Способ получения энантиомерных соединений или их производных - RU2068849C1
Код документа: RU2068849C1
Чертежи

Описание
Изобретение относится к аналогам нуклеозидов пурина, содержащим ненасыщенное карбоциклическое кольцо вместо сахарного остатка, их фармацевтически приемлемым производным и к их применению в медицинской терапии, в частности для лечения некоторых вирусных заболеваний.
СПИД (синдром приобретенного иммунодефицита) представляет собой иммуносуппрессивное или иммунодеструктивное заболевание, которое предрасполагает субъектов к фатальным внезапным инфекциям. Типично СПИД ассоциируется с постепенным истощением Т-клеток, в особенности поднабора индуктор-помощник, содержащего поверхностный маркер ОКТ4.
Вирус иммунодефицита человека (ВИЧ) был выделен воспроизводимым образом от пациентов со СПИДом или с симптомами, которые часто предшествуют СПИДу, ВИЧ является цитопатическим и, как кажется, предпочтительно инфицирует и разрушает Т-клетки, содержащие маркер ОКТ4, и теперь всеми признано, что ВИЧ представляет собой этиологический агент СПИДа.
Так как открытие того, что ВИЧ представляет собой этиологический агент СПИДа, было дано много предложений в отношении химиотерапевтических агентов против ВИЧ, которые могут быть эффективными при лечении потерпевших от СПИДа. Так, например, в патенте США N 4724232, описан 3'-азидо-3'-дезокситимидин, который имеет принятое название зидовудин, его фармацевтически приемлемые производные и их применение при лечении инфекций от тетровируса человека, включая СПИД и связанные с ним клинические состояния. Vince et al. Antiviral Research, 9 (1/2), 120 (1988) описывают некоторые аналоги карбоциклических нуклеозидов и их использование против ВИЧ. На Второй международной конференции по противовирусным исследованиям, Уильямсбург, Виргиния, 10 14 апреля 1988 г. был описан (± )-9-(цис-4-(оксиметил)-2-циклопентенил)-групп (NSC-614846), также известный как карбонир.
Распространенный по всему свету вирус гепатита В (ВГВ) представляет собой другой вирусный патогенный возбудитель с важнейшими последствиями. Он наиболее распространен в азиатских странах и преобладает в присахарской Африке. Этот вирус этиологически связан с первичным печеночно-клеточным раком.
США в настоящее время содержат значительный круг из 500 тыс. 1 млн. носителей инфекции. Хронический активный гепатит разовьется у более чем 25 носителей и часто прогрессирует до цирроза. Оценивается, что 5000 человек умирает ежегодно от связанного с ВГВ цирроза в США и что возможно 1000 умирает от рака печени, связанного с ВГВ. Даже, когда имеется универсальная вакцина против ВГВ, потребность в эффективных соединениях против ВГВ будет продолжать иметь место. Значительный резервуар постоянно инфицированных носителей, оцениваемый в 220 млн. по всему миру, не получит выгоды от вакцинации и будет продолжать иметь место при высоком риске, вызванным ВГВ заболеванием печени. Это население носитель служит источником инфекции склонных к ней лиц, увековечивая заболеваемость, особенно в эндемических зонах или в группах повышенного риска, таких как лица, злоупотребляющие внутривенным приемом наркотиков и гомосексуалисты. Таким образом, существует большая нужда в эффективных противовирусных агентах, как для регулирования хронических инфекций, так и для уменьшения прогрессии в печеночно-клеточный рак.
Клинические эффекты инфицирования вирусом ВГВ находятся в пределах от головной боли, жара, недомогания, тошноты, рвоты, анорексии до болей в желудке. Репликация вируса обычно регулируется иммунной реакцией, с течением выздоровления недели или месяцы у людей, но инфекция может быть более серьезной, приводя к стойкому хроническому заболеванию печени, как подчеркнуто выше.
В описании к Европейскому патенту N 349242 описываются определенные 6-замещенные карбоциклические нуклеозиды пурина и их применение в медицинской терапии, в частности при лечении инфекций ВИЧ и ВГВ. Среди таких нуклеозидов находятся соединения (± )-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/ -2-циклопентен-1-метанол и (±)-цис-4-/2-амино-6-(циклопропилметиламино)-9Н-пурин-9-ил/-2- циклопентен-1-метанол, то есть каждое в виде рацемической смеси своих соответствующих энантиомеров.
Обнаружено, что индивидуальные изолированные энантиомеры обоих вышеупомянутых соединений и их фармацевтические производные имеют полезную противовирусную активность, в частности, против инфекций ВИЧ и ВГВ, в сочетании с низкой цитотоксичностью и/или полезны в качестве промежуточных веществ для получения соединений, имеющих такую активность.
Согласно одному признаку изобретения, созданы энантиомерные соединения общей формулы

где R обозначает циклопропиламино или N-циклопропил-N-метиламиновую группу, а A обозначает 2-циклопентен-1-метанол-4-ильную группу в конфигурации (1S, 4R) или (1R, 4S) и их производных (например, сложных эфиров, солей и солей сложных эфиров), при этом каждое из указанных соединений и их производные находятся в форме энантиомера, практически свободного (например, в количестве менее, чем 10 мас. предпочтительно менее 5 мас.) от соответствующего энантиомера.
Целесообразно, чтобы соединения формулы I содержали соединения, имеющие
следующие конфигурации:

где R имеет вышеуказанные значения.
Энантиомерные
соединения формулы I, т. е. практически свободные от соответствующего энантиомера, таким образом содержат:
1) (1S,
4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол;
2) (1R, 4S)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол;
3) (1S,
4R)-цис-4-/2-амино-6-(N-циклопропил-N-метиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол;
4) (1R,4S)-цис-4-/2-амино-6-(N-циклопропил-N-метиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Вышеуказанные соединения 1) и 3), называемые ниже как энантиомерные соединения (1S, 4R) формулы I, с отрицательным вращением плоскости поляризации света (-), как оказалось, имеют особо мощную активность против ВИЧ и ВГВ-инфекций, и эти соединения и их фармацевтически приемлемые производные обозначают предпочтительные варианты выполнения изобретения. Эти соединения имеют дополнительное преимущество в том, что после введения они способны проникать в гематоэнцефалический барьер для получения высоких уровней соединений или их активных метаболитов в центральной нервной системе, где проявления инфекций ВИЧ особенно расстраивают здоровье. Вышеуказанное соединение 1) является особо предпочтительным с точки зрения его исключительно мощной активности против инфекции ВИЧ и ВГВ. Это соединение, как оказалось, также имеет значительно более низкую токсичность против клеток-предшественников костного мозга, чем вышеуказанный 3'-азидо-3'-дезокситимидин (зидовудин).
Было также обнаружено, что фосфатные производные соединений 2) и 4), приведенных выше, которые ниже называются как энантиомерные соединения (1R, 4S) формулы I, с положительным вращением плоскости поляризации света (+), имеют мощную активность против вирусных инфекций, таких как вышеназванные. Эти фосфатные производные представляют собой, таким образом, дополнительный предпочтительный вариант выполнения изобретения.
Ссылка здесь на "фосфатные производные" энантиомерных соединений (1R, 4S) формулы I обозначает производные, в которых фосфатная группа прикреплена к 1-метанольной группе формулы I и включает моно-, ди- и три-фосфаты.
Родственные энантиомерные соединения (1R, 4S) формулы I и их несодержащие фосфатов производные полезны в качестве промежуточных веществ для получения указанных фосфатных производных.
Вышеназванные энантиомерные соединения (1S, 4R) формулы I и их фармацевтически приемлемые производные и фосфатные производные энантиомерных соединений (1R, 4S) формулы I ниже называются противовирусными соединениями согласно изобретению.
Согласно дополнительным признакам изобретения обеспечивается
получение:
а) противовирусных соединений согласно изобретению для применения в медицинской терапии, в частности для лечения или профилактики ретровирусной инфекции или инфекции вируса
гепатита В;
б) способа лечения или профилактики ретровирусных инфекций и инфекций гепатита В в субъекте, например млекопитающем, таком как человек, который включает лечение субъекта
терапевтически эффективным количеством противовирусного соединения согласно настоящему изобретению;
в) применения противовирусного соединения согласно изобретению при изготовлении
лекарственного средства для лечения или профилактики любых из вышеупомянутых инфекций или состояний.
Примеры ретровирусных инфекций, которые могут лечиться или предупреждаться согласно изобретению, включают ретровирусные инфекции человека, такие как вирус иммунодефицита человека (ВИЧ), ВИЧ-1, ВИЧ-2 и лимфотрофный вирус Т-клеток человека (ЛТВЧ), например инфекции от ЛТВЧ-1 или ЛТВЧ-2. Противовирусные соединения согласно изобретению особо полезны для лечения СПИДа и относящихся к нему клинических состояний, таких как связанный со СПИДом комплекс (АСК), прогрессивная генерализованная лимфоденопатия (ПГЛ), связанные со СПИДом неврологические состояния, такие как множественный склероз или тропический парапарез, и состояния, позитивные к антителам противо-ВИЧ и к ВИЧ для примеры у бессимптомных пациентов, и тромбоцитогеническая пурпура. Эти соединения также могут быть использованы для лечения или предупреждения псориаза.
Под "фармацевтически приемлемым производным" по отношению к энантиомерным соединениям (1S, 4R) формулы I понимают любую фармацевтически приемлемую соль, сложный эфир или соль такого сложного эфира от энантиомерных соединений (1S, 4R) формулы I, или любое другое соединение, которое после введения его реципиенту способно привести к получению (непосредственно или косвенным образом) такого энантиомерного соединения, или его активный в противовирусном плане метаболит или остаток.
Предпочтительные сложные эфиры энантиомерных соединений (1S, 4R) формулы I включают эфиры карбоновой кислоты, в которых несодержащая карбонила часть сложноэфирной группы выбирается из прямого или разветвленного алкила, например, н-пропила, трет-бутила, н-бутила, алкоксиалкила (например, метоксиметила), аралкила (например, бензила), арилоксиалкила (например, феноксиметила), арила (например, фенила, возможно замещенного галогеном, алкилом с C1-4 или алкокси с C1-4 или амино), эфиры сульфокислоты, такие как алкил- или аралкилсульфонил (например, метансульфонил), эфиры аминокислот (например, L-валил или L-изолейцил), и моно-, ди или трифосфатные эфиры.
Фосфатные сложные эфиры соединений формулы I могут быть дополнительно этерифицированы с помощью, например, спирта C1-20 или его реакционноспособного производного, или 2,3-ди-(С6-24 )-ацилглицерина, например кислого фосфата, 2,3-бис-(гексаноилокси)-пропила и производных кислого фосфата 2,3-бис-(гексадеканоилокси)-пропила. В дополнение к таким дополнительно этерифицированным производным фосфата соединения формулы I изобретение дополнительно включает в себя такие производные рацемических соединений формулы I.
Что касается вышеописанных сложных эфиров, если иное не указано, любая алкильная часть в целесообразном варианте, которая присутствует, содержит от 1 до 18 атомов углерода, в особенности от 1 до 4 атомов углерода. Любая арильная часть, которая присутствует в таких сложных эфирах, в целесообразном варианте содержит фенильную группу.
Фармацевтически приемлемые соли присоединения кислот энантиомерных соединений (1S, 4R) формулы I включают одно и двухосновные соли с соответствующей кислотой, например органическими карбоновыми кислотами, такими как уксусная, молочная, винная, малеиновая, изотионовая, лактобионовая и сукциновая кислоты, органические сульфокислоты, такие как метансульфоновая, этансульфоновая, бензолсульфоновая и п-толуолсульфоновая кислоты и неорганические кислоты, такие как соляная, серная, фосфорная и сульфатметансульфонатсульфаминовая кислоты. Соли соляной кислоты (то есть хлористо-водородная соль) являются особенно предпочтительными.
Вышеуказанные противовирусные соединения согласно изобретению могут быть использованы в сочетании с другими терапевтическими агентами для лечения вышеназванных инфекций или состояний. Примеры таких дополнительных терапевтических агентов включают вещества, которые эффективны при лечении вирусных инфекций или связанных с ними состояний, такие как 3'-азидо-3'-дезокситимидин (зидовудин), 2',3'-дидезоксинуклеозиды, такие как 2'3'-дидезоксицитидин, 2', 3'-дидезоксиаденозин и 2', 3'-дидезоксиинозин, ациклические нуклеозиды (например, ацикловир), интерфероны, такие как интерферон, ингибиторы почечных выделений, такие как пробеницид, ингибиторы переноса нуклеозидов, такие как дипиридамол, дилазен, мио-, лидо- или солуфлазин, или гексобендин, иммуномодуляторы, такие как интерлейкин II и стимулирующие факторы колонки макрофагов гранулоцитов, их производные, растворимые CD2 или гепатически измененные методами инженерии, и фосфономуравьиная кислота. Соединения компонентов такой комбинационной терапии могут быть введены одновременно, либо в отдельных или скомбинированных рецептурах, или в различные периоды времени, например, с такой последовательностью, что комбинированное воздействие достигается.
Противовирусные соединения согласно изобретению, также называемые здесь активные компоненты, могут быть введены для терапевтического лечения любым подходящим путем, включая орально, ректально, назально, локально (включая буквально и под язык), вагинально и парентерально (включая подкожно, внутримышечно, внутривенно и внутрикожно). Необходимо отметить, что предпочтительный путь будет меняться от состояния и возраста реципиента, природы инфекции и выбранного активного ингредиента.
Обычно подходящая доза для каждого вышеназванных состояний (например, СПИД), будет лежать в диапазоне от 3,0 до 120 мг на килограмм веса тела реципиента (например, человека) в день, предпочтительно в диапазоне от 6 до 90 мг на килограмм веса тела в день и более предпочтительно в пределах от 15 до 60 мг на килограмм веса тела в день. Желательная доза предпочтительно представлена в виде двух, трех, четырех, пяти, шести или более поддоз, вводимых с соответствующими интервалами в течение всего дня. Эти поддозы могут вводиться в виде единичных дозировок, например, содержащих от 10 до 1500 мг, предпочтительно от 20 до 1000 мг и более предпочтительно от 50 до 700 мг активного ингредиента на единичную дозировку препарата.
В идеальном случае активный ингредиент должен быть введен для достижения пиковых концентраций в плазме активного соединения от примерно 1 до примерно 75 мкМ, предпочтительно примерно от 2 до 50 мкМ, более предпочтительно примерно от 3 до 30 мкМ. Это может быть достигнуто, например, при внутривенной инъекции раствора активного ингредиента с концентрацией от 0,1 до 5 возможно в физиологическом растворе или при оральном введении в виде пищевого шарика, содержащего примерно от 1 до примерно 100 мг/кг активного ингредиента. Желательные уровни в крови могут быть поддержаны непрерывным вливанием для обеспечения примерно от 0,01 до примерно 5,0 мг/кг/ч или путем периодических вливаний, содержащих от примерно 0,4 до примерно 15 мг/кг активного ингредиента.
В то время как можно для активного ингредиента быть вводимым без добавок, предпочитают представлять его в виде фармацевтических составов. Составы согласно изобретению содержат по крайней мере один активный ингредиент, как указано выше, вместе с по крайней мере одним фармацевтически приемлемым носителем или эксципиентом. Рецептуры включают вещества, приспособленные для орального, ректального, назального, локального (включая в рот и под язык), вагинального или парентерального (включая подкожно, внутримышечно, внутривенно и внутрикожно) введения. Рецептуры могут быть удобно представлены в виде единичной дозировки и могут быть приготовлены любыми методами, хорошо известными в фармацевтическом искусстве. Такие методы включают этан соединения активного ингредиента с носителем, который представляет собой один или более дополнительных ингредиентов. Обычно рецептуры готовят путем равномерного и однородного соединения активного ингредиента с жидкими носителями или с мелко измельченными твердыми носителями или с теми и другими, а затем, при необходимости, придают продукту форму.
Рецептуры согласно изобретению, предназначенные для орального введения, могут быть представлены как отдельные единицы, такие как капсулы или таблетки, каждая из которых содержит заранее заданное количество активного ингредиента, в виде порошка или гранул, в виде раствора или суспензии в водной или неводной жидкости или в виде жидкой эмульсии типа "масло в воде" или жидкой эмульсии типа "вода в масле". Активный ингредиент может также быть представлен в виде пищевого шарика, электуария или пасты.
Таблетка может быть получена путем прессования или формования возможно с использованием одного или более вспомогательных ингредиентов. Спрессованные таблетки могут быть приготовлены путем прессования в подходящей машине активного ингредиента в свободно текучей форме, такой как порошок или гранулы, возможно смешанные со связующим (например, повидон, желатин, оксипропилметилцеллюлоза), смазочное вещество, инертный разбавитель, консервант, агент расщепления (например, щелочной крахмальный гликолят, сшитый повидон, сшитая щелочная карбоксиметилцеллюлоза), поверхностно-активное вещество или диспергатор.
Сформированные таблетки могут быть приготовлены путем формования в соответствующей машине смеси из порошкообразного соединения, увлажненного инертным жидким разбавителем. Таблетки могут возможно получать покрытие или процарапываться и могут иметь такую рецептуру, чтобы обеспечить медленное или регулируемое высвобождение в ней активного ингредиента, используя, например, оксипропилметилцеллолозу в изменяющихся пропорциях для обеспечения желательного профиля высвобождения. Таблетки возможно могут быть снабжены энтеросолюбильным покрытием для обеспечения высвобождения в частях кишки, иных, чем желудок.
Рецептуры, предназначенные для локального применения во рту, включают лепешки, содержащие активный ингредиент в основе с приданным приятным вкусом, обычно сахарозе и гуттаперче или трагаканте, пастилки, включающие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахароза и жидкости для полоскания рта, содержащие активный ингредиент в соответствующем жидком носителе.
Рецептуры, приспособленные для ректального введения, могут быть представлены с подходящей основой, содержащей, например, масло какао или силицилат.
Рецептуры, предназначенные для вагинального введения, могут быть представлены в виде вагинальных суппозиториев, тампонов, кремов, гелей, паст, рецептур в виде пены или аэрозоля, содержащих, в дополнение к активному ингредиенту, такие носители, которые пригодны, как это известно в данной области.
Составы, предназначенные для парентерального введения, включают водные и неводные изотопические стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостатические факторы и растворенные вещества, которые делают этот состав изотоническим с кровью конкретного реципиента; и водные и неводные стерильные суспензии, которые могут включать агенты суспендирования и загустители. Эти составы могут быть представлены в единичной дозе или в многодозовых герметизированных контейнерах, например ампулах и флаконах, и могут храниться в высушенном на морозе (лиофилизованном) состоянии, требующем только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Растворы и суспензии для немедленной инъекции могут быть приготовлены из стерильных порошков, гранул и таблеток вышеописанного типа.
Предпочтительные рецептуры с единичной дозировкой это такие, которые содержат дневную дозу или единицу, дневную часть дозы, как описано выше, или ее соответствующую часть от активного ингредиента.
Необходимо понимать, что в дополнение к особо упомянутым выше ингредиентам, составы согласно изобретению могут включать другие агенты, обычно применяемые в данной области, относящиеся к типу состава, о котором идет речь, например составы, предназначенные для орального применения, могут включать такие дополнительные вещества, как подслащивающие вещества, загустители и вещества, придающие приятный вкус.
Изобретение дополнительно включает следующий способ приготовления энантиомерных соединений формулы I, указанных выше, и их производных. Энантиомерные исходные материала и предшественники для
таких материалов, которые используются, как описано ниже в отношении данного способа, представлены, каждый, в виде энантиомера, практически свободного (например, в вышеуказанной степени в отношении
соединений формулы I) от другого энантиомера. Способ согласно изобретению включает либо:
А) обработку энантиомерного соединения формулы:
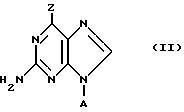
в которой A имеет вышеуказанные значения, а Z обозначает группу-предшественник для указанной группы R, как определено в формуле I),
или его производного с помощью средства или при условиях, обеспечивающих превращение группы-предшественника Z в требуемую R-группу, либо
Б) введение в реакцию энантиомерного соединения формулы:
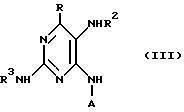
в которой A и R имеют вышеуказанные значения; R2 обозначает водород или формильную группу; R3 обозначает защитную аминогруппу, например ацильную группу, такую как алканоильная С1-6-группа, например формил, ацетил или изобутирил, или ее производное с веществом, служащим для осуществления образования имидазольного кольца в требуемом соединении формулы I, с последующим удалением защитной аминогруппы R2, или
В) введение в реакцию энантиомерного соединения формулы:

в которой A и R имеют вышеуказанные значения; R3 обозначает защитную аминогруппу, например, как описано выше по отношению к формуле III,
или ее производное с веществом, служащим для обеспечения удаления защитной аминогруппы R3, и возможно осуществление одного или обоих нижеследующих преобразований в любом требуемом порядке:
I) в случае образования соединения формулы I, преобразование названного соединения в его производное, или
II) в случае образования производного соединения формулы I, преобразование указанного производного в родственное соединение формулы I или в дополнительное такое производное.
Способ А может быть осуществлен обычным образом, например путем обработки соединения формулы II, в которой Z обозначает покидающую группу (например, галогруппу, такую как хлорогруппа), соответствующим амином, то есть циклопропиламином или N-циклопропил-N-метиламином, предпочтительно в избытке, для введения аминогруппы Р, как определено выше, целесообразно при кипячении с обратным холодильником или при температуре выше 50oC, предпочтительно в присутствии органического растворителя, например метанола или этанола.
Способ Б может быть осуществлен, например, путем введения в реакцию соединения формулы III с муравьиной кислотой или с реакционноспособным производным муравьиной кислоты (например, триэтилортоформиат или диэтоксиметилацетат), возможно с совместным растворителем, такие как диметилацетамид или диметилформамид, при повышенной температуре, предпочтительно при 75 90oC. Эта реакция обычным образом осуществляется путем добавления несколько большего, чем один эквивалент, количества сильной безводной кислоты, например с помощью 1,1 экв. этансульфокислоты на эквивалент соединения формулы III, в этом случае используют более низкие температуры (например, 25oC).
Способ В может быть осуществлен, например, путем введения в реакцию энантиомерного соединения формулы IV с кислотным агентом, например разбавленной водной соляной кислотой.
Соединения формулы II, используемые в качестве исходных материалов в процессе А, могут быть получены,
например, аналогичным образом по процессу Б, то есть путем введения в реакцию соответствующего энантиомерного соединения формулы:

в которой A, Z, R2 и R3 имеют вышеупомянутые значения,
или его производного с веществом, служащим для образования имидазольного кольца в необходимом соединении формулы II и для осуществления удаления аминозащитной группы R3. Эта реакция может быть осуществлена с использованием веществ и условий, описанных выше для процесса Б.
Соединения формулы III, используемые в качестве исходных материалов в процессе Б, могут быть приготовлены, например, путем обработки энантиомерного соединения формулы V, приведенного выше, веществом или условиями, обеспечивающими преобразование группы-предшественника Z в желаемую группу R, аналогичным вышеописанному в процессе А образом.
Соединения
вышеуказанной формулы IV могут быть приготовлены, например, путем введения в реакцию энантиомерного соединения формулы:
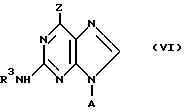
в которой А, Z и R3 имеют вышеуказанные значения,
с веществом или при условиях, служащих для преобразования группы-предшественника Z в требуемую группу R, то есть аналогичным образом с вышеописанным для процесса А.
Соединения вышеприведенной формулы VI могут быть получены, например, путем введения в реакцию энантиомерного соединения формулы IV, приведенной выше, с агентом, служащим для осуществления образования имидазольного кольца в желаемом соединении формулы VI, например, путем обработки муравьиной кислотой или реакционноспособным производным муравьиной кислоты, как описано выше в отношении процесса Б.
Энантиомерные соединения формул II, III, IV, V и VI, приведенные выше, представляют собой дополнительные признаки изобретения, в частности соединения, в которых R2 обозначает формильную группу, или R3 обозначает алканоильную C1-6-группу, в частности ацетил или изобутирил, или Z обозначает галогруппу, такую как хлорогруппа.
Особенно предпочтительными промежуточными соединениями для приготовления (1S,
4R)-цис-4-/2-амино-6-циколпропиламино)-9Н-пурин-9-ил/-2- циклопентен-1-метанола, то есть предпочтительного вышеназванного соединения I, являются включающие:
a) (1R,
4S)-цис-N-/6-(циклопропиламино)-9- (4-(оксиметил)-2-циклопентен-1-ил)-9Н-пурин-2-ил/изобутиламид;
б) (1R,
4S)-цис-N-/4-хлор-5-формамидо-6- ((4-(оксиметил)-2-циклопентен-1-ил)-амино)-2-пиримидинил/-изобутирамид;
в) (1R,
4S)-цис-N-/4-хлор-5-формамидо-6- ((4-(оксиметил)-2-циклопентен-1-ил)-амино)-2-пиримидинил/-ацетамид;
г) (1S,4R)-цис-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентен-1-метанол;
д) (1R,
4S)-цис-N- /6-хлор-9-(4-оксиметил)-2-циклопентен-1-ил)-9Н-пурин-2-ил/-изобутирамид.
Энантиомерные соединения формулы V, используемые в качестве исходных материалов, как описано выше,
могут быть приготовлены, например, путем введения в реакцию соединения формулы:

в которой Z, R2 и R3 имеют вышеуказанные значения; R4 обозначает покидающую группу, например галогруппу, такую как хлорогруппа,
или его производного с энантиомерным соединением формулы (VIIIA) или (VIIIB):
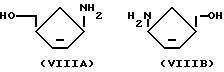
или его производным.
Последнюю упомянутую реакцию целесообразно проводить в присутствии основания, такого как третичный амин, например триэтиламин или триметиламин, целесообразно в органическом растворителе, таком как диметоксиметил или этанол.
Соединения формулы VIIIA или VIIIB, имеющие соответствующую энантиомерную конфигурацию, могут быть получены путем перевода в комплексное соединение соответствующего рацемического соединения, то есть (±)-4-амино-2-циклопентен-1-метанола, с оптически активной карбоновой кислотой (например, дибензоилвинной кислотой), а затем фракционной кристаллизацией полученных диастереомерных солей. В качестве альтернативы может быть использовано растворение с помощью ферментов, как описано, например, в J. Med. Chem. 1987, 30, 746 и J. Med. Chem. 1985, 28, 1385.
Энантиомерные соединения формулы VIIIA или VIIIB и их производные, в частности их соли, с оптически активными карбоновыми кислотами, такими как дибензоил-D-винная кислота, например (1S, 4R)-4-амино-2-циклопентен-1-метанол, и ее дибензоил-D-тартрат обеспечивают дополнительный признак изобретения.
Соединения формулы VII, используемые в качестве
исходных материалов, как указано выше, могут быть приготовлены обычным образом, например, путем восстановления соединения формулы:

в которой Z, R3 и R4 имеют вышеуказанные значения,
для осуществления преобразования NO2-группы в группу NH2 и возможно превращения полученной группы NH2 в группу формамидо, например, обработкой муравьиной кислотой / уксусным ангидридом.
Соединения формулы IX могут быть
приготовлены обычным образом. Те соединения, в которых обозначает галогруппу, например хлорогруппу, могут быть получены, например, галогенированием, например, с использованием хлорокиси фосфора,
соответствующего соединения формулы:

в котором R3 и R4 имеют вышеуказанные значения.
Соединения формулы Х могут быть также приготовлены обычным образом, например путем введения в реакцию соединения формулы:
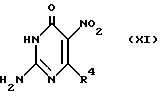
в которой R4 имеет вышеуказанные значения,
с соответствующим веществом, служащим для введения защитной аминогруппы, например, путем реакции с соответствующей карбоновой кислотой или ее функциональным производным, например изомасляным ангидридом. Соединение формулы XI может быть приготовлено нитрацией соответствующего соединения формулы:

в которой R4 имеет вышеуказанное значение.
Соединения формул VII, IX, X и XI обозначают дополнительные признаки изобретения, в частности такие, в которых Z обозначает галогруппу, такую как хлорогруппа, и/или R3 обозначает алканоил C1-6-группу, в частности ацетил или изобутирил, и/или R4 обозначает галогруппу, такую как хлорогруппа.
Особо предпочтительные соединения
формул VII, IX и Х согласно изобретению включают:
N-(4,6-дихлор-5-формамидо-2-пиримидинил)-изобутирамид;
N-(4,6-дихлор-5-нитро-2-пиримидинил)-изобутирамид;
N-(4-хлор-1,
6-дигидро-5-нитро-6-оксо-2-пиримидинил)-изобутирамид.
Соединение А из формулы I может обычно быть преобразовано в его сложный эфир путем реакции с соответствующим этерифицирующим веществом, например галоидангидридом или ангидридом. Соединение формулы I, включая его сложные эфиры, может быть преобразовано в его соли обычным образом, например путем обработки соответствующей кислотой. Сложный эфир или соль соединения формулы I могут быть превращены в родственное соединение, например, путем гидролиза.
Таким образом, О-монофосфат соединения формулы I может быть приготовлен путем обработки родственного соединения соответствующим фосфорилирующим веществом, например хлорокисью фосфора, как описано M. Joshikawa, T. Kato and T. Takenishi, Bulletin Chem. Soc. Japan 1969, 42, 3505. Соответствующие О-ди- и О-трифосфаты могут быть приготовлены методами, описанными в Nucleotide Analogs авторами K. H. Sheit, John Willey and Sons, New Jork, 1980, pp. 211 215 и авторами D.b. Hoard and D.G. Ott, J. Am. Chem. Soc. 1965, 87, 1785, например путем получения имидазолятного производного соответствующего О-монофосфата и путем последующей реакции этого производного с фосфатом для получения О-дифосфата, или с пирофосфатом для получения О-трифосфата. Для приготовления этерифицированных производных фосфата, указанных выше, родственное соединение формулы I может быть обработано соответствующим производным ди-алканоил-фосфатидилхолина в присутствии соответствующей фосфолипазы, например фосфолипазы D, как описано в работе S. Shuto et al. Nucleic Acid Research, 1988, 30, с. 35, или путем введения в реакцию соединения формулы I с соответствующим фосфорилирующим веществом, таким как хлорокись фосфора, за чем следует обработка соответствующим спиртом, как описано авторами A. Rosowsky и S. Kim, Nucleic Acid Chemistry. Pert 3, L.B. Townsend и R.S. Tipson (Editors), John Wiley and Sons, New Jork, 1936, 255.
Энантиомеры соединений формулы I могут быть растворены или выделены обычным образом, например путем хроматографического разделения диастереомерных сложных эфиров, приготовленных ацилированием гидроксила на циклопентенильной части; с помощью соответствующих оптически активных производных карбоновой кислоты, как, например, с помощью напроксена (J. Org. Chem. 1986, 51, 1287).
Нижеследующие примеры предназначены только для иллюстрации и не предназначены для ограничения объема изобретения никоим образом. В примерах значения вращения плоскости поляризации света были установлены по отношению к линии плотности натрия (589 нм) при 20oC. Выражение "активный ингредиент", как оно используется в примерах от A до G, означает противовирусное соединение согласно изобретению, в частности вышеназванное соединение I.
Пример 1.
(±)-цис-4- (2-амино-4-хлор-6-пиримидинил)-амино/-2-циклопентен-1-метанол.
цис-4-ацетамидоциклопент-2-енметилацетат (патент США N 4.268.672) (14,88 г, 0,073 моль) и октагидрат гидроокиси бария (46,19 г 0,146 моль) кипятили с обратным холодильником в воде (300 мл) в атмосфере азота в течение 18 ч. Полученный раствор нейтрализовали двуокисью углерода. Осадок промывали водой, затем этанолом. Слитый вместе фильтрат-промывку выпаривали до сиропа (11,16 г), который конденсировали с 2-амино-4,6-дихлорпиримидином (23,91 г, 0,146 моль) и триэтиламином (30,5 мл, 0,219 моль) при кипячении с обратным холодильником 1-бутанола (100 мл) в течение 1,5 ч. После добавления 1 н. NaOH (73 мл), полученную смесь выпаривали до сухости и остаточное твердое вещество переводили в суспензию в СНCl3 (200 мл). Непрореагировавший 2-амино-4,6-дихлорпиримидин отфильтровывали и промывали хлороформом (100 мл). Фильтрат-промывку с хлороформом концентрировали и пропускали через хроматографическую колонку с силикагелем. Дополнительный исходный материал пиримидин элюировали с 2,5 метанолом-хлороформом. Целевое соединение элюировали с 3,5 метанолом-хлороформом в виде твердой пены нечистого белого цвета (15,90, 91).
1H-ЯМР: (Me2SO d6), δ: 1,15 1,28 и 2,26 2,41 (2М, 2, СН2), 2,60 2,71 (м, 1, 1'-H), 3,4 (м перекрывает H2O, CH2OH), 4,625 (т, J 5,3, 1, CH2OH), 4,95 (brs, 1, CH-N), 5,67 5,87 (m, 2, CH CH), 6,38 (brs, 1, NH2), 7,12 (brs, 1, NH), MS (Cl) M + 1, 241, 243.
Анализ: рассчитано для C10H13N4OCl • 0,2 H2O: C 48,99; H 5,56; N 22,85; Cl 14,46.
Найдено: C 49,10; H 5,56; N 22,81; Cl 14,40.
Пример 2.
(± )-цис-4-/(2-амино-6-хлор-5-/4-хлорфенил)-азо/-4-пиримидинил/ -амино/-2-циклопентен-1-метанол.
(±)-цис-4-/(2-амино-4-хлор-6-пиримидинил)-амино/- 2-циклопентен-1-метанол из примера 1 (11,58 г, 48, 1 ммоль) и тригидрат ацетата натрия (97 г) растворяли в ледяной уксусной кислоте (225 мл) и в воде (225 мл). Холодный раствор (0 5oC) 4-хлорбензолдиазонийхлорида готовили из 4-хлораналина (6,74 г, 52,8 ммоль), концентрированной соляной кислоты (14,7 мл), воды (52 мл) и нитрита натрия (4,01 г, 58,2 ммоль в 47 мл воды). Этот холодный раствор добавляли по каплям в течение 5 мин к первому раствору. Полученный желтый осадок фильтровали спустя 18 ч, промывали водой и экстрагировали этанолом для получения целевого соединения в виде темно-желтого порошка (12,56 г, 69), т. пл. 218 220oC.
1H-ЯМР: (Me2SO d6), d, 10,25 (d, 1, NH), 7,69 и 7,54 (оба, d, J 8, 9, C6H4) перекрывает 7,6 (br, 6, NH2), 5,80 5,95 (m, 2, CH=CH), 5,24 (m, 1, CH), 4,75 (t, 1, CH2OH), 3,41 (t, 2, CH2OH), 2,75 (m, 1, CH), 2,41 (m, 1, CH), 1,44 1,53 (m, 1, CH).
Анализ: рассчитано для C16H16N6Cl2: C 50,57; H 4,25; N 22,16; Cl 18,70.
Найдено: C 50,59; H 4,29; N 22,10; Cl 18,66.
Пример 3.
(±)-цис-4-/(2,5-диамино-4-хлор-6-пиримидинил)-амино/- 2-циклопентен-1-метанол.
Целевое соединение из примера 2 (11,67 г) суспендировали в этаноле (235 мл) ледяной уксусной кислоте (30 мл) и воде (235 мл). Смесь нагревали до кипения с обратным холодильником в атмосфере азота. Небольшими порциями добавляли цинковую пыль (13,5 г) в течение 30 мин, в это время соединение растворялось. Реакцию нагревали еще в течение 20 мин, а затем избыточный цинк отфильтровывали от горячего раствора, и его промывали этанолом. Фильтраты выпаривали и остаток очищали на колонке с силикагелем, элюируя хлороформом (1 л) и хлороформом метанолом 1/4 1 (1,8 л). Фракции, содержащие продукт, соединяли и растворитель удаляли при пониженном давлении до получения целевого соединения в виде красно-оранжевого масла (11,2 г, 100 выход). Чистая проба была получена в течение другой реакции малого масштаба для получения продукта в виде светло-желтого твердого вещества с 76-ным выходом.
1H-ЯМР: (Me2SO d6), d, 1,29 и 2,39 (m, 2, CH2), 2,69 (t, 1, 1'-H), 3,37 (d, 2, CH2OH), 3,91 (br, 2, NH2 ), 4,60 (br, 1, CH2OH), 5,02 (m, 1, CHNH), 5,56 (brs, 2, NH2), 5,74 (m, 1,CH), 5,86 (m, 1,CH), 6t,36 (d, 1, CHNH).
Пример 4.
(± )-цис-4-/2-амино-6-хлор-9Н-пурин-9-ил/-2-циклопентен-1-метанол.
Целевое соединение из примера 3 (примерно 9,7 г) растворяли в диэтоксиметилацетате (100 г) и кипятили с обратным холодильником в течение двух дней. Растворитель удаляли под глубоким вакуумом при 50oC и добавляли диоксан (40 мл) и 0,5 н. HCl (60 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,25 ч и затем резко охлаждали. Реакцию нейтрализовали до рН 7 холодной 5 н. гидроокисью натрия, а затем экстрагировали хлороформом метанолом 1/3 1 несколько раз. Органические слои сушили сульфатом магния, фильтровали и выпаривали. Остаток очищали хроматографией на колонке с силикагелем, элюируя с 2 МеOH-CHCl3 до получения 3,7 г (46-ный выход) целевого соединения, т. пл. 138 139oC.
1H-ЯМР: (Me2SO d6), d, 1,63 и 2,61 (m, 2, CH2), 2,87 (m, 1, 1'-H), 3,44 (d, 2, CH2OH), 5,44 (m, 1, CH-N), 5,89 (m, 1,CH), 6,14 (m, 1, CH), 6,82 (brs, 2, NH2), 8,02 (s, 1, 8-H), (CH2OH не виден под пиком H2O). УФ: рН Iλmax 315 (ξ 7370), 218 (26200), lsh 239,5 (5650). рН 7,4 λmax 307 (ξ 8000), 245,5 (4600), 223 (26400), M (EI) 265, 267 (m) (Cl) 266, 268 (m + 1).
Анализ: рассчитано для C11 H12N5C12 • 2H2O: C 43,79, H 5,35, N 23,21, Cl 11,75.
Найдено: C 43,67, H 5,29, N 23,05, Cl 11,70.
Пример 5.
(±)-цис-4-/2-амино-5-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Целевое соединение из примера 4 (0,50 г) растворяли в этаноле (40 мл) и добавляли циклопропиламин (0,65 мл, 5 экв.). Реакционную смесь кипятили с обратным холодильником в атмосфере азота в течение 6 ч. Добавляли дополнительно 0,65 мл циклопропиламина, а затем реакционную смесь кипятили с обратным холодильником в течение еще 5,5 ч. Растворители выпаривали и добавляли хлороформ (25 мл) и насыщенный раствор бикарбоната натрия (5 мл). Водный слой экстрагировали несколько раз с помощью СНCl3 для получения сырого продукта. Этот продукт очищали на колонке с силикагелем, элюируя с помощью 30 метанолэтилацетата для получения 0,43 г (80) (± -цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/ -2-циклопентен-1-метанола. Последний перекристаллизовывали из ацетонитрила для получения 0,30 г белого порошка: т. пл. падает при 93 130o C, расплавление при 165oC.
1H-ЯМР: (Me2SO d6), d: 0,56 и 0,63 (2m, 4, 2-циклопропил CH2), 1,56 и 2,60 (2m, 2, циклопентенил-СН2), 2,85 (m, 1, 1'-H), 3,02 (m, 1, циклопропил СH-NH), 3,43 (m, 2, CH2OH), 4,71 (t, 1, CH2OH), 5,40 (m, 1, 4'-H), 5,77 (s, 2, NH2) перекрывание 5,84 (m, 1, CH2), 6,09 (m, 1,CH), 7,23 (d, 1, NH-CH), 7,58 (s, 1, пурин-8-H), ms (Cl) 287 (m + 1). УФ: рН I: lmax 296 (ξ 14000), 255 (10700), рН 7,0 lmax 284 (15900), 259 (9200), рН 13 λmax 284 (15800), 259 (9100).
Анализ: рассчитано: для C14H14N60 • 0,25H2O: C 57,82; H 6,41; N 28, 90.
Найдено: C 57,84; H 6,45; N 28,86.
Пример 6.
(±)-цис-4-(2-амино-6-(циклопропилметиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанол.
(±)-цис-4-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентен-1-метанол (0,53 г, 2 ммоль) из примера 4, N-метил-N-циклопропиламин (KarI Industries Aurora, OH, 0,8477 г, 12 ммоль) и метанол (20 мл) помещали в сосуд Парра и нагревали при 62oC в течение 5 ч. Раствор концентрировали и затем разбавляли этанолом до доведения до рН 12 добавлением 1,0 н. NaOH. Этот раствор концентрировали и остаток очищали элюированием из колонки с силикагелем с помощью 3 метанола-хлороформа (0,547 г, 91,2). Кристаллизация такой пробы из воды-этанола давала белый порошок, т. пл. 130 - 131oC.
1H-ЯМР (Me2SO d6), δ: 7,61 (s, 1H, пурин Н-8), 6,10 (m, 1H, CH=), 5,84 (m, 1H, CH=), 5,7 (brs, 2H, NH2), 5,40 (m, 1H, CH), 4,70 (brt, 1H, OH), 3,43 (m, 2H, CH2OH), 3,24 (br, 4H, CH3, NH циклопропил), 2,85 (m, 1H, CH), 2,66 2,57 и 1,61 1,51 (m, 2, циклопентил CH2), 0,90 0,65 (m, 4H, 2CH2 из циклопропила).
Анализ: рассчитано для C15H20N60 • 0,5H2O: C 58,24; H 6,84; N 27,16.
Найдено: C 58, 15; H 6,86; N 27,14.
Пример 7.
(±)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Целевое соединение из примера 5 (0,600 г, 2,00 ммоль) растворяли в 1,3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидинона (AIoг с, 12 мл). Добавляли фосфорилхлорид (0,76 мм 8,0 ммоль) и перемешиваемому, охлажденному (-10oC) раствору. Спустя 3 мин добавляли холодную воду (100 мл) и полученный раствор нейтрализовали гидроокисью аммония 3 М. Нейтрализованный раствор разбавляли до 1 л водой и подавали в колону 2,5 х 20 см Sephadex А25 с диэтиламиноэтилом (Pharmacia, которая была предварительно уравновешена с помощью 50 мМ бикарбоната аммония). Колонку вначале промывали с использованием 4 л 50 мМ бикарбоната аммония. О-монофосфат из (+)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанола затем элюировали при 2-литровом градиенте из 50 до 300 мМ бикарбоната аммония. Фракции, содержащие нуклеотид (то есть вышеуказанный О-монофосфат) выпаривали до белого порошка с целью удаления бикарбоната аммония. 71 рассчитан по поглощению в УФ, один пик с помощью ВЭЖХ (см. ниже), 5'-нуклеотидазу из змеиного яда (ЕС 3.1.3.5) от Crotalus atrox (1000 международных единиц, Sigma) добавляли к 1,4 ммоль нуклеотида, растворенного в воде (20 мл). Раствор инкубировали при 37oC в течение 22 ч, в это время добавляли дополнительный штамм (1000 международных единиц). Инкубирование продолжали в течение еще 3 дней. Анализ с помощью ВЭЖХ (0,4 х 10 см обменной колонки с сильным анионом Whatman Partisil 10, элюирование с помощью градиента от 20 мМ до 1 М фосфата аммония, рН 5,5, содержание метанола 5 детектирование в УФ при 284 нм в этой точке показало, что 50 исходного нуклеотида было фосфорилировано до родственного нуклеозида. Эту смесь вновь вводили в колонку Sephadex с диэтиламиноэтилом вышеописанного типа. Элюирование с использованием 4 л 50 мМ бикарбоната аммония давало фракции, содержащие целевое соединение. Выпаривание воды составляло белый порошок. Этот материал далее очищали хроматографией на силикагеле с MeOH CHCl3 / 1 9 до получения бесцветного стекла. Это стекло отверждали в ацетонитриле для получения (+)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/ -2-циклопентен-1-метанола в виде белого клейкого твердого вещества, которое высушивали до твердой пены при 0,5 мм рт. ст. при 68oC (260 мг, 86 из рацетама).
1H-ЯМР в ДМСО -d6 и масс-спектр, идентичные с аналогичными рацемата (целевое соединение из примера 4), (α) -59,7o, (α) -127,8o, (α)- -218,1o, (с 0,15, метанол).
Анализ: рассчитано для C14H18N60 • 8H2O: C 55,91; H 6,57; N 27,94.
Найдено: С 56,95; Н 6,55; N 27,88.
Непрерывное элюирование последней упомянутой колонки Sephadex с 2-литровым градиентом от 50 до 300 мМ бикарбоната аммония давало О-монофосфат (из (+) энантиомера, соответствующего целевому соединению), который был стабилен к 5'-нуклеотидазе, приготовление этого монофосфата описано более подробно в примере 9.
Пример 8.
(-)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол-Л-монофосфат.
Целевое соединение из примера 7 (0,35 г, 1,2 ммоль), растворяли и 1,3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидинона (Aldrich, 5 мл). Добавляли фосфорилхлорид (Aldrich 0,43 мл, 4,6 ммоль) к перемешиваемому, охлаждаемому (-10oC) раствору. Спустя 3 мин добавляли холодную воду (20 мл) и полученный раствор нейтрализовали с помощью гидроокиси аммония 3 М. Ионообменная хроматография, как описано в примере 7, дала нуклеотид в виде диаммонийной соли после выпаривания воды, в виде белого порошка (выход 95 количество определено с помощью УФ), анализ с помощью ВЭЖХ, как в примере 7, показал один пик, УФ: λmax, нМ (0,1 М NaOH); 259, 284. Соотношение основание / фосфат было 1,0/1,3, как определено по методу B. Ames. Methods in Engymology 8 115, 1966, (α) -89,9o; (α) 73,0o; (α) -84,0o (c 0,52, MeOH H2O / 4 1).
Пример 9.
(+)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил-/- 2-циклопентен-1-метанол-Л-монофосфат.
Элюирование колонки Sephadex с диэтиламиноэтилом, описанное в примере 7, после инкубирования 5'-нуклеотидазы с 2-литровым градиентом от 50 до 300 мМ бикарбоната аммония давало нуклеотидсодержащие фракции, после выпаривания воды, давало целевое соединение, такое как соль диаммония, белый порошок (56 от целевого соединения из примера 5), анализ с помощью ВЭЖХ, как в примере 7, показывает один пик, УФ: λmax, нМ (0,1 М HCl): 254, 297, (pH 7, фосфатный буфер): 259, 284 (0,1 M NaOH) 259, 284. Соотношение основание / фосфат было 1,0/0,98. (α) +62,0o; (α) +65,2o; (α) + 75,2o (c 0,54, MeOH H2O / 4 1).
Пример 10.
(+)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/ -2-циклопентен-1-метанол.
Целевое соединение из примера 9 (0,67 ммоль) растворяли в воде (20 мл) и добавляли щелочную фосфатазу (ЕС 3.1.3.1) из кишки теленка (3000 международных единиц Boehringer Manheim). Раствор инкубировали при 37oC в течение 19 ч, в этой точке анализ с ВЭЖХ, как описано в примере 7, показал, что весь нуклеотид был дефосфорилирован. Раствор выпаривали до сухости и остаточные твердые вещества извлекали этанолом в режиме кипения с обратным холодильником (100 мл). Растворимый в этаноле материал поглощали на силикагель и подавали на колонку для силикагеля. Целевое соединение элюировали метанолом хлороформом / 1 9. Выпаривание раствора ацетонитрил-этанол давало белую твердую пену (164 мг, 79).
1H-ЯМР в ДМСО-d6 и масс-спектр, идентичные с аналогами у рацемата (целевое соединение из примера 5), (α) +58,7o; (α) +126, 2o; (α) +217,5o (c 0,10, метанол).
Анализ: рассчитано для C14H18N6O • 0,60H2O •15 EtOH: C 56,49; H 6,6; N 27,64).
Найдено: C 56,60; H 6,63; N 27,55.
Пример 11.
(-)-цис-4-/2-амино-6-циклопропилметиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Целевое соединение из примера 6 (2,00 г, 6,50 ммоль) растворяли в 1, 3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидиноне (Aldrich 20 мл). Добавляли фосфорилхлорид (2,28 мл, 24,0 ммоль) к перемешиваемому, охлаждаемому (-10oC) раствору. Спустя 3 мин добавляли холодную воду (80 мл). Раствор экстрагировали хлороформом (3 х 80 мл). Водный слой разбавляли этанолом (400 мл) и рН регулировали до 6 с помощью насыщенного водного NaOH. Осажденные неорганические соли были отфильтрованы, фильтрат далее разбавляли этанолом до объема в 1 л и рН регулировали до 8 дополнительным количеством NaOH. Полученный осадок фильтровали и высушивали для получения О-монофосфата (±)-цис-4-/2-амино-6-(циклопропилметиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанола в виде белого порошка (4,0 ммоль, 62 подсчитаны количественно с помощью поглощения в УФ), анализ с помощью ВЭЖХ, как с примере 7, показывает один пик. Этот рацемический О-монофосфат растворяли в воде (200 мл) в 5'-нуклеотидазе из яда змей (ЕС 3.1.3.5) от Crotatus (5,000 международных единиц, Sigma) добавлялась потом. После инкубирования при 37oC в течение 10 дней анализ с помощью ВЭЖХ, как описано в примере 7, показал, что 50 исходного нуклеотида было дефосфорилировано до нуклеозида. Их отделяли на колонке 5 х 14 см Sephadex А25 с диэтиламиноэтилом (Pharmacia), которая была предварительно уравновешена с помощью 50 мМ бикарбоната аммония. Выпаривание воды дало белый порошок, который был растворен в метаноле, его абсорбировали на силикагеле и наносили на колонку с силикагелем. Целевое соединение элюировали с помощью метанола хлороформа / 1 9 в виде бесцветного стекла. Раствор ацетонитрила выпаривали до получения белой твердой пены, сушили при 0,3 мм рт. ст. над P2O5, 649 мг (72 из рацемата).
1H-ЯМР в ДМСО d6 и масс-спектра идентичны аналогам у рацемата (целевое соединение из примера 6), (α) -48,0o; (α) -97,1o; (α) -149o (c 0,14 метанол).
Анализ: рассчитано для C15H20N6 O-0.10CH3C: C 59,96; H 6,72; N 28,06.
Найдено: C 59,93; H 6,76; N 28,03.
Непрерывное элюирование из колонки Sephadex с помощью 2 л 100 мМ бикарбоната аммония затем с помощью 2 л 200 мМ бикарбоната аммония давало О-монофосфат (+) энантиомера, соответствующего целевому соединению, который был стабилен к 5'-нуклеотидазе.
Пример 12.
(+)-цис-4-/2-амино-6-(циклопропилметиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Фракции, содержащие О-монофосфат (+) энантиомера, алюированного из колонки Sephadex из примера 11, соединяли и добавляли щелочную фосфатазу (ЕС 3.1.3.1) из желудка теленка (4800 международных единиц, Boehringer Manheim). Раствор инкубировали при 25oC в течение 18 ч, в этот момент анализ с помощью ВЭЖХ показал, что весь нуклеотид был дефосфорилизирован. Раствор выпаривали до сухости и остаточные твердые вещества экстрагировали с помощью этанола, который кипятили с обратным холодильником (100 мл). Этанолрастворимый материал адсорбировали на силикагеле и наносили на колонку с силикагелем. Целевое соединение элюировали метанолом хлороформом / 1 9 в виде бесцветного стекла. Раствор ацетонитрила выпаривали, получая белую твердую пену, сушили при 0,3 мм рт. ст. над H2O5, 659 мг (73 из рацемата).
1H-ЯМР в ДМСО d6 и масс-спектр идентичны аналогичным рацемата (целевое соединение из примера 6), (α) +47,0o; (α) +93,0o; (α) +141,3o (c 0,11, метанол).
Анализ: рассчитано для C15H20 N6O-0.1CH2CN: C 59,95; H 6,72; N 28,06.
Получено: C 59,92; H 6,80; N 27,96.
Пример 13.
(1S, 4R)-4-амино-2-циклопентен-1-метанолдибензоил-D-атартрат.
(±)-цис-1-ацетамидоциклопент-2-енметилацетат (патент США N 4.268.672) (14,88 г, 0,073 моль) и октагидрат гидроокиси бария (46,19 г, 0,146 моль) кипятили с обратным холодильником в воде (300 мл) в атмосфере азота в течение 18 ч. Полученный раствор нейтрализовали двуокисью углерода. Осадок промывали водой, затем этанолом. Собранный вместе фильтрат с промывкой выпаривали до сиропа (уксуснокислая соль (±)-4-амино-2-циклопентен-1-метанола), который преобразовывали в свободный амин при перемешивании с избытком смолы Amberlite IRA-400 (OH-) в воде. Смолу отфильтровали, промывали водой и фильтрат-промывку выпаривали до бледно-желтого сиропа, который сушили выпариванием порций этанола. Такую пробу амина (2,26 г, 20,0 ммоль) и дибензоил-D-винную кислоту (Aldrich, 3,62 г, 10,0 ммоль в виде 99) растворяли в горячем абсолютном этаноле (35 мл). Кипящий с обратным холодильником ацетонитрил (примерно 150 мл) добавляли до температуры помутнения и раствору давали остыть медленно до комнатной температуры. Белые иглы, которые образовывались, перекристаллизовывали в три раза из того же сочетания растворителей для получения целевого соединения в виде белых пластинок (1,07 г, 37), т. пл. 160 162o, (α) +66,9o; (α) +165o ; (α) +325o (c 0,28, метанол). Кристаллография в рентгеновских лучах этой соли способствовала фиксации абсолютной конфигурации катиона известной конфигурации дианиона-D-дибензоилвинной кислоты. Эта соль кристаллизовалась в пространственной группе С2 с одним катионом C6H12NO и половиной дианиона C18H14O8 в виде асимметричной единицы.
Анализ: рассчитано для C6H11NO-1/2(C18H14O8): C 61,63; H 6,21; N 4,79.
Найдено: C 61,56; H 6,24; N 4,74.
Пример 14.
(1R, 4S)-4-амино-2-циклопентен-1-метанолдибензоил-L-тартрат.
Эта соль была образована и кристаллизована как описано в примере 13, за исключением того, что использовалась дибензоил-L-винная кислота. Три кристаллизации из этанола-ацетонитрила давали целевое соединение в виде белых пластинок (1,00 г, 34), т. пл. 160 162o, (α) -68,2o; (α) -169o; (α) -333o; (c 0,24, метанол).
Анализ: рассчитано: для C6H11NO-1/2(C17H14O8): C 61,63; H 6,21; N 4,79.
Найдено: C 61,59; H 6,21; N 4,76.
Пример 15.
(±)-цис-N-/4-хлор-5-формамидо-6-//4-(оксиметил)-2-циклопентен-1-ил/- амино/-2-пиримидинил/ацетамид.
N-(5-амино-4,6-дихлорпиримидин-2-ил)-ацетамид (J. Org. Chem. 1975, 40, 3141) формилировали добавлением 96-ной муравьиной кислоты (20 мл) к раствору в (0,75 г, 3,4 ммоль), растворенному в уксусном ангидриде (20 мл). Полученный раствор перемешивали при 25oC в течение одного часа и затем выпаривали до получения N-(4,6-дихлор-5-формамидо-2-пиримидинил)-ацетамида в виде рыжевато-коричневого порошка (0,77 г, 91), структура подтверждена с помощью1 Н-ЯМР и масс-спектра. Этот рыжевато-коричневый порошок (840 мг, 3,37 ммоль), (±)-цис-4-амино-2-циклопентен-1-метанол (940 мг, 8,2 ммоль) и триэтиламин (0,80 г, 8,0 ммоль) нагревали в этаноле (50 мл) в атмосфере азота в течение 50 мин и выпаривали до темного масла, который подвергали хроматографии на силикагеле. Целевое соединение элюировали с помощью 5 метанола-хлороформа в виде твердой пены персикового цвета (840 мг). Кристаллизация из метанола давала белые гранулы (575 мг, 52), т. пл. 189 - 193o.
1H-ЯМР (ДМСО d6), δ: 10,23 (br 1,0, NHAC), 9,3 (br, 1,0, NHCHO), 8,15 и 7,90 (оба синглет, всего 1,0 НС=О из двух агентов конформации, пики коалесцируют при 60oC), 7,42 и 7,22 (оба дублет, J 8,3, всего 1,0, СН-NH из двух агентов конформации, пики коалесцируют при 60oC), 5,9 и 5,7 (оба m, 2,0, CH= CH), 5,05 (m, 1, CH-N) 4,73 (m, 1, OH), 3,39 (m, 2, CH2OH), 2,72 (m, 1, CH), 2,40 (m, 1, 1/2 CH2), 1,36 (m, 1, 1/2 CH2).
Анализ: рассчитано для C13H16N5O3Cl: C 47,98; H 4,95; N 21,50; Cl 20,88.
Найдено: C 47,99; H 4,96; N 21,42; Cl 10,96.
Пример 16.
(±)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Целевое соединение из примера 15 (0,91 г, 2,79 ммоль) растворяли в сухом ДМФ (1 мл). Добавляли триэтилортоформиат (10 мл) и этансульфокислоту (0,29 мл, 3,4 ммоль) и раствор нагревали при 65o C в течение 24 ч. Раствор выпаривали до сиропа. Сироп растворяли в 1 н. НCl (15 мл) и перемешивали в течение 3 ч. рН регулировали до 7 с помощью 5 н. гидроокиси натрия и полученную смесь (в виде масла) экстрагировали i-пропанолом хлороформом / 1 3 (3 x 100 мл). Собранные вместе органические слои сушили (MgSO) и выпаривали до красного стекла (0,93 г). Раствор этого стекла в метаноле (20 мл) нагревали с циклопропиламином (2 мл) в сосуде Парра при 70oC в течение 18 ч. Полученный раствор выпаривали до темного стекла, которое адсорбировали на силикагеле. Элюирование с помощью 7 метанол-этилацетата давало целевое соединение (148 мг, 19) в виде белого порошка, после растирания с ацетонитрилом.
1H-ЯМР (ДМСО d6) идентично с аналогичными показателями целевого соединения из примера 5.
Пример 17.
(+)-(1R, 4S)-цис-N-/4-хлор-5-формамидо-6-/4-(оксиметил)-2-циклопентен-1-ил/амино}-2-пиримидинил/-ацетамид.
(1S, 4R)-4-амино-2-циклопентен-1-метанолдибензоил-D-тартрат готовили как описано в примере 13 (2,76 г, 9,02 ммоль), растворяли в воде (20 мл) и наносили на колонку из 65 мл анионообменной смолы Amberlite 1A-400 (форма OH-). Колонку промывали водой. Базовые фракции собирали вместе и выпаривали до остаточного масла, которое высушивали выпариванием абсолютного спирта и затем на 0,5 мм для получения (1S,4R)-4-амино-2-циклопентен-1-метанол (1,2 г) в виде бледно-желтого масла (быстро темнеет на воздухе), которое использовали немедленно. Это масло растворяли в этаноле (5 мл) и добавляли к раствору N-(4,6-дихлор-5-формамидо-2-пиримидинил)-ацетамида (2,07 г, 8,31 ммоль), полученного как описано в примере 15, и триэтиламино (2,50 г, 24,8 ммоль). Полученный темный раствор нагревали (масляная ванна 75 80oC) в атмосфере азота в течение 50 мин. Этот раствор выпаривали до сиропа, который вводили в колонку с силикагелем. Целевое соединение элюировали 3 5 метанолом-хлороформом в виде бледно-желтой твердой пены (1,59 г, 54),1H-ЯМР идентично аналогичным показателям кристаллизованной пробы. Такую пробу кристаллизовали из этанола для получения белых гранул, т. пл. 194 - 195oC.
1H-ЯМР (ДМСО d6) идентично показателям целевого соединения из примера 16, (α) +2,7o; (α) +3,6o; (α) +2,9o; (α) -2, 5o; (α) -41,2o (c 0,238, метанол).
Пример 18.
(-)-(1S,4R)-цис-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентен-1-метанол.
Целевое соединение из примера 17 (1,15 г, 3,53 ммоль) осторожно кипятили с обратным холодильником
в диэтоксиметилацетате (45 мл) в атмосфере азота в течение 3,5 ч. Полученный бледно-желтый раствор концентрировали при 0,5 мм рт. ст. до желтого сиропа. Сироп перемешивали в 1 н. НСl (50 мл) в течение
1 ч. Этот раствор нейтрализовали бикарбонатом натрия и выпаривали до сухости. Остаточные твердые вещества экстрагировали метанолом и растворимый в метаноле материал вводили в колонку силикагеля.
Элюирование колонки с помощью 10 -ного метанол-этилацетата давало целевое соединение в виде бледно-желтой твердой пены (730 мг), 78
1H-ЯМР (ДИСО d6): идентично с
показателями рацемата (целевое соединение из примера 4): (α) -114,9o (c 0,26,
МeOH).
Пример 19.
(-)-(1S, 4R)-цис-4-/2-амино-6-циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
Целевое соединение из примера 18 (560 мг, 2,11 ммоль) в метаноле (12 мл) нагревали с циклопропиламином (2,4 мл) в сосуде Парра при 78oC в течение 17 ч. Растворитель выпаривали и остаток хроматографировали на силикагеле. Целевое соединение элюировали с 5 7 метанол-этилацетата в виде бесцветной твердой пены (367 мг, 59).
1H-ЯМР (ДМСО d6) идентично показателям из примера 7, (α) -59,0oC (c 0,28, МeOH), подтверждает абсолютную конфигурацию целевого соединения из примера 7.
Пример 20.
(1S,4R)-4-амино-2-циклопентен-1-метанолдибензоил-D-тартрат.
2-азабицикло-(2.2.1)-гепт-5-ен-3-он (Daluge and vince, J. Org. Chem. 1978, 43, 2311 и патент США N 4.268.672) (44,0 г, 0,400 моль) перемешивали в 2 н. НСl в метаноле (0,5 л) при 25oC в течение 1,5 ч. Летучие вещества выпаривали, оставляя (± )-цис-метил-4-амино-2-циклопентен-1-карбоксилатгидрохлорид в виде неопределенно-белого порошка (71,7 г). Растирание такой пробы простым диэтиловым эфиром давало белый порошок, т. пл. 92,5 95oC (J. Org. Chem. 1981, 46, 3271, т. пл. 82 83oC).
1H-ЯМР (ДМСО d6), δ: 8,25 (brs, 3, NH), 6,1 и 5,9 (оба m, 2, CH=CH), 3,64 (S) перекрывает 3,75 3,6 (m, всего 4, ОМе и СН), 2,65 - 2,45 и 2,05 1,85 (оба m, 2, СH2).
Анализ: рассчитано для C7H11NO2-HCl: C 47,33; H 6,81; N 7,89; Cl 19,96.
Найдено: C 47,41; H 6,84; N 7,85; Cl 19,89.
(±)-цис-метил-4-амино-2-циклопентен-1-карбоксилатгидрохлорид (17,7 г, 0,100 моль) и диизобутилалюминийгидрид (0,500 моль в виде раствора 1 М в гексане) кипятили с обратным холодильником в гексане (200 мл) в течение 6 ч. Полученный раствор охлаждали и 10 мл водного хлората аммония и затем метанол (200 мл) были добавлены. Эту смесь кипятили с обратным холодильником в течение 30 мин и добавляли MgSO4 (10 г). Твердые вещества отфильтровывали и промывали дополнительным количеством метанола. Фильтрат-промывку выпаривали до температурного масла (15,5 г).
1H-ЯМР (ДМСО d6) идентично показателям (±)-4-амино-2-циклопентен-1-метанола, приготовленного как описано в примере 13. Такой образец после очистки хроматографией на силикагеле (EtOH CHCl3 NH4OH / 10 90 1) кристаллизовали с помощью дибензоил-L-винной кислоты для получения целевого соединения.
Пример 21.
/цис-4-(2-амино-6-циклопропиламино)-9Н-пурин-9-ил)-2-циклопентен-1-ил/метил R-2,3-бис-(гексадеконоилокси)-пропил-кислый фосфат.
Раствор L-α-дипалмитиоилфосфатидилхолина (150 мг, 0,2 ммоль, Sigma) в 5 мл хлороформа добавляли в сосуд, содержащий (±)-цис-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанол (300 мг, 1,03 ммоль), фосфолипазу D, тип VII (из Streptomyces 1,0 мг, удельная активность 185 ед/мг, Sigma) и буфер с рН 4,5 (1,5 мл, 250 мM и СаCl2, 250 мМ в NaOAc, с регулированием рН до 4,5 добавлением 0,1 н. НСl). Полученную бифазу перемешивали при 45oС (масляная ванна) в течение 1 ч. Слои разделяли и водный слой экстрагировали хлороформом (3 х 6 мл). Собранные вместе органические слои промывали 1 н. НСl, высушивали и концентрировали. Такую пробу очищали элюированием из 2 колонок с силикагелем с 12 метанол-хлороформа, получая целевое соединение, 120 мг (47%). Этот материал отверждали с использованием этилацетата-ацетонитрила для получения легкого желтого порошка, т. пл. 155 157oC.
1H-ЯМР (CD3CD-CDCl3), d: 7,78 (s, перекрывающий растворитель, пурин Н-8), 612 и 5,88 (m, 2, HC=CH), 5,53 (m, 1, CHN циклопентен), 5,22 (m, 1, CO2CH), 4,37 (dd, J 3,12, 1, 0,5 POCH2 глицерин), 4,12 (m, 1, 0,5 POCH2 глицерин), 3,42 (m, 4, OCH2 глицерин, OCH2), 3,11 (br, m, 1, CH), 2,90 (m, 1, CH), 2,78 (m, 1, 0,5 CH2 циклопентен), 2,27 (m, 4, 2CH2CO2), 1,70 (m, 1, 0,5 CH2), 1,56 (m, 4, 2CH2CH2CO2), 1,27 (m, 38, 24 CH2), 0,88 (m, 6, 2CH3), 0,83 (m, 2, CH2 циклопропил), 0,60 (m, 2, CH2 циклопропил).
Анализ: рассчитано для C49H85N6O8P • 2,4H2O: C 61,28; H 9,42; N 8,75; P 3,22.
Найдено: C 60,97; H 9,12; N 8,78; N 2,96.
Пример 22.
(цис-4-(2-амино-6-циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-ил/метил/R-2, 3-бис-(гексаноилокси)-пропил-кислый фосфат.
Раствор L- a-дикапроилфосфатидилхолина (300 мг, 0,66 ммоль, Sigma) в 15 мл СНCl3 добавляли в сосуд, содержащий (± )-цис-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанол (378 мг, 1,32 ммоль), фосфолидазу D, тип VII (из Streptomyxes 1,04 мг, удельная активность 185 ед/мг, Sigma), буфер с рН 4, 5 (4,5 мл, 250 мМ в СaCl2, 20 мМ в NaOAc, с регулировкой рН до 4,5 добавлением HCl) и CHCl2 (3 мл). Полученную бифазу перемешивали при 45oC (масляная баня) в течение 4 ч. Слои разделяли и органический слой промывали в 1 н. НСl (2 х 4 мл). Собранные вместе водные слои промывали обратной промывкой хлороформом (10 мл). Собранные вместе органические слои высушивали (MgSO4) и концентрировали. Остаток помещали в колонку с силикагелем и целевое соединение элюировали 16 метанолом-хлороформом и концентрировали до получения мелкого желтого порошка. Этот материал растворяли в этаноле и концентрировали (3 х 50 мл) перед сушкой под глубоким вакуумом для получения 103 мг (выход 21) легкого желтого порошка, т. пл. 182 185oC.
1H-ЯМР: (ДМСО d6), d: 7,61 (s, 1, пурин Н8), 7,22 (brs 1, NH), 6,09 (m, 1, 0,5 CH=CH), 5,89 (m, перекрывание brs при 5,83, 3, 0,5 СН=СН, NH2), 5,41 (brm, 1, CHN), 5,09 (brm, 1, CO2CH), 4,30 (dd, J 2, 7, 12, 1, 0,5 POCH2 глицерин), 4,08 (m, 1, 0,5 POCH2 глицерин), 3,80 (brm перекрывание brm при 3,75, 4, OCH2 глицерин, OCH2), 3,02 (brm, 2, CH, NCH циклопропил), 2,65 (m, 1, 0,5 CH2 циклопентен), 2,23 (+, J 7,5, 4, 2CH2CO2), 1,48 (brm, 5, 2CH2CH2CO2, 0,5 CH2 циклопентен), 1,23 (brm, 8, 2(CH2)2), 0,84 (m, 6, 2 CH3), 0,67 и 0,58 (m, 4, 2CH2 циклопропил).
Анализ: рассчитано для C29H45N6O8P-3.9H2OCHCl3, 0,05 EtOH: C 48,00; H 7,33; N 11,46; Cl 2,9.
Найдено: С 48,56; Н 6,61; N 10, 81; Cl 2,5.
Предыдущий пример представляет собой адаптацию методики, описанной Satashi Stuto et al. Tetrahedron Letters, Vol. 28, N 2, с. 199 202, 1987.
Пример 23.
N-(4-хлор-1,6-дигидро-5-нитро-6-оксо-2-пиримидинил)-изобутирамид.
6-хлор-5-нитроизоцитозин (J. Chem. Soc. 1960, 5041, J. Org. Chem. 1975, 40, 3141) защищали разогревом желтого твердого вещества (14,88 г, 78,09 ммоль) до 100oC в течение 1 ч в изомасляном ангидриде (250 мл) и в концентрированной серной кислоте (3 4 капли). Полученный раствор обрабатывали безводным метанолом (100 мл), перемешивали при 50oC в течение получаса, концентрировали до одной трети от исходного объема и целевое соединение (14,97 г, 74 ) собирали фильтрованием в виде бледно-желтых кристаллов, т. пл. 196 199oC.
1H-ЯМР (ДМСО d6), d: 1,12 (d, J 6,9 Hz, 6H, (CH3)2CH), 2,75 (m, J 6,9 Hz, 1H, (CH2)2CH), 12,41 (brs, 1H).
Анализ: рассчитано для C8H9N4O4Cl: C 36,8; H 3,48; N 21,50; Cl 13,60.
Найдено: C 36,94; H 3,48; N 21,44; Cl 13,53.
Пример 24.
N-(4,6-дихлор-5-нитро-2-пиримидинил)-изобутирамид.
Целевое соединение из примера 23 (10,0 г, 38,87 ммоль) нагревали до кипения с обратным холодильником в хлорокиси фосфора (200 мл) и N,N-диэтиламине (3 4 капли) в течение 5 ч в атмосфере азота. Раствор затем охлаждали до комнатной температуры, концентрировали до сухости и сироп растворяли в холодном (≈ -10oC) метиленхлорида (200 мл). Органический слой обрабатывали насыщенным водным бикарбонатом натрия (100 мл) с сильным перемешиванием и температуру поддерживали ниже 5oC, когда добавляли порциями твердый бикарбонат натрия для повышения рН в пределах от 5 до 7. Слои отделяли и водную фазу экстрагировали метиленхлоридом. Собранные вместе органические слои фильтровали из фазоразделительной бумаги, концентрировали и высушивали под вакуумом для получения целевого соединения (7,71 г, 72) в виде желто-белого твердого вещества, достаточно чистого для использования на следующей стадии. Перекристаллизация твердого вещества из гексана/метиленхлорида давала аналитическую пробу, т. пл. 166 169oC.
1H-ЯМР (ДМСО d6), d: 1,09 (d, J 6,9 Hz, 6H, (CH2)2CH), 2,79 (m, J 6,9 Hz, 1H, (CH3)2CH), 11,61 (s, 1H).
Анализ: рассчитано для C8H8N4O3Cl2: C 34,43; H 2,89; N 20,08; Cl 26,41.
Найдено: C 34,53; H 2,89; N 20,02; Cl 25, 40.
Пример 25.
N-(4,6-дихлор-5-формамидо-2-пиримидинил)-изобутирамид.
Целевое соединение из примера 24 (6,77 г, 24,26 моль) помещали в сосуд Парра, содержащий 220 мл абсолютного ЕtОН и 10,0 г (влажный вес) никелевого катализатора Ренея, который предварительно встряхивали в атмосфере водорода (40 пси 2,8 кг/см2) в течение 10 мин. Смесь встряхивали в атмосфере водорода (40 пси 2,8 атм) в течение часа, фильтровали на целите и фильтрат концентрировали до желто-белого твердого вещества, которое высушивали под вакуумом всю ночь. Это твердое вещество перемешивали в 1,2-дихлорметане (250 мл) при 0oC). Добавляли уксусный ангидрид (30 мл), затем следовала муравьиная кислота (30 мл), по каплям в атмосфере азота. Полученную смесь перемешивали при комнатной температуре в течение 2 ч, концентрировали до половины исходного объема и переводили в азеотропную смесь с толуолом для удаления остаточных муравьиной/уксусной кислот. Сырое твердое вещество растирали с метанолом для получения целевого соединения (4,92 г, 73) в виде твердого вещества неопределенного белого цвета, т. пл. 206 209oC (дес.).
1H-ЯМР (ДМСО d6), d: 1,08 (d, J 6,8 Hz, 6,0 (CH3)2CH), 3,74 (m, J 5,8 Hz, 1,0 (CH3)2CH), 8,18 (d, J 10,3 Нz) и 10,26 (brs) (всего 1,0 NHCHO из двух агентов конформации), 11,17 (brs 1,0).
Анализ: рассчитано для C9H10N4O2Cl2: C 39,01; H 3,64; N 20,22; Cl 25,59.
Найдено: C 39,13; H 3,68; N 20,12; Cl 25,67.
Пример 26.
(+)-(1R, 4S)-цис-N-/4-хлор-5-формамидо-6-{ /4-(оксиметил)-2- циклопентен-1-ил/амино}-2-пиримидинил/-изобутирамид.
(1S, 4R)-4-амино-2-циклопентен-1-метанолдибензоил-D-тартрат (2,44 г, 8,15 ммоль), приготовленный согласно описанному в примере 13, растворяли в 90 этаноле (20 мл) и этот раствор добавляли в колонку со смолой Amberlite 1RA-400 (OH-) (30 мл), которую предварительно промывали тем же самым растворителем. Элюирование с 90 этанолом давало базовые фракции, которые после концентрирования и выпаривания порций толуола-этанола оставляли (1S,4R)-4-амино-2-циклопентен-1-метанол в виде бледно-желтого масла (1,4 г), которое немедленно конденсировали с N-(4,6-дихлор-5-формамидо-2-пиримидинилизобутирамидом (2,26 г, 8,15 ммоль), приготовленным согласно описанному в примере 25 в 1, 2-диметоксиэтане (100 мл) с триэтиламином (2,3 мл), 16,3 ммоль) при 95 110oC в течение 1,5 ч. Полученный раствор выпаривали до темно-желтого сиропа, который подвергали хроматографии на силикагеле. Элюирование колонки с 5 7,5 метанолом-хлороформом давало целевое соединение в виде бледно-желтого твердого вещества (2,45 г, 84). Кристаллизация такой пробы из ацетонитрила давала целевое соединение в виде мелких белых кристаллов, т. пл. 194,5 195,5oC.
1H-ЯМР (ДМСО d6), d: 10,21 (s, 1, NHCOCHMe2), 9,29 (s, 1, NHCHO), 8,12 (s, 1, CHO), 7,18 (d, J 7,9, 1, CHNH), 5,8 и 5,7 (оба m, 2, CH=CH), 5,08 (m, 1, CHN), 4,71 (t, J 5,06, 1, OH), 3,37 (m, 2, CH2OH), 2,9 2,6 (m, 2, CHME2 и CH), 2,40 (m, 1, 0,5 CH2), 1,33 (m, 1, 0,5 CH2), (α) +4,4o, (α) -20,7o (c 0,337, MeOH).
Анализ: рассчитано для C15H20N6ClO3: С 50,92; Н 5,70; N 19,79; Cl 10,02.
Найдено: C 50,67; H 5,78; N 19,82; Cl 9,92.
Пример 27.
(-)-(1R, 4S)-цис-N-/6-(циклопропиламино)-9-(4-оксиметил)- 2-циклопентен-1-ил)-9Н-пурин-2-ил/изобутирамид.
(+)-(1R, 4S)-цис-N-/4-хлор-5-формамидо-6-{ /4-(оксиметил)- 2-циклопентен-1-ил/амино} -2-пиримидинил/изобутирамид (1,949 г, 5,44 ммоль), приготовленный согласно описанному в примере 26, перемешивали с триэтилортоформиатом (30 мл) в водоледяной бане, в то время как концентрированную соляную кислоту (2,0 мл) добавляли по каплям в течение двух минут. Полученный светлый раствор перемешивали всю ночь при окружающей температуре. Летучие вещества удаляли под вакуумом и остаточный сироп (содержащий (1R,4S)-цис-N-/6-хлор-9-(4-оксиметил)-2-циклопентен-1-ил)-9Н-пурин-2-ил/ изобутирамид-ортоэфирконъюгат) кипятили с обратным холодильником в этаноле (30 мл) с циклопропиламином (10 г) в течение 2,5 ч. Выпаривание дало сироп, который растворяли в 10 изопропаноле-хлороформе (200 мл). Этот раствор сильно перемешивали с насыщенным водным бикарбонатом натрия (25 мл). Органический слой отделяли и водный слой промывали дополнительным количеством 10 изопропанола-хлороформа. Собранные вместе органические слои высушивали (MgSO4). Выпаривание дало бледно-желтое стекло (2,4 г), которое подвергали хроматографии на силикагеле. Целевое соединение элюировали 2 3 метанол-этилацетатом в виде белого твердого вещества (1,02 г, 53), перекристаллизация такой пробы из метанола-ацетонитрила давала целевое соединение в виде белых иголок, т. пл. 197,5 198,5oC.
1H-ЯМР (ДМСО d6) δ 9,75 (s, 1, NHCO), 7,93 (s, 1, пурин H-8), 7,82 (brs, 1, NH-циклопропил), 6,12 и 5,92 (оба m, 2 CH=CH), 5,50 (m, 1, CH-N, 4,73 (t, J 5,3, 1, OH), 3,46 (m, 2, CH2-O), 3,25 3,00 (m, 2, CHME2 и CH), 2,91 (m, 1, CH), 2,75 2,6 (m, 1, 0,5 CH2), 1,7 - 1,6 (m, 1, 0,5 CH2), 1,07 (d, J 6,8, 6, CHMe2), 0,75 0,6 (m, 4, 2 циклопропил, CH2), (α) -70,7o, (α) -159,0o (c 1,02, MeOH).
Анализ: рассчитано для C18H24N6O2: C 60,66; H 6,79; N 23,58.
Найдено: C 60,62; H 6,83; N 23,51.
Продолжаемое элюирование колонки с помощью 5 метанола-этилацетата давало дополнительное количество целевого соединения, загрязненного приблизительно 10 -ными (-)(-(1S, 4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанола в виде бледно-желтой твердой пены (928 мг).
Пример 28.
(-)-(1S, 4R)-цис-/4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол.
(-)-(1R, 4S)-цис-N-/6-(циклопропиламино)-9- (4-(оксиметил)-2-циклопентен-1-ил)-9Н-пурин-2-ил/изобутирамид (1,33 г, 3,73 ммоль), приготовленный как описано в примере 27, перемешивали с соляной кислотой 1 н. (40 мл) в течение 2 дней при окружающей температуре. Величина рН регулировалась до 7,0 с помощью гидроокиси натрия и смесь выпаривали до сухости. Остаточные твердые вещества перетирали с EtOH (3 x 25 мл). Этанол выпаривали, получая в остатке желтое стекло, которое хроматографировали на силикагеле. Целевое соединение было элюировано 3 метанолом-этилацетатом в виде бесцветной твердой пены (857 мг, 80).
1H-ЯМР и (α) идентичны аналогичным показателям целевого соединения из примера 19.
Пример 29.
(-)-(1S, 4R)-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанолгидрохлорид.
(-)-(1S, 4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол (1,90 г, приблизительно 6,3 ммоль при1H-ЯМР) растворяли в соляной кислоте 1 н. (7,0 мл) в этаноле. Раствор выпаривали до сухости и остаток вновь растворяли в этаноле (15 мл). Этилацетат добавляли медленно до полного объема 80 мл. Порошок неопределенного белого цвета, который образовывался, отфильтровывали и высушивали под вакуумом для получения целевого соединения (2,07 г, 97), т. пл. коллапоирует при 125 130oC, дес. выше 138oC, (α) -27,1o, (α) -52,3o (c 0,199, MeOH).
Анализ: рассчитано для C14H18N6OHCl • 0,8H2O: C 49,87; H 6,16; N 24,92; Cl 10, 51.
Найдено: C 49,91; H 6,16; N 24,96; Cl 10,52.
Пример 30.
(-)-(1S,4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н -пурин-9-ил/-2-циклопентен-1-метанолдигидрохлорид.
(-)-(1S, 4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол (857 мг, 3,00 ммоль) растворяли в этаноле-этилацетате и добавляли эфирной соляной кислоты 1 н. (12 мл). Мелкий белый осадок промывали этилацетатом и высушивали под вакуумом для получения целевого соединения (642 мг, 75), т. пл. 176 180o дес.
Анализ: рассчитано для C14H18N6O • 2HCl: C 46,81; H 5,61; N 23,39; Cl 19,74.
Найдено: C 46,82; H 5,64; N 23,33; Cl 19,67.
Пример 31.
(1R, 4S)-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанол-О-дифосфат.
(+)-(1R, 4S)-4-(2-амино-6-(циклопропиламино)-9Н/пурин-9-ил/- 2-циклопентен -1-метанол-О-монофосфат, приготовленный согласно описанному в примере 9, преобразовывали в соль триэтиламмония путем использования раствора, содержащего 0,5 ммоль монофосфата в качестве соли аммония, соединив его с 10 мл бикарбоната триэтиламмония 0,5 М и с сушкой в вакууме, затем добавили 10 мл дикарбоната триэтиламмония 0,5 М, а затем сушили. После этого трижды добавляли по 10 мл ацетонитрила и высушивали в вакууме. Затем растворяли в 10 мл, 1,3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидинона (Aldrich), затем 0,43 г 1, 1'-карбонилдиимидазола (Aldrich, 2,6 ммоль) добавляли и перемешивали в течение 2 ч при комнатной температуре. Добавляли метанол (0,18 мл, 4,5 ммоль) и перемешивали в течение 30 мин. Добавляли трибутиламмоний фосфат (Sigma 1,2 г, 2,6 ммоль), перемешивали в течение 18 ч при комнатной температуре, затем добавляли 1 г дополнительно трибутиламмонийпирофосфата (2,2 моль) и перемешивали 8 ч при 40oC, затем 50 мл воды было добавлено. Были получены как О-дифосфат, так и О-трифосфат, так как трибутиламмонийпирофосфат содержал примесь ортофосфата.
Продукты реакции были разделены с помощью ионообменной хроматографии Sephadex с диэтиламоноэтилом в колонке 2,5 х 18 см Sephadex A25 (Pharmacia) с диэтиламоноэтилом, которая была уравновешена с помощью 50 мМ бикарбоната аммония. Колонку промывали с 1 л 50 мМ бикарбоната аммония (БКА), затем с 2 л линейного градиента от 50 до 800 мМ БКА для элюирования целевого соединения, за чем следовал трифосфат, как более подробно описано в примере 32. Фракции, содержащие дифосфат, собирали вместе, высушивали в вакууме, вновь растворяли в воде, а затем вновь сушили, получая аммониевую соль целевого соединения (0, 077 ммоль, выход 15). Сканирование в УФ: в 0,1 М HCl λmax 254 и 298 нм, при рН 7 λmax 259 и 284 нм, в NaOH 0,1 M λmax 259 и 284 нм.
Аликвотное количество дифосфата обрабатывали щелочной фосфатазой (желудок теленка, Boehringer Manheim), отбирали несколько раз пробы и проявляли на тонкослойной хроматографии (полиэтиленимин-целлюлоза, Бринкман, 1 M LiCl / 1 M муравьиная кислота 1 1). Отмечали последующее превращение дифосфата в монофосфат нуклеозида. Конечное количество высвобожденного фосфата определяли с помощью метода Бенчини (Bencini D. A. Wild I. R. и O'Donovan, G. A. Analytical Biochemistry 132: 254 258, 1983) и соотношение основание/фосфат определяли равным 1,0/11,5, что указывает на присутствие неорганического фосфата. Чистота в УФ составляла 99,8 на аналитической ВЭЖХ (обменная колонка с сильным анионом, элюируемая градиентом от 10 мМ до 1 М фосфата аммония, рН 5,5).
Пример 32.
(+)-(1R, 4S)-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол-О-трифосфат.
Непрерывное элюирование колонки, описанной в примере 31, давало после выпаривания аммониевую соль целевого соединения. Эту соль преобразовывали в натриевую соль пропусканием через колонку со смолой Dowex AG 50W-X8 (Bio-Rad) (в форме натрия, 20 мл). Фракции, содержащие нуклеотид, концентрировали в вакууме для получения 0,31 ммоль (61). Сканирование в УФ: в 0,1 М HCl λmax 254 и 299 нм, при рН 7 λmax 259 и 284 нм: в 0,1 М NaOH λmax 259 и 284 нм. Значение вращения плоскости поляризации света в воде при 2,83 г/100 мл составляло (α) +43,2o при 589 нм. Чистота в УФ составляла 99,1 на аналитической ВЭЖХ (обменную колонку с сильным анионом элюировали с градиентом от 10 мМ до 1 М аммонийфосфата, рН 5,5) с 0,9 присутствующего дифосфата. Аликвотное количество трифосфата обрабатывали щелочной фосфатазой (желудок теленка), отбирали пробы несколько раз и проявляли на тонкослойной хроматографии (полиэтилениминцеллюлоза, Брингман, 1 M LiCl / 1M муравьиной кислоты 1 1). Последовательное преобразование трифосфата в дифосфат в монофосфат в нуклеозид было отмечено. Конечное количество фосфата, выделенного при этом, определялось по методу Бенчини Bencini D. A. Wild I. R. и O'Donowan H. A. Analytical Biochemistry 132: 254 258 (1983) и соотношение основание/фосфат было определено как 1,0/2,7.
Пример 33.
(1S, 4R)-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанол О-дифосфат.
О-монофосфат (-)-(1S,4R)-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-циклопентен-1-метанола, приготовленный как описано в примере 8, преобразовали в соль триэтиламмония путем использования раствора, содержащего 0,49 ммоль монофосфата в качестве соли аммония, соединяли с 5 мл бикарбоната триэтиламмония 0,5М и сушили в вакууме, за чем следовало еще 5 мл бикарбоната триэтиламмония 0,5 М, затем повторяли дважды. После этого три раза добавляли 5 мл ацетонитрила и высушивали в вакууме. Растворяли в 7 мл 1,3-диметил-3,4,5,6-тетрагидро-2-(1Н)-пиримидинона (Aldrich) затем 0,39 г 1, 1'-карбонилдиимидазола (Aldrich, 2,4 ммоль) добавляли и перемешивали в течение 30 мин при комнатной температуре. Добавляли метанол (0,16 мл, 4,0 ммоль) и перемешивали в течение 30 мин. Трибутиламмонийпирофосфат (полученный путем обмена с солью тетранатрийпирофосфата на водород на ионообменной смоле, затем нейтрализовали трибутиламином и сушили, 2,4 ммоль), перемешивали в течение 18 ч при комнатной температуре, затем добавляли 50 мл воды. Как O-дифосфат, так и О-трифосфат были получены, так как пирофосфат трибутиламмония содержал примесь ортофосфата.
Продукты реакции разделяли с помощью ионообменной хроматографии Sephadex с диэтиламиноэтилом в колонке 2,5 х 18 см Sephadex A25 (Pharmacia) с диэтиламиноэтилом, которая была уравновешена с помощью 50 мМ бикарбоната аммония (БКА). Колонку промывали с помощью 1 л 100 мМ БКА, затем с 2 л линейного градиента от 100 до 800 мМ БКА для элюирования целевого соединения, за чем следовала трифосфатата, как более подробно описано в примере 34. Фракции, содержащие дифосфат, собирали вместе, высушивали в вакууме, вновь растворяли в воде, а затем дважды повторяли до получения выхода аммонийной соли целевого соединения (0,032 ммоль) выход 6). Сканирование в УФ: в 0,1 М HCl λmax 254 и 298 нм, при рН 7 λmax 259 и 284 нм: в 0,1 М NaOH λmax 258 и 284 нм.
Аликвотное количество дифосфата было обработано щелочной фосфатазой (желудок теленка, Boehringer Manheim), отбирались несколько раз пробы и проявлялись на тонкослойной хроматографии (полиэтилениминцеллюлоза, Бринкман, 1 M LiCl / 1M муравьиной кислоты 1 1). Последующее превращение дифосфата в монофосфат в нуклеозид было отмечено. Конечное количество высвобожденного фосфата определяли по методу Бенчини (Bencini D. A. Wild I. R. и O'Donnovan, C. A. Analytical Biochemistry 132: 254 258, 1983), и соотношение основание/фосфат, как было определено, составило 1,0/4,7, что указывает на присутствие неорганического фосфата. Чистота в УФ составила 97 на аналитической ВЭЖХ (обменная колонка с сильным анионом, элюируемая градиентом от 10 мМ до 1 М аммонийфосфата, рН 5, 5).
Пример 34.
(1S,4R)-4-(2-амино-6-(циклопропиламино)-9Н-пурин-9-ил)- 2-(циклопентен-1-метанол-О-трифосфат.
Предложение элюирования колонки, описанное в примере 33, давало после выпаривания аммонийную соль целевого соединения. Эту соль превращали в натриевую соль путем пропускания через колонку со смолой Dawex AG-50W-X8 (Bio-Rad) (форма натрия, 20 мл). Фракции, содержание нуклеотид, концентрировали в вакууме, что давало 0,4 ммоль (81). Сканирование в УФ: в 0,1 М HCl λmax 254 и 299 нм, при рН 7 λmax 259 и 284 нм: в 0,1 М NaOH λmax 259 и 284 нм. Значение вращения плоскости поляризации света в воде при 6,14 г/100 мл составляло (α) -47,1o при 589 нм. Чистота в УФ составила 99,5 на аналитической ВЭЖХ (обменная колонка с сильным анионом, элюируемая градиентом от 10 мМ до 1 М аммонийфосфата, рН 5,5) с 0,5 присутствующего дифосфата. Аликвотное количество трифосфата обрабатывали щелочной фосфатазой (желудок теленка, Boehringer Manheim), отбирали пробы несколько раз и проявляли на тонкослойной хроматографии (полиэтилениминцеллюлоза, Бринкман, 1 M LiCl/ 1 М муравьиной кислоты 1 1). Последующее превращение трифосфата в дифосфат в монофосфат в нуклеозид было отмечено. Конечное количество высвобожденного фосфата определяли по методу Бенчини (Bencini D. A. Wild I. R. и O'Donovan G. A. Analytical Biochemistry. 132: 254 258, 1983) и соотношение основание/фосфат было определено равным 1,0/2,8.
Пример 35.
(1S,4R)-4-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентен-1-метанол.
(1S, 4R)-4-(2-амино-6-хлор-9Н-пурин-9-ил)-2-циклопентен-1-метанол (274 мг, 1,00 ммоль), N-циклопропил-N-метиламин (0,71 н, 10 ммоль) и абсолютный этанол (6 мл). Остаток подвергали хроматографии на силикагеле. Целевое соединение элюировали с 10 метанолом-хлороформом в виде бесцветного стекла. Выпаривание раствора этанола и сушка пятиокисью фосфора при 0,2 мм рт. ст. давала целевое соединение в виде белой твердой пены (93 мг, 98).
1H-ЯМР и
Пример A.
Составы в виде таблеток.
Нижеследующие составы А, В и С готовят гранулированием в увлажненном состоянии ингредиентов с раствором повидона, затем следует добавление стеарата магния и прессование. Составы A и B приведены в таблице.
Состав С мг/таблетку
Активный ингредиент 100
Лактоза 200
Крахмал 50
Повидон 5
Стеарат магния 4 389
Нижеследующие составы D и Е готовят прямым прессованием смешанных ингредиентов. Лактоза в составе Е
представляет собой лактозу типа непосредственного прессования (Diary Crest "Zeparox").
Состав D мг/таблетку
Активный ингредиент 250
Предварительно желатинизированный
крахмал NF15 150 400
Состав E мг/таблетку
Активный ингредиент 250
Лактоза 150
Avicel 100 500
Состав F (состав с регулируемым высвобождением).
Этот состав готовят гранулированием во влажном состоянии ингредиентов, приведенных ниже, с раствором повидона, за чем следует добавление стеарата магния и прессование.
Состав F мг/таблетку
а) Активный ингредиент 500
б) Оксипропилметилцеллюлоза (Mefhocel K4M Premium) 112
в) Лактоза В. Р. 53
г) Повидон В. Р. 28
д) Стеарат
магния 7 700
Выделение лекарственного препарата происходит в течение периода примерно 6 8 ч и завершается по истечении 12 ч.
Пример B.
Составы в виде капсул.
Cостав A.
Состав в капсуле готовят смешиванием ингредиентов состава D в примере A, приведенном выше, и заполнением в желатиновую капсулу из двух частей. Состав B (infra) готовят подобным образом.
Состав B мг/капсулу
а) Активный ингредиент 250
б) Лактоза В. Р. 143
в) Щелочной крахмальный гликолят 25
г) Стеарат магния 2 420
Состав C мг/капсулу
а) Активный ингредиент 250
б) Macrogjl 4000 B. P. 350 600
Капсулы состава C готовят расплавлением Macrogol 4000 ВР,
диспергированием активного ингредиента в расплаве и заполнением расплавом твердой желатиновой капсулы из двух частей.
Состав D мг/капсулу
Активный ингредиент 250
Лецитин 100
Арахисовое масло 100 450
Капсулы состава D готовят диспергированием активного ингредиента в лецитине и арахисовом масле и заполнением этой дисперсии в мягкие, эластичные
желатиновые капсулы.
Состав E (капсула регулируемого высвобождения).
Нижеследующий состав капсулы с контролируемым высвобождением получают экструдированием ингредиентов а, б и в с использованием экструдера, за чем следует придание шарообразной формы экструдату и сушка. Высушенные гранулы затем покрывают мембраной контролируемого высвобождения (г) и заполняют в твердую желатиновую капсулу из двух частей.
Состав E мг/капсулу
а) Активный ингредиент 250
б) Микрокристаллическая целлюлоза 125
в) Лактоза В.
Р. 125
г) Этилцеллюлоза 13 513
Пример C.
Состав для инъекций.
Cостав A.
Активный ингредиент 0,200 г
Раствор соляной
кислоты 0,1 или раствор гидроокиси натрия, 0,1 М, к-во, достаточное для рН 4,0 7,0
Стерильная вода, к-во достаточное до 10 мл
Активный ингредиент растворяют в большой части воды (35
40oC) и рН регулируют до диапазона от 4,0 до 7,0 с помощью соляной кислоты или гидроокиси натрия (по необходимости). Порцию затем доводят до объема с помощью воды и фильтруют через
стерильный микропористый фильтр в стерильный сосуд из янтарного стекла (тип I) и герметизируют стерильными пробками и средствами перегерметизации.
Состав B.
Активный
ингредиент 0,125 г
Стерильный, свободный от пирогена, фосфатный буфер с рН 7, к-во, достаточное до 25 мл
Пример D.
Внутримышечное инъецирование.
Активный ингредиент 0,20 г
Бензоловый спирт 0,10 г
Гликофурол 75 1,45 г
Вода для инъекций, к-во, достаточное до 3,00 мл
Активный ингредиент растворяют в
гликофуроле. Бензоловый спирт затем добавляют и растворяют и воду добавляют до 3 мл. Смесь затем фильтруют через стерильный микропористый фильтр и герметизирует в стерильных (3 мл) сосудах из
янтарного стекла (тип I).
Пример E.
Сироп.
Активный ингредиент 0,25 г
Раствор сорбитола 1,50 г
Глицерин 2,00 г
Бензоат
натрия 0,005 г
Корригент, Peach 17,42.3169 0,0125 мл
Очищенная вода, к-во, достаточное до 5,00 мл
Активный ингредиент растворяют в смеси глицерина и большей части очищенной
воды. Водный раствор бензоата натрия затем добавляют к этому раствору, за чем следует добавление раствора сорбитола и, наконец, корригента. Объем доводят до нужного очищенной водой и хорошо
смешивают.
Пример F.
Суппозиторий.
Состав мг/суппозиторий
Активный ингредиент 250
Твердый жир, ВР (W-tepsol H15 Dynamit Nobel) 1770
2020.
Одну пятую от Witepsol Н15 расплавляют в котле с паровой рубашкой при не выше 45oC. Активный компонент просевают через сито 200 мкм и добавляют к расплавленной основе со смешиванием, используя аппарат Сильферсона, снабженный режущей головкой, до получения мягкой дисперсии. Поддерживая температуру смеси 45oС, добавляют остальную часть Witepsol Н15 к суспензии и перемешивают для обеспечения гомогенной смеси. Всю суспензию пропускают через сито 250 мкм из нержавеющей стали и при постоянном перемешивании дают ей остыть до 40oC. При температуре от 38 до 40oC 2,02 г смеси заполняют в подходящие пластмассовые формы 2 мл. Суппозитории составляют остывать до комнатной температуры.
Пример G.
Вагинальные суппозитории.
Состав мг/суппозитории
Активный ингредиент 250
Ангидридная декстроза 380
Картофельный крахмал 363
Стеарат магния 7
1000
Вышеуказанные ингредиенты смешивают непосредственно и вагинальные суппозитории готовят путем прямого прессования полученной смеси.
Противовирусная активность.
а) Активность против ВИЧ.
(1S, 4R)-цис-4-/2-амино-6-(циклопропиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол испытывали на активность против ВИЧ в клетках MT4 согласно методу, описанному Averett D. R. J. Virol. Methods, 23, 1989, 263 276, и как сказалось, имеет значение IC50 и 4,0 ± 1,4 мкМ (среднее из 10 измерений).
б) Активность против ВГВ.
Клетка-продуцент ВГВ человека, линия HepG 2,2,2.2.15, описанная и охарактеризованная Sells et al. PNAS 84: 1005, 1987 и J. Virol. 62: 2836, 1988, как было показано, разделяет многие характеристики хронически инфицированного гепатоцита ВГВ. Она является инфекционной, как подтверждено, способностью вызывать болезнь у шимпанзе.
Для испытания соединений в отношении активности против ВГВ однослойные культуры обрабатывали испытываемым соединением: 50 200 мкМ на десять дней.
Всплывшую среду, содержащую внеклеточный вирион ДНК (частицы Дейна), собирали на дни: три, шесть и десять, обрабатывали протеиназой К (1 мг/мл) и додецилсульфатом натрия (1) и инкубировали при 50oC в течение одного часа. ДНК экстрагировали равными объемами фенола, за чем следовал хлороформ, и после этого осаждали ацетатом аммония и пропанолом. Осадок ДНК растворяли и собирали на нитроцеллюлозе, используя процедуру Schleicher и Schuell (Sands, 10 Optical Ave. Keene, NH 03431, Publication 700, 1987), и обрабатывали как описано Southern, J. Mol. Biol. 98, 503, 1975. Клетки собирали в виде урожая и получали внутриклеточную ДНК после лизиса клеток с помощью гуанидинизотиоцианата. Внутриклеточную ДНК обрабатывали таким же образом, как и экстраклеточную ДНК. После осаждения ацетатом аммония и пропанолом осадок внутриклеточной ДНК растворяли, разрывали рестрикционной эндонуклеазой, Н по III, наносили на агаровый гель и затем обрабатывали, как описано Southern для определения количества репликативных промежуточных форм. Противовирусный эффект лекарственного средства определяли измерением по крайней мере 100-кратного уменьшения количества частиц Дейна, экструдированных в культуральную среду, и подобного снижения количества внутриклеточных репликативных промежуточных веществ.
(1S, 4R)-цис-4-/2-амино-6-(N-циклопропил-N-метиламино)-9Н-пурин-9-ил/- 2-циклопентен-1-метанол испытывали согласно вышеназванной процедуре и, как оказалось, он имеет мощную активность против ВГВ при 100 мкМ.
Реферат
Использование: в качестве терапевтических агентов при лечении вирусных заболеваний. Сущность изобретения: продукт -энантиомерные нуклеозиды ф-лы I, где R - циклопропиламин или N-циклопропил-N-метил-аминогруппа, A - 2-циклопентен-1-метанол-4-ил в конфигурации (1S,4R) или (1R,4S) или их производные, выход 74 - 95 %. Условия реакции: реагент 1 - этантиомер ф-лы II, где Z - отщепляемая группа, A - указано выше. Реагент 2: соответствующий алкиламин, с последующим переводом продукта I в его соль, сложный эфир, монофосфат или этерифицированный фосфат. 4 з. п. ф-лы, 1 табл.
Структура соединений ф-л I и II:

Формула

где R циклопропиламино- или N-циклопропил-N- метиламиногруппа;
А 2-циклопентен-1-метанол-4-ильная группа либо в конфигурации (1S, 4R), либо (1R, 4S),
или их производных, причем указанные соединения или их производные находятся в виде энантиомера, свободного от соответствующего энантиомера, отличающийся тем, что осуществляют обработку энантиомерного соединения формулы II

где А имеет указанные значения;
Z удаляемая группа,
или его производного агентом или в условиях, обеспечивающих превращение группы Z в требуемую группу R, и полученный монофосфат или этерифицированный фосфат соединения I превращают в соединение I или один энантиомер соединения I переводят в другой энантиомер соединения I.
Комментарии