Азотсодержащие гетероциклические производные, фармацевтическая композиция на их основе и способ лечения или профилактики тромботических заболеваний - RU2156763C2
Код документа: RU2156763C2
Чертежи
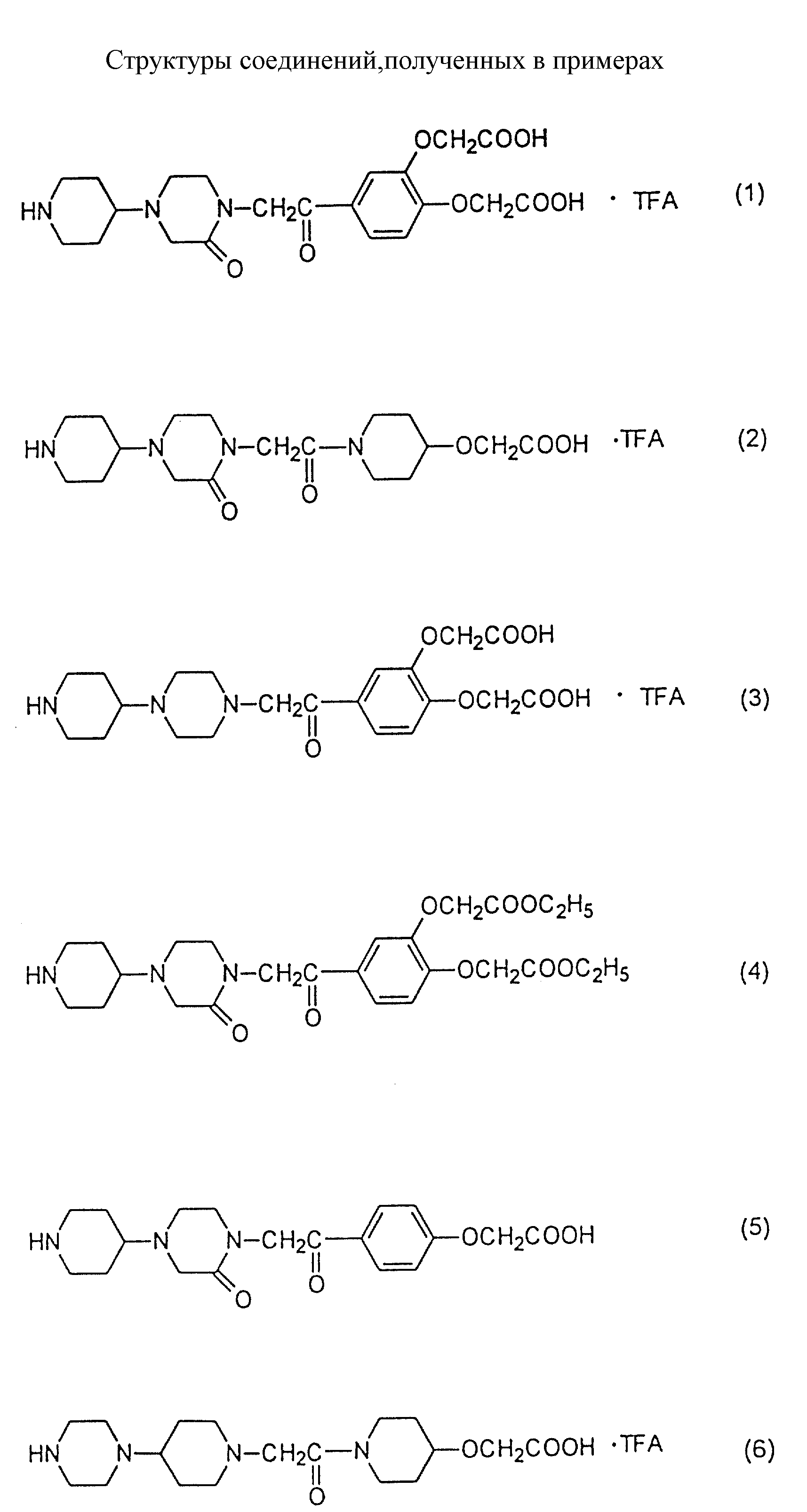
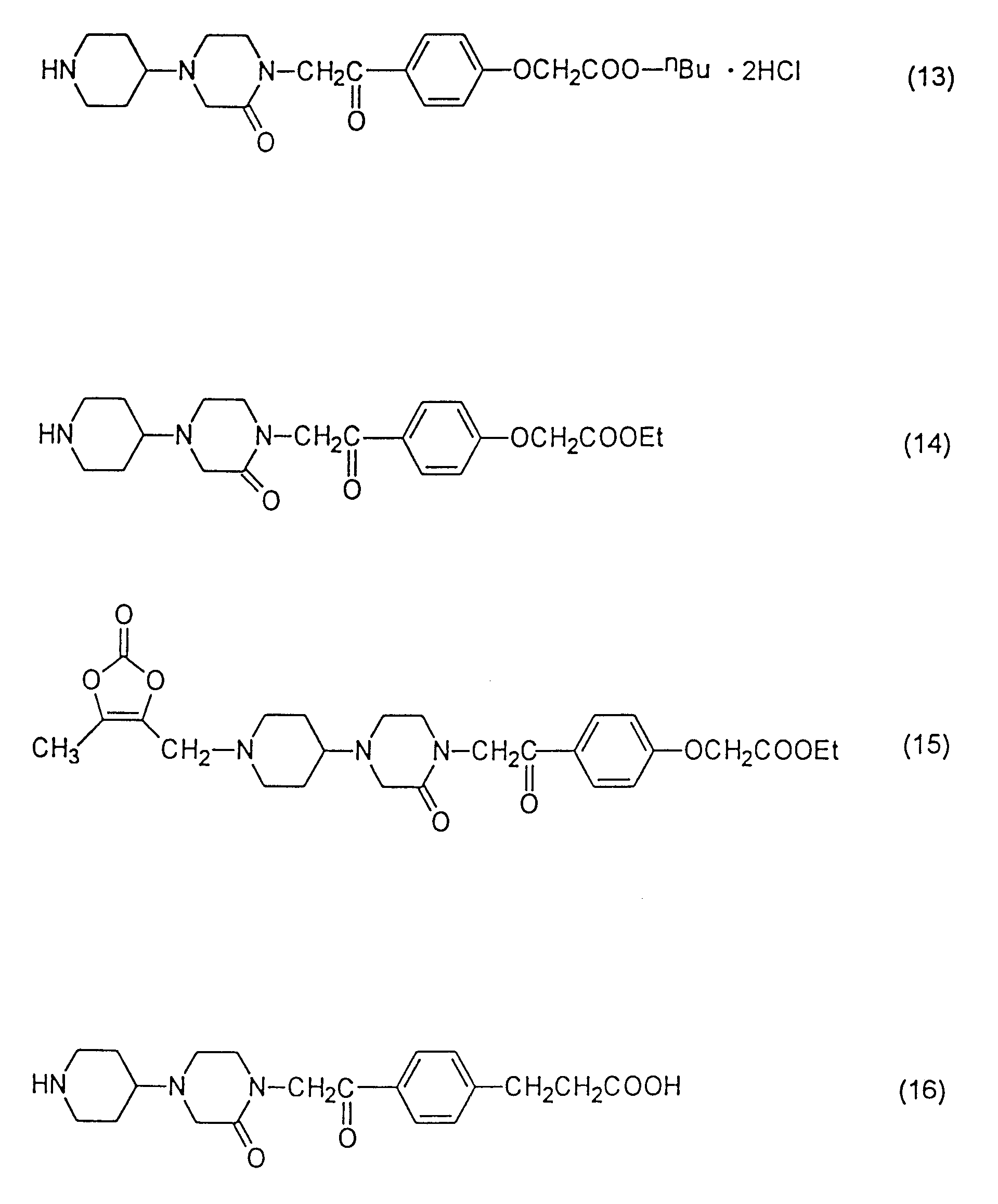
Описание
Настоящее изобретение относится к азотсодержащим гетероциклическим производным, способным ингибировать агрегацию тромбоцитов, и к фармацевтическим композициям для лечения и профилактики тромботических заболеваний, содержащим в качестве активного ингредиента, по крайней мере, одно из этих производных.
Предпосылки изобретения
Количество сердечно-сосудистых заболеваний возрастало в связи с изменением
образа питания и удлинения продолжительности жизни. Почти пятьдесят процентов этих заболеваний могут быть вызваны тромбами.
Тромбоциты в плазме, главным образом, связаны с образованием в организме тромбов. Для лечения и профилактики тромботических заболеваний в клинической практике, использовали лекарства, которые подавляют функции тромбоцитов или ингибируют агрегацию тромбоцитов, например, аспирин, который ингибирует циклооксигеназу, и тиклопидин, который активирует аденилциклазу.
В последние годы интенсивно исследовали гликопротеины на мембране тромбоцитов. В результате было выявлено, что рецептором фибриногена является гликопротеин, называемый GPIIb/IIIa. Это привело к предположению, что антагонисты GPIIb/IIIa могут выступать в качестве ингибиторов агрегации тромбоцитов с новым механизмом действия, который можно эффективно использовать для лечения и профилактики тромботических заболеваний (Trends in Pharmacоlogical Science 13, 413, 1992). Соединения-антагонисты GP11b/IIIa включают моноклональные антитела (Ann. New Jork Acad. Sci, 614, 193, 1991), трипептидные производные, содержащие аргинин-глицин-аспарагиновую кислоту (J.med. Chem. , 35, 2040, 1992), производные амидинофенила (J. med. Chem., 35, 4393, 1992, японская патентная выложенная публикация N 264068/1992, 334351/1992, EP-483667, EP-502536, EP-525629, EP-29858, EP-537980, WO-9307867 и WO-9402472), производные тирозина (J. med. Chem., 35, 4640, 1992) и производные пиперидина (EP-512831, EP-5403344 и EP-578535).
Желательно также создать такое лекарство, которое не имело бы таких побочных эффектов, как кровоизлияния, и отличалось бы весьма селективным действием в качестве терапевтического или профилактического агента для борьбы с тромботическими заболеваниями.
Сущность изобретения.
Авторы настоящего изобретения обнаружили, что некоторый тип соединений является антагонистом GPIIb/IIIa.
Таким образом, объектом настоящего изобретения являются новые соединения, ингибирующие агрегацию тромбоцитов.
Другим объектом изобретения является фармацевтическая композиция, содержащая новое соединение, обладающее вышеуказанным действием.
Следующим объектом изобретения является способ лечения или способ профилактики тромботических заболеваний, который включает введение нового соединения, обладающего указанной активностью.
Еще одним объектом изобретения является применение нового соединения, обладающего указанной активностью, для получения фармацевтической композиции, используемой для лечения или профилактики тромботических нарушений.
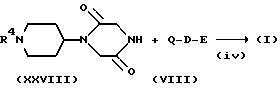
Соединение настоящего изобретения представлено формулой (I):
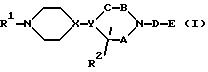
или его фармацевтически приемлемой солью или сольватом, где A, B и C, независимо, представляют CH2 или C=O;
X и Y отличны друг от друга и каждый из них представляет CH или N;
D представляет -(CH2)k - или -(CH2)m -CO (где k является целым числом от 1 до 4, а m является целым числом от 0 до 3),
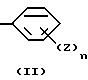
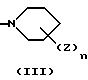
E представляет следующую группу (II) или (III):
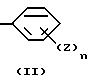
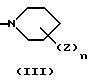
где n является целым числом от 1 до 3, а Z представляет -W-(CH2)p-COOR3 (где W представляет -O- или связь, P является целым числом от 1 до 4, а R3 представляет водород, низший алкил или сложноэфирный фрагмент, который может быть удален в физиологических условиях),
R1 представляет водород или низший алкил, в котором, по крайней мере, один атом водорода может быть замещен гидроксилом, галогеном, амино, низшим алкиламино или имино, или низшей алкилзамещенной-2-оксодиоксол-4-ил группой,
R2 представляет водород, низший алкил, в котором, по крайней мере, один атом водорода может быть замещен гидроксилом, галогеном, амино, карбоксилом, низшим алкокси, низшим алкиламино или низшим алкоксикарбонилом; фенил, в котором, по крайней мере, один атом водорода может быть замещен гидроксилом, амино, галогеном, карбоксилом, низшим алкокси, низшим алкиламино, низшим алкоксикарбонилом или галоген низшим алкилом; или фенил-низший алкил, в котором, по крайней мере, один атом водорода этой фенильной группы может быть замещен гидроксилом, галогеном, амино, карбоксилом, низшим алкокси, низшим алкиламино, низшим алкоксикарбонилом или галоид-низшим алкилом.
Подробное описание изобретения
Соединение формулы (I)
Под термином "низший алкил" в качестве группы или части группы подразумевается здесь разветвленная или неразветвленная
алкильная цепь, содержащая от 1 до 6, предпочтительно, от 1 до 4 атомов углерода. Под термином атом галогена подразумевается фтор, хлор, бром или иод. Кроме того, под термином "галоидалкил"
подразумевается алкильная группа, в которой один или более из атомов водорода замещен атомом или атомами галоида.
В формуле (I) A, B и C, каждый, представляет CH2 или C = О. В соответствии с предпочтительным вариантом настоящего изобретения, соединение, в котором A представляет C = О, а остальные представляют CH2 группы, является предпочтительным. В соответствии с другим вариантом предпочтительным является соединение, в котором два из A, B и C представляют группы C = О, а оставшийся является CH2.
В формуле (I), X и Y отличаются друг от друга, и каждый из них представляет CH или N. Предпочтительным является соединение, в котором X представляет CH, а Y представляет N.
В формуле (I) D представляет -(CH2)k - (где k представляет целое число от 1 до 4, предпочтительно, 1 или 2), или -(CH2)m -CO- (где m является целым числом от 0 до 3, предпочтительно, от 1 до 3, более предпочтительно, 1 или 2). D, предпочтительно, представляет -(CH2)m -CO-, и более предпочтительно, -CH2-CO.
E представляет группу (II) или (III). В группе (II) или (III) n, предпочтительно, равно 1 или 2. То есть, количество заместителей Z предпочтительно, равно 1 или 2. Положение заместителя Z, предпочтительно, является пара-положением или в мета-положением по отношению к D.
В группе -W-(CH2)p-COOR3, представленной Z, W, предпочтительно, представляет -O-, p, предпочтительно, равно 1 или 2, а R3, предпочтительно, является атомом водорода или C1-4-алкилом (например, метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, или трет-бутил). Предпочтительные примеры сложноэфирного остатка, представленного R3, который может быть удален в физиологических условиях, включает пивалоилоксиметил, 1-(циклогексилоксикарбонил)этил и (5-метил-2-оксо-1,3-диоксол-4-ил)метил.
В формуле (I) R1 представляет атом водорода или группу низшего алкила. По крайней мере, один атом водорода низшей алкильной группы может быть замещен. Предпочтительные примеры таких заместителей включают гидроксильную группу, группу галогена (предпочтительно, хлор, бром или фтор), аминогруппу, низшую алкиламиногруппу (предпочтительно, метиламино, этиламино, пропиламино, диметиламино или диэтиламино), иминогруппу или 5-низшая алкильная группа замещенный -2-оксодиоксон-4-ил.
R2 представляет атом водорода, низшую алкильную группу, фенильную группу или фенил-низшую алкильную группу. По крайней мере, один атом водорода в этой низшей алкильной группе может быть замещен. Предпочтительные примеры такого заместителя включают группу гидроксила, атом галогена (предпочтительно, хлор, бром или фтор), амино группу, карбоксильную группу, низшую алкоксигруппу (предпочтительно, метокси, этокси, н-пропокси или изопропокси), низшую алкиламиногруппу (предпочтительно, метиламино, этиламино, пропиламино, диметиламино или диэтиламино), или низшую алкоксикарбонильную группу (предпочтительно, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил или изопропоксикарбонил). Кроме того, по крайней мере, один атом водорода фенильной группы может быть замещен. Предпочтительно примеры такого заместителя включают гидроксильную группу, атом галогена (предпочтительно, хлор, бром, или фтор), аминогрупппу, карбоксильную группу, низшую алкоксигруппу (предпочтительно, метокси, этокси, н-пропокси или изопропокси), низшую алкиламиногруппу (предпочтительно, метиламино, этиламино, пропиламино, диэтиламино или диметиламино), низкую алкоксикарбонильную группу (предпочтительно, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил или изо-пропоксикарбонил), или галоген-низшую алкильную группу (предпочтительно, трифторметил или трифторэтил). Кроме того, по крайней мере, один атом водорода фенильного радикала фенил-низшая алкильная группа (предпочтительно, бензил, 2-фенилэтил, 3-фенилпропил) может быть замещен. Предпочтительные примеры такого заместителя включают гидроксильную группу, атом галогена (предпочтительно, хлор, бром или фтор), аминогруппу, карбоксильную группу, низшую алкоксигруппу (предпочтительно, метокси, этокси, н-пропокси или изопропокси), низшую алкиламиногруппу (предпочтительно, метиламино, этиламино, пропиламино, диметиламино или диэтиламино), или низшую алкоксикарбонильную группу (предпочтительно, метоксикарбонил, этоксикарбонил, н-пропоксикарбонил или изо-пропоксикарбонил) или галоген-низшую алкильную группу (предпочтительно, трифторметил или трифторэтил).
Предпочтительные примеры соединения,
представленного формулой (I) включают:
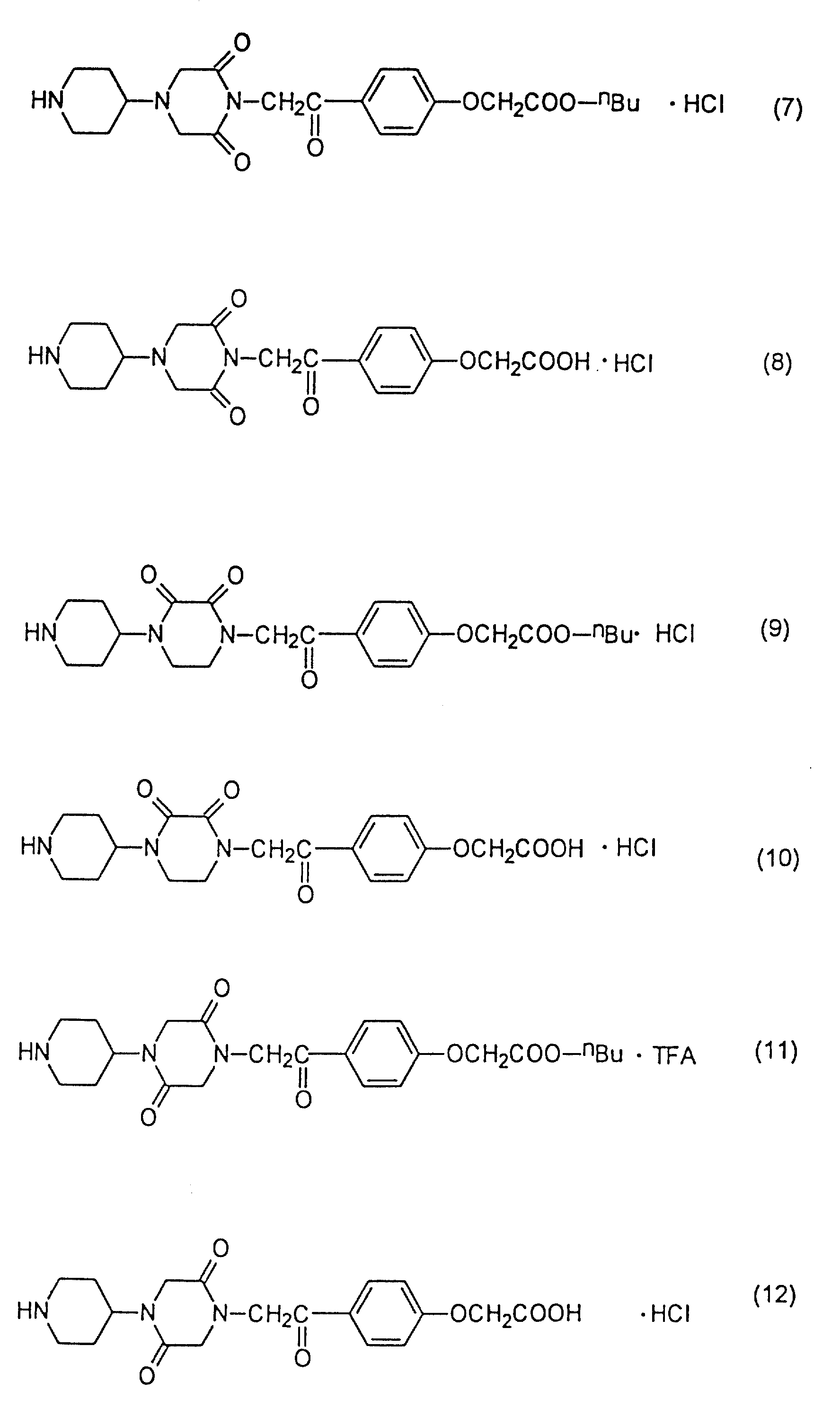
[[4-[[4-пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]-о-фенилен]- диокси] диуксусную кислоту,
[[4-[[4-пиперазин-4-ил)-пиперидин-1-ил]ацетил]-о-фенилен]-диокси]диуксусную кислоту,
4-[[4-пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]феноксиуксусную кислоту,
диэтил[[4-[[4-пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил] -о-фенилен] диокси]диацетат,
н-бутил-4-[[4-[пиперидин-4-ил)-2,6-диоксопиперазин-1-ил]ацетил]-феноксиацетат,
4-[[4-пиперидин-4-ил)-2,6-диоксопиперазин-1-ил] ацетил] феноксиуксусную кислоту,
н-бутил-4-[[4-(пиперидин-4-ил)-3,3-диоксопиперазин-1-ил]ацетил]феноксиацетат,
4-[[4-пиперидин-4-ил)-2,
3-диоксопиперазин-1-ил] ацетил] феноксиуксусную кислоту,
н-бутил-4-[[4-(пиперидин-4-ил)-2,5-диоксопиперазин-1-ил]ацетил]феноксиацетат,
4-[[4-пиперидин-4-ил)-2,5-диоксопиперазин-1-ил]
ацетил] феноксиуксусную кислоту,
н-бутил-4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]феноксиацетат-дигидрохлорид и
этил-4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]-феноксиацетат.
Соединение по настоящему изобретению может существовать в форме соли. Такая соль включает фармакологически приемлемую нетоксичную соль. Предпочтительные примеры соли включают неорганические соли, такие как соль натрия, соль калия, соль магния и соль кальция, соли присоединения кислот, такие как трифторацетат, гидрохлорид, оксалат и метансульфонат, цитрат и соли аминокислот, такие как глютамат и аспартат.
Соединение по настоящему изобретению может быть в виде сольвата. Сольват, предпочтительно, включает гидрат и этанолят.
Получение соединения, представленного формулой (I)
Соединение по настоящему изобретению может быть получено следующими
способами.
В следующих далее способах можно использовать защитные группы для аминогруппы, которые обычно применяют в пептидном синтезе. Предпочтительные примеры защитной группы включают трет-бутоксикарбонил, бензилоксикарбонил, п-метоксибензилоксикарбонил, 2,2,2-трихлорэтоксикарбонил, трифторацетил, аллилоксикарбонил и тритил. Кроме того, в нижеследующих процессах моно применять защитные группы для карбоксильной группы, которые обычно используют в пептидном синтезе. Предпочтительные примеры защитной группы включают метил, этил, трет-бутил, бензил, п-метоксибензил, р-нитробензил, аллил и бензгидрил.
Способ (А)
Соединение, представленное формулой (I), где X представляет CH, а Y представляет N, можно получить в соответствии со стадиями,
представленными на схеме A.
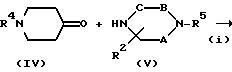
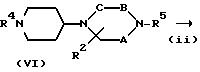
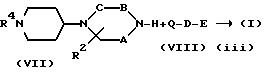
где R4 является R1 или защищенную аминогруппу,
R5 представляет атом водорода или защитную группу для аминогруппы, но отличается от R4,
Q представляет атом галогена (например, хлор, бром или иод), низшую алкилсульфонилоксигруппу (например, метансульфонилокси), трифторметансульфонилокси или арилсульфонилокси группу (например, п-толуолсульфонилокси), и
A, B, C, R2, D, Z и n имеют указанные выше значения.
На стадии (i) соединение формулы (IV) и соединение формулы (V) подвергают восстановительному алкилированию в инертном растворителе (например, ТГФ, дихлорметан, 1,2-дихлорэтан, диоксан или ДМФ) с получением соединения формулы (VI). Поэтому восстановительному алкилированию можно использовать гидриды металлов, такие, как цианборгидриднатрия, цианборгидридлития, боргидриднатрия, боргидридлития и триацетоксиборгидриднатрия. Можно также использовать каталитическое восстановление с катализатором, таким как палладий-на-угле, палладиевая чернь, гидроксид палладия, оксид платины и никель Ренея. Реакцию можно проводить при температуре от - 20oC до 100oC, предпочтительно, от 0 до 70oC, в течение от 0,5 до 48 часов, предпочтительно, от 1 до 24 часов.
Если R5 в соединении формулы (VI) представляет защитную группу для аминогруппы, эту защитную группу удаляют на стадии (ii).
На стадии (iii) соединение формулы (I) получают путем взаимодействия соединения формулы (VII) с соединением формулы (VIII) в присутствии основания в инертном растворителе (например, ТГФ, толуол, ксилол, 1,2-дихлорэтан, ДМФ или диоксан), и, при необходимости, удаления защитной группы. Примеры оснований включают металлический натрий, гидрид натрия, гидрид кальция, гидрид лития, амид натрия, карбонат калия, гидроксид натрия, гидроксид калия, н-бутиллитий и литийдиизопропиламид. При необходимости, вместе с основанием можно использовать катализатор межфазного переноса (например, тетраалкиламмонийгалогенид). Реакцию можно проводить при температуре от -80oC до 150oC, предпочтительно, от -50oC до 120oC в течение от 0,5 до 72 часов, предпочтительно, от 0,5 до 36 часов.
Соединение, представленное формулой (V), можно синтезировать в соответствии со способом, описанным в Bull. Chem. Soc. Jap., Vol. 46, p. 3612 (1973), и в ЕР 529858.
Способ (B)
Соединение, представленное формулой (I), где X представляет N и Y представляет CH, можно получить в соответствии со стадиями, представленными на
схеме B.
Схема В
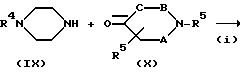
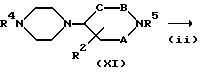
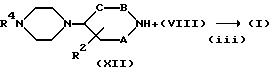
где R4 и R5 имеют указанные ранее значения, а A, B, C и R2 также определены выше. Взаимодействие соединения формулы (IX) с соединением формулы (X) на стадии (i) можно проводить в тех же условиях, что и на стадии (i) схемы A. Кроме того, реакции стадий (ii) и (iii) можно осуществлять тем же способом, что и реакции стадий (iii) и (iii) схемы A.
Способ (C)
Соединение, представленное формулой (I),
где Z представляет группу (III), можно получить в соответствии со стадиями, представленными на схеме C.
Схема C
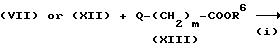
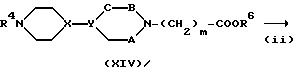
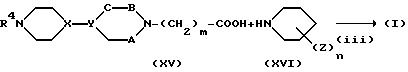
где R6 представляет R3 или защитную группу для карбоксильной группы, A, B, C, m, Z и n имеют значения, указанные для формулы (I), и R4 имеет значения, указанные для схемы A.
На стадии (i) взаимодействия соединения формулы (VII) или соединения формулы (VII) с соединением формулы (VIII) можно осуществлять в присутствии основания тем же способом, что и реакцию стадии (iii) схемы A.
Если R6 в полученном соединении формулы (XIV) представляет защитную группу для карбоксильной группы, эту защитную группу удаляют на стадии (ii).
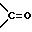
На стадии (iii) соединение формулы (XV) подвергают взаимодействию с соединением формулы (XVI) в присутствии агента, активирующего карбоновую кислоту для получения амидной связи. Применение агентов, активирующих карбоновую кислоту, включают тионилхлорид, оксихлорид фосфора, дициклогексилкарбодиимид, 1-гидроксибензотриазол, бензотриазол-1-илокси-трис(диметиламино)фосфоний и гексафторфосфат.
Способ (D)
Соединение, представленное формулой (I), где X представляет CH, Y представляет N, A и B, каждый,
представляют C = О, а C представляет CH2, можно получить в соответствии со следующими стадиями схемы D:
Схема D
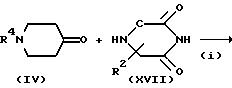
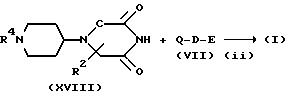
где R4 имеет указанные для схемы A значения.
На стадии (i) соединение формулы (IV) и соединение формулы (XVII) подвергают восстановительному алкилированию в присутствии кислоты (или без нее) в инертном растворителе (например, в ТГФ, дихлорметане, 1,2-дихлорэтане, диоксане или ДМФ) до получения соединения формулы (XVIII). В этом способе можно использовать реагенты восстановительного алкилирования, которые используют на стадии (i) схемы A. Реакцию можно вести при температуре от -20oC до 100oC, предпочтительно, от 0 до 70oC в течение от 0,5 до 48 часов, предпочтительно, от 1 до 24 часов.
На стадии (ii) соединение формулы (I) получают при взаимодействии соединения формулы (XVIII) с соединением формулы (VIII) в присутствии основания в инертном растворителе (например, ДМФ, ацетон, ацетонитрил, дихлорметан, ТГФ или диоксан), и, при необходимости, удаляют группу. В этом способе можно также использовать основания, которые используют на стадии (iii) схемы A. Реакцию ведут при температуре от -78oC до 100oC, предпочтительно, от -30oC до 50oC в течение от 0,5 до 48 часов, предпочтительно, от 0,5 до 12 часов.
Соединение, представленное формулой (XVII) можно синтезировать в соответствии со способом, описанным в J.Chem. Soc., p. 3874 (1953).
Способ E
Соединение, представленное
формулой (I), где X представляет CH, Y представляет N, A представляет CH, а B и C, каждый, представляют C=O, можно получить в соответствии со следующими стадиями схемы E:
Схема Е
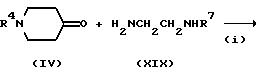
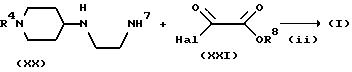
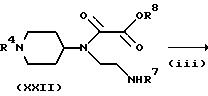
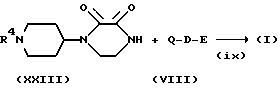
где R7 представляет защитную группу аминогруппы,
R8 представляет атом водорода или группу низшего алкила (например, метил, этил, н-пропил или н-бутил),
Hal представляет атом галогена (например, хлор, бром или иод), и R4 имеет указанные для схемы A значения.
На стадии (i) взаимодействие соединения формулы (IV) с соединением
формулы (XIX) можно вести тем же способом, что и на стадии (i) схемы D
На стадии (ii) соединение формулы (XX) подвергают взаимодействию с соединением формулы (XXI) в присутствии основания
(или без него) в инертном растворителе (например, дихлорметан, ТГФ, диоксан или ацетонитрил) с получением соединения формулы (XXII). Примеры оснований, которые можно использовать в этой реакции,
включают пиридин, триэтиламин, N-метилморфолин и диметиламинопиридин. Реакцию можно при температуре от -50oC до 100oC, предпочтительно, от -20oC до 80oC в
течение промежутка времени от 10 минут до 24 часов, предпочтительно, от 10 минут до 12 часов.
На стадии (iii) соединение формулы (XXII) циклизуют, с удалением защитной группы аминогруппы с получением соединения формулы (XXIII). Реакцию можно проводить при температуре от -20oC до 100oC, предпочтительно от 0 до 80oC в течение 0,5 - 48 часов, предпочтительно, от 1 до 24 часов.
Затем полученное таким образом соединение формулы (XXIII) можно подвергнуть взаимодействию с соединением формулы (VIII) тем же способом, что и на указанной ранее стадии (i) с получением соединения формулы (I).
Соединение формулы (XIX) можно синтезировать в соответствии со способом, описанным в synthesis, p. 1032 (1984).
Способ (F)
Соединение, представленное формулой (I), где X представляет CH, Y представляет N, A и B, каждый, представляют C=O, а B представляет CH, можно получить в соответствии со
способами, представленными на следующих стадиях схемы F.
Схема F
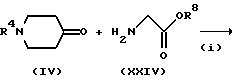
где L представляет атом галогена (например, хлор, бром или иод), низшую алкилсульфонилоксигруппу (например, метансульфонилокси) или арилсульфонилоксигруппу (например, р-толуолсульфонилокси), и R7 и R8 имеют указанные для схемы E значения.
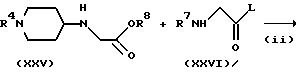
На стадии (i) взаимодействие соединения формулы (IV) с соединением формулы (XXIV) можно проводить тем же способом, что и способ стадии (i) схемы D.
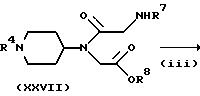
На стадии (ii) соединение формулы (XXV) подвергают взаимодействию с соединением формулы (XXVI) в присутствии основания (или без него) в инертном растворителе (например, ДМФ, дихлорметан, ацетонитрил, ТГФ или диоксан) с получением соединения формулы (XXVII). Примеры основания, которое можно использовать в этой реакции, включают те, которые можно использовать на стадии (iii) схемы A. Реакцию можно проводить при температуре от -20oC до 100oC, предпочтительно, от 0 до 80oC в течение от 0,5 до 48 часов, предпочтительно, от 1 до 24 часов.
Соединение формулы (XXVII) может быть подвергнуто взаимодействию таким же образом, что и на стадии (iii) схемы E, для получения соединения формулы (XXVIII).
Полученное таким образом соединение формулы (XXIII) может затем взаимодействовать с соединением формулы (VIII) таким же образом, что и на стадии (iv) схемы E для получения соединения формулы (I).
В каждом из указанных способов специалистам должно быть ясно, что последовательность синтеза можно выбрать таким образом, чтобы не происходило побочных реакций с функциональными группами, которые не относятся к основным реакциям, и что функциональные группы можно защитить соответствующими защитными группами, чтобы исключить такие нежелательные реакции.
Применение соединения/фармацевтической композиции
Соединение по настоящему изобретению
ингибирует агрегацию тромбоцитов путем ингибирования связывания мембранного протеина GPIIb/IIIa тромбоцита и фибриногена. Таким образом, соединение по настоящему изобретению и его фармакологически
приемлемая соль эффективны при лечении и для профилактики тромботических заболеваний, вызванных агрегацией тромбоцитов, особенно при инфарктах мозга, инфарктах миокарда, стенокардии или окклюзии
периферических артерий.
Фармацевтическая композиция, содержащая соединение по настоящему изобретению или его фармакологически приемлемую соль в качестве активных ингредиентов, может вводиться людям или животным любым способом, например, перорально или парентерально, например, путем внутривенной инъекции, внутримышечной инъекции, подкожно, ректально или через кожу.
Поэтому фармацевтическую композицию, содержащую соединение по настоящему изобретению, получают в виде подходящих дозированных форм, в зависимости от способа применения, и она может быть, в частности получена в виде препаратов, которые, главным образом, включают препараты для инъекций или препараты для перорального введения, такие, как капсулы, таблетки, гранулы, порошки, пилюли, гранулы или пастилки, препараты для ректального введения, масляные суппозитории или водные суппозитории.
Такие препараты можно приготовить обычными способами с обычными добавками, такими, как наполнитель, волокна, связующее, увлажнитель, дисперсант, поверхностно-активный агент, замасливатель, буфер, консервант и агенты, способствующие растворению, антисептики, отдушки, анальгетики или стабилизаторы. Вышеперечисленные приемлемые и нетоксичные добавки включают, например, лактозы, фруктозу, глюкозу, крахмал, желатин, карбонат магния, синтетический силикат магния, стеарат магния, метилцеллюлозу или их соли, гуммиарабик, полиэтиленгликоль, сироп, вазелин, этанол, пропиленгликоль, лимонная кислота, хлорид натрия, сульфит натрия и фосфат натрия.
Соединение по настоящему изобретению представлено в фармацевтической композиции в количестве, которое зависит от дозированных форм, и обычно составляет от около 1 до 70 вес.%, предпочтительно, от около 5 до 50 вес.% всей композиции.
Дозу обычно определяют с учетом применения, возраста, пола и тяжести состояния пациента. Обычно доза бывает в интервале от около 0,1 до 1000 мг, предпочтительно, от 1 до 200 мг в день для взрослых пациентов при лечении тромботических заболеваний. Эти дозы можно вводить сразу или в виде нескольких порций в день.
Далее настоящее изобретение описывается более подробно со ссылкой на примеры, но эти примеры никоим образом не ограничивают объем изобретения.
Пример 1
[[4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]-о-фенилен]-диокси/диуксусной кислоты трифторацетат
(a) Смесь 1-трет-бутоксикарбонил-4-оксопиперидина (2,985 г) и 2-оксопиперазина (1,5
г) растворяют в 25 мл метанола, а затем добавляют молекулярные сита 3 ангстрем (2,5 г) и 1 н. этанол-соляную кислоту (2,5 мл), затем перемешивают в течение 45 минут. 945 мл цианоборнатрийгидрида
разделяют на три части и их последовательно добавляют к раствору при охлаждении льдом, а затем перемешивают в течение 5 часов при комнатной температуре. После того, как нерастворимую часть удаляют с
помощью sellaite, полученный фильтрат концентрируют. К полученному остатку добавляют насыщенный раствор бикарбоната натрия, и экстрагируют этилацетатом. Этилацетатный слой промывают насыщенным
раствором соли, а затем сушат над сульфатом магния. После упаривания растворителя остаток очищают с помощью хроматографической колонки с силикагелем (300 г, элюент хлороформ:метанол 30:1 до 10:1) с
получением 1,448 г 4-/1-трет-бутоксикарбонилпиперидин-4-ил/-2-оксопиперазина.
1H ЯМР (CDCl3) δ : 1,42 (2H, м), 1,46 (9H, с), 1,80 (2H, шир), 2,46 (1H, тт, J = 3,6, 11,2 Гц), 2,7-2,8 (4H), 3,27 (2H, с), 3,35 (2H, тд, J = 3,0, 5,2 Гц), 4,12 (2H, шир.с), 6,06 (1H, с).
E1M (м/Z: 283 (M+) (Масс-спектрометрия электронного удара)
(b) Смесь натрия (35 мг) и толуола (10 мл) нагревают до температуры 110 - 120oC, а затем перемешивают для получения частиц натрия. Затем добавляют соединение (430 мг), полученное
ранее в (a), а полученный раствор нагревают при кипячении с обратным холодильником в течение 3 часов. После охлаждения до около 60oC добавляют
ди-трет-бутил[(4-хлорацетил-о-фенилен)диокси]диацетата (1 г), и раствор нагревают при кипячении с обратным холодильником в течение 3 часов. После охлаждения реакционный раствор разбавляют этилацетатом,
промывают водой, а затем сушат над сульфатом магния. После упаривания растворителя остаток очищают на хроматографической колонке с силикагелем (50 г, хлороформ: метанол = 50:1) с получением 116 мг
ди-трет-бутил-[[4-[[4-(1-трет-бутоксикарбонилпиперидин- 4-ил)-2-оксопиперазин-1-ил] ацетил]-о-фенилен]диокси]диацетата.
1H-ЯМР (CDCl3) δ : 1,46 (9H, с), 1, 47 (9H, с), 1,48 (9H, с), 1,65 (2H, м), 1,82 (2H, шир.д.), 2,47 (1H, м), 2,75 (2H, м), 2,84 (2H, м), 3,36 (4H, м), 4,13 (2H, шир. с), 4,64 (2H, с), 4,68 (2H, с), 4,76 (2H, с), 6,82 (1H, д, J = 8,3 Гц), 7,48 (1H, д, J = 1,9 Гц), 7,60 (1H, дд, J = 1,9, 8,3 Гц).
EIMS (м/Z): 661 (M+).
(c) Соединение (115 мг), полученное ранее в (b), добавляют к смеси трифторуксусной кислоты (1,5 мл) и анизола (0,1 мл), и реакцию проводят при комнатной температуре в течение 2 часов. Добавляют 10 мл диизопропилового эфира при охлаждении льдом, а полученный осадок собирают фильтрацией и затем сушат, в результате чего получают 111 мг указанного в заголовке соединения.
1H-ЯМР (D2O+Na2CO3) δ : 1,61 (2H, кв, J = 3,3 Гц), 2,15 (2H, шир.д), 2,78 (1H, шир.т.), 2,90 (2H, шир.т.), 3,04 (2H, м), 3,39 - 3,51 (6H), 4,64 (2H, с), 4,68 (2H, с), 4,97 (2H, с), 7,01 (1H, д, J = 8,4 Гц), 7,45 (1H, с), 7,743 (1H, д, J = 8,4 Гц).
SIMS (м/Z): 450 (M++1) (Масс-спектрометрия вторичных ионов).
Пример 2
Трифторацетат[[1-[4-[пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил]пиперидин-4-ил]-оксоуксусной кислоты
(a) Повторяют процедуру примера 1 (b), за исключением того, что
ди-трет-бутил-[[4-хлорацетил-о-фенилен)диокси] диацетат заменяют этилбромацетатом с получением [4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2-оксопиперазин-1-ил]ацетата.
1H-ЯМР (CDCl3) δ : 1,28 (3H, т, J=7,2 Гц), 1,40 (2H, м), 1,46 (9H, с), 1,80 (2H, шир.д), 2,45 (1H, м.), 2,74 (2H, шир.т.), 2,82 (2H, шир.т.), 3,33 (2H, с), 3,39 (2H, т, J = 5,4 Гц), 4,12 (2H, с), 4,20 (2H, кв., J = 7,2 Гц).
EIMS (m/z): 369 (M+).
(b) Соединение (360 мг), полученное в (a), растворяют в 5 мл этанола, и добавляют 1 н. раствор гидроксида натрия (1,5 мл), а затем перемешивают при комнатной температуре в течение 3 часов. После того, как реакционный раствор концентрируют, добавляют воду, и pH раствора устанавливают 4,0 с помощью 1 н. соляной кислоты. Затем раствор лиофилизируют с получением гидрохлорида [4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2-оксопиперазин-1-ил] уксусной кислоты (349 мг). Этот продукт используют в последующей реакции без очистки.
1H-ЯМР (D2O) δ : 1,46 (9H, с), 1,60 (2H, кв.д, J=4,3, 12,6 Гц), 2,12 (2H, шир. д), 2,87 (2H, шир.т), 3,42 (1H, шир.т.), 3, 55 (2H, шир.т), 3,67 (2H, шир.т), 3,89 (2H, с), 4,00 (2H, с), 4,18 - 4,27 (2H, шир.с).
(c) Соединение (151 мг), полученное ранее в (b), растворяют в 3 мл метиленхлорида, и к раствору добавляют 1-гидроксибензотриазол и бензотриазол-1-илокситрис(диметиламино)фосфонийгексафторфосфат (195 мг), а затем перемешивают в течение 50 минут. Затем добавляют трет-бутил-4-пиперидинилоксиацетат (90 мг) в 2 мл метиленхлорида, и диизопропилэтиленамин (0,14 мл) и раствор перемешивают при комнатной температуре в течение 3 часов. Реакционный раствор разбавляют этилацетатом, промывают водой, а затем сушат над сульфатом магния. После того, как растворитель выпаривают, полученный остаток очищают на колонке с силикагелем (30 г, хлороформ:метанол=30:1) с получением 170 мг трет-бутил[[1-[4-[1-трет-бутоксикарбонилпиперидин-4-ил)-2- оксопиперазин-1-ил]ацетил]пиперидин-4-ил]оксиацетата.
1H ЯМР (CDCl3) δ : 1,43 (2H, м), 1,46 (9H,
с), 1,48 (9H, с), 1,69 (2H, м), 1,77-1,95 (4H, м), 2,44 (1H, м), 2,74 (2H, шир.т.), 2,82 (2H, м), 3,23-3,45 (6H), 3,62-3,71 (2H, м), 3,84 (1H, м), 3,98 (1H, д, J = 15,2 Гц), 4,02 (1H, д, J = 15,2 Гц),
4,08 (1H, д, J = 15,7 Гц), 4,12 (2H, шир.с.), 4,31 (1H, д, J = 15,7 Гц),
EIMS (m/Z): 538 (M+).
(d) Соединение, полученное ранее в (с), обрабатывают таким же способом, что и в примере 1 (с) с получением указанного в заголовке соединения.
1H ЯМР (D2O) δ : 1,53 - 1,71 (2H, м), 1,97 - 2,10 (4H, м), 2,50 (2H, шир. д),
3,11 - 3,24 (4H, м), 3,31 (1H, м), 3,66 - 3,87 (8H), 4,00 (1H, м), 4,17 (2H, с), 4,28 (2H, с), 4,46 (2H, с),
Пример 3
Трифторацетат
[[4-[[4-(пиперидин-4-ил)пиперазин-1-ил]ацетил]-o-фенилен] диокси]- диуксусной кислоты
(a) По способу примера 1(а), за исключением того, что используют 1-трет-бутоксикарбонил-4-оксопиперидин и
1-бензилпиперазин, получают 1-бензил-4-/1-трет-бутоксикарбонилпиперидин-4-ил/пиперазин.
1H ЯМР (CDCl3) δ : 1,38 (2H, м), 1,45 (9H, с), 1,80 (2H, шир.д.), 2,
35 (1H, м), 2,40 - 2,75 (10H), 3,50 (2H, с), 4,12 (2H, шир.с.), 7,30 (5H, м),
EIMS (м/Z): 359 (M+).
(b) Соединение (2,75 г), полученное в (a), растворяют в 50 мл метиленхлорида, и добавляют к раствору при охлаждении льдом 1,91 г бикарбоната натрия и 2,04 мл бензилхлорформиата. Затем смесь перемешивают при комнатной температуре и добавляют бензилхлорфомиат в количестве 0,8 мл через 4 часа и в количестве 1,0 мл через 9 часов, а затем продолжают перемешивать в течение ночи. Реакционный раствор разбавляют этилацетатом, промывают водой, а затем сушат над сульфатом магния. После того, как растворитель выпаривают, остаток очищают на хроматографической колонке с силикагелем (120 г, хлороформ ---> хлороформ: этилацетат = 1:4) с получением 1-бензилоксикарбони-4-(1-трет-бутоксикарбонилпиперидин-4-ил)пиперазина (2,88 г).
1H ЯМР (CDCl3) δ : 1,39 (2H, м), 1,45 (9H, с), 1,77 (2H, шир.д.), 2,40 (1H, м), 2,51 (4H), 2,69 (2H, шир.т.), 3,51 (4H, м), 4,12 (2H, шир.с), 5,13 (2H, с), 7,37 (5H, м).
(c) Соединение (1,0 г), полученное ранее в (b), растворяют в 5 мл этанола, а затем к раствору добавляют 1 н. раствор этанол-соляной кислоты (5 мл) и палладиевую чернь (50 мг). Каталитическое восстановление ведут при атмосферном давлении в течение 3 часов. После удаления отработанного катализатора путем фильтрования, полученный фильтрат концентрируют. Полученный остаток растворяют в воде и pH раствора доводят до 8 с помощью ионообменной смолы Амберлит JR-45 (OH-). После удаления смолы путем фильтрования фильтрат концентрируют. Полученный остаток очищают с помощью хроматографической колонки с силикагелем (30 г, 1% триэтиламинсодержащая смесь хлороформ:метанол = 20: 1 ---> 2% триэтиламинсодержащая смесь хлороформ:метанол = 10:1) с получением 277 мг 4-/1-трет-бутокикарбонилпиперидин-4-ил)пиперазина.
1H ЯМР (CDCl3) δ
: 1,40 (2H, м), 1,45 (9H, с), 1,76 (2H, шир.д.), 2,46 (1H, м), 2,69 (2H, шир.т.), 2,82 (4H, м), 3,17 (4H, м), 4,12 (2H, шир.с.),
EIMS (m/Z): 269 (M+).
(d) Соединение (135 г), полученное ранее в (c), растворяют в 2 мл ДМФ и к полученному раствору добавляют 138 мг карбоната калия и 275 мг ди-трет-бутил-[[4-хлорацетил-о-фенилен] диокси]диацетата, а затем перемешивают в течение 4 часов. Реакционный раствор разбавляют этилацетатом, промывают водой и затем сушат над сульфатом магния. После испарения этилацетата остаток очищают на хроматографической колонке с силикагелем (хлороформ:метанол = 40:1) с получением 213 мг ди-трет-бутил-[[4-[[4-[1-трет-бутоксикарбонилпиперидин-4-ил]пиперазин- 1-ил]ацетил]-о-фенилен]диокси]диацетата.
1H ЯМР (CDCl3) δ : 1,38 (2H, м), 1,45 (9H, с), 1,47 (9H, с), 1,48 (9H, с), 1,82 (2H, шир. д), 2,37 (1H, м), 2,55 - 2,76 (10H), 3,74 (2H, с), 4,13 (2H, шир. с), 4,64 (2H, с), 4,
67 (2H, с), 6,80 (1H, д, J = 8,5 Гц), 7,53 (1H, д, J= 8,1 Гц), 7,64 (1H, дд, J = 1,8, 8,5 Гц),
EIMS (m/Z): 647 (M+).
(e) Соединение, полученное ранее в (d), обрабатывают тем же способом, что и в примере 1 (c) с получением указанного в заголовке соединения.
1H ЯМР (D2O) δ : 1,97 (2H, м), 2,46 (2H, шир.д), 3,15
(2H, шир.т), 3,51 - 3,72 (9H), 4,85 (2H, шир.с), 4,86 (2H, с), 4,87 (2H, с), 4,91 (2H, с), 7,10 (1H, д, J = 8,6 Гц), 7,51 (1H, с), 7,69 (1H, д, J = 8,6 Гц),
SIMS (m/Z): 435 (M+ +
1).
Пример 4
Диэтил-[[4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил]-о-фенилен] диокси]диацетат
По способу примера 1 (b) и (c), за исключением того, что
ди-трет-бутил-[[4-хлорацетил-o-фенилен] диокси] диацетат заменяют на диэтил-[[4-хлорацетил-o-фенилен)диокси]диацетат, получают указанное в заголовке соединение
1H ЯМР (D2
O) δ : 1,31 (3H, т, J = 7 Гц), 1,32 (3H, т, J = 7 Гц), 2,00 (2H, м), 2,47 (2H, шир.д), 3,18 (2H, шир.т), 3,55-3,76 (7H, м), 4,03 (2H, с), 4,33 (4H, кв., J = 7 Гц), 4,96 (2H, с), 5,01 (2H, с), 5,
05 (2H, с), 7,14 (1H, д, J = 8 Гц), 7,59 (1H, с), 7,79 (1H, д, J = 8 Гц),
SIMS (m/Z): 506 (M+ + 1).
Пример 5
4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]
ацетил]феноксиуксусная кислота
По способу примера 1 (b) и (c), за исключением того, что ди-трет-бутил-[[4-хлорацетил-o-фенилен] диокси] диацетат заменяют на
трет-бутил-4-бромацетилфеноксиацетат, получают указанное в заголовке соединение.
1H ЯМР (CD3OD) δ : 1,85 - 1,95 (2H, м), 2,27 - 2,30 (2H, м), 3,05 - 3,11
(2H, м), 3,15 - 3,21 (1H, м), 3,40 - 3,42 (2H, м), 2,51 - 3,60 (4H, м), 3,72 (2H, с), 4,79 (2H, с), 4,93 (2H, с), 7,06 (2H, д, J = 8,9 Гц), 8,01 (2H, д, J = 8,9 Гц),
SIMS (m/Z): 376 (M+ + 1).
Пример 6
Трифторацетат [[1-[4-(пиперазин-4-ил] пиперидин-1-ил]ацетил]пиперидин-4-ил]оксиуксусной кислоты
По способу примера 2 (b) и (c), за исключением
того, что [4-[1-трет-бутоксикарбонилпиперидин-4-ил)-2-оксопиперазин-1-ил] уксусной кислоты гидрохлорид заменяют на [4-[4-трет-бутоксикарбонилпиперазин-1-ил)пиперидин-1-ил] уксусную кислоту, получают
указанное в заглавии соединение.
1H ЯМР (CD3OD) δ : 1,60 - 1,73 (2H, м), 1,84 - 1,96 (3H, м), 2,10 - 2,20 (2H, м), 2,68 - 2,97 (4H, м), 3,05 - 3,30 (7H, м), 3,42 - 3,48 (2H, м), 3,55 - 3,62 (2H, м), 3,67 - 3,75 (2H, м), 3,80 - 3,90 (2H, м), 4,16 (2H, с), 4,24 (2H, с).
SIMS (m/Z): 369 (M+ + 1).
Пример 7
Гидрохлорид н-бутил-4-[[4-(пиперидин-4-ил)-2,6-диоксопиперазин-1-ил]ацетил]-феноксиацетата
(a) Смесь 1-трет-бутоксикарбонил-4-оксопиперидина (9,06 г) и 2,6-диоксопиперазина (3,99 г)
растворяют в 1,2-дихлорэтане (170 мл), и к полученному раствору добавляют 20 мл уксусной кислоты и 9,64 г триацетоксиборгидрида натрия, и затем перемешивают при комнатной температуре в течение 17
часов. Затем к реакционному раствору добавляют воду при охлаждении льдом и к раствору добавляют бикарбонат натрия для нейтрализации уксусной кислоты. Реакционный раствор выливают в делительную воронку
для разделения водного и органического слоев. Водный слой экстрагируют хлороформом, а затем экстрагированный органический слой объединяют с выделенным ранее органическим слоем. Объединенные
органические слои промывают насыщенным солевым раствором, а затем сушат над сульфатом натрия. После выпаривания растворителя остаток очищают на хроматографической колонке с силикагелем (350 г,
хлороформ ---> хлороформ: метанол = 60:1) до получения кристаллов. Затем к кристаллам добавляют н-гексан, и кристаллы собирают фильтрацией с получением 6,46 г (62%)
4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,6-диоксипиперазина.
1H ЯМР (CDCl3) δ : 1,35 - 1,52 (11H), 1,77 (2H, шир.д), 2,58 (1H, тт, J = 3,6, 11,4 Гц), 2,73 (2H, шир.т), 3,46 (4H, с), 4,15 (2H, шир.с), 8,02 (1H, с).
EIMS (m/Z): 297 (M+).
(b) Соединение (3,57 г), полученное ранее в (a), растворяют в 60 мл диметилформамида, и к полученному раствору добавляют гидрид натрия (60% масляная суспензия, 0,72 г), а затем все это перемешивают в течение 10 минут. К реакционному раствору добавляют н-бутил-4-бромацетилфеноксиацет (4,53 г) в 25 мл диметилформамида при охлаждении льдом, а затем перемешивают в течение 3 часов. К реакционному раствору добавляют воду, и экстрагируют этилацетатом. Полученный экстракт промывают водой и насыщенным солевым раствором, а затем сушат над сульфатом натрия. После того, как растворитель выпаривают, остаток очищают на хроматографической колонке с силикагелем (250 г, гексан: этилацетат = 1:1) с получением 4,67 г (71%) н-бутил-4-[[4-[1-трет-бутокси-карбонилпиперидин-4-ил)-2,6-диоксопиперазин-1-ил] ацетил]феноксиацетата.
1H ЯМР (CDCl3) δ : 0,93 (3H, т, J = 7,4 Гц), 1,30 - 1,53 (13H), 1,56 - 1,70 (2H, м), 1,83 (H, шир.д), 2,64 (1H, тт, J = 3,3, 11,3 Гц), 2,77 (2H, шир.т.), 3,64 (4H, с), 4,16 (2H,
шир.с), 4,22 (2H, т, J = 6,7 Гц), 4,70 (2H, с), 5,14 (2H, с), 6,97 (2H, д, J = 9,0 Гц), 7,95 (2H, д, J = 9,0 Гц),
EIMS (m/Z): 545 (M+).
(c) К соединению (4,67 г), полученному ранее в (b), добавляют 7 мл анизола, 20 мл трифторуксусной кислоты и 10 мл метиленхлорида, а затем перемешивают при комнатной температуре в течение 1 часа. К реакционному раствору добавляют воду, а затем все нейтрализуют бикарбонатом натрия. После выпаривания растворитель к остатку добавляют воду и экстрагируют этилацетатом. Полученный экстракт сушат над сульфатом натрия и растворитель выпаривают. Полученный остаток очищают на хроматографической колонке с силикагелем (200 г, хлороформ: метанол= 10:1 до 7:1) с получением светло-желтых кристаллов (4,15 г). Эти кристаллы используют в гидролитической реакции следующего примера 8. С другой стороны, к 1,02 г этих кристаллов добавляют 6 мл н-бутанола и 18 мл хлороформа, и полученный раствор перемешивают в течение 20 минут при охлаждении льдом, при этом продувая через раствор газообразный хлористый водород. После перемешивания при комнатной температуре в течение еще 1 часа растворитель выпаривают. К полученному остатку добавляют эфир, кристаллы отфильтровывают, в результате чего получают 0,618 г (52%) указанного в заголовке соединения.
1H ЯМР (D2O) δ : 0,76 - 0,85 (3H, м), 1,16 - 1,29 (2H, м), 1,50 - 1,62 (2H, м), 1,65 - 1,79 (2H, м), 2,16 (2H, шир.д), 2,94 - 3,10 (3H), 3,52 (2H, шир. д), 3,74 - 3,87 (4H, м), 4,15 - 4.23 (2H, м), 4,80 - 4,89 (2H, м), 5,20 - 5,28 (2H, м), 6,90 - 7,09 (2H, м), 7,93 - 8,03 (2H, м).
EIMS (m/Z): 445 (M+).
Пример 8
Гидрохлорид 4-[[4-(пиперидин-4-ил)-2,
6-диоксопиперазин-1-ил]ацетил]феноксиуксусной кислоты
К светло-желтым кристаллам (0,312 г), полученные в примере 7 (c), при охлаждении льдом добавляют 7 мл 5 н. соляной кислоты и полученный
раствор перемешивают в течение 16 часов, нагревая до комнатной температуры. После испарения растворителя полученный остаток очищают с помощью колонки высокого давления (150 мл, вода - 5% водный
ацетон), а затем лиофилизируют с получением 0,103 г (35%) указанного в заголовке соединения.
1H-ЯМР (D2O) δ : 2,57 - 2,73 (2H, м), 2,10 (2H, шир.д), 2,84 - 3,03 (3H), 3,45 (2H, шир.д), 3,74 (4H, с), 4,50 (2H, с), 5,21 (2H, с), 6,96 (2H, д, J = 8,8 Гц), 7,94 (2H, д, J = 8,8 Гц).
EIMS (m/Z): 389 (M+).
Пример
9
Гидрохлорид н-бутил-4-[[4-(пиперидин-4-ил)-2,3-диоксопиперазин-1-ил] ацетил]-феноксиацетата
(a) По способу примера 1(a) за исключением того, что 2,6-диоксопиперазин заменяют на
гидрохлорид N-бензилоксикарбонилэтилендиамина получают 14,7 г (87%) N-бензилоксикарбонил-N'-1-трет-бутоксикарбонилпиперидин-4-ил)этилендиамина.
1H-ЯМР (CDCl3) δ : 1,13 - 1,27 (2H, м), 1,45 (9H, с), 1,81 (2H, шир.д), 2,52 - 2,63 (1H, м), 2,44 (1H, м), 2,74 (2H, шир.т), 2,82 (2H, м), 2,70 - 2,86 (4H, м), 3,22 - 3,28 (2H, м), 4,00 (2H, шир.с.), 5,10 (2H, с), 5,19 (1H, шир.с.), 7,29 - 8,29 (5H, м).
EIMS (m/Z): 377 (M+).
(b) Соединение (10,6 г), полученное ранее в (a), растворяют в 140 мл метиленхлорида, и к раствору добавляют 4,7 мл триэтиламина. Затем добавляют 3,1 мл этилхлорглиоксилата при охлаждении льдом, а затем перемешивают в течение 30 минут. К реакционному раствору добавляют 140 мл воды при охлаждении льдом и реакционный раствор выливают в делительную воронку для разделения водного и органического слоев. Водный слой экстрагируют хлороформом и экстрагированный органический слой объединяют с образовавшимся ранее органическим слоем. Объединенные органические слои промывают водой, насыщенным раствором бикарбоната натрия и сушат над сульфатом натрия. После выпаривания растворителя остаток очищают на хроматографической колонке с силикагелем (300 г, гексан:этилацетат = 2:1 до 3:2) с получением 12,3 (93%) этил-N-2-(бензилоксикарбониламино/этил-N-/1-трет-бутоксикарбонилпиперидин- 4-ил/аминоглиоксилата.
1H-ЯМР (CDCl3) δ : 1,36 (3H, т, J = 7,2 Гц), 1,47 (9H с), 1,69 - 1,83 (4H, м), 2,55 - 2,82 (2H, м), 3,37 - 3,50 (5H), 4,09 - 4,30 (2H, м), 4,36 (2H, кв., J = 7,2 Гц), 5,10 (2H, с), 5,21 (1H, шир.с), 7,35 (5H, с).
FDMS (m/Z): 478 (M+ +1) (масс-спектрометрия с полевой десорбцией).
(c) Соединение (8,12 г), полученное ранее в (b), растворяют в 85 мл этанола и к раствору добавляют 270 мг 10% палладия на угле, а затем ведут каталитическое восстановление при атмосферном давлении в течение 4 часов. После того, как катализатор удаляют фильтрованием, растворитель выпаривают, в результате чего получают 6,76 г 4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,3-диоксопиперазина. Этот продукт используют в последующей реакции без дополнительной очистки.
1H-ЯМР (CD3OD) δ :
1,46 (9H, с), 1,60 - 1,74 (4H, м), 2,86 (2H, шир.с), 3,41 - 3,47 (2H, м), 3,49 - 3,55 (2H, м), 4,20 (2H, шир. д), 4,43 - 4,53 (1H, м),
EIMS (m/Z): 297 (M+).
(d) По способу примера 7 (b) за исключением того, что 4-/1-трет-бутоксикарбонилпиперидин-4-ил/-2,6-диоксопиперазин заменяют соединением, полученным ранее в (c), получают 2,59 г н-бутил-4-[[4-[1-трет-бутоксикарбонилпиперидин-4-ил/-2,3-диоксопиперазин-1-ил]-ацетил]феноксиацетата. (Выход продукта из соединения, полученного) ранее в (b), составляет 59%).
1H -ЯМР (CDCl3) δ : 0,93 (3H, т, J = 7,4 Гц), 1,36 (2H, м), 1,47 (9H, с), 1,54 - 1,70 (4H), 1,74 (2H, шир.д), 2,83 (2H, шир.т), 3,51 - 3,62 (4H, м), 4,15 - 4,32 (4H), 4,66 (1H,
тт, J = 3,4, 12,8 Гц), 4,71 (2H, с), 4,93 (2H, с), 6,97 (2H, д, J = 9,0 Гц), 7,94 (2H, д, J = 9,0 Гц),
Масс-спектр с полевой десорбцией (m/Z): 545 (M+).
(e) Соединение, полученное ранее в (d), обрабатывают по способу примера 1 (c) с получением 0,298 г (52%) указанного в заголовке соединения.
1H-ЯМР (D2O) δ : 0, 82 (3H, т, J = 7,3 Гц), 1,25 (2H, м), 1,58 (2H, м), 1,97 - 2,07 (4H, м), 3,07 - 3,19 (2H, м), 3,54 (2H, шир.д), 3,67 (4H, с), 4,21 (2H, т, J = 6,5 Гц), 4,45 - 4,54 (1H, м), 4,86 (2H, с), 5,02 (2H, с), 7,05 (2H, д, J = 9,2 Гц), 7,97 (2H, д, J = 9,2 Гц).
SIMS (m/Z): 446 (M+ + 1).
Пример 10
4-[[4-[пиперидин-4-ил)-2,3-диоксопиперазин-1-ил] ацетил]
феноксиуксусная кислота, гидрохлорид.
Соединение, полученное в примере 9, обрабатывают по способу примера 8 с получением 0,211 г (68%) указанного в заголовке соединения.
1H-ЯМР (CF3COOD) δ : 2,25 - 2,34 (2H, м), 2,36 - 2,51 (2H, м), 3,40 - 3,54 (2H, м), 3,81 - 4,01 (6H), 4,87 (1H, шир.т.), 4,97 (2H, с), 5,23 (2H, с), 7,17 (2H, д), J = 8,8 Гц), 7,38 (1H, шир.с), 8,11 (2H, д, J = 8,8 Гц).
SIMS (m/Z): 390 (M++ 1). Масс-спектрометрия вторичных ионов.
Пример 11
н-Бутил-4-[[4-пиперидин-4-ил)-2,5-диоксопиперазин-1-ил] ацетил] - феноксиацетат-трифторацетат
(a) По способу примера 7 (a), за исключением того, что 2,6-диоксопиперазин заменяют на
гидрохлорид сложного эфира глицинэтил, получают 9,79 г (86%) этил-(1-трет-бутоксикарбонилпиперидин-4-ил)-аминоацетата.
1H-ЯМР (CD3OD) δ : 1,19 - 1,31 (5H), 1,45 (9H, с), 1,86 (2H, шир.д), 2,62 - 2,71 (1H, м), 2,80 (2H, шир.с), 3,43 (2H, с), 4,04 (2H, шир.д), 4,19 (2H, кв., J = 7,2 Гц).
EIMS (m/Z): 286 (M+).
(b) Соединение (9,38 г), полученное ранее в (a), растворяют в 100 мл диметилформамида и к раствору добавляют бензотриазол-1-илокситрис-(диметиламино)-фосфонийгексафторфосфат (26,1 г) и N-метилморфолин (7,6 мл), а затем перемешивают при комнатной температуре в течение 2 часов. К реакционному раствору добавляют при комнатной температуре N-бензилоксикарбонилглицин (6,86 г) в диметилформамиде (60 мл) и полученный раствор продолжают перемешивать в течение 19 часов. Затем к реакционному раствору добавляют 0,2 н. соляную кислоту при охлаждении льдом и полученный реакционный раствор выливают в делительную воронку для разделения водного и органического слоев. Водный слой экстрагируют хлороформом, а экстрагированный органический слой объединяют с полученным ранее органическим слоем. Объединенные органические слои промывают водой, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, а затем сушат над сульфатом натрия. После выпаривания растворителя к остатку добавляют этилацетат и выпавшие в осадок кристаллы удаляют фильтрованием. Полученный фильтрат концентрируют и остаток очищают на хроматографической колонке с силикагелем (350 г, гексан:этилацетат = 2:1) с получением 14,4 г (92%) этил-N-(бензилоксикарбониламиноацетил)-N-(1-трет-бутоксикарбонилпиперидин-4-ил) аминоацетата.
1H-ЯМР (CDCl3) δ : 1,24 - 1,55 (13H), 1,62 - 1,81 (4H, м, 2,65 - 2,84 (2H, м), 3,61 - 3,7, (4,57 - 4,67 (1H, 2хм), 3,86 - 4,00 (3H), 4,05 - 4,32 (5H), 5,12, 5,13 (2H, 2хс), 5,73, 5,80 (1H, шир.с), 7,28 - 7,39 (5H, м).
SIMS (m/Z): 478 (M+ + 1).
(c) Соединение, полученное ранее (b), обрабатывают по способу примера 9 (c) и после перекристаллизации из этилацетата получают 6,31 г
(85%) 4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,5-диоксопиперазина
1H-ЯМР (CDCl3) δ : 1,42 - 1,70 (13H), 2,80 (2H, шир.т), 3,86 (2H, с), 4,23 (2H, шир.с), 4,58
(1H, тт, J = 4,4, 11,8 Гц), 6,41 (1H, шир.с).
EIMS (m/Z): 297 (M+).
(d) Соединение, полученное ранее в (c), подвергают взаимодействию по способу примера 7 (b), а затем к реакционному раствору при охлаждении льдом добавляют 0,9 мл уксусной кислоты, после чего перемешивают в течение 15 минут. Затем к этому добавляют воду при охлаждении льдом для прекращения реакции и реакционный раствор экстрагируют этилацетатом. Полученный экстракт промывают водой, насыщенным раствором бикарбоната натрия и насыщенным солевым раствором, а затем сушат над сульфатом натрия. После выпаривания растворителя остаток очищают на хроматографической колонке с силикагелем (150 г, хлороформ: метанол = 100:1). Затем к полученному кристаллическому веществу добавляют эфир и кристаллы собирают фильтрованием, в результате чего получают 2,11 г (70%) н-бутил-4-[[4-(1-трет-бутоксикарбонилпиперидин-4-ил)-2,5-диоксопиперазин-1-ил] ацетил]феноксиацетата.
1H-ЯМР (CDCl3) δ : 0,93 (3H, т, J = 7,4 Гц), 1,36 (2H, м), 1,47 (9H, с), 1,54 - 1,72 (6H), 2,81 (2H, шир.т), 3,98 (2H, с), 4,10 (2H, с), 4,16 - 4,35 (4H), 4,60 (1H, тт, J = 4,1, 11,8 Гц), 4,70 (2H, с), 4,79 (2H, с), 6,97 (2H, д, J = 7,93 (2H, д, J = 8,7 Гц).
ЕIMS (m/Z): 545 (M+).
(e) К соединению (1,91 г), полученному ранее в (d), при охлаждении льдом добавляют 3,5 мл анизола и 14 мл трифторуксусной кислоты и перемешивание продолжают в течение 1,5 часа. Затем к реакционной смеси добавляют изопропиловый эфир при охлаждении льдом. После декантации растворителя добавляют эфир, чтобы вызвать кристаллизацию. Полученные кристаллы собирают фильтрованием, промывают изопропиловым эфиром и эфиром, а затем сушат с получением 2,11 г (70%) указанного в заголовке соединения.
1H-ЯМР (CD3OD) δ : 0,93 (3H, т, J = 7,4 Гц), 1,37 (2H, м), 1,60 - 1,69 (2H, м), 1,96 - 2, 16 (4H, м), 3,09 - 3,19 (2H, м), 3,52 (2H, шир.д), 4,09 (2H, с), 4,12 (2H, с), 4,21 (2H, т, J = 6,6 Гц), 4,50 (1H, тт, J = 3,9, 11,7 Гц), 4,84 (2H, с), 4,92 (2H, с), 7,05 (2H, д, J = 9,0 Гц), 8,00 (2H, д, J = 9,0 Гц).
EIMS (m/Z): 445 (M+).
Пример 12
Гидрохлорид-4-[[4-(пиперидин-4-ил)-2,5-диоксопиперазин-1Z-ил] ацетил] феноксиуксусной кислоты.
Соединение, полученное в примере 11, обрабатывают по способу примера 8, в результате чего получают 0,417 г (98%) указанного в заголовке соединения.
1H-ЯМР (D2O) δ : 1,92 - 2,01 (4H, м), 3,05 - 3,17 (2H, м), 3,53 (2H, шир. д) 4,11 (4H, с), 4,45 - 4,55 (1H, м), 4,58 (2H, с), 4,91 (2H, с), 7,00 (2H, д, J = 8,8 Гц), 7,93 (2Н, д, J = 8,8 Гц).
SIMS (m/Z) : 390 (M+ + 1).
Пример 13
Дигидрохлорид н-бутил-4-[[4-/пиперидин-4-ил/-2-оксопиперазин-1-ил] ацетил]феноксиацетата
По
способу примера 4, за исключением того, что используют н-бутил-(4-бромацетил)феноксиацетат, получают указанное в заголовке соединение.
1H-ЯМР (CD3OD) δ : 8, 00 (2H, д, J = 9,0 Гц), 7,04 (2H, д, J = 9,0 Гц), 4,87 (2H, с), 4,82 (2H, с), 4,21 (2H, т), J = 6,7 Гц), 3,48 - 3,42 (4H, м), 3,32 - 3,30 (2H, м), 3,04 (2H, дт, J = 2,7 Гц, J = 2,3 Гц), 2,93 (2H, т, J = 5,5 Гц), 2,71 (1H, ддд, J = 3,5 Гц, J = 6,7 Гц, J = 13,8 Гц), 2,15 - 2,10 (2H, м), 1,83 - 1,73 (2H, м), 1,67 - 1,60 (2H, м), 1,37 (2H, дт, J = 7,4 Гц, J = 14,9 Гц), 0,93 (3H, т, J = 7,4 Гц).
Пример 14
Этил-4-[[4-(-пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]феноксиацетат
Повторяют способ примера 4, за исключением того, что используют
этил-/4-бромацетил/феноксиацетат, а результате чего получают указанное в заголовке соединение.
1H-ЯМР (CD3OD) δ : 1,28 (3H, т, J = 7,2 Гц), 1,80 - 1,90 (2H, м), 2,20 - 2,27 (2H, м), 3,02 - 3,10 (3H, м), 3,20 - 3,23 (2H, м), 3,49 - 3,57 (4H, м), 3,61 (2Н, шир. с.), 4,25 (2Н, кв., J = 7,2 Гц), 4,82 (2H, с), 4,91 (2H, с), 7,04 (2H, д, J = 8,9 Гц), 8,00 (2H, д, J = 8,9 Гц).
SIMS (m/Z): 404 (M+ + 1).
Пример 15
Этил-4-[[4-[1-(5-метил-2-оксодиоксол-4-ил)метилпиперидин-4-ил]
-2- оксопиперазин-1-ил]ацетил]феноксиацетат
Соединение, полученное в примере 14 (150 мг), растворяют в 5 мл безводного ДМФ, и к раствору добавляют 98 мг карбоната калия и 55 мг
4-бромметил-5-метил-2-оксодиоксола, а затем все это перемешивают при комнатной температуре в течение 1,5 часа. Затем реакционный раствор разбавляют этилацетатом, дважды промывают водой, а затем сушат
над сульфатом магния. Неорганическую соль отфильтровывают, а полученный фильтрат концентрируют при пониженном давлении. Полученное масло очищают на хроматографической колонке с силикагелем
(дихлорметан: метанол = 20:1). Полученное вещество растворяют в 1,4-диоксане, и к этому добавляют 1 н. HCl. Полученный раствор лиофилизуют в течение ночи с получением 58 мг (выход 40%) указанного в
заголовке соединения.
1H-ЯМР (CDCl3) δ : 1,30 (3H, т, J = 7,2 Гц), 1,57 - 1,65 (2H, м), 1,83 - 1,86 (2H, м), 2,08 - 2,15 (2H, м), 2,11 (3H, с), 2,28 - 2,36 (1H, м), 2,83 - 2,86 (2H, м), 2,92 - 2,94 (2H, м), 3,32 (2H, с), 3,36 - 3,38 (4H, м), 4,28 (2H, кв., J = 7,2 Гц), 4,69 (2H, с), 4,77 (2H, с), 6,95 (2H, д, J = 8,6 Гц), 7,95 (2H, д, J = 8,6 Гц).
Масс-спектр с полевой десорбцией (m/Z): 515 (M+).
Пример 16
Трифторацетат 3-[4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]фенил]пропионовой
кислоты
По способу примера 1 (b) и (c), за исключением того, что используют дифенилметил-3-/4-хлорацетил/фенилпропионат, получают указанное в заголовке соединение.
1 H-ЯМР (D2O) δ : 1,75 - 1,83 (2H, м), 2,29 - 2,33 (2H, м), 2,74 (2H, т, J = 7 Гц), 3,03 - 3,21 (7H, м), 3,55 - 3,77 (6H, м), 5,02 (2H, с), 7,49 (2H, д, J = 8 Гц), 7,98 (2H, д, J = 8 Гц).
Масс-спектр (m/Z): 373 (M+).
В конце текста приведены структуры соединений, полученных в примерах. Номера соединений соответствуют номерам примеров.
Фармакологическое исследование
Ингибирование агрегации тромбоцитов
На PPP человека (platelet rich plasma обогащенная тромбоцитами плазмы) исследуют
действие соединения по настоящему изобретению на ингибирование агрегации тромбоцитов.
Из вены здорового мужчины шприцом, в котором уже есть один объем раствора 3,8% цитрата натрия, отбирают образец - девять объемов крови. Этот образец крови центрифугируют при 170 об/мин при комнатной температуре в течение 10 минут. Полученную надосадочную жидкость выделяют в качестве PRP. Остаток образца крови, из которого уже отобрали PRP, центрифугируют при 2700 об/мин в течение 15 минут. Надосадочную жидкость отбирают в качестве обедненной тромбоцитами плазмы (PRP) (Platelet poor plasma-пер.).
Исследование агрегации тромбоцитов проводят с помощью агглигометра (PAM-8c) изготовитель MEBANICKS Co. Ltd). Исследуемое соединение растворяют в 50% ДМСО физиологическом растворе, 50% метанольном физиологическом растворе или просто в физиологическом растворе. Соединение и PRP предварительно инкубируют в течение 2 минут. В качестве индуктора используют ADP (CHRONO-PAR REAGENTS 384 ADP. CHRONO-LOG Corp.) в виде раствора в физиологическом растворе до конечной концентрации 5 мкМ.
Активность, препятствующую агрегации тромбоцитов, выражают как отношение
ингибирования к агрегации под действием ADP в отсутствии тестируемого соединения следующим образом:

Таблица
N примера - ИК50 (мкМ)
1 - 0,055
2 - 0,08
3 - 0,15
4 - 0,56
5 - 0,032
6 - 2,3
7 - 0,24
8 - 0,019
9 - 1,2
10 - 0,47
11 - 1,1
12 - 1,2
13 - 0,049
14 - 0,25
15 - 0,3
16 - 0,427
Реферат
Описываются азотсодержащие гетероциклические производные общей формулы I, где А, В и С независимо представляют СН2 или С = O, Х и У отличаются друг от друга, и каждый из них представляет СН или N, D представляет -(СН2)k- или -(СН2)m-СО-, где k является целым числом 1-4; m является целым числом 0-3; E представляет группу II или III, где n является целым числом 1-3; Z представляет -W-(СН2 )p-СООR3, где W представляет -O- или связь; р является целым числом 1-4; R3 представляет водород, низший алкил; R1 представляет водород или низший алкил, замещенный -2-оксодиоксол-4-ил; R2 представляет атом водорода, и их фармацевтически приемлемые соли и сольваты, обладающие способностью ингибировать агрегацию тромбоцитов. Описывается фармацевтическая композиция на основе вышеуказанных соединений и способ лечения или профилактики тромботических заболеваний с использованием соединений формулы I. 3 с. и 11 з.п. ф-лы.
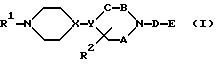
Формула
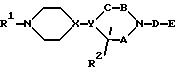
где А, В и С независимо представляют CH2 или С=О;
X и Y отличаются друг от друга и каждый из них представляет CH или N;
D представляет -(CH2)m-CO-, где m является целым числом 0 - 3;
E представляет группу II или III
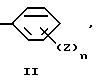
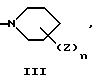
где n представляет целое число 1 - 3;
Z представляет -W-(CH2)p-COOR3, где W представляет -O- или связь, p представляет целое число 1 - 4; R3 представляет водород или низший алкил;
R1 представляет водород или низший алкил, замещенный группой низший алкил - замещенный-2-оксодиоксол-4-ил,
R2 представляет атом водорода,
и их фармацевтически приемлемые соли и сольваты.
[[4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил]-о-фенилен]диокси] диуксусная кислота,
[[4-[[4-(пиперидин-4-ил)пиперазин-1-ил] ацетил] -о-фенилен] диокси]диуксусная кислота,
4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]феноксиуксусная кислота,
диэтил[[4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил] -о-фенилен] диокси]диацетат,
н-бутил 4-[[4-(пиперидин-4-ил)-2,6-диоксопиперазин-1-ил] ацетил] феноксиацетат,
4-[[4-(пиперидин-4-ил)-2,6-диоксопиперазин-1-ил] ацетил] феноксиуксусная кислота,
н-бутил 4-[[4-(пиперидин-4-ил)-2,3-диоксопиперазин-1-ил] ацетил] феноксиацетат,
4-[[4-(пиперидин-4-ил)-2,3-диоксопиперазин-1-ил] ацетил] феноксиуксусная кислота,
н-бутил 4-[[4-(пиперидин-4-ил)-2,5-диоксопиперазин-1-ил] ацетил] феноксиацетат,
4-[[4-(пиперидин-4-ил)-2,5-диоксопиперазин-1-ил] ацетил] феноксиуксусная кислота,
н-бутил 4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил] ацетил]феноксиацетат, дигидрохлорид,
этил 4-[[4-(пиперидин-4-ил)-2-оксопиперазин-1-ил]ацетил]феноксиацетат.
15.07.94 - соединения формулы I, где В и С представляют CH2, Е представляет группы II и III;
02.06.95 - соединения формулы I, где В и С каждый представляет CH2 или
Комментарии