Способ получения дегидропенициллинов - SU683626A3
Код документа: SU683626A3
Описание
(54) СПОСОБ ПОЛУЧЕНИЯ ДЕГИДРОПЕНИДИЛЛИНОВ
Процесс протекает по схеме
s-CKi--cc -а.
s-CK,-cc-r. --.
СН,h
Н
сн.
R
Га
основание,кислота, mepnooSpoBof na m
DCHOloHue,шШа
где X и R имеют значения, указанные выше , а R представляет собой алкил, арил или аралкил.
В синтезе, показанном на схеме, природа грунн X и R исходного соединения И или Па не обязательно должна быть похожей на конечное соединение I, поскольку синтез можно также осуществлять с номощью общепринятых методов модификации таких групн на стадии промежуточного продукта III или 1Па, или после циклизации такого промежуточного соединения в соединение I.
Исходные соединения II или Па можно использовать как в цис-, так и тра«с-форме по отношению к заместителям у Сз и €4 лактамного кольца.
Производные формулы 1, такие как показано на схеме, оптнчески неактивные хнральные продукты, которые могут быть разделены на их энантиомеры.
Предшественники дегидропенициллинов, соединения III и П1а схемы I являются новыми продуктами.
Исходные соединения формул П и Па синтеза являются классом известных продуктов .
Процесс осуществляют следующим образом .
Исходный продукт II или Па подвергается облучению ультрафиолетовой лампой.
-f |Нз
OCHjCOlNH-- -/-снз
СООСНз
IV
В аппарате для фотометрических реакций с иммерсионной лампой, имеюще.м охлаждающий кожух из кварца и водную рубащку , снабженном магнитной мешалкой, капиллярной трубкой для ввода азота в часть и выходом, помещенным сбоку.
4- СНзСОК
СКг
-ь CH,COR -
llta
предпочтительно со средним давлением ртути , снабженной фильтром, предпочтительно фильтром Пирекса или Корекса.
Реакция проходит в атмосфере инертного газа, например азота, в инертном растворителе , таком как ароматический растворитель , или в ацетонитриле, в безводной свободной от кислорода среде нри комнатной температуре. Получают производное П1 или 1Па обычно с высокими выходами. Промежуточное соединение П1 или Ilia зате.м обрабатывают в безводном инертном растворителе , таком как галоидалифатический растворитель или ароматический, количеством , которое может быть также каталитическим , когда III или Ilia является нейтральной молекулой органического основания , предпочтительно триэтиламина, при комнатной температуре. Получают обычно с высоким выходом дегидропенициллин I.
Можно ос)ществлять циклизацию П1 в I Б инертном безводном растворителе, таком как алифатический или ароматический, также с помощью кислоты, предпочтительно силикагеля, или в нейтральной окружающей среде путем простого нагревания до 50-150°С.
Пример 1. Получение 1-(1-метоксикарбонил-2-метил-1-цропенил )-3 - феноксиацетамидо-4-тиоксо-2-азетидинона
и связанной со слоем ртути, загружают з атмосфере азота 0,965 г (2 ммоль) продукта I, растворенного в 150 мл безводного и дегазированного ацетонитрила.
Загрузку осуществляют в течение 5 мин в токе азота, затем проводят облучение при
комнатной температуре при перемешивании в течение 45 мин с помощью лампы Гановия при среднем давлении ртути, 500 Вт энергии, и с фильтром из стекла Пирекс.
Раствор затем переносят в колбу и при комнатной температуре сначала упаривают растворитель под вакуумом (13 мм рт. ст.), а затем преобладающую фракцию образованного ацетофенона под вакуумом 0,1 мм рт. ст. в течение нескольких часов. Стеклянистый остаток 0,645 г (89%) состоит почти полностью из продукта V и содержит следы ацетофенона и незначительное количество (приблизительно 10%) исходного соединения IV. Полученный продукт не кристаллизуется и хроматографически нестабилен , достаточно чист для последуюпхей S-CHi-COPli COOCHiPTi Реакция аналогична получению соединения V, исходя из соединения IV, причем в качестве исходного соединения используют сложный бензиловый эфир VI. Полученный такиА образом продукт VII (выход 86%) в форме стеклянистого твердого вещества является достаточно чистым для последующей реакции и для того, чтобы быть идентифицированным спектрофотометрически . Процедура аналогична процедуре, описанной в примере 1 для приготовления продукта V из IV, но в качестве исходного соединения используют продукт VIII. Полученный.таким образом продукт iX (выход 81%) в форме гомогенной пены, является достаточно чистым для следующей реакции, но может быть очищен снова с по- 3.5 лучением более низких выходов с помощью хроматографии в тонком слое на силикагеле элюированием бензолом, который содержит 5% этилацетата, и экстрагированием из двуокиси кремния хлороформом чистого 40 продукта в форме белой пены.
S-CHiCOPh
СНзСОИН СНз
Е-СН СОСНз СНз COOCHj
реакции и для хроматографической идентификации .
ИК-епектр (СНСЬ): Тмакг 3340 (КП); 3060 и 3040 (плечо, фенил); 1820 (СО беталактама ); 1720 (СО сложного эфира); 1682 (СО-феноксиацетамида); 1635 (плечо, С С); 1595 и 1580 (плечо, фенил); 1528 (NH), 1490 см- (феннл).
ЯМР (CDCls); б 2,06 (ЗН, S); 2,28 (ЗП, 5|(СНз)2С С); 3,80 (ЗН, S, СООСН); 4,36 (2Н, S, ОСНоСО); 4,91 (1П, S, J 8Hz, 3-Н); 6,60-7,60 (5П, М, ароматика); 7,94 (1Н, d, J 8Пг, NH).
Пример 2. Получение 1- (1-бензилоксикарбонил - 2-метил-1-пропенил)-3-феноксиацетамид - 4-тиоксо-2-азетидииона CHj
Щ
--НН
Y СНз
COOCHj
-СНз РЛ-ОСНгСОШ-т СНз ... COOCH,Ph П1 ИК-спектр аналогичен ИК-спектру продукта V. ЯМР (CDCls): б 2,06 (ЗН, S); 2,28 (ЗН, 5(СНз)2С С); 4,36 (2Н, S, ОСН.СО); 4,91 (1Н, d, J 8Hz, ЗН); 5,17 (2Н, S, СООСНа); 6,60-7,60 (ЮН, М, ароматика); 7,94 (Ш, d, J 8Hz, NH). Пример 3. Получение 1-(1-метокеикарбонил-2-метил-1-пропенил ) - 3-трифенилметиламино-4 - тиоксо-2-азетидинона ИК-снектр (СНС1з); Тмак. 3300 (NH); 3050 и 3020 (фенилы); 1810 (СО бета-лактама ); 1720 (СО сложного эфира); 1620 (плечо С С); 1595, 1590 и 1490 см- (фенилы ); 2,80 ,(1Н, d, J, 8Hz, Н);3,67 (ЗН, S, СООСНз); 4,75 (1Н, d, J, 8Hz, 3-Н); 7,10- 7,70 (15Н, М, ароматика). -1,4° (С 1,00 СН1з). Пример 4. Получение 1-(1-метоксикарбонил - 2-метил-1-пропенил)-3-ацетамидо-4тиоксо-2-азетидинона (XII). ЯМР (CDCls): б 1.81 (ЗН, S); 2,26 ЗН, S, (СНз)2С С);
А. Процесс аналогичен описанному в примере 1 для приготовления продукта V из IV, но в качестве исходного продукта используется соединение X.
Полученный таким образом продукт ХП (выход 84,5%) в виде стеклянистого твердого вещества является достаточно чистым для последующей реакции спектрофотометрического идентифицирования.
ИК-спектр (СПСЬ): умяь-- 3320 (МП); 1820 (СО бета-лактама); 1722 (СО сложного эфира); 1675 (СО ацетамида); 1635 (С С); 1530 см-1 (МП).
ЯМР (CDCls): б 2,00 (ЗП, S); 2,32 (ЗП, S, Т(СП,)); 2,08 (ЗН, S, СНзСО); 3,73 (ЗП, S, СООСПя); 4,93 (1П, d, J, ВПг, 3-П); 7,53 (1П, d, J, 8Пг, КШ).
Б. Процесс аналогичен описанному в примере 1 для приготовления продукта V, но в качестве исходного цродукта применяют соединение XI, используют фильтр из стекла Корекс и облучение осуществляют в течение 4 ч. Кроме того, в конце реакции испарение при 0,1 мм рт. ст. не требуется, поS сн,-com сн, li
CHj СООСНз
Процесс аналогичен описанному в примере 1 для приготовления продукта V из VI, но в качестве исходного продукта используют соединение XIII. Продукт XIV, который получают (78,5%) в виде стеклянистого твердого вещества, является достаточно чистым для следующей реакции и для того, чтобы быть идентифицированным спектрофотометрически .
РЛОСНг,СОШ.
сн.
сооенз
А. К 0,645 г (1,78 ммоль) продукта V, приготовленного в соответствии с примером 1, растворенным в 30 мл хлористого метилена , добавляют 0,08 мл триэтиламина и раствор, перемещивают при комнатной температуре в безводной атмосфере (азот) до тех пор, пока не будет наблюдаться в ИКспектре раствора полное исчезновение полосы при 1820 см-, которая является типичной для исходного продукта V (около 4 ч). Смесь затем сущат в вакууме и сырой продукт очищают с помощью хроматографии на силикагельной воронке (2X20 см) при элюировании бензолом и этилацетатом (4 : 1). После элюирования некоторых примесей из последовательных элюатов выдескольку в данном случае побочно образующиГкя кетон представляет собой ацетон, а не ацетофенон.
Пеочищенный смолообразный продукт не кристаллизуется, является хроматографически нестабильным и используется в последующей реакции.
По данным ИК-спектра и ЯМР видно, что неочищенный продукт содержит около 40% продукта ХП и выход чистого продукта составляет около 37%.
В результате опыта по частичной очистке сырого материала с помощью солюбилизации в этиловом эфире и осаждения петролейны .м эфиром получают с низким выходом смолообразный продукт, который является более чистым, ПК- и ЯМР-спектры в сравнении со спектрами продукта, полученного из соединения X, подтверждают структуру поодукта XII.
П р и .м е р 5. Получение 1-(1-метоксикарбонил - 2-метил-2-пропенил)-3-ацетампдо-4тиоксо-2-азетидинона
СНг
,
соосн,
ИК-спектр (пленка): умакс 3300 (МП); 1818 (СО бета-лактама); 1740 (СО сложного эфира); 1670 (СО ацетамида), 1530 см (МП). ЯМР (CDCls): б 1,88 (ЗП, S, СПз, -С
С); 2,07 (ЗП, S, СПзСО-); 3,80 (ЗП, S,
СООСПз); 4,90-5,25 (4П, ,М, СП2 С,
3-П и СПСОО); 7,50 (Ш, d, J, 8Пг, МП).
Пример 6. Приготовление метил 6-феноксиацетамидодегидропенициллаиата
PllOCH2CaNH.
Ы
XV
ляют 0,460 г продукта XV (ыход 71% из V и 63,5% из IV), затем приблизительно 0,05 г исходного продукта IV реакции примера 4.
Дегидропенициллин XV получают в виде аморфного белого твердого вещества, которое является чистым при хрол-атографии в тонком слое, его кристаллизуют из смеси этилового эфира и петролейного эфира; т. пл. 138-139°С.
УФ-спектр (etOn): Ямаке (loge)
220 ( 3,88); 245 (3,87) и 328 ммк (4,41).
ИК-спектр (СНС1з): 7макс 3300 (NH);
3060 и 3020 (фенил); 1742 (СО сложного
эфира); 1720 (СО бета-лактама); 1630 (СО
феноксиацетамида); 1,595 и 1585 (плечо, фенил); 1565 (NH); 1493 см- (фенил).
ЯМР (CDCls): б 1,47 (ЗН, S) и 1,73 {ЗН, S, (СНз) 3,80 (ЗН, S, СООСНз): 4,52 (1Н, S 3-4); 4,80 (2Н, S, ОСНзСО); 6,80- 7,50 (5Н, ароматика); 7,85 (1Н, S, широкая полоса, NH).
Масс-спектр: м/с 362, 269, 209, 199 167, 139, 94, 66.
Вычислено, %: С 56,34; Н 5,01; N 7,73.
C,7Hi8N205S.
Найдено, %: С 55,92; Н 4,88; N 7,52.
Б. К 0,100 г (0,28 ммоль) продукта V, приготовленного в соответствии с примером 1, растворенного в 10 мл хлороформа, добавляют 5 г силикагеля. Смесь перемешивают при комнатной температуре в течение ночи, затем фильтруют, осуществляют повторно промывку кремнезема хлороформом.
Фильтрат, упаренный досуха в вакууме, показывает в ИК-спектре исчезновение полосы при 1820 . Сырой продукт очищаPnoCHjiCONK
снз
H-v.CH3 COOCHiPn
YJI
Процедура аналогична процедуре, описанной в примере 6, А для приготовления XV из IV с триэтиламином, но в качестве исходного вещества используют бензнловый сложный эфир VII, приготовленный в соответствии с примером 2. Продукт, который получают (72,5%) в виде белого аморфного вещества, согласно тонкослойной хроматографии чистый, его можно перекристаллизовывать из смеси этилового эфира и петролейного эфира; т. пл. 129-131°С.
ИК-спектр (CHCls): 3300 (NH); 3040,-3020 (фенилы); 1745 (СО сложного эфира); 1720 (СО бета-лактама); 1630 (СО
-Y JH3 -у.
cnocH,s
Процесс аналогичен описанной в примере 6,А для приготовления соединения XV из V с триэтиламином, но в качестве исходного вещества используют продукт IX, приготовленный в соответствии с примером 3.
В этом случае для достижения исчезновения полосы при 1810 см- требуется больще времени (около 24 ч). Полученный таким образом сырой материал очищают на нейтральной окиси алюминия с элюированием бензолом. Продукт, который получают (выход 23%) в форме пены, является чистым при хроматографии в тонком слое, но он является малостабильным.
ют с пo tolцью .хроматографии на препаративном тонком слое силикагеля с элюированием бензолом и этилацетатом (7: 3) и экстрагированием продукта из кремнезема хлороформом. Получают 0,038 г продукта XV, идентичного продукту, полученному на стадии А.
Выход 38% нз V и 34% из IV.
В. Раствор в 10 мл тетрахлорэтилена 0,100 г (0,28 ммоль) продукта V, приготовленного в соответствии с примером 1, перемешивают в течение 10 ч при 90-100°С. Охлажденный раствор показывает в ИКгпектре исчезновение полосы при 1820 см-. Его сушат в вакууме и огтаток очищают, как описано в пункте Б. Получают 0,036 г продукта XV, идентичного продукту, полученному пункта А.
Выход 36% из V и 32% из IV.
Пример 7. Получение бензил 6-феноксиадетамидодегидропеницнлланата
,/S
СНз
СНз j--и1
Чоосн,рл
XVI
феноксиацетамида); 1600 и 1590 (плечо, фенил ): 1565 (NH); 1465см- (фенилы).
ЯМР-спектр (CDCls): б 1,35 (ЗН, S) и 1,63 {ЗН, S, (СНз) 4,50 (Н, S, 3-4); 4,80 (2Н, S, ОСН2СО); 5,18 (2Н, S, COOCHs); 6,70-7,50 (ЮН, М, ароматика); 7,85 (1Н, S, широкая полоса, NH).
Масс-спектр: м/е 439, 438, 345, 303, 209, 94, 91, 66.
Вычислено, %: С 63,00; Н 5,05; N 6,39.
С2зЕ2зКЬ055.
Найдено, %: С 62,58; Н 5,06; N 6,17. Пример 8. Получение метил 6-трифенилметиламинодегидропеницилланата
СНз CHj
COOCHj
XVll
ИК-спектр (СНС1з): 7мако 3320 (NH); 3060 и 3020 (фенилы); 1750 (СО сложного эфира); 1730 (СО бета-лактама); 1595, 1580 (плечо, фенил) и 1490 см- (фенилы). ЯМР (CDCb): б 1,35 (ЗН, S); 1,72 (ЗН, S, (СНо) 3,20 (1Н, S, широкая полоса, NH); 3,80 (ЗН, S, СООСНз); 4,58 (1Н, S, 3-4); 7, 10-7,68 (15Н, М, ароматика).
Вычислено, %: С 71,47; Н 5,57; N 5,95. CogHoeNoO S.
Найдено, %: С 71,01; Н 5,55; N 7,75.
Пример 9. Получение метил 6-ацетамндодегидропеницилланата
11
12
1т
снз
СНз
-к1
cooCHj
xvni
А. Процесс аналогичен описанному в примере 6,А.
Для приготовления соединения XV из соединения V в Присутствии триэтиламина, но в качестве исходного материала используют продукт VII, приготовленный согласно примеру 4.
Получаемый продукт очищают в первом случае с помощью хроматографии на колонке , как описано в примере 6,А с элюированием , но смесью этилового эфира и этилацетата (2:1) (выход 50,5% из XII и 43% из X) и с помощью хроматографии в препаративном тонком слое силикагеля, но при элюировании смесью этилового эфира и этилацетата (3:1) и экстрагированием из кремнезема хлороформом (выход 24% из XII).
Продукт, получаемый в виде стеклянистого твердого вещества, является чистым п кристаллизоваться из смеси этилового и петролейного эфира; т. пл. 98-100 С.
ИК-спектр (СНС1з): -у-макс 3300 (NH); 1740 (СО сложного эфира); 1715 (СО-бетаРаствор в 20 мл смеси этанола и этилацетата (4:1) 0,438 г (1 ммоль) продукта XVI, приготовленного согласно примеру 7, гидрируют в присутствии 0,430 г 10%-пого палладия на активированном угле при атмосферном давлении и комнатной температуре в течение 30 мин. Катализатор отфильтровывают , к фильтрату добавляют 0,430 г свежего катализатора и гидрирование продолжают еще в течение 40 мин при тех же условиях. Катализатор снова отфильтровывают , и раствор сразу доводят до рН 7,8-8,0 с помощью 5%-ного водного раствора бикарбоната калия и охлаждении ледяной баней. Смесь упаривают досуха в вакууме при комнатной температуре, тверлый осадок забирают в этилацетат, фильтруют и продукт XIX осаждают этиловым эфиром из раствора. ГсОпмают 0,330 г продукта XIX (выход 85% ).
ИК-спектр (КВч): YMaKc 3400 (широкая
лактама); 1630 (СО ацетамида); 1570 см (NH).
ЯМР-спектр (CDCla): б 1,47 (ЗН, S) 1,75
ЗН, S, (СНз) 2,20 (ЗН, S, СНзСО);
3,80 (ЗН, S, СООСНз); 4,48 (1Н, S, 3-4);
7,60 (Ш, S, СООСНз); 4,48 (1Н, S, 3-4);
7,60 (1Н, S, широкая полоса, NH).
Масс-спектр: м/е 270, 238, 211, 199, 139, 38, 39. Вычислено, %: С 48,88; Н 5,22; N 10,36.
CnH,4N204S.
Найдено, %: С 48,55; Н 5,48; N 9,88.
Б. Процесс аналогичен примеру 6,А для приготовления соединения XV из V с триэтиламином , но в качестве исходного продукта используют соединение XIV, приготовленное в соответствии с примером 5. Продукт, иолучаемый (выход 32% из XIV и 25% из XIII) в виде аморфного твердого вещества после тонкослойной преперативР ой хроматографии, идентичен продукту, приготовленному согласно примеру 9,А.
Пример 10. Приготовление 6-феноксиацетамидодегидропеницилланатакалия
FhOCH CONH
COOK
полоса, HjO); 3320 (плечо, NH); 3060 и 3040 (плечо, фенил); 1703 (СО-бета-лактама ); 1620 (СО феноксиацетамида); 1602 (щирокая полоса, СО карбоксилат); 1590 (плечо, фенил); 1555 (NH); 1493 см- (фенил ).
ЯМР (CDsOD): б 1,51 (ЗН, S); 1,76 ЗН, S, (СНз) 4,38 (1Н, S, 3-4); 4,86 (2Н, S, ОСНгСО); 6,85-7,40 (5Н, М, ароматика ).
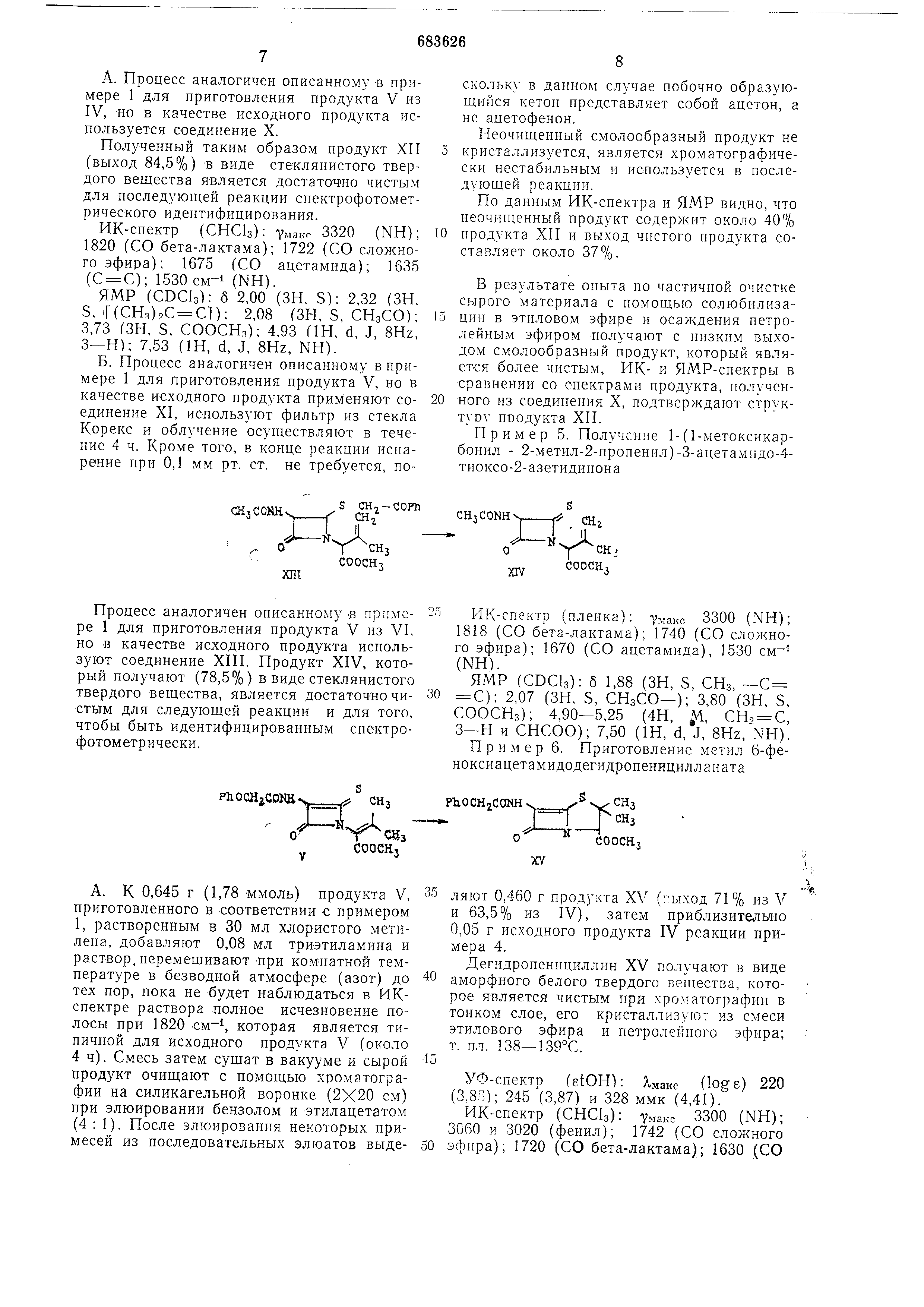
Формула изобретения
1. Способ получения дегидропенициллинов общей формулы
Х-.S
где X - аминогруппа, замещенная ацетилом , феноксиацетилом или тритилом;
13
R - метоксикарбонил, бензилоксикарбоинл или калиевая соль карбоновой кислоты , отличающийся тем, что замещенный 4-ацилметилтио-2-азетидинон подвергают облучению при ком«атной температуре в присутствии безводного инертного растворителя , свободного от кислорода, в атмосфере инертного газа, с последующей циклизацией образующегося продукта путем обработки основанием или кислотой при комнатной температуре или путем нагревания до температуры между 50 и 150°С в при14
сутствии оезводного инертного раствори теля.
Способ по п. 1, о т л и ч а ю щ и и с я тем, что в качестве основания применяют органическое основание, такое как триэтиламин , а в качестве кислоты применяют снликагель .
Источники информации, принятые во внимание при экспертизе 1. Патент США № 3159617, кл. 260- 239.1, опубл. 1964.






Реферат
Формула
Комментарии