Производные пиперидина - RU2099339C1
Код документа: RU2099339C1
Чертежи

Описание
Настоящее изобретение относится к нейрозащитным (противоишемическим и блокирующим стимуляцию аминокислотного рецептора) производным 3-пиперидино-1-хроманола и их аналогам, описывающимися нижеприведенной формулой (1), их фармацевтически приемлемым солям, применению этих соединений при лечении приступов или дегенеративных заболеваний центральной нервной системы, таких болезнь Альцгеймера, болезнь Хантингтона и болезнь Паркинсона, а также к некоторым полупродуктам этих соединений.
Инфенпродил представляет собой рацемическое, так называемое 1-эритро-соединение, имеющее стереохимическую формулу:
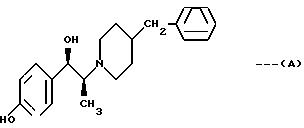
На рынке он продается как гипотензивное средства, и по своим свойствам стоит наравне с рядом близких аналогов (Carron, патент США 3 509 164; Carron, Drug Res. 1971, vol. 21, p. 1992-1999). Недавно было показано, что ифенпродил обладает противоишемической активностью и способностью блокировать стимуляцию аминокислотного рецептора (Gotti и T.Pharm. Exp. Therap. 1988, vol. 247, p. 1211-21; Carter, 1988, р. 1222-32; См. в этом плане также опубликованную европейскую заявку 322361 и патент Франции 2546166). Задачей настоящего изобретения являлось получить соединения, которые обладали бы такой же высокой нейрозащитной активностью, и в то же время не оказывали бы существенного или оказывали более низкое гипотензивное действие.
В литературе имеются сведения, что некоторые близкие по строению 1-фенил-3-/4-арил-4-ацилоксипиперидино/-1-пропанолы также могут использовать как аналгетики (патент США 3294804), а 1-[4-/амино- и гидроксиалкил/фенил]-2-/4-гидрокси-4-толилпиперазино/-1-алканолы обладают гипотензивной, анальгетической, психотропной или противовоспалительной активностью Japanes Kokai 53-02474 (CA 89: 43498y; Derwent Abs. 14858A) и 53-59675 (CA 89:146938w; Derwent Abs. 48671A).
Используемая в настоящем описании номенклатура, как правило, соответствует описанной Rigaudy IUPAC Nomenclature of Organic Chemistry, 1979 Edition, Pergammon Press, Нью-Йорк. Хроманы называют также 3,4-дигидро-1-/2Н/-бензопиранами.
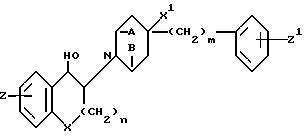
Предметом настоящего изобретения являются соединения формулы:
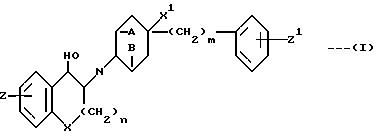
в которой A и B вместе означают -CH2CH2- или A и B означают каждый H;
X означает CH2 или O;
X1 H или OH;
Z H, F, Cl, Br или OH;
Z1 H, F, Cl, Br или /C1-C3/-алкил;
n равно 0 или 1, а m означает целое число, равное 1-6, а также их фармацевтически приемлемые соли.
С точки зрения простоты получения и биологической активности предпочтительными являются те из
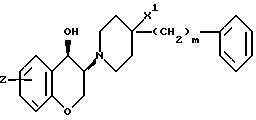
соединений формулы (I), у которых A и B означают каждый атом водорода, Z означает H, F, Cl или OH; Z1 -H, а m равно 0,1 или 2. Если X означает 0, а n равно 1, то наиболее предпочтительные
соединения имеют цисстереохимическую формулу:
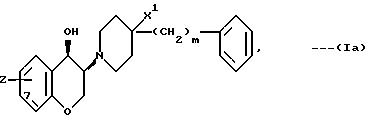
В частности, это соединения, у которых Z означает OH и которые замещены в 7-положении хромановой кольцевой системы. Наиболее предпочтительными соединениями формулы (Ia) являются те из них, у которых m равно 0 или 2. Если X означает CH2, а n равно 1, то наиболее предпочтительными являются соединения, описывающиеся трансстереохимической формулой:
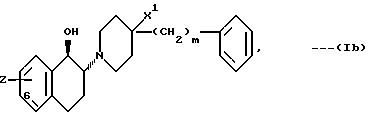
В частности, это соединения, у которых Z означает OH и которые замещены в 6-положении 1,2,3,4-тетрагидронафталиновой кольцевой системы. Наиболее предпочтительными соединениями формулы (Ib) являются те из них, у которых X1 означает OH, а m равно 0. Если n равно 0, то наиболее предпочтительными являются соединения, у которых X означает CH2, X1 OH, X OH, и которые замещены в 5-положении индановой кольцевой системы, а m у них равно 0.
Предметом настоящего изобретения являются также
промежуточные соединения формулы:
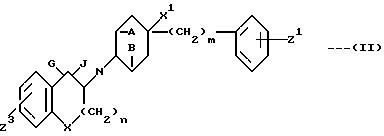
в которой A и B, X, X1, Z1, m и n имеют вышеприведенные определения;
G и J взяты вместе и означают атом кислорода (образующий карбонильную группу) или G и J взяты по отдельности и G означает H, а J гидроксильную группу;
Z3 H, F, Cl, Br или OR;
R переставляет H или обычную защитную группу для гидроксигруппы, при условии, что когда G и H взяты по отдельности, Z3 означает OR1, а R1 обычную защитную группу для гидроксигруппы, а также промежуточные соединения формулы:
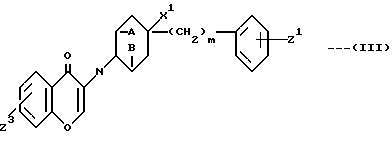
в которой все заместители имеют вышеприведенные определения.
Предметом настоящего изобретения являются далее фармацевтические композиции, содержащие соединение формулы (I), а также способ лечения приступов или дегенеративных заболеваний центральной нервной системы соединением формулы (I).
Под вышеупомянутом выражением "фармацевтически приемлемые соли" во всех случаях без исключения имеются в виду обычные кислые аддитивные соли. Так, соединения формулы (I) содержат аминогруппу, которая является основной, и поэтому они могут образовывать такие соли. Указанные соли включают, но ими однако круг возможных солей не ограничивается, соли с HCl, HBr, HNO3, H2SO4, H3PO4, CH3SO3H, n-CH3C6H4SO3H, CH3CO2H, глюконовой, винной, малеиновой и янтарной кислотами. Обычно их получают обычными методами, например, путем смешения соединения формулы (I) с по меньшей мере одним молярным эквивалентом кислоты в среде подходящего растворителя. Соединения формулы (I), содержащие фенольную оксигруппу, также способны образовывать катионные соли (например, натриевые, калиевые и т.п.), и поэтому выражением "фармацевтически приемлемые соли" охватываются и соли этого типа. Эти соли также могут быть получены обычными способами, например, путем взаимодействия фенольного соединения формулы (I) с одним молярным эквивалентом NaOH или KOH в среде подходящего растворителя.
Как уже отмечалось, получение обладающих фармакологической активностью соединений формулы (I) в соответствии с настоящим изобретением не вызывает никаких затруднений.
В том случае, если в соединении формулы (I) Z означает OH, то непосредственным предшественником его является соответствующее соединение вышеуказанной формулы (II), у которого G и H имеют различные значения, причем G означает атом водорода, а H гидроксильную группу, Z3 означает OR, а R обычную защитную группу для оксигруппы. Эту защитную группу удаляют на последней стадии обычными методами. Указанные группы предпочтительно защищают, переводя соединения в обычные простые силиловые эфиры, т.е. R означает, например, триизопропилсилил или третбутилдиметилсилил. Предпочтительным методом удалений таких силильных групп является обработка соединения 1-1,1 молярными эквивалентами фтористого тетрабутиламмония в среде инертного в условиях проведения реакции растворителя, например, тетрагидрофурана. Реакцию обычно проводят при температуре примерно 0-50oC, предпочтительно при температуре окружающей среды, для того чтобы избежать затрат на нагревание или охлаждение реакционной смеси.
Как в предыдущем абзаце, так и везде в дальнейшем, под выражением "инертный в условиях проведения реакции растворитель" имеется в виду любой растворитель, который не взаимодействует с исходными материалами, реагентами, промежуточными соединениями или продуктами, оказывая какое-либо отрицательное влияние на выход целевого продукта.
Соединения
формулы (I), у которых Z имеет иное, чем OH, значение, а также промежуточные соединения формулы (II), у которых Z3 означает защитную группу для оксигруппы, как правило, получают путем
обычного гидридного восстановления альфа-аминокетона, например:
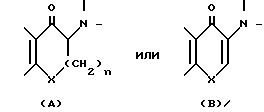
в результате которого обычно образуется смесь цис- и трансизомеров, например, соответственно:
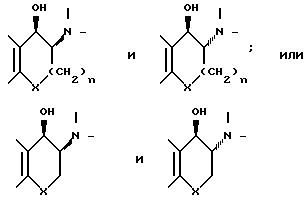
Естественно, в отдельных случаях в смеси будет преобладать один или другой из этих цис- и трансизомеров.
Указанные реакции гидридного восстановления осуществляются с использованием обычных гидридов-восстановителей, например, NaBH4 или LiAlH4. Последний из названных гидридов обычно берется в избытке (например, моль на моль). Реакцию проводят в среде инертного растворителя, например, тетрагидрофурана, при пониженной температуре (например, -15-15oC). По другому варианту промежуточные кетоны, в частности содержащие сложные эфирные группы, восстанавливают с помощью более мягкого гидрида-восстановителя, например, NaBH4, который обычно также берется в избытке. Реакцию в этом случае проводят в среде протонного растворителя, например, метанола или этанола, при несколько повышенной температуре, например, при 15-45oC. Все защитные группы, еще остающиеся после восстановления кетона, удаляют затем вышеописанными способами.
Вышеупомянутые промежуточные соединения типа (A) обычно получают путем взаимодействия соответствующего монобромистого соединения с замещенным соответствующим образом амином
по реакции:
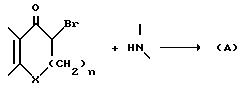
Для специалиста в данной области очевидно, что находящийся в альфа-положении атом брома может быть заменен другой, способной к замещению нуклеофильной группой (например, Cl или OSO2CH3). Эту реакцию проводят в условиях, обычных для реакций нуклеофильного замещения. Если оба реагента примерно одинаковы по доступности, то их можно брать в приблизительно молярном эквивалентном соотношении. Если же один из них более доступен, то его обычно желательно брать в избытке, для того чтобы завершить эту бимолекулярную реакцию за более короткий промежуток времени. Реакцию, как правило, проводят в присутствии по меньшей мере одного молярного эквивалента основания обычно, если оно легко доступно, самого производного пиперидина, чаще однако третичного амина по меньшей мере сравнимого по силе основания с использующимся нуклеофильным пиперидином, а также инертного в условиях проведения реакции растворителя, например этанола. При желании, для ускорения реакции к реакционной смеси добавляют в качестве катализатора до одного молярного эквивалента или более соля йода (например, NaI или KI). Температура не имеет существенного значения, однако обычно ее немного повышают для того, чтобы реакция завершилась за более короткий промежуток времени. Однако она не должна быть настолько высокой, чтобы происходили нежелательные процессы разложения. Обычно достаточной оказывается температура в пределах 50-120oC. Предпочтительной является температура кипения реакционной смеси.
Вышеупомянутые промежутки соединения типа (B) обычно получают путем взаимодействия соответствующего альфа, альфа-дибром-замещенного соединения с
соответствующим образом замещенным амином по реакции:
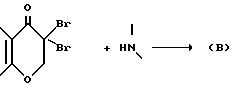
За исключением того, что основание берут в по меньшей мере на один молярный эквивалент большем количестве (для нейтрализации HBr, образующегося при конкурентной реакции дегидрогалогенирования), остальные условия реакции аналогичны вышеприведенным при описании получения соединений типа (A) по реакции нуклеофильного замещения.
Соединения формулы (I) содержит два асимметрических атома углерода, в соответствии с чем имеются два рацемата и четыре оптически активных соединения. Одним из этих рацематов является вышеупомянутый цис-, а другим - трансизомер. Каждый из этих рацематов путем добавления диастереоизомерных кислых аддитивных солей с оптически активной кислотой может разделить на пару энантиомеров. По другому варианту для этой цели рацемический спирт переводят в соответствующие диастереоизомерные сложные эфиры или уретаны с использованием в качестве исходного материала оптически активной кислоты или изоцианата. Такие производные с ковалентными связями могут быть подвергнуты разделению различными методами (например, путем хроматографии). Такие диастереоизомерные эфиры получают из спирта и оптически активной кислоты стандартными методами, обычно включающими активацию кислоты, например, путем перевода ее в хлорангидрид или смешанный ангидрид с помощью алкилхлорформиата или дегидратирующего сочетающего реагента, например, дициклогексилкарбодиимида. После разделения полученных таким образом диастереоизомерных эфиров, например, с помощью хроматографических методов, их гидролизуют обычными способами, например, водным раствором кислоты или основания, получая в результате энантиомерный, оптически активный спирт формулы (1). В намерения заявителя не входит, чтобы настоящая заявка ограничивалась лишь приведенными ниже конкретными примерами рацемических цис- и транссоединений.
Исходные материалы и реагенты, необходимые для синтеза соединений в соответствии с настоящим изобретением, являются легко доступными; они выпускаются промышленностью или могут быть получены описанными в литературе или приведенными ниже в качестве примеров методами.
Заявляемые соединения вышеуказанной формулы (1) обладают селективной нейрозащитной активностью, основанной на их противоишемической активности и способности блокировать стимулирующие аминокислотные рецепторы. В то же время они не оказывают существенного гипотензивного действия или имеют пониженную гипотензивную активность. Противоишемическая активность соединений в соответствии с настоящим изобретением определялась одним или несколькими ранее детально описанными Gotti, Carter (см. выше) или аналогичными методами. Способность соединений в соответствии с настоящим изобретением блокировать стимулирующие аминокислотные рецепторы показана на примере блокирования или индуцированного N-метил-D-аспарагиновой кислотой (NMDA) увеличения цГМФ в мозжечках новорожденных крыс по следующей методике. Иссекали быстро мозжечки у десяти 8-14-дневных крыс породы Wistar и помещали их при 4oC в Krebs (бикарбонатный буфер с pH 7,4, после чего разрезали на кусочки 0,5х0,5 мм с помощью ножа для препаративной обработки тканей McIlvain /The Nickle Laboratory Engineering Co. Gomshall, Surrey, Англия). Приготовленные таким образом кусочки мозжечков переносили в 100 мл Krebs (бикарбонатного буфера при 37oC, через который непрерывно пропускали смесь O2 и CO2 в соотношении 95:5. Кусочки буфера выдерживали в этих условиях в течение 90 мин, три раза за это время меняя буфер, после чего буфер декантировали, а ткань отделяли путем центрифугирования (1 мин, 3200 об/мин) и вновь суспендировали в 20 мл Krebs) бикарбонатном буфере. После этого отбирали аликвотные пробы по 250 мкл (примерно 2 мг) и помещали их в микроцентрифужные пробирки на 1,5 мл. В пробирки добавляли по 10 мкл испытуемого соединения из исходного раствора и после выдержки в течение 10 мин 10 мкл 2,5 мМ раствора NMDA для инициирования реакции. Конечная концентрация NMDA в растворе составляла 100 мкМ. Пробирки выдерживали в течение одной минуты при 37oC на встряхиваемой водяной бане, после чего добавляли в них 750 мкл раствора 50 мМ трис-Cl, 5 мМ ЭДТА для прекращения реакции. После этого пробирки сразу же помещали на кипящую водяную баню и выдерживали на ней в течение 5 мин. Содержимое всех пробирок обрабатывали затем в течение 15 с ультразвуком с помощью ультразвукового зонда при положении переключателя уровня мощности "три". Отбирали из пробирок пробы по 10 мкл и определяли в них содержания белка по методу Lowry, Anal. Biochem. 100: 201-220,1979. Пробирки затем центрифугировали (5 мин, 10000 xg), отбирали пробы надосадочной жидкости по 100 мкл и определяли в них содержание циклического гуанозинмонофосфата (цГМФ) с помощью радиоиммуноанализа на цГМФ New England Nuclear (Bost on, Mass a chusetts) методом suppli er. Данные представляли в виде пМ цГМФ, генерируемого на мг белка. Нежелательную гипотензивную активность также определяли известными методами, например, по методикам Carron (см. выше).
Такая селективная нейрозащитная противоишемическая активность соединений в соответствии с настоящим изобретением и их способность блокировать стимулирующие аминокислотные рецепторы говорят о возможности их использования для лечения дегенеративных расстройств центральной нервной системы, таких как приступы, болезнь Альцгеймера, болезнь Паркинсона и болезнь Хантингтона, не вызывая при этом существенной опасности чрезмерного снижения кровяного давления. При систематическом лечении таких болезней нейрозащитным количеством соединений формулы (I) дозировка их составляет от примерно 0,02 до 10 мг/кг в день (1-500 мг/день) для типичного пациента весом 50 кг, в виде единичной или разделенной на несколько приемов дозы, независимо от способа введения. Естественно, в зависимости от природы соединения и конкретного заболевания, лечащий врач может прописать дозу, выходящую за рамки указанного интервала. Предпочтительным является оральный способ введения лекарства. Однако, если пациент не в состоянии глотать, или оральный прием нежелателен по еще каким-то причинам, предпочтительно является парентеральное введение (внутримышечно, внутривенно) или местное применение.
Соединения в соответствии с настоящим изобретением обычно вводятся в виде фармацевтических композиций, включающих по меньшей мере одно соединение формулы (1) в комбинации с фармацевтически приемлемым носителем или разбавителем. Такие композиции, как правило, получают обычными способами с использованием твердых или жидких носителей или разбавителей в зависимости от способа введения (для орального введения в виде таблеток, твердых или мягких желатиновых капсул, суспензий, гранул, порошков и т.п. для парентерального введения в виде растворов или суспензий для инъекций и т.п. для местного применения в виде растворов, лосьонов, мазей, бальзамов и т.п.).
Нижеследующие примеры иллюстрируют настоящее изобретение. Последнее однако не ограничивается деталями этих примеров.
Все реакции в неводных средах для избежания осложнений и увеличения выхода проводились в атмосфере азота. Все растворители и разбавители высушивались по опубликованным стандартным методикам или покупались уже высушенными. Все реакционные смеси в процессе реакции перемешивались магнитной или механической мешалкой.
ЯРМ-спектры снимались при 300 мГц. Полученные данные приведены в м.д. В качестве растворителя при снятии ЯРМ-спектров, если это специально не оговорено, использовали CDCl3. Данные, полученные при снятии ИК-спектров, приведены в см-1. Как правило, регистрировались только сильные сигналы. Использовались аббревиатуры: ДМФ диметилформамид, ТГФ - тетрагидрофуран, HRMS масс-спектрометрия высокого разрешения.
Пример 1. 3-/4-Окси-4-фенилпиперидино/-7-/триизопропилсилилокси/хромен- 4-он.
3, 3-дибром-7-/триизопропилсилилокси/-4-хроманон /5,0 г, 10,5 ммоля/ растворяли в CH3CN /150 мл/ и добавляли к полученному раствору 4-окси-4-фенилпиперидин (2,2 г, 12,5 ммоля) и триэтиламин (2, 9 мл, 20,8 ммоля). Смесь перемешивали в течение ночи при температуре окружающей среды, после чего концентрировали и остаток распределяли между этилацетатом и водой. Органический слой промывали водой (2•50 мл) и рассолом, высушивали над CaSO4, концентрировали и остаток подвергали хроматографическому разделению с градиентным элюированием смесью этилацетата и гексана, получая в результате целевой продукт в виде белого твердого вещества (2,3 г, 54%). Часть полученного продукта перекристаллизовывали из смеси этанола и этилового эфира. Температура плавления 163-163,5o C; ИК (KBr) 3437, 2950, 2870, 1635, 1615, 1600, 1447, 1285, 1247, 1200, 1185, 703, 690.
Результаты анализа из расчета на формулу C29H39NO4Si.
Рассчитано C, 70,55; H, 7,96; N, 2,84.
Найдено C, 70,44; H, 7,76; N, 2, 84.
Из последующих фракций в процессе хроматографического разделения получали дополнительно 0,61 г продукта, а именно 7-окси-3-/4-оки-4-фенилпиперидино/хромен-4-она, образующегося в результате десилилирования в процессе реакции. Это соединение также можно использовать в качестве промежуточного соединения при получении нижеописанных продуктов аналогичными методами.
Пример 2. Цис- и транс-3-/4-окси-4-фенилпиперидино/-7- /триизопропилсилилокси/-4-хроманол.
Целевой продукт из предыдущего примера (2,0 г, 4,1 ммоля) растворяли в этаноле (75 мл) и добавляли к полученному раствору в один прием NaBH4 (1,5 г, 39,7 ммоля). Смесь перемешивали в течение ночи при комнатной температуре, после чего к ней добавляли еще порцию NaBH4 (0,75 г, 19, 9 ммоля) и продолжали перемешивание в течение еще 5 ч. Затем реакцию прекращали, добавляя к смеси избыток воды, смесь концентрировали и остаток распределяли между этилацетатом и водой. Органический слой промывали водой и рассолом, высушивали над CaSO4 и концентрировали, получая в результате твердое вещество желтоватого цвета, которое перекристаллизовывали из смеси этилового эфира и гексана. В результате получали 1,0 г (50%) целевого продукта (в цис-форме) в виде белого твердого вещества. Температура плавления 145,5-146,5oC. ИК (KBr): 3380, 2940, 2860, 1615, 1280, 1173, 1040.
Результаты анализа из расчета на формулу C29H43NO4Si.
Рассчитано C, 69,98; H, 8,71; N 2,81.
Найдено C, 70,02; H, 8,58; N 2,81.
После хроматографии на силикагеле полученного в результате перекристаллизации фильтрата с градиентным элюированием смесью этилацетата и гексана получали дополнительно 70 мг целевого продукта (в цисформе) и еще 0,27 г (14% ) твердого вещества желтого цвета, которое, как показали данные ЯМР-спектроскопии, представляло собой смесь транс- и цисформ целевого продукта в соотношении 85:15.
Эту смесь использовали без какой-либо обработки для дальнейшего синтеза трансформы.
13C-ЯМР (транс) 156,7, 154,5, 148,2, 128,8, 128,4, 127, 2, 117,2, 113,4, 107,2, 71,4, 64,8, 64,1, 63,4, 48,4, 43,0, 39,0, 17,9, 12,7. HRMS: рассчитано на MH+: 498,3041; получено: 498,3011.
Пример 3. Цис-3-/4-окси-4-фенилпиперидино/-4,7-хромандиол.
К раствору цисформы целевого соединения из предыдущего примера (0,94 г, 1,89 ммоля) в ТГФ добавляли IM раствор фтористого тетрабутиламмония в ТГФ (1,95 мл, 1,95 ммоля). Полученный раствор перемешивали при температуре окружающей среды в течение 1,5 ч, после чего концентрировали и подвергали хроматографическому разделению на силикагеле с градиентным элюированием смесью этилацетата и гексана. В результате получали целевой продукт (0,72 г), который перекристаллизовывали из смеси этанола и этилового эфира, получая 0,54 г (84%) белого твердого вещества. Температура плавления 171,5-172,5oC.
Результаты анализа из расчета на формулу C20H23NO4•0,25H2O.
Рассчитано C, 69,45; H, 6,84; N, 4,05.
Найдено C, 69,26; H, 6,79; N, 3,96.
Пример 4. Транс-3-/4-окси-4-фенилпиперидино/-4,7-хромандиол.
Таким же образом, как и в предыдущем примере, целевой продукт (в трансформе) примера 2 (0,27 г, 0,54 ммоля) с содержанием 15% цисизомера переводили в сырой продукт (0,17 г) в виде маслянистого белого твердого вещества, который перекристаллизовывали из этанола, получая в результате 57 мг (30%) целевого продукта в соответствии с данным примером в виде белого твердого вещества. Температура плавления 192,5-193oC.
Результаты анализа из расчета на 2 формулу C20H23No4.
Рассчитано C, 70,36; H, 6,79; N, 4, 10.
Найдено C, 70,06; H, 6,88; N, 4,04.
Пример 5. 3-/4-Окси-4-фенилпиперидино/-7-/триизопропилсилилокси/-4-хроманон.
7-/триизопропилсилилокси/-4-хромано (2,0 г, 6,2 ммоля) растворяли в CCl4 (45 мл) и добавляли к полученному раствору по каплям при температуре окружающей среды раствор брома (0,3 мл, 6,4 ммоля) в CCl4 (5 мл) в течение 10 мин. Вначале реакционная смесь становилась темно-красной, однако после перемешивания в течение 10 мин цвет ее изменялся на желтоватый. Этот желтый раствор промывали разбавленным, а затем насыщенным раствором NaHCO3 и рассолом, высушивали путем фильтрования через фазоразделительную бумагу и концентрировали, получая в результате маслянистую жидкость коричневого цвета, которая по данным ЯМР представляла собой смесь 3-бром-7-/триизопропилсилилокси/-4-хроманона, 3,3-дибром-7-/триизопропилсилилокси/-4-хроманона и исходного материала в соотношении 2,5:1:1. Эту смесь (2,3 г, 5,5 ммоля) смешивали с 4-окси-4-фенилпиперидином (1,0 г, 5,8 ммоля), триэтиламином (0,9 мл, 6,5 ммоля) и этанолом (50 мл). Реакционную смесь кипятили в течение 3 ч с обратным холодильником, после чего охлаждали и концентрировали. Остаток распределяли между этилацетатом и водой. Органический слой промывали водой и рассолом, высушивали над CaSO4 и остаток подвергали хроматографическому разделению на силикагеле с градиентным элюированием смесью этилацетата и гексана. В результате получали 80 мг (3%) целевого соединения в виде желтого твердого вещества. Температура плавления 132-132, 5oC.
Пример 6. Цис-3-/4-окси-4-фенилпиперидино/-4,7-хромандиол.
Целевой продукт из предыдущего примера (80 мг, 0,16 ммоля) растворяли в этаноле (10 мл) и добавляли к полученному раствору NaBH4 (7 мг, 0,2 ммоля). Смесь перемешивали при температуре окружающей среды в течение 6 ч, после чего реакцию прекращали путем добавления воды и смесь концентрировали. Остаток распределяли между этилацетатом и водой. Органический слой промывали водой и рассолом, высушивали над CaSO4 и концентрировали, получая в результате сырой продукт (цисформа) в соответствии с примером 4 в виде маслянистой жидкости желтого цвета (50 мг, 63%). Полученный продукт подвергали десилилированию таким же образом, как это описано в примере 3, получая в результате соединение (15 мг, 44%), идентичное продукту в соответствии с примером 3.
Примеры 7-13.
Таким же образом,
как это описано в примере 1, используя в качестве
исходных материалов соответствующим образом замещенные 3,3-дибром-4-хроманон и производное пиперидина, получали дополнительно следующие соединения (с
указанными выходами и свойствами):
7.
3-/4-бензил-4-оксипиперидино/-7-/триизопропилсилилокси/ хромен-4-он; 34% Температура плавления 115-116oC (после перекристаллизации из смеси
этилового эфира и гексана);
8.
3-/4-фенилпиперидино/-7-/триизопропилсилилокси/хромен-4-он; температура плавления 99-100oC (после перекристаллизации из смеси этилового эфира и
гексана);
9.
3-/4-бензилпиперидино/-7-/триизопропилсилилокси/хромен-4-он; 38% маслянистая жидкость;13C-ЯМР: 160,7, 157,2, 143,6, 140,5, 137,0,129,2, 128,2, 127,6, 125,8, 118,4,
106,7, 50,7, 43,3, 37,8,
32,1, 17,8, 12,3;
10. 3-[4-окси-4-/2-фенилэтил/пиперидино]-7- /триизопропилсилилокси/хромен-4-он; 2% маслянистая жидкость;13C-ЯМР: 174,7, 160,9, 157,4,
144,3, 142,3, 136,8, 128,5,
128,3, 127,6, 125,9, 118,7, 106,8, 69,5, 46,3, 45,0, 36,8, 29,3, 17,9 12,7;
11. 6-хлор-3-/4-окси-4-фенилпиперидино/-хромен-4-он; 40% температура плавления 191,
5-192oC (после
перекристаллизации из смеси CHCl3 и этилового эфира);
12. 6-фтор-3-/4-окси-4-фенилпиперидино/-хромен-4-он; 40% температура плавления 183,5-184o
C (после
перекристаллизации из смеси CHCl3 и этилового эфира);
13. 3-/4-окси-4-фенилпиперидино/хроман-4-он; 85% температура плавления 168-168,5oC (после
перекристаллизации из
смеси этанола и этилового эфира).
Примеры 14-20.
Таким же образом, как это описано в примере 2, используя в качестве исходного материала продукты
в соответствии с
примерами 7-13, получали следующие соединения (с указанными выходами и свойствами);
14. цис-3-/4-бензил-4-оксипиперидино/-7-/триизопропилсилилокси/ -4-хроманол; 29%
температура плавления 172,
0-172,5oC (после перекристаллизации из смеси этанола и этилового эфира) и смесь (в соотношении 2:1) цис- и трансизомеров
3-/4-бензил-4-оксипиперидино/-7-/триизопропилокси/-4-хроманолов /40%
/, которую можно было разделить на цис- и трансизомеры с помощью колоночной хроматографии;
15.
цис-3-/4-фенилпиперидино/-7-/триизопропилсилилокси/-4- хроманол; 69% температура плавления
148-148,5oC (после перекристаллизации из смеси этанола и этилового эфира);
16.
цис-3-/4-бензиплпиперидино/-7-/триизопропилсилилокси/-4- хроманол; 55% маслянистая жидкость;13C-ЯМР: 157,2, 154,8, 140,4, 131,7 129,1, 128,2, 125,9, 115,3, 113,4, 107,1, 62,3, 61,7, 60,8,
51,5, 49,3, 43,1, 37,8, 32,3, 32,2 17,9, 12,7;
17.
цис-3-[4-окси-4-/2-фенилэтил/пиперидино]-7- /триизопропилсилилокси/-4-хроманол; 25% твердое белое вещество;
18.
цис-6-хлор-3-/4-окси-4-фенилпиперидино/-4-хроманол; 16% температура плавления
185-185,5oC (после перекристаллизации из смеси этанола и этилового эфира) и смесь цис- и транс-изомеров в
соотношении 3:2 (37%), оторую можно разделить на отдельные изомеры с помощью
хроматографии;
19. цис-6-фтор-3-/4-окси-4-фенилпиперидино/-4-хроманол; 28% температура плавления 189-189,5oC (после перекристаллизации из смеси этанола и этилового эфира) и смесь
цис- и транс-изомеров в соотношении 2:1 (28%), из которой путем фракционной кристаллизации получено 80% чистого
трансизомера, температура плавления 164-168oC (общий выход 4%);
20.
цис-3-/4-окси-4-фенилпиперидино/-4-хроманол; 58% температура плавления 187,5-188oC (после
перекристаллизации из смеси этанола и этилового эфира); и полученная путем кристаллизации из
маточного раствора смесь цис- и трансизомеров в соотношении 1:3; 3% температура плавления 170-174oC.
Примеры 21-24.
Таким же образом, как это описано в
примере 3, используя в качестве исходного материала продукты, полученные в соответствии с примерами 7-10,
получали следующие соединения (с указанными выходами и свойствами):
21.
цис-3-/4/бензил-4-оксипиперидино/-4,7-хромандиол; 85% температура плавления 181-182oC (после
перекристаллизации из смеси этанола и этилового эфира);
22. цис-3-/4-фенилпиперидино/-4,
7-хромандиол; 67% температура плавления 195,0-195,5oC (с разложением) (после
перекристаллизации из смеси этанола и этилового эфира);
23. цис-3-/4-бензилпиперидино/-4,7-хромандиол;
31% температура плавления 164,6-165,0oC (после перекристаллизации из смеси
этанола и этилового эфира);
24. цис-3-[4-окси-4-/2-фетилэтил/пиперидино]-4,7-хромандиол; 54% температура
плавления 97-100oC.
Пример 25. 2-/-Окси-4-фенилпиперидино/-6-метокси-1-тетралон.
Таким же образом, как это описано в примере 1, используя в качестве исходного материала 2-бром-6-метокситетралон (2,8 г, 11,5 ммоля), 4-окси-4-фенилпиперидино (2,5 г, 14,1 ммоля) и триэтиламино (4,0 мл, 28,7 ммоля), путем перемешивания смеси в течение ночи в ацетонитриле (75 мл) получали целевое соединение. Сконцентрированный продукт подвергали хроматографическому разделению на силикагеле с градиентным элюированием смесью этилацетата и гексана. В результате получали 1,33 г (33%) целевого соединения. Температура плавления 149,5-150,5oC (после перекристаллизации из смеси этанола и этилового эфира).
Пример 26. Цис- и транс-1,2,3,4-тетрагидро-2-/4-окси-4-фенилпиперидино/-6- метокси-1-нафтол.
Таким же образом, как это описано в примере 2, целевой продукт из предыдущего примера (1,0 г, 2,85 ммоля) переводили в целевые соединения в соответствии с настоящим примером. Разделение осуществляли с помощью хроматографии на силикагеле (с использованием градиентного элюирования смесью этилацетата и гексана), после чего полученные продукты перекристаллизовывали из смеси этанола и этилового эфира.
Трансизомер: 0,13 г (13%), более полярный; температура плавления 155-155,5o C.
Цисизомер: 0,033 г (3% ), менее полярный; температура плавления 159-160oC.
Примеры 27-28.
Таким же образом, как это описано в примере 25,
соответствующим образом замещенные 2-бром-1-тетралоны переводили в
следующие соединения:
27. 2-/4-окси-4-фенилпиперидино/-1-тетралон; 21% температура плавления 148-151oC (с
разлож.) ( после перекристаллизации из смеси этанола и этилового эфира);
28. 2-/4-окси-4-фенилпиперидино/-6-/триизопропилсилилокси/-1- тетралон; температура плавления 151-153oC
(после перекристаллизации из смеси этанола и этилового эфира); 36%
Пример 29. Транс-1,2,3,4-тетрагидро-2-/4-окси-4-фенилпиперидино/-1-нафтол.
Таким же образом, как это описано в примере 26, продукт в соответствии с примером 27 переводили в целевое соединение. Выход: 5% температура плавления 184-184,5oC.
Пример 30. Транс-1,2,3, 4-тетрагидро-2-/4-окси-4-фенилпиперидино/-6-/ триизопропилсилилокси/-1-нафтол.
Продукт в соответствии с примером 28 (0,75 г, 1,61 ммоля) в виде раствора в тетрагидрофуране (25 мл) добавляли по каплям в течение 10 мин к перемешиваемой суспензии LiAlH4 (0,065 г, 1,71 ммоля) в тетрагидрофуране (75 мл). Образующуюся серо-зеленую смесь перемешивали в течение 30 мин при температуре окружающей среды, после чего реакцию прекращали, добавляя к смеси избыток Na2SO4•10H2O. После перемешивания в течение 15 мин, обработанную таким образом смесь высушивали над Na2SO4, концентрировали до 0,65 г и подвергали хроматографическому разделению на силикагеле путем градиентного элюирования смесью этилацетата и гексана. В результате получали 0,45 г (60%) целевого соединения. Температура плавления 171, 0-171,5oC (после перекристаллизации из смеси этанола и этилового эфира).
Пример 31. Транс-1,2,3,4-тетрагидро-2-/4-окси-4-фенилпиперидино/-1,6- нафталиндиол.
Таким же образом, как это описано в примере 3, продукт в соответствии с примером 30 (0,35 г, 0,75 ммоля) переводили в целевое соединение. Выход: 0,12 г (46%); температура плавления 181-183o C (после перекристаллизации из смеси этанола и этилового эфира); ИК (KBr): 3380, 3230, 2950, 2850, 1610, 1495, 1240, 1110, 1045, 7706 705.
Пример 32. 5-/Триизопропилсилилокси/-2-/4-окси-4-фенил-пиперидино/- 1-инданон.
Таким же образом, как это описано в примере 1, 2-бром-5- /триизопропилсилилокси/-1-инданон переводили в целевое соединение. Выход: 41% (в виде пенообразного твердого продукта):13C-ЯМР: 203,3, 163,2, 154,9, 148,1, 129,8, 128,5 128,4, 127,0, 125,9, 124,5, 120,5, 116,7, 71,0, 69,4, 46,2, 44,5, 42,0, 38,2, 37,3, 27,3, 18,0 12,7.
Пример 33. Цис- и транс-5-/триизопропилсилилокси/-2-/4-окси-4- фенилпиперидино/-1-инданол.
Таким же образом, как это описано в примере 2, целевой продукт из предыдущего примера переводили в целевые соединения в соответствии с настоящим примером, которые разделяли с помощью хроматографии на силикагеле с использованием градиентного элюирования смесью этилацетата и гексана.
Цисизомер: выход 27% температура плавления 169,5-170oC (после перекристаллизации из смеси этилового эфира и гексана); ИК (KBr): 3467, 2959, 2894, 2867, 1610, 1490, 1294, 1138, 964, 883, 698.
Трансизомер: выход 43% температура плавления 143-144oC; ИК (KBr): 3321, 2945, 2867, 1613, 1490, 1465, 1291, 1265, 1135, 966, 702, 681.
Примеры 34-35.
Таким же образом, как это описано в примере 3, целевой продукт из предыдущего примера переводили в
34.
цис-2-/4-окси-4-фенилпиперидино/-1,5- индандиол; 54% температура плавления
212,5-213,5oC;13C-ЯМР: 157,7, 150,2, 143,3, 134,8, 127,9, 126,2, 126,1, 124,8, 113,5, 111,2, 71,5, 69,
7, 69,6, 47,8, 47,1 38,0, 37,9, 34,2;
35.
транс-2-/4-окси-4-фенилпиперидино/-1,5-индандиол; 71% температура плавления 196,0-197,0oC;13C-ЯМР: 157,1, 150,3, 140,8, 135,6,
127,8, 126,1, 124,9, 124,8, 113,8, 110,7, 76,7, 75,
2, 69,7, 47,3, 38,1, 33,9.
Методика 1. 7-/Триизопропилсилилокси/-4-хроманон.
7-окси-4-хроманон (1,2 г, 7,3 ммоля; Dann и др. Ann. 587, 16, 1954) и имидазол (1,0 г, 14,7 ммоля) растворяли в ДМФ (10 мл). К полученному раствору добавляли по каплям в течение 10 мин при температуре окружающей среды раствор триизопропилсилилхлорида (1,8 мл, 8,2 ммоля) в ДМФ (2 мл). После перемешивания в течение 3 ч смесь выливали на 100 мл льда и воды и подвергали экстракции диэтиловым эфиром (2•100 мл). Объединенные эфирные экстракты промывали 1 М LiCl и затем рассолом, высушивали над CaSO4 и концентрировали до образования коричневой маслянистой жидкости, которую подвергали очистке путем дистилляции с шариковым дефлегматором (0,5 тор, 70-90oC). При этом удалялась примесная бесцветная вязкая маслянистая жидкость, а в перегонной колбе оставался продукт в виде коричневой маслянистой жидкости (2,0 г, 85%). ИК (KBr) 2945, 2867, 1685, 1605, 1268, 1163. HRMS: рассчитано на МН+: 320, 1807, получено: 320, 1842.
Методика 2. 3, 3-Дибром-7-/триизопропилсилилокси/-4-хроманон.
Целевой продукт из предыдущей методики (7,1 г, 22,1 ммоля) растворяли в четыреххлористом углероде (170 мл) и добавляли к полученному раствору при температуре окружающей среды по каплям в течение 20 мин раствор брома (2,5 мл, 48,5 ммоля) в CCl4 (30 мл). Реакционную смесь перемешивали в течение 0,5 ч до получения темно-красного раствора, который затем последовательно промывали разбавленным раствором NaHCO3 (100 мл), насыщенным раствором NaHCO3 (2•75 мл) и рассолом (100 мл), высушивали путем фильтрования через фазоразделительную бумагу и концентрировали до получения темно-оранжевой маслянистой жидкости (9,9 г, 94%).
13C-ЯМР: 179,0, 164,3, 161,9, 131,3, 116,6, 109,9, 107,5, 78,0, 60,9, 17,8, 12,7. HRMS: рассчитано на MH+: 479,0076; получено: 479,0066.
Методики 3-5.
Таким же способом, как это описано
в предыдущей методике, используя в качестве исходного материала соответствующим образом замещенный 4-хроманон,
получали следующие соединения:
3. 3,3-дибром-6-хлор-4-хроманон; 64% температура
плавления 128-129oC (после перекристаллизации из смеси этанола и диэтилового эфира); ИК (KBr): 3060,
2930, 1710, 1475, 1137, 838;
4. 3,3-дибром-6-фтор-4-хроманон; 70% температура
плавления 90-91oC (после перекристаллизации из смеси диэтилового эфира и гексана); ИК (KBr): 3380,
3080, 1705, 1690, 1485, 1275, 1235, 1170, 1127, 850, 727;
5. 3,
3-дибром-4-хроманон; 90% температура плавления 67-68oC (после перекристаллизации из смеси диэтилового эфира и
гексана); ИК (KBr): 3380, 1705, 1610, 1480, 1300, 818.
Методика 6. 2-Бром-6-метокситетралон.
6-метокситетралон (2,0 г, 11,4 ммоля) и бром (0,6 мл, 11,7 ммоля) кипятили в течение 30 мин в диэтиловом эфире (50 мл) с обратным холодильником. Реакционную смесь затем охлаждали, концентрировали и остаток распределяли между этилацетатом и разбавленным раствором NaHCO3. Органический слой промывали насыщенным раствором NaHCO3 и водой, высушивали над CaSO4 и концентрировали до получения маслянистой жидкости (2,83 г, 100%);1 H-ЯМР: 8,03 /д, J 9,0 Гц, 1Н/, 6,84 /дв. д J1=9,0 Гц, J2=2,7 Гц, 1Н/, 6,69/д, J=2,3 Гц, 1Н/, 4,66 /т, J=4,1 Гц, 1Н/, 3,84 /c, 3H/, 3,20-3,30 /м, 1Н/, 2,82-2,90 /м, 1Н/, 2,34-2,50 /м, 2Н/.
Методика 7. 2-Бромтетралон.
Таким же образом, как и в предыдущей методике, тетралон (2,0 г, 13,7 ммоля) переводили в целевое соединение (2,7 г, 87%) в виде маслянистой жидкости.1H-ЯМР: 8,08 (д, J=7,9 Гц, 1Н), 7,51 /т, J=7,5 Гц, 1Н/, 7,23-7,36 /м, 2Н/, 4,72 /т, J=4,2 Гц, 1Н/ 3,25-3,36 /м, 1Н/, 2,92-2,97 /м, 1Н/, 2,40-2,58 /м, 2Н/.
Методика 8. 1-/Бензилоксикарбонил/-4-окси-4-/2-фенилэтил/пиперидин.
Стружки магния (1,7 г, 70,0 ммолей) суспендировали в диэтиловом эфире (10 мл) и добавляли к полученной суспензии по каплям раствор /2-бромэтил/бензола (11,8 г, 63,8 ммоля) в диэтиловом эфире (15 мл), сначала медленно, до начала реакции, а затем быстрее, для обеспечения выделения тепла. После нагревания реакционной смеси в течение ночи при 60oC ее охлаждали до 0oC, разбавляли диэтиловым эфиром (200 мл) и добавляли к ней по каплям раствор пиперидонбензилкарбамата (14,9 г, 62,9 ммоля) в диэтиловом эфире (100 мл). При этом выпадал белый осадок, и смесь интенсивно перемешивали при комнатной температуре в течение 8 ч, после чего реакцию прекращали путем добавления воды и продолжали перемешивание еще в течение часа. Водный слой затем отделяли, подвергали экстракции этилацетатом (3•100 мл), органические слои объединяли, промывали рассолом, высушивали на CaSO4 и концентрировали, получая в результате прозрачную маслянистую жидкость. Чистый продукт получали с помощью хроматографии на силикагеле (25% этилацетат/гексан), который представлял собой прозрачную маслянистую жидкость (9,2 г, 43%). ИК (CHCl3): 3585, 2939, 1692, 1470, 1429, 1363, 1311, 1275, 1260, 1190.
Методика 9. 4-Окси-4-/2-фенилэтил/пиперидин.
Целевой продукт из предыдущей методики (8,71 г, 25,66 моля) растворяли в атмосфере азота в этаноле (250 мл), добавляли к полученному раствору 10%-ный паладий на угле (936 мг)и смесь гидрировали в аппарате Парра при давлении 45-50 фунтов на кв. дюйм в течение 16 ч. Катализатор затем отделяли путем фильтрования через диатомовую землю, а маточный раствор концентрировали, получая в результате целевое соединение в виде желтого мяслянистого твердого материала (4,96 г, 99%).
ИК /CHCl3/: 3539, 2930, 1715, 1620, 1600, 1452, 1372, 1351, 1322, 1042.
Методика 10. 6-/Триизопропилсилилокси/-1-тетралон.
Таким же образом, как это описано в методике 1, 6-окси- 1-тетралон (5,0 г, 30,83 ммоля; Durden, J. Agr. Food Chem. т. 19, стр. 432, 1971) переводили в целевое соединение в виде маслянистой жидкости (после очистки путем дистилляции с шариковым дефлегматором). ИК /CHCl3/: 2937, 2889, 2862, 1666, 1593, 1349, 1333, 1319, 1274, 1226, 1109, 969, 898.
Методика 11. 2-Бром-7-/триизопропилсилилокси/-1-тетралон.
Таким же образом, как это описано в методике 6, целевой продукт из предыдущей методики (8,3 г, 26,1 ммоля) переводили в целевое соединение в соответствии с настоящей методикой (9,7 г, 94%), которое по данным1Н-ЯМР содержало, кроме того, некоторое количество 2, 2-дибром-производного. Полученный продукт без дополнительной очистки использовали на следующей стадии.
Методика 12. 5-/Триизопропилсилилокси/-1-инданон.
Таким же образом, как это описано в методике 1, 5-окси-1-инданон переводили в целевое соединение со 100% -ным выходом. Температура плавления 63,0-63,5oC.
Методика 13. 2-Бром-5-/триизопропилсилилокси/-1-инданон.
Таким же образом, как это описано в методике 6, целевой продукт из предыдущей методики переводили в целевое соединение в соответствии с настоящей методикой, загрязненное соответствующим дибром-продуктом. Выход 100%1H-ЯМР: 7,72 /д, 1Н/, 6,89 /дв. д, 1Н/, 6,83 /м, 1Н/, 4,62 /дв.д. 1Н/, 3,74 /дв. д, 1Н/, 3,34 /дв. д, 1Н/, 1, 22-1,34 /мБ 3Н/, 1,10 /д. 18Н/. Полученный продукт без дополнительной очистки непосредственно использовали на последующей стадии синтеза (см. выше пример 32).
Пример 36. Цис-3-(4-гидрокси-4-фенилпиперидино)-7-(триизопропилсилилокси)- 4-хроманол.
N-трет-бутоксикарбонил-d-аланин (0,190 г, 1,004 миллимоля) и карбонилдиимидозол (0,165 г, 1,018 миллимоля) растворялись в метиленхлориде (8 мл) и перемешивались в течение 1 ч при комнатной температуре. Добавлялся циспродукт примера 2 (0,250 г, 0,50 миллимоля), и получающаяся смесь перемешивалась всю ночь. Продукт реакции концентрировался, и осадок распределялся между этилацетатом и водой. Органическая фаза промывалась водой и солевым раствором, а затем сушилась над сульфатом магния. Концентрирование с последующей флэш-хроматографией на силикагеле (1х8 дюймов /25х200 мм), градиентное элюирование смесью (диэтиловый эфир/метиленхлорид) дали первый стереоизомер в виде пенообразного белого твердого вещества (0,115 г). Данное вещество перекристаллизовывалось из смеси диэтиловый эфир/петролейный эфир, давая порошкообразное твердое белое вещество (88 мг, 26%), точка плавления 152,5-153o C.1H ЯМР /CDCl3/ 7,46 /д. J 7,3 Гц, 2Н/, 7,34 /т. J 7,4 Гц, 2Н/, 7,22-7,27 /м. 1Н/, 7,15 /шир.д. J 8,2 Гц, 1Н/, 6,42 /дд. J 8,1 Гц, J 2,6 Гц, 1Н/, 6,34 /д. J 2,2 Гц, 1Н/, 6,18 /шир.с. 1Н/, 5,12 /д. J 6,5 Гц, 1Н/, 4,30-4,42 /м. 2Н/, 4,18 /т. J 10,9 Гц, 1Н/, 2,88-2,94 /м. 2Н/, 2,61-2,83 /м. 3Н/, 2,02 /д.кв. J 12,1 Гц, J 4,2 Гц, 2Н/, 1,71-1,79 /м. 2Н/, 1,53 /с. 1Н/, 1,35-1,42 /м. 12Н/, 1,10-1,31 /м. 3Н/, 1,07 /д. J 6,9 Гц, 18Н/.
Анализ. Вычислено для C37H56N2O7Si: C: 66,43, H: 8,44, N: 4,19.
Найдено: C: 66,14, H:8,66, N 4,18. /альфа/D -112,3o, C 1,020 в хлороформе.
Более поздние фракции от хроматографии дали второй стереоизомер в виде пенообразного белого твердого вещества (0,133 г). Данное вещество перекристаллизовывалось из смеси этиловый эфир/петролейный эфир, давая порошкообразное белое твердое вещество (73 мг, 22%), точка плавления 146-147, 5oC.
1H ЯМР /CDCl3/ 7,51 /д. J 7,9 Гц, 2Н/, 7,33 /т. J 7,5 Гц, 2Н/, 7,14 /д. J 8,2 Гц, 1Н/, 6,41 /дд. J 8,1 Гц, J 2,2 Гц, 1Н/, 6,34 /д. J 2,3 Гц, 1Н/, 6,25 /с. 1Н/, 5,03 /д. J 7,8 Гц, 1Н/, 4,36-4,39 /м. 1Н/, 4,36-4,39 /м. 1Н/, 4,22-4,30 /м. 2Н/, 2,95-3,07 /м. 2Н/, 1,59 /с. 1Н/, 1,39 /с. 9Н/, 1,15-1,33 /м. 6Н/, 1,06 /д. J 7,3 Гц, 18Н/, /альфа/D +115,7o, C 1,065 в хлороформе.
Пример 37. Оптический активный (-)-цис-3-(4-гидрокси-4-фенилпиперидино)- 4,7-хромандиол.
(-)-диастереомерный сложноэфирный продукт предыдущего примера (0,080 г, 0,12 миллимоля) растворялся в 10 мл 0,32-норм. раствора метоксида натрия (0,15 г Na, растворенные в 20 мл метанола). Смесь перемешивалась в течение ночи при нормальной температуре и затем концентрировалась. К остатку добавлялась 1 норм. HCl (2 мл) и затем насыщенный NaHCO3 (5 мл). Водная фаза экстрагировалась этилацетатом (3•20 мл). Объединенные экстракты промывались солевым раствором, сушились над сульфатом магния и концентрировались, давая молочное масло (39 мг).
Масло перекристаллизовывалось из смеси абсолютного этанола и этилового эфира, давая продукт в виде твердого вещества кремового цвета (17,9 мг, 44% ), точка плавления 166-168oC (разл.) /альфа/D -87,6o, C 0, 25 в метаноле. Энантиомерный избыток составлял ≥ 98% по данным1H ЯМР, с использованием реагента хирального сдвига, R-(-)-2,2,2-трифтор-1-(9- антрил) этанол.
Пример 38. Оптически активный (+)-цис-3-(4-гидрокси-4-фенилпиперидино)- 4,7-хромандиол.
По методу предыдущего примера /+/-диастереомерный сложноэфирный продукт примера 36 (0,065 г, 0,097
миллимоля) превращался в указанный в заголовке продукт, сначала в виде маслянистого белого твердого вещества (0,039 г), которое перекристаллизовывалось из смеси этанол/эфир, давая очищенный продукт,
указанный в заголовке (16,7 мг) в виде твердого вещества кремового цвета, точка плавления 163,5-166oC, /альфа/D +76,7o, C 0,27 в метаноле. По методу1H ЯМР
предыдущего примера определено, что энантиомерный избыток составил 93%
Нейрозащитное испытание приведено в таблице.
Реферат
Использование: в медицине в качестве нейрозащитных средств. Сущность изобретения: продукты: производные пиперидина ф-лы I, где A и B взяты вместе и представляют -CH2CH2-, или A и B взяты отдельно и каждый представляет H, X-CH2 или O, X1 - H или OH, Z -H, F, Cl или OH, Z1 -H, F, Cl, Br или /C1 -C3/алкил, n-0 или 1, m -0,1 или 2. Реагент: соединение ф-лы 2, где все заместители имеют значения, как указано для соединения I. Реагент 2: восстановитель, например, Na, BH4 или LiAlH4. Условия реакции: в среде инертного растворителя при минус 15- плюс 15oC. 3 с. и 14 з.п. ф-лы, 1 табл.
Формула
в которой A и B взяты вместе и представляют -CH2 CH2- или A и B взяты отдельно и каждый представляет H;
X CH2 или O;
X1 H или OH;
Z H, F, Cl или OH;
Z1 H, F, Cl, Br или C1-C3-алкил;
n 0 или 1;
m 0, 1 или 2.
4. Соединение по п. 3, в котором Z является замещенным в 7-положении хромановой кольцевой системы и представляет OH.
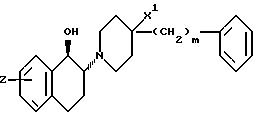
10. Соединение по п. 9, в котором Z является замещенным в 6-положении тетрагидронафталиновой кольцевой системы и представляет собой OH.
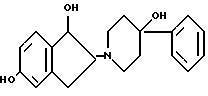
14. Соединение по п.13, имеющее относительную стереохимическую формулу
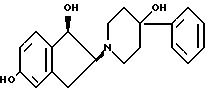
15. Соединение по п.13, имеющее относительную стереохимическую формулу
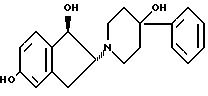
16. Производное пиперидина формулы
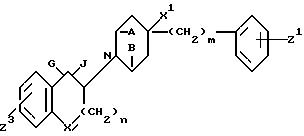
в которой A и B взяты вместе и представляют -CH2CH2- или A и B взяты отдельно и каждый представляет H;
G и J взяты вместе и представляют кислород или G и J взяты отдельно и G представляет водород, а J гидрокси;
X CH2 или O;
X1 H или OH;
Z1 H, F, Cl, Br или C1 C3-алкил;
Z3 H, F, Cl или OR;
R гидрокси защищающая триалкилсилильная группа;
n 0 или 1;
m 0, 1 или 2,
при условии, что, когда G и J взяты отдельно, Z3 OR и R - гидрокси защищающая триалкилсилильная группа.
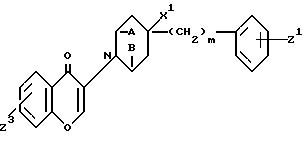
в которой A и B взяты вместе и представляют -CH2CH2- или A и B взяты отдельно и каждый H;
X1 H или OH;
Z1 H, F, Cl, Br или C1 C3-алкил;
Z3 H, F, Cl или OR;
R гидрокси защищающая триалкилсилильная группа;
m 0, 1 или 2.
Комментарии