N-метилгомоцистеины, их применение и способ их получения - RU2305678C2
Код документа: RU2305678C2
Описание
Настоящее изобретение относится к объектам, охарактеризованным в формуле изобретения, а именно к N-метилгомоцистеинам общих формул I и II, к их применению и к способу их получения.
N-метиламинокислоты являются ценными промежуточными продуктами, использующимися в синтетической химии. Прежде всего подобные структурные элементы очень часто используются в фармацевтической химии, поскольку многие высокоэффективные и обладающие избирательным действием фармакологические средства содержат N-метиламинокислоты.
Оптически активные N-метиламинокислоты встречаются в природе в виде отдельных соединений (например, N-метилтриптофаны: Liebigs Ann. Chem. 520, 1935, с.31-34), но они встречаются также в качестве структурных элементов в целом ряде биологически активных природных веществ, таких, например, как доластатины (G.R.Pettit и др., Journ. Org. Chem. 55, 1990, с.2989-2990; Tetrahedron 41, 1993, с.9151-9170) или дидемнины (K.L.Rinehart и др., Journ. Nat. Prod. 51, 1988, с.1-21; Journ. Am. Chem. Soc. 109, 1987, с.6846-6848; Journ. Am. Chem. Soc. 117, 1995. с.3734-3748). В качестве других примеров можно назвать жасплакинолид (Journ. Org. Chem. 56, 1991, с.5196-5202) и некоторые другие цитотоксические пептиды (Journ. Org. Chem. 54, 1989, с.617-627; Tetrahedron 51, 1995, с.10653-10662; Journ. Org. Chem. 54, 1989, с.736-738). Обладающий иммунодепрессантным действием циклоспорин также содержит N-метиламинокислоты (см., например, R.M.Wenger, Helv. Chim. Acta 66, 1983, с.2672-2702; S.L. Schreiber и др. Tetrahedron Lett. 29, 1988, с.6577-6580). N-метиламинокислоты могут также обладать нейрофармакологической активностью (J.C.Watkins, Journ. Med. Pharm. Chem. 5, 1962, с.1187-1199). С целью более детально выявить конформацию и биологическую активность в фармацевтических исследованиях часто используют, например, такой метод, как встраивание N-метиламинокислот в биологически активные пептиды (см., например, Journ. Org. Chem. 46, 1981, с.3436-3440; Int. Journ. Pept. Protein Res. 46, 1995, с.47-55; Int. Journ. Pept. Protein Res. 27, 1986, с.617-632). Обзорный материал о результатах исследований конформации на пептидах и о ее взаимосвязи с биологической активностью опубликован в Angew. Chem. 94, 1982, с.509-520, автор Н. Kessler, и в Ann. Rep. Med. Chem. 13, 1978, с.227-238, авторы G.R.Marshall и др.
С другой стороны, замещение аминокислот в пептидах соответствующими N-метильными соединениями служит повышению эффективности или избирательности действия пептидных лигандов (см., например, Journ. Med. Chem. 37, 1994, с.769-780; Int. Journ. Pept. Protein Res. 46, 1995, с.47-55). N-метилпептидные связи часто проявляют более высокую устойчивость по отношению к протеолизу по сравнению с неметелированными связями, что может способствовать повышению оральной доступности и увеличению продолжительности действия (R.H.Mazur и др., Journ. Med. Chem. 23, 1980, с.758-763). Касательно конструирования пептидов за счет встраивания N-метиламинокислот и других модификаций можно сослаться на следующие публикации: W.F.Degrado, Adv. Protein Chem. 39, 1988, с.51-124, и J.Rizo, L.M.Gierasch, Annu. Rev. Biochem. 61, 1993, с.387-418. В отношении пептидомиметиков можно сослаться на обзорные статьи A. Giannis и др. в Angew. Chem. 105, 1993, с.1303-1326, и J.Gante в Angew. Chem. 106, 1994, с.1780-1802.
В заявке WO 01/44177 описан пептид, который в циклической части 6-5 мерной последовательности содержит N-метилгомоцистеин. Через атом серы гомоцистеина осуществляется соединение последовательности, связывающей ионы металлов. Благодаря хелатированию радионуклидов, таких как99mТс или188Re, создана возможность их применения в качестве радиоизотопных диагностических средств, соответственно радиоизотопных терапевтических средств для диагностики или терапии онкологических заболеваний (см. также патенты US 6056940, US 6022857, US 6017509).
Синтез пептида согласно указанной заявке WO 01/44177 осуществляют N-метилированием в ходе многоступенчатой реакции (см., например, Journ. Am. Chem. Soc. 119, 1997, с.2301-2302). 4-мерный пептид, связанный С-концевым остатком с твердой фазой, взаимодействием с аминокислотой N-Fmoc-S-тритилгомоцистеином (Fmoc обозначает 9Н-флуорен-9-илметилоксикарбонил), которую получают в три стадии из метионина (см. Journ. Med. Chem. 39, 1996, с.1361-1371), превращают в 5-мерный пептид. После отщепления защитной группы концевую аминогруппу гомоцистеина взаимодействием с хлорангидридом 2-нитробензолсульфоновой кислоты превращают в сульфонамид. Затем проводят метилирование с использованием MTBD (1,3,4,6,7,8-гексагидро-1-метил-2Н-пиримидо[1,2-а]пиримидина), после чего отщепляют сульфонильную группу с помощью 2-меркаптоэтанола. Затем продолжают дальнейшее построение пептидной последовательности. Однако описанному синтезу присущ существенный недостаток, заключающийся в том, что не удается достичь полного химического превращения и что требуется проводить дополнительную очистку пептида.
Построение пептидных последовательностей, содержащих N-метиламинокислоты, осуществляют обычно взаимодействием соответствующего остатка последовательности с Fmoc-защищенной N-метиламинокислотой (см., например, Journ. Med. Chem. 39, 1996, с.1361-1371). Однако требуемую для пептидной последовательности в WO 01/44177 аминокислоту N-Fmoc-N-метил-S-тритилгомоцистеин до настоящего времени получить не удалось.
В литературе описан целый ряд методов получения N-метиламинокислот, при этом обычно исходят из соответствующих неметилированных аминокислот, которые в большом количестве предлагаются на рынке в виде хиральных природных веществ.
Синтез нерацемических N-метиламинокислот впервые был предложен Э.Фишером. В этом случае N-тозильные производные аминокислот метилируют взаимодействием с едким натром и метилиодидом. Затем с помощью кипящей соляной кислоты отщепляют тозильную группу (Ann. Chem. 398, 1913, с.96-125; Ber. dt. chem. Ges. 48, 1915, с.360-378). Указывается также, что при осуществлении этого метода происходит частичная рацемизация (А.Н. Cook и др., Journ. chem. Soc., 1949, с.1022-1028).
Согласно другому методу Фишера предлагается осуществлять взаимодействие 2-бромкарбоновых кислот с метиламином (Ber. dt. chem. Ges. 49, 1916, с.1355-1366; информацию о применении см. также в статье А.Н.Cook и др., Journ. chem. Soc., 1949, с.1022-1028). В этом случае также образуются частично рацемические продукты (см. Р.Quitt и др., Helv. Chim. Acta 46, 1963, с.327-333). Данный факт могут подтвердить авторы настоящей заявки, наблюдавшие взаимодействие 2-бром-4-метилсульфанилбутановой кислоты с метиламином. В литературе описано получение N-метил-L-метионина через стадию диазотирования D-метионина с последующим замещением бромидом при удержании, после чего проводят замещение метиламином при инверсии (N.Izumiya, A.Nagamatsu, Kyushu. Mem. Med. Sci. 4, 1953, с.1-16). Энантиомерную чистоту полученного продукта не определяли. Авторам настоящей заявки удалось установить, что даже при использовании в мягких условиях метода диазотирования Эллмана (Synthesis, 1999, с.583-585) в результате реакции образуется энантиомерный избыток, составляющий лишь 75%. Поэтому данный метод непригоден для получения энантиомерочистых N-метилгомоцистеинов.
Одним из наиболее часто применяемых в указанных целях методов является прямое алкилирование по Беноитону (Benoiton) с помощью гидрида натрия и метилиодида в ТГФ или ДМФ (Can. Journ. Chem. 51 (1973), с.1915-1919; Can. Journ. Chem. 55, 1977, с.906-910). Этот метод обычно применяют для получения Z- и Вос-защищенных N-метиламинокислот (см., например, Tetrahedron 51, 1995, с.10653-10662; D.L.Boger и др., Journ. Am. Chem. Soc. 121, 1999, с.6197-6205; H.Waldmann и др., Chem. Eur. Journ. 5, 1999, с.227-236). Взаимодействие Вос-защищенного амида метионина и метилирующих агентов, таких как метилиодид, приводит в рассматриваемом случае к метилированию атома серы с образованием ионов сульфония (см. также S.J.F. Macdonald и др., Journ. Med. Chem. 64, 1999, с.5166-5175). По этой причине данный метод не может использоваться для получения N-метилгомоцистеинов.
Существует целый ряд методов получения N-метиламинокислот, которые исходят из эфиров аминокислот. К таким методам относятся среди прочих избирательное восстановление эфиров N-формиламинокислот с помощью борана (Tetrahedron Lett. 23, 1982, с.3315-3318; Journ. Org. Chem. 56, 1991, с.5196-5202) и обменная реакция между шиффовыми основаниями эфиров аминокислот и диметилсульфатом или метилтрифлатом с последующим гидролизом (M.J.O′Donnell и др., Tetrahedron Lett. 25, 1984, с.3651-3654). Однако при омылении эфиров аминокислот с образованием аминокислот наблюдается частичная рацемизация (S.T.Cheung, N.L.Benoiton, Can. Journ. Chem. 55, 1977, с.906-910). Поэтому данный метод также неприемлем для целей изобретения.
Еще одна возможность состоит в том, чтобы аминокислоты подвергать при восстановительном алкилировании взаимодействию с альдегидом. Образующееся в качестве промежуточного продукта шиффово основание восстанавливают in situ в присутствии водорода и соответствующего катализатора по методу Боумана (R.E.Bowman и др., Journ. Chem. Soc., 1950, с.1342-1345 и 1346-1349) или с помощью борогидрида натрия (К.A.Schellenberg, Journ. Org. Chem. 28, 1963, с.3259-3261). При этом работают как в растворе (см., например, Journ. Org. Chem. 61, 1996, с.3849-3862; Journ. Org. Chem. 60, 1995, с.6776-6784), так и на твердой фазе (см., например, Journ. Org. Chem. 61, 1996, с.6720-6722; Tetrahedron Lett. 38, 1997, с. 4943-4946; Bioorg. Med. Chem. Lett. 5, 1995, с.47-50). С помощью этого метода Боуман первым проводил также перметилирование ди- и трипептидов (Journ. Chem. Soc., 1950, с.1349-1351). Однако избирательное монометилирование с использованием формальдегида по этому методу невозможно, поскольку приводит к получению смесей из неметилированной, монометилированной и диметилированной аминокислоты, которые не поддаются разделению (Quitt и др., Helv. Chim. Acta 46, 1963, с.327-333; см. также R.E.Bowman, H.H.Stroud, Journ. Chem. Soc., 1950, c.1342-1345).
Широкое распространение для синтеза Fmoc-защищенных N-метиламинокислот получил метод Фрейдингера (Journ. Org. Chem. 48, 1983, c.77-81). Этот метод включает две стадии. На первой из них взаимодействием аминокислоты с формальдегидом образуют оксазолидинон, который затем на второй стадии с помощью триэтилсилана в кислых условиях восстанавливают до N-метильного соединения. Фрейдингер утверждает, что в результате взаимодействия Fmoc-L-метионина с формальдегидом с образованием оксазолидинона выход продукта составляет 88%, однако восстановление с помощью триэтилсилана до Fmoc-N-метил-L-метионина даже по истечении пяти дней все еще остается неполным, а выход продукта составляет лишь 22% (Journ. Org. Chem. 48, 1983, c.77-81).
С учетом вышеизложенного в основу настоящего изобретения была положена задача получить соединения, которые могли бы служить для построения пептидных последовательностей, содержащих N-метилированные гомоцистеины, и позволяли бы исключить N-метилирование на твердой фазе, а также разработать способ получения этих соединений.
Указанная задача решается благодаря N-метилгомоцистеинам, которые могут быть представлены как в виде чистых энантиомеров, так и в виде рацематов, общей формулы I

где
X1 обозначает серусодержащую защитную группу, а
Х2 обозначает атом водорода или азотсодержащую защитную группу.
Предпочтительно X1 обозначает бензил (Bn), 4-метоксибензил (Mob), дифенилметил, бис(4-метоксифенил)метил, 4,4′-диметокситрифенилметил (DMT), трифенилметил (тритил), метоксиметил (MOM), 9Н-флуорен-9-илметил или трет-бутилсульфид (T.W.Green, P.G.M.Wuts, Protective Groups in Organic Synthesis, 2-е изд., изд-во J. Wiley & Sons, New York, 1991), более предпочтительно бензил (Bn), 4-метоксибензил (Mob), дифенилметил, бис(4-метоксифенил)метил, 4,4′ -диметокситрифенилметил (DMT) или трифенилметил (тритил), особенно предпочтительно трифенилметил (тритил), а Х2 в случае защитной группы обозначает предпочтительно 9Н-флуорен-9-илметилоксикарбонил (Fmoc), 2,7-дибром-9Н-флуорен-9-илметилоксикарбонил, трет-бутилоксикарбонил (Boc), бензилоксикарбонил (Cbz или Z), трифторацетил, 2,2,2-трихлорэтилоксикарбонил (Troc), 2-триметилсилилэтилоксикарбонил (Теос) или 2-триметилсилилэтилсульфонил (T.W.Green, P.G.M.Wuts, см. выше), более предпочтительно трет-бутилоксикарбонил (Boc), бензилоксикарбонил (Cbz или Z) или 9Н-флуорен-9-илметилоксикарбонил (Fmoc), особенно предпочтительно 9Н-флуорен-9-илметилоксикарбонил (Fmoc).
Предлагаемые в изобретении соединения общей формулы I получают благодаря тому, что оксазолидинон формулы II

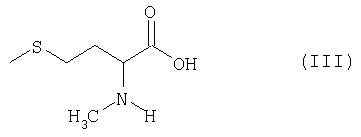
получаемый по известным из литературы методам (Ben-Ishai, Journ. Am. Chem. Soc. 79, 1957, с.5736-5738; М.А.Blaskovich, M.Kahn, Synthesis, 1998, с.379-380; G.V.Reddy и др., Synth. Commun. 29, 1999, с.4071-4077) из метионина (см. пример 1), раскрывают с помощью соответствующего восстановителя в присутствии кислоты с образованием N-метилметионина формулы III

и затем известным образом (см. T.W.Green, P.G.M.Wuts, см. выше) вводят серусодержащую защитную группу, а при необходимости также азотсодержащую защитную группу. Целесообразно использовать в качестве восстановителей алкилсиланы, предпочтительно триэтилсилан. В качестве кислот целесообразно использовать трифторуксусную кислоту, пентафторпропионовую кислоту, трифторметансульфоновую кислоту или метансульфоновую кислоту, предпочтительно трифторуксусную кислоту. Реакцию целесообразно проводить в хлорированных растворителях, диоксане, ТГФ или диметоксиэтане, предпочтительно в дихлорметане или хлороформе. Реакцию проводят при температуре в интервале от -20 до +100°С, предпочтительно в интервале от 0 до 40°С. Продолжительность реакции составляет от 5 минут до 10 часов, предпочтительно от 0,5 до 2 ч. N-метилметионин получают с выходом от 70 до 95%.
Соединения общей формулы I по известным специалистам методам (см. Houben-Weyl, том Е22, Synthesis of Peptids and Peptidomimetics, под ред. М. Gooodman, изд-во Thieme, 2001) используют для построения содержащих N-метилгомоцистеин пептидов и пептидных промежуточных продуктов, предпочтительно для получения Fmoc-(N-CH3)Hcy(Trt)-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-OH, цикло-Tyr-D-Trp-Lys-Thr-Phe-(N-CH3)Hcy и цикло-Tyr-D-Trp-Lys-Thr-Phe-(N-CH3)Hcy(CH2CO-β-Dap-Phe(4-NH2)-Cys-Thr-Ser).
Пример 1
а) трет-Бутил-(S)-4-[2-(метилсульфанил)этил]-5-оксооксазолидинон-3-карбоксилат
Смесь из 100 г N-(трет-бутоксикарбонил)-L-метионина (0,4 моля), 100 г параформальдегида (3,3 моля), 200 г высушенного сульфата магния (1,7 моля) и 4 г паратолуолсульфоновой кислоты (0,02 моля) в 1 л толуола нагревают в течение 3 ч до 90°С. Затем смеси дают остыть до 20°С и при охлаждении льдом добавляют к ней 800 мл насыщенного раствора гидрокарбоната натрия. После этого отфильтровывают и остаток промывают 400 мл этилацетата. Органическую фазу экстрагируют 300 мл воды, сушат над сульфатом натрия и концентрируют досуха в вакууме. Сырой продукт растворяют в 150 мл гексана/этилацетата (соотношение 1:3), фильтруют через силикагель и силикагель промывают 500 мл гексана/этилацетата (в соотношении 1:3). В завершение концентрируют досуха в вакууме, получая в результате 85,3 г (0,33 ммоля, 83% от теории) желтого масла.
Элементный анализ:
рассч.: С 50,56 Н 7,33 N 5,36 S 12,27
обнар.: С 50,38 Н 7,24 N 5,43 S 12,10
ИК: 2980, 2920, 1800, 1715, 1390, 1170, 1050.
ЯМР (CDCl3): 1,5 (9Н), 2,1 (3Н), 2,15-2,35 (2Н), 2,5-2,68 (2Н), 4,33 (1Н), 5,21 (1Н), 5,48 (1Н).
MC (EI): m/z 261, 205, 188, 116, 100, 57.
б) N-метил-L-метионин
К раствору из 19,4 г трет-бутил-(S)-4-[2-(метилсульфанил)этил]-5-оксооксазолидинон-3-карбоксилата (0,074 моля) в 65 мл дихлорметана при 0°С добавляют по каплям 45 мл трифторуксусной кислоты и 40 мл триэтилсилана (0,25 моля). Далее в течение 1 ч перемешивают при охлаждении льдом и в течение 1 ч при 20°С. После этого реакционный раствор концентрируют досуха в вакууме. Остаток трижды вновь растворяют в дихлорметане его порциями по 100 мл и повторно концентрируют досуха в вакууме. Затем остаток растворяют в 100 мл воды и трижды экстрагируют 50 мл метил-трет-бутилового эфира (МТБЭ). Водную фазу концентрируют досуха в вакууме. Остаток вновь трижды растворяют в этаноле его порциями по 50 мл и повторно концентрируют досуха. Остаток смешивают со 150 мл МТБЭ и в течение 2 ч перемешивают при 20°С. 20 Затем отфильтровывают и промывают 50 мл МТБЭ. После сушки получают 9,6 г (0,059 моля, 80% от теории) бесцветного твердого вещества.
Элементный анализ:
рассч.: С 44,15 Н 8,03 N 8,58 S 19,64
обнар.: С 44,01 Н 7,83 N 8,72 S 19,80
ИК: 3440, 3010, 2920, 2830, 2420, 1735, 1675, 1580, 1205, 1140, 800, 720.
ЯМР (ДМСО): 2,01-2,08 (5Н), 2,5-2,65 (5Н), 3,74-3,78 (1Н).
MC(FAB):m/z 164.
CD (вода): 200 нм, Δε 1,64.
в) N-метил-S-тритил-L-гомоцистеин
В реакционный сосуд помещают 7,5 г (0,046 моля) N-метил-L-метионина и по реакции конденсации при охлаждении смесью МеОН/сухой лед вводят 200 мл аммиака. Затем при -35°С порциями добавляют в течение 1 ч 5,2 г (0,23 моля) натрия и перемешивают в течение 2 ч. После этого порциями добавляют 9, 7 г (0,18 моля) хлорида аммония, охлаждающую смесь удаляют и в течение ночи с помощью азота отгоняют аммиак. Далее добавляют 14,0 г (0,054 моля) трифенилметанола. Затем при охлаждении льдом добавляют 50 мл дихлорметана и 90 мл трифторуксусной кислоты. Смесь в течение 1 ч перемешивают при комнатной температуре и концентрируют досуха. Остаток суспендируют в 200 мл воды и с помощью едкого натра значение рН устанавливают на 13. После 1-часового перемешивания проводят вакуум-фильтрацию и фильтрат - твердое вещество - суспендируют в 500 мл воды. Добавлением лимонной кислоты значение рН устанавливают на 4. Далее добавляют 600 мл МТБЭ и перемешивают в течение 30 мин. Затем отфильтровывают вакуум-фильтрацией и дважды промывают МТБЭ его порциями по 100 мл. Остаток смешивают со 100 мл дихлорметана, добавляют 20 мл этанола и затем по каплям добавляют 500 мл МТБЭ. После 1-часового перемешивания отфильтровывают вакуум-фильтрацией и промывают МТБЭ. После сушки получают 12,6 г (0,032 моля, 70% от теории) бесцветного твердого вещества.
Элементный анализ:
рассч.: С 73,62 Н 6,44 N 3,58 S 8,19
обнар.: С 73,40 Н 6,30 N 3,71 S 8,02
ИК: 3440, 3055, 2850, 2400, 1615, 1485, 1445, 1395, 740, 700.
ЯМР (CDCl3): 1,28-1,62 (2Н), 2,12 (3Н), 2,25-2,45 (2Н) 2,84-2,92 (1Н), 7,12-7,41 (15Н).
MC (EI): m/z 243, 165.
г) N-(9Н-флуорен-9-илметилоксикарбонил)-N-метил-S-тритил-L-гомоцистеин
16,6 г (0,042 моля) N-метил-S-тритил-L-гомоцистеина суспендируют в 90 мл воды и 100 мл ТГФ и добавляют 12,58 г (0,12 моля) карбоната натрия. Далее при 10°С по каплям добавляют 14,88 г (0,044 моля) флуоренилметилсукцинимидилкарбоната в 50 мл ТГФ и перемешивают в течение 3 ч. Затем смешивают с 80 мл воды и 130 мл этилацетата, перемешивают в течение 10 мин и с помощью лимонной кислоты значение рН устанавливают на 4. Органическую фазу дважды промывают водой ее порциями по 100 мл и один раз 100 мл раствора поваренной соли. Водные фазы один раз экстрагируют 100 мл этилацетата. Объединенную органическую фазу упаривают с помощью роторного испарителя, смешивают с 260 мл этилацетата и упаривают с помощью роторного испарителя с получением 29,2 г пенистого вещества, которое хроматографируют с использованием смеси дихлорметан/ацетон в соотношении 6:4. В результате получают 22,4 г (0,036 моля, 86% от теории) целевого продукта.
Элементный анализ:
рассч.: С 76,32 Н 5,75 N 2,28 S 5,22
обнар.: С 76,11 Н 5,52 N 2,40 S 5,38
ИК: 3430, 3060, 2950, 1740, 1710, 1175, 740, 700.
ЯМР (CDCl3): 1,46-2,35 (4Н), 2,66-2,73 (3Н), 4,13-4,6 (4Н), 7,13-7,79 (23Н).
MC(FAB): m/z 614, 636.
Пример 2
Соединение, являющееся энантиомером по отношению к соединению, указанному в заголовке примера 1 г, можно получить аналогичным путем, но при условии вместо N-(трет-бутоксикарбонил)-L-метионина использовать N-(трет-бутоксикарбонил)-D-метионин.
а) трет-Бутил-(R)-4-[2-(метилсульфанил)этил]-5-оксооксазолидинон-3-карбоксилат
Элементный анализ:
рассч.: С 50,56 Н 7,33 N 5,36 S 12,27
обнар.: С 50,30 Н 7,51 N 5,50 S 12,41
ИК: 2980, 2920, 1800, 1715, 1390, 1175, 1050.
ЯМР (CDCl3): 1,5 (9Н), 2,08 (3Н), 2,15-2,35 (2Н), 2,5-2,68 (2Н), 4,33 (1Н), 5,21 (1Н), 5,48 (1Н).
MC(EI):m/z 261,205, 188, 116, 100, 57.
6) N-метил-D-метионин
Элементный анализ:
рассч.: С 44,15 Н 8,03 N 8,58 S 19,64
обнар.: С 43,98 Н 7,81 N 8,70 S 19,82
ИК: 3430, 3010, 2920, 2830, 2420, 1630, 1580, 1395.
ЯМР (D2O): 2,13 (3Н), 2,12-2,22 (2Н), 2,55-2,68 (2Н), 2,74 (3Н), 3, 68-3,73 (1Н).
MC (El): m/z 163, 145, 118, 88, 70, 61.
CD (вода): 200 нм, Δε 1,87.
в) N-метил-S-тритил-D-гомоцистеин
Элементный анализ:
рассч.: С 73,62 Н 6,44 N 3,58 S 8,19
обнар.: С 73,45 Н 6,25 N 3,78 S 8,15
ИК: 3440,3055, 2850, 2400, 1615, 1485,1445, 1395, 740, 700.
ЯМР (CDCl3): 1,28-1,62 (2Н), 2,12(3Н), 2,25-2,45 (2Н), 2,84-2,92 (1Н), 7,12-7,41 (15Н).
MC (EI): m/z 243, 165.
г) N-(9Н-флуорен-9-илметилоксикарбонил)-N-метил-S-тритил-D-гомоцистеин
Элементный анализ:
рассч.: С 76,32 Н 5,75 N 2,28 S 5,22
обнар.: С 76,15 Н 5,58 N 2,41 S 5,30
ИК: 3440, 3055, 2950, 2600, 1740,1705, 1445, 1170, 740, 700.
ЯМР (COCl3): 1,45-2,35 (4Н), 2,65-2,73 (3Н), 4,14-4,62 (4Н), 7,13-7,81 (23Н).
MC(FAB): m/z 636, 614.
Пример 3
N-бензилоксикарбонил-N-метил-S-тритил-L-гомоцистеин
16,6 г (0,042 моля) N-метил-S-тритил-L-гомоцистеина (соединение, указанное в заголовке примера 1в) суспендируют в 90 мл воды и 100 мл ТГФ и затем добавляют 12,58 г (0,12 моля) карбоната натрия. При 10°С к суспензии по каплям добавляют 10,97 г (0,044 моля) N-бензилоксикарбонилоксисукцинимида, растворенных в 50 мл ТГФ, и в течение 3 ч перемешивают при 10°С. После этого смешивают с 80 мл воды и 130 мл этилацетата, в течение 10 мин перемешивают при комнатной температуре и с помощью лимонной кислоты значение рН устанавливают на 4. Органическую фазу дважды промывают водой ее порциями по 100 мл и один раз 100 мл раствора поваренной соли. Водные фазы один раз экстрагируют 100 мл этилацетата. Объединенные органические фазы упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 6:4). В результате получают 22,08 г (87% от теории) целевого продукта в виде твердого бесцветного пенистого вещества.
Элементный анализ:
рассч.: С 73,12 Н 5,94 N 2,66 S 6,10
обнар.: С 72,87 Н 6,08 N 2,60 S 5,98
Пример 4
N-трет-бутилоксикарбонил-N-метил-S-тритил-L-гомоцистеин
16,6 г (0,042 моля) N-метил-S-тритил-L-гомоцистеина (соединение, указанное в заголовке примера 1в) суспендируют в 90 мл воды и 100 мл ТГФ и затем добавляют 12,58 г (0,12 моля) карбоната натрия. При 10°С к суспензии по каплям добавляют 9,60 г (0,044 моля) ди-трет-бутилдикарбоната, растворенных в 50 мл ТГФ, и в течение 3 ч перемешивают при 10°С. После этого смешивают с 80 мл воды и 130 мл этилацетата, в течение 10 мин перемешивают при комнатной температуре и с помощью лимонной кислоты значение рН устанавливают на 4. Органическую фазу дважды промывают водой ее порциями по 100 мл и один раз 100 мл раствора поваренной соли. Водные фазы один раз экстрагируют 100 мл этилацетата. Объединенные органические фазы упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 6:4). В результате получают 17,14 г (83% от теории) целевого продукта в виде твердого бесцветного пенистого вещества.
Элементный анализ:
рассч.: С 70,85 Н 6,77 N 2,85 S 6,52
обнар.: С 70,68 Н 6,91 N 2,74 S 6,42
Пример 5
а) N-метил-S-(4,4′-диметокситритил)-L-гомоцистеин
В реакционный сосуд помещают 7,5 г (0,046 моля) N-метил-L-метионина (соединение, указанное в заголовке примера 1б) и по реакции конденсации при охлаждении смесью МеОН/сухой лед вводят 200 мл аммиака. Затем при -35°С порциями добавляют в течение 1 ч 5,2 г (0,23 моля) натрия и перемешивают в течение 2 ч. После этого порциями добавляют 9,7 г (0,18 моля) хлорида аммония, охлаждающую смесь удаляют и в течение ночи с помощью азота отгоняют аммиак. Далее добавляют 17,3 г (0,054 моля) бис(4-метоксифенил)фенилметанола. Затем при охлаждении льдом добавляют 50 мл дихлорметана и 90 мл трифторуксусной кислоты. Смесь в течение 1 ч перемешивают при комнатной температуре и концентрируют досуха. Остаток суспендируют в 200 мл воды и с помощью едкого натра значение рН устанавливают на 13. После 1-часового перемешивания проводят вакуум-фильтрацию и фильтрат - твердое вещество - суспендируют в 500 мл воды. Добавлением лимонной кислоты значение рН устанавливают на 4. Далее добавляют 600 мл МТБЭ и перемешивают в течение 30 мин. Затем отфильтровывают вакуум-фильтрацией и дважды промывают МТБЭ его порциями по 100 мл. Остаток смешивают со 100 мл дихлорметана, добавляют 20 мл этанола и затем по каплям добавляют 500 мл МТБЭ. После 1-часового перемешивания отфильтровывают вакуум-фильтрацией и промывают МТБЭ. После сушки получают 13,92 г (67% от теории) бесцветного твердого вещества.
Элементный анализ:
рассч.: С 69,15 Н 6,47 N 3,10 S 7,10
обнар.: С 68,98 Н 6,63 N 3,01 S 7,02
б) N-(9Н-флуорен-9-илметилоксикарбонил)-N-метил-S-(4,4′-диметокситритил)-L-гомоцистеин
18,97 г (0,042 моля) N-метил-S-(4,4′-диметокситритил)-L-гомоцистеина (соединение, указанное в заголовке примера 5а) суспендируют в 90 мл воды и 100 мл ТГФ и затем добавляют 12,58 г (0,12 моля) карбоната натрия. При 10°С к суспензии по каплям добавляют 14,88 г (0,044 моля) 20 флуоренилметилсукцинимидилкарбоната, растворенных в 50 мл ТГФ, и в течение 3 ч перемешивают при 10°С. После этого смешивают с 80 мл воды и 130 мл этилацетата, в течение 10 мин перемешивают при комнатной температуре и с помощью лимонной кислоты значение рН устанавливают на 4. Органическую фазу дважды промывают водой ее порциями по 100 мл и один раз 100 мл раствора поваренной соли. Водные фазы один раз экстрагируют 100 мл этилацетата. Объединенные органические фазы упаривают досуха в вакууме и остаток хроматографируют на силикагеле (элюент: дихлорметан/ацетон в соотношении 6:4). В результате получают 22,92 г (81% от теории) целевого продукта в виде твердого бесцветного пенистого вещества.
Элементный анализ:
рассч.: С 73,08 Н 5,83 N 2,08 S 4,76
обнар.: С 72,95 Н 5,94 N 2,01 S 4,68
Пример 6
Fmoc-(N-CH3)Hcy(Trt)-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-CITrt на твердой фазе
Полученный по описанной в WO 01/44177 (пример 1, стадии 1-3) методике и связанный с твердой фазой в качестве носителя пептид Fmoc-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-CITrt обрабатывали 75 мл смеси N-МП/ДХМ (в соотношении 1:1), содержавшей 5% пиперидина, в течение 10 мин, после чего подвергали взаимодействию с 75 мл смеси N-МП и 20% пиперидина в течение 15 мин. Твердую фазу последовательно промывали N-МП (трижды порциями по 75 мл в течение 1 мин соответственно) и ДХМ (трижды порциями по 75 мл в течение 1 мин соответственно). Анализ небольшого образца твердой фазы, проведенный с помощью нингидрина, показал, что реакция завершилась, и твердую фазу промывали 75 мл N-МП. Небольшую часть смолы-носителя обрабатывали ГФИПС (гексафторизопропиловый спирт) и анализировали с помощью ЖХВР (см. метод 1 проведения жидкостной хроматографии высокого разрешения, разработанный фирмой Diatide). Пик, относящийся к Fmoc-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-OH, по истечении 21,7 мин не был обнаружен, однако по истечении 12,7 мин был обнаружен пик, относящийся к H-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-OH. Одновременно в отдельном сосуде в 50 мл N-МП растворяли N-α-Fmoc-N-α-Meran-S-тритилгомоцистеин (9,20 г, ммолей), ГАТУ-реагент (гексафторфосфат O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония, 5,70 г, 15 ммолей) и ГОАт (1-гидрокси-7-азабензотриазол, 2,04 г, 15 ммолей). Затем к раствору защищенного N-метилгомоцистеина добавляли ДИЭА (диизопропилэтиламин, 6,96 мл, 40 ммолей). После 1-часового перемешивания этой смеси ее добавляли к твердофазной смеси. Реакцию проводили при встряхивании в течение 4 ч в слабом токе аргона. Затем раствор отделяли и твердую фазу последовательно промывали N-метилпирролидоном (трижды его порциями по 75 мл в течение 1 мин соответственно). Анализ небольшого образца твердой фазы, проведенный с помощью нингидрина, показал, что реакция завершилась. Небольшую часть смолы-носителя обрабатывали ГФИПС и анализировали с помощью ЖХВР (см. метод 1 проведения ЖХВР, разработанный фирмой Diatide). Пик, относящийся к H-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-OH по истечении 12,7 мин не был обнаружен, однако по истечении 26,7 мин был обнаружен пик, относящийся к Fmoc-(N-CH3)Hcy(Trt)-Tyr(трет-Bu)-D-Trp(Boc)-Lys(Boc)-Thr(трет-Bu)-OH.
Реферат
Настоящее изобретение относится к N-метилгомоцистеинам общей формулы I

в которой
X1 обозначает сера-защитную группу, а
Х2 обозначает азот-защитную группу.
Соединения могут быть использованы для построения содержащих N-метилгомоцистеин пептидов и пептидных промежуточных продуктов. Описан также способ получения соединений формулы I. 4 н. и 6 з.п. ф-лы.
Формула


Комментарии