1,4-дизамещенные пиперазины, способ их получения,фармацевтическая композиция и способ лечения нейрогенного воспаления - RU2188197C2
Код документа: RU2188197C2
Чертежи

Описание
Изобретение относится к новым N-замещенным азагетероциклическим соединениям, в которых замещенная алкильная цепь образует часть N-заместителя, или их солям, способам их получения, содержащим их композициям, применению соединений с целью получения композиций для клинической терапии болезненных состоянии, повышенной болевой чувствительности и воспалительных состояний, в которых патофизиологическую роль играют С-волокна, вызывая нейрогенную боль или воспаление, и способам лечения указанных болезненных состояний, повышенной болевой чувствительности и воспалительных состояний. Данное изобретение относится также к применению данных соединений для снижения глюкозы в крови и/или ингибирования секреции, циркуляции или действия инсулин-антагонистических пептидов, таких как CGRP или амилин, известно, что настоящие соединения оказывают негативное влияние содержащим С-волокна, содержащие нейропептиды. Следовательно, настоящие соединения можно использовать при терапии невосприимчивости к инсулину при инсулиннезависимом сахарном диабете (NIDDM) для улучшения толерантности к глюкозе, а также возрастного ожирения.
Нервная система оказывает глубокое действие на воспалительную реакцию. Антидромная стимуляция чувствительных нервов дает ограниченное расширение кровеносных сосудов и повышенную проницаемость сосудов (Janecso и др. Br. J. Pharmacol. 1967, 31, 138-151), аналогичную реакцию наблюдают и после инъекции пептидов, которые, как известно, присутствуют в чувствительных нервах. Таким образом, пептиды, высвобождающиеся из окончаний чувствительных нервов, опосредуют многие воспалительные реакции в таких тканях, как кожа, суставы, мочевые пути, глаза, мозг, желудочно-кишечный тракт и дыхательные пути. Следовательно, ингибирование высвобождения и/или активности пептидов чувствительных нервов может быть полезно в терапии, например, артрита, дерматита, ринита, астмы, цистита, гингивита, тромбофлебита, глаукомы, желудочно-кишечных заболеваний или мигрени.
Кроме того, сильное влияние CGRP на активность гликогенсинтазы скелетных мышц и метаболизм глюкозы мышц вместе с тем, что этот пептид высвобождается из нейромышечного соединения (место соединения) при возбуждении нерва, предполагают, что CGRP может играть физиологическую роль в метаболизме глюкозы скелетных мышц, направляя фосфорилированную глюкозу прочь от хранения гликогена и в гликолитические и окислительные проводящие пути (Rossetti и др. Am. J. Physiol. 264, Е1-Е10, 1993). Этот пептид может выполнять роль важного физиологического модулятора внутриклеточного транспорта глюкозы в физиологических условиях, таких как физические упражнения, а также может вносить свой вклад в ослабление действия инсулина и гликогенсинтазы скелетных мышц в патофизиологических условиях, таких как NIDDM или возрастное ожирение {Меlnyk и др. Obesity Res. 3, 337-344, 1995), когда заметно снижены уровни CGRP в циркулирующей плазме. Следовательно, ингибирование высвобождения и/или активности нейропептида CGRP может быть полезно при лечении невосприимчивости к инсулину, связанной с диабетом типа 2 или старением.
В Патентах США 4,383,999 и 4,514,414 и Европейских Патентах ЕР 236342, а также ЕР 231996 заявлены некоторые производные N-(4,4-дизамещенный-3-бутенил)азагетероциклических карбоновых кислот как ингибиторы поглощения GABA. В Европейских Патентах ЕР 342635 и ЕР 374801 в качестве ингибиторов поглощения GABA заявлены N-замещенные азагетероциклические карбоновые кислоты, в которых оксимэфирная группа и винилэфирная группа, соответственно, образуют часть N-заместителя. Кроме того, в WO 9107389 и WO 9220658 в качестве ингибиторов поглощения GABA заявлены N-замещенные азациклические карбоновые кислоты. Европейский Патент ЕР 221572 раскрывает 1-арилоксиалкилпиридин-3-карбоновые кислоты в качестве ингибиторов поглощения GABA.
Настоящее изобретение относится к соединениям общей формулы I, где X, Y, Z, M1, M2, R1-R12, r и m такие, как определено в подробном описании данного изобретения.
Настоящие соединения полезны для лечения, профилактики, устранения, облегчения или улучшения симптомов, относящихся к болезненным состояниям, повышенной болевой чувствительности и/или воспалительным состояниям, в которых патофизиологическую роль играют С-волокна, например нейрогенной боли, воспаления, мигрени, невропатии, зудящего и ревматоидного артрита, а также симптомов, вызванных или связанных с секрецией и циркуляцией инсулин-антагонистических пептидов, например инсулиннезависимого сахарного диабета (NIDDM) и возрастного ожирения.
В другом аспекте в объем настоящего изобретения включены фармацевтические композиции, содержащие в качестве активного ингредиента, по крайней мере, одно из соединений общей формулы I или его фармацевтически пригодную соль вместе с фармацевтически пригодным носителем или разбавителем.
Еще один аспект настоящего изобретения обеспечивает способ лечения болезненных состояний, повышенной болевой чувствительности и/или воспалительных состояний, в которых патофизиологическую роль играют С-волокна, например нейрогенной боли, воспаления, мигрени, невропатии, зудящего и ревматоидного артрита, а также способ лечения симптомов, вызванных или связанных с секрецией и циркуляцией инсулин-антагонистических пептидов, таких как CGRP или амилин, например инсулиннезависимого сахарного диабета (NIDDM) и возрастного ожирения. Способ лечения можно описать как лечение одного из указанных выше симптомов у нуждающегося в лечении пациента, которое включает введение указанному пациенту нейрологически эффективного количества соединения данного изобретения или его фармацевтически пригодной соли.
Еще один аспект данного изобретения относится к применению соединения данного изобретения для приготовления фармацевтической композиции для лечения болезненных состояний, повышенной болевой чувствительности и/или воспалительных состояний, в которых патофизиологическую роль играют С-волокна, например, нейрогенной боли, воспаления, мигрени, невропатии, зудящего и ревматоидного артрита, а также для лечения симптомов, вызванных или связанных с секрецией и циркуляцией инсулин-антагонистических пептидов, например инсулиннезависимого сахарного диабета (NIDDM) и возрастного ожирения.
Дополнительные задачи будут ясны из следующего описания.
Настоящее изобретение относится к новым 1,4-дизамещенным пиперазинам формулы I:
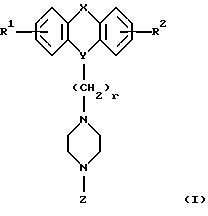
где R1 и R2 независимо являются водородом, галогеном, трифторметилом, гидроксилом, C1-6-алкилом или C1-6-алкокси-группой; и
Х является орто-фениленом, -О-, -S-, -C(R6R7)-, -СН2СН2-, -СН=СН-СН2-, -СН2-СН= СН-, -СН2-(С=O)-, -(С=O)-СН2-, -CH2CH2 CH2-, -СН=СН-, -N(R3)-(С=О)-, -(CO)-N(R8)-, -O-СН2-, -CH2-O-, -OCH2-O, -S-CH2-, -CH2-S-, -(CH2 )N(R8)-, -N(R8) (CH2)-, -N(СН3)SO2-, -SO2N(CH3 )-, -CH (R10) (CH2)-, -CH2CH(R10 )-, -(C= O)-, -N(R9)- или -(S=O)-, где R6, R7, R8 и R9 независимо являются водородом или C1-6-алкилом; и где R10 является C1-6-алкилом или фенилом; и
Y является






где M1 и M2 независимо являются С или N; и
R5 является водородом, С1-6-алкилом, фенилом или бензилом; и
R3 является водородом, галогеном, трифторметилом, нитрогруппой или цианогруппой; и
R4 является водородом, галогеном, трифторметаном, нитрогруппой, цианогруппой, (СН2)mCOR11, (СН2)mОН или (CH2)mSO2R11, где
R11 является гидроксилом, C1-6-алкокси-группой или NHR12, где
R12 является водородом или C1-6-алкилом; и m равно 0, 1 или 2; или
R4 выбирают из

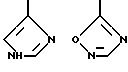

или их фармацевтически пригодных солей.
Соединения формулы I могут существовать в виде геометрических и оптических изомеров, и все выделенные изомеры, чистые и частично очищенные стереоизомеры или их рацемические смеси включены в объем данного изобретения. Изомеры можно выделить стандартными способами, такими как хроматографические способы или фракционная кристаллизация подходящих солей.
Предпочтительны соединения формулы I в виде индивидуальных геометрических или оптических изомеров.
Соединения, соответствующие данному изобретению, могут необязательно существовать в виде фармацевтически пригодных солей присоединения кислот, или (когда карбоксильная кислотная группа не этерифицирована) в виде фармацевтически пригодных солей металлов, или необязательно алкилированных солей аммония.
Примеры таких солей включают неорганические и органические соли присоединения кислот, такие как гидрохлорид, гидробромид, сульфат, фосфат, ацетат, фумарат, малеат, цитрат, лактат, тартрат, оксалат или подобные фармацевтически пригодные соли присоединения неорганических или органических кислот, и включают фармацевтически пригодные соли, перечисленные в Journal of Pharmaceutical Science, 66, 2 (1977), которые известны специалистам.
Включены также гидраты указанных выше солей присоединения кислот, которые способны образовывать настоящие соединения.
Соли присоединения кислот можно получить как непосредственные продукты синтеза соединений. В альтернативном случае можно растворить свободное основание в подходящем растворителе, содержащем подходящую кислоту, и выделить соль путем выпаривания растворителя или посредством осаждения или кристаллизации.
Соединения формулы I можно использовать в виде фармацевтически пригодной соли присоединения кислоты или в виде соли металла или (низший алкил)аммония. Такие соли демонстрируют примерно тот же порядок активности, как свободные основания.
Следующие далее термины имеют указанное значение в приведенных выше структурных формулах и на протяжении всего настоящего описания.
Используемый здесь термин "C1-6-алкил", один или в комбинации, относится к линейной или разветвленной насыщенной углеводородной цепи, имеющей от 1 до 6 атомов углерода, такой как, например, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, 2-метилбутил, 3-метилбутил, 4-метилпентил, неопентил, н-гексил, 1,2-диметилпропил, 2,2-диметилпропил и 1,2, 2-триметилпропил.
Используемый здесь термин "C1-6-алкокси-группа", один или в комбинации, относится к линейному или разветвленному моновалентному заместителю, который включает C1-6-алкильную группу, присоединенную через эфирный кислород, имеет свободную валентность от эфирного кислорода и от 1 до 6 атомов углерода, например метокси-, этокси-, пропокси-, изопропокси-, бутокси-, пентокси-группам.
Термин "галоген" обозначает фтор, хлор, бром или йод.
В предпочтительном варианте данного изобретения R1 и R2 выбирают из водорода, галогена, трифторметила или C1-6-aлкила. Предпочтительно, чтобы R1 и R2 представляли водород, хлор или метил.
В другом предпочтительном варианте данного изобретения Х выбирают из -СН2СН2-, -СН= СН-, -О-СН2 -, -СН2-О-, -OCH2O-, -S-CH2- или -CН2 -S-. Предпочтительно выбирают Х из -СН2СН2-, -OCH2O-, -S-CH2- или -СН2-S-.
Еще в одном предпочтительном варианте данного изобретения Y выбирают из

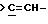

Еще в одном предпочтительном варианте данного изобретения r равно 1 или 2.
В другом предпочтительном варианте данного изобретения Z
выбирают из

где M1 и М2 независимо являются С или N.
Еще в одном предпочтительном варианте данного изобретения R3 является водородом, трифторметилом, нитрогруппой или цианогруппой.
Еще в одном предпочтительном варианте данного изобретения R4 является водородом, трифторметилом, нитрогруппой, цианогруппой или (CH2)mCOR11.
В другом предпочтительном варианте данного изобретения m равно 0 или 1.
Еще в одном предпочтительном варианте данного изобретения R11 является гидроксилом.
Предпочтительные соединения
настоящего изобретения, включают:
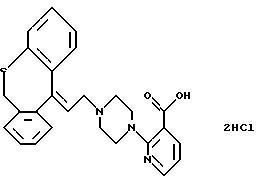
2-(4-(3-(12Н-дибензо[d, g] [1,
3] диоксоцин-12-илиден)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(2,10-дихлор-12Н-дибензо[d, g]
[1,3] диоксоцин-12-илиден)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую
кислоту;
2-(4-(3-(12Н-дибензо[d, g] [1,3,6] диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(2-хлор-12Н-дибензо[d, g] [1,3,6]
диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
1-(3-(10,11-дигидро-5Н-дибензо[a, d]
циклогептен-5-илиден)-1-пропил)-4-(2-пиридил)пиперазин;
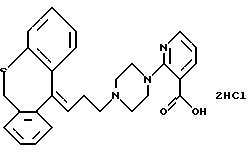
2-(4-(3-(10,
11-дигидро-5Н-дибензо[а, d]циклогептен-5-илиден)-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
2-(4-(2-(10,
11-дигидро-5Н-дибензо[b, f]
азепин-5-ил)-1- этил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
6-(4-(3-(10,11-дигидро-5Н-дибензо[b, f]
азепин-5-ил)-1-пропил)-1-пиперазинил)-2-пиридинкарбоновую кислоту;
2-(4-(3-(10,11-дигидро-5Н-дибензо[b, f] азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(10,11-дигидро-5Н-дибензо[b, f]
азепин-5-ил)-1-пропил)-1-пиперазинил)-5-пиридинкарбоновую кислоту;
2-(4-(3-(3-хлор-10,11-дигидро-5Н-дибензо[b,
f]азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
1-(3-(10,11-дигидро-5Н-дибензо[a, d] циклогептен-5-илиден)-1-пропил)-4-(2-нитрофенил)пиперазин;
2-(4-(3-(10,
11-дигидро-5Н-дибензо[a,
d]циклогептен-5-илиден)-1-пропил)-1-пиперазинил)бензонитрил;
2-(4-(3-(10,11-дигидро-5Н-дибензо[a, d]циклогептен-5-илиден)-1-пропил)-1-пиперазинил)бензойную кислоту;
1-(3-(10,
11-дигидро-5Н-дибензо[a, d] циклогептен-5-илиден)-1-пропил)-4-(3-трифторметил-2-пиридил)пиперазин;
2-(4-(2-(6,11-дигидродибензо[b, е]
тиепин-11-илиден)этил)пиперазин-1-ил)-3-пиридинкарбоновую
кислоту;
2-(4-(3-(6,11-дигидродибензо[b, е] тиепин-11-илиден)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
2-(4-(2-(6,11-дигидродибензо[b, е]
тиепин-11-илокси)этил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
6-(4-(3-(10,11-дигидро-5Н-дибензо[а, d]
циклогептен-5-илиден)-1-пропил)пиперазин-1-ил)-2-пиридинкарбоновую кислоту;
2-(4-(3-(3-метил-10,11-дигидро-5Н-дибензо[b, f]азепин-5- ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
6-(4-(3-(дибензо[d, g] [1,3,
6]диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-пиридин-2-карбоновую кислоту
или их фармацевтически пригодные соли.
Показано, что новые
соединения формулы I ингибируют
нейрогенное воспаление, которое включает высвобождение нейропептидов из периферийных и центральных окончаний чувствительных С-волокон. Экспериментально это можно
продемонстрировать на животных моделях
гистамин-индуцированного отека лапы (Europ. J. Pharmacol. 279, 227-231, 1995), где новые соединения формулы I демонстрируют сильное ингибирующее действие.
Соединения формулы I можно использовать для
лечения всех болезненных состояний, повышенной болевой чувствительности и/или воспалительных состояний, в которых патофизиологическую роль играют С-волокна,
вызывая нейрогенную боль или воспаление, а
именно:
острых болезненных состояний, примерами которых являются мигрень, послеоперационная боль, ожоги, ушибы, постгерпетическая боль
(опоясывающий лишай) и боль, обычно обусловленная острым
воспалением; хронических болезненных и/или воспалительных состояний, примерами которых являются различные типы невропатии (диабетическая,
посттравматическая, токсическая), невралгия, ревматоидный
артрит, спондилит, подагра, воспалительное заболевание кишечника, простатит, раковая боль, хроническая головная боль, кашель, астма, зуд,
хронический панкреатит, воспалительные кожные заболевания,
включая псориаз и аутоиммунный дерматоз, боль при остеопорозе.
Кроме того, показано, что соединения общей формулы I улучшают толерантность к глюкозе у диабетических ob/ob мышей и что это может быть результатом пониженного высвобождения CGRP из периферических нервных окончаний. Следовательно, соединения общей формулы I можно использовать при лечении NIDDM, а также возрастного ожирения. Экспериментально это продемонстрировано путем подкожного введения глюкозы ob/ob мышам с предварительным оральным введением соединения общей формулы I (или без этого).
Соединения формулы I можно получить следующим способом:
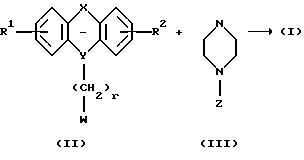
Соединение формулы II, где R1, R2, X, Y и г такие, как определено выше, и W является удобной удаляемой группой, такой как галоген, паратолуолсульфонат или мезилат, может реагировать с азасоединением формулы III, где Z такое, как определено выше. Эту реакцию алкилирования можно проводить в растворителе, таком как ацетон, дибутиловый эфир, 2-бутанон, метилэтилкетон, этилацетат, тетрагидрофуран (ТГФ) или толуол, в присутствии основания, например гидрида натрия, и катализатора, например йодида щелочного металла, при температуре до температуры кипения использованного растворителя с обратным холодильником в течение 1-120 часов. Если получены сложные эфиры, в которых R11 является алкокси-группой, то соединения формулы I, где R11 является ОН, можно получить путем гидролиза эфирной группы, предпочтительно при комнатной температуре, в смеси водного раствора гидроксида щелочного металла и спирта, такого как метанол или этанол, в течение, например, 0,5-6 часов.
Соединения формулы II и III можно легко получить способами, известными специалистам.
При определенных обстоятельствах может быть необходимо защитить промежуточные продукты, использованные в указанных выше способах, например соединение формулы III, подходящими защитными группами. Карбокислатную группу можно, например, этерифицировать. Введение и удаление таких групп описано в "Protective Groups in Organic Chemistry", редактор J.F.W. McOrnie (New York, 1973).
ФАРМАКОЛОГИЧЕСКИЕ МЕТОДЫ
I. Гистамин-индуцированный отек лапы
Исследование гистамин-индуцированного отека лапы у крыс проводят в основном так, как описано в работе Amann и др.
(Europ. J. Pharmacol. 279, 227-231, 1995). Кратко говоря, самцам Sprague-Dawley крыс
весом 250-300 г проводят анестезию пентобарбиталом натрия и помещают их на стол, нагретый до 32oС.
Через десять минут в правую заднюю лапу вводят гистамин (50 микролитров, 3 мг/мл) и через
20 минут после этого определяют опухание лапы путем плетизмографии воды (Uqo Basile). Исследуемые соединения
вводят внутрибрюшинно за 15 минут до анестетика.
II. Пониженное
высвобождение CGRP
Самкам ob/ob мышей (возраст 16 недель] делают подкожную инъекцию глюкозы (2 г/кг). После
этого определяют глюкозу в крови хвостовой вены способом с использованием
глюкозоксидазы (glucose oxidase method). В конце исследования животных обезглавливают и собирают кровь. Определяют
иммунореактивный CGRP в плазме посредством радиоиммунологического исследования.
Изучают две группы животных. Одну группу обрабатывают наполнителем, тогда как другой группе дают с питьевой водой
соединение формулы I (100 мг/л) в течение пяти дней до тестирования.
В таблице приведены значения реакции ингибирования гистамин-индуцированного отека для некоторых типичных соединений.
ФАВМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ
Настоящее изобретение относится
также к фармацевтическим композициям, включающим соединение формулы I или его фармацевтически
пригодную соль, обычно такие композиции содержат также фармацевтический носитель или разбавитель.
Композиции, содержащие соединения данного изобретения, можно получить обычными способами и составлять
в виде обычных форм, например в виде капсул, таблеток, растворов или суспензий.
Используемый фармацевтический носитель может быть общеизвестным твердым или жидким носителем. Примерами твердых носителей являются лактоза, молотый гипс, сахароза, тальк, желатин, агар, пектин, аравийская камедь, стеарат магния и стеариновая кислота. Примерами жидких носителей являются сироп, масло земляного ореха, оливковое масло и вода.
В аналогичном случае носитель или разбавитель может включать любой материал, замедляющий высвобождение активного ингридиента, известный из уровня техники, такой как глицерилмоностеарат или глицерилдистеарат, один или в смеси с парафином.
Способ введения может быть любым способом, который эффективно транспортирует активное соединение к подходящему или необходимому месту действия, таким как оральный, через нос, легочный или парентеральный, например ректальный, депо, трансдермальный, подкожный, внутриносовой, внутримышечный, локальный, внутривенный, внутриуретральный, в виде офтальмологического раствора или мази, предпочтителен оральный способ приема.
Если используют твердый носитель для орального приема, то препарат можно таблетировать, поместить в твердые желатиновые капсулы в виде порошков или гранул, или он может быть в виде пастилки или лепешки. Количество твердого носителя будет меняться в широких пределах, но обычно составляет примерно от 25 мг до 1 г. Если используют жидкий носитель, препарат может быть в виде сиропа, эмульсии, мягкой желатиновой капсулы или стерильной жидкости для инъекций, такой как водные или неводные жидкие суспензии или растворы.
Препарат для носового применения может содержать соединение формулы I, растворенное или суспендированное в жидком носителе, в частности в водном носителе для аэрозолей. Носитель может содержать добавки, такие как солюбирующие агенты, например пропиленгликоль, поверхностно-активные соединения, усилители абсорбции, такие как лецитин (фосфатидилхолин) или циклодекстрин, или консерванты, такие как парабены.
Для парентерального применения особенно подходят растворы или суспензии для инъекций, предпочтительно водные растворы с активным соединением, растворенным в полигидроксилированном касторовом масле.
Таблетки, драже или капсулы, содержащие тальк и/или карбогидрат в качестве носителя, или связующего, или подобного, особо подходят для орального применения. Предпочтительные носители для таблеток, драже или капсул включают лактозу, кукурузный крахмал и/или картофельный крахмал. В случаях, когда можно применять подслащенный носитель, можно использовать сироп или эликсир.
Типичная таблетка, которую можно
приготовить обычным способом таблетирования, может содержать:
Ядро:
Активное соединение (как свободное соединение или его соль) - 100 мг
Коллоидный диоксид кремния
(Aerosil®) - 1,5 мг
Целлюлоза микрокристаллическая (Avicel®) - 70 мг
Модифицированная целлюлозная смола (Ac-Di-Sol®) - 7,5
мг
Стеарат магния
Оболочка:
НРМС приблизительно - 9 мг
*Mywacett® 9-40 Т приблизительно - 0,9 мг
* Ацилированный моноглицерид используют
как пластификатор для пленочной оболочки.
Соединения данного изобретения можно вводить млекопитающим, в особенности человеку, нуждающемуся в таком лечении, профилактике, устранении, облегчении или улучшении симптомов, относящихся ко всем болезненным состояниям, повышенной болевой чувствительности и/или воспалительным состояниям, в которых патофизиологическую роль играют С-волокна, таким как нейрогенная боль, воспаление, мигрень, невропатия, зудящий и ревматоидный артрит, а также симптомов, вызванных или связанных с секрецией и циркуляцией инсулин-антагонистических пептидов, таких как инсулиннезависимый сахарный диабет (NIDDM) или возрастное ожирение. Такие млекопитающие включают также животных: и домашних животных, например животных, содержащихся в квартире, и недомашних животных, таких как дикие животные.
Соединения данного изобретения можно вводить в виде их солей щелочных или щелочноземельных металлов, совместно, последовательно или вместе с фармацевтически пригодным носителем или разбавителем, в особенности и предпочтительно в виде их фармацевтических композиций в эффективном количестве.
Для указанных выше симптомов дозировка в значительной степени зависит от используемого соединения формулы Т, способа приема и необходимого лечения. Однако в основном удовлетворительные результаты получают при дозе примерно от 0,5 мг до 1000 мг, предпочтительно примерно от 1 мг до 500 мг соединений формулы I, принимаемых обычно 1-5 раз в день, необязательно в виде препарата с длительным выделением активного компонента. Обычно дозированные формы, подходящие для орального приема, включают примерно от 0,5 мг до 1000 мг, предпочтительно примерно от 1 мг до 500 мг, соединений формулы I в смеси с фармацевтическим носителем или разбавителем.
Подходящие диапазоны дозировки обычно меняются, как указано выше, в зависимости от конкретного способа приема, принимаемой формы, симптомов, против которых направлен прием, пациента и его веса и предпочтений и опыта лечащего врача или ветеринара.
Обычно соединения данного изобретения разделяют на стандартные дозированные формы, включающие 50-200 мг активного ингредиента/на одну дозу в фармацевтически пригодном носителе или вместе с ним.
Обычно дозированные формы, подходящие для орального, носового, легочного или трансдермального применения, включают примерно от 0,5 мг до 1000 мг, предпочтительно от 1 мг до 500 мг, соединений формулы I в смеси с фармацевтически пригодным носителем или разбавителем.
Любые описанные здесь отличительные признаки или комбинации отличительных признаков считают неотъемлемой частью данного изобретения.
ПРИМЕРЫ
Способ получения соединений формулы I и содержащих их препаратов дополнительно проиллюстрирован следующими примерами, которые,
однако, не ограничивают области изобретения.
Далее сокращение ТСХ обозначает тонкослойную хроматографию, СDСl3 - дейтерохлороформ, а ДМСО-d6 - гексадейтеро-диметилсульфоксид. Структуры соединений подтверждены либо элементным анализом, либо ЯМР, где представленные пики, отнесенные к характеристическим протонам указанных в заголовке соединений, соответствуют структурам. Химические сдвиги1H ЯМР (δн) даны в миллионных долях (м.д.). Т.пл. - это температура плавления, дана вoС и не уточнялась. Колоночную хроматографию проводили, используя способ, описанный W.C. Still и др., J. Org. Chem. (1978), 43, 2923-2925, на силикагеле Merck silica gel 60 (Art. 9385). Соединения, использованные в качестве исходных материалов, являются либо известными соединениями, либо соединениями, которые можно легко получить известными способами.
ПРИМЕР 1
Гидрохлорид 2-(4-(3-(12Н-дибензо[d, g] [1,3] диоксоцин-12-или-ден)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновой
кислоты

2,2'-Дигидроксибензофенон (10,0 г, 46,7 ммоля) и дийод-метан (13,1 г, 49 ммолей) растворяют в сухом N,N-диметил-формамиде (180 мл). Добавляют высушенный тонкодисперсный порошок карбоната калия (9,2 г, 66,7 ммоля) и смесь нагревают при 105oС в течение 16 часов. После охлаждения до комнатной температуры реакционную смесь выливают в ледяную воду (500 мл). Осадок собирают фильтрованием через 0,5 часа, промывают водой на фильтре и растворяют в смеси этанола (80 мл) и 4N гидроксида натрия (20 мл). Раствор перемешивают при температуре кипения с обратным холодильником в течение 1 часа, охлаждают и разбавляют водой (300 мл). Образовавшийся кристаллический осадок отфильтровывают, промывают водой (50 мл) и сушат в вакууме, получая 12Н-дибензо[d, g] [1,3] -диоксоцин-12-он в виде твердого вещества (9,5 г, 90%). Т.пл. 93-95oС.
Раствор циклопропилмагнийбромида в сухом тетрагидрофуране (полученный из циклопропилбромида (24,2 г, 0,2 моля), магниевых стружек (4,86 г, 0,2 моля) и сухого тетрагидрофурана (70 мл) помещают в атмосферу азота. Добавляют по капле раствор указанного выше кетона (9,05 г, 40 ммолей) в сухом тетрагидрофуране (50 мл). Реакционную смесь перемешивают при 40oС в течение 1,5 часов, охлаждают и добавляют к охлажденной льдом смеси насыщенного хлорида аммония (400 мл) и эфира (200 мл). Органический слой отделяют, а водную фазу экстрагируют эфиром (50 мл). Объединенные органические экстракты промывают водой (2•100 мл) и соляным раствором (50 мл), сушат (МgSO4), выпаривают в вакууме и отгоняют с толуолом (2 х 25 мл). Это дает 11,2 г 12-циклопропил-12Н-дибензо[d,g][1, 3]диоксоцин-12-ола.
1Н ЯМР (200 МГц, CDCl3): δн 0,50 (д, 2Н); 0,75 (д, 2Н); 2,00 (м, 1Н); 5,14 (с, 2Н); 6,9-7,4 (м, 6Н); 7,81 (д, 2Н).
К раствору полученного выше спирта (6,21 г, 22 ммоля) в сухом дихлорметане (225 мл) добавляют триметилсилилбромсилан (3,71 г, 24,2 ммоля). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре и выливают в охлажденный льдом насыщенный раствор гидрокарбоната натрия (75 мл). Органическую фазу отделяют, промывают ледяной водой (2•75 мл) и соляным раствором (75 мл), сушат (МgSO4) и выпаривают в вакууме. Это дает 7,95 г сырого 12-(3-бром-1-пропилиден)-12Н-дибензо[d, g] [1,3] диоксоцина, который используют на следующей стадии без дополнительной очистки.
Смесь полученного выше бромида (5,0 г, 15 ммолей), этилового эфира 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (3,54 г, 15 ммолей, полученного, как описано в J. Med. Chem. 31, 618-624 (1988)), йодида натрия (2,23 г, 15 ммолей) и карбоната калия (5,1 г, 37 ммолей) в 2-бутаноне (70 мл) нагревают при температуре кипения с обратным холодильником в течение 5 часов. После охлаждения смесь выливают в диэтиловый эфир (170 мл) и воду (170 мл). Органический слой отделяют, промывают водой (2•100 мл), подкисляют 2N соляной кислотой и промывают водой (2•50 мл). Объединенные водные слои подщелачивают, используя насыщенный раствор гидрокарбоната натрия, и экстрагируют дихлорметаном (200 мл). Органический экстракт сушат (MgSO4) и выпаривают в вакууме. Остаток (4,35 г) чистят хроматографически на силикагеле, используя в качестве элюента смесь бензола и этилацетата и получая этиловый эфир 2-(4-(3-(12Н-дибензо[d, g][1, 3]диоксоцин-12-илиден)-1-пропил)пиперазин-1-ил)-3-пиридинкарбоновой кислоты (3,71 г, 51%).
Смесь полученного выше эфира (0,99 г, 2 ммоля) и 20% гидроксида натрия (1,5 мл) в этаноле (13 мл) перемешивают при комнатной температуре в течение 4 часов. Выливают смесь в дихлорметан (100 мл), подкисляют 2N соляной кислотой и промывают водой (10 мл). Органический раствор сушат (MgSO4) и добавляют активированный уголь. Смесь перемешивают в течение 10 минут, фильтруют и удаляют растворитель в вакууме. Остаток растирают с ацетоном и твердый продукт покрывают этилацетатом, перемешивают 15 минут, отфильтровывают и промывают этилацетатом. После сушки получают указанное в заголовке соединение (0,80 г, 74%). Т.пл. 225-229oС.
Рассчитано для С27Н27N3O4, НСl, 0,25 C4H8O2,
0,25 С2Н5ОН: С 64,89%; Н 6,02%; С1 6,72%; N 7,96%;
Найдено: С 64,
95%; Н 5,86%; С1 6,73%: N 7,94%.
ПРИМЕР 2
Дигидрохлорид 2-(4-(3-(2,
10-дихлор-12H-дибензо[d, g] [1, 3]-диоксопин-12-илиден)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновой
кислоты.
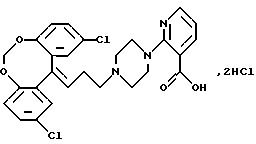
2,2'-Дигидрокси-5,5'-бензофенон (12,1 г, 0,042 моля, полученный, как описано в JACS 77, 543 (1955)) и дийодметан (11,9 г, 0,044 моля) растворяют в сухом N, N-диметилформамиде (226 мл). Сушат и добавляют порошок карбоната калия (8,3 г) и смесь нагревают при 105oС в течение 5 часов и оставляют на ночь при комнатной температуре. Реакционную смесь выливают на лед (220 г). Осадок собирают фильтрованием и растворяют в диэтиловом эфире (500 мл). Органический раствор промывают 5% гидроксидом натрия (50 мл), сушат (МgSО4) и выпаривают в вакууме. Это дает 12 г (96%) 2,10-дихлор-12Н-дибензо[d,g]-[1,3]диоксоцин-12-она в виде твердого вещества.
К раствору циклопропилмагнийбромида в сухом тетрагидрофуране (полученному из циклопропилбромида (15,7 г, 0,130 моля), магниевых стружек (3,15 г, 0,130 моля) и сухого тетрагидрофурана (45 мл) добавляют за 5 минут при охлаждении раствор указанного выше кетона (7,65 г, 0,026 моля) в сухом тетрагидрофуране (30 мл). Реакционную смесь перемешивают при 38-42oС в течение 3 часов, охлаждают на ледяной бане и добавляют смесь насыщенного хлорида аммония (260 мл) и диэтилового эфира (130 мл). Затем реакционную смесь фильтруют и отделяют органический слой, а водную фазу экстрагируют диэтиловым эфиром (35 мл). Объединенные органические экстракты промывают водой (2•70 мл) и соляным раствором (70 мл), сушат (MgSO4) и выпаривают в вакууме. Сырой продукт чистят колоночной хроматографией на силикагеле (140 г), используя в качестве элюента бензол. Это дает 8,75 г (98%) 2,10-дихлор-12-циклопропил-12Н-дибензо[d,g][1,3]диоксоцин-12-ола в виде твердого вещества.
К раствору полученного выше спирта (8,75 г, 0,027 моля) в сухом дихлорметане (245 мл) добавляют триметилсилил-бромид (4,02 г, 0,026 моля). Реакционную смесь перемешивают в течение 1 часа при комнатной температуре и выливают в охлажденный льдом насыщенный раствор гидрокарбоната натрия (80 мл). Органическую фазу отделяют, промывают водой (2•80 мл) и соляным раствором (80 мл), сушат (MgSO4) и выпаривают в вакууме. Это дает 9,12 г масла, которое чистят способом колоночной хроматографии на силикагеле (250 г), используя в качестве элюента смесь циклогексана и бензола (3:1). Это дает 6,61 г (62%) 2, 10-дихлор-12-(3-бром-1-пропилиден)-12Н-дибензо[d,g][1,3]-диоксоцина в виде масла, которое кристаллизуется при стоянии.
Смесь полученного выше бромида (3 г, 0,008 моля), этилового эфира 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (1,76 г, 0,0075 моля), карбоната калия (3,1 г, 0,022 моля) и йодида натрия (1,1 г, 7,3 ммолей) в 2-бутаноне (35 мл) нагревают при температуре кипения с обратным холодильником в течение 6 часов. После охлаждения до комнатной температуры смесь разбавляют ацетоном, фильтруют и выпаривают в вакууме. Твердый остаток чистят колоночной хроматографией на силикагеле (100 г), используя в качестве элюента хлороформ. Это дает 2,7 г (65%) этилового эфира 2-( 4-(3-(2,10-дихлор-12Н-дибензо [d, g] [1,3] диоксоцин-12-илиден)-1-пропил) пиперазин-1-ил; -3-пиридинкарбоновой кислоты в виде масла.
Полученный выше эфир (2,7 г, 4,87 ммоля) растворяют в этаноле (30 мл) и добавляют раствор гидроксида натрия (0,74 г) в воде (2,8 мл). Смесь перемешивают при комнатной температуре в течение 48 часов. Добавляют концентрированную соляную кислоту (2,8 мл), а затем дихлорметан (100 мл). Органический слой отделяют, сушат (MgSO4) и выпаривают в вакууме. Полученную пену перемешивают с горячим ацетоном (150 мл), отфильтровывают продукт и промывают ацетоном, получая 1,8 г (62%) указанного в заголовке соединения.
1H ЯМР (250 МГЦ, ДМСО-d6): δн 8,31 (дд, J=1,9 Гц и 4,7 Гц, 1Н); 8,04 (дд, J= 1,9 Гц и 7,5 Гц, 1Н); 7,53 (д, J=2,5 Гц, 1Н); 7,32 (дд, J=2,5 Гц и 8,5 Гц, 1Н); 7,25 (дд, J=2,5 Гц и 8,5 Гц, 1Н); 7,21 (д, J=2,5 Гц, 1Н); 7,10 (д, J= 8,5 Гц, 1H); 6,97 (д, J=8,5 Гц); 6,95 (дд, J=4,7 Гц и 7,5 Гц, 2Н); 6,15 (т, J=7,5 Гц, 1Н); 5,86 (с, 2Н); 3,73 (ш.с, 4Н); 3,25 (ш.с, 6Н); 2, 50 (кв, J=7,5 Гц, 2Н).
Рассчитано для C27H25Cl2N3O4, 2 НСl, 0,5
H2O: С 53,30%; Н 4,64%; N 6,91%; Cl 23,31%;
Найдено: С 53,41%; Н 4,60%: N 6,73%: Cl 22,71%.
ПРИМЕР 3
Дигидрохлорид 2-(4-(3-(12Н-дибензо[d, g] [1,3,6]
диоксазоцин-12-ил)-1-пропил)пиперазин-1-ил)-3-пиридинкарбоновой
кислоты

N-(2-Гидроксифенил)формамид (16,0 г, 130 ммолей) растворяют в 99,9% этаноле (65 мл). Метилат натрия (7,0 г, 130 ммолей) суспендируют в 99.9% этаноле (70 мл) и добавляют по капле за 30 минут. Полученную смесь перемешивают 30 минут. Добавляют по капле за 15 минут 1-бром-2-хлорметокси-бензол (26,1 г, 118 ммолей, синтез описан в J. Heterocycl. Chem., 11, 1974, 331-337). Реакционную смесь перемешивают в течение 2,5 часов при комнатной температуре, нагревают при температуре кипения с обратным холодильником в течение 2 часов и перемешивают при комнатной температуре в течение ночи. Смесь фильтруют и фильтрат выпаривают. Остаток растворяют в толуоле (500 мл) и промывают насыщенным раствором карбоната натрия (2•200 мл). Органическую фазу сушат (MgSO4) и выпаривают. Остаток суспендируют в этаноле (40 мл), фильтруют и промывают этанолом (3•10 мл). После сушки получают N-(2-(2-бромфеноксиметокси)фенил)формамид (14,1 г, 37%).
Полученный выше формамид (6,8 г, 21 ммоля) суспендируют в даутерме (75 мл) и добавляют карбонат калия (3,9 г, 28 ммолей), а затем медь (1,1 г, 17 ммолей) и бромид меди (1,5 г, 11 ммолей). Реакционную смесь нагревают при 180oС в течение ночи. После охлаждения смесь фильтруют и промывают осадок на фильтре дихлорметаном. Отгоняют даутерм и растворитель и добавляют к остатку этанол (200 мл), смесь оставляют на ночь. Добавляют 4М гидроксид натрия (14 мл) и смесь нагревают при температуре кипения с обратным холодильником в течение 1 часа. После охлаждения смесь фильтруют и выпаривают. Остаток суспендируют в этилацетате (200 мл) и воде (100 мл). Фазы разделяют и органическую фазу промывают водой (2•75 мл). Водные фазы экстрагируют этилацетатом (100 мл) и объединенные органические экстракты выпаривают. Остаток суспендируют в теплом циклогексане (100 мл) и оставляют охлаждаться при перемешивании. Осадившееся твердое вещество отфильтровывают и сушат, получая 12Н-дибензо[d,g][1,3, 6]диоксазоцин (4,57 г, 50%).
Полученный выше диоксазоцин (4,0 г, 19 ммолей) растворяют в сухом N,N-диметилформамиде (150 мл). Добавляют частями гидрид натрия (1,13 г, 28 ммолей, 60% дисперсия в масле) и полученную смесь перемешивают в течение 30 минут при комнатной температуре. Медленно по капле добавляют 1-бром-3-хлорпропан (4,6 мл, 47 ммолей) и перемешивают реакционную смесь при комнатной температуре в течение ночи. Добавляют еще гидрид натрия (0,56 г, 14 ммолей) и продолжают перемешивание в течение 6 часов. Добавляют еще гидрид натрия (0,56 г, 14 ммолей) и продолжают перемешивание в течение ночи. Добавляют хлорид аммония (3,2 г) и смесь перемешивают в течение 30 минут. Добавляют воду (300 мл) и экстрагируют смесь дихлорметаном (2•250 мл). Объединенные органические экстракты сушат (MgSO4) и выпаривают. Остаток чистят колоночной хроматографией на силикагеле, используя в качестве элюента смесь гептана и этилацетата (6: 1). Это дает 12-(3-хлорпропил)-12Н-дибензо[d,g] [1,3,6]-диоксазоцина (2,18 г, 40%).
Полученный выше хлорид (135 г, 4,66 ммоля) и йодид калия (5,0 г, 30 ммолей) в метилэтилкетоне (150 мл) нагревают при температуре кипения с обратным холодильником в течение 4,5 часов. Добавляют этиловый эфир 2-(1-пиперазинил)-3- пиридинкарбоновой кислоты (3,0 г, 12 ммолей), а затем карбонат калия (2,25 г, 16,3 ммоля). Реакционную смесь нагревают при температуре кипения с обратным холодильником в течение 19 часов. После охлаждения смесь фильтруют, осадок на фильтре промывают метилэтилкетоном и фильтрат выпаривают. Оставшееся масло чистят колоночной хроматографией на силикагеле (800 мл), используя в качестве элюента смесь гептана и этилацетата (1:3). Это дает этиловый эфир 2-(4-(3-(12Н-дибензо-[d, g][1,3,6]диоксазоцин-12-ил)-1 -пропил)пиперазин-1-ил)-3-пиридинкарбоновой кислоты (0,71 г, 32%) в виде масла.
Полученный выше эфир (0,50 г, 1,22 ммоля), растворенный в растворе гидроксида натрия (0,24 г, 6 ммолей) в этаноле (30 мл) и воде (3 мл), перемешивают при комнатной температуре в течение 25 часов. Доводят рH смеси до 3, добавляя IN соляную кислоту (6 мл). Смесь экстрагируют дихлорметаном (2•40 мл), объединенные органические фазы промывают соляным раствором (50 мл), сушат (MgSO4) и выпаривают растворитель в вакууме. Остаток растирают с изопропилацетатом (15 мл) и отфильтровывают твердый продукт. Часть продукта (275 мг) суспендируют в ацетоне (160 мл), выпаривают в вакууме и сушат, получая указанное в заголовке соединение (0,23 г, 75%).
Рассчитано для C26H28N4O4, 2 НСl, 0,5 H2O, 0,5 С5H10О2: С 57,67%; Н 6,07%; N 9,44%;
Найдено: С 57,78%; Н 6,02%; N 9,42%.
HPLC время удерживания
(высокоэффективная жидкостная
хроматография) =18,42 минуты (колонка 5 мкм С184•250 мм), элюирование с
20-80% градиентом 0,1% трифторуксусной кислотой/ацетонитрилом и 0,1%
трифторуксусной кислотой/водой в течение 30 минут при 30oС.
ПРИМЕР 4
Дигидрохлорид
2-(4-(3-(2-хлор-12Н-дибензо[d, g][1,3,6]
диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновой кислоты

Суспензию 2-хлор-12Н-дибензо[d,g][1,3,6]диоксазоцина (10,65 г, 43 ммоля, описанного в J. Mol. Struct., 131, 1985, 131-140) и 3-хлорпропионилхлорида (6,55 г, 51, 6 ммоля) в сухом толуоле (100 мл) нагревают при температуре кипения с обратным холодильником в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь промывают насыщенным раствором бикарбоната натрия (50 мл). Органический слой сушат (МgSO4) и выпаривают в вакууме, получая 2-хлор-12-(3-хлорпропионил)-12Н-дибензо[d, g][1,3,6]диоксазоцин (12,85 г, 88%).
К охлажденной суспензии литийалюминийгидрида (3, 0 г, 79 ммолей) в сухом тетрагидрофуране (80 мл) добавляют по капле концентрированную серную кислоту (3,87 г, 39,5 ммоля) с такой скоростью, чтобы поддерживать температуру <12oС. Смесь перемешивают при комнатной температуре 1,5 часа. Добавляют по капле раствор указанного выше амида (12,8 г, 37,8 ммоля) в сухом тетрагидрофуране (80 мл) и продолжают перемешивание в течение 2 часов. Реакцию гасят, осторожно добавляя этилацетат (100 мл), а затем воду (5,7 мл). Фильтрование и выпаривание смеси в вакууме дают 2-хлор-12-(3-хлорпропил)-12Н-дибензо[d,g][1,3,6]диоксазоцин в виде пены.
Смесь полученного выше хлорида (1,14 г, 3,5 ммоля), метилового эфира 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (0,78 г, 3,5 ммоля), сухого карбоната калия (1,45 г, 10,5 ммоля), йодида натрия (0,53 г, 3,5 ммоля) и 2-бутанона (15 мл) нагревают при температуре кипения с обратным холодильником в течение 48 часов. Реакционную смесь выпаривают в вакууме. Остаток растворяют в толуоле (25 мл), промывают водой (2•25 мл) и выпаривают в вакууме. Сырой продукт (1,15 г) чистят колоночной хроматографией на силикагеле, используя в качестве элюента смесь этилацетата и толуола (1:1), содержащую триэтиламин (2,5%). Это дает метиловый эфир 2-(4-(3-(2-хлор-12Н-дибензо[d,g] [1,3, 6]диоксазоцин-12-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты (1,10 г, 62%) в виде масла.
Полученный выше эфир (1,10 г, 2,16 ммоля) растворяют в этаноле (10 мл) и добавляют 2N гидроксид натрия (3,37 мл, 7,13 ммоля). Смесь перемешивают при комнатной температуре в течение 16 часов. Этанол выпаривают в вакууме, а остаток разбавляют водой (25 мл). Доводят рН до 6, добавляя 6N соляную кислоту и экстрагируют водный раствор дихлорметаном (3•15 мл). Объединенные органические экстракты сушат (MgSO4) и выпаривают в вакууме. Остаток растворяют в тетрагидрофуране (25 мл) и добавляют по капле 2,5N раствор хлористого водорода в эфире (1,67 мл, 4,18 ммоля). Смесь перемешивают в течение 3 часов, осадок отфильтровывают и сушат, получая 0,86 г (75%) указанного в заголовке соединения. Т.пл. 165-173oС.
Рассчитано
для C26H27ClN4O4, 2 НС1: С 54,99%; Н 5,15%; N 9,87%;
Найдено: С 54,8%; Н 5,2%; N 9,65%.
ПРИМЕР 5
Дигидрохлорид 1-(3-(10,
11-дигидро-5Н-дибензо [a, d] циклогептен-5-илиден)-1-пропил)-4-(2-пиридил)пиперазина

Смесь 5-(3-бром-1-пропилиден)-10,11-дигидро-5Н-дибензо-[а,d]циклогептена (3,13 г, 10 ммолей, полученного, как описано в WO 9518793), 1-(2-пиридил)пиперазина (2,5 г, 15 ммолей, (описано в J. Org. Chem. 1953, 18, 1484), карбоната калия (2,8 г, 20 ммолей) и N,N-диметилформамида (40 мл) перемешивают при комнатной температуре в течение 16 часов и при 50-55oС в течение 8 часов. После охлаждения смесь разбавляют бензолом (150 мл) и промывают водой (3•100 мл). Органический слой экстрагируют 3N соляной кислотой (100 мл). Кислый водный слой подщелачивают 5N гидроксидом натрия (60 мл) до рН 12 и экстрагируют бензолом (100 мл). Бензольный раствор сушат (К2СО3) и фильтруют через силикагель (12 г). Выпаривание в вакууме дает масло (2,98 г, 75%). Его растворяют в этаноле (20 мл), подкисляют при помощи хлористого водорода в диэтиловом эфире (3 ммоля/мл, 8 мл) и осаждают диэтиловым эфиром (20 мл). Осадок отфильтровывают, промывают диэтиловым эфиром и сушат, получая 2,1 г (44%) указанного в заголовке соединения. Т.пл. 152-155o С.
Рассчитано для C27H29N3, 2HCl, Н2О: С 66,66%; Н 6,84%; N
8,64%; Cl 14,58%;
Найдено: С 67,27%; Н 6,52%; N 8,76%; Cl 14,19%.
ПРИМЕР 6
Гидрохлорид 2-(4-(3-(10,11-дигидро-5Н-дибензо[a, d]
циклогептен-5-илиден)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты
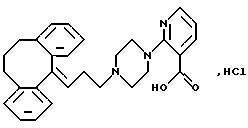
Смесь этилового эфира 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (4,6 г, 0,019 моля), 3-(10,11-дигидро-5Н-дибензо[a, d]циклогептен-5-илиден)-1-пропил метансульфоната (5,0 г, 0,015 моля) и карбоната калия (4,8 г) в N, N-диметилформамиде (50 мл) нагревают до 60oС в течение 5 часов. После стояния в течение ночи отфильтровывают твердое вещество и промывают бензолом. Фильтрат разбавляют бензолом (200 мл) и промывают водой (4•100 мл). Органический раствор сушат (MgSO4) и выпаривают растворитель в вакууме. Остаток (7,8 г) чистят колоночной хроматографией на силикагеле, используя в качестве элюентов бензол и смесь бензола-этилацетата и получая этиловый эфир 2-(4-(3-(10, 11-дигидро-5H-1-дибензо [a, d] циклогептен-5-илиден; -1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты (3,4 г, 48%) в виде масла.
Смесь полученного выше эфира (2,1 г, 4,5 ммоля) и 20% гидроксида натрия (3,0 мл) в этаноле (21 мл) перемешивают при комнатной температуре в течение 16 часов. Раствор выливают в дихлорметан (250 мл) и подкисляют 2N соляной кислотой. Органическую фазу промывают водой (10 мл), сушат (MgSО4) и выпаривают растворитель в вакууме. Остаток в виде пены отгоняют с ацетоном и кристаллизуют из ацетона, получая 1,5 г (70%) указанного в заголовке соединения. Т.пл. 224-230oС.
Рассчитано для С28H29N3О2, НС1, 0,25 Н2О, 0,5 С3Н6О: С 69,53%; Н 6,
63%; С1 6,96%; N 8,25%;
Найдено: С 69,76%; Н 6,76%; С1 7,00%; N 8,17%.
ПРИМЕР
7
Гидрохлорид 2-(4-(2-(10,11-дигидро-5Н-дибензо[b, f]
азепин-5-ил)-1-этил)-1-пиперазинил)-3-пиридинкарбоновой кислоты
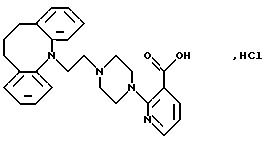
5-(2-Гидроксиэтил)-10, 11-дигидро-5Н-дибензо[b, f] азепин (2,5 г, 0,0104 моля, полученный, как описано в Fr. Pat. 1,215 599) и триэтиламин (2,8 г, 0,028 моля) растворяют в бензоле (90 мл). Добавляют по капле в течение 10 минут раствор метансульфонилхлорида (1,66 г, 0,0145 моля) в бензоле (10 мл) при 15-20oС, охлаждая на бане с холодной водой. Когда добавление завершено, реакционную смесь перемешивают 1 час при комнатной температуре. Добавляют воду (35 мл), органический слой отделяют, промывают водой (25 мл), сушат (МgSO4) и выпаривают в вакууме. Растворяют в ацетоне (55 мл) метансульфонат (3, 2 г, 97%) и добавляют этиловый эфир 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (2,64 г, 0,0112 моля) и карбонат калия (4,0 г). Реакционную смесь нагревают при 50-55oС в течение 28 часов. Смесь фильтруют и удаляют растворитель путем выпаривания в вакууме. Сырой остаток (5,7 г) чистят колоночной хроматографией на силикагеле (65 г), используя в качестве элюента смесь бензола и хлороформа. Это дает 2,0 г (43%) этилового эфира 2-(4-(2-(10,11- дигидро-5Н-дибензо[b, f]азепин-5-ил)-1-этил)-1-пиперазинил)-3-пиридинкарбоновой кислоты в виде масла.
Полученный выше эфир растворяют в этаноле (5 мл) и добавляют 4N гидроксид натрия (5 мл). Добавляют дихлорметан (200 мл), а затем концентрированную соляную кислоту (3 мл). Твердое вещество отфильтровывают и промывают ацетоном, перемешивают с водой (2•25 мл), фильтруют и промывают ацетоном. Сырой продукт растворяют в теплом N,N-диметилформамиде, добавляют бензол и смесь охлаждают в течение ночи. Осадок отфильтровывают, промывают бензолом и сушат, получая 0,9 г (44%) указанного в заголовке соединения.
Т.пл. 240-245oС.
Рассчитано для С26Н28N4O2, НСl, 0,5 H2O: С 65,87%; Н 6,38%; N
11,82%; Cl 7,48%;
Найдено: С 65,82%; Н 6,23%; N 11,66%; Cl 7,90%.
ПРИМЕР 8
Гидрохлорид 6-(4-(3-(10,11-дигидро-5Н-дибензо[b, f]
aзепин-5-ил)-1-пропил)-1-пиперазинил)-2-пиридинкарбоновой кислоты

К суспензии 6-хлорпиридин-2-карбоновой кислоты (8,3 г, 0,0525 моля, полученной, как описано в Сhеm. Веr. 45, 2456 (1912)) в диоксане (25 мл) добавляют тионилхлорид (9,4 мл, 0,13 моля) и полученную смесь перемешивают при 70oС в течение 4 часов. Реакционную смесь концентрируют в вакууме и добавляют смесь диоксана (8,3 мл) и этанола (16,6 мл). Реакционную смесь нагревают до 70oС в течение 2 часов, добавляют триэтиламин (8,3 мл), этанол (4,1 мл) и воду (8,3 мл) и реакционную смесь снова концентрируют. Остаток распределяют между диэтиловым эфиром (28 мл) и водой (18 мл) и фазы разделяют. Водный слой экстрагируют диэтиловым эфиром (30 мл) и объединенные органические слои сушат (МgSО4) и выпаривают в вакууме. Это дает 8,82 г (91%) этилового эфира 6-хлорпири-дин-2-карбоновой кислоты в виде масла.
Смесь полученного выше эфира (8,6 г, 0,046 моля), безводного пиперазина (41 г, 0,476 моля) и йодида натрия (0,46 г) в толуоле (145 мл) нагревают при температуре кипения с обратным холодильником в течение 2,5 часов. После охлаждения примерно до 80oС отфильтровывают пиперазин, а фильтрат смешивают с водой (250 мл) и диэтиловым эфиром (100 мл). Органический слой отделяют и промывают золой (2•50 мл) и соляным раствором (50 мл), сушат (MgSO4) и выпаривают, получая 4,6 г масла. Водный слой экстрагируют хлороформом (5•50 мл), объединенные органические фазы промывают водой и соляным раствором (как выше), сушат и выпаривают, получая еще 3,2 г масла. Оба полученных соединения чистят колоночной хроматографией на силикагеле (200 г), используя в качестве элюента смесь хлороформа и этанола (9:1). Это дает 5 г (46%) этилового эфира 6-(1-пиперазинил)-2-пиридинкарбоновой кислоты в виде масла.
Смесь полученного выше эфира (2,20 г, 9,35 ммоля), (10,11-дигидро-5Н-дибензо[b, f] азепин-5-ил)-1-пропил)метан сульфоната (2,82 г, 8,51 ммоля) и карбоната калия (3,3 г, 0,0239 моля) в ацетоне (47 мл) нагревают при температуре кипения с обратным холодильником в течение 16 часов. Реакционную смесь фильтруют и концентрируют в вакууме. Остаток чистят колоночной хроматографией на силикагеле (100 г), используя в качестве элюента хлороформ. Это дает 1,21 г (30%) этилового эфира 6-(4-(3-(10, 11-дигидро-5Н-дибензо[b,f] азепин-5-ил)-1-пропил)-1-пиперазинил)-2-пиридинкарбоновой кислоты в виде масла.
Смесь полученного выше эфира (1,06 г, 2,25 ммоля), этанола (10 мл), раствора гидроксида натрия (0,342 г) и воды (1,3 мл) перемешивают при комнатной температуре в течение 48 часов. Добавляют концентрированную соляную кислоту (1,25 мл), а затем дихлорметан (60 мл). Фазы разделяют и органическую фазу сушат (МgSO4) и выпаривают в вакууме. Остаток повторно отгоняют с ацетоном (30 мл) и перемешивают горячим этанолом (30 мл). После сушки это дает 0,45 г (42%) указанного в заголовке соединения.
Рассчитано для С27Н30N4О2, НСl: С 67,70%; Н 6,52%; N 11,70%; Cl 7,40%;
Найдено: С 67,26%; Н 6,
70%; N 11,63%; Cl 7,31%.
ПРИМЕР 9
Дигидрохлорид 2-(4-(3-(10,
11-дигидро-5Н-дибензо[b, f] aзепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты
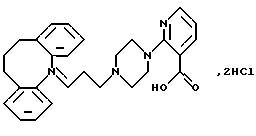
Смесь 5-(3-хлорпропил)-10,11-дигидро-5Н-дибензо [b,f]-азепина (4,54 г, 16,7 ммоля), метилового эфира 2-пиперазинилникотиновой кислоты (3,7 г, 16,7 ммоля), сухого карбоната калия (6,92 г, 50,1 ммоля), йодида натрия (2,5 г, 16,7 ммоля) и 2-бутанона (50 мл) нагревают при температуре кипения с обратным холодильником в течение 60 часов. После охлаждения до комнатной температуры добавляют толуол (100 мл) и воду (100 мл). Органический слой отделяют, промывают водой (2•50 мл) и выпаривают в вакууме. Сырой продукт чистят колоночной хроматографией на силикагеле, используя в качестве элюента смесь толуола и этилацетата (3:1), содержащую триэтиламин (2,5%). Это дает 2,3 г (30%) метилового эфира 2-(4-(3-(10,11-дигидро-5Н-дибензо[b,f]азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты в виде масла.
Полученный выше эфир (2,3 г, 5,04 ммоля), растворенный в смеси 2N гидроксида натрия (8,32 мл, 16,63 ммоля) и этанола (25 мл), перемешивают при комнатной температуре в течение 16 часов. Этанол выпаривают в вакууме, а остаток разбавляют водой (50 мл). Добавляют 2N соляную кислоту (20 мл) и промывают раствор диэтиловым эфиром (25 мл). Водные фазы экстрагируют дихлорметаном (3•30 мл). Объединенные органические экстракты сушат (МgSO4 ) и выпаривают в вакууме, получая пену. Ее кристаллизуют из тетрагидрофурана, фильтруют и сушат, получая 1,20 г (50%) указанного в заголовке соединения. Т.пл. 168-171oС.
Рассчитано для С27Н30N4О2, 1,75 НС1: С 64,04%; Н 6,32%;
N 11,06%; Cl 12,25%;
Найдено: С 63,72%; Н 6,42%; N 10,68%; Cl 11,99%.
ПРИМЕР 10
Гидрохлорид 2-(4-(3-(10,11-дигидро-5Н-дибензо[b, f]
азепин-5-ил)-1-пропил)-1-пиперазинил)-5-пиридинкарбоновой кислоты
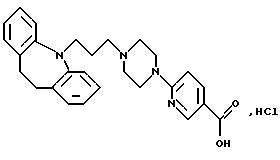
Смесь метилового эфира 2-бромпиридин-5-карбоновой кислоты (1,40 г, 6,5 ммоля) и пиперазина (5,59 г, 65 ммолей) растворяют в ацетонитриле (50 мл) и нагревают при температуре кипения с обратным холодильником в течение 2,5 часов. Реакционную смесь концентрируют в вакууме, растворяют в дихлорметане (50 мл) и экстрагируют водой (3•50 мл). Органический раствор сушат (MgSO4) и выпаривают в вакууме, получая 0,80 г, (56%) метилового эфира 2- (1-пиперазинил)-5-пиридинкарбоновой кислоты. Т.пл. 91-92oС.
Смесь полученного выше эфира (0,73 г, 3,3 ммоля), 5-(3-хлорпропил)-10, 11-дигидро-5Н-дибензо[b, f] азепина (0,9 г, 3,3 ммоля), сухого карбоната калия (1,37 г, 9,9 ммоля), йодида натрия (0,5 г, 3,3 ммоля) и 2-бутанона (10 мл) нагревают при температуре кипения с обратным холодильником в течение 24 часов. После охлаждения до комнатной температуры к реакционной смеси добавляют дихлорметан (25 мл) и воду (25 мл). Органический слой отделяют, промывают водой (2•10 мл) и выпаривают в вакууме, сырой остаток чистят колоночной хроматографией на силикагеле, используя в качестве элюента смесь толуола и этилацетата (1:1), содержащую триэтиламин (2,5%). Это дает 0,65 г (43%) метилового эфира 2-(4-(3-(10,11-дигидро-5Н-дибензо[b, f]азепин-5-ил)-1-пропил)-1-пиперазинил)-5-пиридинкарбоновой кислоты в виде масла.
Полученный выше эфир (0,65 г, 1,25 ммоля), растворенный в смеси 2N гидроксида натрия (2,06 мл, 4,13 ммоля) и этанола (10 мл), перемешивают при комнатной температуре в течение 16 часов. Этанол выпаривают в вакууме, а остаток разбавляют водой (30 мл). Добавляют 2N соляную кислоту (20 мл) и промывают раствор диэтиловым эфиром (25 мл). Водную фазу экстрагируют дихлорметаном (3•15 мл). Объединенные органические экстракты сушат (МgSO4) и концентрируют в вакууме до 20 мл. Осадок отфильтровывают и сушат, получая 0,33 г (60%) указанного в заголовке соединения.
Т.пл. 240-244oС.
Рассчитано для С27Н30N4
О2, НСl, 0,5 Н2O: С 66,45%; Н 6,61%; N 11,48%; Cl 7,26%;
Найдено: С 66,39%; Н 6,64%; N 11,22%; Cl 7,
08%.
ПРИМЕР 11
Дигидрохлорид
2-(4-(3-(3-хлор-10,11-дигидро-5Н-дибензо [b,f]азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты

К раствору 3-хлор-10,11-дигидро-5Н-дибензо[b,f]азепина (5,0 г, 22 ммоля) в толуоле (20 мл; добавляют по капле 3-хлорпропионилхлорид (3,32 г, 26 ммолей) в толуоле (8 мл). Реакционную смесь перемешивают при 95oС в течение 2 часов и оставляют перемешиваться при комнатной температуре в течение ночи. Добавляют 2N гидроксид натрия (25 мл), а затем толуол (50 мл) и разделяют фазы. Толуольную фазу промывают 2N гидроксидом натрия (2•25 мл), водой (3•30 мл) и соляным раствором (30 мл). Органическую фазу сушат (МgSO4) и выпаривают в вакууме. Остаток отгоняют с метанолом (30 мл) и остаток суспендируют в этилацетате (8 мл), перемешивают и отфильтровывают.Твердое вещество промывают этилацетатом (10 мл) и сушат. Это дает 4,0 г (57%) 3-хлор-1-(3-хлор-10,11-дигидро-5Н-дибензо [b,f]азепин-5-ил)пропан-1-она.
1,0 М раствор литийалюминийгидрида в тетрагидрофуране (26,2 мл, 26 ммолей) вводят под азотом в 250 мл сухую трехгорлую колбу. При охлаждении (ледяная баня) добавляют по капле концентрированную серную кислоту (0,7 мл). Смесь перемешивают на ледяной бане 15 минут, а потом 45 минут при комнатной температуре. Полученный выше амид (4,2 г, 13 ммолей) растворяют в тетрагидрофуране (40 мл) и добавляют медленно по капле. Реакционную смесь оставляют перемешиваться на 1,5 часа. Добавляют по капле воду (1 мл), а затем 4N гидроксид натрия (1 мл) и после этого воду (3 мл). Смесь оставляют перемешиваться на 30 минут, фильтруют (hyflo) и выпаривают. Остаток растворяют в этилацетате (50 мл), сушат и выпаривают, получая 3,5 г (88%) 3-хлор-5-(3-хлорпропил)-10,11-дигидро-5Н-дибензо[b,f]азепина.
Йодид калия (2,8 г, 17 ммолей) и метилэтилкетон (70 мл) нагревают при 80oС в течение 1 часа. Полученный выше хлорид (0,8 г, 3 ммоля) растворяют в метилэтилкетоне (10 мл) и добавляют. Смесь нагревают при температуре кипения с обратным холодильником в течение 50 минут. Добавляют карбонат калия (1,25 г, 9 ммолей), а затем метиловый эфир 2-(1-пиперазинил)-3-пиридинкарбоновой кислоты (1,0 г, 4 ммоля) в метилэтилкетоне (10 мл). Реакционную смесь перемешивают при 78oС в течение ночи. После охлаждения смесь фильтруют (hyflo) и выпаривают. Остаток чистят колоночной хроматографией на си-ликагеле, используя в качестве элюента смесь гептана и этилацетата (1:1). Это дает этиловый эфир 2-(4-(3-(3-хлор-10,11-дигидро-5Н-дибензо [b,f] азепин-5-ил) -1-про пил) -1-пиперазинил)-3-пиридинкарбоновой кислоты.
ТСХ: Rf=0,17 (SiO2 этилацетат/гептан = 1:1).
Полученный выше этиловый эфир (0,33 г, 0,67 ммоля) растворяют в этаноле (8 мл). Добавляют 4N гидроксид натрия (0,34 мл) и реакционную смесь перемешивают в течение 2 часов при комнатной температуре и оставляют в холодильнике на ночь. Продолжают перемешивание при комнатной температуре в течение 24 часов. Добавляют еще 4N гидроксид натрия (0,34 мл), перемешивание продолжают в течение 2 часов, добавляют еще 4N гидроксид натрия (0,34 мл) и перемешивание продолжают в течение 3,5 часов. Добавляют еще 4N соляную кислоту (1,18 мл), а затем воду (20 мл) и дихлорметан (100 мл). Фазы разделяют и органическую фазу сушат (МgSO4) и выпаривают. Остаток отгоняют с ацетоном (2•30 мл), перемешивают с изопропилацетатом (3-5 мл) в течение 5 минут, отфильтровывают и сушат. Выход 0,18 г (51%) указанного в заголовке соединения в виде аморфного порошка.
Рассчитано для C27H29ClN4
O2, 2HCl, 0,25 C5H9O2: С 62,98%; Н 6,03%; N 10,40%;
Найдено: С 63,31%: Н 6,36%; N 10,16%.
HPLC время удерживания (высокоэффективная жидкостная хроматография)= 23,82 минуты (колонка 5 мкм С184•250 мм), элюирование с 20-80% градиентом 0,1% трифторуксусной кислотой/ацетонитрилом и 0,1% трифторуксусной кислотой/водой в течение 30 минут при 30oС.
ПРИМЕР 12
Гидрохлорид 1-(3-(10,
11-дигидро-5Н-дибензо[a,
d]циклогептен-5-илиден)-1-пропил)-4-(2-нитрофенил)пиперазина
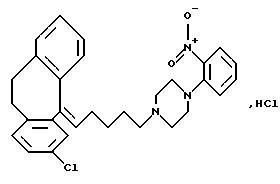
2-Фторнитробензол (11,4 г, 81 ммоль) растворяют в диметилсульфоксиде (200 мл) и добавляют пиперазин (56,4 г, 0,65 моля) и карбонат натрия (20,6 г, 0,2 моля). Смесь перемешивают при 130oС в течение 16 часов. После охлаждения добавляют толуол (300 мл) и промывают смесь водой (3•300 мл) и 1N гидроксидом натрия (300 мл). Органическую фазу сушат (МgSO4) и концентрируют в вакууме. Остаток чистят флэш-хроматографией на силикагеле, элюируя сначала смесью этилацетата, гептана и триэтиламина (5:5:0,25), потом смесью этилацетата и триэтиламина (9:1) и, наконец, смесью метанола и триэтиламина (40: 1). Это дает 1-(2-нитрофенил)-пиперазин (8,08 г, 48%).
ТСХ: Rf=0,27 (SiO2: этилацетат/метанол/триэтиламин 1:1:0,25).
Полученный выше пиперазин (1,51 г, 7,3 ммоля) растворяют в метилэтилкетоне (50 мл), добавляют 5-(3-бром-1-пропилиден)-10,11-дигидро-5Н-дибензо [a,d] циклогептен (2,28 г, 7,3 ммоля), йодид калия (0,96 г, 15 ммолей) и карбонат калия (6,0 г, 44 ммоля) и полученную смесь перемешивают при температуре кипения с обратным холодильником в течение 24 часов. После охлаждения добавляют этилацетат (100 мл) и смесь промывают водой (2•100 мл), сушат (МgSO4) и концентрируют в вакууме. Остаток чистят способом хроматографии с мгновенным испарением на силикагеле, используя в качестве элюента смесь этилацетата и гептана (1:2) и получая 2,18 г (68%) свободного основания. Его растворяют в диэтиловом эфире (30 мл) и добавляют 1N соляную кислоту в диэтиловом эфире (5 мл). Осадившееся твердое вещество выделяют фильтрованием и сушат в вакууме, получая 1,83 г (53%) указанного в заголовке соединения.
Т.пл. 204-206oС.
Рассчитано для C28
H29N3O2, HCl: С 70,65%; Н 6,35%; N 8,83%;
Найдено: С 70,50%; Н 6,51%; N 8,61%.
ПРИМЕР 13
2-(4-(3-(10,11-Дигидро-5Н-дибензо[a,
d]циклогептен-5-илиден;
1-пропил)-1-пиперазинил)бензонитрил
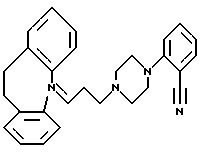
Пиперазин (6,0 г, 69 ммолей) растворяют в диметилсульфоксиде (50 мл), добавляют 2-фторбензонитрил (0,95 мл, 9 ммолей) и карбонат калия (2,2 г, 17 ммолей) и смесь перемешивают при комнатной температуре 16 часов. Добавляют воду и экстрагируют смесь толуолом (2•100 мл). Объединенные органические экстракты промывают 1N гидроксидом натрия (3•75 мл), сушат (МgSO4) и концентрируют в вакууме, получая 1,48 г (88%) 2-(1-пиперазинил)бензонитрила в виде масла. ТСХ: Rf=0,12 (SiO2: этилацетат/триэтиламин = 4:1).
Полученный выше бензонитрил (4,89 г, 26 ммолей) растворяют в метилэтилкетоне (100 мл) и добавляют карбонат калия (21,7 г, 157 ммолей), йодид калия (3,45 г, 52 ммоля) и 5-(3-бром-1-пропилиден;-10,11-дигидро-5Н-дибензо[a, d] циклогептен (8,18 г, 26 ммолей). Полученную смесь перемешивают при температуре кипения с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры добавляют этилацетат (200 мл) и смесь промывают водой (2•150 мл), сушат (МgSO4)и концентрируют в вакууме. Остаток чистят флэш-хроматографией на силикагеле, элюируя сначала смесью этилацетата и гептана (1:4) и затем смесью этилацетата и гептана (1:2). Это дает 5,60 г указанного в заголовке соединения в виде твердого вещества.
Т.пл. 112-114oС.
Рассчитано для С29Н29
N3: С 83,02%; Н 6,97%; N 10,02%;
Найдено: С 83,25%; Н 7,14%; N 9,93%.
ПРИМЕР 14
Гидрохлорид 2-(4-(3-(10,11-дигидро-5Н-дибензо[a, d]
циклогептен-5-илиден)-1-пропил)-1-пиперазинил бензойной кислоты

2-(4-(3-(10, 11-дигидро-5Н-дибензо [a, d] циклогептен-5-илиден)-1-пропил)-1-пиперазин)бензонитрил (0,30 г, 0,72 ммоля, полученный, как описано в примере 13) добавляют к смеси воды (0,6 мл), серной кислоты (0,6 мл) и уксусной кислоты (0,6 мл). Полученную смесь нагревают при температуре кипения с обратным холодильником в течение 24 часов и 3 дня перемешивают при комнатной температуре. Добавляют 1N гидроксид натрия (20 мл) и промывают смесь диэтиловым эфиром (3•20 мл). Водную фазу подкисляют 5N соляной кислотой до рН 1. Смесь экстрагируют дихлорметаном (2•50 мл), объединенные экстракты сушат (МgSO4) и выпаривают в вакууме, получая 0,21 г (56%) указанного в заголовке соединения в виде аморфного твердого вещества.
Рассчитано для С29H23
N2O2, НСl, 1,5 Н2O:
С 69,38%; Н 6,83%; N 5,58%;
Найдено: С 69,69%; Н 6,69%; N 5,06%.
Масс-спектрометрия с электронной ионизацией
(70
эВ):
m/е=438 (М+,0, 1%).
ПРИМЕР 15
Дигидрохлорид 1-(3-(10,11-дигидро-5Н-дибензо[a, d]
циклогептен-5-илиден)-1-пропил)-4-(3-трифторметил-2-пиридил)пиперазина
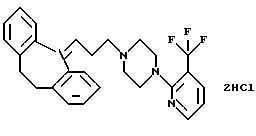
Смесь 5-(3-бром-1-пропилиден)-10,11-дигидро-5Н-дибензо-[а, d] циклогептена (1,10 г, 3,5 ммоля, полученного, как описано в WO 9518793), 1-(3-(трифторметил)-2-пиридил)пиперазина (0,81 г, 3,5 ммоля), карбоната калия (1,45 г, 10,5 ммоля) и йодида натрия (0,53 г, 3,5 ммоля) в сухом 2-бутаноне (10 мл) нагревают при температуре кипения с обратным холодильником в течение 40 часов. Растворитель удаляют в вакууме, а остаток растворяют в толуоле (25 мл) и воде (25 мл). Органический слой отделяют, сушат (МgSO4) и концентрируют в вакууме. Остаток чистят способом хроматографии с мгновенным испарением на силикагеле, используя в качестве элюента смесь толуола и этилацетата (3:1), содержащую 2,5% триэтиламина, и получая 1,54 г (95%) свободного основания. Его растворяют в тетрагидрофуране (25 мл) и добавляют 1N хлористый водород в диэтиловом эфире (7 мл). Осадившееся твердое вещество выделяют фильтрованием и сушат в вакууме, получая 1,45 г (77%) указанного в заголовке соединения в виде твердого вещества.
Т.пл. 225-227oС.
Рассчитано для C28H28N3F3, 2HCl: С 62,69%; Н 5,64%; N 7,83%;
Найдено: С 61,
99%; Н 5,82%; N 7,60%.
ПРИМЕР 16
Дигидрохлорид 2-(4-(2-(6,11-дигидродибензо[b,e]тиепин-11-илиден)этил)-пиперазин-1-ил)-3-пиридинкарбоновой кислоты
Смесь 11-(2-бромэтилиден)-6,11-дигидродибензо[b, e] тиепина (2,5 г, 7,9 ммоля, полученного, как описано в Coll. Czech. Chem. Comm. 52, 1566 (1987)), этил 2-(1-пиперазинил)-3-пиридинкарбоксилата (1,86 г, 7,9 ммоля), карбоната калия (1,45 г, 10,5 ммоля) и N,N-диметилформамида (15 мл) перемешивают и нагревают при 150oС в течение 10 часов. Смесь разбавляют водой и экстрагируют бензолом (50 мл). Органическую фазу сушат (карбонатом калия), фильтруют и выпаривают растворитель в вакууме. Маслянистый остаток чистят на силикагеле (50 г), используя в качестве элюента этилацетат и получая основание, которое растворяют в диэтиловом эфире и нейтрализуют щавелевой кислотой в ацетоне. Это дает 2,7 г (54%) полугидрата этил 2-(4-(2-(6,11-дигидродибензо[b, e] тиепин-11-илиден)этил)-1-пиперазинил)-3-пиридин карбоксилат гидрооксалата.
ТСХ: Rf=0,5 (SiO2 : хлороформ/этанол/аммиак = 40:2:1).
Полученный выше эфир (2,25 г, 4,77 ммоля) растворяют в этаноле (40 мл) и добавляют 5N гидроксид натрия (3 мл). Смесь перемешивают при 40oС в течение 20 часов, выпаривают этанол в вакууме и добавляют воду (40 мл), а потом уксусную кислоту (2 мл). Экстрагируют смесь дихлорметаном (50 мл), органическую фазу сушат (МgSO4 ) и выпаривают растворитель в вакууме. Остаток растворяют в ацетоне и нейтрализуют хлористым водородом в диэтиловом эфире, получая 2,13 г (85%) указанного в заголовке соединения в виде полугидрата.
Т.пл. 188-195oС.
Рассчитано для C26H25N3O2S, 0,5 H2O, 2HCl:
С 59,42%; Н 5,37%;
N 8,00%; Cl 13,
49%; S 6,10%;
Найдено: С 56,94%; Н 5,23%; N 8,10%; Cl 13,44%; S 5,80%.
ПРИМЕР 17
Дигидрохлорид 2-(4-(3-(6,11-дигидроцибензо[b, е]
тиепин-11-илиден)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты
Раствор циклопропилмагнийбромида в сухом тетрагидрофуране (полученный из циклопропилбромида (3,7 г, 0,031 моля), магниевых стружек (0,8 г, 0,033 моля) и сухого тетрагидрофурана (50 мл) в атмосфере азота) добавляют по капле к раствору 6,11-дигидродибензо[b,е]тиепин-11-она (3,5 г, 0,016 моля, полученного аналогично тому, как описано в Chem. Pharm. Bull. 39, 1991, 2564) в сухом тетрагидрофуране (50 мл). Когда добавление завершено, смесь нагревают при 50oС в течение 2 часов. Реакционную смесь охлаждают на ледяной бане и осторожно добавляют хлорид аммония (50 мл) и воду (50 мл). Смесь экстрагируют диэтиловым эфиром (2•100 мл), сушат (МgSO4), фильтруют и выпаривают растворитель в вакууме, получая 4,4 г сырого 11-циклопропил-6, 11-дигидро-11Н-дибензо [b, е] -тиепин-11-ола в виде масла.
Полученный выше спирт (4,0 г) растворяют в дихлорметане (50 мл) и добавляют по капле при комнатной температуре раствор триметилсилилбромида (2,1 мл, 0, 016 моля). Когда добавление завершено, смесь перемешивают при комнатной температуре в течение 1,5 часов и добавляют воду (50 мл). Фазы разделяют и органическую фазу промывают водой (50 мл), сушат (МgSO4) и выпаривают растворитель в вакууме, получая 4,1 г (83%) сырого 1-бром-3-(6,11-дигидродибензо[b,е]тиепин-11-илиден)пропана в виде твердого вещества.
ТСХ: Rf=0,55 (SiO2: бензол/циклогексан = 1:3).
Смесь полученного выше бромида (3,65 г, 0,011 моля), этилового эфира 2-пиперазинил-3-пиридинкарбоновой кислоты (3,11 г, 0, 0132 моля), карбоната калия (4,55 г, 0,033 моля) и йодида натрия (1,61 г, 0,011 моля) в 2-бутаноне (53 мл) нагревают при температуре кипения с обратным холодильником в течение 6 часов. После стояния в течение ночи реакционную смесь фильтруют и фильтрат выпаривают в вакууме. Маслянистый остаток чистят колоночной хроматографией на силикагеле (70 г), используя в качестве элюента хлороформ. Это дает 4, 87 г (91%) этилового эфира 2-(4-(3-(6,11-дигидро-дибензо[b,е]тиепин-11-илиден)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты в виде масла.
ТСХ: Rf=0,42 (SiO2: этилацетат/н-гексан = 1:1).
Полученный выше эфир (2,77 г, 0,0057 моля) растворяют в этаноле (46 мл) и добавляют 4N гидроксид натрия (6,34 мл, 0,024 моля). Смесь оставляют на ночь при комнатной температуре. Добавляют концентрированную соляную кислоту (4,76 мл) и дихлорметан (300 мл) и органический слой отделяют, сушат (МgSO4) и выпаривают в вакууме. Остаток отгоняют с дихлорметаном (3•80 мл). После перемешивания со смесью ацетона (100 мл) и диэтилового эфира (100 мл) получают 0,75 г (24%) указанного в заголовке соединения.
Т.пл. 161-166oС.
Рассчитано для С27Н27N3О2S, 2НСl, Н2O:
С 59,12%; Н 5,70%; N 7,66%; S 5,85%;
Найдено: С
59,27%: Н 5,61%; N 7,54%; S 6,13%.
ПРИMEP 18
2-(4-(2-(6,11-дигидродибензо[b, е] тиепин-11-илокси)этил)-1-пиперазинил)-3-пиридинкарбоновая кислота

6,11-Дигидродибензо[b, е] тиепин-11-ол (13,2 г, 0,0578 моля, полученный аналогично тому, как описано в Ceskoslov. Farm. 11, 404 (1962)) растворяют в бензоле (160 мл) и добавляют 2-бромэтанол (10,8 г, 0,086 моля) и концентрированную серную кислоту (2 мл). Смесь перемешивают при комнатной температуре в течение 2 часов, после стояния в течение ночи добавляют ледяную воду и разделяют фазы. Органическую фазу промывают 5% бикарбонатом натрия, сушат (МgSO4), выпаривают растворитель в вакууме, получая остаток, который кристаллизуют из смеси бензола и циклогексана. Это дает 13,5 г (70%) 11-(2-бромэтокси)-6,11-дигидродибензо-[b,e]тиепина в виде твердого вещества.
ТСХ: Rf=0,08 (SiO2: циклогексан/этилацетат = 5:1).
Смесь полученного выше бромида (2,85 г, 8,5 ммоля), этил 2-(1-пиперазинил)-3-пиридинкарбоксилата (2,0 г, 8,5 ммоля), карбоната калия (1,65 г, 12 ммолей) и N,N-диметилформамида (15 мл) перемешивают и нагревают при 150oС в течение 10 часов. Смесь разбавляют водой и экстрагируют бензолом (50 мл). Органическую фазу сушат (карбонатом калия), фильтруют и выпаривают растворитель в вакууме. Маслянистый остаток чистят на силикагеле (50 г), используя в качестве элюента этилацетат и получая свободное основание, которое растворяют в диэтиловом эфире и нейтрализуют щавелевой кислотой в ацетоне, получая 3,75 г (74%) гидрата этил 2-(4-(6,11-дигидродибензо[b, е]тиепин-11-илоксиэтил)-1-пиперазинил)-3-пиридинкарбоксилат гидрооксалата в виде кристаллов. При помощи водного аммиака из указанного выше оксалата извлекают свободное основание и выделяют его путем экстракции диэтиловым эфиром. ТСХ: Rf=0,55 (SiO2: хлороформ/этанол/аммиак = 40:2:1).
Полученный выше свободный эфир (2,65 г, 5,4 ммоля) растворяют в этаноле (40 мл) и добавляют 5N гидроксид натрия (4 мл). Смесь перемешивают при 40oС в течение 20 часов, выпаривают этанол в вакууме и добавляют воду (40 мл), а затем уксусную кислоту (3 мл). Смесь экстрагируют дихлорметаном (50 мл). Отфильтровывают продукт, кристаллизующийся из этого раствора. Из маточника выпаривают дихлорметан и путем растирания с ацетоном получают второй урожай продукта, общий выход составляет 2,4 г (95%) указанного в заголовке соединения.
Т.пл. 217-218,5oС.
Рассчитано для С26Н27N3О3S, 0,25 Н2О: С 67,00%; Н 5,95%; N 9,02%; S 6,88%;
Найдено: С 67,18%; Н 5,84%; N 8,99%; S 6,64%.
ПРИМЕР 19
Гидрохлорид 6-(4-(3-(10,
11-дигидро-5Н-дибензо[a,d]циклогептен-5-илиден; -1-пропил)пиперазин-1-ил-2-пиридинкарбоновой кислоты

К раствору этилового эфира 6-пиперазинил-2-пиридин-карбоновой кислоты (7,0 г, 0,03 моля) в 2-бутаноне (150 мл) добавляют 3-(10,11-дигидро-5Н-дибензо[а, d] циклогептен-5-илиден)-1-пропил метансульфоната (6,8 г, 0,0207 моля) и карбонат калия (8,0 г). Реакционную смесь нагревают при 40oС в течение 3 часов. После стояния в течение ночи при комнатной температуре продолжают нагревание в течение 9 часов при 60oС и 9 часов при температуре кипения с обратным холодильником. Твердое вещество отфильтровывают, промывают ацетоном и выпаривают фильтрат в вакууме. Остаток чистят колоночной хроматографией на силикагеле (200 г), используя в качестве элюента хлороформ. Это дает 6,63 г (68%) этилового эфира 6-(4-(3-(10,11-дигидро-5Н-дибензо[а, d]циклогептен-5-илиден)-1-пропил)-пиперазин-1-ил)-2-пиридинкарбоновой кислоты в виде масла.
TCX: Rf=0,32 (SiO2: петролейный эфир/этилацетат = 3:2).
К раствору полученного выше эфира (3,74 г, 8,0 ммолей) в этаноле (38 мл) добавляют раствор гидроксида натрия (1,3 г) в воде (4,9 мл) и оставляют реакционную смесь при комнатной температуре на 60 часов. Добавляют концентрированную соляную кислоту (4,9 мл), а затем дихлорметан (230 мл). Смесь перемешивают 5 минут, разделяют фазы и органическую фазу сушат (МgSO4) и выпаривают в вакууме. Пенящийся остаток растворяют в ацетоне (500 мл) и выпаривают до трети своего объема. Образовавшееся твердое вещество отфильтровывают, получая 1,8 г (47%) указанного в заголовке соединения.
Т.пл. 233-243oС.
Рассчитано для C28H29N3O2, HCl, 0,25 Н2О: С 69,98%; Н 6,40%; Сl 8,74%;
N 7,38%;
Найдено: С 69,90%; Н 6,42%; Сl 8,47%; N 7,52%.
ПРИМЕР 20
Гидрохлорид 2-(4-(3-(3-метил-10,11-дигидро-5Н-дибензо[b, f]
-азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты

Под азотом и при перемешивании магнитной мешалкой на масляной бане нагревают натрий (3,14 г, 0,136 моля) при 120-130oС до плавления и затем добавляют по капле метанол (6,1 мл, 0,15 моля). Так как остается некоторое количество не прореагировавшего натрия, добавляет еще метанол (1 мл). Алкоголят нагревают при той же температуре в течение часа и охлаждают белое рыхлое твердое вещество на бане со льдом-солью до 10-15oС. Добавляют по капле смесь диэтил 2-бром-фенилметилфосфоната (27,94 г, 0,091 моля) и 4-метил-2-нитробензальдегида (15,02 г, 0,091 моля, полученного аналогично тому, как описано в Zh. Org. Khim. 1968, 5, 953 или в Ann. 1956, 627, 218) в N, N-диметилформамиде (60 мл). Когда добавление завершено, реакционную смесь перемешивают еще 20 минут при той же температуре, выдерживают еще 1 час при комнатной температуре под светом при охлаждении на водяной бане и оставляют на ночь в холодильнике. Добавляют воду (150 мл) при охлаждении на ледяной бане, реакционную смесь еще разбавляют водой (450 мл) и экстрагируют этилацетатом (5•100 мл). Органический слой промывают водой (100 мл) и сушат (МgSO4). Остаток (32,5 г) перемешивают с циклогексаном, твердое вещество отфильтровывают, растворяют в теплом циклогексане и декантируют горячий раствор с масла, которое отделяют в колбе, и охлаждают. Осадившееся твердое вещество отфильтровывают, промывают циклогексаном и сушат (8,9 г) (продукт А).
Фильтрат объединяют с отделенным ранее маслом, смесь выпаривают в вакууме и остаток (19,25 г) чистят колоночной хроматографией на силикагеле (200 г), используя в качестве элюента смесь циклогексана и бензола (10:1) и получая в качестве побочного продукта 1,2-бис-(2-бромфенил)этилен (3,26 г, 21%).
Изменяя элюент на смесь циклогексана и бензола (1:1), получают дополнительную порцию (6,70 г) твердого вещества, идентичного продукта. Общий выход выделенного, как показано выше, из реакционной смеси (циклогексан) продукта составляет 13,60 г (29%) Z-(2-(2-бромфенил)винил)-4-метил-2-нитробензола.
ТСХ: Rf=0,75 (SiO2: циклогексан/этилацетат = 5:1).
Раствор полученного выше нитробензола (9,68 г, 30,3 ммоля) в этаноле (110 мл) и метаноле (40 мл) гидрируют 2 часа при 3 МПа в присутствии морфолина (0,6 мл) и катализатора Rh/C (КО 319) (2 г). Катализатор отфильтровывают, фильтрат выпаривают в вакууме и остаток отгоняют с толуолом (3•20 мл). После стояния при комнатной температуре маслянистый остаток кристаллизуется. После промывания твердого вещества петролейным эфиром и сушки получают 2-(2-(2-бромфенил)этил)-5-метиланилин (8,15 г, 92%).
Т.пл. 67-69oС.
Рассчитано для C15H16BrN: С
62,08%; Н 5,56%; Вr 27,54; N 4,83%;
Найдено: С 62,45%; Н 5,70%; Вr 27,25; N 5,06%.
Смесь полученного выше амина (10,4 г, 0,036 моля) и формиата натрия (4,9 г, 0,072 моля) в муравьиной кислоте (70 мл) нагревают при температуре кипения с обратным холодильником в течение 3 часов и оставляют стоять на ночь.
Реакционную смесь выливают в воду (500 мл) и перемешивают в течение 30 минут. Твердое вещество отфильтровывают, промывают водой и затем этанолом. После сушки на воздухе получают N-(2-(2-(2-бромфенил)этил)-5-метилфенил)формамид 8,3 г, 72%). Т.пл. 167-169oС.
Смесь полученного выше формильного производного (6,50 г, 20 ммолей), карбоната калия (3,39 г, 24 ммоля), меди (0,81 г, 13 ммолей) и бромида меди(1) (1,02 г, 7 ммолей) в диметилсульфоксиде (40 мл) нагревают до 160oС, охлаждают, добавляют 20% гидроксид натрия (4 мл) и нагревают смесь до 75oС за 30 минут. Смесь выливают в воду (400 мл) и этилацетат (500 мл), отфильтровывают твердое вещество, отделяют органический слой, промывают 1N соляной кислотой, 10% идрокарбонатом натрия и водой (2•100 мл) и сушат (МgSO4). Остаток (4,88 г) чистят колоночной хроматографией на силикагеле, используя в качестве элюента смесь циклогексана и бензола (1:1). Это дает 2,26 г (54%) 3-метил-10,11-дигидро-5Н-дибензо [b, f] азепина. ТСХ: Rf=0,75 (SiO2: циклогексан/этилацетат = 5:1).
К раствору полученного выше азепина (2,09 г, 0,01 моля) в бензоле (25 мл) добавляют суспензию амида натрия (1, 01 г, 0,013 моля, 50% суспензия в толуоле, разбавленная 5 мл бензола) и смесь нагревают до 70oС в течение 1,5 часов в атмосфере азота. Затем добавляют 1-тетрагидропиранил-3-бромпропанол (2, 90 г, 0,013 моля) и смесь нагревают до 70oС в течение 19 часов. После охлаждения реакцию гасят водой (20 мл), добавляют бензол (10 мл), делят слои и органический слой сушат (МgSO4 ). Остаток (3,73 г) растворяют в метаноле (20 мл), добавляют 6N соляную кислоту (6 мл) и нагревают смесь при температуре кипения с обратным холодильником в течение 30 минут. Затем выпаривают метанол в вакууме, остаток растирают с бензолом (20 мл) и водой (10 мл), еще раз промывают водный слой бензолом (10 мл) и объединенные бензольные экстракты сушат (МgSO4). Растворитель выпаривают в вакууме и остаток (2,65 г) чистят колоночной хроматографией на силикагеле (50 г), используя в качестве элюента смесь бензола и этилацетата (10:1). Это дает 1,83 г (68%) 5-(3-гидроксипропил)-3-метил-10, 11-дигидро-5Н-дибензо[b,f]азепина в виде масла.
ТСХ: Rf=0,25 (SiO2: хлороформ).
К раствору полученного выше спирта (1,70 г, 6,3 ммоля) и триэтиламина (1,9 г, 18 ммолей) в бензоле (25 мл) добавляют по капле за 15 минут раствор метансульфонилхлорида (0,91 г, 7,9 ммоля) в бензоле (8 мл) при 18-20oС, охлаждая на ледяной бане. Реакционную смесь перемешивают при комнатной температуре в течение 2 часов. Выделившийся триэтиламин-гидрохлорид отфильтровывают, промывают бензолом (15 мл), объединенные бензольные слои промывают водой (2•25 мл), а затем солевым раствором (20 мл) и сушат (МgSO4). После выпаривания растворителя в вакууме используют маслянистый метансульфонат (2,07 г, 94%) на следующей стадии без дальнейшей очистки.
TCX: Rf=0,50 (SiO2: хлороформ).
Смесь полученного выше метансульфоната (2,07 г, 6 ммолей), этил 2-(1-пиперазинил)-3-пиридинкарбоксилата (1,41 г, 6 ммолей), йодида калия (1,00 г, 6 ммолей) и карбоната калия (2,49 г, 18 ммолей) в 2-бутаноне (40 мл) нагревают при температуре кипения с обратным холодильником в течение 7 часов. Смесь охлаждают, добавляют воду (75 мл) и эфир (75 мл), органическую фазу промывают водой (20 мл) и сушат (МgSO4). Растворитель удаляют в вакууме, получая этиловый эфир указанного в заголовке соединения (3,07 г) в виде масла. TCX: Rf=0,35 (SiO2: насыщенный аммиаком хлороформ/этанол =50:1).
К раствору полученного выше эфира (2,90 г) в 2-пропаноле (15 мл; добавляют раствор дигидрата щавелевой кислоты (0,85 г) в 2-пропаноле (5 мл), смесь осторожно нагревают и затем охлаждают. Выделившуюся соль отфильтровывают, еще промывают 2-пропанолом и эфиром и сушат в вакууме, получая 2,97 г (83%) оксалата этилового эфира 2-(4-(3-(3-метил-10,11-дигидро-5Н-дибензо[b, f]-азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновой кислоты.
Из полученного выше оксалата (2,85 г) высвобождают свободный эфир (1,91 г) в дихлорметановой суспензии с гидроксидом аммония и получают его в виде масла.
К раствору полученного выше эфира (1,83 г, 3,8 ммоля) в этаноле (22 мл) добавляют 20% гидроксид натрия (2,5 мл) и смесь перемешивают в течение 5 часов при комнатной температуре. Основную часть метанола удаляют в вакууме, остаток растворяют в дихлорметане (180 мл) и подкисляют 2N соляной кислотой, водный слой отделяют, органический слой промывают водой (10 мл), сушат (МgSO4)и удаляют растворитель в вакууме. Пенящийся остаток выпаривают с ацетоном (3•20 мл) и растворяют аморфное твердое вещество (1,81 г) в ацетоне. При помощи фильтрования получают кристаллическое нерастворимое в ацетоне соединение, указанное в заголовке и осадившееся из раствора (1,26 г, 67%).
Т.пл. 200-206o С.
Рассчитано для C28H32N4O2, HCl: С 68,21%; Н 6,75%; Сl 7,19%; N 11,36%;
Найдено: С 68,05%; Н 6,83%; Сl 7,28%; N 10,95%.
ПРИМЕР 21
Гидрохлорид 6-(4-(3-(дибензо[d, g][1,3,6]диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-пиридин-2-карбоновой кислоты
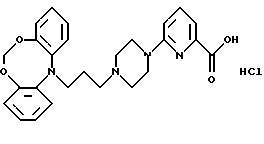
12Н-Дибензо[d, g] [1,3,6] диоксазоцин (1,7 г, 8 ммоля, полученный аналогично тому, как описано в Примере 3) растворяют в сухом бензоле (25 мл) при 45oС. В атмосфере азота добавляют суспензию амида натрия в толуоле (1,1 г, 50%, 14 ммолей) и смесь перемешивают при 70oС в течение 2,5 часов. Добавляют свежеперегнанный 1-тетрагидропиранилокси-3-бромпропан (2,88 г, 13 ммолей, 72oС/70 Па) и смесь нагревают при температуре кипения с обратным холодильником в течение 14 часов и оставляют на ночь при комнатной температуре. К реакционной смеси добавляют бензол (25 мл) и воду (25 мл), перемешивают 15 минут и разделяют. Органический экстракт сушат (сульфатом натрия) и выпаривают растворитель в вакууме. Остаток (4,9 г) растворяют в метаноле (20 мл), добавляют 6N соляную кислоту (6,7 мл, 40 ммолей), смесь перемешивают при 22oС в течение 40 минут и подщелачивают 2N карбонатом натрия (20 мл). Продукт экстрагируют бензолом (80 мл), органическую фазу промывают водой (30 мл), сушат (сульфатом натрия) и выпаривают в вакууме (3,5 г). Сырой продукт чистят хроматографически на силикагеле (80 г), используя в качестве элюента смесь бензола и этилацетата (10:1). Получают 12-(3-гидроксипропил)дибензо[d, g][1,3,6]диоксазоцин (1,36 г, 47%) в виде масла.
ТСХ: Rf =0,45 (SiO2: бензол/этилацетат = 10:1).
Раствор метансульфонилхлорида (0,98 г, 8,6 ммоля) в сухом толуоле (10 мл) добавляют по капле за 20 минут к перемешиваемому раствору полученного выше производного пропанола (1,36 г, 5 ммолей) и триэтиламина (1,6 г, 15 ммолей) в сухом толуоле (40 мл) в атмосфере азота при комнатной температуре. Происходит слегка экзотермическая реакция. Реакционную смесь перемешивают при 24oС в течение 3 часов и затем при 36oС в течение 1,5 часов. Разбавляют реакционную смесь толуолом (50 мл), промывают водой (4• 30 мл) и соляным раствором (25 мл), сушат (сульфатом натрия) и выпаривают в вакууме. Маслянистый мезилат (1,8 г, 100%) используют на следующей стадии без дополнительной очистки.
ТСХ: Rf=0,36 (SiO2: бензол/этилацетат = 10:1).
Полученный выше мезилат (1,8 г, 5 ммолей), этил 6-(пиперазин-1-ил)пиридин-2-карбоксилат (1,9 г, 8 ммолей), сухой карбонат калия (1,6 г, 12 ммолей) и йодид калия (0,15 г, 1 ммоль) в сухом ацетоне (30 мл) и N,N-диметилформамид (10 мл) перемешивают при температуре кипения с обратным холодильником в течение 5 часов. Отгоняют ацетон, остаток растворяют в бензоле (150 мл) и промывают водой (4•50 мл) и соляным раствором (30 мл), сушат (сульфатом натрия) и выпаривают в вакууме. Маслянистый остаток чистят хроматографически на силикагеле (70 г), используя в качестве элюентов сначала бензол, а потом этилацетат. Это дает этиловый эфир указанного в заголовке соединения (1,44 г, 59%) в виде масла.
ТСХ: Rf=0,62 (SiO2: бензол/этилацетат = 10:1).
Раствор полученного выше эфира (1,44 г, 2,95 ммоля) в этаноле (15 мл) и 5N гидроксид натрия (20 мл) перемешивают 15 минут при 50-55oС и затем 21 час при 22oС, добавляют дихлорметан (200 мл) и подкисляют смесь до рН 1 2,5N соляной кислотой. Органический слой отделяют, сушат (сульфатом натрия) и выпаривают в вакууме, получая 1,14 г твердого остатка, который растирают со смесью ацетона (15 мл) и эфира (20 мл). Продукт отфильтровывают и промывают смесью ацетона-эфира (2:3, 3•10 мл) и эфиром (5 мл), получая 0,9 г (79%) указанного в заголовке соединения.
Т.пл. 224-227oС.
Рассчитано для С26
H29N4О4, НСl, 0,5 H2O: С 61,71%; Н 5,98%; N 11,07%;
Найдено: С 61,60%; Н 6,05%; N 10,70%.
Реферат
Изобретение относится к 1,4-дизамещенным пиперазинам общей формулы I, способу их получения, содержащим их композициям и их применению для клинической терапии болезненных состояний, повышенной болевой чувствительности и/или воспалительных состояний, в которых патофизиологическую роль играют С-волокна, вызывая нейрогенную боль или воспаление. 4 с. и 30 з.п. ф-лы, 1 табл.
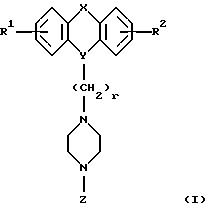
Формула

где R1 и R2 независимо являются водородом, галогеном, С1-6алкилом;
Х является -СН2СН2-, -ОСН2O-, -SCH2-, -CH2-S-;
Y является

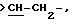
r=1, 2 или 3;
Z выбирают из

где М1 и М2 независимо являются С или N;
R3 является водородом;
R4 является водородом или (СН2)m СОR11, где R11 является гидроксилом, С1-6алкокси группой или NHR12, где R12 является водородом или С1-6алкилом и m=0, 1, 2,
или их фармацевтически приемлемые соли.
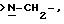

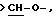
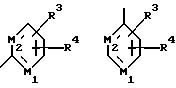
где M1 и М2 независимо являются С или N.
2-(4-(3-(12H-дибензо[d, q] [1,3] диоксоцин-12-илиден)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(2,10-дихлор-12Н-, дибензо[d, q](1, 3)диоксоиик-12-илиден)-1-пропил)-пипераэин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(12Н-дибензо[d, q)(1,3,6] диоксазоиин-12-ил)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(2-хлор-12Н-дибензо[d, q] [1,3,6] диоксазоцин-12-ил)-1-пропил)-пиперазин-1-ил)-3-пиридинкарбоновую кислоту;
1-(3-(10,11-дигидpo-5H-дибeнзo[a, d] циклoгenтeн-5-илиден)-1-пропил)-4-(2-пиридил)пиперазин;
2-(4-(3-(10, 11-дигидро-5Н-дибензо[a, d] циклогептен-5-илиден)-пропил-)-1-пиперазинил)-3-пиридинкарооновую кислоту;
2-(4-(2-10, 11-дигидро-5Н-дибензо[b, f] азепин-5-ил)-1-этил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
6-(4-(3-(10,11-дигидро-5Н-дибензо[b, f] азепин-5-ил)-1-пропил)-1-пиперазинил)-2-пиридинкарбоновуго кислоту;
2-(4-(3-(10,11-дигидро-5Н-дибензо(b, f] азепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(10,11-дигидро-5Н-дибензо[b, f] aзепин-5-ил)-1-пропил)-1-пиперазинил)-5-пиридинкарбоновую кислоту;
2-(4-(3-3-хлор-10,11-дигидро-5Н-дибензо[b, f] aзепин-5-ил)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
1-(3-(10,11-дигидро-5Н-дибензо[а, d] циклогептен-5-илиден)-1-пропил-4-(2-нитрофенил) пиперазин;
2-(4-(3-(10, 11-дигидро-5Н-дибензо[а, d]циклогептен-5-илиден)-1-пробил)-1-пиперазинил)бензонитрил;
2-(4-(3-, 10,11-дигидро-5H-дибензо[а,с]циклогептен-5-илиден)-1-пропил)-1-пиперазинил)бензойную кислоту;
1-(3-(10, 11-дигидро-5Н-дибензо[а, d] циклогептен-5-илиден)-1-пропил)-4-(3-трифторметил-2-пиридил)пиперазин;
2-(4-(2-{ 6,11-дигидродибензо(b, е] тиепин-11-илиден)этил)-пипераэин-1-ил)-3-пиридинкарбоновую кислоту;
2-(4-(3-(6,11-дигидродибензо[b, е] тиепин-11-илиден)-1-пропил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
2-(4-(2-(6,11-дигидродибензо[b, е] тиепин-11-илокси)этил)-1-пиперазинил)-3-пиридинкарбоновую кислоту;
6-(4-(3-(10,11-дигидро-5Н-дибензо[а, d] циклогептен-5-илиден)-1-пропил)пиперазин-1-ил)-2-пиридинкарбоновую кислоту;
2-(4-(5-3-метил-10,11-дигидро-5Н-дибензо[b, f] азепин-5-ил-1-пропил-1-пиперазипил) -3-пирилинкарбоновую кислоту;
6-(4-(3-дибензо[d, g] [1,3,6] диоксазсцин-12-ил)-1-пропил)-пиперазин-1-ил)-пиридин-2-карбоновую кислоту
или их фармацевтически приемлемых солей.

где R1, R2, X, Y, r такие, как определено выше;
W является подходящей удаляемой группой, такой как галоген, паратолуол-сульфонат или мезилат,
с соединением формулы III

где Z такое, как определено выше,
с образованием соединения формулы 1.
Комментарии