Производное n-замещенного 1-гексил-4-фенил-4-пиперидинкарбоксамида или его фармацевтически приемлемая соль и способ его получения - RU2039043C1
Код документа: RU2039043C1
Чертежи


Описание
Изобретение направлено на новые соединения, обладающие как местным анестезирующим, так и анальгетическим действием, на их использовании в производстве фармацевтических препаратов и на способ их получения.
Петидин является часто используемым анальгетиком. Он обладает также слабым местным анастетическим действием. Анестетическое/анальгетическое действие петидина после спинального введения часто является недостаточным в отношении обоих указанных действий. Вместо этого обычно используются сочетания бупивакаина и фентанила или морфина. Наркотические анальгетики имеют ряд серьезных недостатков, таких как, например, развитие толерантности, наркомании, риск угнетения дыхания. Таким образом, существует необходимость в агентах, дающих местную анестезию с остаточным анальгетическим действием. Такие агенты должны использоваться после спинальных или эпидуральных инъекций в качестве местных анестеризующих средств. После этого соединения обычно дают хорошее послеоперационное облегчение или снятие болей.
Наrdy D. G. с сотрудн. описывают в I.Мed.Chem. 8, стр. 847-851 (1965) взаимосвязь между структурой и активностью некоторых аналогов петидина, которые оказывают анальгетическое действие.
Шведский патент 96980 описывает 1-метил-1-фенил-пиперидин-4-карбоновую кислоту и два ее амида. В данном документе не описано никакого конкретного фармацевтического действия, а только отмечается, что соединения могут использоваться в производстве новых лекарственных средств. Из патента FR 2156470 известны производные 1-(3,3-дифенилпропил)-пиперидина, которые благодаря своей высокой липидной растворимости могут быть активными только в качестве анальгетиков, но не в качестве местных анестетиков. В работе Асta Роl. Рharm. 1979, 36(4), стр. 439-4, (Сhem. Abstracts 93 (1980) 7970 v) описываются некоторые амиды 1-бутил-4-фенил-4-пиперидинкарбоновой кислоты. Эти амиды, похоже, обладают анальгетическим, но не местным анестетическим действием.
Обнаружено, что соединения, соответствующие представленной формуле, или их фармацевтически приемлемые соли, дают не только неожиданно хороший эффект в качестве спинальных и эпидуральных анестетиков, но также оказывают дополнительное анальгетическое действие, которое продолжается в течение длительного времени после того, как снижается анестетический эффект. Таким образом, нет необходимости в сочетании активных соединений и, следовательно, можно избежать упомянутого риска. Соединения, соответствующие изобретению, определяются следующей формулой.
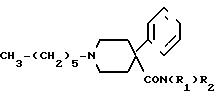
Предпочтительными соединениями в соответствии с изобретением являются такие, в которых R1 и R2 являются алкильными группами.
Особенно предпочтительным является соединение, в котором группа R1 является метилом или этилом, а R2 является этилом.
Предпочтительными солями в соответствии с изобретением являются фармацевтически приемлемые соли. Особенно предпочтительным является гидрохлорид.
Соединения формулы IV,
приведенной выше, могут быть получены в
соответствии со следующей общей схемой:
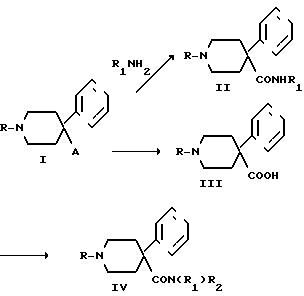

где R гексил, А СN или -СО2С2Н5, и имеют определенные выше значения.
Соединения формулы I получаются из соответствующего вторичного амина (R= Н). Соединения формулы II (которые также являются соединениями изобретения) получаются непосредственно из соединения I, в котором А является группой -СО2С2Н5 (см. пример 1) взаимодействием с алкиламином или получаются таким же образом, как соединения формулы IV. Они получаются с помощью сначала гидролиза соединений формулы I с получением карбоновых кислот формулы III, которые затем подвергаются взаимодействию с оксалилхлоридом и соответствующим амином, давая соединение формулы IV.
Детальное описание процесса приготовления.
Примеры, имеющие буквенные обозначения, описывают промежуточные соединения для получения соединений формулы IV.
Соединение I.
П р и м е р А. Этил 1-гексил-4-феенил-4-пиперидин карбоксилат гидрохлорид.
Норпетидин (23,5 г, 01,0 мол), гексилйодид (23,5 г, 0,11 мол), безводный Nа2СО3 (11,7 г, 0,11 мол) и ацетонитрил (250 мл) нагревались с обратным холодильником и перемещались в течение 1,5 ч. Смесь фильтровалась и растворитель удалялся. Остаток растворялся в СН2Сl2, раствор промывался 100 мл 1 н. раствора NаОН, затем водой и наконец высушивался (К2СО3). Затем добавлялся к раствору НСl в диэтиловом эфире, после растворители удалялись, а остаток перекристаллизовывался из этилацетата. Выход составил 22,5 г гидрохлорида с т.пл. 156-158оС.
Т. пл. в соответствии с J.Мed.Chem. 8, стр.847-851 (1965) составляет 158оС.
Соединение III.
П р и м е р В. Гидрохлорид 1-гексил-4-фенил-4-пиперидинкарбоновой кислоты.
Смесь этилового эфира (22,5 г, 64 ммол), 20% соляной кислоты (225 мл) и уксусной кислоты (70 мл) нагревалась в течение 30 ч с обратным холодильником. После охлаждения смесь выливались в 200 мл ледяной воды, кислота отфильтровывалась и высушивалась на воздухе. Выход 12,9 г. Фильтрат выпаривался, а на остаток действовали ацетонитрилом, получая еще 4 г кислоты. Перекристаллизация из ацетонитрила дала 16,9 г с т.пл. 193-195о С. Кислота содержит растворитель кристаллизации.
Соединение II.
П р и м е р 1. N-Этил-1-гексил-4-фенил-4-пиперидинкарбоксамид.
Это соединение получалось из этил 1-гексил-4-фенил-4-пиперидинкаpбоксилат гидрохлорида (3,54 г, 10 ммол) и этиламина (2,25 г, 50 ммол) при нагревании в автоклаве при 180оС. Время реакции 2 дня. Реакционная смесь разделялась встряхиванием между 10 мл 1 н. NаОН и диэтиловым эфиром и эфирные экстракты сушились (МgSО4). Растворитель испарялся, а остаток хроматографивался на окиси алюминия с использованием в качестве элюента этилацетата. Неочищенный продукт (1,0 г) перекристаллизовывался из диизопропилового эфира, давая 0,81 г продукта с т.пл. 92-94оС. Гидрохлорид имеет т.пл. 221-223оС (из 2%-ного водного ацетона).
Соединение IV.
Общий метод получения. Оксалилхлорид (4 мл) добавлялся по каплям при перемешивании к раствору пиперидинкарбоновой кислоты соединения III (5-6 ммол) в СН2Сl2 (20 мл). Реакционная смесь перемешивалась в течение 2 ч при 50оС. Растворитель удалялся, добавлялось несколько мл толуола, затем растворитель снова удалялся. Остаток растворялся в СН2Сl2 (10 мл), раствор по каплям при перемешивании добавлялся к раствору соответствующего аминам (35-42 ммол) в СН2Сl2 (20 мл), охладился в ледяной воде. Затем реакционная смесь в течение нескольких часов перемешивалась при комнатной температуре. Затем она встряхивалась в 1 н. NаОН (20 мл), один раз с водой, сушилась (К2СО3) и растворитель выпаривался.
Перед превращением в гидрохлорид неочищенное основание в нескольких случаях подвергалось дополнительной очистке, например, с помощью хроматографии.
В табл.1 даются некоторые соединения, соответствующие изобретению.
Для приготовления фармацевтических препаратов новое соединение растворяется в жидком разбавителе, который является подходящим для инъекции. Используемые препараты представляют собой водные растворы, которые содержат 2, 5-40,0 мг/мл активного соединения в расчете на гидрохлоридную соль.
Биологические исследования.
Спинальная анестезия.
Соединения, соответствующие изобретению, испытывались на спинальную анестезию на мышах. В каждой группе испытанию подвергались шесть животных. В качестве контрольных соединений испытывались петидин и исходный материал для соединений 2 (-8), а именно, этил 1-гексил-4-фенил-4-пиперидин карбоксилат гидрохлорид (пример АМ).
Результаты представлены в табл.2.
Как можно видеть из табл.2, соединения, соответствующие изобретению, дают лучший локальный анестетический эффект, чем известный анальгетик петидин. В связи с тем, что анестетическое действие сочетается с хорошим анальгетическим эффектом, соединения изобретения являются более ценными, чем петидин. Они могут также заменять сочетание одного анальгетического и одного анестетического агента с хорошим результатом.
Как видно из представленного, наилучшими соединениями изобретения являются соединения 6 или 7.
Реферат
Использование: в качестве местного анальгезирующего и анальгетического препарата и в качестве промежуточного продукта в синтезе фармацевтических препаратов. Сущность изобретения: N-замещение 1-гексил-4-фенил-4-пиперидинкарбоксамида ф-лы I: CH2 CH2N(C4H9)CH2CH2C(C6H5)CONR1R2, где R1 и R2 одинаковы или различны и каждый представляет C1-C4 -алкил, или R1 и R2 вместе образуют цепь (CH2)5 или один из R1 и R2 является водородом, а другой C1-C4 -алкил. Реагент 1: этиловый эфир 1-гексил-4-фенил-4-пиперидинилкарбоновая кислота. Реагент 2: R1NH3. Условия реакции: с получением соединения общей ф-лы I, где R2 водород. 2 с. и 3 з.п. ф-лы, 2 табл.
Формула

где R1 и R2, одинаковые или различные, и каждый С1 - С4-алкил, или R1 и R2 вместе образуют цепь (CH2)5, или один из R1 и R2 водород, а другой С1 С4-алкил,
или его фармацевтически приемлемая соль.

где R1 и R2, одинаковые или различные, и каждый С1 - С4-алкил, или R1 и R2 вместе образуют цепь (CH2)5, или один из R1 и R2 водород, а другой - С1 С4-алкил,
или его фармацевтически приемлемой соли, отличающийся тем, что этиловый эфир 1-гексил-4-фенил-4-пиперидинилкарбоновой кислоты общей формулы II
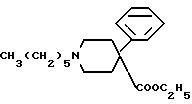
либо подвергают гидролизу до соответствующей кислоты, которую затем подвергают взаимодействию с оксалилхлоридом и затем с соответствующим амином общей формулы III
R1R2 NH,
где R1 и R2 имеют указанные значения,
с получением соединения общей формулы I, либо подвергают взаимодействию с амином общей формулы IV
R1NH2,
где R1 имеет указанные значения,
с получением соединения общей формулы I, где R2 водород.
Комментарии