Производные n-замещенных 3-азабицикло(3.2.0) гептанов - RU2160254C2
Код документа: RU2160254C2
Описание
Изобретение относится к новым производным азабициклоалканов, обладающих биологической активностью, в частности к производным N- замещенных 3-азабицикло[3.2.0]гептанов.
Известны производные N-замещенных азабициклогептанов, обладающих антипсихотической активностью (см. заявку DE N 42 19 973, кл. С 07 D 209/52, A 61 К 31/40, 23.12.1993 г.).
Задачей изобретения является расширение арсенала производных N-замещенных 3-азабицикло[3.2.0]гептанов, обладающих антипсихотической активностью.
Поставленная задача решается предлагаемыми производными N- замещенных 3-азабицикло[3.2.0]гептанов формулы (I)

где R означает нафтильную или фенантрильную группу, которые могут моно- или дизамещены атомами галоида,
A означает остаток формулы

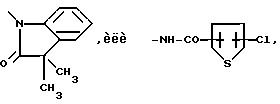
или нафтильную группу, которая может быть замещена галоидом, или их солями с физиологически приемлемыми кислотами.
Соединения формулы I можно получать тем, что соединения формулы (II)
Nu-(CH2)2-A,
в которой А имеет указанные выше значения и Nu является нуклеофильной отходящей группой, подвергают взаимодействию с производным 3-азабицикло- [3.2.0]гептана формулы (III)

где R означает нафтильную или фенантрильную группу, которые могут быть моно- или дизамещены галоидом,
и полученное соединение при необходимости переводят в соль обработкой физиологически приемлемой кислотой.
Нуклеофильной отходящей группой Nu являются предпочтительно атомы галоида, в частности бром или хлор.
Взаимодействие целесообразно проводить в присутствии инертного основания, такого как триэтиламин или карбонат калия, в качестве связывающего кислоту средства, и в инертном растворителе, таком как циклический насыщенный простой эфир, в частности тетрагидрофуран или диоксан, или углеводород бензольного ряда, такой как толуол или ксилол.
Взаимодействие происходит, как правило, при температуре от 20 до 150oC, в частности от 80 до 140oC, и заканчивается обычно в течение 1-10 ч.
Соединения формулы (1) согласно изобретению очищают или перекристаллизацией из обычных органических растворителей, предпочтительно низших спиртов, таких как этанол, или колоночной хроматографией.
Рацематы разделяют на энантиомеры простым способом классического расщепления с оптически активными карбоновыми кислотами, например производными винной кислоты, в инертном растворителе, например низших спиртах.
Свободные производные 3-азабицикло [3.2.0.] гептана формулы (I) могут быть переведены обычным образом в соль присоединения фармакологически переносимой кислоты, предпочтительно смешиванием раствора с эквивалентом соответствующей кислоты. Фармацевтически переносимыми кислотами являются, например, соляная кислота, фосфорная кислота, серная кислота, метан-сульфокислота, амидосульфокислота, малеиновая кислота, фумаровая кислота, щавелевая кислота, винная кислота или лимонная кислота.
Используемые для синтеза новых соединений соединения формулы (II) известны.
Соединения формулы (III) получают тем, что амин формулы (IV),

где R имеет указанные выше значения и R1 означает водород, ацетил, бензил или трифторацетил, подвергают фотохимическому [2+2] циклоприсоединению и при необходимости отщепляют ацильную или бензильную группу.
Фотореакция хорошо удается в инертном растворителе, предпочтительно ацетоне, при температуре от 20 до 80oC. В качестве источника света хорошо подходит ртутная лампа высокого давления. Иногда является предпочтительным проводить фотоциклизацию в кварцевой аппаратуре в атмосфере азота с добавкой около одного моля соляной кислоты на моль амина.
Фотоциклизация протекает в большинстве случаев с высокой степенью диастереоселективности к бициклическим соединениям (III) с экзоконфигурацией
относительно R:

Расщеплением рацемата, например, с оптически активными производными винной кислоты получают отдельные энантиомеры.
Ацильную группу R1 целесообразно отщеплять известными способами. Это же является действительным для удаления бензильной группы.
Амины формулы (IV) известны из литературы или получаются тем, что альдегид R-CHO подвергают взаимодействию с винилмагнийхлоридом до получения аллилового спирта формулы (V)

затем подвергают перегруппировке с хлористым водородом до аллилового хлорида формулы (VI)

и затем подвергают взаимодействию с соответствующим аллиламином формулы (VII),

или же коричный альдегид формулы (VIII)

непосредственно подвергают восстановительному аминированию с аллиловым амином формулы (VII).
Благодаря высокому сродству к субтипу D4 -допаминового рецептора соединения вышеприведенной формулы (I) проявляют антипсихотическую активность и поэтому могут применяться в качестве нейролептических средств (в частности, нетипичных), антидепрессантов, успокаивающих средств, снотворных, защищающих центральную нервную систему средств или мышечных релаксантов.
Сродство соединений вышеприведенной формулы (I) к субтипу 4,2 D4-допаминового рецептора иллюстрируется следующим опытом.
Опыт по определению сродства к субтипу 4.2 D4-допаминового рецептора
Лизированные клетки
(Cos-клетки, проявляющие стабильную экспрессию клонированного человеческого D4.2-рецептора) повторно суспендировали в инкубационном буфере (50 ммоль Трис-HCl, pH 7,4), содержащим 5 ммоль
ЭДТУК, 1,5 ммоль хлористого калия, 120 нмоль хлористого натрия и 5 ммоль хлористого магния. При этом концентрация клеток составляла 105/опыт. Инкубацию проводили при 25oC в
присутствии или отсутствии исследуемого соединения с применением 50 пмоль [125J] спиперона. Неспецифичное связывание определяли с помощью галоперидола, взятого в концентрации 10-6
моль. После 60-минутной инкубации связанную и свободную радиоактивность разделяли путем фильтрации с помощью стекловолокнистого фильтра GF/B фирмы Вуотманн, GB, с применением прибора Скатрон для
сбора клеток. Фильтр промывали холодным буфером Трис-HCl, pH 7,0 (20 ммоль Трис, 20 ммоль хлористого магния, 7% полиэтиленгликоля с молярной массой 7000). Связанную радиоактивность определяли
жидкостной сцинтилляцией с применением прибора Пакард 2200 CA. Значения Кi(в нмоль/л) исследуемых соединений определяли нелинейным регрессионным анализом с помощью программы Лаганд.
Результаты опыта:
Средство к субтипу 4.2D4-допаминового рецептора
Соединение примера N - Ki (нмоль/л)
1 - 6,7
2 - 4,5
3 - 19
4 - 1,8
6 - 5,1
7 - 4,3
8 - 4,4
10 - 31
Соединения согласно изобретению относятся к категории малотоксичных веществ.
Соединения согласно изобретению могут вводиться обычным образом орально, парентерально, внутривенно или внутримышечно.
Дозировка зависит от возраста, состояния и веса пациента, а также от вида введения. Как правило, дневная доза активного вещества составляет от 1 до 100 мг/кг веса при оральном введении и от 0,1 до 10 мг/кг веса при парентеральном введении.
Соединения согласно изобретению могут использоваться в виде обычных твердых или жидких фармацевтических форм, например в виде таблеток, таблеток, покрытых оболочкой, капсул, порошка, гранул, драже, свечей, растворов, мазей, кремов или аэрозолей. Эти формы изготавливают обычным способом. Активные вещества перерабатывают при этом с обычными, применяемыми в фармацевтике вспомогательными средствами, такими как наполнители, консерванты, разрыхлители, средствами для регулирования текучести, пластификаторами, смачивающими средствами, диспергаторами, эмульгаторами, растворителями, замедляющими выделение активного вещества средствами, антиоксидантами и/или рабочими газами (см. Н. Sucker и др., Pharmazeutische Technologie, изд. Tieme, Штутгарт, 1978).
Полученные таким образом формы содержат обычно активное вещество в количестве от 1 до 99 вес.%.
Следующие примеры служат для пояснения изобретения.
А. Получение исходных соединений
aa)
1-(1-нафтил)-аллиловый спирт
В двухлитровую колбу заливают в атмосфере азота 277 мл (360 мМ) 1,3 М раствора винилмагнийхлорида в тетрагидрофуране. Затем добавляют при перемешивании в
атмосфере азота 50 г (320 мМ) 1-нафтальдегида, растворенного в 250 мл тетрагидрофурана, в течение 60 мин при 30-35oC. Реакционную смесь перемешивают еще 4,5 ч в атмосфере азота при
комнатной температуре. Затем при перемешивании и охлаждении льдом прибавляют 90 мл насыщенного раствора хлорида аммония, отсасывают и остаток на фильтре трижды промывают с помощью 150 мл
тетрагидрофурана. Фильтраты объединяют, сушат над сульфатом натрия и сгущают. Получают 58,3 г (99%) сырого продукта в виде коричневого масла;
аб) 3-(1-нафтил)-аллилхлорид 58,3 г (317 мМ)
1-(1-нафтил)- аллилового спирта растворяют при перемешивании в 400 мл дихлорметана. После этого пропускают до насыщения хлористый водород, причем температура повышается до 37oC. Затем
перемешивают один час. После промывки с помощью 200 мл ледяной воды сушат органическую фазу над сульфатом натрия и сгущают. Получают 59,2 г (92%) коричневатого твердого вещества;
ав)
N-аллил-N-[3-(1-нафтил)-аллил]-амин
К 167 г (2,9 М) аллиламина прибавляют 59,2 г (0,29 М)3-(1-нафтил)-аллилхлорида, растворенного в 250 мл толуола, при нагревании с обратным холодильником в
течение одного часа. Смесь продолжают перемешивать два часа при температуре обратного потока. После этого реакционную смесь сгущают, остаток поглощают 250 мл воды и устанавливают значение pH 12 с
помощью 50%-ной натриевой щелочи. Водную фазу экстрагируют дихлорметаном, органическую фазу сушат над сульфатом натрия и сгущают.
Выход: 67,6 г (97%) темно-коричневого масла;
аг) экзо-6-(1-нафтил)-3-азабицикло[3.2.0]гептан
50,0 г (193 мМ) N-аллил-N-[3-(1-нафтил)-аллил]аммонийхлорида растворяют в 1600 мл ацетона и смешивают с 210 мл 10%-ной соляной кислоты.
Прозрачный желтый раствор облучают в атмосфере азота ртутной лампой высокого давления мощностью 700 Вт в кварцевой аппаратуре в течение четырех часов при комнатной температуре. После этого реакционный
раствор сгущают, остаток поглощают водой и устанавливают значение рН 12 с помощью 50%-ной натриевой щелочи. Перемешивают 30 мин и дважды экстрагируют трет.-бутил-метиловым эфиром. Объединенные
органические фазы сушат над сульфатом натрия и сгущают.
Темно-коричневый маслянистый остаток (43,2 г) растворяют в 150 мл изопропанола и смешивают с 25,5 г (220 мМ) малеиновой кислоты, растворенной в 220 мл изопропанола. Выпавший малеинат отсасывают, промывают изопропанолом и сушат в вакуумном сушильном шкафу при 40oC в течение ночи.
Выход: 43,9 г (67%) бесцветного порошка с т.пл. 162-164oC (малеинат).
Аналогичным способом получают следующие соединения:
ад) экзо-6-(2-нафтил)-3-азабицикло[3.2.0]гептан,
т.пл.: 145-147oC (малеинат)
ае) экзо-6-(6-хлор-2-нафтил)-3-азабицикло[3.2.0]гептан,
т.пл.: 164-165oC.
Б. Получение целевых соединений
Пример 1
М-[2-(экзо-6-(1-нафтил)-3-азабицикло[3.2.0]гептан-3-ил)- этил]-бензамид
4,0 г (17,8 мМ) экзо-6-(1-нафтил)-3-азабицикло[3.2.0]гептана в 70 мл толуола смешивают с 6,6 г (35,2
мМ) N-(2-хлорэтил)- бензамида, а также с 2,5 г (18,1 мМ) измельченного в тонкий порошок карбоната калия и 0.5 г иодида калия и кипятят с обратным холодильником при хорошем перемешивании в течение 6 ч.
После охлаждения сгущают на ротационном испарителе и остаток распределяют между метиленхлоридом и водой. Водную фазу дважды экстрагируют метиленхлоридом и сгущают органическую фазу после сушки
сульфатом натрия. Сырой продукт (8,9 г) очищают колоночной хроматографией (силикагель, подвижная фаза дихлорметан/метанол в соотношении 96:4). Свободное основание (3,0 г) растворяют в 100 мл
трет.-бутил-метилового эфира и смешивают при охлаждении льдом с избыточным количеством эфирного раствора соляной кислоты. Выпавший гидрохлорид отсасывают в атмосфере азота, промывают большим
количеством трет.-бутил-метилового эфира и сушат на нутче в потоке азота. Получают 2,6 г продукта в виде гидрохлорида с т.пл. 184-186oC (выход 35%).
Аналогично получают
следующие соединения:
2. N-12-(экзо-6-(2-нафтил)-3-азабицикло[3.2.0]гептан-3- ил)этил]-бензамид, т.пл. 233-235oC (гидрохлорид),
3.
3-[2-(1-нафтил)этил]-экзо-6-(1-нафтил)-3- азабицикло[3.2.0]гептан, т. пл. 227-229oC (гидрохлорид),
4. 3-[2-(1-нафтил)этил]-экзо-6-(2-нафтил)-3- азабицикло[3.2.0]гептан, т. пл.
208-210oC (гидрохлорид),
5. 1-[2-(экзо-6-(1-нафтил)-3-азабицикло[3.2.0]гептан-3- ил)этил]-1H-бенэо-[cd]индол-2-он, т.пл. 174-176oC (гидрохлорид),
6.
1-[2-(экзо-6-(2-нафтил)-3-азабицикло[3.2.0]гептан-3- ил)этил]-1H-бензо-[cd]индол-2-он, т.пл. 258-260oC (гидрохлорид),
7. 3,3-диметил-1-[2-(экзо-6-(2-нафтил)-3- азабицикло[3.2.0]
гептан-3-ил)этил]-1,3-дигидроиндол-2-он, т.пл. 124-125oC,
8. амид 5-хлор-М-[2-(экзо-6-(1-нафтил)-3- азабицикло[3.2.0]гептан-3-ил)-этил]-2-тиофенкарбоновой кислоты, т.пл.
160-162oC,
9. N-[2-(экзо-6-(6-хлор-2-нафтил)-3-азабицикло[3.2.0]гептан- 3-ил)-этил] -бензамид, т.пл. 102-104oC,
10.
N-[2-(экзо-6-(9-фенантрил)-3-азабицикло[3.2.0]гептан-3-ил)-этил]-бензамид, т.пл. 110-112oC (гидрохлорид).
Реферат
Описываются новые производные N-замещенных 3-азабицикло[3.2.0]гептанов формулы I, где R означает нафтильную или фенантрильную группу, которые могут моно- или дизамещены атомами галоида, А означает остаток формул или нафтильную группу, которая может быть замещена галоидом, или их соли с физиологически приемлемыми кислотами, которые могут быть использованы в качестве нейролептических средств. 1 з.п.ф-лы.


Формула

где R означает нафтильную или фенантрильную группу, которые могут моно- или дизамещены атомами галоида,
А означает остаток формулы


или нафтильную группу, которая может быть замещена галоидом, или их соли с физиологически приемлемыми кислотами.
Комментарии