2,4-дисульфонил-α-фенил-трет-бутилнитрон, его фармацевтически приемлемые соли, его форма свободной кислоты или солевая форма, фармацевтические композиции и способы лечения - RU2159231C2
Код документа: RU2159231C2
Чертежи











Описание
Настоящее изобретение относится к производному нитрона, его солям и их применению в качестве фармацевтических нитронных средств улавливания свободных радикалов.
Альфа-фенил-трет-бутилнитрон (

В конце 70-х - начале 80-х годов медики начали сосредоточивать внимание на роли, которую играют свободные радикалы при таких заболеваниях, как сердечные приступы, удары и при подобных заболеваниях. ФБН все чаще применяют in vitro, чтобы получить аналитические данные о присутствии свободных радикалов при таких состояниях. Позднее его вводят животным моделям in vivo, вновь в качестве вспомогательного средства при попытках обнаружить свободные радикалы при моделировании ишемии и подобных заболеваний.
В середине 80-х появились первые предположения о возможном терапевтическом действии ФБН, когда эксперименты на животных с тяжелой травматической ишемией показали, что животные, которых лечили ФБН, имеют больше шансов на выживание, чем контрольные.
В 1991 г. , 2 мая, опубликована заявка PCT WO 91-05552. В этой заявке, которая частично
соответствует выданным в настоящее время патентам США N 5825032 и 5036097, описывается ФБН и ряд производных ФБН, определяемых формулой

где X представляет собой фенил или

где R представляет собой H,


и n представляет собой целое число от 1 до 5, или
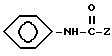
и Y представляет собой трет-бутил или гидроксилированный или ацетилированный трет-бутил, или замещенный фенил. Такие соединения описываются как фармацевтические средства для лечения последствий удара и других состояний, которые описываются как ассоциируемые с вредным влиянием свободных радикалов.
В 1992 г. была подана другая заявка PCT, относящаяся к ФБН и родственным соединениям и применению их в медицине. Эта заявка, основанная на предшествующей заявке США (сер. N 716952 с датой подачи 18 июня 1991), опубликована 23 декабря 1992 под номером WO 92/22290. В этой публикации 1992 г. приводятся два очень широких описания общего характера. Во-первых, делается попытка описать все возможные болезненные состояния, которые ассоциируются со свободными радикалами. Диапазон этих состояний начинается от нарушений ЦНС (удар, старение, мигрень и т.п.), проходит через заболевания периферических органов (включая атеросклероз, пролежни, раны и мышечное перенапряжение), через воздействие УФ-излучения к упоминанию, но лишь мельком, о воздействии яркого освещения. Во-вторых, делается попытка привести перечень всех возможных спиновых ловушек.
Помимо целого ряда
веществ, не относящихся к ФБН, в этой заявке весьма подробно дается определение потенциально полезным соединениям ФБН, к которым относятся ФБН и его производные формулы

где X представляет собой фенил, имидазолил, фенотиазинил или (R2)n

n = 1-5, предпочтительно, 1-3,
R2 = независимо (может изменяться в пределах молекулы) галоген, алкил, оксиалкил, алкенил, оксиалкенил, OH, NH2, NHZ, NZ, NO,



или
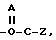
A = O или S, и
Z представляет собой C1-C6-линейную, разветвленную, алкильную или циклическую группу, и
Y представляет собой трет-бутильную группу, которая может быть гидроксилированной или ацетилированной в одном или нескольких положениях, фенил или

Утверждается, что в настоящий момент PBN является наиболее предпочтительным соединением, причем сообщается, что он не имеет ощутимого действия на нормальные или поврежденные клетки, и также утверждается, что и некоторые производные являются полезными, в том числе гидроксипроизводные, особенно 2-, 3- или 4-гидроксифенил-трет-бутилнитрон и фенил (моно-, ди- или тригидрокси)-трет-бутилнитрон, сложные эфиры ФБН, в особенности эфиры, которые высвобождают 2-, 3- или 4-гидроксифенил-трет-бутилнитрон, такие как ацетоксипроизводное, 2-, 3- или 4-карбоксифенил-трет-бутилнитрон, фенилгидроксибутилнитрон, алкоксильные производные, в особенности алкоксильные производные, которые высвобождают 2-, 3- или 4-гидроксифенил-трет-бутилнитрон, например, 2-, 3- или 4-метоксифенильные производные ФБН, и ацетамидные производные, в особенности ацетамидные производные, которые высвобождают 2-, 3- или 4-аминофенил-трет-бутилнитрон, дифенилнитрон ДФН и аналогичные производные дифенилнитрона, N-трет-бутил- α -(4-нитрофенил)нитрон, и N-трет-бутил- α -(2-сульфофенил)нитрон.
Обнаружено, что одно из производных ФБН и его соли обладают превосходными фармакологическими свойствами. Хотя это производное (2,4-дисульфонил-ФБН-) входит в обширную группу веществ, описанных в общем виде в вышеупомянутой публикации WO 92/022290, подробно это соединение не описывается. Не сообщается и о его преимущественных свойствах.
Ожидается, что это соединение с его двумя сульфонатными группами будет обладать повышенной растворимостью, но также ожидается, что оно будет плохо преодолевать гематоэнцефалический барьер по причине своего лиофобного характера. Однако, когда это соединение получают и испытывают in vivo, оно обнаруживает неожиданное возрастание эффективности при сравнении с ФБН. Такое возрастание эффективности имеет место наряду с возрастанием активности - по сравнению с ФБН. Прямой противоположностью этому заметному возрастанию активности и эффективности является заметное и весьма существенное снижение токсичности по сравнению с ФБН.
Такие результаты оказались неожиданными, поскольку в литературе по корреляции строения и активности среди определенных групп соединений терапевтическая активность, как правило, является ковариантной токсичности. Таким образом, наиболее близкие родственные соединения сохраняют свое соотношение терапевтической силы и токсичности. В противоположность этому, соединение настоящего изобретения отклоняется от такого ожидаемого соотношения, когда его активность возрастает, а его токсичность уменьшается при сравнении с ближайшими аналогами.
Соответственно, настоящее изобретение, с одной стороны, раскрывает
ФБН-дисульфонильное производное

и его фармацевтически приемлемые соли.
В соответствии со вторым своим аспектом, настоящее изобретение относится к фармацевтическим композициям, содержащим это соединение или его соль в качестве активного ингредиента, которые можно вводить парентерально, например внутривенно, и перорально.
В своем третьем аспекте настоящее изобретение относится к способу лечения пациента, который страдает от заболевания, включающего острое окислительное поражение центральной нервной системы, такого пациента, как пациент, который перенес удар, причем при упомянутом способе лечения вводят парентерально, например внутривенно, фармацевтическую композицию на основе упомянутого соединения или его соли.
В-четвертых, настоящее изобретение относится к способу лечения пациента, страдающего от заболевания, характеризуемого длительным низким окислительным давлением на центральную нервную систему и прогрессирующей потерей функции центральной нервной системы. При упомянутом способе вводят парентерально, например внутривенно, или, предпочтительно, перорально фармацевтическую композицию на основе упомянутого соединения или его соли.
В-пятых, настоящее изобретение относится к способу снижения или уменьшения интенсивности побочных действий, возникающих из-за окислительного нарушения, происходящего у пациента при лечении рака. По этому способу вводят парентерально, например внутривенно, или перорально фармацевтическую композицию на основе упомянутого соединения или его соли.
Подробное описание настоящего изобретения состоит из перечисленных далее разделов.
Краткое описание чертежей.
Соединение и его соли.
Получение соединения.
Фармацевтические композиции.
Состояния, которые лечат, и схемы лечения.
Примеры.
Краткое описание чертежей.
В настоящем разделе описываются прилагаемые чертежи.
На фиг. 1 дается схема последовательности реакций при получении соединения настоящего изобретения.
Фиг. 2 (A, B и C) и фиг. 3 (A, B и C) представляют собой два набора графиков, иллюстрирующих нежелательное изменение способности к терморегулированию организма животного, которое проявляется в виде зависимости от дозы известного нитронного соединения-ловушки радикалов и, в противоположность этому, отсутствие такого нежелательного токсичного действия в случае заявленного соединения.
Фиг. 4 (A, B, C и D) представляет собой набор из четырех графиков, демонстрирующих превосходство соединения изобретения над ближайшим известным нитронным соединением при сравнении их для лечения постепенной нейродегенерации (таких как болезнь Альцгеймера), что иллюстрируется их относительной способностью препятствовать бета-амилоидбелковой инактивации ключевых ферментов в растворе.
На фиг. 5 приводится диаграмма, иллюстрирующая эффективность соединения изобретения в снижении конечной зоны инфаркта, наблюдаемого после закупорки средней артерии головного мозга у крыс.
На фиг. 6 (A, B и C) приводятся три диаграммы, иллюстрирующие способность соединения изобретения снижать побочное действие высоких концентраций противораковых средств у животных.
Соединение и его соли
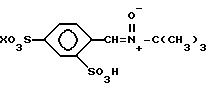
Соединение настоящего изобретения
представляет собой 2,4-дисульфонил- α -фенил-трет-бутилнитрон. Оно также упоминается в настоящем описании как "2,4-дисульфонил-ФБН" или как "ФБН-2,4-дисульфонат". Это соединение существует в
форме кислоты

в виде твердого вещества и в растворе с низким pH. Оно также существует при более высоких значениях pH в форме ионизированной соли, которую можно представить как

или в виде
где X представляет собой фармацевтически приемлемый катион. Чаще всего этот катион представляет собой одновалентную частицу, такую как натрий, калий или аммоний, но он также может представлять собой многовалентный катион в сочетании с фармацевтически приемлемым одновалентным анионом, например, катион кальция с хлорид-, бромид-, иодид-, гидроксил-, нитрат-, сульфонат-, ацетат-, тартрат-, оксалат-, сукцинат-, пальмоат-анионом или с подобным анионом, катион магния в сочетании с такими же анионами, катион цинка с такими же анионами или подобное. Когда в настоящем описании в структурных формулах показываются такие сочетания поливалентного катиона с одновалентным анионом, одновалентный анион изображается как "Y".
Среди этих веществ наиболее предпочтительными являются свободная кислота и простые натриевая, калиевая и аммониевая соли, наряду с солями кальция и магния, которые также являются предпочтительными, но в меньшей степени.
Получение
Как подробно показывается на фиг. 1 и описывается в примере 1, соединение
настоящего изобретения может быть получено при проведении реакции в две последовательные стадии. На первой стадии промышленный трет-бутилнитрат (2-метил-2-нитропропан) превращают в соответствующий
н-гидроксиламин, применяя подходящий катализатор, такой как активированный цинк с уксусной кислотой или ртутная амальгама алюминия. Эта реакция может быть осуществлена в течение 0,5-12 часов, в
основном в течение 2-6 часов или около этого, при температуре от 15 до 100oC в жидкой реакционной среде, такой как смесь спирта с водой в случае цинкового катализатора, или смесь эфира с
водой в случае амальгамы алюминия.
На второй стадии свежеполученный гидроксиламин вводят во взаимодействие с 4-формил-1,3-бензолдисульфоновой кислотой, причем, как правило, используют небольшой избыток амина. Эта реакция может быть осуществлена при аналогичных температурных условиях. Эта реакция обычно завершается за время от 10 до 24 часов.
Полученный таким образом продукт представляет собой свободную кислоту и имеет молекулярную массу 89 г/моль. Он представляет собой вещество в виде белого порошка, которое разлагается при нагревании. Это вещество отличается растворимостью в воде свыше 1 г/мл, и имеет1H ЯМР-спектр в D2O 8,048 м.д. (дд, 8,4, 1,7 Гц), 8,836 ppm (д, 8,4 Гц), 8,839 ppm (д, 1,7 Гц), 8,774 ppm (с).
Различные соли можно легко получить путем смешивания свободной кислоты в водной среде с двумя эквивалентами соответствующего основания, например KOH, чтобы получить калиевую соль, и т.п.
Фармацевтические композиции
Соединение (в том числе его соли) может быть введено в состав фармацевтических композиций, пригодных для перорального или парентерального, например путем
внутривенной или внутримышечной инъекции, введения.
Композиции для перорального введения могут принимать форму жидких растворов или суспензий, порошков, таблеток, капсул или подобную. В таких композициях ФБН-2,4-дисульфонат или его соли составляют, как правило, небольшую часть (от 0,1 до, скажем, 50 мас.%), причем остальную часть составляют различные наполнители или носители и вспомогательные вещества, полезные при формировании нужной лекарственной формы. Жидкая форма может включать в себя подходящий водный или неводный наполнитель, содержащий буферы, суспендирующие и диспергирующие вещества, красители, корригенты и подобные добавки.
Твердая форма может включать, например, любой из перечисленных далее ингредиентов или соединение аналогичной природы: связующее, такое как микрокристаллическая целлюлоза, трагакантовая смола или желатин; эксципиент, такой как крахмал или лактоза; дезинтегрирующее средство, такое как альгиновая кислота, примогель (Primogel) или кукурузный крахмал; смазывающее вещество, такое как стеарат магния; облегчающее скольжение вещество, такое как коллидальный диоксид кремния; смачиватель, такой как сахароза или сахарин, или корригирующий агент, такой как перечная мята, сахар, метилсалицилат или апельсиновая заправка.
В случае композиций для инъекций основу составляет, как правило, стерильный солевой раствор для инъекций или солевой раствор с фосфатным буфером, либо другие известные носители для инъекций. Активный нитрон вновь, как правило, является компонентом, присутствующим в небольшом количестве, часто - от 0,05 до 10 мас.%, причем остальную часть составляют носитель, пригодный для инъекций, и подобные вещества.
Заболевания и схемы лечения
Заболевания,
которые лечат с помощью 2,4-дисульфонил-ФБН, делятся на три группы. Первая включает острое интенсивное окислительное нарушение в области центральной нервной системы. Примерами таких состояний являются
удар, состояния, ассоциируемые с ударом, контузия и субарахноидальное кровотечение. При таком состоянии соединение вводят способом, который предназначен для непосредственной подачи лекарственного
средства в кровоток пациента настолько быстро насколько это возможно. Обычно такой способ включает внутривенное введение.
Дозы для внутривенного введения при лечении таких состояний колеблются от 0,1 мг/кг/час до по крайней мере 10 мг/кг/час, и вводятся за время 1 до 120 часов, в основном от 24 до 96 часов. Может быть также назначен предварительный болюс от 10 до 500 мг, чтобы добиться адекватного динамического равновесия.
Хотя внутривенное введение является предпочтительным способом, также могут использоваться другие формы парентерального введения, такие как внутримышечная инъекция. Неожиданным и ключевым преимуществом 2,4-дисульфонил-ФБН является тот факт, что его можно вводить при значительно более высоких концентрациях, чем те, которые возможны для самого ФБН. Как будет показано в примерах, возможно введение доз до 1000 мг/кг/час и выше или внутривенных болюсных доз от 10 до 2500 мг/кг для 2,4-дисульфонил-ФБН или его солей, в то время как для самого ФБН такие дозы приводят к смерти или острой токсичности. Для 2,4-дисульфонил-ФБН наблюдается неожиданное позитивное продолжение кривой доза/отклик при этих высоких дозах при четком обнаружении того, что введение таких интенсивных доз сразу после удара или других травм может во многих случаях дать основной положительный импульс к выздоровлению.
Второй группой заболеваний, которые благоприятно реагируют на лечение с помощью 2,4-дисульфонил-ФБН, являются заболевания, характеризуемые длительным низким оксидативным напряжением центральной нервной системы и постепенной потерей функций центральной нервной системы. К таким состояниям относятся болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз (ALS), мультиинфарктная деменция, ретинопатия и подобные состояния. Каждое из этих состояний характеризуется прогрессирующей потерей функции. При пероральном или парентеральном, например внутривенном введении 2,4-дисульфонил ФБН или его солей, можно замедлить потерю этой функции и, возможно, восстановить ее. Если желательно парентеральное, например внутривенное введение, обычно используют дозировки, подобные применяемым при острых состояниях, но в более узких пределах.
В таких случаях схема лечения может растянуться на многие месяцы или годы, поэтому оральное введение предпочтительно ввиду удобства для пациента и своей толерантности. При пероральном введении требуется от одной до трех доз в сутки, от 0,02 до 50 мг/кг каждая, причем предпочтительные дозы составляют от 0,04 до 5,0 мг/кг.
Конечно, 2,4-дисульфонил-ФБН можно вводить как единственное активное средство, или его можно вводить в сочетании с другими средствами. Такой подход ведет к третьему варианту применения этого соединения.
Третьей группой заболеваний, которые отвечают на лечение 2,4-дисульфонил-ФБН, являются побочные действия, которые появляются у пациентов из-за окислительного нарушения, вызываемого лечением рака (опухолевого заболевания). Лечение, которое вызывает окислительное нарушение (и, таким образом, побочные эффекты), включает лучевую терапию (например, гамма-излучением) и лечение соответствующими химиотерапевтическими средствами. Примерами таких средств являются антибиотики, такие как даунорубицин, доксорубицин и блеомицин, прокарбазин, азотистые иприты, такие как ифосфамид, мелфалан и хлорамбуцил, алкилирующие средства, антиметаболиты, гормоны и антагонисты.
Введение 2,4-дисульфонил-ФБН может снизить ощущение дискомфорта у пациента во время такого лечения. Кроме того, введение соединения настоящего изобретения может увеличить способность пациента переносить такое лечение. Часто побочное действие при лечении удлиняет продолжительность этого лечения или препятствует введению оптимально высоких доз или частой повторяемости такого лечения. В некоторых случаях побочное действие является настолько деструктивным, что ведет к сердечной недостаточности и потере основной функции. При испытаниях на животных наблюдали, что заявленное соединение может увеличить переносимость пациентом назначенного лечения.
При таком лечении соединение настоящего изобретения может быть введено до, во время и после того, как назначается облучение или химиотерапия. Введение может быть осуществлено парентеральным, или пероральным, или любым другим способом, который позволит 2,4-дисульфонил-ФБН попасть в кровь пациента.
Наблюдалось позитивное соотношение доза-отклик. По существу, имея в виду тяжесть побочного действия и преимущества обеспечения максимально возможного снижения интенсивности побочного действия, в некоторых случаях может потребоваться введение больших количеств 2,4-дисульфонил-ФБН, как для лечения состояний с острым интенсивным окислительным поражением ЦНС. При других заболеваниях могут быть использованы меньшие дозы, назначаемые для лечения прогрессирующих нейронных заболеваний.
Примерами характерных схем введения являются следующие схемы. При монотерапии (только адриамицин) две комбинации представляют собой 10-600 мг соединения I на кв.м площади плюс 60 мг адриамицина на кв.м площади поверхности при введении адриамицина, которое осуществляют каждые семь суток или двадцать одни сутки. Соединение I может быть введено до адриамицина, например за 60 минут перед введением адриамицина, одновременно с ним или после его введения, спустя несколько часов или несколько суток. Педиатрическая доза, как правило, ниже для обоих лекарственных препаратов. Для лечения опухолей, резистентных к многократным дозам, могут быть использованы более высокие дозы.
ПРИМЕРЫ
Пример 1
Синтез 2,4-дисульфонил-N-трет-бутилнитрона (в
последующих примерах - соединение I)
Предлагаемый способ синтеза основан на работе авторов R.H. Hinton and E. G. Janzen (J. Org. Chem., 57: 2646-2651, 1992). Как показано на фиг. 1, этот
способ включает конденсацию альдегида с гидроксиламином. Гидроксиламин является неустойчивым соединением, и его используют свежеприготовленным в день применения, используя в качестве катализатора
активированный цинк. Синтез проводят следующим образом.
Необходимые реактивы
1. Этанол 95%
2. 2-Метил-2-нитропропан
3. Цинковая пыль
4. Ледяная
уксусная кислота
5. Диэтиловый эфир
6. Насыщенный раствор хлорида натрия
7. Сульфат магния, твердый, безводный
8. Гидрат динатриевой соли 4-формил-1,
3-бензолсульфоновой кислоты (ММ 310,21 г/моль)
9. Метанол
10. Дихлорметан
Методика
A. Получение N-трет-бутилгидроксиламина
1. Трехгорлую 500-мл колбу
снабжают магнитной мешалкой, держателем термометра, термометром и капельной воронкой.
2. В колбу загружают 95%-ный этанол (350 мл) и охлаждают до 10oC на ледяной бане.
3. Добавляют в один прием 2-метил-2-нитропропан (6,18 г, 0,060 моль) и цинковую пыль (5,89 г, 0,090 моль).
4. В капельную воронку наливают ледяную уксусную кислоту (10,8 г, 0, 180 моль), и добавляют ее к реакционной смеси по каплям с такой скоростью, чтобы при энергичном перемешивании температура оставалась ниже 15oC.
5. Удаляют ледяную баню, и смесь перемешивают в течение 3 часов при комнатной температуре.
6. Растворитель удаляют из смеси, оставляя трет-бутил-гидроксиламин, ацетат цинка и воду.
7. Добавляют дихлорметан (50 мл), и смесь фильтруют через воронку Бюхнера.
8. Оставшийся на фильтре ацетат цинка промывают дихлорметаном (2 х 25 мл).
9. В делительной воронке от фильтрата отделяют воду, и органический слой сушат над сульфатом магния.
10. Сульфат магния удаляют фильтрацией через складчатый бумажный фильтр, затем из фильтрата удаляют дихлорметан на роторном испарителе.
11. Продукт (выход 100% = 5,34 г), имеющий вид вязкой жидкости, растворяют в метаноле (50 мл) для использования на стадии B.
B. Получение 2,
4-дисульфофенил-N-трет-бутилнитрона
1. Круглодонную 250-мл трехгорлую колбу снабжают магнитной мешалкой, газоподводящей трубкой, капельной воронкой и холодильником Фридрихса (Friedrichs),
охлаждаемым циркуляционной смесью воды со льдом.
2. В колбу загружают 200 мл метанола, 4-формил-1,3-бензолдисульфоновую кислоту (9,31 г, 30 ммоль) и N-трет-бутил-гидроксиламин (25 мл раствора в метаноле продукта со стадии A, теор. 30 ммоль).
3. Реакционную смесь нагревают до температуры перегонки с помощью нагревателя при перемешивании и пропускании через нее азота.
4. Смесь кипятят с обратным холодильником в течение 2 часов.
5. Добавляют остальной гидроксиламин со стадии A.
6. Кипячение с обратным холодильником при пропускании азота продолжают по крайней мере в течение 18 часов, но не более 24 часов.
7. Горячую реакционную смесь фильтруют на воронке Бюхнера, и твердое вещество промывают горячим метанолом.
8. Метанол удаляют на роторном испарителе, и получают желтое вязкое масло.
9. Добавляют горячую смесь этанола с ацетоном (1:1, 200 мл), и смесь нагревают для растворения масла.
10. Для кристаллизации продукта раствор охлаждают.
11. Продукт собирают на воронке Бюхнера и сушат в вакууме в течение ночи.
12. Реакция, как правило, приводит к получению соединения I в виде белого порошка с выходом 75%.
Пример 2
Альтернативный синтез 2,
4-дисульфонил-N-трет-бутилнитрона (соединение I)
Он представляет собой ранее разработанный способ, который используют для получения образцов соединения, применяемого в некоторых примерах
настоящего описания. Продукт, который получают в этом примере, по всем параметрам идентичен продукту примера 1. Этот синтез осуществляют следующим образом.
Необходимые реактивы
1. Алюминиевая фольга, нарезанная на широкие полоски и скатанная в цилиндрики диаметром прибл. 1 см
2. Хлорид ртути (II) (9,68 г в 476 мл воды)
3. Этанол
4. Эфир (6 л)
5. Чистая вода
6. 2-Метил-2-нитропропан
7. Гидроксид натрия, 2М (80 г в 1 л воды)
8. Сульфат магния, твердый, безводный
9. 4-Формил-1,3-бензолсульфоновая
кислота (ММ 310,21 г/моль)
Методика
A. Получение N-трет-бутилгидроксиламина
1. Цилиндры из алюминиевой фольги погружают в раствор HgCl2 на 15-30 секунд, затем их
погружают в этанол, затем погружают в эфир, и затем помещают в 5 л колбу, содержащую 500 мл диэтилового эфира и 21,4 мл воды.
2. Колбу снабжают механической мешалкой, трубкой для ввода азота и холодильником Фридрихса, охлаждаемым оборотной смесью воды со льдом.
3. Смесь перемешивают в течение 10 минут.
4. Добавляют 2-метил-2-нитропропан (71,68 г, 75,5 мл), используя капельную воронку, с такой скоростью, чтобы поддерживалось энергичное обратное отекание.
Примечание. Добавка должна быть завершена менее, чем за 20 минут, или существенно падает выход.
5. В добавление вводят эфир порциями по 500 мл. Это делается для того, чтобы сохранить высокую концентрацию продукта, по возможности без образования геля. Можно добавить до 2 л эфира без отрицательного влияния на выход продукта.
6. Как только добавление 2-метил-2-нитропропана завершено, реакционную смесь перемешивают еще в течение 30 минут.
7. Получающуюся в результате серую суспензию фильтруют с отсосом в три загрузки, чтобы удалить соли алюминия.
8. Каждый раз осадок на фильтре промывают 1 л эфира.
9. Объединенные эфирные слои промывают 300 мл 2М NaOH, затем сушат (MgSO4) и концентрируют в вакууме, получают в остатке низкоплавкое белое твердое вещество.
10. Твердое вещество плавится при температуре чуть выше комнатной, но его можно еще посушить в вакуумной печи (не более нескольких минут), причем остается 38-45 г твердого вещества.
11. Твердое вещество можно использовать в том виде, какое оно получено, или очистить перекристаллизацией из пентана.
12. Молекулярная масса 89 г/моль.
B.
Получение 2,4-дисульфонилфенил-N-трет-бутилнитрона
1. Колбу емк. 250 мл снабжают мешалкой и холодильником Фридрихса, охлаждаемым оборотной смесью воды со льдом.
2. В колбу загружают 71,8 мл метанола, 14,5 г 4-формил-1,3-бензолдисульфоновой кислоты (46,7 ммоль, 1 экв.) и 5,0 г N-трет-бутилгидроксиламина (56,2 ммоль, 1,2 экв.).
3. Смесь кипятят с обратным холодильником в течение ночи.
4. Продукт реакции переносят в круглодонную колбу и упаривают досуха на роторном испарителе.
5. Твердый остаток растирают с эфиром, и эфир декантируют (желтого цвета).
6. Повторяют стадию 5.
7. Продукт ("соединение I") кристаллизуют из метанола, осуществляя горячее фильтрование метанольного раствора для удаления нерастворимого осадка, и дважды перекристаллизовывают из метанола.
Пример 3
Выполняют ряд экспериментов, чтобы сравнить in vivo эффективность 2,4-дисульфонил-ФБН
- ("соединение I"), ФБН и двух моносульфонатов ФБН в качестве средств для защиты от потери нейронов после ишемии головного мозга и реперфузионного повреждения. Процедура испытаний соответствует
описанной статье в W. Cao, J.M. Carney, A. Duchon, R.A. Floyd and M.Chevion, "Oxygen free radical involvement in ischemia and reperfusion injury to brain", Neuroschcience Letters, 88 (1988), 233. В
этих экспериментах проверяемое соединение вводят методом i.p. (интраперитонеальным) песчанкам в группах по шесть особей, в виде единичной дозы за 30 мин до билатерального 5-минутного закрытия сонной
артерии. Измеряют плотность нейронных ядер на протяжении 100 мкм. Присутствуют две контрольных группы: контрольная группа, которая не получает проверяемого соединения, и контрольная группа, которая не
получает проверяемого соединения и не испытывает ишемии головного мозга. Как видно из табл. 1, соединение изобретения показывает неожиданные преимущества, при сравнении с соединениями, известными в
технике. Во-первых, видно, что при низких уровнях дозы, таких как 3,2 мг/кг, соединение I в 2-3 раза действеннее для предотвращения нейронной потери. Выясняется, что при более высоких дозах соединение
I способно обеспечить полную защиту от потери нейронов, так как нейронная плотность у экспериментальных животных идентична показателям контрольной группы, не подвергавшейся ишемии. Соединения,
известные в технике, являются либо токсичными при таких дозах, либо обеспечивают защиту в значительно меньшей степени. Эти результаты показывают четкое возрастание эффективности соединения I для
нейронной защиты, по сравнению с ФБН и двумя близкими аналогами, и неожиданное снижение токсичности по сравнению с ФБН.
Пример 4
Проводят ряд экспериментов, при которых
соединение I сравнивают с ФБН и двумя аналогами-сульфонатами при постишемическом лечении. Применяют общий способ, описанный в примере 1, но проверяемые соединения вводят i.p. в виде единичной дозы
через 30 минут после реперфузии, следующей за 5-минутной ишемией. Результаты сводятся в табл. 2. Табл. 2 показывает, что соединение I вновь является более сильным при малых дозах, и более действенным
и менее токсичным при высоких дозах. Также вновь токсичность препятствует возможности применения таких высоких доз соединений, известных в технике, при которых соединение изобретения обеспечивает
наглядно эффективное лечение.
Пример 5
Чтобы определить относительную эффективность для защиты от потери нейронов, соединение I сравнивают с ФБН при введении i.v. через 60 мин
после начала реперфузии, следующей за 5-минутной ишемией у песчанок, используя общий метод испытаний, описанный в примере 1. Результаты сводятся в табл. 3, и они показывают, что соединение I является
значительно более полезным средством при лечении состояния, следующего за повреждением головного мозга.
Ни ФБН, ни соединение I не влияют на плотность нейронов у контрольных песчанок без повреждения головного мозга.
Пример 6
Повреждение головного мозга может само проявляться как изменения поведенческих реакций. В этом эксперименте подвергают испытанию
молодых взрослых песчанок (возраст 3-4 месяца), чтобы определить их способность выполнять тест через 24 часа после события ишемии, как оно описано в примере 1. При сравнении с животными, у которых
ишемия не вызывалась, когда животных не лечат, они совершают значительно больше ошибок. Некоторым из подопытных животных вводят ФБН и соединение I. Как подробно описано в таблице 4, песчанки,
получившие высокие дозы соединения I, имеют уровень ошибок неотличимый от уровня ошибок животных без ишемии. ФБН является менее эффективным. Это свидетельствует о том, что соединение I может защитить
от потери временной/пространственной кратковременной памяти, следующей за ишемией (спустя 24 часа), проявляющейся в ошибках в испытании у молодых песчанок после 5-минутной ишемии.
Пример 7
Определяют способность соединения изобретения снижать инфарктный объем после ишемического события. Как подробно показано в таблице 5, наблюдают, что хотя ФБН и соединение I оба
являются эффективными при малых дозах, при высоких дозах соединение I дает наилучшую защиту, а ФБН является токсичным. В таблице 5 приводится инфарктный объем, отслеживаемый в случае, когда
проверяемое соединение вводят i. v. спустя 60 мин после средней церебральной закупорки, и затем в течение 24 часов, у
мышей C57BL/6J.
Пример 8
При этом исследовании
соединение I и ФБН сравнивают по их способности защищать от летальности (% выживающих) старых песчанок (возраст 18-24 месяцев, n = 12/группу) после 10-минутной ишемии, когда препарат дают за 30 минут
до ишемии. Как показано в таблице 6, соединение I является превосходным при всех уровнях дозы, и обеспечивает полную защиту при высоких уровнях, в то время как ФБН является лишь частично
эффективным.
Пример 9
Важным преимуществом соединения настоящего изобретения, при сравнении с известным в технике соединением - ФБН, является его заметно уменьшенная
токсичность. Как показано в таблице 7, у мышей C57BL/6L наблюдают сильную летальность, вызванную изменением величины единичных i.p. доз нитрона. ФБН показывает значительную токсичность при уровне дозы
560 мг/кг. Соединение I не проявляет токсичности при дозах примерно в двадцать раз больших.
Пример 10
Еще одним нежелательным побочным действием, которое наблюдают in vivo
для нитронных ловушек радикалов, является снижение температуры тела. Такая токсичность может иметь серьезные последствия для здоровья, и также может усложнить диагностику других состояний. Как
показывается на фиг. 2 и 4, соединение настоящего изобретения вводят мышам и песчанкам при таких высоких уровнях как 1000 мг/кг без заметного падения температуры. Напротив, известное соединение ФБН
дает снижение температуры тела на 8oC при дозах, составляющих только 500 мг/кг
Пример 11
Проводят испытания соединения изобретения для определения его эффективности при
лечении состояний, характеризуемых длительным низким окислительным давлением на центральную нервную систему и постепенной прогрессирующей потерей функции центральной нервной системы. Эффективность
проверяют на модели болезни Альцгеймера ("AD"). Такая модель основана на следующем. Последние исследования показали, что существуют связанные с возрастом увеличение окисления белков и потеря
ферментативной активности в головном мозгу пожилых индивидуумов. Как тканевые культуры фибробластов пожилых индивидуумов, так и эритроциты индивидуумов разного возраста показывают экспоненциальное
возрастание содержания протеинкарбонила (мера окисления белков) и снижение отметки ферментативной активности. Окисление белков головного мозга постепенно увеличивается с продолжительностью жизни
индивидуума.
Роль обработки аномального амилоидного предшественника белка и метаболизма при AD также объясняется на ряде различных моделей. Исследования in vitro с применением эмбриональных гиппокампальных нейронных и нейронных/глиальных культур показали, что ВАР 1-40 продуцирует цитотоксичность свыше продолжительного периода коинкубации. Когда такой пептид инфузируют в головной мозг крысы, продуцируются повреждения. Некоторые из названных фрагментов расщепления ВАР являются также нейротоксичными /например, ВАР (25-35)/. Оказывается, что нейротоксичность переносится как через механизм глутаматных рецепторов, так и через механизм неглутаматных рецепторов. Исследования нейронных культур методами софокусной спектроскопии показали, что воздействие ВАР (1-40) приводит в результате к окислительному стрессу /дихлорфлуоросцеин и возрастание внутриклеточного свободного кальций - Fura-2/.
Показано, что фрагменты ВАР могут непосредственно инактивировать глутаминсинтетазу (GS) и креатинкиназу (CK) в экстрактах тканей и в культивированных гиппокампальных нейронах и глиях (см. фиг. 4, A и B). На фиг. 4, A и B, приводится связанная с дозой инактивация AP (25-35) глутаминсинтетазы и креатинкиназы. Получают цитозольные фракции из неокортекса песчанки, и определяют ферментную активность. Образцы перед испытаниями инкубируют в присутствии различных концентраций пептида в течение 10 мин. Заштрихованные значки представляют действие встречающегося в природе фрагмента 25-35. Незаштрихованные кружки показывают, что обратная последовательность (32-25) не влияет на ферментативную активность. Незаштрихованные треугольники показывают, что смешанная (scvambled) аминокислотная последовательность также не влияет на ферментативную активность, по сравнению с действием 25-35. Каждая точка представляет собой среднее (+/- ст.откл.) из результатов 5 измерений. Производное ВАР и другие клеточные источники являются важным определяющим фактором инициирования и развития AD.
Как показывает фиг. 4, C и D, каждое из соединений I и ФБН демонстрирует способность защищать GS и CK от действия фрагментов ВАР. Фиг. 4, C и D, представляет защитное действие коинкубации цитозольных фракций с ВАР 25-35 (0,4 мг/мл) в сочетании с различными концентрациями ФБН (незаштрихованные кружки) или соединением I (заштрихованные кружки). Каждая точка представляет собой среднее (+/- ст.откл.) из результатов 3 измерений. Как можно видеть на фиг. 4, C и D, соединение I дает полную защиту и фактически даже может реверсировать действие окисления. Напротив, эффективность ФБН является весьма ограниченной, асимптотически выравниваясь, по существу, при неполной степени защиты.
Пример 12
Проводят эксперименты, чтобы
показать еще эффективность соединения изобретения при защите центральной нервной системы при повреждении, вызываемом ударом.
Результаты, полученные при фокальной ишемии у крыс
Проводят два опыта, чтобы определить эффективность соединения I при фокальной ишемии на крысиной модели. В этой модели крыс Sprague Dawley (200-300 г) длительно подвергают перманентной закупорке
средней артерии головного мозга (МСАО), чтобы индуцировать фокальный удар. Соединение I вводят после перманентной закупорки сначала в виде интраперитонеальной (i.p.) болюсной дозы, а затем путем
непрерывной инфузии в течение времени, оставшегося до 24 часов после удара. Используемые дозы соединения I составляют либо 100 мг/кг, i. p., и затем 4,2 мг/кг/ч, i.v., либо 10 мг/кг, i.p., и затем 0,
42 мг/кг/ч, i.v.
Крыс скарифицируют через 3 суток после удара, ткани обрабатывают гистологически, используя методику с окрашиванием трифенилтетразолием, и количественно определяют инфарктный объем и область полного омертвления клеток, используя анализ подобия. Результаты этих экспериментов даются в графическом изображении на фиг. 5 и показывают, что соединение I обеспечивает существенную защиту, приблизительно 70%, в обоих опытах.
Сравнение с результатами, приведенными в литературе
О подобных данных для соединения, не являющегося соединением
изобретения, а именно, для ФБН, недавно сообщалось Cao и Phillis (Brain Research 664: 267-272, 1994). При этих исследованиях крыс подвергают перманентной закупорке средней артерии головного мозга
(МСАО) и закупорке общей сонной артерии. ФБН вводят i.p. при дозе 100 мг/кг в различное время после удара. Крыс скарифицируют через 2 суток после удара, и определяют количественно инфарктный объем,
используя трифенилтетразолиевый краситель.
Когда ФБН вводят через 0,5, 5, 17, 29 и 41 час после удара, или через 5, 17, 29 и 41 час после удара, инфарктный объем в каждом случае уменьшается приблизительно на 50%. Кумулятивная доза введенного ФБН для достижения 50%-ной защиты как минимум в 4 раза выше количества соединения I, требуемого для 70%-ной защиты. Таким образом, соединение I значительно превосходит ФБН по возможности защиты на крысиной модели фокальной ишемии, вызванной МСАО.
Пример 13
В этом примере проводят оценку соединения
настоящего изобретения (соединение I) на его способность уменьшать вызванные окислением побочные действия при противораковой терапии.
Эксперименты с адриамицином
Адриамицин
является широко применяемым противораковым средством. Известно, что он является весьма эффективным, но также известно, что он дает тяжелые побочные эффекты, проявляющиеся в силу его склонности
вызывать окислительное нарушение. К таким побочным эффектам относятся серьезные нарушения сердечной деятельности при высоких уровнях дозировки. Такие побочные действия часто ограничивают применение
этого средства или ограничивают уровни дозировки, которые можно использовать, до уровней, которые ниже требуемых для достижения максимальной эффективности при опухолевом заболевании.
Осуществляют эксперименты, чтобы показать, что соединение настоящего изобретения является эффективным для снижения побочного действия таких противораковых средств, как адриамицин, и позволяет животным переносить более высокие дозы адриамицина.
Для проверки острого летального действия адриамицина и предупреждения острой летальности посредством предварительной обработки дозами соединения I используют самцов мышей C57BL/6J и DBA/2J (35-40 г). Мышам дают инъекции либо солевого раствора, либо соединения I, за 30 минут до введения адриамицина. Все инъекции интраперитонеальные. Острая летальность адриамицина находится в интервале от 10 до 30 мг/кг. В этих испытаниях находят, что LD50 для адриамицина составляет 25 мг/кг для обоих родов. Соединение I при дозе до 300 мг/кг без лечения адриамицином не влияет на выживаемость мышей того и другого рода.
Предварительное введение от 30 до 100 мг/кг соединения I дает соотнесенные с дозой сдвиги на диаграмме зависимости летальности от дозы адриамицина. Фиг. 6 показывает результаты, полученные на мышах DBA/2J. Доза соединения I в 100 мг/кг вызывает 5-кратный сдвиг вправо (в направлении уменьшенной летальности). Таким образом, сочетание соединения I с адриамицином дает в результате заметное увеличение максимально переносимой дозы. Такие более высокие дозы находятся в интервале, в котором будут эффективно уничтожаться опухоли, резистентные ко многим лекарственным средствам.
Сравнительные испытания
Предварительная обработка ФБН приводит в результате к
небольшому сдвигу вправо на диаграмме зависимости действия от дозы адриамицина. В то время, как дозу соединения I в сочетании с адриамицином можно увеличить до 300 мг/кг, для такого сочетания с ФБН
существует верхний предел. Доза ФБН в 100 мг/кг дает небольшой седативный эффект, и доза в 300 мг/кг дает заметный седативный эффект и некоторую общую токсичность (летальность 10- 20%). Соединение I с
адриамицином не дает никакой токсичности при дозе соединения I до 300 мг/кг.
Пример 14
Испытания на безопасность
Проверяют соединение I и ФБН на их острую
токсичность на самцах крыс Sprague Dawley (200-300 г). Соединения вводят i.p. при дозе 1000 мг/кг группам крыс по 6 особей. Через 3 суток проверяют летальность. Соединение I летальности не вызывает, в
то время как ФБН является летальным для 5 из 6 крыс, используемых при этом испытании. Эти данные подтверждают те результаты, полученные для песчанок, что соединение I намного безопаснее ФБН.
Реферат
Описывается новое соединение 2,4-дисульфо-альфа-фенил-трет-бутилнитрон формулы

и его фармацевтически приемлемые соли. Эти вещества пригодны в качестве фармацевтических средств для перорального или парентерального, например внутривенного, введения пациентам, страдающим от острого окисления центральной нервной системы, которое происходит при ударе, или от постепенного окисления центральной нервной системы, которое может обнаруживать себя как прогрессирующая потеря функции центральной нервной системы. Эти вещества также применяются для ослабления интенсивности побочных эффектов при лечении заболеваний, которое вызывает окислительное нарушение. Описываются также фармацевтическая композиция и способы лечения с использованием соединений общей формулы I. 17 с. и 4 з.п. ф-лы, 6 ил., 7 табл.

Формула

или его фармацевтически приемлемые соли
2. 2,4-Дисульфонил-α-фенил-трет-бутилнитрон в форме свободной кислоты

3. 2,4-Дисульфонил-α-фенил-трет-бутилнитрон в фармацевтически приемлемой ионизированной солевой форме

4. Соединение по п.3, отличающееся тем, что имеет следующую формулу

где X выбирают из группы, состоящей из Na, K, NH4, Ca, Mg, CaY и MgY,
и Y представляет собой фармацевтически приемлемый одновалентный анион.
Комментарии