Нитроксисоединения и фармацевтическая композиция на их основе, имеющие противовоспалительную, анальгетическую и антитромбоцитарную активности - RU2145595C1
Код документа: RU2145595C1
Чертежи



Описание
Настоящее изобретение относится к новым продуктам, имеющим противовоспалительную, анальгетическую и антитромбоцитную активности.
В частности, это относится к ингибиторам циклооксигеназы (COX).
Известно, что противовоспалительная и антитромбоцитная активность, но главным образом толерантность NSAID (нестероидных противовоспалительных лекарственных средств), также известных как FANS, по-видимому значительно определяется их циклооксигеназной (COX)-ингибирующей активностью в воспаленном участке так же хорошо, как в здоровой ткани. Смотри, например, FACEB Journal 1, 89, 1987; Bioch. Biophys. Acta 1083, 1, 1991. В основном предполагается, что наиболее сильнодействующий COX-ингибитор является наиболее эффективным.
Недостатком этих продуктов является их токсичность.
Кроме того, также известно, что COX-ингибирующие свойства определяются несколькими факторами, связанными с физико- химическими и структурными характеристиками их молекул, таких, как, например, кислотная функция. Смотри, например, J. Pharmacol. Exp. Therap. 196, 226, 1976; Arch. Toxicol. 60, 261, 1987.
Известные ингибиторы циклооксигеназы - в основном
кислоты, которые могут быть сведены к общим структурам, включающим:
- карбоновые кислоты, или их ацетилированные производные, например, аспирин или трифлюсал, или неацетилированные, такие как,
например, салицилат, дифлюнисал, салсалат;
- уксусные кислоты, например, диклофенак, индометацин, толметин, сулиндак, этодолак, кеторолак:
- пропионовые кислоты, такие как, например,
ибупрофен, напроксен, пирпрофен, тиапрофеновая кислота, локсопрофен, индопрофен, оксапрозин, кетопрофен, фенопрофен, фенбуфен, фторбипрофен, карпрофен, супрофен:
- энольные кислоты, такие
как, например, оксифенбутазон, фенилбутазон, пироксикам, судоксикам, теноксикам, изоксикам, мелоксикам.
Смотри патенты USP 3558690; USP 3755427; USP 3641127; FR 2112111; USP 4035376; USP 3997669; USP 3784701; USP 3896145; USP 3600437; USP 3843681; USP 3904682; USP 3228831; USP 4161538; USP 4233299; USP 3591584; DE 2537070; USP 3161654; USP 4061779; USP 4556672; USP 4089969.
Недостаток этих продуктов в том, что они очень эффективны, но высокотоксичны.
Важность кислотной функции доказывается тем фактом, что маскировка этой функции в COX-ингибиторах приводит фактически к полной потере их простаноид-ингибирующих свойств. Смотри Drugs, 35, 504, 1988.
Также известны продукты, которые высокоэффективны в ингибировании циклооксигеназы и даже имеют низкую токсичность, хотя они не содержат кислотной функции в своих молекулах.
Эти продукты известны как эфиры азотной кислоты с некислотными концами. Смотри, например, патенты PCT WO 94/04484, который описывает определенную группу соединений, включающую хорошо известный коммерческий продукт диклофенак; PCT/EP 93/03193, который описывает другую специфическую группу соединений, включающую коммерческие продукты фторбипрофен и индопрофен.
Заявитель неожиданно обнаружил, что другие соединения, имеющие концевую группу -NO2, когда X1=-YO-, как показано здесь ниже, имеют противовоспалительную, анальгетическую и антитромботическую активности при использовании в качестве лекарственных средств с высокой эффективностью в ингибировании циклооксигеназы и имеют низкую токсичность.
Следующей целью изобретения является то, что известные продукты, как описано в PCT WO 94/04484 и PCT/EP 93/03193, и новые соединения, найденные заявителем, имеющие X1=-YO-, имеют фармакологические недостатки. Фактически, в биохимических тестах, оценивающих ингибирующую активность циклооксигеназы, эксперименты, проведенные заявителем, показали высокую реакционную вариабельность, порядка 10-40%.
В большинстве случаев эти результаты случайного и непредсказуемого действия, и таким образом, определение точной дозы затруднено.
На практике, чтобы ограничить вышеуказанную вариабельность, должны быть применены более высокие дозы. Недостатком этого является повышение риска побочных эффектов.
Другой недостаток этих продуктов заключается в трудности рецептуры с той точки зрения, что оральные или парентеральные препараты более трудны для приготовления, чем традиционные препараты, основанные на кислотных FANS.
Молекулярная растворимость, как известно, является одним из наиболее важных свойств, определяющих молекулярную фармокинетику и динамику процессов.
Например, для парентерального введения, особенно внутривенно, лекарства должны быть представлены в растворимой форме.
Аналогично, при оральном введении процесс растворения является решающим для всасывания и взаимодействия с эффектором.
В этом отношении выбор особых растворителей и/или наполнителей, включая сурфактанты, также является решающим с точки зрения токсикологии. Например, внутривенное применение не должно вызывать гемолизис или несовместимость с компонентами крови.
Между тем, имеется большое количество фактов, которые показывают, что сурфактанты и неполярные растворители могут обладать раздражающими свойствами. Смотри, например, J. Pharm. Science 72, 1014, 1983.
Испытания, проведенные заявителем, с использованием 0.1% Твин 80 и 1%диметилсульфоксида с суспендированным нитроксибутилфторбипрофеном, показали, что этот растворитель раздражает слизистую желудка.
Однако, было неожиданно найдено, что при использовании NO-фторбипрофена производного, как описано ниже, который является частью объекта настоящего изобретения, количества Твин 80 и диметилсульфоксида, необходимого для приготовления суспензии, были меньше, благодаря чему не возникло раздражающего эффекта, хотя растворимость при этом оказалась аналогичной.
Было неожиданно и непредсказуемо обнаружено после множества исследований, что возможно получить противовоспалительные продукты, как описано ниже, имеющие высокую ингибирующую циклооксигеназную активность в сочетании с низкой токсичностью и удовлетворительной фармакокинетикой и имеющие очень ограниченную вариабельность ответных реакций со средним коэффициентом около половины того, что у известных продуктов, и более легких в приготовлении как для приема внутрь, так и для парентерального введения.
Это было неожиданно и непредсказуемо также в отношении факторов, которые определяют противовоспалительное и антитромботическое действие NSAIDS, зависимое от разных параметров. Следовательно, невозможно предсказать фармакокинетику, например, продуктов фракционного всасывания, фармакодинамическую активность, токсичность и COX-ингибиторные свойства, и более всего, нельзя прогнозировать или ограничивать вариабельность результата.
Объектом настоящего
изобретения являются соединения или их композиции общей формулы:
A - X1 - NO2
или их соли, для использования в качестве лекарственных средств, в частности
противовоспалительных и антитромбоцитарных агентов, где:
A=R(COXu)t, где t означает ноль или 1; u означает ноль или 1:
X=О, NH, NR1C, где R1C означает линейный или разветвленный алкил, имеющий от 1 до 10 углеродных атомов; R выбирают из следующих групп:
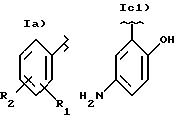
- группа I), где t = 1 и u = 1

где R1 означает OCOR3 группу, где R3 означает метил, этил или линейный или разветвленный C3-C5 алкил, или остаток гетероцикла с одним кольцом, имеющим 5 или 6 атомов, которое может быть ароматическим, частично или полностью гидрированным, содержащим один или более гетероатомов, независимо выбранных из О, N и S;
R2 означает водород, гидрокси, галоген, линейный или, когда возможно, разветвленный алкил, имеющий от 1 до 4 C атомов, линейный или, когда возможно разветвленный алкоксил, имеющий от 1 до 4 углеродных атомов, линейный или, когда возможно, разветвленный перфторалкил, имеющий от 1 до 4 C атомов, например, трифторметил, нитро, амино, моно- или ди-(C1-4)алкиламино:
R1 и R2 вместе означают диоксиметиленовую группу, при условии, что когда X=NH, тогда X1 означает этилен и R2=H; R1 не может быть OCOR3 в положении 2, когда R3 означает метил: nI является 0 или 1.
Предпочтительно, в Ia) X равен O или -NH, R1
означает ацетокси, предпочтительно в орто-положении по отношению к -CO-, X1 означает (CH2-CH2-O)2, R2 - водород, наиболее предпочтительны
следующие A-X1-NO2 соединения: 3-ацетокси-N-(2-нитроксиэтил)-бензамид, 4- ацетокси-N-(2-нитроксиэтил)-бензамид, 3-ацетокси-N-(2- нитроксипентил)-бензамид;
2-ацетокси-н-(5-нитроксипентил)- бензамид, N-2-(5-нитроксиэтил)-2-пропионокси-бензамид, 2-ацетокси- 2-нитрокси-этилбензоат, 2-ацетокси-N-(цис-2-нитроксициклогексил)- бензамид,
2-ацетокси-4-хлор-N-(2-нитроксиэтил)-бензамид, N-(2- нитроксиэтил)-бензамид, N-(2-нитроксиэтил)-((4- тиазолилдинил)карбонилокси)-бензамид гидрохлорид, 2- никотиноилокси-N-(2-нитроксиэтил)-бензамид,
2-ацетокси-5- нитропентилбензоат;
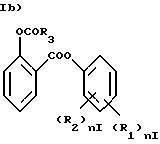
предпочтительно, в Ib) R3=CH3, nI=0;
X равен 0, X1 означает этилен: в этом случае Ib) означает остаток
ацетилсалициловой кислоты;
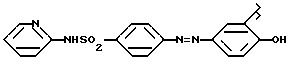
Соединения Ic) класса Ic1) производные 5-аминосалициловой кислоты (5-амино-2-гидроксибензойная кислота), известны как месаламин, когда валентность
насыщена -COOH.
В соединениях Ic2) по крайней мере одна -COOH реагирует с образованием соединений изобретения. Когда реагируют обе -COOH; получают бифункциональные соединения. Когда соединение насыщено -COOH, оно известно как олсалазин.
Соединение Ic3) известно, когда исходный радикал имеет -COOH, как сульфалазин: 2-гидрокси-5-[(4-[(2- пиридиниламино)сульфонил]фенил)азо]бензойная кислота.
Предпочтительные соединения Ic) имеют X=О и u=1 и x1 отличается от -YO-,
- группа II)
где t = 1, u = 1

где RII5 означает водород, линейный или разветвленный C1-C3алкил, когда возможно, RII6 имеет те же значения, как RII5, или, когда RII5 означает водород, он может быть бензилом;
RII1, RII2 и RII3 независимо друг от друга означают водород, линейный или, когда возможно, разветвленный C1-C6 алкил, или C1-C6 алкокси, или Cl, F, Br;
RII4 означает RII1 или бром;
предпочтительны соединения, где RII1, RII2 и RII4 означают водород и RII3 означает хлор, и RII3 находится в орто-положении по отношению к NH;
RII5 и RII6 означают H, X равен O и X1 означает (CH2-CH2 -O)2;
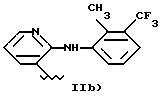
IIb) означает остаток 2-[(2-метил-3-(трифторметил)фенил)амино]- 3-пиридинкарбоновой кислоты, и когда -COOH присутствует, известно как флюниксин.
Предпочтительны соединения, в которых u=1 и X=О;
- группа III), где t = 1, u = 1 и R означает:

где: R2a и R3a означают H, линейный или, когда возможно, разветвленный, замещенный или незамещенный C1-C12 алкил, аллил, при условии, что когда одна из двух групп означает аллил, другая означает водород; предпочтительно R2a означает H, алкил, имеющий от 1 до 4 C, R3a означает H;
R1a выбирают из

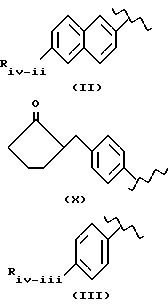
III D) имеет следующие соединения:
где значения следующие:
- в соединении формулы (IV) остаток Кетопрофена;
RIII1 означает H, SRIII3, где RIII3 содержит от 1 до 4 C атомов, линейные или, когда возможно, разветвленные;
RIII2 означает H, гидрокси;
предпочтительные соединения, где RIII1 и RIII2 означают H, R3a означает H и R2a означает метил, X=О;
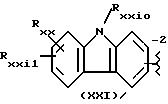
в соединениях формулы (XXI), остаток карпрофена:
Rxxi0 означает H, линейный или, когда возможно, разветвленный алкил, имеющий от 1 до 6 C атомов, C1-C6 алкоксикарбонил, связанный с C1-C6 алкилом, C1-C6 карбоксилалкил, C1-C6 алканоил, необязательно замещенный галогенами, бензил или галоидбензил, бензоил или галоидбензоил;
Rxxi означает H, галоген, гидрокси, CN, C1-C6 алкил, необязательно содержащий OH группы, C1-C6 алкокси, ацетил, бензилокси, SRxxi2, где Rxxi2 означает алкил C1-C6; перфторалкил, имеющий от 1 до 3 C атомов, C1-C6 карбоксиалкил, необязательно содержащий OH группы, NO2, амино, сульфамоил, диалкилсульфамоил с алкилом, имеющим от 1 до 6 C атомов, или дифторалкилсульфонил с алкилом, имеющим от 1 до 3 C атомов:
Rxxi1 означает галоген, CN, C1-C6 алкил, содержащий одну или более OH группы, C1-C6 алкокси, ацетил, ацетамид, бензилокси, SRIII3 как показано выше, перфторалкил, имеющий от 1 до 3 C, гидрокси, карбоксиалкил, имеющий от 1 до 3 C, гидрокси, карбоксиалкил, имеющий от 1 до 6 C, NO2, амино, моно- или диалкиламино, имеющий от 1 до 6 C, сульфамоил, диалкил сульфамоил, имеющий от 1 до 6 C, или дифторалкилсульфамоил, как показано выше; или Rxxi вместе с Rxxi1 означают алкилендиокси, имеющий от 1 до 6 C;
предпочтительные соединения, где Rxxi0 означает H, связывающий мостик находится в положении 2, Rxxi означает H, Rxxi1 означает хлор в параположении по отношению к азоту;
R3a означает H, R2a означает метил и X означает O;
- в соединениях формулы (XXXV), остаток тиапрофеновой кислоты:
Ar означает фенил, гидроксифенил, необязательно моно- или полизамещенный галогеном, алканоил и алкокси, имеющий от 1 до 6 C, триалкил, имеющий от 1 до 6 C, предпочтительно от 1 до 3 C, циклопентил, циклогексил, циклогептил, гетероарил, предпочтительно тиенил, фурил, необязательно содержащий OH, пиридил;
предпочтительные (XXXV) соединения такие, где Ar означает фенил, R3a означает H, R2a означает метил и X означает O;
- в соединении формулы (II) остаток супрофена, из которых один предпочтительный показан, где R3a означает H, R2a означает метил и X=O; его эквиваленты, как описано и получено в USP 4035376, который приведен здесь полностью как эталон, можно также использовать;
- в соединении формулы (VI)
одно из которых предпочтительно индопрофен, когда R2a означает CH3 и индобуфен, когда R2a равен H, R3a=-CH3 и X=O, было показано;
его эквиваленты, как описано и получено в соответствии с USP 3997669, который приведен здесь полностью как эталон, можно также использовать;
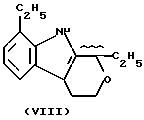
- в соединениях формулы (VIII), из которых одно предпочтительно этодолак, где R2a=R3a=H и X=O, было показано; его эквиваленты, как описано и получено в соответствии с USP 3843681, который приведен здесь полностью, как эталон, можно также использовать;
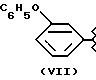
- в соединениях формулы (VII), из которых одно предпочтительно фенопрофен, где R3a=X, R2a=-CH3 и X=O, было показано; его эквиваленты, как описано и получено в соответствии с USP 3600437, который приведен здесь полностью как эталон, можно также использовать,
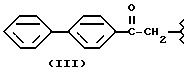
- в соединениях формулы (III), из которых предпочтителен фенбутен, где R2a= R3a= H и X=O был показан; его эквиваленты, как описано и получено в соответствии с USP 3784701, который приведен здесь полностью как эталон, можно также использовать;
- в соединениях формулы (IX), остаток фторбипрофена, где R3a означает H, R2a означает -CH3 и X=O;
- в соединениях формулы (X), остаток толметина, где R2a=R3a=H и X=O; его эквиваленты, как описано и получено в соответствии с патентом FR 1574570, который приведен здесь полностью как эталон, может быть также использован;
В классе III D) значения следующие:
III а) когда оно содержит -CH(CH3)-COOH, показано как пранопрофен: α-метил-5H-[1]бензопиран[2, 3-b]пиридин-7-уксусная кислота.
В предпочтительном соединении R2a=H, R3a=CH3, u=1 и X=O.
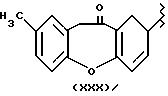
Остаток (XXX), который содержит -CH(CH3)-COOH, известен как бермопрофен: дибензо[b,f]оксепин-2-уксусная кислота.
Предпочтительное соединение имеет u=1, X=O, R2a=H, R3a=CH3.
Остаток (XXXI) показан как CS-670: 2-[4-(2-оксо-1- циклогексилиденметил)фенил]пропионовая кислота, когда радикал означает -CH(CH3)-COOH.
Предпочтительное соединение имеет R2a=H, R3a=CH3, u=1, X=O.
Остаток (XXXII) - производное из известного пемедолака, который содержит -CH2COOH группы.
Предпочтительное соединение имеет R2a=R3a=H, u=1 и X=O.
Этот остаток (XXXIII) известен как пиразолак, когда он насыщен -CH2COOH:
4-(4-хлорфенил)-1-(4-фторфенил)-3-пиразолил производные кислоты. Предпочтительные соединения имеют R2a=R3a=H, u=1 и X=O.
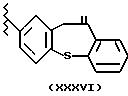
Группа, когда R1a является остатком (XXXVI), R2a=H, R3a=CH3, t=1, u=1 и X=O, известна как залтопрофен.
Когда остаток насыщен гидрокси или аминогруппой или солями кислоты, соединения известны как дибензотиепин производные.
Предпочтительные продукты имеют R2a=H, R3a=CH3, u=1, X=O.
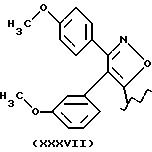
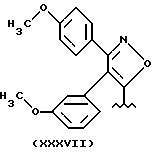
Остаток (XXXVII) - производное из известного мофезолака: 3,4-ди(п-метоксифенил)изоксазол-5-уксусная кислота, когда остаток -CH2-COOH.
Предпочтительные соединения R2a=R3a =H, t=1, X=O.
группа IV), в которой t=1, u=1 и R означает

где: RIVd и RIVd1 означают по крайней мере одна H, а другая - линейный или, когда возможно, разветвленный C1-C6 алкил, предпочтительно C1 и C2, или дифторалкил с алкилом, имеющим от 1 до 6 C, C1 предпочтителен, или RIVd и RIVd1 вместе образуют метиленовую группу;
RIV имеет следующие значения:
где соединения группы IV) имеют следующие значения:
в соединениях формулы (II):
Riv-ii означает 1-6 C алкил, циклоалкил, имеющий от 3 до 7 C, алкоксиметил, имеющий от 1 до 7 C, трифторалкил, имеющий от 1 до 3 C, винил, этинил, галоген, алкокси, имеющий от 1 до 6 C, дифторалкокси с алкилом, имеющим от 1 до 7 C, алкоксиметилокси, имеющий от 1 до 7 C, алкилтиометилокси с алкилом, имеющим от 1 до 7 C, алкилметилтио с алкилом, имеющим от 1 до 7 C, циано, дифторметилтио, фенил- или фенилалкил, замещенный алкилом, имеющим от 1 до 8 C;
предпочтительно Riv-ii означает -CH3O, Rivd означает H и Rivd1 означает -CH3, и известен как радикал напроксена;
X= NH и X1 равен -(CH2-CH2-O)2; также предпочтительным является такое же соединение, где X равен O;
в соединениях формулы (X),
в которых показан остаток локсопрофена, радикалы, описанные в USP 4161538, который приведен здесь полностью как эталон, могут быть использованы как эквиваленты. Предпочтительны соединения, в которых Rivd означает H и Rivd1 означает -CH3, X=NH и X1 равен (CH2-CH2-O); также предпочтительным является такое же соединение, где X равен O;
- в соединениях формулы (III):
Riv-iii означает C2-C5 алкил, даже разветвленный когда возможно, C2 и C3 алкилокси, аллилокси, фенокси, фенилтио, циклоалкил, имеющий от 5 до 7 C атомов, необязательно замещенный в позиции 1 C1-C2 алкилом;
предпочтительным является соединение, где Riv-iii означает
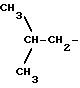
и Rivd= H, Rivd1 означает -CH3, соединение известно как остаток ибупрофена;
X=NH и X1 равен (CH2-CH2-O)2; также предпочтительно такое же соединение, где X равен O;
группа V)

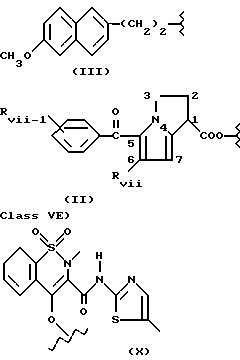

В группе V) соединения имеют следующие значения:
- в соединениях формулы (II)
Rvii означает H или линейный или, когда возможно, разветвленный алкил, имеющий от 1 до 4 C;
Rvii-i означает Rvii или линейный или, когда возможно, разветвленный алкокси, имеющий от 1 до 4 C; Cl, Br, F; положение Rvii-i в о-, м- или п-;
предпочтительным является радикал, известный как кеторолак, где Rvii и Rvii-i означают H, и A=R и t=0
- в соединениях формулы (V),
в котором остаток известного тенидапа показан, их эквиваленты, описанные и полученные в USP 4556672, который приведен здесь полностью как эталон, можно также использовать:
в этих соединениях формулы (V) A=R и t=0,
- в соединениях формулы (VII),
в которых остаток известного теноксикама показан, A означает RCO и t=1 и u=0 или A означает R и t=0: их эквиваленты, описанные и полученные в патенте DE 2537070, который приведен здесь полностью как эталон, можно также использовать;
- в соединениях формулы (IX)
где A= R и t=0, или A=RCO с t=1 и u=0, в которых остаток известного пироксикама показан; их эквиваленты, описанные и полученные в USP 3591584, который приведен здесь полностью, можно использовать;
- в соединениях формулы (III),
где A= RCOO, t= 1 и u=0 или 1; или t=0 и A=R, остаток представлен известным набуметоном, их эквиваленты описаны и получены в USP 4061779, который представлен здесь полностью как эталон, можно также использовать:
- в соединениях формулы (IV),
где A= RCOO, t=1 и u=1, остаток представлен известным индометацином, их эквиваленты, описанные и полученные в USP 3161654, который представлен здесь полностью как эталон, можно также использовать.
- в соединениях формулы (X):
остаток (X) известен как мелоксикам.
Предпочтительными являются соединения с t=0.
- Остаток (XI) известен как ампироксикам, концевой группой является -COOC2H5.
Предпочтительные соединения имеют u=1 и X=O; или t=0.
- Радикал (XII), насыщенный -CH2COO-, означает, как известно бромфенак.
Предпочтительные соединения имеют u= 1, X=O и R2a=R3a=H; или t=0.
Группу XIII) получают из известного Лорноксикама, когда валентность насыщена H.
Предпочтительные соединения имеют t=0.
X1 в формуле A-X1
-NO2 означает бивалентный связывающий мостик, выбранный из следующего:
-YO-
где Y означает:
линейный или, когда возможно, замещенный C1-C20
алкилен, предпочтительно имеющий от 2 до 5 углеродных атомов, исключая связывающий мостик, когда R:
радикал группы I), исключая класс Ib) и Ic);
радикал группы II), исключая IIb);
радикал группы III), исключая класс соединений из IIID)
радикал группы IV);
радикал группы V), исключая X) и включая -(CH2)4- для соединений формул (III)
и (IV);
- или циклоалкилен, имеющий от 5 до 7 углеродных атомов, необязательно замещенных;

где n3 означает 0 или целое число от 1 до 3
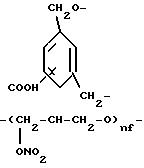
где nf означает целое число от 1 до 6, предпочтительно от 1 до 3;
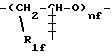
где R1f=H, -CH3 и nf означает целое число от 1 до 6, предпочтительно от 2 до 4.
Соединения, содержащие R из группы I типа Ia) описаны в патенте WO92/01668, где также описан метод получения. Этот патент приведен здесь полностью как эталон. Соединения типа Ib) получают, например, используя метод, описанный в Merck Index, XI Ed., 1989, p. 16, n.95, для остатка ацетилсалицилсалициловой кислоты. Изменения в соединениях формулы Ib) могут быть получены, при использовании способа, описанного в патенте WO 92/01668.
Соединения Ic) класса Ic1), в которых радикал означает производное 5-аминосалициловой кислоты (5-амино-2-гидроксибензойная кислота), известное как месаламин, когда начальный радикал содержит -COOH, получены восстановлением м-нитробензойной кислоты Zn пылью и HCl (смотри H. Weil et al., Ber. 55В, 2664 (1992)); или электролитическим восстановлением: Le Guyader, Peltier, Compt. Rend. 253, 2544 (1961). Эти публикации приведены здесь как эталон.
Начальный радикал Ic2), когда он содержит -COOH, известен как олсалазин: 3,3'-азобис(6-гидроксибензойная кислота); его получают согласно EP 36636 или USP 4528367, здесь приведены оба как эталон.
Соединения Ic3) получают согласно USP 2396145, здесь приведенному как эталон. Эквивалентные соединения у Ic1), Ic2) и Ic3) содержат заместители, указанные в вышеприведенных ссылках.
Продукты настоящего изобретения, имеющие главную формулу
A-X1-NO2
со связывающими мостиками
X1, как приведено выше по отношению к соединениям группы I), могут быть получены при использовании вышеуказанных известных методов или изменяя известные методы по введению мостиков X1, описанных в вышеуказанных патентах.
Соединения, где R означает группу II), описаны в патентах WO94/04484 и USP 3558690, где методы получения также описаны. Эти патенты приведены здесь полностью как эталон.
Начальное соединение из IIb), когда валентность насыщена -COOH (флюниксин), получено согласно USP 3337570 и USP 3689653 здесь приведены как эталон. Соединения, содержащие заместители, указанные в вышеприведенных патентах, эквивалентны флюнексину.
В соответствии с соединениями группы II), связывающие мостики X1, как определено выше, могут быть получены при использовании вышеуказанных известных методов или изменяя известные методы по введению мостиков X1, описанных в вышеуказанных патентах.
Соединения, где R из группы III), описаны и получены способами, представленными в следующих патентах: заявка на изобретение PCT/EP/93 03193; для соединений формулы (IV) также смотри USP 3641127; для соединений формулы (XXI) также смотри USP 3896145: для соединений формулы (IX), остаток фторбипрофена, также смотри USP 3755427; для соединений формулы (II) также смотри USP 4035376; для соединений формулы (VI) также смотри USP 3997669; для соединений формулы (VIII) также смотри USP 3843681: для соединений формулы (VII) также смотри USP 3600437; для соединений формулы (III) также смотри USP 3784701. Все эти патенты приведены здесь полностью как эталон.
Способы получения соединений класса IIID) следующие:
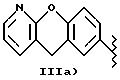
IIIa) остаток получают обработкой кислотным
соединением, согласно USP 3931205, валентность насыщена -CH(CH3)-COOH. Соединения, содержащие заместители, указанные в вышеприведенном патенте, эквивалентны пранопрофену.
Остаток (XXX) получен через соединение с -CH(CH3)- COOH (бермопрофен) согласно USP 4238620, здесь приведен как эталон.
Другие эквивалентные продукты перечислены в вышеуказанном патенте.
Остаток (XXXI) получают, начиная с соответствующей кислоты -CH(CH3)-COOH, согласно USP 4254274. Эквивалентные соединения перечислены в том патенте.
Остаток (XXXII) получают согласно EP 238226, здесь приведен как эталон, когда валентность насыщена -CH2COOH. Эквивалентные продукты приведены в вышеупомянутом патенте как замещенный 1,3,4,9 тетрагидропиран[3,4-b]индол-1-уксусные кислоты.
Остаток (XXXIII) получают из пиразолака (валентность насыщена - CH2COOH), как показано в EP 54812, здесь приведен как эталон. Эквивалентные продукты перечислены в вышеупомянутом патенте.
Остаток (XXXVI) получают согласно патенту UK 2035311, здесь приведен как эталон, начиная с залтопрофена, имеющего конец -CH(CH3)-COO-. Эквивалентные соединения перечислены в вышеупомянутом патенте.
Способ получения радикала XXXVII) начинают с Мофезолака и его получают согласно EP 26928. Эквивалентные продукты приведены там.
По отношению к соединениям группы III), связывающие мостики X1, как выше указано, можно получить, используя вышеупомянутыми методами, или изменяя известные методы при введении мостиков X1, когда они отличаются от связывающих мостиков, описанных в вышеуказанных патентах.
Соединения, где R означает группу IV), описаны в английской заявке на изобретение 93205995, где методы получения также описаны. Этот патент приведен здесь полностью как эталон.
В группе IV) соединения могут быть также получены: для соединений формулы (II), используя патент USP 3904682; для соединений формулы (X), согласно патенту USP 4161538; для соединений формулы (III), согласно патенту 3228831. Эти патенты полностью включены в настоящую заявку как эталон.
По отношению к соединениям группы IV), связывающие мостики X1, как указано выше, могут быть получены, используя вышеуказанные известные методы или изменяя известные методы введения мостиков X1, когда они отличаются от связывающих мостиков, описанных в вышеупомянутых патентах.
Соединения, где R означает группу V), описаны в итальянском патенте М194Ф 000916, где методы получения также описаны. Этот патент приведен здесь полностью как эталон. В группе V) соединения могут быть также получены: для соединений формулы (II), используя патент USP 4089969, который приведен здесь полностью как эталон: соединения формулы (V) могут быть получены согласно патенту USP 4556672, который приведен здесь полностью как эталон.
Остаток (X) получают согласно немецкому патенту 2756113. Эквивалентные продукты перечислены в вышеупомянутом патенте.
Остаток (XI) получают согласно патенту EP 147177, приведен здесь как эталон, начиная с ампироксикама, имея концевую группу -COOC2H5.
Эквивалентные продукты перечислены в вышеупомянутом патенте.
Остаток (XII) получают согласно J. Medicinal Chem., vol. 27, N 11, Nov. 1984, Walsh et al, Antiinflammatory Agents. 3. Synthesis and Pharmacological Evaluation of 2-Amino-3-Benzoylphenilacetic Acid and Analogues, здесь приведен как эталон. Эквивалентные продукты перечислены в вышеупомянутой публикации.
Радикал (XIII) получают, начиная с Лорноксикама, где валентность насыщена H. Его готовят согласно GBP 2003877. Эквивалентные продукты описаны в вышеупомянутом патенте.
Что касается соединений группы V), связывающие мостики X1, как указано выше, могут быть получены при использовании известных методов или изменяя известные методы введения мостиков X1, когда они отличны от связывающих мостиков, описанных в вышеуказанных патентах.
Связь между A и X1 является, как мы видим, главным образом сложноэфирного или амидного типа (NH или NR1c, как определено в X), когда R выбран из групп I), II), III), IV). Все хорошо известные пути для образования этих связей могут быть использованы для получения этой связи.
В случае сложных эфиров группы I), III) и IV) главное направление пути синтеза включает реакцию ацилхлоридов R-CO-Cl с галогенированными спиртами типа HO-Y-Cl, HO-Y-Br, HO-Y-I в известных экспериментальных условиях.
Реакционные продукты формулы R-CO-O-Y-Cl(Br, I) могут быть также получены для класса II реакцией солей натрия или калия вышеупомянутых R-CO-OH кислот с дигалоген производными основной формулы YCl2, YBr2 или YI2.
Реакционные продукты превращают в конечные продукты реакцией с AgNO3 в ацетонитриле в соответствии с литературными источниками.
Основные пути для групп I), III), IV) следующие:
R-CO-Cl+HO-Y-Br ---> R-CO-O-Y-Br+AgNO3 --->
A-X1-NO2, где X1=YO.
Основной путь для группы II следующий:
R-CO-ONa + Br2Y ---> R-CO-O-Y-Br + AgNO3 --->
A-X1-NO2, где X1=YO.
В случае амидов путь синтеза включает реакцию таких же ацилхлоридов RCOCl с аминоспиртами главной формулы NH2-Y-OH,
NHR1c-Y-OH для получения амидов главной формулы:
R-CO-NH-Y-OH и R-CO-NR1c-Y-OH
в соответствии с известными методами.
Реакция вышеупомянутых
амидов с галогенирующими агентами, такими как, например, PCl5, PBr3, SOCl2 и т.д., приводит к галогенпроизводным основной формулы:
R-CO-NH-Y-Br(Cl) и
R-CO-NR1c-Y-Br(Cl).
Они, реагируя с AgNO3 в ацетонитриле согласно известным из литературы методам, приводят к конечным продуктам A-X1-NO2 .
Этот путь может быть в общих словах следующим:
PCl5
R-CO-Cl + NHR1c-Y-OH ---> R-CO-NR1c-Y-OH ---> R-CO-NR1c
-Y-Cl + AgNO3 ---> R-CO-NR1c-Y-ONO2
где YO означает Xx1.
Альтернативным путем образования сложных эфиров является реакция
солей натрия или калия кислот с нитроэфирами галогенированных спиртов общей формулы:
NO2-O-Y-Cl(Br,I)
получая прямо продукт изобретения.
Путь реакции
следующий:
R-CO-ONa + Br-Y-ONO2 ---> R-CO-O-Y-ONO2
где YO означает X1.
Пути синтеза подобные этим, описанные выше, могут быть использованы для продуктов Va и Vb группы V), где дигалогенпроизводные Br2Y реагируют с енолятами, например, теноксикама или пироксикама. Продукты реакции затем превращаются в ацетонитриле, реагируя с AgNO3 согласно вышеуказанной реакции.
Основной путь, показанный ниже, относится к пироксикаму формулы IX в группе V).
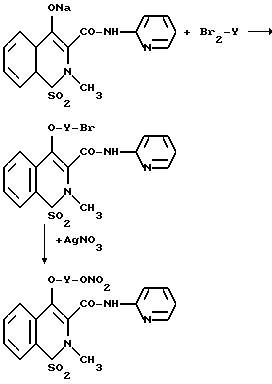
Указанные выше продукты в различных группах используют для противовоспалительных, анальгетических и антитромбоцитарных активностей. Для группы I) нет необходимости исключать значения X1.
Для групп II), III), IV) и V) значения X1 ограничены, как указано выше, для тех способов, когда X1=-YO- для некоторых соединений.
Следующим объектом изобретения является неожиданное открытие, что продукты изобретения, содержащие -ONO2 группы способны иметь эффект ингибирования воспаления, вызванного липополисахаридом (ЛПС), и, следовательно, могут быть использованы при септическом шоке.
Это было неожиданно, так как хорошо известно, что обычно противовоспалительная активность не изменяется значительно активностью нитросинтетазы, вызванной липополисахаридами у крыс и, таким образом, не может использоваться для септического шока.
Продукты, которые могут быть использованы для такого фармацевтического применения, являются продуктами общей формулы:
A-X1-NO2
описанные
выше, где бивалентный связывающий мостик X1 не имеет ограничений в этом случае, т.е. известные связывающие мостики не могут быть исключены, как и те, что были описаны в предыдущих патентах
для этой цели.
Нужно понимать, что когда соединения различных групп содержат по крайней мере один асимметрический углерод, продукты могут использоваться в рацемической форме или как отдельные изомеры. Хорошо известен факт, что при терапевтическом использовании изобретения вообще изомерная форма более активна, чем другие.
Следующие примеры даны для пояснения, но они не ограничивают настоящее изобретение.
Примеры
Пример 1: Химические примеры - полученный продукт.
Пример 1a:
Получение соединения A-X1
-NO2, где R относится к классу I, X1 означает -(CH2-CH2-O)2-; здесь обозначен как ASA.NO-DEG, и имеет общую формулу:
2-ацетокси-бензоат
2-[2-(нитрокси)этокси]этила

Получение промежуточного продукта формулы:
2-ацетокси-бензоат 2-[2-(хлор)этокси]этила
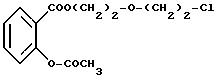
1.0 г гидрида натрия (NaH) (80% суспензия в светлом минеральном масле) было добавлено порционно к раствору:
ацетилсалициловая кислота - 5.6 г и
диметилформамид - 20 мл
выдержали при 0oC в токе азота.
Смесь перемешивали один час и затем добавляли по каплям около 5 часов в перемешиваемый раствор
2,2'-дибромодиэтилэфир - 10 г и
диметилформамид - 15 мл
при 25oC. Смесь перемешивали 3 дня, затем высушивали при пониженном давлении. Остаток обрабатывали:
вода - 50 мл и
дихлорметан - 50 мл.
Фазы разделили и водную фазу проэкстрагировали дихлорметаном 10 мл.
Объединенные органические фазы промыли водой (3х25 мл), высушили (MgSO4), обесцветили животным углем (1 г) и поместили сушить в вакуум.
Остаток (11.2 г) использовали сырым для следующей реакции.
Получение ASA-NO-DEG:
8.6 г нитрата серебра добавили к раствору
ASA-(CH2)-O-(CH2)2Cl - 11.2 г и
ацетонитрил - 25 мл
выдержали при температуре окружающей среды и в отсутствие света. После перемешивания в течение двух дней 2.2
г нитрата серебра добавили.
После следующих двух дней в тех же условиях, нерастворимые соли отфильтровали и фильтрат был освобожден от растворителя при пониженном давлении.
Остаток 7.0 г был получен и хроматографирован на колонке с силикагелем (500 г двуокиси кремния), элюируя смесью толуол/этилацетат 95/5 об/об.
Фракции, которые были найдены однородными по ТСX (тонкослойная хроматография) объединили и высушили.
Выход 3.0 г ASA-NO-DEG.
1H-ЯМР анализ (CDCl3) (80 МГц) дал следующие
результаты:
2.28 (3H, с); 3.7 (4H,м); 4.35 (2H, т); 4.52 (2H, т); 7.3 (3H, м); 7.98 (1H, дд).
ИК-анализ (нуйол) показал следующие результаты.
νOCO=1780 см-1; νCOO=1725 см-1;
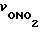
Масс-спектрометрия показала молекулярный вес 313.
Пример 1b:
Получение соединения A-X1-NO2, где R относится к классу II), X1
означает -(CH2-CH2-O)2-, здесь обозначен как Диклофенак - NO-DEG и имеет формулу:
2-{N-[2-6-(дихлор)фенил]амино}фенилацетат 2-[2-(нитрокси)этокси] этила

Получение промежуточного продукта, имеющего формулу:
2-{N-[2, 6-(дихлор)фенил]амино}фенилацетат 2-[2-(бром)этокси]этила

Раствор
Диклофенак натриевая соль - 13.3 г и
диметилформамид - 25 мл
добавили к раствору
2,2'-дибромдиэтилэфир - 12.3 г и
диметилформамид - 15 мл
выдерживали при температуре окружающей среды в токе азота.
Смесь оставили реагировать на два дня, а затем растворитель был удален при пониженном давлении. Остаток обработали этилацетатом (50 мл), промыли 5% раствором карбоната калия (2х10 мл), затем водой (20 мл), высушили над безводным сульфатом натрия. Растворитель удалили при пониженном давлении. Остаток весил 16 г и был использован для следующей реакции без очистки.
Получение Диклофенак-NO-DEG:
Нитрат серебра - 8 г в
ацетонитриле - 16 мл
добавили к раствору Диклофенак-(CH2)2
-O- (CH2)2-Br - 16 г и
ацетонитрил - 30 мл
выдержали при комнатной температуре и отсутствии света.
Смесь перемешивали при температуре окружающей среды 3 дня.
Нитрат серебра - 3 г после 1 дня
Нитрат серебра - 3 г после 2 дней
затем добавили.
Смесь перемешивали еще 2 дня. Нерастворимые соли были затем отфильтрованы и растворитель удален из фильтрата при пониженном давлении. Остаток обработали этилацетатом (50 мл), нерастворимые соли затем отфильтровали и выбросили. Растворитель удалили из фильтрата при пониженном давлении. Был получен остаток 16.2 г и хроматографирован на колонке с силикагелем (700 г двуокиси кремния), элюирован сначала толуолом, затем смесью толуол/этилацетат 99/1 об/об, в конце - смесью толуол/этилацетат 98/2 об/об.
Фракции, которые были найдены однородными с помощью ТСX анализа (тонкослойная хроматография) объединили и высушили, выход 4.38 г Диклофенака-NO-DEG.
1H-ЯМР анализ (CDCl3) (300 МГц) показал следующие результаты:
3.69 (4H, т); 3.87 (2H, с); 4,3 (2H, м): 4.52
(2H, т); 6.55 (1H, д); 6.88 (1H, широкий синглет для D2O, NH): 6.97 (2H, т); 7.11 (2H, д); 7.23 (2H, д); 7.35 (2H, д).
Масс-спектрометрия показала молекулярный вес 588.
Пример 1c:
Получение соединения A-X1-NO2, где R относится к классу III) и представляет остаток соединения формулы IV, X1 означает -C6
H5CH2-, здесь обозначен как кетопрофен -NO-DEG, и имеет формулу:
2-(3-бензоил)фенилпропионат 3-(нитроксиметил)фенила

Получение промежуточного продукта, имеющего формулу:
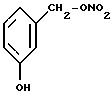
Используемые ниже реагенты в указанных количествах реагировали, как описано ниже:
3-гидроксибензиловый спирт - 10 г
48% HBr по весу - 50 мл
CH2Cl2 - 30 мл
AgNO3 - 13.7 г
CH3CN - 70 мл
3-Гидроксибензиловый спирт в CH2Cl2 реагировал с HBr 4 часа при температуре окружающей среды.
CH2Cl2 затем выпарили при пониженном давлении при 30oC после промывки водным раствором 5% NaHCO3 и высушили над безводным Na2SO4.
Маслянистый остаток растворили в CH3CN (50 мл) и раствор AgNO3 в оставшемся количестве CH3CN добавили по каплям. Колба была изолирована от света.
После 8 часов осадок AgBr отфильтровали и органическую фазу выпарили при пониженном давлении.
Маслянистый осадок, полученный таким образом, растворили в толуоле (45 мл), и раствор фильтровали на колонке с силикагелем (400 г). Элюат высушили при пониженном давлении при 30oC и получили 20 г 3-нитроксиметилфенола.
Получение промежуточного продукта Кетопрофен -COCl:
Хлорид 2-(3-бензоил)фенил пропионовой кислоты.
Кетопрофен - 20 г
Тионилхлорид - 50 мл
смешали и раствор кипятили с обратным холодильником 45 минут. Тионилхлорид выпарили при пониженном давлении. Получили маслянистый желтый остаток весом 21 г и использовали
без очистки.
Получение Кетопрофен-Ar-NO2.
Ниже использовали реагенты в следующих количествах:
Кетопрофен - COCl - 5.45 г
3-нитроксиметилфенол - 3.9 г

K2CO3 и AcOEt были добавлены вместе;
Кетопрофен хлорид затем был добавлен под азотом при t=0 в течение 30 минут. Все оставили реагировать на 5 часов при температуре окружающей среды, затем разбавили водой (50 мл). Органическую фазу промыли 5% NaOH (2х10 мл) и выпарили при пониженном давлении. Полученный маслянистый остаток хроматографировали на окиси кремния, используя смесь толуол/EtOAc 9.5/0.5 об/об в качестве элюента. Выпаривание элюата дало Кетопрофен-Ar-NO2 с выходом 85%.
1H-ЯМР анализ (CDCl3) (300 МГц) показал следующие результаты:
1.63 (3H, д): 4.00 (1H,
q): 5.37 (2H, с); 7.01 - 7.89 (м, 13H).
Масс-спектрометрия показала молекулярный вес 405.
Пример 1d:
Получение соединения A-X1-NO2,
здесь обозначен как Ибупрофен-NO-DEG, где R относится к группе IV; X1 означает -(CH2-CH2-O)2-, A=RCOO, R остаток Ибупрофена, имеющего формулу:
-(CH3)2CHCH2C5H4-CH(CH3)-
Следовали процедуре примера 1a, используя указанный выше R, остаток Ибупрофена, вместо остатка R
группы I как показано в примере 1a.
Пример 1e:
Получение соединения A-X1-NO2, здесь обозначен как Фторбипрофен-NO-DEG, где R относится к группе III,
X1 означает -(CH2-CH2-O)2-, A=RCOO, R3a=H, R2a=CH3, R имеет формулу:
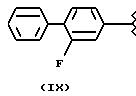
Следовали процедуре примера 1a, используя указанный выше R, остаток фторбипрофена, вместо остатка R группы I как показано в примере 1a.
Пример 1f:
Получение соединения A-X1-NO2, Кеторолак-NO-DEG, где R относится к группе V; X1 означает -(CH2-CH2-O)2
-;
A=R, R формулы II, имеющего формулу:

Следовали процедуре примера 1a, используя указанный выше R, остаток Кеторолака, вместо остатка R группы I как показано в примере 1a.
Пример 1g:
Получение соединения A-X1-NO2, Тиапрофеновая
кислота NO-DEG, где R относится к группе III, X1 означает -(CH2-CH2-O)2-, A=RCOO, R означает остаток формулы XXXV, где R означает:
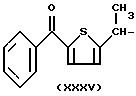
Следовали процедуре примера 1a, используя указанный выше R, остаток тиапрофеновой кислоты, вместо остатка R в примере 1a.
Пример 1h:
Получение соединения A-X1-NO2, Напроксен NO-DEG, где R относится к группе IV, X1 означает -(CH2-CH2-O)2-, A=RCOO, R означает остаток формулы II Напроксена, имеющий формулу:
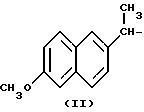
Следовали процедуре примера 1a, используя указанный выше R, остаток Напроксен, вместо остатка R группы I, как показано в примере 1a.
Пример 2: Фармакологические примеры.
Примеры, используемые выше, были охарактеризованы фармакологически.
Пример 2a: ASA-NO-DEG, полученный в примере 1a;
Пример 2b: Диклофенак-NO-DEG, полученный в
примере 1b;
Пример 2c: Кетопрофен-NO-DEG, полученный в примере 1c;
Пример 2d: Ибупрофен-NO-DEG, полученный в примере 1d;
Пример 2e: Фторбипрофен-NO-DEG, полученный в примере
1e;
Пример 2f: Кеторолак-NO-DEG, полученный в примере 1f;
Пример 2g: Тиапрофеновая кислота NO-DEG, полученная в примере 1g;
Пример 2h: Напроксен NO-DEG, полученный в примере
1h.
Токсичность.
Острая токсичность была оценена при оральном введении отдельных доз 1, 3, 10, 100 мг/кг продукта в группах из 10 мышей.
Смертельные дозы и начало симптомов интоксикации были описаны для периода 14 дней. Даже после применения дозы в 100 мг/кг животные не проявляли явных симптомов токсичности.
Противовоспалительная активность.
Противовоспалительная активность была изучена при использовании метода воздействия на индуцированный каррагином отек, как это описано Winter и др. (Proc. Soc. Exp. Biol. Med. 111, 544, 1962) на крысах.
Анальгетическая активность.
Анальгетическая активность была изучена на мышах породы Свисс, как это описано Hendershot и др. (J. Pharm. Exp. Therap. 125, 237, 1959).
Толерантность.
Желудочная толерантность определялась при оральном введении на крысах, оценивая тяжесть гастропатии в соответствии с критериями, описанными Wallace и др. (Am. J. Physiol. 259, G642, 1990).
Антитромбоцитарная активность.
Антитромбоцитарная активность была изучена in vitro на человеческих тромбоцитах, стимулированных тромбином в соответствии с методом, описанным Bertele и др. (Science 220, 517, 1983).
Сосудорасширяющая активность.
Сосудорасширяющая активность была определена на изолированной аорте крысы путем измерения ингибирования сокращения, вызванного эпинефрином в ткани, препарированной в соответствии с методом, описанным Reynolds и др. (J. Pharmacol. Exp. Therap. 252, 915, 1990).
COX ингибирование.
Активность ингибирования циклооксигеназы была определена на изолированных клетках. Эндотелиальные клетки бычьей аорты были использованы в качестве источника COX, и макрофаги линии J.774.2 - в качестве источника COX-2. Такие условия, описанные Mitchell и др. (Proc. Nat. Acad. Sci., 90, 1 1693. 1993) для тестирования роста и жизнеспособности, были использованы.
Вкратце, клетки были выдержаны в термостате в течение 30 мин с тестируемым веществом в определенных концентрациях и затем в течение следующих 15 мин в термостат добавляли субстрат (арахидоновая кислота). Активность фермента определялась радиоиммунологически измерением образования 6-кето-PGF 1 альфа. В случае клеток линии J.774.2, клетки были инкубированы на 12 часов с эндотоксином для стимулирования образования COX-2.
Ингибирование нитросинтетазы ЛПС.
Активность ингибирования нитросинтетазы, индуцированного липополисахаридом (ЛПС) определялась на крысиных нейтрофилах и желудке после введения одного из тестируемых веществ и сравнивалась с таковой, полученной после обработки лишь суспензией носителя.
Вкратце, крысам породы Вистар за 24 часа перед обработкой орально вводили тестируемый продукт (10 мг/кг) и внутривенно (хвостовая вена) вводили ЛПС (5 мг/кг).
Через 4 часа животные были убиты и взяты: кровь - для изолирования нейтрофилов, и желудок.
Активность фермента определяли в соответствии с методом, описанным Assreuy и др. (Br. J. Pharmacol., 108, 833, 1993).
Результаты:
Полученные результаты описаны ниже.
Как можно видеть из данных, показанных в таблицах с 1 по 4, фармакодинамика активности (I и II в таблице 1; Таблица 2) и толерантность (Таблица 1, колонка III) нитропроизводных показала лучший баланс по сравнению с природными продуктами.
Таблица 4 также показывает, что подобно диклофенак нитробутиловому эфиру, диклофенак нитропроизводное, которое является объектом этого патента, способно к прямому ингибированию циклооксигеназы COX-1 и COX-2, но со значительно более низкой вариабельностью.
Таблица 1 (Фармакология кол. I и II; Токсикология кол.III).
Изучение противовоспалительных (I) и анальгетических (II) свойств (фармакодинамика) и гастроинтестинальной толерантности (III) (токсичность) тестируемых соединений после орального введения доз от 3 до 30 мг/кг в карбоксиметилцеллюлозных суспензиях и построение кривой зависимости от дозы. Показанные результаты означают соотношение активности по сравнению со стандартом.
Активности, выраженные как соотношение по сравнению с природными продуктами, использовали как единицу стандарта.
Нитропроизводное, которое взято из показанных примеров, и соответствующее природное соединение как эталон.
Таблица 2 (Фармакодинамическая активность).
Примеры анти-циклооксигеназных (I), антитромботических (II) и сосудорасширяющих свойств (III) тестируемы соединений выполнялись in vitro при молярной концентрации от 10-5 до 10-7 продукта в среде вода/спирт с добавлением небольших количеств ДМСО (диметилсульфоксида).
Активности, выраженные как соотношение по сравнению с природными продуктами, использовали как единицу стандарта, как изложено в Таблице 1.
Таблица 3 (Биохимия: действие на NOS для септического шока).
Изучение ингибиторных свойств активности нитросинтетазы (NOS), вызванных липосахаридом (ЛПС) у крыс с использованием оральных доз от 5 до 20 мг/кг в карбоксиметилцеллюлозном основании.
Таблица 4 (COX - ингибиторная активность).
Изучение анти-циклооксигеназных (COX-1/COX-2) свойств в изолированных клетках.
Ответная экспрессия как % контролей с относительной ответной вариабельностью.
Способ получения пероральной дозированной формы:
В ступке смешивают 10 г соединения,
имеющего лабораторное название Диклофенак-NO-DEG (пример Ib со стр. 35 описания) с 3 г крахмала, 1,5 г стеарата полиэтиленгликоля, 2 г талька и 1 г стеарата магния. Порошок перетирают в ступке до
получения тонко измельченной консистенции. К порошку в ступке порциями добавляют раствор 4 г натрийкарбоксиметилцеллюлозы в 50 мл воды при перемешивании, с промежуточной сушкой при 60oC в
вакууме, измельчают и опять сушат. Полученные гранулы продавливают через решетку с отверстием 6-меш. Единичная доза указанного гранулята, содержащего указанное выше соединение данного изобретения,
имеет следующий состав:
Диклофенак-NO-DEG - 25 мг
Стеарат полиэтиленгликоля - 3,75 мг
Кукурузный крахмал - 75 мг
Тальк - 5 мг
Стеарат магния - 2,5 мг
Натрийкарбоксиметилцеллюлоза - 10 мг
Пример 1i
Получение соединения A-X1-NO2, где R относится к группе I и представляет собой остаток соединения формулы
Ic1, X1 является YO, где Y является линейным C4 алкиленом. A=RCOO, R - остаток мезаламина, имеющего формулу:
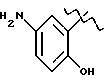
обозначенного здесь как Мезаламин-NO-DEG и имеющего общую формулу: 5-амино-2-гидроксибензоат 4(нитроокси)бутила

Получение промежуточного соединения Boc-Мезаламин
5-трет-бутоксикарбониламино-2-гидрокси бензойная кислота
Мезаламин - 15 г
Ди-трет-бутил бикарбонат - 25.65 г
Триэтиламин - 24.6 мл
Диоксан - 100 мл
Вода - 150 мл
К смеси 5-амино-6-гидрокси бензойной кислоты в диоксане и воде добавляют триэтиламин и ди-трет-бутил бикарбонат. Через 4 дня раствор концентрируют в вакууме, обрабатывают HCl 5% до pH 2 и, после добавления NaCl экстрагируют этилацетатом. Органический слой обезвоживают сульфатом натрия и выпаривают в вакууме. Получают неочищенный остаток весом в 23 г и используют без дальнейшей очистки.
Получение промежуточного соединения формулы:
хлорид 5-трет-бутоксикарбониламино-2-гидрокси бензойной кислоты
5-Трет-бутоксикарбониламино-2-гидрокси бензойная кислота - 23 г
Тионил хлорид - 7.15 мл
Пиридин - 0.25 мл
Метилен хлорид - 200 мл
К смеси 5-трет-бутоксикарбониламино-2-гидроксибензойной кислоты в метилен хлориде, охлажденной до
температуры 0oC, добавляют пиридин и тионил хлорид. Смесь кипятят с обратным холодильником в течение 1 часа, растворитель выпаривают в вакууме. Неочищенный остаток (24.5 г) используют на
следующей стадии.
Получение промежуточного соединения формулы:
5-трет-бутоксикарбониламино-2-гидроксибензоат-4-(хлор)бутила.
5-Трет-бутоксикарбониламино-2-гидроксибензоил хлорид - 24.8 г
4-Хлорбутанол - 14 мл
Триэтиламин - 25.4 мл
Метиленхлорид - 150 мл
К смеси
5-трет-бутоксикарбониламино-2-гидрокси бензоил хлорида в метиленхлориде, охлажденной до температуры 0oC, добавляют триэтиламин и 4-хлорбутанол. Смесь перемешивают при комнатной температуре
в течение 4 часов, промывают водой. Органический слой обезвоживают сульфатом натрия и растворитель выпаривают в вакууме. Остаток очищают хроматографией на колонке с силикагелем, элюент
н-гексан/этилацетат 8/2, с получением 21.6 г соединения в виде аморфного твердого вещества.
Получение промежуточного соединения формулы:
5-трет-бутоксикарбониламино-2-гидрокси бензоат 4-(нитроокси)бутила
5-Трет-бутоксикарбониламино-2-гидрокси бензоат 4-(хлор)бутил - 21 г
Нитрат серебра - 16.4 г
Ацетонитрил
- 200 мл
К раствору 5-трет-бутоксикарбониламино-2-гидрокси бензоата 4-(хлор)бутила в ацетонитриле добавляют нитрат серебра при комнатной температуре и полученную смесь кипятят с обратным
холодильником в темноте в течение 2 дней. Осадок отфильтровывают и растворитель выпаривают в вакууме. Остаток очищают хроматографией на колонке с силикагелем, элюент н-гексан/этилацетат 7/3, с
получением 19 г соединения в виде белого твердого вещества. Т.пл.= 107-109oC.
Получение хлорида Мезаламина-NO-DEG:
5-Трет-бутоксикарбониламино-2-гидрокси бензоат
4-(нитроокси) бутила - 19 г
Этилацетат/HCl 4.8 М - 41 мл
Этилацетат - 25 мл
К раствору 5-трет-бутоксикарбониламино-2-гидрокси бензоата 4-(нитроокси)бутила в этилацетате при
температуре 0oC по каплям добавляют этилацетат/HCl 4.8 М. Смесь перемешивают при комнатной температуре в течение 4 часов. Осадок собирают, промывают н-гексаном с
получением 13.5 г хлорида
Мезаламина-NO-DEG в виде белого твердого вещества. Т.пл.=136-140oC.
1H-ЯМР анализ (ДМСО)(200 МГц) показал следующие результаты:
1.80 (4H, м); 4.34 (2H,т);
4.60 (2H,т); 7.11 (1H,д); 7.51 (1H,дд); 7.80 (1H,д); 10.60 (3H,с).
Получение нитрата Мезаламина-NO-DEG:
Хлорид Мезаламина-NO-DEG - 7 г
Нитрат серебра - 3.88 г
Ацетонитрил - 500 мл
К раствору хлорида Мезаламина-NO-DEG в ацетонитриле добавляют нитрат серебра. Смесь перемешивают при комнатной температуре. Через 20 минут хлорид серебра
отфильтровывают, раствор концентрируют в вакууме (250 мл) и охлаждают при температуре 0oC. Осадок фильтруют, промывают н-гексаном с получением 6.15 г нитрата Мезаламина-NO-DEG в виде белого
твердого вещества. Т.пл. = 141- 145oC.
Пример 1j
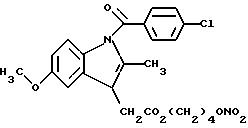
Получение соединения A-X1-NO2, где R относится к группе V и является остатком соединения
формулы IV, X1 является YO, где Y является линейным C4 алкиленом, A=RCOO, R - остаток индометацина, имеющий формулу:

обозначенного здесь как Индометацин-NO-DEG и имеющего общую формулу:
Получение Индометацин-COCl:
Хлорид 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-уксусной кислоты.
Индометацин - 5 г
Оксалил
хлорид - 3.5 г
Толуол - 70 мл
N,N-диметилформамид - 1 мл
К раствору индометацина в толуоле и N,N-диметилформамиде, охлажденном до 0oC, добавляют оксалил хлорид.
Смесь перемешивают при комнатной температуре в течение 1 дня. Растворитель выпаривают в вакууме и остаток (5.2 г) используют неочищенным в следующей стадии. Получение промежуточного соединения,
имеющего формулу: 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-ацетат 4(хлор)бутила.
Индометацин COCl - 5.2 г
4-Хлорбутанол - 2.2 мл
Триэтиламин - 3.9 мл
Метиленхлорид - 60 мл
К раствору индометацина COCl в метиленхлориде добавляют триэтиламин и 4-хлорбутанол. Раствор перемешивают при комнатной температуре в течение 6 часов, промывают водой.
Органический слой обезвоживают сульфатом натрия и выпаривают в вакууме. Остаток очищают хроматографией на колонке с силикагелем, элюент н-гексан/этилацетат 9/1 с получением 3.74 г соединения в виде
аморфного твердого вещества.
Получение Индометацина-NO-DEG:
1-(4-Хлорбензоил)-5-метокси-2-метил-1H-индол-3-ацетат 4(хлор) бутила - 3.74 г
Нитрат серебра - 1.77 г
Ацетонитрил - 35 мл
К раствору 1-(4-хлорбензоил)-5-метокси-2-метил-1H-индол-3-ацетата 4(хлор)бутила в ацетонитриле добавляют нитрат серебра при комнатной температуре и полученную смесь
кипятят с обратным холодильником в темноте в течение 2 дней. Осадок отфильтровывают и растворитель выпаривают в вакууме. Остаток очищают хроматографией на колонке с силикагелем, элюент
н-гексан/этилацетат 8/2, с получением 3 г соединения в виде желтого твердого вещества. Т.пл. = 56-61oC.
1H-ЯМР анализ (CDCl3) (200 МГц) показал
следующие результаты:
1.80 (4H, м); 2.39 (3H,с); 3.35 (2H,т); 3.65 (2H,с); 3.82 (3H,с); 4.10 (2H,т); 6.66 (1H,дд); 6.84 (1H,д); 6.95 (1H.д); 7.45 (2H,м); 7.65 (2H,м).
Пример
1k
Получение соединения A-X1-NO2, где R относится к группе IV и является остатком соединения формулы X, X1 является (CH2-CH2-О)2, A=RCOO, R - остаток локсопрофена, имеющий формулу:

обозначенного здесь как Локсопрофен-NO-DEG и имеющего общую формулу:
α- метил-4-[(2-оксоциклопентил)метил]бензол ацетат 2-[2- (нитрокси)этокси]этила.

Следуют методике примера 1a, используя указанный выше R, остаток локсопрофена, вместо остатка R группы I, как указано в примере 1a.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.23 (3H,д); 1.43-2.42 (7H,м); 2.47 (1H,дд); 3.07 (1H,дд); 3.65 (1H,кв); 3.75 (4H,м) 4.29 (2H,т);
4.34 (2H,т); 7.19-7.06 (4H,м).
Пример 1l
Получение соединения A-X1-NO2, где R относится к группе V и является остатком соединения формулы IX, X1 является C6H4-CH2-O, R является остатком пироксикама, имеющим формулу:

обозначенного здесь как Пироксикам-NO-DEG и имеющего общую формулу:

Получение промежуточного соединения, имеющего формулу Пироксикам-CО-C6H4-CH2Cl.
Пироксикам - 5 г
3-(Хлорметил)бензоил хлорид
- 4.2 мл
Триэтиламин - 4.2 мл
Тетрагидрофуран - 60 мл
К раствору пироксикама в тетрагидрофуране добавляют триэтиламин и 3-(хлорметил)бензоил хлорид. Через 3 часа
растворитель выпаривают в вакууме и остаток обрабатывают водой и экстрагируют этилацетатом. Органический слой обезвоживают сульфатом натрия и выпаривают. Остаток очищают хроматографией на колонке с
силикагелем, элюент н- гексан/этилацетат 6/4, с получением 4.5 г соединения.
Получение промежуточного соединения, имеющего формулу Пироксикам-NO-DEG:
Пироксикам-CО-C6H4-CH2-Cl - 4.5 г
Нитрат серебра - 2.37 г
Ацетонитрил - 100 мл
К раствору Пироксикама-CО-C6H4-CH2-Cl в
ацетонитриле добавляют нитрат серебра при комнатной температуре и полученную смесь кипятят с обратным холодильником в темноте в течение 2 часов. Осадок отфильтровывают и растворитель выпаривают в
вакууме. Остаток очищают хроматографией на колонке с силикагелем, элюент н- гексан/этилацетат 6/4, с получением 3.1 г Пироксикама-NO-DEG.
1H-ЯМР анализ (CDCl3)
(80 МГц) показал следующие результаты:
3.44 (3H,с); 5.40 (2H,с); 7.35-8.40 (12H,м).
Пример 1m
Получение соединения A-X1-NO2, где R относится к
группе V и является остатком соединения формулы VII, X1 является C6H4-CH2, A=RCO, K является остатком теноксикама, имеющим формулу
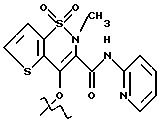
обозначенного здесь как Теноксикам-NO-DEG и имеющего общую формулу:
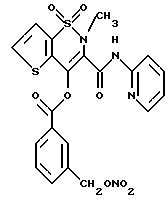
Следуют методике примера 1l, используя указанный выше R, остаток теноксикама, вместо остатка R группы V, как описано в примере 1l.
1H-ЯМР анализ (ДМСО) (200 МГц) показал следующие результаты:
8.35 (1H, д); 8.17-8.11 (3H,м); 8.04 (1H,д); 7.87 (1H,д); 7.72( 2H,м); 7.46 (1H,д); 7.33 (1H,т);
5.76 (2H,с); 3.43 (3H,с).
Пример 1n
Получение соединения A-X1-NO2, где R относится к группе V и является остатком соединения формулы XIII, X1
является C6H4-CH2-О, A=RCO, R является остатком лорноксикама, имеющим формулу

обозначенного здесь как Лорноксикам-NO-DEG и имеющего общую формулу:

Следуют методике примера 1l, используя указанный выше R, остаток лорноксикама, вместо остатка R группы V, как показано в примере 1l.
1H-ЯМР анализ (ДМСО)(200
МГц) показал следующие результаты:
8.35 (1H, д); 8.17-8.11 (3H,м); 7.87 (1H,д); 7.72 (2H,м); 7.50 (1H,с); 7.33 (1H,т); 5.76 (2H,с); 3.45 (3H,с).
Пример 1o
Получение
соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы XXXIII, X1 является -(CH2-CH2-О)2, A=RCOO, R
является остатком пиразолака, имеющим формулу

обозначенного здесь как Пиразолак-NO-DEG и имеющего общую формулу:
4-(4-хлорфенил)-1-(4-фторфенил)-1H-пиразол-3ацетат 2- [2-(нитрокси)этокси]этила

Следуют методике примера 1a, используя указанный выше R, остаток лороксофена, вместо остатка R группы I, как показано в примере 1a.
1
H-ЯМР анализ (CDCl3)(80 МГц) показал следующие результаты:
3.60 (4H, м); 3.98 (2H,с); 4.60 (2H,т): 4.17 (2H,т); 7.20-7.40 (5H,м); 7.50 (4H,м).
Пример 1p
Получение соединения A-X1-NO2, где R относится к группе IIId и является остатком соединения формулы IIIa, X1 является -(CH2-CH2-О)2,
A=RCOO, R является остатком пранопрофена, имеющим формулу

обозначенного здесь как Пранопрофен-NO-DEG и имеющего общую формулу:
α- метил-5H-[1]бензопирано[2,3-b]пиридин-7-ацетат 2-[2-(нитрокси)этокси] этила

Следуют методике примера 1a, используя указанный выше R, остаток пранопрофена, вместо остатка R группы I, как показано в примере 1a.
1H-ЯМР анализ (CDCl3)(80 МГц) показал следующие результаты:
1.51 (3H, д); 3.60 (5H,м); 4.16 (4H,м); 4.61 (2H,т); 6.90- 7.00 (3H,м); 7.35 (1H,дд); 7.40 (1H,с);
8.36 (1H,дд).
Пример 1q
Получение соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы VIII, X1 является
-(CH2-CH2-O)2, A=RCOO, R является остатком этодолака, имеющим формулу
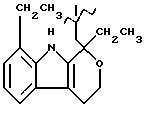
обозначенного здесь как Этодолак-NO-DEG и имеющего общую формулу:
1,8-диэтил-1,3,4,9-тетрагидропирано[3,4-b]индол-1-ацетат 2-[-(нитрокси)этокси]этила
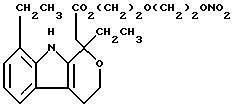
Следуют методике примера 1a, используя указанный выше R, остаток этодолака, вместо остатка R группы I, как показано в примере 1a.
1H-ЯМР анализ (ДМСО)(80 МГц) показал следующие результаты:
0.75 (3H,т); 1.38 (3H,т); 1.65-1.95 (2H,м); 2.80- 2.95 (4H,м); 3.55-3.78 (8H,м);
4.16 (2H,т); 4.58 (2H,т); 6.95- 7.05 (2H,м); 7.15 (1H,д).
Пример 1r
Получение соединения A-X1-NO2, где R относится к группе V и является остатком
соединения формулы X, X1 является YO, где Y является линейным C3 алкиленом, A=RCO, R является остатком мелоксикама, имеющим формулу
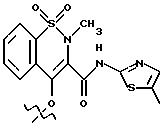
обозначенного здесь как Мелоксикам-NO-DEG и имеющего общую формулу:
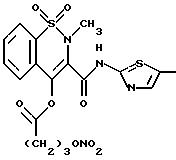
Получение промежуточного соединения Мелоксикам-CО(CH2)3Br
Мелоксикам - 5 г
4-Бромбутурил хлорид - 3.29 мл
Триэтиламин - 3.97 мл
Тетрагидрофуран - 200 мл
К раствору мелоксикама в тетрагидрофуране добавляют триэтиламин и 4-бромбутурил хлорид. Через 7 часов растворитель выпаривают в вакууме и остаток обрабатывают водой и экстрагируют этилацетатом. Органический слой обезвоживают сульфатом натрия и выпаривают. Остаток очищают хроматографией на колонке с силикагелем, элюент н-гексан/этилацетат 1/1, с получением 5.5 г соединения.
Получение промежуточного соединения, имеющего формулу Мелоксикам-NO-DEG
Meлoкcикaм-CO(CH2)3Br
- 5.5 г
Нитрат серебра - 2.8 г
Ацетонитрил - 100 мл
К раствору Мелоксикам-CО(CH2)3Br в ацетонитриле добавляют нитрат серебра при комнатной температуре
и полученную смесь кипятят с обратным холодильником в темноте в течение 8 часов. Осадок отфильтровывают и растворитель выпаривают в вакууме. Остаток очищают хроматографией на колонке с силикагелем,
элюент н-гексан/этилацетат 1/1, с получением 3.4 г Мелоксикама-NO-DEG.
1H-ЯМР анализ (ДМСО)(80 МГц) показал следующие результаты:
2.02 (2H, м); 2.30 (3H,с): 3.45
(3H.с): 4.17 (2H,т); 4.38 (2H,т); 7.16 (1H,с); 7.50 (1H,дд); 7.79 (1H,д); 7.90-8.05 (2H,м); 11.35 (1H,с).
Пример 1s
Получение соединения A-X1-NO2, где R
относится к группе III и является остатком соединения формулы VII, X1 является -C6H4-CH3-О-, A=RCOO, R является остатком фенопрофена, имеющим формулу
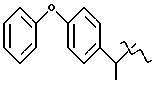
обозначенного здесь как Фенопрофен-NO-DEG и имеющего общую формулу:
α - метил-4-феноксибензол ацетат 3 (нитроксиметил)фенила
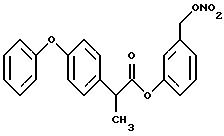
Следуют методике примера 1c, используя указанный выше R, остаток фенопрофена, вместо остатка R группы III, как показано в примере 1c.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.61 (3H,д); 3.90 (1H,кв); 5.41 (2H,c); 6.89-7.14 (4H,м); 7.35-7.80 (9H, м).
Пример 1t
Получение соединения A-X1-NO2, где R относится к группе II
и является остатком соединения формулы IIb, X1 является YO, где Y является линейным C4 алкиленом, A=RCOO, R является остатком флуниксина, имеющим формулу
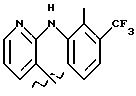
обозначенного здесь как Флуниксин-NO-DEG, и имеющего общую формулу:
2[[2-метил-3-(трифторметил)фенил] амино] -3-пиридинкарбоксилат 4-(нитрокси)бутила.

Следуют методике примера 1i, используя указанный выше R, остаток флуниксина, вместо остатка R группы I, как показано в примере 1i.
1H-ЯМР анализ (CDCl3) (80 МГц)
показал следующие результаты:
1.65 (4H, м); 1.80 (3H,с); 4.59 (2H,7); 4.25 (2H,т); 6.45 (1H,д); 7.25 (1H,д); 7.48 (1H,д); 7.75 (1H,м); 7.93 (1H,д); 8.50 (1H.д); 8.90 (1H,с).
Пример 1u
Получение соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы II, X1 является -(CH2-CH2
-O)2, A=RCOO, R является остатком супрофена, имеющим формулу

обозначенного здесь как Супрофен-NO-DEG, и имеющего общую формулу:
α- метил-4-(2-тиенилкарбонил)бензол ацетат 2-[2-(нитрокси) этокси]этила

Следуют методике примера 1a, используя указанный выше R, остаток супрофена, вместо остатка R группы I, как показано в примере 1a.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.51 (3H, д); 3.62 (5H,м); 4.20 (2H,т); 4.60 (2H,т); 6.96 (1H,д); 7.50 (2H,м); 7.65 (2H,м); 7.94
(2H,м).
Пример 1v
Получение соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы XXI, X1 является -C6H4-CH2-O, A=RCOO, R является остатком капрофена, имеющим формулу
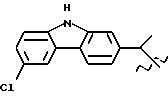
обозначенного здесь как Капрофен-NO-DEG, и имеющего общую формулу:
6-хлор α- метил-9Н-карбазол-2-ацетат 3-(нитроксиметил)фенила.

Следуют методике примера 1c, используя указанный выше R, остаток капрофена, вместо остатка R группы III, как показано в примере 1c.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.60 (3H, д); 3.2 (1H,кв); 5.41 (2H.с); 6.89-7.14 (5H,м); 7.30 (1H,д); 7.39 (1H,д); 7.55 (1H,с);
7.60 (1H,с); 8.20 (1H.с).
Пример 1w
Получение соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы VI, X1
является -C6H4-CH2-О, A=RCOO, R является остатком индопрофена, имеющим формулу
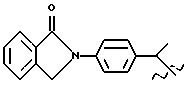
обозначенного здесь как Индопрофен-NO-DEG, и имеющего общую формулу:
4-(1,3-дигидро-1-оксо-2H-изоиндол-2-ил) α- метилбензолацетат 3-(нитроксиметил)фенила.
Следуют методике примера 1c, используя указанный выше R, остаток индопрофена, вместо остатка R группы III, как показано в примере 1c.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.51 (3H,д);
4.60 (1H,кв); 4.70 (2H,дд); 5.43 (2H,с); 6.91-7.19 (7H,м); 7.40 (1H,дд); 7.52-7.64 (4H,м).
Пример 1y
Получение соединения A-X1-NO2, где R относится к
группе III и является остатком соединения формулы X, X1 является -(CH2-CH2-O)2, A=RCOO, R является остатком толметина, имеющим формулу

обозначенного здесь как Толметин-NO-DEG, и имеющего общую формулу:
1-метил-5-(4-метилбензоил)-1H-пиррол-2-ацетат 2-[2- (нитрокси)этокси] этила
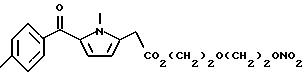
Следуют методике примера 1a, используя указанный выше R, остаток толметина, вместо остатка R группы I, как показано в примере 1a.
1Н-ЯМР анализ (CDCl3) (80 МГц) показал следующие
результаты:
2.10 (3H, с); 3.18 (3H,с); 3.55-3.63 (4H,м); 3.84 (2H,с); 4.62 (2H,т); 4.15 (1H,т); 6.32 (1H,д); 6.60 (1H,д); 7,18 (2H,д); 7.85 (2H,д).
Пример 1z
Получение соединения A-X1-NO2, где R относится к группе III и является остатком соединения формулы III, X1 является -(CH2-CH2-О)2,
A=RCOO, R является остатком фенбуфена, имеющим формулу

обозначенного здесь как Фенбуфен-NO-DEG, и имеющего общую формулу:
γ- оксо-[1,1'-бифенил]-4-бутаноат 2-[2-(нитрокси)этокси]этила
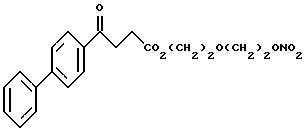
Следуют методике примера 1a, используя указанный выше R, остаток фенбуфена, вместо остатка R группы I, как показано в примере 1a.
1
H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
2.71 (2H, т); 3.32 (2H,т); 3.57 (4H,м); 4.20 (2H,т); 4.61 (2H.т); 7.22 (3H,м); 7.40-7.55 (4H,м); 7.70 (2H,м).
Пример 1aa
Получение соединения A-X1-NO2, где R относится к группе IIId и является остатком соединения формулы XXX, X1 является YO, где Y является
линейным C4 алкиленом, A=RCOO, R является остатком бермопрофена, имеющим формулу

обозначенного здесь как Бермопрофен-NO-DEG и имеющего общую формулу:
10,11-дигидро -α,8- диметил-11-оксодибенз[b, f] оксепин-2-ацетат 4-(нитрокси)бутила.
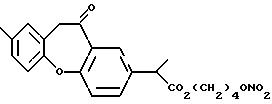
Следуют методике примера 1i, используя указанный выше R, остаток бермопрофена, вместо остатка R группы I, как показано в примере 1i.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.52 (3H,д); 1.59-1.67 (4H,м); 2.10 (3H,c); 3.61 (1H,кв); 4.04
(2H,дд); 4.10 (2H, т); 4.30 (2H,т); 6.71-6.95 (3H,м); 7.14 (1H,д); 7.51 (1H,д); 7.80 (1H,с).
Пример 1ab
Получение соединения A-X1-NO2, где R относится к
группе IIId и является остатком соединения формулы XXXII, X1 является YO, где Y является линейным C4 алкиленом, A=RCOO, R является остатком пемедолака, имеющим формулу

обозначенного здесь как Пемедолак-NO-DEG и имеющего общую формулу:

Следуют методике примера 1i, используя указанный выше R, остаток пемедолака, вместо остатка R группы I, как показано в примере 1i.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
0.76 (3H,т); 1.60-1,66 (4H,м); 1.75-1.95 (2H,м); 2.35-2.55 (2H,м); 3.40 (2H, дд);
3.70-4.10 (5H,м); 4.18 (2H.т), 4.31 (2H,т); 7.00 (2H,м); 7.10-7.35 (7H,м); 7.90 (1H,с).
Пример 1ac
Получение соединения A-X1-NO2, где R относится к
группе IIId и является остатком соединения формулы XXXVI, X1 является YO, где Y является линейным C4 алкиленом, A=RCOO, R является остатком залтопрофена, имеющим формулу

обозначенного здесь как Залтопрофен-NO-DEG и имеющего общую формулу:
10,11-дигидро-α -метил-10-оксодибенз[b,f]тиепин-2-ацетат 4-(нитрокси)бутила.

Следуют методике примера 1i, используя указанный выше R, остаток залтопрофена, вместо остатка R группы I, как показано в примере 1i.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие
результаты:
1.50 (3H,д); 1.60 (4H,м); 3.60 (1H,кв); 4.00 (2H,дд); 4.30 (2H,т); 4.60 (2H,т); 6.90-7.05 (2H,м); 7.30-7.45 (4H,м); 7.71 (1H,д).
Пример 1ad
Получение
соединения A-X1-NO2, где R относится к группе IIId и является остатком соединения формулы XXXVII, X1 является -(CH2-CH2-O)2, A=RCOO, R
является остатком мофезолака, имеющим формулу
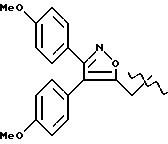
обозначенного здесь как Мофезолак-NO-DEG, и имеющего общую формулу:
3,4-бис(4-метоксифенил)-5-изоксазолацетат 2-[2-(нитрокси)этокси] этила.
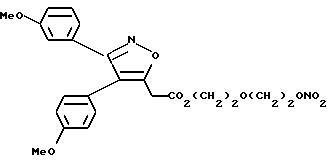
Следуют методике примера 1a, используя указанный выше R, остаток мофезолака, вместо остатка R группы I, как показано в примере 1a.
1
H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
3.57-3.62 (1H, м); 3.81 (2H,с); 4.05 (6H,с): 4.18 (2H,т); 4.64 (2H,т); 6.70-6.86 (4H,м); 7.03 (2H,д); 7.38 (2H,д).
Пример 1ae
Получение соединения A-X1-NO2, где R относится к группе Ve и является остатком соединения формулы XI, X1 является C6H4-CH2, A=RCO, R является остатком ампироксикама, имеющим формулу

обозначенного здесь как Ампироксикам-NO-DEG, и имеющего общую формулу:

Следуют методике примера 1m, используя указанный выше R, остаток ампироксикама, вместо остатка R группы V, как показано в примере 1m.
1H-ЯМР анализ (ДМСО) (200 МГц) показал следующие
результаты:
1.45 (3H,д); 3.45 (3H,с); 5.41 (2H,с); 6.25 (1H,кв); 6.91- 7.19 (4H,м); 7.30 (1H,т); 7.52 (1H,дд); 7.75 (2H,м); 8.00 (2H,м); 8.16 (1H,т); 8.35 (1H, д).
Пример
1af
Получение соединения A-X1-NO2, где R относится к группе IIId и является остатком соединения формулы XXXI, X1 является YO, где Y является линейным C4 алкиленом, A=RCOO, R является остатком залтопрофена, имеющим формулу
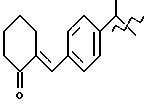
обозначенного здесь как CS-670-NO-DEG, и имеющего общую формулу:
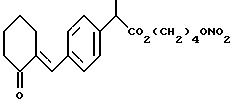
Следуют методике примера 1i, используя указанный выше R, остаток залтопрофена, вместо остатка R группы I, как показано в примере 1i.
1H-ЯМР анализ (CDCl3) (80 МГц) показал следующие результаты:
1.56 (3H, с); 1.65 (4H,м); 1.70-2.55 (8H,м); 3.87 (1H,кв); 4.25 (2H,т); 4.31 (2H,т); 7.45 (2H,д); 7.57 (2H.д); 7.70 (1H,с).
Пример 1ag
Получение соединения A-X1
-NO2, где R относится к группе IV и является остатком соединения формулы XII, X1 является -(CH2-CH2-O)2, A=RCOO, R является остатком бромофенака,
имеющим формулу
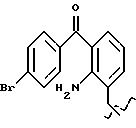
обозначенного здесь как Бромофенак-NO-DEG и имеющего общую формулу:
2-амино-3-(4-бромбензоил)бензол ацетат 2-[2-(нитрокси)этокси] этила.

Следуют методике примера 1a, используя указанный выше R, остаток бромофенака, вместо остатка R группы I, как показано в примере 1a.
1H-ЯМР анализ (CDCl3) (80 МГц) показал
следующие результаты:
3.80 (2H, с), 3.57 (4Н,м), 4.20 (2H,л), 4,61 (2H,т), 7,20 (1H,дд), 7,35 (1H,д), 7,45 (3H,м), 7,85 (2H,д).
Пример 1ah
Получение соединения
A-X1-NO2, где R относится к группе I и является остатком соединения формулы I C3, X1 является -(CH2-CH2-O)2, A= RCOO, R
является остатком бромофенака, имеющим формулу:

обозначенного здесь как Сульфасалазин-NO-DEG, и имеющего общую формулу:
2-гидрокси-5-[[4-4 [(2-пиридиниламино)сульфонил] фенил] азо]бензоат 2-[2-(нитрокси)этокси]этила.
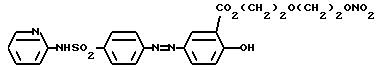
Следует методике примера 1a, используя указанный выше R, остаток сульфасалазина, вместо остатка R группы I, как показано в примере 1a.
1H-ЯMP анализ (ДМСО) (80 МГц) показал следующие результаты
3.57 (4H,м), 4.20 (2H,т), 4,61 (2H,т) 7.00-8.60 (11H,м),
Пример 1a
Получение соединения
A-X1-NO2, где R относится к группе I и представляет остаток соединения формулы Ib, X1 представляет YO, где Y является линейным C2 алкиленом, A=RCOO, R
представляет остаток ацетилсалицилсалициловой кислоты, имеющей формулу:

и относится при этом к Ацетилсалициловой-NO-DEG и имеет общую формулу:
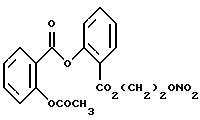
Получение промежуточного продукта (I), имеющего формулу ацетилсалицилсалициловой -CO2(CH2)2Br
Ацетилсалицилсалициловая кислота - 9,3 г
N,N-диметилформамид - 80 мл
Гидрид натрия (80% суспензии в виде белого минерального масла) - 1 г
1,2 -Дибромэтан - 8,1 г
К раствору ацетилсалицилсалициловой кислоты в DMF (50 мл) при температуре 0oC добавляли гидрид натрия. Смесь перемешивали в течение 2 часов и затем добавляли каплями в течение более 6 часов в раствор 1,2-дибромэтана в DMF (30 мл) при комнатной температуре. Раствор перемешивали в течение 24 часов и концентрировали в условиях вакуума. Остаток обрабатывали дихлорметаном и водой. Выделяли органический слой и концентрировали в условиях вакуума. Остаток очищали с помощью хроматографической колонны с силикатным гелем, элуент n-гексан/этилацетат 1/1, чтобы получить 11 г промежуточного соединения (I).
Получение ацетилсалицилсалициловой-NO-DEG
Промежуточный продукт
- 11 г
Нитрат серебра - 6,9 г
Ацетонитрил - 200 мл
Нитрат серебра добавляли в раствор промежуточного продукта (I) ацетонитриле и смесь орошали в темноте в течение 4 дней.
Хлорид серебра фильтровали и раствор концентрировали в условиях вакуума. Остаток очищали в хроматографической колонне с силикатным гелем, элуент n-гексан/этилацетат 1/1, чтобы получить 11,5 г
ацетилсалицилсалициловой - NO-DEG.
А1H-NMR анализ (CDCl3) (80 MHz), получая следующие данные: 2,3 (3H,s), 4,25 (2H,t), 4,50 (2H,t) 7,30 (6H,m) 7,67 (1H,m), 8,20 (1H,d).
Пример 1aj
Получение соединения A-X1-NO2, где R относится к группе IIID и представляет остаток соединения формулы IIIa, X является YO, где Y
является разветвленным C2алкиленом, A= RCOO, R является остатком пранопрофена, имеющего формулу:

и имеющего общую формулу:

Получение промежуточного продукта I
L-метил-5H-(1) бензопирано(2,3-b)пиридин-7-уксусная кислота 1-хлор-2-метил-2-пропил эфир.
Пранопрофен - 5,1 г
1-Хлор-2-метил-2-пропанол - 2,2 г
N,
N'-дициклогексилкарбодиимид - 4,2 г
4-Диметиламинопиридин - 0,25 г
Хлороформ - 100 мл
N,N-диметилформамид - 20 мл
К раствору пранопрофена в хлороформе и DMF
добавляли при комнатной температуре 1-хлор-2-метил-2-пропанол, N, N'-дициклогексилкарбодиимид и 4-диметиламинопиридин. Смесь перемешивали в течение 24 часов. Твердую массу фильтровали и раствор
промывали водой. Выделяли органический слой и концентрировали в условиях вакуума. Остаток очищали в хроматографической колонне с силикатным гелем, элуент n-гексан/этилацетат 7/3, чтобы получить 4,84 г
промежуточного продукта 1.
Получение нитрата:
L-метил-5H-(1) бензопирано[2,3-b] пиридин-7-уксусная кислота 1- нитрокси-2-метил-2-пропил эфир.
Промежуточный
продукт - 4,5 г
Нитрат серебра - 2,6 г
Ацетонитрил - 100 мл
Нитрат серебра добавляли в раствор промежуточного продукта 1 в ацетонитриле и смесь орошали в темноте в течение 2
дней. Хлорид серебра фильтровали и раствор концентрировали в условиях вакуума. Остаток очищали в хроматографической колонне с силикатным гелем, элуент - гексан/этилацетат 7/3, чтобы получить 3 г
соединения.
А1H-NMR анализ (CDCl3) (80 MHz), получая следующие данные: 1,29 (6H,s), 1,51 (3H, d), 3,50 (1H,q) 4,15 (4H, m), 6,90-7,00 (3H,m) 7,35 (1H,dd), 7,40 (1H,s) 8,36 (1H,dd).
Реферат
Изобретение относится к нитроксисоединениям общей формулы А-Х1-NО2 или их солям, где А и Х1 имеют значения, указанные в формуле изобретения, а также к фармацевтической композиции на их основе. Соединения и композиция могут быть использованы в медицине, как продукты, обладающие противовоспалительной, анальгетической и антитромботической активностями, а также активностью против септического шока. 2 c. и 2 з.п. ф-лы, 4 табл.
Формула
А - Х1 - NO2
или их соли,
где A = R(COXu)t, где t = 0 или 1, u = 0 или 1; Х = О, NH; R выбирают из следующих групп:
группа I), где t = 1, u = 1,
где R1 означает OCOR3, где R3 - метил, R2 означает водород, гидрокси;
группа II), где t = 1, u = 1,

где RII1, RII2, RII3 независимо друг от друга означают водород или Сl, RII4 означает RII1, RII5 означает водород, RII6 означает водород;
группа III), где t = 1, u = 1 и R означает
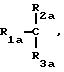
где R2а и R3а означают Н, линейный или, когда возможно, разветвленный, замещенный или незамещенный С1-С12алкил; предпочтительно R2а означает Н, алкил, имеющий от 1 до 4 С,
R3а означает Н;
R1а выбирают из соединений формул






IIID) имеют следующие соединения:



в соединении формулы (IV), остаток кетопрофена:
RIII1 означает Н, RIII2 означает Н, предпочтительные соединения, где R3а означает Н, R2а означает метил, Х = O; R3а означает водород;
в соединениях формулы (XXI), остаток карпрофена:
Rxxio означает Н, Rxxi означает Н, Rxxi1 означает галоген, предпочтительные соединения, где Rxxio означает Н, связывающий мостик в положении 2, Rxxi1 означает хлор в параположении к азоту; R2а означает метил и Х = O;
в соединениях формулы (XXXV), остаток тиапрофеновой кислоты:
Ar означает фенил, предпочтительные соединения, где R2а означает метил и Х = O;
R3а означает водород;
Rxxi1 означает галоген, CN, C1-C6 алкил, содержащий одну или более ОН группы, C1-C6 алкокси, ацетил, ацетамид, бензилокси, SRIII3 как показано выше, перфторалкил, имеющий от 1 до 3 С, гидрокси, карбоксиалкил, имеющий от 1 до 6 С, NO2, амино, моно- или диалкиламино, имеющий от 1 до 6 С, сульфамоил, диалкил сульфамоил, имеющий от 1 до 6 С, или дифторалкилсульфамоил, как показано выше; или Rxxi вместе с Rxxi1 означают алкилендиокси, имеющий от 1 до 6 С;
предпочтительные соединения, где Rxxio означает Н, связывающий мостик в положении 2, Rxxi означает Н, Rxxi1 означает хлор в параположении по отношению к азоту;
R3а означает Н, R2а означает метил и Х означает О;
в соединениях формулы (XXXV), остаток тиапрофеновой кислоты:
Ar означает фенил, гидроксифенил, необязательно моно- или полизамещенный галогеном, алканоил и алкокси, имеющий от 1 до 6 С, триалкил, имеющий от 1 до 6 С, предпочтительно от 1 до 3 С, циклопентил, циклогексил, циклогептил, гетероарил, предпочтительно тиенил, фурил, необязательно содержащий ОН, пиридил;
предпочтительные (XXXV) соединения такие, где Аr означает фенил, R3а означает Н, R2а означает метил и Х означает О;
в соединении формулы (II) остаток супрофена, где R3а означает Н, R2а означает метил и Х = О, или его эквиваленты;
в соединении формулы (VI) остаток индопрофена, где R2а означает СН3, и остаток индобуфена, где R2а означает Н, R3а = -СН3, и Х = О и его эквиваленты;
в соединениях формулы (VIII) остаток этодолака, где R2а = R3а = Н и Х = О и его эквиваленты;
в соединениях формулы (VII) остаток фенопрофена, где R3а = Х, R2а = -СН3 и Х = О и его эквиваленты;
в соединениях формулы (III) остаток фенбуфена, где R2а = R3а = Н и Х = О и его эквиваленты;
в соединениях формулы (IX) остаток фторбипрофена, где R3а означает Н, R2а означает -СН3 и Х = О;
в соединениях формулы (Х) остаток толметина, где R2а = R3а = Н и Х = О или его эквиваленты;
соединение IIIa), когда оно содержит -СН(СН3)-СООН, пранопрофен остаток: aльфа-метил-5Н-[1]бензопиран[2,3b]-пиридин-7-уксусная кислота;
предпочтительное соединение имеет R2а = Н, R3а = -СН3, u = 1 и Х = О;
соединение (ХХХ), когда оно содержит -СН(СН3)-СООН, бермопрофен остаток: дибензо[b,f]оксепин-2-уксусная кислота;
предпочтительное соединение имеет u = 1, Х = О, R2а = Н, R3а = СН3;
соединение (XXXI)-CS-670 остаток: 2-[4-(2-оксо-1-циклогексилиденметил)фенил] пропионовая кислота, когда радикал означает - СН(СН3)-СООН; предпочтительное соединение имеет R2а = Н, R3а = СН3, u = 1, Х = О;
соединение (XXXII) производное пемедолака, который содержит - СН2СООН группы; предпочтительное соединение имеет R2а = R3а = H, u = 1, Х = О;
соединение (XXXIII) означает пиразолак остаток, когда насыщен - СН2СООН: 4-(4-хлорфенил)-1-(4-фторфенил)-3-пиразолил производное кислоты; предпочтительные соединения R2а = R3а = H, u = 1, Х = О;
группа, когда R1а является остатком (XXXVI), R2а = Н, R3а = СН3, t = 1, u = 1 и Х = О, известна как залтопрофен, когда насыщена гидрокси, или аминогруппой, или солями кислоты, или соли кислоты, то означает одно из производных дибензотиепина, предпочтительные продукты имеют R2а = Н, R3а = СН3, u = 1, Х = О;
соединение (XXXVII) производное из мофезолака: 3,4-ди(п-метоксифенил)изоксазол-5-уксусная кислота, когда остаток означает -СН2-СООН, предпочтительные соединения R2а = R3а = H, t = 1, X = О;
группа IV) в которой t = 1, u = 1 и R означает

где RIVd и RIVd1 означают по крайней мере одна Н, а другая линейный или, когда возможно, разветвленный С1-С6 алкил, предпочтительно С1 и С2, или дифторалкил с алкилом, имеющим от 1 до 6 С, С1 предпочтителен, или RIVd и RIVd1 вместе образуют метиленовую группу;
RIV имеет следующие значения:
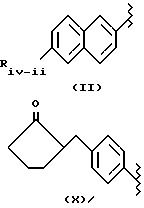
группа 1V), в которой t = 1, u = 1, R означает

где RIVd и RIVd1 означают, по крайней мере, одна Н, а другая линейный или, когда возможно, разветвленный С1 -С6-алкил, предпочтительно С1 и С2, или RIVd и RIVd1 вместе образуют метиленовую группу;
RIV имеет следующие значения: (II), (X), (III)

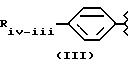
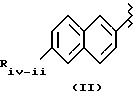
где соединения группы 1V) имеют следующие значения:
в соединениях формулы (II) RIV-ii означает -СН3О, предпочтительно RIVd означает Н и RIVd1 означает - СН3, X = NH или О, X1 = -(СН2-СН2 -О)2;
в соединениях формулы (X), остаток локсопрофена,
или его эквиваленты; предпочтительно соединения, в которых RIVd означает водород и RIVd1 означает -СН3, Х = NH или О и Х1 = (СН2-СН2-О)2;
в соединениях формулы (III) RIV-iii означает
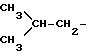
и RIVd = Н, RIVd1 означает - СН3, X = NH или О, X1 = -(СН2-СН2-О)2;
группа V) - значения (VII), (IX), (IV), (II), (X), (XI), (XII), (XIII) -


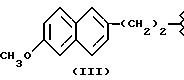


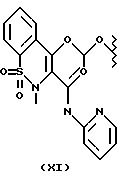
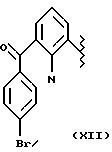

где в соединениях формулы (II) RVii означает Н, RVii-1 означает RVii

где соединения группы IV) имеют следующие значения:
в соединениях формулы (II) RIV-II означает 1 - 6 С алкил, циклоалкил, имеющий от 3 до 7 С, алкоксиметил, имеющий от 1 до 7 С, трифторалкил, имеющий от 1 до 3 С, винил, этинил, галоген, алкокси, имеющий от 1 до 6 С, дифторалкокси с алкилом, имеющим от 1 до 7 С, алкоксиметилокси, имеющий от 1 до 7 С, алкилтиометилокси с алкилом, имеющим от 1 до 7 С, алкилметилтио с алкилом, имеющим от 1 до 7 С, циано, дифторметилтио, фенил- или фенилалкил, замещенный алкилом, имеющим от 1 до 8 С;
предпочтительно Riv-ii означает -СН3О, Rivd означает Н и Rivdi означает CН3, который означает остаток напроксена;
Х = NH или О и Х1 = -(СН2-СН2-О)2;
в соединениях формулы (Х) остаток локсопрофена или его эквивалентов; предпочтительные соединения, в которых RIVd означает Н и RIVd1 означает -СН3, Х = NH или О и Х1 = -(СН2-СН2-О)2;
в соединениях формулы (III): RIV-iii означает С2-С5-алкил, разветвленный, когда возможно, С2 и С3 алкилокси, аллилокси, фенокси, фенилтио, циклоалкил, имеющий от 5 до 7 С атомов, необязательно замещенный в положении 1 С1-С2-алкилом:
предпочтительные соединения, где RIV-iii означает
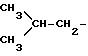
Х1 в формуле А-Х1-NO2 означает бивалентный связывающий мостик и выбран из следующих:
-YO-,
где Y означает циклоалкилен, имеющий от 5 до 7 углеродных атомов; разветвленный С2 алифатический связывающий мостик, имеющий две метиловые группы с углеродным атомом, связанные в отношении эфирной функции, исключая такую мостиковую связь, когда R означает радикал группы I), исключая Iс; радикал группы II), исключая IIб; радикал группы III), исключая класс соединений из IIID); радикал группы IV), радикал группы V), исключая X) и включая -(СН2)4- для соединений формулы (IV); группу
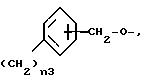
где n3 = 0 или 1; группу

где R1f = H, nf означает целое число 1 - 6, предпочтительно 2 - 4.
Х означает О, R1 означает ацетокси, предпочтительно в ортоположении по отношению к -СО-; Х1 означает (СН2-СН2-О)2, R2 означает водород;
в группе II: где RII1, RII2 и RII4 означают водород, RII3 означает хлор и RII3 в ортоположении по отношению к NH; RII5 и RII6 означают Н; Х = О и Х1 означает (СН2-СН2-О)2.
10.05.94 - по п.1: соединения, где R в А представляют группу V) формулы (VII), (IX), (IV), (III), (II) с Х1, являющимся линейным или разветвленным алкилом С1-С10;
09.08.94 - по п.1: соединения формулы А-Х1 -NO2, где R в А выбран из групп: группа 1): формула 1а), 1в); группа II); группа III): формула (II), (XXI), (IV), (XXXV), (VI), (VII), (VIII), (IX), (X), (III); группа IV): формула (II), (X), (III); группа V): формула (VII), (IX), (IV), (V), (III), (II), имеющая бивалентный связывающий мостик;
09.08.94 - по пп.2 - 4.
Комментарии