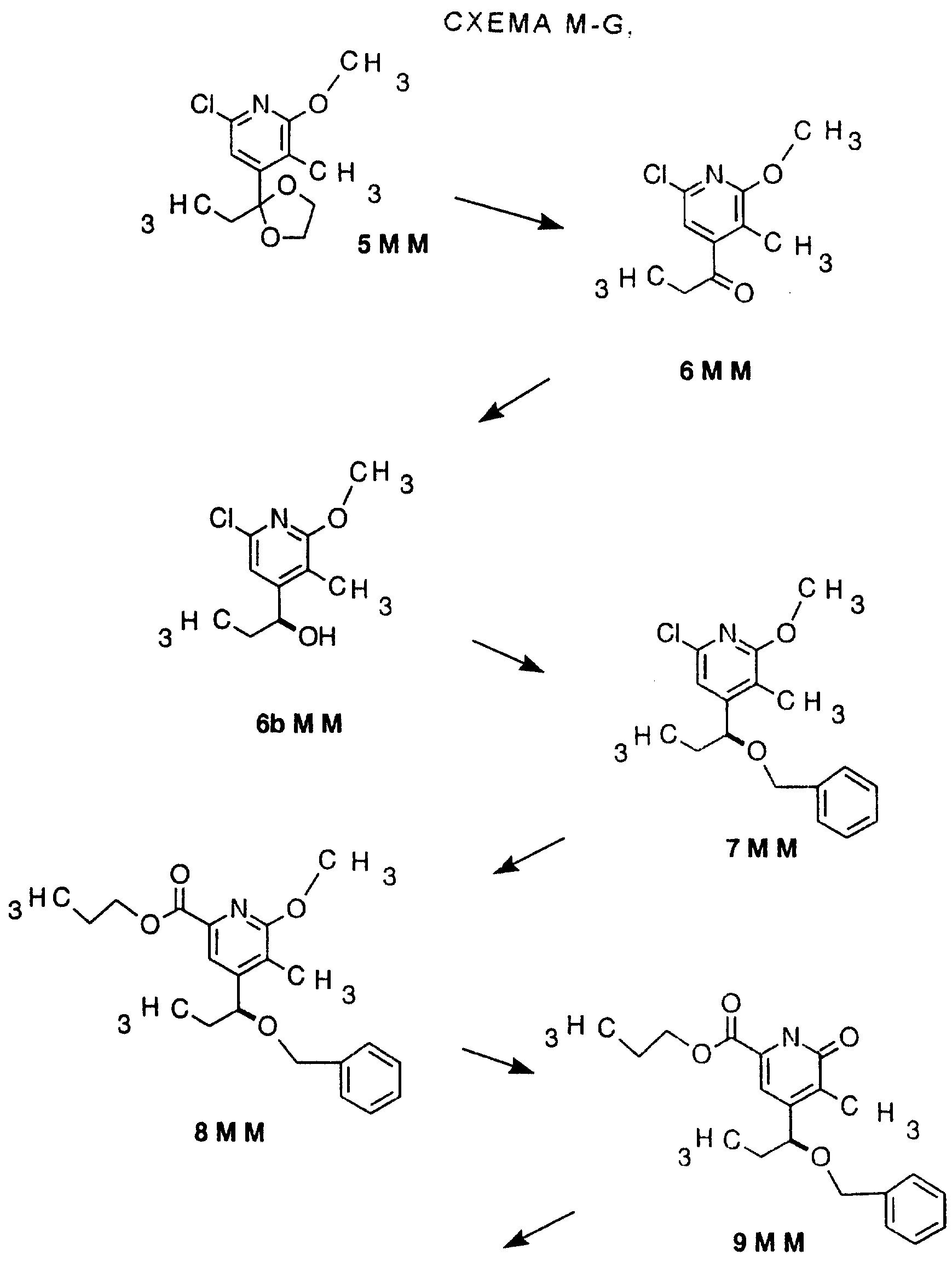
Другие конкретные соединения по изобретению выбраны из соединений, описанных и обозначенных в описании как 5ММ, 6ММ, 7ММ, 8ММ, 9ММ, 10ММ, 11ММ или 12ММ.
Кроме соединений в этом изобретении описываются и заявляются также различные способы, обозначенные как СТАДИИ. Эти СТАДИИ включают СТАДИИ, описанные и обозначенные в описании как СХЕМА G, включающая СТАДИЮ 2, или СТАДИЮ 3, или СТАДИЮ 4, или СТАДИЮ 5, или СТАДИЮ 5а, или СТАДИЮ 5b, или СТАДИЮ 6, или СТАДИЮ 7GG, или СТАДИЮ 7GA, или СТАДИЮ 8GG, или СТАДИЮ 8GA, или СТАДИЮ 8GB, или СТАДИЮ 9GG, или СТАДИЮ 9GA, или СТАДИЮ 9GB, или СТАДИЮ 10GG, или СТАДИЮ 10GA, или расщепление на энантиомеры СТАДИИ 10, или СТАДИЮ 11, или СТАДИЮ 12, или СТАДИЮ 13, или СТАДИЮ 14, или любое их сочетание, содержащее две или более СТАДИЙ.
Кроме того, описываются и заявляются СТАДИИ, описанные и обозначенные в описании как СХЕМА CPT, включающая СТАДИЮ 7G или СТАДИЮ 7А, или СТАДИЮ 8G, или СТАДИЮ 8А, или СТАДИЮ 8В, или СТАДИЮ 9G, или СТАДИЮ 9А, или СТАДИЮ 9В, или СТАДИЮ 10G, или СТАДИЮ 10А, или СТАДИЮ 11, или СТАДИЮ 12, или СТАДИЮ 13, или СТАДИЮ 14, или любую комбинацию их, содержащую две или более СТАДИЙ.
Кроме того, описываются и заявляются СТАДИИ, описанные и обозначенные в описании как СХЕМА M-G, включающая СТАДИЮ 5, или СТАДИЮ 6, или СТАДИЮ 7, или СТАДИЮ 8, или СТАДИЮ 9, или СТАДИЮ 10, или СТАДИЮ 11, или СТАДИЮ 12, или СТАДИЮ 13, или любую комбинацию их, содержащую две или более СТАДИЙ.
Кроме того, описываются и заявляются СТАДИИ, описанные и обозначенные в описании как СХЕМА М-М, включающая СТАДИЮ 5, или СТАДИЮ 6, или СТАДИЮ 7, или СТАДИЮ 8, или СТАДИЮ 9, или СТАДИЮ 10, или СТАДИЮ 11, или СТАДИЮ 12, или СТАДИЮ 13, или любую комбинацию их, содержащую две или более СТАДИЙ.
Дополнительное описание изобретения и описание предпочтительных осуществлений изобретения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Соединения по данному
изобретению идентифицируют двумя способами: описательными названиями и указанием структур, показывающих различные химические структурные элементы. В подходящих случаях присущую им стереохимию
описывают также в названии или отображают в структурах. В некоторых случаях, когда молекула имеет два хиральных центра, указывают стереохимию только одного хирального центра, если стереохимия другого
хирального центра не рассматривается, стереохимия другого хирального центра представлена нерасщепленной или рацемической смесью. Все температуры приводятся в градусах по шкале Цельсия независимо от
того, указаны ли они как "o" или "oC" или нет. Минуты могут быть написаны как м или мин. Час может быть написан как Ч или ч. Аббревиатура в описании стандартная или понятная
химику, если не оговорена особо. Когда соединения добавляют или подвергают любым образом воздействию других соединений, можно сказать, что их "смешивают" с этими соединениями. Обычно целью смешивания
соединений является промотирование химических реакций среди одного или нескольких смешанных соединений. Можно также применять следующие термины.
НЕОБЯЗАТЕЛЬНО ЗАМЕЩЕННЫЙ. Термин "замещенный" или "необязательно замещенный" обычно обозначает прежде всего "C1-8 алкил", но следует учесть, что он включает все варианты всех групп. Термин может обозначать группу или радикал, который замещен галогеном, низшим алкилом, моно- или ди(низший алкил)замещенным низшим алкилом, (низший алкил)тио, галогензамещенным низшим алкилом, аминозамещенным низшим алкилом, моно- или ди(низший алкил)замещенной амино, низшим алкенилом, низшим алкинилом, галогеном, низшим алкокси, арилокси, арил(низшим алкилом), гидрокси, циано, амино, моно- и ди(низший алкил)амино или нитро и тому подобное. Химик, являющийся специалистом в данной области, обычно должен знать, когда и как провести такие очевидные замещения.
АЛКИЛ. Заключенный в скобки термин (Cn-Cm алкил) включает такие значения, что соединение (C1-C8) должно включать соединения, содержащие от 1 до 8 углеродов, и их изомерные формы. Разные углеродные остатки являются алифатическими углеводородными радикалами и включают разветвленные и неразветвленные формы, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, н-гексил, изогексил, н-гептил, изогептил и н-октил и его изомерные формы.
н-АЛКИЛ. Заключенный в скобки термин (Cn-Cm н-алкил) имеет такие значения, чтобы соединение (C1-C8) включало соединения, содержащие от 1 до 8 атомов углерода и имело неразветвленную форму цепи.
НИЗШИЙ АЛКИЛ. Термин "низший алкил" относится к разветвленным или неразветвленным насыщенным углеводородным радикалам, имеющим от одного до шести атомов углерода. Представителями таких групп являются метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, изомерные формы пентила, изомерные формы гексила и тому подобное.
(НИЗШИЙ АЛКИЛ)ТИО. Термин "(низший алкил)тио" относится к низшей алкильной группе, как определено выше, присоединенной к исходному молекулярному остатку через атом серы. Типичные (низший алкил)тиогруппы включают метилтио, этилтио, пропилтио, изопропилтио и тому подобное.
АЛКОКСИ. Алкокси, представляемый формулой -OR1, где R1 представляет (C1-C8)алкил, относится к алкильному радикалу, который присоединен к остальной части молекулы посредством кислорода и включает разветвленные или неразветвленные формы, такие как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси, трет-бутокси, н-пентокси, изопентокси, н-гексокси, изогексокси, н-гептокси, изогептокси и н-октокси и тому подобное.
НИЗШИЙ АЛКОКСИ. Термин "низший алкокси" обозначает алкильную группу, как определено выше, присоединенную к исходному молекулярному остатку через атом кислорода. Представители таких групп включают метокси, этокси, бутокси и тому подобное.
АЛКЕНИЛ. Алкенил относится к радикалу алифатического ненасыщенного углеводорода, имеющего по меньшей мере одну двойную связь, и включает как разветвленные, так и неразветвленные формы, например этенил (-CH=CH2), 1-метил-1-этенил, 1-пропенил(-CH2-CH= CH2), 2-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-метил-1-бутенил, 1-пентенил, аллил, 3-пентенил, 4-пентенил, 1-метил-4-пентенил, 3-метил-1-пентенил, 3-метилаллил, 1-гексенил, 2-гексенил, 3-гексенил, 4-гексенил, 1-метил-4-гексенил, 3-метил-1-гексенил, 3-метил-2-гексенил, 1-гептенил, 2-гептенил, 3-гептенил, 4-гептенил, 1-метил-4-гептенил, 3-метил-1-гептенил, 3-метил-2-гептенил, 1-октенил, 2-октенил или 3-октенил и тому подобное.
АЛКИНИЛ. Термин алкинил относится к одновалентному разветвленному или неразветвленному углеводородному радикалу, содержащему по меньшей мере одну углерод-углеродную тройную связь, например этинилу, пропинилу и тому подобное.
ЦИКЛОАЛКИЛ. Заключенный в скобки термин (Cn-m циклоалкил) включает такие значения, чтобы соединение (C3-10) включало радикалы насыщенного циклического углеводорода, содержащего от 3 до 10 углеродов в его циклической цепи. Термин может включать также алкилзамещенный циклоалкил, такой как циклопропил, 2-метилциклопропил, 2,2-диметилциклопропил, 2,3-диэтилциклопропил, 2-бутилциклопропил, циклобутил, 2-метилциклобутил, 3-пропилциклобутил, циклопентил, 2,2-диметилциклопентил, циклогексил, циклогептил, циклооктил и циклодецил и тому подобное. Каждый из этих остатков, когда это приемлемо, может быть замещен.
ГЕТЕРОАЛКИЛ. Термин "гетероалкил" относится к алкилам, как описано выше, только где один, два или три несоседних атома углерода замещены гетероатомами, например азотом, серой и кислородом.
АРИЛ. Термин (C6-12)арил относится к базовой структуре, содержащей от 6 до 12 атомов углерода, одному или двум конденсированным или неконденсированным ароматическим кольцам, которые могут быть необязательно замещены или замещены 1-3 группами гидрокси, C1-C3 алкокси, C1-C3 алкил, трифторметил, фтор, хлор или бром. Примерами "арила" являются: фенил, м-метилфенил, п-трифторметилфенил, α-нафтил, β-нафтил, (о-, м-, п-)толил, (о-, м-, п-)этилфенил, 2-этилтолил, 4-этил-о-толил, 5-этил-м-толил, (о-, м- или п-)пропилфенил, 2-пропил-(о-, м- или п-)толил, 4-изопропил-2,6-ксилил, 3-пропил-4-этилфенил, (2,3,4-, 2,3,6- или 2,4,5- )триметилфенил, (о-, м- или п-)фторфенил, (о-, м- или п-трифторметил)фенил, 4-фтор-2,5-ксилил, (2,4-, 2,5-, 2,6-, 3,4- или 3,5-)дифторфенил, (о-, м- или п-)хлорфенил, 2-хлор-п-толил, (3-, 4- , 5- или 6-)хлор-о-толил, 4-хлор-2-пропилфенил, 2-изопропил-4-хлорфенил, 4-хлор-3-фторфенил, (3- или 4-)хлор-2-фторфенил, (о-, м- или п-)трифторфенил, (о-, м- или п-)этоксифенил, (4- или 5-)хлор-2-метоксифенил и 2, 4-дихлор-(5- или 6-)метилфенил и тому подобное. Каждый из этих остатков, когда это уместно, может быть замещен.
АЛКИЛАРИЛ. Термин арилалкил относится к алкильным цепям, содержащим от 1 до 8 атомов углерода, и их изомерным формам, которые замещены арильными группами, содержащими от 6 до 12 атомов углерода, как описывается выше.
ГЕТЕРОЦИКЛЫ. Примеры гетероциклов включают: (2-, 3- или 4-)пиридил, имидазолил, индолил, Nin-формилиндолил, Nin-C2-C5 алкил-C(O)-индолил, [1,2,4] -триазолил, (2-, 4-, 5-)пиримидинил, (2-, 3-) тиенил, пиперидинил, пирролил, пирролинил, пирролидинил, пиразолил, пиразолинил, пиразолидинил, имидазолил, имидазолинил, имидазолидинил, пиразинил, пиперазинил, пиридазинил, оксазолил, оксазолидинил, изоксазолил, изоксазолидинил, морфолинил, тиазолил, тиазолидинил, изотиазолил, изотиазолидинил, хинолинил, изохинолинил, бензимидазолил, бензотиазолил, бензоксазолил, фурил, пурил, феназил, карбазолил, тиенил и бензотиенил, тиенил, индолил, изохинолил и тому подобное. Каждый из этих остатков, когда это уместно, может быть замещен.
ГЕТЕРОАРИЛ. Гетероарил относится к моно- или бициклической структуре, содержащей в кольцах 5-12 атомов, где как минимум одно кольцо ароматическое, но только где один, два или три несоседних атома углерода замещены гетероатомами, такими как атом азота, серы и кислорода. Примеры их могут включать пиридин, тиофен, фуран, пиримидин, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидинил, 4-пиримидинил, 5-пиримидинил, 3-пиридазинил, 4-пиридазинил, 3-пиразинил, 2-хинолил, 3-хинолил, 1-изохинолил, 3-изохинолил, 4-изохинолил, 2-хиназолинил, 4-хиназолинил, 2-хиноксалинил, 1-фталазинил, 2-имидазолил, 4-имидазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 3-пиразолил, 4-пиразолил, 5-пиразолил, 2-оксазолил, 4-оксазолил, 5-оксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-индолил, 3-индолил, 3-индазолил, 2-бензоксазолил, 2-бензотиазолил, 2-бензимидазолил, 2-бензофуранил, 3-бензофуранил, 2-фуранил, 3-фуранил, 2-тиенил, 3-тиенил, 2-пирролил, 3-пирролил, 1,2,4-оксадиазол-3-ил, 1,2,4-оксадиазол-5-ил, 1,2,4-тиадиазол-3-ил, 1,2,4-тиадиазол-5-ил, 1,2,4-триазол-3-ил, 1,2,4-триазол-5-ил, 1,2,3,4-тетразол-5-ил, 5-оксазолил, 1-пирролил, 1-пиразолил, 1,2,3-триазол-1-ил, 1,2,4-триазол-1-ил, 1-тетразолил, 1-индолил, 1-индазолил, 2-изоиндолил, 1-пуринил, 3-изотиазолил, 4-изотиазолил и 5-изотиазолил. Каждый из этих остатков, когда это уместно, может быть замещен.
ХИРАЛЬНОСТЬ. Специалистам данной области очевидно, что соединения по этому изобретению могут содержать один или несколько хиральных центров и могут существовать в оптически активных формах, включая цис-/транс- и/или R- и S-изомерные формы и их смеси. Объем этого изобретения включает все эти формы, энантиомерные или диастереомерные формы соединений, включая оптически активные формы, в чистой форме или в виде смеси энантиомеров или диастереомеров, включая цис-/транс-изомерные формы. Терапевтические свойства соединений могут в большей или меньшей степени зависеть от стереохимии отдельного соединения. Расщепление на энантиомеры можно выполнять при помощи расщепляющих на энантиомеры средств, например оптически активной дибензоилвинной кислоты, камфорасульфокислоты, бис-о-толуоилвинной кислоты, винной кислоты и диацетилвинной кислоты.
ОПТИЧЕСКАЯ ЧИСТОТА иногда обозначается как "% ее" (% энантиомерного избытка).
Способы и соединения изобретения
Приведенные ниже способы относятся к соединениям и
формулам, идентифицированным в СХЕМАХ.
СПОСОБЫ, РЕАКЦИИ И СОЕДИНЕНИЯ СХЕМЫ G
Общее описание реакций
Все значения радикалов для способов, описанных ниже, определяются
выше в кратком изложении сущности изобретения и в определениях. Ниже раскрываются более предпочтительные заместители.
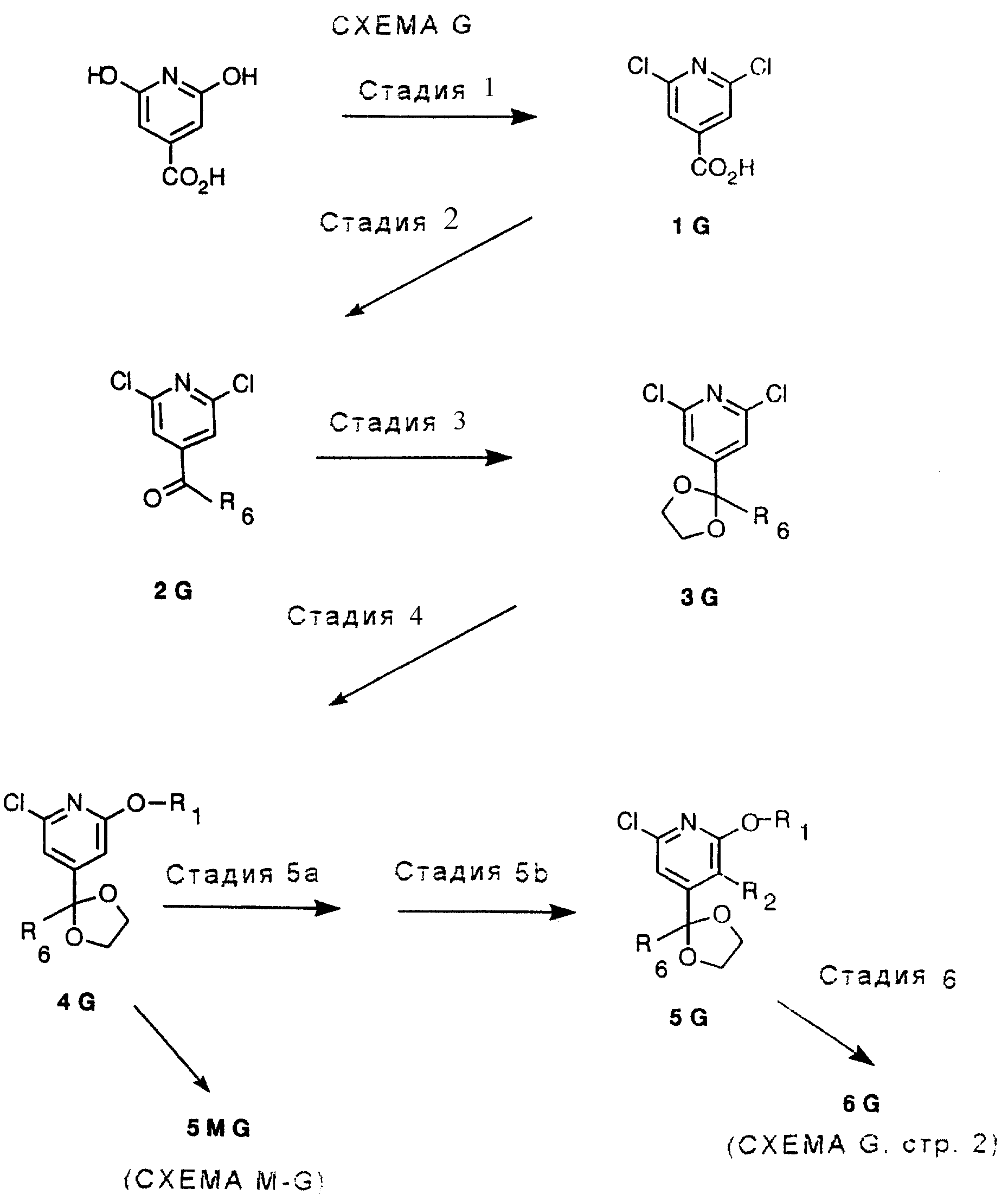
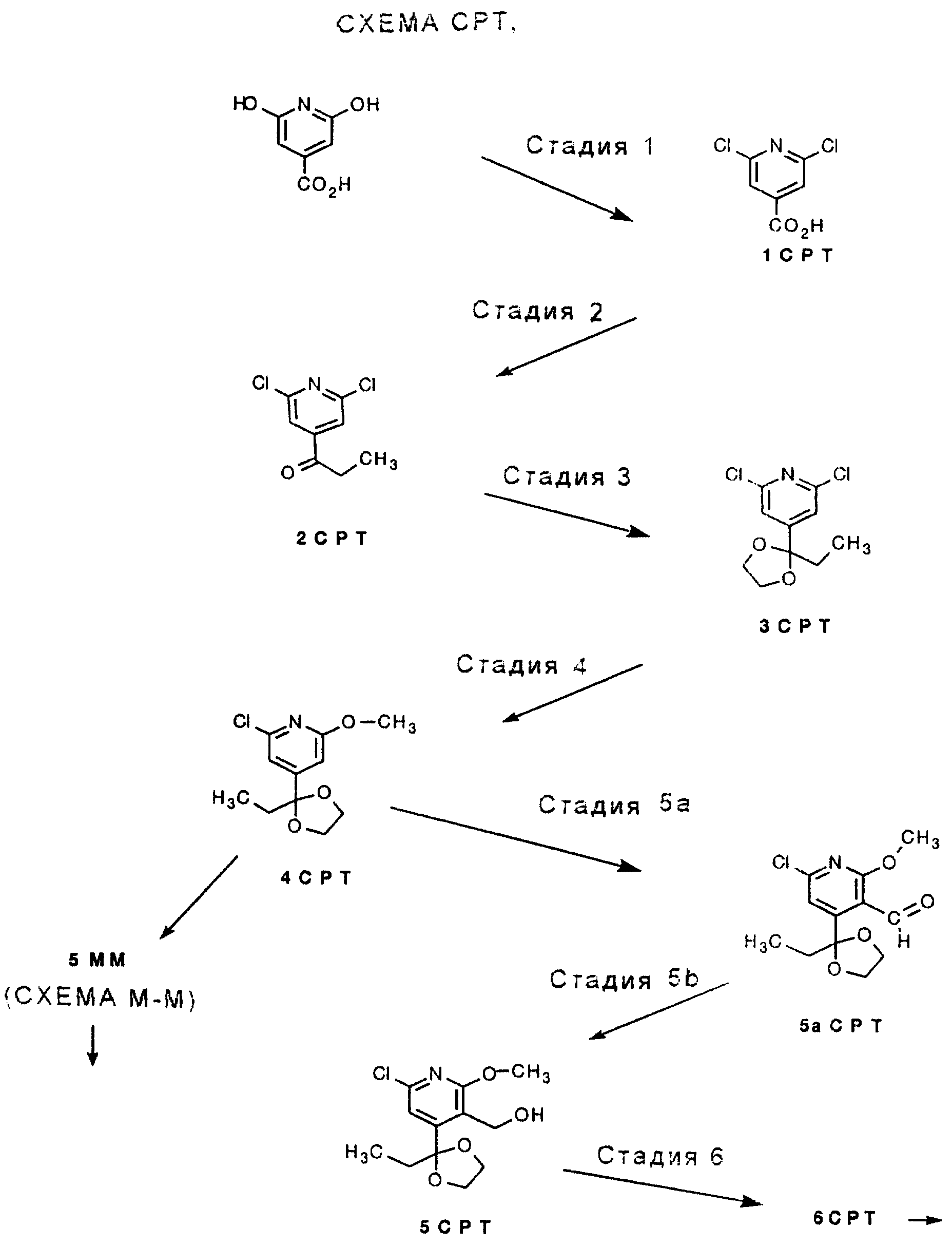
СТАДИЯ 1. (Цитразиновая кислота ---> 1 G)
Исходный
материал, дихлоризоникотиновая кислота, является известным соединением и ее легко получить из коммерчески доступной цитразиновой кислоты.
Способы получения и диапазон приемлемых условий известны и указываются в следующей ссылке: М. Е. Baizer, М. Dub, S. Gister, N.G. Steinberg, J. Am. Pharm. Assoc., 1956, 45, 478-480. Применение солей тетраалкиламмония и третичных аминов в реакции этого типа описывается в Chemical Abstracts, CA 97, 216024, и East German patent, DD 154538 by E. Schroetter, H. Schick, H. Niedrich, P. Oehme and L. Piesche. Смотри также W.H. Levelt and J.P. Wibaut, Rec. Trav. Chem., 1929, 44, 466.
По предпочтительному способу цитразиновую кислоту нагревают с оксихлоридом фосфора и хлоридом тетраалкиламмония или гидрохлоридом третичного амина, наиболее предпочтительно с хлоридом тетраметиламмония, при температуре между 120o и 140o в течение приблизительно от 12 до 24 часов. Смесь затем подвергают реакции с водой, получая продукт 1CPT.
СТАДИЯ 2. (1 G ---> 2 G)
2,6-Дихлоризоникотиновую кислоту растворяют или суспендируют в эфирном растворителе, таком как диэтиловый эфир,
тетрагидрофуран или 1,2-диметоксиэтан, и подвергают взаимодействию с избытком этилмагнийгалогенида или этиллития в растворе диэтилового эфира или тетрагидрофурана при температуре между около - 30o и +10o. Избыток этилмагнийгалогенида или этиллития разлагают реакцией с разбавленной кислотой, такой как соляная кислота, или реакцией сначала со сложным эфиром, таким как
метилформиат, или кетоном, таким как ацетон, и последующей реакцией с разбавленной кислотой, например соляной кислотой.
В соответствии с другим вариантом 2,6-дихлоризоникотиновую кислоту можно превратить в ее хлорангидрид реакцией с тионилхлоридом или пентахлоридом фосфора и затем превратить в амид Weinreb. Смотри S. Nahm and S.M. Weinreb, Tet. Lett, 1981, 3815-3818. Амид Weinreb затем растворяют в используемом в реакции эфирном растворителе, например диметиловом эфире, тетрагидрофуране или 1,2-диметоксиэтане, и подвергают взаимодействию с избытком этилмагнийгалогенида или этиллития в растворе диэтилового эфира или тетрагидрофурана при температуре между около -30o и около +10o. Продукт затем выделяют после реакции промежуточного комплекса с разбавленной кислотой, такой как соляная кислота. Предпочтительный R6 представляет низший алкил, включая C1-4 алкил и этил, арил и замещенный арил, алкиларил и замещенный алкиларил, включая бензил и замещенный бензил, C3-10 циклоалкил, гетероарил или замещенный гетероарил, предпочтительно C1-4 алкил, этил, бензил.
СТАДИЯ 3. (2 G
---> 3G)
Алкилкетон, обозначенный в СХЕМЕ G (см. в конце описания) как 2G, подвергают взаимодействию со спиртом или диолом в присутствии триметилхлорсилана. Спирты могут быть диолами
(двухатомными спиртами), например этиленгликолем, 1,3-пропандиолом или 2,2-диметил-1,3-пропандиолом, или одноатомными спиртами, например метанолом. Предпочтительным спиртом является этиленгликоль.
Когда применяют этиленгликоль, получают этиленкеталь, другие кетали можно получить при использовании других спиртов. Можно добавлять растворитель, например метиленхлорид. Реакцию проводят при
температуре между около 0o и около 60o, предпочтительно при около 40o. Предпочтительный R6 представляет низший алкил, включая C1-4 алкил и этил,
арил и замещенный арил, алкиларил и замещенный алкиларил, включая бензил и замещенный бензил, C3-10 циклоалкил, гетероарил или замещенный гетероарил, предпочтительно C1-4 алкил,
этил, бензил.
СТАДИЯ 4. (3 G ---> 4G)
Соединение в СХЕМЕ G (см. в конце описания), обозначенное 3G, подвергают взаимодействию с алкоксидом натрия или калия либо в
избытке спирта, либо растворителя, такого как тетрагидрофуран или 1,2-диметоксиэтан. Реакцию можно проводить при температуре между около 20o и 80o. Алкоксидом, или
предпочтительной группой R1 СХЕМЫ G, может быть любая из ранее определенных низших алкильных, циклоалкильных, C3-10 циклоалкильных, алкенильных, арильных и арилалкильных групп,
включая бензил и замещенный бензил. Наиболее предпочтительными группами R1 являются метил и бензил.
СТАДИЯ 5а (необязательная) и СТАДИЯ 5b. (4G ---> 5G)
СТАДИЯ 5а. Обзор орто-направленных реакций металлирования был опубликован, смотри V. Snieckus, Chem. Rev., 1990, vol. 90, pp. 879-933, он включен здесь в качестве ссылки.
Соединение в СХЕМЕ G (см. в конце описания), обозначенное как 4 G, растворяют в растворителе и подвергают взаимодействию с алкиллитиевым основанием или ариллитиевым основанием для образования пиридиланиона. Получаемый анион затем подвергают взаимодействию с электрофилом и продукт выделяют после дальнейшей реакции с разбавленной кислотой. Подходящими растворителями для реакции являются простые эфиры, такие как диэтиловый эфир, тетрагидрофуран или 1,2-диметоксиэтан, или углеводороды, такие как толуол, гексан, гептан, циклогексан или изооктан, или смеси любых из этих или подобных растворителей.
Алкиллитием может быть метиллитий, н-бутиллитий, втор-бутиллитий или трет-бутиллитий. Температура реакции может быть между около -40o и около +50o. Электрофилом может быть алкилгалогенид, такой как метилиодид, диметилсульфат, хлорметилметиловый эфир, бензилхлорметиловый эфир или бензилбромид: альдегиды или кетоны, такие как формальдегид, ацетон, бензальдегид или другие подобные соединения; или амиды, такие как формамиды, включая диметилформамид, N-формилпиперидин или N-формилморфолин или N-метилформанилид или подобные формамиды. Кислотой, используемой для выделения продукта, может быть соляная кислота, уксусная кислота, серная кислота или другие кислоты от средней силы до сильных.
Предпочтительным растворителем является гептан, предпочтительным основанием является н-бутиллитий и предпочтительным амидом является N-формилпиперидин. Реакцию предпочтительно проводят при температуре между около -5o и около +5o. Очистку продукта можно проводить кристаллизацией, хроматографией или через образование бисульфитного аддитивного соединения, которое можно разложить реакцией либо с кислотой, либо с основанием.
Заметьте, что СТАДИЯ 5а может быть пропущена, для получения 5G СТАДИЮ 5b можно проводить без СТАДИИ 5а.
СТАДИЯ 5b.
Альдегид СТАДИИ 5а восстанавливают в спирт гидридным восстановителем, таким как борогидрид натрия. Реакцию можно проводить с использованием спирта, например метанола или 2-пропанола, в качестве растворителя, или ее можно проводить в двухфазной среде с использованием воды и органической фазы, состоящей из гептана, метиленхлорида или метил-трет-бутилового эфира или смеси этих и подобных растворителей. Можно добавлять катализатор межфазового переноса, такой как хлорид тетрабутиламмония, но это необязательно.
СТАДИЯ 5а и СТАДИЯ 5b (4G ---> 5G)
Предпочтительным R2,
показанным в СХЕМЕ G, может быть H или а) любой необязательно замещенный C1-8 алкил, алкиларил, C1-8 алкиларил, включая C1-8 алкил-C6 арил, замещенный
бензил и незамещенный бензил: б) -C(O)-R3 или в) -C(R7)2-O-R3, где каждый R7 не зависит от другого; и где R3 и R7
определены выше в кратком изложении сущности изобретения. За этой последовательностью реакций следует непосредственно ниже указанная СТАДИЯ 6 СХЕМЫ G, см. в конце описания, только когда R2
представляет б) -C(O)-R3 или в) -C(R7)2-O-R3, где каждый R7 не зависит от другого. Когда R2 представляет H или любой необязательно
замещенный C1-8 алкил, алкиларил, C1-8 алкиларил, включая C1-8 алкил-C6 арил, замещенный бензил и незамещенный бензил, то реакции идут в соответствии со
СХЕМОЙ M-G и СХЕМОЙ М-М и могут привести к получению маппицина или аналогов маппицина.
СТАДИЯ 6. (5 G ---> 6 G) (CXEMA G - продолжение)
Спирт подвергают взаимодействию
с основанием и алкилирующим агентом в
подходящем растворителе с получением продукта. Основаниями могут быть гидриды, такие как гидрид натрия или гидрид калия, или алкоксидные основания, такие как
трет-бутоксид калия.
Подходящими растворителями являются эфирные растворители, такие как тетрагидрофуран или 1,2-диметоксиэтан, или спирты, такие как трет-бутанол. Температура может быть между около 15o и около 80o. Предпочтительным основанием является трет-бутоксид калия и предпочтительными растворителями являются ТГФ или МТБЭ (метил-трет-бутиловый эфир) при температуре предпочтительно между около 20o и около 40o.
В соответствии с другим вариантом реакцию можно проводить в условиях переноса фаз, используя воду и органический растворитель, такой как метиленхлорид или углеводороды, например гексан, гептан или толуол, или подобные растворители. Основанием может быть гидроксид, такой как гидроксид натрия или калия, или карбонат натрия или калия. Можно добавлять катализатор межфазового переноса, например хлорид тетрабутиламмония, и предпочтительный диапазон температуры находится между около 10o и около 30o.
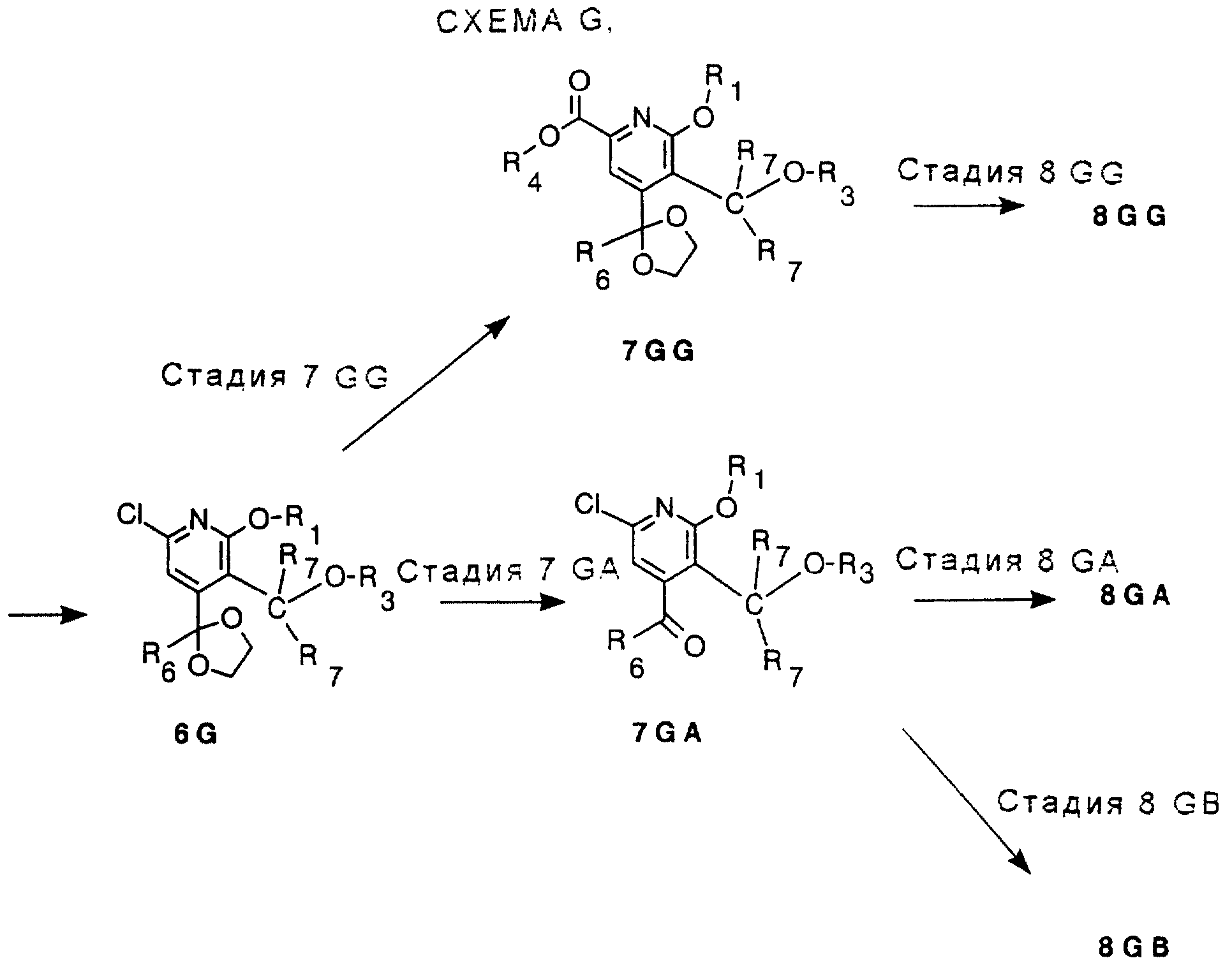
АЛЬТЕРНАТИВНЫЕ СТАДИИ
Имеются 2 различные реакции Стадии 7, ряды 7GG и 7GA; 3 различные реакции Стадии 8, ряды 8GG, 8GA, 8GB: 3 различные реакции
Стадии 9, ряды 9GG, 9GA, 9GB и 2 различные реакции Стадии 10, ряды 10GG и 10GA, за которыми следует процедура расщепления на энантиомеры Стадии 10. Смотри Схему G в конце описания.
СТАДИЯ 7 GG и СТАДИЯ 10GA. (6G ---> 7 GG) и (9GA ---> 10 G).
Реакции карбонилирования арилгалогенидов, катализируемые палладием с нулевой валентностью, хорошо известны, смотри J.К. Stille and P.К. Wong, J. Org. Chem., 1975, 40, 532-534, но арилхлориды обычно имеют низкую реакционную способность в этих реакциях. Известно, что в противоположность простым арилхлоридам, 2-хлорпиридины легко претерпевают реакции внедрения с участием палладия с нулевой валентностью. Известны различные реакции сочетания 2-хлорпиридинов, катализируемые палладием с нулевой валентностью, но в литературе не описаны реакции карбонилирования 2-хлорпиридинов, катализируемые палладием с нулевой валентностью.
Соединения, обозначенные 6G, подвергают реакции с оксидом углерода и спиртом в присутствии растворимой соли палладия II (такой как ацетат палладия), фосфинового лиганда (такого как 1,3-бисдифенилфосфинопропан) и основания, такого как ацетат натрия или калия, карбонат натрия или калия, триэтиламин или три-н-бутиламин, в полярном апротонном растворителе, таком как диметилформамид или ацетонитрил.
Предпочтительной группой R3, приведенной в СХЕМЕ G (см. в конце описания), может быть любая из ранее определенных, H, низших алкильных, циклоалкильных, алкенильных, арильных и арилалкильных групп, включая бензильную и замещенную бензильную группы. Более предпочтительными группами R3 являются метил и бензил.
Предпочтительной группой R4 спирта, приведенного в СХЕМЕ G (см. в конце описания), может быть любая из ранее определенных, H, низших алкильных, циклоалкильных, алкенильных, арильных и арилалкильных групп, включая бензильную и замещенную бензильную группы. Более предпочтительной группой R4 является н-пропил. Диапазон температур находится между около 50o и около 100o.
Предпочтительная группа R7 независимо представляет H, низший алкил, арил, алкиларил, замещенный арил, замещенный алкиларил или две группы R7 могут быть соединены с образованием циклопентана или циклогексана или их замещенных производных.
Некоторые ссылки, описывающие реакции внедрения, указанные в приведенной выше СТАДИИ 7, следующие: а) К. Isobe and S. Kawaguchi, Heterocycles, 1981, 16, 1603-1612; б) N. Sato, A. Hayakaws and R. Takeuchi, J. Het. Chem., 1990, 503-506; в) M. Ishikura, М. Kamada and M. Terashima, Synthesis, 1984, 936-938 и г) К. Isobe, К. Nanjo, Y. Nakamura and S. Kawaguchi, Bul. Chem. Soc. Japan, 1986, 59, 2141-2149.
СТАДИЯ 7 GA и СТАДИЯ 8 GG. (6 G ---> 7 GA), также для (7 GG ---> 8 GG)
Кеталь гидролизуют реакцией с водой в присутствии сильной кислоты, такой
как трифторуксусная кислота. Концентрация трифторуксусной кислоты может быть между около 50% и 90% и температура реакции между около 15o и около 30o. В соответствии с другим
вариантом кеталь можно удалить реакцией обмена с кетоном, таким как ацетон или 2-бутанон, катализируемой сильной кислотой, такой как п-толуолсульфокислота или кислотная ионообменная смола, например
смола амберлист А-15. Для реакции обмена предпочтительна температура около температуры флегмы кетона.
СТАДИЯ 8GA. (7GA ---> 8GA)
Соединение 8GA растворяют в
растворителе и подвергают реакции с виниллитием или винилмагнийгалогенидом. Подходящими растворителями являются простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, 1,2-диметоксиэтан или МТБЭ,
либо взятый отдельно, либо в виде смесей, или в виде смесей с углеводородами, например толуолом, гептаном или циклогексаном. Температура реакции может быть между около -78oC и около 25o. Продукт выделяют после последующей реакции с разбавленной кислотой, например соляной, серной или уксусной кислотой. Предпочтительным реагентом является винилмагнийбромид в тетрагидрофуране,
применяемом в качестве растворителя, при температуре от около -40o до около 25o, после этого реакцию гасят соляной кислотой. Предпочтительный R5 независимо
представляет H, низший алкил, арил, замещенный арил или две группы R5 могут быть соединены с образованием циклопентана или циклогексана или их замещенных производных.
СТАДИЯ
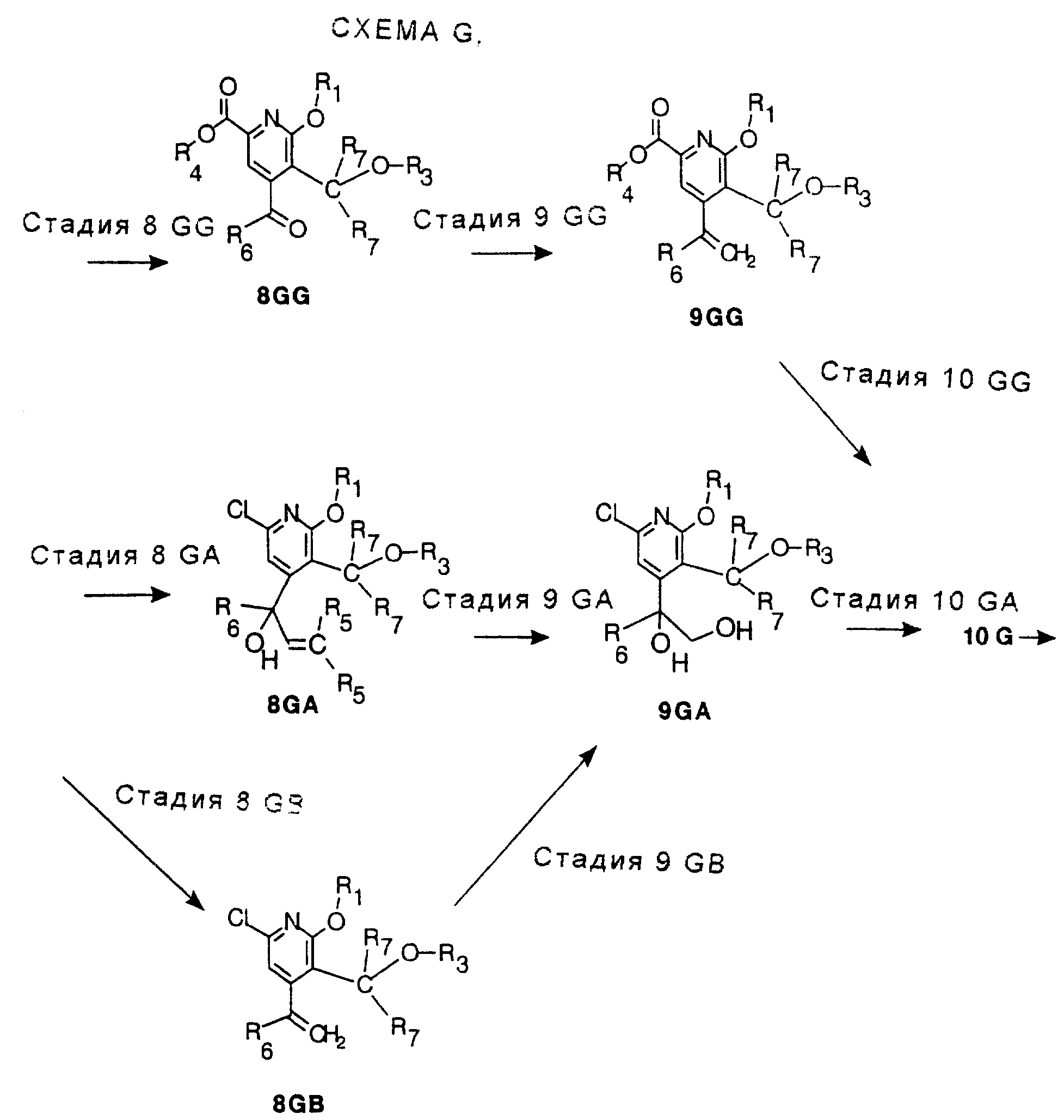
8GB и СТАДИЯ 9GG. (7 GA ---> 8GB и 8 GG ---> 9 GG)
Реакцию Виттига проводят путем взаимодействия кетона с раствором илида, полученным из соли метилтрифенилфосфония,
предпочтительно бромида, и сильного основания, такого как н-бутиллитий, трет-бутоксид калия или бистриметилсилиламид калия, в растворителе, например диэтиловом эфире, тетрагидрофуране, 1,
2-диметоксиэтане или ДМФ. Предпочтительным основанием является бистриметилсилиламид калия, предпочтительным растворителем ДМФ. Реакцию проводят при температуре между около -5o и около
25o. Время реакции между около 5 мин и около 2 часов.
СТАДИЯ 9GA. (8GA ---> 9GA)
9GA растворяют в растворителе и подвергают взаимодействию с озоном для
получения промежуточного продукта. В зависимости от состава растворителя этот промежуточный продукт может быть озонидом или смесью гидропероксидов. Этот промежуточный продукт подвергают взаимодействию
с подходящим восстановителем для получения продукта либо непосредственно, либо постадийно через образование промежуточного альдегида. Температура реакции может быть между около -78o и около
25o. Подходящими растворителями для реакции являются хлорированные углеводороды, такие как метиленхлорид, хлороформ, тетрахлорид углерода, 1,2-дихлорэтан или другие полихлорированные
производные этана или этилена, либо взятые по отдельности, либо в виде смесей, или в виде смесей со спиртами, например метанолом. Предпочтительным растворителем является смесь метиленхлорида и
метанола при температуре от около -78o до около -40o для первоначальной реакции с озоном и температуре от около 0o до 25o для восстановления промежуточного
продукта. Предпочтительным восстановителем является борогидрид натрия.
СТАДИЯ 9GB и СТАДИЯ 10GG. (8 GB ---> 9 GA и 9 GG ---> 10 GG)
Алкен превращают в диол
осмилированием в стандартных условиях, смотри V. VanRheenen, R.C. Kelley and D.Y. Cha, Tet. Lett., 1976, 1973, каталитическим тетроксидом осмия и стехиометрическим соокислителем, например N-оксидом
триметиламина или N-оксидом N-метилморфолина, либо в водном ТГФ, либо, предпочтительно, в трет-бутаноле, применяемом в качестве растворителя. Температура реакции может быть между около 15o
и около 50o, предпочтительно около 40o, в течение около 12-48 часов.
Альтернативой рацемическому осмилированию является использование асимметрического
осмилирования, как описывается Sharpless, для превращения 9CРT непосредственно в 10G (R) или (S). Конкретные ссылки для асимметрического осмилирования по Sharpless следующие:
G. A. Crispino,
A. Makita, Z.-M. Wang, К.В. Sharpless, Tet. Lett., 1944, 543-546.
G. A. Crispino, K.-S. Jeong, H.C. Kolb, Z.-M. Wang, D. Xu, K.B. Sharpless, J. Org. Chem., 1993, 3785-3786 и многие ссылки, перечисленные в этой статье.
K.B. Sharpless, W.K. Amberg, U.S. Patent 5227543.
K. B. Sharpless, M. Beller, В. Blackburn, Y.Kawanami, H.-L. Kwong, Y. Ogino, T. Shibata, T. Ukita, L. Wang, PCT WO 92/20677.
J. Hartung, K.B. Sharpless, PCT WO 93/07142.
СТАДИЯ 10. Расщепление на энантиомеры (10 G ---> 10 G (R или S))
Рацемический диол, подобный 10 G, можно обработать ацетилирующим реагентом, подобным винилацетату, изопропенилацетату, уксусному ангидриду или этилацетату, в органическом растворителе в
присутствии липазы. Возможные растворители включают простой эфир или гексан, липазой может быть cepaica, подобная Pseudomonas cepaica. При использовании этого способа можно получить индивидуальный
ацетатный изомер и индивидуальный диоловый изомер. Реакцию обычно проводят при температуре между 25o и 45oC при концентрации субстрата 15-40 мг/мл. Продукты реакции можно
разделить кристаллизацией с использованием обычных органических растворителей или обычной хроматографией на силикагеле. Оптическую чистоту (% ее) каждого энантиомера можно определить анализом ЯМР с
хиральными шифт-реагентами или хиральной ВЭЖХ.
СТАДИИ 11-14
Следующие реакции можно проводить с индивидуальным энантиомером, или с рацемическими смесями, или смесями с другими
отношениями энантиомеров. Продукт реакций будет зависеть от исходных материалов. СХЕМА G (см. в конце описания) и приведенные ниже стадии относятся для удобства к индивидуальному энантиомеру и
приводятся в качестве примера. Индивидуальный энантиомер обычно обозначают заглавной буквой "R" или "S". Одним примером этого энантиомера является "10 G (R)". Рацемическую смесь обычно обозначают
числом, за которым следует заглавная буква "G". Одним примером рацемической смеси является "10G". Смотри СХЕМУ G. Реакции по этому изобретению, конечно, не ограничиваются теми, которые приводятся в
Схемах, например СХЕМА G не показывает СТАДИИ от 11 до 13 реакции для рацемических смесей, но они подразумеваются в СХЕМЕ и описываются здесь. Аналогично ряд "R" не показан полностью как ряд "S".
СХЕМЫ являются только наглядными вспомогательными средствами и не представляют полное изобретение.
СТАДИЯ 11. (10G ---> 11G)
Диол можно окислить в гидроксиальдегид,
применяя окисление в условиях типа Swern, например ДМСО, оксалилхлорид и триэтиламин в апротонном растворителе, таком как метиленхлорид, при температуре в диапазоне от около -78o до около
25o. В соответствии с другим вариантом окисление можно проводить раствором гипохлорита натрия, катализируемым TEMPO или замещенным TEMPO, таким как 4-ацетокси-ТЕМРО, в двухфазной системе,
состоящей из воды и апротонного растворителя, например метиленхлорида. Предпочтительна температура реакции между около -5o и около +25o и время реакции между около 30 мин и около
2 часов.
Условия типа Swern описываются в A. J. Mancuso and D. Swern Synthesis, 1981, 165-185. Двухфазная система, состоящая из воды и апротонного растворителя, описывается в P. L. Anelli, C. Biffi, F. Montanari and S. Quici, J. Org. Chem. , 1987, 52, 2559-2562. Эти публикации вводятся здесь в качестве ссылки.
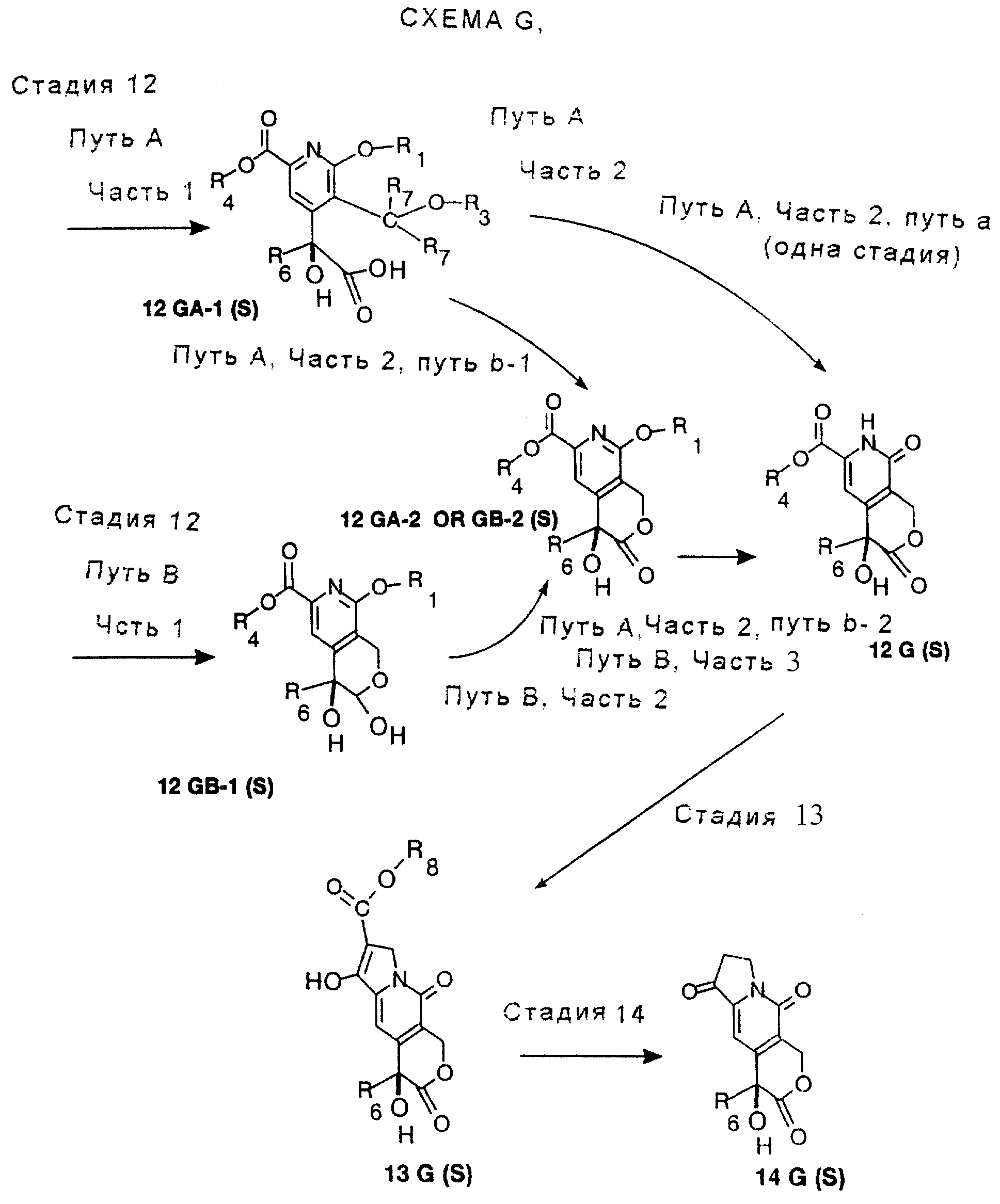
СТАДИЯ 12. (11G ---> 12G)
Для
превращения гидроксиальдегида, 11G, в 12G применяли несколько вариантов. В первом способе гидроксиальдегид, 11G, окисляют в гидроксикислоту, 12GA-1, хлоритом натрия. Гидроксикислоту можно затем
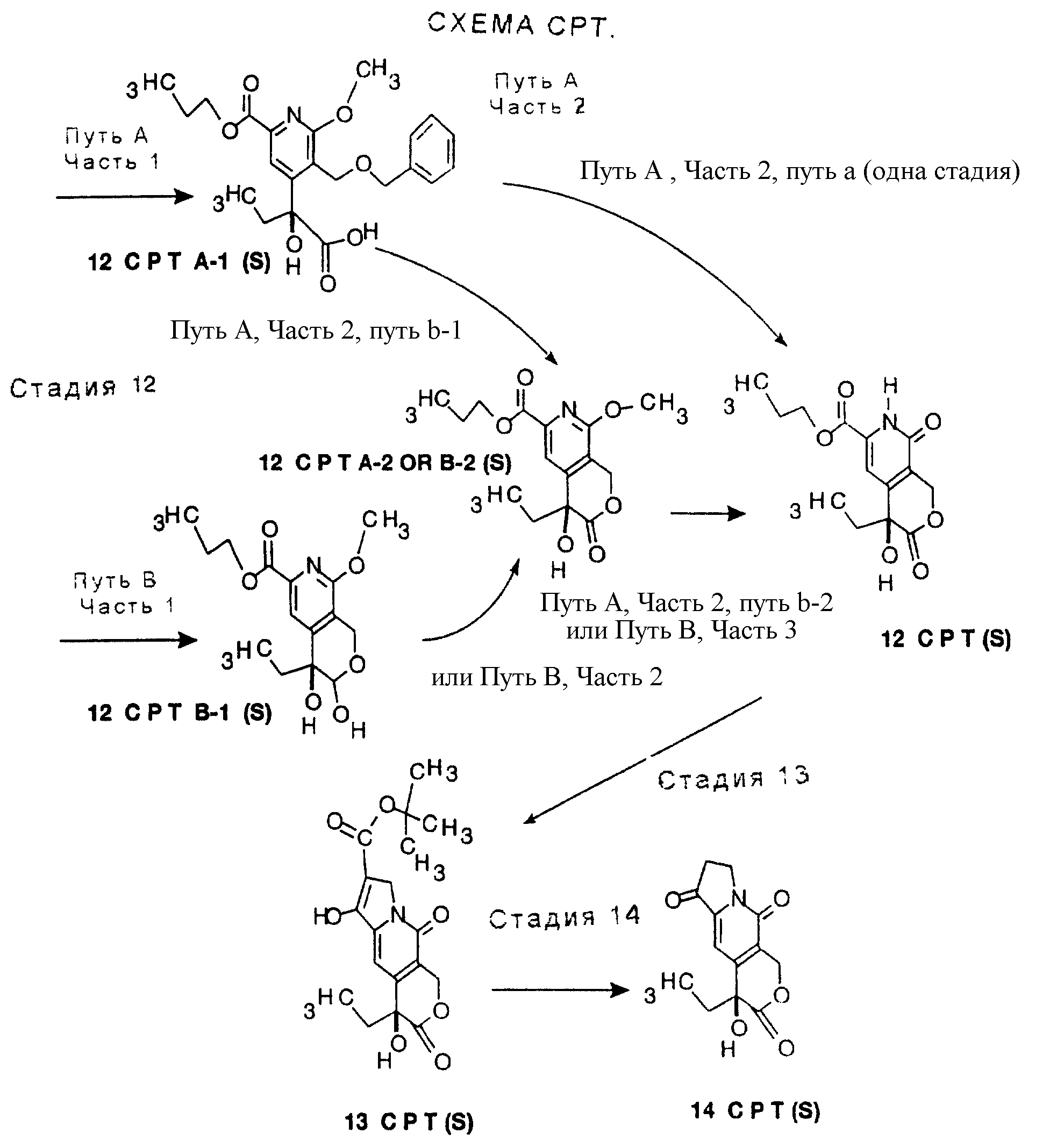
превратить в 12G реакцией с триметилсилилиодидом в одном сосуде. Преимущество этого способа в одностадийном превращении 11G в 12G. См. СХЕМУ G в конце описания, СТАДИЯ 12, Путь А, Часть 2, путь а.
Недостатком этого одностадийного превращения является относительно низкий выход и непостоянное время реакции.
По способу с более высоким выходом сначала удаляют бензиловую группу либо гидрированием, либо реакцией с трибромидом бора и затем удаляют метоксигруппу реакцией с триметилсилилиодидом. См. СХЕМУ G в конце описания, СТАДИЯ 12, Путь А, Часть 2, путь b-1 и b-2. Очевидно, что порядок стадий удаления защитных групп можно поменять на обратный.
Второй способ превращения 11G в 12G изменяет порядок стадий окисления и удаления защитных групп. См. СХЕМУ G в конце описания, СТАДИЯ 12, Путь В. Бензиловую группу удаляют гидрированием, получая лактол. Лактол затем окисляют гипохлоритом натрия, катализируемым TEMPO. Отщепление метоксигруппы проводят, как и раньше, триметилсилилиодидом. См. СХЕМУ G в конце описания, СТАДИЯ 12, Путь В, Части 1, 2 и 3. Преимуществом этой последовательности является отсутствие стадии окисления хлоритом натрия и связанной с ней опасностью.
Пути А и В описываются ниже более подробно, Путь В предпочтителен. Смотри СХЕМУ G в конце описания.
СТАДИЯ 12. Путь А. (11G ---> 12G, Путь А)
Путь А имеет две части, часть 1 и часть 2. Часть 2 следует за частью 1. Часть 2 Пути А имеет также 2 пути, путь а и путь b. Путь а Пути А, часть 2, имеет только одну стадию. Путь b Пути А, часть 2,
имеет две стадии. См. СХЕМУ G в конце описания, показан только один стереоизомер, другие стереоизомеры и рацемические смеси предполагаются.
Часть 1
Окисление с целью
образования гидроксикислоты предпочтительно проводят хлоритом натрия с использованием условий, описанных в литературе. Смотри B. S. Bal, W.E. Childers, H.W. Pinnick, Tetrahedron, 1981, 2091-2096. Для
предотвращения образования диоксида хлора также использовали другие добавки, например пероксид водорода или сульфаминовую кислоту. Таким образом получают 12 GA-1.
Часть 2
Путь
а
Одностадийное удаление бензильной и метильной групп проводят триметилсилилиодидом, либо полученным предварительно, либо генерированным in situ из триметилсилилхлорида и иодида натрия в
метиленхлориде или ацетонитриле. Смотри T. Morita, Y. Okamoto, H. Sakurai, J.C.S. Chem. Comm. 1978, 874-875, и M.E. Jung and M.A. Lyster, J. Am. Chem. Soc., 1977, 99, 968. Можно добавлять пиридин, но
это не требуется. Температура реакции между около 15o и 50o в течение времени между 12 и 48 часами. Таким образом получают 12 G.
Путь b
Часть 1 пути b-1.
Двухстадийное удаление бензиловой и метиловой групп можно проводить двумя путями. Бензиловую группу удаляют гидрированием над катализатором, предпочтительно палладиевым катализатором, осажденным на
угле или другом пористом субстрате, или палладиевой чернью. В качестве растворителя предпочтительно применяют спирт, более предпочтительно метанол. Реакцию проводят при температуре от около 15o до около 40o в атмосфере водорода при давлении от около 1 атмосферы до около 4 атмосфер в течение около 2-4 часов.
В соответствии с другим вариантом бензиловую группу можно удалить реакцией с трибромидом бора в растворителе, таком как метиленхлорид, при температуре от около -5o до около 20o в течение от около 30 минут до около 2 часов. Таким образом получают продукт 12 GA-2, по-другому обозначаемый 12 GB-2.
Часть 2 пути b-2. Отщепление метоксигруппы с получением 12G можно проводить триметилсилилиодидом, как описано выше. (Эта стадия такая же, как третья стадия приведенного ниже Пути В).
СТАДИЯ 12. Путь В. (11G ---> 12G, Путь В)
Путь В имеет 3 стадии.
Часть 1. Бензиловую или другую подходящую группу удаляют гидрированием над катализатором, предпочтительно палладиевым катализатором, осажденным на угле или другом пористом субстрате, или палладиевой чернью. В качестве растворителя предпочтителен спирт, наиболее предпочтителен метанол. Реакцию проводят при температуре от около 15o до около 40o в атмосфере водорода при давлении от около 1 атмосферы до около 4 атмосфер в течение от около 12 до около 96 часов. Таким образом получают 12 GB-1.
Часть 2. Лактол затем окисляют в таких же условиях для образования гидроксиальдегида, применяя любое окисление в условиях Swern, например ДМСО, оксалилхлорид и триэтиламин в апротонном растворителе, например метиленхлориде, при температурном пределе от -78 до около 25.
В соответствии с другим вариантом окисление проводят раствором гипохлорита натрия, катализируемым ТЕМПО или замещенным ТЕМПО, таким как 4-ацетокси-ТЕМПО, в двухфазной системе, состоящей из воды и апротонного растворителя, такого как метиленхлорид. Температура реакции между около -5o и около +25o и время реакции между около 30 минутами и 2 часами. Таким образом получают 12 GB-2, по другому обозначенный 12 GB-1.
Часть 3. Удаление метильной группы проводят триметилсилилиодидом, либо полученным предварительно, либо генерированным in situ из триметилсилилхлорида и иодида натрия, в метиленхлориде или ацетонитриле. Условия описываются выше. Таким образом получают 12 G.
СТАДИЯ 13. (12G --->13G)
12G
подвергают реакции с акрилатным эфиром, например метил-, этил- или трет-бутилакрилатом, в присутствии основания, таких как гидрид калия, гидрид натрия, трет-бутоксид калия, карбонат натрия, карбонат
калия, карбонат цезия или третичные амины, например диизопропилэтиламина, в полярном апротонном растворителе, таком как диметилсульфоксид, ДМФ или ацетонитрил, при температуре между около 20o и 100o. Смотри СХЕМУ G в конце описания. Предпочтительными условиями является реакция с трет-бутилакрилатом и карбонатом цезия в ДМСО при около 50o. Продукт можно
выделить в виде сольвата с толуолом. Таким образом получают кетоэфир, соединения 13G.
СТАДИЯ 14. (13G ---> 14G)
Кетоэфир, который может существовать в основном или
исключительно в виде енола, преобразуют в 14G реакцией с сильной кислотой, такой как трифторуксусная кислота, при температуре от около 80o до около 110o в течение от около 10
минут до около 6 часов. Можно добавить растворитель, например толуол. Предпочтительными условиями являются смесь толуола и три-фторуксусной кислоты при 100-110o в течение 1-4 часов.
Все ссылки, перечисленные в описании Схем, включены здесь в качестве ссылок. С использованием описанных выше способов и заменой подходящих исходных материалов любой средний специалист в данной области может получить соединения и проводить реакции по этому изобретению. Одно из осуществлений этого изобретения описывается реакциями, способами и структурами СХЕМЫ CPT-11. Это осуществление только иллюстрирует данное изобретение и не должно ограничивать его никоим образом.
СПОСОБЫ, РЕАКЦИИ И СОЕДИНЕНИЯ СХЕМЫ CPT-11
СТАДИЯ 1. (цитраниловая кислота
1CPT)
Цитраниловую кислоту (152,0 г, 0,98 моль) и хлорид тетраметиламмония (107,71 г, 1,02 моль) суспендируют в оксихлориде фосфора (450 г, 273 мл, 2,9 моль) и нагревают на бане с
температурой 130oC. Твердые материалы растворяются со слабым экзотермическим эффектом, когда внутренняя температура достигает около 75oC, образуя прозрачный коричневый раствор.
Реакционную смесь нагревают при 130oC в течение 18 часов, затем нагревают до 145oC в течение 2 часов. Смесь охлаждают до комнатной температуры, выливают на 2 кг льда и
перемешивают в течение 2 часов. Твердую часть растворяют в 1,5 л этилацетата. Органический раствор сушат над сульфатом натрия, фильтруют и выпаривают, получая 146,9 г (78%) светло-коричневого твердого
продукта.
Т. пл. 195-197oC (разложение). (Литер.1 т.пл. 205-207oC).
1H ЯМР (300,13 МГц, DMSO-d6) δ 7,80 (с, 2Н).
13C ЯМР (75,47 МГц, DMSO-d6) δ 122,87, 144,60, 150,13, 163,66.
Номинальный масс-спектр: вычисленный m/z 192, найденный m/z 192.
Ссылки:
1. M. E. Baizer, M. Dub, S. Gister, N.G. Steiberg, J. Am. Pharm. Assoc, 1956, 45, 478-480.
2. Применение солей тетраалкиламмония в этом типе реакций описывается в патенте ГДР 154538.
СТАДИЯ 2. (1PCT ---> 2CPT)
1CPT (6,6 г, 0,034 моль) смешивают с 82 мл ТГФ и смесь охлаждают до -40oC. В течение
около 15 мин добавляют этилмагнийхлорид (52 мл, 104 ммоль, 2М раствор в ТГФ), поддерживая внутреннюю температуру реакции при менее чем -30oC. Охлаждающую баню убирают и получаемой
темной-коричневой смеси дают нагреться до 0oC и перемешивают при 0o в течение одного часа. Реакционную смесь снова охлаждают до -25oC и добавляют метилформиат (3,2 мл,
52 ммоль). После выдерживания в течение 15 мин при -25oC добавляют 20 мл 6 М соляной кислоты и смеси дают нагреться до комнатной температуры. Фазы разделяют и нижнюю водную фазу
экстрагируют 3 х 10 мл ТГФ. Объединенные фазы ТГФ промывают 2 раза смесью по 15 мл 1 н NaOH и 15 мл насыщенного раствора NaCl и затем один раз 15 мл насыщенного раствора NaCl. Органическую фазу сушат
над сульфатом натрия и затем концентрируют до получения масла. Добавляют толуол (50 мл) и смесь концентрируют до получения масла и эту процедуру повторяют, получая 6,01 г (84%) коричневого масла,
которое кристаллизуется в вакууме.
Т. пл. 60-63oC.
1H ЯМР (300,13 МГц, CDCl3) δ 1,17 (т, J = 7,1 Гц, 3Н), 2,88 (кв, J = 6,6 Гц, 2Н), 7,61 (с, 2Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,50, 32,61, 120,88, 147,66, 151,83, 197,15.
Номинальный масс-спектр: вычисленный m/z 204,
найденный m/z
204.
СТАДИЯ 3. (2CPT ---> 3CPT)
2CPT (90,2 г, 0,44 моль), этиленгликоль (650 мл) и триметилсилилхлорид (140 мл, 1,1 моль) смешивают и
перемешивают при комнатной температуре. В смеси постепенно образуются белые кристаллы. Через около 12 часов реакция завершается. Реакционную смесь нейтрализуют добавлением 1 л 1 н раствора NaOH и
экстрагируют 3 х 250 мл смеси этилацетат/гептан (1:1). Органические экстракты объединяют, сушат над сульфатом натрия и выпаривают. Кристаллический остаток сушат в высоком вакууме, получая 109,71 г
(100%) продукта.
Т. пл. 91oC.
1H ЯМР (300,13 МГц, CDCl3) δ 0,80 (т, J = 7,4 Гц, 3Н), 1,78 (кв, J = 7,4 Гц, 2Н), 3,72 (т, J = 7, 0 Гц, 2Н), 3,99 (т, J = 7,0 Гц, 2Н), 7,27 (с, 2Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,45, 32,77, 65,10, 108,94, 120,30, 150,57, 158,06.
Номинальный масс-спектр: вычисленный m/z 248, найденный m/z 248.
СТАДИЯ 4. (3CPT ---> 4CPT)
3CPT (57,5 г, 0,23 моль) растворяют в метаноле (170 мл). Добавляют метоксид
натрия (80 мл, 0,35 моль, 25 мас.% раствор в метаноле) и реакционную смесь кипятят с обратным холодильником на масляной бане с 85oC. Через 20 часов реакционной смеси дают охладиться до
комнатной температуры и затем тушат 250 мл воды. Двухфазную смесь разбавляют 200 мл метиленхлорида и разделяют. Водную фазу экстрагируют двумя порциями более чем по 100 мл метиленхлорида. Органические
экстракты объединяют, сушат над MgSO4, фильтруют и концентрируют до образования масла янтарного цвета, которое кристаллизуется при внесении затравки, получая 50,43 г (89%) светло-желтого
твердого продукта.
Т. пл. 47oC.
1H ЯМР (300,13 МГц, CDCl3) δ 0,88 (т, J = 7,4 Гц, 3Н), 1,85 (кв, J = 7,5 Гц, 2Н), 3,78 (т, J = 6,9 Гц, 2Н), 3,93 (с, 3Н), 4,02 (т, J = 7,1 Гц, 2Н), 6,73(с, 1Н), 6,98 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,62, 32,53, 54,04, 64,79, 106,25, 109,23, 113, 83, 148,33, 157,27, 163,94.
Номинальный масс-спектр: вычисленный m/z 243, найденный m/z 244 (M + 1).
СТАДИЯ 5. (4CPT ---> 5CPT)
4CPT (73,05 г, 0,299
моль) растворяют в 1400 мл гептана и охлаждают до -10oC. В течение 10 мин добавляют н-бутиллитий (294 мл, 0,588 моль, 2,5 М раствор в гексане), поддерживая внутреннюю температуру <
5oC. После завершения добавления бутиллития оранжевую смесь перемешивают при 0oC в течение 30 мин. Смесь затем охлаждают до -30oC и добавляют N-формилпиперидин (66,0
мл, 0,588 моль). Смеси дают нагреться до 0oC и перемешивают при 0oC в течение 1 ч. Темно-красную смесь тушат добавлением 600 мл 1 н HCl. Фазы разделяют и водную фазу экстрагируют
2 х 250 мл МТБЭ. Органические фазы объединяют, получая раствор 5аCPT. Часть этого раствора хроматографируют на диоксиде кремния с применением смеси гексан/этилацетат (4:1), получая очищенный образец
5аCPT для идентификации.
К раствору 5аCPT добавляют воду (250 мл), хлорид тетрабутиламмония (8,3 г, 0,029 моль) и борогидрид натрия (11,3 г, 0,29 моль) и смесь энергично перемешивают при комнатной температуре. Приблизительно через 18 часов восстановление заканчивается. Добавляют 20 мл ацетона и смесь перемешивают при комнатной температуре в течение 30 мин. Водную фазу удаляют и органическую фазу промывают один раз 500 мл воды. Органическую фазу выпаривают до получения масла. Масло хроматографируют на 800 г диоксида кремния, применяя смесь гексан/этилацетат (4:1). Выход продукта 57,30 г, 71%. Выделяют также 15,0 г (20%) по существу чистого 4CPT.
5аCPT
1H ЯМР (300,13 МГц, CDCl3) δ 0,96 (т, J = 9,0 Гц, 3Н), 2,03 (кв,
J = 9,0 Гц, 2Н), 3,75 (м, 2Н), 4,00 (м, 2Н), 4,00 (с, 3Н), 7,13 (с, 1Н), 10,44 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,32, 33,28, 54,81, 64,78, 109,66, 114,67, 117,20, 150,83, 157,52, 161,75, 190,80.
Номинальный масс-спектр: вычисленный m/z 271, найденный m/z 271.
5CPT
Т. пл. 49-56oC.
1H ЯМР (300,13 МГц, CDCl3) δ 0,84 (т, J = 7,5 Гц, 3Н), 1,87 (кв, J = 7,0 Гц, 2Н), 3,74 (м, 2Н), 3,92 (с, 3Н), 3,97 (м, 2Н), 4,72 (с, 1Н), 7,05 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,46, 33,01, 54,50, 56,16, 64,98, 110,25, 114,53, 119,15, 147,39, 154,50, 163,00.
Номинальный масс-спектр: вычисленный m/z 273, найденный m/z 273.
СТАДИЯ 6. (5CPT ---> 6CPT, СХЕМА CPT см. в конце описания)
5CPT (503,98 г, 1,841 моль) растворяют в 1330 мл ТГФ в колбе на 12 л,
снабженной механической мешалкой, капельной воронкой и термопарой с соединительным устройством. В колбу добавляют 1188 мл 20% раствора трет-бутоксида калия в ТГФ, поддерживая внутреннюю температуру
ниже 30o. Смесь перемешивают в течение 30 мин, затем через капельную воронку добавляют бензилбромид (230,0 мл, 2,117 моль), поддерживая внутреннюю температуру ниже 30o. После
окончания добавления бензилбромида смесь перемешивают при 20-30o в течение 1 часа. Через один час добавляют 38 мл 40% водного раствора диметиламина и смесь перемешивают при 20-30o
в течение 30 мин. Добавляют 276 мл 1 н HCl и 2 л этилацетата и фазы разделяют. Органическую фазу промывают 3 х 1 л воды и затем выпаривают до получения масла. Выход продукта: 663,5 г, 99,3%.
1H ЯМР (300,13 МГц, CDCl3) δ 0,75 (т, J = 7,4 Гц, 3H), 1,82 (кв, J = 7,4 Гц, 2Н), 3,61 (м, 2Н), 3,82 (с, 3H), 3,85 (м, 2Н), 4,48 (с, 2Н), 4,57 (с, 2Н), 6,97 (с, 1Н), 7,23 (м, 5Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,50, 32,96, 54,47, 62,83, 64,73, 73,20, 110,12, 114,8, 116,42, 127,54, 127,76, 128,24, 138,43, 147,91, 155, 62, 163,74.
Номинальный масс-спектр: вычисленный m/z 363, найденный m/z 364 (М+1).
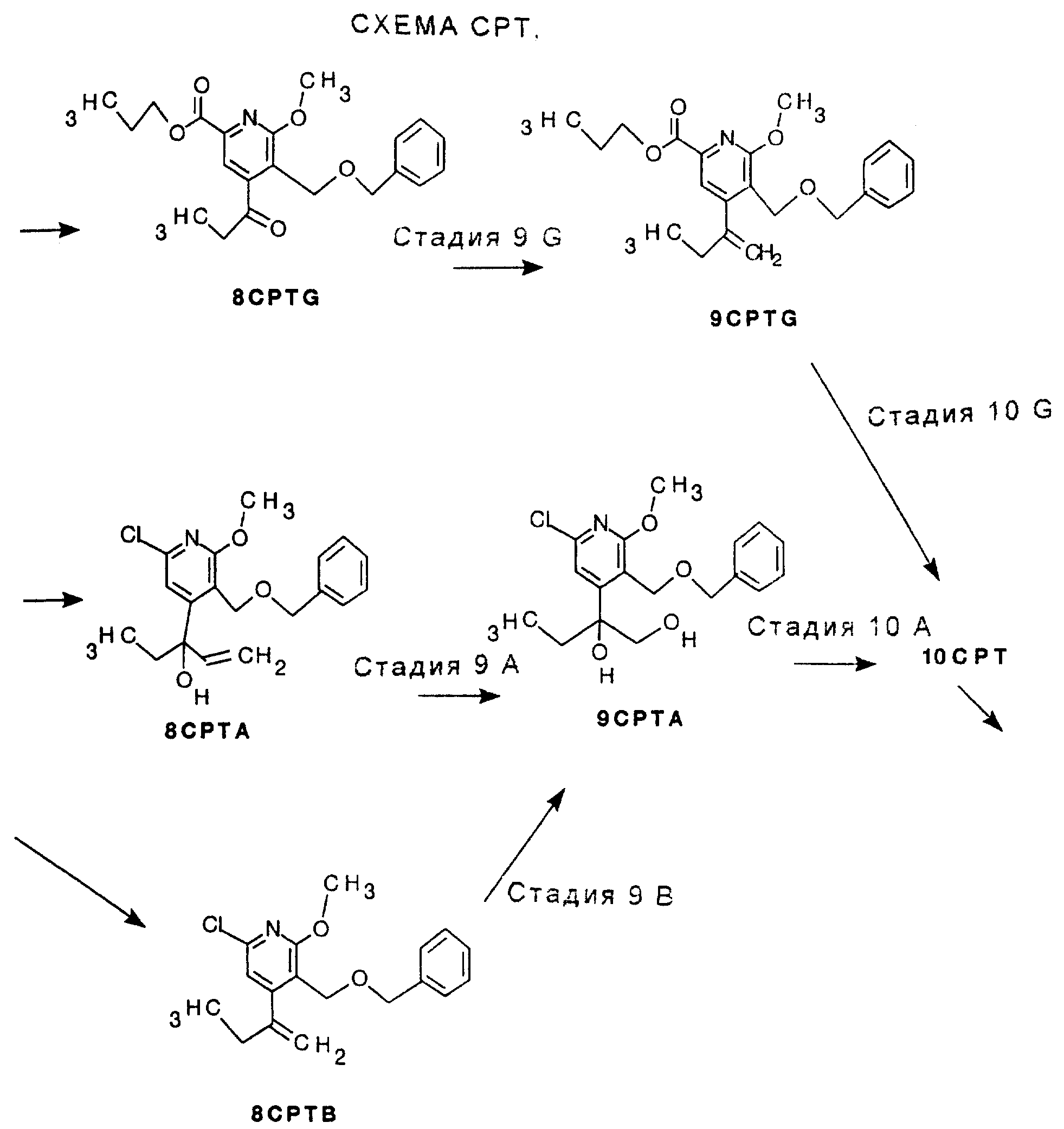
Имеется две разные возможные реакции СТАДИИ 7, ряды G и A, и три разные возможные реакции СТАДИИ 8. Смотри Схему CPT в конце описания.
СТАДИЯ 7G. (6CPT ---> 7CPTG)
6CPT (66,45 г, 183 ммоль), ацетат палладия (2,05 г, 9,13 ммоль), DPPP (4,14 г, 10,0
ммоль), карбонат калия (37,86 г, 274 ммоль), н-пропанол (665 мл) и ДМФ (332 мл) загружают в колбу. Колбу продувают азотом и затем оксидом углерода. Смесь нагревают до 90oC в атмосфере
оксида углерода в течение 16 часов. Реакционную смесь охлаждают и вентилируют. Твердую часть удаляют фильтрованием через целит и целит промывают 350 мл ТГФ. Объединенные фильтраты и промывную жидкость
концентрируют до объема около 400 мл. Добавляют воду (700 мл) и МТБЭ (700 мл). Водную фазу отделяют и экстрагируют 350 мл МТБЭ. Объединенные растворы МТБЭ экстрагируют 4 х 350 мл воды, сушат над
сульфатом натрия и выпаривают, получая 68,03 г (выход 89%) светло-оранжевого масла после колоночной хроматографии (силикагель: 230-400 меш, элюент: смесь гептан/этилацетат, 80:20).
1H ЯМР (300,13 МГц, CDCl3) δ 10,87 (т, J = 7,4 Гц, 3Н), 0,98 (т, J = 7,4 Гц, 3Н), 1,77 (м, 2Н), 1,93 (кв, J = 7,4 Гц, 2Н), 3,71 (м, 2Н), 3,94 (м, 2Н), 3,99 (с, 3Н), 4,26 (т, J = 6,7 Гц, 2Н), 4,59 (с, 2Н), 4,74 (с, 2Н), 7,29 (м, 5Н), 7,82 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,5, 10,42, 22,02, 33,08, 54,07, 63,08, 64,72, 66, 98, 73,29, 110,26, 117,05, 122,14, 127,51, 127,99, 128,22, 138,45, 144,70, 153,62, 163,88, 165,29.
Номинальный масс-спектр: вычисленный m/z 415, найденный m/z 416 (М + 1).
СТАДИЯ 7A. (6CPT ---> 7CPTA)
6CPT (50,0 г 0,137 моль) растворяют в 50% водной трифторуксусной кислоте (250 мл) и перемешивают при комнатной температуре в течение 48 часов.
Добавляют воду (200 мл) и этилацетат (200 мл). Фазы разделяют и водную фазу экстрагируют этилацетатом (3 х 200 мл). Объединенные органические слои промывают насыщенным раствором бикарбоната натрия
(500 мл) до тех пор, пока не будет удалено остаточное количество трифторуксусной кислоты, и затем промывают водой (200 мл). Органический слой сушат над безводным сульфатом магния, фильтруют и
концентрируют, получая 42,6 г (97%) продукта.
1H ЯМР (300,13 МГц, CDCl3) δ 1,04 (т, 7,2 Гц, 3Н), 2,71 (кв, 7,2 Гц, 2Н), 3,95 (с, 3Н), 4,47 (с, 2Н), 4,56 (с, 2Н), 6,77 (с, 1Н), 7,29 (м, 5Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,39, 36,15, 54,56, 63,16, 73,43, 113,35, 115,73, 127,86, 127,97, 128,51, 137,50, 147,81, 153,07, 161,38, 204,47.
Номинальный масс-спектр: вычисленный m/z 319, найденный m/z 320 (М+1).
Имеются 3 возможные разные реакции СТАДИИ 8, ряды G, A и B, смотри Схему CPT в конце описания.
СТАДИЯ 8G. (7CPTG ---> 8CPTG)
7CPTG (68,02 г, 163,7 ммоль) растворяют при комнатной температуре в 384 мл 50% водной трифторуксусной
кислоты. Смесь перемешивают при комнатной температуре в течение 21 часа. Добавляют 880 мл воды и смесь экстрагируют 2 х 500 мл этилацетата. Органические фазы объединяют и промывают 2 х 500 мл воды и
затем нейтрализуют насыщенным раствором бикарбоната натрия. Органическую фазу затем сушат над сульфатом натрия и выпаривают, получая 59,86 г (98,4%) продукта в виде светло-желтого масла.
1H ЯМР (300,13 МГц, CDCl3) δ 0,96 (м, 6Н), 1,72 (м, 2Н), 2,68 (кв, J = 7,2 Гц, 2Н), 3,96 (с, 3Н), 4,23 (т, J = 6,7 Гц, 2Н), 4,42 (с, 2Н), 4,58 (с, 2Н), 7,24 (м, 5Н) 7,48 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,55, 10,41, 21,99, 36,21, 54,13, 63,83, 67,22, 73,56, 115,50, 121,49, 127,86, 127,97, 128,19, 128,37, 137, 32, 144,87, 150,96, 161,31, 164,54.
Номинальный масс-спектр: вычисленный m/z 371, найденный m/z 372 (М + 1).
СТАДИЯ 8А. (7CPTA ---> 8CPTA)
7CPTA (1,00
г, 3,13 ммоль) растворяют в 5 мл ТГФ и охлаждают до -40oC в атмосфере азота. Добавляют винилмагнийбромид (2,9 мл, 4,4 ммоль, 1,5 М раствор в ТГФ). Реакционную смесь выдерживают при -40oC в течение одного часа и затем дают согреться до комнатной температуры. После выдерживания 1 час при комнатной температуре реакционную смесь тушат насыщенным водным раствором хлорида аммония
(10 мл) и разбавляют этилацетатом (10 мл). Водный слой экстрагируют 10 мл этилацетата, экстракт объединяют с предыдущим органическим слоем и сушат над сульфатом магния. После фильтрования и
концентрирования получают 1,098 г (выход 100%) масла цвета светлого янтаря.
1H ЯМР (300,133 МГц, CDCl3) δ 0,87 (т, J = 7,32 Гц, 3Н), 1,79 - 2,00 (м, 2Н), 3, 93 (с, 3Н), 4,54 (с, 2Н), 4,83 (с, 2Н), 5,16 (дд, J = 0,99 Гц, 10,59 Гц, 1Н), 5,25 (дд, J = 0,99, 17,23 Гц, 1Н), 6,01 (дд, J = 10,59, 17,23 Гц, 1Н), 6,94 (с, 1Н), 7,30 - 7,37 (м, 5Н).
13C ЯМР (75,468 МГц, CDCl3) δ 7,7, 34,2, 54,5, 62,6, 72,4, 78,0, 114,0, 115,6, 115,9, 127,9, 128,0, 137,2, 143,0, 148,2, 159,2, 163,1.
Номинальный масс-спектр; вычисленный m/z 347, найденный m/z 348 (М+1).
Имеются три разные возможные реакции СТАДИИ 9, ряды G, A и B, смотри СХЕМУ CTP в конце описания.
СТАДИЯ 9G.
(8CPTG ---> 9CPTG)
Бромид метилтрифенилфосфония (2,14 г, 6,0 ммоль) растворяют в 15 мл ДМФ и перемешивают при комнатной температуре. Добавляют раствор бистриметилсилиламида калия (10
мл, 5,0 ммоль, 0,5 М раствор в толуоле) и желтый раствор с суспендированным белым твердым веществом перемешивают при комнатной температуре в течение 10 мин. Сразу добавляют весь раствор 8CPTG (1,48 г,
4,0 ммоль) в 5 мл ТГФ, получая смесь темно-красного цвета, которая быстро превращается в коричневую смесь. Смесь перемешивают в течение 10 мин. Добавляют дополнительное количество раствора илида до
тех пор, пока не израсходуется весь 8CPTG. Реакцию тушат добавлением 10 мл 1 н HCl. Добавляют 20 мл МТБЭ и фазы разделяют. Водную фазу экстрагируют 2 х 20 мл МТБЭ. Объединенные органические фазы
промывают 3 х 20 мл воды, сушат над сульфатом натрия и выпаривают до объема около 15 мл (имеет место слабая кристаллизация оксида трифенилфосфина). Раствор хроматографируют на диоксиде кремния (около
20 г) с применением смеси гексан/этилацетат (4:1), получая 1,39 г продукта (выход 92%).
1H ЯМР (300,13 МГц, CDCl3) δ 0,85 (м, 6Н), 1,59 (м, 2Н), 2,20 (кв, J = 7,4 Гц, 2Н), 3,89 (с, 3Н), 4,12 (т, J = 6,7 Гц, 2Н), 4,33 (с, 2Н), 4,42 (с, 2Н), 4,89 (с, 1Н), 5,06 (с, 1Н), 7,17 (м, 5Н), 7,35 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 10,43, 12,07, 22,02, 30,23, 53,95, 63,79, 67,00, 73,03, 114,66, 118,67, 121,40, 127,60, 127,90, 128,26, 138,21, 144,49, 147,58, 155,33, 163,11, 165,25.
Номинальный масс-спектр: вычисленный m/z 369, найденный m/z 369.
СТАДИЯ 9A. (8CPTA ---> 9CPTA)
8CPTA (0,500 г, 1,43 ммоль) растворяют в 40 мл смеси метанол:метиленхлорид (1: 1) и
охлаждают до -70oC и затем продувают кислородом в течение 15 минут. Через раствор до превращения его в синий цвет пропускают поток озона из генератора озона Welsbach. Раствор затем
продувают кислородом в течение пяти минут для удаления избытка озона и затем продувают в течение десяти минут азотом. При -78oC раствор затем обрабатывают борогидридом натрия (0,250 г, 6,61
ммоль) в виде раствора в 5 мл 50% водного метанола. Через пятнадцать минут реакционной смеси дают нагреться до комнатной температуры в течение часа. После выдерживания один час при комнатной
температуре реакционную смесь тушат 1 М раствором HCl (10 мл) и разделяют. Водную фазу экстрагируют порциями по 20 мл и 10 мл метиленхлорида, экстракты объединяют с первоначальным органическим слоем и
сушат над сульфатом натрия. После фильтрования и концентрирования получают 0,491 г (выход 99%) 9CPTA.
1H ЯМР (300,133 МГц, CDCl3) δ 0,82 (т, J = 7,20 Гц, 3Н), 1,86 (дд, J = 7,20 Гц, 14,71 Гц, 2Н), 3,69 (с, 2Н), 3,96 (с, 3Н), 4,19 - 4,31 (м, 2Н), 4,28 (с, 2Н), 4,59 (с, 2Н), 7,20 (с, 1Н), 7,40 - 7,29 (м, 5Н).
13C ЯМР (75,468 МГц, CDCl3) δ 7,61, 35,44, 54,50, 62,97, 73,40, 75,26, 84,72, 113,71, 114,13, 127,91, 128,18, 128,35, 137,48, 148,56, 158,01, 163,46.
Номинальный масс-спектр: вычисленный m/z 351, найденный m/z.
СТАДИЯ 9В (8CPTB ---> 9CPTA)
Применение реагентов и условий, сходных с описанными в СТАДИИ 9CPTG.
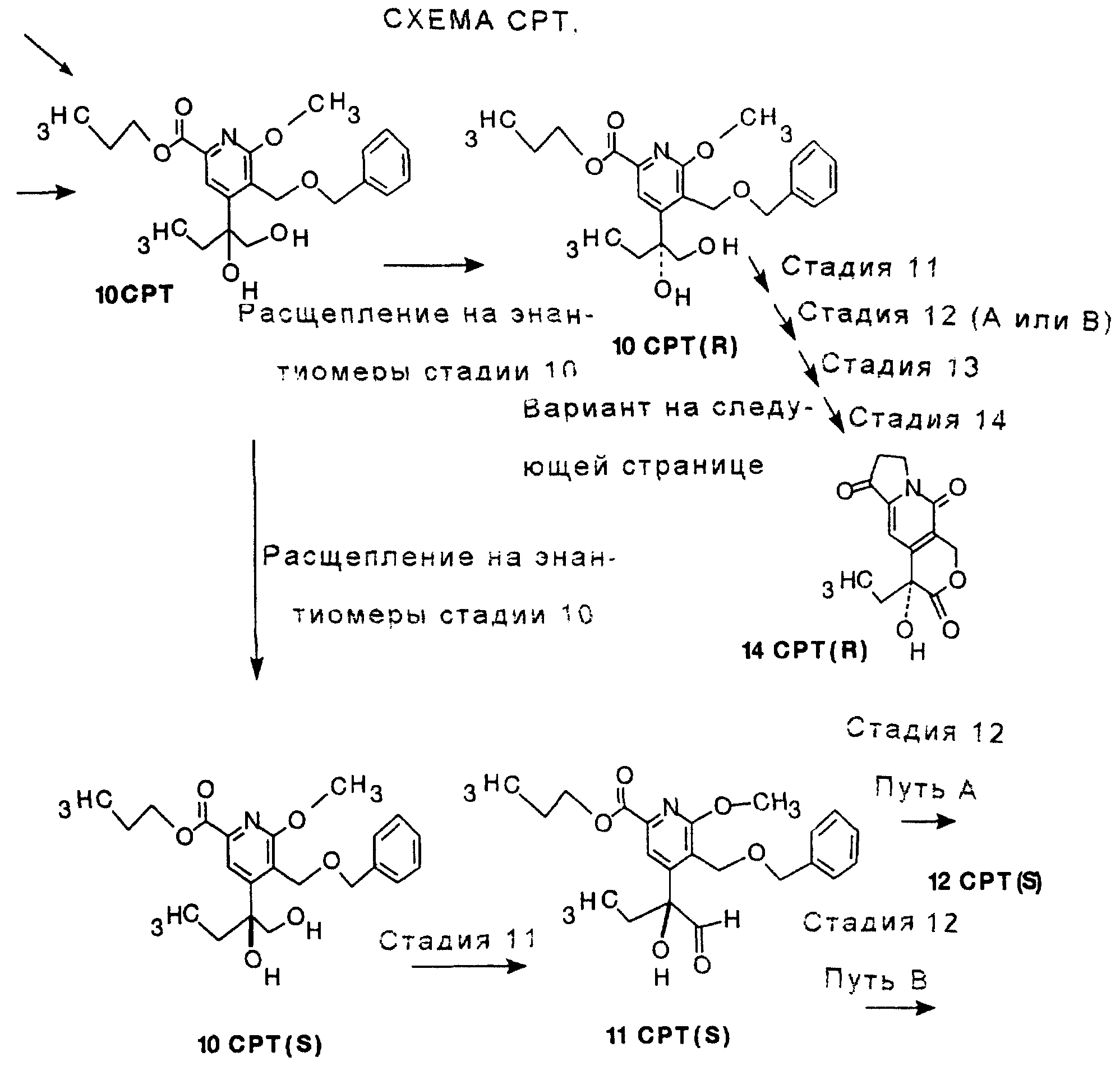
СТАДИЯ 10G.
(9CPTG ---> 10 CPTG)
9CPTG (100,0 г, 0,271 моль), дигидрат N-оксида триметиламина (90,24 г, 0,81 моль) и тетроксид осмия (0,68 г, 2,7 ммоль) и 300 мл трет-бутанола загружают в колбу.
Смесь нагревают до 40o. Через 24 часа смесь охлаждают до 20-25o. Добавляют 300 мл воды и 110 г метабисульфита натрия и смесь перемешивают в течение 30 мин при комнатной
температуре. Смесь экстрагируют 4 х 200 мл этилацетата. Органические фазы объединяют и перемешивают с 50 г диоксида кремния 70-230 меш в течение 1 часа. Диоксид кремния отделяют фильтрованием и
промывают 100 мл этилацетата. Фильтрат перемешивают со 100 г магнезола в течение 30 мин и затем суспензию фильтруют через 50 г магнезола. Фильтраты объединяют и концентрируют до образования масла.
Добавляют 200 мл толуола и 800 мл гептана и смеси дают кристаллизоваться при -20o в течение 18 часов. Твердую часть отделяют фильтрованием и промывают 200 мл гептана. Выход 10CPT 83,5 г. Из
фильтратов и промывных жидкостей можно выделить хроматографией дополнительное количество 10CPT.
1H ЯМР (300,13 МГц, CDCl3) δ 0,74 (т, J = 7,4 Гц, 3Н), 1,03 (т, J = 7,4 Гц, 3Н), 1,80 (м, 4Н), 3,69 (д, J = 11,2 Гц, 1Н), 3,86 (д, J = 11,2 Гц, 1Н), 4,01 (с, 3Н), 4,31 (т, J = 6,7 Гц, 2Н), 4,88 (д, J = 10,7 Гц, 1Н), 4,96 (д, J = 10,7 Гц, 1Н), 7,33 (м, 5Н), 7, 64 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,55, 10,41, 22,01, 31,71, 54,16, 62,95, 67,13, 70,86, 72,69, 80,12, 117,83, 122,25, 128,00, 128,42, 137,14, 144, 74, 155,82, 163,16, 165,23.
Номинальный масс-спектр: вычисленный m/z 403, найденный m/z 404 (М + 1).
СТАДИЯ 10A. (9CPTA ---> 10CPTA)
9CPTA (2,13 г, 6,
0 ммоль) растворяют в 1-пропаноле (25 мл) и ДМФ (50 мл) в колбе, снабженной линией для продувки и магнитной мешалкой. В колбу загружают твердый карбонат калия (1,24 г, 9,0 ммоль), ацетат палладия (II)
(67 мг, 0,3 ммоль) и DPPP (124 мг, 0,3 ммоль) и затем продувают оксидом углерода и нагревают до 85oC в течение 15 часов. Реакционную смесь затем охлаждают до комнатной температуры и
продувают азотом. Раствор фильтруют через целит и целит промывают этилацетатом (3 х 50 мл). Объединенный фильтрат и промывную жидкость концентрируют в вакууме до образования масла. Масло разбавляют
этилацетатом (100 мл) и получаемый раствор промывают водой (50 мл) и затем концентрируют в вакууме. Продукт выделяют колоночной хроматографией (силикагель, 230-400 меш, элюентом является смесь
этилацетат:гексан, 1:4), получая 1,40 г (58%) 10CPT.
После проведения СТАДИЙ 10 можно проводить расщепление продукта на оптические изомеры (энантиомеры), это называют в СХЕМАХ как расщепление на оптические изомеры СТАДИИ 10, смотри Схему CPT в конце описания.
СТАДИЯ 10 - Расщепление на оптические изомеры
К 10CPT (8,0 г, 20 ммоль), суспендированному в
200 мл метил-трет-бутиловом эфире, добавляют 8,0 г катализатора PS-30 (липаза Pseudomonas cepaica, иммобилизованная на равном по массе целите 521) и 1,85 мл (20 ммоль) винилацетата. Получаемую
суспензию перемешивают магнитной мешалкой при комнатной температуре в течение 24 ч. Катализатор удаляют фильтрованием, промывают метил-трет-бутиловым эфиром (3 х 100 мл), органический растворитель
концентрируют в вакууме приблизительно до объема раствора 25 мл. Раствор сохраняют при 0-5oC, получаемый твердый продукт собирают фильтрованием и промывают гексаном (3 х 25 мл), что дает 2,
75 г 10CPT (s-энантиомер), [α]5 = +3,25o в хлороформе (ее > 99%
по ВЭЖХ на колонке Chirapak AD, гексан/изопропанол, 90: 10, 1 мл/мин, 254 нм).
СТАДИЯ 11. (10CPT ---> 11CPT)
10CPT (0,565 г, 1,4 моль), 4-ацетокси-ТЕМПО (0,006 г, 0,
028 ммоль), бромид калия (0,0167 г, 0,14 ммоль) и бикарбонат натрия (0,0153 r, 0,182 ммоль) загружают в колбу. Добавляют метиленхлорид (7 мл) и воду (1 мл) и смесь перемешивают при комнатной
температуре в течение 5 мин. В течение около 40 мин через шприц добавляют раствор гипохлорита натрия (1,6 мл, 0,95 М). После окончания добавления реакцию тушат добавлением 5% водного раствора
метабисульфита натрия. Водную фазу отделяют и экстрагируют 2 х 5 мл метиленхлорида. Объединенные органические фазы сушат над сульфатом натрия и выпаривают, получая 0,601 г коричневого сиропа.
Химический выход по существу составляет 100%.
1H ЯМР (300,13 МГц, CDCl3) δ 0,91 (т, J = 7,5 Гц, 3Н), 1,03 (т, J = 7,5 Гц, 3Н), 1,83 (м, 2Н), 2,10 (м, 2Н), 4,02 (с, 3Н), 4,35 (т, J = 6,6 Гц, 2Н), 4,55 (с, 2Н), 4,68 (д, J = 11,7 Гц, 1Н), 4,87 (д, J = 11,7 Гц, 1Н), 7,35 (м, 5Н), 7,78 (с, 1Н), 9,62 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,24, 10,43, 22,02, 29,72, 54,30, 63,2, 67,24, 73,12, 82,37, 117,45, 122,48, 128,23, 128,55, 136,67, 145,05, 150,55, 162,88, 164,93, 200,14.
Номинальный масс-спектр: вычисленный m/z 401, найденный m/z 402 (М + 1).
Альтернативные реакции
Существует два различных пути реакции для СТАДИИ 12, названные Путем A или B. Путь А имеет
две части. Вторая часть пути A, Часть 2, имеет два пути реакций, путь а, одностадийный способ, и путь b, двустадийный способ. Путь В имеет всего три части. Второй промежуточный продукт, полученный
через путь A, Часть 2, путь b-1, 12 GA-2, такой же, как второй промежуточный продукт, полученный через Путь В, Часть 2, 12 GB-2. Третья часть Пути B такая же, как вторая стадия Пути A, Часть 2, путь
b-2. Смотри СХЕМУ CPT в конце описания.
СТАДИЯ 12, Путь A, Часть 1. (11CPT ---> 12CPT A-1)
Раствор 11CPT (0,206 г, 0,5 ммоль) в 6 мл трет-бутанола смешивают с
раствором NaH2PO4 (0,035 г) в 2 мл воды и охлаждают до 0o. Добавляют 50% раствор пероксида водорода (0,043 мл) и затем сразу все количество раствора хлорита натрия (0,
076 г, 0,675 ммоль) в 0,5 мл воды. Через 5 мин реакцию тушат добавлением 1,8 мл 10% водного раствора метабисульфита натрия. Смесь распределяют между водой и метиленхлоридом и водную фазу экстрагируют
2 раза метиленхлоридом. Объединенные органические фазы выпаривают, получая 0,200 г (93%) продукта 12 CPT A-1.
1H ЯМР (300,13 МГц, CDCl3) δ 1,02 (м, 6H), 1, 82 (м, 2H), 2,23 (м, 2H), 3,99 (с, 3Н), 4,32 (т, J = 6,9 Гц, 2Н), 4,53 (д, J = 11,7 Гц, 1Н), 4,62 (д, J = 11,7 Гц, 1Н), 4,68 (д, J = 11,7 Гц, 1Н), 4,97 (д, J = 11,7 Гц, 1Н), 7,32 (м, 5Н), 7,90 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,83, 10,41, 22,01, 32,15, 54,36, 62,62, 67,31, 72,95, 79,21, 117,39, 121,82, 128,21, 128,52, 136,52, 145,25 152,55, 162, 97, 165,01, 176,06.
Номинальный масс-спектр: вычисленный m/z 417, найденный m/z 418 (М+1).
СТАДИЯ 12, Путь A, Часть 2, путь а (одна стадия) (12CTA A-1 --->
12CPT)
Раствор 12A-1 CPT (0,17 г, 0,40 ммоль) и пиридина (0,05 мл, 0,6 ммоль) в 5 мл ацетонитрила перемешивают при комнатной температуре. Добавляют триметилсилилиодид (0,2 мл, 1,4 ммоль) и
смесь перемешивают в течение ночи при комнатной температуре, затем нагревают при 45o в течение 48 часов. Добавляют соляную кислоту (5 мл, 6 н) и смесь перемешивают при комнатной температуре
в течение 15 мин. Смесь экстрагируют 3 х 5 мл этилацетата и объединенные экстракты промывают 5% раствором бисульфита натрия. Раствор этилацетата сушат над сульфатом натрия и выпаривают. Остаток
хроматографируют на диоксиде кремния с применением смеси метиленхлорид/метанол (95:5), получая 0,083 г (69%) продукта в виде светло-желтого масла.
1H ЯМР (300,13 МГц, CDCl3) δ 1,02 (м, 6Н), 1,80 (м, 4Н), 4,36 (т, J = 6,0 Гц, 2Н), 5,22 (д, J = 16,5 Гц, 1Н), 5,60 (д, J = 16,5 Гц, 1Н), 7,40 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,66, 10,33, 21,84, 31,88, 66,07, 68,68 72,32, 107,10, 124,45, 134,41, 149,99, 159,80, 173,26, 176,63.
Номинальный масс-спектр: вычисленный m/z 295, найденный m/z 296 (М + 1).
СТАДИЯ 12, Путь A, Часть 2, путь b-1. (12 CPT A-1 ---> 12CPT A-2)
Раствор гидроксикислоты 12 CPT A-1 (2,64 г, 6,3 ммоль) в 50 мл метанола
перемешивают с 10% палладием на угле (0,264 г) в атмосфере водорода при атмосферном давлении в течение 2 часов при комнатной температуре. Катализатор удаляют фильтрованием через целит и промывают 10
мл метанола. Объединенный фильтрат и промывную жидкость выпаривают, получая продукт (1,82 г, 93%) в виде светло-желтого очень вязкого масла.
1H ЯМР (300,13 МГц, CDCl3) δ d 0,88 (т, J = 7,5 Гц, 3Н), 0,97 (т, J = 7,6 Гц, 3Н), 1,76 (м, 4Н), 4,0 (с, 3Н), 4,25 (т, J = 6,9 Гц, 2Н), 5,23 (д, J = 16,2 Гц, 1Н), 5,52 (д, J = 16,2 Гц, 1Н), 7,85 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,49, 10,32, 21,89, 31,88, 54,08, 65,53, 67,22, 72,72, 114,79, 115,22, 146,01, 148,91, 158,50, 164,51, 173,53.
СТАДИЯ 12, Путь A, Часть 2, путь b-2 (12 CPT A-2 ---> 12 CPT)
Раствор гидроксилактона 12CPT A-2 (1,93 г, 6,2 ммоль) и иодида натрия (1,86 г, 12,4 ммоль) в 30 мл ацетонитрила
перемешивают при 0o. Добавляют триметилсилилиодид (1,6 мл, 12,4 ммоль) и смесь перемешивают и дают нагреться до комнатной температуры в течение 12 часов. Добавляют дополнительное количество
иодида натрия (0,9 г, 6,2 ммоль) и триметилсилилхлорида (0,8 мл, 6,2 ммоль) и перемешивание продолжают в течение более чем 6 часов. Добавляют 1 н соляную кислоту (10 мл) и метабисульфит натрия (0,6 г)
и смесь перемешивают при комнатной температуре в течение 1 часа. Добавляют этилацетат (30 мл) и водную фазу экстрагируют дополнительными 30 мл этилацетата. Объединенные органические фазы промывают
водой, сушат над сульфатом натрия и выпаривают, получая продукт в виде светло-желтого твердого вещества (1,84 г, 100%).
1H ЯМР (300,13 МГц, CDCl3) δ 1,02 (м, 6Н), 1,80 (м, 4Н), 4,36 (т, J = 6,0 Гц, 2Н), 5,22 (д, J = 16,5 Гц, 1Н), 5,60 (д, J = 16,5 Гц, 1Н), 7,40 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,66, 10, 33, 21,84, 31,88, 66,07, 68,68, 72,32, 107,10, 124,45, 134,41, 149,99, 159,80, 173,26, 176,63.
Номинальный масс-спектр: вычисленный m/z 295, найденный m/z 296 (М + 1).
СТАДИЯ 12, Путь B, Часть 1 (11 CPT ---> 12 CPT B-1)
Гидроксиальдегид 11CPT (2,62 г, 6,6 моль) растворяют в 30 мл метанола и перемешивают с 10% палладием на угле (0,26 г) в атмосфере
водорода при атмосферном давлении. Реакция завершается через 96 часов. Катализатор удаляют фильтрованием через целит и промывают 10 мл метанола. Объединенный фильтрат и промывную жидкость выпаривают,
получая 1,97 г (96%) продукта в виде белого твердого вещества.
1H ЯМР (300,13 МГц, CDCl3) δ 0,84 (т, J = 7,5 Гц, 3Н), 0,95 (т, J = 7,4 Гц, 3Н), 1,73 (м, 4Н), 3,89 (с, 3Н), 4,24 (т, J = 6,7 Гц, 2Н), 4,57 (д, J = 17,2 Гц, 1Н), 4,73 (д, J = 17,2 Гц, 1Н), 7,86 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,53, 10,36, 21,94, 31,70, 53,69, 58,31, 67,04, 70,68, 93,26, 116,57, 120,38, 143,54, 148,98, 158,48, 165,34.
СТАДИЯ 12, Путь B, Часть 2 (12CPT B-1 ---> 12CPT B-2)
Раствор лактола
12CPT В-1 (1,94 г, 6,2 ммоль) в 37 мл метиленхлорида перемешивают при комнатной температуре с раствором TEMPO (0,04 г, 0,25 ммоль), бикарбоната натрия (0,081 г, 0,96 ммоль) и бромида калия (0,088 г, 0,
74 ммоль) в 3 мл воды. В течение 30 мин по каплям добавляют раствор гипохлорита натрия (12%, приблизительно 12 мл). Для разложения избытка гипохлорита натрия добавляют бисульфит натрия (1,0 г). Водную
фазу экстрагируют метиленхлоридом (10 мл) и объединенные органические фазы промывают один раз водой (10 мл) и сушат над сульфатом натрия. Растворитель выпаривают, получая продукт (1,90 г, 99%) в виде
масла, которое отверждается при стоянии.
1H ЯМР (300,13 МГц, CDCl3) δ d 0,88 (т, J = 7,5 Гц, 3Н), 0,97 (т, J = 7,6 Гц, 3Н), 1,76 (м, 4Н), 4,0 (с, 3Н), 4,25 (т, J = 6,9 Гц, 2Н), 5,23 (д, J = 16,2 Гц, 1Н), 5,52 (д, J = 16,2 Гц, 1Н), 7,85 (с, 1Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,49, 10,32, 21,89, 31,88, 54,08, 65, 53, 67,22, 72,72, 114,79, 115,22, 146,01, 148,91, 158,50, 164,51, 173,53.
СТАДИЯ 12, Путь B, Часть 3. (12 CPT В-2 ---> 12 CPT)
Эта стадия идентична СТАДИИ 12 и
применяемые способы такие же, как в СТАДИИ 12.
СТАДИЯ 13, Путь A, Часть 2, путь b-2. (12 CPT ---> 13 CPT)
12 CPT (10,1 г, 0,339 моль), карбонат цезия (22,0 г, 0,067
моль), трет-бутилакрилат (25 мл, 0,169 моль) и ДМСО (150 мл) перемешивают при 47-50o в течение 19 часов. Смесь охлаждают и добавляют 20 мл концентрированной соляной кислоты и 180 мл воды.
Смесь экстрагируют 4 раза, применяя всего 500 мл смеси толуола и этилацетата (4:1, об./об.). Объединенные экстракты промывают три раза водой и затем выпаривают до получения масла. Добавляют 200 мл
толуола и раствор концентрируют, получая 13CPT в виде кристаллического сольвата 1:1 с толуолом (11,5 г, 67%).
1H ЯМР (300,13 МГц, CDCl3) δ 0,92 (т, J = 7,4 Гц, 3Н), 1,50 (с, 9Н), 1,71-1,79 (м, 2Н), 2,28 (с, 3Н), 4,59 (с, 2Н), 5,16 (д, J = 17,8 Гц, 1Н), 5,61 (д, J = 17,8 Гц, 1Н), 6,94 (с, 1Н), 7,0-7,2 (м, 5Н).
13C ЯМР (75,47 МГц, CDCl3) δ 7,64, 21,38, 28,20, 31,41, 49,27, 66,13, 72,50, 83,55, 97,80, 105,69, 118,59, 125,22, 128,14, 128,95, 137,78, 143,82, 149,48, 156,84, 159,26, 166,02, 173,60.
СТАДИЯ 14. (13CPT ---> 14CPT)
Сольват 13CTP-толуол (70,3 г, 0,153 моль) растворяют в 1400 мл толуола и 140 мл трифторуксусной кислоты и нагревают при 110o в
течение 2 часов. Раствор охлаждают и концентрируют в вакууме приблизительно до 350 мл. Добавляют этилацетат (1 л) и смесь охлаждают до -20o. После фильтрования получают 14 CPT в виде
светло-коричневого кристаллического твердого продукта (37,92 г, 93,4%).
1H ЯМР (300,13 МГц, CDCl3) δ 0,98 (т, J = 7,5 Гц, 3Н), 1,80 (кв, J = 6,0 Гц, 2Н), 2, 96 (м, 2Н), 4,36 (т, J = 6 Гц, 2Н), 5,24 (д, J = 15 Гц, 1Н), 5,66 (д, J = 15 Гц, 1Н), 7,27 (с, 1Н).
13C ЯМР (300,13 МГц, DMSO-d6) δ 0,80 (т, J = 7,3 Гц, 3Н), 1,81 (м, 2Н), 2,89 (т, J = 6,3 Гц, 2Н), 4,13 (т, J = 6,3 Гц, 2Н), 5,34 (д, J = 17,1 Гц, 1Н), 5,41 (д, J = 17,1 Гц, 1Н), 6,86 (с, 1Н).
13C ЯМР (75,47 МГц, DMSO-d6) δ 7,52, 30,31, 33,71, 42,56, 65,20, 71,92, 98,49, 123,81, 140,19, 149,05, 156,97, 172,03, 197,93.
Номинальный масс-спектр: вычисленный m/z 263, найденный m/z 264 (M + 1).
ДОПОЛНИТЕЛЬНЫЕ ОПИСАННЫЕ РЕАКЦИИ
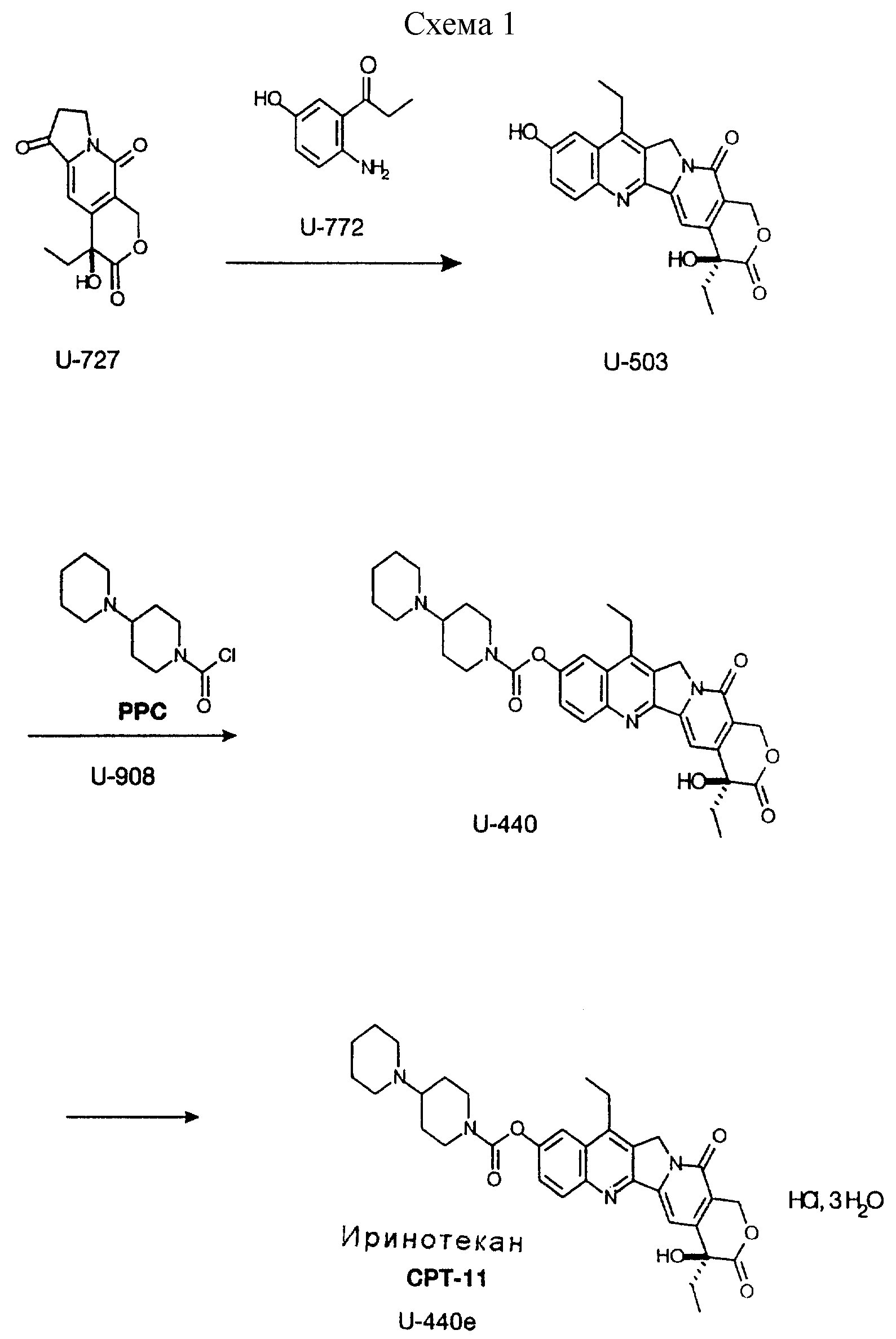
В данное изобретение включаются также следующие реакции, способы и формулы, содержащиеся в Схеме 1 (см. в конце описания)
Следующие ссылки могут быть полезными для понимания приведенных выше дополнительных описанных реакций. Получение U-503 из природного камптотецина описывается в патенте США 4473692 (25 сентября, 1984),
T. Miyasaka, S. Sawada, K. Nokata, M. Mutai. Связанное с ним получение U-440 из U-503 описывается в патенте США 4604463 (5 августа, 1986), T. Miyasaka, S. Sawada, K. Nokata, E. Sugino, M. Mutai.
Превращение U-440 в CPT-11 описывается в: S. Sawada, S. Okajima, R. Aiyama, K. Nokata, T. Furuta, T. Yokohura, E. Siguno, K. Yamaguchi, T. Miyasaka, Chem. Pharm. Bull., 1991, volume 39, pp.
1446-1454.
Реакции, приведенные в указанной выше Схеме Дополнительных Раскрытых Реакций, описываются ниже.
Получение U-503 и U-440
U-727 и U-772 подвергают
реакции при температуре от 95o до 100o в смеси толуола и уксусной кислоты в течение около 18-24 часов. Толуол и уксусную кислоту удаляют отгонкой, получая U-503, который
превращают без очистки в U-440.
Неочищенный U-503 растворяют в пиридине и подвергают реакции при температуре от 20o до 25o с 4-пиперидинопиперидинкарбамоилхлоридом, растворенным в метиленхлориде. Метиленхлорид и пиридин удаляют отгонкой и сырой U-440 снова растворяют в метиленхлориде и обрабатывают насыщенным водным раствором бикарбоната натрия. U-440 затем хроматографируют на силикагеле, элюируя смесью метиленхлорида и метанола, и выделяют в виде кристаллического твердого продукта кристаллизацией из смеси метиленхлорида и этанола.
U-503. U-727 (1,05 г, 4,0 ммоль), U-772 (0,62 г, 3,8 ммоль) и моногидрат п-толуолсульфокислоты (0,02 г) смешивают с толуолом (10 мл) и уксусной кислотой (10 мл) и нагревают в течение 18-24 часов при температуре от 95o до 100o. В течение реакции постепенно осаждается U-503. Когда реакция заканчивается, толуол и уксусную кислоту удаляют отгонкой при пониженном давлении, получая U-503 в виде твердой массы.
U-440. К неочищенному U-503 добавляют пиридин (15 мл) и смесь перемешивают в течение 15 минут при температуре от 20o до 25o для растворения U-503. Добавляют раствор 4-пиперидинопиперидинкарбамоилхлорида (1,32 г, 5,7 ммоль) в метиленхлориде (5 мл). Смесь перемешивают при 20-25o в течение 2 часов для завершения реакции. Смесь перегоняют досуха при пониженном давлении. Добавляют толуол (20 мл) и смесь перегоняют почти досуха при пониженном давлении.
Неочищенный U-440 растворяют в метиленхлориде (25 мл), добавляют насыщенный водный раствор бикарбоната натрия (5 мл) и смесь перемешивают при комнатной температуре в течение 5 мин. Фазам дают отстояться и фазу метиленхлорида удаляют. Водную фазу экстрагируют метиленхлоридом (10 мл). Фазы метиленхлорида объединяют и перегоняют, получая сырой твердый U-440.
Сырой твердый U-440 растворяют в смеси метиленхлорид/метанол (95:5, об. /об. , 10 мл) и хроматографируют на колонке, наполненной 30 г диоксида кремния 230-400 меш, элюируя смесью метиленхлорид/метанол (95:5, об./об.). Фракции, содержащие продукт, объединяют и перегоняют до объема около 10 мл при атмосферном давлении. В конце перегонки может иметь место некоторая кристаллизация продукта. Добавляют этанол (15 мл) и суспензии дают стоять при -20oC в течение 24 часов. Продукт фильтруют, промывают этанолом (10 мл) и сушат, получая 1,34 г (выход 62%, считая на 16CPT) U-440.
СПОСОБЫ, РЕАКЦИИ И СОЕДИНЕНИЯ СХЕМ М-М И M-G
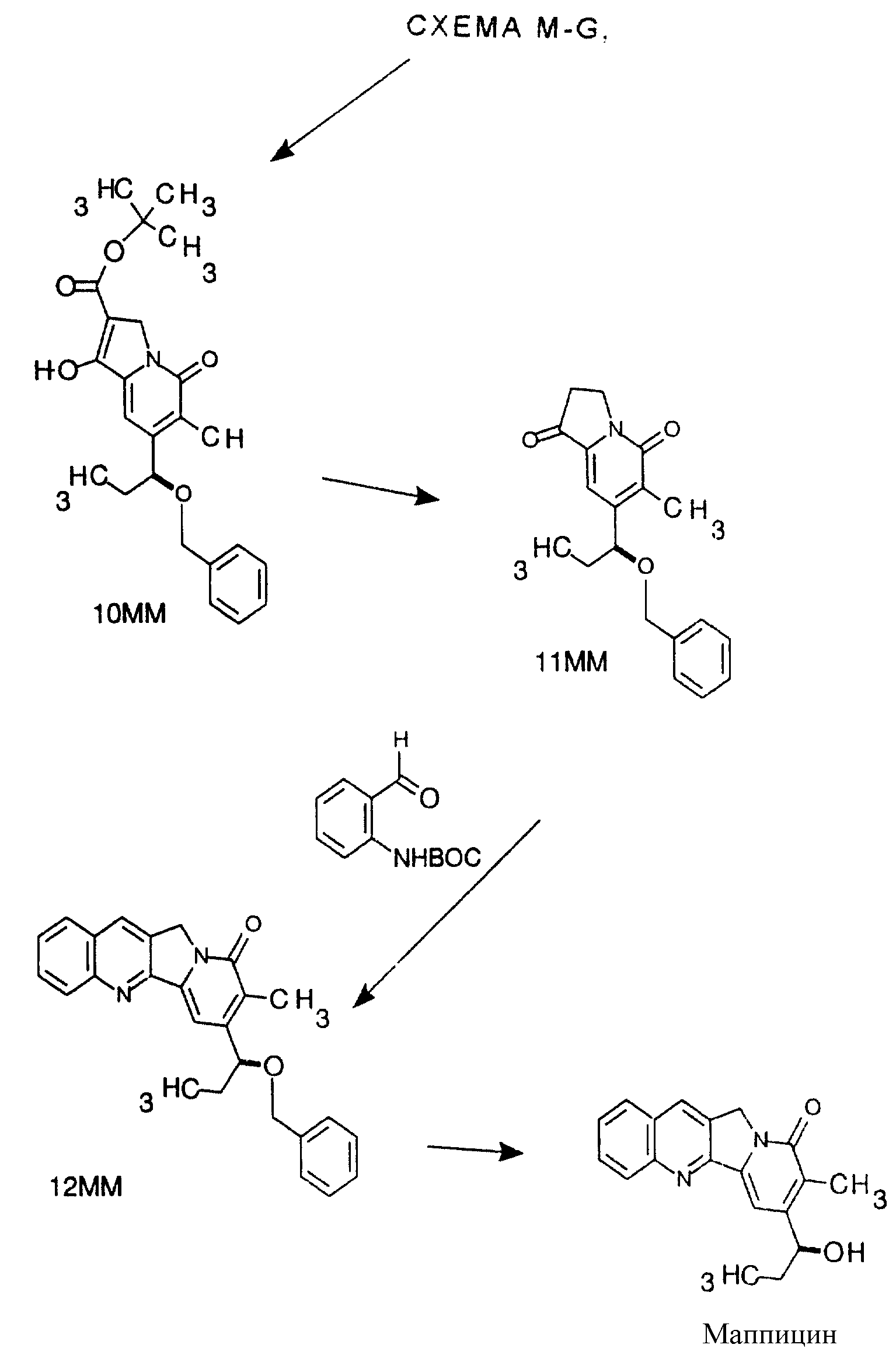
Хиральное восстановление для синтеза маппицина и родственных соединений показано в СХЕМАХ M-G и М-М. Предшественники этих соединений описываются
ранее указанными реакциями СХЕМЫ G.
Существует ряд реагентов, доступных для восстановления кетонов для получения хиральных вторичных спиртов. Арилалкилкетоны, подобные по структуре промежуточным продуктам, показанным в СХЕМАХ для маппицина, являются особенно подходящими субстратами для хирального восстановления. Среди реагентов, которые эффективны для этого типа восстановления, имеется комплекс1 Noyori бинафтол-литийалюминийгидрид, комплексы боранов и хиральных аминоспиртов, разработанные Itsuno2, борановые восстановители, катализируемые хиральными оксазаборолидинами3 и комплексы литийалюминийгидрида и спирта darvon4.
1. R. Noyori, I. Tomino and Y. Tanimoto, J. Am. Chem. Soc, 1979, 101, 3129: R. Noyori. U.S. patent 4284581.
2. S. Itsuno, К. Ito, A. Hirao and S. Nakahama, J. Chem. Soc. Chem. Comm. , 1983, 469. S. Itsuno, M. Nakano, K. Miyazaki, H. Masuda, K. Ito, A. Hirao and S. Nakahama. J. Chem. Soc. Perkin I, 1985, 2039.
3. E.J. Corey, R.K. Bakshi, S. Shibate. J. Am. Chem. Soc., 1987, 5551.
4. N. Cohen, R.J. Lopresti, C. Neukom, G. Saucey. J. Org. Chem., 1980, 45, 582.
Указанные выше продукты реакции и промежуточные продукты можно затем подвергать реакции по очевидным вариантам конденсации типа Фридландера для получения целевых продуктов, таких как продукты, показанные в СХЕМАХ ниже.
Ниже приводится один конкретный пример, показывающий подробно условия реакций, показанных в СХЕМАХ M-G и M-M.
5ММ. 4CPT (10,0 г, 41,0 ммоль) растворяют в 500 мл гептана. Раствор охлаждают до 0oC и добавляют 24,4 мл н-BuLi в гексане (2,10 М, 51,2 ммоль), поддерживая температуру реакции
0oC. Ярко-оранжевую суспензию перемешивают при 0oC в течение 1,75 часа. Добавляют диметилсульфат (4,8 мл, 51,2 ммоль), поддерживая температуру реакции ниже 10oC.
Реакционную смесь перемешивают при 0oC в течение 2 ч и затем обрабатывают 1,5 мл концентрированного раствора NH4OH до перемешивания в течение дополнительного 1 ч. Добавляют воду
(40 мл) и этилацетат (EtOAc, 75 мл). Через 15 мин фазы разделяют и водную фазу экстрагируют 3 раза порциями по 50 мл EtOAc. Органические экстракты объединяют, сушат над Na2SO4
фильтруют и концентрируют до получения красного масла. Очисткой флэш-хроматографией (CH2Cl2) получают 5ММ (6,97 г, 66%) в виде прозрачного, бесцветного масла: MS (электронная
ионизация, ЭИ) m/z 257, 259: MS (химическая ионизация, ХИ)) m/z (-NH3+) 258, 260;
1H ЯМР (300,14 МГц, CDCl3) δ 7,08 (с, 1Н), 4,05-4,01 (м,
2Н), 3,97 (с, 3Н), 3,80-3,75 (м, 2Н), 2,28 (с, 3Н), 1,93 (дд, J = 7,4, 14,9 Гц), 0,91 (т, J = 7,4 Гц);13C ЯМР (75,47 МГц) δ 162,9, 153,3, 144,9, 116,9, 114,4, 110,1, 64,5, 54,2, 31,
5, 12,0, 7,4.
6MM. 5ММ (12,0 г, 46 ммоль) растворяют в 25 мл водной трифторуксусной кислоты (64%, об. /об. ) и нагревают до 40oC. Через 4 ч реакционную смесь охлаждают и
тушат 50 мл H2O и 75 мл смеси EtOAc:гептан (2:1, об./об.). Фазы разделяют и водную фазу экстрагируют 3 раза порциями по 40 мл смеси EtOAc/гептан (2: 1). Органические экстракты объединяют и
нейтрализуют промыванием 200 мл 9% (мас./об.) водного NaHCO3. Фазы разделяют и водную фазу экстрагируют 3 раза порциями по 50 мл EtOAc. Органические фазы объединяют, сушат (Na2
SO4), фильтруют и концентрируют, получая 10 г желтоватого масла. Сырой продукт вводят непосредственно в следующую реакцию. Небольшую часть его очищают для его идентификации: MS (ЭИ) m/z 213,
215; MS (ХИ) m/z (-NH3+) 214, 216:
1H ЯМР (300,14 МГц, CDCl3) δ 6,88 (с, 1Н, 3,99 (с, 3Н), 2,82 (дд, J = 7,2, 14,5 Гц), 2,16 (с, 3Н), 1,19
(т, J = 7,2 Гц);13C ЯМР (75,47 МГц) δ 204,2, 162,6, 150,5, 145,5, 116,3, 113,0, 54,4, 35,9, 11,8, 7,6.
6bММ. Сырой 6MM (10 г, приблизительно 46 ммоль) растворяют в 100 мл MeOH и охлаждают до 0oC. Сразу добавляют все количество свежеполученного раствора 2,18 г NaBH4 (58 ммоль) в 20 мл 50% водного MeOH. Через 20 мин реакцию тушат 50 мл водного HCl (1 М, 50 ммоль) и затем разбавляют 100 мл CH2Cl2 и 10 мл воды. Фазы разделяют и водную фазу экстрагируют 3 раза порциями по 50 мл CH2Cl2. Органические экстракты концентрируют до получения белого твердого материала. Этот твердый материал перекристаллизуют из гексана, получая 6bММ (8,54 г, 85% от 5ММ) в виде длинных игл: т. пл. = 97,0 - 97,5oC; MS (ЭИ) m/z 215, 217: MS (ХИ) m/z (-NH3+) 216, 218;1H ЯМР (300,14 МГц, CDCl3) δ 7,05 (с, 1Н), 4,85 - 4,81 (м, 1Н), 3,95 (с, 3Н), 2,08 (с, 3Н), 1,76 - 1,63 (м, 2Н), 0,98 (т, J = 7,4 Гц);13C ЯМР (75,47 МГц) δ 161,8, 155,6, 145,4, 115,0, 113,1, 71,0, 54,1, 30,4, 10,5, 9,8.
7MM 6bММ (4,00 г, 18, 5 ммоль) и гидрида натрия (1,55 г, 64,6 ммоль) перемешивают с 40 мл ТГФ в течение 30 мин. Добавляют бензилбромид (2,3 мл, 18,9 ммоль) и смесь перемешивают в течение 8 часов при комнатной температуре. Добавляют насыщенный раствор хлорида аммония (10 мл), 10 мл воды и 20 мл CH3Cl2. Фазы разделяют и нейтральное значение pH водной фазы устанавливают при помощи 1 М HCl до экстракции 3 раза порциями по 20 мл CH2Cl2. Органические фазы объединяют, сушат (NaSO4), фильтруют и концентрируют до образования желтого масла. После флэш-хроматографии на силикагеле получают 7ММ (5,09 г, 90%) в виде прозрачного масла: MS (ЭИ) m/z 305, 307; MS (ХИ) m/z (-NH3+) 306, 308;1H ЯМР (300,14 МГц, CDCl3) δ 7,38 - 7,32 (м, 5Н), 7,07 (с, 1Н), 4,53 - 4,46 (м, 2Н), 4,26 (д, J = 11,7 Гц, 1Н), 4,00 (с, 3Н), 2,09 (с, 3Н), 1,83 - 1,62 (м, 1Н), 0,98 (т, J = 7,3 Гц, 3Н);13C ЯМР (75, 47 МГц) δ 162,0, 154,0, 145,6, 137,9, 128,4, 127,8, 127,7, 116,3, 113,8, 78,0, 71,0, 54,2, 29,5, 10,5, 10,1.
8MM 7ММ (4,00 г, 13,1 ммоль), ацетат калия (1,92 г, 19,6 ммоль), ацетат палладия (0,147 г, 0,65 ммоль) и DPPP (0,268 г, 0,65 ммоль) растворяют в 80 мл ДМФ и 40 мл н-пропанола. Колбу продувают CO и затем нагревают до 85o в атмосфере CO. Через 25 мин смесь охлаждают и продувают азотом, раствор фильтруют через целит и фильтрат концентрируют и затем распределяют между 80 мл воды и 160 мл МТБЭ. Водную фазу далее экстрагируют 3 раза порциями по 50 мл МТБЭ. Органические экстракты объединяют, промывают 4 раза порциями по 25 мл воды, сушат (Na2SO4), фильтруют и концентрируют. После очистки флэш-хроматографией с использованием в качестве элюента CH2Cl2 получают 8MM (4,14 г, 89%) в виде прозрачного, бесцветного масла: MS (ЭИ) m/z 358; MS (ХИ) m/z (-NH3+) 358, 360;1H ЯМР (300,14 МГц, CDCl3) δ 7,88 (с, 1Н), 7,38 - 7,28 (м, 5Н), 4,57 - 4,53 (м, 1Н), 4,48 (д, J = 11,6 Гц, 1Н), 4,36 - 4,32 (м 2Н), 4,25 (д, J = 11,6 Гц, 1Н), 4,09 (с, 3Н), 2,18 (с, 3Н), 1, 90 - 1,80 (м, 3H), 1,78 - 1,64 (м, 2Н), 1,06 (т, J = 7,4 Гц), 0,97 (т, J = 7,4 Гц):13C ЯМР (75,47 МГц) δ 165,5, 162,3, 151,5, 142,8, 138,0, 128,3, 127,8, 127,7, 122,9, 116,5, 78,1, 70,9, 66,8, 53,8, 29,5, 22,0, 11,3, 10,4, 10,2.
9MM Раствор иодида натрия (1,89 г, 12,6 ммоль) и 8MM (3,00 г, 8,4 ммоль) и 30 мл CH3CN охлаждают до 0oC и
добавляют триметилсилилхлорид (1,6 мл, 12,6 ммоль). Через 15 мин реакционной смеси дают нагреться до комнатной температуры. Через 24 ч реакцию тушат последовательным добавлением 4,2 мл 6 М HCl, 5,3 мл
насыщенного раствора хлорида натрия, 10,6 мл H2O, 0,4 мл 38% Na2S2O5 (водный) и 20 мл EtOAc. После перемешивания при комнатной температуре в течение 30 мин
фазы разделяют и водную фазу экстрагируют 3 раза порциями по 10 мл EtOAc.
Органические растворы объединяют и промывают 7 мл насыщенного NaHCO3 и 0,25 мл 38% водного раствора бисульфита
натрия. После перемешивания в течение 15 мин фазы разделяют и органический раствор промывают 2 раза порциями по 10 мл насыщенного водного раствора хлорида натрия. Раствор сушат над Na2
SO4, затем фильтруют и концентрируют, получая 2,80 г (97%) 9ММ в виде воскообразного желто-белого твердого вещества: MS (ЭИ) m/z 343, 344; MS (ХИ) m/z (-NH3+) 344, 345;1H ЯМР (300,14 МГц CDCl3) δ 9,82 (шир. с), 7,39 - 7,29 (м, 6Н), 4,51 - 4,46 (м, 1Н), 4,35 - 4,26 (м, 2Н), 2,14 (с, 3Н), 1,87 - 1,75 (м, 3Н), 1,71 - 1,57 (м, 1Н), 1,05 - 0,
95 (м, 6Н);
13C ЯМР (75,47 МГц) δ 162,42, 161,28, 150,41, 137,63, 133,04, 130,54, 128,40, 127,87, 108,01, 77,76, 71,13, 67,95, 28,80, 21,84, 12,04, 10,29, 10,06.
10MM. Смесь 9MM (3,22 г, 9,4 ммоль), карбоната цезия (6,12 г, 18,8 ммоль), трет-бутилакрилата (13,5 мл, 92,3 ммоль) и 50 мл ДМСО нагревают до 65oC в атмосфере азота. Через 3 часа реакционную смесь охлаждают до 0oC и затем медленно тушат 60 мл 0,5 М HCl, поддерживая на всем протяжении внутреннюю температуру 15oC или ниже. Смесь разбавляют 30 мл смеси EtOAc/толуол (1:4, об./об.) и фазы разделяют. Водную фазу экстрагируют 2 раза порциями по 30 мл указанного выше растворителя. Органические экстракты объединяют и промывают 3 раза порциями по 30 мл воды, сушат над Na2SO4, фильтруют и концентрируют до получения 4,57 г желтого масла. После очистки колоночной хроматографией получают 3,28 г 10ММ в виде пенообразного беловатого твердого вещества (85%): MS (ЭИ) m/z 411, 412; MS (ХИ) m/z (-NH3+) 412, 413;1H ЯМР (300,14 МГц CDCl3) δ 9,91 (шир. с), 7,39 - 7,29 (м, 5Н), 6,91 (с, 1Н), 4,67 (с, 2Н), 4,52 - 4,48 (м, 2Н), 4,26 (д, J = 11,8 Гц, 1Н), 2,18 (с, 3Н), 1,87 - 1,78 (м, 1Н), 1,73 - 1,53 (м, 2Н), 1,59 (с, 9Н), 0,97 (т, J = 7,4 Гц, 3Н):13C ЯМР (75,47 МГц) δ 166,61, 160,77, 160,28, 150,69, 140,25, 137,89, 128,35, 127,77, 127,69, 126,63, 103,99, 99,13, 82,95, 78,02, 70,88, 49,08, 29,01, 28,25, 27,87, 11,84, 10,07.
11MM Раствор 10ММ (0,25 г, 0,61 ммоль), трифторуксусной кислоты (0,45 мл) и толуола (18 мл) нагревают до 75oC. Через 24 ч раствор концентрируют роторным испарением до образования густого масла. Масло разбавляют 20 мл толуола и концентрируют до образования густого масла. Масло очищают флэш-хроматографией (5% MeOH в CH2Cl2), получая 0,138 г 11MM в виде пенообразного желтого твердого вещества (73%): MS (ЭИ) m/z; MS (ХИ) m/z (-NH3+);1H ЯМР (300,14 МГц CDCl3) δ 7,27 - 7,18 (м, 5Н), 7,04 (с, 1Н), 4,43 - 4,38 (м, 2Н), 4,23 - 4,13 (м, 3Н), 2,80 (т, J = 6,9 Гц), 2,09 (с, 3Н), 1,75 - 1,47 (м, 2Н), 0,86 (т, J = 7,4 Гц, 3Н);13C ЯМР (75,47 МГц) δ 196,91, 161,48, 150,48, 137,67, 136,95, 132,94, 128,36, 127, 71, 102,40, 77,77, 70,97, 41,92, 33,69, 28,90, 12,41, 9,98.
12MM Раствор 11MM (0,135 г, 0,43 ммоль), N-Boc-о-аминобензальдегида (0,14 г, 0,63 ммоль), п-толуолсульфокислоты (0,010 г, 0, 06 ммоль), ледяной уксусной кислоты (5 мл) и толуола (25 мл) нагревают до 100oC. Через 36 ч раствор концентрируют в вакууме досуха. Остаток растворяют в 25 мл толуола и затем концентрируют до получения 0,333 г красно-коричневого твердого материала. Материал очищают флэш-хроматографией (2% MeOH в CH2Cl2), получая 0,123 г 12ММ в виде пенообразного желтого твердого продукта (72%): MS (ЭИ) m/z 396, 398; MS (ХИ) m/z (-NH3+) 397, 399;1H ЯМР (300,14 МГц CDCl3) δ 8,33 (с, 1Н), 8,22 (д, J = 8,5 Гц, 1Н), 7,90 (д, J = 8,1 Гц, 1Н), 7,80 (т, J = 7 Гц, 1Н), 7,64 - 7,57 (м, 2Н), 7,38 - 7,29 (м, 5Н), 5,28 (с, 2Н), 4,64 - 4,54 (м, 2Н), 4,32 (д, J = 12 Гц, 1Н), 2,25 (с, 3Н), 1,99 - 1,67 (м, 1Н), 1,02 (т, J = 7,3 Гц);13C ЯМР (75,47 МГц) δ 161,51, 153,51, 151,30, 148,78, 142,67, 138,12, 130,67, 130,12, 129,56, 128,64, 128,33, 127,98, 127,76, 127,60, 127,29, 126,87, 99,51, 78,18, 70,81, 49,91, 29,08, 11, 99, 10,19.
СХЕМЫ
Схемы, которые можно использовать для описания этого изобретения, описываются здесь кратко и приводятся в конце описания. Подробное описание дается выше.
СХЕМА G представляет общее описание, показывающее родовые структуры, включенные в реакции. После получения соединения, обозначенного 4G, имеются два совершенно разных пути реакций, по которым можно
действовать. Один путь продолжается по СХЕМЕ G и в конечном счете приводит к получению камптотецина или родственных соединений. Другой путь, СХЕМА M-G, в конечном счете приводит к получению маппицина
или родственных соединений.
СХЕМА CPT-11 представляет собой конкретное воплощение одной разновидности СХЕМЫ G, которая показывает конкретные реакции и промежуточные продукты, приводящие к получению камптотецина. СХЕМА М-М представляет собой конкретное воплощение одной разновидности СХЕМЫ М, которая показывает конкретные реакции и промежуточные продукты, приводящие к получению маппицина.
Стадия 10 в СХЕМАХ G и CPT-11 показывает расщепление на энантиомеры. Хотя показан только один энантиомер, можно выделить также другой энантиомер с использованием подходящих исходных материалов и разработкой необходимых модификаций, которые должны быть очевидны специалисту данной области. Данные способы, кроме того, должны быть пригодны, независимо от того, что желательно получить, энантиомер или смеси энантиомеров.
Когда присутствует только один асимметрический центр, эти способы, когда необходимо с подходящими модификациями, можно применять для получения любого энантиомера. Когда присутствуют два асимметрических центра, может быть показана стереохимия только одного из двух центров. Когда в молекуле имеются два асимметрических центра, способы здесь обычно приводят к расщеплению только по одному асимметрическому центру, расщепление по второму центру обычно не происходит. Разработкой подходящих модификаций к способам данного изобретения в комбинации со способами, доступными для обычного специалиста данной области, можно достичь полного разделения всех четырех стереоизомеров для молекул, имеющих два асимметрических центра.
СХЕМЫ M-G и G-G показывают один энантиомер жирной линией, показывающей ориентацию, однако, другой энантиомер можно с таким же успехом получить и выделить с использованием способов, известным специалисту данной области. Эти способы должны быть пригодны вне зависимости от того, выбран ли энантиомер или смеси энантиомеров. Другие энантиомеры СХЕМ M-G и G-G приводятся в некоторых пунктах формулы изобретения, где ориентация показана жирной сплошной линией или пунктирной линией.
Атомы водорода и соединяющие их связи в следующих СХЕМАХ или в любой применяемой здесь формуле обычно не изображены. Иногда атомы углерода обозначаются только связями, а не буквой "с".
Ниже приводятся различные схемы.
Реферат
Описываются новые промежуточные продукты камптотецина общей формулы (I), где Ra представляет собой атом хлора, низшую алкоксигруппу, Rb представляет собой атом водорода, С1-8алкил, гидроксиС1-8алкил, группу -С(O)Н или группу (a), Rc представляет собой группу, выбранную из ряда формул (II-IХ) или Rb и Rc вместе образуют остаток формулы -СН2-O-С(O)-С(ОН)(R6)- или -СН2-О-СН(ОН)-С(ОН)(R6)-, Rd представляет собой атом хлора или группу C(O)OR4, R3 представляет собой атом водорода или бензил, R4 представляет собой С1-8алкил, R5 представляет собой атом водорода, R6 представляет собой С1-8алкил, R7 представляет собой атом водорода. Соединения являются эффективными противораковыми лекарственными средствами. Описывается также способ получения производных камптотецина (СРТ-11) и родственных соединений.
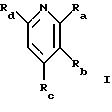
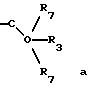

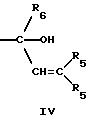
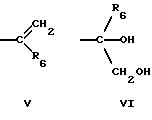

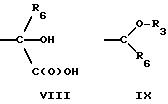
6 с. и 50 з.п. ф-лы.
Формула
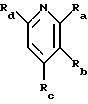
где Ra представляет собой атом хлора, низшую алкоксигруппу;
Rb представляет собой атом водорода, С1-8алкил, гидроксиС1-8алкил, группу -С(O)Н или группу
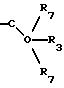
Rc представляет собой группу, выбранную из следующего ряда:

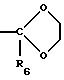
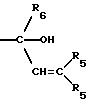


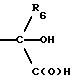

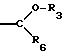
или Rb и Rc вместе образуют остаток формулы
-CH2-O-C(O)-C(OH)(R6)- или -CH2-O-CH(ОН)-С(ОН)(R6)-;
Rd представляет собой атом хлора или группу С(O)ОR4;
R3 представляет собой атом водорода или бензил;
R4 представляет собой С1-8алкил;
R5 представляет собой атом водорода;
R6 представляет собой С1-8алкил;
R7 представляет собой атом водорода.
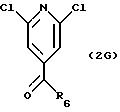
где R6 представляет собой C1-8алкил,
например, представленное формулой, показанной ниже
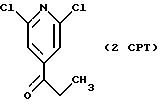
3. Соединение по п.1, представленное формулой, показанной ниже

где R6 представляет собой С1-8алкил,
например, представленное формулой, показанной ниже

4. Соединение по п.1, представленное формулой, показанной ниже
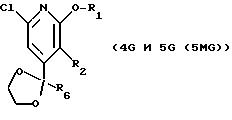
где R1 представляет собой С1-8алкил;
R2 представляет собой атом водорода, C1-8алкил, гидроксиС1-8алкил или группу С(O)R3;
R6 представляет собой C1-8алкил;
R3 представляет собой атом водорода,
например, представленные формулой, показанными ниже
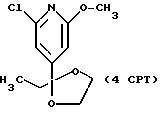
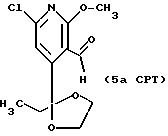
или бисульфатный аддукт соединения формулы 5а СРТ;
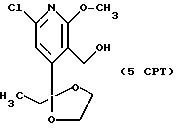
или
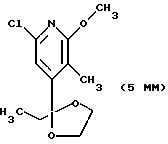
5. Соединение по п.1, представленное формулой, показанной ниже
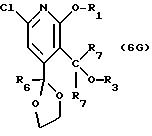
где R1 представляет собой С1-8алкил;
R3 представляет собой бензил;
R6 представляет собой С1-8алкил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
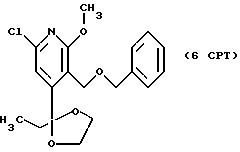
6. Соединение по п.1, представленное формулой, показанной ниже,
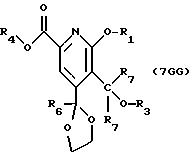
где R1 представляет собой C1-8алкил;
R3 представляет собой бензил;
R4 представляет собой С1-8алкил;
R6 представляет собой C1-8алкил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
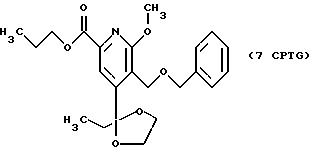
7. Соединение по п.1, представленное формулой, показанной ниже

где R1 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже

8. Соединение по п.1, представленное формулой, показанной ниже
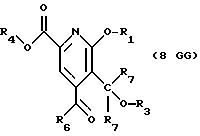
где R1 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
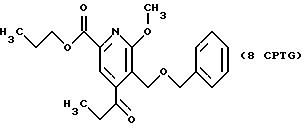
9. Соединение по п.1, представленное формулой, показанной ниже
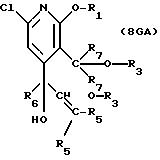
где R1 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R5 и R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
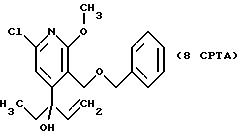
10. Соединение по п.1, представленное формулой, показанной ниже

где R1 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
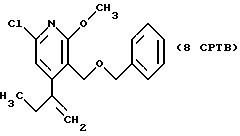
11. Соединение по п.1, представленное формулой, показанной ниже

где R1, R4 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
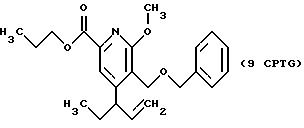
12. Соединение по п.1, представленное формулой, показанной ниже
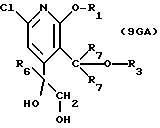
где R1 и R6 представляют С1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже

13. Соединение по п.1, представленное формулой, показанной ниже

где R1, R4 и R6 представляют C1-8алкил,
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленное формулой, показанной ниже
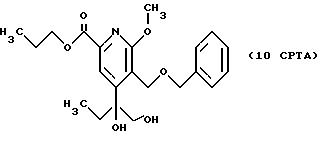
и оптические изомеры соединения формулы 10 СPТ, представленные формулами, показанными ниже

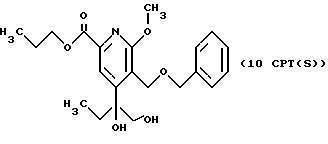
14. Соединения по п.1, представленные формулой, показанной ниже

где R1, R4 и R6 представляют C1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленные формулами, показанными ниже

и его оптические изомеры
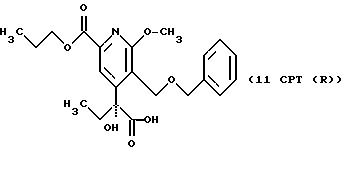
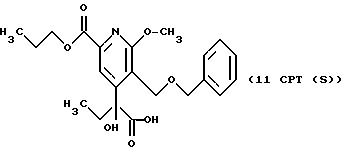
15. Соединения по п.1, представленные формулой, показанной ниже
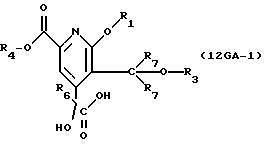
где R1, R4 и R6 представляют C1-8алкил;
R3 представляет собой бензил;
R7 представляет собой атом водорода,
например, представленные формулами, показанными ниже

и его оптические изомеры
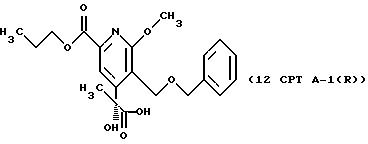
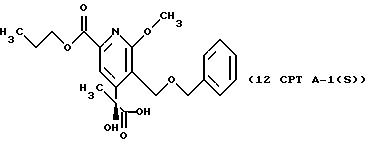
16. Соединение по п.1, представленное формулой, показанной ниже
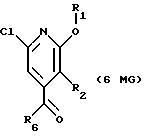
где R1, R2 и R6 представляют С1-8алкил,
например, представленное формулой, показанной ниже
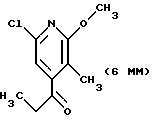
17. Соединение по п.1, представленное формулой, показанной ниже
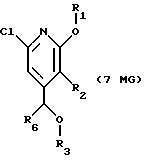
где R1, R2 и R6 представляют C1-8алкил;
R3 представляет собой бензил, водород,
например, представленное формулами, показанными ниже

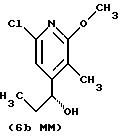
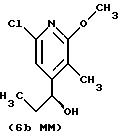
или формулами 7MM
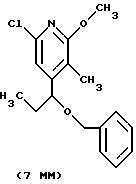

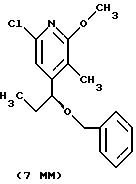
18. Соединение по п.1, представленное формулой, показанной ниже

где R1, R2, R4 и R6 представляют С1-8алкил;
R3 представляет бензил,
например, представленные формулами, показанными ниже

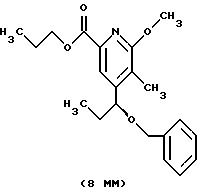

19. Соединения по п.1, представленные формулой, показанной ниже
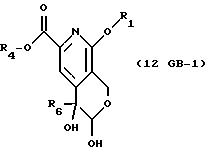
где R1, R4 и R6 представляют C1-8алкил,
например, представленные формулами, показанными ниже
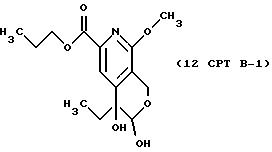
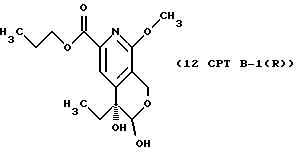
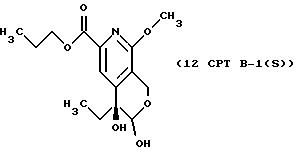
20. Соединения по п.1, представленные формулой, показанной ниже

где R1, R4 и R6 представляют С1-8алкил,
например, представленные формулами, показанными ниже

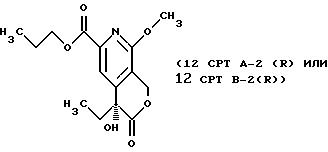

21. Промежуточные производные камптотецина общей формулы II
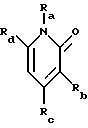
где Ra представляет собой атом водорода;
Rb представляет собой C1-8алкил;
Rc представляет собой группу

Rd представляет собой группу
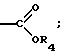
или Ra и Rd вместе образуют остаток формулы
-CH2-C(C(O)OR8)=C(OH)-
или
-СН2-СН2-С(О)-,
Rb и Rc вместе образуют остаток формулы
-СН2-O-С(O)-С(ОН)(R6)-
или
R3 представляет собой бензил,
R4, R6 и R8 представляют собой С1-8алкил.
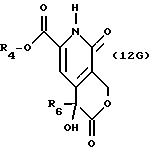
где R4 и R6 представляют С1-8алкил,
например, представленное формулой, показанной ниже
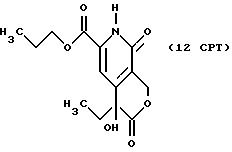
и его оптические изомеры
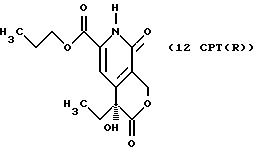
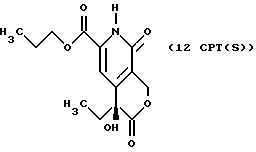
23. Соединение по п.21, представленное формулой, показанной ниже

где R6 и R8 представляют С1-8алкил,
например, представленное формулой, показанной ниже
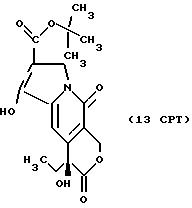
и его оптические изомеры
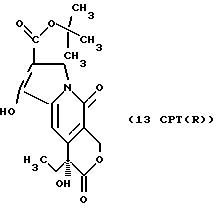
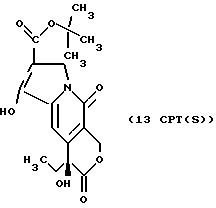
24. Соединение по п.21, представленное формулой, показанной ниже
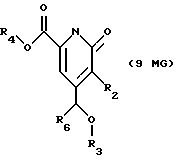
где R2, R4 и R6 представляют С1-8алкил;
R3 представляет бензил,
например, представленное формулой, показанной ниже
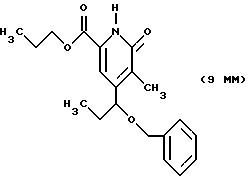
и его оптические изомеры
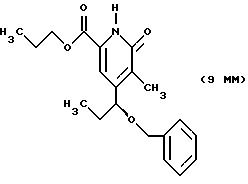
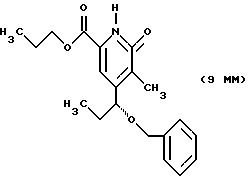
25. Соединение по п.21, представленное формулой, показанной ниже
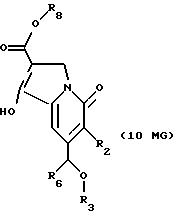
где R2, R6 и R8 представляют С1-8алкил;
R3 представляет бензил,
например, представленное формулой, показанной ниже
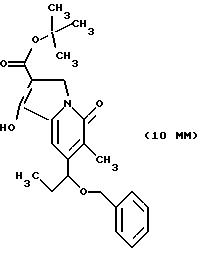
и его оптические изомеры
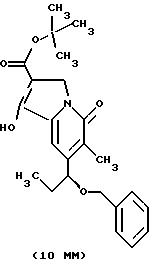
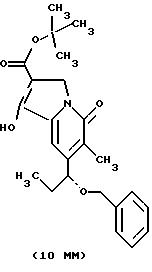
26. Соединение по п.21, представленное формулой, показанной ниже

где R2 и R6 представляют С1-8алкил;
R3 представляет бензил,
например, представленное формулой, показанной ниже
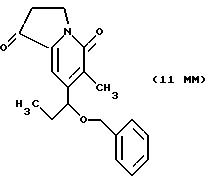
и его оптические изомеры

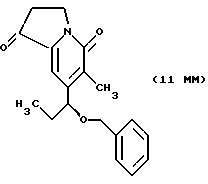
27. Промежуточные производные камптотецина общей формулы III
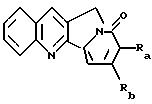
где Ra представляет собой С1-8алкил;
Rb представляет собой группу

R3 представляет собой бензил;
R6 представляет собой C1-8алкил.
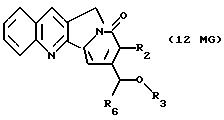
где R2 и R6 представляют C1-8алкил;
R3 представляет бензил,
например, представленное формулой, показанной ниже

и его оптические изомеры:

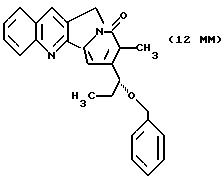
29. Способ получения соединений формулы I по п.1, заключающийся в том, что либо 1) смешивают 2,6-дихлоризоникотиновую кислоту с нуклеофилом, который либо является реактивом Гриньяра, либо алкиллитием, и эфирным растворителем с последующей обработкой продуктов реакции разбавленной кислотой, либо 2) преобразуют 2,6-дихлоризоникотиновую кислоту в ее хлорангидрид с последующим превращением в амид Weinreb и затем смешивают с нуклеофилом, который представляет собой либо реактив Гриньяра, либо алкиллитий, и эфирным растворителем, с последующей обработкой продуктов реакции разбавленной кислотой, что приводит к получению соединения формулы 2G, которое, если необходимо, смешивают с диолом в присутствии триметилхлорсилана, что приводит к получению соединения формулы 3G, которое, если необходимо, смешивают с алкоксидом натрия или калия в растворителе, что приводит к получению соединения формулы 4G, где R2 представляет H, которое, если необходимо, смешивают с растворителем и алкиллитиевым основанием или ариллитиевым основанием для образования пиридил-аниона, который затем смешивают с электрофилом, продукт этого смешивания затем смешивают с кислотой и затем выделяют конечный продукт формулы 5G, в свободном виде или смешивают с бисульфитом натрия и выделяют в виде бисульфитного аддукта, либо соединение формулы 4G, где R2 представляет H, или соединение формулы 5G смешивают с восстановителем, что приводит к получению других соединений формул 4G или 5G (5MG), и, если необходимо, соединение формулы 5G а) смешивают с основанием и алкилирующим агентом в растворителе или б) смешивают в условиях переноса фаз с использованием воды и органического растворителя, что приводит к получению соединения формулы 6G, которое, если необходимо, смешивают с оксидом углерода и спиртом в присутствии растворимой соли палладия II, фосфинового лиганда и основания в полярном апротонном растворителе, что приводит к получению соединения формулы 7GG, или в соединении формулы 6G, если необходимо, удаляют кеталь а) смешиванием этого продукта с водой в присутствии сильной кислоты или б) использованием реакции обмена для удаления кеталя, что приводит к получению соединения формулы 7GA, или, если необходимо, в соединении формулы 7GG удаляют кеталь a) смешиванием с водой в присутствии сильной кислоты или б) использованием реакции обмена для удаления кеталя, что приводит к получению соединения формулы 8GG, или, если желательно, соединение формулы 7GA растворяют в растворителе и смешивают с виниллитием или винилмагнийгалогенидом, что приводит к получению соединения формулы 8GA, или, если желательно, соединение формулы 7GA смешивают с раствором илида и растворителем для проведения реакции Виттига, что приводит к получению соединения формулы 8GB, или, если желательно, соединение формулы 8GG смешивают с раствором илида и растворителем для проведения реакции Виттига, что приводит к получению соединения формулы 9GG, или, если желательно, соединение формулы 8GA смешивают с растворителем и озоном для получения промежуточного продукта, который восстанавливают, либо непосредственно, либо через промежуточный продукт, в 9GA, или, если желательно, соединение формулы 9GG превращают в диол формулы 10G осмилированием в стандартных условиях, или, если желательно, соединение 10G получают смешиванием соединения формулы 9GA с оксидом углерода и спиртом в присутствии растворимой соли палладия II, фосфинового лиганда и основания в полярном апротонном растворителе, или, если желательно, соединение формулы 10G может быть разделено на энантиомеры обработкой рацемического диола ацетилирующим реагентом, или, если желательно, соединение формулы 10G подвергают окислению до гидроксиальдегида либо а) в условиях типа Swern, либо б) двухфазной системе, содержащей воду и апротонный растворитель, что приводит к получению соединения формулы 11G, которое, если необходимо, подвергают окислению, что приводит к получению соединения формулы 12GA-1, или в котором удаляют уходящую группу, что приводит к получению соединения формулы 12GB-1, или, если желательно, в соединении формулы 12GA-1 удаляют уходящую группу, что приводит к получению соединения формулы 12GA-2 или 12GB-2, или, если желательно, лактол формулы 12GB-1 окисляют, что приводит к получению соединения формулы 12GA-2 или 12GB-2, и, если желательно, соединение формулы 5MG декетализируют, что приводит к получению соединения формулы 6MG, которое, если необходимо, восстанавливают с получением соединения формулы 7MG, где R3 представляет H, или формулы 6bММ, рацемата или любого изомера, или, если желательно, соединение формулы 7MG, где R3 представляет H, смешивают с основанием и алкилирующим агентом в растворителе, что приводит к получению соединения формулы 7MG, где R3 не представляет H, или формулы 7ММ, рацемата или любого изомера, или, если желательно, соединение формулы 7MG смешивают с оксидом углерода и спиртом в присутствии растворимой соли палладия II, фосфинового лиганда и основания в полярном апротонном растворителе, что приводит к получению соединения формулы 8MG или формулы 8ММ, либо изомера, либо рацемата.
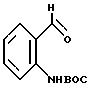
что приводит к получению соединения формулы 12MG или формулы 12ММ, рацемата или обоих изомеров.
Комментарии