Пиперазиновые соединения - RU2133744C1
Код документа: RU2133744C1
Чертежи

Описание
Данное изобретение касается в основном соединений, имеющих диарилметилпиперазиновую природу и находящих применение в терапии главным образом в качестве рецептор-связывающих соединений, т.е. в качестве агонист/антагонист составляющих пар конъюгатов, для проверки/анализа действия рецепторов и нейротрансмиттеров. Соединения, предусмотренные настоящим изобретением, включают вещества диарилметилпиперазиновой природы, которые могут быть, с одной стороны, использованы в качестве мю и/или дельта-рецепторных опиоидных соединений, опосредующих аналгезию, а с другой - найти применение при лечении болей различной природы, при борьбе с наркоманией и алкоголизмом, при лечении состояний, вызванных передозировкой лекарственных средств, психических заболеваний, недержания мочи, лечении кашля, отека легких, диареи, депрессии и когнитивных, дыхательных и желудочно-кишечных расстройств. Изобретение также касается фармацевтических составов, включающих такие соединения, методов лечения некоторых расстройств с помощью указанных соединений и процессов получения таких соединений.
Изучение биохимии опиоидных соединений привело к идентификации большого числа разнообразных соединений этого класса как эндогенного, так и экзогенного происхождения. При этом значительная часть научных исследований была сфокусирована на выяснении механизма действия лекарств опиоидной природы, в особенности того, каким образом они взаимодействуют с опиатными рецепторами клеток и дифференцированных тканей.
Обычно лекарства опиоидной природы классифицируют по селективности их связывания с рецепторами клеток и дифференцирующихся тканей, с которыми специфический вид лекарства связывается как лиганд. Такие рецепторы включают мю ( μ ) -, дельта ( δ ) -, сигма ( δ )- и каппа ( æ ) - рецепторы.
Широко известные наркотические опиаты, такие как морфин и его аналоги, селективны к опиатным мю-рецепторам. Мю-рецепторы являются посредниками при аналгезии, угнетении дыхания и ингибировании прохождения содержимого по желудочно-кишечному тракту. Каппа-рецепторы опосредуют аналгезию и седативный эффект. Сигма-рецепторы опосредуют разнообразные биологические активности.
Существование опиоидного дельта-рецептора было открыто относительно недавно вслед за выделением и исследованием характеристик эндогенных энкефалиновых пептидов, являющихся лигандами к этому виду рецептора. Исследования последнего десятилетия позволили получить важную информацию о дельта-рецепторе, однако в настоящее время все еще нет ясной картины его действия. Дельта-рецепторы опосредуют аналгезию, но, по-видимому, не являются ингибиторами кишечного транзита подобно мю-рецепторам.
Обычно опиоидные агенты характеризуются как агонисты, либо антагонисты. Агонисты и антагонисты являются агентами, которые узнают и связываются с рецепторами, вызывая (либо инициацией, либо блокированием) биохимические/физиологические последствия; этот процесс известен как трансдукция. Агонисты ингибируют или подавляют выход нейротрансмиттеров в ткани, содержащие рецепторы, например при ингибировании болевых реакций или воздействии на другие явления, связанные с их выходом. Антагонисты также связываются с рецепторами, но не ингибируют выхода нейротрансмиттеров. Таким образом, антагонисты связываются с теми же рецепторными сайтами, что и агонисты, и блокируют связывание последних.
Что касается специфичных к рецептору лигандов, то ранее различие между дельта-рецепторными агонистами и антагонистами делалось на основании их активности в анализах с использованием стимулированных электрическим током семявыводящих сосудов мыши, которые обычно рассматривались в качестве подходящей ткани для диагностики дельта-рецепторов. В противоположность этому мю-рецепторные агонисты в основном характеризовали по их активности в анализе стимулированной электрическим током подвздошной кишки морской свинки.
Известно только относительно небольшое число полученных в чистом виде агентов, селективных к дельта-рецептору. За исключением дельта-рецепторных опиоидных антагонистов и агонистов, описанных в патентах (патент США 4,816,586; международная заявка на патент WO93/15062), все известные селективные к дельта-рецептору опиоидные соединения, представляют собой пептиды, включая эндогенные энкефалины и другие эндорфины, равно как и аналоги экзогенных пептидов. Все ранее синтезированные аналоги экзогенных пептидов обладают многочисленными общими недостатками в плане их стабильности, возможных подходящих путей их доставки в качестве лекарственных агентов и их распределения in vivo по тканям.
Изучены различные физиологические воздействия известных опиоидных лигандов пептидной природы, такие как: аналгезия, угнетение дыхания, желудочно-кишечные эффекты, воздействие на психический, эмоциональный и когнитивный функциональные виды деятельности и опосредование/модуляция других физиологических процессов.
В работе (патент США 4,518,711) описываются циклические конформационно-напряженные аналоги энкефалинов. В число этих соединений входят как агонисты, так и антагонисты дельта-рецептора.
В дополнение к указанным выше сноскам касательно опиоидных соединений, к той же области, что и соединения по настоящему изобретению, принадлежат соединения полиарилпиперазиновой природы, описанные в указанных ниже работах.
S.Goenechea и др. описывает оральное применение соединения полиарилпиперазиновой природы при изучении метаболизма меклозина в человеческом организме (Goenechea S. et. al. Investigation of the biotransformation of meclozine in the human body. J. Clin. Chem. Clin. Biochem., 26(2), 105-115, (1988)).
В работе (Meuldermans W. et. al. Plasma levels, biotransformation, and excretion of oxatomide in rats, dogs, and man. Xenobiotica, 15(6), 445-462, (1984)) проведен метаболический анализ содержания в плазме, биотрансформации и экскреции оксатомида.
T. Iwamoto и др. (Iwamoto Т. et. al. Effects of KB-2796, a new calcium antagonist, and other diphenylpiperazines on [3 H]nitrendipine binding. Jpn. J. Pharmacol. , 48(2), 241-247, (1988)) описывает действие отдельного соединения полиарилпиперазиновой природы в качестве антагониста кальция.
K. Natsuka и др. описывает рацематы и энантиомеры замещенных по первому положению 4-[2-(3-оксифенил)-1-фенилэтил]пиперазиновых производных (Natsuka К. et. al. Synthesis and structure-activity relationships of 1-substituted 4-(1, 2-diphenylethyl)piperazine derivatives having narcotic agonist and antagonist activity. J. Med. Chem., 30(10), 1779-1787, (1987)).
В работе (заявка на Европейский патент N 458,160) описаны замещенные производные дифенилметана, которые используют в качестве аналгезирующих или противовоспалительных средств, включая соединения, отличающиеся тем, что метиленовая группа, связывающая две фенильных половины молекулы, может иметь в качестве заместителя при своем атоме углерода пиперидильную или пиперазинильную группы.
В работе (заявка на патент Южной Африки N 8604522) описаны N-замещенные арилалкил- и арилалкилензамещенные амино-гетероциклические соединения, включая производные пиперидина, которые представлены как полезные сердечно-сосудистые, антигистаминные и антисекреторные агенты.
В работе (заявка на Европейский патент N 133,323) описаны некоторые соединения дифенилметилпиперазиновой природы, полезные в качестве неседативных антигистаминов.
Потребность в улучшенных опиоидных соединениях, особенно в не оказывающих вредных побочных действий, которые присущи таким традиционным опиатам, как морфин и петидин, существует постоянно.
Настоящее изобретение, в частности, касается соединений диарилметилпиперазиновой природы, отвечающих формуле:
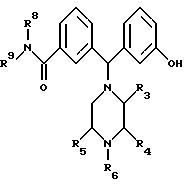
где один из радикалов R8 и R9 представляет собой фенил, возможно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, C1-C3алкокси, C1-C3алкил и трифторметил, а другой из радикалов R8 и R9 представляет собой водород, C1-C6алкил, C3-C6циклоалкил, C1-C6алкенил или C3-C6 алкинил; любые два из радикалов R3, R4 и R5 представляют собой метил, а другой радикал представляет собой водород; и R6 представляет собой водород, C1-C6алкил, C3-C6циклоалкил, C3-C6алкенил или C3-C6алкинил; а также его фармацевтически приемлемые простой эфир, сложный эфир, соль или его физиологически функциональное производное.
Термин "алкил" в рамках настоящего изобретения подразумевает широкое толкование, включающее как алкильные группы с неразветвленной цепью, так и алкильные группы, имеющие разветвленную структуру цепи.
Под "физиологически-функциональным производным" понимаются фармацевтически приемлемые соль, сложный или простой эфиры или соль этих эфиров соединения, отвечающего формуле (I), или другое соединение, которое будучи введенным реципиенту способно образовывать (прямо или косвенно) вышеуказанное соединение формулы (I) или его активный метаболит или остаток. Фенольные C1-C6алкиловые эфиры представляют собой подкласс физиологически-функциональных производных соединений формулы (I).
Энантиомерные формы соединений, предусмотренных изобретением, представляют собой индивидуальные энантиомеры соединений формулы (I) как в чистом виде, свободном от других энантиомеров, так и в форме, которая может содержать примеси (в смеси энантиомерных пар и/или в смеси многочисленных разновидностей энантиомеров).
Подклассом соединений, описываемых формулой (I), являются соединения, в которых R6, R8 или R9 представляет собой C1-C6алкил или C3 -C6циклоалкил.
Подклассом соединений, описываемых формулой (I), являются соединения, в которых R3 и R5 оба представляют собой метил, а R4 - водород.
Одним из предпочтительных
подклассов соединений, охватываемых настоящим изобретением, являются соединения, отвечающие формуле:

где R6, R8 и R9 такие же, как определены выше.
Следующим подклассом соединений, описываемых формулой (I), являются соединения, в которых R6 представляет собой C3-C6алкенил или C3-C6алкинил и предпочтительно аллильную группу.
Еще одним подклассом соединений, описываемых формулой (I), являются соединения, в которых либо R8, либо R9 является фенильной группой, которая может быть замещена одним из следующих заместителей: атом галогена, C1-C3алкокси и трифторметильная группа. Предпочтительно, чтобы атом галогена являлся атомом хлора или фтора и/или группа C1-C6 - метоксигруппой.
Еще одним подклассом соединений, описываемых формулой (I), являются соединения, в которых один из R8 и R9 представляет собой незамещенную фенильную группу.
Следующим подклассом соединений, описываемых формулой (I), являются соединения, в которых другой из R8 и R9 представляет собой атом водорода, C1-C6 алкил, C3-C6циклоалкил, или аллильную группу. Предпочтительно, чтобы C3-C6алкил представлял собой, например, метильную, этильную или пропильную группу (включая н-, изо- и цикло-пропильную группу).
С точки зрения специфичности и предпочтительности диарилметилпиперазиновых
производных, описываемых вышеуказанной формулой, соединение
NR8R9 может быть выбрано, например, из группы, состоящей из:
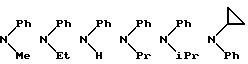
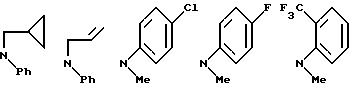
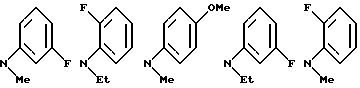
Примеры бензамидных соединений, отвечающих указанной выше общей формуле (I) и охватываемых изобретением, приведены ниже.
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-фторфенил)-N-метилбензамид;
± 3-( (α
R)-α
- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил-N-метил-N-фенилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-хлорфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-М-фенилбензамид;
(-) - 3 ( (αR)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-фенилбензамид;
3-(
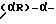
3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил-N-метил-N-(2,4,6-трихлорфенил)бензамид;
3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-(3-фторфенил)-N-метилбензамид;
± 3- ( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-фенил-N-пропилбензамид;
± 3-( (αR)-α- (((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- (((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-(2-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(4-фторфенил)бензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-аллил-N-фенилбензамид;
±-3-( (αR)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-(циклопропил)метил-N-фенилбензамид;
3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-изопропил-N-фенилбензамид;
3-( (αR)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-циклопропил-N-фенилбензамид;
3-( (αR)-α- ((2S, 5R) -4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-(3-фторфенил)-N-пропилбензамид;
3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(3-фторфенил)бензамид;
3-( (αR)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(2-фторфенил)бензамид;
3-( (αR)-α- (2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-метоксифенил)-N-пропилбензамид;
3-( (αR)-α- (2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-(4-метоксифенил)-N-пропилбензамид;
3-( (αR)-α- (2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-(4-метоксифенил)-N-бензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)-N-метил-N-фенилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- метоксибензил)-N-(3-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- метоксибензил)-N-этил-N-(4-фторфенил)-бензамид;
3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- (оксибензил)-N-(4-метоксифенил)-N-пропилбензамид;
3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- (N-(3-фторфенил)-N-метилкарбамоил)бензил)фенилмонофосфат; а также их фармацевтически приемлемые простой и сложный эфиры, соли или их физиологически функциональные производные.
Особенно предпочтительными соединениями из приведенного выше списка соединений, предусмотренных изобретением, являются:
± 3-( (αR)-α- ((2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-метил-N-фенилбензамид;
± 3-((αR)-α- (2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-фенилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-фенил-N-пропилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-метоксифенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(2-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(3-фторфенил)-N-метилбензамид;
± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(4-фторфенил)-бензамид; а также их фармацевтически приемлемые простой и сложный эфиры, соли или физиологически функциональные производные.
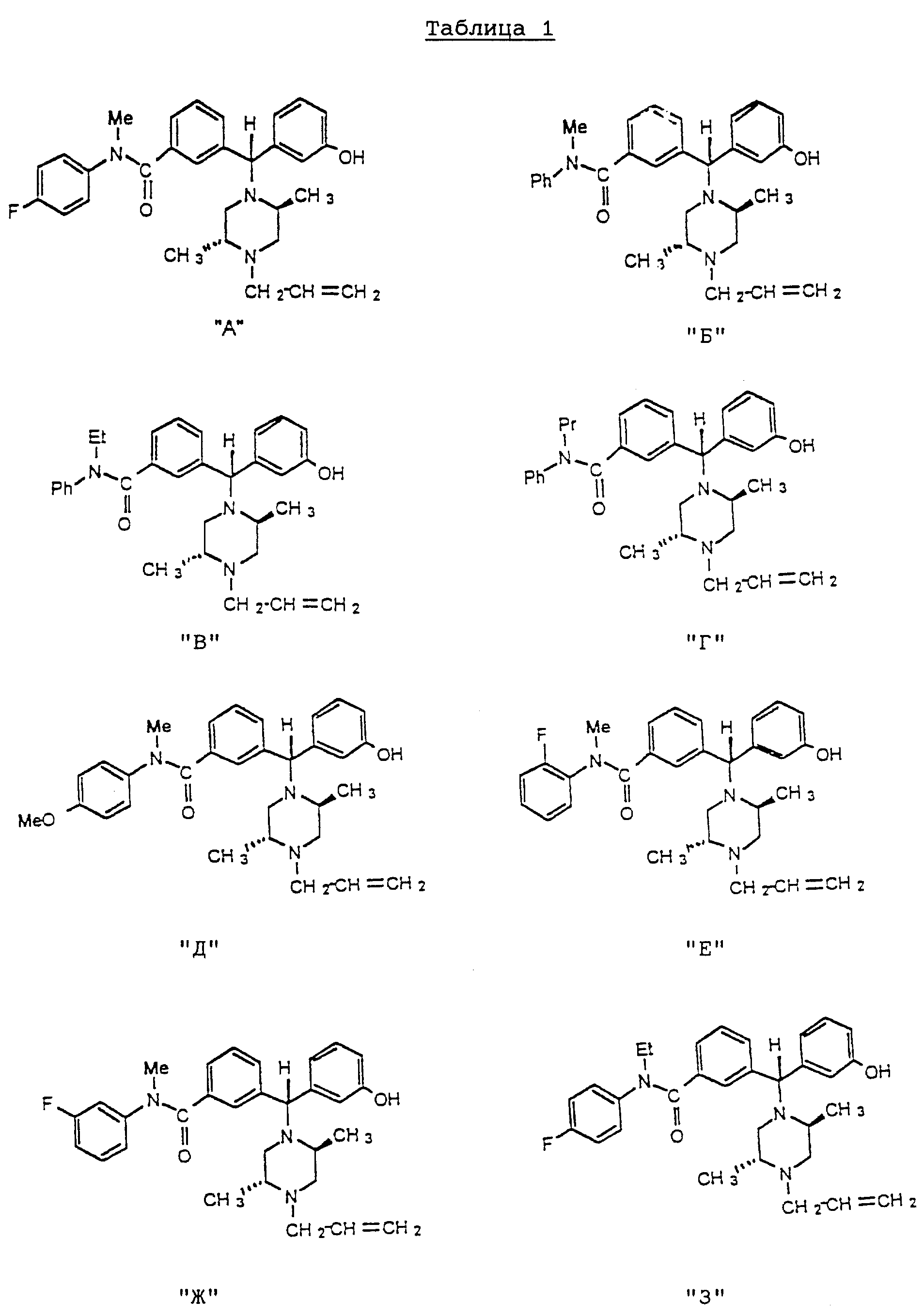
В таблице 1, приведенной ниже, показана химическая структура восьми вышепредставленных особо предпочтительных соединений по настоящему изобретению, обозначенных здесь как соединения "А", "Б", "В", "Г", "Д", "Е", "Ж" и "З", соответственно.
Соединения, описываемые вышеприведенной общей формулой (I), проявляют селективность в отношении связывания с рецептором(ами). В зависимости от структуры и стереоспецифичности соединений, описываемых частными случаями формулы (I), они оказываются способными связываться с тем или иным рецептором(ами) из числа дельта-рецепторов, мю-рецепторов, каппа-рецепторов, сигма-рецепторов и комбинациями подобных рецепторов.
Различные соединения, описываемые общей формулой (I), проявляют дельта-рецепторную агонистическую активность, включая опосредование аналгезии. Другие соединения, отвечающие этой же общей формуле, демонстрируют дельта-рецепторную антагонистическую активность, что описано ниже более полно. Еще одни соединения, предусмотренные общей формулой (I), показывают мю-рецепторную активность и в некоторых случаях - смешанную мю-рецепторную/дельта-рецепторную активность.
Примерами фармацевтически приемлемых сложных эфиров, описываемых настоящим изобретением, являются: 1) эфиры карбоновых кислот по гидроксильной группе соединений формулы (I), в которых некарбонильная часть карбоксильного участка эфирной группировки представлена следующими заместителями: неразветвленной или разветвленной алкильной (например, н-пропильной, т-бутильной, н-бутильной), алкоксиалкильной (например, метоксиметильной), арилалкильной (например, бензильной), арилоксиалкильной (например, феноксиметильной) и арильной (например, фенильной) группами; 2) эфиры аминокислот (например, L-валильная или L-изолейцильная группы); 3) эфиры дикарбоновых кислот (например, гемисукцинатная группа); 4) карбонатные эфиры (например, этоксикарбонильная группа); 5) карбаматные эфиры (например, диметиламинокарбонильная, (2-аминоэтил)аминокарбонильная группы); и 6) неорганические эфиры (например, моно-, ди- и трифосфаты).
Примерами фармацевтически приемлемых солей соединений формулы (I) и их физиологически функциональных производных являются соли, образованные из соответствующих оснований щелочных металлов (например, натрия, калия), щелочноземельных металлов (например, кальция, магния), аммония и NX4+ (где X представляет собой C1-C4алкильную группу). Фармацевтически приемлемые соли, образованные по аминогруппе, включают соли: органических карбоновых кислот, таких как уксусная, молочная, винная, яблочная, лактобионовая, фумаровая и янтарная кислоты; органических сульфоновых кислот, как например метансульфоновая, этансульфоновая, изэтионовая, бензолсульфоновая и п-толуолсульфоновая кислоты; и неорганических кислот, таких как соляная, бромистоводородная, серная, фосфорная и sulfamic кислоты. Фармацевтически приемлемые соли соединения, имеющего гидроксильную группу, состоят из аниона вышеуказанного соединения в сочетании с подходящим катионом, таким как Na+, NH4+ или NX4+ (где X представляет собой, например, С1-4алкильную группу).
Использование здесь при изложении настоящего изобретения термина "арил" подразумевает широкое толкование: как имеющее отношение к карбоциклическим, равно как и гетероциклическим ароматическим группам.
Для фармацевтического использования соли соединений формулы (I) должны быть фармацевтически приемлемыми, другими словами, это должны быть соли фармацевтически приемлемых кислот или оснований. Тем не менее соли кислот или оснований, не являющихся фармацевтически приемлемыми, могут также найти применение, например, при получении или очистке фармацевтически приемлемого соединения . Все соли как образованные из фармацевтически приемлемых кислоты и основания, так и из не являющихся ими, охватываются настоящим изобретением.
Соединения, предусмотренные настоящим изобретением, находят применение в качестве экзогенных соединений, взаимодействующих с рецепторами, другими словами, соединений, использующихся для связывания либо с отдельными рецепторами, таким как дельта-, мю-, сигма-, каппа-рецепторы, либо с двумя или более рецепторами такого типа. Взаимодействующее соединение может выступать в качестве агонист/антагонист-компонента пары конъюгата, что возможно использовать для анализа проводящей функции нейротрансмиттера в клеточных системах или системах дифференцированных тканей. В дополнение к использованию соединений по настоящему изобретению в рецепторном анализе, дифференциальном связывании и особых случаях для клеточного, гистологического мониторинга и мониторинга на уровне организма в целом, а также для произведения оценок эти соединения проявляют специфическую биологическую активность, что делает возможным их использование в качестве лекарственных средств в терапии различных физиологических и патологических состояний.
Соединения, предусмотренные настоящим изобретением, включают виды агонистов, полезные при лечении болей различной природы, диареи, депрессии, недержания мочи, психических расстройств, кашля, отека легкого, желудочно-кишечных расстройств, повреждения позвоночника и привыкания к чрезмерному употреблению лекарственных средств.
Соединения по настоящему изобретению также включают виды антагонистов, которые, как было сказано, полезны в исследованиях нейтротрансмиттеров, равно как и виды антагонистов, которые можно применять при лечении алкоголизма, заболеваний, вызванных передозировкой лекарственных средств опиатной природы или лекарственных средств, представляющих собой другую разновидность агонистов.
Кроме этого, когда деградация или дисфункция опиоидных рецепторов имеет место или обуславливает болезненное состояние, распространяющееся на ткань или отдельные клеточные локусы, в целях диагностики и визуализации находят применение изотопно меченные варианты опиоидных соединений, предусмотренных изобретением, например в диагностических методах, заключающихся в получении сканограмм мозга с помощью позитронной эмиссионнной томографии (ПЭТ).
Как было указано выше, опиоидные рецепторные сайты представляют собой локусы на клетках, узнающие и связывающие опиаты и опиоидные лекарственные средства, что в свою очередь может иметь (инициирование/блокирование) биохимические/физиологические последствия (трансдукция).
В случае опиоидных средств непептидной природы, рассмотренных настоящим изобретением, характер проявления зависимости между структурой и функцией в кругу различных соединений, отвечающих общей формуле (I), сильно различается, и даже такие неуловимые отличия, как изменения в стереохимии, могут приводить к различным трансдукционным эффектам. Вследствие этого формула (I) охватывает как разновидности агонистов, так и антагонистов.
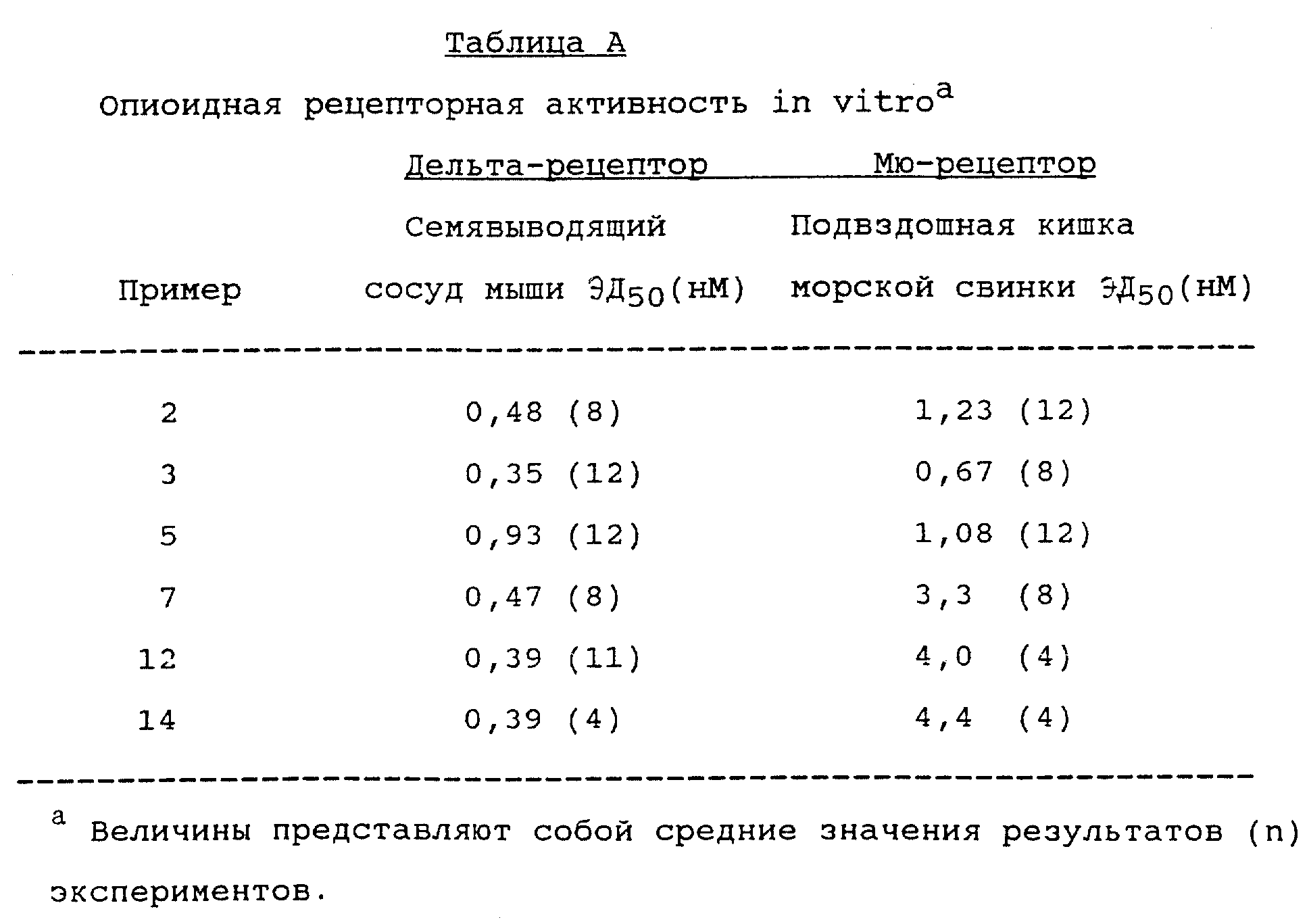
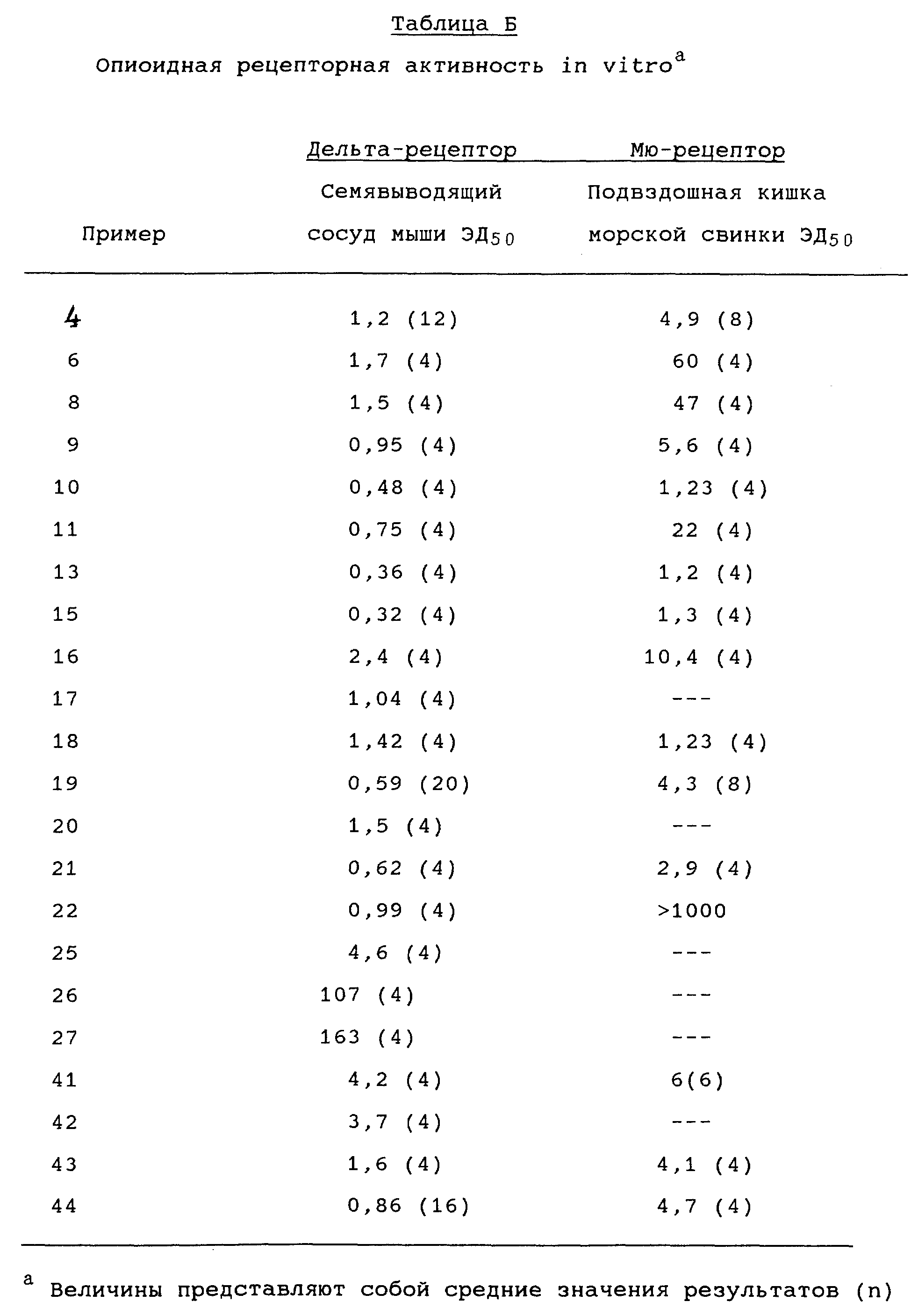
Что касается дельта-рецепторных агонистов, то их активность в основном устанавливают и измеряют, используя для анализа семявыводящие сосуды мыши, стимулированные электрическим током.
Кроме этого, в результате практического применения соединений по настоящему изобретению были получены строгие доказательства существования в мозгу подтипа дельта-рецепторов, отличающихся от дельта-рецепторов семявыводящих сосудов мыши.
Вследствие этого для предсказания активности в качестве агонистов и антагонистов соединений, предусмотренных изобретением, могут быть использованы и другие методы анализа связывания с рецепторами или методиками скрининга, например тесты на аналгезию.
Активность мю-рецепторных агонистов в основном устанавливают и измеряют на стимулированной электрическим током подвздошной кишке морской свинки.
Соединения А, Б, В, Г, Д, Е, Ж и З представляют собой разновидности высокоселективных опиоидных рецепторных лигандов. Все они являются эффективными средствами для опосредования аналгезии. В общем, спектр применимости соединений диарилметилпиперазиновой природы, предусмотренных изобретением, в качестве обезболивающих препаратов может быть без труда выяснен путем простых тестов по связыванию с рецепторами без чрезмерной экспериментальной работы. С этой точки зрения и единственно в качестве иллюстрации, те соединения диарилметилпиперазиновой природы, предусмотренные настоящим изобретением, которые преимущественно являются агонистами мю-рецепторов, могут быть использованы, например для опосредования аналгезии в хирургии. Предусмотренные изобретением соединения диарилметилпиперазиновой природы, являющиеся преимущественно агонистами дельта-рецепторов, можно использовать для эпидурального обезболивания, а соединения, представляющие собой смешанные мю-/дельта-опиоидные агонисты, т. е. соединения А, Б, В, Г, Д, Е, Ж и З - например для хирургического и/или после операционного обезболивания.
Смешанный мю-/дельта-рецепторный характер соединений, охватываемых настоящим изобретением, обуславливает их существенное преимущество над уже известными различными мю-рецепторными соединениями, применяемыми в настоящее время для обезболивания.
Подавляющее большинство используемых в настоящее время высокоэффективных обезболивающих средств, включая морфин, фентанил, меперидин, суфентанил и кодеин, являются соединениями, связывающимися с мю-рецепторами. Хорошо известно, что эти соединения, эффективно опосредуя аналгезию, вызывают побочные эффекты, такие как потеря координации, ослабление остроты восприятия, мышечная ригидность и угнетение дыхания, и могут являться причиной тошноты, рвоты, лихорадки и обморочного состояния. Подобные побочные эффекты обычно отсутствуют или, по меньшей мере, не столь сильны при использовании для опосредования аналгезии соединений, связывающихся с дельта-рецепторами. Соответственно, применение смешанного вида мю-/дельта-рецепторных соединений по настоящему изобретению может ослаблять или даже убирать побочные явления, в норме сопутствующие действию мю-рецепторсвязывающихся соединений.
Для использования в фармацевтике и диагностике соединений по изобретению желательно, чтобы они были получены в существенно чистом в плане энантиомерного состава виде; подобная энантиомерная чистота (энантиомерный избыток, ЭИ) должна составлять по меньшей мере 90%, предпочтительно - 95%, еще более предпочтительно - 98% и наиболее предпочтительное значение ЭИ составляет по меньшей мере 99%. Величина энантиомерного избытка является количественной мерой избытка основного изомера над минорным, присутствующим вместе с ним, и может быть без труда измерена подходящими для этой цели методами, хорошо известными специалистам, такими например, как высокоэффективная жидкостная хроматография (ВЖХ), газовая хроматография (ГХ), ядерный магнитный резонанс (ЯМР) с использованием реагентов, вызывающих хиральный химический сдвиг, и т.д.
Соединения А, Б, В, Г, Д, Е, Ж и З являются энантиомерно чистыми обезболивающими агентами, проявляющими агонизм в отношении как мю-, так и дельта-рецепторов. В тестах на грызунах, например, эти соединения демонстрируют аналгезию, сравнимую с обезболиванием, вызываемым морфином, но в гораздо меньшей степени усиливают мышечную ригидность и угнетение дыхания. Кроме этого, тесты на грызунах показали, что эти соединения не вызывают судорог, как это бывает в случаях структурно чистых дельта-агонистов.
Несмотря на то, что на первый взгляд может показаться, что все соединения по изобретению, являющиеся агонистами дельта-рецепторов, будут проявлять похожие свойства in vivo с активностью, соответствующей измеряемой в опытах на семявыводящих сосудах мыши, это не обязательно так.
Соединения диарилметилпиперазиновой природы, предусмотренные настоящим изобретением, включают соединения, которые могут служить важным инструментом при проведении рецепторного анализа (на мозге крысы), среди них соединения, предпочтительно активные в отношении одного или другого дельта-рецепторных подтипов, и соединения, обладающие мю-рецепторной или смешанной мю-рецепторной/дельта-рецепторной активностью.
Результаты анализа связывания и теста на обезболивание показывают, что соединения по настоящему изобретению по-разному опосредуют аналгезию в отношении разнообразных раздражителей и физиологических расстройств. Это, в свою очередь, служит доказательством высокого уровня сложности действия нейротрансмиттеров и ответов, вызываемых раздражителями, связанными с различными опиодными рецепторами, включая мю-рецепторы, дельта-рецепторы и рецепторы дельта-подтипов.
Ряд соединений по настоящему изобретению, описываемых формулой (I), или их предшественников (которые также во многих отношениях представляют собой новые соединения и таким образом рассматриваются в качестве предмета изобретения) обнаруживают дополнительно к опиоидной активности и другие виды биологической активности, в том числе биологическую активность, заключающуюся в связывании с сигма-рецепторами и резистентности к множественным лекарственным средствам.
Как следует из приведенного выше обсуждения, соединения по настоящему изобретению находят широкое применение для лечения большого числа разнообразных физиологических состояний и расстройств. Изобретение, таким образом, предполагает использование подобных соединений в производстве лекарственных средств для лечения или профилактики этих физиологических состояний и расстройств. В дополнение к этим уже упомянутым возможным терапевтическим применениям другие возможности применения соединений, предусмотренных настоящим изобретением, включают лечение бронхиальных расстройств, таких как астма, эмфизема и удушье.
Кроме этого, в связи с различными расстройствами ЦНС, такими как компульсивное поведение, депрессия, психоз и т.д., были обнаружены эндогенные опиоиды, такие как энкефалины и эндорфины, и их нейрологические системы, а разновидности агонистов или антагонистов, охватываемые формулой (I) настоящего изобретения находят применение в борьбе с такими заболеваниями.
Различные разновидности как агонистов, так и антагонистов среди соединений формулы (I) также находят применение в лечении состояний, связанных со злоупотреблением/привыканием к лекарствам (опиоидной/наркотической природы), и таким образом могут применяться в качестве замены метадона или других традиционных опиатных средств в реабилитационных программах в силу того, что традиционные лекарственные средства оказывают побочные эффекты или обладают другими недостатками, служащими в качестве противопоказаний или ограничивающими их использование.
Что касается лечения привыкания к лекарствам с помощью широкого круга эффективных соединений, охватываемых настоящим изобретением, отмечается, что метадон представляет собой мю-рецепторный опиат с действием, аналогичным действию морфина, т.е. применение метадона влечет за собой привыкание и злоупотребление им. Метадон используют в качестве агента для "терапии поддержания" наркоманов, позволяя подобным индивидуумам сохранять функциональное состояние в период устранения их привыкания к наркотикам безопасным и некриминальным способом. В этом отношении соединения, предусмотренные изобретением, могут найти применение взамен или совместно с традиционно используемыми средствами для лечения состояний привыкания к лекарствам, такими как naltrexone, метадон, clonidine и т.д.
Некоторые соединения, охватываемые настоящим изобретением, как говорилось выше, применяются в качестве местных обезболивающих средств, как например при аналгезии на позвоночнике, а также соединения по изобретению могут найти применение в качестве средств для понижения аппетита и подобных им средств.
Соединения, предусмотренные настоящим изобретением, включают различные соединения, являющиеся дельта-опиоидными агонистами рецепторов дельта-подтипа семявыводящих сосудов мыши, равно как и соединения, являющиеся антагонистами подобного подтипа дельта-рецепторов. Кроме этого, они включают соединения, представляющие собой агонистов и антагонистов дельта-рецепторов мозга, которые, как было показано экспериментально, относятся к подтипу дельта-рецепторов, отличающихся от таковых из семявыводящих сосудов мыши. Значительное число соединений, описываемых вышеуказанной формулой (I) настоящего изобретения, обладает как агонистической, так и антагонистической активностью по отношению к обоим подтипам дельта-рецепторов. Ряд этих соединений обладает высокой активностью по отношению к мю-опиоидным рецепторам, являясь либо чисто мю-рецепторсвязывающимися соединениями, либо смешанными мю-/дельта-рецепторсвязывающимися, а другие соединения, охватываемые настоящим изобретением, обладают значительной аффинностью по отношению к сигма-рецепторам.
В тестах in vitro на активность в качестве агониста/антагониста, таких как тесты на аффинность к рецептору, и в тестах на ингибирование сокращения стимулированной электрическим током мышцы, соединения по настоящему изобретению являются эффективными в зависимости от конкретного соединения в интервале от наномолярных до микромолярных концентраций.
Соединения по настоящему изобретению обладают фармацевтической активностью, включающей, inter alia, обезболивающую активность, и применяться на млекопитающих, например людях, в условиях, когда необходимо обезболивание.
Способ получения обезболивающего ответа в нуждающихся в этом субъектах, заключается во введении животному необходимого для обезболивания количества соединения формулы (I).
Кроме этого, различные соединения по настоящему изобретению, находящие терапевтическое применение, могут быть успешно использованы для лечения таких состояний, как наркомания и хронический алкоголизм, чрезмерное употребление лекарств и алкоголя; психические, эмоциональные и когнитивные расстройства; кашель; отек легких; желудочно-кишечные заболевания. Соответственно, настоящее изобретение предполагает способ лечения животных субъектов в таком состоянии(ях) и нуждающихся в подобном лечении, заключающийся во введении таким животным необходимого количества соединения по настоящему изобретению, терапевтически эффективного для вышеуказанного состояния.
Субъекты, при лечении которых могут применяться способы, предусмотренные настоящим изобретением, включают как человека, так и животных (например, собак, кошек, коров, лошадей), а также птиц, и преимущественно подразумевают млекопитающих, и особо преимущественно, людей.
В зависимости от специфики лечимого состояния соединения формулы (I) могут вводиться в любой подходящей терапевтически эффективной и безопасной дозе, которая без чрезмерного экспериментирования может быть определена специалистом.
В целом, в то время как эффективная терапевтическая доза соединений, предусмотренных изобретением, может широко варьироваться в зависимости от специфики применения, состояния или характера заболевания, что легко определяется специалистами в данной области, подходящие терапевтические дозы соединений формулы (I) для каждого вида описанных здесь составов и для достижения терапевтической пользы в лечении любого из описанных здесь состояний будут находиться в интервале от 1 микрограмма (мкг) до 100 миллиграммов (мг) на килограмм живого веса реципиента в день, предпочтительно в интервале от 5 мкг до 75 мг и наиболее предпочтительно - в интервале от 10 мкг до 50 мг на килограмм веса в день. Необходимая доза разделяется преимущественно на две, три, четыре, пять, шесть и более поддоз, вводимых через соответствующие интервалы времени в течение дня. Эти поддозы могут вводится в виде отдельных лекарственных форм, например содержащих от 10 мкг до 1000 мг, предпочтительно от 50 мкг до 500 мг, более предпочтительно от 50 мкг до 250 мг и наиболее всего предпочтительно от 50 мкг до 10 мг активного ингредиента в лекарственной форме. В альтернативном случае, если того требует состояние реципиента, дозы могут вводиться путем непрерывной инфузии.
Способ введения и вид лекарственных форм без сомнения будут оказывать влияние на определение терапевтического количества соединений, необходимого и эффективного для данного терапевтического использования.
Например, орально вводимые дозы по меньшей мере вдвое, а именно в 2-10 раз, больше уровня доз, применяемых при парентеральном способе введения, для одного и того же активного ингредиента. Порядок доз соединений по изобретению, обладающих сродством к мю-рецепторам, при введении оральным способом с целью обезболивания составляет от 5 до 200 мг в расчете на 70 кг веса в день. Уровень доз при использовании капсул в общем составляет примерно 10% от уровня парентерального введения. Для лекарственных форм в виде таблеток типичный порядок уровня дозы активного ингредиента, достаточный для обезболивания, составляет 10-100 мг на таблетку.
Соединения формулы (I) могут применяться per se, равно как и в форме их фармацевтически приемлемых простых и сложных эфиров, солей и других физиологически функциональных производных.
Настоящее изобретение также рассматривает технологии приготовления лекарственных средств как для ветеринарии, так и для медицины, которые включают в качестве активного агента одно или более соединение(й) по изобретению, и фармацевтически приемлемый носитель. Изобретение, кроме того, предусматривает использование соединения по изобретению, как например соединения, охватываемого вышеобсуждаемыми формулами (I) и (II), в производстве лекарственных средств для лечения и профилактики состояний и расстройств, широко в нем описанных.
В подобных технологиях изготовления лекарственных средств активный агент используется преимущественно совместно с одним или более фармацевтически приемлемым носителем(ями), а также может использоваться с другими терапевтическими ингредиентами. Носитель(и) должен быть фармацевтически приемлемым в смысле его совместимости с другими составляющими лекарственного средства и безвредности для реципиента. Активный агент вводится, как описано выше, в количествах, обеспечивающих эффективное достижение желаемого фармакологического эффекта и необходимую дневную дозу.
Лекарственные средства включают средства, пригодные как для парентерального, так и для не парентерального применения, и специфическими способами их введения являются оральное, ректальное, местное, назальное, офтальмийное, подкожное, внутримышечное, введение через кожу и с помощью капсул, внутрисуставное, внутриартериальное, подарахноидное, бронхиальное, лимфатическое и внутриматочное введение. Предпочтительными являются лекарственные средства, пригодные для парентерального введения.
В случаях, когда активный агент используется в составе жидкого лекарственного средства, такое средство удобно вводить парентерально. Когда же лекарственное средство представляет собой суспензию или порошок в смеси с биосовместимым носителем, для его введения удобно использовать оральный, ректальный или бронхиальный способы.
При использовании непосредственно активного агента в форме порошкообразного твердого материала, его удобно вводить орально. В альтернативном случае он может быть введен бронхиально путем распыления порошка в газе-носителе для образования газовой дисперсии, которая при использовании специального оборудования для ингаляции (подходящего распыляющего устройства) вдыхается пациентом.
В некоторых случаях может быть полезно использовать активный агент в "векторной" форме, например, инкапсулируя его в липосому или другую подходящую для инкапсуляции среду или фиксируя активный агент с помощью, например, ковалентного связывания, хелатирования или ассоциативной координации на подходящих биомолекулах, таких как белки, липопротеины, гликопротеины и полисахариды.
Подходящей формой лекарственных средств, включающих активный агент, предусмотренный настоящим изобретением, являются единичные дозовые лекарственные формы, которые могут быть приготовлены хорошо известными фармацевтическими способами. Такие способы обычно включают этап объединения активного соединения(ий) с носителем, составляющим один или более дополнительный ингредиент. В типичном случае лекарственное средство готовится путем однообразного и однородного объединения активного соединения(ий) с жидким носителем, тонко измельченным твердым носителем или обоими типами этих носителей с последующим, при необходимости, приданием продукту формы желаемого лекарственного средства.
Лекарственные средства по настоящему изобретению, пригодные для орального введения, могут иметь вид отдельных единичных лекарственных средств, таких как желатиновые и крахмальные капсулы, таблетки или пастилки, каждое из которых содержит определенное количество активного ингредиента в виде порошка или гранул; либо иметь вид суспензий в воде или неводной жидкости в форме сиропа, эликсира, эмульсии, либо иметь вид жидкого лекарства.
Таблетка может быть приготовлена прессованием либо формованием совместно с одним или более дополнительными ингредиентами. Штампованные таблетки можно изготавливать, используя подходящее оборудование, с подачей активного соединения в форме порошка или гранул, которое может быть смешано со связующим, измельчителем, смазывающим, инертным разбавителем, поверхностно-активным агентом или выводящим агентом. Формованные таблетки, состоящие из смеси порошкообразного активного соединения с подходящим носителем, могут быть изготовлены формованием на подходящем оборудовании.
Сироп можно получить добавлением активного соединения к концентрированному водному раствору сахара, например сахарозы, в который может быть добавлен также и дополнительный ингредиент(ы).
Такой дополнительный ингредиент(ы) может включать ароматизаторы, подходящий консервант, агенты, замедляющие кристаллизацию сахара, и агенты, увеличивающие растворимость любого другого ингредиента, такие как полигидроксиспирты, например глицерин или сорбит.
Лекарственные средства для парентерального введения обычно представляют собой стерильный водный препарат активного соединения, преимущественно изотоничный с кровью реципиента (например, в физиологическом растворе). Подобные лекарственные средства могут включать суспендирующие и загущающие агенты, липосомы или другие системы микрочастиц, обеспечивающие доставку соединения к компонентам крови либо к одному или более органам. Лекарственные средства могут быть приготовлены в виде одноразовой и многоразовой дозовых форм.
Лекарственные средства для назального применения включают очищенные водные растворы активных соединений с консервирующими агентами и агентами, обеспечивающими изотоничность. Значение pH и изотоническое состояние таких лекарственных средств предпочтительно следует установить равными с таковыми для слизистой оболочки носовой полости.
Лекарственные средства для ректального введения могут представлять собой суппозитории с использованием подходящего носителя, такого как кокосовое масло, насыщенные жиры или насыщенные жирные карбоновые кислоты.
Глазные лекарственные средства могут быть приготовлены тем же методом, что и назальный раствор, за исключением того, что pH и изотонический фактор в этом случае должны быть приближены к таковым в глазах.
Лекарственные средства для местного применения включают активное соединение, растворенное или суспендированное в одной или большем числе сред, таких как минеральное масло, нефть, полигидроксиспирты, либо в других основах, используемых для приготовления лекарственных средств местного применения.
Лекарственные средства для введения через кожу можно приготовить путем включения активного агента в тиксотропный или гелеобразный носитель, такой как носитель на основе целлюлозы, например метилцеллюлозы или оксиэтилцеллюлозы, с последующим помещением полученного лекарственного средства в приспособление, обеспечивающее надежный контакт с кожей при его ношении.
В дополнение к вышеупомянутым ингредиентам лекарственные средства, предусмотренные настоящим изобретением, могут, кроме того, включать один или более добавочный ингредиент(ы) из числа растворителей, буферных растворов, ароматизирующих агентов, связующих, измельчителей, поверхностно-активных агентов, загустителей, смазывающих веществ, консервантов (включая антиоксиданты) и тому подобное.
Настоящее изобретение также предполагает способ
получения определенного выше соединения формулы (I), а также
его фармацевтически приемлемых простого и сложного эфиров, соли или другого
физиологически функционального производного, при этом
указанный способ включает методику синтеза, выбранную из
нижеследующих методик (А), (Б) и (В):
(А) алкилирование пиперазина формулы (IV)
алкилирующим агентом формулы (III),
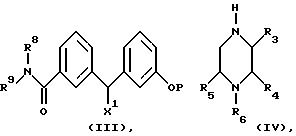
где заместители от R3 до R6 и R8 и R9 определены так же как в любом из предыдущих мест описания, P представляет собой атом водорода или гидроксилзащитную группу, а X1 является уходящей группой; и, когда заместитель R6 является водородом, возможно алкилирование полученного соединения формулы (I) алкилирующим агентом формулы R6-X1, где R6 является C1-C6алкилом, C3-C6циклоалкилом, C3-C6алкенилом или C3-C6алкинилом; а X1 представляет собой уходящую группу, или возможно алкилирование полученного соединения формулы (I) путем восстановительного аминирования C1-C6альдегидом в присутствии восстанавливающего агента;
(Б) взаимодействие соединения формулы (V),
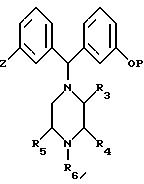
где заместители от R3 до R6 и P определены так же, как и ранее, и подходящими в качестве заместителя Z могут быть атомы брома, иода или трифторметилсульфонильная группа, с
(а) в случае, когда Z является атомом брома или иода; алкилом металла или подходящим реакционноспособным металлом, в результате чего может происходить замещение на переходный металл в образующемся содержащем металл соединении с образованием другого металлсодержащего соединения; взаимодействие полученного металлсодержащего соединения с двуокисью углерода с превращением образовавшейся карбоновой кислоты в соответствующий хлорид, ангидрид или сложный эфир и взаимодействие полученного хлорида, ангидрида или эфира с амином формулы HNR8R9, где R8 и R9 определены как и ранее, или взаимодействие полученного металлсодержащего соединения с аминокарбонилхлоридом формулы ClCONR8R9, где R8 и R9 определены как и ранее; или
(б) в случае, когда Z является атомом брома, иода или трифторметилсульфонильной группой; цианирующим реагентом с гидролизом образующегося нитрила щелочью или водным раствором минеральной кислоты, превращением образующейся карбоновой кислоты в соответствующий хлорид, ангидрид или эфир и взаимодействием образующегося хлорида, ангидрида или эфира с амином формулы HNR8R9, где R8 и R9 определены как и ранее; или
(в) в случае, когда Z является атомом брома, иода или трифторметилсульфонильной группой; избытком амина формулы HNR8R9, где R8 и R9 определены как и ранее, и окисью углерода в присутствии катализатора в виде переходного металла с образованием соединения формулы (I), где R8 и R9 определены как и ранее; или
(В) взаимодействие соединения формулы (VI) с фенилметаллосодержащим соединением формулы (VII):
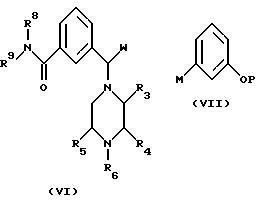
где заместители от R3 до R6 и R8 и R9 определены как и ранее, P представляет собой атом водорода или гидроксилзащитную группу, V является атомом металла, a W - бензотиазолил- или трихлорти- таноксигруппу; (Katritzky A.R., Yannakopoulou К., Lue P., Rasala D., Urogdi L. J. Chem. Soc. , Perkin Trans. 1, 1139, (1989); Seebach D., Betcart С., Schiess M. Helv. Chim. Acta, 67, 1593, (1984)) и, когда P является гидроксилзащитной группой, снятие защиты с гидроксильной группы; при необходимости превращение полученного соединения формулы (I) в его фармацевтически приемлемые простой и сложный эфиры или соль или его физиологически функциональное производное.
Методика А
Реакция
между алкилирующим агентом формулы (III)
и пиперазином формулы (IV)
может быть осуществлена в таком растворителе как толуол или ацетонитрил.
Алкилирующие агенты формулы R6 -X1 коммерчески доступны или могут быть получены с использованием опубликованных в литературе методик. В качестве альтернативы алкилированию алкилирующим агентом формулы R6-X1 может быть применен метод восстановительного аминирования, в котором подходящий, коммерчески доступный C1-C6альдегид восстанавливают восстанавливающим агентом, таким как цианборгидрид натрия в растворителях типа спиртов или эфиров.
Методика Б
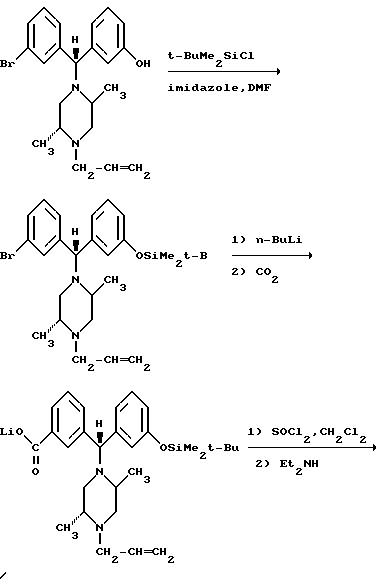
(а) Соединение формулы (I) может быть получено из соединения формулы (V), в котором Z является атомом
брома или иода и P
- гидроксилзащитной группой,
такой как трет-бутилдиметилсилил, путем низкотемпературного (например, от -60oC до -78oC) обмена реактивного галогена на
органометаллический реагент,
такой как н-бутиллитий,
или на активированную форму металла, такого как литий или магний, приводящего к образованию промежуточного металлсодержащего соединения с
последующей реакцией с диоксидом
углерода, результатом
которой является образование карбоновой кислоты в безводном растворителе, таком как тетрагидрофуран, в атмосфере инертного газа (например,
азота). Карбоновая кислота может быть
после этого
превращена в карбоксамид формулы (I) методами, описанными ниже.
Альтернативно промежуточное металлсодержащее соединение, образованное из соединения формулы (V), можно обработать подходящим карбамоилхлоридом (ClCONR8R9) для получения соединения формулы (I).
(б) Соединение формулы (I) также может быть получено из соединения формулы (V), в котором Z является атомом брома, йода или triflate (трифторметилсульфонильной) группой путем обработки цианирующим реагентом, таким как цианид меди, в подходящем растворителе, таком как диметилформамид или N-метилпирролидон, приводящей к получению соответствующего соединения формулы (V), в котором Z представляет собой нитрильную группу, которая далее может быть гидролизована щелочью или водным раствором минеральной кислоты до соответствующей карбоновой кислоты. Карбоновая кислота может быть после этого превращена в соединение формулы (I) различными хорошо известными специалистам способами, такими как образование хлорида (например, с помощью тианилхлорида или оксалилхлорида), или образование смешанного ангидрида (например, с помощью изобутилхлорформиата), или образование активированного эфира с помощью традиционных реагентов пептидного синтеза (например, дициклогексилкарбодиимида или гексафторфосфата бензотриазол-1-илокси-трис(диметиламино)фосфония), причем любое из этих промежуточных соединений может быть превращено в желаемый карбоксамид формулы (I) с помощью реакции с соответствующим амином (HNR8R9) в подходящем растворителе, таком как дихлорметан или диметилформамид.
(в) Соединение формулы (I) также может быть получено из соединения формулы (V), в котором Z является атомом брома, йода или triflate группой путем обработки при наличии катализатора переходного металла, такого как тетракис(трифенилфосфин)палладий, избытком амина или оксида углерода в таком растворителе, как тетрагидрофуран или ацетонитрил.
Методика В
Соединение формулы (VI) может быть получено в качестве реакционноспособного промежуточного соединения
сочетанием альдегида
формулы (VIII) с пиперазином формулы (IV),
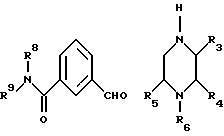
в которых заместители от R3 до R6, а также R8 и R9 определены как и ранее, в присутствии тетрахлорида титана или бензотриазолила в подходящем растворителе, таком как толуол или дихлорметан, или для промежуточного соединения формулы (VI), в котором W представляет собой бензотриазольную группу, при этом, если необходимо, активное промежуточное соединение может быть выделено кристаллизацией или другими подходящими способами.
Соединение формулы (I) может быть получено в виде одной энантиомерной формы путем классического разделения с помощью энантиомерно чистой кислоты, такой как миндальная, или путем образования легко разделяемых диастереомеров с помощью энантиомерно чистого дериватизирующего агента, или путем хроматографического разделения, или с помощью ферментативного разделения соединения формулы (I) либо его подходящего производного, или путем получения соединения формулы (I) из энантиомерно чистых предшественников, которые сами могут быть получены в виде единичной энантиомерной формы аналогичными способами.
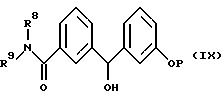
Соединения формулы (III) могут быть получены из подходящих спиртов формулы (IX), в которых фенол защищен подходящей защитной группой P такими методами, как галогенирование с помощью тионилхлорида или тетрабромида трифенилфосфина/углерода или реакция с металлсульфонилхлоридом или толуолсульфонилхлоридом в таком растворителе, как дихлорметан.
Пиперазины формулы (IV) коммерчески доступны или могут быть получены с использованием опубликованных в литературе методик или их модификаций, в которых R6 изменен соответствующим алкилированием с помощью агента R6-X1 .
Соединения формулы (V) могут быть получены алкилированием пиперазина формулы (IV) алкилирующим агентом формулы (X) так же, как было описано выше для случая алкилирования пиперазина. Алкилирующие агенты формулы (X) аналогичным образом получают из спиртов формулы (XI) методами, сходными с описанными выше для соединений формулы (III).
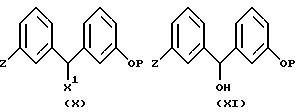
Спирты формулы (IX) или (XI) можно получить добавлением при низкой температуре (например, от -60oC до -78oC) замещенных арилметаллических соединений, приготовленных из соединений формулы (XII), в которых Z представляет собой реакционноспособный атом галогена (например, йода или брома), к замещенным бензальдегидам формулы (XIII), используя вышеописанные методы.
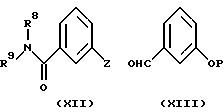
И наоборот, соединения формулы (IX) или (XI) могут также быть образованы в схожей реакции упомянутых ранее замещенных фенилметаллорганических соединений (VII) с бензальдегидами формулы (VIII).
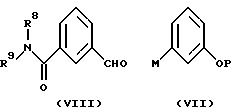
Соединения (VII), (VIII), (XII) и (XIII) и их соответствующие замещенные производные можно получить из коммерчески доступных материалов с помощью стандартных, описанных в литературе методик.
Соединение формулы (I) может быть превращено в фармацевтически приемлемый эфир в реакции с подходящим агентом, например кислым галогенидом или ангидридом. Соединение формулы (I), в том числе и его эфиры, может быть превращено общепринятыми способами в его фармацевтически приемлемые соли, например путем обработки подходящей кислотой. Эфир или соль соединения формулы (I) можно превратить в исходное соединение, например, посредством гидролиза. Фенольные эфиры соединения формулы (I), в которых P представляет собой C1-C6алкильную группу, могут быть приготовлены таким же образом, как было описано здесь ранее.
На основании как вышеизложенных, так и общих соображений, касающихся путей синтеза, должно быть понятно, что для получения диарилметилпиперазиновых соединений по настоящему изобретению могут быть полезны разнообразные методы синтеза, что должно быть очевидным даже для специалистов средней квалификации. Иллюстративные методы синтеза соединений, относящихся к обширной области соединений, охватываемых настоящим изобретением, изложены ниже в виде примеров; становится очевидным, что соединения, предусмотренные изобретением, могут быть произведены с использованием других, отличных методов и путей синтеза, известных специалистам, и что приведенные ниже иллюстративные методы синтеза не должны быть поэтому истолкованы ограничивающе в отношении сферы изобретения. Кроме этого, должно быть понятно, что новые типы соединений по настоящему изобретению охватывают различные новые типы промежуточных соединений, предшественников, про-лекарств, аналогов и производных соединений, отнесенных здесь к области изобретения.
В тех случая, когда в результате применения методик синтеза соединений, предусмотренных изобретением, в качестве промежуточных продуктов реакции получаются рацемические смеси, они могут быть разделены с использованием хорошо известных и принятых в этой области методов и средств, таких например, как образование диастереомерных солей с энантиомерно чистыми карбоновыми кислотами, хроматографическое разделение стереоизомеров, ферментативное разделение и других подходящих общепринятых методов.
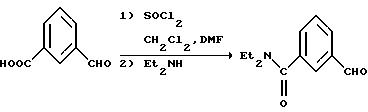
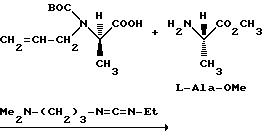
Ниже изложены иллюстративные схемы синтеза рацемического (± )-3-( (αR*)-α- ((2S*,5R*)-4-аллил-2,5-диметил-1- пиперазинил)-3-оксибензил)-N,N-диэтилбензамида, далее обозначенного как Соединение (± )-1, которое может быть получено в виде составляющих его энантиомеров с применением классического разделения или стереонаправленных методов синтеза конечных продуктов либо подходящих промежуточных соединений. Подобные методы дополнительно проиллюстрированы в случае получения энантиомера (+)-3-(
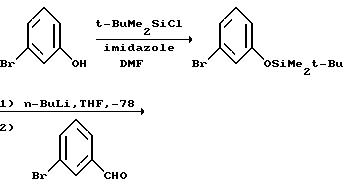
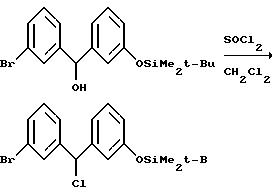
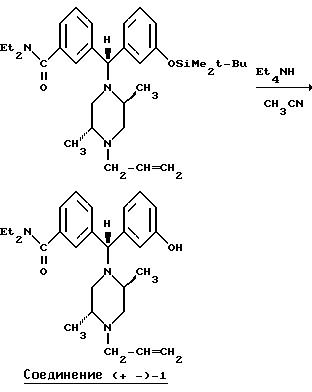
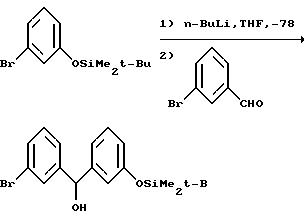
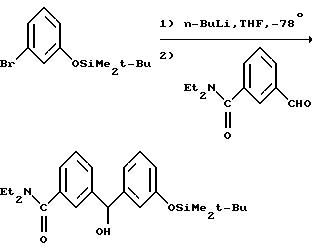
Что касается предшествующей схемы синтеза, то исходный бензгидриловый спирт может быть получен из 3-(т-бутилдиметилсилилокси)бромбензола по следующей схеме:
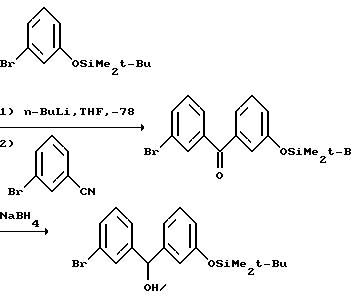
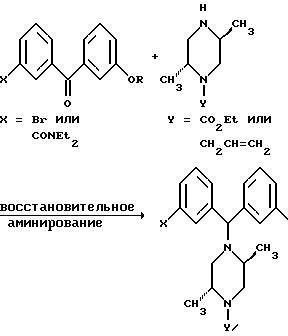
Промежуточное соединение можно также получить через бензофенон, который в свою очередь может быть синтезирован добавлением органометаллического соединения к 4-бромбензонитрилу:
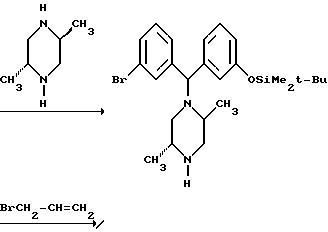
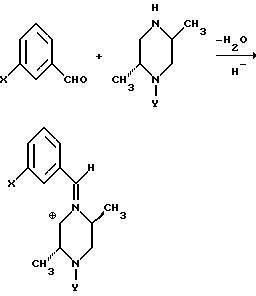
Другие способы получения промежуточных соединений включают конденсацию подходящего замещенного пиперазина с карбонильным соединением. Конденсация с бензальдегидом может дать иминную соль, которая в присутствии ариллития может образовывать бензгидрилпиперазиновые соединения, в которых X = CONEt2, Y = CH2CH = CH2 или X = Br, Y = CH2CH = CH2, в виде смеси их диастереомеров или защищенных предшественников этих соединений.
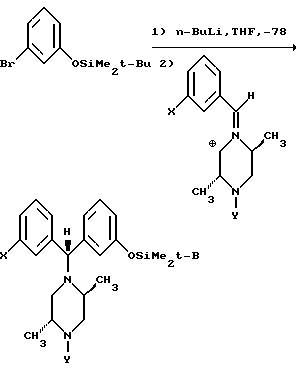
Похожим образом восстановительное аминирование подходящего бензофенона соответствующим пиперазином может привести к прямому получению желаемых соединений.
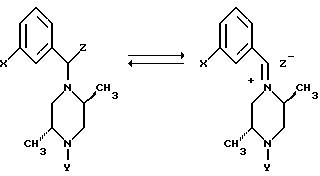
Аналогично "замаскированное иминное" соединение, в котором Z
является удобной уходящей группой (например, бензотриазольной группой или трихлоридом оксотитана), может быть обработано
арилметаллическими соединениями (например, ариллитием или бромидом арилмагния),

после чего бензилпиперазин может диссоциировать с образованием требуемого иминного иона in situ.
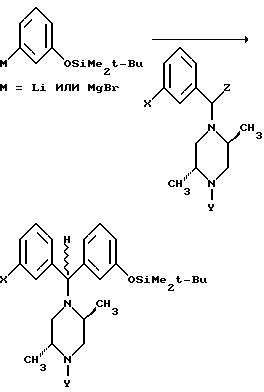
Сходным образом восстановительное аминирование подходящего бензофена соответствующим амперазином может непосредственно привести к получению желаемых соединений.
Соединение (±)-1 может также быть синтезирован альтернативным путем синтеза, приведенным ниже.
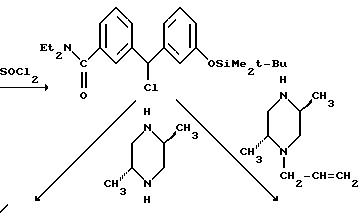
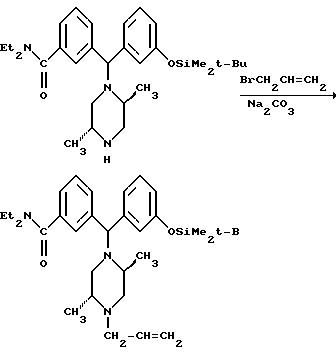
Транс-1-аллил-2,5-диметилпиперазиновый реагент, используемый в вышеуказанной схеме синтеза, может быть образован соответственно в результате следующего процесса синтеза.
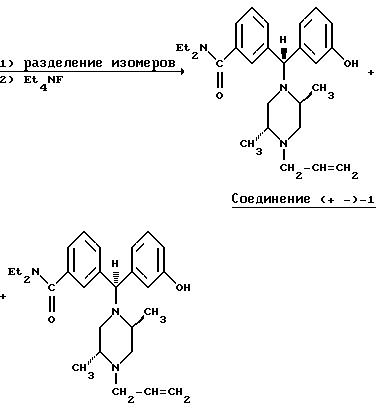
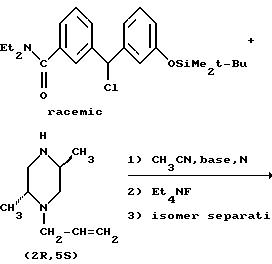
Рацемический транс-1-аллил-2,5-диметилпиперазин может быть разделен на составляющие его энантиомеры классическим методом разделения с использованием энантиомерно чистой карбоновой кислоты, в результате чего получается хиральное промежуточное соединение (2R, 5S)-1-аллил-2,5-диметилпиперазин, пригодное для получения (+)-антипода Соединения I.
(2R, 5S)-1-Аллил-2,5-диметилпиперазин может быть также получен в энтатиомерно чистой форме с использованием иллюстративного пути синтеза, представленного ниже.
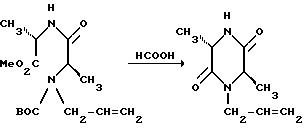
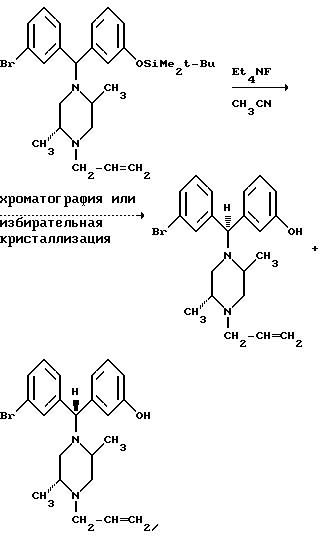
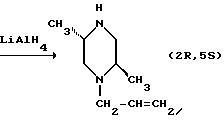
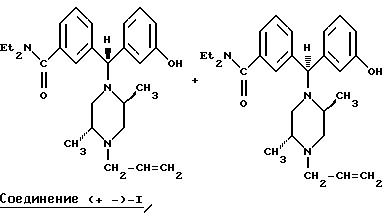
Когда энантиомерно чистый (2R,5S)-1-аллил-2,5-диметилпиперазин обрабатывают рацемическим бензгидрилхлоридом, получающийся продукт представляет собой смесь двух энантиомерно чистых диастереомеров, которые могут быть разделены общепринятыми методами, такими как хроматография или избирательная кристаллизация.
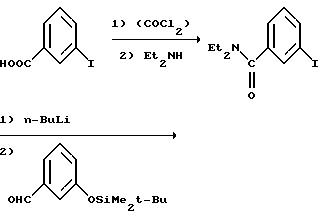
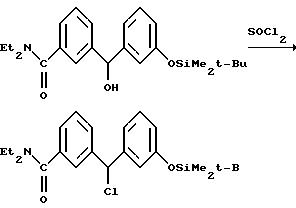
В дополнение к упомянутому выше, Соединения I или (±)-1 могут быть синтезированы через нитрильный путь синтеза, использующий цианид меди в качестве цианирующего агента, как показано ниже.
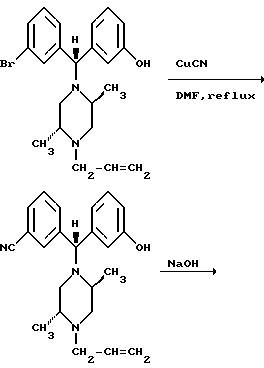
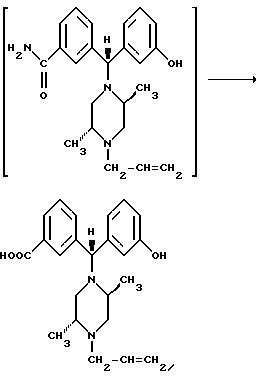
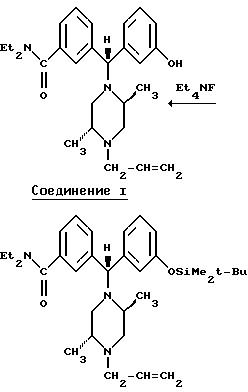
Альтернативные схемы синтеза Соединения I из соответствующего галогенированного соединения приведены ниже.
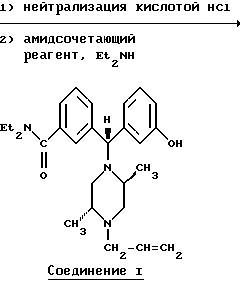
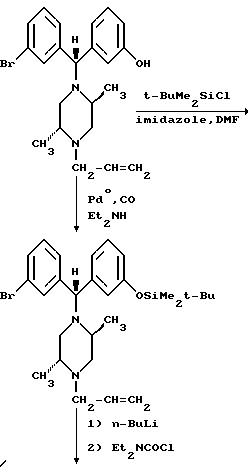
Представленный выше материал иллюстративно продемонстрировал примеры методик синтеза, которые могут быть полезны для образования соединений, таких как Соединение I или (±)-1, равно как и соединений бензгидрилпиперазиновой природы, предусмотренных настоящим изобретением, с использованием соответствующих или аналогичных реагентов.
Особенности и преимущества настоящего изобретения более полно показаны в следующих не лимитирующих его применение примерах.
Конкретные детали и методы, общие для большинства следующих примеров, касающиеся химических синтезов, описаны в ближайшем параграфе.
Точки плавления были измерены с помощью аппарата Thomas-Hoover и не корректировались. Все химические реактивы, если специально не оговариваются, были закуплены в Aldrich Chemical Company, Milwaukee, Wisconsin. Коммерческие растворители использовали без дальнейшей очистки за исключением тетрагидрофурана, который перегоняли над металлическим калием. Спектры ядерного магнитного резонанса (ЯМР) получали, используя спектрометры моделей Perkin-Elmer R-24, Varian XL-200 или XL-300. ВЖХ-анализы осуществляли с помощью жидкостной хроматографической системы фирмы Waters, оборудованной спектральной системой 700 Satellite WISP, контроллером 600 и фотодиодным кристаллическим детектором 991, используя либо колонку Cyclobond I (4,6 • 250 мм. Advanced Separations Technologies, Whippany, New Jersey), либо колонку
(Ссылка) Пример 1
(+)-3-( μ- ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)
-3-оксибензил)-N, N-диэтилбензамид
3-Иодбензойную кислоту (55,5 г, 0,224
моля) растворяли в
тетрагидрофуране (220 мл) и оксалилхлориде (22 мл, 0,252 моля). После добавления 4 капель
диметилформамида в качестве катализатора раствор перемешивали в течение 1 часа при
комнатной температуре и
растворитель удаляли под вакуумом. Остаток растворяли в 220 мл петролейного эфира (диапазон
кипения 35-60oC) и охлаждали в ледяной бане до 0oC. Затем по
каплям добавляли
диэтиламин (55 мл, 0,532 моля) в течение 15 минут. Реакционную смесь перемешивали в ледяной бане
последующие 15 минут, далее разбавляли этилацетатом (100 мл) и промывали насыщенным
раствором хлорида
натрия (50 мл). Органический слой отделяли, высушивали над сульфатом магния и концентрировали in
vacuo до достижения приблизительно половины начального объема. Затем раствор
фильтровали через
небольшую набивку из силикагеля, используя этилацетат для промывания набивки. Все летучие компоненты
удаляли in vacuo, а продукт высушивали под высоким вакуумом до получения 65,69 г
(97%) N,
N-диэтил-3-иодобензамида в виде масла янтарной окраски. ЯМР (300 МГц, CDCl3): (αR)-α
- 1,11 (br s, 3H); 1,21 (br s, 3H); 3,23 (br s, 2H); 3,51 (br s, 2H); 7,13 (ddd
J1 = 0,8 Гц, J2 = 7,6 Гц, J3 = 7,6 Гц, 1H); 7,32 (ddd, J1 = 1,3 Гц, J2 = 1,3 Гц, J3 = 7,5 Гц, 1H); 7,71 (d, J = 1,2 Гц, 1H); 7,72
(ddd,
J1 = 1,3 Гц, J2 = 1,3 Гц, J3 = примерно 8,0 Гц (частично перекрыт), 1H).
Масс-спектр (Cl-CH4) m/e: 304 (МM1, 100%). Расч. для C11H14
NOI: C, 43,58; H, 4,56; N, 4,62; I, 41,86. Обнаружено: C, 43,68; H, 4,64; N, 4,64; I, 41,92.
К 250 мл диметилформамида добавляли 3-оксибензальдегид (70 г, 0,57 моля), трет-бутилдиметилсилилхлорид (92 г, 0,61 моля) и имидазол (92 г, 1,35 моля). Полученную смесь перемешивали в атмосфере азота в течение 1 часа. Раствор выливали в воду (1,5 л) и экстрагировали петролейным эфиром (2 • 500 мл, диапазон кипения 35-60oC). Органическую фазу промывали насыщенным раствором хлорида натрия (100 мл), высушивали над сульфатом магния, обрабатывали силикагелем (20 г), фильтровали и концентрировали in vacuo. Остаток дополнительно высушивали под высоким вакуумом, получая 126,6 г (94%) чувствительного к действию воздуха и света 3-((трет-бутилдиметилсилил)окси)бензальдегида в виде масла янтарной окраски. ЯМР (300 МГц, CDCl3): δ 0,22 (s, 6Н); 0,99 (s, 9H); 7,10 (ddd, J1 = 1,2 Гц, J2 = 2, 5 Гц, J3 = 7,9 Гц, 1Н); 7,32 (dd, J1 = 1,5 Гц, J2 = 2,4 Гц, 1Н); 7,39 (t, J = 7,8 Гц, 1Н); 7,47 (ddd, J1 = 1,3 Гц, J2 = 1,3 Гц, J3 = 7, 6 Гц, 1Н); 9,95 (s, 1Н). Масс-спектр (Cl-CH4) m/e: 237 (М+1, 100%). Рассчитано для C13H20 O2Si: C, 66,05; H, 8,53. Обнаружено: C, 65,95; H, 8,56.
н-Бутиллитий в гексане (280 мл 2,5 М раствора) добавляли с помощью капельной воронки к 1,4 л тетрагидрофурана, находящегося при температуре -78oC в атмосфере азота. После охлаждения раствора н-бутиллития до -78oC к нему медленно в течение 20 минут добавляли раствор N, N-диэтил-3-иодбензамида (106 г, 0,35 моля) в тетрагидрофуране (350 мл). В течение добавления внутренняя температура поднималась до -65oC. По окончании добавления раствор перемешивали 10 минут и к нему медленно в течение 7 минут добавляли раствор 3-((трет-бутилдиметилсилил)окси)бензальдегида (88 г, 0,37 моля) в тетрагидрофуране (90 мл). Реакционную смесь перемешивали дополнительно 5 минут при -78oC и оставляли для нагревания до -10oC. Смесь выливали в 875 мл петролейного эфира (диапазон кипения 35-60oC) и раствора двухосновного фосфата натрия (300 мл 2М раствора), встряхивали и отделяли органическую фазу. Органическую фазу высушивали над сульфатом магния и концентрировали in vacuo. Остаток растворяли в смеси этилацетат:петролейный эфир (1:3, 90 мл), наносили на колонку с силикагелем (1 кг) и промывали с помощью смеси этилацетат: петролейный эфир (1:3) для удаления легко элюируемых примесей. Элюирование этилацетатом дало после концентрирования in vacuo 115,9 г (80%) 3-(3-((трет-бутилдиметилсилил)окси) δ -оксибензил)-N, N-диэтилбензамида в виде вязкого масла янтарной окраски. ЯМР (300 МГц, DMCO-d6): -α- 0,13 (s, 6Н); 0,92 (s, 9H); 0,98 (br s, 3H); 1,11 (br s, 3H); 3,10 (br s, 2Н); 3,39 (br s, 2H); 5,69 (d, J = 4,1 Гц, 1Н); 5,96 (d, J = 4,2 Гц, 1Н); 6,68 (dd, J1 = 1,9 Гц, J2 = 7,7 Гц, 1Н); 6,84 (s, 1H); 6,97 (d, J = 7,7 Гц, 1Н); 7,16 (d, J = примерно 8 Гц (частично скрытый), 1H); 7,17 (t, J = 7,7 Гц, 1H); 7,28 (s, 1H); 7,35 (t, J = 7,8 Гц, 1H); 7,42 (d, J = 7,6 Гц, 1H). Масс-спектр (Cl-CH4) m/e: 414 (М+1, 11%), 178 (32%). Расч. для C24H35NO3Si: C, 69,69; H, 8,53; N, 3,39. Обнаружено: C, 69,65; H, 8,56; N, 3, 40.
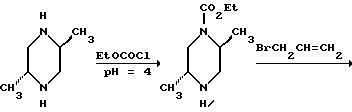
Трехгорлую круглодонную колбу объемом 12 л заполняли транс-2,5-диметилпиперазином (767 г, 6,72 моля), перекристаллизованным из толуола (температура плавления 115-119o C), и 600 мл воды. Колбу охлаждали в ледяной бане и к ее содержимому медленно при перемешивании добавляли раствор метансульфоновой кислоты (1290 г, 13,4 моля) в 600 мл воды, поддерживая температуру ниже 40oC. Раствор охлаждали до 20o C и к нему добавляли 800 мл этанола. Дополнительную воронку объемом 500 мл наполняли 60% водным раствором ацетата калия из 2-х литрового резервуара с этим раствором и ацетат калия добавляли к реакционной смеси до достижения значения pH, равного 4,0. Вторую дополнительную воронку заполняли раствором этилхлорформиата (642 мл, 6,71 моля) в 360 мл тетрагидрофурана. Растворы этилхлорформиата и ацетата калия одновременно добавляли по каплям, согласуя скорость их добавления так, чтобы поддерживать в реакционной смеси значение pH 4, 0 ± 0,1, при необходимости охлаждая реакционный сосуд и обеспечивая температуру 25oC. По окончании добавления этилхлорформиата реакционную смесь перемешивали в течение 1 часа, продолжая добавлять раствор ацетата калия для поддержания значения pH, равным 4,0. Органические растворители отгоняли под вакуумом. Оставшийся водный раствор промывали 1500 мл этилацетата для удаления примеси бис-карбамата. Этилацетатную промывку экстрагировали двумя порциями по 500 мл 1М соляной кислоты для выделения из них продукта. Кислотные экстракты объединяли с исходным водным раствором и, добавляя 10М раствор гидроксида натрия, подводили pH до значения 11 с одновременным охлаждением для поддержания температуры ниже 40oC. Водный раствор экстрагировали двумя порциями этилацетата по 1500 мл, объединенные экстракты высушивали над сульфатом магния и удаляли растворитель, получая 927 г (74%) этил-транс-2,5-диметил-1-пиперазинкарбоксилата в виде масла желтого цвета.
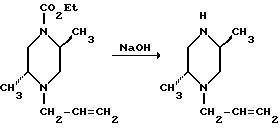
Смесь этил-транс-2, 5-диметил-1-пиперазинкарбоксилата (643 г, 3, 45 моля), аллилбромида (328 мл, 3,80 моля) и карбоната натрия (440 г, 4,15 моля) в 2500 мл ацетонитрила кипятили с обратным холодильником в течение 1,5 часа. Реакционную смесь охлаждали до комнатной температуры, фильтровали и удаляли из нее растворитель под вакуумом. Остаток растворяли в 4000 мл дихлорметана и промывали двумя порциями по 500 мл 1М гидроксида натрия. Раствор дихлорметана высушивали над сульфатом магния и удаляли растворитель, получая 630 г (81%) этил-транс-4-аллил-2, 5-диметил-1-пиперазинкарбоксилата в виде масла.
Этил-транс-4-аллил-2, 5-диметил-1-пиперазинкарбоксилат (630 г, 2,78 моля) добавляли к 87% раствору гранулированного гидроксида калия (2970 г, 46 моль) в 4300 мл 95% этанола и кипятили с обратным холодильником в течение 1,5 часа. В течение первых 0,5-1 часа нагревания наблюдали выделение двуокиси углерода. Реакционную смесь охлаждали до температуры ниже температуры кипения, после чего к ней осторожно добавляли 2000 мл толуола. Этанол удаляли азеотропной дистилляцией при 105oC, добавляя дополнительные 4000 мл толуола в реакционный сосуд в течение отгонки. После сбора 9000 мл дистиллята реакционную смесь охлаждали до 100oC и к ней осторожно добавляли 1000 мл толуола. Раствор медленно охлаждали до 5oC и выдерживали при этой температуре в течение 30 минут. После этого раствор фильтровали, промывая отфильтровальный осадок дополнительными 1500 мл толуола. Фильтрат промывали 1000 мл воды, высушивали над сульфатом магния и удаляли растворитель, получая 296 г (69%) транс-1-аллил-2,5-диметилпиперазина в виде жидкости темной окраски. ЯМР (300 МГц, DMCO-d6): δ 0,87 (d, J = 6,3 Гц, 3H); 0,92 (d, J = 6,3 Гц, 3H); 1, 63 (t, J = 11 Гц, 1H); 2,05 (m, 1H); 2,30 (t, J = 11 Гц, 1H); 2,6-2,8 (m, 4H); 3,33 (dd, J1 = 5 Гц, J2 = 14 Гц, 1H); 5,09 (d, J = 8,7 Гц, 1H); 5,13 (d, J = 14 Гц, 1H); 5,8 (m, 1H).
Ди-п-толуоил-D-винную кислоту (Schweizerhall, Inc., South Plainfield, New Jersey) (1,25 кг, 3,2 моля) растворяли в горячем (примерно 60oC) 95% этаноле (16 л) и к раствору за несколько приемов добавляли рацемический транс-1-аллил-2,5-диметилпиперазин (500 г, 3,2 моля) (осторожно: экзотермическая реакция). В горячий раствор добавляли затравку кристаллов диастереоизомерно чистой соли (полученной в предварительном мелкомасштабном разделении) и раствор охлаждали до комнатной температуры за 2-3 часа. Раствор медленно перемешивали в течение двух дней при комнатной температуре. Образовавшуюся соль собирали фильтрацией, дважды промывали 95% этанолом и высушивали в вакууме, получая 826,5 г твердого вещества белого цвета (47%). Повторение процесса со второй порцией ди-п-толуоил-D-винной кислоты и рацемическим транс-1-аллил-2,5-диметилпиперазином дало 869 г (50%) вещества.
Вся полученная соль (1695 г) была разделена на три порции, каждую из которых перекристаллизовывали дважды описанным далее образом. Соль растворяли в кипящем 95% этаноле (примерно 2,7 л на 100 г соли) в колбе с обратным холодильником и приблизительно половину объема этанола удаляли дистилляцией. (Примечание: необходимо энергичное перемешивание раствора во время дистилляции во избежание кристаллизации на стенках сосуда). В горячий раствор добавляли затравку диастереоизомерно чистой соли, охлаждали до комнатной температуры и медленно перемешивали в течение 2 дней перед сбором соли фильтрацией. (Примечание: последующий эксперимент показал, что время кристаллизации может быть уменьшено от 2 дней до 8 часов). Общий выход продукта составил 1151 г. Соль растворяли в 3 л 2 М водного раствора гидроксида натрия и водный раствор экстрагировали с помощью четырех порций дихлорметана по 1 л каждая. Органические экстракты объединяли, высушивали над сульфатом натрия и растворитель удаляли с помощью роторного испарителя при температуре менее 20o C, получая 293 г (29% из расчета на рацемический вес) (2R,5S)-1-аллил-2,5-диметилпиперазина в виде прозрачного масла. δ =-55,1o (абс. этанол, с= 1,2). Трифторацетамид продукта, полученный с помощью трифторуксусного ангидрида и проанализированный посредством капиллярной газовой хроматографии (колонка 20 м • 0,32 мм, Chiraldex B-PH, Advanced Separation Technologies Inc. , Whippany, NJ, 120oC), показал стереочистоту более 99% ее (время задержки искомого энантиомера 11,7 мин; другого энантиомера 10,7 мин).
3-(3-((трет-Бутилдиметилсилил)окси) [α] -оксибензил)-N, N-диэтилбензамид этилбензамид (115, 9 г, 0,280 моля) растворяли в тетрагидрофуране (560 мл) и к раствору добавляли тионилхлорид (24,5 мл, 0,336 моля). Реакция была заметно экзотермической. Смесь перемешивали в течение 15 минут и концентрировали in vacuo (осторожно в начале процесса из-за быстрого газовыделения). После удаления всех летучих компонентов неочищенный 3-(3-трет-бутилдиметилсилил)окси)- -α- хлорбензил)-N, N-диэтилбензамид растворяли в ацетонитриле (560 мл). К раствору добавляли иодид натрия (42 г, 0,280 моля), диизопропилэтиламин (73 мл, 0, 42 моля) и (2R,5S)-1-аллил-2,5-диметилпиперазин (52,5 г, 0,280 моля). Смесь кипятили с обратным холодильником в атмосфере азота при перемешивании в течение 2,5 часов. Ацетонитрил убирали дистилляцией в атмосфере азота продолжительностью в несколько часов. После охлаждения реакционную смесь выливали в этилацетат (1,1 л) и раствор карбоната калия (350 мл 2 М водного раствора) и встряхивали. Органическую фазу отделяли, высушивали над твердым карбонатом калия и концентрировали in vacuo. Остаток растворяли в смеси этилацетат:петролейный эфир (1:1, 150 мл) и наносили на колонку с силикагелем (3 кг). Элюирование смесью этилацетат:петролейный эфир (1: 1) дало желаемый изомер первым из двух элюируемых эпимеров. Раствор элюата концентрировали до небольшого объема и оставляли на 12 часов. Осевшую кристаллическую примесь удаляли фильтрацией и фильтрат концентрировали досуха.
Остаток растворяли в смеси тетрагидрофуран:петролейный эфир (1:1, 125 мл) и экстрагировали 350 мл 0,75 М соляной кислоты. Водную фазу, содержащую желаемый продукт, перемешивали при комнатной температуре в течение 24 часов для расщепления силилового эфира. После этого раствор промывали смесью этилацетат:петролейный эфир (1:1, 2 • 100 мл). Водный раствор перемешивали с этилацетатом (100 мл), добавляя порциями с соблюдением осторожности (бурное газовыделение) твердый бикарбонат натрия (38 г). После 15 минут дополнительного перемешивания слои разделяли и водный слой экстрагировали этилацетатом (100 мл). Две этилацетатные порции объединяли, высушивали над сульфатом натрия, концентрировали in vacuo и сушили под высоким вакуумом, получая 37, 3 г (30%) ± 3-(( -α- -((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)- N, N-диэтилбензамида в виде твердого вещества беловатого оттенка. (αR)-α- = +20o (метанол, с=2). ЯМР (400 МГц, DMCO-d6): [α] 0,91 (d, J = 6,2 Гц, 3H); 0,99 (br s, 3H); 1, 05 (d, J = 6,2 Гц, 3H); 1,09 (br s, 3H); 1,84 (dd, J1 = 7,3 Гц, J2 = 10,9 Гц, 1Н); 2,06 (dd, J1 = 7,3 Гц, J2 = 10,9 Гц, 1H); 2,48 (m, 1H); 2,51 (dd, J1 = 2,7 Гц, J2 = 10,9 Гц, 1H); 2,58 (br s, 1H); 2,70 (dd, J1 = 2,7 Гц, J2 = 10,9 Гц, 1H); 2,81 (dd, J1 = 7,0 Гц, J2 = 13,9 Гц, 1H); 3,12 (br s, 2H); 3,15 (dd, J1 = 5,1 Гц, J2 = 13,9 Гц, 1H); 3,38 (br s, 2H); 4,97 (br s, 1H); 5,07 (d, J = 10,2 Гц, 1H); 5,14 (d, J = 16,9 Гц, 1H); 5,70 - 5,82 (m, 1H); 6,64 (dd, J1 = 2,1 Гц, J2 = 8,0 Гц, 1H); 6,65 (s, 1H); 6,68 (d, J = 7,7 Гц, 1H); 7,11 (t, J = 8,0 Гц, 1H); 7,14 (d, J = 7,6 Гц, 1H); 7,30 (s, 1H); 7,33 (t, J = 7,6 Гц, 1H); 7,39 (d, J = 8,0 Гц, 1H); 9,31 (s, 1H). Масс-спектр (Cl-CH4) m/e: 436 (М+1, 53%). Расч. для C27H37N3O2 • 0,5 H2O: C, 72,94; H, 8,61; N, 9,45. Обнаружено: C, 73,00; H, 8,57; N, 9,40. Свободный амин (32,2 г) растворяли в 200 мл абсолютного этанола и титровали этанольным хлористым водородом (7 М и 1 M) до значения pH 3,95. Растворитель удаляли и остаток перерастворяли в 50 мл дихлорметана. При энергичном перемешивании добавляли диэтиловый эфир (900 мл), вызывая осаждение смолистого продукта, который отвердевал в процессе перемешивания в течение ночи в атмосфере азота. Продукт собирали фильтрацией и высушивали под вакуумом при 55oC, получая 33,06 г (выход 91%) моногидрохлорида. Расч. для C27H37N3O2 • H2O: C, 66,17; H, 8,23; N, 8,57; Cl, 7,23. Обнаружено: C, 66,40; H, 8,17; N, 8,48; Cl, 7,28.
Пример 2
± 3-(
δ ((2S,
5R)-4-Аллил-2,5-диметил-1-пиперазинил)
-3-оксибензил)-N-метил-N-фенилбензамид
Смесь, состоящую из 1400 г (8,1 моля) 3-бромфенола, 1218 г (8,1 моля) трет-бутилхлордиметилсилана
и 1376 г (20,2
моля) имидазола в 1600 мл N,
N-диметилформамида перемешивали при комнатной температуре в атмосфере азота в течение 18 часов. Реакционную смесь выливали в водный буферный раствор pH 8 и
экстрагировали
диэтиловым эфиром. Эфирные
экстракты промывали водой и солевым раствором, высушивали над сульфатом натрия и растворитель упаривали под вакуумом, получая 2314 г неочищенного
3-бромфенил-трет-бутилдиметилсилилового эфира в виде
масла оранжевого цвета. ЯМР (CDCl3 200 МГц) d: 0,2 (s, 6H); 0,95 (s, 9H); 6,8 (m, 1H); 7,0-7,1 (m, 3H).
Силиловый эфир (1771 г, 6,17 моля) растворяли в 4 л сухого тетрагидрофурана, дополнительно высушивали над молекулярными ситами, переносили в 12 л реакционную колбу в атмосферу азота и охлаждали до -78oC. В колбу при перемешивании в атмосфере азота добавляли н-бутиллитий (2400 мл 1,6 М раствора в гексане) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже -70oC. Перемешивание при -78oC продолжали в течение 2 часов. К смеси добавляли раствор 3-бромбензальдегида (1119 г, 6,05 моля) в 600 мл сухого тетрагидрофурана со скоростью, обеспечивающей поддержание температуры реакционной смеси ниже -70oC. После перемешивания при температуре -78oC в течение 2 часов реакцию останавливали с помощью 1400 мл насыщенного водного раствора хлорида аммония и реакционную смесь оставляли для нагревания до комнатной температуры. Смесь фильтровали для удаления твердых частиц и производили разделение слоев. Органическую фазу промывали солевым раствором, высушивали над сульфатом натрия и упаривали, получая 2500 г неочищенного (αR)-α- (3-бромфенил)-3-(трет-бутилдиметилсилилокси)бензилового спирта в виде масла желтого цвета. Хроматография на силикагеле 1 кг неочищенного продукта в системе растворителей гексан: дихлорметан (градиент от 90:10 до 75:25 с последующей сменой растворителей на дихлорметан:этилацетат/90:10) дала 692,3 г α - (3-бромфенил)-3- (трет-бутилдиметилсилилокси)бензилового спирта в виде масла желтого цвета. ЯМР (CDCl3, 200 МГц) α- 0,2 (s, 6Н); 0,95 (s, 9Н); 2,3 (br s, 1H) ; 5,7 (s, 1H); 6,75 (d, J = 8 Гц, 1H); 6,8 (s, 1H); 6,9 (d, J = 8 Гц, 1H); 7,2 (m, 2H); 7,3 (d, J = 8 Гц, 1H); 7,4 (d, J = 8 Гц, 1H); 7,5 (s, 1H).
К раствору бензгидрилового спирта (160 г, 0,41 моля) в 1 л дихлорметана по каплям добавляли тионилхлорид (38 мл, 0,51 моля) и полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель удаляли под вакуумом, остаток перерастворяли в толуоле, после чего растворитель опять отгоняли под вакуумом с целью удаления избытка тионилхлорида, получая неочищенный δ: (3-бромфенил)-3-(трет-бутилдиметилсилилокси)бензилхлорид в виде масла коричневого цвета. ЯМР (CDCl3, 200 МГц) α- 0,2 (s, 6H); 0,95 (s, 9H); 6,0 (s, 1H); 6,8-7,0 (m, 3H); 7,2-7,6 (m, 5H).
Смесь бензгидрилхлорида и (-)-(2R, 5S)-1-аллил-2,5-диметилпиперазина (137,6 г, 0,89 моля из примера 1, infra) в 1500 мл ацетонитрила кипятили с обратным холодильником в течение 48 часов, концентрировали in vacuo и остаток растворяли в этилацетате. Смесь промывали 0,25 М водным раствором гидроксида натрия, высушивали над сульфатом натрия и концентрировали in vacuo, получая 202,6 г масла темной окраски, которое растворяли в ацетонитриле (1л) и смешивали с двухводным фторидом тетраэтиламмония (88,9 г, 0,48 моля). После перемешивания при комнатной температуре в течение ночи растворитель удаляли под вакуумом. Остаток растворяли в дихлорметане (2 л), промывали буферным раствором с pH 8, высушивали над сульфатом натрия и концентрировали до состояния масла темной окраски, которое перемешивали в ацетонитриле (700 мл) при 25oC в течение 72 часов, получая осадок рыжевато-коричневой окраски. Перекристаллизация из ацетонитрила (2 л) дала 35,3 г единственного диастереомера: ± 3-( δ: ((2S,5R)-4-аллил-2,5-диметил-1- пиперазинил)-3-бромбензил)фенола в виде твердого вещества белого цвета. ЯМР (DMCO-d6, 200 МГц) (α R)-α- 0,95 (d, J=6 Гц, 3H); 1,03 (d, J = 6 Гц, 3H); 1,8 (dd, J1 = 6 Гц, J2 = 10 Гц, 1Н); 2,1 (dd, J1 = 6 Гц, J2 = 10 Гц, 1Н); 2,4-2,6 (m, 3H); 2,7 (d, J = 11 Гц, 1H); 2,8 (dd, J1 = 7 Гц, J2 = 14 Гц, 1Н); 3,2 (dd, J1 = 6 Гц, J2 = 13 Гц, 1H); 4,9 (s, 1H); 5,1 (d, J = 10 Гц, 1H); 5,2 (d, J=18 Гц, 1H); 5, 7-5, 9 (m, 1H); 6,6-6,8 (m, 3H); 7,0-7,4 (m, 4H); 7,55 (s, 1H); 9,35 (s, 1H). Маточную жидкость упаривали, получая 127 г твердого вещества коричневого цвета. Часть (11 г) этого материала очищали хроматографией на силикагеле в системе растворителей дихлорметан:этанол (0-2,5%). Собирали первый элюируемый с колонки изомер, получая 2,32 г 3-( δ: ((2S,5R)-4-аллил-2, 5-диметил-1- пиперазинил)-3-бромбензил)фенола в виде твердого светло-желтого вещества. ЯМР (DMCO-d6, 200 МГц) (αS)-α- 0,95 (d, J=6 Гц, 3H); 1,05 (d, J=6 Гц, 3H); 1,85 (dd, J1=7 Гц, J2= 9 Гц, 1H); 2,1 (dd, J1=6 Гц, J2=9 Гц, 1H); 2,5 (m, 3H); 2,7 (dd, J1=2 Гц, J2=8 Гц, 1H); 2,9 (dd, J1=7 Гц, J2=7 Гц, 1H); 3,1 (dd, J1=5 Гц, J2= 9 Гц, 1H); 4,95 (s, 1H); 5,1 (d, J=10 Гц, 1H); 5,2 (d, J=17 Гц, 1H); 5,8 (m; 1H); 6,6 (d, J=8 Гц, 1H); 6,8 (m, 2Н); 7,1 (t, J=8 Гц, 1H); 7,3 (m, 2H); 7,5 (m, 2H); 9,3 (s, 1H).
± 3-( δ: ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)- 3-бромбензил)фенол (147,3 г, 0,355 моля) растворяли в 1 л N-метил-2-пирролидона с цианидом меди (63,6 г, 0,71 моля) и реакционную смесь помещали на 170oC на 30 часов. После охлаждения до комнатной температуры содержимое реакционной смеси выливали в 7 л водного 14% раствора цианида натрия. Смесь перемешивали в течение ночи и экстрагировали этилацетатом. Этилацетатные экстракты объединяли, промывали водой, высушивали над сульфатом натрия и концентрировали in vacuo, получая 133,3 г твердого вещества коричневого цвета. Хроматография на силикагеле с использованием этанола (2-7%) в дихлорметане дала 97,8 г неочищенного ± 3-( (α R)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)бензонитрила. После перекристаллизации из ацетонитрила было получено 74,2 г (58%) чистого ± 3-( (α R)-α- ((2S, 5R)-4-аллил- 2,5-диметил-1-пиперазинил)-3-оксибензил)бензонитрила в виде твердого вещества белого цвета.
Бензонитрил (78,8 г, 0,22 моля) объединяли с 60 г гранулированного гидроксида натрия в 1 л 95% этанола и кипятили с обратным холодильником в течение 72 часов. Для удаления этанола смесь концентрировали in vacuo. Остаток растворяли в воде и pH раствора доводили концентрированной соляной кислотой до значения 5. Растворитель удаляли in vacuo, получая 138,8 г 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-оксибензил)бензойной кислоты в виде смеси с хлоридом натрия. Часть (5,0 г) неочищенной кислоты перемешивали с 50 мл воды. Образовавшуюся суспензию фильтровали, промывали твердый материал на фильтре тремя порциями воды и сушили в вакууме в течение трех часов, получая 2,02 г ± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- оксибензил)бензойной кислоты в виде твердого вещества светло-бежевой окраски. ЯМР (DMCO-d6,200 МГц) (αR)-α- 0,95 (d, J=6 Гц, 3H); 1,1 (d, J=6 Гц, 3H); 1,9 (ddd, J1=3 Гц, J2=7 Гц, J3=10 Гц, 1Н); 2,1 (dd, J1=8 Гц, J2=10 Гц, 1Н); 2,5 (m, 2H); 2,7-2,9 (m, 2H); 3,2 (m, 2H); 5,05 (d, J=12 Гц, 1Н); 5,2 (d, J=18 Гц, 1H); 5,8 (m, 1H); 6,7 (m, 3H); 7,1 (t, J=8 Гц, 1H); 7,4 (t, J=8 Гц, 1H); 7,65 (d, J=8 Гц, 1H); 7,8 (d, J=8 Гц, 1H); 8,0 (s, 1H); 9,4 (s, 1H).
3-( [α] ((2S, 5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)бензойную кислоту (25, 9 г 50% по весу смеси с хлоридом натрия, 34,0 ммоля) растворяли в 40 мл диметилформамида, содержащего 12,8 г (84,9 ммоля) трет-бутилхлордиметилсилана и 11,5 г (169,1 ммоль) имидазола и перемешивали при комнатной температуре в течение ночи. Реакционную смесь выливали в 500 мл ледяной воды и экстрагировали 500 мл диэтилового эфира. Эфирный экстракт промывали дважды 250 мл воды и затем 125 мл насыщенного раствора хлорида натрия, после чего высушивали над сульфатом натрия и удаляли растворитель, получая 20,8 г неочищенного трет-бутилдиметилсилил 3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)-бензоата.
Неочищенный силил эфир-силиловый эфир (20,7 г, ≤ 33,9 ммоля на основании предыдущей реакции) растворяли в 60 мл дихлорметана и охлаждали в атмосфере азота до 0oC. К раствору по каплям добавляли оксалилхлорид (3,7 мл, 42,4 ммоля). Поддерживая температуру смеси на уровне 0oC, к ней медленно добавляли 10 капель диметилформамида в качестве катализатора. Во время добавления диметилформамида наблюдалось выделение газа. Температуру смеси поддерживали равной 0oC в течение 30 минут, вслед за чем смесь оставляли для нагревания до комнатной температуры. Раствор перемешивали при комнатной температуре в атмосфере азота в течение 24 часов. Все летучие соединения удаляли упариванием под пониженным давлением, получая 29,76 г неочищенного 3-( (αR)-α - ((2S, 5R)- 4-аллил-2,5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси) бензил)бензоилхлорида в виде твердого вещества желто-коричневой окраски. Полученный хлорид использовали без дальнейшей очистки.
Метод получения бензамида
3-( (α
R)-α- (2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3- трет-бутилдиметилсилилокси)бензил)бензоилхлорид (2,33 г
неочищенного, примерно 1,44 г основного вещества, 2,81 ммоля, исходя из 3-(
(α
R)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил) бензойной кислоты) растворяли в 12 мл
дихлорметана при комнатной температуре в атмосфере азота. К раствору добавляли 0,
5 мл
триэтиламина. Далее добавляли по каплям N-метиланилин (0,46 мл, 4,3 ммоля) (реакция экзотермична) и реакционную
смесь перемешивали в течение ночи при комнатной температуре. Все летучие
соединения
удаляли упариванием под пониженным давлением, получая твердое смолистое вещество коричневого цвета.
Неочищенное твердое вещество растворяли в ацетонитриле (8 мл) в атмосфере азота при комнатной температуре. К раствору добавляли фторид тетраэтиламмония гидрат (1,19 г, 6,42 ммоля) и раствор перемешивали при комнатной температуре в течение 1 часа. После удаления растворителя остаток очищали хроматографией на силикагеле (4 см • 12 см) в системе растворителей 0,5- 2% этанол в дихлорметане, получая 0,368 г (28% за 4 этапа от 3-( (αR)-α - ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)бензойной кислоты) ± 3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- (оксибензил)-N-метил-N-фенилбензамида в виде твердого вещества светло-желтой окраски. ЯМР (300 МГц, DMCO-d6): (αR)-α- 0,89 (d, J=6,0 Гц, 3H); 0, 96 (d, J=6,0 Гц, 3H); 1,66 (dd, J1=7,3 Гц, J2=11,4 Гц, 1Н); 2,01 (dd, J1=7,8 Гц, J2 =10,6 Гц, 1H); 2,26 (br d, J=10,6 Гц, 1H); 2,37-2,54 (m, 2H); 2,66 (br d, J=11,0 ГЦ, 1H); 2,82 (dd, J1=7,0 Гц, J2=13,9 Гц, 1H); 3,17 (dd, J1=4,8 Гц, J2=13,9 Гц, 1H); 3,34 (s, 3H); 4,77 (s, 1H); 5,10(d, J=10,1 Гц, 1H); 5,16 (d, J=17,3 Гц, 1Н); 5,70 -5,82 (m, 1H); 6,41 (d, J=7,4 Гц, 1H); 6,54 (s, 1H); 6,64 (d, J=8,0 Гц, 1H); 7,05-7,26 (m, 10H); 9,31 (s, 1H). Масс-спектр (Cl-CH4) m/e: 470 (М+1, 100%), 376 (81%), 316 (45%), 153 (97%). δ = +12,3o (этанол, с=1,2). Свободный амин (0,339 г) растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,0 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая 0,321 г (88% выход) моногидрохлорида в виде гигроскопичного порошка светло-желтой окраски. Расч. для C30H35N3O2 • HCl • H2O: C, 68,75; H, 7,31; N, 8,02; Cl, 6,76. Обнаружено: C, 68,86; H, 7,42; N, 8,00; Cl, 6,84.
Пример 3
± 3-(
[α] ((2S, 5R) -4-Аллил-2,5-диметил-1-пиперазинил)
-3-оксибензил)-N-(4-фторфенил)-N-метилбензамид
Следуя общему методу восстановительного алкилирования,
описанному в литературе (Krishnamurty S. Tetrahedron Lett., 23, 3315, (1982)),
уксусно-муравьиный ангидрид получали путем медленного добавления муравьиной кислоты (7,5 мл) к уксусному ангидриду при
0oC. После перемешивания в течение 5 минут при 0oC смесь
нагревали до 55oC и выдерживали при этой температуре в атмосфере азота в течение 1,75 часа. Смесь охлаждали
до 0oC и использовали без очистки. К уксусно-муравьиному ангидриду
(12,
5 мл, 88 ммолей) при температуре 0oC добавляли 4-фторанилин (3,1 мл, 32,8 ммоля) в тетрагидрофуране (10
мл). Реакционную смесь перемешивали в течение 25 минут и удаляли под вакуумом
все
летучие соединения для получения формамида в виде твердого вещества коричневого цвета. Часть полученного неочищенного
продукта (2,39 г, 17,2 ммоля) растворяли в тетрагидрофуране (8 мл) и
охлаждали до
0oC. К раствору по каплям добавляли боран в тетрагидрофуране (40 мл 1,0 М раствора). В течение первой
половины процесса добавления наблюдали выделение газа. По окончании
процесса добавления
раствор кипятили с обратным холодильником в течение 3 часов. Раствор охлаждали до 0oC и к нему
осторожно добавляли 10 мл метанола. После перемешивания в течение 10
минут к реакционной смеси
добавляли этанольный раствор хлористого водорода (7 мл 7,1 М раствора) и смесь перемешивали в течение
ночи. После удаления всех летучих соединений in vacuo получали
неочищенный N-метил-4-фторанилин в
виде твердого вещества светло-пурпурной окраски. ЯМР (200 МГц, DMCO-d6): (α
R)-α- 2,65 (s, 3H); 5,54 (s, 1H); 6,51 (dd, J1=4,7
Гц, J2=8,8 Гц, 2Н); 6,93
(dd, J1=8,9 Гц, J2= 8,8 Гц, 2Н).
3-( δ ((2S, 5R)-4-Аллил-2, 5-диметил-1-пиперазинил)-3-трет- бутилдиметилсилилокси)бензил)бензоилхлорид (Пример 2, infra, 2, 03 г неочищенного, примерно 1,29 г основного вещества, 2,51 ммоля, исходя из 3-( (α R)-α - ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)бензойной кислоты) растворяли в 8 мл дихлорметана при комнатной температуре в атмосфере азота. К раствору добавляли 0,5 мл триэтиламина. Далее добавляли по каплям 4-фтор-N-метиланилин (0,478 мг, 3,82 ммоля) в дихлорметане (5 мл) (реакция экзотермична) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Все летучие соединения удаляли упариванием под пониженным давлением, получая твердое смолистое вещество желто-коричневой окраски.
Неочищенное твердое вещество растворяли в ацетонитриле (8 мл) в атмосфере азота при комнатной температуре. К раствору добавляли фторид тетраэтиламмония гидрат (1,06 г, 5,7 ммоля) и раствор перемешивали при комнатной температуре в течение ночи. После удаления растворителя остаток очищали хроматографией на силикагеле (4 см • 14 см) в системе растворителей 0,25-3,5% этанол в дихлорметане, получая 0,419 г (34% за 4 этапа от 3-( (α R)-α- ((2S, 5R)-4-аллил-2,5- диметил-1-пиперазинил)-3-оксибензил) бензойной кислоты) ± 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3- (оксибензил)-N-(4-фторфенил)-N-метилбензамида в виде желтого порошка. ЯМР (300 МГц, DMCO-d6): (αR)-α- 0,88 (d, J=6,0 Гц, 3H); 0,96 (d, J=6,0 Гц, 3H); 1,68 (dd, J1= 7,7 Гц, J2= 10,8 Гц, 1Н); 2,02 (dd, J1=7,1 Гц, J2=10,7 Гц, 1Н); 2,28 (br d, J=10,7 Гц, 1Н); 2,35-2, 52 (m, 2H); 2,66 (br d, J=10,6 Гц, 1Н); 2,82 (dd, J1= 7,4 Гц, J2=13,9 Гц, 1Н); 3,16 (dd, J1=4,6 Гц, J2=14,0 Гц, 1Н); 3,32 (s, 3H); 4,77 (s, 1H); 5,10 (d, J=10,3 Гц, 1Н); 5,16 (d, J=17,3 Гц, 1Н); 5,70- 5,84 (m, 1H); 6,43 (d, J= 7,4 Гц, 1H); 6,56 (s, 1H); 6,64 (d, J=8,0 Гц, 1H); 7,02-7,22 (m, 9H); 9,31 (s, 1H). Масс-спектр (Cl-CH4) m/e: 488 (М+1, 100%), 334 (11%), 153 (68%). δ +6,9o (этанол, с=1,6). Свободный амин (0,390 г) растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,3 с последующим осаждением диэтиловым эфиром из дихлорметана, получая 0,327 г (78% выход) моногидрохлорида в виде гигроскопичного порошка светло-желтой окраски. Расч. для C30H34N3 O22F • HCl • H2O: C, 66,47; H, 6,88; N, 7,75; F, 3,50; Cl, 6,54. Обнаружено: C, 66,36; H, 6,74; N, 7,82; F, 3,27; Cl, 6,62.
Пример 4
± 3-( [α]= ((2S,5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3-оксибензил-N- (4-хлорфенил)-N-метилбензамид
4-Хлор-N-метиламин получали из 4-хлоранилина, соединяли с 3-( (αR)-α- ((2S, 5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3-(трет- бутилдиметилсилилокси)бензил)бензоилхлорид, убирали защитные
группы и очищали с помощью методов, описанных в примере 3, получая ± 3-( (αR)-α
- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил-N- (4-хлорфенил)-N-метилбензамид в виде
порошка светло-желтой окраски. ЯМР (300 МГц, DMCO-d6): (αR)-α- 0,89 (d, J= 6,
2 Гц, 3H); 0,93 (d, J=6,1 Гц, 3H); 1,65 (dd, J1=7,6 Гц, J2=10,8 Гц, 1H); 2,
01 (dd, J1=7,6 Гц, J2=10,4 Гц, 1H); 2,27 (dd, J1=1,5 Гц, J2
= 11,4 Гц, 1H); 2,35-2,52 (m, 2H); 2,65 (br d, J =10,8 Гц, 1H); 2,82 dd, J1=7,6 Гц,
J2=13,5 Гц, 1H); 3,16 (dd, J1=4,5 Гц, J2=14,6 Гц, 1H); 3,33 (s, 3H); 4,
77 (s, 1H); 5,10 (d, J= 10,2 Гц, 1H); 5,16 (d, J=17,2 Гц, 1H); 5,70-5,86 (m, 1H); 6,42 (d, J=8,
1 Гц, 1H); 6,56 (s, 1H); 6,64 (d, J=7,5 Гц, 1H); 7,04-7,25 (m, 5H); 7,13 (d, J= 8,5 Гц, 2H); 7,29 (d, J=8,
5 Гц, 2H); 9,31 (s, 1H). Масс-спектр (Cl-CH4) m/e 504 (35Cl, M+1, 86%),
350 (28%), 153 (100%). δ = +10,2o (этанол, c = 1,6). Моногидрохлорид готовили так же,
как в примере 3, получая гигроскопический порошок светло-желтой окраски. Расч. для C30H34N3O2Cl • HCl • 0,75H2O: C, 65,04; H, 6,64;
N, 7,58; Cl, 12,80. Обнаружено: C,6504; H, 6,71; N 7,49; Cl, 12,83.
Пример 5
± 3-([α] ((2S,5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3-оксибензил)-N-этил-N- фенилбензамид
3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- (третбутилдиметилсилилокси)бензил)бензоилхлорид
(пример 2, infra, 2,81 г неочищенного, приблизительно 1,74 г основного вещества, 3,39 ммоля, исходя из 3-( (α
R)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3 -оксибензил)бензойной
кислоты) растворяли в 10 мл дихлорметана при комнатной температуре в атмосфере азота. К раствору добавляли 0,5 мл
триэтиламина. Далее добавляли по каплям N-этиланилин (0,780 мл, 6,2 ммоля) (реакция
экзотермична) и реакционную смесь перемешивали в течение ночи при комнатной температуре. Все летучие соединения
удаляли упариванием под пониженным давлением, получая густое масло коричневого
цвета.
Неочищенное масло растворяли в ацетонитриле (10 мл) в атмосфере азота при комнатной температуре. К раствору добавляли фторид тетраэтиламмония гидрат (1,5 г, 8,1 ммоля) и раствор перемешивали при комнатной температуре в течение 1 часа. После удаления растворителя остаток очищали хроматографией на силикагеле (4 см • 15 см) в системе растворителей 0,5 - 3% этанол в дихлорметане, получая 0,508 г (31% за 4 этапа от 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1- пиперазинил)-3-оксибензил)бензойной кислоты ± 3-( (αR)-α - ((2S, 5R)-4- аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)-N-этил-N-фенилбензамида в виде твердого вещества белого цвета. ЯМР (300 МГц, DMCO-d6): (αR)-α- 0,89 (d, J=6,1 Гц, 3H); 0, 96 (d, J=6,1 Гц, 3H); 1,07 (t, J=7,0 Гц, 3H); 1,67 (dd, J1=7,4 Гц, J2=10,4 Гц, 1H); 2,02 (dd, J1=7,4 Гц, J2=10,6 Гц, 1H); 2,27 (dd, J1=1,4 Гц, J2=10,6 Гц, 1H); 2,36-2,52 (m, 2H); 2,66 (br d, J= 10,4 Гц, 1H); 2,82 (dd, J1=7,8 Гц, J2=13,5 Гц, 1H); 3,16 (dd, J1=4,0 Гц, J2= 13,9 Гц, 1H); 3, 83 (q, J=7,0 Гц, 2H); 4,75 (s, 1H); 5,09 (d, J=9,9 Гц, 1H); 5,16 (d, J=17,2 Гц, 1H); 5,70-5,84 (m, 1H); 6, 41 (d, J=7,6 Гц, 1H); 6,54 (s, 1H); 6,63 (d, J=8,2 Гц, 1H); 7,03-7,29 (m, 10H); 9,30 (s, 1H). Масс-спектр (Cl-CH4) m/e: 484 (M+1, 100%), 330 (57%), 153 (66%). δ +10,4o (этанол, c= 1,2). Моногидрохлорид готовили из 0,473 г свободного амина так же, как в примере 3, получая 0,389 г (76% выход) гигроскопичного порошка белого цвета. Расч. для C27H37 N3O2 • HCl • H2O: C, 66,19; H, 7,49; N, 7,81; Cl, 6,59. Обнаружено: C, 69,41; H, 7,52; N, 7,73; Cl, 6,48.
Пример 6
(-)-3-(
[α]= ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3- оксобензил)-N-фенилбензамид
Это соединение было получено в виде твердого вещества светло-желтой
окраски из анилина и 3-(
(αR)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-(т-бутилдиметилсилилокси)
бензил)бензоилхлорида (пример 2, infra) с использованием метода получение
бензамида, описанного в примере
2. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,99 (d, J=5,7 Гц, 3H); 1,10 (d,
J=5,8 Гц, 3H); 1,91 (dd, J1= 7,0 Гц, J2= 10,5 Гц,
1H); 2,14 (dd, J1=6,0
Гц, J2=10,4 Гц, 1H); 2,51-2,81 (m, 2H); 2,88 (dd, J1=6,8 Гц, J2
=13,9 Гц, 1H); 3,18 (dd, J1=5,4 Гц, J2=13,8 Гц,
1H); 5,06 (d, J=15,6 Гц, 1H); 5,
14 (s, 1H); 5,19 (d, J=18,1 Гц, 1H); 5,75 (m, 1H); 6,73 (m, 3H); 7,10 (d, J=7,8 Гц, 1H); 7,17 (d,
J=8,0 Гц, 1H); 7,30-7,59 (m, 3H); 7,65 (d, J=7,6 Гц, 1H); 7,71-7,83
(m, 3H); 7,93 (s, 1H); 9,37 (s,
1H); 10,21 (s, 1H). Масс-спектр (Cl-CH4) m/e: 456 (M+1, 100%), 302 (41%), 153 (77%); .
δ -4,44o (этанол, c=1,4). Моногидрохлорид готовили
так же, как в примере 2, получая
гигроскопичный порошок светло-желтой окраски. Расч. для C29H33N3
O2 • HCl • 0,75H2O: C, 68,90; H,
7,08; N, 8,31; Cl, 7,01.
Обнаружено: C, 69,00; H, 7,06; N, 8,32; Cl, 6,95.
Пример 7
± 3-( [α
]= ((2S, 5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3- оксобензил-N-(3-фторфенил)-N-метилбензамид
1-(3-Фтор-)-N-метиланилин получали из 3-фторанилина,
используя модифицированный
метод восстановительного аминирования. На первом этапе получали 1-оксиметилбензотриазол путем добавления 37%
водного раствора формальдегида к бензотриазолу при 40oC в отношении 1:1 и
охлаждения до комнатной температуры с целью осаждения продукта реакции. После фильтрации оксиметиленбензотриазол
(125 г) нагревали до кипения в колбе с обратным
холодильником, содержащей толуол и
3-фторанилин (92,2 г). Воду удаляли в виде азеотропной смеси, используя ловушку Dean-Stark. После трех часов
кипячения смесь охлаждали до комнатной температуры,
затем помещали на несколько часов в
холодильник для достижения полного осаждения. Твердое вещество в виде кристаллов белого цвета собирали
фильтрацией, получая 174,2 г (86,6%)
1-(3-фторанилин)метил-1H-бензтриазола.
1-((3-Фторанилин)метил-1H-бензотриазол (173,9 г) суспендировали в безводном тетрагидрофуране. К смеси при комнатной температуре порциями добавляли боргидрид натрия (32,5 г). По окончании добавления смесь кипятили с обратным холодильником в течение 4 часов. Раствор охлаждали и медленно выливали в 400 мл 5 М соляной кислоты со льдом и перемешивали 1 час при комнатной температуре. pH раствора подводили до 9 - 10, используя 10 М раствор гидроксида натрия. Продукт экстрагировали диэтиловым эфиром. Эфирные экстракты последовательно промывали 1 М раствором гидроксида натрия, насыщенным раствором хлорида натрия и водой. Органическую фазу высушивали над сульфатом натрия упаривали под пониженным давлением, получая 87,5 г (97%) 3-фтор-N-метиланилина в виде бесцветного масла. [ЯМР (200 МГц, DMCO-d6): (αR)-α- 2,76 (s, 2H); 3,41 (br s, 1H); 6,59-6,92 (m, 3H); 7,27 (q, J=8,0 Гц, 1H)].
3-Карбоксибензальдегид (Alfredo Inc., Monroe, Ohio; 2,0 г) суспендировали в тионилхлориде (6 мл). На колбу помещали обратный холодильник, снабженный хлоркальциевой трубкой для улавливания влаги. Реакционный сосуд помещали в масляную баню и нагревали, поддерживая температуру бани ниже 100oC. Смесь кипятили с обратным холодильником до получения прозрачного раствора и еще в течение дополнительных 5-10 минут перед охлаждением до комнатной температуры. Раствор разбавляли безводным толуолом и все летучие соединения удаляли под вакуумом.
Неочищенный хлорид растворяли в дихлорметане и охлаждали в водяной бане со льдом. К раствору из капельной воронки добавляли сначала 6 мл триэтиламина, а затем 1,83 г N-метил-3-фторанилина в дихлорметане. Мутный раствор оставляли на 1 час для нагревания до комнатной температуры. К раствору добавляли воду и продукт экстрагировали дихлорметаном. Органический слой промывали водой и насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и растворитель удаляли под вакуумом. N-(3-Фторфенил)-3-формил-N-метилбензамид (3,20 г) получали в виде масла светло-золотистой окраски (93% выход без хроматографии). [ЯМР (300 МГц, DMCO-d6): δ 3,38 (s, 3H); 6, 94-7,02 (m, 2H); 7,18-7,29 (m, 2H); 7,46 (t, J=7,7 Гц, 1H); 7,55 (d, J=7,6 Гц, 1H); 7,81 (m, 2H); 9,90 (s, 1H)].
В 12 литровую 3-х горловую круглодонную колбу помещали транс-2, 5-диметилпиперазин (767 г, 6,72 моля), перекристаллизованный из толуола и имеющий температуру плавления 115 - 119o C, и 600 мл воды. Колбу охлаждали в ледяной бане и к ее содержимому медленно с перемешиванием и охлаждением для поддержания температуры ниже 40oC добавляли раствор метансульфоновой кислоты (1290 г, 13,4 моля) в 600 мл воды. Раствор охлаждали до 20oC и к нему добавляли 800 мл этанола. Капельную воронку объемом 500 мл наполняли 60% водным раствором ацетата калия из 2 л резервуара с этим раствором и ацетат калия добавлялся в реакционный сосуд для подведения pH до 4,0. Вторую капельную воронку заполняли раствором этилхлорформиата (642 мл, 6,71 моля) в 360 мл тетрагидрофурана. Растворы этилхлорформиата и ацетата калия одновременно добавляли по каплям с согласованной скоростью для поддержания значения pH раствора реакции равным 4,0 ± 0,1 при необходимости охлаждая его для поддержания температуры 25oC. По окончании добавления этилхлорформиата реакционную смесь перемешивали в течение 1 часа, продолжая добавлять раствор ацетата калия для поддержания значения pH 4,0. Органические растворители удаляли дистилляцией под вакуумом. Оставшийся водный раствор промывали 1500 мл этилацетата для удаления всех бис-карбаматных примесей. Этилацетатную промывку экстрагировали двумя порциями по 500 мл 1 М соляной кислоты, извлекая продукт. Кислотные экстракты объединяли с основным водным раствором и pH подводили до 11, добавляя 10 М гидроксид натрия с охлаждением для поддержания температуры ниже 40oC. Водный раствор экстрагировали двумя 1500 мл порциями этилацетата, объединенные экстракты высушивали над сульфатом магния и растворитель удаляли, получая 927 г (74%) этил-транс-2, 5-диметил-1-пиперазинкарбоксилата в виде масла желтого цвета.
Смесь этил-транс-2, 5-диметил-1-пиперазинкарбоксилата (643 г, 3,45 моля), аллилбромида (328 мл, 3,80 моля) и карбоната натрия (440 г, 4,15 моля) в 2500 мл ацетонитрила кипятили с обратным холодильником в течение 1,5 часа. Реакционную смесь охлаждали до комнатной температуры, фильтровали и растворитель удаляли под вакуумом. Остаток растворяли в 4000 мл дихлорметана и промывали двумя порциями по 500 мл 1 М раствора гидроксида натрия. Раствор дихлорметана высушивали над сульфатом магния и растворитель удаляли, получая 630 г (81%) этил-транс-4-аллил-2,5-диметил-1-пиперазинкарбоксилата в виде масла.
Этил-транс-4-аллил-2, 5-диметил-1-пиперазинкарбоксилат (630 г, 2,78 моля) добавляли к раствору 87% гранулированного гидроксида калия (2970 г, 46 молей) в 4300 мл 95% этанола и кипятили с обратным холодильником в течение 1, 5 часа. Во время первых 0,5 - 1 часа наблюдали выделение углекислого газа. Реакционную смесь охлаждали до температуры ниже температуры кипения и к ней аккуратно добавляли 2000 мл толуола. Этанол отгоняли в виде азеотропной смеси при температуре 105oC, добавляя в процессе дистилляции дополнительные 4000 мл толуола в реакционный сосуд. После сбора 9000 мл дистиллята реакционную смесь охлаждали до температуры 100oC и в нее аккуратно добавляли 1000 мл толуола. Раствор медленно охлаждали до 5oC и выдерживали при этой температуре в течение 30 минут. Раствор фильтровали и отфильтрованный осадок промывали дополнительными 1500 мл толуола. Фильтрат промывали водой (1000 мл), высушивали над сульфатом магния и удаляли растворитель, получая 296 г (69%) транс-1-аллил-2,5-диметилпиперазина в виде темной жидкости. ЯМР (300 МГц, DMCO-d6): δ 0,87 (d, J=6,3 Гц, 3H); 0,92 (d, J=6,3 Гц, 3H); 1,63 (t, J= 11 Гц, 1H); 2,05 (m, 1H); 2,30 (t, J=11 Гц, 1H); 2,6-2,8 (m, 4H); 3,33 (dd, J1= 5 Гц, J2=14 Гц, 1H); 5,09 (d, J=8,7 Гц, 1H); 5,13 (d, J=14 Гц, 1H); 5,8 (m, 1H).
Ди-п-толуол-D-винную кислоту (Schweizerhall, Inc., South Plainfield, New Jersey) (1,25 кг, 3,2 моля) растворяли в горячем (примерно 60oC) 95% этаноле (16 л) и к раствору за несколько приемов добавляли рацемический транс-1-аллил-2,5-диметилпиперазин (500 г, 3,2 моля) (осторожно: экзотермическая реакция). В горячий раствор добавляли затравку кристаллов диастереоизомеров чистой соли (полученной в предварительном мелкомасштабном разделении) и раствор охлаждали до комнатной температуры за 2 - 3 часа. Раствор медленно перемешивали в течение двух дней при комнатной температуре. Образовавшуюся соль собирали фильтрацией, дважды промывали 95% этанолом и высушивали в вакууме, получая 826,5 г твердого вещества белого цвета (47%). Повторение процесса со второй порцией ди-п-толуол-D-винной кислоты и рацемическим транс-1-аллил-2,5-диметилпиперазином дало 869 г (80%) вещества.
Вся полученная соль (1695 г) была разделена на три порции, каждую из которых перекристаллизовывали дважды описанным далее образом. Соль растворяли в кипящем 95% этаноле (примерно 2,7 л на 100 г соли) в колбе с обратным холодильником и приблизительно половину объема этанола удаляли дистилляцией. (Примечание: необходимо энергичное перемешивание раствора во время дистилляции во избежание кристаллизации на стенках сосуда). В горячий раствор добавляли затравку диастереоизомерно чистой соли, охлаждали до комнатной температуры и медленно перемешивали в течение 2 дней сбором соли фильтрацией. (Примечание: последующий эксперимент показал, что время кристаллизации может быть уменьшено от 2 дней до 8 часов). Общий выход продукта составил 1151 г. Соль растворяли в 3 л 2 М водного раствора гидроксида натрия и водный раствор экстрагировали с помощью четырех порций дихлорметана по 1 л каждая. Органические экстракты объединяли, высушивали над сульфатом натрия и растворитель удаляли с помощью роторного испарителя при температуре менее 20oC, получая 293 г (29% из расчета на рацемический вес) (2R,5S)-1-аллил-2,5-диметилпиперазина в виде прозрачного масла. δ = -55,1o (абс. этанол, c= 1,2). Трифторацетамид продукта, полученный с помощью трифторуксусного ангидрида и проанализированный посредством капиллярной газовой хроматографии (колонка 20 М • 0,32 мм, Chiraldex B-PH, Advanced Separation Technolohies Inc. , Whippany, NJ, 120oC), показал стереочистоту более 99% ее (время задержки искомого энантиомера 11,7 мин; другого энантиомера 10,7 мин).
(2R, 5S)-1-аллил-2,5-диметилпиперазин (6,13 г), бензотриазол (4,79 г) и N-(3-фторфенил)-3-формил-N-метилбензамид (10,23 г) перемешивали в безводном толуоле, содержащем одну каплю триэтиламина. Смесь помещали в масляную баню с температурой 140oC (температура бани). Колбу соединяли с ловушкой Dean-Stark для азеотропного удаления воды в токе азота. Смесь кипятили с обратным холодильником 2-3 часа и большую часть толуола под пониженным давлением. Неочищенный аддукт может быть выделен на этой стадии кристаллизацией, дающей 3-(((2R, 5S)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(1H-бензотриазол-1- ил)метил)-N-(3-фторфенил)-N-метилбензамид в виде смеси эпимеров, но из-за чувствительности его к воде, как правило, удобно использовать в последующих реакциях материал без очистки. (Реакционная смесь в толуоле обычно пригодна для дальнейшей стадии).
Раствор 3-бромфенола (500 г, 2,89 моля), трет-бутилхлордиметилсилана (436 г, 2,89 моля) и имидазола (500 г, 7,22 моля) в 500 мл диметилформамида перемешивали в течение ночи при комнатной температуре. Реакционный раствор выливали в 3000 мл воды и экстрагировали двумя порциями по 2000 мл диэтилового эфира. Объединенные экстракты высушивали над сульфатом натрия и удаляли растворитель, получая 846 г 3-(бромфенокси)-трет-бутилдиметилсилана в виде бледно-желтой жидкости. ЯМР (300 МГц, CDCl3): [α] 0,2 (s, 6H); 1,0 (s, 9H); 6,75 (m, 1H); 7,0 (br s, 1H); 7, 1 (m, 2H).
3-(Бромфенокси)-трет-бутилдиметилсилан (17,12 г) растворяли в безводном тетрагидрофуране (150 мл) и охлаждали до -78oC в атмосфере азота. К раствору медленно с помощью шприца добавляли н-бутиллитий в гексане (23,88 мл 2,5 М раствора). В процессе перемешивания в течение 40 минут при -78oC раствор становился белым и отчасти мутным. Раствор переносили с помощью раздвоенной иглы в колбу, содержащую etherate бромида магния (16,5 г) в тетрагидрофуране (150 мл) и перемешивали при комнатной температуре в течение 1 часа. Неочищенный безотриазольный аддукт с предыдущей стадии, содержащий в основном 3-(((2R,5S)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(1H-бензотриазол-1- ил)метил-N-(3-фторфенил)-N-метилбензамид, растворяли в тетрагидрофуране и добавляли к только что приготовленному реагенту бромида арилмагния. Раствор слегка нагревался в процессе добавления и приобретал мутную желто-коричневую окраску. После перемешивания при комнатной температуре в течение 2 часов к раствору аккуратно добавляли 0,5 М водный раствор соляной кислоты до тех пор, пока его pH не достигал значения, равного 6. Продукт экстрагировали с помощью 250 мл этилацетата и растворитель удаляли под вакуумом.
Трет-бутилметилсилиловую защитную группу удаляли, растворяя остаток в 175 мл тетрагидрофурана и добавляя 85 мл водного HCl при комнатной температуре. Раствор нагревался при добавлении кислоты. Смесь перемешивали 40 минут при комнатной температуре. После добавления диэтилового эфира производили отделение водного кислотного слоя. Водный слой промывали второй раз диэтиловым эфиром и подводили в нем pH до значения, равного 8-9, с помощью водного раствора гидроксида натрия. Продукт экстрагировали этилацетатом. Этилацетатные экстрагенты объединяли и промывали разбавленным раствором гидроксида натрия для удаления оставшегося бензотриазола. После этого органический слой промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и упаривали под пониженным давлением. Продукт (10,85 г, 56%) получали в виде смеси двух диастереомеров в соотношении 91:9 в пользу нужного диастереомера, как было установлено с помощью ВЖХ-анализа. ВЖХ проводили на колонке δ -Bondapak C-18 (125 A, 3,9 • 300 мм, Waters Chromatohraphy Division, Millipore Corparation, Milford, MA), используя 60% метанол и 40% 0,1 М водный ацетат аммония при скорости протекания 1 мл/мин. Смесь диастереомеров перекристаллизовывали из этилацетата/гексана, получая ± 3-( μ ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)-N- (3-фторфенил)-N-метилбензамид в виде твердого кристаллического вещества белого цвета (т. пл. 144 - 145o C) с чистотой относительно изомерного состава в 99% (по данным ВЖХ). ЯМР (200 МГц, DMCO-d6): (α R)-α- 0,84 (d, J=6,0 Гц, 3H); 0,97 (d, J= 5,9 Гц, 3H); 1,69 (dd, J1 =7,7 Гц, J2=10,7 Гц, 1H); 2,01 (dd, J1=7,4 Гц, J2=10,7 Гц, 1H); 2,28 (br d, J=8,3 Гц, 1Н); 2,40-2,52 (m, 2H); 2,67 (br d, J=10,5 Гц, 1Н); 2,82 (dd, J1=7,6 Гц, J2=13,2 Гц, 1Н); 3,17 (br d, J=14,0 Гц, 1Н); 3,34 (s, 3H); 4,80 (s, 1Н); 5,10 (d, J=10,1 Гц, 1Н); 5, 17 (d, J= 17,3 Гц, 1Н); 5,70-5,84 (m, 1Н); 6,42 (d, J=7,1 Гц, 1Н); 6,56 (s, 1Н); 6, 65 (d, J= 8,3 Гц, 1Н); 6,90-7,32 (m, 9H); 9,31 (s, 1Н). Масс-спектр (Cl-CH4) m/e: 488 (m+1, 100%), 334 (39%), 153 (87%). δ = +4,9o (абс. этанол, с= 1,2). Свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,7 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопичного порошка беловатой окраски. Расч. для C30H34N3O2F • HCl • 1,25H2O: C, 65,92; H, 6,92; N, 7,69; Cl, 6,49. Обнаружено: C, 66,07; H, 6,95; N, 7,53; Cl, 6,54.
Пример 8
3-( [α] ((-2S,5R)-4-Аллил=2,5-диметил-1-пиперазинил)-3- оксибензил)-N-метил-N-(2,4,6-трихлорфенил)бензамид
N-Метил-2,4,
6-трихлоранилин [ЯМР (200 МГц, CDCl3): (αR)-α
- 2,82 (s, 3H); 5,11 (s, 1H); 7,46.(s, 2H)], полученный из 2,4,6-трихлоранилина, связывали с 3-(( δ R)- αR -((2S,
5R)-4- аллил-2,5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)
бензил)бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая ± 3-( α ((2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-метил-N-(2,4,
6-трихлорфенил)бензамид в виде порошка беловатой окраски. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,90 (d, J= 6,1
Гц, 3H); 0,98 (d, J=6,0 Гц, 3H); 1,65 (dd, J1=7,4 Гц,
J2=10,6 Гц, 1H); 2,03 (dd, J1=7,5 Гц, J2=10,2 Гц, 1H); 2,35 (d, J=11,7 Гц, 1H); 2,38-2,51 (m, 2H); 2,
65 (br d, J=10,6 Гц, 1H); 2,80 (dd, J1=7,0 Гц, J2=13,3 Гц, 1H); 3,12 (m, 1H); 3,18 (s, 3H); 4,80 (s, 1H); 5,11 (d, J=11,0 Гц, 1H); 5,18 (d, J= 16,8 Гц, 1H); 5,66-5,87 (m, 1H); 6,
48 (d, J=8,4 Гц, 1H); 6,56 (s, 1H); 6,64 (d, J=8,6 Гц, 1Н); 7,
16 (t, J=8,0 Гц, 1H); 7,22-7,28 (m, 3H); 7,38 (s, 1H); 7,69 (d, J= 2,2 Гц, 1H); 7,72 (d, J=2,2 Гц, 1H); 9,31 (s, 1H). Масс-спектр
(Cl-CH4) m/e: 572 (М+1, 14%), 153 (100%).
Пример 9
3-(δ ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-метил-N-(2-(трифторметил)фенил)бензамид
N-Метил-2-(трифторметил)анилин [ЯМР (200 МГц, DMCO-d6): (αR)-α- 2,75 (S, 3H); 3,40
(S, 1Н); 6,70 (t, J=8,0 Гц, 1Н); 6,94-7,16 (br m, 2H); 7,38 (d, J= 7,3 Гц, 1H)] полученный из 2-(трифторметил)анилина, связывали с 3-( δ
((2S, 5R)-4-аллил-2,
5- диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил) бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая ± 3-( (α
R)-α- ((2S,
5R)-4-аллил- 2,5-диметил-1-пиперазинил)-3-оксибензил)-N-метил-N-(2-трифтор- метил)фенил)бензамид в виде желтого порошка. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,
90 (d, J=6,0 Гц,
3H);
0,97 (d, J=6,0 Гц, 3H);1,64 (m, 1H); 2,05 (m, 1H); 2,27 (br d, J=10,5 Гц, 1H); 2,40-2,84 (m, 4H); 3,18 (br d, J=13,5 Гц, 1H); 3,29 (s, 3H); 4,79 (s, 1H); 5,11 d, J=10,2 Гц, 1H);
5,18 (d, J=17,0
Гц,
1H); 5,70-5,82 (m, 1H); 6,42 (d, J=7,6 Гц, 1Н); 6,65 (d, J=7,7 Гц, 1Н); 6,67 (s, 1H); 7,04-7,83 (m, 9H); 9,32 (s, 1H). Масс-спектр (Cl-CH4) m/e: 538 (М+1, 82%), 384
(13%), 153
(100%).
Пример 10
3-( δ ((2S,5R)-4Аллил-2,5-диметил-1-пиперазинил)-3-оксибензил) -N-метил-N-фенилбензамид
3-( (αS)-α- ((2S,
5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- бромбензил)фенол (2,30 г, 5,5 ммоля, пример 2, infra) смешивали с трет-бутилхлордиметилсиланом (1,67 г, 11 ммолей) и имидазолом (0,94 г, 13,8 ммоля) в 30 мл
диметилформамида при комнатной температуре в течение ночи в атмосфере азота. Реакционную смесь выливали в воду со льдом и экстрагировали диэтиловым эфиром. Эфирные слои промывали водой и солевым
раствором, высушивали над сульфатом натрия и концентрировали досуха. Остаток очищали с помощью хроматографии на силикагеле в системе растворителей гексан:этилацетат (0-50%), получая 2,36 г
силилового
эфира в виде желтого масла.
Силиловый эфир (2,25 г, 4,2 ммоля) растворяли в 80 мл сухого тетрагидрофурана, дополнительно высушивали над молекулярными ситами, переносили в реакционную колбу в атмосферу азота и охлаждали до -78oC. В колбу при перемешивании в атмосфере азота добавляли н-бутиллитий (2,6 мл 1,6 М раствора в гексане) с такой скоростью, чтобы поддерживать температуру реакционной смеси ниже -70oC. Углекислый газ пропускали через реакционную смесь в течение 2-3 минут. Смесь нагревали до комнатной температуры с постоянным перемешиванием для поддержания дегазации растворенного углекислого газа. Растворитель упаривали, остаток перерастворяли в толуоле, после чего опять под вакуумом убирали растворитель, удаляя весь н-бромбутан. Остаток растворяли в дихлорметане (50 мл), добавляли тионилхлорид (0,46 мл, 6,3 ммоля) и смесь перемешивали при комнатной температуре в течение 40 минут. После этого добавляли триэтиламин (2,3 мл, 16,8 ммоля) и N-метиланилин (0,5 мл, 4,6 ммоля) и перемешивание при комнатной температуре продолжали в течение ночи. Реакционную смесь промывали водой, высушивали над сульфатом натрия и растворитель удаляли под вакуумом, получая 2,68 г коричневого масла. Неочищенный продукт растворяли в ацетонитриле и подвергали действию 1,2 г (6,3 ммоля) фторида тетраэтиламмония дигидрата в течение 10 минут при комнатной температуре. Растворитель упаривали и остаток очищали хроматографией на силикагеле в системе растворителей дихлорметан:этанол (0-3,5%), получая 0,92 г ± 3-( (αS)-α - ((2S, 5R)-4-аллил-2,5-диметил- 1-пиперазинил)-3-оксибензил)-N-метил-N-фенилбензамида в виде твердого вещества светло-бежевой окраски. ЯМР (DMCO-d6, 200 МГц): (αS)-α- 0,9 (d, J=6 Гц, 3H); 0,95 (d, J=6 Гц, 3H); 1,7 (dd, J1=6 Гц, J2=8 Гц, 1H); 2,0 (dd, J1=7 Гц, J2=10 Гц, 1H); 2,1 (m, 1H); 2,4-2,7 (m, 3H); 2,85 (dd. J1 =7 Гц, J2=14 Гц, 1H); 3,15 (dd, J1=7 Гц, J2=15 Гц, 1H); 3,4 (s, 3H); 4,7 (s, 1Н); 5,1 (d, J=10 Гц, 1Н); 5,2 (d, J=17 Гц, 1Н); 5,8 (m, 1Н); 6,6 (m, 2H); 6,8 (s, 1Н); 7, 0 (t, J=8 Гц, 1Н); 7,1- 7,3 (m, 9Н); 9,4 (s, 1Н). δ = +4o (абс. этанол, с=2,7). Продукт растворяли в абсолютном этаноле и титровали этанольным раствором хлористого водорода до pH 3. Раствор концентрировали и к нему добавляли диэтиловый эфир с целью осаждения моногидрохлорида, который сушили под вакуумом, получая 0,617 г порошка светло-бежевой окраски. Расч. для C30 H35N3O2 • HCl • 0,70 H2O: C, 69,47; H, 7,27; N, 8,10; Cl, 6,84. Обнаружено: C, 69,76; H, 7, 27; N, 7,74; Cl, 6,60.
Пример
11
± 3-( [α]) ((2S,
5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-фенил-N-пропилбензамид
N-Пропиланилин получали из анилина и пропионового ангидрида, связывали с 3-( (αR)-α- (2S,
5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3-(трет- бутилдиметилсилилокси)бензилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая ± 3-( (αR)-α
- ((2S,
5R)-4аллил-2,
5-диметил-1-пиперазинил)-3-оксибензил)-N-фенил-N- пропилбензамид в виде порошка светло-желтой окраски. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,87 (t, J=7,4 Гц,
3H); 0,
91 (d, J=5,9
Гц, 3H); 0,98 (d, J=6,0 Гц, 3H); 1,51 (m, 2H); 1,69 (dd, J1= 7,2 Гц, J2=10,9 Гц, 1Н); 2,06 (dd, J1=7,0 Гц, J2=10,5 Гц, 1Н); 2,30 (d,
J= 10,3
Гц, 1Н); 2,39
-2,54 (m, 2H); 2,65 (br d, J=10,3 Гц, 1Н); 2,85 (dd, J1= 7,4 Гц, J2=14,5 Гц, 1Н); 3,16 (dd, J1=5,1 Гц, J2=14,2 Гц, 1Н); 3,79 (t, J=
7,6 Гц,
2H); 4,77 (s,
1H); 5,12 (d, J=10,2 Гц, 1H); 5,18 (d, J=16,0 Гц, 1Н); 5,71-5,84 (m, 1Н); 6,43 (d, J=7,6 Гц, 1Н); 6,57 (s, 1Н); 6,64 (d, J=8,0 Гц, 1Н); 7,02-7,33 (m, 10H); 9,32 (s, 1H).
Масс-спектр
(Cl-CH4
) m/e: 498 (М+1, 100%), 344 (23%), 153 (80%). δ +8,9o (этанол, с=1,1). Свободный амин (0,585 г) растворяли в этаноле и титровали этанольным раствором
хлористого
водорода до pH 4,0 с
последующим осаждением с помощью диэтилового эфира из дихлорметана, получая 0,479 г моногидрохлорида в виде гигроскопичного порошка беловатой окраски. Расч. для C32
H39N3O2 • HCl • 0,75 H2O: C, 70,18; H, 7,64; N, 7,67; Cl, 6,47. Обнаружено: C, 70,16; H, 7,73; N, 7,59; Cl, 6,51.
Пример
12
(+)-3-(
[α] ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(4-фторфенил)бензамид
4-Фтор-N-этиланилин [ЯМР (200 МГц, DМСО-d6):
Свободный амин (0,313 г) растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,95 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая 0,263 г моногидрохлорида в виде гигроскопичного белого порошка. Расч. для C31H36N3O2F • HCl • H2O: C, 66,95; H, 7,07; N, 7,56; Cl, 6,38. Обнаружено: C, 66,97; H, 7,10; N, 7,47; Cl, 6, 41.
Пример 13.
(+)-3-( [α] ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-метоксифенил)-N-метилбензамид
4-Метокси N-метиланилин связывали с 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3-(трет- бутилдиметилсилилокси)бензил)бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая (+)-3-( (αR)-α- ((2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)-N- (4-метоксифенил)-N-метилбензамид в виде порошка светло-пурпурной окраски, ЯМР (200 МГц, DМСО-d6): (αR)-α- 0,89 (d, J
=
6,0 Гц, 3H); 0,96 (d, J = 6,1 Гц, 3H); 1,66 (dd, J1 = 6,5 Гц, J2 = 11,0 Гц, 1H); 2,00 (dd, J1 = 7,1 Гц, J2 = 10,4 Гц, 1H); 2,27 (br d, J = 11,4 Гц, 1H);
2,
36-2,54 (m, 2H); 2,64 (d, J = 11,6 Гц, 1H); 2,82 (dd, J1 = 6,9 Гц, J2 = 13,6 Гц, 1H); 3,18 (dd, J1 = 5,4 Гц, J2 = 12,8 Гц, 1H); 3,30 (b, 3H); 3,68 (s,
3H);
4,76 (s, 1H); 5,11 (d, J = 10,6 Гц, 1H); 5,18 (d, J = 17,1 Гц 1H); 5,66-5,88 (m, 1H); 6,42 (d, J = 7,4 Гц, 1H); 6,58 (s, 1H); 6,63 (d, J = 7,4 Гц, 1H); 6,78 (d, J = 8,8 Гц, 2H); 6,97-7,24 (m,
7H); 9,
34 (s, 1H). Масс-спектр (Cl-CH4) m/e: 500 (M+1, 79%(, 346 (49%), 153 (100%). δ = + 96o (абс. этанол, c = 1,0). Свободный амин растворяли в этаноле и титровали
этанольным
раствором хлористого водорода до pH 4,0 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопичного порошка светло-пурпурной
окраски. Расч.
для
C31H37N3O3 • HCl • H2O: C, 67,19; H, 7,28; N, 7,58; Cl, 6,40. Обнаружено: C, 67,01; H 7,30; N, 7,53; Cl, 6,
42.
Пример 14
(+)-3-( [α] ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(2-фторфенил)-N-метилбензамид
2-Фтор-N-метиланилин [ЯМР (200 МГц, DМСО-d6): (αR)-α- 2,89 (s, 3H); 3,87 (br s, 1H); 6,59-6,
78
(m, 2H); 6,91-7,10 (m, 2H)] готовили из 2-фторанилина, связывали с 3-( δ ((2S,5R)-4-аллил- 2,5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил) бензоилхлоридом, снимали
защитные
группы и очищали методами, описанными в примере 3, получая 3-( (αR)-α- ((2S,5R)-4-аллил-2,5- диметил-1-пиперазинил)-3-оксибензил)-N-(2-фторфенил)-N-метилбензамид в виде
порошка
беловатой
окраски. ЯМР (200 МГц, DМСО-d6): (αR)-α- 0,92 (d, J = 6,1 Гц, 3H); 0,99 (d, J = 6,1 Гц. 3H); 1,69 (dd, J1 = 6,7 Гц, J2 = 10,8 Гц, 1H);
2,05
(dd, J1 = 7,6 Гц, J2 = 11,1 Гц, 1H); 2,30 (br d, J = 11,5 Гц, 1H); 2,41-2,52 (m, 2H); 2,68 (br d, J = 10,4 Гц, 1H); 2,83 (dd, J1 = 7,2 Гц, J2 = 13,8
Гц,
1H); 3,20 (dd,
J1 = 6,1 Гц, J2 = 14,2 Гц, 1H); 3,30 (s, 3H); 4,82 (s, 1H); 5,12 (d, J = 9,7 Гц, 1H); 5,18 (d, J = 15,8 Гц, 1H); 5,72-5,86 (m, 1H); 6,45 (d, J = 7,4 Гц, 1H);
6,56
(s, 1H); 6,66
(d, J = 8,0 Гц, 1H); 7,05-7,38 (m, 9H); 9,33 (s, 1H). Масс-спектр (Cl-CH4) m/e: 488 (M+1, 100%), 334 (45%), 153 (86%). δ = +2,02o (абс. этанол, с = 1,
1).
Свободный амин
растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 4,0 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в
виде
гигроскопического бежевого порошка. Расч. для C30H34N3O2F • HCl • 0,75H2O: C, 67,03; H 6,84; N, 7,82; Cl, 6,59. Обнаружено:
C, 67,
05; H, 6,86; N, 7,77; Cl, 6,67.
Пример 15
± 3-( [α] ((2S,
5R)-4-Аллил-2,5-диметил-1-пиперазинил) -3-оксибенил)-N-аллил-N-фенилбензамид
N-Аллиланилин [ЯМР (200 МГц, DMCO-d6): (αR)-α- 3,68 (t, J=5,2 Гц, 2Н); 5,
10 (d, J=10,2
Гц,
1Н); 5,23 (d, J=17,2 Гц, 1Н); 5,78 (br s, 1Н); 5,75-5,97 (m, 1Н); 6,52 (t, J=7,3 Гц, 2Н); 6,56 (d, J=7,8 Гц, 2H); 7,06 (t, J=7,3 Гц, 2H)] был приготовлен из анилина и аллобромида
через
трифторацетанилид с использованием общего метода, описанного в (Harland P.A., Hodge P., Maughan W. , Wildsmith E. Synthesis, 941, (1984).
N-Аллиланилин связывали с 3-( δ ((2S, 5R)-4-аллил-2,5-диметил-1- пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил)-бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая 3-( (α R)-α - ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)-N-аллил-N-фенилбензамид в виде порошка беловатой окраски. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,91 (d, J=6,3 Гц, 3H); 0,97 (d, J=5,8 Гц, 3H); 1,67 (dd, J1=6,7 Гц, J2= 10,6 Гц, 1H); 2,03 (dd, J1=7,0 Гц, J2=10,3 Гц, 1H); 2,29 (d, J=11,9 Гц, 1Н); 2,39-2,53 (m, 2Н); 2,67 (br d, J=11,2 Гц, 1Н); 2,83 (dd, J1=6,8 Гц, J2= 14,4 Гц, 1Н); 3,17 (dd, J1=5,2 Гц, J2=14,0 Гц, 1Н); 4,45 (d, J=5,5 Гц, 2Н); 4,78 (s, 1H); 5,11 (d, J=7,4 Гц, 1H); 5, 12 (d, J=8,5 Гц, 1H); 5,17 (d, J= 11,9 Гц, 1Н); 5,18 (d, J=15,3 Гц, 1Н); 5,71-5,98 (m, 2H); 6,42 (d, J=7,6 Гц, 1Н); 6,56 (s, 1Н); 6,65 (d, J=7,8 Гц, 1Н); 7,02-7,33 (m, 10H); 9, 33 (s, 1H). Масс-спектр (Cl-CH4) m/e: 496 (М+1, 45%), 342 (22%), 153 (100%). δ +6,0o (абс. этанол, с=1,1). Свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,8 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопичного беловатого порошка. Расч. для C32H37N3 O2 • HCl • H2O: C, 69,86; H, 7,33; N, 7,64; Cl, 6,44. Обнаружено: C, 69,94; H, 7,24; N, 7,62; Cl, 6,52.
Пример
16
± 3-( [α
] ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(циклопропил)метил-N-фенилбензамид
N-(Циклопропилметил) анилин [ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,21 (m, 2H); 0,51 (m, 2H);
1,01 (m, 1Н); 3,63 (d, J=7,3 Гц, 1Н); 3,80 (br s, 1Н); 5,78 (br s, 1Н); 7,18 (t, J=7,2 Гц, 1Н); 7,25 (d, J=7,8 Гц, 2H); 7,42 (t, J= 7,3 Гц, 2H)] был приготовлен из анилина и (бромметил)циклопропана
через трифторацетанилид с использованием общего метода, описанного в (Harland P. A., Hodge P., Vaughan W., Wildsmith E. Synthesis, 941, (1984)).
N-(Циклопропил)метиланилин связывали с 3-( δ ((2S,5R)-4-аллил- 2,5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил) бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая 3-( (αR)-α- ((2S,5R)-4-аллил-2,5-диметил- 1-пиперазинил)-3-оксибензил)-N-(циклопропил)метил-N-фенилбензамид в виде порошка беловатой окраски.
ЯМР (200 МГц, DMCO-d6 ): (αR)-α- 0,09 (m, 2H); 0,39 (m, 2H); 0,92 (d, J= 6,3 Гц, 3H); 0,96 (d, J=6,2 Гц, 3H); 1,28 (m, 1H); 1,69 (dd, J1=7,4 Гц, J2= 11,5 Гц, 1H); 2,04 (dd, J1 =6,6 Гц, J2=11,0 Гц, 1H); 2,30 (br d, J1=12,1 Гц, 1H); 2,40-2,54 (m, 2H); 2,67 (br d, J=9,8 Гц, 1H); 2,85 (dd, J1=7,4 Гц, J2= 13,7 Гц, 1H); 3, 16 (dd, J1=4,5 Гц, J2=14,7 Гц, 1H); 3,72 (d, J=7,0 Гц, 2H); 4,77 (s, 1H); 5,12 (d, J=10,0 Гц, 1H); 5,18 (d, J=15,6 Гц, 1H); 5,70-5,85 (m, 1H); 6,44 (d, J=7,3 Гц, 1H); 6,57 (s, 1H); 6, 65 (d, J=8,0 1H); 7,02-7,33 (m, 10H); 9,33 (s, 1H). Масс-спектр (Cl-CH4) m/e: 510 (M+1, 61%), 356 (42%), 153 (100%). δ = +8,9o (абс. этанол, c=1,1). свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,75 с последующим осаждением с помощью диэтилового эфира из дихлорэтана, получая моногидрохлорид в виде гигроскопического беловатого порошка. Расч. для C33H39N3O2 • HCl • 1,25H2O: C, 69,70; H, 7,53; N 7,39; Cl, 6,23. Обнаружено: C, 69, 82; H 7,52; N 7,36; Cl, 6, 28.
Пример 17
3-( [α] ((2S,
5R)-4-Аллил-2,
5-диметил-1-пиперазинил)-3-оксибензил)- N-изопропил-N-фенилбензамид
N-изопропиланилин [ЯМР (200 Мгц, DMCO-d6): (αR)-α- 1,13 (d, J=6,3 Гц, 6Н); 3,58 (m,
1H); 5,30 (d,
J=8,0 Гц, 1H); 6,49 (t, J=7,2 Гц, 1H); 6,55 (d, J= 7,8 Гц, 2H); 7,06 (t, J=7,6, 2H)] был приготовлен из анилина и ацетона с использованием восстановительного аминирования по общему
методу, описанному
в (Schellenberg К.A. J. Org. Chem., 28., 3259, (1963)).
N-Изопропиланилин связывали после этого с 3-( δ ((2S,5R)-4- аллил-2, 5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил) бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая 3-( (αR)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-оксибензил)- N-изопропил-N-фенилбензамид в виде порошка беловатой окраски. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,92 (d, J=6,1 Гц, 3H); 0, 99 (d, J=5,9 Гц, 3H); 1,11 (d, J=6,9 Гц,6H); 1,70 (dd, J1=7,2 Гц, J2=11,1 Гц, 1H); 2,07 (dd, J1=7,6 Гц, J2= 10,6 Гц, 1H); 2,33 (br d, J=9,9 Гц, 1H); 2, 42-2, 54 (m, 2H); 2,68 ( br d, J= 10,4 Гц, 1H); 2,85 (dd, J1=6,5 Гц, J2=13,9 Гц, 1H); 3,16 (dd, J1=4,9 Гц, J2= 14,1 Гц, 1H); 4,75 (s, 1H); 4,85 (m, 1H); 5, 10 (d, J=10,2 Гц, 1H); 5, 18 (d, J=16,8 Гц, 1H); 5,70-5,84 (m, 1H); 6,50 (d, J=7,1 Гц, 1Н); 6,59 (s, 1Н); 6,65 (d, J=8,2 Гц, 1Н); 7,03-7,32 (m, 10Н); 9,33 (s, 1Н). Масс-спектр (Cl-CH4) m/e: 498 (M+1, 100%), 344 (43%), 153 (76%). δ = +6,4o (абс. этанол, с= 1,4). Свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 4,0 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопического беловатого порошка. Расч. для C32H39N3O2 • HCl • 0, 5 H2O : C, 70,76; H, 7,61; N, 7,74; Cl, 6,53. Обнаружено: C, 71,01; H 7,83; N, 7,49; Cl, 6,41.
Пример 18
3-( [α] ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3-оксибензил) -N-циклопропил-N-фенилбензамид
N-Циклопропиланилин
получали с использованием подхода Бэртона для арилирования аминов (Darton D.H., Finet J.-P., Khamsi J. Tetrahedron Lett., 28, 887, (1987)). Циклопропиламин (1,0 г, 17,5 ммоля)
добавляли к
трифенилвисмуту (9,25 г, 21,0 ммоля) и ацетату меди (1,6 г, 8,75 ммоля) в дихлорметане (30 мл) при комнатной температуре в атмосфере азота. Смесь перемешивали в течение 18 часов,
фильтровали для
удаления всего нерастворимого материала через набивку из целита небольшой высоты и очищали хроматографией на колонке с силикагелем (4 см • 10 см), используя для элюирования
систему
растворителей гексан/этилацетат (95/5). Для получения N-циклопопиланилина (0,8 г) из фракции, содержащей продукт, удаляли под вакуумом все летучие соединения. ЯМР (200 МГц, DMCO-d6
):
(αR)-α- 0,37 (m, 2H); 0,68 (m, 2H); 2,30 (m, 1H); 63,3 (br s, 1H); 6,56 (t, J=7,4 Гц, 1H); 6,70 (d, J=8,2 Гц, 2H); 7,09 (t, J= 7,8 Гц, 2Н).
N-Циклопропиланилин связывали после этого с 3-( δ ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3, получая 3-( (αR)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)- N-циклопропил-N-фенилбензамид в виде желтого порошка. ЯМР (200 МГц, DMCO-d6): (α R)-α- 0,44 (m, 2H); 0,70 (m, 2H); 0,93 (d, J=6,1 Гц, 3H); 1,01 (d, J=5,7 Гц, 3H); 1,74 (dd, J1=7,7 Гц, J2=11,8 Гц, 1H); 2,05 (dd, J1= 6,8 Гц, J2=11,1 Гц, 1H); 2,39 (br d, J=10,5 Гц, 1H); 2,41-2,54 (m, 2H); 2,69 (br d, J=11,8 Гц, 1H); 2,83 (dd, J1=6,6 Гц, J2=13,6 Гц, 1H); 3,5-3,36 (m, 2H); 4,83 (s, 1H); 5,10 (d, J=9,8 Гц, 1H); 5,17 (d, J= 17,4 Гц, 1H); 5,70-5,86 (m, 1H); 6,57 (d, J=7,1 Гц, 1H); 6,63 (s, 1H); 6,65 (d, J= 8,2 Гц, 1H); 7,03-7,38 (m, 10H); 9,34 (s, 1H). Масс-спектр (Cl-CH4) m/e: 496 (М+1, 100%), 342 (45%), 153 (90%). δ +7,1o (абс. этанол, с=1,1). Свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 3,95 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопичного оранжевого порошка. Расч. для C32H37N3O2 • HCl • 1,50 H2O: C, 68,74; H, 7,39; N, 7,51; Cl, 6,34. Обнаружено: C, 68,56; H, 7,49; N 7,26; Cl, 6,37.
Пример 19 ± 3-( [α]= ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3- оксибензил)N-этил-N-(3-фторфенил)бензамид
3-Фтор-N-этиланилин [ЯМР
(DMCO-d6, 200
МГц): (αR)-α- 1,18 (t, 3=7,2 Гц, 3H); 3,02 (dq, J1= 7,2 Гц, J2=7,2 Гц, 2Н); 5,86 (br m, 1H); 6,24-6,42 (m, 3H);
7,07 (q, J=7,8 Гц, 1Н)]
готовили из 3-фторанилина и
уксусного ангидрида, связывали с 3-( δ ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)бензоилхлоридом,
снимали защитные группы и
очищали методами, описанными
в примере 3, получая ± 3-( (αR)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)
-N-этил-N-(3-фторфенил)бензамид в виде твердого
вещества белого цвета. ЯМР (200 МГц,
DMCO-d6): (αR)-α- 0,92 (d, J=6 Гц, 3H); 0,96 (d, J=6 Гц, 3H); 1,05 (t, J=7 Гц, 3H); 1,7
(m, 1H); 2,05 (m, 1H); 2,3 (m, 1H); 2,5 (m, 2Н);
2,7 (m, 1H); 2,9 (m, 1H); 3,2 (m, 1H);
3,9 (q, J=7 Гц, 2Н); 4,8 (s, 1H); 5,1 (d, J=10 Гц, 1H); 5,2 (d, J=16 Гц, 1H); 5,8 (m, 1H); 6,45 (d, J=8 Гц,
1Н); 6,6 (s, 1H); 6,65 (d, J= 8 Гц, 1Н); 6,9 (d, J=8
Гц, 1Н); 7,0- 7,2 (m, 3H); 7,2-7,4 (m,
5H); 9,35 (s, 1H). δ = +4,3o (абс. этанол, с=3,9). Расч. для C31H36
FN3O2 • HCl • 0,
5H2O: C, 68,06; J, 7,00; N, 7,
68; Cl, 6,48. Обнаружено: C, 68,10; H, 7,04; N, 7,63; Cl, 6,42. Масс-спектр (Cl-CH4) m/e: 502
(М+1, 39%), 501 (M, 9%), 348 (29%), 153 (100%).
Пример 20
±
3-( [α] ((2S, 5R)-4-Аллил-2,
5-диметил-1-пиперазинил)
-3-оксибензил)-N-(2-фторфенил)-N-пропилбензамид
2-Фтор-N-пропиланилин [ЯМР (DMCO-d6, 200 МГц): (αR)-α
- 0,93 (t, J=7,4 Гц, 3H); 1,59 (m, 2H);
3,04 (q, J=6,5 Гц, 2Н); 5,
33 (br m, 1H); 6,47-6,58 (m, 1H); 6,70 (t, J=8,1 Гц, 1Н); 6,93-7,05 (m, 2H)] готовили из 2-фторанилина и пропионового ангидрида,
связывали с 3-( δ ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)бензоилхлоридом, снимали защитные группы и очищали методами, описанными в примере 3,
получая (± 3-( (αR)-α- ((2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)- N-(2-фторфенил)-N-пропилбензамид в виде твердого вещества белого цвета. ЯМР (DMCO-d6,
200 МГц): (αR)-α- 0,9-1,05 (m, 9H);
1,5 (m, 2H); 1,7 (m, 1H); 2,05 (m, 1H); 2,3 (m, 1H); 2,5 (m, 2H); 2,7 (m, 1H); 2,85 (m, 1H); 3,2 (m, 1H); 3,7 (m, 2H); 4,8 (br s, 1H); 5,1 (d, J=10
Гц, 1H); 5,2 (d, J=16 Гц, 1H); 5,8 (m, 1H); 6,5 (d,
J=8 Гц, 1H); 6,6 (s, 1H); 6,65 (d, J=8 Гц, 1H); 7,0-7,4 (m, 9H); 9,3 (s, 1H). δ = +1,8o (абс. этанол, с=2,8). Расч. для C32H38FN3O2
• HCl • 0,25H2O: C, 69,05; H, 7,15; N, 7,55; Cl, 6,37. Обнаружено: C, 68,94; H, 7,19; N, 7,57; Cl, 6,41. Масс-спектр
(Cl-CH4) m/e: 516 (М+1, 93%), 515 (M, 29%),
362
(26%), 153 (100%).
Пример 21
± 3-( [α] ((2S,
5R)-4-Аллил-2,5-диметил-1-пиперазинил) -3-оксибензил)-N-этил-N-(2-фторфенил)бензамид
2-Фтор-N-этиланилин [ЯМР (DMCO-d6,
200 МГц): (αR)-α- 1,16 (t, J=7,1 Гц, 3H); 3,
11
(dq, J1= 7,2 Гц, J2=6,5 Гц, 2H); 5,30 (br m, 1H); 6,48-6,59 (m, 1H); 6,70 (t, J=8,5 Гц, 1H); 6,92-7,06 (m, 2H))]
готовили из 2-фторанилина и уксусного ангидрида, связывали с
3-(
δ ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)бензоилхлоридом, снимали защитные группы и
очищали методами, описанными в примере 3, получая ±
3-(
(αR)-α- ((2S,5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)-N-этил-N- (2-фторфенил)бензамид в виде воска
светло-желтой окраски. ЯМР (DMCO-d6, 200 МГц): (α
R)-α- 0,9 (d, J=6 Гц, 3H); 0,95 (d, J=6 Гц, 3H); 1,1 (J=7 Гц, 3H); 1,7 (m, 1H); 2,1 (m, 1H); 2,3 (m, 1H); 2,5 (m, 2H); 2,7
(m, 1H); 2,85 (m, 1H); 3,8 (br m, 2H);4,8 (br s, 1H); 5,1 (d, J=10 Гц,
1H); 5,2 (d, J=17 Гц, 1H); 5,8 (m, 1H); 6,45 (m, 1H); 6,5 (s, 1H); 6,65 (m, 1H); 7,0-7,4 (m, 9Н); 9,35 (s, 1H). δ = + 3,
4o (абс. этанол, с=2,04). Расч. для C31H36FN3O2 • HCl • H2O: C, 66,95; H, 7,07; N, 7,56; Cl, 6,38. Обнаружено: C, 66,
61; H, 7,14; N, 7,53; Cl, 6,40. Масс-спектр (Cl-CH4) m/e:
502 (M+1, 89%), 501 (M, 17%), 348 (36%), 153 (100%).
Пример 22
± 3-( [α]\ ((2S, 5R)-4-Аллил-2,5-диметил-1-пиперазинил) -3-оксибензил)-N-(3-фторфенил)-N-пропилбензамид
3-Фтор-N-пропиланилин [ЯМР (DMCO-d6, 200 МГц): (αR)-α- 0,
96 (t, J=7,3 Гц, 3H); 1,56 (m, 2H); 2,97 (q, J=6,9 Гц, 2H); 5,93 (br m, 1H); 6,22-6,43 (m, 3H); 7,06 (q, J=7,8 Гц,
1H)] готовили из 3-фторанилина и пропионового ангидрида, связывали с 3-( δ
((2S,5R)- 4-аллил-2,5-диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси) бензил)бензоилхлоридом, снимали защитные
группы и очищали методами, описанными в примере 3, получая ± 3-( (α
R)-α- ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-оксибензил)-N-(3- фторфенил)-N-пропилбензамид в виде
твердого вещества светло-бежевой окраски. ЯМР (DMCO-d6, 200 МГц): (α
R)-α- 0,9-1,05 (m, 9H); 1,5 (m, 2H); 1,7 (m, 1H); 2,05 (m, 1H); 2,3 (m, 1H); 2,5 (m, 2Н); 2,7 (m, 1H); 2,
85 (m, 1H); 3,8 (m, 2Н); 4,8 (s, 1H); 5,1 (d, J=10 Гц, 1H); 5,2 (d, J=16 Гц, 1H); 5,8
(m,
1H); 6,45 (d, J= 8 Гц, 1H); 6,6 (s, 1H); 6,7 (d, J=8 Гц, 1H); 6,9 (d, J=8 Гц, 1H); 7,0-7,4 (m, 9H); 9,3 (s,
1H). δ = +4,3o (абс. этанол, с=1,5). Расч. для C32H38FN3O2 • HCl • 0,75 H2O: C, 67,95; H, 7,22; N, 7,43; Cl, 6,
27. Обнаружено: C, 67,72; H, 7,19; N 7,49; Cl, 6,30. Масс-спектр (Cl-CH4)
m/e: 516 (M+1, 100%), 515 (M, 22%), 362 (30%), 153 (73%).
Примеры 23-24
Следующие
соединения могут быть приготовлены путем синтеза подходящего замещенного анилина (который
может быть получен из исходного анилина и соответствующего ангидрида карбоновой кислоты так же, как описано
в примере 3), связывания с 3-(( [α] ((2S, 5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- (трет-бутилдиметилсилилокси)бензил)бензоилхлоридом, снятия защитных групп и очистки методами,
описанными в примере 3. Моногидрохлориды, как и в примере 3, могут быть
получены с использованием этанольного раствора хлористого водорода.
3-( (αR)-α- ((2S,
5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(4-метоксифенил)-N-пропилбензамид
3-( (αR)-α- ((2S, 5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-этил-N-(4-метоксифенил)бензамид
Пример 25
3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- N-этил-(3-фторфенил)-N-метилкарбамоил)бензил)фенилмонофосфат ± 3-( (αR)-α- ((2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- оксибензил)-N-(3-фторфенил)-N-метилбензамид (пример 7, 0,05 г, 1,03 ммоля) растворяли в сухом пиридине (8 мл) в атмосфере азота. Раствор охлаждали до -10oC в
бане, содержащей метанол со льдом. К холодному раствору медленно добавляли 394 мг фосфорилхлорида. Реакционную смесь оставляли для нагревания до комнатной температуры и перемешивали в течение ночи в
атмосфере азота.
К раствору по каплям добавляли 2 мл воды. Раствор перемешивали в течение пятнадцати минут и все летучие соединения удаляли под вакуумом. С помощью ионной спрей масс-спектрометрии было установлено, что полученный неочищенный твердый остаток преимущественно представляет собой 3-( (αR)-α- ((2S,5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(N-фторфенил)- N-метилкарбамоил)бензил)фенилмонофосфат. (ISMS М+H пик = 568,1). Фосфат может быть выделен в виде моноаммониевой соли с помощью ионообменной хроматографии.
Пример 26
± 3-((αR)-α- ((2S,5R)-4-Аллил-2,5-диметил-1-пиперазинил)-3-метоксибензил)- N-(3-фторфенил)-N-метилбензамид
Неочищенный
3-(((2R,
5S)-4-аллил-2,5-диметил-1-пиперазинил)-3- (1Н-бензотриазол-1-ил)метил)-N-(3-фторфенил)-N-метилбензамид получали из
(2R,5S)-1-аллил-2,5-диметилпиперазина (1,89 г), бензотриазола (1,39 г) и
N-(3-фторфенил)-3-формил-N-метилбензамида (3,0 г) в толуоле так же, как описано в примере 7.
3-Броманизол (4,36 г) растворяли в сухом тетрагидрофуране (40 мл) и охлаждали в атмосфере азота до -78oC. К раствору медленно с помощью шприца добавляли н-бутиллитий в гексане (9,2 мл 2,5 М раствора). В процессе перемешивания в течение 25 минут при -78oC раствор становился белым и отчасти мутным. Раствор переносили с помощью раздвоенной иглы в колбу, содержащую etherate бромида магния (6,02 г) в тетрагидрофуране (60 мл) и перемешивали при комнатной температуре в течение 1 часа. Неочищенный 3-(((2R,5S)-4-аллил-2,5-диметил-1-пиперазинил)-3-(1H-бензотриазол-1- ил)метил)-N-(3-фторфенил)-N-метилбензамид в толуоле добавляли к только что приготовленному реагенту - бромиду арилмагния. Раствор слегка нагревался в процессе добавления и приобретал мутную желто-коричневую окраску. После перемешивания при комнатной температуре в течение 2, 5 часа к раствору аккуратно добавляли 0,5 М водный раствор соляной кислоты до тех пор, пока его pH не достигал значения, равного 5. Продукт экстрагировали с помощью 100 мл этилацетата и растворитель удаляли под вакуумом. Остаток растворяли в 25 мл 3 H водного раствора соляной кислоты при комнатной температуре, добавляли диэтиловый эфир и отделяли кислотный водный слой. Водный слой промывали второй раз диэтиловым эфиром и подводили его pH до значения 10 с помощью водного раствора гидроксида натрия. Продукт экстрагировали этилацетатом. Этилацетатные порции объединяли, промывали разбавленным раствором гидроксида натрия для удаления всех остатков бензотриазола, промывали насыщенным раствором хлорида натрия, высушивали над сульфатом натрия и упаривали при пониженном давлении. Полученный неочищенный продукт очищали с помощью хроматографии на колонке с силикагелем, используя 1% этанол в дихлорметане в качестве элюента и получая 1,71 г ± 3-(( (αR)-α - ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-метоксибензил)-N- (3-фторфенил)-N-метилбензамида в виде твердого кристаллического вещества белого цвета с чистотой относительно изомерного состава, превышающей 98% (по данным ВЖХ, проведенной на колонке m-Bondapak C-18 (125 A, 3,9 • 300 мм, Waters Chromatography Division, Millipore Corporation, Milford, Vassachusetts), используя 70% метанол и 30% 0,1 М водный ацетат аммония при скорости протекания 1 мл/мин. ЯМР (200 МГц, DMCO-d6): (αR)-α- 0,91 (d, J=6,0 Гц, 3H); 1,00 (d, J=6,2 Гц, 3H); 1,69 (dd, J1=7,1 Гц, J2=11,0 Гц, 1H); 2,05 (dd, J1= 7,5 Гц, J2=11,0 1H); 2,31 (br d, J=9,3 Гц, 1H); 2,42-2,53 (m, 2H); 2,69 (br d, J=11,2 Гц, 1H); 2,85 (dd, J1 -7, 0 Гц, J2=14,1 1H); 3,18 (dd, J1= 5,5 Гц, J2=13,5 Гц, 1H); 3,37 (s, 3H); 3,74 (s, 3H); 4,88 (s, 1H); 5,12 (d, J= 10,0 Гц, 1H); 5,18 (d, J=15,7 Гц, 1H); 5,70 -5,83 (m, 1H); 6,58 (d, J=7,6 Гц, 1H); 6,70 (s, 1H); 6,84 (d, J=8,2 Гц, 1H); 6,94 (t, J=7,8 Гц, 1H); 7,02-7,14 (m, 2H); 7,18-7,14 (m, 6H); 9,31 (s, 1H). Масс-спектр (Cl-CH4) m/e: 502 (m+1, 100%), 348 (81%), 153 (12%). δ = +7,73o (абс. этанол, с=1,1). Свободный амин растворяли в этаноле и титровали этанольным раствором хлористого водорода до pH 4,0 с последующим осаждением с помощью диэтилового эфира из дихлорметана, получая моногидрохлорид в виде гигроскопического порошка светло-желтой окраски. Расч. для C13H36N3O2F • HCl • 0,5H2O: C, 68,06; H, 7,00; N, 7,68; Cl, 6,48. Обнаружено: C, 68,13; H, 7,12; N, 7,55; Cl, 6,35.
Пример 27
± 3-( [α
] ((2S,5R)-4Аллил-2,5-диметил-1-пиперазинил)
-3-метоксибензил)-N-этил-N-(4-фторфенил)бензамид
Соединение
получали из неочищенного 3-(((2R,5S)-4-аллил-2,
5- диметил-1-пиперазинил)-3-(1Н-бензотриазол-1-ил)метил)-N-этил-N- (4-фторфенил)бензамида (пример 12, infra) и 3-броманизола с помощью
методов, описанных
в примере 7. ЯМР (200 МГц, DMCO-d6
): (αR)-α- 0,91 (d, J=6,2 Гц, 3H); 0,99 (d, J=6,3 Гц, 3H); 1,08(t, J=7,0 Гц, 3H); 1,71 (dd, J1=7,0 Гц, J2=11,1 Гц, 1H);
2,03 (dd, J1= 7,1 Гц, J2=10,9 Гц, 1Н); 2,31 (d, J=11,2 Гц, 1Н); 2,40-2,57 (m, 2Н); 2,67 (d, J=11,5 Гц, 1Н); 2,84 (dd, J1=6,6 Гц, J2
=13,9 Гц, 1Н); 3,17 (dd,
J1=5,5 Гц, J2
=13,9 Гц, 1Н); 3,74 (s, 3H); 3,83 (q, J=7,0 Гц, 2H); 4,83 (s, 1H); 5,11 (d, J=10,2 Гц, 1H); 5,18 (d, J=16,4 Гц, 1H); 5,63-5,85 (m, 1H); 6,
60 (d, J=7,4 Гц, 1Н); 6,71
(s, 1H); 6,84 (d, J=8,2 Гц, 1Н);
7,02-7,28 (m, 9H). Масс-спектр (Cl-CH4) m/e: 516 (М+1, 38%), 362 (100%), 153 (16%).
Пример 28
Отдельные соединения,
предусмотренные настоящим изобретением,
идентифицированные ниже со ссылками на примеры с их синтезом, были оценены на их опиоидную рецепторную активность в опытах in vitro в
различных рецепторных
системах, включающую дельта-рецепторный
агонизм на сосуде семявыводящего протока мыши (Семявыводящий сосуд мыши ЭД50) и мю-рецепторный агонизм на подвздошной кишке
морской свинки
(Подвздошная кишка морской свинки ЭД50
).
Методы анализа, использованные в ходе таких определений рецепторной активности, излагаются ниже.
Биоанализы in vitro: Семявыводящий сосуд извлекали из мыши и подвешивали между платиновыми электродами с натяжением 0,5 г в орган-омываемых камерах, содержащих модифицированный буферный раствор Кребса, имеющий следующий состав (миллимолярный): NaCl, 118; KCl, 4,75; CaCl2, 2,6; KH2PO4, 1,20; NaHCO3, 24,5 г и глюкоза, 11. Буферный раствор насыщали смесью 95% O2 /5% CO2 и термостатировали при 37oC. Ткани стимулировали сверхвысоким напряжением в виде серий импульсов частотой 10 Гц продолжительностью в 400 мсек; интервал между сериями - 10 секунд; длительность импульса - 0,5 мсек. Интактные подвздошные кишки (длиной примерно 3 см) извлекали из морской свинки и подвешивали с натяжением 1 г в омываемой камере так же, как было описано для семявыводящего сосуда. Модифицированный буферный раствор Кребса дополнительно содержал в этом случае MgSO4 (1,20 мМ). Кишки стимулировали электрическими прямоугольными волновыми импульсами частотой 0,1 Гц, длительностью 0,5 мсек при сверхвысоком напряжении. Определяли процент ингибирования индуцируемого электричеством мышечного сокращения при различных значениях концентраций соединений, предусмотренных изобретением. Величины ЭД50 находили путем экстраполяции кривых, выражающих зависимость величины ответа от дозовой концентрации (Lord J. A.H., Waterfield A.A., Hughes J., Kosterlitz H.W. Nature, 267, 495 (1977)).
Результаты приведены ниже в таблице А (см. в конце описания).
Пример 29
Обезболивающую активность
измеряли с
помощью анализа на ущемление хвоста крыс (мужские особи линии Spraque-Dawley CD, массой приблизительно 300 г) после внутривенной инъекции в хвостовую вену. Группе
из 6-8 животных
внутривенно вводили
соединение, предусмотренное изобретением, в концентрации 1-5 мг/мл в стерильном 5% растворе декстрозы. Через пять минут после инъекции на хвост примерно в 1 дюйме
от его конца
помещали на короткий
промежуток времени (максимум 20 секунд) артериальный зажим (Fisher Scientific Co., самозакрывающийся артериальный зажим, каталожный номер # 08-905), вызывая за счет
сдавливания
болезненные ощущения.
Ноцицептивный ответ оценивали по признакам, выражающим дискомфорт, таким как беготня, пищание или кручение на месте с целью укусить зажим. Для каждого из соединений,
предусмотренных изобретением,
строили кривую зависимости ответа от дозы введенного соединения. Обезболивающую активность (половинную максимальную эффективную дозу, ЭД50) определяли как
дозу,
при введении которой
половина животных не показывала никакого ноцицептивного ответа на введение на 20 секунд артериального зажима. Антиноцицептивные ЭД50 дозы составляли 0,03 мг/кг
для
соединений из примеров
7 и 12.
Фармацевтические препараты
В нижеследующих примерах на составы препаратов термин "Активный ингредиент" может означать любое из соединений
по
изобретению, например
соединение, отвечающее формулам (I) и (II).
Пример 30
Препарат в форме таблеток
Препараты А, Б и В (см. рецепты в конце описания) готовили
посредством влажной
грануляции ингредиентов с раствором повидона с последующим добавлением стеарата магния и штампованием.
Нижеследующие препараты Г и Д готовили посредством прямого штампования перемешанных ингредиентов.
Рецептура Г - мг/на таблетку
Активный ингредиент - 100
Преджелатинизированный крахмал NF15 - 50/150
Рецептура Д
- мг/на
таблетку
Активный ингредиент - 100
Лактоза - 150
Авицель - 100/350
Рецептура Е (с контролируемым временем выделения)
Лекарственное средство
готовили
посредством влажной грануляции нижеследующих ингредиентов с раствором повидона с последующим добавлением стеарата магния и штампованием. - мг/на таблетку
(а) Активный ингредиент
- 500
(б) Гидроксипропилметилцеллюлоза (Метоцель K4M Premium - 112
(в) Лактоза В.Р. - 53
(г) Повидон В.Р.С. - 28
(д) Стеарат магния - 7/500
Лекарство
выделяется в
течение 6-8 часов и полностью процесс выделения завершается после 12 часов.
Пример 31
Фармацевтический препарат в форме капсул
Рецептура А
Препарат в
форме капсул готовят путем перемешивания ингредиентов описанного выше препарата Г из примера 30 и заполнения ими состоящих из двух частей твердых желатиновых капсул.
Рецептура Б
- мг/на капсулу
(а) Активный ингредиент - 100
(б) Лактоза В.Р. - 143
(в) Натриевая соль гликолата крахмала - 25
(г) Стеарат магния - 2/270
Капсулы
готовят путем перемешивания указанных выше ингредиентов и заполнения ими состоящих из двух частей твердых желатиновых капсул.
Рецептура В - мг/на капсулу
(а)
Активный
ингредиент - 100
(б) Макрогель 4000 ВР - 350/450
Капсулы готовят путем расплавления Макрогеля 4000 ВР, диспергирования активного ингредиента в полученном расплаве и
заполнения
расплавом состоящих из двух частей твердых желатиновых капсул.
Рецептура Г - мг/на капсулу
(а) Активный ингредиент - 100
(б) Лецитин - 100
(в)
Арахисовое
масло - 100/300
Капсулы готовят путем диспергирования активного ингредиента в лецитине и арахисовом масле и заполнения полученной дисперсией мягких, эластичных желатиновых
капсул.
Рецептура Д (капсула с контролируемым временем выделения)
Нижеследующее лекарственное средство в форме капсулы с контролируемым временем выведения готовят с помощью
выдавливания
ингредиентов (а), (б) и (в) с использованием экструдера с последующим приобретением экструдатом сферической формы и его высушиванием. Высушенные шарики покрывают мембраной (г),
контролирующей
высвобождение, и наполняют ими состоящих из двух частей твердые желатиновые капсулы. - мг/на капсулу
(а) Активный ингредиент - 250
(б) Микрокристаллическая целлюлоза
- 125
(в) Лактоза В.Р. - 125
(г) Этилцеллюлоза - 13/513
Пример 32
Препараты для инъекций
Рецептура А
Активный ингредиент - 5,0 мг
Раствор соляной
кислоты, 0,1 М - количество, необходимое для подведения pH в интервал 4,0 - 7,0
Раствор гидроксида натрия, 0,1 М - количество, необходимое для подведения pH в интервал 4,0-7,
0
Стерильная вода - до 10 мл
Активный ингредиент растворяют в большей части воды (35-40oC) и подводят значение pH раствора в интервал от 4,0 до 7,0, используя раствор
соляной
кислоты
либо гидроксида натрия. Смесь доводят водой до нужного значения объема и фильтруют через стерильный микропористый фильтр в стерильные виалы желтого стекла вместимостью 10 мл,
укупоривают
стерильными
пробками и сверху герметизируют крышками.
Рецептура Б
Активный ингредиент - 12,5 мг
Стерильный апирогенный фосфатный буферный раствор, pH 7
- до 25
мл
Пример 33
Препарат для внутримышечных инъекций
Активный ингредиент - 4,0 мг
Бензиловый спирт - 0,1 г
Гликофурал 75 - 1,45 г
Вода для
инъекций
- до 4,00
мл
Активный ингредиент растворяют в гликофурале. После этого добавляют и растворяют бензиловый спирт и раствор доводят водой до объема 4 мл. Полученную смесь фильтруют
через
стерильный
микропористый фильтр и герметизируют в стерильные виалы желтого стекла.
Пример 34
Сироп:
Активный ингредиент - 0,025 г
Раствор сорбита - 0,
10
г
Глицерин - 2,00 г
Бензоат натрия - 0,005 г
Ароматизатор, Персик 17.42.3169 - 0,0125 мл
Очищенная вода - до 5,00 мл
Активный ингредиент растворяют в
смеси глицерина
и большей части очищенной воды. После этого к раствору добавляют водный раствор бензоата натрия с последующим добавлением раствора сорбита и ароматизатора. Смесь доводят очищенной
водой до нужного
объема и хорошо перемешивают.
Пример 35
Суппозиторий: - мг/на суппозиторий
Активный ингредиент - 30
Твердый жир, ВР (Витепзол H15
- Динамит Нобеля)
- 1970/2000
Одну пятую часть Витепзола Н15 расплавляют в емкости с рубашкой для пара при температуре до 45oC. Активный ингредиент просеивают через 200 мм сито и
добавляют к
расплавленной основе, перемешивая с помощью Silverson, снабженного режущей головкой, до тех пор, пока не образуется однородная дисперсия. Продолжая поддерживать температуру смеси на
уровне 45oC, добавляют оставшуюся часть Витепзола Н15 и перемешивают до получения гомогенной смеси. Готовую суспензию пропускают через 250 мм сито нержавеющей стали и охлаждают при
постоянном
перемешивании до 40oC. При температуре от 38oC до 40oC по 2 г смеси вводят в подходящие пластиковые формы объемом по 2 мл. Суппозитории оставляют для
охлаждения до
комнатной температуры.
Пример 36
Ниже приведен пример препаратов в форме пессариев, содержащих по меньшей мере одно из диарилметилпиперазиновых соединений по
настоящему
изобретению.
Пессарии: - мг/на пессарий
Активный ингредиент - 30
Безводная декстроза - 490
Картофельный крахмал - 473
Стеарат магния
- 7/1000
Перечисленные выше ингредиенты перемешиваются непосредственно и из полученной смеси прямым штампованием получают пессарии.
Пример 37
Ниже приведены
дополнительные
примеры препаратов, в которых могут быть полезными соединения, предусмотренные изобретением, включая дозированные препараты в виде суспензий для орального введения, суспензий для
инъекций, распыляемых
суспензий, препараты в виде аэрозолей, в виде порошков для ингаляций и в виде назальных капель.
Таблетка: - 25,0 мг
Соединение формулы (I) - 25,0
мг
Лактоза ВР - 48,
5 мг
Микрокристаллическая целлюлоза ВР ("Авицель pH 101") - 10,0 мг
Слабо замещенная оксипропилцеллюлоза ВР (LHHC LH-11") - 10,0 мг
Натриевая
соль
гликолата крахмала ВР
("Explotab") - 3,0 мг
Повидон ВР ("K 30") - 3,0 мг
Стеарат магния ВР - 0,5 мг - 100,0 мг
Суспензия для орального введения:
Соединение
формулы
(I) - 50,0 мг
Авицель RC 591 - 75,0 мг
Сахарный сироп - 3,5 мл
Метилоксибензоат - 5,0 мг
Краситель 0,01% в/о
Вишневый ароматизатор - 0,1% о/о
Твин 80 - 0,2% о/о
Вода до 5,0 мл
Суспензия для инъекций:
Соединение формулы (I) - 1,5 мг
Поливинилпирролидон (ПВП) - 170,0 мг
Твин 80 - 0,2%
о/о
Метилоксибензоат - 0,1%
в/о
Вода для инъекций - до 3,0 мл
Препарат в форме капсул:
Соединение формулы (I) - 1,5 мг
Крахмал 1500 - 150,0 мг
Стеарат магния
- 2,5 мг
Наполните описанным выше препаратом капсулу из твердого желатина.
Суспензия для распыления:
Соединение формулы (I), стерильное - 1,0 мг
Вода для
инъекций - до 10,0
мл
Диспергируйте соединение формулы (I) в воде для инъекций, предварительно простерилизованное в стерильном контейнере. Заполните смесью в стерильных
условиях стеклянные
ампулы (10 мл на
ампулу) и заплавьте каждую из ампул.
Препарат в форме аэрозоля:
Соединение формулы (I), измельченное - 1,0 мг
Аэрозольный
диспергатор - до 5,0
мл
Суспендируйте измельченное на микронной мельнице соединение формулы (I) в аэрозольном диспергаторе. Заполните суспензией под давлением заранее приготовленные
аэрозольные канистры (5 мл на
канистру)
через отверстие крана.
Порошок для ингаляций:
Соединение формулы (I), измельченное - 1,0 мг
Лактоза - 29,0 мг
Разотрите в порошок и смешайте
измельченное на
микронной мельнице соединение формулы (I) и лактозу. Заполните полученным порошком капсульные оболочки из твердого желатина (30 мг на капсулу).
Назальные капли
Соединение
формулы (I) - 20,0 мг
Метилоксибензоат - 10,0 мг
Вода для инъекций - до 10,0 мл
Диспергируйте соединение формулы (I) и
метилоксибензоат в воде для
инъекций. Заполните
полученной суспензией подходящие бутылки с капельницами (10 мл на бутылку) и закройте, соединив капельницу с крышкой бутылки.
Пример
38
Нижеследующий
препарат, содержащий в
качестве активного ингредиента по меньшей мере одно из соединений, предусмотренных изобретением, может быть применен для микроинфузного вливания.
Препарат для
микроинфузного
вливания
Активный ингредиент - 10,0 мг
Хлорид натрия - 16,0 г
Раствор соляной кислоты, 0,1 М - количество, необходимое для
подведения pH в интервал от 4,0 до
7,0
Раствор гидроксида натрия, 0,1 М - количество, необходимое для подведения pH в интервал от 4,0 до 7,0
Стерильная вода - до 20 мл
Активный ингредиент и хлорид натрия
растворяют в
большей части воды (35-40oC) и подводят значение pH раствора в интервал от 4,0 до 7,0, используя раствор соляной кислоты либо гидроксида
натрия. Смесь доводят водой до нужного
объема и
фильтруют через стерильный микропористый фильтр в стерильные виалы желтого стекла вместимостью 20 мл, укупоривают стерильными пробками и сверху
герметизируют крышками.
Пример 39
Трансдермальное введение
Составы, содержащие в качестве активного ингредиента соединения формулы (I), могут применяться для
трансдермального введения, например как
трансдермальные
пластыри.
Пластыри, пропитанные или несущие каким-либо другим способом препараты для трансдермального применения помещают на теле таким образом, чтобы они оставались в контакте с кожей пациента в течение длительного периода времени.
Подобные пластыри содержат активный ингредиент (1) в забуференном оптимальным образом водном растворе, (2) растворенный и/или диспергированный в связующем, обеспечивающем адгезию, или (3) диспергированном в полимере.
Подходящая концентрация активного ингредиента составляет приблизительно от 1% до 35%, предпочтительно от 3% до 15%.
Одним из примеров применения может быть введение активного соединения с пластыря путем электротранспорта или ионофореза, как описано в общем виде в литературе (Pharmaceutical Research, 3(6), 318, (1986)).
Пример 40
Отдельный пример препарата, содержащего
соединение, отвечающее изобретению, для
трансдермального применения
приводится ниже.
Препарат для трансдермального применения
Активный ингредиент - 200,0 мг
Спирт
USP - 0,1 мл
Оксиэтилцеллюлоза
Из активного
ингредиента и спирта USP с помощью оксиметилцеллюлозы образуют гель, из которого формируют приспособление для трансдермального введения
с площадью поверхности
10 см2.
Пример 41
(+)-3-( δ ((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- гидроксибензил)-N-бензил-N-метилбензамид
Это
соединение получают из
3-( (αR)-α- ((2S,
5R)-4-аллил-2,5- диметил-1-пиперазинил)-3-(трет-бутилдиметилсилилокси)бензил) бензоилхлорида и N-бензил-N-метиламина методом получения бензамида,
описанным в примере 2.
ЯМР (300 МГц, DМСО-d6
, 121oC): d 0,93 (d, J = 6,2 Гц, 3H); 1,06 (d, J = 6,2 Гц, 3H); 1,96 (dd, J1 = 6,7 Гц, J2 = 11,0 Гц, 1H); 2,
12 (dd, J1 = 7,
0 Гц, J2 = 11,0 Гц,
1H); 2,58 (dd, J1 = 2,9 Гц, J2 = 11,0 Гц, 1H); 2,67-2,84 (m, 3H); 2,86 (s, 3H); 2,89 (dd, J1 = 6,6 Гц, J2 = 13,5 Гц, 1H); 3,
18 (dd, J1 = 4,0 Гц,
J2 = 14,1 Гц, 1H); 4,58 (s, 2H); 4,98 (s, 1H); 5,08 (d, J = 10,2 Гц, 1H); 5,16 (d, J = 17,3 Гц, 1H); 5,74-5,89 (m, 1H); 6,62-6,74
(m, 3H); 7,10 (t, J = 7,8
Гц, 1H); 7,21-7,50 (m, 9H); 8,76
(s, 1H). Масс-спектр (Cl-CH4) m/e: 484 (M+1,88%), 330 (33%), 153 (100%). (αR)-α- = +14.3o (этанол, c =
1,25). Свободный амин
растворяют в этаноле и титруют
этанольным хлороводородом до pH 3,4, а затем осаждают диэтиловым эфиром из дихлорметана с получением (+)-3-( [α] (2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N- бензил-N-метилбензамида моногидрохлорида в виде
гигроскопического
светло-желтого порошка. Расч. для C31H37N3O2 HCl 0,75 H2O: C, 69,78; H, 7,46; N, 7,87; Cl, 6,64. Найдено: C, 69,76; H, 7,48;
N, 7,69; Cl, 6,
74.
Пример 42
(+)-3-( (αR)-α- (2S,5R)-4-н-пропил-2,5-диметил-1-пиперазинил)-3- гидроксибензил)-N-(3-фторфенил)-N-метилбензамид
Это
соединение получают
путем каталитического гидрирования
соединения из примера 7 в атмосфере водорода в присутствии катализатора Линдлера (Lindler's) в толуольной растворяющей среде.
Пример 43
(+)-3-( (αR)-α- (2S,
5R)-4-аллил-2,5-диметил-1-пиперазинил)-3- гидроксибензил)-N-(3,4,5-трифторфенил)-N-метилбензамид
Это соединение получают способом из
примера 7, но с
использованием 3,4,5-трифторанилина вместо
3-фторанилина.
Пример 44
(+)-3-( (αR)-α- (2S,5R)-4-аллил-2,
5-диметил-1-пиперазинил)-3- гидроксибензил)-N-(3,
5-дифторфенил)-N-метилбензамид
Это
соединение получают способом из примера 7, но с использованием 3,5-дифторанилина вместо 3-фторанилина.
Пример 45
Отдельные
соединения, предусмотренные настоящим изобретением,
идентифицированные ниже со ссылками на примеры с их синтезом, были оценены на их опиоидную рецепторную
активность в опытах in vitro в различных
рецепторных системах, включающую дельта-рецепторный
агонизм на сосуде семявыводящего протока мыши (семявыводящий сосуд мыши ЭД50) и мю-рецепторный
агонизм на подвздошной кишке морской
свинки (подвздошная кишка морской свинки ЭД50
).
Методы анализа, использованные в ходе таких определений рецепторной активности, излагаются ниже.
Биоанализы in vitro: Семявыводящий сосуд извлекали из мыши и подвешивали между платиновыми электродами с натяжением 0,5 г в орган-омываемых камерах, содержащих модифицированный буферный раствор Кребса, имеющий следующий состав (миллимолярный): NaCl, 118; KCl, 4, 75; CaCl2, 2,6; KH2PO4, 1,20; NaHCO3, 24,5 и глюкоза, 11. Буферный раствор насыщали смесью 95% O2/5% CO2 и термостатировали при 37oC. Ткани стимулировали сверхвысоким напряжением в виде серий импульсов частотой 10 Гц продолжительностью в 400 мсек; интервал между сериями - 10 секунд; длительность импульса - 0,5 мсек. Интактные подвздошные кишки (длиной примерно 3 см) извлекали из морской свинки и подвешивали с натяжением 1 г в омываемой камере так же, как было описано для семявыводящего сосуда. Модифицированный буферный раствор Кребса дополнительно содержал в этом случае MgSO4 (1,20 мМ). Кишки стимулировали электрическими прямоугольными волновыми импульсами частотой 0,1 Гц, длительностью 0,5 сек при сверхвысоком напряжении. Определяли процент ингибирования индуцируемого электричеством мышечного сокращения при различных значениях кумулятивных концентраций. Величины ЭД50 находили путем экстраполяции кривых, выражающих зависимость величины ответа от дозовой концентрации (J.A.H. Lord, A.A. Waterfield, J. Hughes, H.W. Kosterlitz, Nature 267, 495, (1977)).
Результаты приведены ниже в Таблице Б. экспериментов.
Пример 46
Обезболивающую активность
измеряли с помощью анализа на ущемление хвоста крыс (мужские особи штамма
Sprague-Dawley CD, масса приблизительно 300 г) после подкожной инъекции. Группе из 6-8 животных
подкожно инъецировали
соединение в концентрации 1-5 мг/мл в стерильном 5%-ном растворе декстрозы. Через
пять минут после инъекции на хвост примерно в 1 дюйме от его конца помещали на короткий
промежуток времени (максимум
20 секунд) артериальный зажим (Fisher Scientific Co., самозакрывающийся
артериальный зажим. Каталожный номер # 08-905), вызывая за счет сдавливания болезненные ощущения.
Ноцицептивный ответ оценивали
по признакам, выражающим дискомфорт, таким как беготня, пищание или
кручение на месте с целью укусить зажим.
Результаты
Соединения из примеров
2, 3 и 5 продуцировали сильный
аналгезирующий эффект после подкожной инъекции в дозе 5 мг/кг массы
тела.
Пример 47
Процедуру из примера 46 повторяют с соединением из
примера 41 с тем исключением, что
делают не подкожную, а внутривенную инъекцию. Аналгезирующую активность
(полумаксимальная эффективная доза, ЭД50) определяют по дозе, при которой
половина животных не демонстрирует
никакого ноцицептивного ответа на сдавливание артериальным зажимом в течение 20
секунд. Антиноцицептивная ЭД50 доза составляет 0,12 мг/кг для соединения
из примера 41.
Способы осуществления изобретения
Преимущественный способ осуществления
изобретения включает в себя синтез и использование предпочтительных соединений по
изобретению (произведенных любым
подходящим методом синтеза, как например описанный выше бензотриазольный путь
синтеза), например, соединения из группы, включающей в себя соединения, обозначенные А,
Б, В, Г, Д, Е, Ж и З, и его
фармацевтически приемлемых эфиров, солей и других физиологических значимых
производных для лечения таких состояний и расстройств, как: физиологическая боль, диарея,
недержание мочи, психические
заболевания, хронический алкоголизм и привыкание к чрезмерному употреблению
лекарств, отек легких, депрессия, астма, эмфизема и удушье, когнитивные и желудочно-кишечные
расстройства.
В рамках предшествующего изложения типичным способом осуществления изобретения в смысле использования соединений по изобретению является их введение в фармацевтически безопасной и эффективной дозе и подходящей дозовой форме объектам животного происхождения, например человеку, с целью вызывания у таких объектов аналгезии.
Наиболее предпочтительным видом из соединений, предусмотренных изобретением, является соединения Ж, ± 3-( (α R)-α- ((2S,5R)-4-аллил- 2, 5-диметил-1-пиперазинил)-3-оксибензил)-N-(3-фторфенил)- N-метилбензамид.
Промышленная применимость
Соединения по настоящему изобретению
представляют собой соединения, с
высокой селективностью связывающиеся с опиодными рецепторами, полезные в этом качестве,
например как агонист/антагонист-составляющие пар конъюгатов для
проверки/анализа действия рецепторов и
нейротрансмиттеров.
Соединения, предусмотренные изобретением, включают бензгидрилпиперазиновые соединения, которые могут быть использованы в качестве способствующих аналгезии, равно как и соединения, способные найти применение при лечении таких состояний, как привыкание к чрезмерному употреблению лекарственных средств, хронический алкоголизм, передозировка лекарств, психические заболевания, недержание мочи, диарея, отек легких, кашель и расстройства дыхания.
Наиболее предпочтительное соединение в рамках настоящего изобретения, ± 3-( (α R)-α- ((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-оксибензил)-N- (3-фторфенил)-N-метилбензамид, является смешанным дельта/мю-опиоидным агонистом, обладающим существенным преимуществом над различными известными мю-рецепторными соединениями, употребляемыми в качестве аналгетиков.
Реферат
Описываются новые пиперазиновые соединения общей формулы I, где один из R8 и R9 представляет собой фенил, который может быть замещен одним или более заместителями, выбранными из группы, включающей галоген, С1 - С3-алкокси, С1-С3-алкил и трифторметил, а другой из R8 и R9 представляет собой водород, С1-С6-алкил, С3 - С6-циклоалкил, С3-С6-алкенил или С3-С6 -алкинил; любые два из R3, R4 и R5 представляют собой метил, а другой радикал - водород; R6 представляет собой атом водорода, С1-С6 -алкил, С3-С6-циклоалкил, С3-С6-алкенил, или С3-С6-алкинил; а также его фармацевтически приемлемые простой, сложный эфиры, соль или его физиологически функциональное производное. Соединение формулы 1 могут использоваться в качестве агонист/антагонист-составляющих пар коньюгатов в целях мониторинга трансдукции и исследования функции нейротрансмиттеров, а также они демонстрируют разнообразное терапевтическое применение, включая опосредование аналгезии, и возможность использования при лечении диареи, недержания мочи, психических заболеваний, наркомании и хронического алкоголизма, состояний, вызванных передозировкой лекарственных средств и алкоголя, отека легких, депрессии, астмы, эмфиземы и удушья, когнитивных расстройств и заболеваний желудочно-кишечного тракта. 3 табл.
Формула
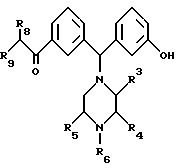
где один из радикалов R8 и R9 представляет собой фенил, возможно замещенный одним или несколькими заместителями, выбранными из группы, включающей галоген, С1-С3-алкокси, С1-С3-алкил и трифторметил, а другой из радикалов R8 и R9 представляет собой водород, С1-С6-алкил, С3-С6-циклоалкил, С3-С6 -алкенил или С3-С6 -алкинил;
любые два из радикалов R3, R4 и R5 представляют собой метил, а другой радикал представляет собой водород;
R6 представляет собой водород, С1-С6-алкил, С3-С6-циклоалкил, С3-С6-алкенил или С3-С6-алкинил;
или его фармацевтически приемлемые простой эфир, сложный эфир, соль или его физиологически функциональное производное.
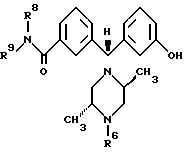
где R8, R9 и R6 определены в п.1.
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(4-фторфенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-метил-N-фенилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(4-хлорфенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-этил-N-фенилбензамид;
(-)-3-((αR)-α-((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N-фенилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-метил-N-(2-(трифторметил)фенил)бензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-метил-N-(2,4, 6-трихлорфенил)бензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(3-фторфенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(гидроксибензил)-N-фенил-N-пропилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(гидроксибензил)-N-(4-метоксифенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(2-фторфенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-этил-N-(4-фторфенил)бензамид;
(+)-3-((αR)-α-(2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N-аллил-N-фенилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(циклопропил)метил-N-фенилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-изопропил-N-фенилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N-циклопропил-N-фенилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2,5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(3-фторфенил)-N-пропилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-этил-N-(3-фторфенил)бензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(2-фторфенил)-N-пропилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-этил-N-(2-фторфенил)бензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-(4-метоксифенил)-N-пропилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-гидроксибензил)-N-этил-N-(4-метоксифенил)бензамид;
(+)-3-((αS)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(гидроксибензил)-N-метил-N-фенилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-метоксибензил)-N-(3-фторфенил)-N-метилбензамид;
(+)-3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-метоксибензил)-N-этил-N-(4-фторфенил)бензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(гидроксибензил)-N-(4-метоксифенил)-N-пропилбензамид;
3-((αR)-α-((2S, 5R)-4-аллил-2, 5-диметил-1-пиперазинил)-3-(N-(3-фторфенил)-N-метилкарбамоил)бензил)фенилмонофосфат,
или его фармацевтически приемлемые простой эфир, сложный эфир, соль или его физиологически функциональное производное.
30.07.93 - все пункты формулы изобретения;
17.12.93 - материалы приоритетной заявки использованы в описании.
Комментарии