Полициклические аминосодержащие соединения или их соли, их оптически чистые изомеры, способ получения полициклических аминосодержащих соединений, циклические аминосодержащие соединения или их соли, их оптически чистые изомеры и фармацевтическая композиция - RU2083574C1
Код документа: RU2083574C1
Чертежи
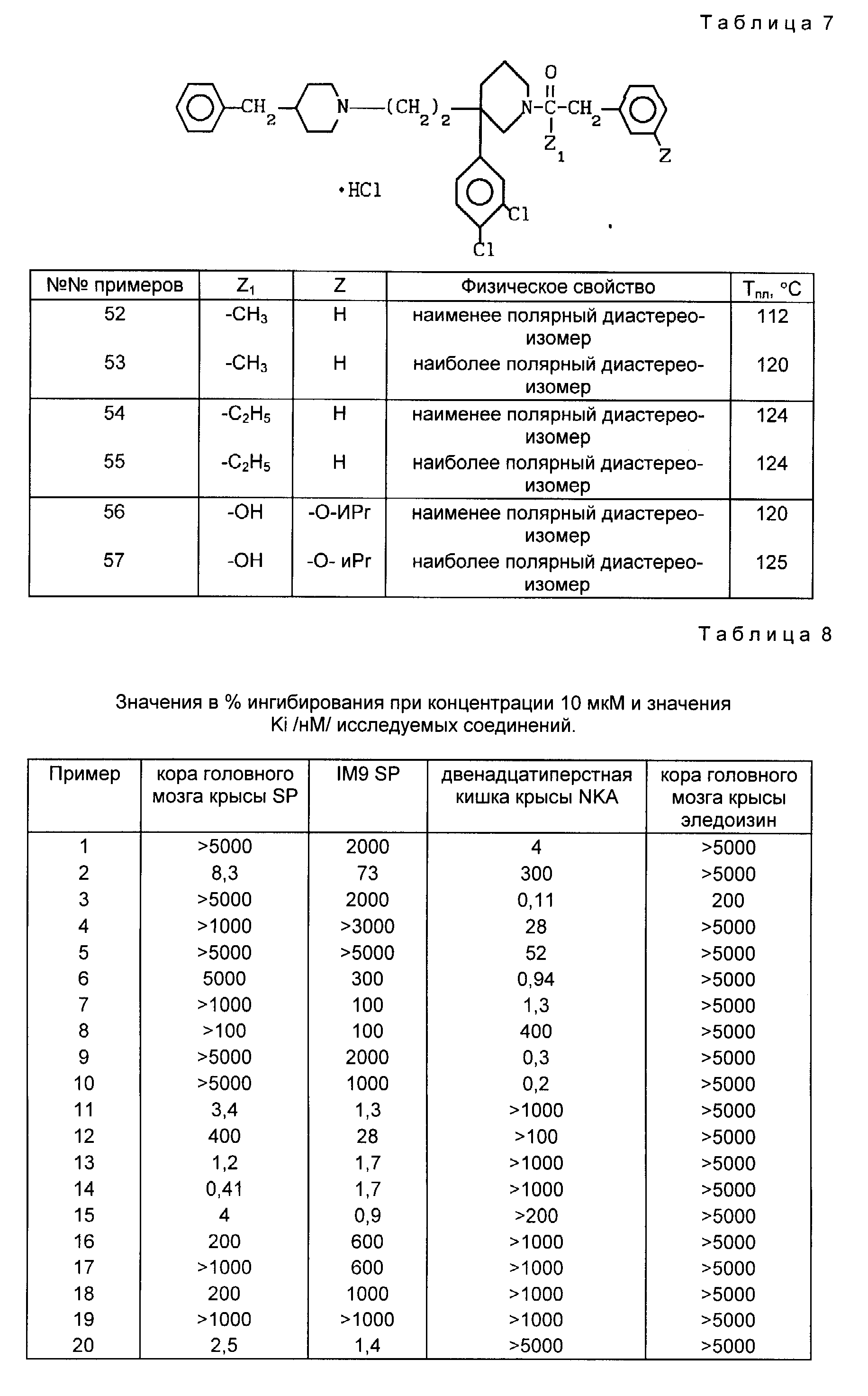
Описание
Изобретение относится к полициклическим аминосодержащим соединениям, к их оптически чистым энантиомерам, к способу их получения, к фармокомпозиции на их основе, а также к новым промежуточным соединениям синтеза полициклических соединений.
Новые полициклические соединения можно применять в терапевтических целях, в частности при патологических явлениях, в которых затронута система нейрокининов, как, например, боль (D. Regoli и comp. Life Sciences, 1987, 40, 109-117), аллергия и воспаление (J.E. Morlay и comp. Life Sciences, 1987, 41, 527-544), недостаточность кровообращения (J. Losay и comp. Substance P, Von Eulez, U.S. Pernow ed. Raven Press, New York, 1977, 287-293), желудочно-кишечные нарушения (D. Regoli и comp. Trend Pharmacol. Sci. 1985, 6, 481-484), распираторные нарушения (J. Mizrahi и comp. Pharmacology, 182, 25, 39-50).
Известны эндогенные лиганды с рецепторами нейрокининов, таких как вещество P (SP), нейрокинин A (NKA) (S. J. Bailey и comp. Substance P, P. Skrabanck ed. Boole Press, Dublin, 1983, 16-17) и нейрокинин B(NKB) (S.P. Watson, Life Sciences, 1983, 25, 797-808).
Рецепторы нейрокининов находятся во многих препаратах и в настоящее время они классифицируются на три типа: NK1, NK2 и NK3. Большинство изученных до настоящего времени препаратов имеют несколько типов рецепторов, например препараты из подвздошной кишки морской свинки содержат NK1, NK2 и NK3, некоторые препараты имеют только один тип рецепторов, например препараты из сонной артерии собаки содержат NK1, из лишенной эндотелия легочной артерии кролика содержит NK2 и из воротной вены крысы содержит NK3 (D. Regoli et al. Trends Pharmacol. Sci. 1988, 9, 290-295 и Pharmacology, 1989, 38, 1-15).
Более точное описание различных рецепторов стало возможным благодаря синтезу селективных агонистов. Так, [Sar9, Met-(O2)11SP, [NIe10 ]NKA4-10 и Me Phe7 -NKB обладают селективностью соответственно для рецепторов NK1, NK2 и NK3 (см. работу D. Regoli 1988 и 1989).
В настоящее время было обнаружено, что новые полициклические аминосодержащие соединения обладают ценными фармакологическими свойствами, в частности антагонистической активностью к рецепторам нейрокининов, и могут быть пригодны для лечения любой патологии, зависимой от вещества P и нейрокинина.
Таким образом,
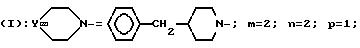
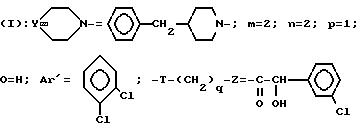
изобретение относится к полициклическим аминосодержащим
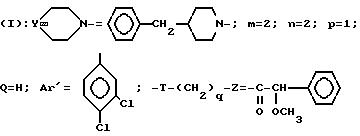
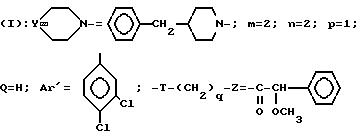
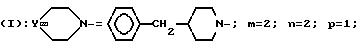
соединениям формулы
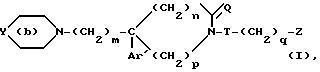
где
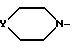
Y представляет собой
Ar представляет собой фенил,
x равно 0 или 1,
X представляет собой водород, гидроксил, ацетоксигруппу или ациламиногруппу,
m равно 2 или 3,
Ar' представляет собой фенил, замещенный метилом или двумя атомами галогена, или нафтил,
n равняется 0, 1, 2 или 3,
p равняется 1 или 2 при условии, что когда p равно 2, то n равно 1 и Q является двумя атомами водорода,
Q представляет собой кислород или два атома водорода,
T представляет собой группу, выбранную из -C(O)- и -CH2-,
q равно 0 или 1,
Z представляет собой фенил, который может быть одно- или дизамещен C1 -C4-алкоксигруппой или галогеном, или моногалогензамещенный нафтил, при условии, что когда T является группой -C(O)-, то -(CH2)q-Z может представлять собой бензильную группу, которая замещена в алифатическом остатке гидроксилом, C1-C4-алкоксигруппой или C1-C4 алкилом и, возможно, замещена на ароматическом цикле галогеном или C1-C4-алкоксигруппой или к их солям с минеральными или органическими кислотами или четвертичным солям аммония или N-оксидным производным по азоту (b) пиперидина.
Соли соединения формулы (l) включают как соли с минеральными или органическими кислотами, которые позволяют осуществить подходящие разделение или кристаллизацию соединений формулы (l), с такими как пикриновая кислота или щавелевая кислота, или оптически активная кислота, например, миндальная или камфосульфоновая кислота, так и соли с кислотами, которые образуют фармацевтически приемлемые соли, такие как хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метаносульфонат, метилсульфанат, малеат, фумарат, 2-нафталинсульфонат, гликолят, глюконат, цитрат, изатионат.
Когда
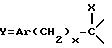
В соответствии
с другим аспектом изобретение относится к способу получения
соединений формулы (l) и их солей, отличающемуся тем, что
а) соединение
общей формулы
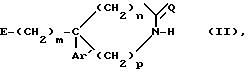
в которой
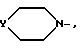
m, Ar, n, p и Q являются такими, как определено выше, а E представляет собой гидроксил или, возможно, O-защищенную группу, такую как, например, тетрагидропиранил-2-оксигруппа, или группу
в которой
Y представляет собой группу
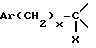
обрабатывают
либо функциональным производным кислоты общей формулы
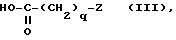
в которой
q и z являются такими, как определено выше, если хотят получить соединения формулы (l), где T является группой -CO-;
либо галогенсодержащим производным общей формулы
Hal-(CH2)q+1-Z (lV)
в которой
q и Z являются такими, как определено выше, и Hal представляет собой галоген, предпочтительно атом брома или хрома, если хотят получить соединения формулы (l), где T является группой -CH2-;
получают соединение общей формулы
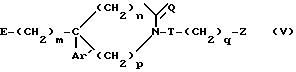
b) затем, когда E представляет собой тетрагидропиранилоксигруппу, отщепляют тетрагидропиранилогсигруппу под действием кислоты;
c) полученный таким образом спирт общей формулы
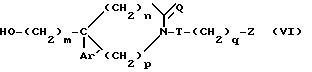
обрабатывают метансульфонилхлоридом;
d) полученный мезилат общей формулы
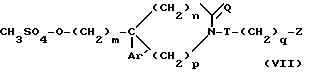
обрабатывают вторичным амином формулы
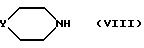
в которой
Y является таким, как определено выше;
e) после возможного снятия защиты с гидроксила X превращают в случае необходимости полученный таким образом продукт в одну из его солей с минеральной или органической кислотой, или в соль четвертичного аммония, или в N-оксидное производное по азоту (в) пиперидина.
Соли четвертичного аммония получают в
результате реакции соединений l в виде свободных оснований, в которых другие аминофункции, возможно
присутствующие,
являются N-защищенными любой N-защищающей группой, с избытком алкилирующего агента
формулы
A Q'
в которой A представляет собой удаляемую группу и является такой, как
хлорид, бромид,
иодид, метансульфонат или паратолуолсульфонат, предпочтительно хлорид или иодид, а
Q' является алкилом C1-C6 или бензилом,
затем нагревают реакционную
смесь в
растворителе, выбираемом, например, среди дихлорметана, хлороформа, ацетона или ацетонитрила
при температуре, заключенной между температурой окружающей среды и температурой флегмы, в течение
времени
одного до нескольких часов, чтобы получить после обработки в соответствии с обычными методами
и после возможного снятия защиты смесь аксиальных и экваториальных диастереоизомеров солей
четвертичного
аммония.
A⊖ представляет собой предпочтительно иодид,
который может быть заменен другим анионом или фармакологически приемлемым анионом, например
хлоридом, в
результате элюирования соединений (l) через ионообменную смолу, например, Амберлит IPA68® или Дуолит A375®
Диастереоизомеры разделяются в
соответствии с
обычными методами, например в результате хроматографии или в результате перекристаллизации.
N-оксидные производные получаются путем взаимодействия с пероксидным производным, например метахлорпербензойной кислотой или перекисью водорода, в соответствии с обычными методами.
В качестве функционального производного кислоты (III) используют саму кислоту, активированную подходящим образом, например, циклогексилкарбодиимидом или гексафторфосфатом бензотриазолил-N-окситрисдиметиламинофосфония (БОФ), или же одно из функциональных производных, которые реагируют с аминами, например ангидрид, смешанный ангидрид, хлорангидрид кислоты или активированный сложный эфир.
Если в качестве исходного продукта используют соединение
формулы (II), где E
представляет собой группу
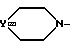
то способ по изобретению может быть представлен и детально проиллюстрирован по схеме 1.
Схема 1.
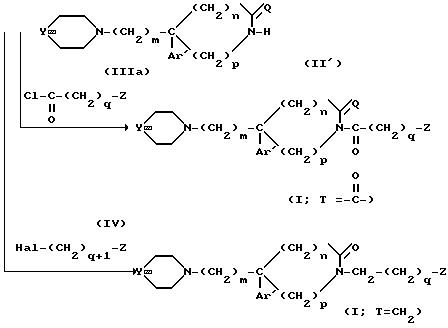
В формуле (IIIa) хлорангидрид кислоты рассматривают в качестве реакционноспособного функционального производного кислоты (III). Однако можно использовать другое функциональное производное или можно исходить из свободной кислоты (III), осуществляя взаимодействие соединения (II') с БОФ, затем прибавляя кислоту (III) в присутствии органического основания, такого как, например, триэтиламин, в растворителе, таком как дихлорметан или диметилформамид, при температуре окружающей среды. Полученные соединения (I) выделяются и очищаются в соответствии с обычными методами, такими как, например, хроматография или перекристаллизация.
Если в качестве исходного продукта используют соединение формулы (II), где E представляет собой тетрагидропиранилоксигруппу (ТГП-O-), то способ по изобретению может быть изображен и проиллюстрирован на схеме 2.
Реакция соединения (II) с реагентами (IIIa) и (IV) протекают, как описано выше, причем хлорангидрид кислоты (IIIa) может быть заменен другим функциональным производным или свободной кислотой, активированной, например, посредством БОФ.
Полученное таким образом промежуточное соединение (V) подвергают снятию защиты путем мягкого кислотного гидролиза, что приводит к свободному гидроксилсодержащему соединению (VI), из которого получают мезилат (VII), затем обрабатывают его вторичным амином формулы (VIII) и получают окончательно соединения (I) согласно изобретению.
Схема 2.
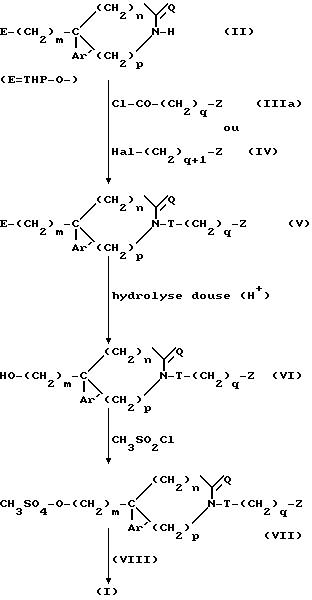
Полученные таким образом соединения формулы (I) выделяют в виде свободного основания или соли в соответствии с классическими методами.
Когда соединение формулы (I) получается в виде свободного основания, то солеобразование осуществляется в результате обработки выбранной кислотой в органическом растворителе. В результате обработки свободного основания, растворенного, например, в спирте, таком как изопропанол, раствором выбранной кислоты в том же самом растворителе, получают соответствующую соль, которая выделяется при помощи классических методов. Так получают, например, хлоргидрат, бромгидрат, сульфат, гидросульфат, дигидрофосфат, метансульфонат, оксалат, малеат, фумарат, 2-нафталинсульфонат.
По окончании реакции соединения формулы (I) могут выделяться в виде одной из их солей, например хлоргидрата или оксалата; в этом случае, если это необходимо, свободное основание может быть получено в результате нейтрализации указанной соли минеральным или органическим основанием, таким как гидроксид натрия или триэталамин, или карбонатом или бикарбонатом щелочного металла, таким как карбонат или бикарбонат натрия или калия.
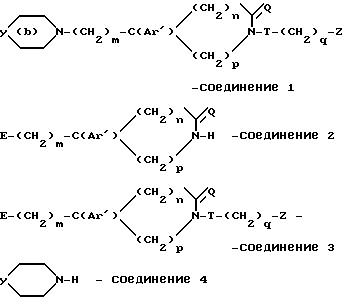
Исходные соединения с формулой (II) получаются, исходя из нитрилов, коммерческих или полученных в соответствии с известными методами.
Разделение рацемических смесей (I)
позволяет выделять энантиомеры (I*) формулы
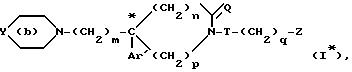
в которой
"*" означает, что атом углерода, отмеченный таким образом, имеет определенную абсолютную конфигурацию (+) или (-);
Y, m, Az', n, p, Q, T, q и z являются такими, как определено выше для производных формулы (l); или одну из их солей с минеральными или органическими кислотами или образованные с атомом азота (в) одну из солей четвертичного аммония или N-оксидные производные.
Указанные соли или N-оксидные производные получаются, как указано выше для солей и производных, соответствующих производным формулы (I).
Энантиомеры формулы (l*) являются новыми продуктами, которые составляют часть изобретения.
Можно также осуществлять разделение путем фракционной кристаллизации рацемических смесей продуктов формулы (II), в которых m, Az', n и p являются такими, как определено для соединений (l), E представляет собой гидроксил, а O представляет собой водород, с целью получить знантиомеры (l*) продуктов формулы (I).
Разделение рацематов осуществляют на промежуточных соединениях (II), способных давать соли с оптически активными кислотами. В таком случае знантиомеры разделяют при помощи классических методов, таких как кристаллизация пли препаративная хиральная хроматография высокого давления.
Полученный таким образом оптически чистый аминоспирт является новым соединением общей
формулы
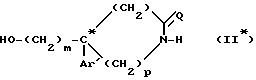
в которой
"*" означает, что атом углерода, отмеченный таким образом, имеет определенную конфигурацию (+) или (-).
Соединения по изобретению были объектом биохимических испытаний.
Соединение (l) и его соли проявили антагонистические свойства в отношении связывания вещества P в испытаниях, проведенных на оболочках коры головного мозга крыс и лимфобластических клетках IM9 в соответствии с работами M. A. Cascieri и comp. j. Biol. Chem. 1983, 258, 5158-5164 и D.D. Paya и comp. j. Immunol. 1984, 133, 3260-3265.
Те же самые соединения и их соли проявили антагонистические свойства по отношению к связыванию NKA в опытах, проведенных на оболочках двенадцатиперстной кишки крыс в соответствии с работой L. Bergstom и comp. Md. Pharmacol. 1987, 32, 764-771.
Те же самые соединения и их соли проявили антагонистические свойства по отношению к связыванию эледойзина в опытах, проведенных на оболочках крыс в соответствии с A.C. Foster и comp. Br. j. Pharmacol. 1988, 94, 602-608.
Эледойзин представляет собой пептид, выделенный из земноводных, который эквивалентен нейрокинину B.
Соединения в соответствии с изобретением являются антагонистами вещества P, нейрокинина A или нейрокинина B.
Так, соединение 2 из примера 2 проявляет антагонизм к связыванию вещества P при Ki, равном 8,3 нмоля, соединение 7 из примера 7 проявляет антагонизм к связыванию нейрокинина A при Ki, равном 1,3 нмоля, и соединение 3 из примера 3 проявляет антагонизм к связыванию элейдозина при Ki, равном 200 нмолей.
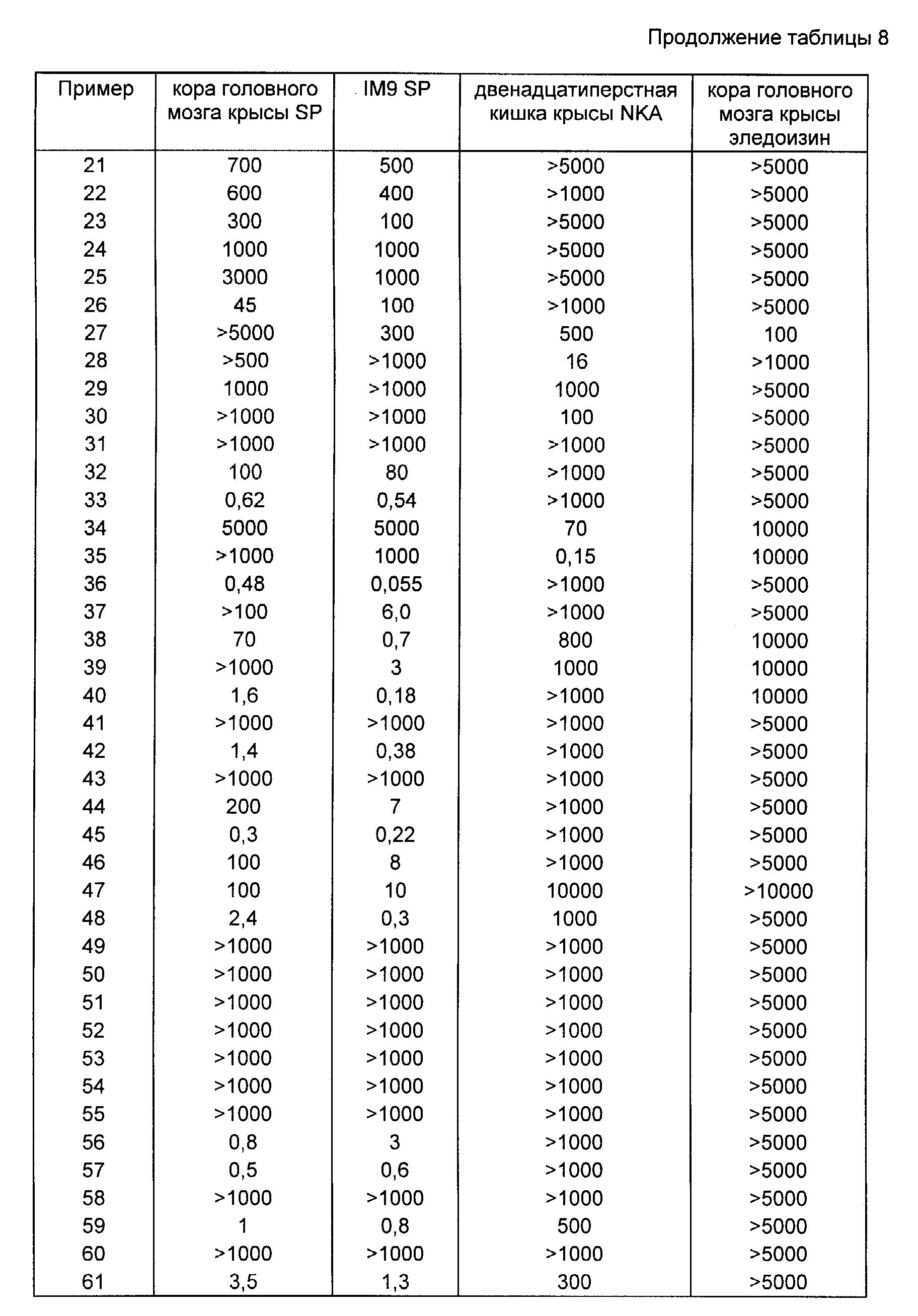
Результаты испытаний соединений, проведенных в соответствии с указанными методиками, сведены в табл. 8.
Соединения по изобретению обычно вводятся в единичных дозах в виде фармацевтических композиций, в которых активный компонент смешан с фармацевтическим индифферентным веществом.
Таким образом, изобретение относится также к фармацевтическим композициям, содержащим в качестве активного компонента соединение формулы (l) или одну из его фармацевтически приемлемых солей.
В фармацевтических композициях для орального, подъязычного, подкожного, внутримышечного, трансдермического, местного или ректального введения активные компоненты могут находиться в единичных дозах в смеси с классическими фармацевтическими носителями. Единичные дозы соответствуют различным формам, таким как таблетки, эелатиновые капсулы, порошки, гранулы для орального введения и оральные растворы или суспензии, формам подъязычного и защечного введения, формам для подкожного, внутримышечного, внутривенного, интраназального или интраокулярного введения и формам для ректального введения.
Когда приготавливают твердую композицию в виде таблеток, то смешивают основной активный компонент с фармацевтическим носителем, таким как желатина, крахмал, лактоза, стеарат магния, тальк, гуммиарабик или аналогичные. Можно покрыть таблетки сахарозой или другими соответствующими веществами или можно их обрабатывать таким образом, чтобы они имели пролангированную активность или наоборот, запаздывающую, и чтобы они непрерывно высвобождали предопределенное количество активного компонента.
Препарат в желатиновых капсулах получают, смешивая активный компонент с разбавителем и заливая полученную смесь в мягкие или твердые желатиновые капсулы.
Препарат в виде сиропа или элексира может содержать активный компонент одновременно с подсластителем, предпочтительно бескалорийным, метилпарабеном и пропилпарабеном в качестве антисептиков, а также с придающим вкус веществом и с соответствующим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный компонент в смеси с диспергирующими агентами или смачивающими агентами или агентами для перевода в суспензию, как, например, поливинилпирролидон, а также с подсластителем или с корректорами вкуса.
Для ректального введения используют свечи, которые приготавливаются со связующими, плавящимися при ректальной температуре, например масло какао или полиэтиленгликоли.
Для парентерального, интраназального введения используют водные суспензии, солевые изотонические растворы или стерильные и инъекционные растворы, которые содержат фармокологически совместимые диспергирующие и/или смачивающие агенты, например пропилиенгликоль или бутиленгликоль.
Для введения путем ингаляции используют аэрозоль, содержащую, например, триолеат сорбитана или олеиновую кислоту, а также трихлорфторметан, дихлорфторметан, дихлортетрафторэтан или любой биологически совместимый газ, служащий для выталкивания.
Активный компонент может также вводиться в рецептуру в виде микрокапсул, в случае необходимости, с одним или несколькими носителями или дополнительными веществами.
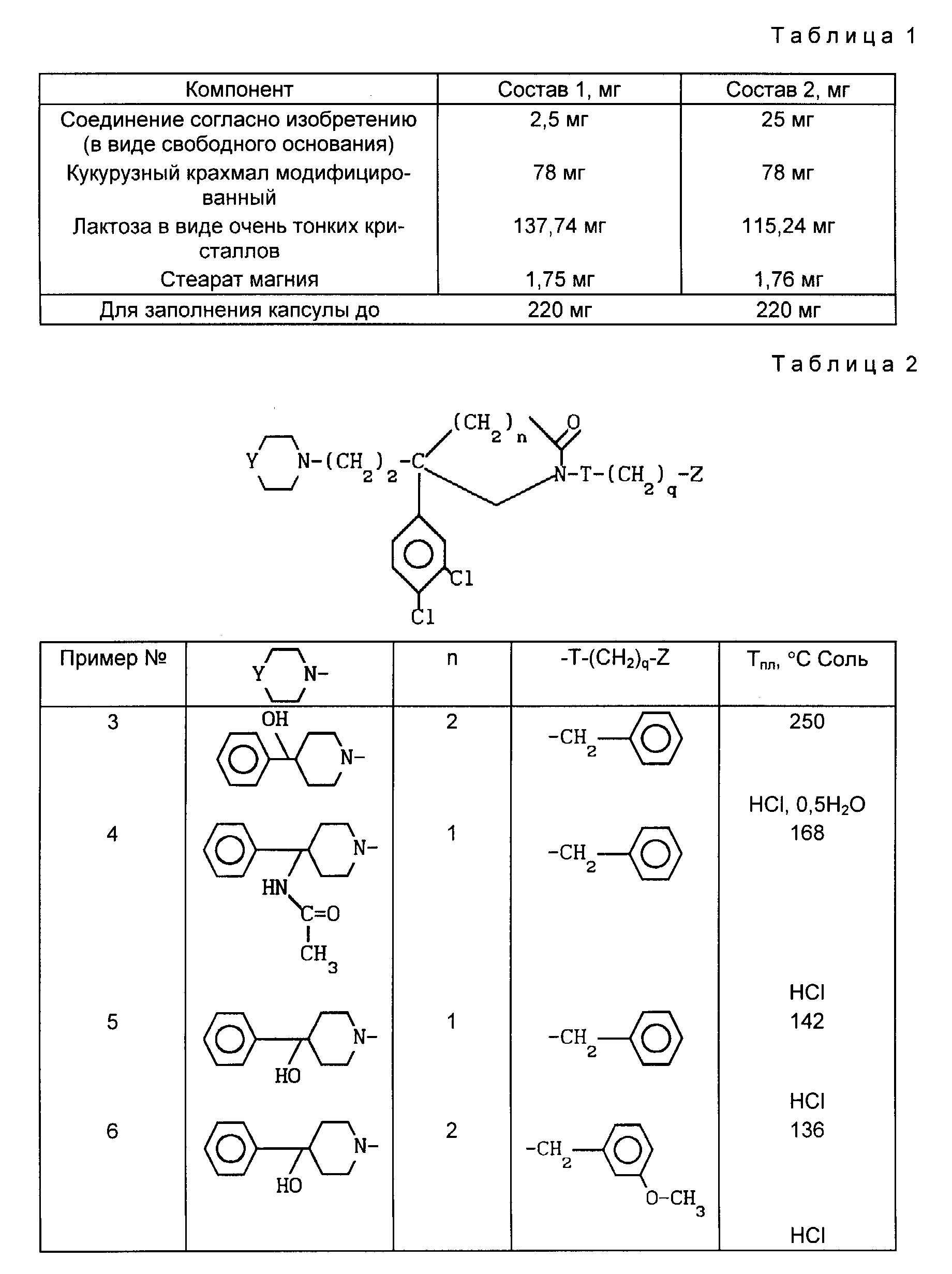
Приведены примеры рецептур фармкомпозиций в виде желатиновых капсул.
1. Желатиновая капсула весом 220 мг представлена в табл. 1.
2. Желатиновая капсула с оксидом весом 200 мг, мг:
Соединение формулы I (в форме основания) 0,25
Кукурузный крахмал 70,0
Лактоза
в виде экстратонких кристаллов 128,0
Стеарат магния 1,75
При определении
токсичности новых соединений получены следующие данные:
DL50 на мыши и крысе составляет
более 2 г/кг.
Соединения согласно изобретению не оказывают токсического эффекта на собаку при дозе 20 мг/кг и на крысу при дозе 30 мг/кг, вводимых в течение 4 недель.
Тесты на мутагенез отрицательные, в частности тест Амеса. Следовательно, соединения согласно изобретению можно отнести к нетоксичным или малотоксичным.
Последующие примеры иллюстрируют изобретение, не ограничивая его.
Температуры плавления и разложения продуктов (Tпл) были измерены на нагревающей установке Коффлера. Спектры ядерного магнитного резонанса13C были получены при 50 МГц в диметилсульфоксиде.
Пример 1. Хлоргидрат 5-2-4(4-бензил-1-пиперидинил)-этил-5-(3,
4-дихлорфенил)-1-бензилпиперидинона
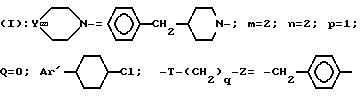
A/ 3,4-Дихлортетрагидропиранилоксиэтил- α -бензолацетонитрил.
Приготавливают суспензию из 20 г гидрида натрия, находящегося в жидком масле с концентрацией 55-60% и 200 мл безводного тетрагидрофурана. При 20oC прибавляют по капле за 30 мин раствор, содержащий 85 г 3, 4-дихлорфенилацетонитрила в 500 мл тетрагидрофурана, затем перемешивают реакционную смесь при температуре окружающей среды в течение 2 ч. Смесь охлаждают до -20oC и прибавляют раствор, содержащий 98 г 2-бромэтокситетрагидропирана в 100 мл тетрагидрофурана, дают смеси вернуться к температуре окружающей среды и спустя 2 ч прибавляют раствор, содержащий 50 г хлорида аммония в 3 л воды. Экстрагируют посредством 1,5 л эфира, промывают насыщенным раствором хлорида натрия, декантируют, сушат на MgSO4 и концентрируют под вакуумом.
Остаток хроматографируется на силикагеле, элюент дихлорметан. Фракции чистого продукта концентрируются под вакуумом, что дает 83,6 г жидкого масла.
B/ g Тетрагидропиранилоксиэтил g- циано-3, 4-дихлорбензилбутаноат этила.
В 100 мл тетрагидрофурана растворяют 21 г нитрила, полученного выше на стадии (A), затем прибавляют по капле при температуре окружающей среды раствор, содержащий 0,067 моль диизопропиламида лития в 100 мл тетрагидрофурана, и перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды. Затем прибавляют 12 г бромпропионата этила и нагревают при 50oC в течение 2 ч. Смесь охлаждают, ее приливают к насыщенному раствору хлорида аммония и экстрагируют эфиром, промывают водой, отделяют эфирную фазу путем декантации, сушат над Na2SO4 и концентрируют под вакуумом. Остаток очищается методом хроматографии на силикагеле, элюент дихлорметан/этилацетат 100/1 (об/об). Концентрирование чистых фракций дает 13 г ожидаемого продукта.
C/ 5-Тетрагидропиранилоксиэтил-5-(3,4-дихлорфенил)-пиперидинон.
Растворяют 13 г полученного перед этим соединения в 250 мл этанола и 40 мл гидрата окиси аммония, затем гидрируют при температуре окружающей среды и под атмосферным давлением в присутствии никеля Ренея. Когда поглощается теоретически рассчитанный объем водорода, фильтруют смесь на целите и концентрируют фильтрат под вакуумом. Остаток поглощают водой, экстрагируют эфиром, затем промывают эфирную фазу водой, сушат над MgSO4 и концентрируют под вакуумом.
m 9 г.
D/ 5-Тетрагидропиранилоксиэтил-5-(3,4-дихлорфенил)-1-бензилпиперидинон.
Прибавляют 2,05 г бензилбромида к раствору, содержащему 4,5 г полученного выше продукта в 60 мл диметилформамида в присутствии 0,3 г гидрида натрия. Реакционную смесь нагревают при 40-50oC, в течение 2 ч концентрируют под вакуумом. Остаток поглощают водой, экстрагируют эфиром, а эфирную фазу промывают водой, сушат над MgSO4 и концентрируют под вакуумом.
Остаток хроматографируют на силикагеле, элюент диметанол 100/1 (об/об).
Фракции чистого продукта концентрируют под вакуумом.
m 2 г.
E/ 5-Метансульфонилоксиэтил-5(3,4-дихлорфенил)-1-бензилпиперидинон.
Растворяют 2 г полученного выше продукта в 40 мл метанола, насыщенного газообразной хлороводородной кислотой, и перемешивают раствор в течение двух часов при температуре окружающей среды. Растворители концентрируют под вакуумом, а остаток поглощают смесью пентан/эфир-50/50, затем отфильтровывают осадок. Осадок растворяют в 50 мл дихлорметана и прибавляют 0,4 г триэтиламина и 0,45 г мезилхлорида, затем перемешивают смесь в течение получаса при температуре окружающей среды. Концентрируют под вакуумом, извлекают остаток в воде, экстрагируют эфиром, промывают эфирную фазу водой, декантируют, сушат над MgSO4 и концентрируют под вакуумом.
m=1,6 г.
K/ Соединение 1.
Растворяют 0,68 г полученного выше продукта и 0,63 г 4-бенозилпиперидина в 2 мл диметилформамида и нагревают смесь при 80oC в течение 2 ч. Охлаждают раствор, приливают к воде, экстрагируют этилацетатом, декантируют органическую фазу, сушат над MgSO4 и концентрируют под вакуумом. Остаток очищается методом хроматографии на силикагеле, элюент дихлорметан/метанол 100/3 (об/об).
Фракции чистого продукта концентрируются под вакуумом, затем получают хлоргидрат, который затвердевает в смеси эфир/пентан 50/50.
m 0, 25 г.
Tпл 115oC.
Пример 2. Хлоргидрат
3-[2(4-бензил-1-пиперидинил)-этил]-3-(3,4-дихлорфенил)-1-фенилацетилпиперидина
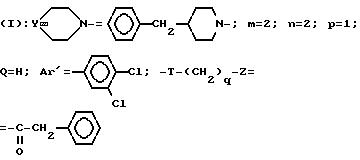
A/ 3-Тетрагидропиранилоксиэтил-3-(3,4-дихлорфенил)-пиперидин.
Растворяют в 50 мл тетрагидрофурана 4,5 г 5-тетрагидропиранилоксиэтил-5-(3,4-дихлорфенил)-пиперидинона, полученного в соответствии с примером 1C, и прибавляют раствор к суспензии, содержащей 0,9 г алюмогидрида лития, которая нагрета до 60oC. Реакционную смесь нагревают в течение 1 ч при 60oC, затем охлаждают. Прибавляют 1 мл воды, 1 мл 4н гидроксида натрия и 3 мл воды. Отделяют минеральный осадок путем фильтрования и концентрируют фильтрат под вакуумом. Остаток извлекают эфиром, сушат над MgSO4 и концентрируют под вакуумом, что дает 3,5 г целевого продукта.
B/ 3-Тетрагидропиранилоксиэтил-3(3,4-дихлорфенил)-1- фенилацетилпиперидин.
Прибавляют 0,75 г хлорангидрида фенилуксусной кислоты к раствору, содержащему 1,7 г полученного перед этим продукта и 0,9 г триэтиламина в 50 мл дихлорметана. Реакционную смесь перемешивают в течение 1 ч при температуре окружающей среды и концентрируют под вакуумом. Остаток извлекают этилацетатом, затем промывают водой, органическую фазу сушат над MgSO4 и концентрируют под вакуумом. Остаток очищают методом хроматографии на силикагеле, элюент дихлорметан/метанол 100/0,5 (об/об).
Концентрирование чистых фракций дает 1 г целевого продукта.
C/ 3-Метансульфонилоксиэтил-3(3,4-дихлорфенил)-1- фенилацетилпиперидин.
Растворяют 0,8 г полученного выше продукта в 40 мл метанола, насыщенного хлороводородной кислотой, и перемешивают смесь в течение 0,5 ч при температуре окружающей среды. Концентрируют под вакуумом и извлекают остаток 40 мл дихлорметана. Прибавляют 0,4 г триэтиламина и 0,23 г мезилхлорида и перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды, затем концентрируют под вакуумом. Остаток извлекают этилацетатом, промывают водой, отделяют органическую фазу путем декантации, сушат над MgSO4 и концентрируют под вакуумом.
m 0,71 г.
I/ Соединение 2.
В течение 3 ч нагревают при 80oC 0,7 г полученного перед этим продукта и 0,52 г 4-бензилпиперидина, растворенных в 2 мл диметилформамида. Охлаждают реакционную смесь, приливают ее в воде, экстрагируют эфиром, промывают эфирную фазу водой, сушат над MgSO4 и концентрируют под вакуумом, затем перекристаллизовывают хлоргидрат в смеси дихлорметан/эфир.
m=0,12 г.
Tпл 210-212o C.
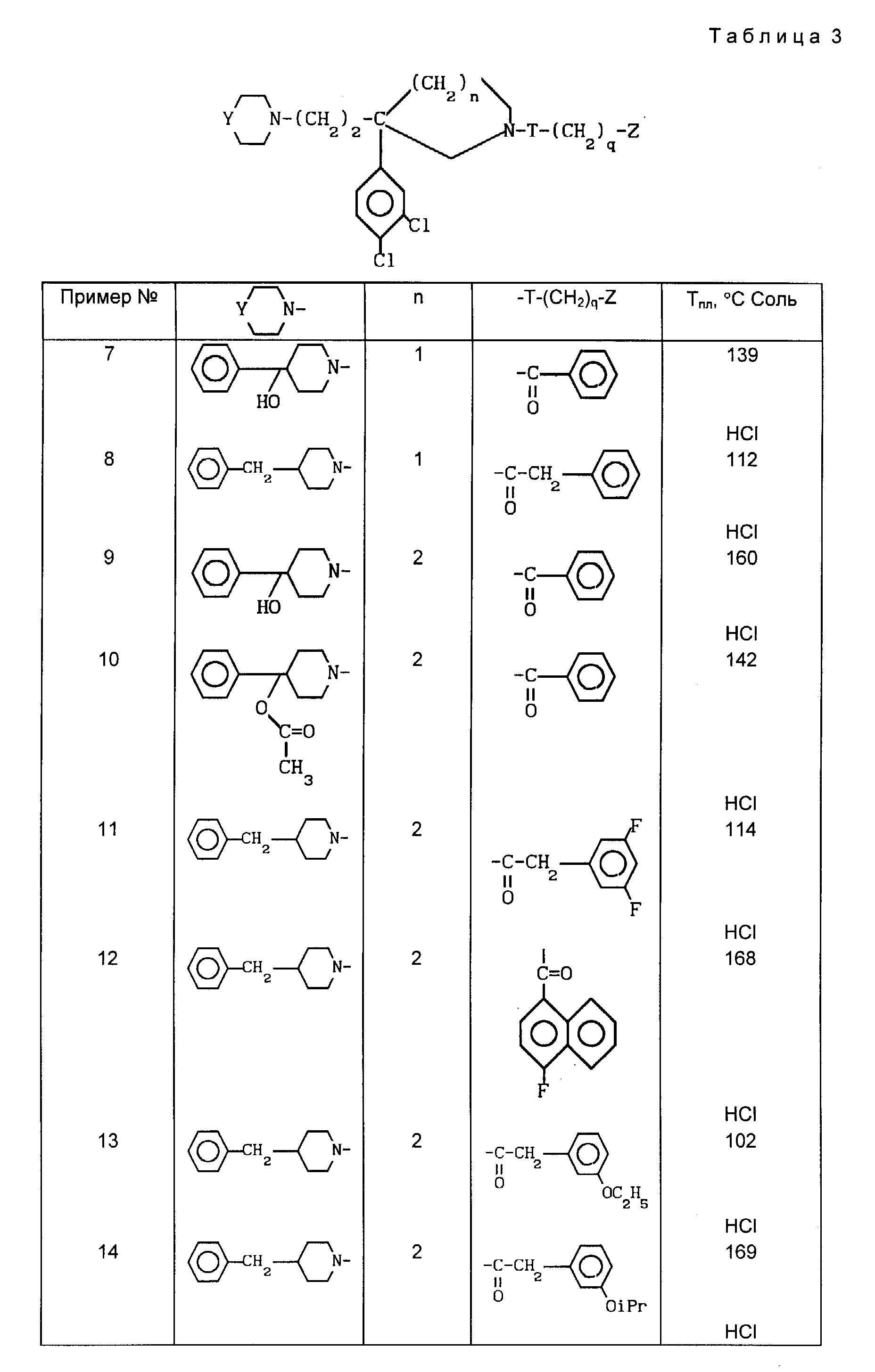
Проводя синтезы в соответствии с примером 1, получают соединения 3-6, описанные в табл. 2
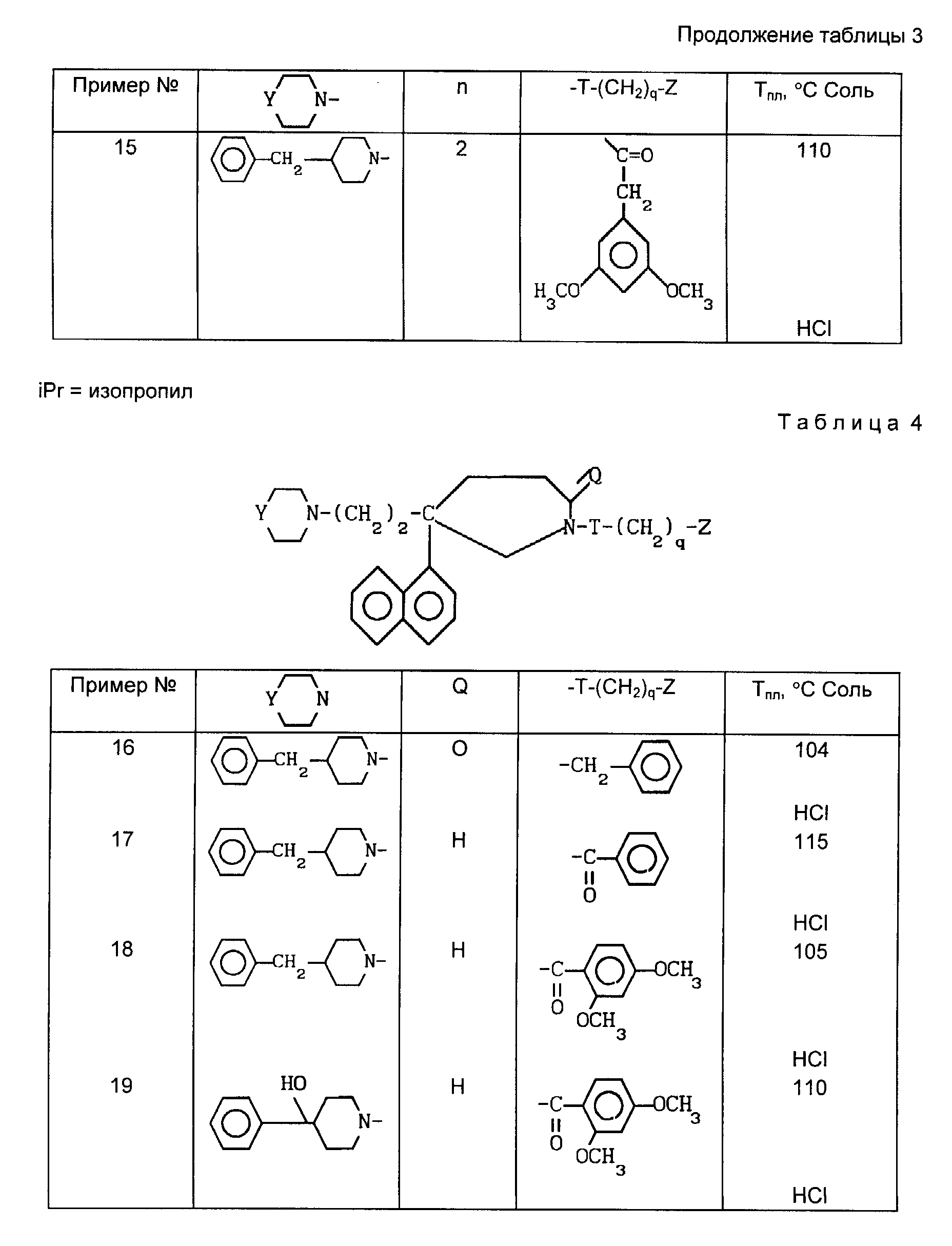
Проводя
синтезы в соответствии с примером 2, получают соединения 7-15, описанные
в табл. 3, приведенной ниже.
Проводя синтезы в соответствии с приведенными выше примерами 1 и 2, но заменяя в них 3,4-дихлорфенилацетонитрил на α -нафтилацетонитрил, получают соединения 16-19, описанные в табл. 4.
Пример 20. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]- 3-(3,
4-дихлорфенил)-1-(3-изопропоксифенил)-ацетилазепина
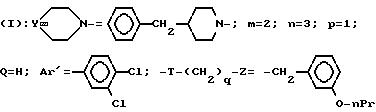
A/ δ -Тетрагидропиранилоксиэтил- d -циано-3,4-дихлорбензилпентаноат этила.
К 36 г 3,4-дихлор- a -тетрагидропиранилоксиэтилбензол-ацетонитрила (полученного в соответствии со стадией A примера 1), растворенного в 100 мл диметилформамида, прибавляют небольшими порциями 4,6 г NaH с концентрацией 60% Перемешивают реакционную смесь в течение 3 ч при температуре окружающей среды, охлаждают до 0oC, затем прибавляют 22,4 г 4-бромбутирата этила, растворенного в 40 мл диметилформамида. Перемешивают реакционную смесь в течение 3 ч при температуре окружающей среды, приливают к воде, экстрагируют эфиром, промывают насыщенным раствором NaCl, сушат над Na2 SO4 и концентрируют под вакуумом. Очищают полученный остаток методом хроматографии на силикагеле, элюент толуол.
m 24 г.
B/ 6-Титрагидропиранилоксиэтил-6-(3, 4-дихлорфенил)-азепинон.
Гидрируют 8 г полученного перед этим продуктом под атмосферным давлением и при температуре окружающей среды в присутствии никеля Ренея в растворе, образованном посредством 120 мл этанола.
Когда поглощается теоретически рассчитанный объем водорода, то отфильтровывают катализатор и концентрируют под вакуумом.
Полученное масло поглощают затем 20 мл ксилола и реакционную смесь нагревают в течение 48 ч при температуре образования флегмы. Выпаривают и очищают полученный остаток методом хроматографии на силикагеле, элюент дихлорметан/метанол- 100/1 (об/об).
Получают таким образом 4 г жидкого масла.
C/ 3 Тетрагидропиранилоксиэтил-3-(3,4 дихлорфенил)-азепин.
Исходя из 2 г полученного перед этим продукта, 0,49 г алюмогидрида лития и проводя синтез в соответствии со стадией А примера 2, получают 1,7 г ожидаемого продукта в виде жидкого масла.
D/ 3-Тетрагидропиранилоксиэтил-3-(3,4-дихлорфенил)-1- изопропоксифенил)-ацетилазепин.
Исходя из 1,7 г полученного перед этим продукта и приводя синтез в соответствии со стадией B примера 2, получают 1,7 г ожидаемого продукта.
E/ 3-Метансульфонилоксиэтил-3-(3, 4-дихлорфенил)-1-(3- изопропоксифенил)-ацетилазепин.
Исходя из 1,7 г полученного перед этим продукта и 0,34 г мезилхлорида и приводя синтез в соответствии со стадией C примера 2, получают 1,5 г ожидаемого продукта.
K/ Соединение 20.
В течение 2 ч нагревают при 80oC 1,5 г полученного перед этим продукта и 1,4 г 4-бензин пиперидина, растворенных в 3 мл диметилформамида. Охлаждают, приливают реакционную смесь к воде, экстрагируют эфиром, промывают ограниченную фазу водой, сушат над Na2SO4 и концентрируют под вакуумом.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/2 (об/об). Концентрируют чистые фракции и получают хлоргидрат в изопропиловом эфире, фильтруют, промывают эфиром и сушат под вакуумом. Получают таким образом 1,3 г ожидаемого продукта.
Tпл 164o C.
Пример 21. Хлоргидрат
3-[2-(4-бензин-1-пиперидинил)-этил]-3(3, 4-дихлорфенил)-1-(3-метоксифенил)-ацетилазетидин
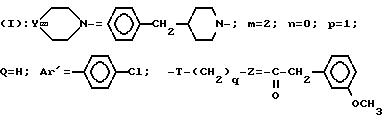
A/ 3-Тетрагидропиранилоксиэтил-3-(3,4-дихлорфенил)-1 -(3-метоксифенил)-ацетилазетидин.
К 1 г 3-тетрагидропиранилоксиэтил-3-(3, 4-дихлорфенил)- азетидина, растворенного в 50 мл дихлорметана в присутствии 1 г триэтиламина и 0,5 г 3-метоксифенилуксусной кислоты, прибавляют 1,5 г БОФ. Перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды, выпаривают досуха, извлекают остаток этилацетатом, промывают водой, разбавленным гидроксидом натрия, буферным раствором с pH 2 и окончательно водой, насыщенной NaCl. Сушат органическую фазу над MgSO4 и выпаривают досуха. Очищают методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH - 100/0,75 (об/об).
Получают таким образом 0,50 г жидкого масла.
B/ 3-Метансульфонилоксиэтил-3-(3,4- дихлорфенил)-1-(3-метоксифенил)-ацетилазетидин.
К 0,50 г полученного перед этим продукта, растворенного в 50 мл метанола, прибавляют эфир, насыщенный хлороводородной кислотой до значения pH 1. Перемешивают раствор при температуре окружающей среды в течение 1 ч, выпаривают досуха, извлекают остаток в воде, экстрагируют посредством AcOEt, промывают водой, сушат на MgSO4 и выпаривают досуха.
Полученное жидкое масло поглощают 30 мл дихлорметана и прибавляют 0,20 г триэтиламина и 0,12 г мезилхлорида. Перемешивают реакционную смесь при температуре окружающей среды в течение 1 ч, выпаривают досуха, извлекают остаток в этилацетате, промывают водой, сушат на MgSO4 и выпаривают досуха.
Получают таким образом 0,50 г жидкого масла.
C/ Соединение 21.
В течение 3 ч нагревают при 80oC 0,50 г описанного перед этим продукта, растворенного в 2 мл диметилформамида с 0,40 г 4-бензилпиперидина. Охлаждают реакционную смесь, приливают к воде, экстрагируют этилцетатом, промывают водой, сушат над MgSO4 и выпаривают досуха.
Очищают полученный таким образом остаток методом хромаграфии на силикагеле, элюент CH2Cl2/CH3OH 100/2,5 (об/об).
Концентрируют чистые фракции под вакуумом и получают хлоргидрат путем прибавления эфира, насыщенного хлороводородной кислотой. Извлекают остаток в дихлорметане и осаждают хлоргидрат в эфире, фильтруют, промывают эфиром и сушат под вакуумом.
Получают таким образом 0,22 г ожидаемого продукта.
Тпл 102oC.
Пример
22. Хлоргидрат 4-[2-(4-бензин-1-пиперидинил)-этил]-4 -(3-метилфенил)-1-(3-хлорфенил)-ацетилпиперидина
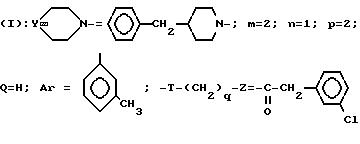
A/ 4-Метансульфонилоксиэтил-4-(3-метилфенил)-N- тритилпиперидин.
К 21 г 4-(2-гидроксиэтил)-4-(3-метилфенил)-N- тритилпиперидина, растворенного в 200 мл дихлорметана и охлажденного до 0oC, прибавляют по капле 3,8 мл метансульфонилхлорида. Реакционную смесь оставляют в течение 0,5 ч при температуре окружающей среды, промывают дважды водой, сушат над MgSO4 и концентрируют под вакуумом.
Получают таким образом 23,5 г пенообразного вещества.
B/ 4-[2-(4-бензин-1-пиперидинил)-этил]
-4-4(3-метилфенил)-N- тритилпиперидин
В течение 4 ч нагревают при 60oC 18,5 г описанного выше мезилата и 13,5 г 4-бензилпиперидина, растворенных в 40 мл диметилформамида.
Прилагают
реакционную смесь к 500 мл ледяной воды, отфильтровывают осадок и промывают водой. Поглощают осадок эфиром, промывают разбавленным NaOH, затем водой, сушат над MgSO4 и
концентрируют
досуха.
Очищают полученный остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/3 (об/об).
Получают таким образом 18 г пенообразного вещества.
C/ Дихлоргидрат 4-[2-(4-бензин-1-пиперидинил)-этил]-4-(3-метилфенил)-пиперидина.
В течение 30 мин нагревают при 60oC 18 г полученного выше продукта, растворенного в 150 мл 50% -ной муравьиной кислоты. Охлаждают, отделяют трифенилкарбонил в результате фильтрования, промывают водой, концентрируют досуха. Остаток поглощают водой, промывают эфиром, подщелачивают раствором NaOH, экстрагируют дихлорметаном, сушат над MgSO4 и концентрируют досуха.
Основание растворяют в дихлорметане, добавляют эфир, насыщенный хлороводородной кислотой, и концентрируют досуха. Перемешивают полученный таким образом хлоргидрат в эфире, отфильтровывают и сушат.
m 12,7
г
Tпл
160oC.
D/ Соединение 22.
К 2 г полученного выше продукта, растворенного в 30 мл дихлорметана с 0,77 3-хлорфенилуксусной кислоты и 2,2 г триэтиламина, прибавляют 2,4 г БОФ. Перемешивают реакционную смесь в течение 30 мин при температуре окружающей среды, концентрируют досуха, поглощают остаток этилацетатом, промывают водой, затем разбавленным раствором NaOH, потом водой, насыщенной NaCl, сушат над MgSO4 и концентрируют под вакуумом. Очищают остаток методом хроматографии на силикагеле, элюент: CH2 Cl2/CH3OH - 100/10 (об/об). Получают хлоргидрат путем прибавления эфира, насыщенного хлороводородной кислоты, и концентрируют досуха. Поглощают остаток изопропиловым эфиром, фильтруют и сушат под вакуумом.
m 2,1 г
T 106oC.
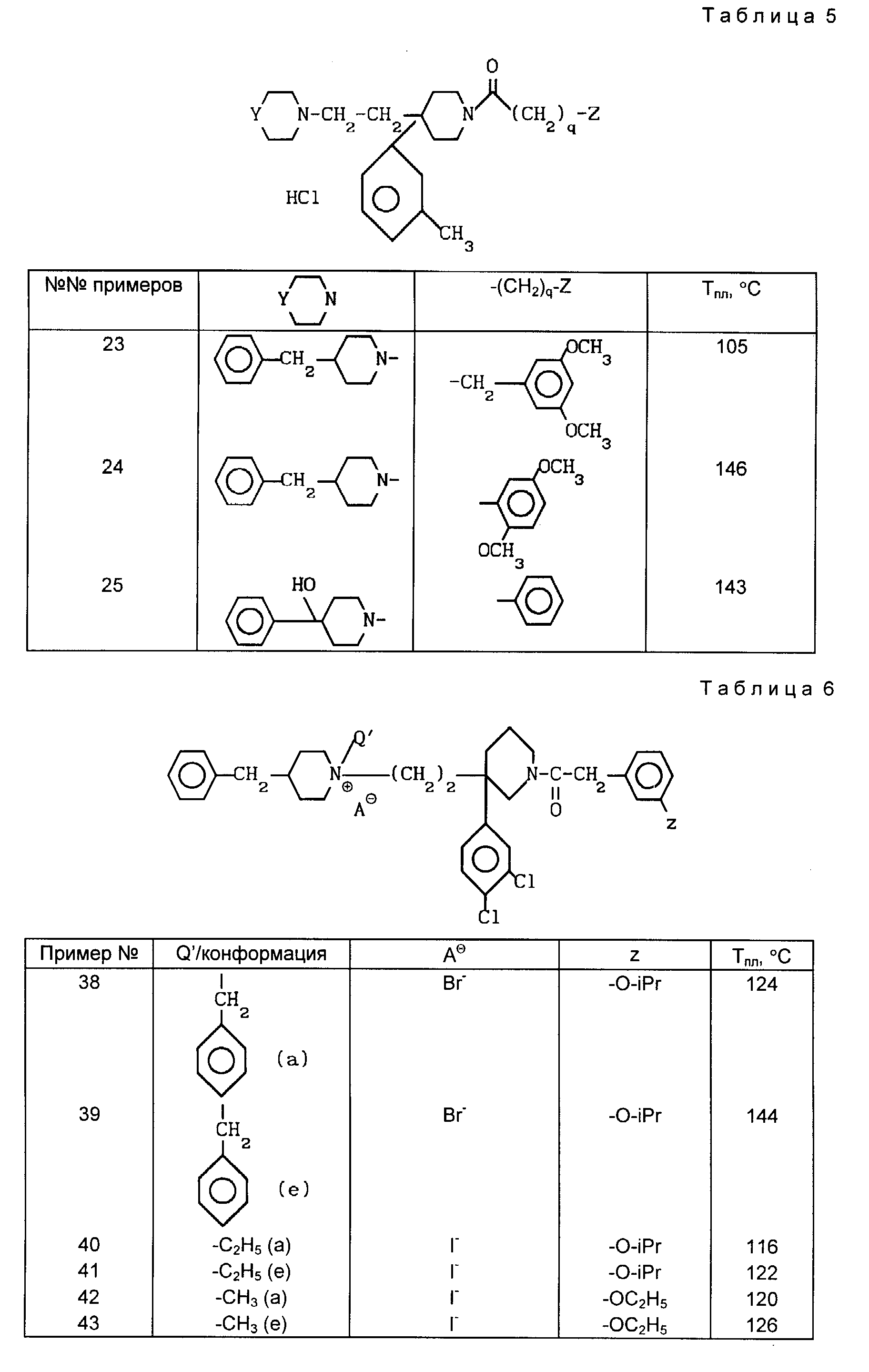
Проводя синтезы, как указано в примере 22, получают соединения, описанные в табл. 5.
Пример 26.
Хлоргидрат 3-[3-(4-бензин-1-пиперидинил)-пропил]-3-(3, 4-дихлорфенил)-1-(3-метоксифенил)-ацетилпиперидина
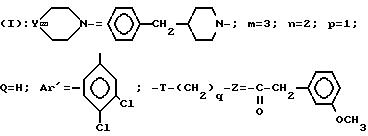
A/ 3,4-Дихлортетрагидропиранилоксипропил- α -бензолацетонитрил
Способом, идентичным стадии A примера 1, и исходя из 37,2 г 3, 4-дихлорфенилацетонитрила и 44,6 г 3-бромпропокситетрагидропирана, получают 35 г ожидаемого продукта.
B/ g -Тетрагидропиранилоксипропил- g -циано-3,4- дихлорбензилбутаноат этила.
Исходя из 35 г полученного на стадии A продукта и 19,2 г бромпропионата этила, проводя синтез идентично стадии B примера 1, получают 28 г ожидаемого продукта.
C/ 5-Тетрагидропиранилоксипропил-5-(3, 4-дихлорфенил)-пиперидон.
Гидрируют 23 г полученного на стадии B продукта, растворенного в 650 мл этанола, под атмосферным давлением и при температуре окружающей среды в присутствии никеля Ренея. Когда поглощается теоретически рассчитанный объем водорода, то отфильтровывают катализатор, выпаривают досуха, извлекают остаток эфиром, промывают водой, буферным раствором с pH 2, сушат над Na2SO4 и выпаривают досуха.
Получают таким образом 18 г ожидаемого продукта.
D/ 3-Тетрагидропиранилоксипропил-3-(3, 4-дихлорфенил)-пиперидин.
Прибавляют по капле 14 г полученного на стадии С продукта, растворенного в 50 мл тетрагидрофурана, к суспензии, содержащей 2,75 г алюмогидрида лития, которая нагрета до 60oC.
Поддерживают температуру 60oC в течение 1 ч. Охлаждают реакционную смесь, гидролизуют путем прибавления 3 мл воды 4н раствора NaOH и 9 мл воды. Отделяют минеральный осадок и выпаривают органическую фазу под вакуумом.
Получают таким образом 12,4 г ожидаемого продукта.
E/ 3-Тетрагидропиранилоксипропил-3-(3, 4-дихлорфенил)-1-(3- метоксифенил)-ацетилпиперидин.
Прибавляют 3,9 г БОФ к раствору, содержащему 3 г полученного на стадии 1 продукта, 2,4 г триэтиламина и 1,3 г 3-метоксифенилуксусной кислоты в 50 мл дихлорметана. Перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды, выпаривают досуха, поглощают AcOEt, промывают водой, сушат над Na2SO4 и выпаривают досуха.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2 Cl2/CH3OH 100/2 (об/об).
Концентрирование частиц фракций дает 3 г ожидаемого продукта.
K/ 3-Метансульфонилоксипропил-3-(3, 4-дихлорфенил)-1-(3-метоксифенил)- ацетилпиперидин.
Исходя из 3 г полученного на стадии E продукта и 0,68 г мезилхлорида, получают ожидаемое соединение, проводя синтез, как описано на стадии C примера 2. После очистки методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/1,5 (об/об), и концентрирования фракций чистого продукта получают 2 г ожидаемого продукта.
Л/ Соединение 26.
В течение 1 ч нагревают при 70oC 2 г полученного на стадии K продукта и 1,6 г 4-бензилпиперидина, растворенных в 3 мл диметилформамида. Охлаждают реакционную смесь, приливают к воде, экстрагируют эфиром, промывают водой и сушат над Na2SO4, фильтруют и концентрируют под вакуумом.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3 100/3 (об/об). Чистые фракции концентрируют под вакуумом, остаток поглощают ацетоном, затем получают хлоргидрат в результате прибавления эфира, насыщенного хлороводородной кислотой. Отфильтровывают хлоргидрат, промывают в пентане и сушат под вакуумом на P2O5.
Получают таким образом 1,1 г ожидаемого продукта.
Tпл=108oC.
Пример 27. Хлоргидрат
3-[3-(4-фенил-4-ацетамидо-1-пиперидинил)-пропил]
-3-(3,4-дихлорфенил)-1- бензоилпиперидина
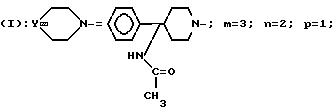

A/ 3-Тетрагидропиранилоксипропил-3-(3,4-дихлорфенил)-1- бензоилпиперидин.
Прибавляют 1,13 г бензоилхлорида к 3 г 3-тетрагидропиранилоксипропил-3-(3,4-дихлорфенил)-пиперидина, полученного в соответствии со стадией 1 примера 26, в присутствии 1,62 г триэтиламина, растворенных в 50 мл дихлорметана. Перемешивают реакционную смесь в течение 30 мин при температуре окружающей среды, выпаривают досуха, поглощают остаток эфиром, промывают водой, сушат над Na2SO4 и выпаривают досуха. Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/1 (об/об).
Получают таким образом 3 г жидкого масла.
B/ 3 Метансульфонилоксипропил-3-(3,4-дихлорфенил)-1-бензоилпиперидин.
К 3 г полученного на стадии A продукта, растворенного в 50 мл метанола, прибавляют эфир, насыщенный хлороводородной кислотой до значения pH 1. Перемешивают реакционную смесь в течение 30 мин при температуре окружающей среды и выпаривают досуха. Извлекают остаток посредством 50 мл дихлорметана и 1,07 г триэтиламина, затем прибавляют 0,72 г мезилхлорида. Перемешивают при температуре окружающей среды в течение 1 ч, выпаривают досуха, извлекают остаток этилацетатом, промывают водой и сушат над Na2SO4, фильтруют и концентрируют под вакуумом. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2 /AcOEt 100/3 (об/об).
Получают таким образом 1,6 г ожидаемого продукта.
C/ Соединение 27.
В течение 4 ч нагревают при 80oC 1,5 г полученного на стадии B продукта и 1,5 г 2-фенил-4-ацетамидопиперидина, растворенных в 5 мл диметилформамида. Охлаждают реакционную смесь, приливают к воде, экстрагируют дихролметаном, промывают водой и сушат над Na2SO4, фильтруют и концентрируют под вакуумом. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/5 (об/об). Концентрируют фракции чистого продукта, извлекают дихлорметаном, получают хлоргидрат в результате прибавления эфира, насыщенного хлороводородной кислотой, выпаривают досуха, поглощают остаток этанолом и осаждают в эфире. Отфильтровывают, промывают осадок в пентане и сушат под вакуумом.
m=0,60 г.
Tпл=184oC.
Пример 28. Хлоргидрат
5-[3-(4-гидрокси-4-фенил-1-пиперидинил)-пропил]-5- (3,4-дихлорфенил)-1-(3-метоксибензил)-пиперидона
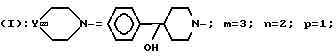
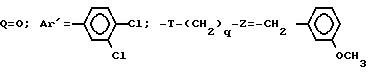
A/ 5-Тетрагидропиранилоксипропил-5-(3, 4-дихлорфенил)-1-(3-метоксибензил)-пиперидон.
Прибавляют 0,66 г NaH с концентрацией 60% к раствору, содержащему 6,4 г 5-тетрагидропиранилоксипропил-5-(3,4-дихлорфенил)-пиперидона, описанного на стадии C примера 26, в 60 мл диметилформамида. Перемешивают реакционную смесь в течение 30 мин при температуре окружающей среды. Затем прибавляют по капле 2,5 г 3-метоксибензилхлорида и нагревают реакционную смесь в течение 1 ч при 80oC. Выпаривают диметилформамид под вакуумом, экстрагируют остаток в дихлорметане, промывают водой, сушат над Na2SO4 и выпаривают досуха.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/AcOEt 100/5 (об/об). Концентрируют фракции чистого продукта, в результате чего получают 6 г жидкого масла.
B/ 5-(3-Гидроксипропил)-5-(3,4-дихлорфенил)-1-(3-метоксибензил)- пиперидон.
Перемешивают 6 г полученного на стадии A продукта, растворенного в 50 мл метанола, насыщенного хлороводородной кислотой, в течение 1 ч при температуре окружающей среды.
Выпаривают досуха, в результате чего получают 4,3 г жидкого масла.
C/ 5-Метансульфонилоксипропил-5-(3,4-дихлорфенил)-1-(3-метоксибензил)-пиперидон.
Прибавляют 1,14 г мезилхлорида к 4,3 г полученного на стадии B продукта в присутствии 2 г триэтиламина, растворенных в 50 мл дихлорметана. Перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды, выпаривают досуха, извлекают остаток в AcOEt, промывают водой, водой, насыщенной NaCl, сушат над Na2SO4 и выпаривают досуха.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/2 (об/об). Концентрируют фракции чистого продукта, в результате чего получают 4 г жидкого масла.
I/ Соединение 28/
В течение 2 ч нагревают при 80oC 4
г полученного на стадии C продукта и 3,1 г 4-гидрокси-4-фенилпиперидина, растворенных в 5 мл диметилформамида. Охлаждают,
приливают к воде, экстрагируют посредством AcOEt, промывают водой, сушат над
Na2SO4 и выпаривают досуха. Извлекают полученное жидкое масло эфиром и получают хлоргидрат в
результате прибавления эфира, насыщенного хлороводородной кислотой. Фильтруют,
промывают эфиром и сушат под вакуумом.
m=4 г.
Tпл=110-117oC.
Пример 29. Дихлоргидрат
3-[3-(4-гидрокси-4-фенил-1-пиперидинил)-пропил] -3- (3,4-дихлорфенил)-1-(3-метоксибензил)-пиперидина
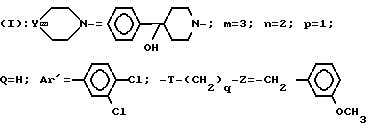
Прибавляют 2 г 5-[3-(4-гидрокси-4-фенил-1-пиперидинил)-пропил] -5- (3,4-дихлорфенил)-1-(3-метоксибензил)-пиперидона к суспензии, содержащей 0, 60 г алюмогидрида лития в 50 мл тетрагидрофурана. Нагревают реакционную смесь в течение 1 ч при температуре окружающей среды, охлаждают, гидрализуют под действием 5 мл воды, отфильтровывают минеральный осадок и выпаривают досуха.
Очищают полученный таким образом остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/5 (об/об), концентрируют фракции чистого продукта и получают хлоргидрат в дихлорметане в результате прибавления эфира, насыщенного хлороводородной кислотой.
Отделяют хлоргидрат путем фильтрования, промывают эфиром и сушат под вакуумом на P2O5.
m= 1,5 г.
Tпл= 160-175oC.
Пример 30.
Дихлоргидрат
3-(4-гидрокси-4-фенил-1-пиперидинил)-этил]-3- (3,4-дихлорфенил)-1-бензилпирролидина
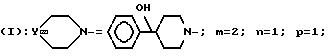
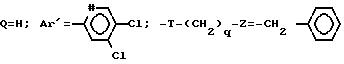
Проводя синтез в соответствии с примером 29, но исходя из продукта, описанного в примере 5, получают ожидаемый продукт.
Tпл= 170oC.
Пример 31. Дихлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3-(1-нафтил)- 1-бензилпиперидина
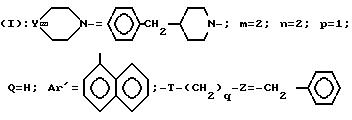
Проводя синтез в соответствии с примером 29, но исходя из продукта, описанного в примере 17, получают указанное выше соединение.
Tпл= 140oC.
Пример 32. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3-(3,
4-дихлорфенил)-1- (3-изопропоксифенил)-ацетилпиперидина (-)
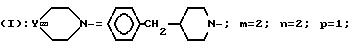
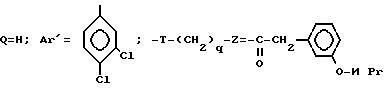
I-получение оптически чистого аминоспирта.
A/ 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-пиперидин.
К 55 г 3-тетрагидропиранилоксиэтил-3-(3,4-дихлорфенил)-пиперидина, растворенного в 200 мл метанола, прибавляют эфир, насыщенный хлороводородной кислотой до значения pH 1. Перемешивают в течение 0,5 ч при температуре окружающей среды, концентрируют досуха, извлекают остаток водой, подщелачивают раствором гидроксида натрия, экстрагируют дихлорметаном, промывают насыщенным раствором NaCl, сушат над Na2SO4 и выпаривают досуха. Получают жидкое масло.
Извлекают посредством 200 мл смеси изопропиловый эфир/ эфир 50/50 (об/об). Перемешивают, фильтруют, промывают эфиром и сушат под вакуумом на P2O5.
m=45 г.
Тпл=122oC.
B/ 3-(2-гидроксиэтил)-3-(3,
4-дихлорфенил)-пиперидин (+)
К 43 г полученного на стадии A продукта, растворенного в 250 мл 100o-го этанола при температуре образования флегмы, прибавляют 23,54 г L-(+)-винной
кислоты, растворенной в 750 мл 100o-го этанола. Нагревают реакционную смесь при температуре образования флегмы в течение получаса, дают вернуться к температуре окружающей среды,
отфильтровывают полученные кристаллы, промывают в 100o-м этаноле и сушат под вакуумом при 50oC на P2O5.
m=31 г.
Затем перекристаллизовывают в 540 мл 100o-го этанола, отфильтровывают, промывают эфиром и сушат под вакуумом на P2O5.
m=25 г.
(α
)= +8,5° (c=1, H2O)
Затем тартрат извлекают водой,
нейтрализуют раствором NaOH, экстрагируют дихлорметаном, промывают водой, сушат над Na2SO4 и выпаривают досуха. Поглощают масло смесью эфир/изопропиловый эфир, отфильтровывают
кристаллы, промывают эфиром и сушат под вакуумом при 50oC.
m=13,5 г.
Tпл=138oC.
(α)= +8,2° (c=1, CH3OH)
C/ 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-пиперидин (-).
Проводя синтез, как указано выше, исходя из D (-) винной кислоты, получают энантиомер (-).
Tпл=139oC.
(α)= -8,4° (c=1, CH3OH)
II получение соединения 32.
A/ 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-1-бутилкарбамоилпиперидин.
К 13 г 3-(2-гидрокси)-3-(3,4-дихлорфенил)-пиперидина (+), растворенного в 100 мл диоксана, прибавляют 12,4 г дитрет. бутилдикарбоната. Затем перемешивают в течение 1 ч при 40oC. Выпаривают досуха, извлекают остаток эфиром, промывают водой, затем буферным раствором с pH 2 и окончательно водой. Сушат над Na2SO4, фильтруют и выпаривают досуха. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/2) об/об). После концентрирования чистых фракций получают таким образом 16,7 г ожидаемого продукта в виде масла.
B/ 3-Метансульфонилоксиэтил-3-(3,4-дихлорфенил)-1-трет. бутилкарбамоилпиперидин.
К 16,5 г полученного на стадии A продукта, растворенного в 100 мл дихлорметана в присутствии 4,9 г триэтиламина, прибавляют по капле 5,5 г мезилхлорида. Перемешивают в течение получаса при температуре окружающей среды, выпаривают досуха, извлекают остаток в эфире, промывают водой, сушат над Na2SO4 и концентрируют под вакуумом. Получают таким образом 19 г жидкого масла.
C/ 3-2-(4-Бензил-1-пиперидинил)-этил-3-(3,4-дихлорфенил)-1-трет. бутилкарбамоилпиперидин
В течение 3 ч нагревают при 80oC 18 г полученного на
стадии B
продукта и 14 г 4-бензилпиперидина, растворенных в 40 мл диметилформамида. Затем выпаривают диметилформамид, извлекают остаток в воде, экстрагируют эфиром, промывают водой, сушат над Na2
SO4 и концентрируют под вакуумом. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/3 (об/об). Чистые фракции
концентрируются
под
вакуумом.
m=15 г.
D/ Дихлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3-(3,4- дихлорфенил)-пиперидина (-).
Перемешивают 15 г полученного на стадии C продукта, растворенного в 75 мл метанола, 60 мл концентрированной хлороводородной кислоты и 15 мл воды, при температуре окружающей среды в течение 1 ч. Выпаривают досуха, извлекают остаток посредством 100 мл дихлорметана и осаждают в эфире. Отфильтровывают осадок, промывают эфиром и сушат под вакуумом.
m=11,5 г.
Tпл =175oC.
(α)= -2,2° (c=1, CH3
OH)
E/ Соединение
32.
Прибавляют 10,6 г БОФ к 11 г полученного на стадии 1 продукта 6,09 г триэтиламина и 4,65 г 3-изопропоксифенилуксусной кислоты, растворенных в 100 мл дихлорметана. Перемешивают при температуре окружающей среды в течение 1 ч, выпаривают досуха, извлекают остаток в этилацетате, промывают водой, сушат над Na2SO4, фильтруют и концентрируют под вакуумом. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/5 (об/об). Чистые фракции концентрируются под вакуумом, хлоргидрат получают обработкой CH2Cl2 после прибавления эфира, насыщенного хлороводородной кислотой, выпаривают досуха, кристаллизируют в изопропиловом эфире, фильтруют, промывают эфиром и сушат под вакуумом.
m=11,4 г.
Tпл=105oC.
(α)
= -2,9° (c=1, CH3OH)
Пример 33. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3-(3,
4- дихлорфенил)-1-(3-изопропоксиэтил)-ацетилпиперидина (+)
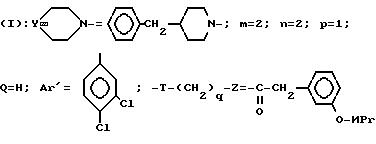
Проводя синтез в соответствии с примером 32 и используя в качестве исходного соединения энантиомер 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-пиперидина (-), получают указанное выше соединение 33 в виде энантиомера (+).
Tпл=105oC.
(α)= +3,0°
(c=1, CH3OH).
Пример 34. Хлоргидрат
3-[2-(4-гидрокси-4-фенил-1-пиперидинил)-этил]-3- (3,4-дихлорфенил)-1-бензоилпиперидина (-)
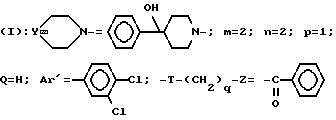
A/ 3-[2-(4-гидрокси-4-фенил-1-пиперидинил)-этил]-3-(3,4- дихлорфенил)-1-трет.бутил-карбамоилпиперидин.
В течение 2 ч нагревают при 80oC 0,9 г 3-метансульфонилоксиэтил-3-(3, 4-дихлорфенил)-1-трет. бутил-карбамоилпиперидина, полученного в соответствии со стадией B примера 32, и 0,88 г 4-гидрокси-4-фенилпиперидина, растворенных в 3 мл диметилформамида. Выпаривают досуха, извлекают остаток в воде, экстрагируют посредством AcOEt, промывают водой, насыщенной NaCl, сушат над MgSO4 и выпаривают досуха. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/2 (об/об). Концентрируют чистые фракции, в результате чего получают 0,8 г масла.
B/ 3-[2-(4-гидрокси-4-фенил-1-пиперидинил)-этил]-3-(3,4- дихлорфенил)-пиперидин.
Перемешивают 0,8 г полученного на стадии A продукта, растворенного в 5 мл метанола, 4 мл концентрированной хлороводородной кислоты и 1 мл воды, в течение 1 ч при температуре окружающей среды. Затем выпаривают досуха и используют остаток в таком виде, как он есть, на следующей стадии.
m=0,77 г.
C/ Соединение 34.
К 0,77 г полученного на стадии B продукта и 0,3 г триэтиламина, растворенных в 30 мл дихлорметана, прибавляют 0,26 г бензоилхлорида. Перемешивают реакционную смесь в течение 1 ч при температуре окружающей среды, выпаривают досуха, извлекают остаток в этилацетате, промывают водой, сушат над Na2SO4 и выпаривают досуха. Очищают методом хроматографии на силикагеле, элюент CH2Cl2/CH3OH 100/3 (об/об). Концентрируют чистые фракции, извлекают в CH2 Cl2 и получают хлоргидрат в результате прибавления эфира, насыщенного хлороводородной кислотой. Выпаривают досуха, кристаллизуют остаток в эфире, отфильтровывают, промывают эфиром и сушат под вакуумом.
m=0,2 г.
Tпл=176oC.
(α)= -32,0°(c=1, CH3OH).
Пример 35. Хлоргидрат
3-[2-(4-гидрокси-4-фенил-1-пиперидинил)-этил]-3-(3,4- дихлорфенил)-1-бензоипиперидина (+)
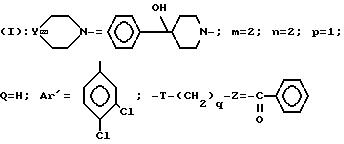
Проводя синтез в соответствии с примером 34, исходя из 3-(2-гидроксиэтил)-3-(3, 4-дихлорфенил)-пиперидина (-), получают указанный выше энантиомер (+).
Tпл=175o C.
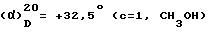
Пример 36. Иодид N(a)-метил-3-[2-(4-бензил-1-пиперидиний)-этил] -3-(3, 4-дихлорфенил)- 1-(3-изопропоксифенил)-ацетилпиперидина
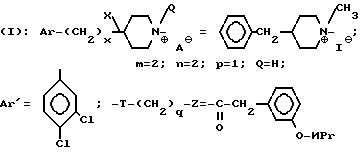
Перемешивают 1 г продукта, описанного в примере 14, растворенного в 10 мл метилиодида в течение 24 ч при температуре окружающей среды. Затем концентрируют под вакуумом. Остаток хроматографируется на силикагеле, элюент CH2 Cl2/CH3OH 100/3 (об/об). Первый элюированный продукт соответствует соединению, у которого метил на азоте (b) 4-бензилпиперидина находится в аксиальном положении.
m=0,35 г.
Спектр ЯМР13C:

Пример 37. Иодид N(e)-3-[2-(4-бензил-1-пиперидиний)-этил]-3-(3, 4- дихлорфенил)-1-(3-изопропоксифенил)-ацетилпиперидина
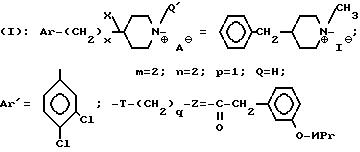
Приводя синтез как в примере 36, описанном выше, и собирая фракцию, элюируемую во вторую очередь, получают продукт, у которого метил на азоте (b) 4-бензилпиперидина находится в экваториальном положении.
m=0,15 г.
Спектр ЯМР13C:
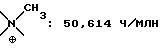
Проводя синтезы в соответствии с примерами 36 и 37, приведенными выше, получают соли четвертичного аммония, описанные в табл. 6.
Пример 44. Хлорид N
(a)-метил-3-[2-(4-бензил-1-пиперидиний)-этил] -3-(3,
4-дихлорфенил)-1-(3-изопропоксифенил)-ацетилпиперидина (-)
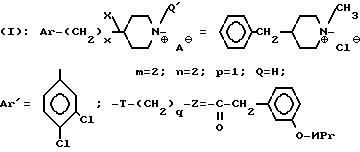
A/ Получение иодидного производного.
Перемешивая 10 г описанного в примере 32 продукта, растворенного в 50 мл метилиодида, в течение 2 ч при температуре окружающей среды. Выпаривают досуха и хроматографируют остаток на силикагеле: CH2Cl2/CH3OH - 100/3 (об/об). Конформер, который элюируется первым, соответствует соединению, у которого метил находится в аксиальном положении при азоте (в) 4-бензилпипеидина.
B/ Получение хлоридного производного.
Иодид-ион обменивается затем на хлорид-ион при элюировании продукта через ионообменную смолу Амбелит IRA68®.
Получают таким образом 5,6 г хлорида четвертичного аммония.
Тпл 103oC.
Пример 45. Хлорид N(a)-метил-3-[2-(4-бензил-1-пиперидиний)-этил]- 3-(3, 4-дихлорфенил)-1-(3-изопропоксифенил)-ацетилпипеидина (+)
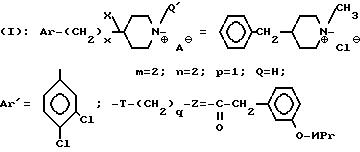
Проводя синтез способом, идентичным примеру 44, исходя из продукта, описанного в примере 33, получают 8,9 г ожидаемой соли четвертичного аммония.
Тпл 104oC.
(α
) = + 13,0° (c=1, CH3OH)
Пример 46. Иодид
N(e)-метил-3-[2-(4-бензил-1-пиперидиний)-этил]-3-(3,4- дихлорфенил)-1-(3-изопропоксифенил)-ацетилпиперидина (-)
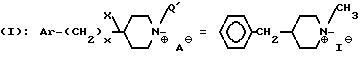
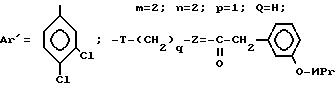
Проводя синтез в соответствии с примером 44А и собирая фракцию, элюируемую во вторую очередь, получают энантиомер, у которого метил на азоте (в) 4-бензилпиперидина находится в экваториальном положении. Получают таким образом 2,6 г соли четвертичного аммония.
Тпл 110oC.
(α)= -0,1°
(c=1, CH3OH)
Пример 47. Иодид N(e)-метил-3-[2-(4-бензил-1-пиперидиний)-этил] - 3-(3,
4-дихлорфенил)-1-(3-изопроксифенил)-ацетилпиперидина (+)
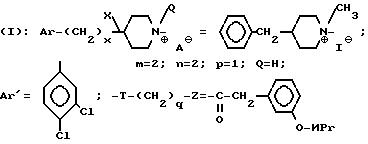
Проводя синтез в соответствии с примером 46, исходя из продукта, описанного в примере 33, получают ожидаемое соединение.
Тпл 110oC
(α) = + 0,1° (c=1,
CH3OH)
Пример 48. N-Оксид3-[2-(4-бензил-1-пиперидиний)-этил]-3-(3,
4-дихлорфенил)- 1-(3-изопропоксифенил)-ацетилпиперидина
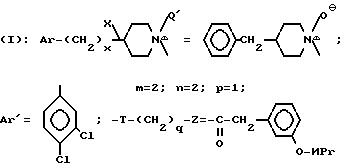
Растворяют 2 г свободного основания соединения по примеру 14 в 20 мл тетрагидрофурана. Прибавляют 1,1 г метахлорпербензойной кислоты и перемешивают реакционную смесь в течение 2 ч при температуре окружающей среды. Концентрируют под вакуумом до объема величиной 5 мл и разбавляют остаток в 10 мл дихлорметана. Промывают дважды этот раствор насыщенным раствором NaHCO3, декантируют, сушат над MgSO4 и концентрируют под вакуумом. Хроматографируют остаток на силикагеле, элюент CH2Cl2 /CH3OH 100/5 (об/об). Концентрируют фракции чистого продукта под вакуумом и кристаллизируют остаток в изопропиловом эфире.
m 1,47 г.
Тпл 135o C.
Пример 49. Дихлоргидрат
3-[2-(4-бензил-1-пиперидинил)-этил] -3-(3,4- дихлорфенил)-пиперидина
Промежуточное соединение синтеза формулы (ll)
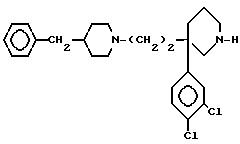
A/ 4-(4-бензил-1-пиперидинил)-2-(3,4-дихлорфенил)-бутиронитрил
К 94 г 3,4 дихлорфенилацетонитрила, растворенного в 500 мл безводного эфира, прибавляют небольшими порциями 23,5 г амида натрия. Затем перемешивают в течение 1 ч при температуре окружающей среды, потом в течение 3 ч при температуре образования флегмы. Охлаждают смесь до 0oC и прибавляют по капле 129 г 2-(4-бензил-1-пиперидинил)-1-хлорэтана, растворенного в 300 мл эфира. Дают вернуться реакционной смеси к температуре окружающей среды, затем нагревают в течение 3 ч при температуре образования флегмы. Охлаждают, приливают к 600 мл воды, декантируют органическую фазу, промывают водой и экстрагирруют два раза по 500 мл 15%-ного раствора HCl. Перемешивают водную фазу и осаждают продукт в виде хлоргидрата. Отфильтровывают, промывают водой и сушат под вакуумом. Перекристаллизовывают остаток в 600 мл изопропанола. Получают таким образом 95 г.
Продукт извлекается в воде, а раствор нейтрализуется раствором NaOH. Экстрагирует эфиром, промывают водой, сушат над Na2SO4 и выпаривают досуха. Получают 87 г масла.
B/ γ -[2-(4-бензил-1-пиперидинил)-этил] g - циано-3,4-дихлорбензилбутаноат этила
В течение 24 ч нагревают при 80oC
87 г описанного на
стадии А
продукта, 28 г этилакрилата и 2,5 мл тритона B, растворенных в 45 мл диоксина. Охлаждают, извлекают эфиром, промываю водой, сушат над Na2SO4 и
выпаривают досуха.
Получают 109,5
г масла.
C/ 5-[2-(4-бензил-1-пиперидинил)-этил]-5-(3,4-дихлорфенил)- пиперидон.
Гидрируют 100 г полученного на стадии B продукта, растворенного в 1,5 л этанола, при 60oC и под атмосферным давлением в присутствии никеля Ренея. После поглощения объема водорода отфильтровывают катализатор, выпаивают досуха, извлекают в дихлорметане, промывают водой и сушат над Na2SO4. Затем получают хлоргидрат, который перекристаллизовывают в 220 мл изопропанола. Отфильтровывают и сушат под вакуумом. Продукт извлекают водой, нейтрализуют раствором NaOH, экстрагируют эфиром и сушат над Na2SO4. Получают 44 г масла.
D/ Соединение 49.
Прибавляют по капле 44 г полученного на стадии C продукта, растворенного в 200 мл тетрагидрофуана, к суспензии, содержащей 9,4 г алюмогидрида лития в 250 мл тетрагидрофурана, которая нагрета до 60oC. Продолжают нагревать при температуре образования флегмы в течение 3 ч. Охлаждают льдом и прибавляют последовательно 10 мл воды, 10 мл 4н NaOH и 30 мл воды. Отфильтровывают минеральный осадок и выпаривают фильтрат досуха, извлекают в дихлорметане и получают хлоргират. Выпаривание досуха, осаждают в виде порошка в пентане, отфильтровывают и сушат под вакуумом.
m 35 г.
Тпл 170oC.
Пример 50. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-3-(3,4-дихлорфенил) -1-(2-фенил-2-метокси)-ацетилпиперидина.
Наименее полярной диастереоизомер.
Перемешивают в течение 2 ч 1,5 г диамина, полученного в примере 49, 1,06 г триэтиламина, 0,55 г (±)- α метоксифенилуксусной кислоты и 1,6 г БОФ в 25 мл дихлорметана. Выпаривают досуха, извлекают остаток в этилацетате, промывают водой, сушат над Na2SO4 и выпаривают досуха. Очищают остаток методом хроматографии на силикагеле, элюент CH2Cl/CH3OH 100/0,5 (об/об). Продукт, который элюируется первым, является ожидаемым соединением. Фракции концентрируется под вакуумом, извлекают в CH2Cl2, получают хлоргидрат, выпаривают досуха, осаждают в виде порошка в пентане, отфильтровывают и сушат под вакуумом.
m 0,5 г.
Tпл 134oC.
Пример 51. Хлоргидат 3-[2-(4-бензил-1-пиперидинил)-этил]-3 -(3, 4-дихлорфенил)-1-(2-фенил-2-метокси)-ацетилпиперидина (наиболее полярный диастереоизомер).
Проводят синтез в соответствии с примером 50 и осуществляя элюирование смесью CH2Cl2/CH3OH 100/2 (об/об), получают наиболее полярный диастереизомер. Хлоргидрат получают в дихлорметане, выпаривают досуха, извлекают в пентане.
m 0,50 г.
Тпл 118oC.
Проводя синтезы в соответствии с примерами 50 и 51, получают пары диастереоизомеров 52, 53, 54, 55 и 56, 57, описанных в табл. 7.
α Гидрокси-3-изопропоксифенилуксусная кислота, используемая в качестве реагента для получения соединений из примеров 56 и 57, которая является новым продуктом, может быть получена, как указано ниже.
a -Гидрокси-3-изопропоксифенилуксусная кислота.
Стадия 1.
К раствору, содержащему 50 г 3-гидроксибензальдегина в 250 мл ДМФ, прибавляют 60 г K2CO3, затем 60 мл 2-иодпропана.
Нагревают реакционную смесь при 50oC в течение 18 ч. Полученная смесь приливается в 2,5 л воды. Экстрагируют эфиром, промывают разбавленным раствором NaOH, затем водой. Сушат над MgSO4, выпаривают растворитель, в результате чего получают 53,5 жидкого остатка.
Стадия 2.
Прибавляют 53 г продукта, полученного в соответствии с предыдущей стадией 1, к раствору, содержащему 38 г бисульфита натрия в 120 мл воды. Перемешивают в течение 20 ч, затем при 20oC прибавляют раствор, содержащий 44,2 г цианида калия в 90 мл воды.
Спустя 2 ч экстрагируют эфиром, промывают водой, осушат над MgSO4 и выпаривают растворитель под вакуумом. Остаток хроматографируют на силикагеле, элюент гептан/этилацетат 100/30 (об/об). Выделяют 57 г продукта в виде жидкого масла.
Стадия 3.
Прибавляют 46 г продукта, полученного в соответствии с предыдущей стадией 2, к 50 мл воды и 50 мл концентрированной HCI. Нагревают при 110oC в течение 1 ч. После охлаждения экстрагируют эфиром, промывают водой. Кислоту экстрагируют разбавленным раствором NaOH. Подкисляют водную фазу, экстрагируют эфиром, сушат над MgSO4, выпаривают растворители. Кислоту кристаллизируют в смеси толуол/пентан 1/2 (об/об). m 27,5 г (табл.7).
Пример 58. Хлоргидрат
3-[2-(4-бензил-1-пиперидинил)-этил]-3 -(3,4-дихлорфенил)-1-[2-(3-хлорфенил)-2-гидрокси]-ацетилпиперидина (+)
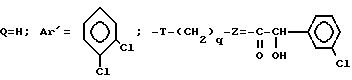
Перемешивают в течение 2 ч 0,67 г дихлоргидрата 3-[2-(4-бензил-1-пиперидинил)-этил] -3-(3,4-дихлорфенил)-пиперидина (-), описанного на стадии 1 примера 32, 0,17 г триэтиламина, 0,32 г S-(+)- альфа-гидрокси-3-хлорфенилуксусной кислоты и 0,82 г БОФ, растворенных в дихлорметана. Выпаривают досуха, поглощают остаток этилацетатом, промывают водой, сушат над Na2SO4, концентрируют под вакуумом. Очищают остаток методом хроматогррафии на силикагеле, элюент CH2Cl2/CH3OH 100/1,5 (об/об). Концентрируют чистые фракции под вакуумом, извлекают в дихлорметане, получают хлоргидрат, выпаривают досуха и извлекают пентаном. Фильтруют, промывают эфиром и сушат под вакуумом.
m 0,40 г.
Тпл 122oC.
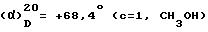
Пример 59. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3 -(3, 4-дихлорфенил)-1-[2-(3-хлорфенил)-2-гидрокси]-ацетилпиперидина (-)
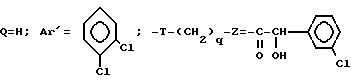
Проводя синтез в соответствии с примером 58, исходя из 3-[2-4(бензил-1-пиперидинил)-этил] 3-(3,4-дихлорфенил)-пиперидина (+), полученного в соответствии со стадией I примера 32, исходя из 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-пиперидина (-), описанного на стадии C получения оптически чистого аминоспирта, при взаимодействии с R (-) α -гидрокси-3-хлорфенилуксусной кислотой, получают ожидаемый продукт.
m=0,50 г.
Tпл=122oC.
(α)= -74 (c =1, CH3OH)
Пример 60. Хлоргидрат
3-[2-(4-бензил-1-пиперидинил)-этил]-3-(3,
4-дихлорфенил)- 1-[2-(3-хлорфенил)-2-гидрокси]-ацетилпиперидина (-)
Перемешивают при температуре окружающей среды в течение 2 ч 0,67 г 3-[2-(4-бензил-1-пиперидинил)-этил] -3-дихлорфенил)-пиперидина (-), 0,32 г R (-) α -гидрокси-3-хлорфенилуксусной кислоты, 0,17 г триэтиламина и 0,82 г БОФ, растворенных в 50 мл дихлорметана. Затем, проводя синтез, как в примере 58, получают ожидаемый продукт.
m=0, 40 г.
Тпл=128oC.
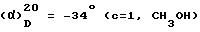
Пример 61. Хлоргидрат 3-[2-(4-бензил-1-пиперидинил)-этил]-3-(3, 4- дихлорфенил)-1-[2-(3-хлорфенил)-2-гидрокси]-ацетилпиперидина (+)
Проводя синтез в соответствии с примером 58, исходя из S (+) α -гидрокси-3-хлорфенилуксусной кислоты и из 3-[2-(4-бензил-1-пиперидинил)-этил] -3-(3, 4-дихлорфенил)-пиперидина (+), полученного в соответствии со стадией D примера 32 и исходя из 3-(2-гидроксиэтил)-3-(3,4-дихлорфенил)-пиперидина (-), описанного на стадии C получения оптически чистого аминоспирта, получают ожидаемый продукт.
m=0, 4 г.
Tпл=127oC.
(α) = +35° (c=1, CH3OH)
Все примеры даны в табл. 8.
Реферат
Использование: в медицине в качестве антагонистов рецепторов нейрокинина. Продукты: полициклические аминосодержащие соединения ф-лы l, где
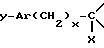
9 с. и 4 з.п. ф-лы, 8 табл.
Формула
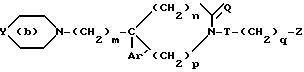
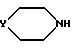
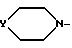
где Y группа
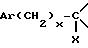
где Ar фенил, x 0 или 1, X водород, гидроксил, ацетокси- или ациламиногруппа;
m 2 или 3;
Ar' фенил, замещенный метилом или двумя атомами галогена, или нафтил;
n 0 3 целое число;
p 1 или 2, при условии, что когда p 2, то n 1 и Q является двумя атомами водорода;
Q кислород или два атома водорода;
T группа, выбранная из -С(О)- и -СН2-;
q 0 или 1;
Z фенил, который может быть одно- или дизамещен С1 - С4-алкоксигруппой или галогеном, или моногалогензамещенный нафтил, при условии, что когда T является группой -С(О)-, то (СН2)q Z может представлять собой бензильную группу, которая замещена в алифатическом остатке гидроксилом, С1 С4-алкоксигруппой или С1 - С4-алкилом и, возможно, замещена на ароматическом цикле галогеном или С1 С4 -алкоксигруппой,
или их соли с минеральными или органическими кислотами, или четвертичные соли аммония, или N-оксидные производные по азоту (b)пиперидина.
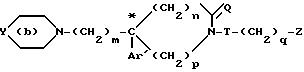
где С* атом углерода, имеющий определенную абсолютную конфигурацию (+) или (-);
Y, m, Ar', n, p, Q, T, q и Z имеют значения, указаные в п.1,
или их соли с минеральными или органическими кислотами, или четвертичные соли аммония, или N-оксидные производные по азоту (b)пиперидина.
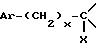
Ar, x, X, m, Ar', n, p, Q, T, q и Z имеют значения по п.1,
в виде соли четвертичного аммония или N-оксидного производного по азоту (b)пиперидина, где группа
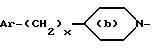
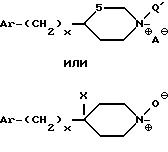
где A⊖ анион, выбираемый среди хлорида, бромида, иодида, ацетата, метансульфоната или паратолуолсульфоната;
Q1 C1 C6-алкил или бензил.
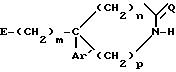
где m, Ar', n, p и Q определены в п.1;
Е гидроксил или в случае необходимости О-защищенная группа, например тетрагидропиранил-2-оксигруппа или группа
где Y группа
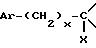
где X гидроксил или защищенный гидроксил,
обрабатывают либо функциональным производным кислоты общей формулы III
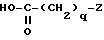
где q и Z определены в п.1,
для получения соединений формулы I, где T группа -C(O)-, либо галогенсодержащим производным общей формулы IV
Hal-(CH2)q+1 -Z
где q и Z определены в п.1;
Hal галоген, предпочтительно бром или хлор,
для получения соединений формулы I, где T группа -CH2-, получают соединение общей формулы V
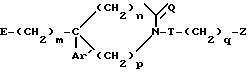
в) с последующим в случае необходимости отщеплением тетрагидропиранилоксигруппы под действием кислоты, с) образующийся спирт общей формулы VI
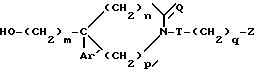
обрабатывают метансульфонилхлоридом, d) полученный продукт общей формулы VIII
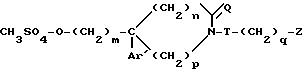
обрабатывают вторичным амином общей формулы VIII
где Y определен в п.1,
е) после в случае необходимости снятия защиты с гидроксила Х превращают в случае необходимости полученный продукт в одну из его солей с минеральной или органической кислотой, или четвертичную соль аммония, или N-оксидное производное по азоту (b)пиперидина.
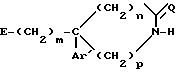
где m, Ar', n, p и Q определены в п.1;
Е гидроксил или возможно О-защищенная группа, такая, как тетрагидропропиранил-2-оксигруппа или группа
при условии, что если Е гидроксил, m 2, n 1, p 1 и Ar' - 3,4-дихлорфенил или 4-метилфенил, то Q является кислородом.
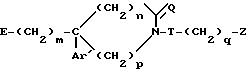
где Е гидроксил или О-защищенная группа, такая, как 2-тетрагидропиранилоксигруппа, или метансульфонильная группа, или группа
m, Ar', n, p, Q, T и Z определены в п.1,
или их соли с минеральными или органическими кислотами.
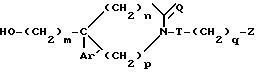
где m, Ar', Q, n, p, T, q и Z определены в п.1,
или их соли с минеральными или органическими кислотами.
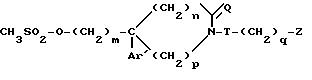
где m, Ar', n, p, Q, T и Z определены в п.1,
или их соли с минеральными или органическими кислотами.
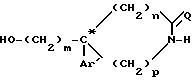
где m, Ar', n и p определены в п.1;
Q водород,
C* атом углерода, имеющий абсолютную конфигурацию (+) или (-).
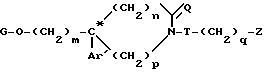
где G водород или метансульфонильная группа;
Q водород;
m, Ar', n, p, q, T, Z и C* имеют указанные значения.
Комментарии