Способ получения полиолефиновых основ синтетических масел - RU2287552C2
Код документа: RU2287552C2
Описание
Изобретение относится к способу получения полиолефиновых основ синтетических масел путем катионной олигомеризации олефинового сырья и может быть использовано в нефтехимической промышленности.
Получаемые по патентуемому способу продукты могут использоваться в качестве основы синтетических полиолефиновых (олигоолефиновых) масел разнообразного назначения: моторных (автомобильных, авиационных, вертолетных, тракторных, танковых); трансмиссионных, редукторных, вакуумных, компрессорных, холодильных, трансформаторных, кабельных, веретенных, медицинских, в составе различных смазок, а также в качестве пластификаторов для пластмасс, каучуков, твердых ракетных топлив; сырья для получения присадок, эмульгаторов, флотореагентов, пенообразователей, компонентов смазочно-охлаждающих и гидравлических жидкостей; высокооктановых добавок к топливам и т.п.
Известны способы получения полиолефиновых основ синтетических масел путем катионной олигомеризации высших олефинов, содержащие стадию подготовки олефинового сырья и растворов компонентов каталитической системы, стадию олигомеризации олефинового сырья, стадию выделения из олигомеризата отработанного катализатора методом водно-щелочной и последующей водной отмывки, стадию разделения очищенного олигомеризата на фракции и стадию гидрирования выделенных целевых фракций.
Наиболее существенно известные способы получения полиолефиновых основ синтетических масел различаются между собой составами применяемых в них катионных катализаторов.
В соответствии с известными способами катионную олигомеризацию олефинов С3-С14 (т.е. олефинов, содержащих от 3 до 14 атомов углерода) инициируют (катализируют) с помощью: протонных кислот (кислот Бренстеда); апротонных кислот (кислот Льюиса); алкилалюминий- (или бор) галогенидов; солей стабильных карбкатионов R+А-; природных и синтетических алюмосиликатов, цеолитов или гетерополикислот в Н-форме; различных двух- и трехкомпонентных комплексов, включающих мономер; полифункциональных катализаторов Циглера-Натта; металлоценовых катализаторов; физических методов стимулирования химических реакций [1. Дж.Кеннеди. Катионная полимеризация олефинов. М.: Мир, 1978. 430 С.; 2. J.P.Kennedy, E.Marechal. Carbocationic Polymerization. N.-Y., 1982. 510 P.]. Наиболее широкое промышленное применение в качестве катализаторов катионной олигомеризации олефинов и других мономеров нашли каталитические системы, включающие кислоты Льюиса (BF3, AlCl3, AlBr3, TiCl4, ZrCl4 и др.), алкилалюминий- (или бор) галогениды Rn МХ3-n (где R - алкил Cl-С10-, арил-, алкенил- и другие группы; М-Al или В; Х-Cl, Вг, I) и природные или синтетические алюмосиликаты, цеолиты и гетерополикислоты в Н-форме. При получении моторных ПАОМ на основе линейных альфа-олефинов (ЛАО) С6-С14 (преимущественно - на основе децена - 1) обычно используют каталитические системы, включающие кислоты Льюиса или алкилалюминийгалогениды.
Так, известно большое число способов получения полиолефиновых основ синтетических масел, в соответствии с которыми в качестве катализаторов олигомеризации ЛАО С6-С14 используют системы, включающие трехфтористый бор и различные протонодонорные сокатализаторы - воду, спирты, карбоновые кислоты, ангидриды карбоновых кислот, кетоны, полиолы и их смеси [3-20] [3. Пат. США 3780128 от 18.12.1973; Мкл. С 07 С 5/02; Нкл. 585-12; 260-683.9; 4. Пат. США 3997621 от 14.12.1976; Мкл. С 07 С 3/18; Нкл. 585-255; 260-683.15 В; 5. Пат. США 4 032591 от 28.06.1977; Мкл. С 07 С 5/24, 5/18; Нкл. 585-643; 260-683.65; 6. Пат. США 4376222 от 08.03.1983. Мкл. С 07 С 2/74; Нкл. 585/255; 7. Пат. США 4 225739 от 30.09.1980; Мкл. С 07 С 3/18; Нкл. 585/525; 8. Пат. США 4263465 от 21.04.1981; Мкл. С 07 С 9/00; Нкл. 585/18; 9. Пат. США 4263467 от 21.04.1981; Мкл. С 07 С 9/00; Нкл. 585/18; 10. Пат. США 4409415 от 11.10.1983. Мкл. С 07 С 3/02; Нкл. 585/525; 11. Пат. США 4956512 от 11.09.1990. Мкл. С 07 С 2/024; Нкл. 585/521; 12. Пат. США 4436947 от 13.03.1984. Мкл. С 07 С 3/18; Нкл. 585/525; 13. Пат. США 4451689 от 29.05.1984. Мкл. С 07 С 3/0; Нкл. 585/525; 14. Пат. США 4454366 от 12.06.1984. Мкл. С 07 С 7/12; Нкл. 585/525; 15. Пат. США 4587368 от 06.05.1986. Мкл. C 10 L 1/16; С 07 С 2/02; Нкл. 585/12; 16. Пат. США 4910355 от 20.03.1990. Мкл. С 07 С 2/74; Нкл. 585/255; 17. Пат. США 5254784 от 20.12.1991. Мкл. С 07 С 2/22; Нкл. 585/525; 18. Пат. США 5191140 от 02.03.1993. Мкл. С 07 С 2/02; Нкл. 585/525; 19. Пат. США 5420373 от 30.05.1995. Мкл. С 07 С 2/08; Нкл. 858/525; 20. Пат. США 5550307 от 27.08.1996. Мкл. С 07 С 2/14; Нкл. 585/525.]. Полиолефиновые основы синтетических масел в соответствии с этими способами получают путем олигомеризации олефинов С6-С14 под действием упомянутых борфторидных катализаторов при температурах 20-90°С в массе в течение 2-5 часов. Концентрацию трехфтористого бора в реакционной среде варьируют в пределах от 0.1 до 10 мас.%. Конверсия исходных олефинов изменяется в пределах от 80 до 99 мас.%. В результате олигомеризации, например, децена-1, образуется смесь ди-, три-, тетра-меров и более высокомолекулярных олигомеров. Суммарное содержание ди- и тримеров в продуктах изменяется в пределах от 30 до 70 мас.%.
Главным недостатком всех способов получения полиолефиновых основ синтетических масел этого типа является то, что они основаны на использовании катализаторов, включающих дефицитный, легколетучий, ядовитый, коррозионно-активный трехфтористый бор. Кроме того, из-за относительно низкой активности катализаторов этого типа в олигомеризации ЛАО процесс протекает в течение 2-5 часов. При промышленной реализации этих способов используются дорогостоящие большие по объему и по металлоемкости реакторы смешения в антикоррозионном исполнении.
Известно [21-32] [21. Пат. США 3725498 от 03.04.1973. Мкл. С 07 С 3/18; Нкл. 585-532; 22. Пат. США 3952071 от 20.04.1976. Мкл. С 07 С 3/18; Нкл. 585-532; 23. Пат. США 3997622 от 14.12.1976. Мкл. С 07 С 3/18; Нкл. 585-532; 24. Пат. США 3997623 от 14.12.1976; Мкл. С 07 С 3/18; Нкл. 585-532; 25. Пат. США 4006199 от 01.02.1977. Мкл. С 07 С 3/18; Нкл. 585-532; 26. Пат. США 4031158 от 21.06.1977. Мкл. С 07 С 3/18; Нкл. 585-532; 27. Пат. США 4031159 от 21.06.1977. Мкл. С 07 С 3/18; Нкл. 585-532; 28. Пат. США 4066715 от 03.01.1978. Мкл. С 07 С 3/18; Нкл. 585-532; 29. Пат. США 4113790 от 12.09.1978. Мкл. С 07 С 3/10; Нкл. 585-532; 30. Пат. США 4167534 от 11.09.1979. Мкл. С 07 С 5/04; Нкл. 585-532; 31. Пат. США 4219691 от 26.08.1980. Мкл. С 07 с 3/18; Нкл. 585-532; 32. Пат. США 5196635 от 23.03.1993. Мкл. С 07 С 2/22; Нкл. 585-532] также большое число способов получения полиолефиновых основ синтетических масел, в соответствии с которыми олигомеризацию олефинов проводят под действием катионных катализаторов, включающих галогениды алюминия и протонодоноры - воду, спирты, карбоновые кислоты, простые или сложные эфиры, кетоны (например, диметиловый эфир этиленгликоля, этиленгликольдиацетат), галоидалкилы [28, 32] [28. Пат. США 4066715 от 03.01.1978. Мкл. С 07 С 3/18; Нкл. 585-532; 32. Пат. США 5196635 от 23.03.1993. Мкл. С 07 С 2/22; Нкл. 585-532]. В некоторых способах эти катализаторы используют в комбинации с соединениями никеля [33. Пат. США 5489721 от 06.02.1996. Мкл. С 07 С 2/20; Нкл. 585-532]. Добавки соединений никеля в применяемые в соответствии с этими способами катализаторы обеспечивают регулирование строения и фракционного состава получаемых олигоолефинов.
Получение полиолефиновых основ синтетических масел олигомеризацией альфа-(С4-С14) или внутренних Cl10-Cl15 олефинов (полученных путем дегидрирования парафинов) по способу [29. Пат. США 4113790 от 12.09.1978. Мкл. С 07 С 3/10; Нкл. 585-532] осуществляют под действием катализаторов AlX3+протонодонор при температурах 100-140°С в течение 3-5 часов. Концентрацию AlX3 варьируют в пределах от 0.1 до 10 мол.% в расчете на олефины, мольное соотношение протонодонор/Al варьируют в пределах от 0.05 до 1.25. С повышением этого соотношения от 0.05 до 1.25 конверсия олефинов снижается от 99 до 12 мас.%.
Способы этого типа характеризуются следующими общими недостатками:
- сложной процедурой приготовления катализаторов, включающей много операций - возгонка и размол AlCl3, приготовление комплекса;
- получаемые по этим способам катализаторы являются вязкими, клейкими веществами, плохо растворимыми в олефинах, из-за высокой адгезии к охлаждаемым стенкам реакторов они плохо выгружаются из реакторов после завершения олигомеризации;
- низкой активностью используемых катализаторов в процессе олигомеризации, что требует применения больших по объему металлоемких реакторов смешения;
- высокими расходными коэффициентами по AlX3 в расчете на получаемые продукты.
Главным общим недостатком способов этого типа является то, что использование их приводит к получению в основном высокомолекулярных и высоковязких содержащих в своем составе до 1 мас.% хлора продуктов.
Разработано несколько способов получения полиолефиновых основ синтетических масел, основанных на использовании бифункциональных комплексных катализаторов, включающих соединения переходных металлов (TiCl4, ZrCl4 и алкилалюминийгалогениды Rn AlX3-n (смотри, например, [34-46]. [34. Пат.США 4214112 от 22.07.1980. Мкл. С 07 С 3/18; Нкл. 585-532; 35. J.Skupmska. Oligomerization of alpha-olefins to higher oligomers // Chem. Rev. 1991. V.91. N 4. P.613-648; 36. Пат. США 3168588. 12.03.1975. Мкл. С 07 С 2/22; Нкл. 260-683.15; 37. Пат. США 3884988. 20.05.1975. Мкл. С 07 С 3/10; Нкл. 584-524; Франц. Заявка 2221467. 1974. Мкл. С 08 F 1/32, 15/40; 38. Пат. США 4384089. Мкл. С 07 F 4/64; Нкл. 526-159; 39. Пат. США 4510342. 09.04.1985. Мкл. С 07 С 3/02; Нкл. 584-524; 40. Пат. США 4579991. 01.04.1986. Мкл. С 07 С 2/22; Нкл. 585-524; 41. Пат. США 4855526. 08.08.1989. Мкл. С 07 С 2/02; Нкл. 585-524; 42. Пат. Великобритании 1430497. Мкл. С 07 С 2/22; Пат. Великобритании 1522129. Мкл. С 07 С 2/22. Нкл. СЗР; 43. А.С. СССР 1073279. Мкл. С 07 С 2/32; Нкл. СЮМ 3/12; 44. Японский пат.52-11350. 1977 (РЖХим. 1978. 2 С 27 0 П). Мкл. С08 F 10/14; 45. А.С. СССР 1075500. 17.03.1982. Мкл. С 07 С 2/22. Нкл. В 01 J 31/14; 46. А.С. СССР 1192346. 12.08.1983. Мкл. С 07 С 2/22. Нкл. С 10 М 107/02]). В применяемых в соответствии с этими способами бифункциональных каталитических системах типа TiCl4 - RnAlX3-n образуется два типа активных центров - катионные и анионно-координационные. Из-за этого олигомеризация олефинов С3-С14 под действием катионных активных центров практически во всех случаях сопровождается полимеризацией олефинов С3-С14 под действием анионно-координационных активных центров в нерастворимые трудноудаляемые из реактора высокомолекулярные полиолефины. При этом под действием бифункциональных комплексных катализаторов во всех случаях образуются высокомолекулярные высоковязкие олигоолефины, которые не могут использоваться в качестве основы наиболее широко потребляемых моторных масел. Это является главным недостатком способов такого типа.
В соответствии с некоторыми способами в катионных процессах полимеризации, олигомеризации и алкилирования широко применяются также двухкомпонентные растворимые монофункциональные каталитические системы, включающие алкилалюминийгалогенид Rn AlX3-n и галоидорганическое соединение R'X при мольном соотношении R'Х/RnAlX3-n=1.0-5.0 (где R - СН3, C2H5, С3Н7 или изо-C4H9; Х - хлор, бром или иод; n=1.0; 1.5 или 2.0; R' - Н [47. Пат. США 4952739. 28.08.1990. Мкл. С 07 С 2/18; Нкл. 585/18; 585/511], первичный, вторичный или третичный алкил, аллил или бензил [48-51] [48. Пат. США 4041098. 09.08.1977. Мкл. С 07 С 3/10; Нкл. 585-524; 49. Пат. Великобритании 1535324, 1535325. 1978. Мкл. С 07 С 3/21; Нкл. В 1 Е, С 5 Е; 50. 3аявка ФРГ 2526615. 13.06.1975. Мкл. С 07 С 3/21; Пат. ФРГ 2304314. 1980. Мкл. С 08 F 110/20; 51. А.С. СССР 1723101. 20.04.1989. Мкл. С 07 С 2/30. Нкл. С 10 М 107/10]). В каталитических системах этого типа RnAlX3-n является основой катализатора, a R'X - сокатализатором.
В соответствии с этими способами каталитические системы RnAlX3-n-R'X используются для инициирования катионной олигомеризации индивидуальных или смесей линейных альфа-олефинов от пропилена до тетрадецена включительно в полиальфа-олефиновые основы синтетических масел в среде исходных олефинов или смесей их с продуктами олигомеризации и парафиновыми, ароматическими или галоидсодержащими углеводородами при температурах до 250°С.
Катионные активные центры ([R'+(RnAlX4-n)] и R'+) в каталитических системах RnAlX3-n-R'X образуются в соответствии со следующей упрощенной схемой:
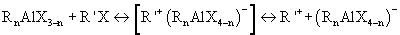
Образование катионных активных центров в рассматриваемых каталитических системах происходит с очень высокой скоростью. Благодаря этому сразу же после смешения растворов компонентов рассматриваемых каталитических систем достигается высокая концентрация катионных активных центров и процесс олигомеризации протекает без индукционного периода с очень высокой начальной скоростью. При этом 95-98%-ная процентная конверсия исходных олефинов в олигомерные продукты при температурах 20-200°С достигается в течение шести-одной минут, соответственно. Такой характер кинетики олигомеризации линейных альфа-олефинов (ЛАО) под действием рассматриваемых каталитических систем обеспечивает возможность проведения процесса олигомеризации в форсированном изотермическом режиме в реакторах вытеснения трубчатого типа при временах пребывания от 1 до 10 минут [51. А.С. СССР 1723101. 20.04.1989. Мкл. С 07 С 2/30. Нкл. С 10 М 107/10; Пат. РФ 2201799. 29.09.2000. Мкл. 7 В 01 J 8/06, С 08 F 110/10; Бюл. Изобр. 2003. № 10].
При получении олиго-олефиновых основ синтетических масел в соответствии с этими способами в ходе олигомеризации ЛАО в массе или в среде парафиновых углеводородов под действием каталитических систем RnAlX3-n-R'X образуются высокоразветвленные застывающие при низких температурах олигомеры, содержащие одну ди-, три- или тетраалкилзамещенную двойную связь и до 0.2 мас.% монохлоролигоолефинов, а при олигомеризации в среде или в присутствии ароматических углеводородов (бензол, толуол, нафталин) образуются олигоалкилароматические маслообразные продукты (теломеры), не содержащие двойных связей [51. А.С. СССР 1723101. 20.04.1989. Мкл. С 07 С 2/30. Нкл. С 10 М 107/10; Пат. РФ 2199516 от 18.04.2001 г. МКН 7 С 07 С 2/22. Бюл. Изобр. № 6 от 27.02.2003 г.].
Главным недостатком способов получения олиго-олефиновых основ синтетических масел путем олигомеризации олефинов под действием каталитических систем RnAlX3-n-R'X является то, что применение их в процессе олигомеризации ЛАО (в частности - децена-1) приводит к образованию преимущественно высокомолекулярных продуктов с широким молекулярно-массовым распределением и с низким (менее 20 мас.%) содержанием целевых низкомолекулярных фракций (димеров и тримеров децена-1).
Другим недостатком способов, основанных на использовании каталитических систем этого типа, является то, что получаемые по этим способам димеры децена-1 являются линейными и после гидрирования имеют температуру застывания, превышающую минус 20°С.
Для устранения этого недостатка, т.е. для повышения селективности и улучшения технико-экономических показателей способа, разработан способ получения олиго-олефиновых основ синтетических масел, в соответствии с которым не гидрированные димеры децена-1 направляют в рецикл на соолигомеризацию их с деценом-1 в широко потребляемые три- и тетрамеры децена [46. Пат. США 4263467. 02.04.1981]. Соолигомеризацию димеров децена-1 (43.8 мас.% в шихте) с деценом-1 (40 мас.% децена-1 и 15.4 мас.% декана в шихте) по этому способу осуществляют под действием системы BF3/SiO2 (D=0.8-2.0 мм)+Н2О (65 м.д. в шихте) при температурах 15-30°С, давлении 1-6.9 ат при расходе шихты 2.5 л в час на 1 л катализатора. Содержание димеров децена-1 на выходе из реактора уменьшается от 43.8 до 20.7 мас.%, а содержание тримеров децена-1 возрастает до 41.8 мас.%. Это решение позволяет квалифицированно использовать не потребляемые (застывающие при высоких температурах) линейные димеры децена-1. Недостатком этого решения является резкое снижение производительности процесса, который основан на этом способе.
Третьим общим недостатком всех способов, основанных на использовании каталитических систем RnAlX3-n-R/X, является то, что применяемые каталитические системы включают горючее, самовоспламеняющееся на воздухе, опасное при производстве, при транспортировке и при использовании алюминийорганического соединения RnAlX3-n.
И, наконец, четвертым общим недостатком всех способов, основанных на использовании каталитических систем RnAlX3-nR/X, является то, что под действием этих каталитических систем образуются продукты, содержащие до 1,0 мас.% хлора в виде монохлоролигоолефинов.
Известны также способы катионной полимеризации, олигомеризации олефинов и алкилирования ароматических углеводородов олефинами под действием каталитических систем, включающих металлический алюминий. Сам по себе металлический алюминий не является катализатором упоминавшихся процессов. Для обеспечения этим системам каталитической активности алюминий обычно используют в комбинации с сокатализатором. Так, например, известны способы полимеризации, олигомеризации и теломеризации олефинов, а также алкилирования ароматических углеводородов олефинами, включающие металлический алюминий и галоидорганическое соединение [53-58] [53. АС СССР 541494. Нкл. В 01 J 37/00. 1975; 54. Пат. США 3303230. Нкл. 260-671. 1967; 55. Пат. США 3343911. Нкл. 260-683.15. 1969; 56. А.С. СССР 430581. Мкл. С 07 С 3/10. 1974; 57. А.С. СССР 732229. Мкл. С 07 С 15/02. 1975; 58. RU 2212935 от 27.09.03; МПК B 01 J 27/06].
Наиболее близким по технической сущности и достигаемому результату к разработанному в соответствии с настоящим изобретением способу получения полиолефиновых основ синтетических масел является способ олигомеризации и полимеризации олефинов под действием каталитической системы, включающей металлический алюминий и четыреххлористый углерод [58. RU 2212935 от 27.09.03 г.; МПК B01J 27/06]. Катализатор для олигомеризации и полимеризации олефинов по этому способу получают путем взаимодействия металлического алюминия с четыреххлористым углеродом при температурах 40-80°С и массовом соотношении алюминия к четыреххлористому углероду, равном 1:(20-80) в среде четыреххлористого углерода в отсутствие олефинов в инертной атмосфере [58. RU 2212935 от 27.09.03 г.; МПК В 01J 27/06]. В соответствии с этим способом вначале в отсутствие олефинов в инертной атмосфере получают твердый черный продукт неизвестного состава, который далее используется в качестве катализатора олигомеризации альфа-олефинов и полимеризации изобутилена. Мы рассматриваем этот способ в качестве прототипа для разработанного нами способа получения полиолефиновых основ синтетических масел.
Недостатком способа прототипа является использование в этом способе четыреххлористого углерода в составе применяемой каталитической системы при высоком соотношении CCl4/Al(0). Это приводит к вхождению в состав продуктов большого количества (до 3,0 масс.%) трудноудаляемого из них хлора.
Другим недостатком способа-прототипа [58. RU 2212935 от 27.09.03 г.; МПК B 01 J 27/06] является низкая активность, низкая производительность и низкая селективность по целевым продуктам применяемой по этому способу каталитической системы Al(0)-CCl4.
Недостатком способа-прототипа является также многостадийность и высокая трудоемкость приготовления и использования катализатора олигомеризации олефинов и полимеризации изобутилена из алюминия и CCl4.
Общей задачей настоящего технического решения является устранение всех вышеуказанных недостатков известных способов.
Основной конкретной задачей настоящего изобретения являлась разработка способа получения полиолефиновых основ синтетических масел с использованием усовершенствованной каталитической системы для катионной олигомеризации линейных альфа-олефинов (ЛАО) С3-С14, которая характеризовалась бы повышенной активностью и повышенной производительностью, обеспечила бы повышение управляемости процессом олигомеризации, в частности, позволила бы регулировать скорость олигомеризации, повысить выход целевых низкомолекулярных фракций олигомеров (например, димеров и тримеров децена-1), повысить разветвленность цепи продуктов олигомеризации и снизить температуру их застывания, а также повысила бы безопасность использования ее в процессе олигомеризации олефинов. Второй задачей настоящего изобретения являлось упрощение способа приготовления и использования каталитической системы олигомеризации олефинов, включающей металлический алюминий.
Сформулированные задачи решены в настоящем изобретении в результате совершенствования всех основных стадий способа получения полиолефиновых основ синтетических масел.
Раскрытие сущности изобретения. Разработанный согласно настоящему изобретению способ получения полиолефиновых основ синтетических масел, так же как и любой другой аналогичный способ содержит стадию подготовки олефинового сырья и растворов компонентов катионной каталитической системы, стадию изомеризации высших линейных альфа-олефинов, стадию олигомеризации олефинового сырья под действием катионной алюминийсодержащей каталитической системы, стадию выделения из олигомеризата отработанного катализатора, стадию разделения олигомеризата на фракции и стадию гидрирования выделенных фракций. Кроме того, он после стадии олигомеризации и/или после стадии выделения отработанного катализатора из олигомеризата, дополнительно содержит стадию дехлорирования присутствующих в олигомеризате монохлорсодержащих олигомеров, а после стадии разделения олигомеризата на фракции он содержит стадию деполимеризации высокомолекулярных продуктов, выделенных из олигомеризата в виде кубового остатка на стадии разделения олигомеризата на фракции (пункт 1 формулы изобретения). Эти стадии предназначены для улучшения технико-экономических показателей способа, для решения специфических химических проблем и для повышения гибкости разработанного способа по продуктам. В частности, стадия дехлорирования образующихся в ходе олигомеризации и содержащихся в олигомеризате монохлоролигодеценов предназначена для превращения ковалентно связанного с углеродом в монохлоролигодеценах так называемого, "органического" хлора в ионносвязанный с металлами, так называемый, "ионный" хлор. Полученный из хлорсодержащих олигодеценов "ионный" хлор вместе с отработанным катионным катализатором, как и в других способах катионной олигомеризации олефинов или алкилирования, далее удаляется из олигомеризата методом водно-щелочной отмывки.
В соответствии с настоящим изобретением полиолефиновые основы синтетических масел получают путем олигомеризации высших олефинов в смесях олигомеризуемых высших олефинов с продуктами их олигомеризации или в смесях олигомеризуемых высших олефинов с продуктами их олигомеризации и с ароматическими углеводородами под действием трехкомпонентной, безопасной при транспортировке, хранении и использовании, устойчивой на воздухе, доступной катионной каталитической системы Al(0)-HCl-(СН3)3CCl при температурах от 110 до 180°С, концентрациях Al(0) от 0.02 до 0.08 г-атом/л, мольных соотношениях HCl/Al(0), изменяющихся в пределах от 0.002 до 0.06 и мольных соотношениях RCl/Al(0), изменяющихся в пределах от 1.0 до 5.0, где Al(0) - высокодисперсный порошкообразный алюминий с размерами частиц, изменяющимися в пределах от 1 до 100 мкм, например, Al(0) марки ПА-1, ПА-4, АСД-4, АСД-40, АСД-Т (пункт 2 формулы изобретения). Индивидуальные компоненты системы Al(0)-HCl-(СН3)3CCl катализаторами олигомеризации высших олефинов не являются. Предшественники (пресуркоры) катионных активных центров и сами катионные активные центры олигомеризации высших олефинов в этой системе образуются в последовательности многих химических реакций между компонентами системы. Применяемый в составе рассматриваемой каталитической системы металлический высокодисперсный алюминий состоит из частиц алюминия, покрытых плотной не реакционноспособной алюминийоксидной оболочкой. Из-за этого он устойчив на воздухе и при температурах 20-110°С практически не реагирует HCl и с (СН3)3CCl. Реакция Al(0) с HCl и с (СН3)3CCl начинается только при температурах, превышающих 110°С. В соответствии с разработанным способом получения полиолефиновых основ синтетических масел в разработанной каталитической системе алюминий в первую очередь реагирует с хлористым водородом. HCl, по крайней мере, частично разрушает алюминийоксидную пленку на поверхности частиц алюминия и обеспечивает возможность протекания реакции алюминия с третбутилхлоридом. Другими словами, хлористый водород в рассматриваемой системе является активатором металлического алюминия. Реакция алюминия с третбутилхлоридом протекает по следующей упрощенной схеме:
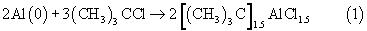
Образующийся сесквитретбутилалюминийхлорид (1), так же как и HCl, реагирует с алюминийоксидной оболочкой на поверхности частиц алюминия. Это ведет к ускорению процесса образования (1) и к полному растворению металлического алюминия. Активация алюминия обеспечивается очень малым количеством хлористого водорода, который растворяют в третбутилхлориде (ТБХ) в процессе его получения по реакции изобутилена с хлористым водородом. Мольное соотношение HCl/Al(0) в каталитической системе Al(0)-HCl-(СН3)3CCl варьируют в интервале от 0.002 до 0.06 путем изменения концентрации HCl в ТБХ от 0.015 до 0.5 мас.%.
Концентрацию алюминия в реакционной среде в процессе олигомеризации олефинового сырья варьируют в интервале от 0.02 до 0.08 г-атом/л. При концентрациях алюминия ниже 0.02 г-атом/л олигомеризация не протекает из-за ингибирующего действия присутствующих в олефинах примесей, а при концентрациях алюминия выше 0.08 г-атом/л резко возрастает удельный расход компонентов катализатора. Оптимальная концентрация алюминия в реакционной среде изменяется в пределах от 0.03 до 0.04 г-атома/л.
Мольное соотношение ТБХ/Al(0) варьируют в пределах от 1.0 до 5.0, оптимальное мольное соотношение ТБХ/Al(0) равняется 3.5. При мольных соотношениях ТБХ/Al(0) менее 3.5 растворяется только часть содержащегося в системе Al(0)-HCl-(СН3)3CCl металлического алюминия, а при мольных соотношениях ТБХ/Al(0) выше 4.0 резко возрастает содержание хлора в олигомерах. Первичные катионные активные центры в рассматриваемой каталитической системе образуются по схеме:
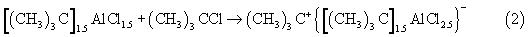
ТБХ при этом выполняет функции сокатализатора.
Олигомеризация олефинов под действием системы Al(0)-HCl-(СН3)3CCl с высокой скоростью и с высокой конверсией олефинов в продукты (выше 95 мол.%) протекает при температурах от 110 до 180°С. В ходе олигомеризации альфа-олефинов под действием системы Al(0)-HCl-(СН3)3CCl происходит частичная изомеризация альфа-олефинов в смесь позиционных и геометрических изомеров олефинов с внутренним расположением двойных связей, которые соолигомеризуются с исходным альфа-олефином. Это приводит к повышению разветвленности молекул получаемых продуктов и к снижению температуры их застывания. В случае олигомеризации децена-1 применение системы Al(0)-HCl-(СН3)3CCl обеспечивает снижение доли высокомолекулярных олигодеценов С60+ от 50 (прототип) до 8 мас.%. Образующиеся под действием этой системы олигодецены содержат от 4300 до 9970 м.д. хлора (таблица 1). Использование системы Al(0)-HCl-(СН3)3CCl в процессе олигомеризации обеспечивает решение проблемы регулирования фракционного состава и разветвленности продуктов олигомеризации деценов. Расход компонентов этой каталитической системы по массе не превышает соответствующих показателей лучших известных (в том числе борфторидных) катализаторов.
Практически важной особенностью разработанного способа является то, что взаимодействие алюминия с активатором (HCl) и сокатализатором (ТБХ) проводят непосредственно в процессе олигомеризации в среде смесей олигомеризуемых олефинов с продуктами олигомеризации и дополнительно добавляемыми ароматическими углеводородами (бензолом, толуолом, нафталином). Именно это решение исключает работу с концентрированными высокореакционными предшественниками и продуктами реакций образования активных центров и повышает безопасность способа.
В соответствии с настоящим изобретением в качестве олигомеризуемых высших олефинов берут смеси линейных или разветвленных альфа-олефинов с изо-олефинами и с олефинами с внутримолекулярным расположением двойной связи (с "внутренними" олефинами), содержащие от 3 до 14 (преимущественно - 10) атомов углерода, при следующем соотношении ингредиентов, мас.%: альфа-олефины 0.5-99.0; изо-олефины 0.5-5.0; "внутренние" олефины - остальное до 100 мас.% (таблица 2) (пункт 3 формулы изобретения). Из приведенных в таблице 2 данных видно, что при прочих одинаковых условиях увеличение числа атомов углерода в молекулах олефинов приводит к уменьшению разветвленности молекул олигоолефинов и к повышению их индекса вязкости.
Наиболее распространены способы получения полиолефиновых основ синтетических масел, в которых в качестве олефинового сырья используется децен-1. Это обусловлено тем, что выделяемые из олигомеров децена-1 тримеры децена-1 после гидрирования характеризуются уникальным комплексом физических свойств (кинематическая вязкость при 100° С равна 3.9 сСт, индекс вязкости=130, температура застывания=минус 60°С, температура вспышки=215-220°С). Такое сочетание свойств обеспечивает возможность использования их в качестве основы широко потребляемых синтетических и полусинтетических моторных (автомобильных, авиационных, вертолетных, тракторных, танковых) и других масел. Поэтому, наилучшим сырьем для получения полиолефиновых основ синтетических масел является децен-1. В соответствии с настоящим способом наличие в исходном децене-1 примесей изоолефинов (от 0.5 до 11.0 мас.%) и олефинов с внутримолекулярным расположением двойной связи (от 0.5 до 5.0 мас.%) практически не влияет на физико-химические характеристики выделенных из продуктов не гидрированных и гидрированных фракций. В ходе катионной олигомеризации децен-1 перед вхождением в олигомерный продукт под действием каталитической системы Al(0)-HCl-(СН3)3CCl и других содержащих соединения алюминия каталитических систем изомеризуется в смесь позиционных и геометрических изомеров децена с внутримолекулярным расположением двойных связей. Децены с внутримолекулярным расположением двойных связей (включая индивидуальный децен-5) под действием каталитической системы Al(0)-HCl-(СН3)3CCl так же легко (но более медленно) олигомеризуются, как и децен-1. В ходе олигомеризации деценов с внутримолекулярным расположением двойных связей образуются более разветвленные, чем в случае децена-1 (и поэтому застывающие при более низких температурах) молекулы олигодеценов.
Из классического стадийного механизма процессов катионной олигомеризации олефинов под действием хлорсодержащих (в том числе - и алюминийорганических) катионных катализаторов следует [1. Дж.Кеннеди. Катионная полимеризация олефинов. М.: Мир, 1978. 430 С.; 2. J.P.Kennedy, E.Marechal. Carbocationic Polymerization. N.-Y., 1982. 510 P.], что образующиеся в этих процессах олигомеры могут содержать "органически" связанный хлор (т.е. хлор, связанный с атомом углерода молекул олигодеценов в составе олигомеризата). Теоретические расчеты и экспериментальные данные показывают, что от 2 до 10% молекул олигодеценов, полученных под действием применяемых катионных алюминийсодержащих катализаторов, содержат в своем составе по одному атому хлора, а 90-98% молекул олигодеценов содержат по одной двойной связи. После дезактивации и удаления из олигомеризата методом водно-щелочной отмывки при температуре +95°С отработанного катионного катализатора, олигомеризат содержит около от 1000 до 10000 м.д. (0.1-1.0 мас.%) хлора, связанного с атомами углерода молекул олигодецена. Хлор в олигодецены попадает в актах истинного (практически необратимого) обрыва цепи в результате захвата растущим карбкатионом аниона хлора из анионного фрагмента катионного активного центра (АЦ). Схема этого процесса на примере простейшего катализатора RCl+AlCl3 (R+AlCl4-) имеет следующий вид (3):
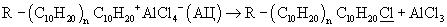
Содержащийся в олигомеризате и в целевых фракциях хлор вызывает коррозию соответствующего оборудования не только на всех стадиях процесса получения олигодеценов, но и в процессе использования олигодеценовых основ синтетических масел. Из-за этого хлор необходимо удалять не только из основных фракций олигодеценов, но и из олигомеризата на наиболее ранних стадиях их получения.
В соответствии с настоящим изобретением дехлорирование присутствующих в олигомеризате монохлорсодержащих молекул олигомеров (RCl) осуществляют как после стадии олигомеризаации, так и после стадии выделения из олигомеризата отработанного катализатора Al(0)-HCl-(СН3)3CCl (пункт 1 формулы изобретения). При разработке настоящего способа получения полиолефиновых основ синтетических масел разработано 5 вариантов решения проблемы дехлорирования.
По первому варианту настоящего способа дехлорирование RCl производят высокодисперсным порошкообразным металлическим алюминием - Al(0) с размерами частиц, изменяющимися в пределах от 1 до 100 мкм (например, марок ПА-1, ПА-4, АСД-4, АСД-40, АСД-Т) при мольных соотношениях Al(0)/RCl, изменяющихся в пределах от 0.5 до 2.0, в интервале температур от 110 до 180°С в течение от 30 до 180 минут (пункт 4 формулы изобретения) (таблица 3). Из-за высокой энергии связи С-Cl, хлор из хлоралканов, содержащих фрагменты -СН2Cl, с помощью химических реагентов удаляется с большим трудом. Наиболее легко из хлоралканов удаляется хлор, содержащийся в фрагментах R3CCl. Поэтому тест-индикатором скорости и глубины дехлорирования хлоралканов использовали 1-хлор-додекан, содержащий фрагмент -СН2Cl. Из таблицы 3 видно, что при температурах, не превышающих 95°С, под действием алюминия марки ПА-4 дехлорирование 1-хлор-додекана и хлорсодержащих олигодеценов не происходит. Повышение температуры до 120°С и выше приводит к полному дехлорированию упомянутых хлоралканов. Реакция дехлорирования 1-хлордодекана алюминием в среде децена-1 приводит к почти 100%-ной олигомеризации децена-1. Это свидетельствует о том, что реакция алюминия с 1-хлордодеканом протекает таким же образом, как и реакция алюминия с ТБХ (т.е. с промежуточным образованием катионных активных центров олигомеризации). При этом реакция алюминия с RCl приводит к превращению ковалентно-связанного с углеродом хлора в ионно-связанный с металлом хлор, который на стадии водно-щелочной отмывки удаляется из олигомеризата.
В соответствии со вторым вариантом дехлорирования по патентуемому способу присутствующие в олигомеризате монохлорсодержащие олигодецены (RCl) дехлорируют после стадии олигомеризации триэтилалюминием (ТЭА) при мольных соотношениях ТЭА/RCl, изменяющихся в пределах от 0.5 до 2.0, в интервале температур от 95 до 150°С в течение от 30 до 180 минут (пункт 5 формулы изобретения).
Дехлорирование RCl триэтилалюминием в указанных выше условиях протекает в соответствии со следующей схемой (4):
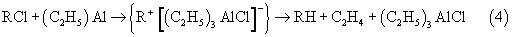
Из таблиц (3, 4) видно, что дехлорирование 1-хлор-додекана алюминием и триэтилалюминием происходит только при температурах 130-164°С, как в отсутствие отработанного катионного катализатора, так и в присутствии продуктов его эволюции в ходе олигомеризации. Видно также, что конверсия децена-1 в продукты олигомеризации в ходе реакции дехлорирования RCl алюминием и триэтилалюминием возрастает. Это согласуется со схемами (3, 4), в соответствии с которыми реакции RCl с алюминием и триэтилалюминием протекают через стадию образования катионного активного центра {R+[(C2H5)3 AlCl]-), который инициирует олигомеризацию децена-1.
Общим достоинством первого и второго вариантов дехлорирования RCl с помощью Al(0) и ТЭА, в соответствии с настоящим изобретением, является то, что реакции RCl с упомянутыми дехлорирующими реагентами проводят непосредственно после олигомеризации в присутствии отработанных, но не выделенных из олигомеризата, продуктов превращения применяемой катионной каталитической системы Al(0)-HCl-ТБХ. Образующиеся при дехлорировании алкил-алюминий хлориды выделяются из олигомеризата одновременно с отработанным катализатором на стадии водно-щелочной отмывки. Это упрощает технологическое оформление этой стадии, но требует повышенного расхода дисперсного алюминия или использования ТЭА.
Первичные и вторичные хлоралканы в обычных условиях (т.е. при температурах, не превышающих 100°С) не гидролизуются водой и водными растворами гидроксидов натрия или калия. В соответствии с третьим вариантом настоящего способа получения полиолефиновых основ синтетических масел, дехлорирование образующихся в ходе олигомеризации и присутствующих в олигомеризате монохлорсодержащих олигомеров (RCl) осуществляют до или после стадии выделения отработанного катализатора спиртовым (бутанольным или гексанольным - ROH) раствором гидроксида калия или натрия (МОН) при мольных соотношениях MOH/RCl, изменяющихся в пределах от 1.1 до 2.0, в интервале температур от 120 до 160°С в течение от 30 до 240 минут (таблица 5) (пункт 6 формулы изобретения). Вместо упомянутых ROH в качестве растворителя KOH можно использовать моноэтиловый эфир этиленгликоля. Концентрацию МОН в ROH варьируют в пределах от 1 до 5 мас.%. Из таблицы 5 видно, что при прочих одинаковых условиях скорость реакции и конверсия дехлорирования резко снижаются при замене KOH на NaOH. По этой причине KOH является предпочтительным. Реакция дехлорирования RCl спиртовыми растворами МОН даже при 120°С протекает медленно. Повышение температуры от 120 до 150°С приводит к резкому повышению скорости реакции. При 150°С реакция завершается в течение 60 минут.Дехлорирование RCl по этому варианту способа протекает по схеме, включающей промежуточное образование алкоксидов щелочных металлов, которые реагируют далее с хлоралканами RCl:
KOH+С4Н9 OH→С4Н9OK+Н2O
С4Н9OK+RCl→KCl+С4Н9OR
Хлориды натрия и калия в упомянутых спиртах в условиях реакции дехлорирования хлоралканов не растворимы, что позволяет выделять их из реакционной массы осаждением с последующим растворением осадка соли MCl водой.
Наиболее простым из разработанных вариантов дехлорирования хлорсодержащих олигодеценов является вариант, в соответствии с которым дехлорирование присутствующих в олигомеризате монохлорсодержащих олигомеров (RCl) осуществляют после стадии выделения отработанного катализатора путем термического дегидрохлорирования RCl в интервале температур от 280 до 350°С и давлениях 1-2 ат в течение от 30 до 180 минут при отдувке выделяющегося хлористого водорода азотом, диоксидом углерода, метаном (природным газом) или перегретым водяным паром (таблица 6) (пункт 7 формулы изобретения). Термическое дегидрохлорирование по этому варианту способа осуществляют в подогревателе-испарителе атмосферной колонны при интенсивном перемешивании олигомеризата отдувочным газом или перегретым водяным паром. Этот вариант дехлорирования хлорсодержащих олигомеров децена имеет и достоинства и недостатки: он обеспечивает глубокую степень дегидрохлорирования олигомеризата (на 97-98%), не требует применения новых реагентов, но приводит к усложнению оформления атмосферной колонны.
Термическое дегидрохлорирование хлорсодержащих соединений начинается при температурах, превышающих 250° С. Скорость термического дегидрохлорирования существенно зависит от строения хлорсодержащего соединения. В случае хлорсодержащих соединений, имеющих бэта-С-Н связи по отношению к связям С-Cl, наиболее термоустойчивыми являются первичные галоидалкилы, наименее термоустойчивыми являются третичные галоидалкилы. Термическое дегидрохлорирование R3CCl с заметной скоростью протекает даже при 100°С. Из состава исходных реагентов и стадийного механизма олигомеризации децена-1 следует, что в олигомеризате могут присутствовать все теоретически возможные для каждого конкретного случая типы хлорсодержащих соединений (содержащие и не содержащие бэта-С-Н связи первичные, вторичные и третичные галоидалкилы). Скорости, и при прочих одинаковых условиях, степени дегидрохлорирования содержащих бэта-С-Н связи первичных, вторичных и третичных галоидалкилов обычно возрастают с повышением температуры.
При температурах 250-300°С дегидрохлорирование хлорсодержащего олигомеризата протекает относительно медленно и, видимо, обратимо в течение 6-10 часов. Выделяющийся в актах дегидрохлорирования хлористый водород сразу же может присоединяться к молекулам олигодеценов, содержащих двойные связи. Этому благоприятствует относительно высокая концентрация двойных связей различных типов в молекулах олигодеценов.
При прочих одинаковых статических условиях скорость и степень дегидрохлорирования олигомеризата возрастают с повышением температуры от 300 до 330°С. Деполимеризация олигодеценов при этом не протекает. При 330° С и времени пребывания два часа почти весь содержащийся в олигомерах органически связанный хлор удаляется из олигомеризата в виде HCl. В олигомеризате после термообработки остается около 20 м.д. (ppm) органически связанного хлора (т.е. около 0.002 мас.%). В условиях отдувки хлористого водорода и парообразных компонентов из олигомеризата азотом при 330°С и времени пребывания один час в олигомеризате остается только 3 м.д. (ppm) органически связанного хлора. Из полученных данных следует, что степень удаления хлорсодержащих углеводородов из олигомеризата=99,94 мас.%. Содержание хлорорганических соединений в рециклируемой децен-декановой фракции не превышает 160 м.д. (что соответствует 0.016 мас.%). Эта фракция может быть смешана со свежим поступающим на установку деценом, а полученная смесь может использоваться в качестве исходного сырья для получения олигодеценов.
Термическое дегидрохлорирование хлорсодержащих молекул олигодеценов при температурах 300-330°С и общем времени пребывания два часа протекает почти количественно по следующей схеме:
R-(Cl0H20)x-1-Cl10H21 Cl→HCl+R-(Cl10H20)x-1-Cl10H20(=)
Выделяющийся при этом хлористый водород отдувается азотом или перегретым водяным паром и вместе с парами воды и углеводородов поступает вначале в конденсатор, а из него направляется в скруббер для нейтрализации HCl водным раствором гидроксида натрия.
С целью исключения выделения хлористого водорода в свободном состоянии в паро-газовую фазу, дегидрохлорирование присутствующих в олигомеризате монохлорсодержащих олигомеров (RCl), в соответствии с настоящим изобретением (пункт 8 формулы изобретения) осуществляют в интервале температур от 300 до 330°С в присутствии сухих гидроксидов щелочных металлов (МОН) при мольных соотношениях MOH/RCl, изменяющихся в пределах от 1.1 до 2.0 (таблица 7). Для обеспечения максимально возможной степени диспергирования частиц не растворимых в олигомеризате сухих гидроксидов щелочных металлов их получают в соответствии с настоящим изобретением (пункт 9 формулы изобретения) непосредственно в олигомеризате путем отгонки воды при нагревании смеси освобожденного от отработанного катализатора олигомеризата и 5-40%-ного водного раствора гидроксида щелочного металла при температурах, изменяющихся в интервале температур от 100 до 200°С (таблица 7). При повышении температуры от 100 до 200°С происходит испарение и полное удаление воды из реакционной массы. Последующее дегидрохлорирование хлорсодержащих олиго-деценов проводят в течение одного часа при температурах 300, 310, 320 и 330°С. Из таблицы 7 видно, что при прочих одинаковых условиях остаточное содержание хлора в олигомере с повышением температуры от 300 до 330°С резко снижается и при 330°С достигает 8 м.д. (8 ppm) при степени дегидрохлорирования 95.56 мас.%. Хлористый водород в газовую фазу не выходит - он полностью реагирует с гидроксидом калия и обнаруживается в виде KCl в твердом осадке, который полностью растворяется в воде. Промывка дегидрохлорированного олигомеризата водой после выгрузки его из реактора и анализ промывной воды на хлор свидетельствуют о том, что некоторая часть (менее 0.5 мас.%) образовавшегося хлористого калия удерживается во взвешенном состоянии в дегидрохлорированном олигомере.
Разработанные варианты дехлорирования молекул хлорсодержащих олигодеценов (т.е. - хлоралканов) являются универсальными и могут использоваться самостоятельно для решения аналогичных задач в других химических процессах, в нефтепереработке, а также при дехлорировании моно-, ди- и полихлорсодержащих алифатических и ароматических углеводородов, олигомеров, полимеров, нефтяных фракций и различных жидких и твердых хлорсодержащих органических отходов (пункт 10 формулы изобретения).
Для примера в таблице 8 приведены данные о дегидрохлорировании жидких полихлорпарафинов, содержащих 44 мас.% хлора, бутанольным раствором KOH. Это решение используется, в частности, при получении синтетической олифы и олигомеров ацетилена.
Патентуемый способ получения полиолефиновых основ синтетических масел предусматривает утилизацию не находящих квалифицированного применения фракций высокомолекулярного олигодецена путем их деполимеризации. Стадия деполимеризации высокомолекулярных олигодеценов предназначена для корректировки молекулярно-массового распределения и фракционного состава олигодеценов, получаемых на стадии олигомеризации. Деполимеризацию высокомолекулярных продуктов, выделенных в виде кубового остатка на стадии разделения олигомеризата на фракции, в соответствии с разработанным способом (пункт 11 формулы изобретения), проводят нагреванием их при температурах от 330 до 360°С и давлениях от 1.0 до 10.0 мм рт.ст. в течение от 30 до 120 минут с непрерывным удалением продуктов из реактора деполимеризации в систему атмосферной и двух вакуумных колонн для разделения олигомеризата на фракции.
Разделение деценового олигомеризата на фракции и деполимеризацию выделенного кубового остатка, содержащего высокомолекулярные олигодецены, производили на вакуумной установке французской компании "GECIL". Использовавшаяся компьютеризованная автоматизированная установка (model "minidist С") включает две колонны, два электрообогреваемых куба объемом 10 и 22 литра, стеклянный сборник, около 10 приемников для выделяемых фракций, вакуумный пост, вакуумный насос, вакуумметр и два устройства для измерения температуры в кубе и в верхней части колонны. Сборник был оснащен автоматической системой контроля скорости отбора фракций. Вакуумная система этой установки обеспечивает возможность фракционирования олигомеризата при строго заданном остаточном давлении. Первая колонна с регулярной сеточной насадкой и с орошением может функционировать при атмосферном либо пониженном давлении (вплоть до 2.0 мм рт.ст.). Вторая - вакуумная колонна (без насадки и без орошения) может функционировать при глубоком вакууме (даже когда остаточное давление равно 1.0-0.01 мм рт.ст.) при температурах до 370°С. Рассматриваемая дистилляционная установка оснащена также автоматическим узлом определения массы фракций. Вся информация о деталях разделения олигомеризата поступает в компьютер, где она накапливается и обрабатывается. В частности, компьютер по специальной программе, исходя из реальных значений температуры куба и верха колонны и остаточного давления в колонне определяет виртуальную температуру процесса при атмосферном давлении (Тв), которая совпадает с условной температурой фракционирования (Ту), найденной с помощью известной номограммы Т - Р. Разделение олигомеризата на фракции производили на первой колонне с регулярной насадкой (15 теоретических тарелок) с кубом около 20 литров. Деполимеризацию высокомолекулярных олигодеценов проводили на второй (вакуумной) колонне без регулярной насадки и без орошения с кубом около 10 литров.
В электрообогреваемый куб первой колонны загружали от 5 до 10 кг очищенного от катализатора и от органически связанного хлора олигомеризата. Разделение олигомеризата на фракции производили в режиме медленного подъема температуры от 20 до 300°С. Легко кипящие компоненты, а также ПАО-2 и ПАО-4 из олигомеризата выделяли на первой колоне с насадкой и с орошением. ПАО-6 и ПАО-8 выделяли на второй вакуумной системе.
Было установлено, что в процессе разделения деценового олигомеризата на узкие фракции при температурах, превышающих 330°С, наблюдается термическая деполимеризация олигодеценов. В интервале температур 300-330°С при остаточном давлении менее 5 мм рт.ст. из олигомеризата выделяются оставшиеся там тримеры и тетрамеры децена. При 360°С в течение трех часов при остаточном давлении в колонне менее 3 мм рт.ст. в кубе происходит практически полная деполимеризация высокомолекулярных олигомеров децена. Полученные при этом продукты были разделены на фракции. Установлено, что наиболее легкая фракция, выделенная при температурах 20-150°С, из продуктов деполимеризации высокомолекулярных олигодеценов состоит из смеси олефинов (преимущественно деценов) с винильными (53.6%), трансвиниленовыми (18.1%) и винилиденовыми (28.3%) двойными связями. Выделенные в интервалах температур 150-240 и 240-300°С продукты представляют собой димеры и тримеры децена, соответственно. Это подтверждено методом газовой хроматографии и совпадением основных свойств этих фракций со свойствами стандартных образцов димеров и тримеров деценов. В кубовом остатке после разделения продуктов деструкции на фракции содержались не деполимеризованные высокомолекулярные олигомеры деценов.
Описанный состав продуктов свидетельствует о том, что в процессе термообработки высокомолекулярных олигодеценов происходит (видимо, статистическая) их деполимеризация. Обнаруженный эффект имеет большое практическое значение, т.к. позволяет, в случае необходимости, простым способом перерабатывать высокомолекулярные олигодецены в ди-, три- и тетрамеры деценов. Результаты разделения деценового олигомеризата на фракции в условиях частичной деполимеризации высокомолекулярных компонентов приведены в таблице 9.
Деполимеризация высокомолекулярных олигомеров децена (ПАО-10-ПАО-20) с кинематической вязкостью при 100° С=10-20 сСт целенаправленно проводилась при температурах 330-360°С при остаточном давлении на верху колонны деполимеризатора 1 мм рт.ст., а в кубе колонны 3-5 мм рт.ст.
Для лучшего понимания работы деполимеризатора приводим описание одного из типичных опытов. В куб деполимеризатора было загружено 3000 г олигомера децена с температурой кипения при остаточном давлении на верху колонны 1 мм рт.ст. выше 360°С. В результате деполимеризации этого олигодецена при температуре 350°С в течение трех часов было отогнано 2258 г (75.27 мас.%) продуктов деполимеризации. Выделенные через верх реактора-колонны продукты деполимеризации олигодецена-1 в общем количестве 2258 г были повторно разделены на следующие фракции (см. таблицу А):
При деполимеризации олигодеценов с температурой кипения более 300° С при температуре куба 360°С, температуре стенок деполимеризатора 350°С и остаточном давлении в конденсаторе 1 мм рт.ст. были получены следующие результаты (см. таблицу Б):
Для выявления оптимальных условий функционирования деполимеризатора изучено влияние температуры на кинетику термической деполимеризации высокомолекулярного олигодецена с температурой кипения выше 330°С.
Из полученных данных следует:
1. Деполимеризатор выходит на заданный стационарный температурный режим в течение 10-20 минут.
2. После выхода деполимеризатора на заданный температурный режим термическая деполимеризация высокомолекулярного олигодецена протекает с постоянной скоростью.
3. Скорость деполимеризации, выход продуктов деполимеризации и конверсия высокомолекулярного олигодецена в целевые продукты с повышением температуры от 340 до 360°С монотонно возрастают.
4. Наблюдаемая энергия активации деполимеризации равна 27.5 ккал/моль, lg A=9.9073.
5. При 360°С в течение часа достигается более чем семидесятипроцентная конверсия высокомолекулярного олигодецена в продукты деполимеризации, которые при указанной температуре и при остаточном давлении в кубе не более 3 мм рт.ст. отгоняются из деполимеризатора и направляются в колонну для повторного разделения.
6. Продукты деполимеризации отличаются от продуктов олигомеризации децена-1 не только тем, что наряду с молекулами с внутренними (виниленовыми) и винилиденовыми двойными связями они содержат значительное количество (около 30 мол.%) молекул с винильными двойными связями, но и физико-химическими свойствами (Таблицы 9, 10).
7. Термическая деполимеризация высокомолекулярных олиго-олефинов представляет собой химический эндотермический процесс. Для обеспечения деполимеризации 1000 кг высокомолекулярных олигодеценов в час должен быть предусмотрен нагреватель общей мощностью 116 кВт.
Тепловой баланс деполимеризатора таков:
Поступление тепла - 116 кВт/час.
Расход тепла:
- обогрев высокомолекулярных олигодеценов до 360° С - 30 кВт/час;
- эндотермическая реакция деполимеризации при 360°С - 33 кВт/час;
- испарение образовавшихся продуктов - 28 кВт/час;
- тепловой резерв - 11 кВт/час;
- потери тепла - 14 кВт/час.
Важно отметить, что условия процесса (температура, давление) оказывают существенное влияние на состав продуктов деполимеризации и на их характеристики (таблицы 9, 10). Особенно большое влияние на состав и характеристики продуктов деполимеризации оказывает скорость удаления их из реактора. При повышении температуры и давления, а также при уменьшении времени отгонки продуктов из деполимеризатора глубина деполимеризации олигодеценов резко повышается.
Вид хроматограмм димеров и тримеров, а также высокие значения температур их застывания, свидетельствуют о том, что в ходе термической деполимеризации высокомолекулярных олигодеценов при высоком остаточном давлении (более 10 мм рт.ст.) на колонне с регулярной насадкой и с орошением происходит их парафинизация. Указанные условия термической деполимеризации, когда продукты первичной деполимеризации не удаляются из зоны реакции, а многократно возвращаются в куб, видимо, благоприятствуют протеканию цепного распада олигодеценов. Это заключение подтверждается примером, который осуществлялся на колонне без регулярной насадки и без орошения при остаточном давлении 1.0-0.6 мм рт.ст. В этом примере в качестве исходного олигомеризата использовался кубовый остаток, выделенный из деценового олигомеризата после водно-щелочного дегидрохлорирования. Он содержал около 30 м.д. хлора и имел следующий состав (см. таблицу В):
В куб колонны-деполимеризатора было загружено 1484.6 г охарактеризованного выше дегидрохлорированного высокомолекулярного олигодецена. Вначале в кубе при перемешивании олигомеризата мощной электромагнитной мешалкой производился постепенный разогрев олигомеризата до 360°С. Деполимеризация высокомолекулярного олигодецена начиналась при температуре 330°С и осуществлялась в основном при 360°С. Реальная и виртуальная температура куба и верха колонны определялись теплопотерями, расходом тепла на разогрев олигомеризата и продуктов деполимеризации, на термическую деполимеризацию олигодеценов и на испарение продуктов деполимеризации. На термограмме протекание всех этих эндотермических процессов проявилось в виде нескольких эндо-эффектов. Скорость деполимеризации (точнее - скорость отгонки продуктов термической деполимеризации) во времени изменялась сложным образом. Вначале скорость процесса по мере подъема температуры от 330 до 360°С монотонно возрастала до примерно 13 мл продуктов в минуту. Однако на 170 минуте совершенно неожиданно произошло скачкообразное возрастание (почти в 3 раза) скорости отгонки продуктов термической деполимеризации. Поскольку температура и остаточное давление при этом оставались неизменными, то можно предположить, что отмеченное ускорение процесса определяется его вырожденно-цепным характером.
В результате деполимеризации высокомолекулярных олигодеценов в указанных выше условиях в ловушке собран конденсат, выделено две фракции продуктов и получен кубовый остаток. Конденсат состоял в основном (98.3 мас.%) из углеводородов С4-С12. Доля углеводородов (парафинов и олефинов) в конденсате монотонно снижалась от С4 до С12. Такой состав конденсата, видимо, отражает соотношение различных ответвлений в молекулах олигодеценов. Если это предположение соответствует действительности, то можно сделать вывод о том, что в молекулах олигодеценов больше всего содержится бутильных ответвлений. Первая фракция продуктов термической деполимеризации, выделенная в количестве 657.7 г при виртуальных температурах от 209 до 575°С, имеет очень сложный состав (см. таблицу Г):
Из приведенной таблицы видно, что эта фракция представляет собой смесь 1.43 мас.% низкомолекулярных продуктов термической деполимеризации, а также исходных и полученных в результате деполимеризации ди-, три-, тетра- и более высокомолекулярных олигодеценов. Температура застывания этой фракции = -39°С. Она снижается до -49°С после выделения из нее низкомолекулярных C4-Cl18 углеводородов.
Вторая фракция продуктов термической деполимеризации высокомолекулярных олигодеценов, выделенная в количестве 295.1 г при виртуальных температурах 575-534°С (при остаточном давлении 0.6 мм рт.ст.), так же как и первая фракция, представляет собой сложную смесь низкомолекулярных продуктов термической деполимеризации, а также исходных и полученных в результате деполимеризации ди-, три-, тетра- и более высокомолекулярных олигодеценов. Температура застывания этой фракции = -33°С, но после выделения из нее низкомолекулярных C4-Cl18 углеводородов снижается до -45°С. Содержание низкомолекулярных продуктов деполимеризации (3.0 мас.%) во второй фракции более чем в два раза превышает содержание низкомолекулярных продуктов деполимеризации (1.43 мас.%) в составе первой фракции. Это указывает на повышение глубины деполимеризации. Общая конверсия исходных олигодеценов в продукты деполимеризации превышала 70 мас.%. Суммарный выход второй и третьей фракций (952.8 г)=64.18 мас.%.
Кубовый остаток (458.8 г=30.9 мас.%) состоит из смеси исходных и деполимеризованных высокомолекулярных олигодеценов (см. таблицу Д):
Он имеет температуру застывания=-30°С.
Совокупность полученных данных позволяет сделать следующие общие выводы:
- высокомолекулярные олигодецены методом термической деполимеризации при температурах 330-360°С и остаточном давлении 1.0 мм рт.ст. можно переработать с высокой конверсией в смесь низкомолекулярных продуктов;
- из смеси термической деполимеризации высокомолекулярных продуктов методом вакуумной дистилляции можно выделить фракции продуктов, по фракционному составу и по вязкостным свойствам приближающиеся к ПАО-2, ПАО-4 и ПАО-6;
- температуры застывания выделенных фракций превышают температуры застывания ПАО-2, ПАО-4 и ПАО-6, выделенных из исходного олигомеризата, но являются существенно ниже, чем температуры застывания соответствующих по вязкостным свойствам не компаундированных фракций минеральных масел.
В случае необходимости это позволяет рекомендовать их после гидрирования к использованию в смеси с соответствующими основными продуктами в качестве основ синтетических и полусинтетических масел. Наличие стадии деполимеризации обеспечивает возможность переработки всех ограниченно потребляемых высокомолекулярных олигодеценов с кинематической вязкостью при 100°С, изменяющейся в пределах от 10 до 20 сСт в широко потребляемые полиолефины с кинематической вязкостью при 100°С, изменяющейся в пределах от 2 до 8 сСт (т.е. в ПАО-2, ПАО-4, ПАО-6 и ПАО-8, соответственно).
Разделение олигомеризата на узкие фракции по патентуемому способу, как уже отмечалось, производится так же, как и в других известных способах, за исключением того, что глубокий вакуум в колоннах разделения обеспечивается оригинальной вакуумной паро-эжекторной системой.
Выделяемые на стадии разделения олигомеризата на фракции продукты во всех случаях представляют собой смеси ненасыщенных и хлорсодержащих молекул олигодеценов. Остаточное содержание хлора в выделяемых фракциях не превышает 10 м.д. (10 ppm). Для повышения термоокислительной стабильности получаемых продуктов во всех известных способах их гидрируют.
Гидрирование выделенных из олигомеризата узких фракций олигоолефинов в соответствии с настоящим изобретением проводят под действием палладиевого нанесенного на оксид алюминия катализатора (преимущественно - Pd (0.2 мас.%)/Al2O3), модифицированного безводным KOH, который берут в количестве от 30 до 100 мас.% в расчете на катализатор гидрирования, при температурах, изменяющихся в интервале температур от 150 до 250°С при давлении водорода 20 ат. Температура и давление на стадии гидрирования по патентуемому способу значительно ниже, чем в случае других известных способов. Гидрирование узких фракций олигодеценов в указанных выше условиях позволяет гидрировать не только С=С, но и С-Cl связи олигодеценов. Осуществление гидрирования олигодеценовых фракций в присутствии безводного KOH обеспечивает повышение активности и производительности катализатора гидрирования в результате нейтрализации хлористого водорода, образующегося при гидрировании С-Cl связей оставшихся в гидрируемых фракциях хлорсодержащих олигодеценов. Это решение, кроме того, устраняет коррозию реакторов гидрирования и вспомогательного оборудования и предотвращает ускоренную дезактивацию хлористым водородом палладиевого катализатора гидрирования.
При разработке способа получения полиолефиновых основ синтетических масел использовались следующие реактивы: децен-1, выделенный из продуктов олигомеризации этилена под действием триэтилалюминия на ОАО "Нижнекамскнефтехим" (ТУ 2411-057-05766801-96), а также децен-1, выделенный из продуктов олигомеризации этилена под действием триэтилалюминия в г.Нератовице (Чехия) компанией "Сполана". Используемый децен-1 производства ОАО "НКНХ" имел следующий групповой состав: СН2-=СН - 83.4 мол.%; транс-СН=СН - 5.44 мол.%; СН2=С<11.2 мол.%. Перед употреблением децен-1 сушили над прокаленными при 600°С молекулярными ситами NaX; н-гептан, марки "эталонный", применяемый в качестве растворителя для приготовления растворов компонентов катализаторов, сушили перегонкой над натриевой проволокой; АОС общей формулы RnAlCl3-n, (R-С2Н5; n=1.5, 3.0) очищали перегонкой при пониженном давлении. Подготовленные олефины, растворители и АОС хранили в инертной атмосфере в герметичных сосудах. АОС использовали в виде разбавленных н-гептановых растворов.
Олигомеризацию децена-1 осуществляли под действием систем Al (0)-HCl-(СН3)3CCl (ТБХ) и Al(0)-(C2H5)1.5AlCl1.5 (ЭАСХ)-(СН3)3CCl, в которых HCl и ЭАСХ выполняли функции активаторов алюминия марок АСД-4, АСД-40, ПА-1 и ПА-4. При разработке патентуемого способа в основном использовали алюминий марки ПА-4. Скорость олигомеризации децена-1 под действием упомянутых каталитических систем лимитируется скоростью растворения алюминия третбутилхлоридом. Этот процесс предшествует образованию активных центров олигомеризации децена-1. Включение в упомянутые каталитические системы активаторов алюминия сокращает или устраняет индукционный период.
В качестве сокатализатора в обеих упомянутых системах использовали трет-бутилхлорид - (СН3)3CCl, который был получен по реакции изобутилена с сухим HCl. ТБХ содержал 0.5 мас.% HCl (ТУ 6-09-07-1338-83).
В предварительных исследованиях было установлено, что по совокупности характеристик лучшим активатором алюминия является HCl, а лучшим сокатализатором в процессе олигомеризации децена-1 является ТБХ. Поэтому основные исследования выполнены с применением каталитической системы Al(0) марки ПА-4-HCl-ТБХ.
Олигомеризацию децена-1 проводили в термостатированном высушенном стеклянном или металлическом реакторах смешения в атмосфере сухого аргона при непрерывном интенсивном перемешивании реакционной массы с помощью электромагнитной мешалки. Тестирование каталитических систем в процессе катионной олигомеризации децена-1 и других олефинов производили следующим образом: в термостатированный до заданной температуры реактор последовательно загружали алюминий, децен-1 или другой олефин, а затем раствор HCl в ТБХ. Al загружали в реактор в виде заранее подготовленной в инертной атмосфере навески, хранившейся в стеклянной запаянной ампуле. Вслед за загрузкой в реактор всех упомянутых реагентов (обычно сразу же) начиналась олигомеризация олефина, что сопровождалось заметным повышением температуры реакционной среды (олигомеризата). Реакцию продолжали в течение запланированного времени, а затем обрывали путем введения в реактор водного раствора щелочи (NaOH) либо этанола. Полученный олигомеризат из реактора выгружали далее в промежуточную емкость. Отработанный (дезактивированный) катализатор из олигомеризата удаляли методом водно-щелочной и водной отмывки.
Содержание децена-1 (или другого исходного олефина) в реакционной массе (олигомеризате) в ходе и после олигомеризации (т.е. текущую и общую конверсию) определяли методом газовой хроматографии на приборах ЛХМ-8-МД, ЛХМ-2000 и "Hewlett-Packard 5880А" (внутренний стандарт - пентадекан) и методом ИК-спектроскопии на приборе "Specord M-80". Для этого в заданный момент времени из реактора под аргоном отбирали часть реакционной массы, которую сразу же смешивали с этанолом или 5%-ным водным раствором NaOH в условиях интенсивного перемешивания. Реакционную смесь после прекращения олигомеризации многократно промывали дистиллированной водой в закрытой воронке.
Непрореагировавший децен-1, а также ди-, три- и тетрадецены из олигомеризата выделяли на вакуумной колонне с электрообогреваемым до 360°С кубом при остаточном давлении от 1 до 10 мм рт.ст.
Фракционный состав олигомеризата определяли на хроматографах ЛХМ-8-МД, ЛХМ-2000 и "Hewlett-Packard 5880A" с ионизационно пламенными детекторами в режиме программирования температуры от 20 до 350°С со скоростью подъема температуры 8-10 град/мин. При хроматографировании олигодеценов использовали колонки из нержавеющей стали (0.4×70-0.4×200 см), заполненные хроматоном NAWDMCS с 3.0% силикона OV-17, хромосорбом W-AW с 3% Dexil - 300 или Паропаком-Q. Среднеэквивалентный диаметр частиц упомянутых носителей 0.200-0.250 мм. Скорость подачи предварительно очищенного газа-носителя (гелий, азот) составляла ˜40, водорода ˜30, воздуха ˜300 мл/мин. Пробу в испаритель хроматографа вводили с помощью микрошприца (0.2-1.0 мкл) после того, как температура испарителя достигала 350°С. Продолжительность анализа 50 мин. Идентификацию хроматографических пиков проводили методом добавки реперных углеводородов (пентадекана) и путем сравнения с хроматограммами смесей углеводородов известного состава. Количественную обработку хроматограмм осуществляли по интегральным площадям пиков, которые определяли с помощью компьютерного интегратора или методом триангуляции. Фракционный состав олигомеризата определяли также методом фракционирования его на вакуумной ректификационной колонне. Результаты этих количественных анализов совпадали с точностью ±3 мас.%.
Ненасыщенность олигомеров (т.е. содержание в молекулах олигомеров двойных связей) определяли методом озонолиза на анализаторе двойных связей АДС-4М.
Строение не превращенных деценов и продуктов олигомеризации количественно определяли методом ИК-спектороскопии на приборе "Specord M-80", а также методами спектроскопии ПМР и ЯМР13С. Спектры ПМР и ЯМР13С регистрировали при комнатной температуре на импульсном спектрометре ЯМР АС-200Р (200 МГц) фирмы "Bruker". Для снятия спектров ПМР и ЯМР13С готовили 10-20%-ные растворы продуктов в дейтерохлороформе. В качестве стандарта использовали тетраметилсилан.
Примеси ионного хлора в олигомерах определяли аргентометрическим методом по Фольгарду путем титрования водного экстракта. Ковалентно-связанный с молекулами олигомеров хлор вначале переводили в ионную форму путем мокрого сжигания пробы в кварцевом реакторе или с помощью бифенил натрия по методу UOP 395-66 с последующим аргентометрическим титрованием его по Фольгарду. В некоторых случаях содержание ковалентно-связанного с молекулами олигомеров хлора определяли рентгенофлюоресцентным методом на спектрометре "SPECTRO XEPOS" по калибровочной кривой.
Для лучшего понимания данного изобретения в качестве иллюстраций в таблицах 1-10 приведены примеры реализации разработанного согласно изобретению способа получения полиолефиновых основ синтетических масел. Эти примеры демонстрируют, но не исчерпывают возможности изобретения.
Совокупность решений по различным аспектам патентуемого изобретения позволяет утверждать, что разработан новый способ получения полиолефиновых основ синтетических масел. Разработанный способ содержит стадии подготовки олефинового сырья, приготовления и дозирования в реактор растворов и суспензии компонентов каталитической системы Al(0)-HCl-ТБХ, изомеризации альфа-олефинов и олигомеризации высших олефинов и их смесей под действием каталитической системы Al(0)-HCl-ТБХ, выделения отработанного катализатора, разделения олигомеризата на фракции и гидрирования выделенных фракций под действием катализатора Pd (0.2 мас.%)/Al2O3 +NaOH. Изобретение обеспечивает совершенствование всех стадий разработанного способа.
С целью устранения коррозионной активности продуктов способ дополнительно содержит стадию дехлорирования присутствующих в олигомеризате хлор содержащих олигоолефинов металлическим алюминием, триэтилалюминием, спиртовыми растворами KOH или термическим дегидрохлорированием хлор содержащих полиолефинов в отсутствие или в присутствии KOH. Для улучшения технико-экономических показателей способа за счет повышения выхода целевых фракций полиолефинов с кинематической вязкостью 2-8 сСт при 100°С способ дополнительно содержит стадию термической деполимеризации ограниченно потребляемых высокомолекулярных полиолефинов с кинематической вязкостью 10-20 сСт при 100°С в целевые полиолефины с кинематической вязкостью 2-8 сСт при 100°С.
Все решения по настоящему изобретению подтверждены не только в лабораторных условиях, но и на опытных установках и на промышленных установках в цехах Нижнекамского завода синтетических масел.
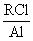
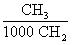
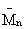
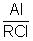
Реферат
Изобретение относится к способу получения полиолефиновых основ синтетических масел путем катионной олигомеризации олефинового сырья и может быть использовано в нефтехимической промышленности. Разработанный способ содержит стадии подготовки олефинового сырья, приготовления и дозирования в реактор растворов и суспензии компонентов каталитической системы Al(0)-HCl-(СН3)3CCl (ТБХ), изомеризации альфа-олефинов и олигомеризации высших олефинов и их смесей под действием каталитической системы Al(0)-HCl-ТБХ, выделения отработанного катализатора, разделения олигомеризата на фракции и гидрирования выделенных фракций под действием катализатора Pd (0.2 мас.%)/Al2О3+NaOH. Изобретение обеспечивает совершенствование всех стадий разработанного способа. Для устранения коррозионной активности продуктов способ дополнительно содержит стадию дехлорирования присутствующих в олигомеризате хлор содержащих олигоолефинов металлическим алюминием, триэтилалюминием, спиртовыми растворами KOH или термическим дегидро-хлорированием хлор содержащих полиолефинов в отсутствие или в присутствии KOH. Для улучшения технико-экономических показателей способа за счет повышения выхода целевых фракций полиолефинов с кинематической вязкостью 2-8 сСт при 100°С способ дополнительно содержит стадию термической деполимеризации ограниченно потребляемых высокомолекулярных полиолефинов с кинематической вязкостью 10-20 сСт при 100°С в целевые полиолефины с кинематической вязкостью 2-8 сСт при 100°С. 11 з.п. ф-лы. 15 табл.
Комментарии