Аналог витамина д - RU2037484C1
Код документа: RU2037484C1
Чертежи
Описание
Изобретение относится к неизвестному классу соединений, который обнаруживает иммуномодулирующий эффект, а также высокую активность в снижении дифференциации и предотвращении нежелательной пролиферации определенных клеток, включая раковые клетки и кожные клетки, к фармацевтическим препаратам, содержащим эти соединения, и стандартным дозам таких препаратов и к их использованию при лечении и профилактике аутоиммунных расстройств, включая сахарный диабет, артериальную гипертонию, воспалительные заболевания, такие, как ревматоидный артрит и астма, а также болезни, характеризуемые патологической клеточной дифференциацией и/или пролиферацией, и/или дисбалансом в иммунной системе.
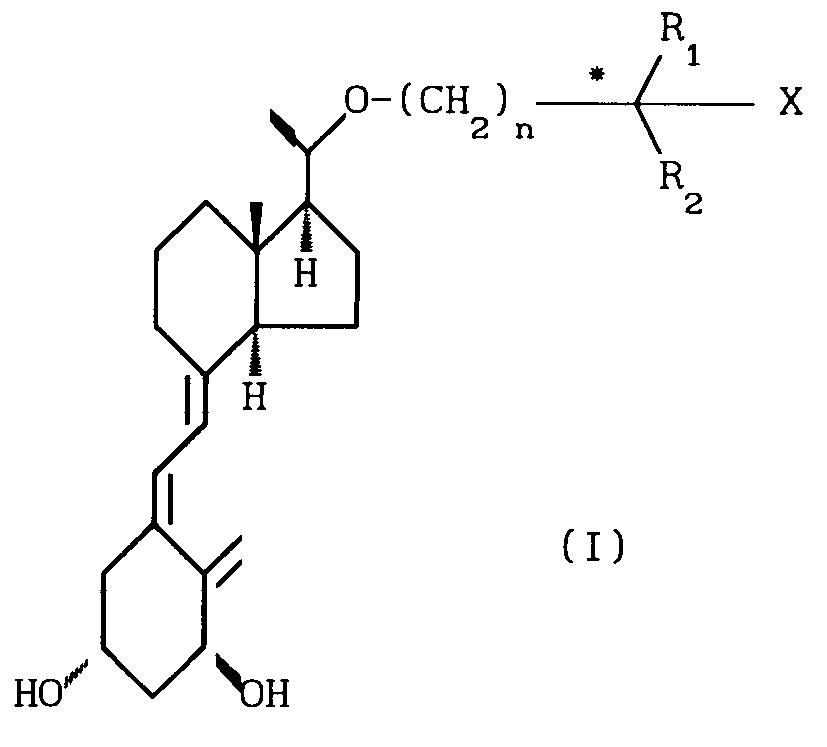
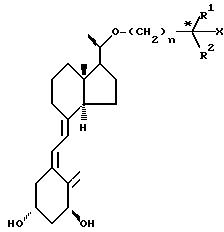
Соединения изобретения представлены
общей формулой I
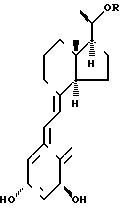
(I) в которой формула R обозначает алкильную группу, содержащую от 4 до 12 атомов углерода, по выбору замещенных гидроксильной группой.
Предпочтительно R представляет собой
группу формулы II,
(CH2)n
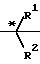
(II) где n является целым числом от 1 до 7, R1 и R2, которые могут быть одинаковыми или различными, обозначают водород, низший алкил, низший циклоалкил или объединенный вместе с углеродным атомом (помеченным звездочкой в формуле II), образуя группу Х, R1 и R2 могут образовывать циклическое углеводородное кольцо С3-С8, Х обозначает водород или гидроксил.
В контексте изобретения выражение "низший алкил" указывает прямую или разветвленную, насыщенную или ненасыщенную углеродную цепь с длиною от 1 до 5 углеродных атомов, а выражение "низший циклоалкил" насыщенное или ненасыщенное карбоциклическое кольцо С3-С7.
Как видно из формулы I и II, в зависимости от значений R, Х, R1 и R2 соединения изобретения могут включать несколько диастереоизомерных форм (например,


Выражение "биопревращаемые производные или пролекарства формулы I" включает, но не ограничивается производными соединениями формулы I, в которых одна или большее число гидроксильных групп превращены в -0-ацильные или -0-гликозильные группы или в фосфатный эфир и такие превращенные группы являются гидролизуемыми in vivo.
Соединения формулы I, в которых группа R не замещена гидроксилом, являются пролекарством другого типа. Такие соединения относительно неактивны in vivo, но превращаются в активные соединения формулы I посредством энзиматической гидролизации после приема пациентом.
В последнее время было показано, что 1 α, 25-дигидроксивитамин D3(1,25(OН)2D3) оказывается эффективным и/или создающим interleukins, что выявляет потенциальную возможность использования этого соединения для лечения болезней, характеризуемых дисфункцией иммунной системы, например аутоиммунных расстройств и отторжением трансплантатов. Кроме того, с помощью 1,25(OН)2D3 могут лечиться другие состояния, характризуемые созданием паталогического интерлейкин-1, например воспалительные заболевания, такие как ревматоидный артрит.
Было показано также, что 1,25(OН)2D3 способен побуждать дифференциацию клеток и ингибировать чрезмерную клеточную пролиферацию, и было высказано предположение, что это соединение могло бы быть полезным при лечении болезней, характеризуемых патологической клеточной пролиферацией и/или клеточной дифференциацией такими, как рак и псориаз.
Предложено также использование 1,25(OН)2D3 для лечения артериальной гипертонии и сахарного диабета.
Однако терапевтические возможности такого использования 1,25(OН)2D3существенно ограничены хорошо известным потенциальным эффектом этого гормона на кальциевый обмен; повышенные кровяные концентрации могут привести к резкому подъему гиперкальцемии. Таким образом, это соединение и его потенциальные синтетические аналоги не полностью удовлетворяют всем требованиям для использования их в качестве лекарств при лечении, например, псориаза, рака или иммунных заболеваний, которые могут требовать непрерывного приема лекарства в относительно высоких дозах.
Известен ряд окса-аналогов витамина D3 [1] 1 α 25-дигидрокси-20- окси-21-норвитамин D3 и 1α гидрокси-20-окси-21-норвитамин D3, 1 α, 25-дигидрокси-22-оксивитамин D3 и 25-гидрокси-22-оксивитамин D3 и 1α, 25-дигидрокcи-23-окcивитамин D3.
Эксперименты in vitro показали, что некоторые из этих соединений могут иметь преимущества по сравнению с 1α, 25(OН)2D3. Так, 1α 25-дигидрокси-22-оксивитамин D3 выявляет только 1/14 часть сродства по сравнению с 1α 25(OН)2D3 по отношению к кишечным цитозольным рецепторам цыплят, более слабое сходство по сравнению с 1,25(OН)2D3 в отношении рецепторов клеточной линии (HL-60) человеческой миелоидной лейкемии и высокую активность в снижении дифференциации в HL-60 клетках.
В противоположность соединениям данного изобретения упомянутые 22-оксисоединения имеют S-конфигурацию в 20 позициях.
Полезность аналога витамина D в приведенных использованиях зависит не только от благоприятного отношения связывающего сродства в отношении соответствующих рецепторов по сравнению с кишечным рецептором, но также и от судьбы соединения в организме.
В настоящее время установлено, что соединения данного изобретения выявляют благоприятную селективность в отношении связывания рецептора и в то же самое время показывают высокую биологическую пригодность, а также химическую и метаболическую стабильность.
Селективность соединений иллюстрируется тем фактом, что наряду с тем, что они имеют более высокое сродство в отношении рецептора в опухолевых клетках (аналогичное или значительно большее, чем соответствующий показатель для 1,25(OН)2D3), необходимая концентрация для снижения клеточной дифференциации в человеческой моноцитовой опухолевой клеточной линии та же или значительно ниже, чем необходимое количество 1,25(OН)2D3 для достижения того же эффекта, и их связывающее сродство в отношении кишечного рецептора ниже, чем соответствующий показатель для 1,25(OН)2D3. В опытах in vivo на крысах соединения являются менее активными, чем 1, 25(OН)2D3 в снижении гиперкальцинирования мочи и гиперкальцемии.
Это делает соединения изобретения особенно подходящими как для локального, так и систематического лечения и профилактики как заболеваний человека, так и животных тех, которые характеризуются патологической клеточной пролиферацией и/или клеточной дифференциацией, такие как определенные дерматологические нарушения, включая псориаз и определенные формы рака, например лейкемию и миелофиброз, и болезни, характеризуемые нарушением баланса в иммунной системе, например аутоиммунные болезни или СПИД, и для того, чтобы получить нужное иммуноподавление, как это имеет место при трансплантации, а также при лечении акне, сахарного диабета, артериальной гипертонии и воспалительных заболеваний, таких как ревматоидный артрит и астма. Поскольку соединения данного изобретения могут вызывать дифференцию клеток волосяного фолликула, они могут использоваться для лечения алопеции (облысения).
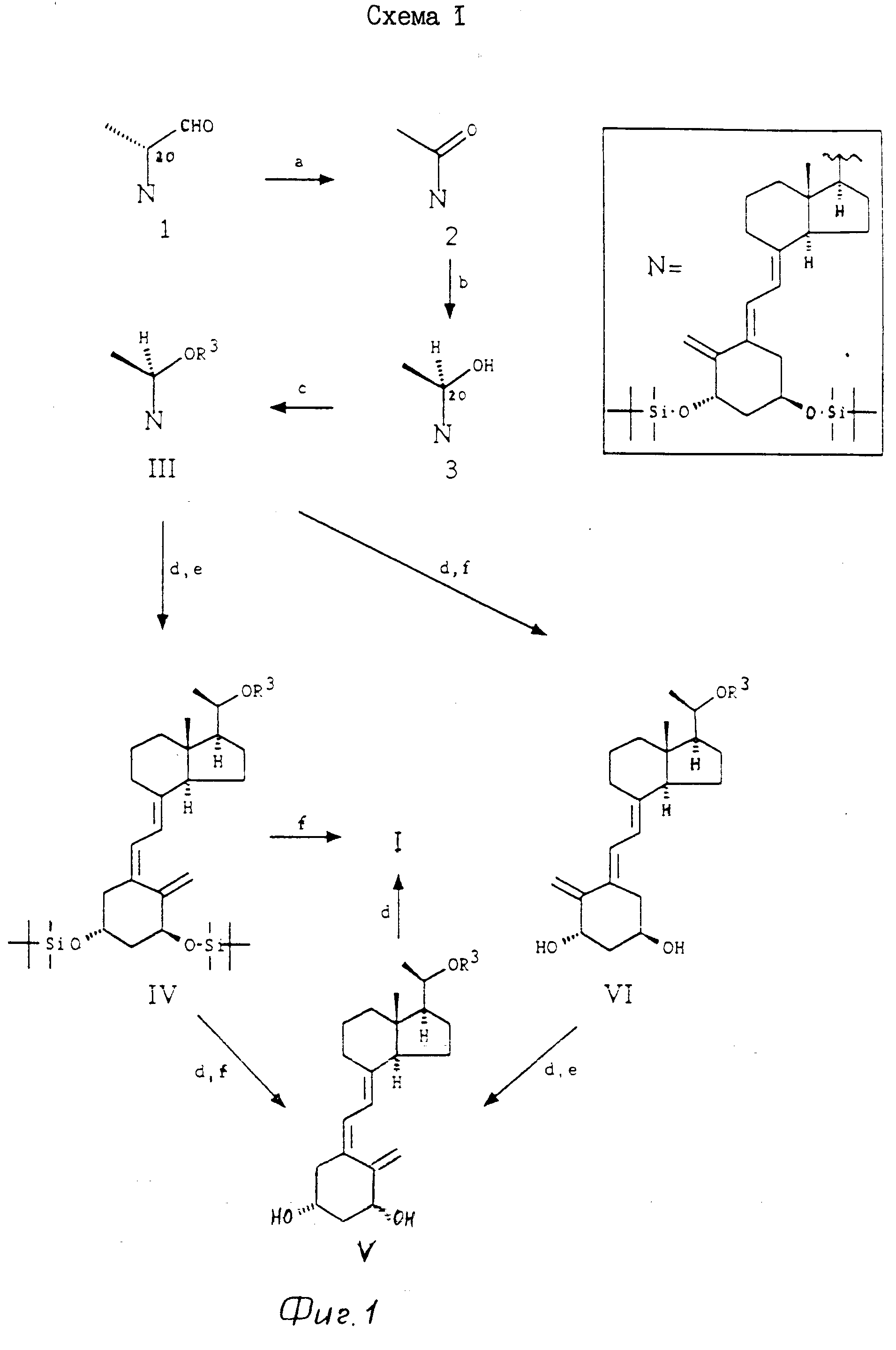
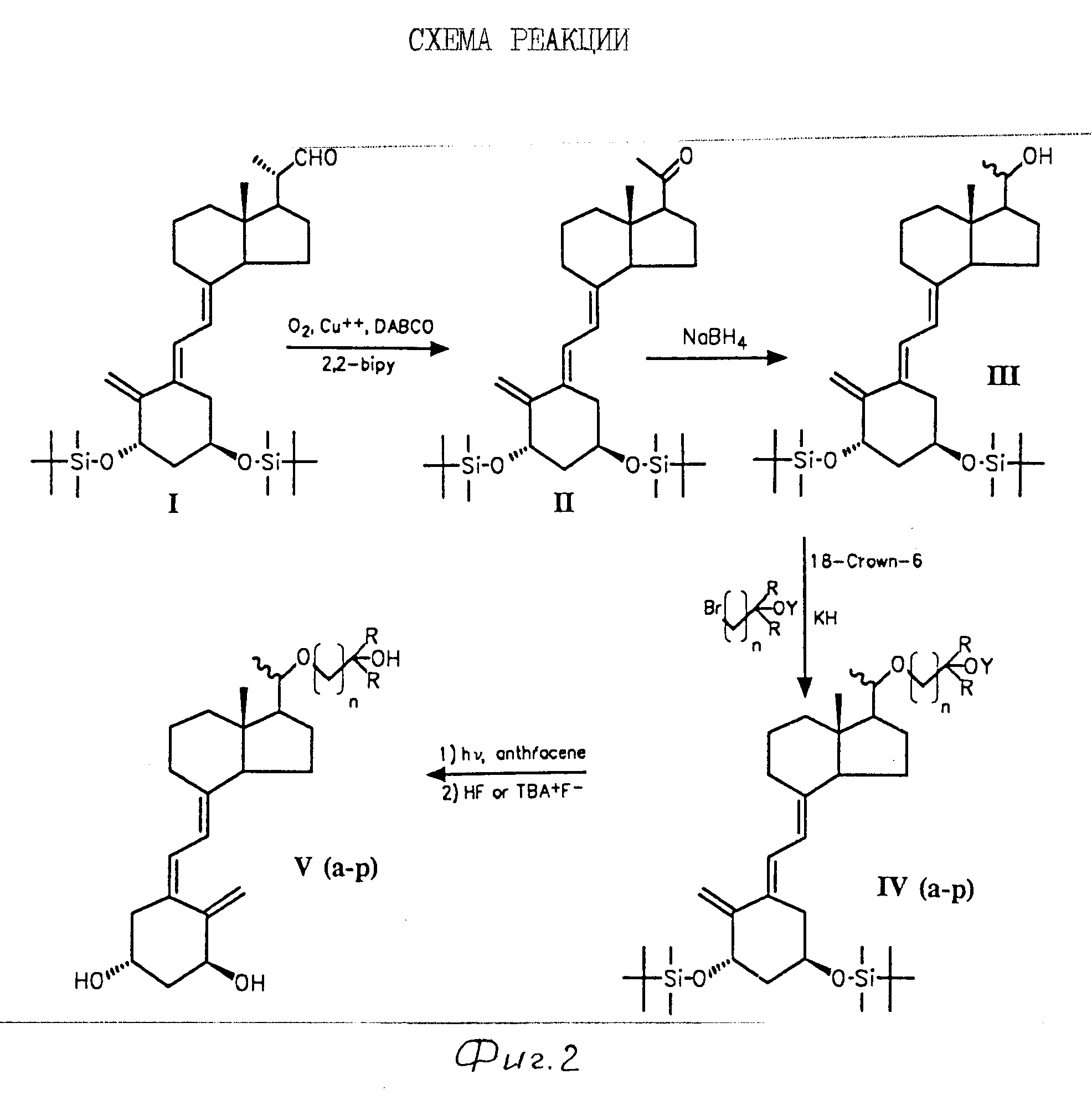
Соединения формулы I могут быть изготовлены обычным способом из D-производного 1 (или его изомера 20 R) [2] по методу, описанному в схеме 1 (см. на фиг. 1). Например, окисление 1 с использованием метода van Rheenen [3] дает кетон 2, который восстанавливается до 20R-спирта При использовании подходящего хирального восстановительного агента 3 может быть приготовлен с очень высокой стереоселективностью, но 3 обычно готовится восстановлением соединения 2 с помощью NaBН4 и выделением минимального количества соответствующего 20S-спирта хроматографическим методом. О-алкилирование соединения 3 с образованием III достигается воздействием в щелочных условиях боковой цепью строящегося блока общей формулы Z-R3, в которой Z является остающейся (концевой) группой, такой как галоген (Сl, Br или I) или п-толуолсульфонилокси или метансульфонилокси, и R3 является R (формулы I) или по выбору радикалом, который может быть превращен в R любой удобной последующей стадией (или посредством нескольких стадий). Сле- довательно, R3 в соединениях III, IV, V и VI не обязательно имеет то же самое значение в частной синтетической последовательности. Превращение R3 в R вполне может включать несколько стадий и возможно включение временной защиты чувствительной триеновой системы молекулы. Помимо любой необходимой модификации внутри боковой цепи (R3) конверсия соединения III в соединение I включает стадию фотоизомеризации и стадию десилирования, аналогично стадиям, используемым на последних этапах синтеза других аналогов витамина D.
Боковые цепи строящихся блоков R3Z являются известными соединениями. R3 обычно идентично с формулой II, в которой Х является защищенной ОН группой, например тетрагидропиранилокси или триалкилсилилокси.
В изобретении используются следующие стандартные аббревиатуры: Ме-метил, Et-этил, Pr- н-пропил, Pr i-изопропил, But-трет-бутил, THP-тетра-гидро-4Н-пиран-2-ил, THF-тетрагидрофуран, T-п-толуолсульфонил, ТВA-тетра-(н-бутил)-аммоний.
Примечания к схеме 1.
а). Окисление, например, с использованием О2, Сu(AcO)2, 2,2'-бипиридил и 1,4-диазабицикло [2,2,2] октана в качестве катализатора.
b). Восстановление (например, с использованием NaBH4).
с). Алкилирование с использованием фрагмента боковой цепи R3-Z в присутствии основания (например, KOН, KOBut или KН c катализатором или без катализатора (например, 18-Краун-6) в растворителе, например в тетрагидрофуране.
d). Необязательная (дополнительная) модификация функциональных групп в боковой цепи.
e). Изомеризация с использованием h ν -триплетного сенсибилизатора, например антрацена.
f). Cнятие защиты с использованием TBA+F- или HF.
Следует отметить, что хотя показанные промежуточные соединения могут иметь гидроксильные группы, замещенные как, например, трет-бутилдиметилсилиловыми эфирами, изобретение не включает использование альтернативных гидроксильных защищающих групп, хорошо известных в данной области наряду с альтернативными реакциями для снятия защиты.
Данные соединения предполагается применять в фармацевтических композициях, которые используются для лечения человеческих болезней и в ветеринарии.
Требуемое количество соединения формулы I (в дальнейшем ссылаются на активный ингредиент) для достижения терапевтического эффекта, конечно, изменяется для данного соединения в зависимости от пути приема и питания при лечении. Соединения изобретения могут приниматься парентерально, внутрисуставно, энтерально или местным введением. Они хорошо абсорбируются при энтеральном приеме и этот путь является предпочтительным при приеме для лечения систематических расстройств. При лечении дерматологических заболеваний, подобных псориазу, предпочтительными являются местное или энтеральное введение.
При лечении респираторных болезней, таких как астма, предпочтительной формой является аэрозоль.
Хотя активный ингредиент может приниматься один (сам по себе как химическое соединение), более желательно, чтобы он находился в виде фармацевтической композиции. Обычно, активный ингредиент содержит от 1 части/миллион до 0,1% в массе композиции.
Термином "стандартная доза" обозначается унитарность, то есть единичная доза, которая может быть принята пациентом и которая может быть быстро принята и упакована (расфасована), оставаясь физически и химически стабильной стандартной дозой, содержащей активное вещество как таковое или смесь его с твердыми или жидкими фармацевтическими разбавителями или носителями.
Композиции изобретения, используемые как в ветеринарии, так и для лечения людей, содержат активный ингредиент в сочетании с фармацевтически приемлемым носителем и, по выбору, с другими терапевтическими ингредиентами. Носитель(и) должен быть совместимым с другими ингредиентами композиций и не должен быть вредным для принимающего его.
Композиции, например, включают эти вещества в форме, подходящей для орального, ректального, парентерального (включая подкожный, внутримышечный и внутривенный), внутрисуставного и местного приема.
Обычно композиции могут быть
представлены в форме стандартной дозы и могут быть приготовлены любым из методов, хорошо известных в
фармации. Все эти методы включают этапы введения активного ингредиента в сочетании с носителем,
который замещает один или большее число вспомогательных ингредиентов. Обычно, композиции готовятся
равномерным и тщательным введением активного ингредиента в сочетание с жидким носителем или тон- ко
измельченным твердым носителем или с двумя носителями одновременно и
затем, если это
необходимо, продукту придают желаемую форму.
Композиции изобретения, предназначенные для орального приема, могут существовать в форме дискретных элементов, таких как капсулы, порошки, таблетки или лепешек, каждый из которых содержит предусмотренное количество активного ингредиента; в форме порошков или гранул; в форме раствора или суспензии в водной или неводной жидкости; или в форме эмульсии типа масло в воде, или эмульсии типа вода в масле. Активный ингредиент может также приниматься в виде болюсов (шариков), электуарий (кашки) или пасты.
Таблетки могут быть приготовлены прессованием или расплавлением активного ингредиента выборочно с одним или с большим числом необходимых ингредиентов. Спрессованные таблетки могут быть приготовлены прессованием на соответствующей машине активного ингредиента в свободно-текучем (движущемся) виде, таком как порошок или гранулы, по выбору в смешанном виде со связующим, смазкой, инертным разбавителем, поверхностно-активным или диспергирующим агентом.
Композиции для ректального приема могут быть изготовлены в виде суппозитория, включающего активный элемент и носитель, такой как масло какао, или в виде клизмы.
Композиции, предназначенные для парентерального приема, обычно содержат стерильные масляные или водные составы активного ингредиента, которые, желательно, являлись бы изотоничными по отношению к крови реципиента.
Композиции, предназначенные для внутрисуставного введения, могут существовать в виде стерильного водного раствора активного ингредиента, который может быть в микрокристалличной форме, например в форме водной микрокристалличной суспензии. Могут использоваться также липосоматические композиции или биоразлагаемые полимерные системы для введения активного ингредиента как внутрисуставно, так и для офтальмологического приема.
Композиции, предназначенные для местного использования, включают жидкие или полужидкие составы, такие как линименты, лосьоны, компрессы (примочки), эмульсии типа масло в воде или вода в масле, такие как кремы, мази или пасты; или растворы и суспензии, такие как капли.
Для лечения астмы могут быть использованы ингаляции порошком, самопроникающие или распыляемые композиции, диспергируемые напылением с использованием распылителя или пульверизатора. При диспергировании желательно, чтобы композиции имели размер частиц от 10 до 100 мкм.
Композиции являются наиболее предпочтительными: в форме тонко измельченного порошка для легочного приема в отличие от механизма порошковой ингаляции или самопроникающих порошковых дисперсий. В случае композиций самопроникающих растворов и напылений эффект достигается либо при использовании клапана, имеющего нужные характеристики напыления (то есть способного создавать напыление с необходимым размером частиц), либо введением активного ингредиента в виде суспензированного порошка с контролируемым размером частиц. Такие самопроникающие композиции могут быть либо самодиспергирующимися композициями, либо композициями, диспергирующими активный ингредиент в виде капель раствора или суспензии.
Самопроникающие диспергирующие порошковые композиции содержат, желательно, диспергированные частицы твердого активного ингредиента и жидкий пропелент, имеющий температуру кипения ниже 18oC при атмосферном давлении. Жидкий пропелент может быть любым из известных пропелентов, известных в качестве применяемых в медицине для внутреннего приема, и может содержать один или большее число С1-С6-алкильных углеводородов или галогенированных С1-C6-алкильных углеводородов, или их смесь; особенно желательны хлорированные или фторированные С1-С6-алкильные углеводороды. Обычно пропелент составляет от 45 до 99,9 мас. композиции, в то время как активный ингредиент составляет от 1 части/миллион до 0,1 мас. композиции.
Помимо приведенных ингредиентов, композиции изобретения могут включать один или большее число дополнительных ингредиентов, таких как разбавители, буфферы, ароматизирующие агенты, связующие, поверхностно-активные агенты, загустители, смазки, защитные вещества (стабилизаторы), например метилгидроксибензоат (включающий антиоксиданты), эмульгирующие агенты и т.п.
Кроме того, композиции могут содержать другие терапевтически активные соединения, обычно применяемые при лечении упомянутых патологических расстройств.
Изобретение касается также метода для курса лечения пациентов, страдающих от одного из приведенных выше патологических состояний, при этом названный метод состоит в приеме пациентом при необходимом лечении эффективного количества одного или большего числа соединений формулы I, одних или в комбинации с одним или большим числом других терапевтически активных соединений, обычно используемых при лечении названных патологических состояний.
Лечение представленными соединениями и/или дополнительными терапевтически активными соединениями может быть одновременным или с промежутками времени.
При лечении систематических расстройств ежедневные дозы приема могут составлять от 0,1 до 100 мкг (10-6 г), предпочтительно от 0,2 до 25 мкг соединения формулы I. При местном лечении дерматологических заболеваний принимаются мази, кремы или лосьоны, содержащие от 0,1 до 500 мкг/г, предпочтительно от 1 до 100 мкг/г cоединения формулы I. Оральные композиции готовятся предпочтительно в виде таблеток, капсул или капель, содержащих от 0,05 до 50 мкг, преимущественно от 0,1 до 25 мкг соединения формулы I на стандартную дозу.
Приготовления и примеры.
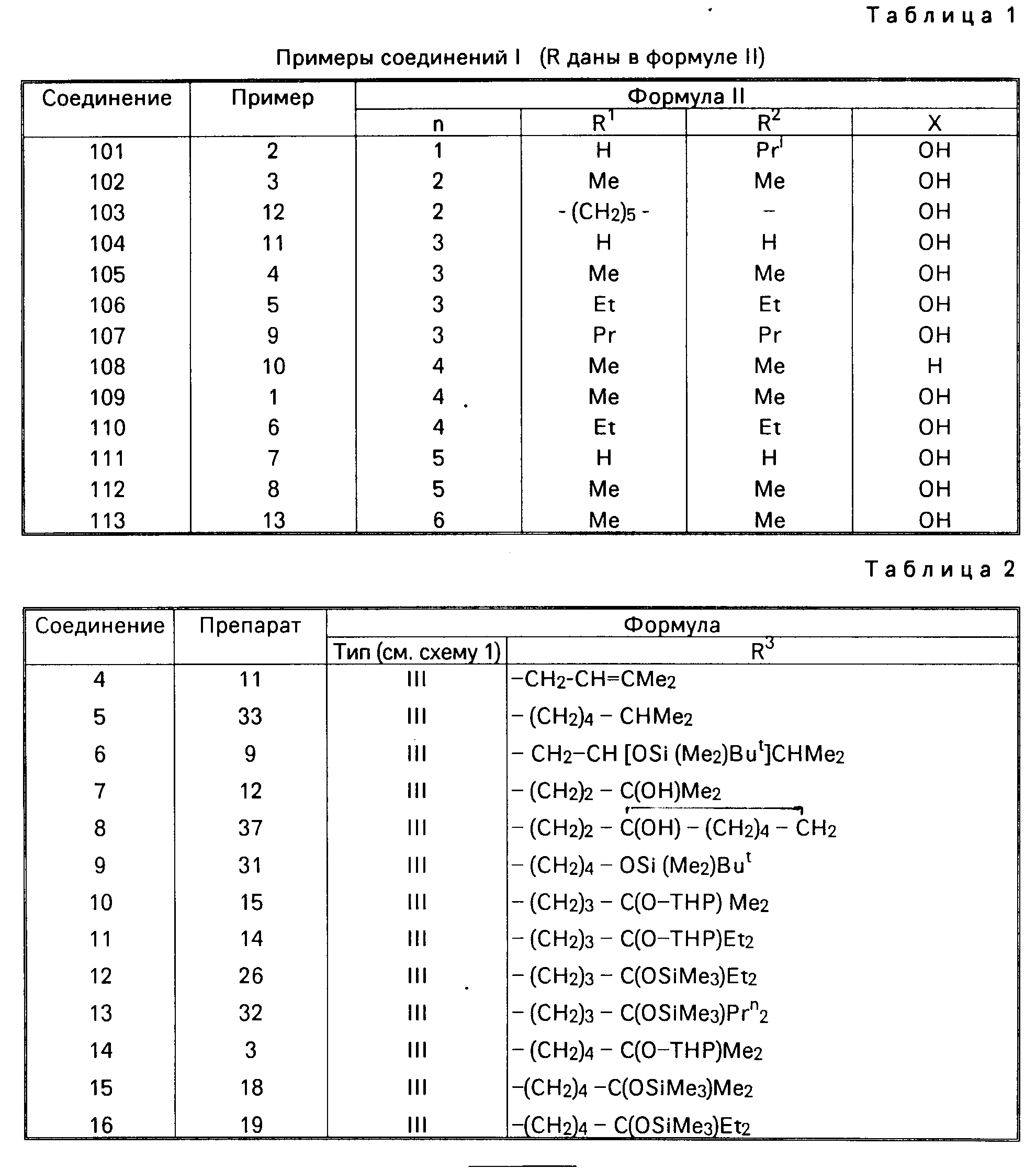
Приведенные примеры соединений I перечислены в табл.1.
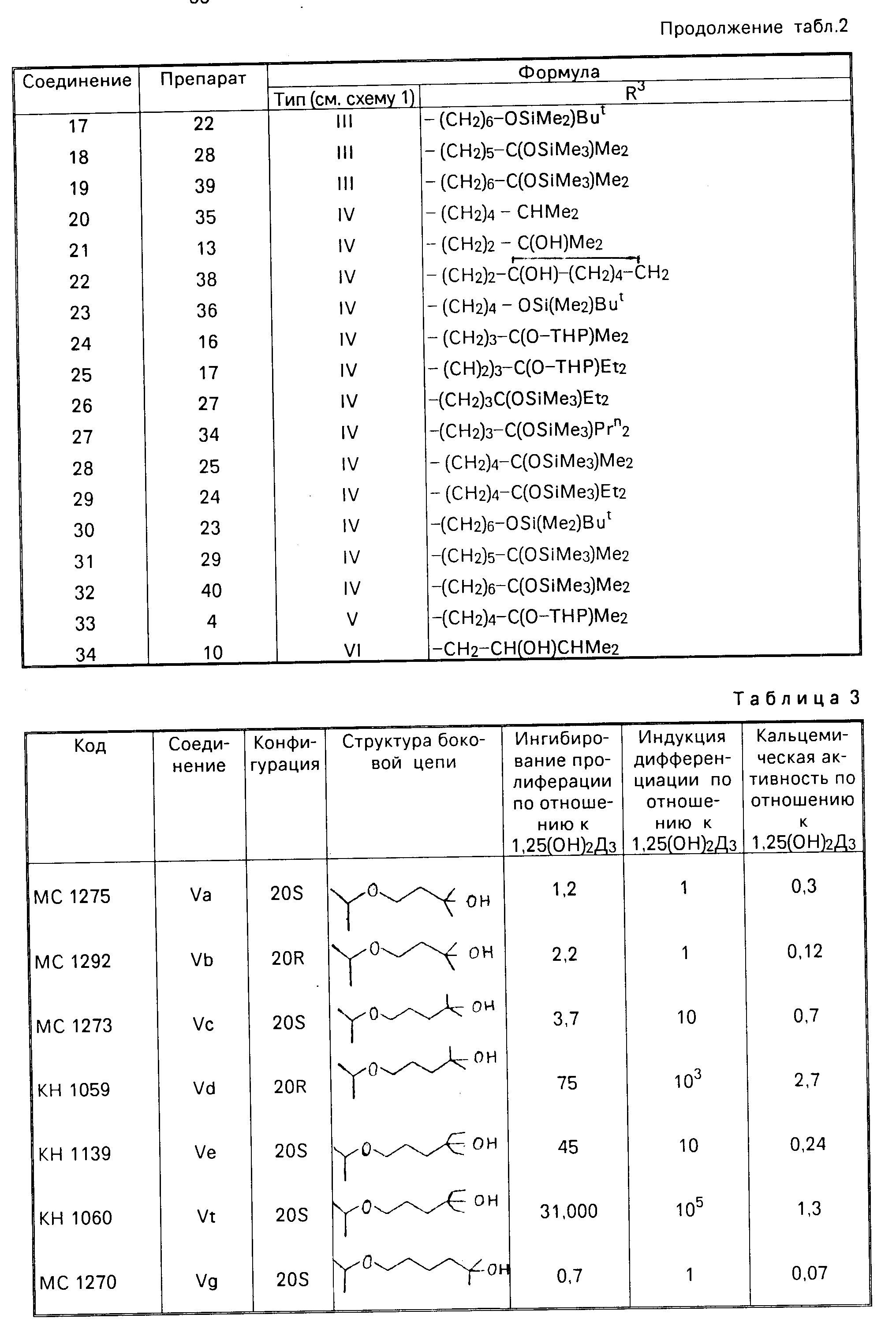
Промежуточные соединения в схеме 1, относящиеся к приготовлениям, должны быть идентифицированы перечисленными соответствующими формулами в табл.2.
Для спектра ядерного магнитного резонанса (300 МГц) значения химических сдвигов (δ) относятся к растворам дейтерированного хлороформа по отношению к внутримолекулярному тетраметилсилану ( δ= 0) или хлороформу ( δ= 7,25). Даны значения для мультиплета или для любого определенного (дублета (д), триплета (т), квартета (кв) или неопределенного (м) в приблизительно средней точке, если не сделана ссылка на интервал (с синглет, b уш). Константы спаривания (резонанса) (
Эфир представляет собой диэтиловый эфир, он был высушен над натрием. ТГФ был высушен над бензофеноном натрия. Петролейный эфир относится к пентановой фракции. Реакции проводились при комнатной температуре, если не отмечено особо. Разработанный метод включает разбавление специфичным растворителем (иначе говоря реакция в органическом растворителе), экстракцию водой и затем раствором хлористого кальция, высушивание над безводным MgSO4 и концентрирование в вакууме до образования осадка.
Приготовление I: Соединение 2.
К раствору 1(S), 3(R)-бис-(трет-бутилдиметилсилилокси)- 20(S)-формил-9,10-втор-прегна-5(Е), (7Е), 10(19)-триена (3,44 г, 6 ммоль) (I) в N, N-диметилформамиде (150 мл) добавлялись 1,4-диазабицикло [2,2,2] октан (600 мг, 5,3 ммоль), моногидрат ацетата меди (90 мг, 0,45 ммоль) и 2,2'-бипиридил (72 мг, 0,45 ммоль). В течение 6 дней через хорошо перемешиваемый раствор при 40оС осуществлялось пропускание (продувка) воздуха.
Реакционная смесь разбавлялась этилацетатом (500 мл), экстрагировалась водой (2х100 мл) и насыщалась водным раствором хлорида натрия (3х50 мл), затем высушивалась над MgSO4. Этилацетат удалялся выпариванием, а твердый остаток очищался хроматографически (силикагель, 10%-ный эфир в петролейном эфире как разбавитель) с образованием названного соединения.
ЯМР: δ= 0,037 (С, 3Н), 0,043 (с, 3Н), 0,056 (с, 6Н), 0,49 (С, 3Н), 0,84 (с, 9Н), 0,89 (С, 9Н), 1,5-2,30 (м,13Н), 2,13 (С, 3Н), 2,55 (дд, 1Н), 2,70 (т, 1Н), 2,89 (уш.д. 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,94 (м, 1Н), 4,98 (м, 1Н), 5,83 (д, 1Н), 6,43 (д, 1Н) частей/миллион.
Приготовление 2: Соединение 3 и его 20S-изомера
Соединение 2 (приг.1) (3,10 г, 5,5 ммоль) растворялось в тетрагидрофуране (140 мл) и добавлялся борогидрид
натрия (0,35 г, 3,3 ммоль). Затем
по каплям в течение 15 мин добавлялся метанол. Реакционная смесь перемешивалась в течение 20 мин, затем разбавлялась этилацетатом (560 мл). Раствор экстрагировался
водой (5х150 мл) и насыщенным
водным раствором хлорида натрия (150 мл), высушивался над MgSO4 и выпаривался, образуя бесцветное масло. Маслянистый остаток очищался хроматографически
(силикагель, 15%-ный этилацетат в
петролейном эфире в качестве разбавителя) и кристаллизовался из метанола с образованием 3.
ЯМР: δ= 0,05 (м, 12Н), 0,62 (с, 3Н), 0,86 (с, 9Н), 0, 89 (с, 9Н), 1,10-2,10 (м, 14Н), 1,15 (д, 3Н), 2,30 (уш, д. 1Н), 2,53 (дд, 1Н), 2,89 (м, 1Н), 2,89 (м, 1Н), 3,71 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,81 (д, 1Н), 6,45 (д, 1Н) частей/миллион.
Остающиеся фракции более полярного 20S-изомера выпаривались с образованием бесцветного остатка, который кристаллизовался из метанола:
ЯМР, δ= 0,052
(уш. д, 12Н), 0,54 (с,
3Н), 0,85 (с, 9Н), 0,89 (с, 9Н), 1,22 (д, 3Н), 1,20-2,10 (м, 14Н), 2,30 (уш. д, 1Н), 2,55 (дд, 1Н), 2,87 (м, 1Н), 3,72 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,94 (бс, 1Н), 4,98
(м, 1Н), 5,82 (д, 1Н),
6,44 (д, 1Н) частей/миллион.
Приготовление 3: соединение 14 (R3-5-(тетрагидро-4-Н-пиран-2-илокси)-5-метил-1- гексил).
К раствору соединения 3 (5,61 мг, 1 ммоль) в сухом тетрагидрофуране (10 мл) добавлялись гидроокись калия (0,70 г, 10 ммоль), 18-Crown-6 (40 мг) и 2-(6-бром-2-метил-2-гексилокси)тетрагидро-4Н-пиран (приготовление 5а) (2,7 г, 10 ммоль). Смесь интенсивно перемешивалась в течение вик-энд (от субботы до понедельника), реакционная смесь фильтровалась и фильтрат выпаривался в вакууме.
Остаток очищался хроматографически (силикагель, 10%-ный эфир в петролейном эфире в качестве разбавителя) с образованием 14 в виде бесцветного масла.
ЯМР, δ= 0,054 (м, 12Н), 0,54 (с, 3Н), 0,86 (с, 9Н), 0,88 (с, 9Н), 1,07 (д, I 6, 3Н), 1,17 (с, 3Н), 1,19 (с, 3Н), 1,15-1,95 (м, 23Н), 2,02 (т, 1Н), 2,20 (ушд, 1Н), 2,30 (ушд. 1Н), 2,53 (дд, 1Н), 2,85 (м, 1Н), 3,10-3,30 (м, 2Н), 3,40 (м, 1Н), 3,55 (м, 1Н), 3,93 (м, 1Е), 4,20 (м, 1Н), 4,51 (м, 1Н), 4,69 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,79 (д, I 11, 1Н), 6,45 (д, I Н, 1Н) частей/миллион.
Приготовление 4: 1(S), 3(R)-Дигидрокси-20(R)-[5'- (тетрагидро-4Н-пиран-2"-илокси)-5'-метил-1'-гексилокси] -9,10-втор- прегна- -5(Z), 7(Е), 10(19)-триен (соединение 33).
Раствор соединения 14 (400 мг, 0,5 ммоль), антрацен (200 мг, 1,1 ммоль) и триэтиламин (1 капля) в дихлорметане (15 мл) в атмосфере азота в пайрексовой колбе подвергались световому облучению с использованием ультрафиолетовой лампы высокого давления, тип ТQ 150Z2 (Hanau) при комнатной температуре в течение 30 мин. Раствор фильтровался и концентрировался в вакууме с образованием неочищенного промежуточного продукта (Соединение IV), Схема 1, R3=-5-(тетрагидро-4-Н-пиран-2-илокси)-2- метил-1-гексил. Это соединение растворялось в тетрагидрофуране (ТНF) (15 мл) и добавлялся тетра-n-тригидрат бутиламмония (1,05 г, 3,7 ммоль). Раствор нагревался в течение часа при 60oC в атмосфере азота. После охлаждения реакционная смесь разделялась между этилацетатом (50 мл) и насыщенным водным раствором гидрокарбоната (10 мл). Органический слой промывался водой (10 мл), высушивался и концентрировался. Остаток очищался хроматографически (100 г, силикагель, 50%-ный этилацетат в петролейном эфире в качестве разбавителя). С образованием нужного соединения.
ЯМР, δ 0,56 (с, 3Н), 1,07 (д, 3Н), 1,18 (с, 3Н), 1,20 (с, 3Н), 1,1-2,05 (м, 24Н), 2,17 (ушд, 1Н), 2,30 (дд, 1Н), 2,57 (дд, 1Н), 2,81 (м, 1Н), 3,10-3,30 (м, 2Н), 3,42 (м, 1Н), 3,56 (м, 1Н), 3,93 (м, 1Н), 4,22 (м, 3Н), 4,41 (м, 1Н), 4,70 (м, 1Н), 5,00 (ушс, 1Н), 5,33 (ушс, 1Н), 5,99 (д, 1Н), 6,39 (д, 1Н) частей/миллион.
Приготовление 5а: 2-(6-Бром-2-метил-2-гексилокси)тетрагидро-4-Н- пиран.
К перемешиваемому, охлаждаемому льдом раствору этил-5-бромпентаноат (18,7 мл) в высушенном эфире (100 мл) по каплям в течение 1 ч добавлялся профильтрованный раствор реактива Гриньяра, приготовленный из магния (10 г) и йодистого метила (25 мл) в высушенном эфире (200 мл). После пребывания в течение 30 мин на ледяной бане реакционной смеси дали нагреться в течение 30 мин до комнатной температуры, после чего приливали ее к охлаждаемому льдом раствору хлорида аммония (30 г) в воде (200 мл). После прекращения энергичной реакции эфирный слой отделяли, а водный слой дополнительно экстрагировали эфиром. Соединенные эфирные слои промывались последовательно водой и насыщенным водным раствором хлорида натрия, высушивались и концентрировались в вакууме с образованием неочищенного промежуточного продукта (6-бром-2-метил-2-гексанол) в виде палевого желтого масла. Этот продукт растворяли в дихлорметане (100 мл), затем при комнатной температуре добавлялись 3,4-дигидро-2Н-пиран (8,9 мл) и п-толуолсульфонат пиридиния (0,8 г). Через 1 ч реакционный раствор разбавляли эфиром (250 мл) и экстрагировали последовательно насыщенным водным раствором гидрокарбоната натрия (150 мл), водой (100 мл) и насыщенным водным раствором хлорида натрия (100 мл). После высушивания и удаления растворителя в вакууме продукт очищали хроматографически методом (150 г силикагеля, 10%-ный эфир в петролейном эфире в качестве разбавителя) с образованием необходимого соединения в виде бесцветного масла.
ЯМP: δ 1,20 (с, 3Н), 1,22 (с, 3Н), 1,40-1,95 (м, 12Н), 3,42 (т, 2Н), 3,94 (м, 1Н), 3,45 (м, 1Н), 4,72 (м, 1Н) частей/миллион.
Приготовление 5в: 2-(6-бром-3-этил-2-гексилокси) тетрагидро-4-Н-пиран.
При использовании метода, аналогичного описанному для приготовления 5а, названное соединение было получено из 4-бромбутаноата и реактива Гриньяра, образованного из этилйодида. ЯМР согласуется со структурой.
Приготовление 6: 5-Гидрокси-2,2,6-триметил-3(Е)-гептен.
К раствору диэтилизобутирилметилфосфоната (22 г), тетрабутиламмоний бромиду (4 г) и пивальдегиду (13 мл) в дихлорметане (340 мл) добавлялся 4 н. водный раствор гидроокиси натрия (140 мл). Смесь перемешивалась в течение ночи и после разбавления водой органическая часть отделялась. Промежуточный 5-оксо-2,2,6-триметил-3(Е)-гептан затем выделялся дистилляцией (т.кип. 45-48оС/0,1 миллибара). На перемешиваемый, охлаждаемый льдом раствор этого соединения (5 г) в 0,4 М растворе хлорида церия III в метаноле (90 мл) действовали порционно борогидридом натрия (1,4 г). Спустя 10 мин смесь обрабатывали (этилацетат) с образованием названного соединения в виде масла.
ЯМР δ 0,87 (д, 3Н, J 6,8), 1,02 (с, 9Н), 1,50 (шир. с, 1Н), 1,70 (м, 1Н), 3,77 (шир. т. 1Н), 5,36 (дд, 1Н, I 7,4 и 15,7), 5,65 (дд, 1Н, J 15,7 и 0,8).
П р и м е ч а н и е: Это рацемическое соединение разрешалось с использованием известного разрешающего кинетического метода, давая либо S-форму (используя (-) диизопропилтартрат) или R-форму (используя (+) диизопропилтартрат). Эти разрешающие формы могли быть использованы в качестве исходного материала для дальнейших стадий в описанной здесь последовательности для перехода рацемата в строящийся блок боковой цепи и затем для получения целевого соединения примера 2.
Приготовление 7: 3-Метил-2-(трет-бутилдиметилсилилокси) бутанал.
Раствор 5-гидрокси-2,2,6-триметил-3(Е)-гептен (приготовление 6) (4,5 г), имидазол (5 г) и трет-бутилдиметилсилил хлорид (5 г) в диметилформамиде (50 мл) перемешивался в течение 1 ч. Выделение (эфир) и перегонка дали промежуточный 5-(трет-бутилдиметилсилилокси)-2,2,6-триметил-3(Е)-гептен в виде масла (т. кип. 65-69о С/0,03 миллибара). Раствор этого соединения (7 г) в метаноле (100 мл) и дихлорметане (320 мл) при 70оС обрабатывали озонированным кислородом до установления окончания реакции (тсх-анализ) (40 мин), после чего добавлялся трифенилфосфин (9 г) и реакционной смеси давали нагреться до комнатной температуры. Обработки (дихлорметан) и перегонка дали названное в заглавии соединение в виде масла, т.кип. 45-48оС/1 миллибар.
ЯМР δ 0,04 (с, 6Н), 0,90 (д, 3Н), 0,92 (с, 9Н), 0,95 (д, 3Н), 2,01 (м, 1Н), 3,70 (дд, 1Н, J 4,8 и 2,1), 9,58 (д, 1Н, J 2,1).
Приготовление 8: 3-Метил-2-(трет-бутилдиметилсилилокси)-1- (трифторметансульфонилокси)бутан.
На перемешиваемый, охлаждаемый льдом раствор 3-метил-2-(трет-бутилдиметилсилилокси)бутанал (приготовление 7) (0,5 г) в ТГФ (4 мл) и этаноле (8 мл) действовали борогидридом натрия (0,1 г). Спустя 20 мин реакционную смесь обрабатывали (этилацетат) с образованием промежуточного 3-метил-2-(трет-бутилдиметилсилилокси)-1-бу- танола в виде масла. Его растворяли в дихлорметане (5 мл), охлаждали до 0оС и обрабатывали пиридином (0,5 мл) и трифторметансульфоновым ангидридом (0,5 мл). После перемешивания в течение часа реакционная смесь обрабатывалась (эфир) с образованием названного в заголовке соединения в виде масла.
ЯМР: δ= 0,07 (с, 3Н), 0,08 (с, 3Н), 0,90 (с, 9Н), 0,90 (д, 3Н), 0,93 (д, 3Н), 1,84 (м, 1Н), 3,74 (м, 1Н), 4,34 (дд, 1Н, J 9,9 и 6,8), 4,43 (д, 1Н, J 9,9 и 3,8).
Приготовление 9: 1(S), 3(R)-Бис-(трет-бутилдиметилсилилокси) -20(R)-(3'-метил-2'-трет-бутилдиметилсилилокси-1'-бутокси)-9; 10-втор-прегна -5(Е), 7(Е),10(19)-триен (соединение 6).
Перемешиваемый раствор соединения 3 (0,24 г), 18-Сrown-6 (40 мг) и трет-бутоксид калия (0,15 г) в высушенном ТГФ (4 мл) обрабатывался 3-метил-2-трет-бутилдиметилсилилокси)-1- трифторметансульфонилокси)бутаном (приготовление 9) (0,3 г). Через 15 мин реакционная смесь обрабатывалась (эфир) и остаток очищался хроматографическим методом (силикагель, 2%-ный эфир в петролейном эфире в качестве разбавителя) с образованием озаглавленного соединения в виде приблизительно равной смеси диастереоизомеров (эпимеры в положении 2').
ЯМР δ 0,0-0,12 (м, 18Н), 0,53 и 0,54 (2с, 3Н), 0,60-2,65 (м, 52Н), 2,87 (м, 1Н), 3,17 (м, 1Н), 3,23 (м, 1Н), 3,44 (м, 1Н), 3,55 (м, 1Н), 4,21 (м, 1Н), 4,53 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,80 (д, 1Н, J 11,4), 6,46 (д, 1Н, J 11,4).
Приготовление 10: 1(S), 3(R)-дигидрокси-20(R)-(2'-гидрокси-3' -метил-1'-бутокси)-9,10-вторпрегна-5(Е), 7(Е), 10(19)-триен (соединение 34).
Перемешиваемый раствор 1(S), 3(R)-бис(трет-бутилдиметилсилилокси)- 20(R), (3'-метил-2'-трет- бутилдиметилсилилокси-1'-бутокси)-9,10- вторпрегна-5(Е), 7(Е), 10(19)-триена (соединение 6) (0,2 г) и тетрабутиламмоний (фторид (0,7 г) в ТГФ (5 мл) нагревался при 60oC в течение часа в атмосфере азота. После охлаждения реакционная смесь обрабатывалась (экстрагировалась) (этилацетат). Очистка хроматографическим методом (силикагель, этилацетат в качестве разбавителя) дала названное в заглавии соединение.
ЯМР: δ 0,58 и 0,60 (2с, 3Н), 0,92 (д, 3Н, J 6,9), 0,98 (д, 3Н, J 6,9), 1,05-2,70 (м, 20Н), 2,86 (м, 2Н), 3,13-3,63 (м, 5Н), 4,22 (м, 1Н), 4,48 (м, 1Н), 4,97 (м, 1Н), 5,12 (м, 1Н), 5,87 (д, 1Н, J 11,4), 6,57 (д, 1Н, J 11,4).
Приготовление II: 1(S), 3(R)-Бис-[трет-бутил(диметилсилил)окси] 20(R)-(3-метилбут-2-ен-1-илокси)-9,10-втор-прегна-5(Е), 7(Е), 10(19)-триен (соединение N 4).
К раствору соединения 3 (0,61 г) в высушенном ТГФ (10 мл) добавлялись порошкообразная гидроокись калия (1,2 г (18-Сrown-6-(80 мг) и 3,3-диметилаллилбромид (2,2 г). После перемешивания в течение 24 ч при комнатной температуре смесь разделялась между эфиром и водой. Эфирный слой промывался хлористым кальцием, высушивался и концентрировался в вакууме с образованием масла. Хроматографическая очистка (силикагель; 2-5%-ный эфир в петролейном эфире в качестве разбавителя) с последующей кристаллизацией из метанола дали соединение 4 в виде игольчатых кристаллов.
ЯМР: δ 0,05 (уш, с, 12Н), 0,55 (с, 3Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,10 (д, 3Н), 1,65 (м, 3Н), 1,72 (м, 3Н), 1,05-1,82 (м, 10Н), 1,90 (м, 1Н), 2,03 (уш. т, 1Н), 2,14 (м, 1Н), 2,30 (м, 1Н), 2,54 (дд, 1Н), 2,87 (м, 1Н), 3,30 (м, 1Н), 3,78 (м, 1Н), 4,06 (м, 1Н), 4, 21 (м, 1Н), 4,52 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,33 (м, 1Н), 5,80 (д, 1Н, J 11,5), 6,46 (д, 1Н, J 11,5).
Приготовление 12: 1(S), 3(R)-Бис-[трет-бутил(диметилсили) окси]-20(R)-(3-гидрокси-3-метил-1-бутокси-9,10-втор-прегна- 5(Е), 7(Е), 10(19)-триен (соединение 7).
NB: Это приготовление иллюстрирует защиту триеновой системы III в виде SO2-аддукта для проведения эффективной модификации функциональной группы в боковой цепи.
На раствор соединения 4 (100 мг) в нескольких каплях эфира действовали при -10оС жидкой двуокисью серы (3 мл). Перемешиваемой смеси давали произвольно нагреться в медленном потоке азота, и спустя 30 мин остаток летучего вещества удаляли на ротационном выпаривателе. Остаток растворяли в ТГФ (2 мл) и воздействовали на него смесью, приготовленной добавленем ТГФ (1 мл) к раствору ацетата ртути II (100 мг) в воде (1 мл). Реакционную смесь перемешивали при 5oC в течение 18 ч и затем действовали на нее 3 н. NaOH (3 мл), затем раствором NaBH4(0,05 г) в 3 н. NaOH (2 мл). Затем добавлялся этилацетат и смесь фильтровалась через целит. Органический слой промывался хлористым кальцием, сушился и концентрировался в вакууме, образуя смолу. Смола растворялась (диспергировалась в 96% -ном этаноле (4 мл) вместе с бикарбонатом натрия (0,2 г), и перемешиваемая смесь нагревалась с обратным холодильником в атмосфере азота в течение 80 мин. После охлаждения добавлялся этилацетат и смесь экстрагировалась водой. Органический слой промывался водой. Органический слой промывался хлористым кальцием, высушивался и концентрировался в вакууме с образованием осадка.
Хроматографическая очистка (силикагель, 5-30%-ный раствор эфира в петролейном эфире в качестве разбавителя) дали 7.
ЯМР: δ= 0,05 (м, 12Н), 0,54 (с, 3Н), 0,85 (с, 9Н), 0,89 (с, 9Н), 1,13 (д, 3Н), 1,22 (с, 3Н), 1,23 (с, 3Н), 1,00-2,20 (м, 15Н), 2,30 (уш, 1Н), 2,53 (дд, 1Н), 2,86 (м, 1Н), 3,27 (м, 1Н), 3,45 (м, 1Н), 3,55 (с, 1Н), 3,83 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,79 (д, 1Н, J 11,4), 6,45 (д, 1Н, J 11, 4).
Приготовление 13: 1(S), 3(R)-Бис-[трет-бутил(диметилсилил) окси] -20(R)-(3-гидрокси-3-метил-1-бутокси-9', 10-втор-прегна-5(Z), 7,(Е), 10(19)-триен (соединение 21).
Раствор соединения 7 (40 мг) в дихлорметане (4 мл), содержащий антрацен (20 мг) и триэтиламин (50 мкл) в пайрексовой колбе в атмосфере азота подвергался световому облучению с использованием ультрафиолетовой лампы высокого давления (тип TQ150Z2 (Hanau) в течение 30 мин при 15oC. Реакционная смесь фильтровалась и концентрировалась в вакууме с образованием остатка.
Очистка хроматографическим методом (силикагель, 30% эфира в петролейном эфире в качестве разбавителя) дали 21.
ЯМР находится в согласовании со структурой.
Приготовление 14: соединение 11 (R3 =4-(тетрагидро-4Н-пиран-2- илокси)-4-этил-1-гексил).
К раствору соединения 3 (561 мг, 1,0 ммоль) в сухом тетрагидрофуране (10 мл) добавлялись трет-бутоксид калия (0,4 г, 3,6 ммоль), 18-Сrown-6 (80 мг) и 2-(6-бром-3-этил-3-гексилокси) тетрагидро-4Н-пиран (приготовление 5в) (1,08 г, 3,68 ммоль). Смесь перемешивалась в течение ночи и разбавлялась этилацетатом (60 мл), затем промывалась водой (3х10 мл) и насыщалась водным раствором хлорида натрия (10 мл), сушилась над MgSO4 и концентрировалась в вакууме. Затем соединение очищалось хроматографическим методом (150 г силикагеля, 10% эфира в петролейном эфире в качестве разбавителя) с образованием нужного соединения в виде бесцветного масла.
ЯМР: δ 0,05 (м, 12Н), 0,55 (с, 3Н), 0,82 (м, 6Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н, J 6), 1,0-2,1 (м. 25Н), 2,03 (уш. т. 1Н), 2,18 (уш, д. 1Н), 2,30 (ушд. 1Н), 2,54 (дд, 1Н), 2,87 (уш.д. 1Н), 3,12 (м, 1Н), 3,25 (м, 1Н), 3,42 (м, 1Н), 3,55 (м, 1Н), 3,95 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,68 (м, 1Н), 4,92 (уш.с, 1Н), 4,98 (уш.с, 1Н), 5,79 (д, 1Н, J 11), 6,46 (д, 1Н, J 11) частей/миллион.
Приготовление 15: соединение 10 (R3=4-(тетрагидро-4Н-пиран-2-илокси)-4-этил-1-пентил).
Последовательными процедурами приготовления 14 и заменой соединения 2-(6-бром-3-этил-3-гексилокси)тетрагидро-4Н-пирана соединением 2-(5-бром-2-метил-2-пентилокси)тетрагидро-4Н-пиран было получено необходимое соединение в виде бесцветного масла.
ЯМР: δ 0,05 (м, 12Н), 0,55 (с, 3Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н, J 6), 1,19 (с, 3Н), 1,20 (с, 3Н), 0,9-2,0 (м, 21Н), 2,03 (м, 1Н), 2,16 (уш. д. 1Н), 2,30 (уш.д.1Н), 2, 55 (дд, 1Н), 2,87 (уш.д,1Н), 3,15 (м, 1Н), 3,25 (м, 1Н), 3,43 (м, 1Н), 3,55 (м, 1Н), 3,93 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,71 (м,1Н), 4,93 (уш.с, 1Н), 4,98 (уш.с.1Н), 5,80 (д, 1Н, J 11), 6,46 (д, 1Н, J 11) частей/миллион.
Приготовление 16: соединение 24 (R3=4-(тетрагидро-4Н-пиран -2-илокси)-
4-метил-1-пентил).
Раствор соединения 10, синтезированный по приготовлению 15 (200 мг, 0,27 ммоль), антрацен (200 мг, 1,1 ммоль) и триэтиламин (1 капля) в дихлорметане (15 мл) в атмосфере азота в пайрексовой колбе облучался (светом) с использованием ультрафиолетовой лампы высокого давления, типа ТQ150Z2 (Hanau) в течение 30 мин при температуре около 10oC. Реакционная смесь отфильтровывалась, концентрировалась в вакууме и очищалась хроматографически (30 г силикагеля, 50% эфира в петролейном эфире в качестве разбавителя), давая нужное соединение в виде бесцветного масла.
ЯМР: δ= 0,05 (м, 12Н), 0, 53 (с, 3Н), 0,87 (м, 18Н), 1,06 (д, 3Н, J 6), 1,18 (с, 3Н), 1,20 (с, 3Н), 1,0-1,9 (м, 12Н), 1,98 (уш.т. 1Н), 2,16 (м, 2Н), 2,43 (дд, 1Н), 2,82 (уш.д. 1Н), 3,18 (м, 1Н), 3,24 (м, 1Н), 3,43 (м, 1Н), 3, 53 (м, 1Н), 3,93 (м,1Н), 4,18 (м, 1Н), 4,36 (м, 1Н), 4,70 (м, 1Н), 4,85 (уш. с, 1Н), 5,16 (уш. с, 1Н), 5,99 (д,1Н, J 11), 6,24 (д, 1Н, J 11) частей/миллион.
Приготовление 17: соединение 25 (R3=4-(тетрагидро-4Н-пиран -2-илокси)-4-этил-1-гексил).
Последовательными процедурами приготовления 16 и заменой соединением 11, приготовленным в соответствии с приготовлением 14, соединения 10, приготовленного в соответствии с приготовлением 15, было получено необходимое соединение в виде бесцветного масла.
ЯМР: δ= 0,05 (м, 12Н), 0, 53 (с, 3Н), 0,82 (м, 6Н), 0,87 (с, 18Н), 1,06 (д, 3Н, J 6), 1,0-1,9 (м, 25Н), 1,98 (уш.т.1Н), 2,19 (м, 2Н), 2,44 (дд, 1Н), 2,82 (ущ. д, 1Н), 3,12 (м, 1Н), 3,25 (м, 1Н), 3,43 (м, 1Н), 3,55 (м, 1Н), 3, 93 (м, 1Н), 4,18, (м, 1Н), 4,36 (м, 1Н), 4,69 (м, 1Н), 4,85 (уш.с, 1Н), 5,16 (уш.с, 1Н), 5,99 (д, 1Н, J 11), 6,24 (д, 1Н, J 11) частей/миллион.
Приготовление 18: Cоединение 15 (R3=5-триметилсилилокси-5- метил-1-гексил).
К раствору 3 (561 мг, 1,0 ммоль) в сухом тетрагидрофуране (10 мл) добавлялись трет-бутоксид калия (0,65 г, 5,8 ммоль), 18-Сrown-6 (120 мг) и 6-бром-2-метил-2-триметилсилилоксигексан (1,4 мл, 5,0 ммоль). Реакционная смесь перемешивалась в течение 2 ч и обрабатывалась (экстрагировалась) (эфир). Неочищенный продукт подвергался хроматографической очистке (40 г силикагеля, 2% эфира в петролейном эфире в качестве разбавителя) с образованием бесцветного масла, которое кристаллизовалось из метанола.
Т.пл. 75, 5-77,5оС.
ЯМР: δ 0,05-0,09 (м, 21Н), 0,55 (с, 3Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,13 (с, 6Н), 1,15-2,0 (м, 17Н), 2,02 (т, 1Н), 2,17 (д, 1Н), 2,31 (д, 1Н), 2,55 (д.д,1Н), 2,85 (уш.д, 1Н), 3,15 (м, 1Н), 3,26 (м, 1Н), 3,56 (м, 1Н), 4,21 (м, 1Н), 4,53 (м, 1Н), 4,93 (уш.с,1Н), 4,99 (уш.с.1Н), 5,79 (д,1Н), 6,46 (д,1Н), частей/миллион.
Приготовление 19: Соединение 16 (R3=5-триметилсилил- окси-5-этил-1-гептил).
К раствору соединения 3 (561 мг, 1,0 ммоль) в сухом тетрагидрофуране (10 мл) добавлялись трет-бутоксид калия (0,45 г, 4,0 ммоль) 18-Сrown-6 (80 мг) и 7-бром-3-этил-3- триметилсилилоксигептан (0,44 мл, 1,5 моль). Реакционная смесь перемешивалась в течение 4 ч и экстрагировалась (этилацетат). Сырой (неочищенный) продукт очищался хроматографически (100 г силикагеля, 5% эфира в петролейном эфире в качестве разбавителя) с образованием бесцветного масла, которое кристаллизовалось из метанола.
Т.пл. 70,5-72,5оС.
ЯМР: δ 0,04-0,1 (м, 21Н), 0,55 (с, 3Н), 0,80 (дт, 6Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,43 (д.кв,4Н), 1, 0-1,96 (м, 17Н), 2,04 (ушт, 1Н), 2,17 (уш. д,1Н), 2,30 (уш.д, 1Н), 2,55 (дд, 1Н), 2,86 (ушд, 1Н), 3,15 (м, 1Н), 3,26 (м, 1Н), 3,58 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,80 (д, 1Н, J 11,3), 6,46 (д, 1Н, J 11,3) частей/миллион.
Приготовление 20: 1-(трет-бутилдиметилсилилокси)-6-хлоргексан.
К раствору 6-хлоргексан-1-ола (6,8 мл, 75,4 ммоль) в сухом дихлорметане (100 мл) добавлялись трет-бутилдиметилсилилхлорид (12,5 г, 83 ммоль) и имидазол (10,21 г, 150 ммоль), и реакционная смесь перемешивалась в течение ночи при комнатной температуре. Экстракция (дихлорметан) и дистилляция дали озаглавленное соединение в виде масла.
Т.кип. 130-134оС/12 миллибар.
ЯМР: δ 0-03 (c, 6Н), 0, 88 (с, 9Н), 1,27-1,60 (м, 6Н), 1,77 (м, 2Н), 3,52 (т, 2Н), 3,59 (т, 2Н) частей/миллион.
Приготовление 21: 1-(трет-бутилдиметилсилилокси)-6-йодгексан.
Раствор йодида натрия (13,5 г, 90 ммоль) и 1-(трет-бутилдиметилсилилокси)-6-хлоргексан (приготовление 20) (8,35 г, 22 ммоль) в ацетоне (70 мл) перемешивались в течение ночи с обратным холодильником. Реакционная смесь охлаждалась до комнатной температуры и фильтровалась. Фильтрат экстрагировался (гексан) с образованием нужного соединения в виде желтого масла.
ЯМР: δ 0-03 (c, 6Н), 0,88 (с, 9Н), 1,22-1,60 (м, 6Н), 1,82 (м, 2Н), 3,18 (т, 2Н), 3,59 (т, 2Н) частей/миллион.
Приготовление 22: Соединение 17 (R3 =6-(трет-бутилдиметилсилилокси)-1- гексил.
К раствору соединения 3 (516 мг, 0,9 ммоль) в сухом тетрагидрофуране (8 мл) добавлялись трет-бутоксид калия (0,65 г, 5,8 ммоль), 18-Сrown-6 (100 мг) и 1-трет-бутилдиметилсилилокси)-6-йодгексан (приготовление 21) (1,70 мл, 5 ммоль). Смесь перемешивалась в течение ночи и экстрагировалась (эфир). Сырой продукт очищался хроматографически (100 г силикагеля, 30% толуола в петролейном эфире в качестве разбавителя) с образованием бесцветного масла, которое кристаллизовалось из метанола.
Т.пл. 84-87оС.
ЯМР: δ= 0,03 (с, 6Н), 0,06 (м, 12Н), 0,54 (с, 3Н), 0,86 (с, 9Н), 0,87 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,10-1,82 (м, 18Н), 1,92 (м, 1Н), 2,03 (уш. т, 1Н), 2,14 (уш.д,1Н), 2,30 (уш.д, 1Н), 2, 52 (дд, 1Н), 2,87 (м, 1Н), 3,22 (м, 2Н), 3,55 (м, 1Н), 3,58 (т, 2Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,80 (д, 1Н, J 11,4), 6,46 (д, 1Н, J 11,4) частей/миллион.
Приготовление 23: Cоединение 30 (R3=6-(трет-бутилдиметилсилилокси)-1- гексил).
Раствор соединения 17, приготовленный в соответствии с приготовлением 22 (238 мг, 0,3 ммоль), антрацен (150 мг, 0,8 ммоль) и триэтиламин (2 капли) в дихлорметане (12 мл) облучали в атмосфере азота в пайрексовой колбе светом с использованием ультрафиолетовой лампы высокого давления, тип ТQ150Z2 (Hanau) в течение 30 мин при 15оС. Реакционную смесь фильтровали, концентрировали в вакууме и очищали хроматографически (40 г силикагеля, 10% эфира в петролейном эфире в качестве разбавителя) с образованием нужного соединения в виде бесцветного масла.
ЯМР: δ 0,03 (с, 6Н), 0,04 (м, 6Н), 0,05 (с, 6Н), 0,53 (с, 3Н), 0,86 (с, 9Н), 0,87 (с, 9Н), 0,88 (с, 9Н), 1,06 (д, 3Н), 1,00-2,30 (м, 22Н), 2,44 (дд, 1Н), 2,82 (уш. д, 1Н), 3,20 (м, 2Н), 3,55 (м, 1Н), 3,58 (т, 2Н), 4,18 (м, 1Н), 4,36 (м, 1Н), 4,86 (м, 1Н), 5,16 (м, 1Н), 5,99 (д, 1Н, J 11,3), 6,24 (д, 1Н, J 11,3) частей/миллион.
Приготовление 24: Соединение 29 (R3=5-триметилсилилокси)-5-этил-1-гептил).
Раствор соединения 16, приготовленного в соответствии с приготовлением 19 (300 мг, 0,4 ммоль), антрацен (300 мг, 1,7 ммоль) и триэтиламин (1 капля) в дихлорметане (15 мл) подвергались световому облучению в атмосфере азота в пайрексовой колбе, получаемому с помощью ультрафиолетовой лампы высокого давления, тип ТQ150Z2 (Hanau) в течение 45 мин при 15oC. Реакционная смесь фильтровалась, концентрировалась в вакууме, очищалась хроматографическим методом (15 г силикагеля, 30% толуола в петролейном эфире в качестве разбавителя), давая необходимое соединение в виде бесцветного масла.
ЯМР: δ 0,05 (с, 6Н), 0,06 (с, 6Н), 0, 08 (с, 9Н), 0,54 (с, 3Н), 0,80 (дс, 3Н), 0,87 (с, 18Н), 1,07 (д, 3Н), 1,43 (уш.кв, 4Н), 1,00-2,25 (м, 20Н), 2,45 (дд, 1Н), 2,82 (уш.д,1Н), 3,15 (м, 1Н), 3,24 (м, 1Н), 3,57 (м, 1Н), 4,18 (м, 1Н), 4,35 (м, 1Н), 4,86 (м, 1Н), 5,16 (м, 1Н), 5,99 (д, 1Н, J 11,3), 6,24 (д, 1Н, J 11,3) частей/миллион.
Приготовление 25: Cоединение 28 (R3=5-триметилсилилокси
-5-метил-1-гексил)
Раствор соединения 15, приготовленного в соответствии с приготовлением 18 (3,50 г, 4,7 ммоль), антрацен
(2,2 г, 12 ммоль) и триэтиламин (0,5 мл) в дихлорметане (175 мл)
подвергались в пайрексовой колбе в атмосфере азота действию светового излучения, получаемого при использовании ультрафио- летовой
лампы высокого давления, тип TQ150Z2 (Hanau) в течение 2 ч при 15оС. Реакционная смесь фильтровалась, концентрировалась в вакууме и очищалась хроматографически (75 г силикагеля, 5% эфира в
петролейном эфире в качестве разбавителя), давая нужное соединение в
виде бесцветного масла.
ЯМР: δ 0,05-0,10 (м, 21Н), 0,54 (с, 3Н), 0,87 (с 18Н), 1,06 (д, 3Н), 1,18 (с, 6Н), 1,15-1,90 (м, 17Н), 1,99 (т,1Н), 2,15 (м, 1Н), 2,17 (м, 1Н), 2,44 (дд, 1Н), 2,81 (м, 1Н), 3,20 (м, 2Н), 3,56 (м, 1Н), 4,18 (м, 1Н), 4,36 (м, 1Н), 4,86 (уш.д, 1Н), 5,16 (уш.с, 1Н), 5,98 (д, 1Н), 6,23 (д, 1Н), частей/миллион.
Приготовление 26: Cоединение 12 (R3=4-триметилсилилокси-4-этил-1-гексил).
Раствор трет-бутоксида калия (1,95 г, 17 ммоль) в сухом тетрагидрофуране (15 мл) добавлялся по каплям в течение 40 мин через шприц (капельницу) к раствору соединения 3 (1,68 г, 3 ммоль) 18-Сrown-6 (600 мг) и 6-бром-3-этил-3-триметилсилилоксигексану (2,53 мл, 9 ммоль) в сухом тетрагидрофуране (20 мл), перемешанному в атмосфере азота. Получающийся раствор перемешивался в течение 45 мин и экстрагировался (гексан). Неочищенный продукт очищался хроматографически (140 г силикагеля, 30% толуола в петролейном эфире в качестве разбавителя), давая бесцветное масло, которое кристаллизовалось из метанола.
Т.пл. 52-57o C.
ЯМР: δ 0,05-0,1 (м, 21Н), 0,55 (с, 3Н), 0,80 (дт, 6Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,10-2,05 (м, 20Н), 2,18 (д, 1Н), 2,30 (д, 1Н), 2,54 (дд, 1Н), 2,86 (уш.д, 1Н), 3,12 (м, 1Н), 32,5 (м, 1Н), 3,55 (м, 1Н), 4,21 (м, 1Н), 4,52 (м, 1Н), 4,93 (уш.с,12Н), 4,98 (уш.с,1Н), 5,79 (д,1Н), 6,46 (д, 1Н) частей/миллион.
Приготовление 27: Соединение 26 (R3-4-триметилсилилокси)-4- этил-1-гексил).
Раствор соединения 12, приготовленного в соответствии с приготовлением 26 (1,0 г, 1,3 ммоль), антрацен (1,0 г, 5,6 ммоль) и триэтиламин (3 капли) в дихлорметане (70 мл) в пайрексовой колбе в атмосфере азота подвергались действию светового облучения, получаемого с помощью ультрафиолетовой лампы высокого давления, тип TQ150Z2 (Hanau) в течение 55 мин при температуре 15оС. Реакционная смесь фильтровалась, концентрировалась в вакууме и очищалась хроматографически (35 г силикагеля, 2% эфира в петролейном эфире в качестве разбавителя), давая желаемое соединение в виде бесцветного масла.
ЯМР: δ 0,05-0,10 (м, 21Н), 0,54 (с, 3Н), 0,80 (дт, 6Н), 0,87 (с, 18Н), 1,06 (д, 3Н), 1,0-2,05 (м, 20Н), 2,16 (д, 1Н), 2,20 (м, 1Н), 2,43 (дд, 1Н), 2,81 (дд, 1Н), 3,12 (м, 1Н), 3, 24 (м, 1Н), 3,55 (м, 1Н), 4,18 (м, 1Н), 4,35 (м, 1Н), 4,85 (уш, д, 1Н), 5,16 (уш. с, 1Н), 5,98 (д, 1Н), 6,23 (д, 1Н) частей/миллион.
Приготовление 28: Соединение 18 (R3 -6-метил-6-триметилсилилокси- 1-гептил).
Раствор 18-Crown-6 (264 мг, 1 ммоль) в сухом тетрагидрофуране (4 мл) добавлялся по каплям с помощью шприца (капельницы) к смеси соединения 3 (561 мг, 1 ммоль), 7-бром-2-метил-2-триметилсилилоксигепта- на (1,5 мл, 4 ммоль) и гидрида калия (0,6 мл, 20% суспензия в масле) при перемешивании в атмосфере азота. Получающийся раствор перемешивался в течение 3 ч и экстрагировался (эфир). Сырой (неочищенный) продукт очищался хроматографически (75 г силикагеля, 5% эфира в петролейном эфире в качестве разбавителя),давая бесцветное масло, которое кристаллизовалось из метанола.
ЯМР: δ= 0,06 (м, 12Н), 0,08 (с, 9Н), 0,54 (с, 3Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,18 (с, 6Н), 1,00-1,83 (м, 18Н), 1,90 (м, 1Н), 2,03 (уш. т, 1Н), 2,15 (уш. д, 1Н), 2,31 (уш.д, 1Н), 2,55 (дд, 1Н), 2,87 (уш.д, 1Н), 3,20 (м, 1Н), 3,53 (м, 1Н), 4,21 (м, 1Н), 4,53 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,80 (д, 1Н, I 11,4), 4,46 (д, 1Н, I 11,4) части/миллион.
Приготовление 29: Соединение 31 (R3 -6-метил-6-триметилсилилокси- 1-гептил).
Раствор соединение 18, приготовленного в соответствии с приготовлением 28 (400 мг, 0,152 ммоль), антрацен (300 мг, 1,7 ммоля и триэтиламин (3 капли) в дихлорметане (20 мл) подвергались световому облучению в пайрексовой колбе в атмосфере азота с использованием ультрафиолетовой лампы высокого давления, тип TQ150Z2 (Hanau) в течение 30 мин при 15оС. Реакционная смесь фильтровалась, концентрировалась в вакууме, очищалась хроматографически (35 г силикагеля, 5% эфира в петролейном эфире в качестве разбавителя), давая нужное соединение в виде бесцветной смолы.
ЯМР: δ= 0,05 (м, 12Н), 0,08 (с, 9Н), 0,53 (с, 3Н), 0,87 (с, 18Н), 1,06 (д, 3Н), 1,18 (с, 6Н), 1,00-2,30 (м, 22Н), 2,44 (дд, 1Н), 2,81 (уш.д, 1Н), 3,21 (м, 2Н), 3,54 (м, 1Н), 4,18 (м, 1Н), 4,37 (м, 1Н), 4,86 (м, 1Н), 5,17 (м, 1Н), 5,99 (д, 1Н, I 11,3), 6,24 (д, 1Н, I 11,3) части/миллион.
Приготовление 30: 1-(трет-бутилдиметилсилилокси)-4-хлорбутан.
К раствору 4-хлорбутан-1-олу (10 мл, 100 ммоль) в сухом дихлорметане (100 мл) добавлялись трет-бутилдиметилсилилхлорид (20,8 г, 120 ммоль) и имидазол (13,61 г, 200 ммоль), и реакционная смесь перемешивалась при комнатной температуре в течение ночи. Обработка (этилацетат) дистилляция дала озаглавленное соединение в виде масла.
Т.кип. 89-92оС/12 миллибар.
ЯМР: δ= 0,04 (с, 6Н), 0,88 (с, 9Н), 1,65 (м, 2Н), 1,84 (м, 2Н), 3,56 (т, 2Н), 3,63 (т, 2Н) частей/миллион.
Приготовление 31. Соединение 9 (R3-4-(трет-бутилдиметилсилилокси) 1-бутил).
Раствор 18-Crown-6 (264 мг, 1 ммоль) в сухом тетрагидрофуране (4 мл) добавлялся по каплям с помощью шприца (капельницы) в течение 2 мин к смеси соединения 3 (561 мг, 1 ммоль) 4-хлор-1-трет-бутилдиметилсилилоксибутана (приготовление 30) (1,5 мл, 6 ммоль) и гидрида калия (0,6 мл, 20% суспензия в масле), перемешиваемых в атмосфере азота. Получающийся раствор перемешивался в течение 3 ч 30 мин и экстрагировался (эфир). Неочищенный продукт очищался хроматографически (75 г силикагеля, 5% эфира в петролейном эфире в качестве разбавителя), давая бесцветную смолу, которая кристаллизовалась из метанола.
Т.кип. 91-96оС.
ЯМР: δ= 0,03 (м, 6Н), 0,06 (с, 12Н), 0,54 (с, 3Н), 0,86 (с, 9Н), 0,88 (с, 9Н), 0,89 (с, 9Н), 1,06 (д, 3Н), 1,00-1,83 (м, 14Н), 1,92 (м, 1Н), 2,03 (уш. т, 1Н), 2,14 (уш.д, 1Н), 2,30 (уш.д, 1Н), 2,54 (дд, 1Н), 2,86 (уш.д, 1Н), 3,23 (м, 2Н), 3,57 (дд, 1Н), 3,61 (т, 2Н), 4,21 (м, 1Н), 4,53 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,79 (д, 1Н, I 11,4), 6,46 (д, 1Н), I 11,4) частей/миллион.
Приготовление 32: соединение 13 (R3-4-триметилсилилокси-4-(пропил)-1-гептил).
Соединение готовилось в соответствии с методом, описанным в приготовлении 31, за исключением того, что 4-хлор-1-трет- бутилдиметоксибутан был заменен 7-бром-4-(1-пропил)-4-триметилсилилокси гептаном.
ЯМР: δ= 0,05 (м, 12Н), 0,07 (с, 9Н), 0,55 (с, 3Н), 0,86 (с, 9Н), 0, 87 (м, 6Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,00-1,85 (м, 22Н), 1,91 (м, 1Н), 2,03 (уш. т, 1Н), 2,19 (уш.д, 1Н), 2,30 (уш.д, 1Н), 2,55 (дд, 1Н), 2,87 (уш.д, 1Н), 3,11 (м, 2Н), 3,25 (дд, 1Н), 3,55 (т, 1Н), 4,22 (м, 1Н), 4,53 (м, 1Н), 4,93 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1Н), 5,80 (д, 1Н, I 11,3), 6,46 (д, 1Н, I 11,3) частей/миллион.
Приготовление 33: Соединение 5 (R3 -5-метил-1-гексил).
Соединение готовилось в соответствии с методом, описанным в приготовлении 31, исключая то, что 4-хлор-1-трет-бутилдиметилсилилокси бутан был заменен на 1-бром-5-метилгексан.
Т.пл. 79,5-81оС.
ЯМР: δ 0,06 (м, 12Н), 0,55 (с, 3Н), 0,85 (с, 6Н), 0,86 (с, 9Н), 0,89 (с, 9Н), 1,07 (д, 3Н), 1,00-1,85 (м, 17Н), 1,91 (м, 1Н), 2,03 (уш.т, 1 Н), 2,16 (уш. д, 1Н), 2,31 (уш.д, 1Н), 2,55 (дд, 1Н), 2,87 (уш.д, 1Н), 3,16 (м, 2Н), 3,24 (м, 1Н), 3,55 (м, 1Н), 4,21 (м, 1Н), 4,53 (м, 1Н), 4,93 (м, 1Н), 4,98 (м, 1), 5,80 (д, 1Н, I 11,4), 6,46 (д, 1Н, I 11,4) частей/миллион.
Приготовление 34: Соединение 27 (R3-4-триметилсилилокси-4-(1- пропил)-1-гептил).
Соединение готовилось в соответствии с методом, описанным в приготовлении 23, за исключением того, что соединение 17, полученное в приготовлении 22, заменялось соединением 13, полученным в приготовлении 32.
ЯМР: δ 0,04 (м, 6Н), 0,05 (с, 6Н), 0,07 (с, 9Н), 0,54 (с, 3Н), 0,80-0,93 (м, 24Н), 1,06 (д, 3Н), 1,00-2,07 (м, 24Н), 2,19 (м, 2Н), 2,45 (дд, 1Н), 2,82 (уш. д, 1Н), 3,12 (м, 1Н), 3,24 (м, 2Н), 3,55 (м, 1Н), 4,18 (м, 1Н), 4,36 (м, 1Н), 4,86 (м, Н), 5,17 (м, 1Н), 5,99 (д, 1Н, I 11,2), 6,24 (д, 1Н, I 11,2) частей/миллион.
Приготовление 35: Соединение 20 (R3- 5-метил-1-гексил).
Соединение готовилось в соответствии с методом, описанным в приготовлении 23, за исключением того, что соединение 17, полученное в приготовлении 22, заменялось соединением 5, полученным в приготовлении 33.
ЯМР: δ 0,05 (с, 6Н), 0,06 (с, 6Н), 0,53 (с, 3Н), 0,85 (д, 6Н), 0,87 (с, 18Н), 1,06 (д, 3Н), 1,00-1,92 (м, 18Н), 1,98 (уш.т, 1Н), 2,18 (м, 2Н), 2,44 (дд, 1Н), 2,82 (уш.д, 1Н), 3,18 (м, 2Н), 3,55 (м, 1Н), 4,17 (м, 1Н), 4,36 (м, 1Н), 4,86 (м, 1Н), 5,16 (м, 1Н), 5,99 (д, 1Н, I 11,3), 6,24 (д, 1Н, I 11,3) частей/миллион.
Приготовление 36: Соединение 23 (R3-4-трет-бутилдиметилсилилокси- 1-бутил).
Соединение готовилось в соответствии с методом, описанным в приготовлении 23, за исключением того, что соединение 17, полученное в приготовлении 22, было заменено соединением 9, полученным в приготовлении 31.
ЯМР: δ 0,05 (м, 18Н), 0,53 (с, 3Н), 0,87 (м, 27Н), 1,06 (д, 3Н), 1,00-2,30 (м, 18Н), 2,44 (дд, 1Н), 2,82 (уш.д, 1Н), 3,22 (м, 2Н), 3,57 (м, 1Н), 3,61 (т, 1Н), 4,18 (м, 1Н), 4,36 (м, 1Н), 4,86 (м, 1Н), 5,17 (м, 1Н), 5,99 (д, 1Н, I 11,3), 6,24 (д, 1Н, I 11,3) части/миллион.
Приготовление 37: Соединение 8 (R3 -2-(1-гидроксициклогексил)этил).
При использовании метода приготовления 11, но заменяя 2-циклогексилиден-1-бромэтан (2,5 г) на 3,3-диметилаллилбромид, было получено промежуточное соединение III (R3=CH2-CH=
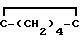
ЯМР в соответствии со структурой.
Приготовление 38: Соединение 22 (R3- 2-(1-гидроксициклогексил)этил).
Было получено соединение с использованием метода приготовления 13, но заменой соединения 8 соединением 7.
ЯМР согласуется со структурой.
Приготовление 39: Соединение 19 (R3-7-метил-7-триметилсилилокси- 1-октил).
Было получено соединение с использованием метода приготовления 28, но с заменой 7-бром-2-метил-2-триметилсилоксигептана 8-бром-2-метил-2-триметилсилоксиоктаном.
ЯМР согласуется со структурой.
8-Бром-2-метил-2-триметилсилилокси-октан, использованный в приготовлении, был получен аналогично известному способу для более низких гомологов.
Т.кип. 92-95о С/0,1 мм рт.ст.
ЯМР: δ 0,09 (с, 9Н), 1,18 (с, 6Н), 1,2-1,5 (м, 8Н), 1,85 (м, 2Н), 3,40 (х, т, 2Н).
Приготовление 40: Соединение 32 (R3 -7-метил-7-триметилсилилокси- 1-октил).
Соединение было получено с использованием метода приготовления 29, но заменой соединения 18 соединением 19.
ЯМР согласуется со структурой.
П р и м е р 1. 1(S), 3(R)-Дигидрокси-20(R)-(5'-гидрокси-5'- метил-1'-гексилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение 109).
Соединение 33, полученное в приготовлении 4, (60 мг, 0,11 ммоль) растворялось в этилацетате (0,5 мл) и добавлялся ацетонитрил (5 мл). Добавлялся 5% раствор фтористоводородной кислоты в ацетонитриле/вода 8:1 (0, 5 мл), и раствор перемешивался в атмосфере азота в течение 1 ч. Добавлялся этилацетат (50 мл) и смесь экстрагировалась насыщенный водным раствором гидрокарбоната натрия (10 мл) в воде (10 мл), высушивался и выпаривался в вакууме. Остаток очищался хроматографически (силикагель, этилацетат в качестве разбавителя), давая соединение 109.
ЯМР: δ= 0,56 (с, 3Н), 1,07 (д, 3Н), 1,20 (с, 6Н), 1,10-2,05 (м, 24Н), 2,15 (уш. д, 1Н), 2,30 (дд, 1Н), 2,60 (дд, 1Н), 2,72 (м, 1Н), 3,20 (м, 2Н), 3,57 (м, 1Н), 4,21 (м, 1Н), 4,42 (м, 1Н), 5,00 (уш.с, 1Н), 5,32 (уш.с, 1Н), 5,99 (д, 1Н), 6,38 (д, 1Н) части/миллион.
То же соединение было получено, когда вместо исходного соединения 33 было использовано соединение 28 (приготовление 25).
П р и м е р 2. 1(S), 3(R)-Дигидрокси-20(R)-(3'-гидрокси-3-метил- 1-бутокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение 102).
Раствор соединения 21 (35 мг) в ацетонитриле (4 мл) и 40%-ный раствор фтористоводородной кислоты (0,2 мл) перемешивался в течение 1 ч при комнатной температуре в атмосфере азота. Затем добавлялся этилацетат и смесь экстрагировалась насыщенным раствором бикарбоната натрия, далее хлористым кальцием. Этилацетатный раствор сушился и концентрировался в вакууме, давая остаток, который очищался хроматографически (силикагель, этилацетат в качестве разбавителя) с образованием названного в заглавии соединения.
ЯМР: δ 0,54 (с, 3Н), 1,12 (д, 3Н), 1,21 (с, 3Н), 1,23 (с, 3Н), 1,35-2,20 (м, 17Н), 2,30 (дд, 1Н), 2,57 (дд, 1Н), 2,81 (м, 1Н), 3, 25 (м, 1 Н), 3,44 (м, 1Н), 3,55 (с, 1Н), 3,82 (м, 1Н), 4,21 (м, 1Н), 4,42 (м, 1 Н), 4,98 (м, 1Н), 5,31 (м, 1Н), 5,98 (д, 1Н, I 11,3), 6,37 (д, 1Н, I 11,3).
П р и м е р 3. 1(S), 3 (R)-Дигидрокси-20(R)-(4'-гидрокси-4'- метил-1'-пентилокси)-9,10- вторпрегна-5(Z), 7(E), 10(19)-триен.
Соединение 24, полученное по приготовлению 16 (128 мг, 0,2 ммоль) растворялось в этилацетате (0,2 мл) и при энергичном перемешивании добавлялся ацетонитрил (4,4 мл). Затем добавлялся раствор 5%-ной фтористоводородной кислоты в ацетонитриле/воде 8:1 (1,94 мл) и реакционная смесь перешивалась в течение 45 мин при комнатной температуре в атмосфере азота. Реакционная смесь обрабатывалась (этилацетат) и очищалась хроматографически (35 г силикагеля, 80% этилацетата в петролейном эфире в качестве разбавителя с образованием нужного соединения в виде бесцветного масла).
ЯМР: δ 0,54 (с, 3Н), 1,08 (д, 3Н), 1,20 (с, 6Н), 1,05-2,50 (м, 21Н), 2,59 (дд, 1Н), 2, 81 (уш.д, 1Н), 3,25 (м, 2Н), 3,54 (м, 1Н), 4,21 (м, 1Н), 4,42 (м, 1Н), 4,99 (м, 1Н), 5,31 (м, 1Н), 5,98 (д, 1Н, I 11,3), 6,37 (д, 1Н, I 11,3) части/миллион.
П р и м е р 4. 1(S)-3(R)-Дигидро-20(R)-(4'-гидрокси-4'-этил-1'- гексилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен.
Выполнением метода, приведенного в примере 4, и заменой соединения 24 соединением 25 или 26 (приготовление 17 или 27), было получено нужное соединение в виде бесцветной смолы.
ЯМР: δ= 0,56 (с, 3Н), 0,85 (дт, 6Н), 1,09 (д, 3Н), 1,47 (уш.кв. 1Н), 1, 00-2,22 (м, 20Н), 2,31 (дд, 1Н), 2,61 (уш.д, 1Н), 2,83 (уш.д, 1Н), 3,25 (м, 2Н), 3,55 (м, 1Н), 4,23 (м, 1Н), 4,43 (м, 1Н), 5,00 (м, 1Н), 5,31 (м, 1Н), 6,00 (д, 1Н, I11,3), 6,39 (л, 1Н, I 11,3) части/миллион.
П р и м е р 5. 1(S), 3(R)-Дигидрокси-20(R)-(5'-гидрокси-5'-этил- 1'-гептилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение 110).
Соединение 29 в приготовлении 24 (40 мг, 0,085 ммоль) растворялось в этилацетате (0,1 мл) и добавлялся ацетонитрил (2,3 мл). Затем добавлялся 5% -ный раствор фтористоводородной кислоты в ацетонитриле/воде 8:1 (1, 05 мл) и реакционная смесь перемешивалась в течение 40 мин в атмосфере азота при комнатной температуре. Реакционная смесь обрабатывалась (этилацетат) и очищался хроматографически (30 г силикагеля, 50% этилацетата в петролейном эфире в качестве разбавителя), образуя нужное соединение.
ЯМР: δ 0,56 (с, 3Н), 0,85 (т, 6Н), 1,08 (д, 3Н), 1, 45 (кв. 4Н), 1,02-2,09 (м, 21Н), 2,17 (уш.д, 1Н), 2,32 (дд, 1Н), 2,60 (дд, 1Н), 2,83 (уш.д, 1Н), 3,20 (м, 2Н), 3,59 (М, 1Н), 4,23 (м, 1Н), 4,42 (м, 1Н), 5,00 (м, 1Н), 5,31 (м, 1Н), 6,00 (д, 1Н, I 11, 3), 6,39 (д, 1Н, I 11,3) части/миллион.
П р и м е р 6. 1(S),3(R)-Дигидрокси-20(R)-(6'-гидрокси-1'- гексилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение III).
Соединение 30, полученное в приготовлении 23 (233 мг, 0,3 ммоль), растворялось в этилацетате (0,6 мл) и при энергичном перемешивании добавлялся ацетонитрил (8 мл). Затем добавлялся 5% -ный раствор фтористоводородной кислоты в ацетонитриле/воде 8:1 (4 мл) и реакционная смесь перемешивалась при комнатной температуре в атмосфере азота в течение 90 мин. Реакционная смесь обрабатывалась (этилацетат) и очищалась хроматографически (40 г силикагеля, 80%-ный этилацетат в петролейном эфире в качестве разбавителя) с образованием нужного соединения в виде бесцветной смолы.
ЯМР: δ= 0,55 (с, 3Н), 1,07 (д, 3Н), 1,00-2,22 (м, 24Н), 2,31 (дд, 1Н), 2,60 (дд, 1Н), 2,83 (уш.д, 1Н), 3,22 (м, 2Н), 3,55 (м, 1Н), 3,64 (т, 2Н), 4,23 (м, 1Н), 4,43 (м, 1Н), 5,00 (м, 1Н), 5,32 (м, 1Н), 6,00 (д, 1Н, I 11,3), 6,39 (д, 1Н, I 11,3) части/миллион.
П р и м е р 7. 1(S),3(R)-Дигидрокси-20(R)-(4'-гидрокси-4'-(1"- пропил)-1'-гептилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение 107).
Соединение было приготовлено в соответствии с методом, описанным в примере 4, за исключением того, что соединение 24, полученное приготовлении 16, заменялось соединением 27, полученным в приготовлении 34.
ЯМР: δ= 0,55 (с, 3Н), 0,91 (т, 6Н), 1,09 (д, 3Н), 1,1-2,05 (м, 25Н), 2,15 (уш.д, 1Н), 2,32 (дд, 1Н), 2,60 (уш.д, 1Н), 2,82 (м, 1Н), 3,22 (м, 2Н), 3,56 (м, 1Н), 4,23 (м, 1Н), 4,43 (м, 1Н), 5,00 (уш.с, 1Н), 5,32 (уш.с, 1Н), 5,99 (д, 1Н), 6,48 (д, 1Н) части/миллион.
П р и м е р 8. 1(S), 3(R)-Дигидрокси-20(R)-(5'-метил-1'- гексилокси)-9,10-вторпрегна-5(Z), 7(E), 10(19)-триен (соединение 108).
Соединение было приготовлено в соответствии с методом, описанным в примере 4, за исключением того, что соединение 24, полученное в приготовлении 16, было заменено соединением 20, полученным в приготовлении 35.
ЯМР: δ= 0,56 (с, 3Н), 0,86 (д, 6Н), 1,07 (д, 3Н), 1,00-2,07 (м, 21Н), 2,16 (уш. д, 1Н), 2,31 (дд, 1Н), 2,60 (уш.д, 1Н), 2,82 (уш.д, 1Н), 3,20 (м, 2Н), 3,55 (м, 1Н), 4,23 (м, 1Н), 4,43 (м, 1Н), 5,00 (уш.с, 1Н), 5,32 (м, 1Н), 6,00 (д, 1Н, I 11,3), 6,39 (д, 1Н, I 11,3) частей/миллион.
СИНТЕЗ И БИОЛОГИЧЕСКАЯ АКТИВНОСТЬ АНАЛОГОВ 22-ОКСА ВИТАМИНА D.
Было обнаружено, что 22-окса аналог 1 α 25(ОН)2 D3 является более активным, чем сам 1α, 25(ОН2)D3 при подавлении пролиферации и индуцировании дифференциации злокачественных клеток. Для соединений, являющихся эффективными регуляторами роста в сочетании с низким кальцемическим эффектом, были синтезированы два ряда 1α-гидроксилированных производных 22-окса витамина D: ряд с повышенным количеством атомов углерода в боковой цепи и соответствующие соединения с эпимерической стереоконфигурацией в положении 20.
Синтез. Альдегид (I), хорошо известный промежуточный продукт для синтеза антипсориатического агента, кальципотриола (2), окисляют кислородом воздуха в условиях, известных из химии стероидов (3), чтобы получить кетон (II). Кетон восстанавливают NaBH4, чтобы получить смесь двух эпимерных форм, 20S и 20R, спирта (III). Спирты являются 0-алкилированными, как подчеркнуто в реакционной схеме (см. на фиг.2).
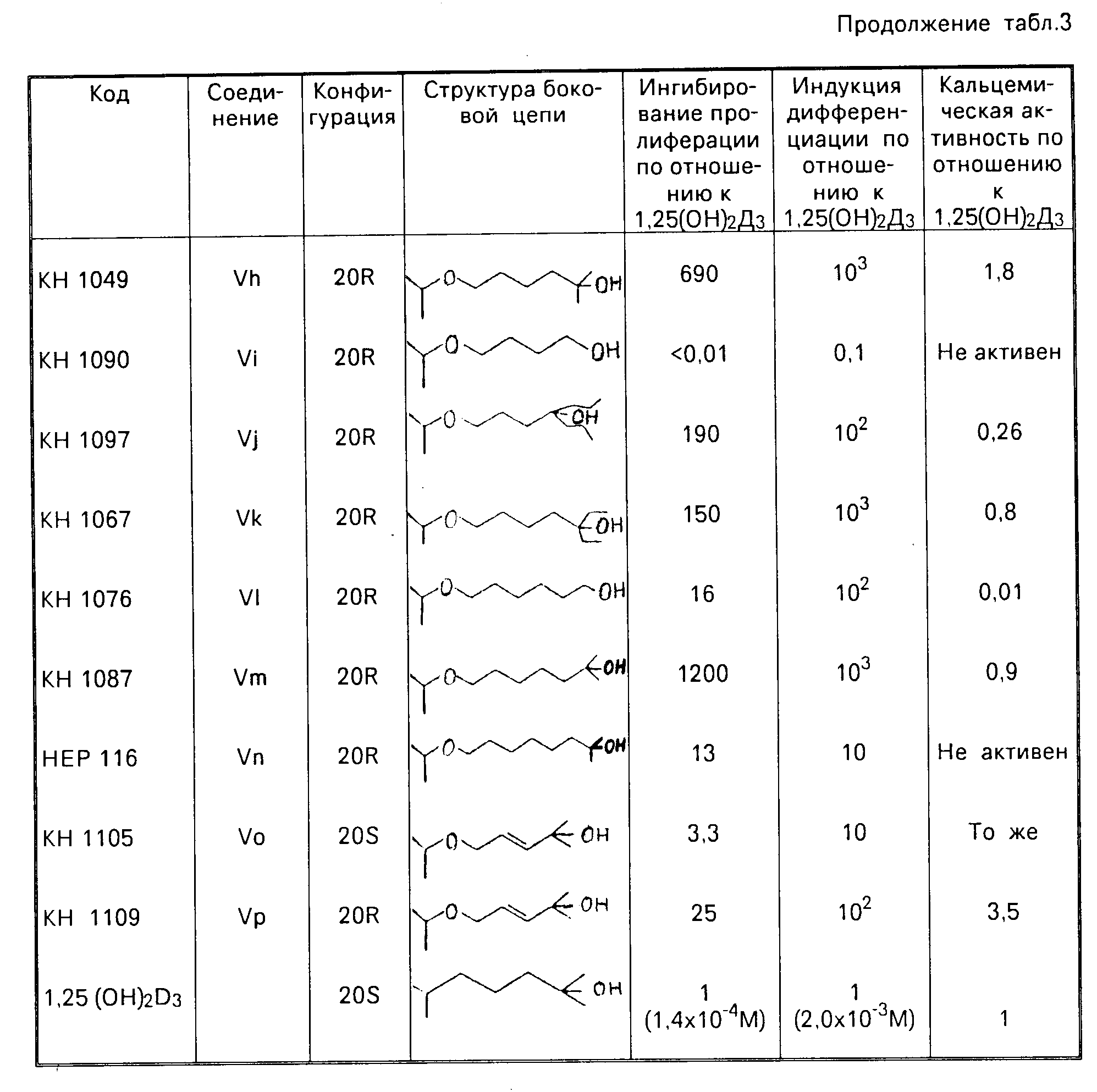
Биологическая активность. Биологическая активность была обнаружена в результате испытаний по методике [4] Во всех экспериментах в качестве стандартного соединения использовали 1 α, 25(ОН)2D3, и данные величины рассчитаны относительно тех, что получены с 1α 25(ОН)2D3.
Сравнение биологических активностей аналогов с различными боковыми цепями показано в табл.3. Сравнение соединений с нормальной 20S конфигурацией показывает, что удлинение боковой цепи на один атом углерода приводит к незначительному улучшению рострегулирующего эффекта (МС 1273), а замещение С26 метильных групп этильными группами приводит к повышению активности (КН 1139). Сравнение пар 20 эпимеров показало, что аналоги с 20R эпимерной конфигурацией являются намного более мощными, чем соответствующие соединения с нормальной 20S стереохимией. В ряду 20R соединений с 8-10 атомами углерода в боковой цепи показывают самую высокую активность. В обеих сериях 24, 26, 27-трисгомо-аналог (КН 1139 и КН 1060) является наиболее активным, в особенности 20R аналог (КН 1060).
Значение > 1 указывает на то, что аналог является более мощным,
чем 1,
25(ОН)2D3
Аналоги 20-эпи-22-оксавитамина D являются на несколько порядков величин более активными в качестве регуляторов пролиферации и дифференциации клеток, чем
1
α, 25(ОН)2D3, несмотря на практически неизменную кальцемическую активность. Наиболее активное соединение КН 1060, которое ингибирует пролиферацию клеток на 50% при 5
˙10-13 М и вызывает дифференциацию клеток при концентрациях до 10-14 М, выбрано для дальнейшего изучения.
Реферат
Использование: в качестве препарата широкого спектра действия. Сущность изобретения: продукт-аналог витамина D ф-лы I, где n - целое число от 1 до 6 R1 и R2 - одинаковые или различные H, CH, CH, i-CH или вместе с атомом углерода, помеченным звездочкой и радикалами R1, R2 и X образуют циклогексановое кольцо, X-H или OH. Предпочтительные соединения 1 (S), 3 (R)-дигидрокси-20(R)- 4′ -гидрокси- 4′ -этил- 1′ -гексилокси-9, 10-вторпрегна-5 (Z), 7 (E), d (19)-триен, 1 (S), 3 (R)-дигидрокси- 20(R)′-(6′ -гидрокси- 1′ -гексилокси 9, 10-втор-прегна-5 (Z), 7 (E), 10 (19)-триен, 1 (S), 3 (R)-дигидрокси-20 (R)- 5′ -гидрокси- 5′ -этил- 1′ -гептилокси, 9, 10-втор-прегна-5 (Z), 7 (E), 10 (19)-триен, 1 (S), 3 (R)-дигидрокси-20 (R)- 5′ -гидрокси- 5′ -этил- 1′ -гептилокси, 9, 10-втор-прегна-5 (Z), 7 (E), 10 (19)-триен, 1 (S), 3 (R)-дигидрокси-20 (R)- 5′ -метил- 5′ -этил- 1′ -гексилокси 9, 10-втор-прегна-5 (Z) 7 (E), 10 (19)-триен. Структура ф-лы I (см. чертеж). 1 з.п. ф-лы, 3 ил., 3 табл.
Формула
где n целое число от 1 до 6;
R1 и R2 могут быть одинаковыми или различными водород, метил, этил, пропил, или изопропил, или вместе с атомом углерода, помеченным звездочкой и радикалами R1, R2 и X, образуют циклогексановое кольцо;
X водород или гидроксил.
a) 1(S), 3(R) Дигидрокси-20(R) (4'-гидрокси-4' этил-1-гексилокси) - 9,10-втор-прегна -5 (Z), 7(Е), 10(19)-триена;
b) 1(S), 3(R)-Дигидрокси-20(R) (6'-гидрокси-1' гексилокси)-9,10-втор прегна-5(Z), 7(Е), 10(19)-триена;
c) 1(S), 3(R)-Дигидрокси-20(R) (5'-гидрокси- 5'-этил-1' - гептилокси)-9,10 втор-прегна-5(Z), 7(Е), 10(19)-триена;
d) 1(S)3-(R) Дигидрокси- 20 (R)-(5'-гидрокси-5'- метил-1'- гексилокси)-9,10 втор-прегна-5(Z), 7(Е), 10(19)-триена;
e) 1(S), 3(R)Дигидрокси 20(R)-(5'- метил-1'- гексилокси)-9,10- втор-прегна-5(Z), 7(Е), 10(19) триена;
f) 1(S), 3(R)-Дигидрокси 20(R)-(4' гидрокси-4' (1''-пропил) - 1'-гептилокси) -9,10-втор-прегна-5(Z), 7(Е), 10(19)-триена;
g) 1(S), 3(R)-Дигидрокси 20(R)-(4' гидрокси-4'- метил-1' - гептилокси)-9,10- втор-прегна-5(Z), 7(Е), 10(19)-триена;
h) 1(S), 3(R)-Дигидрокси 20(R)-3' гидрокси-3' метил-1' - бутилокси)-9,10-втор-прегна-5(Z), 7(Е), 10(19)-триена.
Комментарии