Новые замещенные тетрациклические производные тетрагидрофурана, пирролидина и тетрагидротиофена - RU2401257C2
Код документа: RU2401257C2
Описание
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к новым замещенным тетрациклическим производным тетрагидропирана, пирролидина и тетрагидротиофена, обладающим аффинностями связывания по отношению к рецепторам серотонина, в частности рецепторам 5-HT2A и 5-HT2C, и по отношению к рецепторам допамина, в частности рецепторам допамина D2, и обладающим свойствами ингибирования обратного захвата норэпинефрина, к фармацевтическим композициям, содержащим соединения согласно изобретению, их применению в качестве лекарственных средств, в частности для профилактики и/или лечения ряда психиатрических и неврологических расстройств, в частности некоторых психотических, сердечно-сосудистых и гастрокинетических расстройств, и к способам их получения.
УРОВЕНЬ ТЕХНИКИ
В WO 97/38991, опубликованной 23 октября 1997 (Janssen Pharmaceutica N.V.), описаны замещенные тетрациклические производные тетрагидрофурана, которые могут быть использованы в качестве терапевтических средств для лечения или профилактики расстройств ЦНС, сердечно-сосудистых расстройств или желудочно-кишечных расстройств. В частности, соединения проявляют аффинность по отношению к рецепторам серотонина 5-НТ2, в частности по отношению к рецепторам 5-HT2A и 5-НТ2С.
В WO 99/19317, опубликованной 22 апреля 1999 (Janssen Pharmaceutica N.V.), описаны замещенные тетрациклические производные тетрагидрофурана со специфичной картиной замещения галогенами в цикле дибензоазепина, дибензооксепина, дибензотиепина или дибензосуберана. Соединения применимы для лечения или профилактики расстройств ЦНС, сердечно-сосудистых расстройств или желудочно-кишечных расстройств и проявляют более быстрое начало действия по сравнению с соединениями, которые описаны в WO 97/38991.
В обеих заявках на выдачу патента WO 03/048146, опубликованной 12 июня 2003 (Janssen Pharmaceutica N. V.), и WO 03/048147, опубликованной 12 июня 2003 (Janssen Pharmaceutica N. V.), описаны способы получения каждого из 4 диастереомеров цис- и соответственно транс-конденсированных производных 3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фурана в стереохимически чистой форме из одного энантиомерно чистого предшественника. Соединения проявляют аффинность по отношению к рецепторам серотонина 5-HT2A, 5-HT2C и 5-HT7 и рецепторам H1 (pIC50 = 7,15-7,89), рецепторам D2 и/или D3 и по отношению к переносчикам обратного захвата норэпинефрина (pIC50 = 6,01-7,34).
В WO 03/040122, опубликованной 15 мая 2003 (Janssen Pharmaceutica N.V.), описаны соли, образованные миндальной кислотой и соединениями согласно заявкам на выдачу патента WO 97/38991 и WO 99/19317. Неожиданно было обнаружено, что указанные соли являются более стабильными при повышенной температуре и относительной влажности, чем соединения, описанные в WO 97/38991 и WO 99/19317.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является получение новых аналогов тетрациклических производных тетрагидропирана согласно заявкам на выдачу патента WO 97/38991 и WO 99/19317, которые отличаются от таких производных тем, что они в общем проявляют большую избирательность по отношению к переносчику обратного захвата норэпинефрина, чем рецепторы 5-НТ2А/5-HT2C и рецептор допамина D2, с получением в результате соединений, которые в отношении своих антипсихотических свойств обладают более выраженным антидепрессивным действием. Соединения формулы (I), приведенной ниже, в которых атом азота основания в положении С-2 встроен в циклическую систему, оказывают мощное антагонистическое действие, направленное против рецепторов 5-НТ2А, 5-НТ2С и рецептора допамина D2.
Цель достигается благодаря предлагаемым новым соединениям согласно формуле (I):

их N-оксидной форме, фармацевтически приемлемой аддитивной соли или стереохимически изомерной форме, где:
пунктирная линия означает необязательную связь;
i и j означают целые числа, независимо друг от друга равные нулю, 1, 2, 3 или 4;
А и В каждый независимо друг от друга означают бензо-, нафтогруппу или радикал, выбранный из группы, состоящей из фуро-; тиено-; пирроло-; оксазоло-; тиазоло-; имидазоло-; изоксазоло-; изотиазоло-; оксадиазоло-; триазоло-; пиридино-; пиридазино-; пиримидино-; пиразино-; индоло-; индолизино-; изоиндоло-; бензофуро-; изобензофуро-; бензотиено-; индазоло-; бензимидазоло-; бензтиазоло-; хинолизино-; хинолино-; изохинолино-; фталазино-; хиназолино-; хиноксалино-; хромено- и нафтиридиногруппы;
каждый R9 независимо друг от друга выбран из группы, состоящей из водорода; галогена; цианогруппы; гидроксигруппы; карбоксила; нитрогруппы; аминогруппы; моно- или ди(алкил)аминогруппы; алкилкарбониламиногруппы; аминосульфонила; моно- или ди(алкил)аминосульфонила; алкила; алкилоксигруппы; алкилкарбонила и алкилоксикарбонила;
X означает CR6R7, O, S, S(=O), S(=O)2или NR8;
где R6 и R7 каждый независимо выбраны из группы, состоящей из водорода, гидроксигруппы, алкила и алкилоксигруппы; или
R6 и R7 вместе взятые могут образовывать радикал, выбранный из группы, состоящей из метилена (=CH2); моно- или ди(циано)метилена; двухвалентного радикала формулы -(CH2)2-, -(CH2)3-, -(CH2)4-, -(CH2)5-, -O(CH2)2O-, -O(CH2)3O-; или вместе с атомом углерода, с которым они связаны, могут образовывать карбонил;
R8 выбран из группы, состоящей из водорода; алкила; алкилкарбонила; арилкарбонила; арилалкила; арилалкилкарбонила; алкилсульфонила; арилсульфонила и арилалкилсульфонила;
C означает группу формулы (c-1), (c-2), (c-3) или (c-4);

где Y1 означает S; S(=O); S(=O)2 или NR10; где R10 выбран из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила, алкилоксиалкилкарбонила, арилкарбонила, арилалкила, арилалкилкарбонила, алкилсульфонила, арилсульфонила и арилалкилсульфонила;
Y2 означает Y1 или O;
R10 и R11 могут вместе образовывать двухвалентный радикал (e-1), (e-2) или (e-3);
каждый двухвалентный радикал (e-1), (e-2) и (e-3) необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы, алкила и алкилтиогруппы;
R12 означает водород или алкил;
R13 означает водород или алкил;
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1);
R11 означает группу формулы (d-1);

где n означает ноль, 1, 2, 3, 4, 5 или 6;
R1 и R2 каждый независимо означают водород; алкил; алкилкарбонил; алкилоксиалкил; алкилкарбонилоксиалкил; алкилоксикарбонилалкил; арилалкил; арилкарбонил; алкилоксикарбонил; арилоксикарбонил; арилалкилкарбонил; алкилоксикарбонилалкилкарбонил; моно- или ди(алкил)аминокарбонил; моно- или ди(арил)аминокарбонил; моно- или ди(арилалкил)аминокарбонил; моно- или ди(алкилоксикарбонилалкил)аминокарбонил; алкилсульфонил; арилсульфонил; арилалкилсульфонил; моно- или ди(алкил)аминотиокарбонил; моно- или ди(арил)аминотиокарбонил; моно- или ди(арилалкил)аминотиокарбонил; моно-, ди- или три(алкил)амидиногруппу; моно-, ди- или три(арил)амидиногруппу и моно-, ди- или три(арилалкил)амидиногруппу; или
R1 и R2 вместе взятые с атомом азота, с которым они связаны, могут образовывать радикал формулы (a-1), (a-2), (a-3), (a-4), (a-5) или (a-6);
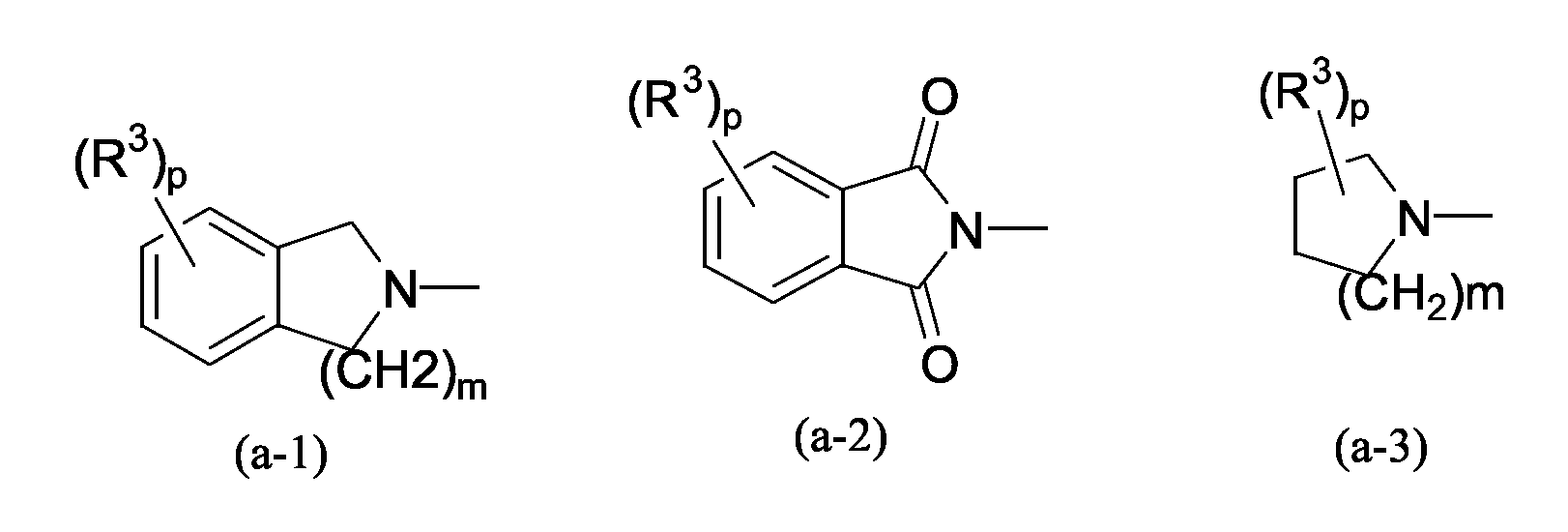

где p равно нулю, 1, 2, 3 или 4;
q равно 1 или 2;
m равно нулю, 1, 2 или 3;
каждый R3 независимо выбран из группы, состоящей из водорода; галогена; гидроксигруппы; цианогруппы; алкила; алкилоксиалкила; арилоксиалкила; моно- или ди(алкил)аминоалкила; гидроксикарбонилалкила; алкилоксикарбонилалкила; моно- или ди(алкил)аминокарбонилалкила; моно- или ди(арил)аминокарбонилалкила; моно- или ди(алкил)аминокарбонилоксиалкила; алкилоксикарбонилоксиалкила; ариламинокарбонилоксиалкила; арилалкиламинокарбонилоксиалкила; арила; алкилоксигруппы; арилоксигруппы; алкилкарбонилоксигруппы; арилкарбонилоксигруппы; арилалкилкарбонилоксигруппы; алкилкарбонила; арилкарбонила; арилоксикарбонила; гидроксикарбонила; алкилоксикарбонила; алкилкарбониламиногруппы; арилалкилкарбониламиногруппы; арилкарбониламиногруппы; алкилоксикарбониламиногруппы; аминокарбониламиногруппы; моно- или ди(арилалкил)аминокарбониламиногруппы; алкилсульфонилалкиламинокарбониламиногруппы; или два радикала R3 вместе могут образовывать двухвалентный радикал
где R5 выбран из группы, состоящей из водорода, галогена, гидроксигруппы, алкилоксигруппы и алкила;
R4 выбран из группы, состоящей из водорода; алкила; арилалкила; алкилоксиалкила; алкилкарбонилоксиалкила; алкилоксикарбонилалкила; арилкарбонилалкила; алкилсульфонилоксиалкила; арилоксиарила; алкилоксикарбониларила; алкилкарбонила; арилалкилкарбонила; алкилоксикарбонилалкилкарбонила; арилкарбонила; алкилоксикарбонила; арилоксикарбонила; арилалкилоксикарбонила; моно- или ди(алкил)аминокарбонила; моно- или ди(арил)аминокарбонила; моно- или ди(арилалкил)аминокарбонила; моно- или ди(алкилоксикарбонилалкил)аминокарбонила; алкилоксиалкиламинокарбонила; моно-, ди- или три(алкил)амидиногруппы; моно-, ди- или три(арил)амидиногруппы; моно-, ди- или три(арилалкил)амидиногруппы; алкилсульфонила; арилалкилсульфонила или арилсульфонила;
арил представляет собой фенил или нафтил; каждый радикал необязательно замещен 1, 2 или 3 заместителями, выбранными из группы, состоящей из галогена, нитрогруппы, цианогруппы, гидроксигруппы, алкилоксигруппы или алкила;
алкил означает насыщенный углеводородный радикал с неразветвленной или разветвленной цепью, имеющий от 1 до 10 атомов углерода, циклический насыщенный углеводородный радикал, имеющий от 3 до 8 атомов углерода или насыщенный углеводородный радикал, содержащий неразветвленный или разветвленный остаток, имеющий от 1 до 10 атомов углерода, и циклический остаток, имеющий от 3 до 8 атомов углерода, необязательно замещенный одним или несколькими радикалами: галогеном, цианогруппой, оксогруппой, гидроксигруппой, формилом, карбоксилом или аминогруппой; и
галоген означает фтор, хлор, бром и йод.
Более конкретно изобретение относится к соединению согласно формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству, где каждый из A и B представляет собой бензогруппу, необязательно замещенную фтором. Предпочтительно A незамещен, а B замещен фтором в положении 11.
Более конкретно изобретение относится к соединению согласно формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству, где C означает группу формулы (c-1) или (c-2); где
Y1означает S; S(=O); S(=O)2 или NR10; где R10 выбран из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила и алкилоксиалкилкарбонила;
соседние R10 и R11 вместе могут образовывать двухвалентный радикал (e-1), (e-2) или (e-3);
каждый радикал необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы, алкила и алкилтиогруппы; и
R12 означает водород.
Более конкретно изобретение относится к соединению согласно формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству, где C означает группу формулы (c-3) или (c-4); где
Y2 означает O;
R12 означает водород;
R13 означает водород; и
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1).
Более конкретно изобретение относится к соединению согласно формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству, где (d-1) определяют как соединение, в котором:
n равно нулю или 1;
R1 и R2 каждый независимо означают водород; алкил или алкилоксикарбонилалкил; или R1 и R2 вместе взятые с атомом азота, с которым они связаны, могут образовывать радикал формулы (a-3), (a-5) или (a-6);
где p равно нулю или 1;
q равно 1;
m равно 1;
каждый R3 независимо выбран из группы, состоящей из водорода и гидроксигруппы; и
R4 означает алкил.
Более конкретно, изобретение относится к соединению согласно общей формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству,
где i и j являются целыми числами, независимо друг от друга равными нулю или 1;
A и B каждый независимо друг от друга означают бензогруппу, необязательно замещенную фтором;
каждый R9 независимо друг от друга выбран из группы, состоящей из водорода и галогена;
X означает CH2 и O;
C означает группу формулы (c-1), (c-2), (c-3) или (c-4);
где Y1 означает S; S(=O); S(=O)2 или NR10; где R10 выбран из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила и алкилоксиалкилкарбонила;
Y2 означает O;
соседние R10 и R11 вместе могут образовывать двухвалентный радикал (e-1), (e-2) или (e-3); каждый радикал необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы, алкила и алкилтиогруппы;
R12 означает водород;
R13 означает водород;
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1);
R11 означает группу формулы (d-1);
где n равно нулю или 1;
R1 и R2 каждый независимо означают водород; алкил или алкилоксикарбонилалкил; или R1 и R2 вместе взятые с атомом азота, с которым они связаны, могут образовывать радикал формулы (a-3), (a-5) или (a-6);
где p равно нулю или 1;
q равной 1;
m равно 1;
каждый R3 независимо выбран из группы, состоящей из водорода и гидроксигруппы; и
R4 означает алкил.
Предпочтительно алкил представляет собой метил, этил или пропил, необязательно замещенный одним или несколькими радикалами: галогеном, цианогруппой, оксогруппой, гидроксигруппой, формилом, карбоксилом или аминогруппой. Предпочтительно алкил необязательно замещен гидроксигруппой.
Предпочтительно арил представляет собой фенил, необязательно замещенный 1, 2 или 3 заместителями, выбранными из группы, состоящей из галогена, нитрогруппы, цианогруппы, гидроксигруппы, алкилоксигруппы или алкила. Предпочтительно арил незамещен.
Предпочтительно галоген представляет собой фтор.
Предпочтительными соединениями также являются такие конкретные соединения согласно изобретению, в которых атомы водорода на атомах углерода 3a и 12b имеют трансконфигурацию, и соединения, имеющие (2α, 3aα, 12bβ) стереохимическую конфигурацию.
Наиболее предпочтительными соединениями также являются такие соединения согласно изобретению, которые выбраны из группы, состоящей из соединений, определяемых номерами соединений, приведенных в таблицах 1-4.
Подробное описание изобретения
В рамках настоящей заявки алкил определяют как одновалентный неразветвленный или разветвленный насыщенный углеводородный радикал, имеющий от 1 до 6 атомов углерода, например метил, этил, пропил, бутил, 1-метилпропил, 1,1-диметилэтил, пентил и гексил; алкил кроме того означает одновалентный циклический насыщенный углеводородный радикал, имеющий от 3 до 6 атомов углерода, например циклопропил, метилциклопропил, циклобутил, циклопентил и циклогексил. Определение алкила также включает алкильный радикал, который необязательно замещен на одном или нескольких атомах углерода одним или несколькими радикалами: фенилом, галогеном, цианогруппой, оксогруппой, гидроксигруппой, формилом и аминогруппой, например гидроксиалкил, в частности гидроксиметил и гидроксиэтил, и полигалогеналкил, в частности дифторметил и трифторметил.
В рамках настоящей заявки галоген является общим названием фтора, хлора, брома и йода.
В рамках настоящей заявки термин «соединения согласно изобретению» означает соединение согласно общей формуле (I), его фармацевтически приемлемые кислотно- или основно-аддитивные соли, его стереохимически изомерные формы, его N-оксидную форму и его пролекарство.
В рамках настоящей заявки элемент, в частности при упоминании в связи с соединением согласно формуле (I), включает все изотопы и изотопные смеси такого элемента, либо встречающиеся в природе, либо полученные синтетически, либо преобладающую в природе форму, либо обогащенную по изотопу форму. В частности, в том случае, когда указан водород, следует понимать, что он относится к1H,2H,3H и их смесям; если указан углерод, то следует понимать, что он относится к11C,12C,13C,14C и их смесям; если указан азот, то следует понимать, что он относится к13N,14N,15N и их смесям; если указан кислород, то следует понимать, что он относится к14O,15O,16O,17O,18O и их смесям; и если указан фтор, то следует понимать, что он относится к18F,19F и их смесям.
Следовательно к соединениям согласно изобретению также относятся соединения с одним или несколькими изотопами одного или нескольких элементов и их смеси, включая радиоактивные соединения, также называемые радиоактивно мечеными соединениями, в которых один или несколько нерадиоактивных атомов были заменены одним из его радиоактивных изотопов. Под термином «радиоактивно меченое соединение» подразумевают любое соединение согласно формуле (I), его N-оксидную форму, его фармацевтически приемлемую аддитивную соль или стереохимически изомерную форму, которая содержит по меньшей мере один радиоактивный атом. Например, соединения могут быть мечены радиоактивными изотопами, испускающими позитроны или гамма-излучение. В случае способов связывания радиоактивного лиганда (анализ мембранных рецепторов) предпочтительным атомом для замены является атом3H или атом125I. Для визуализации наиболее широко используемыми испускающими позитроны (PET) радиоактивными изотопами являются11C,18F,15O и13N, которые все получают с помощью ускорителей и которые имеют период полураспада 20, 100, 2 и 10 минут соответственно. Поскольку периоды полураспада таких радиоактивных изотопов настолько малы, их использование возможно только в институтах, которые имеют ускоритель для их получения, что ограничивает их применение. Наиболее широко используемыми из них являются18F,99mTc,201Tl и123I. Обращение с такими радиоактивными изотопами, их получение, выделение и включение в молекулу известны специалисту.
В частности радиоактивный атом выбран из группы, состоящей из водорода, углерода, азота, серы, кислорода и галогена. Предпочтительно радиоактивный атом выбран из группы, состоящей из водорода, углерода и галогена.
В частности, радиоактивный изотоп выбран из группы, состоящей из3H,11C,18F,122I,123I,125I,131I,75Br,76Br,77Br и82Br. Предпочтительно радиоактивный изотоп выбран из группы, состоящей из3H,11C и18F.
В определение фармацевтически приемлемых солей включены терапевтически активные нетоксичные кислотно-аддитивные формы солей, которые способны образовывать соединения согласно формуле (I). Указанные соли могут быть получены обработкой основания соединений формулы (I) подходящими кислотами, например неорганическими кислотами, например галогенводородной кислотой, в частности хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, азотной кислотой и фосфорной кислотой; органическими кислотами, например уксусной кислотой, гидроксиуксусной кислотой, пропионовой кислотой, молочной кислотой, пировиноградной кислотой, щавелевой кислотой, малоновой кислотой, янтарной кислотой, малеиновой кислотой, миндальной кислотой, фумаровой кислотой, яблочной кислотой, винной кислотой, лимонной кислотой, метансульфоновой кислотой, этансульфоновой кислотой, бензолсульфоновой кислотой, пара-толуолсульфоновой кислотой, цикламовой кислотой, салициловой кислотой, пара-аминосалициловой кислотой и памовой кислотой.
Соединения согласно формуле (I), содержащие протоны кислоты, также могут быть превращены в их терапевтически активные нетоксичные формы аддитивных солей металлов или аминов обработкой соответствующими органическими и неорганическими основаниями. Подходящие формы солей, образованных основаниями, включают, например, соли аммония, соли щелочных и щелочно-земельных металлов, в частности соли лития, натрия, калия, магния и кальция, соли, образованные органическими основаниями, например соли бензатина, N-метил-D-глюкамина, гидрамина, и соли аминокислот, например аргинина и лизина.
Наоборот, указанные формы солей могут быть превращены в свободные формы обработкой соответствующим основанием или кислотой.
Термин аддитивная соль, который используют в рамках настоящей заявки, также включает сольваты, которые способны образовывать соединения согласно формуле (I), а также их соли. Такие сольваты представляют собой, например, гидраты и алкоголяты.
Подразумевается, что N-оксидные формы соединений согласно формуле (I) включают такие соединения формулы (I), в которых один или несколько атомов азота окислены до так называемого N-оксида, особенно такие N-оксиды, в которых один или несколько третичных атомов азота (например радикала пиперазинила или пиперидинила) N-окислены. Такие N-оксиды могут быть легко получены специалистом, не имеющим изобретательского опыта, и они очевидно являются альтернативой соединениям согласно формуле (I), так как такие соединения представляют собой метаболиты, которые образуются при окислении в организме человека после поглощения. Общеизвестно, что окисление обычно является первой стадией, вовлеченной в метаболизм лекарственного средства (Textbook of Organic Medicinal and Pharmaceutical Chemistry, 1977, pages 70-75). Также общеизвестно, что форма метаболита соединения также может быть введена человеку вместо соединения как такового почти с таким же эффектом.
Соединения согласно изобретению имеют по меньшей мере 1 окисляемый атом азота (остаток третичных аминов). Поэтому весьма вероятно, что N-оксиды должны образовываться в процессе метаболизма у человека.
Соединения формулы (I) можно превратить в соответствующие N-оксидные формы, следуя известным в данной области способам превращения трехвалентного азота в его N-оксидную форму. Указанная реакция N-окисления в общем может быть осуществлена посредством взаимодействия исходного вещества формулы (I) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, пероксид водорода, пероксиды щелочных металлов или щелочно-земельных металлов, например пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать пероксикислоты, например, такие как бензолкарбопероксикислота или галогензамещенная бензолкарбопероксикислота, например 3-хлорбензолкарбопероксикислота, пероксоалкановые кислоты, например пероксоуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие алканолы, например этанол и тому подобные, углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например дихлорметан, и смеси таких растворителей.
Термин «стереохимически изомерные формы», который использован выше, определяет все возможные изомерные формы, которые могут иметь соединения формулы (I). Если не оговорено или не указано особо, химические названия соединений означают смесь всех возможных стереохимически изомерных форм, при этом указанные смеси содержат все диастереомеры и энантиомеры основной молекулярной структуры. Более конкретно стереогенные центры могут иметь R- или S-конфигурацию; заместители на двухвалентных циклических (частично) насыщенных радикалах могут иметь либо цис-, либо трансконфигурацию. Соединения, содержащие двойные связи, могут иметь E- или Z-стереохимию по указанной двойной связи. Несомненно подразумевается, что стереохимически изомерные формы соединений формулы (I) входят в объем настоящего изобретения.
Следуя правилам номенклатуры CAS, в том случае, когда в молекуле присутствуют два стереогенных центра известной абсолютной конфигурации, R- или S-дескриптор присваивают (на основании правила последовательности Канна-Инголда-Прелога) хиральному центру, имеющему наименьший номер, реперному центру. Каждый из R* и S* указывают оптически чистые стереогенные центры с неопределенной абсолютной конфигурацией. Если используют «α» и «β»: положение наиболее приоритетного заместителя на асимметричном атоме углерода в циклической системе, имеющей наименьшей номер цикла, произвольно всегда находится в «α»-положении от средней плоскости, определяемой кольцевой системой. Положение наиболее приоритетного заместителя на другом асимметричном атоме углерода в кольцевой системе (атом водорода в соединениях согласно формуле (I)) относительно положения наиболее приоритетного заместителя на реперном атоме называют «α», если он находится с той же стороны средней плоскости, определяемой кольцевой системой, или «β», если он находится с другой стороны средней плоскости, определяемой кольцевой системой.
Нумерация тетрациклических кольцевых систем, присутствующих в соединениях формулы (I-a) и (I-b), когда A и B означают бензогруппу, которая определяется номенклатурой Chemical Abstracts, показана ниже.
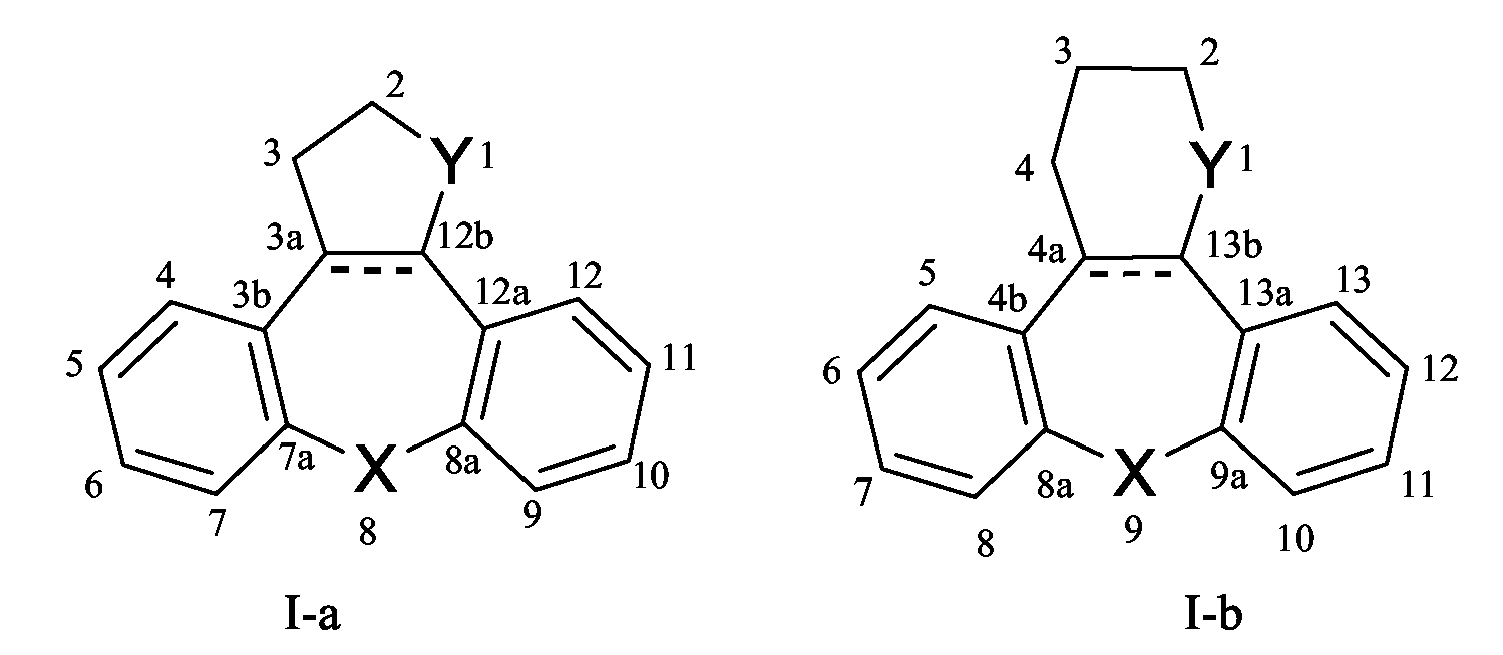
Соединения формулы (I-a) и (I-b) имеют по меньшей мере два асимметричных центра в положениях атома углерода 2 и 3. Указанный асимметричный центр и любой другой асимметричный центр, который может присутствовать (например в положении атома 8 в (I-a) или 9 в (I-b)), указаны дескрипторами R и S. В том случае, когда, например, остаток моноцианометилена присутствует в соединениях формулы (I-a) в положении 8, указанный остаток может иметь E- или Z-конфигурацию.
Изобретение также относится к производным соединениям (обычно называемым «пролекарствами») фармакологически активных соединений согласно изобретению, которые распадаются in vivo, давая соединения согласно изобретению. Пролекарства обычно (но не всегда) обладают более низкой эффективностью по отношению к рецептору-мишени, чем соединения, до которых они распадаются. Пролекарства особенно применимы в том случае, когда требуемое соединение имеет химические или физические свойства, которые делают его введение трудным или неэффективным. Например требуемое соединение может быть только плохо растворимым, оно может плохо транспортироваться через эпителий слизистой оболочки, или оно может иметь нежелательно короткое время полужизни в плазме. Дополнительное обсуждение пролекарств можно найти в Stella, V.J. et al., "Prodrugs", Drug Delivery Systems, 1985, pp.112-176, и Drugs, 1985, 29, pp.455-473.
Формы пролекарств фармакологически активных соединений согласно изобретению, как правило, будут представлять собой соединения согласно формуле (I), его фармацевтически приемлемые кислотно- или основно-аддитивные соли, его стереохимически изомерные формы и его N-оксидную форму, имеющие группу кислоты, которая этерифицирована или амидирована. К таким этерифицированным кислотным группам относятся группы формулы -COORx, где Rx означает C1-6-алкил, фенил, бензил или одну из следующих групп:
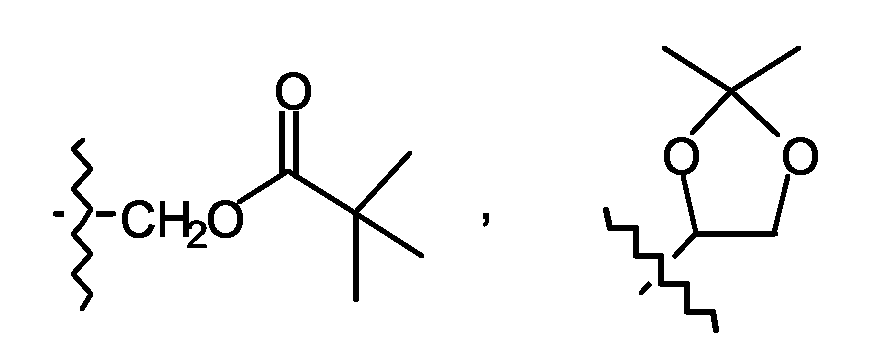
Амидированные группы включают группы формулы - CONRyRz, где Ry означает H, C1-6-алкил, фенил или бензил и Rz означает -OH, H, C1-6-алкил, фенил или бензил. Соединения согласно изобретению, имеющие аминогруппу, могут быть дериватизованы кетоном или альдегидом, таким как формальдегид, с образованием основания Манниха. Такое основание будет гидролизоваться с кинетикой первого порядка в водном растворе.
Соединения формулы (I), которые получают способами, описанными ниже, могут быть синтезированы в форме рацемических смесей энантиомеров, которые могут быть отделены друг от друга известными в данной области способами разделения. Рацемические соединения формулы (I) могут быть превращены в соответствующие формы диастереомерных солей в результате взаимодействия с подходящей хиральной кислотой. Указанные формы диастереомерных солей затем разделяют, например, избирательной или фракционной кристаллизацией, и энантиомеры высвобождают с помощью щелочи. Альтернативный способ разделения энантиомерных форм соединений формулы (I) включает в себя жидкостную хроматографию с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм соответствующих исходных веществ при условии, что реакция происходит стереоспецифично. Предпочтительно, если требуется конкретный стереоизомер, указанное соединение может быть синтезировано стереоспецифичными способами получения. В таких способах преимущественно будут использоваться энантиомерно чистые исходные вещества.
Фармакология
Соединения согласно настоящему изобретению проявляют аффинность по отношению к рецепторам 5-HT2, в частности по отношению к рецепторам 5-HT2A и 5-HT2C (номенклатура соответствует описанию в D.Hoyer "Serotonin (5-HT) in neurologic and psychiatric disorders" под редакцией M.D.Ferrari и опубликованной в 1994 г. Boerhaave Commission of the University of Leiden), и аффинность по отношению к рецептору D2, а также активность, ингибирующую обратный захват норэпинефрина. Антагонистические по отношению к серотонину свойства предлагаемых в изобретении соединений можно продемонстрировать по их ингибирующему действию в тесте на 5-гидрокситриптофан у крыс, который описан в Drug Dev. Res., 13, 237-244 (1988).
Ввиду наличия способности блокировать рецепторы 5-HT2, и в частности блокировать рецепторы 5-HT2A и 5-HT2C, а также рецептор D2, а также наличия активности, ингибирующей обратный захват норэпинефрина, соединения согласно изобретению применимы в качестве лекарственного средства, в частности для профилактического и терапевтического лечения состояний, опосредованных любым из указанных рецепторов.
Поэтому изобретение относится к соединению согласно общей формуле (I), его фармацевтически приемлемым кислотно- или основно-аддитивным солям, его стереохимически изомерным формам, его N-оксидной форме и его пролекарству для применения в качестве лекарственного средства.
Изобретение также относится к применению соединения согласно общей формуле (I), его фармацевтически приемлемых кислотно- или основно-аддитивных солей, его стереохимически изомерных форм, его N-оксидной формы и его пролекарств для производства лекарственного средства для лечения, либо профилактического, либо терапевтического, либо и того и другого, состояний, опосредованных рецепторами 5-HT2 и рецептором D2, а также ингибированием обратного захвата норэпинефрина.
Ввиду указанных фармакологических и физико-химических свойств соединения формулы (I) применимы в качестве терапевтических средств при лечении или профилактике расстройств центральной нервной системы, подобных тревожности, депрессии и мягкой депрессии, биполярных расстройств, расстройств сна и сексуальных расстройств, психоза, пограничного психоза, шизофрении, мигрени, расстройств личности или обсессивно-компульсивных расстройств, социальных фобий или острых тревожных состояний с реакцией паники, органических умственных расстройств, умственных расстройств у детей, таких как ADHD, агрессии, расстройств памяти и нарушений чувства положения тела в пространстве у пожилых людей, наркомании, ожирения, булимии и сходных расстройств. В частности, предлагаемые в изобретении соединения могут быть использованы в качестве анксиолитических средств, антидепрессантов, антипсихотических средств, противошизофренических средств, средств против мигрени и в качестве средств, способных снимать привыкание к наркотическим лекарственным средствам.
Соединения формулы (I) также могут быть использованы в качестве терапевтических средств для лечения моторных расстройств. Может быть предпочтительным применение предлагаемых в настоящем изобретении соединений в комбинации с классическими в случае таких расстройств терапевтическими средствами.
Соединения формулы (I) также могут служить для лечения или профилактики повреждения нервной системы, вызванного травмой, инсультом, нейродегенеративными заболеваниями и тому подобным; сердечно-сосудистых расстройств, подобных высокому кровяному давлению, тромбозу, инсульту, и тому подобных; и желудочно-кишечных расстройств, подобных дисфункции моторики желудочно-кишечной системы, и тому подобных.
Ввиду указанных выше применений соединений формулы (I) следует, что настоящее изобретение также относится к способу лечения теплокровных животных, страдающих от таких заболеваний, при этом указанный способ включает в себя системное введение терапевтического количества соединения формулы (I), эффективного при лечении описанных выше расстройств, в частности при лечении тревожности, психоза, депрессии, мигрени и привыкания к наркотическим лекарственным средствам.
Таким образом, настоящее изобретение также относится к соединениям формулы (I), которые определены выше для применения в качестве лекарственного средства, в частности соединения формулы (I) могут быть использованы для производства лекарственного средства для лечения тревожности, психоза, депрессии, мигрени и привыкания к наркотическим лекарственным средствам.
Специалисты в области лечения таких заболеваний могут определить эффективное терапевтическое суточное количество на основе результатов тестирования, приведенных далее. Эффективное терапевтическое суточное количество может составлять примерно от 0,01 мг/кг до примерно 10 мг/кг массы тела, более предпочтительно примерно от 0,05 мг/кг до примерно 1 мг/кг массы тела.
Изобретение также относится к фармацевтической композиции, содержащей фармацевтически приемлемый носитель и в качестве активного ингредиента терапевтически эффективное количество соединения согласно изобретению, в частности соединения согласно формуле (I), его фармацевтически приемлемых кислотно- или основно-аддитивных солей, его стереохимически изомерных форм, его N-оксидной формы и его пролекарства.
Соединения согласно изобретению, в частности соединения согласно формуле (I), их фармацевтически приемлемые кислотно- или основно-аддитивные соли, их стереохимически изомерные формы, их N-оксидные формы и их пролекарства или их любая подгруппа или комбинация могут быть приготовлены в различных фармацевтических формах в целях введения. В качестве подходящих композиций могут быть перечислены все композиции, обычно используемые в случае системно вводимых лекарственных средств. Чтобы приготовить фармацевтические композиции согласно настоящему изобретению, эффективное количество конкретного соединения, необязательно в форме аддитивной соли, в качестве активного ингредиента объединяют в виде однородной смеси с фармацевтически приемлемым носителем, и такой носитель может иметь широкое множество форм в зависимости от формы препарата, требуемого для введения. Желательно, чтобы указанные фармацевтические композиции имели стандартную дозированную лекарственную форму, подходящую в частности для введения перорально, ректально, чрезкожно, посредством парентеральной инъекции или путем ингаляции. Например, при получении композиции в пероральной дозированной форме можно использовать любые обычные фармацевтические среды, например, такие как вода, гликоли, масла, спирты и тому подобные, в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, разбавители, скользящие вещества, связывающие вещества, дезинтегрирующие средства и тому подобное, в случае порошков, пилюль, капсул и таблеток. Вследствие простоты введения таблетки и капсулы представляют собой наиболее предпочтительные пероральные дозированные лекарственные формы, и в таком случае обычно используют твердые фармацевтические носители. В случае парентеральных композиций носитель обычно будет содержать стерильную воду, по меньшей мере в основном, хотя могут быть включены другие ингредиенты, например способствующие растворимости. Например, могут быть приготовлены инъекционные растворы, в которых носитель содержит раствор соли, раствор глюкозы или смесь раствора соли и глюкозы. Также могут быть приготовлены инъекционные суспензии, в случае которых могут быть использованы подходящие жидкие носители, суспендирующие агенты и тому подобное. Также включены препараты в твердой форме, которые предназначены для превращения непосредственно перед применением в препараты в жидкой форме. В композициях, подходящих для чрезкожного введения, носитель необязательно содержит средство, усиливающее проницаемость, и/или подходящее увлажняющее средство, необязательно вместе с подходящими добавками любой природы в минорных соотношениях, при этом такие добавки не оказывают существенного вредного воздействия на кожу. Указанные добавки могут облегчать введение в кожу и/или могут быть полезными для приготовления требуемой композиции. Такие композиции могут быть введены различными путями, например в виде трансдермального пластыря, в виде локально наносимых пятен, в виде мази. Особенно предпочтительно приготовление указанных выше фармацевтических композиций в дозированной лекарственной форме для простоты введения и единообразия дозирования. Дозированная лекарственная форма в используемом в данном описании смысле относится к физически дискретным единицам, подходящим в качестве одинарных доз, при этом каждая единица содержит предварительно определяемое количество активного ингредиента, рассчитанное для получения требуемого терапевтического эффекта, вместе с требуемым фармацевтическим носителем. Примерами таких дозированных лекарственных форм являются таблетки (включая таблетки с насечками или покрытые оболочками таблетки), капсулы, пилюли, пакетики с порошками, облатки, суппозитории, инъекционные растворы или суспензии и тому подобное, и группы, состоящие из множества отдельных указанных форм.
Так как соединения согласно изобретению являются эффективными при пероральном введении соединениями, то особенно предпочтительны фармацевтические композиции, содержащие указанные соединения, для введения перорально.
Чтобы повысить растворимость и/или стабильность соединений формулы (I) в фармацевтических композициях предпочтительно можно использовать α-, β- или γ-циклодекстрины или их производные, в частности замещенные гидроксиалкилом циклодекстрины, например 2-гидроксипропил-β-циклодекстрин. Также корастворители, такие как спирты, могут улучшить растворимость и/или стабильность соединений согласно изобретению в фармацевтических композициях.
Получение
Походящие схемы получения соединений согласно изобретению описаны ниже.
В тексте использованы следующие сокращения:
Следующие схемы реакций A-D иллюстрируют получение соединений формулы (I), в которых C означает группу формулы (c-1), в которой Y1 означает NH, и R11 означает группу формулы (d-1), представленных формулами Ia и Ib ниже:
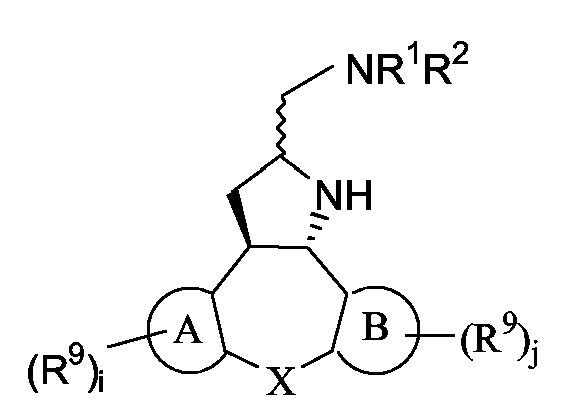
Ia: 2R,3aR,xS, где x означает 12b, если A и B являются бензогруппой;
Ib: 2S,3aR,xS, где x означает 12b, если A и B являются бензогруппой.
Способ A: Получение производных пирролидина
Схема A1: Синтез (2R,3aR,12bS)-промежуточных соединений.

Стадия a): обработка промежуточного соединения 1 кеталь-защищенным (S)-глицеральдегидом (защищенным, например, Me), кислотой Льюиса, такой как галогенид магния, в частности бромид магния, и например t-BuOK в качестве основного катализатора в инертном по отношению к реакции растворителе, таком как толуол или ТГФ, например при комнатной температуре;
Стадия b): гидрирование промежуточного соединения 2 с использованием в качестве катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как i-PrOH, в присутствии основания, такого как третичный амин, в частности Et3N, например при комнатной температуре в течение примерно 4 часов;
Стадия c): восстановление промежуточного соединения 3, например, борогидридом щелочного металла, таким как борогидрид натрия, в фосфатном буфере при максимальном pH 7 (предпочтительно при слабокислом pH) и в инертном по отношению к реакции растворителе, таком как i-PrOH или EtOH, например при 0°C в течение 15 мин с образованием промежуточного соединения 4 в цис-конфигурации;
Стадия d): реакция нуклеофильного замещения промежуточного соединения 4 с DPPA в присутствии основания, такого как DIAD/P(Ph)3 или DBU, в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение примерно 24 часов;
Стадия e): удаление защиты промежуточного соединения 5 с использованием кислоты, такой как хлористоводородная кислота, в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 16 часов;
Стадия f): тритилирование промежуточного соединения 6 с использованием Tr-галогенида, в частности TrCl или TrBr, и катализатора, такого как DMAP, в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 24 часов;
Стадия g): обработка промежуточного соединения 7a MsCl или Ms-ангидридом в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при температуре примерно от -40°C до комнатной температуры в течение примерно 4 часов;
Стадия h): детритилирование промежуточного соединения 7b с использованием сильной кислоты в неводной среде, такой как Amberlyst-15 (макросетчатый сульфонированный полистирол), в инертном по отношению к реакции растворителе, таком как метанол, например примерно при 45°C в течение примерно 2-3 часов;
Стадия i): обработка промежуточного соединения 8 основанием, таким как K2CO3, в инертном по отношению к реакции растворителе, таком как MeOH или EtOH, например при комнатной температуре в течение примерно 2 часов;
Стадия j): гидрирование промежуточного соединения 9 с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, в присутствии основания, такого как Et3N, например при комнатной температуре в течение примерно 3 часов. Полученное в результате промежуточное соединение 10 может быть использовано в качестве исходного вещества, как описано на схеме A3.
Схема A2: Синтез (2S,3aR,12bS)-промежуточного и конечного соединений.

Стадия a): тозилирование промежуточного соединения 6 с использованием TsCl в присутствии основания, такого как Et3N, и катализатора, такого как Bu2SnO, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 16 часов;
Стадия b): обработка промежуточного соединения 11 основанием, таким как K2CO3, в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 10 минут;
Стадия c): гидрирование промежуточного соединения 12 с использованием катализатора палладия на угле (1 атм), в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 16 часов. Полученное в результате соединение 13 может быть использовано в качестве исходного вещества, как описано на схеме A3.
Стадия d): нуклеофильное замещение промежуточного соединения 11 или 12 с использованием азида щелочного металла, такого как азид натрия, в инертном по отношению к реакции растворителе, таком как ДМФА, например при температуре около 90°C;
Стадия e): мезилирование промежуточного соединения 14 MsCl и необязательно DMAP, с использованием третичного аминного основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при температуре примерно от -40°C до комнатной температуры в течение примерно 4 часов;
Стадия f): гидрирование промежуточного соединения 15 с использованием катализатора палладия на угле (1 атм) в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 3 часов приводит к конечному соединению формулы (I-b1), т.е. соединению формулы (I-b), в котором оба радикала R1 и R2 являются водородом.
Стадия g): инверсия Митсунобу промежуточного соединения 14 с использованием DIAD/P(Ph)3 и CbzOH в ТГФ при температуре примерно от 0°C до комнатной температуры в течение примерно 2 часов;
Стадия h) гидролиз промежуточного соединения 14a с использованием, например, K2CO3 в метаноле.
Схема A3: Синтез (2R,3aR,12bS)- и (2S,3aR,12bS)-конечных соединений

Стадия a) обработка промежуточного соединения 10 или 13 CbzCl водным раствором основания, такого как K2CO3, в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение 15 минут;
Стадия b): Способ 1: окисление промежуточного соединения 17 PCC; затем восстановительное аминирование HNR1R2 с использованием восстановителя, такого как NaBH4; или способ 2: мезилирование промежуточного соединения 17 MsCl и DMAP, в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2; затем нуклеофильное замещение с использованием избытка HNR1R2, необязательно в присутствии основания, такого как K2CO3;
Стадия c) удаление защитной группы Cbz гидрированием промежуточного соединения 18 с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, и в присутствии основания, такого как Et3N, например при комнатной температуре в течение примерно 3 часов.
Промежуточное соединение 10 приводит к конечному соединению формулы (Ia); промежуточное соединение 13 приводит к конечному соединению формулы (Ib).
Приведенные ниже способы B-D представляют собой альтернативные пути получения указанных выше конечных соединений формулы Ia и Ib:
Способ B: Синтез (2R,3aR,12bS)- и (2S,3aR,12bS)-конечных соединений

Стадия a): гидрирование промежуточного соединения 14 или 14b с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, и в присутствии основания, такого как Et3N, например при комнатной температуре в течение примерно 3 часов;
Стадия b): обработка промежуточного соединения 19 CbzCl, основанием, таким как K2CO3, в инертной по отношению к реакции смеси растворителей, такой как ТГФ-H2O;
Стадия c): мезилирование промежуточного соединения 20 MsCl и DMAP, в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 16 часов;
Стадия d): обработка промежуточного соединения 21 основанием, таким как t-BuOK, в инертном по отношению к реакции полярном апротонном растворителе, таком как ТГФ;
Стадия e): гидрирование промежуточного соединения 22 с использованием катализатора палладия на угле (1 атм), в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 3 часов;
Стадия f) обработка конечного соединения Ia1 (имеющего 2R(нижнюю)-конфигурацию) альдегидом, таким как формальдегид, или кетон в спиртовом растворителе в присутствии AcOH и восстановителя, такого как водород/палладий на угле или NaCNBH3, приводит к трехзамещенному конечному соединению Ia2.
Способ C: Синтез (2RS,3aR*,12bS*)-конечных соединений

Стадия a): аллилирование промежуточного соединения 1 основанием, таким как NaH, и аллилбромидом в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре около 65°C в течение примерно 2-3 часов;
Стадия b): восстановление промежуточного соединения 23 восстановителем, таким как NaBH4 (в фосфатном буфере при pH 7), в инертном по отношению к реакции растворителе, таком как i-PrOH, например при комнатной температуре приводит к промежуточному соединению 24, содержащему энантиомерную смесь 24a и 24b в конфигурации с обоими заместителями либо в верхнем, либо в нижнем положении;
Стадия c): обработка промежуточного соединения 24 DPPA, DIAD/P(Ph)3 в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение примерно 24 часов;
Стадия d): восстановление промежуточного соединения 25 восстановителем, наиболее предпочтительно LiAlH4, в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от 0°C до комнатной температуры;
Стадия e): защита промежуточного соединения 26 Boc2O с использованием водного раствора основания, такого как K2CO3, в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре;
Стадия f): циклизация промежуточного соединения 27 под действием йода с использованием IPy2BF4 в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре;
Стадия g): аминирование промежуточного соединения 28 с использованием избытка HNR1R2 в водном ТГФ примерно при 135°C в сосуде высокого давления (например в стальном баллоне) в течение примерно 3-6 часов; или альтернативно например с использованием HNMe2 в безводном ТГФ и оксида кальция, чтобы убрать удаляемую группу.
Стадия h): удаление защиты с использованием кислоты, такой как HBr в AcOH или HCl в MeOH, в течение примерно 1-2 часов при температуре образования флегмы или при комнатной температуре.
Способ D: Синтез (2RS,3aR,12bS)-конечных соединений

Стадия a): реакция Митсунобу промежуточного соединения 10 или 13 с DIAD/P(Ph)3 в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение примерно 24 часов;
Стадия b): опосредованное йодтриметилсиланом раскрытие цикла азиридина промежуточного соединения 29 с последующим взаимодействием in-situ с подходящим амином HNR1R2 в кипящем ацетонитриле. Промежуточное соединение 10 приводит к конечному соединению формулы Ia; промежуточное соединение 13 приводит к конечному соединению формулы Ib.
Способ E: Получение производных пирролоимидазола
Следующая схема реакции иллюстрирует получение соединений формулы (I), в которых C является группой формулы (c-1), в которой R11 и R10 образуют конденсированный остаток имидазола, представленной приведенной ниже формулой II.

Схема E

Стадия a): гидрирование конечного соединения I-b1 (имеющего 2S(верхнюю)-конфигурацию) с использованием катализатора палладия на угле (1 атм) и формальдегида в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 3 часов с получением конечного соединения 30;
Стадия b): обработка конечного соединения 30 NaCNBH3/ТФУ в таком растворителе, как MeOH, с получением конечного соединения 31;
Стадия c): обработка конечного соединения I-a1 или I-b1 (имеющих соответственно 2R (нижнюю)- или 2S (верхнюю)-конфигурацию) CS2 в инертном по отношению к реакции растворителе, таком как ДМФА, например при температуре около 50-60°C в течение примерно 30 минут с получением в результате конечного соединения 32;
Стадия d): алкилирование конечного соединения 32, например, алкилгалогенидом в инертном по отношению к реакции растворителе, таком как MeOH или Et3N, например при кипячении с обратным холодильником с получением в результате конечного соединения II.
Способ F: Получение производных пирролопиперазина
Следующие схемы реакций иллюстрируют получение соединений формулы (I), в которых C означает группу формулы (c-1), в которой R11 и R10 образуют конденсированный остаток пиперазина, представленные формулой III ниже, где Rx означает водород или алкил и цикл пиперазина имеет S-конфигурацию (схема F1) или R-конфигурацию (схема F2):

Схема F1
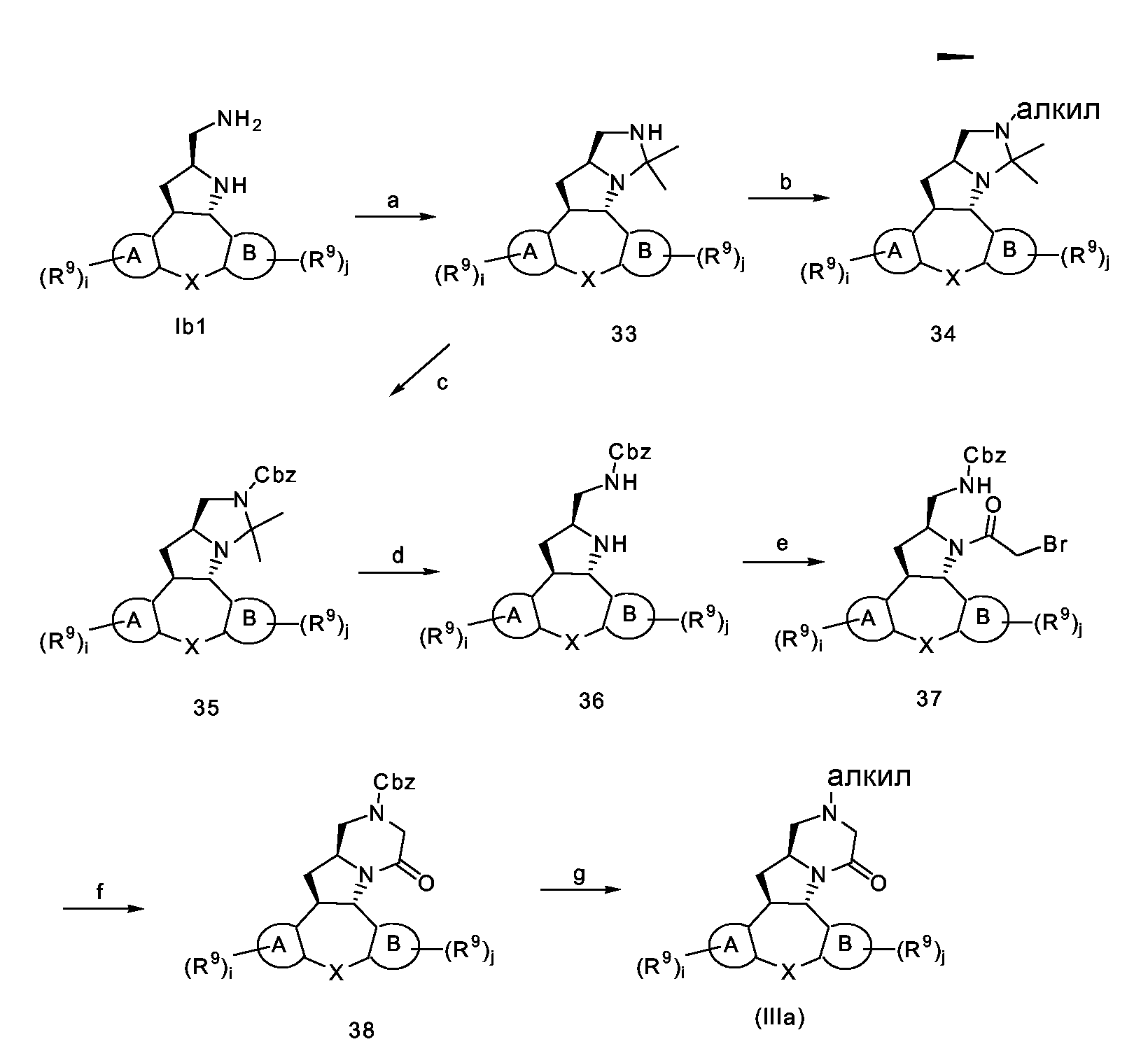
Стадия a): образование аминаля из конечного соединения Ib1 с использованием ацетона в инертном по отношению к реакции растворителе, таком как MeOH, например при температуре около 60°C в течение примерно 4 часов;
Стадия b): транс-аминализация и восстановительное аминирование конечного соединения 33 подходящим альдегидом или кетоном, например формальдегидом, и гидрирование с использованием катализатора палладия на угле (1 атм);
Стадия c): защита конечного соединения 33 CbzCl с использованием основания, такого как K2CO3, в инертной по отношению к реакции смеси растворителей, такой как ТГФ-H2O;
Стадия d): гидролиз промежуточного соединения 35 кислотой, такой как хлористоводородная кислота, в водном ТГФ, например при комнатной температуре в течение примерно 12 часов;
Стадия e): ацилирование промежуточного соединения 36 галогенидом кислоты, в частности BrC(=O)CH2Br, в EtAc в присутствии водного гидроксида натрия;
Стадия f): внутримолекулярная циклизация промежуточного соединения 37 с использованием основания, такого как K2CO3, в инертном по отношению к реакции растворителе, таком как ДМФА;
Стадия g): удаление Cbz-остатка гидрированием промежуточного соединения 38 с использованием катализатора палладия на угле (1 атм) и обработка in-situ альдегидом или кетоном, например формальдегидом, в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 3 часов;
Схема F2

Стадия a): тритилирование промежуточного соединения Ia1 например тритилхлоридом и DMAP обычно в присутствии основания, такого как Et3N, и в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 2 часов;
Стадия b): взаимодействие промежуточного соединения 33a с BrCH2COBr в присутствии основания, такого как гидрокарбонат натрия, и в инертном по отношению к реакции растворителе, таком как CH2Cl2;
Стадия c): взаимодействие промежуточного соединения 34a с муравьиной кислотой в течение примерно 3 часов и затем с EEDQ в инертном по отношению к реакции растворителе, таком как CHCl3, например при комнатной температуре в течение 30 минут;
Стадия d): циклизация промежуточного соединения 35a с использованием основания, такого как t-BuOK, в инертном по отношению к реакции растворителе, таком как ТГФ;
Стадия e): удаление альдегидной группы из промежуточного соединения 36a, например обработкой кислотой, такой как 2 М хлористоводородная кислота в метаноле;
Стадия f): восстановительное аминирование конечного соединения IIIb1 подходящим альдегидом или кетоном и гидрирование с использованием катализатора палладия на угле (1 атм).
Способ G: Получение 8,8-замещенных производных пирролидина
Следующая схема реакции G иллюстрирует получение соединений формулы (I), в которых C означает группу формулы (c-1), и Х означает группу CR6R7, отличную от группы водорода, представленных приведенными ниже формулами IV-VI.
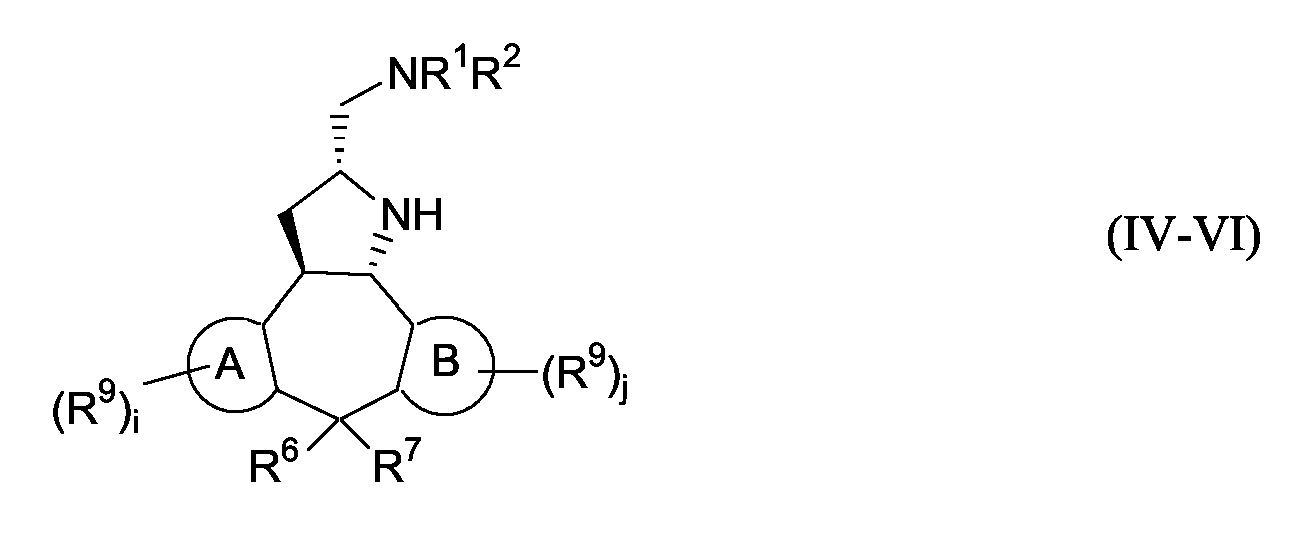
Схема G

Стадия a): обработка промежуточного соединения 10 или 13 Boc2O и основанием, таким как водный KOH или NaOH, в таком растворителе, как ТГФ или диоксан, например при комнатной температуре в течение примерно 6 часов;
Стадия b): обработка промежуточного соединения 39 DIAD/P(Ph)3 в таком растворителе, как ТГФ, например при температуре примерно от -15°C до комнатной температуры;
Стадия c): окисление промежуточного соединения 40 с использованием KMnO4 в присутствии катализатора фазового переноса, такого как n-Bu4NHSO4, в системе растворителей, такой как CH2Cl2-H2O, например при комнатной температуре в течение примерно 16 часов;
Стадия d): гидрирование промежуточного соединения 41 с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре, с последующей обработкой альдегидом или кетоном, таким как формальдегид, в присутствии AcOH с образованием промежуточного соединения, в котором каждый из R1 и R2 означает алкил;
Стадия e) обработка промежуточного соединения 42 50%-ной серной кислотой в диоксане, например при комнатной температуре в течение 3 часов, чтобы удалить защитную Boc-группу с образованием конечного соединения 42a;
Стадия f): конечное соединение 42a подвергают взаимодействию в реакции Гриньяра с бромидом метилмагния в таком растворителе, как ТГФ, например при комнатной температуре с образованием конечного соединения 43;
Стадия g): обработка конечного соединения 43 сульфонилхлоридом и пиридином, например при комнатной температуре в течение 16 часов с образованием конечного соединения 44;
Стадия h): гидрирование конечного соединения 44 с использованием катализатора палладия на угле (1 атм) в таком растворителе, как MeOH, например при комнатной температуре с образованием конечного соединения 45.
Способ H: Получение 3-замещенных производных пирролидина
Следующая схема реакции H иллюстрирует получение соединений формулы (I), в которых C означает группу формулы (c-3), Y1 означает NH, и R11 означает группу формулы (d-1), представленных приведенной ниже формулой VII.

Схема H

Стадия a): окислительное расщепление промежуточного соединения 6 с использованием NaIO4 в фосфатном буфере (при pH 7) в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от 0°C до комнатной температуры в течение примерно 4 часов;
Стадия b): обработка промежуточного соединения 46 CH2(NMe2)I и необязательно AcOH в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 3 часов;
Стадия c): (i) внутримолекулярная циклизация промежуточного соединения 47 связанным с полимером P(Ph)3 в инертном по отношению к реакции растворителе, таком как ТГФ, содержащем следы воды, например при температуре около 40°C в течение примерно 1 часа; с последующим (ii) восстановлением полученного в результате промежуточного соединения с использованием NaCNBH3 и AcOH в инертном по отношению к реакции растворителе, таком как спирт, в частности MeOH, например при комнатной температуре в течение примерно 3 часов;
Стадия d): (i) обработка промежуточного соединения 48 ClCO2Me и водным гидрокарбонатом натрия в инертном по отношению к реакции растворителе, таком как CH2Cl2; (ii) с последующей обработкой полученного в результате промежуточного соединения NaBH4, BF3Et2O в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 24 час; и (iii) последующая обработка полученного в результате промежуточного соединения H2O2 и водным KOH, например при комнатной температуре в течение примерно 3 часов;
Стадия e): обработка промежуточного соединения 49 DIAD/P(Ph)3 и DPPA в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры;
Стадия f): (i) промежуточное соединение 50 подвергают реакции Штаудингера или гидрированию с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре; и затем (ii) осуществляют восстановительное аминирование альдегидом или кетоном, например формальдегидом.
Способ I: Получение тетрагидрофуран-3-замещенных производных
Следующая схема реакции I иллюстрирует получение соединений формулы (I), в которых C означает группу формулы (c-3), Y2 означает O и R11 означает группу формулы (d-1), представленных приведенной ниже формулой VIII.

Схема I

Стадия a): защита спиртовой функции промежуточного соединения 4 с использованием DIAD/P(Ph)3 и 4-нитробензойной кислоты (PNBzOH) в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение подходящего периода времени, например около 15 часов;
Стадия b): обработка промежуточного соединения 51 хлористоводородной кислотой в ТГФ (например в виде смеси 1:1 с использованием 1 н хлористоводородной кислоты) например при комнатной температуре в течение примерно 5 часов;
Стадия c): обработка промежуточного соединения 52 NaIO4 при pH 7 с использованием фосфатного буфера в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от 0°C до комнатной температуры в течение примерно 4 часов;
Стадия d): обработка промежуточного соединения 53 CH2(NMe2)I и AcOH в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 3 часов;
Стадия e): восстановление промежуточного соединения 54 таким восстановителем, как борогидрид натрия, в инертном по отношению к реакции растворителе, таком как метанол, EtOH или i-PrOH, например при комнатной температуре в течение примерно 4 часов;
Стадия f): обработка промежуточного соединения 55a метоксидом натрия в инертном по отношению к реакции растворителе, таком как метанол, например при комнатной температуре в течение примерно 4 часов;
Стадия g): обработка промежуточного соединения 55b смесью DIAD/трибутилфосфин в инертном по отношению к реакции растворителе, таком как толуол, например при комнатной температуре в течение примерно 3 часов;
Стадия h): (i) гидроборирование промежуточного соединения 56 борогидридом натрия и BF3-Et2O в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 24 часов; и (ii) обработка H2O2, водным раствором гидроксида натрия в инертном по отношению к реакции растворителе, таком как ТГФ, например при комнатной температуре в течение примерно 4 часов;
Стадия i): обработка промежуточного соединения 57 DIAD/P(Ph)3, DPPA в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение примерно 15 часов;
Стадия j): (i) промежуточное соединение 58 подвергают реакции Штаудингера или гидрированию с использованием катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 1,5 часов; и (ii) восстановительное аминирование альдегидом или кетоном, например водным формальдегидом в AcOH и метаноле.
Способ J: Получение 3-замещенных производных тетрагидропирана
Следующая схема реакции J иллюстрирует получение соединений формулы (I), в которых C означает группу формулы (c-2), Y2 означает O и R11 означает группу формулы (d-1), представленных приведенной ниже формулой IX. Соединение может представлять собой либо цис- (схема J1), либо транссоединение (схема J2) относительно кислорода.

Схема J1 (цис)

Стадия a): монотозилирование промежуточного соединения 52a (полученного аналогично промежуточному соединению 52) TsCl, Et3N и Bu2SnO, например при комнатной температуре в течение примерно 16 часов в инертном по отношению к реакции растворителе, таком как толуол или CH2Cl2;
Стадия b): обработка промежуточного соединения 59 DHP и CSA в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 3 часов;
Стадия c): деацетилирование промежуточного соединения 60 основанием, таким как K2CO3, в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 3 часов с последующей внутримолекулярной циклизацией с использованием NaH в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от 0°C до комнатной температуры в течение примерно 4 часов;
Стадия d): удаление защиты промежуточного соединения 61 с использованием Dowex в инертном по отношению к реакции растворителе, таком как MeOH/H2O, например при комнатной температуре в течение примерно 2 суток;
Стадия e): мезилирование промежуточного соединения 62 MsCl, DMAP и Et3N в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 4 часов;
Стадия f): обработка промежуточного соединения 63 NaN3 в инертном по отношению к реакции растворителе, таком как ДМФА, например при температуре около 90°C в течение примерно 2 часов;
Стадия g): гидрирование промежуточного соединения 64 с использованием катализатора палладия на угле (1 атм) в инертной по отношению к реакции смеси растворителей, такой как i-PrOH/ТГФ, например при комнатной температуре в течение примерно 3 часов;
Стадия h): гидрирование промежуточного соединения 65 с использованием катализатора палладия на угле (1 атм) в инертной по отношению к реакции смеси растворителей, такой как i-PrOH/ТГФ, и восстановительное аминирование альдегидом или кетоном;
Стадия i): окисление промежуточного соединения 62 с использованием катализатора PCC в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 24 часов;
Стадия j): восстановительное аминирование промежуточного соединения 66 подходящим R1R2NH-соединением и гидрирование с использованием катализатора палладия на угле (1 атм) в присутствии основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре в течение примерно 24 часов.
Схема J2 (транс)
Схема реакции J1 также может быть применена по отношению к трансэпимеру промежуточного соединение 52, приводя к транссоединениям (IX) и (X).

Способ K: Получение 4-замещенных производных тетрагидропирана
Следующая схема реакции K иллюстрирует получение соединений формулы (I), в которых C означает группу формулы (c-2), Y2 означает O, и R11 означает группу формулы (d-1), представленных приведенной ниже формулой X.

Схема K

Стадия a): обработка промежуточного соединения 67 3-пентаноном и CSA, например, при температуре около 50°C в течение примерно 16 часов;
Стадия b): обработка промежуточного соединения 68 PCC в инертном по отношению к реакции растворителе, таком как CH2Cl2, с использованием молекулярных сит (4A), например, при температуре примерно от 0°C до комнатной температуры в течение примерно 75 минут;
Стадия c): взаимодействие промежуточного соединения 1 с промежуточным соединением 69, MgBr2с использованием в качестве катализатора t-BuOK в инертном по отношению к реакции растворителе, таком как PhMe/ТГФ, например при комнатной температуре в течение примерно 23 часов; указанную реакцию необходимо осуществлять в отсутствие кислорода, предпочтительно в атмосфере аргона;
Стадия d): гидрирование промежуточного соединения 71 водородом над катализатором палладий на угле (10%) в инертном по отношению к реакции растворителе, таком как Et3N, j-PrOH или толуол или их смесь, например при комнатной температуре в течение примерно 15 часов;
Стадия e): восстановление промежуточного соединения 72 таким восстановителем, как борогидрид натрия, в фосфатном буфере при pH 7 в инертном по отношению к реакции растворителе, таком как i-PrOH, например при температуре примерно от 0°C до комнатной температуры в течение примерно 1 часа;
Стадия f): обработка промежуточного соединения 73a DIAD/P(Ph)3, 4-нитробензойной кислотой (PNBzOH) в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре примерно от -15°C до комнатной температуры в течение примерно 15 часов;
Стадия g): обработка промежуточного соединения 73b хлористоводородной кислотой (1 н) в ТГФ (1:1), например, при комнатной температуре в течение примерно 5 часов;
Стадия h): тозилирование промежуточного соединения 73c TsCl, Et3N, дибутил(оксо)оловом (Bu2SnO) в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 12 часов;
Стадия i): циклизация промежуточного соединения 73d с использованием метоксида натрия в инертном по отношению к реакции растворителе, таком как метанол, например при комнатной температуре в течение примерно 3 часов;
Стадия j): тозилирование промежуточного соединения 74 TsCl, Et3N и DMAP в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при комнатной температуре в течение примерно 16 часов;
Стадия k): обработка промежуточного соединения 75 соединением формулы HNR1R2 в инертном по отношению к реакции растворителе, таком как ТГФ, в стальном баллоне при температуре около 135°C в течение примерно 15 часов.
Способ L: Получение тетрагидротиофен-2-замещенных производных
Следующие схемы реакций L1-L3 иллюстрируют получение соединений формулы (I), в которых C означает группу формулы (c-1), Y1 означает SO(n), и R11 означает группу формулы (d-1), представленных приведенными ниже формулами XIa-c и XIIa-c.

Схема L1: Синтез промежуточных соединений (2R,3aR,12bS)-тетрагидротиофена

Стадия a): обработка промежуточного соединения 59 азидом натрия и хлоридом аммония в инертном по отношению к реакции растворителе, таком как ДМФА, например при температуре около 90°C;
Стадия b): промежуточное соединение 76 подвергают реакции Митсунобу (обеспечивающей дополнительную инверсию по атому углерода) с использованием DIAD/P(Ph)3 и пара-нитробензойной кислоты (PNBzOH) в инертном по отношению к реакции растворителе, таком как ТГФ, например при температуре от 0°C до комнатной температуры в течение примерно 2 часов;
Стадия c): удаление защиты промежуточного соединения 77 раствором основания, таким как K2CO3/MeOH, например при комнатной температуре в течение примерно 2 часов;
Стадия d): мезилирование промежуточного соединения 78 MsCl и DMAP с использованием основания, такого как Et3N, в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при температуре примерно от 0°C до комнатной температуры в течение примерно 30 минут с последующей обработкой in situ AcSH при температуре примерно от 0°C до комнатной температуры в течение примерно 5 часов;
Стадия e): деацилирование и сопутствующая циклизация промежуточного соединения 79 с использованием раствора основания, такого как K2CO3/MeOH, например при комнатной температуре в течение примерно 2 часов.
Схема L2: Синтез промежуточных соединений (2S,3aR,12bS)-тетрагидротиофена

Стадия a): обработка промежуточного соединения 76 раствором основания, таким как K2CO3/MeOH, например при комнатной температуре в течение примерно 2 часов;
Стадия b): (i) обработка промежуточного соединения 82 (CH3SO2)2O, Et3N, DMAP в инертном по отношению к реакции растворителе, таком как CH2Cl2, например при температуре около 0°C; или (ii) обработка промежуточного соединения 82 MsCl, DMAP и Et3N в инертном по отношению к реакции растворителе, таком как CH2Cl2 при температуре около 0°C с последующей обработкой in situ AcSH примерно при 0°C в течение примерно 5 часов;
Стадия c): деацилирование и сопутствующая циклизация промежуточного соединения 83 основанием, таким как K2CO3/MeOH, например при комнатной температуре в течение примерно 30 минут.
Схема L3: Синтез производных (2RS,3aR,12bS)-тетрагидротиофена

2R,3aR,12bS: XIa (n=0), XIb (n=1), XIc (n=1), XId (n=2)
2S,3aR,12bS: XIIa (n=0), XIIb (n=1), XIIc (n=1), XIId (n=2)
Стадия a): (i) обработка промежуточного соединения 80 или 84 с использованием реакции Штаудингера или гидрирования с применением катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре; и (ii) восстановительное аминирование полученного в результате промежуточного соединения альдегидом или кетоном;
Стадия b): (i) обработка промежуточного соединения 80 или 84 водным пероксидом водорода в инертном по отношению к реакции растворителе, таком как HFIP, например при комнатной температуре в течение примерно 15 минут; (ii) обработка полученного в результате промежуточного соединения с использованием реакции Штаудингера или гидрирования с применением катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре; (iii) восстановительное аминирование полученного в результате промежуточного соединения альдегидом или кетоном;
Стадия c): (i) обработка промежуточного соединения 80 или 84 mCPBA в инертном по отношению к реакции растворителе, таком как CH2Cl2; (ii) обработка полученного в результате промежуточного соединения с использованием реакции Штаудингера или гидрирования с применением катализатора палладия на угле (1 атм) в инертном по отношению к реакции растворителе, таком как MeOH, например при комнатной температуре; и (iii) восстановительное аминирование полученного в результате промежуточного соединения альдегидом или кетоном.
Соединения формулы (I) также могут быть превращены друг в друга, следуя известным в данной области реакциям трансформации. Например,
a) соединение формулы (I), в котором R1 и R2 вместе взятые с атомом азота, с которым они связаны, образуют радикал формулы (a-2), может быть превращено в соответствующий первичный амин обработкой гидразином или водным раствором щелочи;
b) соединение формулы (I), в котором R1 или R2 является трифторметилкарбонилом, может быть превращено в соответствующий первичный или вторичный амин гидролизом водным раствором щелочи;
c) соединение формулы (I), в котором R1 или R2 означает C1-6-алкил, замещенный C1-6-алкилкарбонилоксигруппой, может быть гидрализовано в соединение формулы (I), в котором R1 или R2 означает C1-6-алкил, замещенный гидроксигруппой;
d) соединение формулы (I), в котором оба радикала R1 и R2 являются водородом, может быть моно- или ди-N-алкилировано до соответствующей формы амина;
e) соединение формулы (I), в котором оба радикала R1 и R2являются водородом, R1 или R2 является водородом, могут быть N-ацилированы до соответствующего амида;
f) соединение формулы (I), содержащее C1-6-алкилоксикарбонильную группу, могут быть гидролизованы до соответствующей карбоновой кислоты;
g) соединение формулы (I), в котором R9 означает водород, т.е. i и/или j равно нулю, может быть превращено в соответствующее алкилоксикарбонильное соединение обработкой подходящим ацилирующим агентом, например подходящим алкилоксикарбонилхлоридом в присутствии бутиллития в гексане с использованием органического растворителя, такого как тетрагидрофуран; или
h) соединение формулы (I), в котором R9 является алкилоксикарбонилом, может быть превращено в соответствующее гидроксиметилсоединение восстановлением, например LiAlH4, например в органическом растворителе, таком как тетрагидрофуран.
Описанные выше способы могут быть модифицированы с применением обычных способов, которые будут известны специалистам в данной области для осуществления аналогичных процессов получения соединений формулы (I).
Указанные выше исходные вещества являются либо коммерчески доступными, либо могут быть получены согласно известным в данной области способами. Например, промежуточные соединения 1 могут быть получены согласно способам, описанным в заявках на выдачу патента WO 03/048146 и WO 03/048147, упоминаемых выше, или аналогичными способами.
Чистые стереохимически изомерные формы соединений формулы (I) могут быть получены с применением известных в данной области способов. Диастереомеры могут быть разделены физическими способами, такими как избирательная кристаллизация и хроматографические способы, например противоточное распределение, жидкостная хроматография и тому подобные.
Соединения формулы (I), которые получают описанными выше способами, обычно представляют собой рацемические смеси энантиомеров, которые могут быть отделены друг от друга известными в данной области способами разделения. Рацемические соединения формулы (I), которые обладают достаточными основными или кислотными свойствами, могут быть превращены в формы соответствующих диастереомерных солей в результате взаимодействия с подходящей хиральной кислотой и соответственно с подходящим хиральным основанием. Затем указанные формы диастереомерных солей разделяют, например избирательной или фракционной кристаллизацией, и энантиомеры высвобождают с использованием щелочи или кислоты. Альтернативный способ разделения энантиомерных форм соединений формулы (I) заключается в жидкостной хроматографии с использованием хиральной неподвижной фазы. Указанные чистые стереохимически изомерные формы также могут быть получены из соответствующих чистых стереохимически изомерных форм подходящих исходных веществ, при условии, что реакция происходит стереоспецифично. Предпочтительно, если требуется конкретный стереоизомер, указанное соединение будет синтезировано стереоспецифичными способами получения. В таких способах преимущественно будут использованы энантиомерно чистые исходные вещества.
Следующие примеры предназначены для иллюстрации, но не для ограничения объема настоящего изобретения.
Экспериментальная часть
A. Получение промежуточных соединений
Пример A1
(11R)-11-{[(4R)-2,2-диметил-l,3-диоксолан-4-ил]метил}-8-фтор-5,11-дигидро-10H-дибензо[a,d]циклогептен-10-он (промежуточное соединение 2)

Раствор промежуточного α,β-ненасыщенного кетона 1 (1,00 г, 2,96 ммоль) и Et3N (0,63 мл, 4,50 ммоль) в i-PrOH (30 мл) гидрировали с использованием 10% Pd/C при атмосферном давлении в течение 6 часов. Затем смесь фильтровали через подушку целита и твердые вещества промывали CH2Cl2 (4×20 мл). После выпаривания добавляли i-PrOH (5 мл) и Et3N (1,20 мл) и реакционную смесь перемешивали при 40°C в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры и давали возможность кристаллизоваться. Кристаллы отфильтровывали и сушили в вакууме, получая чистый промежуточный кетон 2 в виде белого кристаллического порошка (0,86 г, 86%); т.пл.: 144-146°C.
Масс-спектр: CI m/z (распределение, относительная интенсивность) 341 (MH+, 2%), 283 (MH+ - ацетон, 100%); EI: m/z (распределение, относительная интенсивность) 340 (M+., 1%), 282 (M+ - ацетон, 79%), 226 (M+ - боковая цепь + H, 100%); Высокое разрешение EI, вычислено C21H21FO3 (M+.): 340,1475, найдено: 340,1479 (1%).
Пример A2
(10R,11R)-11-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 3)

промежуточное соединение 3
К охлажденному на льду раствору промежуточного кетона 2 (0,42 г, 1,23 ммоль) в i-PrOH (15 мл) добавляли раствор фосфатного буфера (pH 7, 5 мл) и затем порциями NaBH4 (0,23 г, 6,16 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем добавляли 10 мл NH4Cl (насыщенный водный раствор), смесь экстрагировали CH2Cl2 (3×15 мл) и органические фазы сушили над MgSO4. После удаления растворителя остаток очищали на колонке с силикагелем, используя смесь эфир/гексан (40:60), получая промежуточное соединение 3 в виде бесцветного масла (0,42 г, 99%).
Масс-спектр: CI m/z (распределение, относительная интенсивность) 325 (MH+ - H2O, 53%), 267 (MH+ - H2O-ацетон, 100%), 249 (MH+ - 2H2O-ацетон, 97%); EI: m/z (распределение, относительная интенсивность) 342 (M+, 3%), 324 (M+ - H2O, 48%), 266 (M+ - H2O-ацетон, 35%), 209 (100%); Высокое разрешение EI вычислено C21H23FO3 (M+): 342,1631, найдено: 342,1627(5%).
Пример A3
(4R)-4-{[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]метил}-2,2-диметил-1,3-диоксолан (промежуточное соединение 4)

промежуточное соединение 4
К охлажденному (-30°C) раствору DIAD (2,43 мл, 33,47 ммоль) в ТГФ (10 мл) добавляли промежуточный спирт 3 (2,30 г, 6,73 ммоль) в ТГФ (18 мл) и Ph3P (3,71 г, 14,07 ммоль). Через 20 минут добавляли дифенилфосфорилазид (DPPA) (3,62 мл, 16,83 ммоль) и реакционной смеси давали возможность нагреться до комнатной температуры. После перемешивания в течение ночи растворитель удаляли в вакууме, получая красное масло. Грубый продукт очищали хроматографией на колонке, используя смесь эфир/гексан (10/90), получая неразделенную смесь промежуточного соединения в виде масла и Ph3PO (3,46 г).
Масс-спектр: CI m/z (распределение, относительная интенсивность) 368 (MH+, 1%), 325 (MH+ -HN3, 9%), 304 (13%), 276 (MH+ -HN3-ацетон, 100%), 248 (20%).
Пример A4
(2R)-3-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-1,2-пропандиол (промежуточное соединение 5)

промежуточное соединение 5
К раствору промежуточного азида 4 (3,68 г, 10,02 ммоль) в ТГФ (30 мл) добавляли 1н HCl (30 мл) и смесь перемешивали при комнатной температуре в течение 8 часов. Добавляли K2CO3 (насыщенный водный раствор) при 0°C, экстрагировали 3 раза CH2Cl2 и сушили над MgSO4. Остаток, полученный при упаривании, очищали хроматографией на колонке с силикагелем, используя смесь Et2O/гептан (30/70), получая маслянистое промежуточное соединение 5 (3,19 г, 91% для 2 стадий, начиная с 3).
Масс-спектр: CI m/z (распределение, относительная интенсивность) 328 (MH+, 2%), 310 (MH+ -H2O, 2%), 300 (MH+ -N2, 5%), 285 (MH+ -HN3, 11%), 267 (MH+ -HN3-H2O, 100%), 249 (MH+ -HN3-2H2O, 33%), 225 (MH+ -HN3-CH2OHCHO, 20%).
Пример A5
(2R)-3-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-2-гидроксипропил-4-метилбензолсульфонат (промежуточное соединение 6)

промежуточное соединение 6
К раствору промежуточного диола 5 (1,1 г, 3,39 ммоль) в безводном толуоле (10 мл) добавляли Bu2SnO (97,6 мг, 0,39 ммоль), Et3N (1,07 мл, 7,74 ммоль) и TsCl (0,739 г, 3,87 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли NH4Cl (насыщенный водный раствор), экстрагировали 3 раза CH2Cl2 и сушили над MgSO4. Остаток очищали хроматографией на колонке с силикагелем, используя EtOAc/гептан (20/80), получая промежуточное соединение 6 в виде масла (1,55 г, 95%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 454 (MH+ -N2, 1%), 421 (MH+ -HN3-H2O, 1%), 282 (MH+ -TsOH-HN3, 20%), 264 (MH+ -TsOH-HN3-H2O, 15%), 173 (TsOH2+, 100%).
Пример A6
(2R)-1-азидо-3-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H- дибензо[a,d]циклогептен-10-ил]-2-пропанол (промежуточное соединение 7)

промежуточное соединение 7
Раствор промежуточного тозилата 6 (2,00 г, 4,15 ммоль) в ДМФА (30 мл) обрабатывали азидом натрия (810,8 мг, 12,47 ммоль) и смесь перемешивали при 90°C в темноте в течение 2 часов. Реакционную смесь разбавляли водой и экстрагировали CH2Cl2. Объединенные экстракты промывали насыщенным раствором соли. После концентрирования органических фаз остаток очищали хроматографией на колонке с силикагелем, используя гептан/EtOAc (80/20), получая промежуточный диазид 7 (1,22 г, 88%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 325 (MH+ -N2, 2%), 310 (MH+ -HN3, 3%), 297 (MH+ - N2-N2, 1%), 282 (MH+ -HN3-N2, 52%), 268 (MH+ -HN3-HN3, 3%).
Пример A7
(1R)-2-азидо-1-{[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]метил}этилметансульфонат (промежуточное соединение 8)

промежуточное соединение 8
К раствору промежуточного соединения диазида 7 (65 мг, 0,18 ммоль) в CH2Cl2 (10 мл) добавляли DMAP (18,5 мг, 0,09 ммоль), Et3N (0,13 мл, 0,63 ммоль) и MsCl (44,5 мкл, 0,40 ммоль). После перемешивания при комнатной температуре в течение 10 минут добавляли 10 мл NH4Cl (насыщенный водный раствор). Экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (20:80) давала промежуточное соединение 8 в виде масла (78,2 мг, 98%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 403 (MH+-N2, 3%), 360 (MH+ -N2-HN3, 43%), 307 (MH+ - MeSO3H-N2, 50%), 264 (MH+ -MeSO3H-HN3-N2, 58%), 250 (MH+ -MeSO3H-HN3, -N3, 21%), 197 (100%).
Пример A8
[(2S,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метанамин (промежуточное соединение 9)

промежуточное соединение 9
Раствор промежуточного соединения диазида 8 (98,2 мг, 0,23 ммоль) в MeOH (10 мл) гидрировали при атмосферном давлении, используя 10% Pd/C в течение 1 ночи. Затем смесь фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После упаривания фильтрата грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH/NH4OH (90/9/1). Очистка давала промежуточное соединение 9 в виде масла (36,4 мг, 56%).
Пример A9
(1S)-2-азидо-1-{[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]метил}этил-4-нитробензоат (промежуточное соединение 10)

промежуточное соединение 10
К охлажденному (0°C) раствору DIAD (4,2 мл, 21,18 ммоль) в ТГФ (50 мл) добавляли Ph3P (5,55 г, 21,18 ммоль). Перемешивали при 0°C в течение 30 минут (осаждение белого твердого вещества). Затем добавляли смесь промежуточного спирта 7 (3,727 г, 10,59 ммоль) и 4-нитробензойной кислоты (3,54 г, 21,18 ммоль) в ТГФ (50 мл). Реакционной смеси давали возможность нагреться до комнатной температуры и после перемешивания в течение 2 часов добавляли MeOH и перемешивание продолжали еще в течение 30 минут. После удаления растворителя грубый продукт очищали хроматографией на колонке, используя смесь EtOAc/гептан (20/80), получая промежуточный сложный эфир 10 в виде масла (4,85 г, 91%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 431 (MH+ -N2-HN3, 36%), 307 (MH+ -N2-p-NO2PHCO2H, 2%), 264 (MH+ -p-NO2PHCO2H-HN3- N2, 58%), 197 (100%), 182 (72%).
Пример A10
(2S)-1-азидо-3-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-2-пропанол (промежуточное соединение 11)

промежуточное соединение 11
Раствор указанного выше промежуточного диазида 10 (78,0 мг, 0,15 ммоль) в MeOH (2 мл) обрабатывали K2CO3 (76,9 мг, 0,47 ммоль) и смесь перемешивали в течение 1 часа. Добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили MgSO4. Остаток очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гептан (20/80), получая промежуточный спирт 11 в виде масла (42,6 мг, 78%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 325 (MH+ -N2, 2%), 310 (MH+ -HN3, 3%), 297 (MH+ -N2-N2, 1%), 282 (MH+ -HN3-N2, 52%), 268 (MH+ -HN3-N3, 3%).
Пример A11
(1S)-2-азидо-1-{[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]метил}этилметансульфонат (промежуточное соединение 12)

промежуточное соединение 12
К раствору промежуточного диазида 11 (42,6 мг, 0,12 ммоль) в CH2Cl2 (5 мл) добавляли DMAP (12,7 мг, 0,06 ммоль), Et3N (0,047 мл, 0,42 ммоль) и MsCl (33,9 мкл, 0,30 ммоль). Перемешивали при комнатной температуре в течение 10 минут. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4; после выпаривания растворителя получали промежуточное соединение 12 в виде масла (53,0 мг, 100%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 403 (MH+ -N2, 3%), 360 (MH+ -N2-HN3, 43%), 307 (MH+ -MeSO3H-N2, 50%), 264 (MH+ -MeSO3H-HN3-N2, 58%), 250 (MH+ -MeSO3H-HN3-N3, 21%), 197 (100 %).
Пример A12
[(1R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метанамин (промежуточное соединение 13)

промежуточное соединение 13
Раствор промежуточного диазида 12 (501,0 мг, 1,16 ммоль) в MeOH (10 мл) гидрировали при давлении в 1 атмосферу, используя 10% палладий на угле, при энергичном перемешивании при комнатной температуре в течение 1 ночи. Затем смесь фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя CHCl3/MeOH/NH4OH (90/9/1). При этом получали промежуточное соединение 13 в виде масла (270,0 мг, 82%).
Пример A13
Бензил[(2S,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метилкарбамат (промежуточное соединение 14)

промежуточное соединение 14
К раствору промежуточного диамина 9 (220,0 мг, 0,78 ммоль) в CH2Cl2 (5 мл) при -20°C добавляли Et3N (0,109 мл, 0,78 ммоль) и бензилхлорформиат (0,112 мл, 0,78 ммоль). Затем смесь перемешивали в течение 1 часа. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Остаток очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гептан (20/80), получая моно-Cbz-промежуточное соединение 14 (128,9 мг, 40%) и ди-Cbz-производное (84,5 мг).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 417 (MH+, 100%), 397 (MH+ -HF, 8%), 311 (MH+ -PhCHO, 7%), 309 (MH+ -PhCH2OH, 32%), 283 (16%), 252 (MH+ -PhCH2OCONHCH3, 24%).
Пример A14
Бензил[(2S,3aR,12bS)-1-(бромацетил)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метилкарбамат (промежуточное соединение 15)

промежуточное соединение 15
К раствору моно-Cbz-промежуточного соединения 14 (32,5 мг, 0,078 ммоль) в EtOAc (3 мл) добавляли 1 мл NaOH (насыщенный водный раствор) и бромацетилбромид (6,8 мкл, 0,078 ммоль). Две фазы энергично перемешивали в течение 1 ночи. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (20/80) давала промежуточное соединение 15 в виде масла (31,4 мг, 62%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 457 (MH+ -HBr, 3%), 413 (MH+ -HBr-CO2, 1%), 365 (MH+ -HBr-PhCH3 1%), 351 (MH+ -PhCHO-HBr, 2%), 323 (MH+ -HBr-PhCHO-CO, 5%), 119 (8%), 91 (100%).
Пример A15
Бензил(5aS,14bR,15aS)-7-фтор-4-оксо-1,3,4,5a,10,14b,15,15a-октагидро-2H-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-a]пиразин-2-карбоксилат (промежуточное соединение 16)
промежуточное соединение 16
К раствору указанного выше промежуточного карбамата 15 (91,7 мг, 0,17 ммоль) в ДМФА (5 мл) добавляли K2CO3 (103,0 мг, 0,75 ммоль) и смесь перемешивали при комнатной температуре в течение 36 часов. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием EtOAc/гептан (30/70) давала полициклическое промежуточное соединение 16 (86,2 мг, 92%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 457 (MH+, 1%), 323 (MH+ -PhCHO-CO, 5%), 279 (MH+ -Cbz-CH2CO, 1%), 91 (10%).
Пример A16
N-{[(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}(трифенил)метанамин (промежуточное соединение 17)

промежуточное соединение 17
К охлажденному на льду раствору диамина 13 (41,6 мг, 0,15 ммоль) в CH2Cl2 (5 мл) добавляли Et3N (42,5 мкл, 0,3 ммоль), DMAP (9,4 мг, 0,07 ммоль) и тритилхлорид (46,1 мг, 0,16 ммоль). Затем смесь перемешивали при 0°C в течение 2 часов. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (20/80) давала кристаллическое промежуточное соединение 17 (52,6 мг, 68%); т.пл.: 58-60°C.
Масс-спектр: -APCI m/z (распределение, относительная интенсивность) 525 (MH+, 38%), 390 (4%), 283 (MH+ -(Tr-H), 15%), 252 (MH+ -CH3NHTr, 27%), 243 (Tr+, 100%), 228 (7%), 165 (29%).
Пример A17
N-{[(2R,3aR,12bS)-l-(бромацетил)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}(трифенил)метанамин (промежуточное соединение 18)

промежуточное соединение 18
Промежуточный диамин 17 (26,7 мг, 0,05 ммоль) добавляли к двухфазной системе, состоящей из 2 мл CH2Cl2 и 0,5 мл Na2CO3 (насыщенный водный раствор), и смесь перемешивали в течение 10 минут. После добавления бромацетилбромида (6,8 мкл, 0,08 ммоль) две фазы энергично перемешивали в течение 3 часов. Экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (20/80) давала промежуточное соединение 18 в виде масла (27,9 мг, 85%), характеризуемого как смесь двух конформеров.
Масс-спектр: -APCI m/z (распределение, относительная интенсивность) 645 (MH+, 39%), 601 (3%), 403 (MH+ -(Tr-H), 7%), 321 (MH+ -TrH-HBr, 21%), 243 (Tr+, 100%), 228 (3%), 165 (15%).
Пример A18
N-{[(2R,3aR,12bS)-11-фтор-1-(метоксиацетил)-1,2,3,3a,8,12b-гексагидродибензо-[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}(трифенил)метанамин (промежуточное соединение 19)

промежуточное соединение 19
К раствору промежуточного соединения 18 (530 мг, 0,82 ммоль) в MeOH (15 мл) добавляли MeSO3H (3 мл) и смесь перемешивали при 60°C в течение 30 минут. После полного выпаривания растворителя остаток растворяли в смеси CH2Cl2/K2CO3 (насыщенный водный раствор) (15/15 мл) и органический слой отделяли. Водный слой экстрагировали CH2Cl2 (3×10 мл) и затем объединенные органические слои сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (20/80) давала промежуточное соединение 19 в виде масла (231,3 мг, 47%), характеризуемое как смесь двух конформеров.
Масс-спектр: -APCI m/z (распределение, относительная интенсивность) 598 (MH+, 1%), 519 (2%), 355 (MH+ -Tr, 13%), 283 (MH+ -Tr-CO=CHOMe, 2%), 271 (10%), 243 (Tr+, 100%), 167 (21%).
Пример A19
[(1R,3aR,12bS)-1-(бромацетил)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метилформамид (промежуточное соединение 20)

промежуточное соединение 20
N-Tr-защищенный промежуточный амин 18 (100 мг, 0,15 ммоль) растворяли в 98% муравьиной кислоте (2 мл) и смесь перемешивали при комнатной температуре в течение 24 часов. После удаления избытка муравьиной кислоты в вакууме остаток растворяли в CHCl3 (2 мл) и добавляли EEDQ (47 мг, 0,19 ммоль). Раствор перемешивали при комнатной температуре в течение 5 часов. После выпаривания растворителя остаток очищали хроматографией на колонке с силикагелем, используя смесь CH2Cl2/MeOH (98/2) в качестве элюента. N-CHO-защищенный промежуточный амин 20 (54,7 мг, 82%) получали в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 431, 433 (MH+, 42%), 353 (MH+ -HBr, 100%), 294 (MH+ -HBr-CH3NHCHO, 9%), 249 (4%), 158 (2%), 130 (7%).
Пример A20
(5aS,14bR,15aR)-7-фтор-4-оксо-1,3,4,5a,10,14b,15,15a-октагидро-2H-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-a]пиразин-2-карбальдегид (промежуточное соединение 21)

промежуточное соединение 21
К раствору N-формил-замещенного промежуточного амина 20 (91 мг, 0,21 ммоль) в безводном ТГФ (10 мл) добавляли раствор t-BuOK (30,3 мг, 0,24 ммоль) в ТГФ (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Затем добавляли воду (10 мл) и смесь экстрагировали CH2Cl2 (10 мл). Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (97/3) давала циклическое промежуточное соединение 21 (47,4 мг, 64%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 351 (MH+, 100%), 331 (MH+ -HF, 5%), 323 (MH+ -CO, 6%), 319 (8%), 219 (2%), 130 (4%).
Пример A21
(10R,11R)-11-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-илацетат (промежуточное соединение 22)

К раствору промежуточного спирта 3 (0,42 г, 1,23 ммоль) в CH2Cl2 (30 мл) добавляли Et3N (0,43 мл, 3,07 ммоль), DMAP (0,15 г, 1,23 ммоль) и ангидрид AcOH (0,29 мл, 3,07 ммоль). Перемешивали при комнатной температуре в течение 1 часа, добавляли NH4Cl (насыщенный водный раствор, 20 мл), экстрагировали CH2Cl2 (3×15 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси эфир/гексан (30:70) давала белое кристаллическое промежуточное соединение 22 (0,45 г, 95%); т.пл.: 147-149°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 385 (MH+, 1%), 325 (MH+ -AcOH, 100%), 267 (MH+ -AcOH-ацетон, 43%), 249 (MH+ -AcOH-ацетон-H2O, 47%); -EI: m/z (распределение, относительная интенсивность) 324 (M+ -AcOH, 46%), 266 (M+ -AcOH-ацетон, 20%), 209 (M+ - AcOH - боковая цепь, 100%); Высокое разрешение EI вычислено C22H21FO2 (M+ -AcOH): 324,1526, найдено: 324,1521 (M+, 72%).
Пример A22
(10R,11R)-11-[(2R)-2,3-дигидроксипропил]-8-фтор-10,11-дигидро-5H-дибензо-[a,d]циклогептен-10-илацетат (промежуточное соединение 23)

промежуточное соединение 23
К раствору промежуточного ацеталя 22 (0,45 г, 1,17 ммоль) в ТГФ (10 мл) добавляли 1н HCl (10 мл). После перемешивания при комнатной температуре в течение 8 часов добавляли 10 мл Na2CO3 (насыщенный водный раствор) при 0°C. Экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гексан (70:30) давала промежуточный диол 23 в виде бесцветного масла (0,39 г, 96%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 345 (MH+, 1%), 327 (MH+ -H2O, 3%), 309 (MH+ -2H2O, 3%), 285 (MH+ -AcOH, 17%), 267 (MH+ -AcOH-H2O, 100%), 249 (MH+ -AcOH-2H2O, 3%); EI: m/z (распределение, относительная интенсивность) 326 (M+ -H2O, 10%), 284 (M+ -AcOH, 13%), 209 (M+ -AcOH - боковая цепь, 100%)); Высокое разрешение EI вычислено C20H19FO3 (M+ -H2O): 326,1318, найдено: 326,1316 (31%).
Пример A23
(10R,11R)-11-[(2S)-2,3-диазидопропил]-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-илацетат (промежуточное соединение 24)

К промежуточному соединению ацетат-диолу 23 (0,59 г, 1,72 ммоль) в CH2Cl2 (15 мл) добавляли Et3N (0,96 мл, 6,86 ммоль), DMAP (209 мг, 1,72 ммоль) и MeSO2Cl (0,53 мл, 6,86 ммоль) при 0°C. Перемешивали при комнатной температуре в течение 1 часа. Обрабатывали добавлением NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (50/50) давала димезилсодержащее соединение в виде масла (0,84 г, 98%). К полученному соединению (182,5 мг, 0,36 ммоль) в ДМФА (10 мл) добавляли NaN3 (95 мг, 1,46 ммоль). Реакционную смесь нагревали при 80°C в течение 3 часов. После охлаждения добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. После упаривания остаток очищали на силикагеле, используя смесь EtOAc/гептан (20/80), получая промежуточное соединение 24 в виде маслянистого продукта (122,3 мг, 85%).
Пример A24
(4S)-4-{[(10R,11R)-2-фтор-11-гидрокси-10,11-дигидро-5H-дибензо[a,d]-циклогептен-10-ил]метил}-2-имидазолидинон (промежуточное соединение 25)

промежуточное соединение 25
Промежуточный диазид 24 превращали через промежуточный диазидоспирт 24a в диамин, который затем превращали в промежуточное соединение 25. К раствору диазида 24 (120,1 мг, 0,30 ммоль) в MeOH (10 мл) добавляли K2CO3 (126,4 мг, 0,91 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси Et2O/гептан (40/60) давала промежуточный диазидоспирт 24a в виде маслянистого продукта (77,5 мг, 72%). Полученное соединение (75 мг, 0,21 ммоль) в MeOH (5 мл) гидрировали при давлении в 1 атмосферу, используя 10% палладий на угле, при энергичном перемешивании при комнатной температуре в течение 1 ночи. Затем смесь фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После выпаривания растворителя грубый продукт растворяли в 5 мл CH3CN и добавляли Et3N (34 мкл, 0,24 ммоль). Реакционную смесь нагревали в атмосфере аргона при 70°C. Через 1 час по каплям добавляли раствор дифенилкарбоната (23 мг, 0,11 ммоль) в CH3CN и смесь перемешивали при 70°C в течение 1 суток. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (90/10), получая промежуточный имидазолидинон 25 в виде масла (34,4 мг, 48%).
Пример A25
(11E)-11-{[(4R)-2,2-диметид-1,3-диоксолан-4-ил]метилен}-8-фтордибензо[b,f]оксепин-10(11H)-он (промежуточное соединение 27)

К суспензии промежуточного соединения 26 (0,228 г, 1 ммоль) и MgBr2 (0,202 г, 1,1 ммоль) в безводном толуоле (5 мл) добавляли ацетонид (S)-глицеральдегида (4 ммоль, 1,5 М раствор в ТГФ) и tBuOK (22,4 мг, 0,2 ммоль) и перемешивали в течение 3 часов при комнатной температуре. Добавляли насыщенный водный раствор NH4Cl (5 мл), органический слой отделяли и выдерживали над безводным MgSO4. Растворитель удаляли при пониженном давлении с последующим отделением α,β-ненасыщенного продукта флэш-хроматографией с использованием смеси EtOAc:гептан (1:9) в качестве элюента, получая промежуточное соединение 27 в виде желтой жидкости с соотношением 85/15 E- и Z-изомеров (85%, 0,289 г).
ВРМС: вычислено 340,1111; найдено 340,1122
Пример A26
a) (10R,11R)-11-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метил}-8-фтор-10,11-дигидродибензо[b,f]оксепин-10-ол (промежуточное соединение 29)

промежуточное соединение 29
К раствору промежуточного соединения 27 (0,340 г, 1 ммоль) в i-PrOH (5 мл) добавляли Et3N (0,21 мл, 1,5 ммоль) и реакционную смесь гидрировали при атмосферном давлении, используя 10% Pd/C (40 мг) в качестве катализатора. После завершения реакции (4 часа) реакционную смесь пропускали через небольшую подушку целита и затем промывали CH2Cl2 (2×5 мл) с последующим выпариванием растворителя и получением неочищенного промежуточного кетона 28.

промежуточное соединение 28
b) Полученное таким образом неочищенное промежуточное соединение 28 растворяли в i-PrOH (10 мл) и к нему добавляли водный раствор фосфатного буфера (3 мл, pH 7). Температуру снижали до 0°C и несколькими партиями добавляли NaBH4 (0,152 г, 4 ммоль) и затем давали возможность перемешиваться еще в течение 15 минут при такой же температуре. Добавляли водный раствор NH4Cl (5 мл) и реакционную смесь экстрагировали, используя Et2O (3×5 мл). После сушки над безводным MgSO4 растворитель удаляли при пониженном давлении и два диастереомерных спирта (1:1) с немного различающейся полярностью разделяли флэш-хроматографией, используя смесь EtOAc:гептан (20:80) в качестве элюента, получая более полярный промежуточный цис-спирт 29 в виде белого твердого вещества (т.пл.: 59-61°C; 49%, 0,16 г).
ВРМС: вычислено 344,1424; найдено 344,1435.
Пример A27
(10S,11R)-11-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метил}-8-фтор-10,11-дигидродибензо[b,f]оксепин-10-илазид (промежуточное соединении 30)

промежуточное соединение 30
К раствору P(Ph)3 (0,524 г, 2 ммоль) в безводном ТГФ (5 мл) при -15°C добавляли раствор DIAD (0,424 г, 2,1 ммоль) в ТГФ (2 мл) и полученный в результате комплекс перемешивали в течение 20 минут с последующим добавлением промежуточного соединения 29 (0,329 г, 1 ммоль), растворенного в ТГФ (2 мл), и раствор DPPA (0,330 г, 1,2 ммоль) в ТГФ (1 мл). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. После добавления MeOH реакционную смесь сушили в вакууме с последующим отделением азида с применением флэш-хроматографии, используя смесь EtOAc:гептан (1:9) в качестве элюента, получая промежуточное соединение 30 в виде бесцветной жидкости (91%, 0,335 г).
ВРМС: вычислено 369,1489; найдено 369,1483.
Пример A28
(2R)-3-[(10R,11S)-11-азидо-2-фтор-10,11-дигидродибензо[b,f]оксепин-10-ил]-1,2-пропандиол (промежуточное соединение 31)

промежуточное соединение 31
К раствору промежуточного соединения 30 (0,369 г, 1 ммоль) в ТГФ (5 мл) добавляли 1 М водный раствор HCl (1 мл) и перемешивали в течение 18 часов. ТГФ удаляли при пониженном давлении и диол экстрагировали с использованием Et2O (3×10 мл). Органический слой обрабатывали водным раствором NaHCO3 (5 мл) с последующей промывкой насыщенным раствором соли (5 мл). После сушки над безводным MgSO4 растворитель удаляли в вакууме, получая промежуточное соединение 31 в виде густой вязкой жидкости (95%, 0,313 г).
ВРМС: вычислено 329,1176; найдено 329,1184.
Пример A29
(2R)-1-[(10R,11S)-11-азидо-2-фтор-10,11-дигидродибензо[b,f]оксепин-10-ил]-3-(тритилокси)-2-пропанол (промежуточное соединение 32)

промежуточное соединение 32
К раствору промежуточного соединения 31 (0,329 г, 1 ммоль) в CH2Cl2 (10 мл) добавляли Et3N (0,28 мл, 2 ммоль), DMAP (0,1 ммоль, 12,2 мг) и TrCl (0,307 г, 1,1 ммоль) и перемешивали в течение 24 часов. Растворитель удаляли при пониженном давлении и неочищенную реакционную смесь подвергали флэш-хроматографии, используя смесь EtOAc:гептан (1:9) в качестве элюента, получая промежуточное соединение 32 в виде белого твердого вещества (т.пл.: 58-59°C; 80%, 0,456 г).
ВРМС: вычислено 571,2271; найдено 571,2286.
Пример A30
(1R)-2-[(10R,11S)-11-азидо-2-фтор-10,11-дигидродибензо[b,f]оксепин-10-ил]-1-[(тритилокси)метил]этилметансульфонат (промежуточное соединение 33)

промежуточное соединение 33
К раствору промежуточного соединения 32 (0,571 г, 1 ммоль) в CH2Cl2 при -10°C добавляли Et3N (0,28 мл, 2 ммоль), DMAP (12,2 мг, 0,1 ммоль) и MsCl (0,126 г, 1,1 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 4 часов. Добавляли воду (3 мл) и органический слой отделяли и сушили над безводным MgSO4 с последующей очисткой флэш-хроматографией с использованием смеси EtOAc:гептан (1:9) в качестве элюента, получая промежуточное соединение 33 в виде белого твердого вещества (т.пл.: 55-56°C; 85%, 0,515 г).
ВРМС: вычислено 649,2047; найдено 649,2064.
Пример A31
(1R)-2-[(10R,11S)-11-азидо-2-фтор-10,11-дигидродибензо[b,f]оксепин-10-ил]-1-(гидроксиметил)этилметансульфонат (промежуточное соединение 34)

промежуточное соединение 34
К раствору промежуточного соединения 33 (0,649 г, 1 ммоль) в MeOH (5 мл), добавляли amberlyst-15 (0,1 г) и реакционную смесь перемешивали при 40°C в течение 3 часов, затем фильтровали, чтобы удалить катализатор. Растворитель удаляли при пониженном давлении и продукт очищали флэш-хроматографией, используя EtOAc:гептан (2:8) в качестве элюента, получая промежуточное соединение 34 в виде густой вязкой жидкости (90%, 0,366 г).
ВРМС: вычислено 407,0951; найдено 407,0975.
Пример A32
(10R,11S)-11-азидо-2-фтор-10-[(2S)-оксиранилметил]-10,11-дигидродибензо[b,f]оксепин (промежуточное соединение 35)

промежуточное соединение 35
Смесь промежуточного соединения 34 (0,407 г, 1 ммоль) и K2CO3 (0,276 г, 2 ммоль) перемешивали в i-PrOH (10 мл) в течение 8 часов, фильтровали, чтобы удалить K2CO3, и растворитель удаляли при пониженном давлении. Продукт очищали флэш-хроматографией, используя смесь EtOAc:гептан (2:8) в качестве элюента, получая промежуточное соединение 35 в виде бесцветной жидкости (78%, 0,242 г).
ВРМС: вычислено 311,1070; найдено 311,1089.
Пример A33
[(2R,3aR,12bS)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метанол (промежуточное соединение 36)

промежуточное соединение 36
К раствору промежуточного соединения 35 (0,311 г, 1 ммоль) в i-PrOH (10 мл) добавляли Et3N (0,140 мл, 1 ммоль). Смесь гидрировали при атмосферном давлении, используя 10% Pd/C (50 мг) в качестве катализатора. После завершения реакции (3 часа) смесь пропускали через небольшую подушку целита и катализатор промывали CH2Cl2 (2×5 мл). Объединенные органические слои упаривали при пониженном давлении и очищали флэш-хроматографией, используя смесь EtOAC:гептан (1:1) в качестве элюента, получая промежуточное соединение 36 в виде белого твердого вещества (т.пл.: 108-109°C; 83%, 0,236 г).
ВРМС: вычислено 285,1165; найдено 285,1172.
Пример A34
Метил-(2R,3aR,12bS)-11-фтор-2-(гидроксиметил)-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-1-карбоксилат (промежуточное соединение 37)

промежуточное соединение 37
К раствору промежуточного соединения 36 (0,14 г, 0,5 ммоль) в CH2Cl2 (4 мл) при 0°C добавляли насыщенный раствор (водный) NaHCO3 (2 мл). После добавления метилхлорформиата (1,5 эквивалента) реакционную смесь энергично перемешивали при 0°C в течение 20 минут, нагревали до комнатной температуры и давали возможность перемешиваться еще в течение 0,5 часа. Органический слой отделяли, сушили над MgSO4 и очищали флэш-хроматографией, используя смесь EtOAc:гептан (4:6) в качестве элюента, получая промежуточное соединение 37 в виде густой вязкой жидкости (83%, 0,14 г).
ВРМС: вычислено 343,1220; найдено 343,1218.
Пример A35
Метил-(2R,3aR,12bS)-2-(аминометил)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-1-карбоксилат (промежуточное соединение 38)

промежуточное соединение 38
К раствору P(Ph)3 (0,26 г, 1 ммоль) в безводном ТГФ (4 мл) при -15°C добавляли раствор DIAD (0,22 г, 1,1 ммоль) в ТГФ (1 мл) и полученный в результате комплекс перемешивали в течение 20 минут. После добавления промежуточного соединения 37 (0,17 г, 0,5 ммоль) растворенного в ТГФ (1 мл), и DPPA (0,14 г, 0,5 ммоль) в ТГФ (1 мл), реакционную смесь нагревали до комнатной температуры и перемешивали в течение 18 часов. К реакционной смеси добавляли избыток P(Ph)3 (5 экв) и воду (0,5 мл) и затем нагревали при 40°C в течение 3 часов, чтобы восстановить азид до функциональности амина. К реакционной смеси добавляли диоксид кремния и растворитель удаляли при пониженном давлении с последующей очисткой продукта флэш-хроматографией с использованием смеси CH2Cl2/MeOH (9:1) в качестве элюента, получая промежуточное соединение 38 в виде густой вязкой жидкости (80%, 0,14 г).
ВРМС: вычислено 342,1380; найдено 342,1376.
Пример A36
{(2R,3aR,12bS)-11-фтор-1-[(2-нитрофенил)сульфонил]-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил}метил-2-нитробензолсульфонат (промежуточное соединение 39)

промежуточное соединение 39
К раствору промежуточного соединения 36 (0,5 ммоль, 0,14 г) Et3N (5 экв) и DMAP (20 моль%) в CH2Cl2 при -20°C добавляли о-нитробензолсульфонилхлорид (3 экв). Реакционную смесь нагревали до комнатной температуры и оставляли для перемешивания в течение ночи. К реакционной смеси добавляли водный раствор NaHCO3 (2 мл) и органический слой отделяли и сушили над MgSO4. После хроматографии (SiO2) с использованием EtOAc:гептан (1:1) в качестве элюента получали промежуточное соединение 39 в виде желтого кристаллического твердого вещества (т.пл.: 88-90°C, 71%, 0,23 г).
Пример A37
a) (10R,11R)-8-фтор-11-((2R)-2-гидрокси-3-{[(4-метилфенил)сульфонил]окси}пропил)-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-илацетат (промежуточное соединение 23a)

промежуточное соединение 23а
К раствору промежуточного ацетат-диола 23 (0,12 г, 0,355 ммоль) в безводном толуоле (10 мл) добавляли n-Bu2SnO (9 мг, 0,036 ммоль), Et3N (0,13 мл, 0,888 ммоль) и TsCl (0,10 г, 0,533 ммоль). Перемешивали при комнатной температуре в течение 24 часов, добавляли NH4Cl (насыщенный водный раствор, 10 мл), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гексан (30:70) давала промежуточное соединение 23a в виде бесцветного масла (0,15 г, 84%).
Масс-спектр: CI m/z (распределение, относительная интенсивность) 481 (MH+ - H2O, 1%), 439 (MH+ -AcOH, 4%), 421 (MH+ -AcOH-H2O, 1%), 267 (MH+ -AcOH-TsOH, 18%), 249 (MH+ -AcOH-TsOH-H2O, 100%); EI: m/z (распределение, относительная интенсивность) 480 (M+ -H2O, 1%), 438 (M+ -AcOH, 36%), 266 (M+ -AcOH-TsOH, 15%), 248 (M+-. -AcOH-TsOH-H2O, 18%); Высокое разрешение EI вычислено C25H23FO4S (M+ -AcOH): 438,1301, найдено: 438,1300 (51%).
b) (10R,11R)-11-[(2R)-3-азидо-2-гидроксипропил]-8-фтор-10,11-дигидро-5H-дибензо2[a,d]циклогептен-10-илацетат (промежуточное соединение 40)

промежуточное соединение 40
К раствору промежуточного тозилата 23a (1,30 г, 2,61 ммоль) в ДМФА (25 мл) добавляли NaN3 (0,51 г, 7,83 ммоль). Реакционную смесь нагревали при 100°C в течение 1 ночи. После охлаждения добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. После упаривания остаток очищали на силикагеле, используя смесь EtOAc/гептан (20/80), получая промежуточное соединение 40 в виде маслянистого продукта (0,79 г, 82%).
Пример A38
(10R,11R)-11-[(2R)-3-азидо-2-гидроксипропил]-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 41)

промежуточное соединение 41
Раствор промежуточного ацетата 40 (454,9 мг, 1,23 ммоль) в MeOH (10 мл) обрабатывали K2CO3 (340,1 мг, 2,46 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Раствор фильтровали и упаривали и остаток очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гептан (30/70), получая промежуточный диол 41 (370,9 мг, 92%).
Пример A39
S-((10S,11R)-11-{(2R)-3-азидо-2-[(метилсульфонил)окси]пропил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил)этантиоат (промежуточное соединение 42)

промежуточное соединение 42
К промежуточному соединению 41 (670 мг, 2,05 ммоль) в CH2Cl2 (25 мл) добавляли Et3N (2,30 мл, 16,4 ммоль), DMAP (0,13 мг, 1,02 ммоль) и (CH3SO2)2O (1,07 г, 6,15 ммоль) при 0°C. Перемешивали при комнатной температуре в течение 1 часа, снова охлаждали до 0°C, добавляли AcSH (0,44 мл, 6,15 ммоль) и перемешивали при комнатной температуре в течение 4 часов. Обрабатывали добавлением NH4Cl (насыщенный водный раствор). 3 раза экстрагировали CH2Cl2. Хроматография на колонке с силикагелем с использованием CH2Cl2 (100%) давала промежуточное соединение 42 в виде масла (0,68 г, 72%).
Пример A40
(2S,3aR,12bS)-2-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиофен (промежуточное соединение 43)

промежуточное соединение 43
К указанному выше промежуточному соединению 42 (0,15 г, 0,33 ммоль) в MeOH (5 мл) добавляли K2CO3 (92 мг, 0,67 ммоль). После перемешивания при комнатной температуре в течение 1 ночи смесь обрабатывали добавлением NH4Cl (насыщенный водный раствор). 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гептан (40/60) давала промежуточное соединение 43 в виде маслянистого продукта (76 мг, 70%).
Масс-спектр: CI m/z (распределение, относительная интенсивность) 326 (MH+, 25%), 298 (MH+ -N2, 60%), 283 (MH+ -HN3, 100%), 269 (MH+ -N2-CH2NH, 12%), 249 (MH+-HN3-H2S, 25%), 235 (MH+ -N2-CH2NH-H2S, 21%), 197 (61%).
Пример A41
(2S,3aR,12bS)-2-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиофен-1,1-диоксид (промежуточное соединение 44)

промежуточное соединение 44
К раствору промежуточного азида 43 (76,1 мг, 0,23 ммоль) в CH2Cl2 (5 мл) добавляли m-CPBA (мета-хлорпербензойную кислоту) (173,2 мг, 0,70 ммоль). Смесь перемешивали при комнатной температуре в течение 15 мин. Добавляли NaHCO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (50/50) давала промежуточный сульфон 44 (73,2 мг, 88%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 358 (MH+, 21%), 340 (MH+ - H2O, 9%), 330 (MH+-N2, 9%), 303 (8%), 265 (24%), 264 (MH+ -N2-H2SO2, 25%), 237(MH+-N2-H2SO2-HCN, 11%), 211 (15%), 197(66%).
Пример A42
(10R,11R)-11-[(2S)-3-азидо-2-гидроксипропил]-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 45)

промежуточное соединение 45
К раствору промежуточного соединения 40 (0,85 г, 2,32 ммоль) в ТГФ (10 мл) добавляли Ph3P (1,22 г, 4,63 ммоль) и DIAD (1,92 мл, 4,63 ммоль). Затем по каплям добавляли раствор паранитробензойной кислоты (0,77 г, 4,63 ммоль) в ТГФ (10 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Обрабатывали добавлением NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гептан (70/30) давала пара-нитробензоат (инвертированная вторичная OH-группа) в виде масла (1,19 г, 99%). К раствору полученного соединения (2,01 г, 4,05 ммоль) в MeOH (50 мл) добавляли K2CO3 (1,12 г, 8,10 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Добавляли NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (30/70) давала маслянистое промежуточное соединение 45 (0,71 г, 98%).
Пример A43
S-((10S,11R)-11-{(2S)-3-азидо-2-[(метилсульфонил)окси]пропил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил)этантиоат (промежуточное соединение 46)

промежуточное соединение 46
К раствору промежуточного диола 45 (1,20 г, 3,66 ммоль) в CH2Cl2 (30 мл) добавляли Et3N (4,10 мл, 29,3 ммоль), DMAP (0,22 мг, 1,83 ммоль) и (CH3SO2)2O (1,92 г, 11,0 ммоль) при 0°C. Перемешивали при комнатной температуре в течение 1 часа. Снова охлаждали до 0°C и добавляли AcSH (0,52 мл, 7,33 ммоль) и перемешивали при комнатной температуре в течение 5 часов. Обрабатывали добавлением NH4Cl (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (30/70) давала промежуточное соединение 46 в виде масла (1,32 г, 78%).
Пример A44
(2R,3aR,12bS)-2-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо-[3,4:6,7]циклогепта[1,2-b]тиофен (промежуточное соединение 47)
промежуточное соединение 47
К раствору указанного выше промежуточного соединения 46 (1,32 г, 2,86 ммоль) в MeOH (30 мл) добавляли K2CO3 (0,79 г, 5,72 ммоль). После перемешивания при комнатной температуре в течение 2 часов, добавляли NH4Cl (насыщенный водный раствор). 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гептан (40/60) давала промежуточное соединение 47 в виде маслянистого продукта (0,82 г, 89%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 326 (MH+, 25%), 298 (MH+ -N2, 60%), 283 (MH+-HN3, 100%), 269 (MH+-N2-CH2NH, 12%), 269 (MH+-HN3-H2S, 25%), 235 (MH+-N2-CH2NH-H2S, 21%), 197 (61%).
Пример A45
(2R,3aR,12bS)-2-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиофен-1,1-диоксид (промежуточное соединение 48)

промежуточное соединение 48
К раствору промежуточного азида 47 (136,1 мг, 0,41 ммоль) в CH2Cl2 (10 мл) добавляли мета-хлорпербензойную кислоту (310,0 мг, 1,26 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли NaHCO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (50/50) давала промежуточный сульфон 48 (146,5 мг, 98%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 358 (MH+, 21%), 340 (MH+ -H2O, 9%), 330 (MH+-N2, 9%), 303 (8%), 265 (24%), 264 (MH+ -N2-H2SO2, 25%), 237(MH+-N2-H2SO2-HCN, 11%), 211 (15%), 197 (66%).
Пример A46
(2S,3aR,12bS)-2-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиофен-1-оксиды (промежуточные соединения 49, 50)

К раствору промежуточного азида 43 (0,34 г, 1,05 ммоль) в гексафторизопропаноле (5 мл) добавляли H2O2 (30%, 0,24 мл, 2,10 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием Et2O (100%) давала промежуточные соединения 49 (110 мг) и 50 (130 мг) с общим выходом 78%.
Масс-спектр: - CI m/z (распределение, относительная интенсивность) 342 (MH+, 100%), 314 (MH+ -N2, 49%), 299 (MH+-HN3, 47%), 264 (17%), 197 (96%).
Пример A47
(2R,3aR,12bS)-2(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[l,2-6]тиофен-1-оксиды (промежуточные соединения 51, 52)

К раствору промежуточного азида 47 (0,21 г, 0,64 ммоль) в гексафторизопропаноле (3 мл) добавляли H2O2 (30%, 0,15 мл, 1,27 ммоль). Смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием Et2O (100%) давала промежуточное соединение 51 (120 мг) и 52 (86 мг) с общим выходом 95%.
Масс-спектр: - CI m/z (распределение, относительная интенсивность) 342 (MH+, 100%), 314 (MH+ -N2, 49%), 299 (MH+ -HN3, 47%), 264 (17%), 197 (96%).
Пример A48
(10S*,11R*)-11-аллил-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-илацетат (промежуточное соединение 56)

Промежуточный спирт 55 (1,72 г, 6,42 ммоль) растворяли в CH2Cl2 (30 мл). Добавляли Et3N (1,79 мл, 12,8 ммоль), DMAP (0,78 г, 6,42 ммоль) и ангидрид AcOH (1,21 мл, 12,8 ммоль). Перемешивали при комнатной температуре в течение 1 часа и добавляли насыщенный водный раствор NH4Cl (15 мл). 3 раза экстрагировали CH2Cl2 (3×20 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гексан (60:40) давала промежуточное соединение 56 в виде масла (1,77 г, 89%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность)311 (MH+, 5%), 251 (MH+ -AcOH, 100%); EI: m/z (распределение, относительная интенсивность) 250 (M+ -AcOH, 16%), 209 (M+ -AcOH-CH2CH2=CH2, 100%); Высокое разрешение EI вычислено C18H15F (M+ -AcOH): 250,1158, найдено: 250,1162 (26%).
Пример A49
a) (2R)-1-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-3-(тритилокси)-2-пропанол (промежуточное соединение 57)

промежуточное соединение 57
Смесь промежуточного диола 5 (6,022 г, 18,40 ммоль), Et3N (5,586 г, 55,2 ммоль), 4-диметиламинопиридина (138 мг, 1,13 ммоль), тритилбромида (9,444 г, 27,6 ммоль) в CH2Cl2 (180 мл) перемешивали при комнатной температуре в атмосфере азота в течение 2 часов, затем гасили насыщенным водным раствором NH4Cl (50 мл). Органическую фазу отделяли, водный слой экстрагировали CH2Cl2 (2×50 мл), объединенные органические фазы промывали водой (3×40 мл), насыщенным раствором соли (40 мл), сушили (MgSO4) и упаривали в вакууме. Очистка флэш-хроматографией (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 5/95 до 10/90) давала промежуточное соединение 57 (8,595 г, 15,09 ммоль, 82%) в виде коричневого полутвердого вещества.
b) (1R)-2-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-1-[(тритилокси)метил]этилметансульфонат (промежуточное соединение 58)

промежуточное соединение 58
Смесь промежуточного спирта 57 (8,500 г, 14,92 ммоль), Et3N (4,529 г, 44,76 ммоль) и DMAP (84 мг, 0,689 ммоль) в CH2Cl2 (200 мл) охлаждали до -78°C в атмосфере азота. Одной порцией добавляли MsCl (2,264 г, 22,38 ммоль), полученный в результате раствор медленно нагревали до комнатной температуры (около 40 мин) и гасили насыщенным водным раствором NH4Cl (50 мл). Органическую фазу отделяли, водный слой экстрагировали CH2Cl2 (3×45 мл), объединенные органические слои промывали водой (3×45 мл) и насыщенным раствором соли (40 мл), сушили (MgSO4) и упаривали в вакууме. Из-за нестабильности промежуточного соединения 58 его использовали сразу же без дополнительной очистки.
c) (1R)-2-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-1-(гидроксиметил)этилметансульфонат (промежуточное соединение 59)
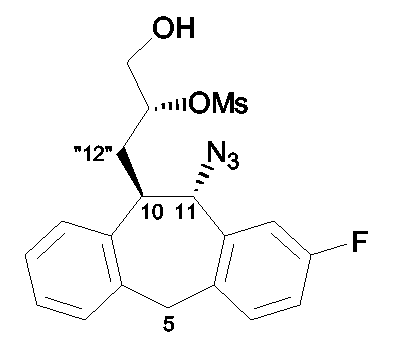
промежуточное соединение 59
Неочищенное промежуточное соединение 58 (неизвестное количество, предположительно 14,92 ммоль), растворяли в MeOH (200 мл), добавляли сухой Amberlyst-15 (15 г) и смесь перемешивали при 45°C в течение 4 часов; за протеканием реакции следили с помощью ТСХ (Kieselgel на стекле; EtOAc-гептан 30/70). Смолу отфильтровывали и промывали MeOH (2×40 мл), метанольный раствор концентрировали в вакууме до 100 мл и промежуточное соединение 59 непосредственно использовали на следующей стадии.
d) (10S,11R)-8-фтор-11-[(2S)-оксиранилметил]-10,11-дигидро-5H-дибензо[a,d]-циклогептен-10-илазид (промежуточное соединение 60)

промежуточное соединение 60
Метанольный раствор промежуточного соединения 59, полученный как указано выше, обрабатывали безводным K2CO3 (4,146 г, 30 ммоль) и перемешивали при комнатной температуре в течение 3 часов. После обработки водой (100 мл) MeOH удаляли в вакууме, продукт экстрагировали Et2O (3×75 мл). Объединенные органические слои промывали водой (3×75 мл) и насыщенным раствором соли (40 мл), сушили (MgSO4) и упаривали в вакууме. Хроматографическая очистка (Kielselgel 60, 70-230 меш, EtOAc-гептан 10/90) давала промежуточное соединение 60 (3,185 г, 10,29 ммоль, 69% из промежуточного соединения 57) в виде бесцветного масла.
ВРМС: вычислено для C18H16FN3O: 309,1277; найдено: 309,1279.
e) [(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метанол (промежуточное соединение 61)
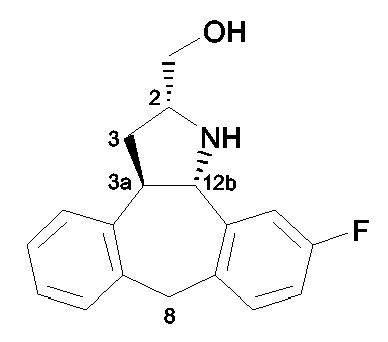
промежуточное соединение 61
Промежуточный эпоксид 60 (3,108 г, 10,04 ммоль) растворяли в MeOH (50 мл), добавляли Et3N (1,012 г, 10 ммоль) и 10% Pd-C (150 мг) и полученную в результате смесь гидрировали при атмосферном давлении в течение 5 часов. Катализатор удаляли фильтрованием через небольшую подушку кизельгура, MeOH и Et3N удаляли в вакууме и остаток очищали хроматографией на колонке (Kieselgel 60, 70-230 меш, EtOH-CH2Cl2 5/95), получая промежуточное соединение 61 (2,333 г, 8,23 ммоль, 82%) в виде желтоватого масла, медленно отверждаемого при стоянии.
ВРМС: вычислено для C18H18FNO: 283,1372; найдено: 283,1380.
f) Метил-(2R,3aR,12bS)-11-Фтор-1-(гидроксиметил)-3,3a,8,12b-тетрагидродибензо-[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (промежуточное соединение 62)

промежуточное соединение 62
Промежуточный пирролидин 61 (567 мг, 2,00 ммоль) растворяли при 0°C в смеси CH2Cl2 (20 мл) и насыщенном водном растворе NaHCO3 (20 мл), затем добавляли метилхлорформиат (0,23 мл, 281 мг, 2,98 ммоль), баню со льдом удаляли и полученную в результате смесь перемешивали в течение 5 часов. Органический слой отделяли, водную фазу экстрагировали CH2Cl2 (40 мл), затем объединенные органические слои промывали водой (2×40 мл), насыщенным раствором соли (20 мл), сушили (MgSO4) и упаривали. Остаток очищали хроматографией на колонке с диоксидом кремния (CH2Cl2-EtOH, 95/5), получая промежуточный карбамат 62 (669 мг, 1,96 ммоль, 98%) в виде желто-коричневого масла, отверждаемого при стоянии.
ВРМС: вычислено для C20H20FNO3: 341,1427; найдено: 341,1435.
g) (2R,3aR,12bS)-11-Фтор-2-(гидроксиметил)-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбальдегид (промежуточное соединение 62a)

промежуточное соединение 62a
Смесь промежуточного соединения 61 (283 мг, 1 ммоль), этилформиата (741 мг, 10 ммоль) и ацетонитрила (10 мл) кипятили с обратным холодильником в течение 18 часов, затем упаривали в вакууме. Остаток очищали хроматографией (Kieselgel 60, 70-230 меш, EtOH-CH2Cl2 5/95), получая 62a (283 мг, 0,91 ммоль, 91%) в виде желто-коричневого твердого вещества.
Продукт представляет собой смесь 2 ротамеров (соотношение 3:2).
CI-МС (CH4) 312 (MH+, 100%); 292 (MH+ -HF, 13%).
Пример A50
(2R,3aR,12bS)-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1,2(2H)-дикарбальдегид (промежуточное соединение 63)

промежуточное соединение 63
Хлорхромат пиридиния (104 мг, 0,48 ммоль) добавляли к раствору промежуточного спирта 62a (100 мг, 0,32 ммоль) в CH2Cl2 (10 мл) и полученную в результате взвесь перемешивали в атмосфере азота в течение 3 часов. После добавления Et2O (20 мл), смесь фильтровали через диоксид кремния, остаток смолы в колбе промывали Et2O (40 мл), снова фильтровали, упаривали досуха в вакууме. Неочищенный промежуточный альдегид 63 (77 мг, 0,25 ммоль, 78%) получали в виде красноватого масла, содержащего следы хрома. Продукт использовали непосредственно без очистки.
CI-МС (CH4) 310 (100%, MH+), 290 (11%, MH+ -HF); 282 (7%, MH+ -CO).
Пример A51
(2aS,11bR,12aR)-4-фтор-1,2a,7,11b,12,12a-гексагидроазирено[1,2-a]дибензо[3,4:6,7]циклогепта[1,2-d]пиррол (промежуточное соединение 64)
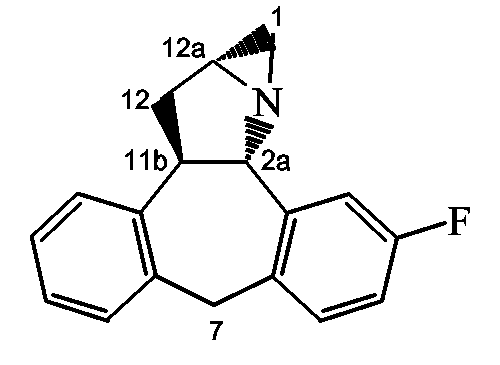
промежуточное соединение 64
Поли(трифенилфосфин) (0,33 г, примерно 1 ммоль Ph3P) подвергали набуханию в атмосфере аргона в безводном CH2Cl2 (10 мл), затем через мембрану добавляли диизопропилазодикарбоксилат (222 мг, 1,1 ммоль) в ТГФ (3 мл) при 0°C. Суспензию перемешивали в течение 30 минут при 0°C с последующим добавлением промежуточного соединение 61 (104 мг, 0,366 ммоль) в ТГФ (4 мл). Охлаждающую баню убирали и реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем добавляли воду (0,1 мл), смолу отфильтровывали и промывали ТГФ (15 мл), объединенные органические слои упаривали и очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-EtOH от 100/0 до 96/4), получая промежуточное соединение 64 (63 мг, 0,238 ммоль, 65%) в виде желтоватого масла.
ВРМС: вычислено для C18H16FN: 265,1267; найдено: 265,1270.
Пример A52
a) {(2R,3aR,12bS)-11-Фтор-1-[(2-нитрофенил)сульфонил]-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил}метил-2-нитробензолсульфонат (промежуточное соединение 65)

промежуточное соединение 65
Раствор промежуточного пирролидина 61 (567 мг, 2,00 ммоль), Et3N (1,012 ммоль, 10,00 ммоль), диметиламинпиридина (40 мг, 0,33 ммоль) в CH2Cl2 (30 мл) обрабатывали 2-нитробензолсульфонилхлоридом (1,330 г, 6,00 ммоль) и полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов, затем гасили насыщенным водным раствором NH4Cl (30 мл). После экстракции CH2Cl2 (3×30 мл) объединенные органические фазы промывали 1н HCl (15 мл), насыщенным водным раствором K2CO3 (40 мл), водой (3×40 мл), насыщенным раствором соли, сушили (MgSO4), упаривали и очищали хроматографией на колонке с диоксидом кремния (гептан-EtOAc, 95/5 → 85/15), получая промежуточное соединение 65 (1,203 г, 1,84 ммоль, 92%) в виде желтых кристаллов, быстро разлагающихся при стоянии.
1H ЯМР (300M Гц, CDCl3) δ 8,26-7,50 (м, 8Н, Ar-H, остатки 2-нозила); 7,20-7,03 (м, 6H, Ar-H, часть дибензосуберона); 6,81 (тд, J=8,3, 2,7 Гц, 1H, Ar-H); 5,40 (д, J=11,0 Гц, 1H, CH-12b); 4,69 (д, J=16,7 Гц, 1H, CH2-8); 4,60 (м, 2Н, CH2ONs); 4,40 (м, 1H, CH-2); 3,73 д, J=16,7 Гц, 1H, CH2'-8); 3,55-3,40 (м, 1Н, CH-3a); 2,80 (дд, J=13,0, 6,2 Гц, CH2-3); 2,33-2,18 (м, 1Н, CH2'-3).
b) 2-[4-({(2R,3aR,12bS)-11-фтор-1-[(2-нитрофенил)сульфонил]-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил}метил)-1-пиперазинил]этанол (промежуточное соединение 66a) и 2-[({(2R,3aR,12bS)-11-фтор-1-[(2-нитрофенил)сульфонил]-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил}метил)(метил)амино]этанол (промежуточное соединение 66b)



Смесь промежуточного биснозила 65 (0,35 г, 0,54 ммоль) и соответствующего амина (3 ммоль) в диоксане (10 мл) кипятили с обратным холодильником в течение 4 часов, охлаждали до температуры окружающей среды, разбавляли водой (100 мл), преципитированный продукт отфильтровывали, промывали водой (100 мл), растворяли в EtOAc, раствор промывали насыщенным раствором соли, сушили (K2CO3), упаривали и использовали на следующей стадии без очистки.
Пример A53
a) 2-[(4S)-2,2-диэтил-1,3-диоксолан-4-ил]этанол (промежуточное соединение 67b)
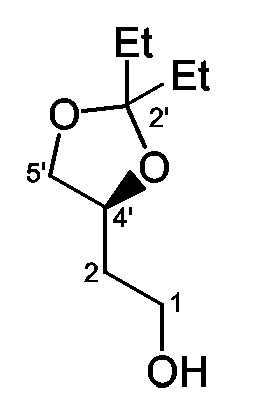
промежуточное соединение 67b
К раствору промежуточного (S)-1,2,4-бутантриола 67a (6,76 г, 63,68 ммоль) в свежем перегнанном пентан-3-оне (320 мл) добавляли пара-толуолсульфоновую кислоту (p-TSA) (6,06 г, 31,84 ммоль). Реакционную смесь перемешивали при 53°C в течение 16 часов, затем добавляли Et3N (10 мл) и реакционную смесь перемешивали при температуре окружающей среды в течение 10 минут. Реакционную смесь концентрировали при пониженном давлении. Градиентная флэш-хроматография (CH2Cl2/MeOH, от 100:0 до 97:3 до 95:5) давала промежуточный защищенный спирт 67b (9,84 г, 89%) в виде бесцветного масла.
b) [(4S)-2,2-диэтил-1,3-диоксолан-4-ил]ацетальдегид (промежуточное соединение 67c)

промежуточное соединение 67c
К раствору промежуточного соединения 67b 2(S)-2-(2,2-диэтил-[1,3]диоксолан-4-ил)этанола (4,00 г, 22,96 ммоль) и молекулярных сит 4A (11,50 г) в CH2Cl2 (200 мл), перемешиваемому при 0°C в течение 5 минут, добавляли хлорхромат пиридиния (PCC) (9,90 г, 45,92 ммоль). Реакционной смеси давали возможность нагреться до температуры окружающей среды и перемешивали в течение 1 часа. Неочищенную реакционную смесь фильтровали через подушку диоксида кремния, промывали Et2O (50 мл) и концентрировали при пониженном давлении, получая промежуточный альдегид 67c (3,56 г, 90%) в виде бесцветного масла.
c) 11-{2-[(4S)-2,2-диэтил-1,3-диоксолан-4-ил]этилиден}-8-фтор-5,11-дигидро-10H-дибензо[a,d]циклогептен-10-он (промежуточное соединение 67d)

промежуточное соединение 67d
MgBr2 (0,733 г, 3,98 ммоль) добавляли к 8-фтор-5,11-дигидро-10H-дибензо[a,d]циклогептен-10-ону (0,75 г, 3,32 ммоль) в толуоле (15 мл) и реакционную смесь перемешивали при комнатной температуре в течение 30 минут. Добавляли промежуточный альдегид 67c (2,05 г, 11,92 ммоль) в ТГФ (10 мл) и одновременно t-BuOK (0,074 г, 0,66 ммоль). Реакционную смесь перемешивали в течение 22 часов при температуре окружающей среды, затем добавляли насыщенный водный раствор NH4Cl (15 мл). Продукт три раза экстрагировали Et2O (3×30 мл), объединенные органические фазы промывали водой (2×35 мл), насыщенным раствором соли (25 мл), сушили над MgSO4. После выпаривания толуола остаток очищали на колонке с силикагелем, используя смесь Et2O/гептан (10/90), получая промежуточное соединение 67d (1,079 г, 86%) в виде желтоватого масла.
ВРМС (EI): вычислено для C24H25FO3: 380,1800; найдено: 380,1785.
d) (11R)-11-{2-[(4S)-2,2-диэтил-1,3-диоксолан-4-ил]этил}-8-фтор-5,11-дигидро-10H-дибензо-[a,d]циклогептен-10-он (промежуточное соединение 67e)

промежуточное соединение 67e
10% Pd-C (200 мг) и Et3N (0,135 мл, 0,97 ммоль) добавляли к промежуточному соединению 67d (0,246 г, 0,647 ммоль) в i-PrOH (25 мл) и толуоле (15 мл) и подвергали гидрированию в течение ночи при комнатной температуре. Реакционную смесь растворяли в CH2Cl2, фильтровали через целит и растворитель выпаривали. Остаток очищали на колонке с силикагелем, используя смесь Et2O/гептан (30/70), получая промежуточное соединение 67e (151 мг, 61%) в виде желтоватого масла.
ВРМС C24H27FO3: 382,1944; найдено: 382,1951.
e) (10R,11R)-11-{2-[(4S)-2,2-диэтил-1,3-диоксолан-4-ил]этил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 67f)

промежуточное соединение 67f
Борогидрид натрия (1,78 г, 46,84 ммоль) добавляли к промежуточному соединению 35 (2,0 г, 5,88 ммоль), растворенному в смеси i-PrOH (80 мл)/фосфатный буфер pH 7 (30 мл) при 0°C. После взаимодействия в течение 1 часа при комнатной температуре добавляли NH4Cl (насыщенный водный раствор) и смесь три раза экстрагировали CH2Cl2. Органическую фазу сушили над MgSO4 и растворитель выпаривали. Продукт очищали на силикагеле, используя смесь Et2O/гептан (30/70), получая промежуточное соединение 67f (1,96 г, 97%) в виде бесцветного масла.
ВРМС: вычислено для C21H23FO3: 342,1631; найдено: 342,1627.
f) (4S)-4-{2-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H -дибензо[a,d]циклогептен-10-ил]этил}-2,2-диэтил-1,3-диоксолан (промежуточное соединение 67g)

промежуточное соединение 67g
Промежуточное соединение 67g получали таким же образом, как описано для промежуточного соединения 30.
g) (2S)-4-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-l,2-бутандиол (промежуточное соединение 67h)
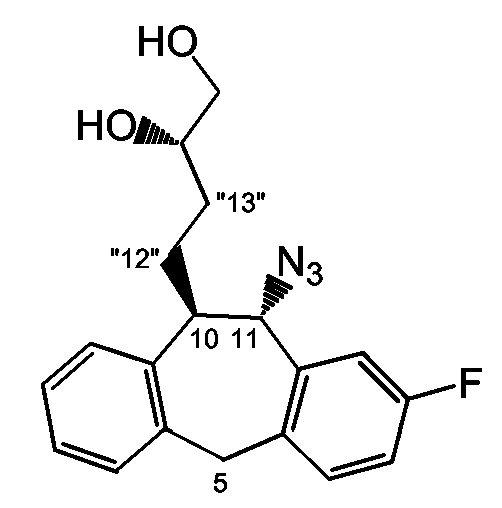
промежуточное соединение 67h
Промежуточное соединение 67h получали таким же образом, как описано для промежуточного соединения 31.
h) (2S)-4-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-2-{[(4-метилфенил)сульфонил]окси}бутил-4-метилбензолсульфонат (промежуточное соединение 67i)

промежуточное соединение 67i
Смесь промежуточного (2S)-4-[(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-1,2-бутандиола 67h (225 мг, 0,66 ммоль), Et3N (506 мг, 5,0 ммоль), диметиламинопиридина (12 мг, 0,1 ммоль) и TsCl (503 мг, 2,64 ммоль) в CH2Cl2 (25 мл) перемешивали при комнатной температуре в атмосфере азота в течение 15 часов. После гашения насыщенным водным раствором хлорида аммония (15 мл) органическую фазу отделяли и водный слой экстрагировали CH2Cl2 (3×20 мл). Объединенные органические фазы промывали водой (3×30 мл), насыщенным раствором соли (25 мл), сушили над сульфатом магния и упаривали в вакууме. Остаток очищали хроматографией на колонке (Kieselgel 60, 70-230 меш, гептан-этилацетат 90/10), получая промежуточное соединение 67i (352 мг, 0,54 ммоль, 82%) в виде бесцветного полутвердого вещества.
i) [(2R,4aR,13bS)-12-фтор-2,3,4,4a,9,13b-гексагидро-1H-дибензо[3,4:6,7]циклогепта[1,2-b]пиридин-2-ил]метил-4-метилбензолсульфонат (промежуточное соединение 67k)
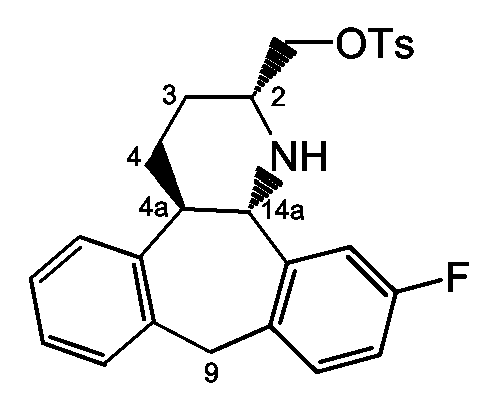
промежуточное соединение 67k
Промежуточный бистозилат 67i (340 мг, 0,52 ммоль) растворяли в MeOH (15 мл), добавляли Et3N (1,012 г, 10 ммоль) и 10% Pd-C (150 мг) и полученную в результате смесь гидрировали при атмосферном давлении в течение 5 часов. Катализатор удаляли фильтрованием через небольшую подушку кизельгура, добавляли безводный K2CO3 (138 мг, 1 ммоль) и полученную в результате взвесь перемешивали при комнатной температуре в течение 5 часов. После отфильтровывания твердых веществ MeOH и Et3N удаляли в вакууме и остаток очищали флэш-хроматографией (Kieselgel 60, 230-400 меш, EtOH-CH2Cl2 от 5/95 до 12/88), получая промежуточное соединение 67k (153 мг, 0,338 ммоль, 65%) в виде желтоватого масла.
CI-МС (CH4) 452 (MH+, 1%); 280 (MH+ -TsOH, 100%).
Пример A54
a) (10S,11R)-11-{2-[(4S)-2,2-диэтил-1,3-диоксолан-4-ил]этил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 610)
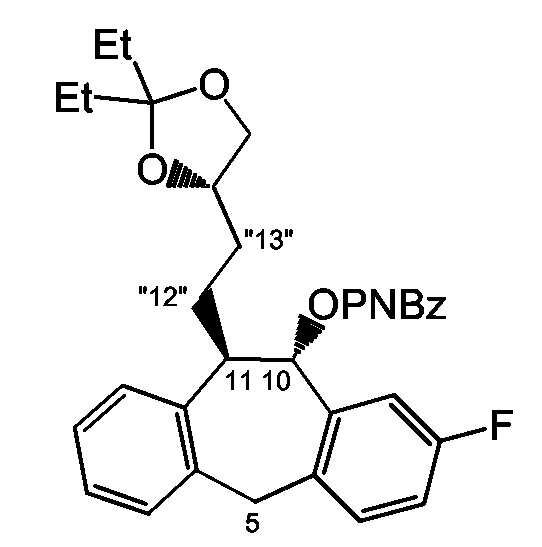
промежуточное соединение 68a
Раствор трифенилфосфина (910 мг, 3,5 ммоль) в безводном ТГФ (25 мл) помещали в двугорлую колбу объемом 100 мл, снабженную мембраной, входным отверстием для аргона и магнитной мешалкой. После охлаждения до -15°C через мембрану добавляли неразбавленный диизопропилазодикарбоксилат (708 мг, 3,5 ммоль) при интенсивном перемешивании. Полученную в результате желтую суспензию перемешивали при указанной выше температуре в течение 30 минут, затем добавляли раствор 4-нитробензойной кислоты (585 мг, 3,50 ммоль) и промежуточного спирта 67f (673 мг, 1,75 ммоль) в ТГФ (25 мл) в течение 10 минут. Полученной в результате желтой суспензии давали возможность нагреться до комнатной температуры и затем перемешивали в течение 12 часов. Добавляли воду (0,3 мл), затем силикагель (Kieselgel 60, 70-230 меш, 4 г), ТГФ удаляли в вакууме и порошок диоксида кремния использовали для флэш-хроматографии (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 5/95 до 15/85), получая промежуточный нитробензоат 610 (795 мг, 1,49 ммоль, 85%) в виде оранжевого полутвердого вещества.
b) (10S,11R)-11-[(3S)-3,4-дигидроксибутил]-8-фтор-10,11-дигидро-5H-дибензо-[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 68b)

промежуточное соединение 68b
Гидролиз промежуточного диоксолана 610 (795 мг, 1,49 ммоль) осуществляли таким же образом, как описано в примере A28, получая промежуточный диол 68b (695 мг, 1,49 ммоль, 100%) в виде оранжевого полутвердого вещества. Продукт использовали без очистки.
c) (10S,11R)-8-фтор-11-((3S)-3-гидрокси-4-{[(4-метилфенил)сульфонил]окси}бутил)-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 68c)

промежуточное соединение 68c
Смесь промежуточного диола 68b (695 мг, 1,49 ммоль), Et3N (607 мг, 6 ммоль), дибутил(оксо)олова (141 мг, 0,566 ммоль) и TsCl (431 мг, 2,26 ммоль) в CH2Cl2 (20 мл) перемешивали при комнатной температуре в атмосфере азота в течение 12 часов. После гашения насыщенным водным раствором NH4Cl (15 мл) органическую фазу отделяли и водный раствор экстрагировали CH2Cl2 (3×30 мл). Объединенные органические фазы промывали водой (3×20 мл), фильтровали через 5 см-слой MgSO4 и упаривали в вакууме, получая неочищенное промежуточное соединение 68c (601 мг, 0,97 ммоль, 65%) в виде желтоватой полутвердой массы, которую подвергали превращению без дополнительной очистки.
d) [(2R,4aR,13bS)-12-фтор-2,3,4a,9,l3b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-2-ил]метанол (промежуточное соединение 68d)

промежуточное соединение 68d
Смесь промежуточного тозилата 68c (601 мг, 0,97 ммоль), метоксида натрия (162 мг, 3,0 ммоль) и MeOH (10 мл) перемешивали при комнатной температуре в течение 3 часов. После обработки водой (100 мл) продукт экстрагировали диэтиловым эфиром (3×30 мл). Объединенные органические фазы промывали водой (3×40 мл) и насыщенным раствором соли (40 мл), сушили (MgSO4) и упаривали в вакууме. Хроматографическая очистка (Kielselgel 60, 70-230 меш, EtOAc-гептан от 10/90 до 25/75) давала промежуточное соединение 68d (211 мг, 0,71 ммоль, 73%) в виде бесцветного масла.
ВРМС: вычислено для C19H19FO2: 298,1369; найдено 298,1350.
e) [(2R,4aR,13bS)-12-фтор-2,3,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-2-ил]метил-4-метилбензолсульфонат (промежуточное соединение 68е)

промежуточное соединение 68e
Смесь промежуточного спирта 68d (211 мг, 0,708 ммоль), Et3N (209 мкл, 287 мг, 2,83 ммоль), DMAP (86,5 мг, 0,708 ммоль) и TsCl (270 мг, 1,42 ммоль) в CH2Cl2 (10 мл) перемешивали при комнатной температуре в атмосфере азота в течение 16 часов. После гашения насыщенным водным раствором NH4Cl (10 мл) органическую фазу отделяли и водный раствор экстрагировали CH2Cl2 (3×15 мл). Объединенные органические фазы промывали водой (3×15 мл), сушили (MgSO4) и упаривали в вакууме, получая неочищенное промежуточное соединение 68e (282 мг, 0,62 ммоль, 88%) в виде желтоватого масла, которое использовали без дополнительной очистки.
Пример A55
a) (10S,11R)-11-{[(4R)-2,2-диметил-1,3-диоксолан-4-ил]метил}-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 69a)

промежуточное соединение 69a
Раствор трифенилфосфина (1049 мг, 4,0 ммоль) в безводном ТГФ (20 мл) помещали в двугорлую колбу объемом 100 мл, снабженную мембраной, входным отверстием и магнитной мешалкой. После охлаждения до -15°C через мембрану добавляли неразбавленный диизопропилазодикарбоксилат (809 мг, 4,0 ммоль) при интенсивном перемешивании. Полученную в результате желтую суспензию перемешивали при указанной выше температуре в течение 30 минут, затем добавляли раствор 4-нитробензойной кислоты (4,0 ммоль) и 11-(2,2-диметил[1,3]диоксолан-4-илметил)-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ола (промежуточное соединение 3) (685 мг, 2,0 ммоль) в ТГФ (25 мл) в течение 10 минут. Полученной в результате желтой суспензии давали возможность нагреться до комнатной температуры и затем перемешивали в течение 12 часов. Добавляли воду (0,3 мл), затем силикагель (Kieselgel 60, 70-230 меш, 4 г), ТГФ удаляли в вакууме и порошок диоксида кремния использовали для флэш-хроматографии (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 5/95 до 10/90), получая промежуточный нитробензоат 69a (875 мг, 1,78 ммоль, 89%) в виде оранжевого полутвердого вещества.
b) (10S,11R)-11-[(2R)-2,3-дигидроксипропил]-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 69b)

промежуточное соединение 69b
Промежуточное соединение 69b было получено из промежуточного ацеталя 69a (860 мг, 1,75 ммоль) таким же образом, как описано для промежуточного соединения 5. Хроматография на колонке (Kieselgel 60, 70-230 меш, EtOAc-гептан, от 35/65 до 50/50) давала промежуточный диол 69b (774 мг, 1,715 ммоль, 98%) в виде желтого полутвердого вещества.
c) (10S,11R)-8-фтор-11-(2-оксоэтил)-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 69c)
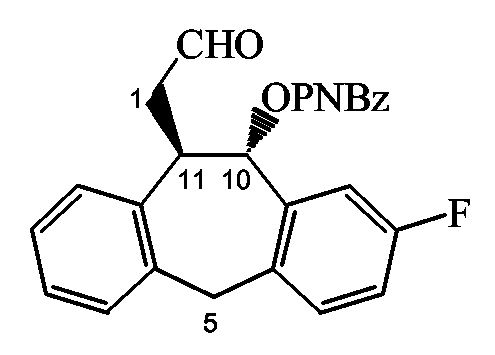
промежуточное соединение 69c
Промежуточный диол 69b (774 мг, 1,715 ммоль) растворяли при 0°C в смеси ТГФ (25 мл) и фосфатного буфера pH 7 (5 мл), затем одной порцией добавляли периодат натрия (642 мг, 3 ммоль) при 0°C, охлаждающую баню убирали и полученную в результате смесь перемешивали при комнатной температуре в течение 4 часов. Добавляли воду (50 мл), продукт экстрагировали диэтиловым эфиром (3×30 мл). Объединенные органические фазы промывали насыщенным водным раствором метабисульфита натрия (50 мл), водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали в вакууме, получая промежуточный альдегид 69c (680 мг, 1,66 ммоль, 97%) в виде желтой пены. Продукт использовали непосредственно без очистки.
d) (10S,11S)-8-фтор-11-(1-формилвинил)-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 69d)

промежуточное соединение 69d
Смесь промежуточного альдегида 69c (680 мг, 1,66 ммоль), AcOH (240 мг, 4,0 ммоль), бис(диметиламино)метана (719 мг, 7,0 ммоль) и ТГФ (30 мл) перемешивали при комнатной температуре в течение 3 часов. Добавляли воду (50 мл), продукт экстрагировали диэтиловым эфиром (3×30 мл). Объединенные органические фазы промывали насыщенным водным раствором бикарбоната натрия (25 мл), водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали в вакууме, получая промежуточный ненасыщенный альдегид 69d (680 мг, 1,58 ммоль, 95%) в виде желтого масла.
e) (10S,11S)-8-фтор-11-[1-(гидроксиметил)винил]-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил-4-нитробензоат (промежуточное соединение 69e)

промежуточное соединение 69e
Борогидрид натрия (190 мг, 5,00 ммоль) добавляли при комнатной температуре в течение 10 минут к раствору промежуточного альдегида 69d (680 мг, 1,58 ммоль) в MeOH (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, гасили насыщенным водным раствором хлорида аммония (20 мл) и экстрагировали Et2O (3×30 мл). Объединенные органические фазы промывали водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали в вакууме, получая промежуточный спирт 69e (582 мг, 1,34 ммоль, 85%) в виде оранжевого масла.
f) (10S,11S)-8-фтор-11-[1-(гидроксиметил)винил]-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 69f)

промежуточное соединение 69f
Смесь промежуточного нитробензоата 69e (582 мг, 1,34 ммоль), метоксида натрия (162 мг, 3,0 ммоль) и MeOH перемешивали при комнатной температуре в течение 4 часов. Добавляли воду (70 мл), продукт экстрагировали EtOAc (3×30 мл). Объединенные органические фазы промывали водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали в вакууме. Остаток очищали флэш-хроматографией (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 35/65 до 60/40), получая промежуточный диол 69f (286 мг, 1,005 ммоль, 75%) в виде бесцветного масла.
g) (3a5,12bS)-11-фтор-3-метилен-3,3a,8,12b-тетрагидро-2H- дибензо[3,4:6,7]циклогепта-[1,2-b]фуран (промежуточное соединение 69g)

промежуточное соединение 69g
Трибутилфосфин (405 мг, 2,0 ммоль) растворяли в толуоле (25 мл) в атмосфере аргона. По каплям добавляли диизопропилазодикарбоксилат (405 мг, 2,0 ммоль) в толуоле (3 мл), затем раствор промежуточного диола 69f (270 мг, 0,95 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 3 часов, затем реакцию гасили водой (1 мл). Добавляли силикагель (Kieselgel 60, 70-230 меш, 1,3 г), толуол удаляли в вакууме и порошок диоксида кремния подвергали флэш-хроматографии (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 5/95 до 12/88), получая промежуточное ТГФ-производное 69g (205 мг, 0,77 ммоль, 81%) в виде бесцветной пены.
h) [(3aR,12bS)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-3-ил]метанол (промежуточное соединение 69h)

промежуточное соединение 69h
Эфират трифторида бора (0,43 мл, 3,54 ммоль) в ТГФ (1 мл) добавляли при комнатной температуре в атмосфере аргона к раствору промежуточного ТГФ 69g (188 мг, 0,66 ммоль), борогидрида натрия (496 мг, 2,64 ммоль) в безводном ТГФ (2 мл). Полученный в результате раствор перемешивали в атмосфере аргона в течение 24 часов, избыток борогидрида осторожно разлагали водой (3,8 мл), добавляли MeOH (1,5 мл), затем 3 М NaOH (3,8 мл) и 30% пероксид водорода (0,55 мл). Реакционной смеси давали возможность перемешиваться в течение 4 часов при комнатной температуре, затем продукт экстрагировали Et2O (3×30 мл). Объединенные органические фазы промывали водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали в вакууме. Остаток очищали флэш-хроматографией (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 5/95 до 20/80), получая промежуточное производное ТГФ 69h (139 мг, 0,49 ммоль, 74%) в виде бесцветного масла.
i) (3aR,12bS)-3-(азидометил)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фуран (промежуточное соединение 69i)

промежуточное соединение 69i
Поли(трифенилфосфин) (0,33 г, примерно 1 ммоль Ph3P) подвергали набуханию при комнатной температуре в атмосфере аргона в безводном ТГФ (10 мл), затем через мембрану добавляли диизопропилазодикарбоксилат (222 мг, 1,1 ммоль) в ТГФ (3 мл) при -15°C. Суспензию перемешивали в течение 30 минут при -15°C, затем одной порцией добавляли промежуточный спирт 69h (139 мг, 0,49 ммоль) в безводном ТГФ (2,5 мл), затем по каплям добавляли дифенилфосфорилазид (160 мг, 0,58 ммоль) в ТГФ (3 мл). Полученную в результате суспензию перемешивали в атмосфере аргона в течение 12 часов. После гашения водой (0,3 мл) смолу отфильтровывали и растворитель удаляли в вакууме. Остаток очищали флэш-хроматографией (Kieselgel 60, 230-400 меш, EtOAc-гептан, 15/85), получая промежуточный азид 69i (136 мг, 0,44 ммоль, 90%) в виде бесцветной пены.
Пример A56
a)(2R)-3-[(10R,11R)-2-фтор-11-гидрокси-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]-1,2-пропандиол (промежуточное соединение 70a)

промежуточное соединение 70a
Промежуточный триол 70a получали из промежуточного соединения 3 (514 мг, 1,50 ммоль) таким же образом, как описано в примере A5. Неочищенное промежуточное соединение 70a (449 мг, 1,485 ммоль, 99%) получали в виде бесцветного масла и использовали без очистки.
b) (3aR,12bR)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-2-ол (промежуточное соединение 70b)

промежуточное соединение 70b
Промежуточное соединение 70b получали из промежуточного триола 70a (449 мг, 1,485 ммоль) таким же образом, как описано для соединения 44. Флэш-хроматография (Kieselgel 60, 230-400 меш, EtOAc-гептан, от 10/90 до 33/67) давала промежуточное соединение 70c (357 мг, 1,32 ммоль, 89%) в виде желто-коричневого твердого вещества.
с) (3aR,12bR)-3-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо-[3,4:6,7]циклогепта[1,2-b]фуран-2-ол (промежуточное соединение 70c)

промежуточное соединение 70c
Реакцию в случае промежуточного соединения 70c (335 мг, 1,24 ммоль) осуществляли таким же образом, как описано для соединения 45. Получали сложную, неразделимую смесь продуктов и использовали на следующей стадии без очистки.
d) (10R,11R)-11-[2-(диметиламино)-1-(гидроксиметил)этил]-8-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ол (промежуточное соединение 70d)

промежуточное соединение 70d
Реакцию с использованием смеси, содержащей промежуточное соединение 70c, осуществляли таким же образом, как описано для соединения 46. Очистка ОФ-ВЭЖХ (Waters Xterra® C18, 19×50 мм, MeOH-вода 50/50, затем чистый MeOH, 4 мл/мин) давала промежуточное соединение 70d (135 мг, 0,41 ммоль, 33%, исходя из промежуточного соединения 70b) в виде желтого масла.
Пример A57
a) [(10R,11S)-11-азидо-2-фтор-10,11-дигидро-5H-дибензо[a,dl]циклогептен-10-ил]ацетальдегид (промежуточное соединение 71a)

промежуточное соединение 71a
Реакцию с использованием промежуточного диола 5 (0,99 г, 3,02 ммоль) осуществляли таким же образом, как описано для соединения 44. Очистка хроматографией на колонке (Kieselgel 60, 230-400 меш, диэтиловый эфир-гептан, 50/50) давала промежуточный альдегид 71a (778 мг, 2,63 ммоль, 87%) в виде бесцветного масла.
b) 2-[(10S,11S)-11-азидо-1-фтор-10,11-дигидро-5H-дибензо[a,d]циклогептен-10-ил]акрилальдегид (промежуточное соединение 71b)

промежуточное соединение 71b
Реакцию с использованием промежуточного соединения 71a (618 мг, 2,09 ммоль) осуществляли таким же образом, как описано для соединения 45. Неочищенный промежуточный альдегид 71b (605 мг, 1,97 ммоль, 94%) получали в виде бесцветного масла и использовали без дополнительной очистки.
c) (3aS,12bS)-11-фтор-3-метилен-1,23,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта-[1,2-b]пиррол (промежуточное соединение 71c)
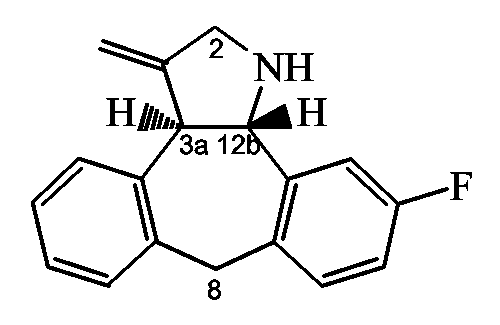
промежуточное соединение 71c
Поли(трифенилфосфин) (1,40 г, примерно 4,2 ммоль Ph3P) подвергали набуханию при комнатной температуре в атмосфере аргона в ТГФ (30 мл), затем добавляли промежуточный альдегид 71b (405 мг, 1,32 ммоль) в ТГФ (10 мл) и воду (0,19 г). Полученную в результате смесь перемешивали в атмосфере аргона при 50°C в течение 1 часа. После указанного периода времени смолу отфильтровывали, ТГФ удаляли в вакууме. Остаток растворяли в MeOH (10 мл), добавляли AcOH (1 мл) и цианоборогидрид натрия (200 мг, 3,2 ммоль) и полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов, затем гасили концентрированной HCl (1 мл), обрабатывали насыщенным водным раствором NaHCO3 (15 мл) и подщелачивали 1н гидроксидом натрия (3 мл). Продукт экстрагировали CH2Cl2 (3×50 мл), объединенные органические фазы промывали водой (2×30 мл), насыщенным раствором соли (30 мл), сушили (MgSO4) и упаривали в вакууме, получая промежуточный пирролидин 71c (258 мг, 0,97 ммоль, 74%) в виде желтой пены. Промежуточное соединение 71c использовали без дополнительной очистки.
d) Метил(3aS,12bS)-11-фтор-3-метилен-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (промежуточное соединение 71d)

промежуточное соединение 71d
Реакцию с использованием промежуточного соединения 71c (258 мг, 0,97 ммоль) осуществляли таким же образом, как описано для соединения 9. Флэш-хроматография (Kieselgel 60, 230-400 меш, гептан-EtOAc от 50/50 до 0/100) давала промежуточное соединение 71d (282 мг, 0,87 ммоль, 90%) в виде желтого масла.
e) Метил(3aR,12bS)-11-фтор-3-(гидроксиметил)-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (промежуточное соединение 71e)
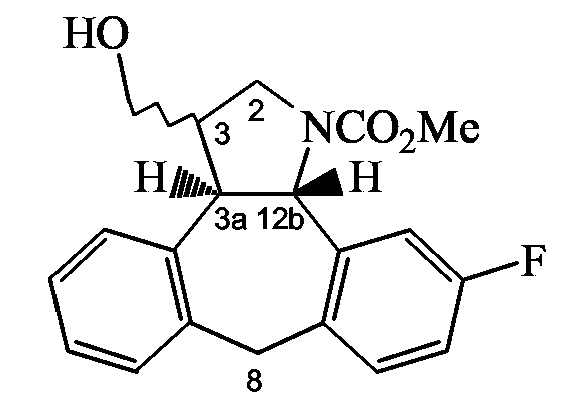
промежуточное соединение 71e
Реакцию с использованием 71d (255 мг, 0,79 ммоль) осуществляли таким же образом, как описано для соединения 49. Флэш-хроматография (Kieselgel 60, 230-400 меш, EtOH-CH2Cl2 от 1/99 до 3/97) давала 71e (215 мг, 0,63 ммоль, 80%) в виде бесцветного масла.
f) Метил(3aR,12bS)-3-(азидометил)-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (промежуточное соединение 71f)

промежуточное соединение 71f
Реакцию с использованием промежуточного соединения 71f (215 мг, 0,63 ммоль) осуществляли таким же образом, как описано для соединения 50. Флэш-хроматография (Kieselgel 60, 230-400 меш, этилацетат) давала промежуточное соединение 71f (194 мг, 0,53 ммоль, 84%) в виде бесцветного масла.
B. Получение конечных соединений
Все соединения, полученные как описано далее, представляют собой смеси изомерных форм, если не оговорено особо.
Пример B1
(4aS,13bR,14aS)-6-фтор-2-метил-1,2,3,4a,9,13b,14,14a-октагидродибензо[3',4':6',7]циклогепта[1',2':4,5]пирроло[1,2-c]имидазол (конечное соединение 1)

конечное соединение 1
К раствору промежуточного диамина 9 (130 мг, 0,3 ммоль) в MeOH (5 мл) добавляли Et3N (126,5 мкл, 0,91 ммоль) и смесь гидрировали при давлении в 1 атмосферу, используя 10% палладий на угле, при энергичном перемешивании при комнатной температуре. Через 1 час добавляли формальдегид (112,8 мкл, 1,5 ммоль) и смесь гидрировали еще в течение часа. Затем суспензию фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (95/5). Очистка давала конечное соединение 1 в виде масла (50,5 мг, 54%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 309 (MH+, 100%), 289 (MH+ -HF, 26%); EI: m/z (распределение, относительная интенсивность) 308 (M+ 68%), 279 (M+ -CH2NH, 4%), 265 (M+ -CH3NCH2, 100%), 197 (23%); Высокое разрешение EI вычислено C20H21FN2 (M+): 308,1689, найдено: 308,1684 (35%).
Пример B2
[(2S,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 2)

конечное соединение 2
Указанное выше соединение 1 (0,114 г, 0,37 ммоль) растворяли в MeOH (10 мл) и добавляли ТФУ (0,071 мл, 0,93 ммоль), NaCNBH3 (0,058 г, 0,93 ммоль) и перемешивали при комнатной температуре в течение 1 часа. Добавляли 10 мл K2CO3 (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (10%) давала конечное соединение 2 в виде масла (0,067 г, 59%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 311 (MH+, 100%), 291 (MH+ -HF, 25%), 282 (MH+ CH2NH), 266 (MH+ NH(CH3)2, 13%), 252 (8%); EI: m/z (распределение, относительная интенсивность) 310 (M+ 26%), 266 (M+ -(CH3)2N, 76%), 252 (M+ -CH3)2NCH2, 70%), 235 (100%), 209 (61%); Высокое разрешение EI вычислено C20H23FN2 (M+): 310,1845, найдено: 310,1820 (5%).
Пример B3
(4aS,13bR,14aS)-6-фтор-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-тион (конечное соединение 3)

конечное соединение 3
К раствору промежуточного диамина 9 (238,6 мг, 0,85 ммоль) в ДМФА (3 мл) добавляли дисульфид углерода (0,076 мл, 1,28 ммоль). Перемешивали при 60°C в течение 20 минут. После выпаривания растворителя остаток очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гептан (50/50), получая соединение 3 в виде полутвердого конечного соединения (124,6 мг, 45%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 325 (MH+, 100%), 252 (1%), 224 (2%).
Пример B4
(5aS,14bR,15aS)-7-фтор-2-метил-1,2,3,5a,10,14b,15,15a-октагидро-4H-дибензо[3',4':6',7']циклогепта-[1',2':4,5]пирроло[1,2-a]пиразин-4-он (конечное соединение 4)

конечное соединение 4
Раствор указанного выше промежуточного карбамата 16 (86,2 мг, 0,19 ммоль) в MeOH (3 мл) гидрировали при давлении в 1 атмосферу, используя 10% палладий на угле, при энергичном перемешивании при комнатной температуре. После взаимодействия в течение 1 часа добавляли формальдегид (70,7 мкл, 0,94 ммоль) и смесь гидрировали еще в течение часа. Суспензию фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После выпаривания растворителя грубый продукт очищали хроматографией на колонке с силикагелем, используя CHCl3, получая конечное соединение 4 (18,6 мг, 29%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 337 (MH+, 100%), 317 (MH+ -HF, 18%), 309 (MH+ -CO, 9%), 161 (9%), 133 (75%), 93 (72%).
Пример B5
[(2R,3aR,12bS)-11-фтор-1-метил-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 5)

конечное соединение 5
К раствору промежуточного диамина 13 (28,7 мг, 0,05 ммоль) в MeOH (2 мл) добавляли Et3N (21,4 мкл, 0,15 ммоль) и формальдегид (18,8 мкл, 0,25 ммоль) и смесь обрабатывали водородом при давлении в 1 атмосферу и используя 10% палладий на угле при энергичном перемешивании при комнатной температуре. После взаимодействия в течение 1 часа суспензию фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (90/10). Очистка давала конечное соединение 5 в виде масла (16,7 мг, 98%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 325 (MH+, 100%), 323 (25%), 305 (MH+-HF, 19%), 280 (MH+-HN(CH3)2, 12%), 266 (MH+ -CH3N(CH3)2, 36%).
Пример B6
(4aS,13bR,14aR)-6-фтор-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-он (конечное соединение 6)

конечное соединение 6
К раствору промежуточного диамина 13 (40,4 мг, 0,14 ммоль) в CH3CN (2 мл) добавляли Et3N (50 мкл, 0,36 ммоль) и смесь нагревали в атмосфере аргона при 70°C. Через 1 час по каплям добавляли раствор дифенилкарбоната (36,6 мг, 0,17 ммоль) в CH3CN и смесь перемешивали при 70°C в течение 2 суток. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гептан (20/80), получая конечное соединение - мочевину 6 в виде масла (23 мг, 52%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 309 (MH+, 100%), 308 (12%), 289 (MH+ -HF, 20%), 279 (3%), 113 (8%).
Пример B7
(4aS,13bR,14aR)-6-фтор-2-метил-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7]циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-он (конечное соединение 7)

конечное соединение 7
К раствору соединения мочевины 6 (29 мг, 0,10 ммоль) в ТГФ (3 мл) добавляли NaH (15,9 мг, 0,31 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. Затем добавляли Me2SO4 (25,4 мг, 0,26 ммоль) и смесь перемешивали дополнительно в течение 30 минут. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (40/60) давала конечное соединение - метилированную мочевину 7 в виде масла (19 мг, 63%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 323 (MH+, 100%), 303 (MH+ -HF, 26%), 209 (2%), 127 (3%).
Пример B8
(4aS,13bR,14aR)-6-фтор-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-тион (конечное соединение 8)

конечное соединение 8
К раствору промежуточного диамина 13 (54 мг, 0,19 ммоль) в ДМФА (3 мл) добавляли дисульфид углерода (17,3 мкл, 0,29 ммоль). После перемешивания при 60°C в течение 20 минут, последующего выпаривания растворителя очистка на колонке с силикагелем (элюент: EtOAc/гептан (50/50)) давала кристаллическое конечное соединение 8 (27,3 мг, 44%); т.пл.: 150-151°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 325 (MH+, 100%), 252 (1%), 224 (2%).
Пример B9
(4aS,13bR,14aR)-6-фтор-3-(метилсульфанил)-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол (конечное соединение 9)

конечное соединение 9
К раствору конечного соединения 8 (140,6 мг, 0,43 ммоль) в MeOH (10 мл) добавляли метилйодид (53,5 мкл, 0,86 ммоль) и Et3N (129 мкл, 0,86 ммоль). После перемешивания при 80°C в течение 2 суток растворитель и реагенты выпаривали. Очистка на колонке с силикагелем (элюент: EtOAc/гептан (40/60)) давала S-метилированное конечное соединение 9 в виде масла (72,7 мг, 49%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 339 (MH+, 100%), 319 (MH+ -HF, 4%), 268 (8%), 266 (3%).
Пример B10
[(2R,3aR,12bS)-11-фтор-1-(метоксиацетил)-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-6]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 10)

конечное соединение 10
К раствору промежуточного соединения 19 (265,3 мг, 0,44 ммоль) в MeOH (20 мл) добавляли MeSO3H (2 мл) и смесь перемешивали при 60°C в течение 30 минут. После выпаривания растворителя добавляли NaHCO3 (насыщенный водный раствор) (15 мл) и смесь экстрагировали CH2Cl2 (3×10 мл). Объединенные органические слои сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (5%) давала аминосоединение (125 мг, 79%). Последнее затем растворяли в MeOH (30 мл). После добавления формальдегида (80 мкл, 1,06 ммоль) смесь гидрировали (1 атм), используя 10% палладий на угле при энергичном перемешивании при комнатной температуре в течение 6 часов. Затем суспензию фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После выпаривания растворителя грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (95/5). Конечное соединение 10 (90,1 мг, 67%) получали в виде масла (смесь конформеров).
Масс-спектр: -APCI m/z (распределение, относительная интенсивность) 383 (MH+, 100%), 369 (4%), 367 (4%), 363 (MH+ -HF, 5%), 354 (2%), 351 (2%).
Пример B11
Метил({[(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}амино)ацетат (конечное соединение 11)

конечное соединение 11
Промежуточное соединение 21 (53,4 мг, 0,15 ммоль) растворяли в насыщенном растворе HCl в MeOH (10 мл) и смесь перемешивали при 60°C в течение ночи. После удаления растворителя добавляли 10 мл K2CO3 (насыщенный водный раствор) и смесь экстрагировали CH2Cl2 (3×10 мл). Очистка на колонке с силикагелем с использованием смеси CHCl3/MeOH (97/3) давала соединение сложного аминоэфира 11 (20 мг, 37%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 355 (MH+, 100%), 335 (MH+ -HF, 14%), 295 (MH+ -CH3OH-CO, 4%), 252 (MH+ -CH3OH-CH2CO-NHCH2, 8%), 169 (5%), 141 (46%); EI m/z (распределение, относительная интенсивность) 354 (M+, 3%), 295 (M+ -CH3OCO, 4%), 252 (M+ -CH3OCOCH2NHCH2, 100%), 235 (M+ -CH3OCOCH2NHCH2-NH3, 68%), 223 (8%), 209 (22%); Высокое разрешение EI вычислено C21H23N2O2F (M+): 354,1744, найдено: 354,1751 (9%).
Пример B12
(5aS,14bR,15aR)-7-фтор-1,2,3,5a,10,14b,15,15a-октагидро-4H-дибензо[3',4':6',7]циклогепта[1',2':4,5]пирроло[1,2-a]пиразин-4-он (конечное соединение 12)

конечное соединение 12
Промежуточное соединение 21 (250 мг, 0,71 ммоль) растворяли в 10 мл HCl в MeOH (насыщенный раствор) и смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили добавлением 10 мл K2CO3 (насыщенный водный раствор). Затем смесь 3 раза экстрагировали 10 мл CH2Cl2. Объединенные органические слои сушили над MgSO4 и упаривали. Очистка на колонке с силикагелем с использованием смеси CHCl3/MeOH (95/5) давала конечное соединение 12 (67,6 мг, 29%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 323 (MH+, 100%), 303 (MH+ -HF, 20%), 295 (MH+ -CO, 2%), 252 (MH+ -COCH2-NHCH2, 1%), 188 (2%), 160 (5%); EI m/z (распределение, относительная интенсивность) 322 (M+, 100%), 252 (M+ -COCH2N=CH2, 40%), 235 (68%), 223 (M+ -COCH2N=CH2- CH2NH, 44%), 207 (13%), 209 (88%), 209 (22%); Высокое разрешение EI вычислено C20H19N2OF (M+.): 322,1481, найдено: 322,1484 (100%).
Пример B13
(5aS,14bR,15aR)-7-фтор-2-метил-1,2,3,5a,10,14b,15,15a-октагидро-4H-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-a]пиразин-4-он (конечное соединение 13)

конечное соединение 13
К раствору конечного соединения 12 (82,3 мг, 0,25 ммоль) в MeOH (10 мл) добавляли формальдегид (96 мкл, 1,22 ммоль) и смесь гидрировали (1 атм), используя 10% палладий на угле, при энергичном перемешивании при комнатной температуре в течение 1 часа. Затем смесь фильтровали через подушку целита и твердые вещества 4 раза промывали CH2Cl2. После упаривания грубый продукт очищали хроматографией на колонке с силикагелем, используя CHCl3/MeOH (3%) в качестве элюента. Конечное соединение 13 (43,4 мг, 50%) получали в виде твердого вещества; т.пл.: 139-141°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 337 (MH+, 100%), 317 (MH+ -HF, 30%), 279 (1%), 251 (1%), 209 (1%); EI m/z (распределение, относительная интенсивность) 336 (M+, 74%), 293 (M+ -COCH3, 13%), 265 (M+ -CO=CHNHCH3, 9%), 233 (18%), 209 (42%), 196 (26%), 57 (100%); Высокое разрешение EI вычислено C21H21N2OF (M+): 336,1638, найдено: 336,1641 (100%).
Пример B14
(5aS,14bR,15aR)-7-фтор-2-метил-1,3,4,5a,10,14b,15,15a-октагидро-2H-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-a]пиразин (конечное соединение 14)

конечное соединение 14
К раствору конечного соединения 13 (34,3 мг, 0,1 ммоль) в ТГФ (10 мл) добавляли BH3·Me2S (100 мкл, 0,2 ммоль) и смесь нагревали при 85°C в течение ночи. После выпаривания растворителя остаток растворяли в 10 мл HCl в MeOH (насыщенный раствор) и смесь кипятили с обратным холодильником в течение 30 минут. После удаления растворителя добавляли 10 мл K2CO3 (насыщенный водный раствор) и раствор 4 раза экстрагировали CH2Cl2. Затем объединенные органические слои упаривали и грубый продукт очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (3%) в качестве элюента. Конечное соединение 14 (15,9 мг, 50%) получали в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 323 (MH+, 73%), 303 (MH+ -HF, 18%), 247 (4%), 219 (3%), 43 (100%); EI m/z (распределение, относительная интенсивность) 322 (M+, 73%), 278 (M+ -N(CH3)2, 44%), 266 (M+-N(CH2)3, 85%), 264 (M+ -CH2CH2NHCH3, 94%), 251 (M+ -CH2CH2- CH2N(CH3), 100%), 209 (68%), 196 (38%); Высокое разрешение EI вычислено C21H23N2F (M+.): 322,1845, найдено: 322,1849 (100%).
Пример B15
(4aS,13bR,14aS)-6-фтор-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-он (конечное соединение 15)

конечное соединение 15
К указанному выше промежуточному соединению 25 (13,5 мг, 0,04 ммоль) в CH2Cl2 (1 мл) добавляли CH3SO3H (1,3 мкл, 0,02 ммоль). После перемешивания при комнатной температуре в течение 1 минуты смесь обрабатывали добавлением Na2CO3 (насыщенный водный раствор). 3 раза экстрагировали CH2Cl2 и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CHCl3/MeOH (95/05) давала конечное соединение 15 в виде маслянистого продукта (10,5 мг, 82%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 309 (MH+, 100%), 289 (MH+ -HF, 17%), 257 (1%).
Пример B16
(4aS,13bR,14aS)-6-фтор-2-метил-1,4a,9,13b,14,14a-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-c]имидазол-3(2H)-он (конечное соединение 16)
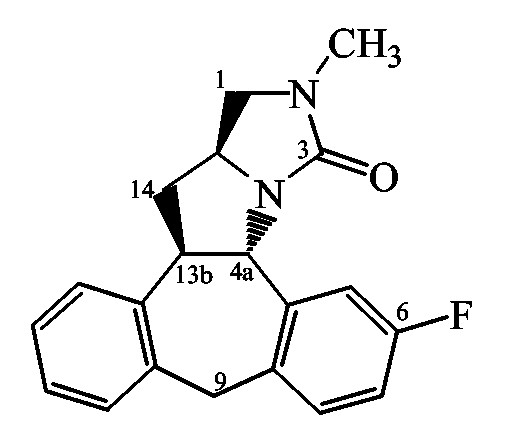
соединение 16
К раствору конечного соединения имидазолона 15 (10 мг, 0,03 ммоль) в ТГФ (1 мл) добавляли NaH (5 мг, 0,1 ммоль) и смесь перемешивали при комнатной температуре в течение 20 минут. Затем добавляли Me2SO4 (8 мкл, 0,08 ммоль) и смесь перемешивали еще в течение 30 мин. Добавляли 10 мл NH4Cl (насыщенный водный раствор), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси EtOAc/гептан (50/50) давала конечное соединение N-метилированный имидазолон 16 (8,4 мг, 80%) в виде масла.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 323 (MH+, 100%), 303 (MH+ -HF, 6%), 257 (11%), 252 (MH+ -CH2N(CH3)CO, 9%), 229 (9%).
Пример B17
[(2R,3aR,12bS)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 17)

соединение 17
К раствору промежуточного соединения 38 (0,17 г, 0,5 ммоль), CH2O (3 экв) и AcOH (3 экв) в MeOH (5 мл) при 0°C несколькими порциями добавляли NaCNBH3 (4 экв). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 6 часов. К реакционной смеси добавляли твердый NaHCO3 (0,5 г) и перемешивали в течение 0,5 часа. Чтобы удалить неорганические комплексы реакционную смесь наносили на короткую фильтрационную колонку и разбавляли смесью CH2Cl2:MeOH (9,5:0,5). Полученное таким образом неочищенное промежуточное соединение

растворяли в i-PrOH (4 мл) и к нему добавляли раствор KOH (56 мг) в воде (0,5 мл) и затем кипятили с обратным холодильником в течение 3 часов. К реакционной смеси добавляли диоксид кремния и растворитель удаляли при пониженном давлении, затем соединение очищали флэш-хроматографией, используя смесь CH2Cl2/MeOH (9:1) в качестве элюента, получая конечное соединение 17 в виде густой вязкой жидкости (60%, 93 мг).
ВРМС: вычислено 312,1638; найдено 312,1633.
Примеры B18-20
a) К раствору промежуточного соединения 39 (0,5 ммоль, 0,33 г) в диоксане (5 мл) добавляли соответствующий аминоспирт (5 экв) и затем кипятили с обратным холодильником в течение 6 часов. Растворитель удаляли при пониженном давлении с последующей хроматографией (силикагель) с использованием смеси CH2Cl2:MeOH (9:1) в качестве элюента, получая промежуточные соединения 39a, 39b и 39c в виде густых вязких жидкостей с общим выходом 40-50%.



b) Смесь соответствующих промежуточных нозиламидов 39a, 39b и 39c (примерно 0,4 ммоль), тиофенола (110 мг, 1,0 ммоль), безводного K2CO3 (138 мг, 1 ммоль) и ДМФА (20 мл) перемешивали при 80°C в течение 4 часов, охлаждали до температуры окружающей среды, разбавляли водой, продукт экстрагировали EtOAc (3×50 мл), объединенные органические фазы промывали водой (4×50 мл), насыщенным раствором соли (35 мл), сушили (K2CO3), упаривали и очищали твердофазной экстракцией на щелочном оксиде алюминия (активность по Брокману II, гептан-этилацетат 50/50, затем этилацетат-MeOH от 100/0 до 96/4 до 90/10), получая конечные соединения 18, 19 и 20.
2-[{[(2R,3aR,12bS)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}(метил)амино]этанол (конечное соединение 18)

конечное соединение 18
ВРМС: вычислено 342,1744; найдено 342,1750.
2-(4-{[(2R,3aR,12bS)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}-1-пиперазинил)этанол (конечное соединение 19)

конечное соединение 19
ВРМС: вычислено 397,2166; найдено 397,2158.
1-{[(2R,3aR,12bS)-11-фтор-2,3,3a,12b-тетрагидро-1H-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}-3-пирролидинол (конечное соединение 20)

конечное соединение 20
ВРМС: вычислено 354,1744; найдено 354,1755.
Пример B21
[(2S,3aR,12bS)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 21)

конечное соединение 21
К раствору указанного выше промежуточного соединения 43 (81 мг, 0,25 ммоль) в ТГФ и воде (3 мл/1 мл) добавляли Ph3P (0,13 г, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли MeOH (5 мл), HCHO (37% масс. водный раствор, 0,20 мл, 2,5 ммоль), AcOH (1 мл) и NaCNBH3 (75 мг, 1,20 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием EtOAc давала конечное соединение 21 в виде маслянистого продукта (70 мг, 86%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 328 (MH+, 100%), 308 (MH+ -HF, 20%), 283 (MH+-Me2NH, 40%), 249 (MH+ -Me2NH- H2S, 12%).
Пример B22
[(2S,3aR,12bS)-11-фтор-1,1-диоксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 22)

конечное соединение 22
К раствору указанного выше промежуточного сульфоназида 44 (133,5 мг, 0,37 ммоль) в ТГФ (8 мл) добавляли воду (67,0 мкл, 3,74 ммоль) и Ph3P (0,13 г, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37% масс. водный раствор, 0,24 мл, 2,98 ммоль), AcOH (0,5 мл) и NaCNBH3 (94 мг, 1,49 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (95/05) давала конечное соединение 22 в виде маслянистого продукта (60,7 мг, 45%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 360 (MH+, 100%), 358 (6%), 340 (MH+ -HF, 12%), 303 (8%), 294 (MH+ -H2SO2, 4%), 250 (1%).
Пример B23
[(2R,3aR,12bS)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 23)

конечное соединение 23
К раствору указанного выше промежуточного соединения 47 (0,15 г, 0,46 ммоль) в ТГФ и воде (5 мл/1 мл) добавляли Ph3P (0,13 г, 0,50 ммоль). После перемешивания при комнатной температуре в течение 1 ночи и выпаривания растворителя добавляли 5 мл MeOH, HCHO (37% масс. водный раствор, 0,20 мл, 2,5 ммоль), AcOH (1 мл) и NaCNBH3 (75 мг, 1,20 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием EtOAc давала конечное соединение 23 в виде маслянистого продукта (70 мг, 86%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 328 (MH+, 100%), 308 (MH+ -HF, 20%), 283 (MH+ -Me2NH, 40%), 249 (MH+ -Me2NH-H2S, 12%).
Пример B24
[(2R,3aR,12bS)-11-фтор-1,1-диоксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 24)

конечное соединение 24
К раствору указанного выше промежуточного сульфоназида 48 (146,5 мг, 0,41 ммоль) в ТГФ (8 мл) добавляли воду (74,0 мкл, 4,10 ммоль) и Ph3P (0,215 мг, 0,82 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37% масс. водный раствор, 0,28 мл, 3,51 ммоль), AcOH (0,5 мл) и NaCNBH3 (110,0 мг, 1,75 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (90/10) давала конечное соединение 24 в виде маслянистого продукта (105,0 мг, 71%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 360 (MH+, 100%), 358 (6%), 340 (MH+ -HF, 12%), 303 (8%), 294 (MH+ -H2SO2, 4%), 250 (1%).
Пример B25
[(2S,3aR,12bS)-11-фтор-1-оксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 25)

конечное соединение 25
К раствору указанного выше промежуточного соединения 49 (107,9 мг, 0,32 ммоль) в ТГФ (5 мл) добавляли воду (57 мкл, 3,16 ммоль) и Ph3P (166,0 мг, 0,63 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37%, 0,26 мл, 3,33 ммоль), AcOH (0,5 мл) и NaCNBH3 (104,7 мг, 1,67 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (95/05) давала конечное соединение 25 в виде маслянистого продукта (80,4 мг, 74%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 344 (MH+, 100%), 328 (MH+ -O, 13%), 326 (MH+ -H2O, 15%), 324 (MH+ -HF, 15%), 182 (14%), 100 (27%).
Пример B26
[(2S,3aR,12bS)-11-фтор-1-оксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 26)

конечное соединение 26
К раствору указанного выше промежуточного соединения 50 (133,4 мг, 0,39 ммоль) в ТГФ (5 мл) добавляли воду (70 мкл, 3,91 ммоль) и Ph3P (205,2 мг, 0,78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37%, 0,24 мл, 2,99 ммоль), AcOH (0,4 мл) и NaCNBH3 (94,0 мг, 1,50 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (95/05) давала конечное соединение 26 в виде маслянистого продукта (85,2 мг, 63%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 344 (MH+, 100%), 328 (MH+ -O, 10%), 327 (12%), 326 (MH+-H2O, 46%), 324 (MH+ -HF, 22%), 283 (12%).
Пример B27
[(2R,3aR,12bS)-11-фтор-1-оксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 27)

конечное соединение 27
К раствору промежуточного соединения 51 (85 мг, 0,25 ммоль) в ТГФ (5 мл) добавляли воду (45 мкл, 2,49 ммоль) и Ph3P (130,8 мг, 0,50 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37%, 0,08 мл, 1,03 ммоль), AcOH (0,3 мл) и NaCNBH3 (32 мг, 0,52 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (95/05) давала конечное соединение 27 в виде маслянистого продукта (35 мг, 41%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 344 (MH+, 100%), 328 (MH+ -O, 4%), 327 (3%), 326 (MH+ -H2O, 10%), 324 (MH+ -HF, 8%), 281 (6%).
Пример B28
[(2R,3aR,12bS)-11-фтор-1-оксидо-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин (конечное соединение 28)

конечное соединение 28
К раствору указанного выше промежуточного соединения 52 (158,5 мг, 0,46 ммоль) в ТГФ (5 мл) добавляли воду (84 мкл, 4,65 ммоль) и Ph3P (243,8 мг, 0,93 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ночи. После выпаривания растворителя добавляли 5 мл MeOH, HCHO (37%, 0,32 мл, 4,05 ммоль), AcOH (0,5 мл) и NaCNBH3 (130 мг, 2,03 ммоль). Перемешивание продолжали при комнатной температуре в течение 1 суток. Добавляли Na2CO3 (насыщенный водный раствор), 3 раза экстрагировали CH2Cl2. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/MeOH (95/05) давала конечное соединение 28 в виде маслянистого продукта (115,7 мг, 72%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 344 (MH+, 100%), 328 (MH+ -O, 3%), 327 (3%), 326 (MH+ -H2O, 13%), 324 (MH+ -HF, 14%), 281 (6%).
Пример B29
(3R,4aR,13bR)-12-фтор-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-ол (конечное соединение 29)

Промежуточное соединение 23a (1,31 г, 2,63 ммоль) растворяли в CH2Cl2 (50 мл). Добавляли дигидропиран (1,20 мл, 13,2 ммоль) и камфорсульфоновую кислоту (6 мг, 0,026 ммоль). Перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали и остаток растворяли в 50 мл MeOH. Добавляли K2CO3 (0,73 г, 5,26 ммоль) и перемешивали при комнатной температуре в течение 1 ночи. Обрабатывали добавлением насыщенного водного раствора NH4Cl (30 мл), экстрагировали CH2Cl2 (3×15 мл) и сушили над MgSO4. Растворитель выпаривали и остаток растворяли в безводном ТГФ (50 мл). Добавляли NaH (0,24 г, 7,78 ммоль) и перемешивали при комнатной температуре в течение 1 суток. Добавляли 30 мл насыщенного водного раствора NH4Cl, экстрагировали CH2Cl2 (3×20 мл) и органические фазы сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси эфир/гексан (35/65) давала масло (0,86 г, 90%, из 2). Полученное масло (0,86 г, 2,34 ммоль) растворяли в 20 мл MeOH/H2O (9/1) и добавляли Dowex 50WX8-100 (1,00 г). Смесь нагревали при 50°C в течение 1 ночи. Фильтровали через фильтр P3, твердые вещества промывали CH2Cl2 (5×15 мл) и растворитель выпаривали. Очистка на колонке с силикагелем с использованием смеси эфир/гексан (70:30) давала конечное соединение 29 в виде масла (0,61 г, 93%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 285 (MH+, 25%), 267 (MH+ -H2O, 100%), 249 (MH+ -2H2O, 36%); EI: m/z (распределение, относительная интенсивность) 284 (M+, 1%), 209 (M+ -CH2CHOHCH2OH, 100%); Высокое разрешение EI вычислено C18H17FO2 (M+): 284,1213, найдено: 284,1204 (2%).
Пример B30
a)(3R,4aR,13bR)-12-фтор-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-илметансульфонат (промежуточное соединение 53)

промежуточное соединение 53
Конечное соединение 29 (0,61 г, 2,16 ммоль) растворяли в CH2Cl2 (50 мл). Добавляли Et3N (0,60 мл, 4,32 ммоль), DMAP (0,13 г, 1,08 ммоль) и MsCl (0,25 мл, 3,24 ммоль). Перемешивали при комнатной температуре в течение 4 часов. Обрабатывали добавлением насыщенного водного раствора NH4Cl (20 мл), экстрагировали CH2Cl2 (3×20 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием CH2Cl2 давала промежуточное соединение 53 в виде масла (0,76 г, 97%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 363 (MH+, 1%), 267 (MH+ -MsOH, 100%), 249 (MH+-MsOH-H2O, 33%); EI: m/z (распределение, относительная интенсивность) 362 (M+, 5%), 266 (M+ -MsOH, 3%), 248 (M+ -MsOH-H2O, 4%), 209 (M+ -CH2CHOMsCH2OH, 100%); Высокое разрешение EI вычислено C19H19FO4S (M+): 362,0988, найдено: 362,0984 (12%).
b) (3S,4aR,13bR)-3-азидо-12-фтор-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран (промежуточное соединение 54)

промежуточное соединение 54
Промежуточный мезилат 53 (0,29 г, 0,79 ммоль) растворяют в ДМФА (10 мл), добавляли NaN3 (0,10 г, 1,58 ммоль) и смесь нагревают при 90°C в течение 2 часов. Добавляли насыщенный водный раствор NH4Cl (10 мл), экстрагировали CH2Cl2 (3×10 мл) и сушили над MgSO4. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гептан (40:60) давала промежуточное соединение 54 в виде кристаллического продукта (0,22 г, 88%); т.пл.: 91-93°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 310 (MH+, 13%), 282 (MH+ -N2, 100%); EI: m/z (распределение, относительная интенсивность) 281 (M+ -N2, 28%), 208 (100%); Высокое разрешение EI вычислено C18H16FNO (M+ -N2): 309,1216, найдено: 309,1223 (40%).
c) (3S,4aR,13bR)-12-фтор-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (конечное соединение 30)

конечное соединение 30
Промежуточное соединение 54 (0,16 г, 0,52 ммоль) растворяли в i-PrOH/ТГФ (2:1, 15 мл). Добавляли 10% Pd-C (примерно 100 мг) и подвергали гидрированию (давление 1 атм) в течение 1 ночи. Фильтровали через подушку целита, твердые вещества промывали CH2Cl2 (5×10 мл) и фильтрат упаривали. Остаток очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (75:25), получая конечное соединение 30 в виде кристаллического продукта (0,14 г, 94%); т.пл.: 74-76°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 284 (MH+, 100%); EI: m/z (распределение, относительная интенсивность) 283 (M+; 5%), 209 (M+. -CH2CHNH2CH2OH, 100%); Высокое разрешение EI вычислено C18H18FNO (M+.): 283,1372, найдено: 283,1370 (43%).
Пример B31
(4aR,13bR)-12-фтор-4,4a,9,13b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3(2H)-он (конечное соединение 31)

конечное соединение 31
Конечное соединение 29 (77 мг, 0,27 ммоль) растворяли в CH2Cl2 (10 мл) и добавляли хлорхромат пиридиния (131 мг, 0,54 ммоль). Перемешивали при комнатной температуре в течение 20 часов. Фильтровали через подушку целита, твердые вещества промывали CH2Cl2 (5×20 мл) и фильтрат упаривали. Очистка на колонке с силикагелем с использованием смеси эфир/гексан (50:50) давала конечное соединение 31 в виде белого кристаллического продукта (61 мг, 80%); т.пл.: 146-148°C.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 283 (MH+, 11%), 265 (MH+ -H2O, 100%), 237 (MH+ -H2O-CO, 22%); EI: m/z (распределение, относительная интенсивность) 282 (M+, 26%), 209 (M+ -CH2COCH2OH, 100%); Высокое разрешение EI вычислено C18H15FO2 (M+): 282,1056, найдено: 282,1057 (40%).
Пример B32
(3S,4aR,13bR)-12-фтор-N,N-диметил-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (конечное соединение 32)

конечное соединение 32
Промежуточное соединение 54 (0,24 г, 0,76 ммоль) растворяли в i-PrOH/ТГФ (2:1, 15 мл). Добавляли 10% Pd-C (примерно 150 мг) и подвергали смесь гидрированию (давление 1 атм) в течение 1 ночи. Добавляли 35% водный раствор CH2O (0,60 мл, 7,6 ммоль) и продолжали гидрирование в течение 2 суток. Фильтровали через целит и промывали CH2Cl2 (5×15 мл). Органические фазы объединяли и сушили над MgSO4. Раствор фильтровали и упаривали и остаток очищали хроматографией на колонке с силикагелем с использованием смеси CHCl3/MeOH (90:10), получая конечное соединение 32 MH-170 в виде масла (0,22 г, 93%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 312 (MH+, 100%); EI: m/z (распределение, относительная интенсивность) 311 (M+, 7%).
Пример B33
(3R,4aR,13bR)-12-фтор-N-метил-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (конечное соединение 33)

конечное соединение 33
Конечное соединение 31 (0,18 г, 0,63 ммоль) растворяли в I-PrOH/ТГФ (2:1, 10 мл). Добавляли 10% Pd-C (примерно 100 мг), Et3N (0,87 мл, 6,3 ммоль) и MeNH2·HCl (0,42 г, 6,3 ммоль). Смесь подвергали гидрированию (давление 1 атм) в течение 1 ночи. Фильтровали через подушку целита и твердые вещества промывали CH2Cl2 (5×15 мл). Раствор фильтровали и упаривали и остаток очищали хроматографией на колонке с силикагелем, используя смесь CHCl3/MeOH (90:10), получая два диастереоизомера (0,18 г, 95%) в соотношении 5:1, при этом основной (3R)-изомер (конечное соединение 33) может быть частично отделен.
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 298 (MH+, 100%); EI: m/z (распределение, относительная интенсивность) 297 (M+, 5%), 266 (M+ -CH3NH2, 19%); Высокое разрешение EI вычислено C19H20FNO (M+): 297,1529, найдено: 297,1528 (3,5%).
Пример B34
(4aR*,13bS*)-12-фтор-4,4a,9,13b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3(2H)-он (конечное соединение 34)

a) Превращение алкена в диастереоизомерные диолы. Промежуточное соединение 56 (1,40 г, 4,52 ммоль) растворяли в ацетоне (30 мл). Добавляли небольшой кристалл OsO4 (каталитическое количество) и N-метилморфолин-N-оксид (0,63 г, 5,42 ммоль). Перемешивали при комнатной температуре в течение 1 суток. Растворитель удаляли при пониженном давлении и остаток очищали хроматографией на колонке с силикагелем, используя смесь EtOAc/гексан (80:20), получая смесь двух диастереоизомерных диолов (масло, 1,45 г, 93%).
b) Избирательное монотозилирование первичной спиртовой группы. Указанные выше диолы (1,45 г, 4,22 ммоль) растворяли в толуоле (50 мл). Добавляли Et3N (1,76 мл, 12,6 ммоль), TsCl (1,05 г, 5,48 ммоль) и Bu2SnO (0,10 г, 0,42 ммоль). Перемешивали при комнатной температуре в течение 1 суток. Добавляли насыщенный водный раствор NH4Cl (30 мл), экстрагировали CH2Cl2 (3×20 мл) и сушили над MgSO4. Раствор фильтровали и упаривали и остаток очищали хроматографией на колонке с использованием смеси EtOAc/гексан (40:60), получая диастереоизомерные производные монотозилата, соответствующие избирательному сульфонилированию первичной OH-группы (масло, 1,74 г, 83%).
c) Защита вторичной спиртовой группы. Монотозилаты (1,74 г, 3,49 ммоль) растворяли в CH2Cl2 (60 мл) и добавляли дигидропиран (1,59 мл, 17,5 ммоль), камфорсульфоновую кислоту (10 мг, 0,035 ммоль). Перемешивали при комнатной температуре в течение 1 часа и удаляли растворитель при пониженном давлении.
d) Удаление защиты и циклизация бензилового спирта. Остаток растворяли в MeOH (50 мл). Добавляли K2CO3 (0,79 г, 6,99 ммоль) и перемешивали при комнатной температуре в течение 1 ночи. Обрабатывали добавлением насыщенного водного раствора NH4Cl (30 мл), 3 раза экстрагировали CH2Cl2 (3×20 мл) и сушили над MgSO4. Растворитель выпаривали и остаток, содержащий бензиловый спирт, растворяли в безводном ТГФ (50 мл). Добавляли NaH (0,21 г, 6,99 ммоль) и перемешивали при комнатной температуре в течение 3 суток, чтобы осуществить циклизацию. Обрабатывали добавлением насыщенного водного раствора NH4Cl (30 мл) и экстрагировали CH2Cl2 (3×20 мл). Сушили над MgSO4 и растворитель выпаривали.
e) Удаление защиты и окисление вторичной спиртовой группы. Остаток (1,70 г) растворяли в 20 мл MeOH/H2O (9:1) и добавляли Dowex 50WX8-100 (1,00 г). Смесь нагревали при 50°C в течение 2 часов. Фильтровали через фильтр P3, твердые вещества промывали CH2Cl2 (5×15 мл) и упаривали. Очистка на колонке с силикагелем с использованием смеси эфир/гексан (70:30) давала масло (два диастереоизомерных спирта) (0,87 г, 88%). Указанное выше масло (0,87 г, 3,06 ммоль) растворяли в CH2Cl2 (40 мл). Добавляли хлорхромат пиридиния (1,32 г, 6,13 ммоль) и перемешивали при комнатной температуре в течение 1 ночи. Фильтровали через подушку целита, твердые вещества промывали CH2Cl2 (5×20 мл) и упаривали. Очистка на колонке с силикагелем с использованием смеси CH2Cl2/гексан (80:20) давала конечное соединение 34 в виде масла (0,66 г, 76%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 283 (MH+, 25%), 265 (MH+ -H2O, 100%); EI: m/z (распределение, относительная интенсивность) 282 (M+, 39%), 209 (M+ -CH2COCH2OH, 100%).
Пример B35
(3S*,4aR*,13bS*)-12-фтор-N-метил-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (конечное соединение 35)

конечное соединение 35
Конечное соединение 34 (0,23 г, 0,83 ммоль) растворяли в i-PrOH (15 мл). Добавляли Et3N (1,15 мл, 8,25 ммоль), MeNH2HCl (0,56 г, 8,25 ммоль) и 10% Pd/C (примерно 150 мг). Подвергали гидрированию (давление 1 атм) в течение 1 ночи. Фильтровали через подушку целита и твердые вещества промывали CH2Cl2 (5×10 мл). Раствор фильтровали и упаривали и остаток очищали хроматографией на колонке с силикагелем с использованием CHCl3/MeOH (90:10), получая конечное соединение 35 в виде почти единственного диастереоизомера (0,23 г, 95%).
Масс-спектр: -CI m/z (распределение, относительная интенсивность) 298 (MH+, 100%); EI: m/z (распределение, относительная интенсивность) 209 (M+ CH2CH(NHMe)CH2OH, 100%).
Пример B36
a) Метил(2R,3aR,12bS)-2-(аминометил)-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (промежуточное соединение 62b), [(2R,3aR,12bS)-1-ацетил-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метанамин (промежуточное соединение 62c) и (2R,3aR,12bS)-2-(аминометил)-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбальдегид (промежуточное соединение 62d)

промежуточное соединение 62b R=OMe
промежуточное соединение 62c R=Me
промежуточное соединение 62d R=H
Типичный способ - синтез метил(2R,3aR,12bS)-2-(аминометил)-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилата (промежуточного соединения 62b): Раствор трифенилфосфина (996 мг, 3,8 ммоль) в безводном ТГФ (20 мл) помещали в двугорлую колбу объемом 100 мл, снабженную мембраной, входным отверстием для аргона и магнитной мешалкой, охлаждали до -15°C. Через мембрану добавляли неразбавленный диизопропилазодикарбоксилат (768 мг, 3,8 ммоль) при интенсивном перемешивании. Полученную в результате желтую суспензию перемешивали при указанной выше температуре в течение 30 минут, затем одной порцией добавляли промежуточный карбамат 62 (650 мг, 1,9 ммоль) в ТГФ (5 мл). После 5-минутного перемешивания по каплям добавляли дифенилфосфорилазид (606 мг, 2,2 ммоль) в ТГФ (3 мл) в течение 3 минут, полученной в результате мутной смеси давали возможность нагреться до комнатной температуры и затем перемешивали в течение 12 часов. После указанного периода времени добавляли воду (0,2 мл) и трифенилфосфин (996 мг, 3,8 ммоль) и раствор перемешивали при 45°C в течение 2 часов. После охлаждения до комнатной температуры добавляли силикагель (Kieselgel 60, 70-230 меш, 4 г), ТГФ удаляли в вакууме и порошок диоксида кремния подвергали флэш-хроматографии (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH, от 100/0 постепенно до 85/15), получая требуемый промежуточный амин 62b (401 мг, 1,18 ммоль, 62%) в виде бесцветного масла, который темнел при стоянии.
Промежуточное соединение 62b
ВРМС: вычислено для C20H21FN2O2: 340,1587; найдено: 340,1588.
Промежуточное соединение 62c: присутствуют два ротамера (в соотношении примерно 2:1)
CI-МС (CH4) 325 (MH+, 100%); 305 (MH+ -HF, 10%).
ВРМС: вычислено для C20H21FN2O: 324,1638; найдено: 324,1644.
Промежуточное соединение 62d: присутствуют два ротамера (в соотношении примерно 5:2)
ВРМС: вычислено для C19H19FN2O: 310,1481; найдено: 310,1480.
b) Метил(2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилат (конечное соединение 36a),
[(2R,3aR,12bS)-1-ацетил-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 36b) и
(2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбальдегид (конечное соединение 36c)

конечное соединение 36a R=OMe
конечное соединение 36b R=Me
конечное соединение 36c R=H
Типичный способ - синтез метил(2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-карбоксилата (конечного соединения 36a)
Промежуточный амин 62b (401 мг, 1,18 ммоль) растворяли в MeOH (30 мл), добавляли AcOH (1 мл) и 35% водный раствор формальдегида (1 г, 11,7 ммоль), затем цианоборогидрид натрия (628 мг, 10 ммоль). Полученную в результате смесь перемешивали при комнатной температуре в течение 4 часов, гасили концентрированной HCl (5 мл), обрабатывали твердым NaHCO3 (8,4 г, 100 ммоль), 1н гидроксидом натрия (15 мл). Преципитированный продукт отфильтровывали, промывали водой (5×25 мл), растворяли в этилацетате, промывали насыщенным раствором соли (30 мл), сушили (K2CO3), упаривали в вакууме и очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH от 95/5 до 90/10 до 85/15), получая конечное соединение 36a (313 мг, 0,85 ммоль, 72%) в виде желтоватого масла.
Конечное соединение 36a:
ВРМС: вычислено для C22H25FN2O2: 368,1900; найдено: 368,1895.
Конечное соединение 36b: два ротамера в соотношении примерно 3:2.
ВРМС: вычислено для C22H25FN2O: 352,1951; найдено: 352,1955.
Конечное соединение 36c: два ротамера в соотношении примерно 5:3.
ВРМС: вычислено для C21H23FN2O: 338,1794; найдено: 338,1790.
Пример 37
[(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 37)
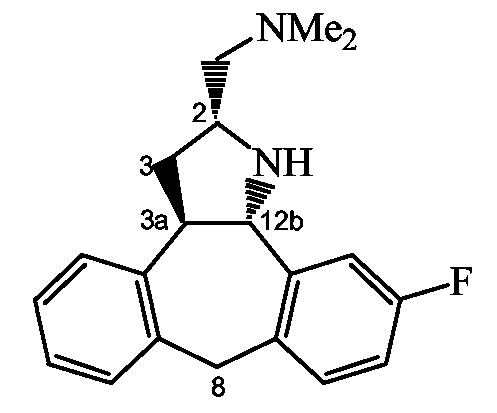
конечное соединение 37
Смесь конечного соединения 36a (100 мг, 0,27 ммоль), i-PrOH (10 мл), гидроксида калия (560 мг, 10 ммоль) и воды (0,1 мл) кипятили с обратным холодильником в атмосфере азота в течение 12 часов (температура масляной бани 135°C), затем охлаждали до комнатной температуры. После разбавления водой (50 мл), экстракции EtOAc (3×40 мл) объединенные органические фазы промывали водой (3×40 мл), насыщенным раствором соли (40 мл), сушили над K2CO3 и упаривали, получая чистое конечное соединение 37 (84 мг, 100%) в виде желтоватого полутвердого вещества, которое превращали в гидрохлоридную соль (конечное соединение 37a).
ВРМС: вычислено для C20H23FN2: 310,1845; найдено: 310,1851.
Пример B38
(2R,3aR,12bS)-11-фтор-2-[(метиламино)метил]-3,3a,8,12b-тетрагидродибензо[3,4:6,7]-циклогепта[1,2-b]пиррол-1(2H)-карбальдегид (конечное соединение 38)

конечное соединение 38
Смесь промежуточного альдегида 63 (50 мг, 0,162 ммоль), гидрохлорида метиламина (218 мг, 3,24 ммоль), Et3N (405 мг, 4,0 ммоль), 10% Pd-C (30 мг) и MeOH (12 мл) гидрировали в течение 2 часов при атмосферном давлении. Реакционную смесь фильтровали через кизельгур, который затем промывали EtOAc (2×10 мл). Объединенные растворы упаривали в вакууме и остаток очищали хроматографией на колонке (Kieselgel 60, 70-230 меш, CH2Cl2/MeOH от 100/0 до 85/15), получая конечное соединение 38 (21 мг, 0,065 ммоль, 40%) в виде коричневого масла.
Присутствуют четыре ротамера (в соотношении 10:6:4:1).
CI-МС (CH4): 325 (100%, M+H+), 305 (12%, -HF). ВРМС: вычислено для C20H21FN2O: 324,1638; найдено: 324,1650.
Пример B39
2-((2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-ил)этанол (конечное соединение 39)

конечное соединение 139
Димер гидроксиацетальдегида (2,5-дигидрокси-1,4-диоксан) (240 мг, 2,0 ммоль) растворяли в MeOH (25 мл) и перемешивали при 40°C в течение 30 минут, затем добавляли амин 37 (124 мг, 0,40 ммоль) и перемешивание при 40°C продолжали еще в течение 30 минут. После охлаждения до комнатной температуры добавляли AcOH (120 мг, 2,0 ммоль), затем цианоборогидрид натрия (188 мг, 3,0 ммоль) и полученную в результате смесь перемешивали в течение 2 часов. После указанного периода времени смесь гасили концентрированной HCl (2 мл), обрабатывали твердым NaHCO3 (2,94 г, 35 ммоль), 1н гидроксидом натрия (3 мл). Примерно 20 мл MeOH удаляли в вакууме, остаток разбавляли водой (30 мл) и экстрагировали EtOAc (3×30 мл). Объединенные органические фазы промывали водой (5×25 мл), насыщенным раствором соли (30 мл), сушили (K2CO3), упаривали в вакууме и очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH от 95/5 до 90/10 до 85/15), получая соединение амина 39 (80 мг, 0,244 ммоль, 61%) в виде бесцветного масла.
ВРМС: вычислено для C22H27FN2O: 354,2107; найдено: 354,2107.
Пример B40
[(2R,3aR,12bS)-11-фтор-1-(2-метоксиэтил)-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 40)
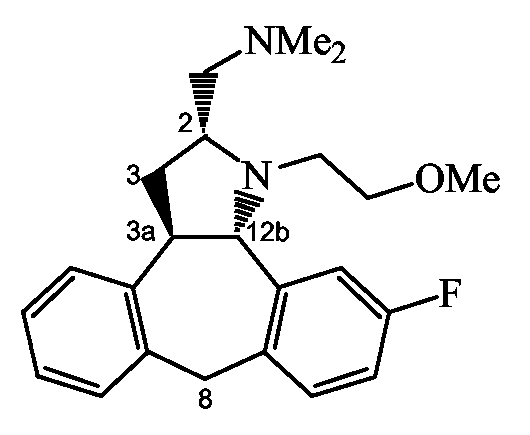
конечное соединение 40
2-((2R,3aR,12bS)-2-[(Диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2H)-ил)этанол, соединение 39 (50 мг, 0,153 ммоль) растворяли в безводном ТГФ (10 мл), затем добавляли 60% дисперсию NaH (8 мг, 0,2 ммоль), затем диметилсульфат (25 мг, 0,2 ммоль). Полученную в результате смесь перемешивали в атмосфере аргона при 60°C в течение 5 часов, затем охлаждали, гасили концентрированным гидроксидом аммония (2 мл), разбавляли водой (40 мл). После экстракции продукта EtOAc (3×25 мл) объединенные органические фазы промывали водой (3×25 мл), насыщенным раствором соли (25 мл), сушили над K2CO3, упаривали в вакууме и остаток очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH от 95/5 до 90/10 до 85/15), получая конечное соединение 40 (39 мг, 0,107 ммоль, 70%) в виде желтоватого масла.
ВРМС: вычислено для C23H29FN2O: 368,2264; найдено: 368,2270.
Пример B41
[(2R,3aR,12bS)-1-циано-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин (конечное соединение 41)

конечное соединение 41
Поли(4-винилпиридин), поперечно сшитый 2% дивинилбензолом (0,5 г) подвергали набуханию в течение 1 часа, используя CH2Cl2 (10 мл), затем одной порцией добавляли конечное соединение 37 (57 мг, 0,184 ммоль) в CH2Cl2 (2 мл) с последующим добавлением цианогенбромида (39 мг, 0,367 ммоль), затем суспензию перемешивали при комнатной температуре в течение 30 минут. Смолу отфильтровывали, фильтрат обрабатывали насыщенным водным раствором K2CO3 (10 мл), органическую фазу отделяли, упаривали в вакууме и остаток очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH от 95/5 до 90/10 → 87/13), получая конечное соединение 41 (24 мг, 0,077 ммоль, 42%) в виде коричневатого масла.
CI-МС (CH4): 308 (100%, M+H+), 288 (8%, -HF).
ВРМС: вычислено для C19H18FN3: 307,1485; найдено: 307,1499.
Пример B42
(2R,3aR,12bS)-11-фтор-2-(4-морфолинилметил)-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол (конечное соединение 42)
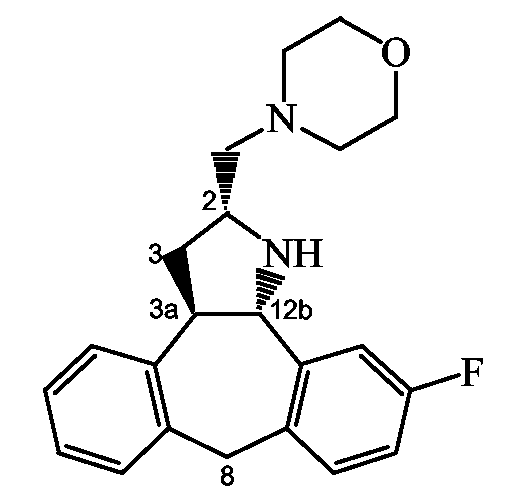
конечное соединение 42
К раствору промежуточного азиридина 64 (63 мг, 0,237 ммоль) в ацетонитриле (1 мл) добавляли йодид натрия (107 мг, 0,711 ммоль) и триметилсилилхлорид (90 мкл, 0,711 ммоль) при комнатной температуре. После перемешивания раствора в течение 2 часов к смеси по каплям добавляли морфолин (44 мг, 0,5 ммоль) в ацетонитриле (0,5 мл). Раствор нагревали до температуры кипения растворителя в течение 2 часов. Темно-коричневую реакционную смесь гасили 1,2н водным раствором HCl и затем обрабатывали насыщенным раствором NaHCO3. Органический слой отделяли и водный слой экстрагировали метиленхлоридом (3×10 мл). Объединенные органические экстракты промывали 20 мл насыщенного раствора соли, сушили над безводным MgSO4, фильтровали и концентрировали в вакууме. Остаток очищали хроматографией на колонке на щелочном оксиде алюминия (активность по Брокману III, EtOAc-MeOH, от 100/0 до 98/2 до 95/5), получая конечное соединение 42 (38 мг, 0,11 ммоль, 45%) в виде коричневатого масла.
CI-МС (CH4): 353 (100%, M+H+); 333 (-HF, 7%).
ВРМС: вычислено для C22H25FN2O: 352,1951; найдено: 352,1966.
Пример B43
2-(4-{[(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}-1-пиперазинил)этанол (конечное соединение 43a) и
2-[{[(2R,3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}(метил)амино]этанол (конечное соединение 43b)



Смесь соответствующего промежуточного нозиламида 66a или 66b (примерно 0,4 ммоль), тиофенола (110 мг, 1,0 ммоль), безводного K2CO3 (138 мг, 1 ммоль) и ДМФА (20 мл) перемешивали при 80°C в течение 4 часов, охлаждали до температуры окружающей среды, разбавляли водой, продукт экстрагировали EtOAc (3×50 мл), объединенные органические фазы промывали водой (4×50 мл), насыщенным раствором соли (35 мл), сушили (K2CO3), упаривали и очищали твердофазной экстракцией на щелочном оксиде алюминия (активность по Брокману II, гептан-этилацетат 50/50, затем EtOAc-MeOH от 100/0 до 96/4 до 90/10), получая соединение 43a (111 мг, 0,28 ммоль, 52% от промежуточного соединения 65) или соединение 43b (80 мг, 0,24 ммоль, 44% от промежуточного соединения 65), оба в виде коричневатого масла.
Соединение 43a (TK-895):
ВРМС: вычислено для C24H30FN3O: 395,2373; найдено: 395,2374.
Соединение 43b (TK-1013):
ВРМС: вычислено для C21H25FN2O: 340,1951; 340,1943.
Пример B44
[(2R,4aR,13bS)-12-фтор-2,3,4,4a,9,13b-гексагидро-1H-дибензо[3,4:6,7]циклогепта[1,2-b]пиридин-2-ил]-N,N-диметилметанамин (соединение 44)
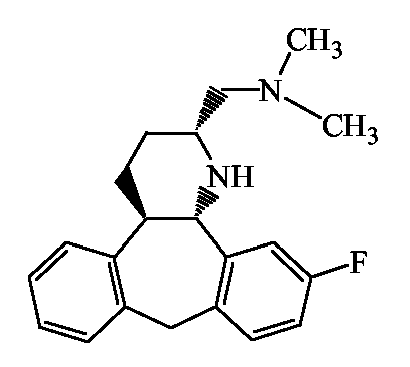
соединение 44
Промежуточный тозилат 67k (153 мг, 0,338 ммоль), 40% водный раствор метиламина (15 мл) и ТГФ (35 мл) нагревали в баллоне из нержавеющей стали при 135°C в течение 15 часов. После охлаждения баллон открывали, ТГФ и метиламин выпаривали в вакууме, остаток экстрагировали CH2Cl2 (4×20 мл). Объединенные органические фазы промывали водой (3×20 мл), сушили (K2CO3), упаривали и очищали хроматографией на колонке (Kieselgel 60, 230-400 меш, CH2Cl2-MeOH от 98/2 до 85/15), получая конечное соединение 44 (32 мг, 0,098 ммоль, 29%) в виде коричневого масла, которое превращали в оксалатную соль (конечное соединение 44a).
ВРМС: вычислено для C21H25FN2: 324,2002; найдено: 324,1995.
Пример B45
Метил(2R,4aR,13bS)-2-[(диметиламино)метил]-12-фтор-2,3,4,4a,9,13b-гексагидро-1H-дибензо[3,4:6,7]циклогепта[1,2-b]пиридин-1-карбоксилат (конечное соединение 45)

конечное соединение 45
Превращение конечного соединения 44 (48 мг, 0,15 ммоль) с использованием метилхлорформиата осуществляли таким же способом, который описан для получения промежуточного соединения 62. Хроматография на колонке (Kieselgel 60, 70-230 меш, MeOH-CH2Cl2 от 3/97 до 15/85) давала конечное соединение 45 (45 мг, 0,118 ммоль, 79%) в виде бесцветного масла.
ВРМС: вычислено для C23H27FN2O2: 382,2057; найдено: 382,2064.
Пример B46
[(2R,4aR,13bS)-12-фтор-2,3,4,4a,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-2-ил]-N-метилметанамин (конечное соединение 46)

конечное соединение 46
Промежуточный тозилат 68e (282 мг, 0,62 ммоль), 40% водный раствор метиламина (25 мл) и ТГФ (35 мл) нагревали в стальном баллоне при 135°C в течение 15 часов. После охлаждения баллон открывали, ТГФ и метиламин выпаривали в вакууме. Остаток экстрагировали CH2Cl2 (4×30 мл) и объединенные органические фазы промывали водой (3×20 мл), сушили (K2CO3) и упаривали. Кристаллизация из смеси CH2Cl2/гексан давала конечное соединение 46 (70 мг, 0,225 ммоль, 36%) в виде бежевого порошка.
ВРМС: вычислено для C20H22FNO: 311,1685; найдено: 311,1700.
Пример B47
[(3aR,12bS)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-3-ил]-N,N-диметилметанамин (конечное соединение 47)
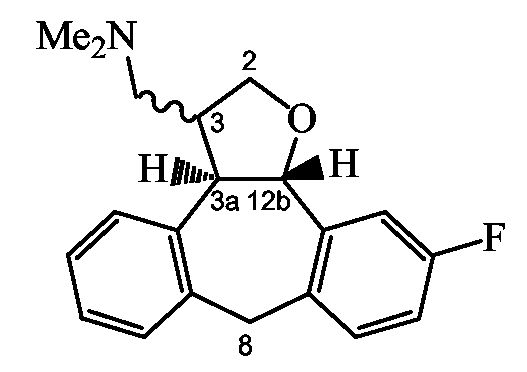
конечное соединение 47
Промежуточный азид 69i (122 мг, 0,39 ммоль) растворяли в MeOH (10 мл), добавляли 10% палладий на угле (40 мг) и смесь подвергали гидрированию при атмосферном давлении в течение 1,5 часа, затем добавляли 35% водный раствор формальдегида (1 г) и AcOH (120 мг, 2 ммоль) и гидрирование продолжали в течение 2 часов. После фильтрования через небольшую подушку целита и добавления EtOAc (45 мл) реакционную смесь промывали насыщенным водным раствором бикарбоната натрия (25 мл), водой (2×50 мл), насыщенным раствором соли (30 мл), сушили над K2CO3 и упаривали в вакууме. Остаток очищали хроматографией на колонке (Kieselgel 60, 70-230 меш, этилацетат-MeOH, от 100/0 до 95/5 до 92/8 до 87/13), получая конечное соединение 47 (77 мг, 0,248 ммоль, 63%) в виде желтого масла. Продукт представляет собой смесь 2 эпимеров (в соотношении 12,8:1).
ВРМС: вычислено для C20H22FNO: 311,1685; найдено: 311,1680.
CI-МС (CH4) 312 (MH+, 100%); 292 (MH+ - HF, 9%).
Пример B48
[(3aR,12bR)-11-фтор-3,3a,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-3-ил]-N,N-диметилметанамин (конечное соединение 48)

конечное соединение 48
Реакцию с использованием промежуточного соединения 70d (100 мг, 0,304 ммоль) осуществляли таким же способом, который описан для конечного соединения 47. Очистка твердофазной экстракцией (Alltech C18 картридж 2г, вода-MeOH, от 100/0 до 50/50 до 0/100) давала соединение 48 (57 мг, 0,18 ммоль, 59%). Продукт представляет собой смесь 2 эпимеров (в соотношении 2:1).
ВРМС: вычислено для C20H22FNO: 311,1685; найдено: 311,1692.
CI-МС (CH4) 312 (MH+, 100%); 292 (MH+ - HF, 12%).
Пример B49
[(3aR,12bS)-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-3-ил]-N,N-диметилметанамин (конечное соединение 49)

конечное соединение 49
Смесь промежуточного соединения 71f (194 мг, 0,53 ммоль) и 10% палладия на угле (50 мг) в MeOH (35 мл) гидрировали при атмосферном давлении в течение 40 минут, затем добавляли 35% водный раствор формальдегида (1 мл) и гидрирование продолжали еще в течение 40 минут. После фильтрования через небольшую подушку целита реакционную смесь упаривали в вакууме. Остаток растворяли в i-PrOH (20 мл), добавляли KOH (560 мг, 10 ммоль) и воду (0,1 мл) и полученный в результате раствор кипятили с обратным холодильником в атмосфере азота в течение 12 часов (температура масляной бани 135°C), затем охлаждали до комнатной температуры. После разбавления водой (50 мл) и экстракции EtOAc (3×40 мл) объединенные органические фазы промывали водой (3×40 мл), насыщенным раствором соли (40 мл), сушили над K2CO3 и упаривали в вакууме. Остаток очищали хроматографией на колонке (щелочной оксид алюминия, степень активности по Брокману I, этилацетат-MeOH от 100/0 до 85/15), получая чистое конечное соединение 49 (102 мг, 0,33 ммоль, 62%) в виде коричневого масла. Продукт представляет собой смесь 2 эпимеров (в соотношении 1:1).
ВРМС: вычислено для C20H23FN2: 310,1845; найдено: 310,1833.
В таблицах 1-3 перечислены соединения формулы (I), которые получены согласно одному из указанных выше примеров.




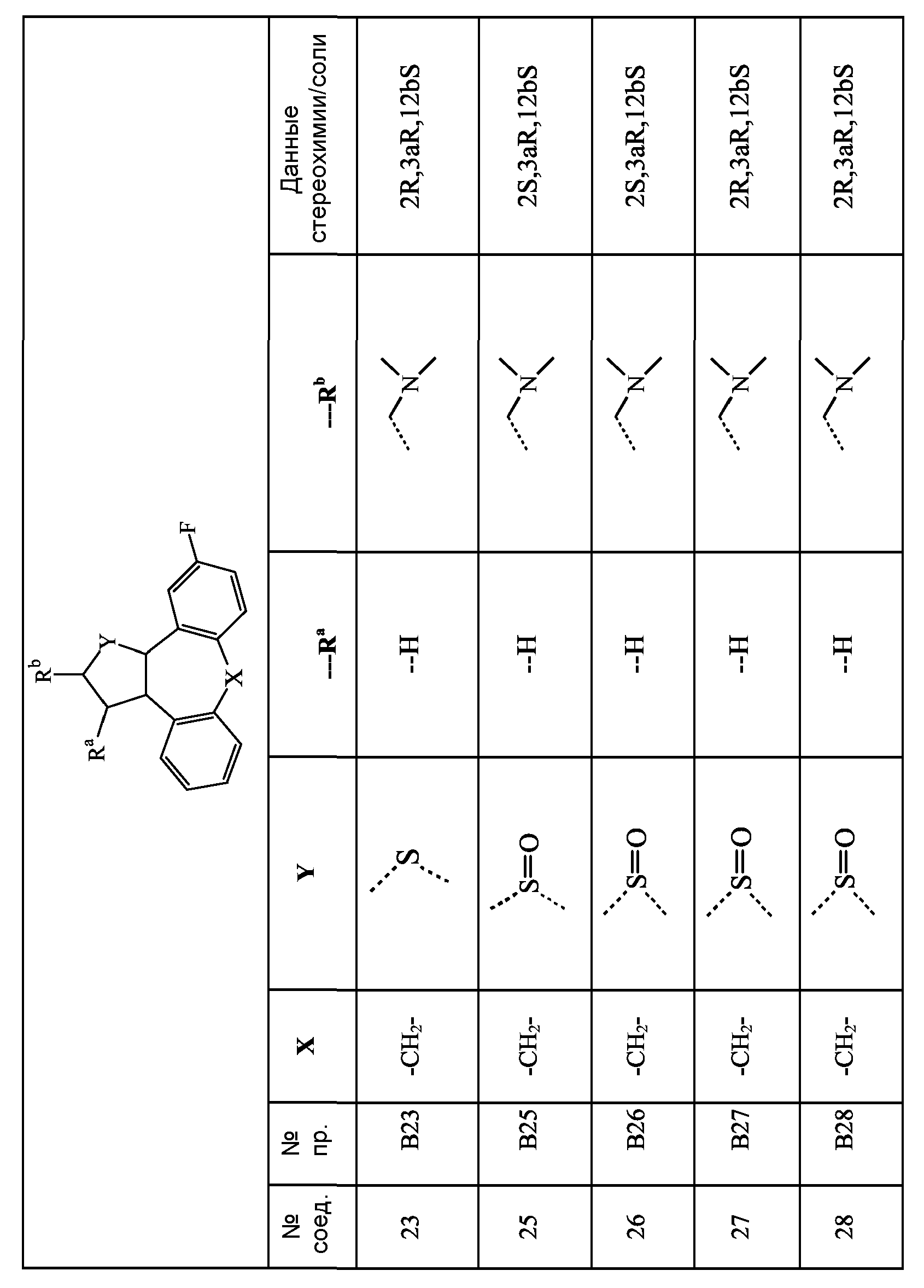







C. Фармакологические примеры
Пример C.1: Аффинность связывания in vitro с рецепторами 5-HT2A и 5-HT2C
Взаимодействие соединений формулы (I) с рецепторами 5-HT2A и 5-HT2C оценивали в экспериментах связывания радиоактивного лиганда in vitro. В общем, радиоактивный лиганд с высокой аффинностью связывания по отношению к рецептору в низкой концентрации инкубируют с образцом препарата ткани, обогащенным конкретным рецептором (1-5 мг ткани) в забуференной среде (от 0,2 до 5 мл). Во время инкубации радиоактивные лиганды связываются с рецептором. Когда достигается равновесие связывания, связанную с рецептором радиоактивность отделяют от несвязанной радиоактивности и подсчитывают связанную с рецептором активность. Взаимодействие тестируемых соединений с рецепторами оценивают в экспериментах по конкурентному связыванию. Различные концентрации тестируемого соединения добавляют к инкубационной смеси, содержащей препарат ткани и радиоактивный лиганд. Связывание радиоактивного лиганда будет ингибироваться тестируемым соединением пропорционально аффинности его связывания и его концентрации. Аффинности соединений по отношению к рецепторам 5-HT2 измеряли с помощью исследований связывания радиоактивного лиганда, проводимых с использованием: (a) клонированного рецептора 5-HT2A человека, экспрессируемого в клетках L929, с использованием [125I]R91150 в качестве радиоактивного лиганда, и (b) клонированного рецептора 5-HT2C человека, экспрессируемого в клетках CHO, с использованием [3H]мезулергина в качестве радиоактивного лиганда.
Пример C.2: Определение ингибирования обратного захвата NET in vitro
Кору головного мозга крыс собирали и гомогенизировали, используя гомогенизатор Ultra-Turrax T25 и гомогенизатор Dual в ледяном буфере для гомогенизации, содержащем трис, NaCl и KCl (50 мМ, 120 мМ и 5 мМ, соответственно, pH 7,4), затем разбавляли до соответствующей концентрации белка, оптимизированной для специфичного и неспецифичного связывания. Связывание осуществляли с радиоактивным лигандом [3H]никсозетином (NEN, NET-1084, удельная активность ~70 кюри/ммоль), разбавленном в ледяном буфере для анализа, содержащем трис, NaCl и KCl (50 мМ, 300 мМ и 5 мМ, соответственно, pH 7,4), в концентрации 20 нмоль/л. Затем приготовленный радиоактивный лиганд (50 мкл) инкубировали (60 мин, 25°C) с препаратами мембран, предварительно разбавленными до соответствующей концентрации белка (400 мкл), и с 50 мкл либо контрольного 10% ДМСО, мазиндола (конечная концентрация 10-6 моль/л), либо представляющего интерес соединения. Связанную с мембранами активность определяли фильтрованием через устройство для сбора Packard Filtermate, используя фильтры GF/B Unifilterplates, промывая ледяным трис-HCl-буфером, содержащим NaCl и KCl (50 мМ, 120 мМ и 4 мМ; pH 7,4; 6×0,5 мл). Фильтрам давали возможность высохнуть в течение 24 час перед добавлением сцинтилляционной жидкости. Сцинтилляционной жидкости давали возможность пропитать фильтры в течение 24 час перед подсчетом в сцинтилляционном счетчике Topcount. Специфичное связывание в процентах и кривые конкурентного связывания рассчитывали, используя компьютерную программу S-Plus (Insightful).
Пример C.3 : Аффинность связывания in vitro с рецептором D2L человека
Замороженные мембраны клеток CHO, трансфицированных рецептором допамина D2L человека, оттаивали, недолго гомогенизировали, используя гомогенизатор Ultra-Turrax T25, и разбавляли в трис-HCl-буфере для анализа, содержащем NaCl, CaCl2, MgCl2, KCl (50, 120, 2, 1 и 5 мМ соответственно, доведенным до pH 7,7 с помощью HCl) до соответствующей концентрации белка, оптимизированной в отношении специфичного и неспецифичного связывания. Радиоактивный лиганд [3H]-спиперон (NEN, удельная активность ~70 кюри/ммоль) разбавляли в буфере для анализа в концентрации 2 нмоль/л. Затем приготовленный радиоактивный лиганд (50 мкл) вместе с 50 мкл либо контрольного 10% ДМСО, бутакламола (конечная концентрация 10-6 моль/л), либо представляющего интерес соединения инкубировали (30 мин, 37°C) с 400 мкл приготовленного раствора мембран. Связанную с мембранами активность фильтровали через устройство для сбора Packard Filtermate, используя фильтры GF/B Unifilterplates, и промывали ледяным трис-HCl-буфером (50 мМ; pH 7,7; 6×0,5 мл). Фильтрам давали возможность высохнуть перед добавлением сцинтилляционной жидкости и подсчитывали с сцинтилляционном счетчике Topcount. Специфичное связывание в процентах и кривые конкурентного связывания рассчитывали, используя компьютерную программу S-Plus (Insightful).
D. Примеры композиций
«Активный ингредиент» (АИ) в используемом в примерах смысле относится к соединению формулы (I), его фармацевтически приемлемой кислотно-аддитивной соли, его стереохимически изомерной форме или его N-оксидной форме.
Пример D.1: ПЕРОРАЛЬНЫЙ РАСТВОР
Метил-4-гидроксибензоат (9 г) и пропил-4-гидроксибензоат (1 г) растворяли в очищенной кипячением воде (4 л). В 3 л полученного раствора сначала растворяли 2,3-дигидроксибутандионовую кислоту (10 г), а затем АИ (20 г). Последний раствор объединяли с оставшейся частью упомянутого выше раствора и к нему добавляли 1,2,3-пропантриол (12 л) и 70% раствор сорбита (3 л). Сахарин натрия (40 г) растворяли в воде (500 мл) и добавляли малиновую эссенцию (2 мл) и эссенцию крыжовника (2 мл). Последний раствор объединяли с упомянутым выше, добавляли воду q.s. до объема 20 л, получая пероральный раствор, содержащий 5 мг активного ингредиента на чайную ложку (5 мл). Полученным в результате раствором заполняли подходящие емкости.
Пример D.2: ПОКРЫТЫЕ ПЛЕНКОЙ ТАБЛЕТКИ
Получение сердцевины таблетки
Смесь АИ (100 г), лактозы (570 г) и крахмала (200 г) хорошо смешивали и затем смачивали раствором додецилсульфата натрия (5 г) и поливинилпирролидона (10 г) в воде (200 мл). Увлажненную смесь порошка просеивали, сушили и снова просеивали. Затем добавляли микрокристаллическую целлюлозу (100 г) и гидрогенизированное растительное масло (15 г). Все хорошо перемешивали и прессовали таблетки, получая 10000 таблеток, каждая из которых содержит 10 мг активного ингредиента.
Покрытие
К раствору метилцеллюлозы (10 г) в денатурированном этаноле (75 мл) добавляли раствор этилцеллюлозы (5 г) в дихлорметане (150 мл). Затем добавляли дихлорметан (75 мл) и 1,2,3-пропантриол (2,5 мл). Полиэтиленгликоль (10 г) расплавляли и растворяли в дихлорметане (75 мл). Последний раствор добавляли к указанному выше и затем добавляли октадеканоат магния (2,5 г), поливинилпирролидон (5 г) и концентрированную суспензию красителя (30 мл) и все гомогенизировали. Сердцевины таблеток покрывали полученной таким образом смесью в устройстве для нанесения покрытия на таблетки.
Пример D.3: ИНЪЕКЦИОННЫЙ РАСТВОР
Метил-4-гидроксибензоат (1,8 г) и пропил-4-гидроксибензоат (0,2 г) растворяли в кипящей воде (500 мл) для инъекций. После охлаждения примерно до 50°C при перемешивании добавляли молочную кислоту (4 г), пропиленгликоль (0,05 г) и АИ (4 г). Раствор охлаждали до комнатной температуры и добавляли воду для инъекций до достижения объема 1000 мл, получая раствор, содержащий 4 мг/мл АИ. Раствор стерилизовали фильтрованием и полученным раствором наполняли стерильные емкости.
Реферат
Настоящее изобретение относится к новым замещенным тетрациклическим производным тетрагидропирана, пирролидина и тетрагидротиофена общей формулы (I) ! ! их фармацевтически приемлемым аддитивным солям, их стереохимически изомерным формам, их N-оксидным формам, в которых все заместители имеют значения, определенные в п.1 формулы изобретения. Эти соединения обладают аффинностями связывания по отношению к рецепторам серотонина, в частности рецепторам 5-HT2A и 5-HT2C, и по отношению к рецепторам допамина, в частности рецепторам допамина D2, и обладают свойствами ингибирования обратного захвата норэпинефрина. Изобретение также относится к фармацевтической композиции, содержащей эти соединения, способу ее получения и применению указанных соединений в качестве лекарственных средств, в частности для профилактики и/или лечения ряда психиатрических и неврологических расстройств. 5 н. и 7 з.п. ф-лы, 3 табл.
Формула

его N-оксидная форма, фармацевтически приемлемая аддитивная соль или стереохимически изомерная форма, где
i и j означают целые числа, независимо друг от друга равные нулю или 1;
А и В означают бензо;
каждый R9 означает галоген;
Х означает CR6R7 или О; где
R6 и R7 каждый означает водород;
С означает группу формулы (с-1), (с-2), (с-3) или (с-4);




где Y1 означает S; S (=O); S(=O)2 или NR10; где R10 выбирают из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила, диалкиламиноалкила и алкилоксиалкилкарбонила;
Y2 означает Y1 или О;
R10 и R11 могут вместе образовывать двухвалентный радикал (е-1), (е-2) или (е-3);



каждый двухвалентный радикал (е-1), (е-2) и (е-3) необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы и алкила;
R12 означает водород;
R13 означает водород;
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1);
R11 означает группу формулы (d-1);

где n равен нулю или 1;
R1 и R2 каждый независимо означают водород; алкил, необязательно замещенный гидроксигруппой; алкилкарбонил; алкилоксиалкил; алкилкарбонилоксиалкил; алкилоксикарбонилалкил; или
R1 и R2 взятые вместе с атомом азота, с которым они связаны, могут образовывать радикал формулы (а-3), (а-5) или (а-6);



где р равен нулю или 1;
q равно 1;
m равно 1;
каждый R3 независимо означает гидроксигруппу;
R4 выбирают из группы, состоящей из водорода или алкила, необязательно замещенного гидроксигруппой;
алкил означает насыщенный углеводородный радикал с неразветвленной или разветвленной цепью, имеющий от 1 до 10 атомов углерода; и галоген означает фтор, хлор, бром и йод;
за исключением соединений формулы (I), где Х=СН2, i=0, j=1, R9=F, С означает группу формулы (с-4), где Y=O, R13=H, и R14 означает оксогруппу, гидроксигруппу или группу формулы (d-1), где n=0, R1= водород и R2= метил.
Y1 означает S; S (=O); S(=O)2 или NR10; где R10 выбран из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила и алкилоксиалкилкарбонила;
соседние R10 и R11 вместе могут образовывать двухвалентный радикал (е-1), (е-2) или (е-3); при этом каждый радикал необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы и алкила; и R12 означает водород.
Y2 означает О;
R12 означает водород;
R13 означает водород; и
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1).
n равно нулю или 1;
R1 и R2 каждый независимо означают водород; алкил или алкилоксикарбонилалкил; или R1 и R2 вместе взятые с атомом азота, с которым они связаны, могут образовывать радикал формулы (а-3), (а-5) или (а-6); где
р равно нулю или 1;
q равно 1;
m равно 1;
каждый R3 независимо выбран из группы, состоящей из водорода и гидроксигруппы; и R4 означает алкил.
i и j являются целыми числами, независимо друг от друга равными нулю или 1;
А и В каждый независимо друг от друга означают бензогруппу, необязательно замещенную фтором;
каждый R9 независимо друг от друга выбран из группы, состоящей из водорода и галогена;
Х означает СН2 и О;
С означает группу формулы (с-1), (с-2), (с-3) или (с-4); где
Y1 означает S; S (=O); S(=O)2 или NR10; где R10 выбран из группы, состоящей из водорода, цианогруппы, алкила, алкилоксиалкила, формила, алкилкарбонила, алкилоксикарбонила и алкилоксиалкилкарбонила;
Y2 означает О;
соседние R10 и R11 вместе могут образовывать двухвалентный радикал (е-1), (е-2) или (е-3); при этом каждый радикал необязательно замещен одним или несколькими заместителями, выбранными из оксогруппы, тиоксогруппы и алкила;
R12 означает водород;
R13 означает водород;
R14 означает водород, гидроксигруппу, оксогруппу или группу формулы (d-1);
R11 означает группу формулы (d-1); где
n равно нулю или 1;
R1 и R2 каждый независимо означают водород; алкил или алкилоксикарбонилалкил; или R1 и R2 вместе взятые с атомом азота, с которым они связаны, могут образовывать радикал формулы (а-3), (а-5) или (а-6); где
р равно нулю или 1;
q равно 1;
m равно 1;
R3 означает гидроксигруппу; и
R4 означает алкил.
(4aS,13bR,14aS)-6-фтор-2-метил-1,2,3,4a,9,13b,14,14a-октагидродибензо[3',4':6',7]циклогепта[1',2':4,5]пирроло[1,2-с]имидазол;
[(2S,3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
(4aS,13bR,14aS)-6-фтор-1,4а,9,13b,14,14а-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-тион;
(5aS,14bR,15aS)-7-фтор-2-метил-1,2,3,5а,10,14b,15,15а-октагидро-4Н-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-а]пиразин-4-он;
[(2R,3aR,12bS)-11-фтор-1-метил-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
(4aS,13bR,14aR)-6-фтор-1,4а,9,13b,14,14а-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-он;
(4aS,13bR,14aR)-6-фтор-2-метил-1,4а,9,13b,14,14а-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-он;
(4aS,13bR,14aR)-6-фтор-1,4а,9,13b,14,14а-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-тион;
[(2R,3aR,12bS)-11-фтор-1-(метоксиацетил)-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
метил({[(2R,3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}амино)ацетат;
(5aS,14bR,15aR)-7-фтор-1,2,3,5а,10,14b,15,15а-октагидро-4Н-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-а]пиразин-4-он;
(5aS,14bR,15aR)-7-фтор-2-метил-1,2,3,5a,10,14b,15,15а-октагидро-4Н-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-а]пиразин-4-он;
(5aS,14bR,15aR)-7-фтор-2-метил-1,3,4,5а,10,14b,15,15а-октагидро-2Н-дибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-а]пиразин;
(4aS,13bR,14aS)-6-фтор-1,4а,9,13b,14,14а-гексагидродибензо-[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-он;
(4aS,13bR,14aS)-6-фтор-2-метил-1,4а,9,13b,14,14а-гексагидродибензо[3',4':6',7']циклогепта[1',2':4,5]пирроло[1,2-с]имидазол-3(2Н)-он;
[(2R,3aR,12bS)-11-фтор-2,3,3а,12b-тетрагидро-1Н-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]-N,N-диметилметанамин;
2-[{[(2R,3aR,12bS)-11-фтор-2,3,3а,12b-тетрагидро-1Н-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}(метил)амино]этанол;
2-(4-{[(2R,3aR,12bS)-11-фтор-2,3,3а,12b-тетрагидро-1Н-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}-1-пиперазинил)этанол;
1-{[(2R,3aR,12bS)-11-фтор-2,3,3а,12b-тетрагидро-1Н-дибензо[2,3:6,7]оксепино[4,5-b]пиррол-2-ил]метил}-3-пирролидинол;
[(2S,3aR,12bS)-11-фтор-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2S,3aR,12bS)-11-фтор-1,1-диоксидо-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2R,3aR,12bS)-11-фтор-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2R,3aR,12bS)-11-фтор-1,1-диоксидо-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2S,3aR,12bS)-11-фтор-1-оксидо-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2S,3aR,12bS)-11-фтор-1-оксидо-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2R,3aR,12bS)-11-фтор-1-оксидо-3,3а,8,12b-тетрагидро-2H-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
[(2R,3aR,12bS)-11-фтор-1-оксидо-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]тиен-2-ил]-N,N-диметилметанамин;
(3R,4aR,13bR)-12-фтор-2,3,4,4а,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-ол;
(3S,4aR,13bR)-12-фтор-2,3,4,4а,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (соединение 30);
(4aR,13bR)-12-фтор-4,4а,9,13b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3(2Н)-он;
(3S,4aR,13bR)-12-фтор-N,N-диметил-2,3,4,4а,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин;
(3R,4aR,13bR)-12-фтор-N-метил-2,3,4,4а,9,13b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиран-3-амин (соединение 33);
метил(2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3а,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2Н)-карбоксилат;
[(2R,3aR,12bS)-1-ацетил-11-фтор-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
(2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3а,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2Н)-карбальдегид;
[(2R,3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
(2R,3aR,12bS)-11-фтор-2-[(метиламино)метил]-3,3а,8,12b-тетрагидродибензо[3,4:6,7]-циклогепта[1,2-b]пиррол-1(2Н)-карбальдегод;
2-((2R,3aR,12bS)-2-[(диметиламино)метил]-11-фтор-3,3a,8,12b-тетрагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-1(2Н)-ил)этанол;
[(2R,3aR,12bS)-11-фтор-1-(2-метоксиэтил)-1,2,3,3a,8,12b-гексагидродибензо[3,4:6,7]-циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
[(2R,3aR,12bS)-1-суано-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]-N,N-диметилметанамин;
(2R,3aR,12bS)-11-фтор-2-(4-морфолинилметил)-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол;
2-(4-{[(2R,3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}-1-пиперазинил)этанол;
2-[{[(2R,3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-2-ил]метил}(метил)амино]-этанол;
[(2R,4aR,13bS)-12-фтор-2,3,4,4а,9,13b-гексагидро-1Н-дибензо[3,4:6,7]циклогепта[1,2-b]пиридин-2-ил]-N,N-диметилметанамин;
метил(2R,4aR,13bS)-2-[(диметиламино)метил]-12-фтор-2,3,4,4а,9,13b-гексагидро-1Н-дибензо[3,4:6,7]циклогепта[1,2-b]пиридине-1-карбоксилат;
[(2R,4aR,13bS)-12-фтор-2,3,4,4а,9,13b-гексагидродибензо[3,4:6,7]циклогепта-[1,2-b]пиран-2-ил]-N-метилметанамин;
[(3aR,12bS)-11-фтор-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-3-ил]-N,N-диметилметанамин;
[(3aR,12bR)-11-фтор-3,3а,8,12b-тетрагидро-2Н-дибензо[3,4:6,7]циклогепта[1,2-b]фуран-3-ил]-N,N-диметилметанамин;
[(3aR,12bS)-11-фтор-1,2,3,3а,8,12b-гексагидродибензо[3,4:6,7]циклогепта[1,2-b]пиррол-3-ил]-N,N-диметилметанамин.
Документы, цитированные в отчёте о поиске
Конденсированное индольное производное или его фармацевтически приемлемая соль, фармацевтическая композиция, проявляющая активность антагониста 5-нт*004-рецептора
Комментарии