Дизамещенные фураноны, тиазолы и пентеноны в качестве ингибиторов циклооксигеназы, способ их получения, фармацевтическая композиция и способ лечения - RU2131423C1
Код документа: RU2131423C1
Чертежи


































Описание
Настоящее изобретение относится к соединениям и фармацевтическим композициям, предназначенным для лечения заболеваний, опосредованных циклооксигеназой, а также к способу лечения этих заболеваний.
Нестероидные противовоспалительные средства в большинстве своем обладают противовоспалительной, болеутоляющей и жаропонижающей активностью, и ингибируют гормонально индуцированные маточные сокращения и рост некоторых типов раковых опухолей посредством подавления простогландин-G/H-синтазы, известной также как циклооксигеназа. До настоящего времени была охарактеризована лишь одна форма циклооксигеназы, которая соответствует циклооксигеназе-1 или конститутивному ферменту, первоначально идентифицированному в бычьих семенных пузырьках. Недавно из таких источников, как куриные, мышиные и человеческие ткани был секвенирован и охарактеризован ген, кодирующий вторую индуцибельную форму циклооксигеназы (циклооксигеназы-2). Этот фермент отличается от циклооксигеназы-1, которая также была клонирована, секвенирована и охарактеризована из овечьей, мышиной, и человеческой ткани. Вторая форма циклооксигеназы, а именно, циклооксигеназа-2, быстро и легко индуцируется рядом агентов, включая митогены, эндоксины, гормоны, цитокины, и факторы роста. Поскольку простагландины играют в организме как физиологическую, так и патологическую роль, мы пришли к заключению, что такой конститутивный фермент, как циклооксигеназа-1, является, в основном, ответственным за базальное эндогенное высвобождение простагландинов, а поэтому он осуществляет важные физиологические функции, такие как поддержание целостности желудочно-кишечного тракта и кровообращение в почках. И напротив, мы пришли в выводу, что индуцибельная форма фермента циклооксигеназы-2, ответственна, главным образом, за патологические действия простагландина, где быстрое индуцирование этого фермента происходит в ответ на действие таких агентов, как воспалительные факторы, гормоны, факторы роста, и цикотины. Так, например, селективный ингибитор циклооксигеназы-2 будет обладать такими же противовоспалительными, жаропонижающими и болеутоляющими свойствами, что и стандартные нестероидные противовоспалительные лекарственные средства, и кроме того, он будет ингибировать гормонально-индуцированные маточные сокращения, и обладать потенциальным противораковым действием, но при этом он будет иметь минимальную способность индуцировать некоторые из побочных эффектов, обусловленных механизмом действия известных ингибиторов. В частности, это соединение должно обладать пониженной желудочно-кишечной токсичностью, пониженной способностью вызывать почечные побочные эффекты, пониженным действием на время кровотечения, и по возможности минимальной способностью индуцировать астматические приступы у восприимчивых к аспирину людей, страдающих астмой.
Краткое описание изобретения
Настоящее изобретение относится к новым соединениям формулы I, предназначенным для лечения заболеваний, опосредованных
действием циклогексигеназы-2.
Настоящее изобретение также относится к некоторым фармацевтическим композициям и к способам лечения заболеваний, опосредованных действием циклооксигеназы-2, предусматривающим использование соединений формулы I.
Подробное описание
изобретения
Настоящее
изобретение относится к новому
соединению формулы I,
предназначенному для лечения заболеваний, опосредованных действием циклооксигеназы-2.
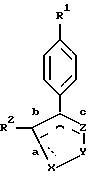
или их фармацевтически приемлемым солям,
где X-Y-Z - выбирают из группы, состоящей из
(a) -CH2CH2 CH2-,
(b) -C(O)CH2CH2 -,
(c) -CH2CH2C(O)-,
(d) -CR5(R5' )-O-C(O)-,
(e) -C(O)-O-CR5(R5' )-.
(f) -CH2NR3-CH2-,
(g) -CR5(R5')-NR3
-C(O)-,
(h) -CR4=CR4'-S-,
(i)
-S-CR4=CR4'-,
(j) -S-N=CH-,
(k) -CH=N-S-,
(l) -N=CR4-O-,
(m) -O-CR4=N-,
(n)
-N=CR4-NH-,
(o) -N=CR4-S-,
(p) -S-CR4=N-,
(q) -C(O)-NR3-CR5(R5'
)-,
(r) -R3-CH=CH-,
при условии, что R1 не
является -S(O)2Me,
(s)
=CH=CH-NR3-, при условии что R1 не является -S(O)2Me,
в случае, когда сторона b
является двойной связью, а стороны
a и c являются простыми связями; и
X-Y-Z - выбирают из группы, включающей в себя
(a) = CH-O-CH=
(b) = CH-NR3-CH=,
(c) = N-S-CH=,
(d) =CH-S-N=,
(e) =N-O-CH=,
(f) =CH-O-N=,
(g) =N-S-N=,
(h) =N-O-N=,
в случае, когда
стороны a и c являются двойными связями, а
сторона b является простой связью; и
R1 выбирают из группы,
включающей в себя
(a) S(O)2CH3,
(b)
S(O)2NH2,
(c) S(O)2NHC(O)CF3,
(d) S(O)(NH)CH3,
(e) S(O)(NH)NH2,
(f) S(O)(NH)NHC(O)CF3,
(g) P(O)(CH3)OH,
и
(h) P(O)(CH3)NH2;
R2 выбирают из
группы, включающей в себя
(a) C1-6-алкил,
(b) C3, C4, C5, C6 и C7
-циклоалкил,
(c) моно-, ди- или
тризамещенный фенил или нафтил, где заместителя выбирают из группы, включающей
в себя
(1) водород,
(2) галоген,
(3) C1-6
-алкокси,
(4) C1-6
-алкилтио,
(5) CN,
(6) CF3,
(7) C1-6-алкил,
(8) N3,
(9) -CO2H,
(10) -CO2-C1-4
-алкил,
(11) -C(R5)(R6)-OH,
(12)
-C(R5)(R6)-O-C1-4-алкил, и
(13) -C1-6
-алкил-CO2-R5;
(d) моно-, ди- или тризамещенный гетероарил, где этот
гетероарил представляет собой моноциклическое
ароматическое кольцо из 5 атомов, причем указанное
кольцо из 5 атомов, причем, указанное
кольцо имеет один гетероатом, являющийся S, O или N, и
необязательно, еще 1, 2 или 3 атома N; либо этот
гетероарил представляет собой моноциклическое кольцо из 6
атомов, причем, указанное кольцо имеет
один гетероатом, N, и необязательно, еще 1, 2, 3
или 4 атома N; а заместителей выбирают из группы,
включающей в себя
(1) водород,
(2) галоген,
включая фтор, хлор, бром и иод,
(3) C1-6-алкил,
(4)
C1-6-алкокси,
(5) C1-6
-алкилтио,
(6) CN,
(7) CF3,
(8) N3,
(9) -C(R5)(R5)-OH, и
(10) -C(R5)(R6)-O-C1-4-алкил;
(e) бензогетероарил, который представляет собой
конденсированные с бензолом аналоги (d);
R3 выбирают из
группы, включающей в себя
(a) водород,
(b) CF3,
(c) CN,
(d) C1-6
-алкил,
(e) гидрокси-C1-6-алкил,
(А)
-C(O)-C1-6-алкил,
(g) необязательно
замещенные
(1) -C1-5-алкил-Q,
(2) -C1-3-алкил-O-C1-3-алкил-Q,
(3) -C1-3
-алкил-S-C1-3-алкил-Q,
(4) -C1-5-алкил-O-Q, или
(5) -C1-5-алкил-S-Q,
где заместитель находится на алкиле, и
этим заместителем является
C1-3-алкил;
(h) -Q;
R4 и R4', каждый независимо выбирают из группы, включающей в
себя
(a) водород,
(b)
CF3,
(c) CN,
(d) C1-6-алкил,
(e)
-Q,
(f) -O-Q,
(g) -S-Q, и
(h) необязательно
замещенные
(1) C1-5-алкил-Q,
(2)
-O-C1-5-алкил-Q,
(3) -S-C1-5
-алкил-Q,
(4) -C1-3-алкил-O-C1-3-алкил-Q,
(5) -C1-3-алкил-S-C1-3
-алкил-Q,
(6) -C1-5-алкил-O-Q,
(7) -C1-5-алкил-S-Q,
где заместитель находится на алкиле, и этим
заместителем является C1-3-алкил; R5, R5', R6, R7 и R8,
каждый независимо выбирают из группы, включающей в себя
(a) водород,
(b) C1-6-алкил,
либо
R5
и R6 или R7 и R8,
взятые вместе с атомом углерода, с которым они связаны, образуют насыщенное
моноциклическое углеродное кольцо из 3, 4, 5, 6 или 7
атомов;
Q представляет собой CO2H, CO2
-C1-4--алкил, тетразолил-5-ил, C(R7)(R8
)(OH), или C(R7)(R8)(O-C1-4
-алкил);
при условии, что если X-Y-Z представляет собой
-S-CR4=CR4', то R4 и R4' не
являются CF3.
В одном из аспектов
своего
осуществления, настоящее изобретение относится к
соединениям формулы I:
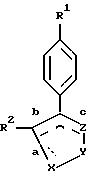
или их фармацевтически приемлемым солям, где X-Y-Z выбирают из группы, состоящей из -C(O)-O-CR5(R5'), в том случае, когда сторона b является двойной связью, а стороны a и c являются простыми связями; и
R1 выбирают из группы, включающей в себя
(a) S(O)2CH3,
(b) S(O)2NH2,
R2 выбирают из группы, включающей в себя
(a) C1-6 -алкил,
(b) C3, C4, C5, C6 и C7, циклоалкил,
(c) гетероарил,
(d) бензогетероарил,
(e) моно- или дизамещенный фенил, где заместителя выбирают из группы, включающей в себя
(1) водород,
(2) галоген,
(3) C1-6 -алкокси,
(4) C1-6-алкилтио,
(5) CN,
(6) CF3,
(7) C1-6-алкил,
(8) N3,
(9) -CO2H,
(10) -CO2-C1-4-алкил,
(11) -C(R5)(R6)-OH,
(12) -C(R5)(R6)-O-C1-4-алкил, и
(13) -C1-6 -алкил-CO2-R5;
R5, R5' и R6, каждый независимо выбирают из группы, включающей в себя
(a) водород,
(b) C1-6 -алкил, либо R5 и R6, взятые вместе с атомом углерода, с которым они связаны, образуют насыщенное моноциклическое углеродное кольцо из 3, 4, 5, 6 или 7 атомов;
Одним из видов в вышеописанном осуществлении изобретения является соединение формулы I,
где X-Y-Z - выбирают из группы, включающей в себя
(a) -CH2 CH2CH2-,
(b) -C(O)CH2CH2 -,
(c) -CH2CH2C(O)-,
(d) -CR5(R5')-O-C(O)-,
(e) -C(O)-O-CR5(R5' )-,
(f) -CH2NR3-CH2-,
(g) -CR5(R5')-NR3-C(O)-,
(h) -CR4 =CR4'-S-,
(i) -N-CR4=CR4'-,
(j) -S-N=CH-,
(k) -CH=N-S-,
(l) -N=CR4-O-,
(m) -O-CR4=N-,
(n) -N=CR4-NH-,
(o) -N=CR4-S-, и
(p) -S-CR4=N,
(q) -C(O)-NR3-CR5(R5'-,
(r) -R3-CH=CH-, при условии, что R1 не является -S(O)2Me,
(s) -CH=CH-NR3-, при условии, что R1 не является -S(O)2Me,
Описанный вид соединений включает в себя подвид соединений формулы I, где
R1 выбирают из группы, включающей в себя
(a) S(O)2 CH3,
(b) S(O)2 NH2,
(c) S(O)2 NHC(O)CF3,
(d) S(O)NHCH3,
(e) S(O)NHNH2,
(f) S(O)NHNHC(O)CF3,
R2 выбирают из группы, включающей в себя
(a) C1-4-алкил,
(b) C3, C4, C5, C6 и C7-циклоалкил,
(c) моно- или дизамещенный фенил, где заместитель выбирают из группы, включающей в себя
(1) водород,
(2) фтор, хлор и бром,
(3) C1-4-алкокси,
(4) C1-4-алкилтио,
(5) CN,
(6) CF3,
(7) C1-4-алкил,
(8) N3,
(9) -CO2H,
(10) -CO2-C1-3-алкил,
(11) -C(R5)(R6)-OH, и
(12) -C(R5)(R6)-O-C1-3-алкил,
(d) моно- или дизамещенный гетероарил, выбранный из группы, включающей в себя
(1) фуранил,
(2) диазинил, трианизил, и тетразинил,
(3) имидазолил,
(4) изооксазолил,
(5) изотиазолил,
(6) оксадиазолил,
(7) оксазолил,
(8) пиразолил,
(9) пирролил,
(10) тиадиазолил,
(11) тиазолил,
(12) тиенил,
(13) триазолил, и
(14) тетразолил,
где указанных заместителей выбирают из группы, включающей в себя
(a) водород,
(b) фтор, хлор, бром,
(c) C1-4-алкокси,
(d) C1-4-алкилтио,
(e) CN,
(f) CF3,
(g) C1-4 -алкил,
(h) N3,
(i) -C(R5 )(R6)-OH,
()j) -C(R5)(R6)-O-C1-4--алкил.
Внутри этого подвида
имеется класс соединений формулы I, где R2 выбирают из
группы включающей в себя
(a) циклогексил, и
(b) моно- или дизамещенный фенил, где заместителей
выбирают из группы,
включающей в себя
(1) водород,
(2) галоген,
(3) C1-4-алкокси,
(4) C1-4-алкилтио,
(5) CN,
(6) CF3,
(7)
C1-4-алкил,
(8) N3, и
(9)
-C(R5)(R6
)-OH;
R3 выбирают из группы, включающей в себя
(a)
водород,
(b)
CF3,
(c) C1-3-алкил и гидрокси
C1-3-алкил,
(d) CN;
R4 и R4', каждый независимо выбирают из
группы, включающей в
себя
(a) водород,
(b) CF3,
(c) C1-3-алкил,
(d) CN,
(e) хлор и фтор; и
R5, R5',
R6, каждый
независимо выбирают из группы, включающей в себя
(a)
водород,
(b) метил или
этил,
либо R5 и R6, взятые вместе с атомом углерода,
с которым они
связаны, образуют насыщенное углеродное кольцо из 4, 5 или 6
атомов.
Внутри этого
класса имеется подкласс соединений формулы I, где X-Y-Z - выбирают из группы, включающей
в себя
(a) -CH2-O-C(O)-,
(b) -C(O)-O-CH2-, и
(c) -CH2-NR3-C(O)-,
R1 выбирают из группы, включающей в себя
(a)
S(O)2CH3,
(b) S(O)2
NH2,
(c)
S(O)NHCH3, и
(d) S(O)NHNH2;
R2 выбирают из группы,
включающей в себя
моно- или дизамещенный фенил, где
заместителей выбирают из группы,
включающей в себя
(1) водород,
(2) галоген, выбранный из фтора, хлора и брома,
(3) C1-3-алкокси,
(4) C1-3
-алкилтио,
(5) CN, и
(6) C1-3-алкил;
R3 выбирают из группы, включающей в себя
(a)
водород,
(b) CF3,
(c) C1-3-алкил и гидрокси C1-3-алкил,
Внутри этого подкласса имеется группа соединений формулы I,
где X-Y-Z
- выбирают из группы, включающей в себя
(a) -CH2-O-C(O)-, и
(b)
-C(O)-O-CH2-, и
R1 выбирают из группы, включающей в себя
(a)
S(O)2CH3,
(b) S(O)2
NH2,
(c)
S(O)NHCH3, и
(d) S(O)NHNH2;
R2 представляет собой
моно- или дизамещенный фенил, где заместителей выбирают из
группы, включающей в себя
(1) водород,
(2) галоген, выбранный из фтора, хлора и брома,
(3) метокси, и
(4)
метил.
Эта группа может быть более
конкретно определена как
соединения формулы I, где
X-Y-Z - выбирают из группы, включающей в себя
(a) -CH2-O-C(O)-,
и
(b) -C(O)-O-CH2-, и
R1 выбирают из
группы, включающей в себя
(a) S(O)2CH3, и
(b) S(O)2NH2;
R2 представляет собой
моно- или
дизамещенный фенил,
где заместителей выбирают из группы, включающей в себя
(1) водород,
(2) галоген, выбранный из фтора,
хлора и
брома.
Внутри этого подвида,
описанного выше, имеется
класс соединений формулы I, где
R2 представляет собой моно- или дизамещенный гетероарил, где
гетероарил
выбирают из группы, включающей в себя
(1)
фуранил,
(2)
диазинил, трианизил, и тетразинил,
(3) имидазолил,
(4) изооксазолил,
(5) изотиазолил,
(6)
оксадиазолил,
(7) оксазолил,
(8)
пиразолил,
(9) пирролил,
(10) тиадиазолил,
(11) тиазолил,
(12) тиенил,
(13) триазолил,
и
(14)
тетразолил,
где заместителей выбирают из
группы, включающей
в себя
(a) водород,
(b) фтор или хлор,
(c) C1-3-алкокси,
(d) C1-6
-алкилтио,
(e) CN,
(f) CF3,
(g)
C1-3-алкил,
(h) -C(R5)(R6)-OH,
(i) -C(R5)(R6
)-O-C1-4-алкил.
Внутри этого класса
имеется подкласс
соединений формулы I, где
R2 представляет собой моно- или дизамещенный гетероарил, где гетероарил
выбирают из
группы, включающей в себя
(1) 2-фуранил,
(2)
3-фуранил,
(3) 2-тиенил,
(4) 3-тиенил,
(5) 3-изоксазолил,
(6) 4-изоксазолил,
(7)
5-изоксазолил,
(8) 3-изотиазолил,
(9)
4-изотиазолил,
(10) 5-изотиазолил,
(11) 2-оксазолил,
(12) 4-оксазолил,
(13) 5-оксазолил,
(14)
2-тиазолил,
(15) 4-тиазолил,
(16)
5-тиазолил,
(17) 1,2,3-тиадиазол-4-ил,
(18) 1,2,3-тиадиазол-5-ил,
(19) 1,2,4-тиадиазол-3-ил,
(20) 1,2,
4-тиадиазол-5-ил,
(21) 1,3,4-тиадиазол-2-ил,
(22) 1,2,
5-тиадиазол-3-ил,
(23) 1,2,3-оксадиазол-4-ил,
(24) 1,2,3-оксадиазол-5-ил,
(25) 1,2,
4-оксадиазол-3-ил,
(26) 1,2,4-оксадиазол-5-ил,
(27) 1,3,
4-оксадиазол-2-ил,
(28) 1,2,5-оксадиазол-3-ил,
(29) пиразол-4-ил,
(30) пиразол-5-ил,
(31) 1,2,
3-триадиазол-4-ил,
(32) 1,2,3-триадиазол-5-ил,
(33)
1,2,4-триадиазол-3-ил,
(34) 1,2,4-триадиазол-5-ил,
(35) 1,2-диазинил,
(36) 1,3-диазинил,
(37)
1,4-диазинил,
(38) 1,2,3,4-тетразин-5-ил,
(39) 1,2,
3,5-тетразин-4-ил,
(40) 1,3,4,5-тетразин-2-ил, и
(41) 1,2,3,5-тетразин-4-ил,
Внутри этого
подкласса
имеется группа соединений формулы I, где гетероарил выбирают
из группы,
включающей в себя
(1) 3-изоксазолил,
(2) 4-изоксазолил,
(3) 5-изоксазолил,
(4)
3-изотиазолил,
(5) 4-изотиазолил,
(6) 5-изотиазолил,
(7)
2-оксазолил,
(8) 4-оксазолил,
(9) 5-оксазолил,
(10) 2-тиазолил,
(11)
4-тиазолил,
(12)
5-тиазолил,
(13) 1,2,3-тиадиазол-4-ил,
(14) 1,2,
3-тиадиазол-3-ил,
(15) 1,2,4-тиадиазол-3-ил,
(16) 1,2,4-тиадиазол-5-ил,
(17) 1,
3,4-тиадиазол-2-ил,
(18) 1,2,5-тиадиазол-3-ил,
(19) 1,2,
3-оксадиазол-4-ил,
(20) 1,2,3-оксадиазол-5-ил,
(21) 1,2,4-оксадиазол-3-ил,
(22) 1,2,
4-оксадиазол-5-ил,
(23) 1,3,
4-оксадиазол-2-ил,
(24) 1,2,
5-оксадиазол-3-ил,
(25)
1,2-диазинил,
(26) 1,3-диазинил, и
(27) 1,4-диазинил.
Указанные гетероарилы могут быть более
конкретно определены как гетероарилы,
выбранные из группы, включающей в
себя
(1) 3-изотиазолил,
(2) 4-изотиазолил,
(3) 5-изотиазолил,
(4) 2-оксазолил,
(5)
4-оксазолил,
(6)
5-оксазолил,
(7) 2-тиазолил,
(8) 4-тиазолил,
(9) 5-тиазолил,
(10) 1,2-диазинил,
(11) 1,
3-диазинил, и
(12) 1,4-диазинил,
и
где
заместителей выбирают из группы, включающей в себя
(1) водород,
(2) фтора или хлор,
(3) C1-3-алкокси,
(4) C1-3-алкилтио,
(5) CN,
(6) C1-3-алкил и
(7) -C(R5)(R6)-OH,
где R5 и R6 независимо
представляют собой водород, метил или
этил.
И кроме того, если учесть эти более конкретные
определения гетероарила, то соединения формулы I могут быть представлены как соединений,
где
X-Y-Z - выбирают из группы,
включающей в себя
(a) -CH2-O-C(O)-,
(b)
-C(O)-O-CH2-, и
(c) -CH2-NR3-C(O)-,
R1 выбирают из группы, включающей
в себя
S(O)2CH3,
(b)
S(O)2NH2,
(c) S(O)NHCH3, и
(d)
S(O)NHNH2;
R3 выбирают из
группы,
включающей в себя
(a) водород,
(b)
CF3,
(c) C1-3-алкил и гидрокси-C1-3
-алкил,
(d) CN.
Вторым видом в
вышеописанном осуществлении изобретения являются соединения
формулы I, где
X-Y-Z - выбирают из группы, включающей в себя
(a)
=CH-O-CH=,
(b) =CH-NR3-CH=,
(c) =N-S-CH=,
(d) =CH-S-N=,
(e) =N-O-CH=,
(f) =CH-O-N=,
(g) =N-S-N=,
(h) =N-O-N=,
Внутри этого вида имеется подвид соединений формулы I,
где
R1 выбирают из группы, включающей в себя
(a) S(O)2CH3,
(b) S(O)2NH2,
(c) S(O)2NHC(O)CF3,
(d) S(O)(NH)CH3,
(e)
S(O)(NH)NH2,
(f) S(O)(NH)NHC(O)CF3;
R2 выбирают из группы, включающей в себя
(a) C1-4-алкил,
(b) C3, C4, C5, C6 и C7, циклоалкил,
(c)
моно- или дизамещенный фенил, где заместителя выбирают
из группы,
включающей в себя
(1) водород,
(2)
фтор, хлор или бром,
(3) C1-4-алкокси,
(4) C1-4-алкилтио,
(5) CN,
(6)
CF3,
(7) C1-4-алкил,
(8) N3,
(9) -CO2H,
(10) -CO2-C1-3-алкил,
(11) -C(R5
)(R6)-OH, и
(12) -C(R5)(R6)-O-C1-3-алкил,
(d) моно- или дизамещенный гетероарил, выбранный
из группы, включающей в себя
(1)
фуранил,
(2) диазинил,
триазинил, и тетразинил,
(3) имидазолил,
(4) изооксазолил,
(5) изотиазолил,
(6)
оксадиазолил,
(7) оксазолил,
(8) пиразолил,
(9) пирролил,
(10) тиадиазолил,
(11)
тиазолил,
(12) тиенил,
(13) триазолил, и
(14)
тетразолил,
где заместителей
выбирают из группы, включающей в себя
(a) водород,
(b) фтор, хлор,
бром,
(c) C1-4алкокси,
(d) C1-4
-алкилтио,
(e) CN,
(f)
CF3,
(g) C1-4-алкил,
(h) N3,
(i) -C(R5)(R6)-OH,
(j) -C(R5)(%6)-O-C1-4
-алкил.
В целях настоящего описания, гетероарилы этого подвида могут быть более конкретно определены как гетероарилы, определенные в любом из вышеописанных вариантов.
Внутри этого подвида имеется класс соединений формулы
I, где
R2
выбирают из группы, включающей в себя
(a) циклогексил, и
(b) моно- или дизамещенный фенил, где
заместителей выбирают из группы, включающей в себя
(1) водород,
(2)
галоген,
(3) C1-4алкокси,
(4) C1-4
-алкилтио,
(5) CN,
(6) CF3,
(7) C1-4-алкил,
(8) N3, и
(9) -C(R5)(R6)-OH;
R3
выбирают из группы, включающей в
себя
(a) водород,
(b) CF3,
(c) C1-3-алкил и
гидрокси-C1-3-алкил,
(d) CN;
R5,
R5', R6,
каждый независимо выбирают из группы, включающей в себя
(a) водород,
(b)
метил или этил,
либо R5 и R6, взятые вместе с
атомом углерода, с которым
они связаны, образуют насыщенное углеродное кольцо и 4, 5 или 6
атомов.
Внутри
этого класса имеется подкласс соединений формулы I, где
X-Y-Z
- выбирают из группы,
включающей в себя
(a) =CH-O-CH=,
(b) =N-S-N=,
(c)
=N-O-N=,
R1 выбирают из группы, включающей в себя
(a) S(O)2
CH3, и
(b) S(O)2NH2;
R2 выбирают из
группы, включающей в себя
моно- или дизамещенный фенил, где заместителей выбирают из группы,
включающей в себя
(1) водород,
(2) галоген, выбранный из фтора, хлора и брома,
(3) C1-3алкокси,
(4) C1-3-алкилтио,
(5) CF3,
(6) C1-3-алкил,
R3 выбирают из группы, включающей в себя
(a) водород,
(b) CF3,
(c) C1-3-алкил и
гидрокси C1-3-алкил,
(d) CN;
R5 и R6, каждый независимо выбирают из
группы, включающей в
себя
(a) водород,
(b) метил или этил,
либо R5,
R5' и R6, взятые вместе с атомом углерода, с которым они связаны,
образуют насыщенное
углеродное кольцо из 5, 6 или 7 атомов.
Используемый в настоящем описании
термин "алкил" означает линейные, разветвленные и циклические структуры, где C1-6-алкил
представляет собой метил, этил, пропил, 2-пропил, втор- и
трет-бутил, бутил, пентил, гексил, 1,
1-диметилэтил, циклопропил, циклобутил, циклопентил, или циклогексил. Аналогично, C1-6
-алкокси означает алкоксигруппы прямой, разветвленной или
циклической конфигурации с 1-6 атомами
углерода. Примерами низших алкоксигрупп являются метокси, этокси, пропокси, изопропокси,
циклопропокси,
циклогексилокси и т.п. Понятие "C1-6
-алкилтио" означает алкилтиогруппы прямой,
разветвленной или циклической конфигурации, имеющие от 1 до 6 атомов углерода. Примерами низших
алкилтиогрупп являются метилтио, пропилтио, изопропилтио,
циклогептилтио и т.п. В качестве
иллюстрации, пропилтио-группа означает
-SCH2CH2CH3.
Гетероарилом являются фуран, тиофен, пиррол, изоксазол, изотиазол, пиразол, оксазол, тиазол, имидазол, 1,2,3-оксадиазол, 1,2,3-тиадиазол, 1,2,3-триазол, 1,3,4-оксадиазол, 1,3,4-тиадиазол, 1,3, 4-триазол, 1,2,5-оксадиазол, 1,2,5-тиадиазол, пиридин, пиридазин, пиримидин, пиразин, 1,2, 4-триазин, 1,3,5-триазин, 1,2,4,5-тетразин и т.п.
Бензогетероарил представляет собой вышеописанные гетероарильные кольца, с которыми может быть конденсировано бензольное кольцо.
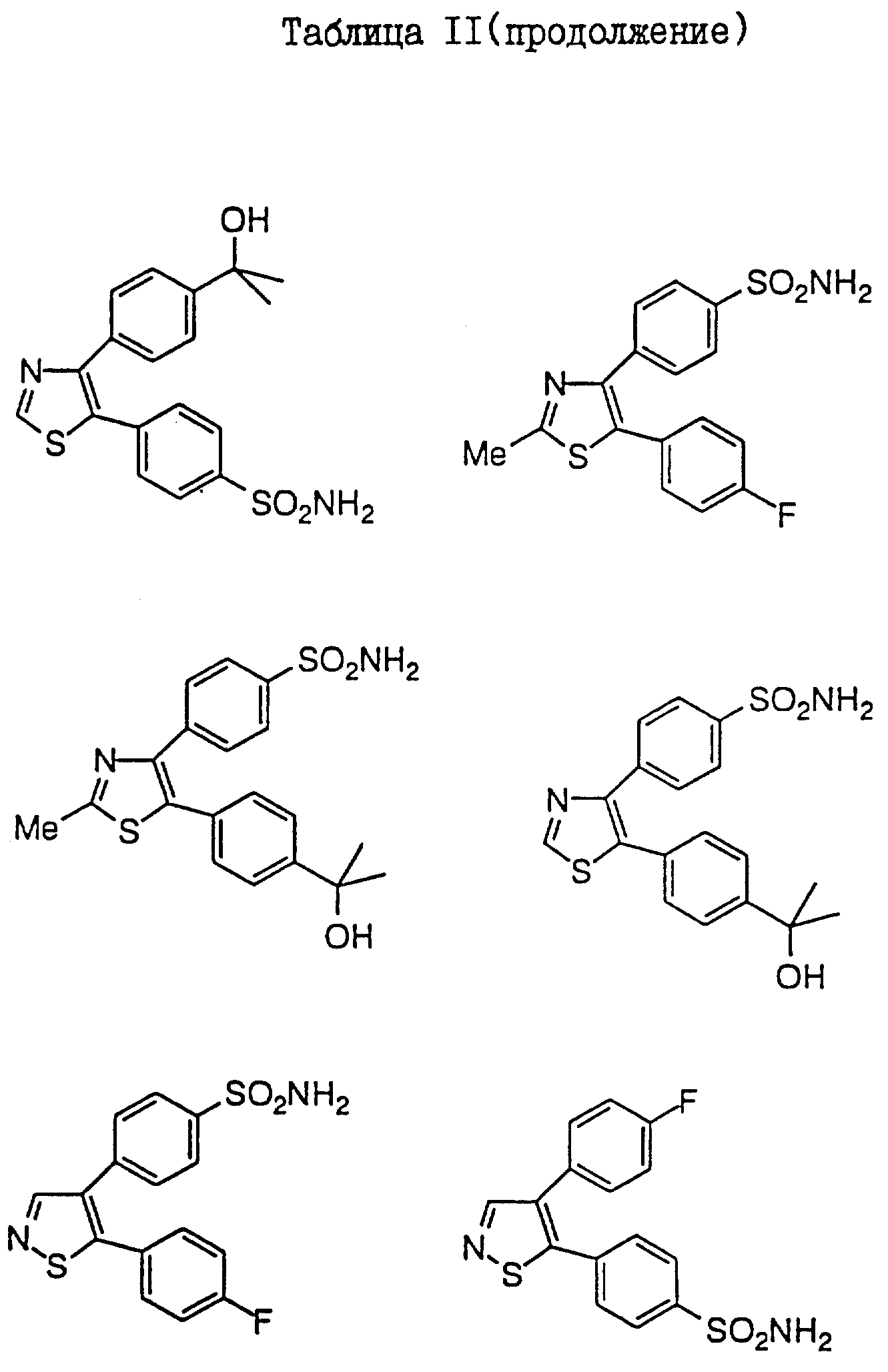
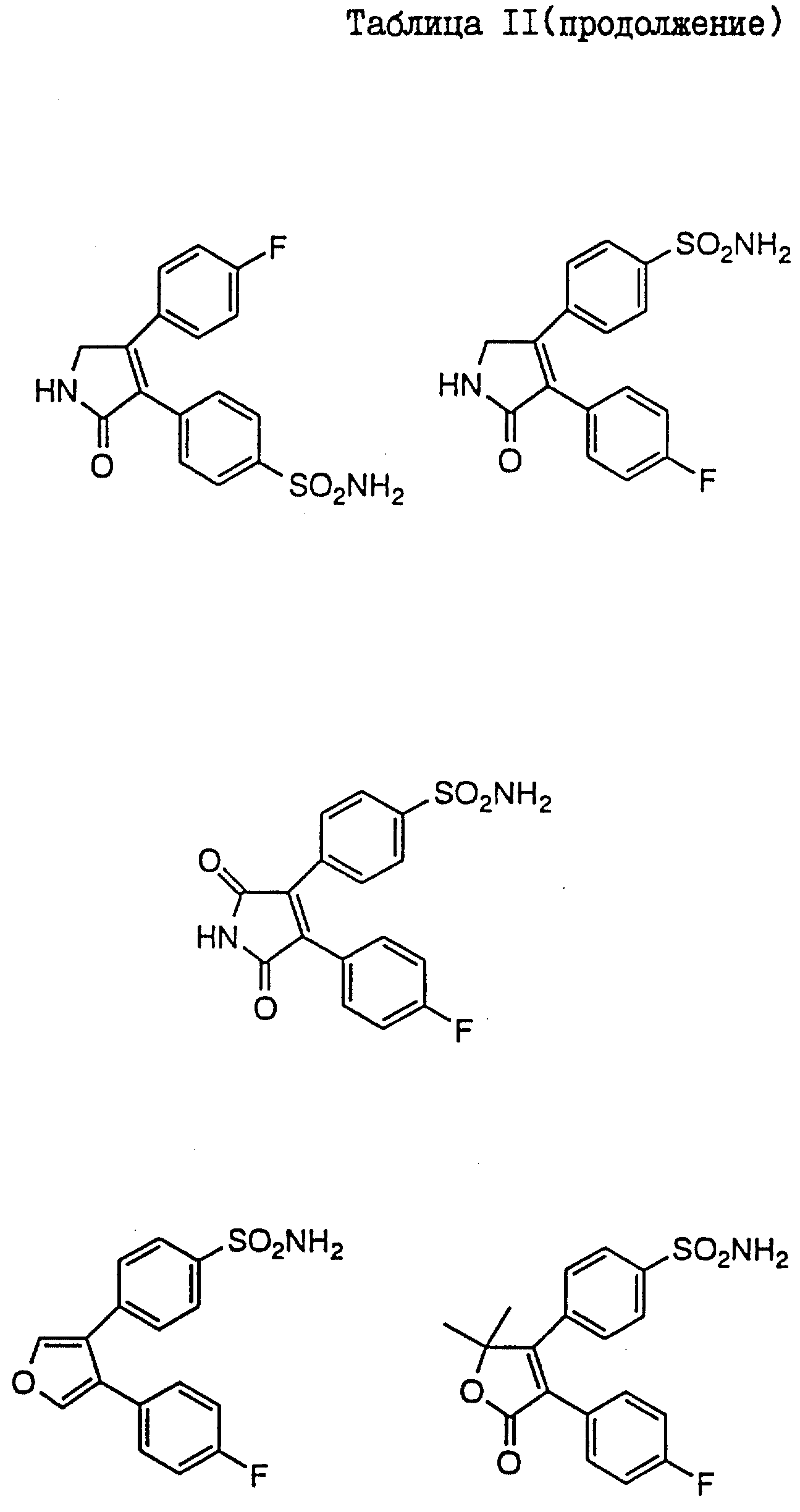
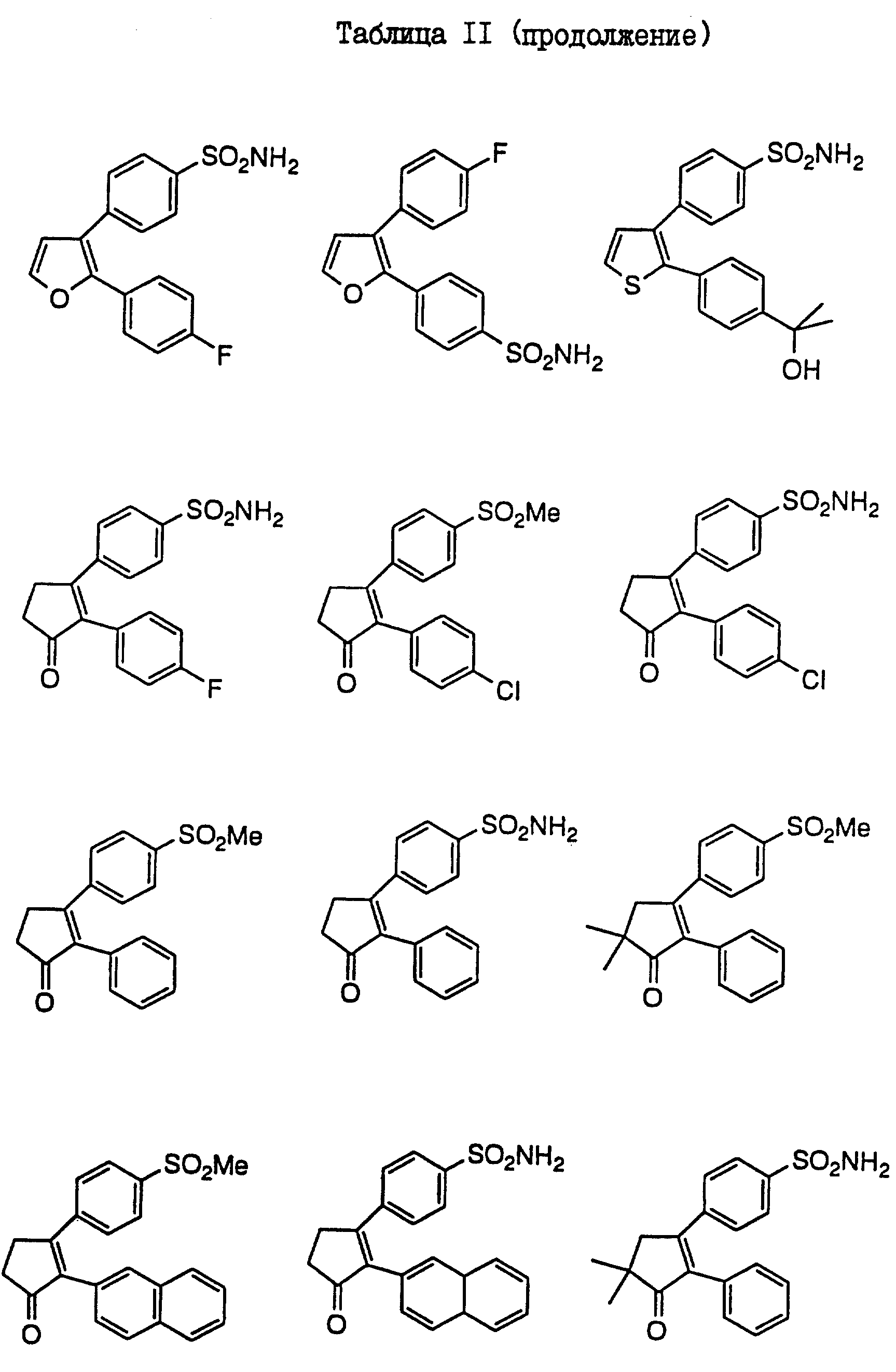
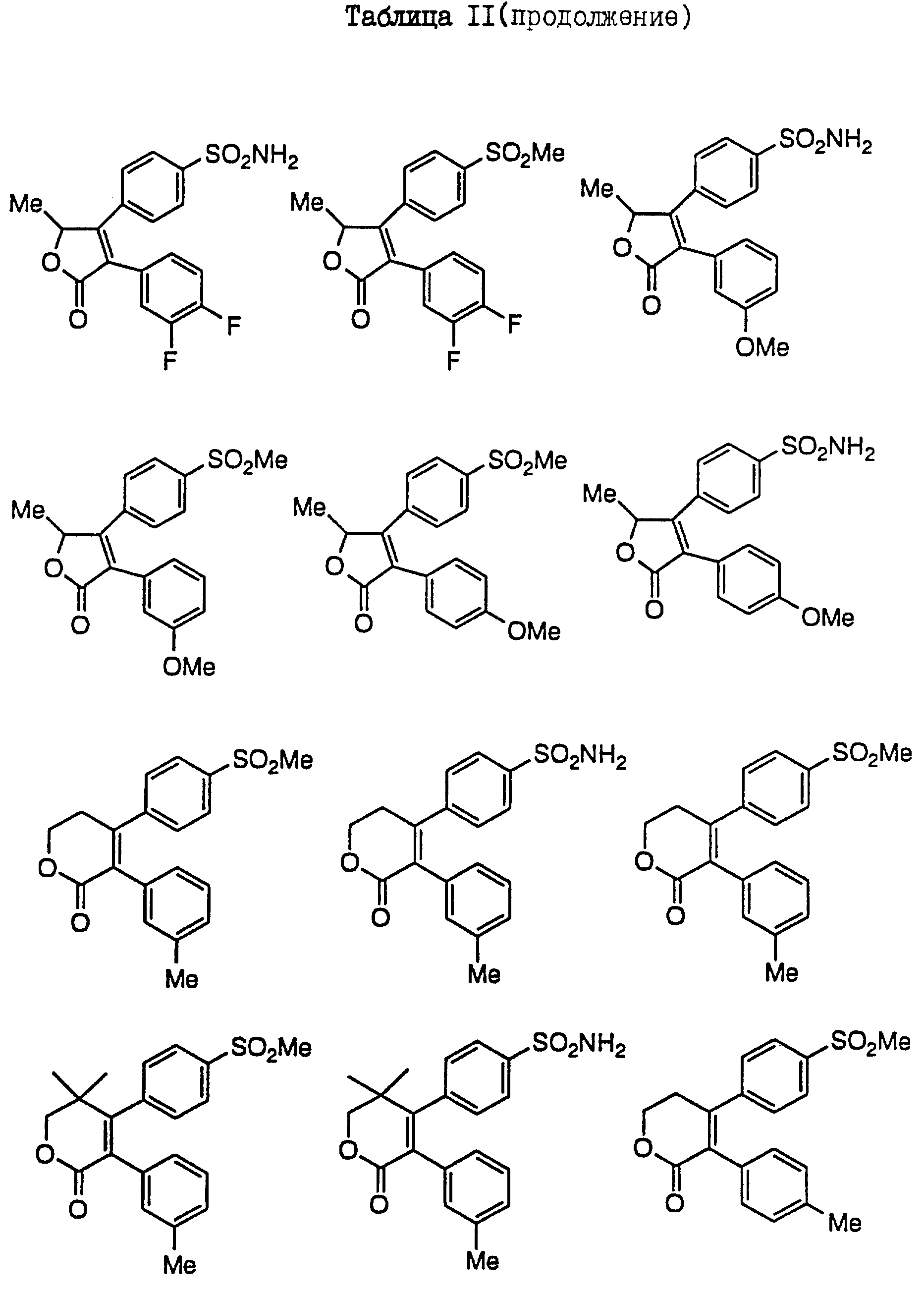
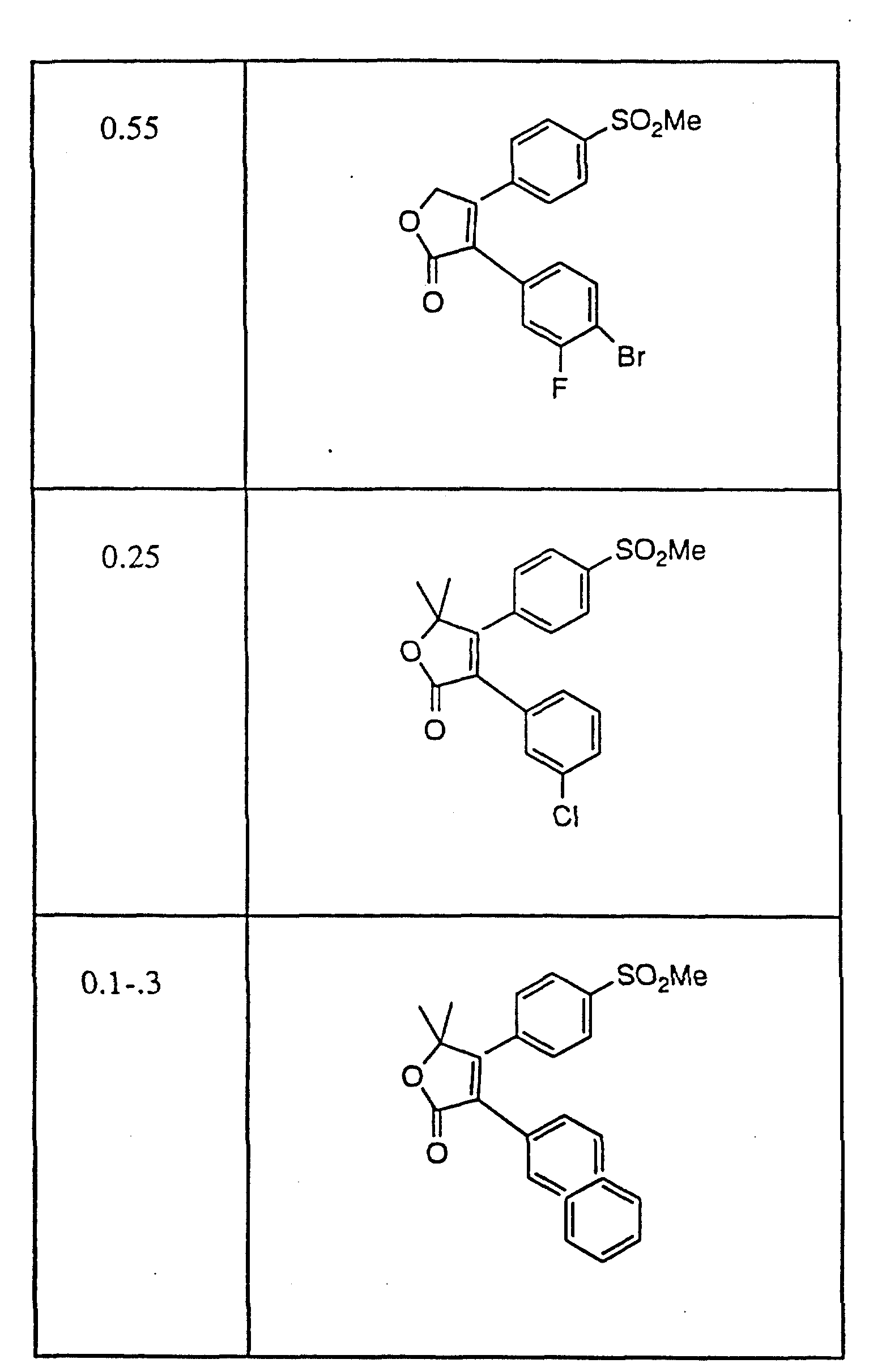
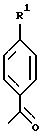
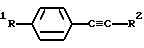
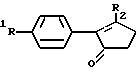
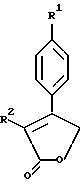
Примерами соединений настоящего изобретения являются:
(a)
3-(4-(Аминосульфонил)фенил)-2-(4-фторфенил)-5- (2-гидрокси-2-пропил)тиофен;
(b)
3-(4-(Аминосульфонил)фенил)-2-(4-фторфенил)тиофен;
(c)
3-(4-(Аминосульфонил)фенил)-2-(4-фторфенил)-5-(2-пропил) тиофен;
(d)
3-(4-(Аминосульфонил)фенил)-2-циклогексилтиофен;
(e) 5-(4-(Карбоксифенил)-4-(4-(метилсульфонил)фенил)
тиофен-2-карбоновая кислота;
(f)
4-(4-(Фторфенил)-2-метил-5-(4-(метилсульфонил)фенил) тиазол;
(g)
2-(4-(Фторфенил)-3-(4-(метилсульфонил)фенил)-2-циклопентен- 1-он;
(h)
4-(4-(Метилсульфонил)фенил)-5-(4-фторфенил)изотиазол;
(i)
3-(4-(Фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)- фуранон;
(j)
3-(4-Фторфенил)-4-(4-(аминосульфонил)фенил)-2-(5H)- фуранон;
(k) 3-(4-Фторфенил)-4-(4-(метилсульфонил)фенил)фуран;
(l) 5,
5-Диметил-3- (4-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон;
(m) 2-(4-(Аминосульфонил)фенил)-3-(4-фторфенил)тиофен;
(n)
3-(4-(Трифторацетиламиносульфонил)фенил)-2-(4-фторфенил) тиофен;
(o) 3-(3-Фторфенил)-4-(4-метилсульфонил)фенил)-2-(5H)- фуранон;
(p) 5,
5-Диметил-3-(3-фторфенил)-4-(4-метилсульфонил)фенил)-2-(5H)- фуранон;
(q) 5,5-Диметил-3-(3-хлорфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(r) 3-(3,
4-Дифторфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(s) 3-(3,4-Дихлорфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(t) 5,5-Диметил-3-(3,
4-дифторфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(u) 5,5-Диметил-3-(3,4-дихлорфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(v) 5,
5-Диметил-3-(4-хлорфенил)-4-(4-метилсульфонил)фенил)-2- (5H)-фуранон;
(w) 3-(2-Нафтил)-4-(4-(метилсульфонил)фенил)-2- (5H)-фуранон;
(x) 5,
5-Диметил-3-(2-нафтил)-4-(4-(метилсульфонил)фенил)-2- (5H)-фуранон;
(y) 3-Фенил-4-(метилсульфонил)фенил)-2-(5H)-фуранон.
Другим примером соединений настоящего изобретения
являются:
(a) 3-(3,4-Дифторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)- фуранон, и
(b) 3-(Фенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
или их фармацевтически
приемлемые
соли
Некоторые из соединений, описанные в настоящей заявке, содержат один или несколько асимметричных центров, а поэтому могут существовать в виде диастереомеров и оптических
изомеров.
Следует отметить, что соединения настоящего изобретения могут присутствовать в виде диастереомеров, их рацемических смесей, и разделенных энантиомерно чистых форм, а также в виде их
фармацевтически
приемлемых солей.
Некоторые соединения, описанные в настоящей заявке, содержат олефиновые двойные связи, и если это не оговорено особо, они могут присутствовать как в виде E-, так и в виде Z-изомеров.
Во втором варианте своего осуществления, настоящее изобретение относится к фармацевтическим композициям, предназначенным для ингибирования циклооксигеназы и для лечения опосредованных циклооксигеназой заболеваний; и содержащим фармацевтически приемлемый носитель и нетоксичное терапевтически эффективное количество соединения формулы I, описанного выше.
В этом варианте настоящее изобретение относится к фармацевтическим композициям, предназначенным для ингибирования циклооксигеназы-2 и для лечения опосредованных циклооксигеназой-2 заболеваний, как описано ниже, и содержащим фармацевтически приемлемый носитель и нетоксичное терапевтическое эффективное количество соединения формулы I, описанного выше.
В третьем варианте своего осуществления, настоящее изобретение относится к способу ингибирования циклооксигеназы и для лечения опосредованных циклооксигеназой заболеваний, эффективно проводимого с использованием активного агента, который избирательно ингибируют циклооксигеназу-2 (СОХ-2), отдавая ей предпочтение перед СОХ-1, как описано ниже; причем, указанный способ предусматривает введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I, описанного выше.
В настоящем описании считается, что соединение селективно ингибирует СОХ-2, отдавая ей предпочтение перед СОХ-1, в том случае, если концентрация IC50 для ингибирования СОХ-1 в 100 или более раз превышает концентрацию IC50 для ингибирования СОХ-2.
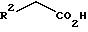
Фармацевтические композиции настоящего изобретения содержат соединение формулы I или его фармацевтически приемлемую соль в качестве активного ингредиента, а также могут содержать фармацевтически приемлемый носитель, и, необязательно, другие терапевтические ингредиенты. Термин "фармацевтически приемлемые соли" относится к солям, полученным из фармацевтически приемлемых нетоксичных оснований, включая неорганические основания и органические основания. Солями, происходящими от неорганических оснований, являются соли алюминия, аммония, кальция, меди, железа(3), железа(2), лития, магния, марганца(3), марганца(2), калия, натрия, цинка и т.п. Особенно предпочтительными являются соли аммония, кальция, магния, калия и натрия. Солями, происходящими от фармацевтически приемлемых органических нетоксичных оснований, являются соли первичных, вторичных и третичных аминов; замещенных аминов, включая природные замещенные амины; циклических аминов, и основных ионообменных смол, таких как аргинин, бетаин, кофеин, холин, N,N-дибензилэтилендиамин, диэтиламин, 2-диэтиламиноэтанол, 2-диметиламиноэтанол, этаноламин, этилендиамин, N-этилморфолин, N-этилпиперидин, глюкамин, глюкозамин, гистидин, гидрабамин, изопропиламин, лизин, метилглюкамин, морфолин, пиперазин, пиперидин, полиаминовые смолы, прокаин, пурины, теобромин, триэтиламин, триметиламин, трипропиламин, трометамин и т.п.
Следует отметить, что в нижеследующем обсуждении способов лечения, при ссылках на соединения формулы I подразумеваются также и их фармацевтически приемлемые соли.
Соединения формулы I могут быть использованы для ослабления боли, снижения температуры, и подавления воспалительных процессов при ряде состояний, включая ревматическую атаку; симптомы, ассоциированные с гриппом или другими вирусными инфекциями; общее переохлаждение организма; боли в нижнем отделе позвоночника и в области шеи; дисменорею; головную боль; растяжение и напряжение связок; миозит; невралгии; синовит; артрит; включая ревматоидный артрит; дегенеративные процессы в суставах (остеоартрит); подагра; и анкилозирующий спондилоартрит; бурсит; ожоги; и повреждения после хирургических и стоматологических операций. Кроме того, указанные соединения могут инбигировать опухолевые трансформации клеток, и метастатический опухолевый рост, а поэтому они могут быть использованы для лечения рака. Соединения формулы I могут быть также использованы для лечения деменции, включая пресенильную деменцию и сенильную деменцию, а частности, деменцию ассоциированную с болезнью Альцгеймера (т.е. деменция Альцгеймера).
Соединения формулы I могут ингибировать простаноид-индуцированное сокращение гладкой мышцы путем предупреждения синтеза сократительных простаноидов, а поэтому они могут быть использованы для лечения дисменореи, предупреждения преждевременных родов и лечения астмы.
Очевидно, что благодаря своей активности, направленной против циклооксигеназы-2 (СОХ-2) и/или своей селективности в предпочтении циклооксигеназы-2 (СОХ-2) циклооксигеназе-1 (СОХ-1), как описано выше, соединения формулы I могут быть использованы в качестве лекарственных средств, альтернативных стандартным нестероидным противовоспалительным средствам (НСПВС), особенно в тех случаях, когда эти нестероидные противовоспалительные средства могут быть противопоказаны, например, пациентам, страдающим пептической язвой, гастритом, региональным энтеритом, язвенным колитом, дивертикулитом, или рецидивирующими поражениями желудочно-кишечного тракта; желудочно-кишечными кровотечениями; нарушениями коагуляции, включая анемию, например, гипопротромбинемию, гемофилию, или другие состояния, связанные с кровотечениями (включая состояния, связанные со снижением или нарушением функции тромбоцитов); заболеваниями почек (например, почечной недостаточности); а также пациентам, подготавливаемым к хирургической операции или принимающим антикоагулянты; и пациентам, восприимчивым, к НСПВС-индуцированной астме.
Аналогично, соединения формулы I могут быть использованы в качестве частичной или полной замены НСПВС в препаратах, где они до настоящего времени использовались совместно с другими агентами или ингредиентами. Таки образом, в других аспектах, настоящее изобретение относится к фармацевтическим композициям, предназначенным для лечения опосредованных циклооксигеназой-2 заболеваний, определенных выше, и содержащим нетоксичное терапевтически эффективное количество соединения формулы I, определенного выше, и один или несколько ингредиентов, таких как другое болеутоляющее средство, включая ацетоминофен или фенацетин; потенцирующее средство, включая кофеин; H2-антагонист, гидроксид алюминия или магния, симетикон, противозастойное или противоотечное средство, включая фенилэприн, фенилпропаноламин, псевдофедрин, оксиметазолин, эпинефрин, нафазолин, ксилометазолин, пропилгекседрин или лево-дезоксиэфедрин; противокашелевое средство, включая кодеин, гидрокодон, карамифен, карбетапентан или декстраметорфан; мочегонное средство; седативные или неседативные антигистаминные средства. Кроме того, настоящее изобретение относится к способу лечения опосредованных циклооксигеназой заболеваний, предусматривающему введение пациенту, нуждающемуся в таком лечении, нетоксичного терапевтически эффективного количества соединения формулы I, которое может быть, но необязательно, введено в сочетании с одним или несколькими из вышеперечисленных ингредиентов.
Соединения настоящего изобретения являются ингибиторами циклооксигеназы-2, а поэтому они могут быть использованы для лечения заболеваний, опосредованных действием циклооксигеназы-2, и перечисленных выше. Активность этих соединений была проиллюстрирована их способностью к избирательному ингибированию циклооксигеназы-2 по сравнению с циклооксигеназой-1. В соответствии с этим, в одном из анализом, способность соединений настоящего изобретения лечить заболевания, опосредованные действием циклооксигеназы, может быть проиллюстрировано путем измерения количества простагландина E2 (PGE2) синтезированного в присутствии арахидоновой кислоты, циклооксигеназы-1 или циклооксигеназы-2, и соединения формулы I. Значения IC50 представляют собой концентрацию ингибитора, необходимую для снижения синтеза PGE2 до уровня, составляющего 50% от уровня, получаемого при неингибированном контроле. При иллюстрации этого аспекта настоящего изобретения, нами было установлено, что Соединения, представленные в Примерах, являются в 100 раз более эффективными при ингибировании СОХ-2, чем при ингибировании СОХ-1. Кроме того, для всех этих соединений, IC50 для СОХ-2 составляет от 1 нМ до 1 мкМ. Для сравнения: ибупрофен имеет IC50 для СОХ-2, равную 1 мкМ, а индометацин имеет IC50 для СОХ-2, равную приблизительно 100 нМ. Для лечения любого из указанных заболеваний, опосредованных циклооксигеназой, соединения формулы I могут быть введены перорально, местно, парентерально, путем ингаляции раствора или ректально в виде унифицированных препаратов, содержащих обычно используемые нетоксичные фармацевтически приемлемые носители, адъюванты и наполнители. Используемый в настоящем описании термин "парентерально" относится к подкожным, внутривенным, внутримышечным, внутригрудинным инъекциям и вливаниям. Помимо лечения теплокровных животных, таких как мыши, крысы, лошади, крупный рогатый скот, овцы, собаки, кошки и т.п., соединения настоящего изобретения являются эффективными для лечения человека.
Как указывалось выше, фармацевтические композиции для лечения вышеперечисленных заболеваний, опосредованных циклооксигеназой-2, могут, но необязательно, содержать один или несколько из вышеуказанных ингредиентов.
Фармацевтические композиции, содержащие активный ингредиент, могут быть изготовлены в виде соответствующих форм, подходящих для перорального введения, например в виде таблеток, пастилок, драже, водных или масляных суспензий, диспергируемых порошков или гранул, эмульсий, жестких или мягких капсул, сиропов или эликсиров. Композиции, предназначенные для перорального введения, могут быть получены любым способом, обычно используемым для изготовления фармацевтических композиций; причем, такие композиции могут содержать один или несколько ингредиентов, выбранных из группы, включающей в себя подслащивающие агенты, ароматизирующие агенты, красители и консерванты, для придания фармацевтическим препаратам эстетичного вида и приятного вкуса. Таблетки содержат активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, подходящими для изготовления таблеток. Указанными наполнителями могут быть, например, инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие и дезинтегрирующие агенты, например кукурузный крахмал или альгиновая кислота; связующие агенты, например крахмал, желатин или аравийская камедь; и замасливающие агенты, например стеарат магния, стеариновая кислота или тальк. Эти таблетки могут быть непокрыми, либо они могут быть покрыты с использованием известной техники в целях замедления дезинтеграции и абсорбции в желудочно-кишечном тракте, и тем самым сообщения этим таблеткам пролонгированного действия. Например, для замедления времени высвобождения может быть использовано такое соединение, как глицерилмоностеарат или глицерилдистеарат. Эти таблетки могут быть также покрыты способом, описанным в патентах США 4256108, 4166452 и 42658741, в результате чего могут быть получены осмотические терапевтические таблетки с регулируемым высвобождением лекарственного средства.
Препараты для перорального введения могут быть также получены в виде жестких желатиновых капсул, где активный ингредиент смешивают с инертным твердым разбавителем, таким как карбонат кальция, фосфат кальция или каолин; либо они могут быть получены в виде мягких желатиновых капсул, где активные ингредиенты смешивают с водной или масляной средой, такой как арахисовое масло, вазелиновое масло или оливковое масло.
Водные суспензии содержат активное вещество в смеси с наполнителями, походящими для изготовления водных суспензий. Такими наполнителями являются суспендирующие агенты, например, натрий-карбоксиметилцеллюлоза, метилцеллюлоза, гидрокси-пропилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и аравийская камедь; диспергирующие или смачивающие агенты, которые могут быть природным фосфатидом, таким как лецитин или продуктами конденсации алкиленоксида с жирными кислотами, например, такими как полиэтиленстеарат, или продуктами конденсации этиленоксида с длинноцепочечными алифатическими спиртами, например, такими как гептадекаэтиленоксицетанол; или продуктами конденсации этиленоксида с неполными сложными эфирами, полученными из жирных кислот и гексита, например, такими как полиоксиэтиленсорбитмоноолеат; или продуктами конденсации этиленоксида с неполными сложными эфирами, происходящими от жирных кислот и сорбитангидридов, например, полиэтиленсорбитанмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например, этил или н-пропил, п-гидроксибензоат, один или несколько окрашивающих агентов, один или несколько ароматизирующих агентов, и один или несколько подслащивающих агентов, таких как сахароза, сахарин или аспартам.
Масляные суспензии могут быть получены путем суспендирования активного ингредиента в растительном масле, например, в арахисовом масле, оливковом масле, кунжутном масле или кокосовом масле, либо в минеральном масле, таком как вазелиновое масло. Масляные суспензии могут содержать загустители, например, пчелиный воск, твердый парафин или цетиловый спирт. Для придания пероральной композиции приятного вкуса могут быть добавлены подслащивающие вещества, указанные выше, и ароматизирующие вещества. Для лучшей сохранности этих композиций может быть добавлен антиоксидант, такой как аскорбиновая кислота.
Диспергируемые порошки и гранулы для получения водной суспензии путем добавления воды изготавливают в виде смеси активного ингредиента с диспергирующим или смачивающим агентом, суспендирующим агентом, и с одним или несколькими консервантами. Примеры подходящих диспергирующих или смачивающих агентов, и суспендирующих агентов были приведены выше. В этих композициях могут также присутствовать и другие добавки, например, подслащивающие вещества, ароматизирующие вещества и красители.
Фармацевтические композиции настоящего изобретения могут быть также изготовлены в виде эмульсий "масло в воде". В качестве масляной фазы может быть использовано растительное масло, например оливковое масло или арахисовое масло, либо минеральное масло, например вазелиновое масло или их смеси. Подходящими эмульгирующими агентами могут быть природные фосфатиды, например соя, лецитин; сложные эфиры или неполные сложные эфиры, происходящие от жирных кислот и гекситангидридов, например сорбитаимоноолета; и продукты конденсации указанных неполных эфиров с этиленоксидами, например полиоксиэтиленсорбитанмолоолеат. Эти эмульсии могут также содержать подслащивающие и ароматизирующие вещества.
Сиропы и эликсиры могут быть получены с использованием подслащивающих веществ, например, таких как глицерин, пропиленгликоль, сорбит или сахароза. Эти композиции могут также содержать средство, уменьшающее раздражение; консервант; ароматизирующее вещество; и краситель. Фармацевтические композиции могут быть изготовлены в виде стерильных инъецируемых водных и масляных суспензий. Эти суспензии могут быть получены известными способами с использованием подходящих диспергирующих или смачивающих агентов, и суспендирующих агентов, упомянутых выше. Стерильный инъецируемый препарат может быть также изготовлен в виде стерильного инъецируемого раствора или суспензии в нетоксичном парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. В качестве приемлемых наполнителей и растворителей могут быть использованы вода, раствор Рингера, и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно используют стерильные жирные масла. В этих целях может быть использовано любое мягкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, при получении инъецируемых растворов могут быть использованы жирные кислоты, такие как олеиновая кислота.
Соединения формулы I могут быть также введены ректально, в виде суппозиториев. Эти композиции могут быть получены путем смешивания лекарственного средства с соответствующим нераздражающим наполнителем, который при обычных температурах является твердым, а при температуре прямой кишки - жидким, в результате чего этот наполнитель, при его введении в прямую кишку, расплавляется, высвобождая тем самым, лекарственное средство. В качестве таких наполнителей могут служить какао-масло и полиэтиленгликоли.
Для местного применения используют кремы мази, гели, растворы или суспензии и т. п., содержащие соединения формулы I. (В настоящей заявке понятие "местное применение" включает в себя полоскание рта и горла).
Для лечения вышеуказанных заболеваний могут быть использованы дозы порядка от около 0,01 мг до около 140 мг/кг веса тела в день, или альтернативно, от около 0,5 мг до около 7 г для одного пациента в день. Так, например, эффективное лечение воспалений может быть проведено путем введения от около 0,01 до 50 мг соединения на 1 кг веса тела пациента в день, либо альтернативно, от около 0,5 мг до около 3,5 г для одного пациента в день, а предпочтительно 2,5 мг - 1 г соединения на пациента в день.
Количество активного ингредиента, которое может быть смешано с ингредиентами носителя в целях получения разовой лекарственной формы, может варьироваться в зависимости от хозяина, подвергающегося лечению, и от конкретного способа введения. Например, композиция, предназначенная для перорального введения человеку, может содержать от 0,5 мг до 5 г активного ингредиента, смешанного с приемлемым и подходящим количеством носителя, которое может варьироваться от около 5 до около 95% от полной массы композиции. В основном, стандартная лекарственная форма содержит примерно от около 1 мг до около 500 мг активного ингредиента, а обычно 25 мг, 50 мг, 100 мг, 200 мг, 300 мг, 400 мг, 500 мг, 600 мг, 800 мг или 1000 мг активного ингредиента.
Однако следует отметить, что конкретная доза для каждого конкретного пациента будет зависеть от ряда различных факторов, например, от возраста, веса тела, общего состояния здоровья, пола и режима питания пациента, а также от схемы и способа введения лекарственного средства, скорости его высвобождения, комбинации лекарственных средств и тяжести конкретного заболевания, подвергаемого лечению.
Методы синтеза
Соединения настоящего изобретения
могут быть получены в соответствии с нижеописанными
методами.
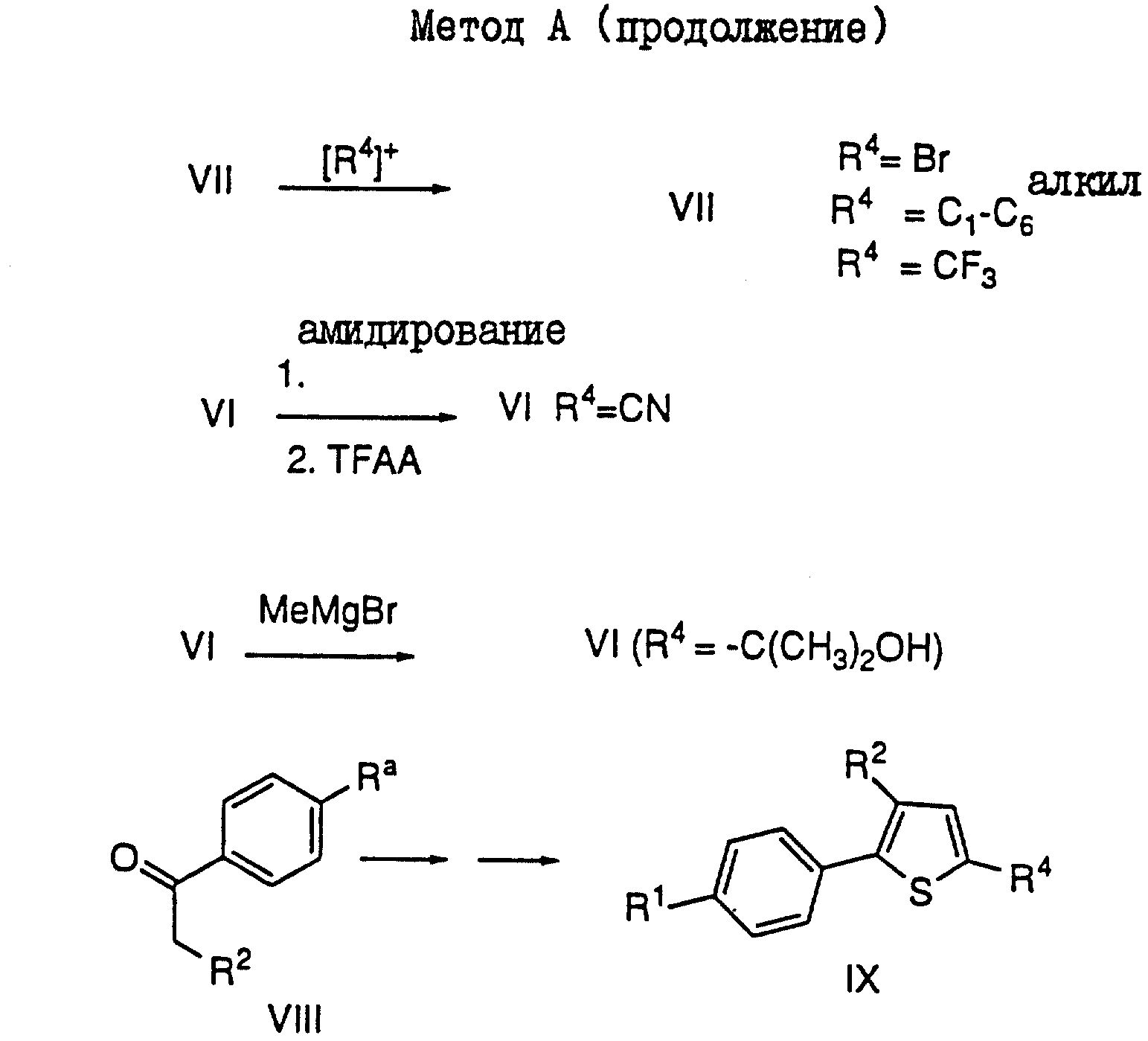
Метод A (см. схему в конце описания).
β- Хлоровинилальдегид III может быть получен из кетона II и реагента Вильсмайера (ДМФ-POCl3) с использованием общего метода, описанного Weissenfels (Z. Chem. 1966, 6, 471). Тиофеновое соединение IV получают из соединения III с использованием общего метода, описанного Weissenfels (Z. Chem. 1973, 13, 57). Тиоловое соединение V может быть получено после окисления соединения IV (Ra = -SMe) одним эквивалентом m-CPBA с последующей обработкой полученного сульфоксида соединением TFAA при нагревании с обратным холодильником. Затем, с помощью метода Kharash (J. Amer. Chem. Soc. 1951, 73, 3240) может быть образована сульфонамидная группа (VI). В результате гидролиза соединения VI и декарбоксилирования с использованием CU-бронзы в хинолине получают соединение VII. Это соединение VII (R4=H) может быть затем обработано галогенирующим агентом, таким как бром в уксусной кислоте, с получением 5-бромтиофена (VII, R4 = Br). Если необходимо получить нитрильную группу у C-5, то это может быть осуществлено путем образования амида с использованием методики Wenireb (Tetrahedron Letters, 1977, 4171) и последующей дегидратации с помощью TFAA. CF3-группа может быть введена в VII y C-5 методом Girard (J. Org. Chem., 1983, 48, 3220).
Введение алкильной группы y C-5 может быть осуществлено с помощью реакции Фриделя-Крафтса между VII (R4=H) и хлористым ацилом, C1-CO-низшим алкилом и катализатором, таким как TiCl4, с последующей реакцией восстановления. Для R4= Me, эта реакция может быть осуществлена из сложного эфира (R4=CO2Me) посредством восстановления DIBAL-H, и с последующим дезоксигенированием с использованием метода Lau (Org. Chem., 1986, 51, 3038). Третичные спирты (R4= -C(CH3)2OH) могут быть получены из VI и MeMgBr. Эти третичные спирты могут быть также подвергнуты дезоксигенированию методом Lau. Аналогичным образом тиофен IX может быть получен из кетона VIII.
Метод B (см. схему в конце описания).
Кетон X может быть превращен в тиофеновое соединение XI с использованием общего метода, уже описанного в Методе A.
Тиофен XII может быть получен путем металлизации XI с использованием n-BuLi, с последующим тушением реакции двухлористым первичным метилфосфином, и добавлением воды или аммиака (X=OH или NH2). Аналогичным образом, из кетона XIII может быть получен другой региоизомер XIV.
Метод C (см. схему в конце описания).
В результате бромирования кетона II получают альфа -бромокетон XV, который затем, после обработки тиоамидом, превращают в тиазол XVI. Аналогичным образом кетон VIII может быть превращен в тиазол XVII.
Метод D (см. схему в конце описания)
После обработки
формамидом с
использованием препарата Brederick et al., Chem. Ber. 1953, стр. 88, кетон XV может быть превращен в
имидазоловое соединение XVIII.
Метод E
Пироловое соединение
XX может быть
получено из дикетона XIX с использованием общей методики Friedman et al., J. Org. Chem. 1965, 30,
p.
854, K. Dimroth et al. , Ber. 1956, 56, 2 2602; K. Dimroth et al., Ann. 1961, 634,
102.
Свободная группа NH может быть ацилирована с использованием C1-CO-низшего алкила в присутствии основания, такого как Et3N. Алкилированные продукты могут быть также получены с использованием в качестве реагентов алкилгалогенидов в присутствии основания, такого как NaH.
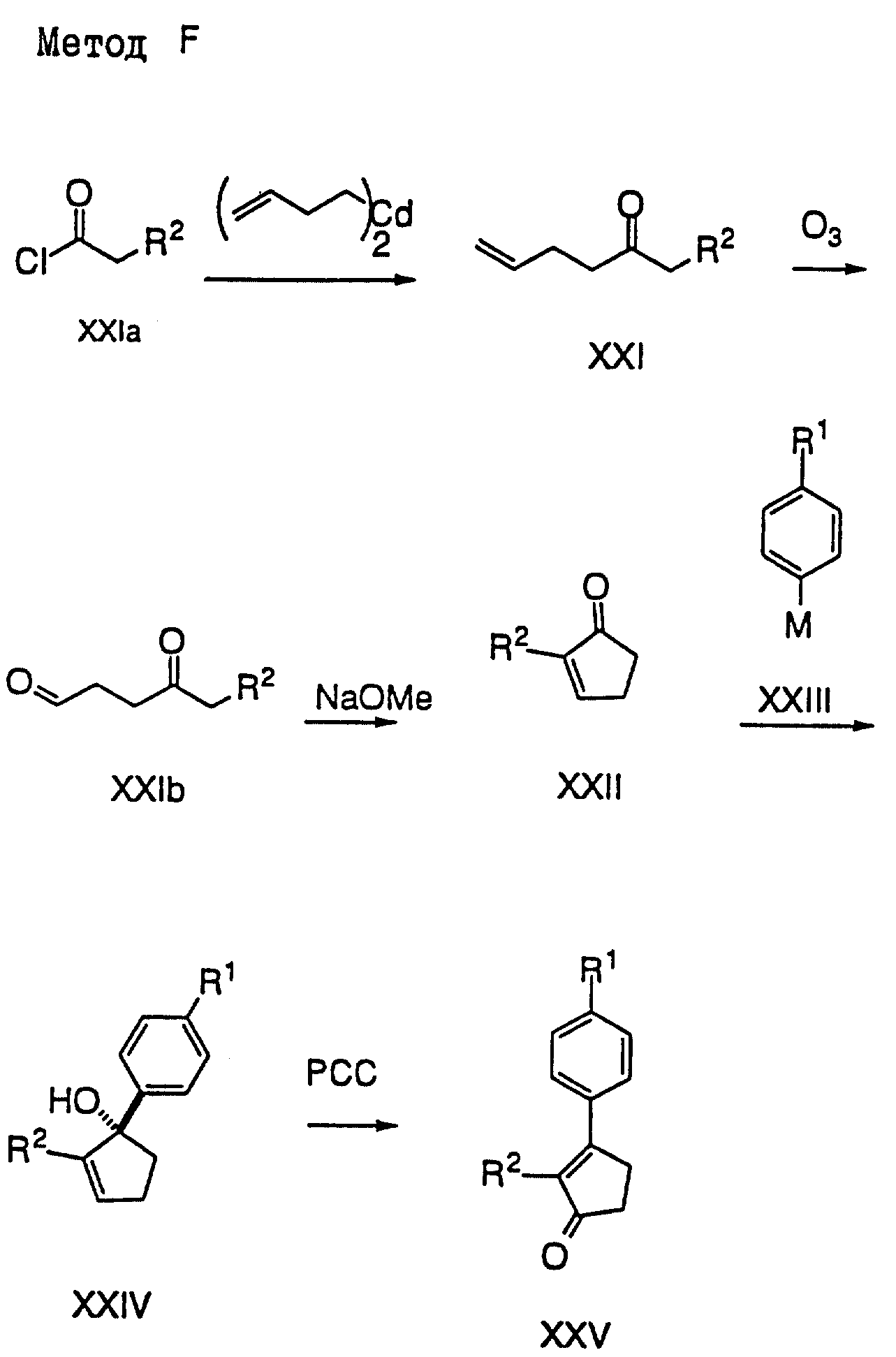
Метод F (см. схему в конце описания)
Соединения типа XXV могут быть
получены из
легкодоступных 4-замещенных фенилацетилхлоридов XXIa. В результате реакции
ди(3-бутенил)кадмия с 4-замещенным
фенилацетилхлоридом получают кетон XXI. После озонирования кетона XXIa
получают
кетоальдегид XXIa, который циклируют с помощью основания с образованием
циклопентенона XXII. Затем к XXII добавляют
бромид арилмагния или ариллитий, в результате чего получают аллильный
спирт XXIV.
После окисления XXIV хлорхроматом пиридиния получают нужный 2,
3-дизамещенный циклопентенон XXV. Для получения
соединения XXV (R1=SO2Me) используют
4-метилтиофениллитий с
последующим окислением магниевой соли монопероксиафталевой кислоты
(MMPP) или хлоропероксибензойной кислоты (mCPBA)
для введения в XXV необходимой метилсульфонильной
группы.
Метод G
(см. схему в конце описания)
Метод G осуществляют в той
же последовательности, что и Метод F, за исключением
того, что в качестве исходного соединения
используют R1, содержащий
хлорангидрид. R2 вводят в более поздней стадии путем
реакции присоединения карбонильной группы с последующим
PCC-окислением.
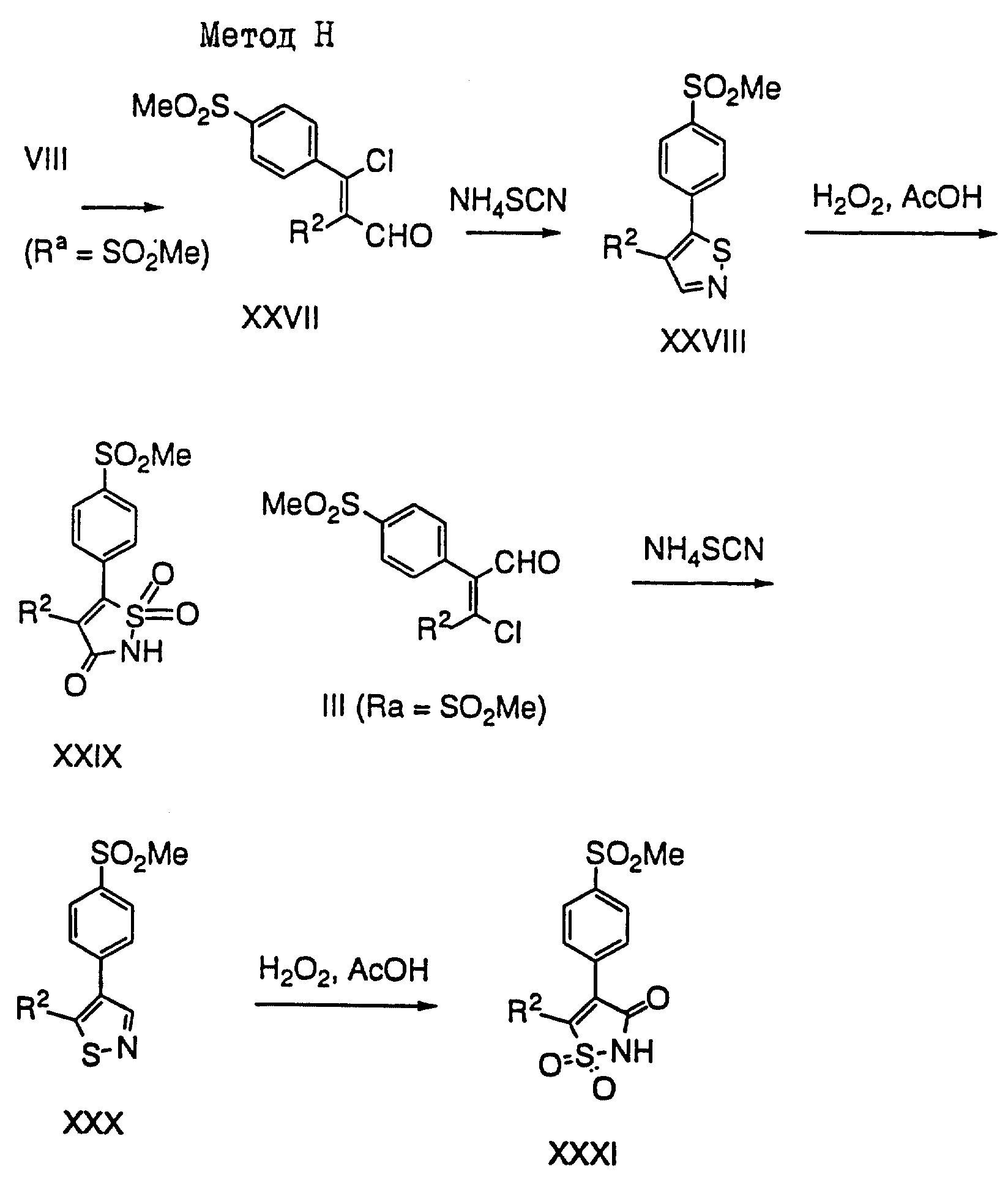
Метод H
(см. схему в конце описания)
4,
5-Дизамещенные изотиазолы и изотиазол-3(2H)-он-1,1-диоксиды
могут быть получены с использованием общего метода,
описанного B. Schulze et al., Helvetica
Chimica Acta, 1991, 74, 1059. Так, например,
альдегид III (Ra=SO2Me) или XXVII
обрабатывают избыточным количеством NH4SCN в
кипящем ацетоне, в результате чего
получают соответствующие 4,5-замещенные изотиазолы
XXX и XXVIII, после окисления которых пероксидом
водорода получают XXXI и XXIX.
Метод I: (см.
схему в конце описания)
Соответствующим образом замещенный арилбромометилкетон
подвергают реакции с соответствующим образом
замещенной арилуксусной кислотой в растворителе таком, как
ацетонитрил, и в присутствии основания
такого, как триэтиламин, а затем обрабатывают 1,
8-диазабицикло[5.4.0]ундек-7-ен (DBU) с получением
либо лактона, либо XXXV.
Метод J: (см. схему
в конце описания)
Либо лактон
XXXIII, либо XXXV в растворителе, таком как ТГФ,
подвергают реакции с восстановителем, таким как
гидрид диизобутилалюминия или борогидрид лития при -78oC, в результате чего получают фуран
XXXVI.
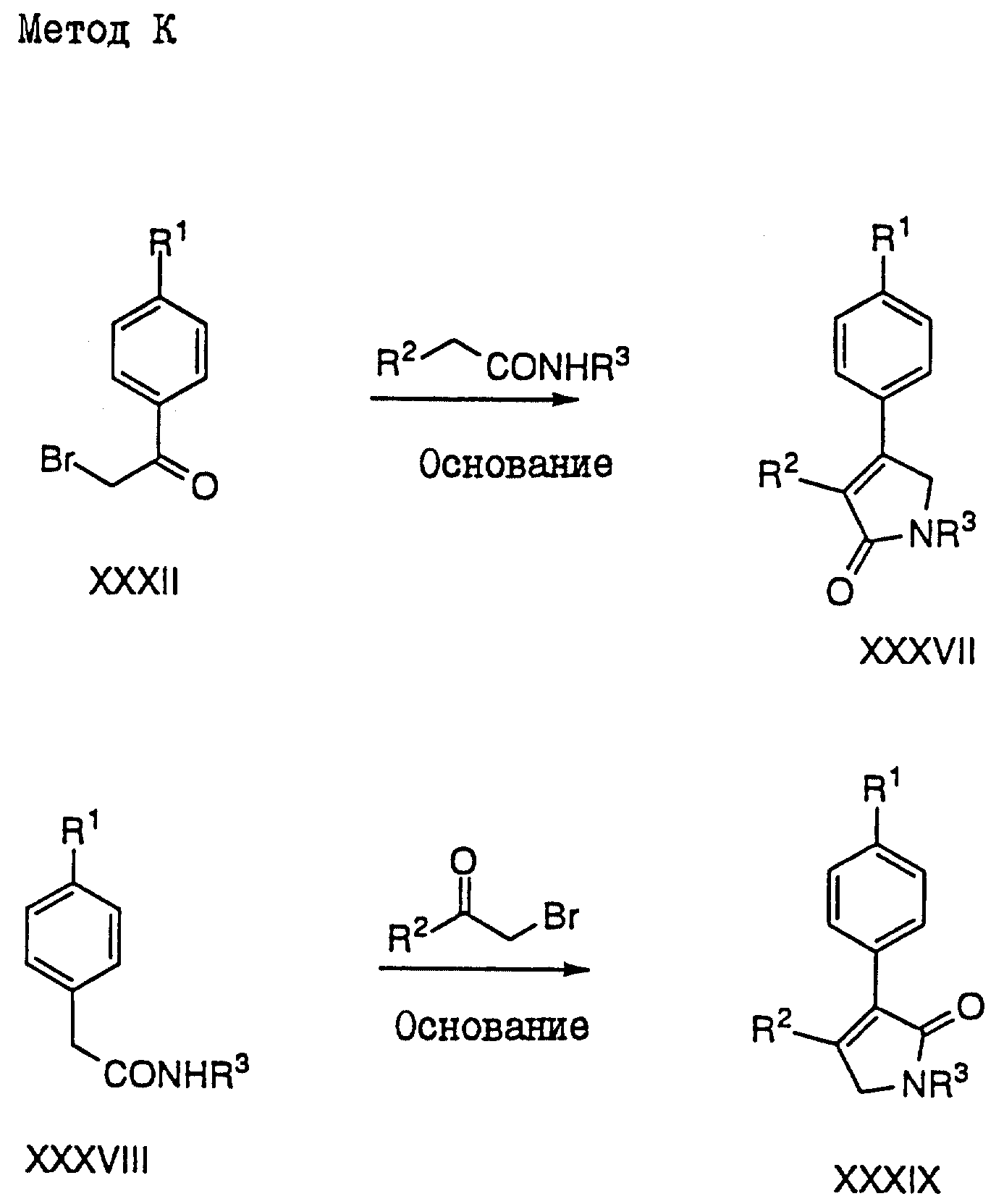
Метод K: (см. схему в конце
описания)
Получение лактамов XXXVII
и XXXIX может быть осуществлено с помощью той же самой
реакции, которая была описана в Методе I, за
исключением того, что в этой реакции был использован
соответствующий амид XXXVIII.
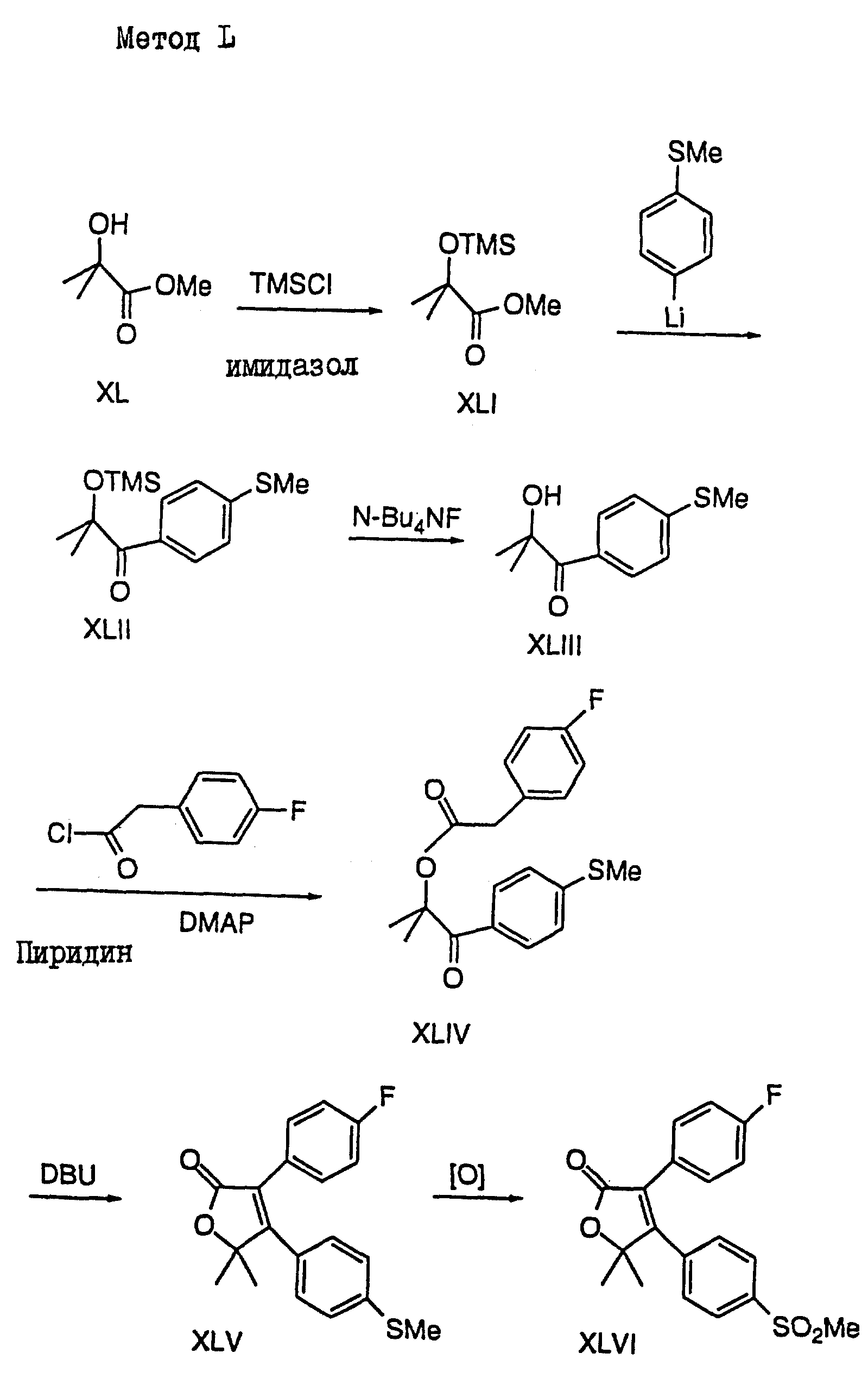
Метод L: (см. схему в конце описания)
Метил-2-гидроксиизобутират подвергают силилированию
с использованием TMSCI, в результате чего получают
соединение TMS или XLI, которое затем
обрабатывают 4-метилтиофениллитием с получением кетона
XLII. После десилилирования с последующим
ацилированием получают кето-эфир XLIV, который может быть затем
циклизован с образованием лактона
XLV путем основного катализа. После окисления XLV
соединением MMPP или mCPBA получают нужный продукт
XLVI.
Альтернативный способ получения гидроксикетона XLIII (см. схему метода M в конце описания) предусматривает окисление известного (J. Org. Chem. 1991 56, 5955-8; Sulfur Lett. 1991, 12, 123-32) кетона XLVII. Смесь соединения XLVII, водного основания, такого как NaOH, органических растворителей, таких как тетрахлорметан/толуол и межфазного катализатора, такого как ALIQUAT 336, размешивают в атмосфере воздуха при комнатной температуре, в результате чего получают XLIII. Соединение также описано в патенте США 4321118 и Org. Coat. 1986, 6, 175-95.
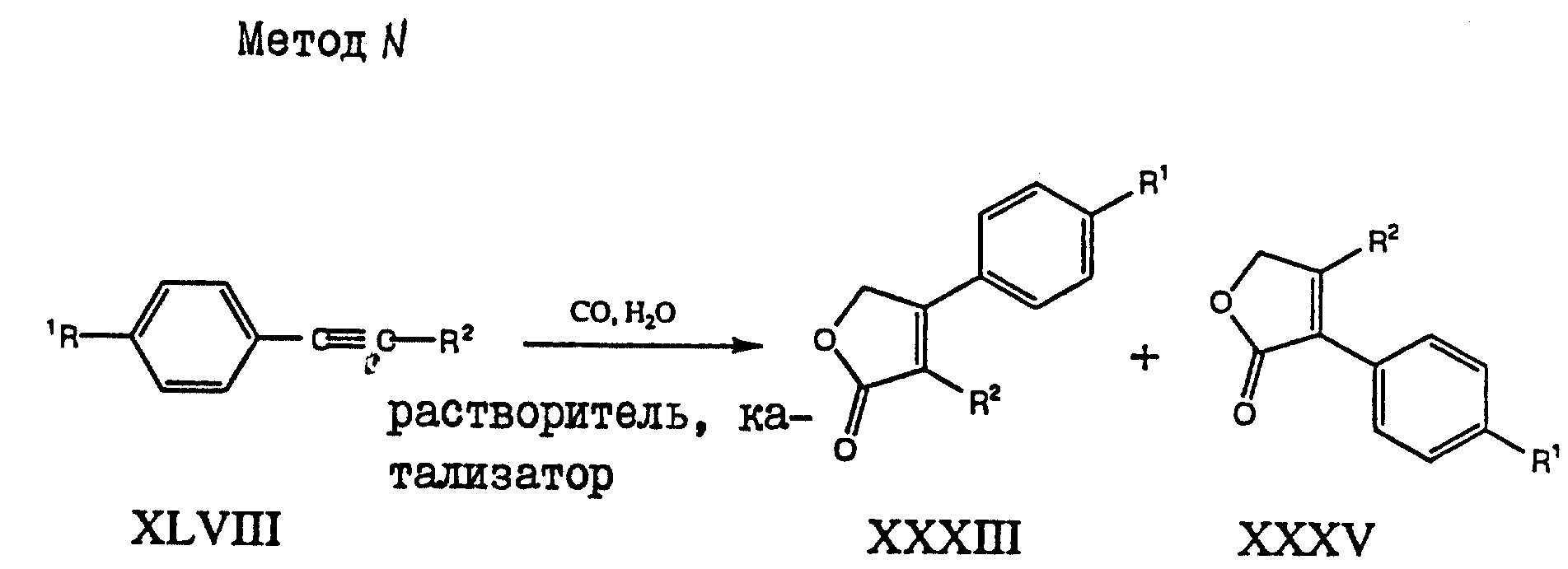
Путем реакции ацетилена XLVIII с окисью углерода и водой (см. схему метода N в конце описания) в присутствии подходящих катализаторов получают смесь соединения XXXIII и его изомера XXXV. Изомеры разделяют с использованием методов, таких как хроматография или кристаллизация. Примерами подходящих катализаторов и условий являются PdCl2 в водной HCl и EtOH, при нагревании при 50-150o C, и при давлении 50-150 атмосфер, либо Ph4(CO)12 (или Ph6(CO)16 в водном ТГФ (или ацетоне, ацетонитриле, бензоле, толуоле, EtOH, MeOH), содержащем триалкиламин, при 50-150oC, и при давлении 20-300 атмосфер. См. Takahashi et al., Organomettallics 1991, 10, 2493-2498; и Tsuji et al., Am. Chem. Soc. 1966, 88, 1289 - 1292.
В результате 1,4-присоединения к XLIX
4-метилтиофениловых
металлорганических реагентов L в присутствии солей меди, и захвата
полученного енолята триалкилсилилхлоридом, таким как TMSCI или
TIPSCI, получают кетенацеталь LI. Этот
кетенацеталь может быть
затем окислен с получением замещенного бутенолида LII методом lto с
использованием каталитических количеств Pd2(OAc)2 и Cu(OAc)2 и O2
в MeOH, либо
методом Magnus с использованием Cu(OAc)2 и O2 в MeOH,
либо методом Magnus с использованием Ph10/TMSN3 и
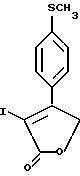
Bu4NH. Введение иода может быть
осуществлено
путем обработки LII иодом I2 в присутствии пиридина и с получением, в
результате, соединения LIII. После катализируемой палладием
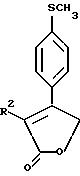
реакции взаимодействия Susuki или Stille
соединения LIII
с соответствующим арильным или алкильным реагентом, таким как бороновая кислота
LIV получают бутенолид LV. Сульфид может быть окислен до
сульфона с помощью окислителей, таких как
надуксусная
кислота, MPPM, MMPP или H2O2 с получением нужного соединения LVI.
См., Y. Ito et al., J. Am. Chem. Soc. 1979, 101, 494 and
P. Magnues et al., Tet. Lett. 1992, 2933
(см. схему
метода O в конце описания)
В соответствии с вышеизложенным, в еще одном своем
аспекте, настоящее изобретение относится к способу
получения соединения формулы XXXII:

предусматривающему
(a1) реакцию в органическом растворителе соединения формулы XXXII
с бромовым реагентом с получением соединения формулы XXXII

(В настоящем описании подразумевается, что органическим растворителем является (но не ограничивается ими) метиленхлорид, хлороформ, тетрахлорметан и уксусная кислота. Аналогично, бромовым реагентом является (но не ограничивается ими) бром, гидробромид пербромида пиридиния, CuBR2 и N-бромоскцинимид).
(a2)
реакцию в безводном полярном растворителе соединения XXXII с соединением
формулы
в присутствии основания, с получением соединения формулы A

(a3) обработку в безводном полярном растворителе соединения формулы A сильным основанием, с получением соединения формулы XXXIII.
В настоящем описании подразумевается, что безводным полярным растворителем является (но не ограничивается ими): ацетонитрил, пропионитрил, ацетон, 2-бутанон и тетрагидрофуран. Аналогично, основание является (но не ограничивается ими): три-C1-3-алкиламин, такой как триэтиламин. Кроме того, сильным основанием является (но не ограничивается ими) амид, гуанидин, диизопропиламид лития и бис-(триметилсилил)амид калия.
Альтернативно, настоящее
изобретение относится
к способу получения соединения формулы XXXIII

предусматривающему
(b1) реакцию ацетиленового соединения формулы XLVIII
с окисью углерода и водой в присутствии подходящего катализатора с получением формулы XXXIII и XXXV
В целях настоящего изобретения, подходящими катализаторами являются (но не ограничивается ими) Ru4(CO)12, CO2(CO)8 или PdCl2 в водном ТГФ или ацетоне, ацетонитриле, бензоле, толуоле, в метиловом спирте или этиловом спирте.
Во втором альтернативном варианте настоящее изобретение относится к способу получения соединения
формулы
XXXIII
предусматривающему
(c1) реакцию соединения формулы LIII
и (c2) окисление соединения формулы LV с получением соединения формулы XXXIII.
В настоящем описании подразумевается, что катализатором является, но не ограничивается им, палладиевый катализатор. Аналогично, растворителем является, но не ограничивается ими, бензол, толуол, ТГФ, MeOH, DME или EtOH.
Во всех
альтернативных вариантах способа, R1 и R2 являются
такими как они были определены выше в
главе "Подробное описание изобретения" и в пунктах формулы изобретения, относящихся к
соединениям формулы I
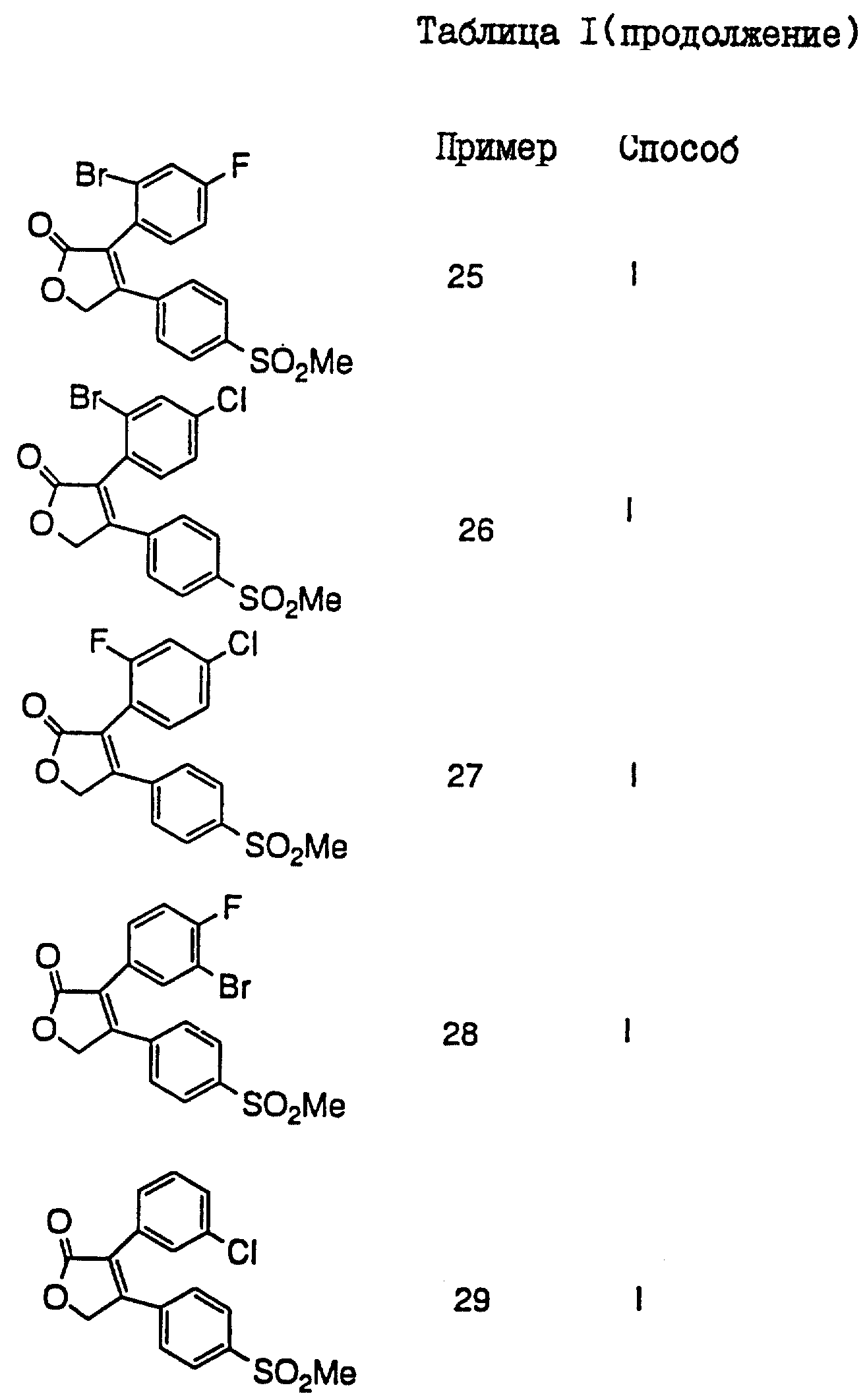
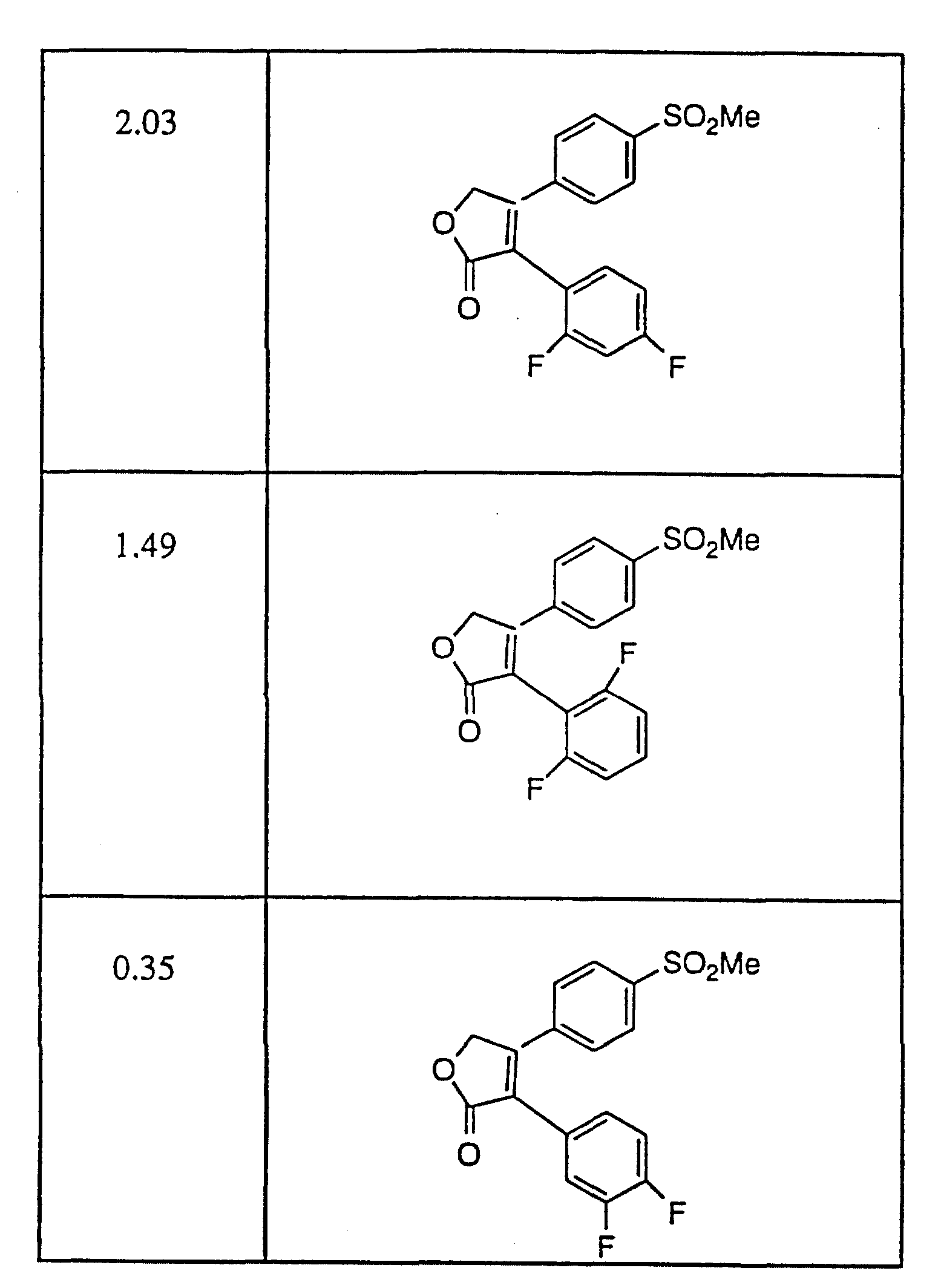
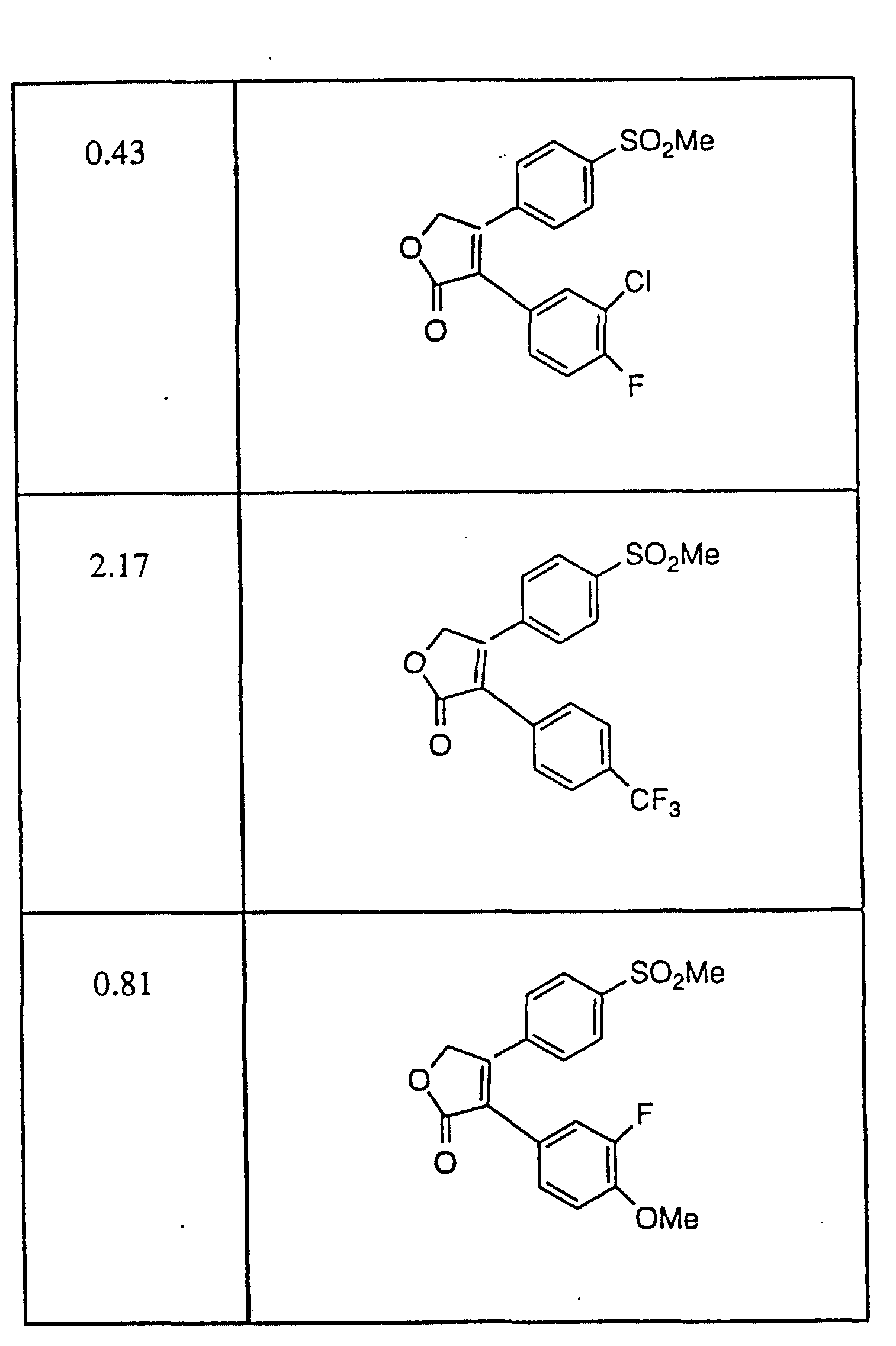
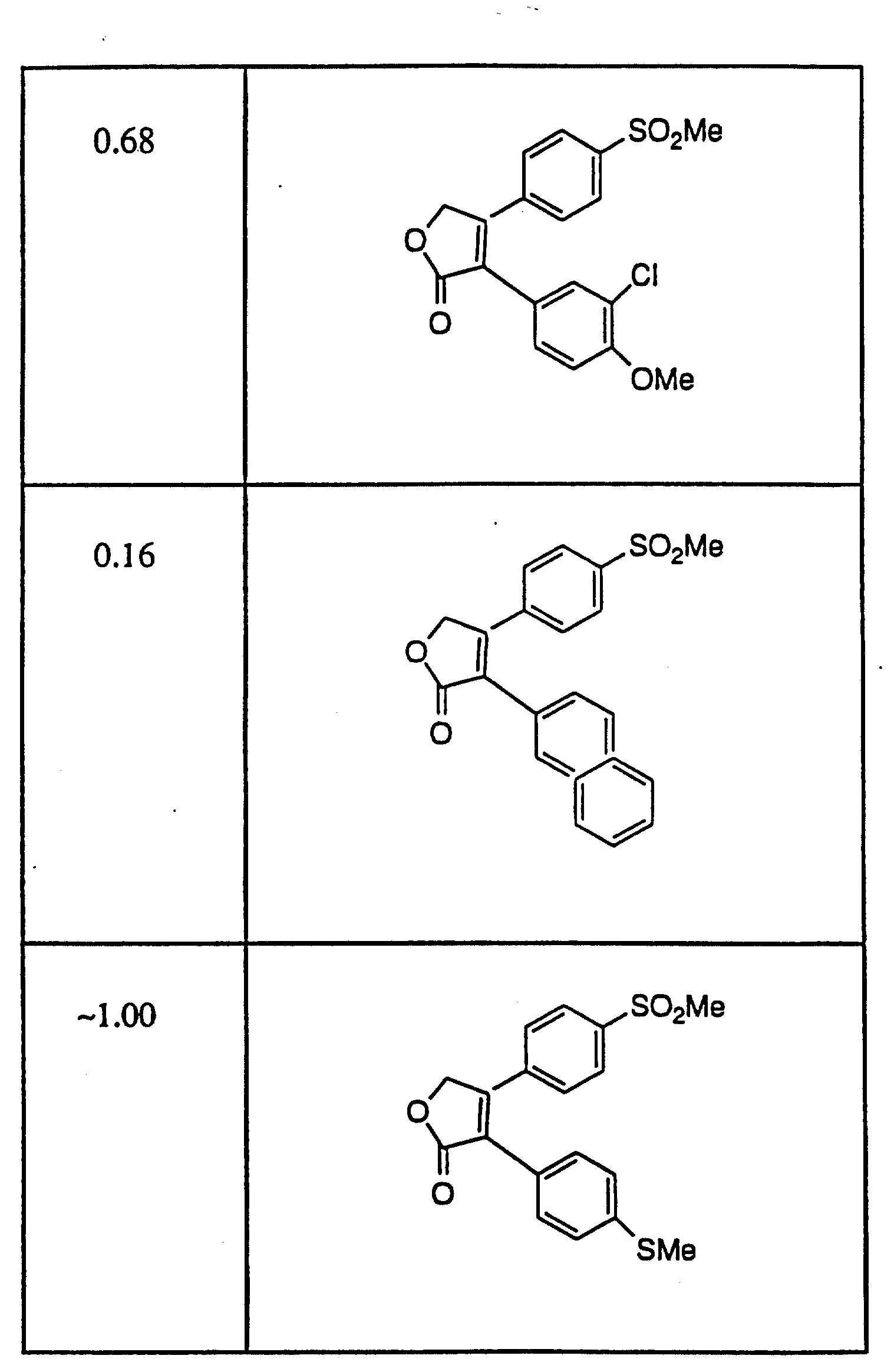
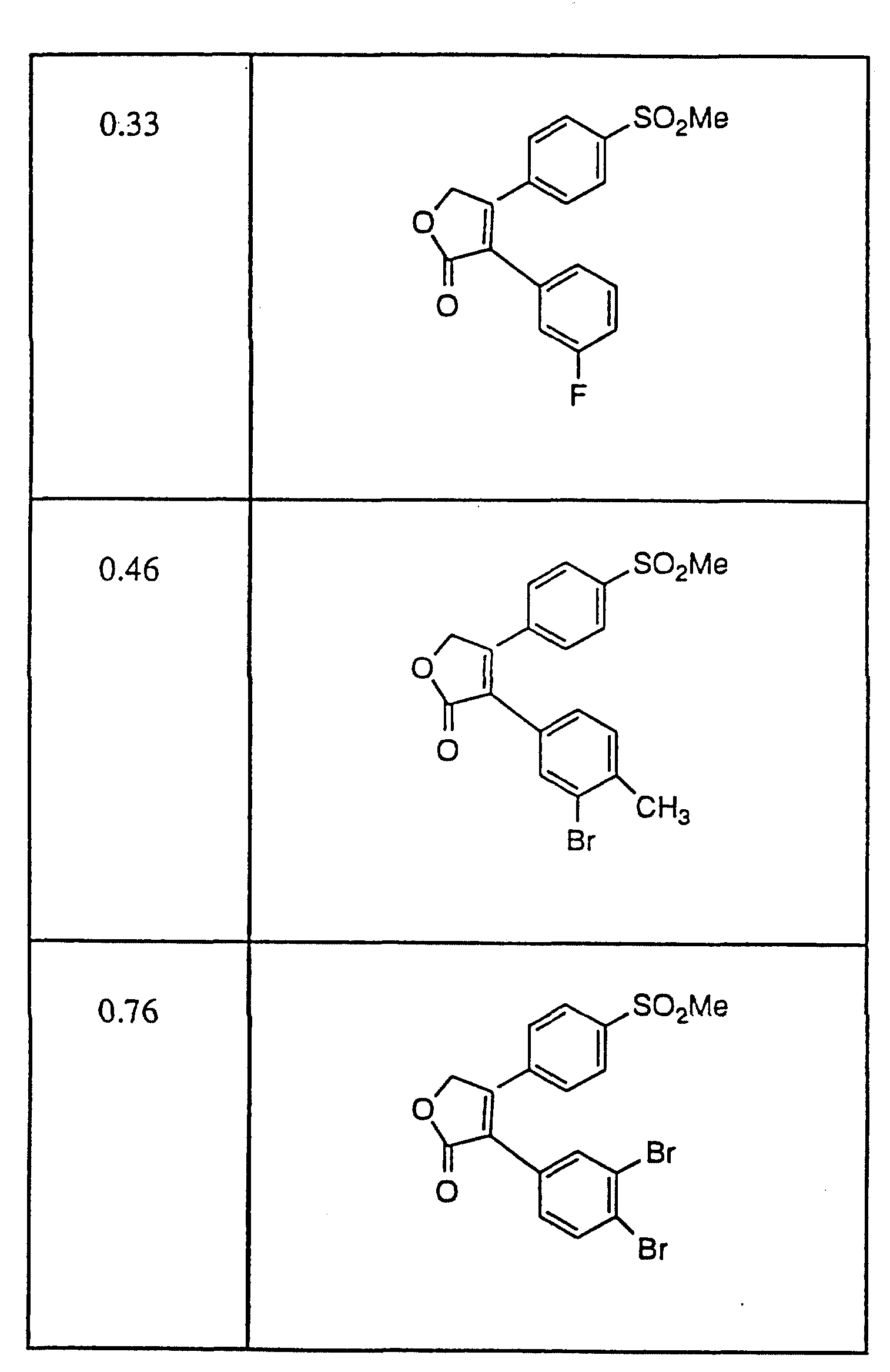
Характерные соединения
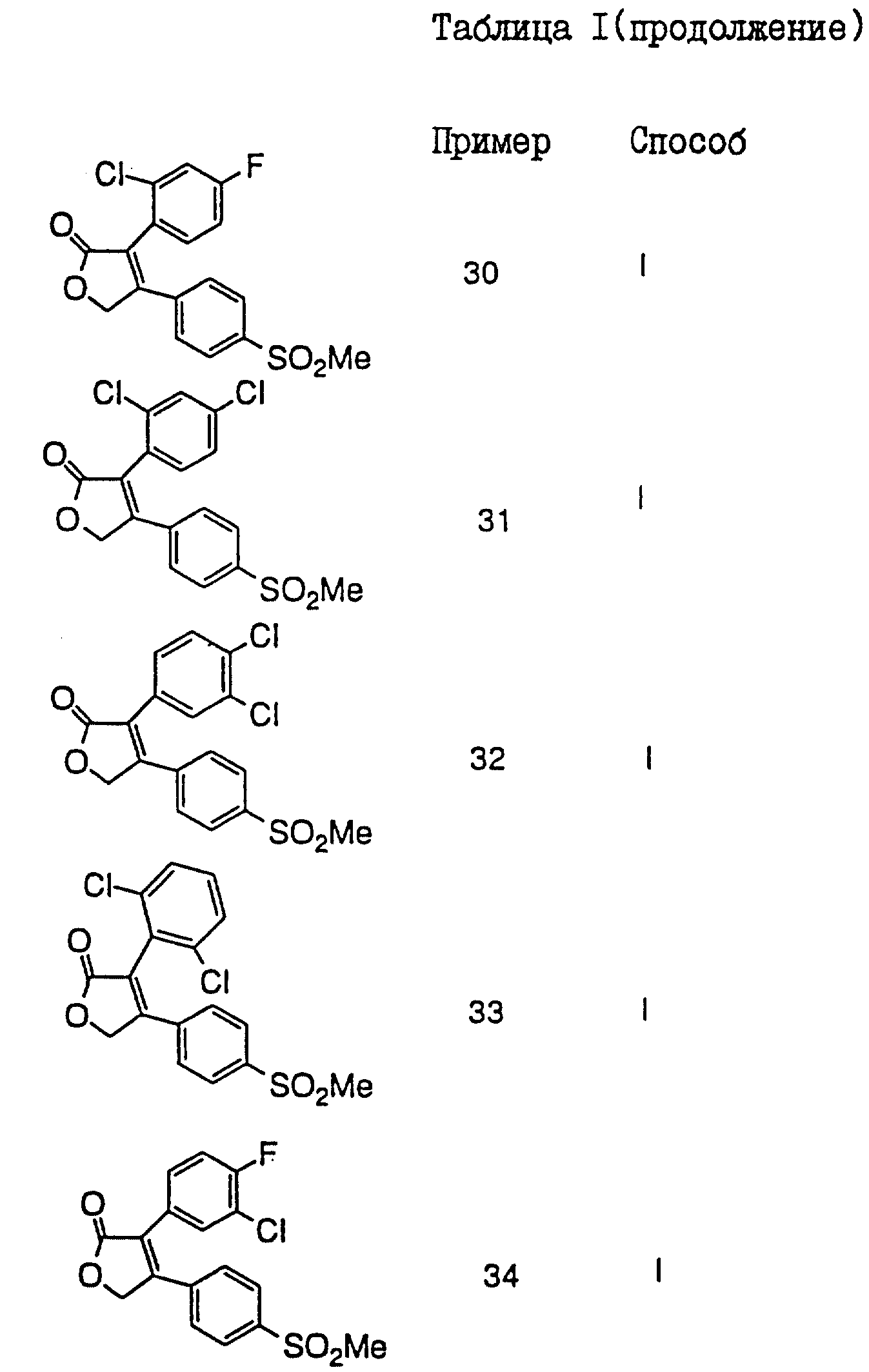
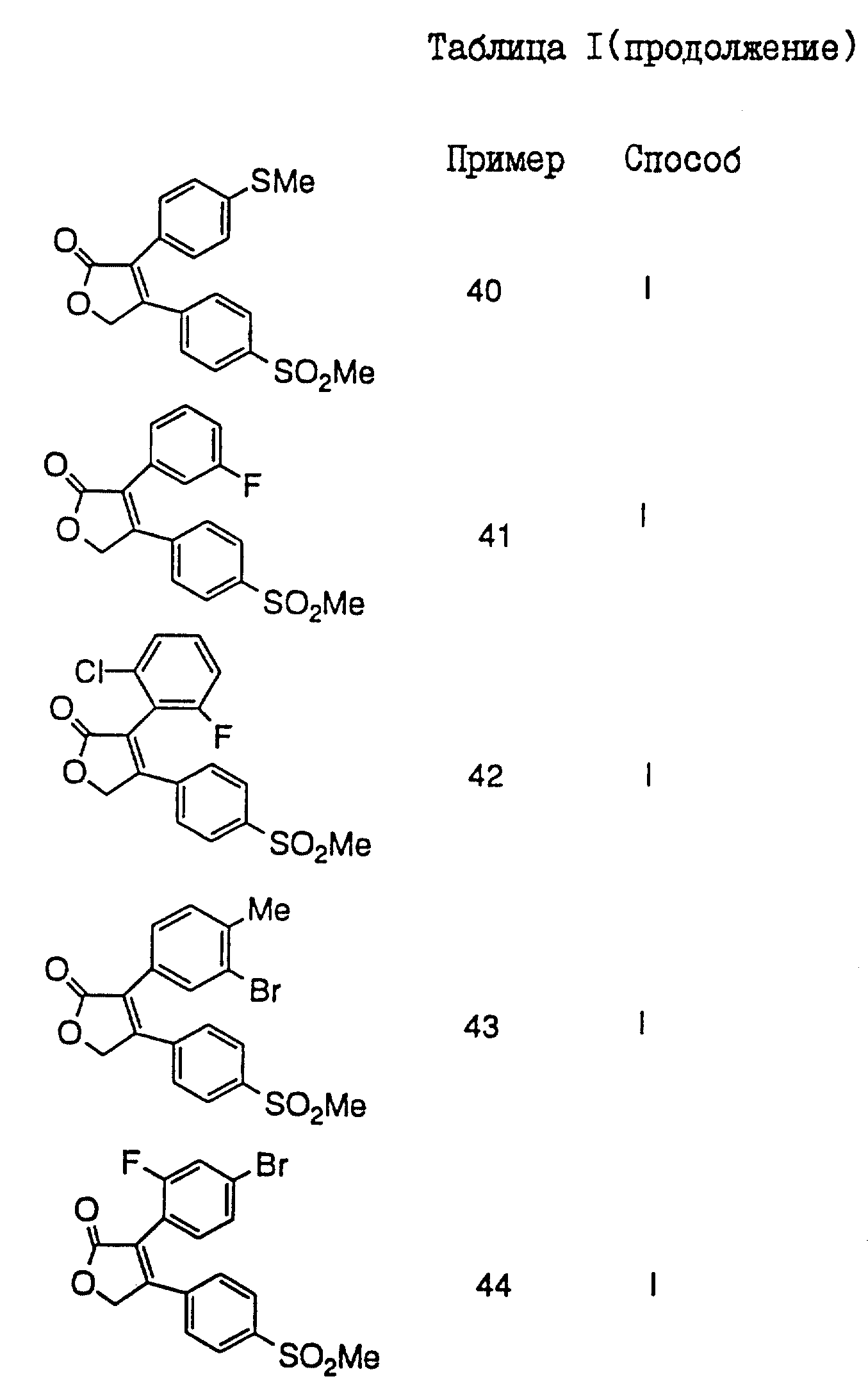
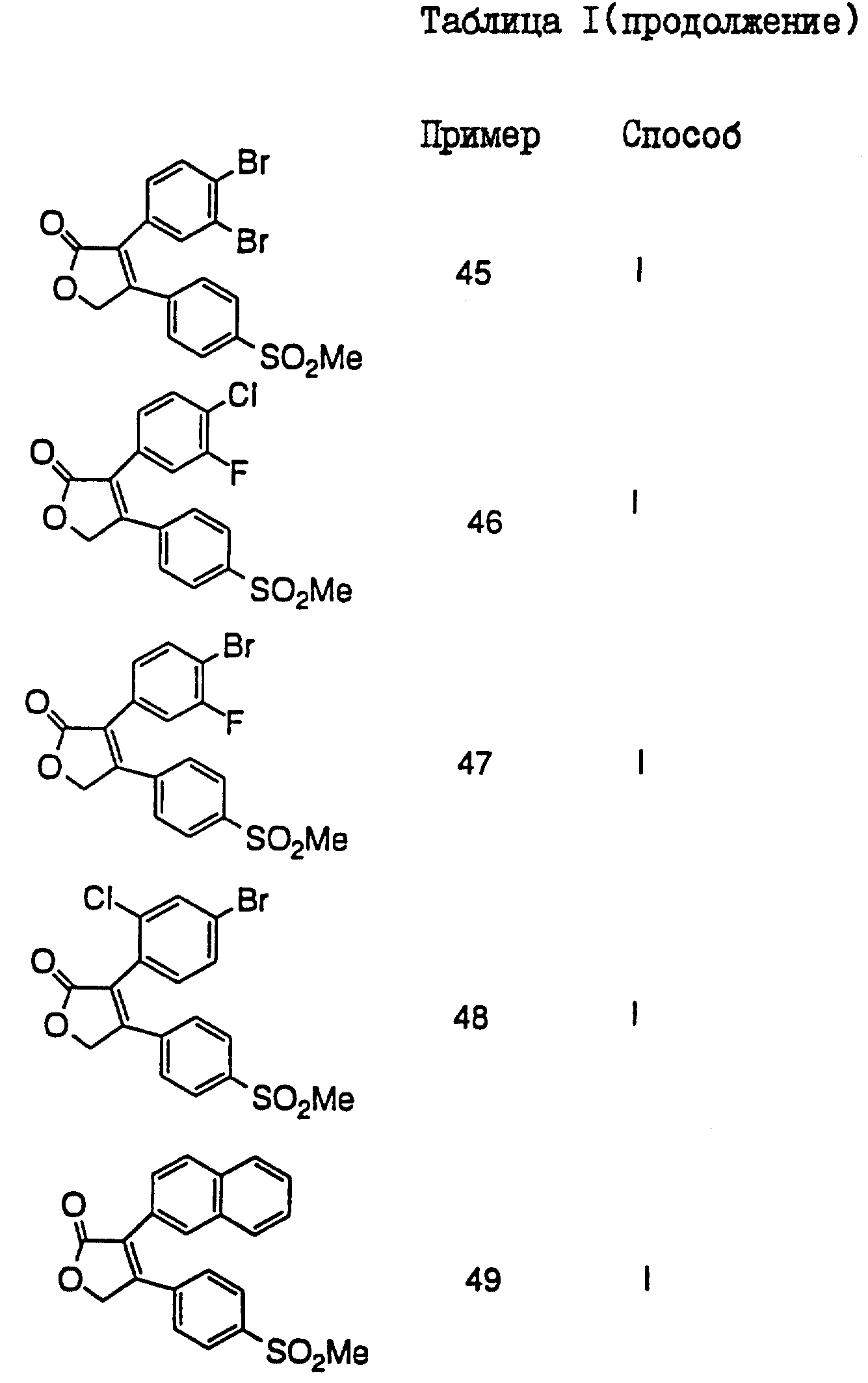
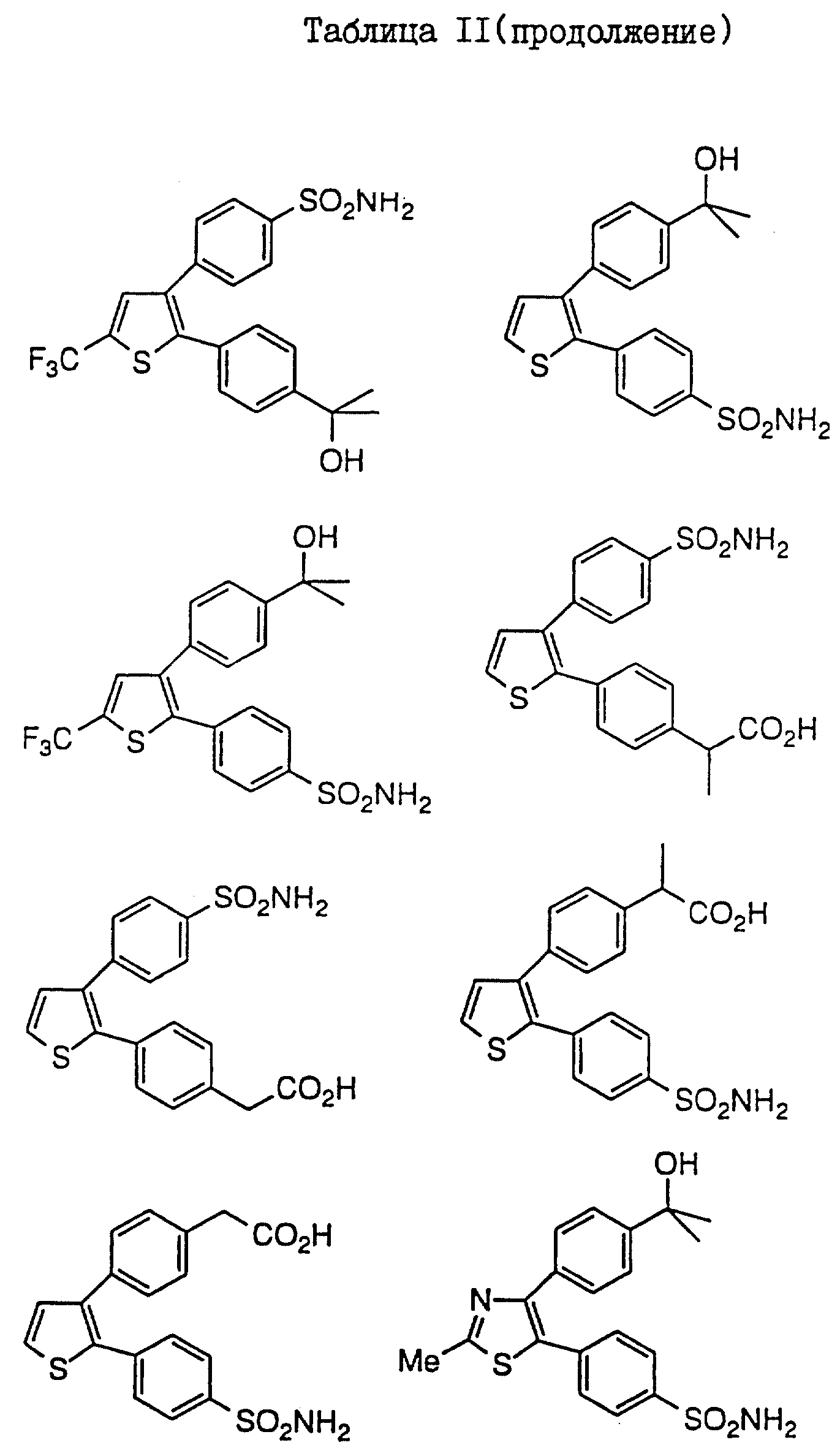
В таблицах I
и II (см. в конце описания)
проиллюстрированы соединения формулы I.
Анализ для определения биологической
активности
Для
определения активности соединений формулы I,
направленной на ингибирование
циклооксигеназы-2, могут быть произведены следующие анализы.
Ингибирование
циклооксигеназной активности
Испытание соединений на ингибирование
циклооксигеназной активности
проводились с помощью анализов на циклооксигеназу, в которых использовали как целые клетки,
так и их микросомные фракции. В
обоих случаях, используя радиоиммуноанализ,
осуществляли оценку синтеза
простагландина E2 (PGE2) в ответ на арахидоновую кислоту. Клетки,
используемые для клеточного анализа, и
клетки, из которых приготовляли микросомы для
микросомного анализа,
представляли собой клетки остеосаркомы человека 143 (специфически продуцирующие
циклооксигеназу-2), и клетки u937 (специфические
продуцирующие циклооксигеназу-1). В этих анализах,
100%-ную активность
определяли как разницу между синтезом простагландина E2 в случае
отсутствия, и в случае добавления арахидоната.
Величины IC50 представляют собой концентрацию
предполагаемого
ингибитора, необходимую для снижения синтеза PGE2 до уровня,
составляющего 50% от уровня, получаемого при
неингибированном контроле. Репрезантивные результаты приведены в
таблице III* (см. в конце описания)
Схема репрезантивного анализа
на отек лап у крыс
Крысам-самцам
Sprague-Dawley (150-200 г) не давали пищи в течение ночи, а в 9-10 час
утра им
перорально вводили либо (5% Твин 80 или 1% Метоцел), либо испытуемое
соединение. Затем, через 1 час, используя
маркировальный карандаш, прочеркивали линию на задней лапе крысы, на уровне выше
лодыжки,
в целях последующего слежения за этой областью лапы. Объем лапы
(voh) измеряли с помощью
плетизмометра (Ugo-Basile, Italy), основанного на принципе вытеснения водой. Затем животным
инъецировали субпланарно (в подошву лапы) 50 мкл 1% раствора
каррагенана в физиологическом растворе (FMC
Corp., Maine), используя при этом для инсулиновых инъекций с иглой 25-калибра (т.е. 500 мкг
каррагенана на лапу). Через 3 часа после этого, измеряли
объем лапы (v3h), и подсчитывали
увеличение объема лапы (v3h-v0h). Затем животных эутанизировали
(безболезненно умерщвляли) посредством CO2-асфиксии, и
оценивали на отсутствие или наличие желудочных
поражений. Эти оценки выражали как сумму всех поражений желудка в мм. Данные,
полученные после измерения отека лапы, сравнивали с данными контрольной
группы (наполнитель), после чего вычисляли
процент ингибирования, принимая значения контрольной группы за 100%. Поскольку
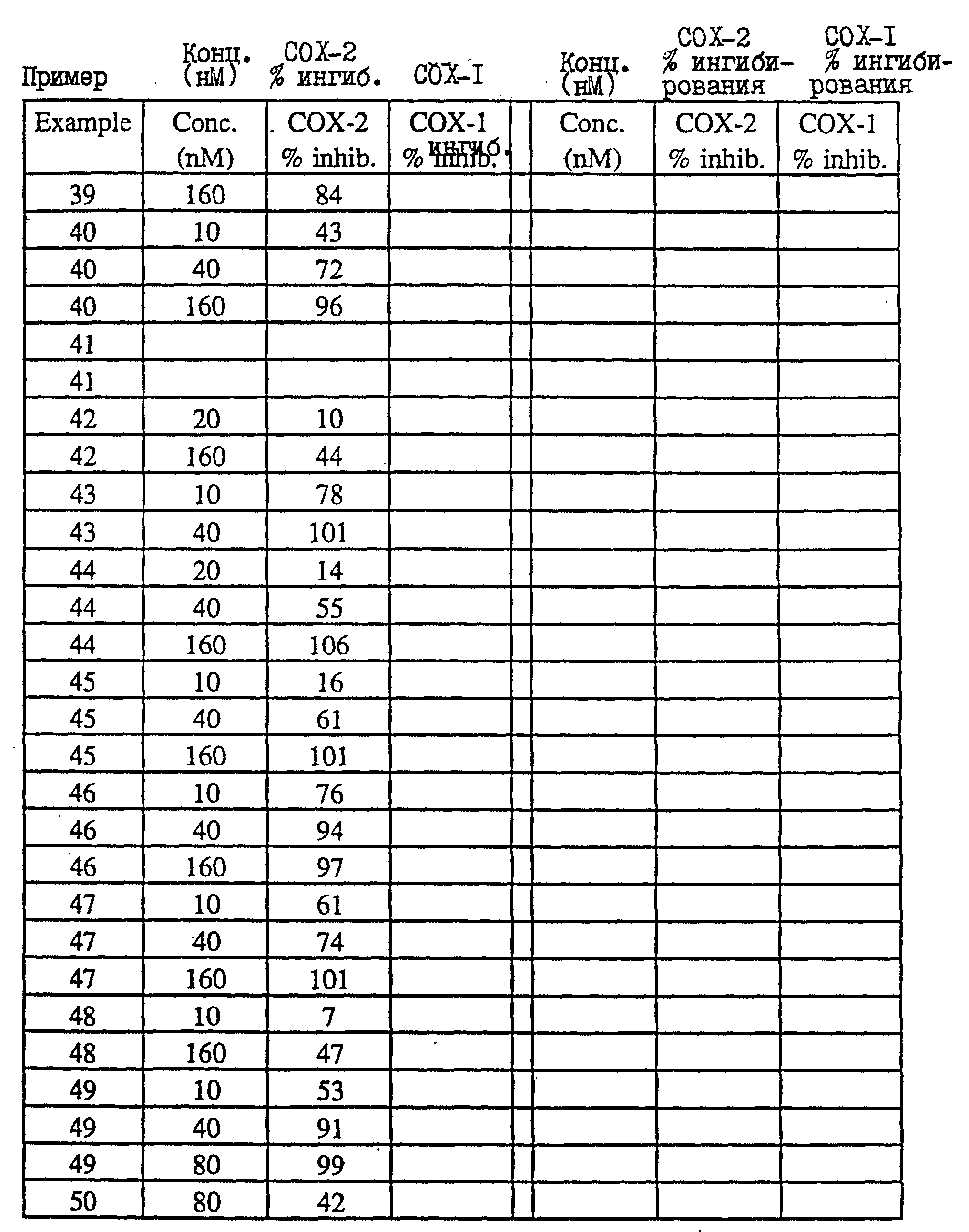
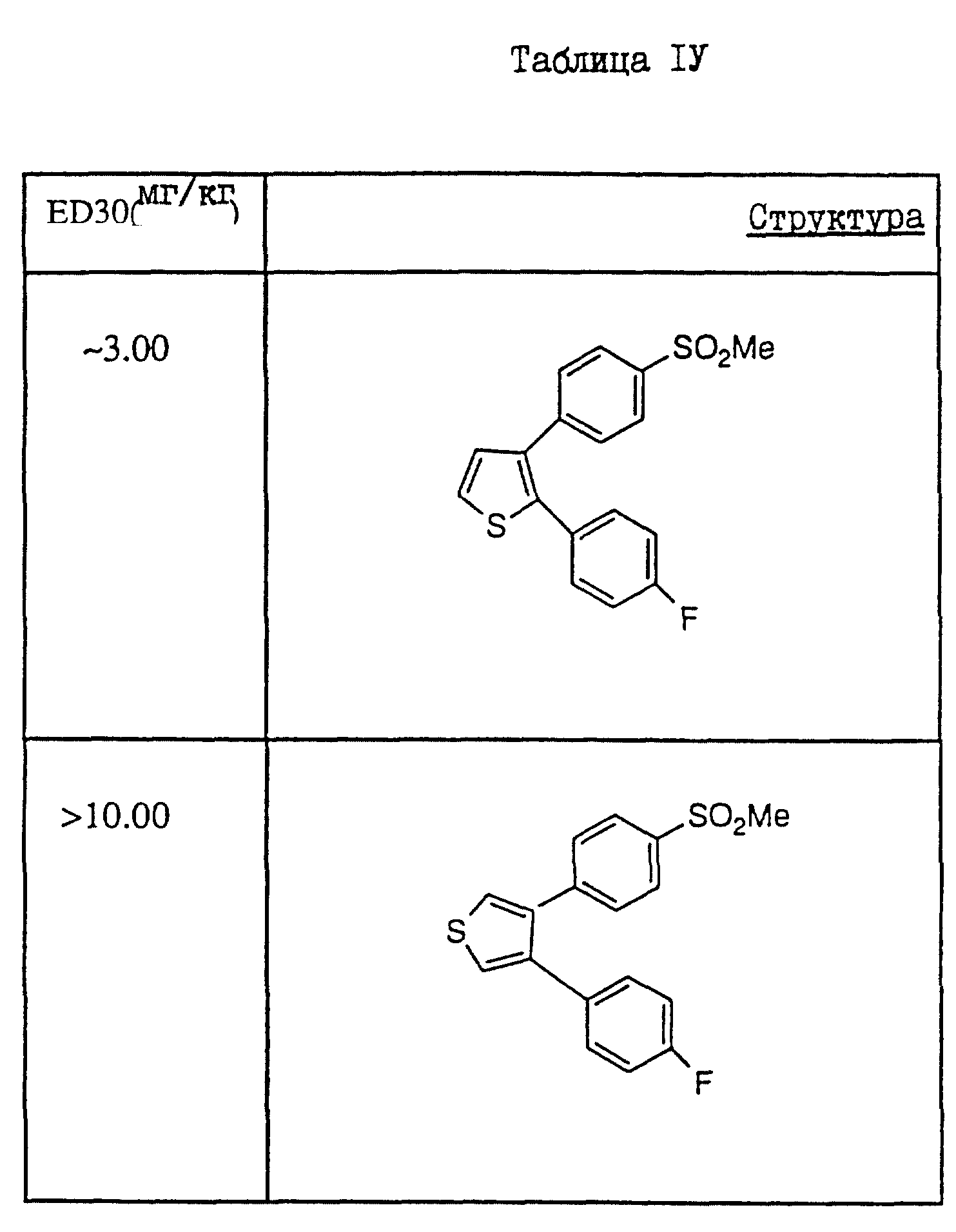
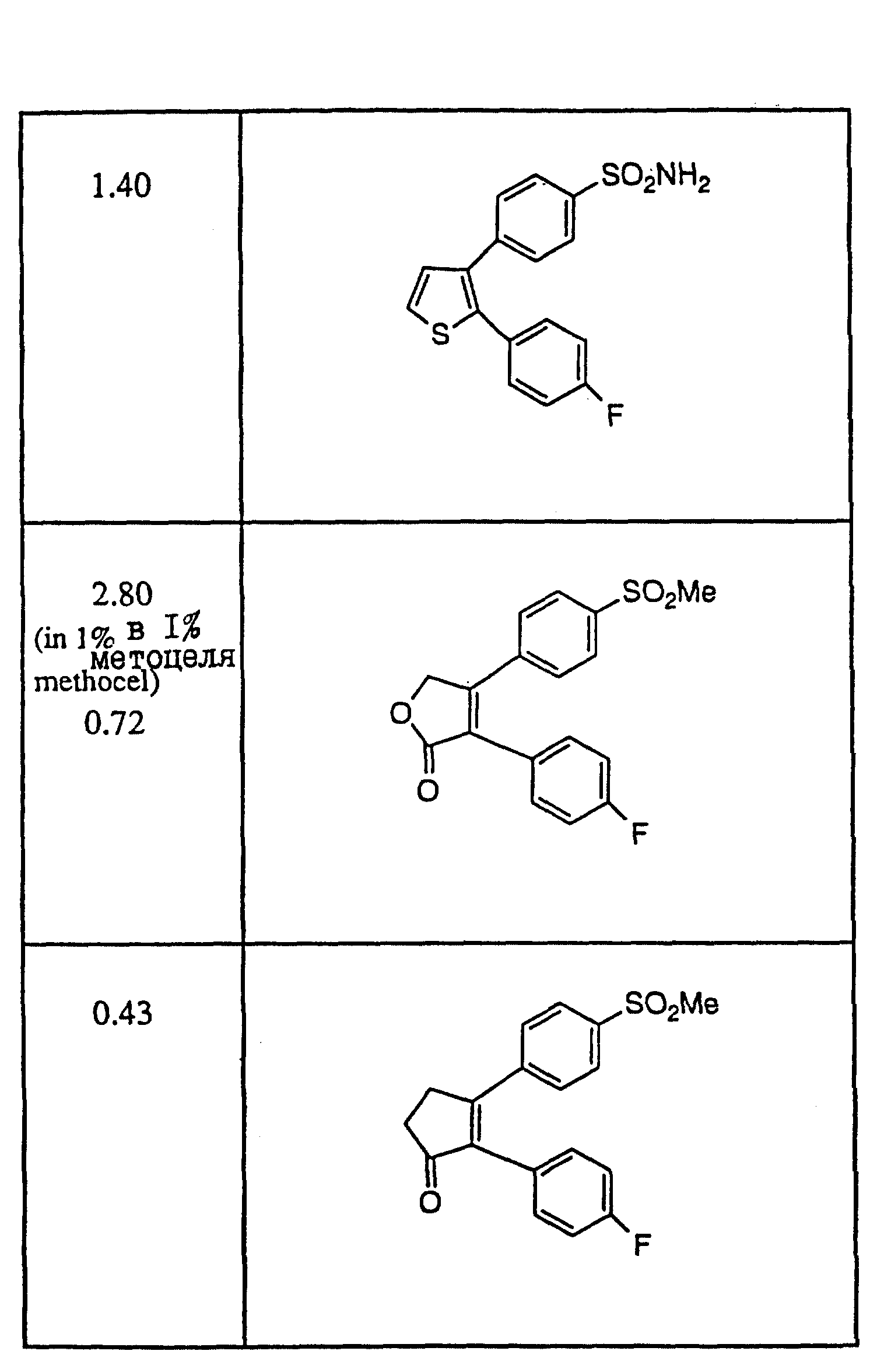
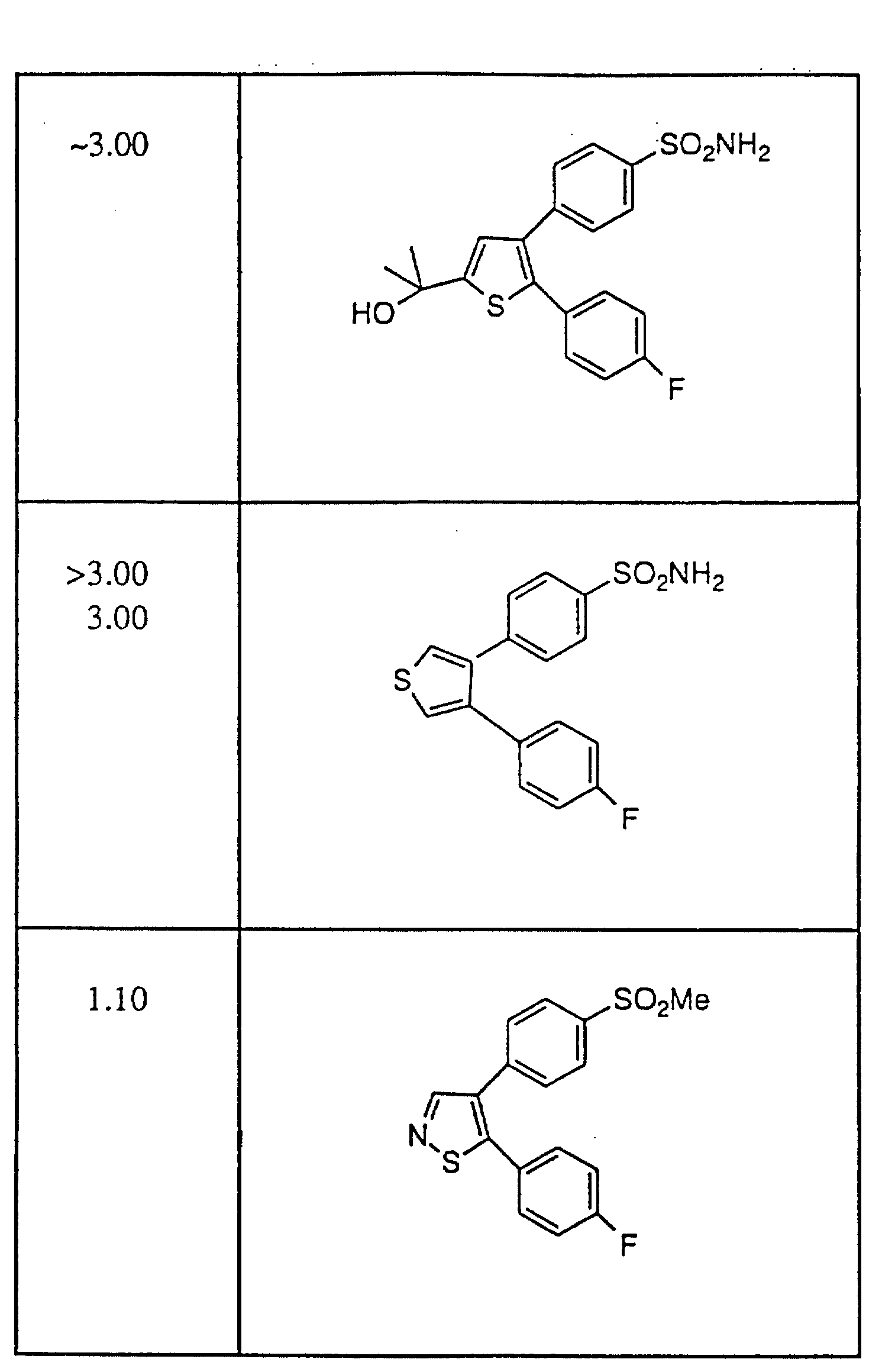
максимум 60-70% ингибирования (отек лапы) был получен с использованием
нестероидных противовоспалительных средств
(НСПВС), то для сравнения были использованы ED3-величины. Все группы
обработанных крыс были закодированы во избежание систематической ошибки
исследователя. В соответствии с описанной
схемой, ED3 для Индометацина составляла 1,0 мг/кг. Репрезантивные
результаты проводятся в Таблице IV (см. в конце описания)
Настоящее
изобретение проиллюстрировано нижеследующими
не ограничивающими изобретение примерами, в которых, если это не оговорено
особо
(i) все операции проводили при комнатной температуре или при
температуре окружающей среды, т.е. при
18-25oC; выпаривание растворителя осуществляли с использованием роторного
испарителя при пониженном давлении (600-40000 Па, 4,5-30 мм рт. ст.), при
этом температура бани составляла до
60oC; за ходом реакции следили с помощью тонкослойной хроматографии (ТСХ),
причем указанное в примерах время реакция носит лишь иллюстративный
характер; точки плавления даны без поправок,
а "d" означает "разложение"; указанные точки плавления представляют собой температуры
плавления соединений, полученных как описано в примере; при
некоторых способах получения полиформизм может
приводить к получению продуктов с различными точками плавления; структура и чистота всех
конечных продуктов были подтверждены, по крайней мере, одним
из следующих способов; ТСХ, масс-спектроскопия,
спектроскопия с использованием ядерного магнитного резонанса (ЯМР), или данными
микроанализа; выходы приводятся лишь в иллюстративных целях; данные
ЯМР, если они имеются, приводятся в виде дельта (
δ )-величины для главных диагностических протонов, и выражены в миллионных
долях (млн.д.) по отношению к тетраметилсилану (TMS),
используемому в качестве внутреннего стандарта, при этом
спектры снимали при частоте 300 МГц или 400 МГц с использованием указанного растворителя;
для обозначения форм сигналов были использованы
следующие сокращения: с - синглет; д - дублет; т - триплет;
м - мультиплет; шир. - широкий; и т.п., "A" означает ароматический сигнал; химические
символы имеют свои обычные значения; при этом были
также использованы следующие сокращения: v (объем), w (масса),
т. кип. (точка кипения, т.пл. (точка плавления); л (литр), мл (миллилитр), г (грамм),
мг (миллиграмм); М (моль), мМ (миллимоль), экв.
(эквивалент).
Указанные ниже аббревиатуры имеют
следующие значения:
Ac - ацетил
Bn - бензил
DBU - 1,
8-диазабицикло 5.4.0 ундек-7-ен
DIBAL
- диизобутилалюминия гидрид
DMAP - 4-(диметиламино)пиридин
DMF (ДМФ) - N,N-диметилформамид
Et3F - триэтиламин
LDA - диизопропиламид лития
m-CPBA
- метахлоропербензоная кислота
MMPP - моноперфталевая
кислота
MPPM - монопероксифталевая кислота, магниевая соль 6H2O
MS - метансульфонил=мезил=SO2
Me
MSO - метансульфонат=мезилат
NSAID (НСПВС)
- нестероидное противовоспалительное средство
OXONER - 2KHSO5 KHSO4 K2SO4
PCC - хлорохромат пиридиния
PDC - дихромат
пиридиния
Ph - фенил
Phe - бензилдиил
Pye - пиридиндиил
r.t. - комнатная температура
rac. - рацемическая
SAM - аминосульфонил или сульфонамид или
SO2NH2
TBAF - фторид тетра-н-бутиламмония
Th
- 2- или 3-тиенил
TFAA - ангидрид
трифторуксусной кислоты
THF - тетрагидрофуран
Thi
- тиофендиил
TCL - тонкослойная хроматография
TMS-CN
- триметилсилилцианид
Tz - 1H (или
2H)-тетразол-5-ил
C3H5 - аллил
Сокращения
алкильных групп
Me - метил
Et - этил
n-Pr
- неразветвленный пропил
i-Pr
- изопропил
n-Bu - неразветвленный бутил
i-Bu - изобутил
s-Bu
- вторичный бутил
t-Bu - третичный бутил
c-Pr
- циклопропил
c-Bu - циклобутил
c-Pen - циклопентил
c-Hex - циклогексил
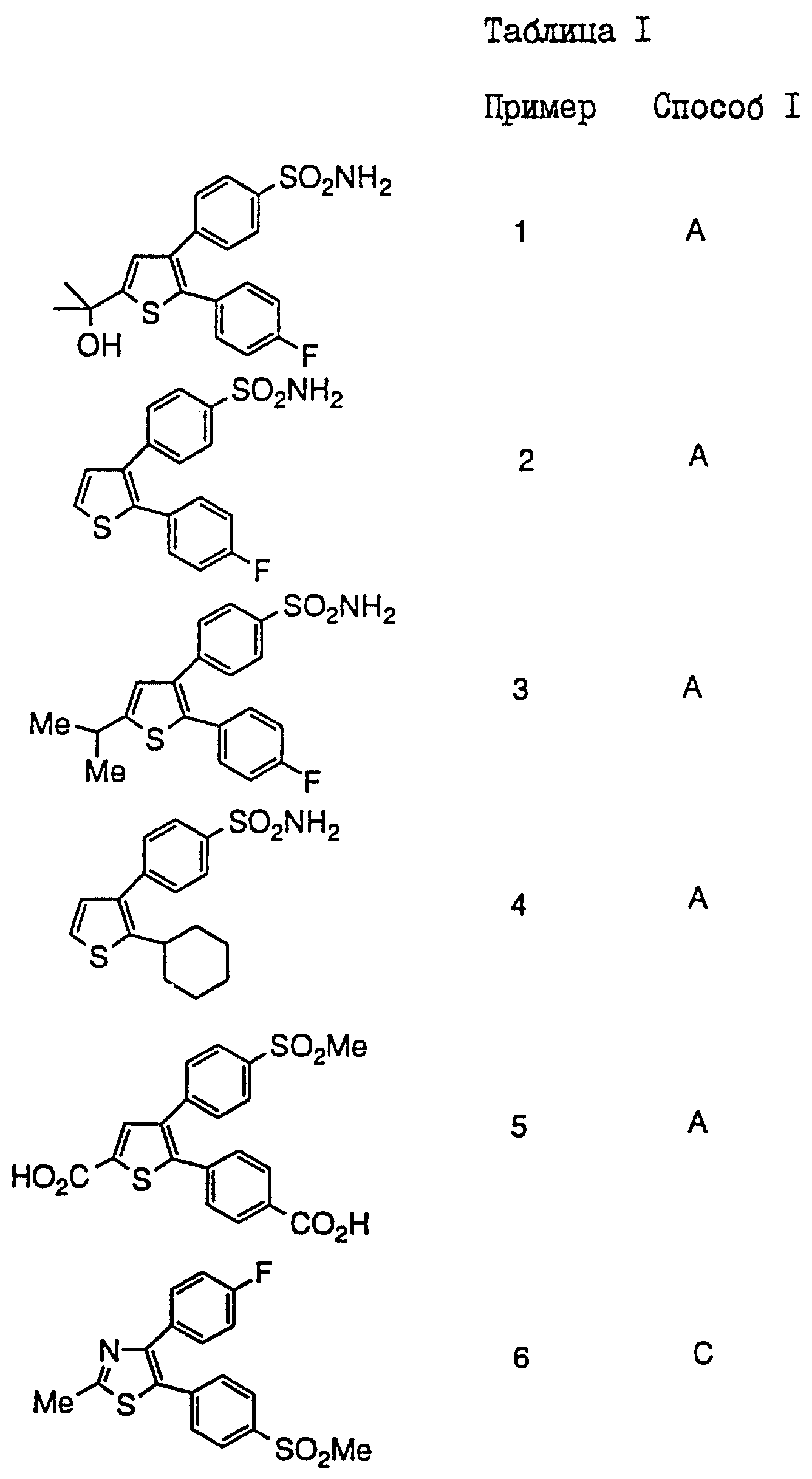
Пример 1
3-(4-Аминосульфонил)фенил-2-(4-фторфенил)-5-(2-гидрокси-2- пропил)тиофен
Стадия 1:
1-(4-фторфенил)-2-(4-(метилтио)фенил)этанон
К 5,40 г 4-фторбензальдегида в 1,2-дихлорэтане (43,50
мл) добавляют TMS-CN (4,32 г) и ZnI2 (44 мг). После выдерживания в течение
получаса при комнатной температуре, растворитель удаляли в вакууме. К полученному триметилсилилцианогидрину (9,
20
г) в тетрагидрофуране (42,0 мл) при температуре -78oC по каплям
добавляли раствор (0,51 М) диизопропиламида лития в тетрагидрофуране (88,9 мл). Через 0,5 часа к смеси по капле в
течение
получаса добавляли тетрагидрофурановый раствор (30,0 мл)
4-(хлорметил)тиоанизола (9,93 г). После выдерживания в течение 18 часов при температуре +5oC, полученную смесь
обрабатывали 57,5
миллилитрами TBAF, а затем 100 миллилитрами 25%-ного
водного раствора NH4OAc, и наконец, экстрагировали этилацетатом (2 х 150 мл). После выпаривания, к неочищенному
кетону добавляли смесь
(10:1) Et2O и гексана (200 мл).
Полученную смесь перемешивали в течение 10 часов и фильтровали, в результате чего получали 2,40 г целевого продукта в виде твердого
вещества.
1H-ЯМР (CD3COCD3): δ 2,45 (3H, с), 4,34 (2H, с), 7,19 - 7,29 (6H, м), 8,14 (2H, кв).
Стадия 2:
Цис- и
транс-3-хлор-3-(4-фторфенил)-2-(4-метилтио)фенил)пропенал
В
раствор 1-(4-фторфенил)-2-(4-метилтио)фенилэтанона (2,50 г) в 1,2-дихлорэтане (27,0 мл) вводили реактив Вильсмайера (каталог
AIdrich, 1992-1993) (3,3 М, 11,6 мл) и DMAP (1,17 г). После выдерживания
в течение 4 часов при температуре 80oC, реакционную смесь экстрагировали этилацетатом и 25%-ным водным раствором
NH4OAc. После выпаривания в вакууме и сушки в течение
нескольких часов был получен целевой продукт, который непосредственно использовали в последующей стадии.
1 H-ЯМР (CD3COCD3): δ 2,40 и 2,48 (3H, 2с), 6,90 - 7,80 (8H, м), 9,55 (1H, с).
Стадия 3: 5-(4-Фторфенил)-4-(4-(метилтио)фенил)тиофен-2- карбоновой кислоты
метиловый сложный эфир
К раствору цис,
транс-3-хлор-3-(4-фторфенил)-2-(4-метилтио) фенил)пропенала (3,00 г) в пиридине (12,0 мл) добавляли метилтиогликолят (1,16 мл) и Et3N (4,09
мл). Полученную смесь нагревали в течение 2
часов при температуре 80oC. После экстрагирования этилацетатом и промывки 3 и соляной кислотой был получен целевой продукт (2,00 г), который
очищали с помощью флэш-хроматографии (элюент:
30% EtOAc в гексане).
1H-ЯМР (CD3COCD3): δ 2,48 3,88 (3H, 2с), 7,11 (2H, т), 7,21 (4H, с), 7,37 (2H, кв), 7,80 (1H, с).
Стадия
4: 5-(4-Фторфенил)-4-(4-(метилтиосульфинил)фенил)тиофен-2- карбоновой кислоты метиловый сложный эфир
К раствору, содержащему 5,60 г метилового
сложного эфира
5-(4-фторфенил)-4-(4-метилтио)фенил)тиофен-2-карбоновой кислоты в метиленхлориде (84,0 мл) при температуре 0oC, порциями добавляли 50-60% м-CPBA (5,39 г). После завершения
реакции, о чем
свидетельствовали ТСХ (50% EtOAc в гексане), реакционную смесь экстрагировали насыщенным гидроксикарбонатом натрия, осушали сульфатом натрия, фильтровали и выпаривали досуха, в
результате чего
получали целевое соединение (5,00 г) в виде белой пены.
1H-ЯМР (CD3COCD3) δ 2,75 (3H, с), 3,92 (3H, с), 7,15 (2H, т), 7,40 (2H, кв), 7,52 (2H, д), 7,66 (2H, д), 7,90 (1H, с).
Стадия 5: 4-(4-Аминосульфонил)фенил)-5-(4-фторфенил)тиофен-2- карбоновой кислоты метиловый сложный эфир
5-(4-Фторфенил)-4-(4-метилсульфонил)фенил)тиофен-2-карбоновой кислоты метиловый сложный эфир (0,500 г) растворяли в TFAA (10,0 мл) и нагревали с обратным холодильником в течение получаса. Затем
растворитель удаляли в вакууме, и полученный остаток 10-кратно выпаривали совместно с раствором Et3N-MeOH (1:1) (100,0 мл) и после накачки в течение нескольких часов получали вязкое
маслообразное вещество. Это маслообразное вещество растворяли в HOAc (10,0 мл) и обрабатывали при температуре +10oC раствором Cl2 в HOAc (1,9 М) (3,5 мл). Через 20 минут
растворитель удаляли при пониженном давлении, и после накачки к полученной массе продукта добавляли 20,0 мл тетрагидрофурана. После барботирования через смесь NH3 в течение нескольких
минут
при температуре 0oC, полученную реакционную смесь перемешивали в течение получаса при комнатной температуре. Затем эту смесь экстрагировали раствором EtOAc в 25% NH4OAc,
и
подвергали флэш-хроматографии (элюент: 30-40% EtOAc в гексане), в результате чего получали целевой продукт (0,210 г) в виде белого твердого вещества.
1H-ЯМР (CD3 COCD3) δ 3,90 (3H, с), 6,55 (2H, шир.с), 7,13 (2H, т), 7,40 (2H, кв), 7,46 (2H, д), 7,83 (2H, д), 7,90 (1H, с).
Стадия 6:
3-(4-Аминосульфонил)фенил)-2-(4-фторфенил)-5-(2- гидрокси-2-пропил)тиофен
К метиловому сложному эфиру 4-(4-аминосульфонил)фенил)-5-(4- фторфеил)тиофен-2-карбоновой кислоты (0,460 г) в
тетрагидрофуране (5,70 мл) при температуре 0oC добавляли MeMgBr (1,4 М) в растворе толуола и ТГФ (5,00 мл). Затем полученную смесь перемешивали в течение нескольких часов при комнатной
температуре. Реакционную смесь гасили путем добавления 25%-ного раствора NH4OAc, экстрагировали этилацетатом, и осушали сульфатом натрия. После очистки с помощью флэш-хроматографии
(элюент:
40-50% EtOAc в гексане) получали 0,300 г целевого соединения.
1H-ЯМР (CD3COCD3) δ 1,65 (6H, с), 4,52 (1H, с), 6,55 (2H, шир.с), 7,09 (3H, м), 7, 34 (2H, дд), 7,30 (2H, м), 7,43 (2H, д), 7,82 (2H, д).
Анализ для C19H18FNO3S2:
Вычислено: C 58,31; H 4,60; N 3,58;
Найдено: C 57,94; H 4,66; N 3,44;
Пример 2
3-(4-Аминосульфонил)фенил)-2-(4-фторфенил)тиофен
Стадия 1:
4-(4-Аминосульфонил)фенил)-5-(4-фторфенил)тиофен-2- карбоновая
кислота
К раствору метилового сложного эфира 4-(4-аминосульфонил)фенил)-5-(4-фторфенил)тиофен-2-карбоновой кислоты (Пример 1,
Стадия 5) (0,210 г) в тетрагидрофуране (2,0 мл) добавляли MeOH
(1,
0 мл), 1 н. NaOH (1,0 мл) и несколько капель 10 н. NaOH. Полученную смесь нагревали в течение 2 часов при температуре 45o
C, а затем, реакционную смесь разделяли между этилацетатом и 3
н.
соляной кислотой, в результате чего получали 0,200 г целевого продукта в виде белого твердого вещества.
1 H-ЯМР (CD3COCD3) δ 6,60 (2H, с), 7, 15 (2H, т), 7,35 (2H, кв), 7,45 (2H, д), 7,82 (2H, д), 7,87 (1H, с).
Стадия 2:
3-(4-(Аминосульфонил)фенил)-2-(4-фторфенил)тиофен
К раствору
3-(4-аминосульфонил)фенил)-2-(4-фторфенил)тиофен-2-карбоновой кислоты (0,280 г) в хинолине (4,0 мл) добавляли Cu бронзу (0,300
г). После выдерживания в течение получаса при температуре 180o
C в атмосфере азота, реакционную смесь экстрагировали этилацетатом и 3 н. соляной кислотой, а затем осушали сульфатом натрия,
и очищали с помощью флэш-хроматографии (элюент: 30% этилацетата в
гексане)
в результате чего получали 0,180 г целевого соединения в виде белого твердого вещества.
1H-ЯМР (CD3COCD3) δ 6,60 (2H, шир.с), 7,15 (2H, т), 7,29 (1H, д), 7,35 (2H, кв), 7,45 (2H, д), 7,60 (1H, д), 7,83 (2H, д).
Анализ для C16H12FNO3S2:
Вычислено: C 57,65; H 3,60;
N
4,20;
Найдено: C 57,62; H 3,59; N 4,15.
Пример 3
3-(4-Аминосульфонил)фенил)-2-(4-фторфенил)-5-(2-пропил)тиофен
1H-ЯМР (CD3COCD3)
δ 1,40 (6H, д), 3,25 (1H, септ.), 6,58 (2H, шир.с), 7,05 (1H, с), 7,
15 (2H, т), 7,32 (2H, дд), 7,46 (2H, д), 7,80 (2H, д).
Анализ для C19H18
FNO2
S2:
Вычислено: C 60,80; H 4,80; N 3,73;
Найдено: C 60,59; H 4,45; N 3,60.
Пример 4
3-(4-Аминосульфонил)фенил)-2-циклогексилтиофен
1H-ЯМР (CD3)2CO): δ 1,24-1,40 (3H,
м), 1,40 - 1,56 (2H, м), 1,65 - 1,85 (3H, м), 1,90 - 2,0 (2H, м), 3,
18 (1H, м), 6,58 (2H, шир.с), 7,05 (1H, д), 7,37 (1H, д), 7,58
(2H, д), 7,97 (2H, д).
Пример 5
5-(4-Карбоксифенил)-4-(4-метилсульфонил)фенил)тиофен-2- карбоновая кислота
Стадия 1: 4-(2-(4-Метилтиофенил)-1-оксоэтил)бензойной
кислоты метиловый сложный эфир
К раствору метил
4-формилбензоата (10,30 г) в 1,2-дихлорэтане при комнатной температуре добавляли
TMS-CN (6,58 мл) и ZnI2 (2,00 г), а после
выдерживания в течение получаса при комнатной температуре
растворитель удаляли в вакууме. К полученному раствору триметилсилилцианогидрида (5,00
г) в тетрагидрофуране (22,0 мл) при температуре -78oC по капле добавляли раствор LDA (0,87 М), в
тетрагидрофуране (26,2 мл). После выдерживания в течение получаса к смеси в течение 0,5
часа по капле добавляли тетрагидрофурановый раствор (10,0
мл) 4-(хлорометил)тиоанизола. Температуру смеси
медленно доводили до -20oC, а затем до 5oC в течение 2 часов, после
чего добавляли 1М TBAF в ТГФ (50,0 мл). После добавления
25%-ного водного раствора NH4OAc
реакционную смесь экстрагировали этилацетатом, осушали сульфатом натрия, выпаривали в вакууме и
очищали с помощью флэш-хроматографии (элюент: 20-30% EtOAc в
гексане), в результате чего получали 7,
00 г целевого соединения в виде белого твердого вещества.
Стадия 2:
4-(1-Оксо-2-(4-метилсульфонил)фенил)этил)бензойной кислоты метиловый сложный
эфир
К 7,10 г метилового
сложного эфира 4-(2-(4-метилтиофенил)-1-оксоэтил)бензойной кислоты в MeOH (100 мл)
добавляли Оксон (21,0 г) в H2O (20,0 мл) при температуре 0oC.
После выдерживания в течение
нескольких часов при комнатной температуре реакционную смесь экстрагировали
этилацетатом и водой, а затем подвергали флэш-хроматографии (элюент: 50-100% EtOAc в гексане),
в результате чего получали
3,20 г целевого продукта в виде белого твердого вещества.
1H-ЯМР (CD3COCD3): δ 3,10 (3H, с), 3,95 (3H, с), 4,65 (2H, с), 7,60 (2H, д), 7,96 (2H, д), 8,20 (4H, кв).
Стадия 3: Цис- и
транс-4-(1-хлор-3-оксо-2-(4-(метилсульфонил) фенил)-1-пропенил)бензойной кислоты метиловый сложный эфир
К
раствору
4-(1-оксо-2-((4-метилсульфонил)фенил)этил)бензойной кислоты (1,70 г) в 1,
2-дихлорэтане (15,0 мл) добавляли реактив Вильсмайера (3,3 М; 6,2 мл) и DMAP (0,624 г). Полученную смесь нагревали в
течение
4 часов при 80oC. Затем реакционную смесь экстрагировали
25%-ным водным раствором NH4OAc и EtOAc. После сушки сульфатом натрия и выпаривания было получено целевое
соединение в
виде маслообразного вещества, которое непосредственно использовали в
последующей стадии.
Стадия 4: 5-(4-(Метоксикарбонил)фенил)-4-(4-(метилсульфонил)
фенил)-тиофен-2-карбоновой
кислоты метиловый сложный эфир
Целевое соединение получали из
метилового сложного эфира 4-(1-хлоро-3-оксо-2-(4-метилсульфонил)фенил)-1-пропенил)бензойной кислоты,
как описано в Стадии 3
Примера 1.
1H-ЯМР (CD3COCD3 ) δ 3,13 (3H, с), 3,85 и 3,92 (6H, 2с), 7,50 (2H, д), 7,55 (2H, д), 7,90 (2H, д), 7,92 (1H, с), 7,92 (2H, д).
Стадия 5:
5-(4-(Карбоксифенил)-4-(4-(метил)сульфонил)фенил- тиофен-2-карбоновая кислота
Целевое соединение получали из метилового сложного эфира
5-(4-(метоксикарбонил)фенил)-4-(4-(метил)сульфонил)фенил)тиофен-2- карбоновой кислоты, как описано в Стадии 1 Примера 2.
1H-ЯМР (CD3COCD3) δ 3, 15 (3H, с), 7,50 (2H, д), 7,62 (2H, д), 7,95 (2H, д), 7,98 (1H, с), 8,05 (2H, д).
Анализ для C19H14O6S2 • 0,1 H2O
Вычислено: C 56,46; H 3,51;
Найдено: C 56,18; H 3,51;
Пример 6
4-(4-Фторфенил)-2-метил-5-(4-метилсульфонил)фенил)тиазол
Стадия 1:
1-(4-Фторфенил)-2-(4-метилсульфонил)фенил)этанон
К 17,9 г 1-(4-Фторфенил)-2-(4-(метилтио)фенил)этанона (Стадия 1 Примера 1) в растворе метиленхлорида и MeOH (272,0 мл/27,0 мл) при
температуре
0oC добавляли MPPM (28,0 г). После удаления охлаждающей бани, реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем при температуре 0oC
добавляли еще
28,0 г MPPM и реакционную смесь выдерживали в течение полутора часов при комнатной температуре. Нерастворившиеся вещества фильтровали, растворители выпаривали, а образовавшийся
остаток
экстрагировали
смесью бикарбоната натрия и метиленхлорида. После выпаривания в вакууме, полученное твердое вещество промывали смесью эфира и гексана (1:1), а затем фильтровали, в результате
чего
получали 16,8 г
целевого соединения.
1H-ЯМР (CD3COCD3): δ 3,13 (3H, с), 3,58 (2H, с), 7,29 (2H, т), 7,55 (2H, д), 7,88 (2H, д), 8,20 (2H, дд).
Стадия 2: 2-Бром-1-(4-фторфенил)-2-(4-метилсульфонил)фенил) этанон
К 1,00 г 1-(4-фторфенил)-2-(4-метилсульфонил)фенил)этанона в метиленхлориде, содержащем CHCl3 (1,0 мл) и CCl4 (1,0 мл), добавляли 0,614 г брома. После 1-часового облучения светом, реакцию гасили путем добавления Na2S2O4, а затем
экстрагировали метиленхлоридом, осушали
сульфатом натрия, и выпаривали, в результате чего получали целевое соединение (1,10 г), которое непосредственно использовали в последующей стадии.
1H-ЯМР (CD3 COCD3): δ 3,10 (3H, с), 7,05 (1H, с), 7,30 (2H, т), 7,87 (2H, д), 7,95 (2H, д), 8,25 (2H, дд).
Стадия 3:
4-(4-Фторфенил)-2-метил-5-(4-метилсульфонил)фенил) тиазол
К раствору 2-бромо-1-(4-фторфенил)-2-(4-метилсульфонил)фенил)- этанона (1,10 г) в этаноле (15,0 мл) добавляли тиоацетамид (0,266 г)
и
пиридин (0,300 мл). После нагревания с обратным
холодильником в течение 2 часов, реакционную смесь экстрагировали этилацетатом и 25% NH4OAc, а затем очищали с помощью
флэш-хроматографии
(элюент: 50% этилацетата в гексане, а затем 90%
Et2O в гексане), в результате чего получали 0,320 г целевого соединения.
1H-ЯМР (CD3COCD3 ): δ 2,72 (3H, с), 3,15 (3H, с), 7,09 (2H, т), 7,52 (2H, дд), 7,60 (2H, д), 7,92 (2H, д).
Анализ для C17H14FNO2S2:
Вычислено:
C 58,78; H 4,03; N 4,03;
Найдено: C 58,
71; H 4,17; N 3,85.
Пример 7
2-(4-Фторфенил)-3-(4-(метилсульфонил)фенил)-2-циклопентен-1-он
Стадия 1:
1-(4-Фторфенил)-5-гексен-2-он
К суспензии, состоящей
из 14,6 г (80 мМ) CdCl2 и 200 мл эфира, и охлажденной до 0oC, по капле добавляли 115 мл (1,3 М)
раствора
3-бутен-1-магнийбромида. Полученную смесь нагревали с обратным
холодильником в течение 1 часа, после чего эфир удаляли путем дистилляции. Затем в эту смесь вводили 500 мл бензола, после
чего
добавляли 17,5 г раствора 4-фторфенилацетилхлорида (100 мМ). После
нагревания с обратным холодильником в течение одного часа реакцию гасили путем добавления 200 мл насыщенного водного раствора
NH4Cl и 50 мл 1 н, соляной кислоты, а затем эту смесь
экстрагировали 200 миллилитрами смеси (1: 1) гексана и EtOAc. Органическую фазу осушали сульфатом магния и концентрировали. Остаток
очищали с
помощью флэш-хроматографии, элюируя смесью (4:1) гексана и
этилацетата, в результате чего получали 15 г целевого продукта.
1H-ЯМР (CDCl3): δ 2, 40 (2H, т), 2,53 (2H, т), 3,63 (2H, с), 4,90 - 4,98 (2H, м), 5,67 - 5, 78 (1H, м), 6,98 (2H, т), 7,13 (2H, м).
Стадия 2: 1-(4-Фторфенил)-5-оксо-2-пентанон
Раствор
1-(4-Фторфенил)-5-гексен-2-она (14 г) в 200 мл смеси (3:1) метиленхлорида и MeOH
охлаждали до температуры -78oC и обрабатывали избыточным количеством озона. Полученную смесь обрабатывали
15 граммами трифенилфосфина и перемешивали при комнатной температуре в течение
одного часа. Затем реакционную смесь концентрировали и подвергали флэш-хроматографии, используя в качестве элюента
смесь
(3:1) гексана/EtOAc, в результате чего получали 8 г целевого продукта
кетоальдегида.
1H-ЯМР (CDCl3): δ 2,72 (4H, с), 3,71 (2H, с), 6,99 (2H, т), 7, 14 (2H, м), 9,73 (1H, с).
Стадия 3:
2-(4-Фторфенил)-2-оксо-циклопентен-1-он
Раствор из 8 г 1-(4-Фторфенил)-5-оксо-2-пентанона в 300 мл MeOH обрабатывали 2 граммами
NaOMe.
Реакционную смесь перемешивали в течение 2 часов, а
затем гасили путем добавления 5 мл HOAc. После выпаривания растворителя остаток очищали с помощью флэш-хроматографии, используя в качестве
элюента
смесь (3:1) гексана/EtOAc, в результате чего получали
7 г целевого продукта кетоальдегида.
1H-ЯМР (CDCl3): δ 2,57 (2H, м), 2,68 (2H, м), 7,04 (2H, J = 8, 8 Гц, т), 7,67 (2H, J = 8,8, 5,5 Гц, дд), 3,71 (2H, с), 7,77 (1H, м).
Стадия 4: 1-(4-(Метилтио)фенил)-2-(4-фторфенил)-2-циклопентен- 1-ол
К раствору 3,86 г (19
мМ)
4-бромтиоанизола в 90 мл Et2O, охлажденному при -78oC, по капле добавляли 22 мл 1,7 М раствора т-бутиллития в пентане (38 мМ). Реакционную смесь перемешивали в течение 15
минут
при температуре -78oC, а затем добавляли раствор 2,23 г
2-(4-фторфенил)-2-циклопентен-1-она в 10 мл Et2O. После перемешивания в течение 15 минут при температуре -78o
C,
реакционную смесь нагревали до 0oC, и гасили путем
добавления 50 мл насыщенного NH4Cl. Полученный продукт экстрагировали 100 миллилитрами этилацетата, осушали
сульфатом
натрия
и очищали с помощью флэш-хроматографии (элюент: смесь (4:1)
гексана/этилацетата), в результате чего получали 3,4 г целевого продукта.
1H-ЯМР (CDCl3): δ 2, 12 (1H, с), 2,34 (2H, м), 2,44(3Н,с), 2,45 - 2,52 (1H, м), 2, 56 - 2,65 (1H, м), 6,37 (1H, м), 6,84 (2H, J = 8,7 Гц, т), 7,17 (2H, J = 8,3 Гц, д), 7,24 - 7,33 (4H, м).
Стадия 5:
2-(4-Фторфенил)-3-(4-(метилтио)фенил)-2-циклопентен-1- он
К суспензии, состоящей из PCC (4,5 г, 20,9 мМ) и 10 г безводных молекулярных сит 4

1H-ЯМР (CDCl3): δ 2,45 (3H, с), 2,68 (2H, м), 3,00 (2H, м), 7,02 (2H, J = 8,6 Гц, т), 7,11 (2H, J = 8,6 Гц, д), 7,15 - 7,23 (4H, м).
Стадия 6:
2-(4-Фторфенил)-3-(4-(метилсульфонил)фенил)-2-циклопентен-1-он
К
раствору 50 мг (0,17 мМ) 2-(4-фторфенил)-3-(4-метилтио)фенил)- 2-циклопентен-1-она в 8 мл
смеси (10:1) CH2Cl2/MeOH добавляли 124 мг (0,2 мМ) MPPM. Реакционную смесь
перемешивали
при комнатной температуре в течение 2 часов, а затем разводили 10 миллилитрами смеси (1:1)
гексана/этилацетата. После
фильтрования и концентрирования остаток очищали с помощью
флэш-хроматографии,
используя в качестве элюента смесь (2:1) EtOAc/гексана, в результате чего получали 45 мг
целевого продукта.
1H-ЯМР (ацетон-d6): δ 2, 67 (2H, м), 3,14 (3H, с), 3,16 (2H, м), 7,05 - 7,10 (2H, м), 7,20 - 7,25 (2H, м), 7,63 (2H, д), 7,93 (2H, д).
Пример
8
4-(4-(Метилсульфонил)фенил)-5-(4-фторфенил)-изотиазол
К раствору 338
мг (1 мМ) цис, транс-3-хлор-3-(4-фторфенил)-2- (4-(метилсульфонил)фенил)пропеналя в 5 мл
ацетона добавляли
230 мг (3 мМ)
NH4SCN. Реакционную смесь нагревали с обратным холодильником в
течение 3 часов, а затем гасили 20 миллилитрами насыщенного бикарбоната натрия. Полученный
продукт
экстрагировали 100
миллилитрами этилацетата, осушали сульфатом натрия, концентрировали и очищали с
помощью флэш-хроматографии, используя в качестве элюента смесь (3:2) гексана/EtOAc, в
результате
чего получали 250 мг
целевого продукта.
1H-ЯМР (CDCl3): δ 8,57 (1H, с), 7,93 (3H, д), 7,50 (2H, д), 7,30 (2H, т), 7,08 (2H, т).
Пример
9
3-(4-Фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Стадия 1:
2-Бром-1-(4-(метилсульфонил)фенил)этанон
К раствору 197 мг 4-(метилтиол)ацетофенона (JACS, 1952,
74
стр. 54-75)
в 700 мл MeOH и 3500 мл метилхлорида добавляли в течение 30 минут 881 г MMPP.
После выдерживания в течение 3 часов при комнатной температуре реакционную смесь фильтровали и полученный
фильтрат
промывали 2 литрами насыщенного водного раствора бикарбоната натрия и 1 литром
солевого раствора. Водную фазу экстрагировали 2 литрами метиленхлорида. Объединенные экстракты осушали
сульфатом
натрия,
концентрировали и получали 240 г 4-(метилсульфонил)ацетофенона в виде белого
твердого вещества.
К охлажденному (-5oC) раствору 174 г 4-(метилсульфонил)ацетофенона в 2,5 л CHCl3 добавляли 20 мг AlCl3, а затем раствор 40 мл Br2 в 300 мл CHCl3. Затем реакционную смесь обрабатывали 1,5 литрами воды, а CHCl3 отделяли. Водный слой экстрагировали 1 литром этилацетата. Объединенные экстракты осушали сульфатом натрия и концентрировали. Неочищенный продукт перекристаллизовывали из смеси EtOAc/гексана (50/50) и получали 210 г 2-бром-1-(4-(метилсульфонил)фенил)этанона в виде белого твердого вещества.
Стадия 2:
К продукту
Стадии 1 (216 мг), растворенному в ацетонитриле (4 мл),
добавляли Et3N (0,26 мл), а затем 4-фторфенилуксусную кислоту (102 мг).
После выдерживания в течение полутора часов при комнатной
температуре к смеси добавляли 0,23 мл DBU. Реакционную
смесь перемешивали еще 45 минут, а затем обрабатывали 5 миллилитрами 1 н. соляной
кислоты. Полученный продукт экстрагировали этилацетатом,
осушали сульфатом натрия и концентрировали. Остаток очищали
с
помощью флэш-хроматографии, используя в качестве элюента 40% этилацетата в
гексане, в результате чего получали 150 мг целевого
соединения в виде твердого вещества.
1 H-ЯМР (CD3COCD3): δ 3,15 (3H, с), 5,36 (3H, с), 7,18 (2H, J = 8,9 Гц, т), 7,46 (2H, м), 7,7 (2H, J = 8, 65 Гц, д), 7,97 (2H, J = 8,68 Гц, д).
Пример
10
3-(4-Фторфенил)-4-(4-(аминосульфонил)фенил)-2-(2H)-фуранон
1H-ЯМР (CD3COCD3):
δ 5,34 (2H, с), 6,67 (2H, шир.д), 7,18 (2H, м), 7,46 (2H, м),
7,61 (2H, м),
7,90 (2H, м).
Т.пл. 5: 187-188oC
(разлож.)
Пример 11
3-(4-Фторфенил)-4-(4-(метилсульфонил)фенил)фуран
Стадия 1:
К
продукту Примера 10
(0,2 г) в тетрагидрофуране (5 мл) и толуоле (3 мл) медленно,
при температуре -78oC,
добавляли раствор DIBAL (0,72 мл 1 М в толуоле). Через 15 минут, раствор нагревали
до температуры 0oC в течение 15 минут. Эту смесь выливали в охлажденный
раствор воды и тартрата
калия-натрия в этилацетате. Органический слой перемешивали в течение получаса с несколькими
кристаллами
камфарсульфоновой кислоты. Затем этот раствор концентрировали и очищали
с помощью
флэш-хроматографии, с получением целевого соединения.
1H-ЯМР (CDCl3 ): δ 3, 1 (3H, с), 7,02 (2H, J = 8,9 Гц, т), 7,18 (2H, м), 7,4 (2H, J = 8,8 Гц, д), 7, 58 (1H, с), 7,68 (1H, c), 7,85 (2H, J = 8,8 Гц, д).
Пример 12
5,
5-Диметил-3-(4-фторфенил)-4-(4-метилсульфонилфенил)-2-(5H)- фуранон
Стадия 1: Метил
2-триметилсилилоксиизобутират
К раствору 1,2 мл (10,4 мМ) метил 2-гидроксиизобутирата в 50 мл
метиленхлорида добавляли 1,2 г (17,6 мМ) имидазола и 2,1 мл (16,6 мМ) TMSCI. Полученную смесь
перемешивали при комнатной температуре в течение полутора часов, а затем гасили путем добавления 20 мл
H2O. Органический слой осушали сульфатом магния, концентрировали и пропускали через
узкую пробку из силикагеля, элюируя гексаном/этилацетатом (9:1). После выпаривания растворителя
получали
1,27 г целевого соединения в виде бесцветного маслообразного вещества.
1H-ЯМР (CD3COCD3): δ 0,08 (9H, с), 1,38 (6H, с), 3,67 (3H, с).
Стадия 2: 2-Триметилсилилокси-4'-(метилтио)изобутирофенон
Раствор 204 мг
(1,
0 мМ) 4-бромтиоанизола в 2,5 мл тетрагидрофурана охлаждали до -78oC и обрабатывали
0,42
миллилитрами (2,5 М) раствора н-бутиллития в гексане. После перемешивания при температуре
-78oC в течение 1 часа к смеси добавляли раствор метил 2-триметилсилилоксиизобутирата (380
мг, 2,0
мМ) в 2 мл тетрагирофурана. Затем смесь перемешивали в течение 2 часов при -78oC,
после чего гасили путем добавления NH4OAc-буфера. Полученный продукт экстрагировали
этилацетатом,
осушали сульфатом магния и концентрировали. Остаток очищали с помощью
флэш-хроматографии,
используя в качестве элюента смесь (19:1) гексана/EtOAc, в результате чего получали 95 мг
целевого
продукта.
1H-ЯМР (CD3COCD3): δ 0,05 (9H, с), 1,52 (6H, с), 2,53 (3H, с), 7,33 (2H, д), 8,12 (2H, д).
Стадия 3:
2-Гидрокси-4'-(метилтио)изобутирофенон
К раствору
2-триметилсилилокси-4'-(метилтио)изобутирофенона (40 мг,
0,14 мМ) в 2 мл ТГФ добавляли 0,2 мл 1 М н-Bu4NF в ТГФ. Полученную
смесь
перемешивали в течение 30 минут, а затем гасили 10
миллилитрами NH4OAc-буфера. Полученный продукт
экстрагировали этилацетатом, осушали сульфатом магния и концентрировали. Остаток
очищали с
помощью флэш-хроматографии, используя в качестве
элюента гексан/этилацетат (4:1), в результате чего получали
25 мг целевого продукта.
1H-ЯМР (CD3 COCD3 ): δ 1,50 (6H, с), 2,54 (3H, с), 4,68 (1H, с), 7,30 (2H, д), 8,15 (2H, д).
Стадия 4:
2-(4-(Фторфенилацетокси)-4-(метилтио)изобутирофенон
К раствору
2-гидрокси-4'-(метилтио)изобутирофенона (72 мг, 0,34 мМ) в 1,
7 мл метиленхлорида добавляли 0,2 мл пиридина и 140 мг (0,81 мМ)
4-фторфенилацетилхлорида. Полученную смесь перемешивали при комнатной
температуре в течение ночи, а затем гасили путем добавления
NH4OAc-буфера. Полученный продукт экстрагировали
этилацетатом, осушали сульфатом магния и концентрировали. Неочищенный продукт
очищали с помощью флэш-хроматографии, элюируя
гексаном/этилацетатом (8: 1), в результате чего получали 95 мг целевого
продукта.
1H-ЯМР (CD3COCD3: δ 1,62 (3H, с), 1,67 (3H, с), 2,48 (3H, с), 3,79 (2H, с), 7,0 - 7,3 (6H, м), 7,78 (2H, д).
Стадия
5: 5,5-Диметил-3-(4-фторфенил)-4-(4-метилтиофенил)-2- (5H)-фуранон
К
раствору 95 мг
2-(4-фторфенилацетокси)-4-(метилтио)- изобутирофенона в 4 мл метиленхлорида добавляли 0,2 мл 1,
8-диазабицикло(5.4.0)ундек-7-ена. Полученную смесь перемешивали в течение 4 часов и
разводили NH4OAc-буфером. Полученный продукт экстрагировали этилацетатом, осушали сульфатом магния и
концентрировали. Остаток очищали с помощью флэш-хроматографии, элюируя смесью (20:1)
толуола/этилацетата, в результате чего получали 75 мг целевого продукта.
1H-ЯМР (CD3COCD3: δ 1,58 (6H, с), 2,50 (3H, с), 7,03 (2H, дд), 7,25 - 7, 35 (4H, м), 7,41 (2H, дд).
Стадия 6: 5,
5-Диметил-3-(4-фторфенил)-4-(метилсульфонил)-2-(5H)- фуранон
К раствору 81 мг 5,
5-диметил-3-(4-фторфенил)-4-(4-метил-тиофенил)-2-оксо-2H-дигидрофурана в 1,8 мл метиленхлорида и 0,2 мл MeOH
добавляли 250 мг MPPM. Реакционную смесь перемешивали при комнатной температуре в
течение
1
часа, а затем гасили водным раствором бикарбоната натрия. Полученный продукт экстрагировали этилацетатом,
осушали сульфатом магния и концентрировали. Неочищенный продукт очищали с помощью
флэш-хроматографии, элюируя гексаном/этилацетатом (1:1), в результате чего получали 73 мг целевого продукта.
1H-ЯМР (CD3COCD3): δ 1,62 (6H, c), 3,15 (3H, c), 7,02 (2H, дд), 7,40 (2H, дд), 7,65 (2H, д), 8,03 (2H, д).
Пример 13
2-((4-Аминосульфонил)фенил)-3-(4-фторфенил)тиофен
1H-ЯМР (CD3
COCD3): δ 6,60 (2H, шир.c), 7,12 (2H, т), 7,25 (1H, д), 7,35 (2H, м), 7,45 (2H, д), 7,65
(1H, д), 7,85 (2H, д).
Анализ для C16H12
FNS2
O2:
Вычислено: C 57,65; H 3,60; N 4,20;
Найдено: C 57,55; H 3,79; N 4,03.
Пример 14
3-(4-(Трифторацетиламиносульфонил)фенил)-2-(4-фторфенил)тиофен
1H-ЯМР (300 МГц CD3COCD3): δ 7,15 (2H, т), 7,30 (3H, м),
7,45 (2H, д), 7,65 (1H, д), 7,
95
(2H, д).
Пример 15
3-(2,4-Дифторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12F2O4S:
Вычислено: C 58,28; H 3,45; N 9,15;
Найдено: C 58,27; H 3,50; N 9,27.
Пример 16
3-(3,
4-Дифторфенил)-4-(4-метилсульфонил)фенил)-2-(5H)-фуранон
К раствору
3,
4-дифторфенилуксусной кислоты (ALDRICH CHIMICAL) (10 г) и 2-бромо-1-(4-(метилсульфонил)фенил)этанона (Пример 9, Стадия
1) (17,3 г) в ацетонитриле (200 мл) при комнатной температуре, медленно
добавляя
20,2 мл триэтиламина. После выдерживания в течение 1 часа при комнатной температуре, смесь охлаждали в ледяной бане и
обрабатывали 17,4 миллилитрами DBU. После выдерживания в течение 2
часов при
температуре 0oC эту смесь обрабатывали 200 миллилитрами 1 н. соляной кислоты, и полученный продукт
экстрагировали этилацетатом, осушали сульфатом натрия и концентрировали.
Остаток наносили
на поверхность пробки из силикагеля (воронки из спеченного стекла), используя в качестве элюента 75%
EtOAc/гексан, а затем, после выпаривания растворителя и энергичной продувки в
этилацетате, получали
10 г целевого соединения.
Анализ для C17H12F2
O4S:
Вычислено: C 58,28; H 3,45; S 9,15;
Найдено: C
58,02; H 3,51; S 9,
35.
Пример 17
3-(2,
6-Дифторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12F2O4
S:
Вычислено:
C 58,28; H 3,45; S 9,15;
Найдено: C
58,18; H 3,50; S 9,44.
Пример 18
3-(2,5-Дифторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17
H12F2O4
S:
Вычислено: C 58,28; H 3,45; S 9,15;
Найдено: C 58,89; H 3,51; S 9,11.
Пример 19
3-(3,
5-Дифторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12F2O4S:
Вычислено: C 58,28; H 3,45; S 9,15;
Найдено:
C
58,27; H 3,62; S 9,32.
Пример 20
3-(4-Бромфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H13BrO4S:
Вычислено: C 51,94; H 3,33; S 8,16;
Найдено: C 51,76; H
3,42; S 8,21.
Пример 21
3-(4-Хлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1
H-ЯМР
(300 МГц CDCl3): δ 7,93 (2H, д), 7,49 (2H, д),
7,35 (4H, м), 5,16 (2H, с), 3,06 (3H, с).
Пример 22
3-(4-Метоксифенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C18H16O5S:
Вычислено: C 62,78; H 4,68; S 9,31;
Найдено: C 62,75;
H
4,72; S 9,39.
Пример 23
3-(Фенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
К
раствору фенилуксусной кислоты (27,4 г, 201 мМ) и
бром-1-(4-(метилсульфонил)фенил)этанола (Стадия 1 Примера 9) (60 г, 216 мМ, 1,075 эквивалентов) в ацетонитриле (630 мл) при 25oC медленно
добавляли триэтиламин (30,8 мл, 1,1 экв.).
Полученную смесь перемешивали в течение 20 минут при комнатной температуре, а затем охлаждали в ледяной бане. После этого к смеси медленно добавляли DBU (60,
1 мл, 3 экв.). После перемешивания в
течение 20 минут, в ледяной бане, реакцию полностью завершали и смесь подкисляли 1 н. соляной кислотой (окраска менялась с темно-коричневой на желтую). Затем к
смеси добавляли 2,4 л льда и воды,
перемешивали в течение нескольких минут, после чего образовавшийся осадок фильтровали и промывали водой (с получением 64 г неочищенного влажного продукта). Этот
твердый продукт растворяли в 750 мл
дихлорметана (осушил сульфатом магния и фильтровали) и добавляли 300 г силикагеля. Растворитель выпаривали почти досуха (немного клейкий силикагель), а остаток
наносили на поверхность силикагелевой
пробки (воронки из спеченного стекла), элюируя 10%-ным этилацетатом/метиленхлоридом, после чего, в результате выпаривания растворителя и энергичной продувки в
этилацетате, получали 36,6 г (58%)
целевого соединения.
Анализ для C17H14O4S:
Вычислено: C 64,95; H 4,49; S 10,20;
Найдено: C 64,63;
H 4,65; S 10,44.
Пример 24
3-(2-Хлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H13ClO4S:
Вычислено:
C 58,54; H 3,76; S 9,
19;
Найдено: C 58,59; H 3,80; S 9,37.
Пример 25
3-(2-Бром-4-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12
BrO4
S:
Вычислено: C 49,75; H 2,93;
Найдено: C 49,75; H 3,01.
Пример 26
3-(2-Бром-4-хлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1
H-ЯМР (300 МГц ацетон-d6): δ 7,95 (2H, д), 7,85 (1H, д), 7,63 (2H, дд), 7,55 (1H, дд), 7,45 (1H,
д), 5,50 (2H, с), 3,15 (3H, с).
Пример 27
3-(4-Хлор-2-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1H-ЯМР (300 МГц ацетон-d6):
δ 8,0 (2H, д), 7,70 (2H, д), 7,50 - 7,30 (3H, м), 5,35 (2H,
с), 3,
15
(3H, с).
Пример 28
3-(3-Бром-4-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12BrFO4S:
Вычислено: C
49,75; H 2,93;
Найдено: C 49,44; H 2,98.
Пример 29
3-(3-Хлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H13ClO4S:
Вычислено: C 58,54; H 3,76;
Найдено: C 58,29; H 3,76.
Пример 30
3-(2-Хлор-4-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ
для C17H12BrFO4S:
Вычислено: C 55,67; H 3,
30;
Найдено: C 55,67;
H
3,26.
Пример 31
3-(2,
4-Дихлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12Cl2
O4S:
Вычислено: C 53,28; H 3,16; S 8,37;
Найдено: C
52,89; H 3,23; S 8,58.
Пример 32
3-(3,
4-Дихлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для
C17H12Cl2O4
S:
Вычислено: C 53,28; H 3,16; S 8,37;
Найдено:
C 53,07; H 3,32; S 8,51.
Пример 33
3-(2,
6-Дихлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12Cl2O4
S:
Вычислено: C 53,28; H 3,16; S 8,37;
Найдено:
C 52,99; H 3,22; S 8,54.
Пример 34
3-(3-Хлор-4-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1H-ЯМР (300 МГц ацетон-d6):
δ 8,0
(2H, д), 7,70 (2H, д), 7,60 (1H, д), 7,25 - 7,40 (2H, м), 5,35
(2H, с), 3,15 (3H, с).
Пример 35
3-(4-Трифторметилфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)- фуранон
1H-ЯМР (300 МГц CD3COCD3):
δ 8,10 (2H, д), 7,82 - 7,93 (4H, м), 7,75 (2H, д), 5,55
(22, с), 3,30 (3H, с).
Пример 36
3-(3-Фтор-4-метоксифенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C18H15FO5S:
Вычислено: C 59,66; H 4,17;
Найдено: C 59,92; H
4,37.
Пример 37
3-(3-Хлор-4-метоксифенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для
C18H15ClO5S:
Вычислено: C
57,07; H 3,99;
Найдено: C 57,29; H
4,15.
Пример 38
3-(3-Бром-4-метоксифенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C18H15BrO5S:
Вычислено: C
51,08; H 3,57;
Найдено: C 51,38;
H 3,62.
Пример 39
3-(2-Фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H13FO4S:
Вычислено: C 61,44;
H
3,94;
Найдено: C 61,13; H 3,85.
Пример 40
3-(4-Фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1
H-ЯМР (300 МГц ацетон-d6):
δ 8,0 (2H, д), 7,70 (2H, д), 7,35 (2H, д), 7,25 (2H, д), 5,35 (2H, д), 3,
15 (3H, с), 2,55 (3H, с).
Пример 41
3-(3-Фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1H-ЯМР (300 МГц CDCl3): δ 7,93 (2H, д),
7,49 (2H, д), 7,35 (1H, м), 7,12 (3H, м), 5,18 (2H, с), 3,06
(3H,
с).
Пример 42
3-(2-Хлор-6-фторфенил)-4-(4-метилсульфонил)фенил)-2-(5H)-фуранон
1H-ЯМР
(300 МГц ацетон-d6): δ 8,0 (2H, д), 7,70
(2H,
д),
7,55 - 7,65 (1H, м), 7,40 (1H, д), 7,30 (1H, м), 5,60 (2H, с), 3,15 (3H, с).
Пример 43
3-(3-Бром-4-метилфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)- фуранон
Анализ для
C18H15BrO4S:
Вычислено: C 53,08; H 3,71;
Найдено: C 53,06;
H 3,83.
Пример 44
3-(4-Бром-2-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12BrFO4S:
Вычислено: C
49,65; H 2,94;
Найдено: C 49,76;
H
3,
00.
Пример 45
3-(3,4-Дибромфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
1H-ЯМР (300 МГц ацетон-d6): δ 8,0 (2H, д), 7,80 (1H,
д),
7,
75 (3H, м), 7,25 (1H, д), 5,35 (2H, с), 3,15 (1H, с).
Пример 46
3-(4-Хлор-3-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17
H12ClFO4S:
Вычислено: C 55,67; H 3,30;
Найдено: C 55,45; H
3,30.
Пример 47
3-(4-Бром-2-фторфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12BrFO4S:
Вычислено: C
49,66; H 2,94; S 7,80;
Найдено: C
49,
79; H 3,01; S 7,51.
Пример 48
3-(4-Бром-2-хлорфенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C17H12BrClO4S:
Вычислено: C 47,74; H 2,83; S 7,50;
Найдено: C 47,92; H 2,84; S 7,42.
Пример 49
3-(2-Нафтил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для
C21H16O4S:
Вычислено: C 69,22; H 4,43;
Найдено: C 69,22; H 4,46.
Пример 50
3-(7-Хинолинил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
Анализ для C20H15NO4S:
Вычислено: C 65,74; H 4,14; S 3,83;
Найдено: C 65,34; H
4,
40; S 3,80.
MC (DCI, CH4): вычислено для M+:365
найдено для M+ + 1:366
Пример 51
3-(3,
4-Дихлорфенил)-4-(4-(аминосульфонил)фенил)-2-(2H)-фуранон
1H-ЯМР (400 МГц CD3COCD3): δ 7,92 (2H, дд), 7,64 (3H, дм), 7,60 (1H, дд), 7,32 (1H, дд), 6,
70
(1H, шир.с), 5,38 (2H, с).
Пример 52
3-(3,4-Дифторфенил)-4-(4-(аминосульфонил)фенил)-2-(2H)-фуранон
1H-ЯМР (400 МГц CD3COCD3):
δ 7,92 (2H, дд), 7,64 (2H, дд), 7,30 - 7,45 (2H, м), 7,22 (1H, м), 6,68 (2H, шир.с), 5,37 (2H, с).
Пример 53
3-(3-Хлор-4-метоксифенил)-4-(4-(аминосульфонил)фенил)-2-(2H)- фуранон
Анализ для C17H14ClNO5S:
Вычислено: C 53,76; H 3,72; N 3,69;
Найдено:
C 53,32; H 3,84; N 3,59.
MC (DCI, CH4): вычислено для M+:379
Найдено для M+ + 1:380
Пример 54
3-(3-Бром-4-метоксифенил)-4-(4-(аминосульфонил)фенил)-2-(2H)- фуранон
Анализ для C17H14BrNO5S:
Вычислено: C 48,13; H 3,33; N 3,30;
Найдено:
C 48,26; H 3,40; N 3,28.
MC (DCI, CH4): вычислено для M+:342
Найдено для M+ + 1:424
Пример 55
3-(Фенил)-4-(4-(метилсульфонил)фенил)-2-(5H)-фуранон
В 20-миллилитровую стеклянную ампулу добавляли 1 г 2-(4-(метилсульфонил)фенил)фенилацетилена, 20 мг Ph4(CO)12, 1,
5
г
триэтиламина, 10 мл ТГФ и 1 мл воды в атмосфере азота, а затем эту ампулу помещали в 100-миллилитровый автоклав из нержавеющей стали. Реакционную смесь три раза продували CO, а затем CO
загружали
при
комнатной температуре до его первоначального давления 100 атмосфер. Реакцию осуществляли при температуре 100oC в течение 5 часов. Полученный раствор разводили 50
миллилитрами бензола
и
промывали солевым раствором, а затем 1 н. соляной кислотой. Раствор бензола осушали сульфатом натрия и концентрировали. Неочищенные продукты отделяли с помощью колоночной
хроматографии на
силикагеле,
используя в качестве элюента смесь (2: 1) EtOAc/гексана, в результате чего получали целевое соединение и его региоизомер.
Пример 56
3-(Фенил)-4-(4-метилсульфонил)фенил)-2-(5H)-фуранон
Стадия 1: 2-Триметилсилилокси-4-(4-(метилтио)фенил)-3,4- дигирофуран
К раствору 4-бромтиоанизола (3,86 г, 19 мМ) в 90 мл Et2O, охлажденному при температуре -78oC, по капле добавляли 22 мл (1,7 М) раствора т-бутиллития в пентане (38 мМ). Реакционную смесь перемешивали в течение 15 минут при температуре
-78oC, после чего добавляли 3,8 г CuI и эту реакционную смесь оставляли для нагревания до -40oC в течение 30 минут. Затем к смеси добавляли 1,7 г 2(5H)-фуранона в 10 мл
тетрагидрофурана. После перемешивания в течение одного часа, к смеси по капле добавляли 2 мл свежедистиллированного TMSCI. Полученную реакционную смесь обрабатывали 2 миллилитрами триэтиламина и 50
миллилитрами насыщенного NaHCO3, а затем экстрагировали 100 миллилитрами эфира. Эфирный слой осушали сульфатом натрия и концентрировали, в результате чего получали неочищенное целевое
соединение, которое использовали в последующей стадии без дополнительной очистки.
Стадия 2: 4-(4-(Метилтио)фенил)-2-(5H)-фуранон
К раствору Pd(OAc)2 (4 г) в 100
мл
ацетонитрила по капле добавляли неочищенный целевой продукт Стадии 1 (5 г) в атмосфере азота при комнатной температуре. После выдерживания в течение 10 часов при комнатной температуре, смесь
конденсировали при пониженном давлении, а образовавшийся остаток очищали с помощью флэш-хроматографии на силикагеле, используя в качестве элюента смесь (2:1) гексана/EtOAc, в результате чего
получали
целевое соединение.
Стадия 3: 3-Иод-4-(4-метилтио)фенил)-2-(5H)-фуранон
К раствору продукта Стадии 2 (3 г) в 30 мл пиридина добавляли 8,7 г I2.
Полученную смесь
перемешивали в течение 24 часов, разводили 200 миллилитрами эфира, а затем промывали 100 миллилитрами 5 н. соляной кислоты и 50 миллилитрами 5 н. Na2S2O3. Эфирный
слой
осушали сульфатом натрия и концентрировали, в результате чего получали целевое соединение.
Стадия 4: 3-(Фенил)-4-(4-(метилтио)фенил)-2-(5H)-фуранон
Смесь, содержащую 4
г
продукта, полученного в Стадии 3 Примера 56, 3,7 г PhB(OH)2, 0,4 г Ph3As, 0,4 г PdCl2(PhCN)2 в 100 мл бензола и 15 мл 2 н. NaOH,
нагревали с обратным
холодильником в течение 6 часов. После добавления эфира (200 мл), смесь промывали 100 миллилитрами насыщенного NaHCO3. Органический слой осушали сульфатом магния и
концентрировали. Остаток
очищали с помощью флэш-хроматографии на силикагеле, используя в качестве элюента смесь (4:1) гексана/EtOAs, в результате чего получали целевое соединение.
Стадия 5:
3-(Фенил)-4-(4-(метилтио)фенил)-2-(5H)-фуранон
К раствору целевого продукта Стадии 4 (3 г) в 80 мл (10:1) CH2Cl2/MeOH добавляли 5,5 г MPPM. Реакционную
смесь
перемешивали в течение 2 часов при комнатной температуре, а затем разводили 100 миллилитрами смеси (1:1) гексана/этилацетата. После фильтрования и концентрирования остаток очищали с помощью
флэш-хроматографии, используя в качестве элюента смесь (2:1) гексана/EtOAc, в результате чего получали целевой продукт.
Реферат
Описывается соединение формулы 1

или его фармацевтически приемлемая соль,
где X-Y-Z выбирают из группы, включающей в себя a) -C(O)CH2CH2-, b) -С(O)-O-CR5(R5)-, c) -S-N= CH, d) -CH=N-S-, когда сторона b) представляет собой двойную связь, а сторона a) и c) представляют собой простые связи; и X-Y-Z представляет группу =N-S-CH=, когда стороны a и c представляют собой двойные связи, а сторона b представляет собой простую связь;
R1 выбирают из группы, включающей в себя a) S(O)2CH3, b) S(O)2NH2, c) S(O)2NHC(O)CF3;
R2 выбирают из группы, включающей в себя a) C3, C4, C5, C6 и C7-циклоалкил, b) моно-, ди- или тризамещенный фенил, в котором заместители выбирают из группы, включающей в себя (1) водород, (2) галоген, (3) C1-6-алкокси, (4) C1-6-алкилтио, (5) -CO2H;
R5 и R6 каждый независимо выбарают из группы, включающей в себя (a) водород, (b) C1-6-алкил.
Описывается также способ получения вышеуказанных соединений, способ лечения и фармацевтическая композиция на основе соединений формулы 1 для лечения воспалительного заболевания, опосредованного циклооксегеназой. 8 с. и 16 з.п.ф-лы, 4 табл.
Формула

или его фармацевтически приемлемая соль,
где X-Y-Z выбирают из группы, включающей в себя a) -C(O)CH2CH2-, b)-C(O)-O-CR5 (R5')-, c) -S-N=CH-, d) -CH=N-S-, когда сторона b представляет собой двойную связь, а стороны a и c представляет собой простые связи, и X-Y-Z представляет группу =N-S-CH=, когда стороны a и c представляет собой двойные связи, а сторона b представляет собой простую связь;
R1 выбирают из группы, включающей в себя a) S(O)2CH3, b) S(O)2NH2, c) S(O)2NHC(O)CF3;
R2 выбирают из группы, включающей в себя a) C3, C4, C5, C6 и C7 - циклоалкил, b) моно-, ди- или тризамещенный фенил, в котором заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, 3) C1-6 - алкокси, 4) С1-6 - алкилтио, 5) -CO2H;
R5 и R5' каждый независимо выбирают из группы, включающей в себя a) водород; b) С1-6 - алкил.
R5 и R5' каждый независимо выбран из группы, включающей a) водород, b) метил или этил.
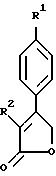
R1 выбирают из группы, включающей в себя a) S(O)2 CH3 b) S(O)2NH2;
R2 представляет собой моно- или дизамещенный фенил, в котором указанные заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, выбранный из группы, состоящей из фтора, хлора и брома, 3) метокси.
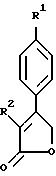
где R2 представляет собой моно- или дизамещенный фенил, где указанные заместители выбирают из группы, включающей в себя 1) водород и 2) галоген, выбранный из группы, состоящей из фтора, хлора и брома.
(1) 4-/4-фторфенил/-2-метил-5-/4-/метилсульфонил/фенил/тиазол;
(2) 2-/4-фторфенил/-3-/4-/метилсульфонил/фенил/-2-циклопентен-1-он;
(3) 2-/3, 5-дифторфенил/-3-/4-/метилсульфонил/фенил/-2-циклопентен-1-он;
(4) 4-/4-/Метилсульфонил/фенил/-5-/4-фторфенил/изотиазол;
(5) 3-/4-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(6) 3-/4-фторфенил/-4-/4-/аминосульфонил/фенил/-2-/5H/-фуранон;
(7) 3-/4-фторфенил/-4-/4-метилсульфонил/фенил фуран;
(8) 5,5-Диметил-3-/4-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(9) 3-/2,4-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(10) 3-/3, 4-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(11) 3-/2,6-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(12) 3-/2, 5-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(13) 3-/3,5-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(14) 3-/Бромфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(15) 3-/4-Хлорфенил/-4-/4-/метилсульфонил/фенил-2-/5H/-фуранон;
(16) 3-/4-Метоксифенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(17) 3-/Фенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(18) 3-/2-Хлорфенил/-4-/4-/метилсульфонил/фенил-2-/5H/-фуранон;
(19) 3-/2-Бром-4-фторфенил/-4-/4-/метилсульфонил/фенил/-2/5H/-фуранон;
(20) 3-/2-Бром-4-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(21) 3-/2-Хлор-2-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(22) 3-/3-Бром-4-фторфенил/-4-/4-/метилсульфонил/-фенил/-2-/5H/-фуранон;
(23) 3-/3-Хлорфенил/-4-/4-/метилсульфонил/фенил-2-/5H/-фуранон;
(24) 3-/2-Хлор-4-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(25) 3-/2,4-Дихлорфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(26) 3-/3, 4-Дихлорфенил/-4-/4-/метилсульфонил/-фенил/-2-/5H/-фуранон;
(27) 3-/2,6-Дихлорфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(28) 3-/3-хлор-4-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(29) 3-/4-Трифторметилфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(30) 3-/3-Фтор-4-метоксифенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(31) 3-/3-Хлор-4-метоксифенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(32) 3-/3-Фтор-4-метоксифенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(33) 3-/2-Фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(34) 3-/4-Метилтиофенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(35) 3-/3-Фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(36) 3-/2-Хлор-6-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(37) 3-/3-Бром-4-метилфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(38) 3-/4-Бром-2-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(39) 3-/3,4-Дибромфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(40) 3-/4-Хлор-3-фторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(41) 3-/4-Бром-3-фторфенил/-4-/4-/метилсульфонил/-фенил/-2-/5H/-фуранон;
(42) 3-/4-Бром-2-хлорфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(43) 3-/2-Нафтил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(44) 3-/7-Хинолинил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон;
(45) 3-/3,4-Дихлорфенил/-4-/4-/аминосульфонил/фенил/-2-/2H/-фуранон;
(46) 3-/3, 4-Дифторфенил/-4-/4-/аминосульфонил/фенил/-2-/2H/-фуранон;
(47) 3-/3-Хлор-4-метоксифенил/-4-/4-/аминосульфонил/фенил/-2-/2H/-фуранон;
(48) 3-/3-Бром-4-метоксифенил/-4-/4-/аминосульфонил/фенил/-2-/2H/-фуранон;
7. Соединение по п.1, представляющее собой
(a) 3-/3, 4-Дифторфенил/-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон/; или
(b) 3-Фенил-4-/4-/метилсульфонил/фенил/-2-/5H/-фуранон.
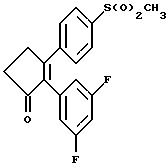
9. Соединение по п.1, имеющее формулу

где X-Y-Z - представляет собой -C(O)-O-CR5 (R5' ).
(1) 5, 5-Диметил-3-/4-фторфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон;
(2) 5, 5-Диметил-3-/3-фторфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон;
(3) 5, 5-Диметил-3-/3-хлорфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон;
(4) 5,5-Диметил-3-/3, 4-дифторфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон;
(5) 5,5-Диметил-3-/3, 4-дихлорфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон;
(6) 5, 5-Диметил-3-/4-хлорфенил/-4-/4-метилсульфонил/фенил-2-/5H/-фуранон.
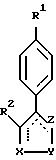
и его фармацевтически приемлемая соль,
где X-Y-Z выбирают из группы, состоящей из -C(O)-O-CR5 (R5')-, при условии, что сторона b представляет собой двойную связь, а стороны a и c представляет собой простые связи;
R1 выбирают из группы, включающей в себя (a) S(O)2CH3, (b) S(O)2 NH2;
R2 выбирают из группы, включающей в себя (a) C3, C4, C5 , C6 и C7 - циклоалкил; (b) монозамещенный или дизамещенный фенил, где указанный заместитель выбирают из группы, включающей в себя (1) водород; (2) галоген; (3) C1-6 - алкокси, (4) C1-6 - алкилтио, (5) -CO2H;
R5 и R5' независимо выбирают из группы, включающей в себя (a) водород; (b) C1-6 -алкил.

или его фармацевтически приемлемой соли,
где R1 выбирают из группы, включающей в себя (a) S(O)2CH3, (b) S(O)2NH2; c) S(O)2NHC(O) CF3;
R2 выбирают из группы, включающей в себя моно- или дизамещенный фенил, и где указанные заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, 3) C1-4 - алкокси, (4) C1-4 - алкилтио,
отличающийся тем, что в безводнном полярном растворителе обрабатывают соединение формулы А

в присутствии сильного основания, с получением соединения формулы XXXIII

16. Способ по п.15, отличающийся тем, что осуществляют реакцию в безводном полярном растворителе соединения формулы XXXII
с соединением формулы

в присутствии основания, с получением соединения формулы А:

с последующей обработкой полученного соединения формулы А в безводном полярном растворителе сильным основанием, с получением соединения формулы XXXIII

где значения R1 и R2 указаны в п.4.
(a1) реакцию в органическом растворителе соединения формулы XXXII
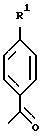
с бромовым реагентом с получением соединения формулы XXXII

(а2) реакцию в безводном полярном растворителе соединения формулы XXXII с соединением формулы

в присутствии основания с получением соединения формулы А
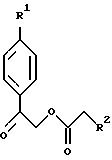
(а3) обработку в безводном полярном растворителе соединения формулы А сильным основанием с получением соединения, формулы XXXIII

где R1 и R2 имеют значения, указанные в п.4.
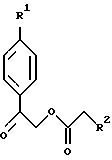
где R1 выбирают из группы, включающей в себя a) S(O)2 CH3, b) S(O)NH2, c) S(O)2NHC(O) CF3;
R2 выбирают из группы, включающей в себя моно- или дизамещенный фенил, и где указанные заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, 3) C1-4 - алкокси, 4) C1-4 - алкилтио.

где R1 выбирают из группы, состоящей из a) S(O)2CH3, b) S(O)2NH2; c) S(O)2NHC(O) CF3;
R2 выбирают из группы, включающей в себя монозамещенный или дизамещенный фенил, и где указанные заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, 3) C1-4 - алкокси, 4) C1-4 - алкилтио,
отличающийся тем, что осуществляют b 1) реакцию ацетиленового соединения формулы XIYIII

с окисью углерода и водой в присутствии подходящего катализатора с получением соединения формулы XXXIII и XXXY
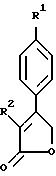
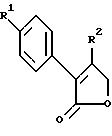
где R1 и R2 имеют значения, указанные выше.
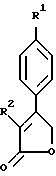
где R1 представляет собой S(O)2 CH3;
R2 выбирают из группы, включающей в себя монозамещенный или дизамещенный фенил, и где указанные заместители выбирают из группы, включающей в себя 1) водород, 2) галоген, 3) C1-4 - алкокси, 4) C1-4 - алкилтио,
отличающийся тем, что осуществляют
(с1) реакцию соединения формулы LIII

с реагентом формулы
(HO)2BR2
в водном растворителе и в присутствии подходящего катализатора с получением соединения формулы LV

где значения R2 указаны выше;
(с2) окисление соединения формулы LV, с получением соединения формулы XXXIII.
(а) 3-/3, 4- Дифторфенил/-4-/4-метилсульфонил/-фенил/-2-/5H/-фуранон или
(b) 3-фенил-4-/4-/метилсульфонил/-фенил-/-2-/5H/-фуранон.
24.06.93 - по пп.1,8,15,19-21,23-24;
10.01.94 - по остальным пунктам;
24.06.93 - соединения 1-8 п.6.
Комментарии