Феноксифенилалкансульфонаты и лекарственное средство на их основе в качестве агониста каннабиноидных рецепторов - RU2278853C2
Код документа: RU2278853C2
Описание
Изобретение относится к новым сложным эфирам сульфоновой кислоты, более конкретно к феноксифенилалкансульфонатам и лекарственному средству на их основе в качестве агониста каннабиноидных рецепторов.
Среди веществ, содержащихся в растениях конопли (Cannabis sativa) самым важным в фармакологическом отношении является Δ9-тетрагидроканнабинол, который обуславливает основные эффекты конопли на центральную нервную систему человека. Потенциальные исторические и современные терапевтические применения препаратов конопли включают наряду с другими анальгезию, рвоту, анорексию, глаукому и нарушения двигательных функций.
До настоящего времени были идентифицированы два подтипа каннабиноидных рецепторов и один вариант с совмещением функций. СВ1- и СВ2-рецепторы имеют семь трансмембранных регионов и относятся к семейству рецепторов, связанных с G-белками.
СВ1-Рецептор и вариант с совмещением функций СВ1а локализованы преимущественно в центральной нервной системе. СВ2-Рецептор обнаруживается преимущественно в периферических тканях, в частности в лейкоцитах, в селезенке и в макрофагах.
До настоящего времени были известны многие структурные классы агонистов канна-биноидных рецепторов: такие классические каннабиноиды, как, например, Δ9-тетра-гидроканнабинол, такие неклассические каннабиноиды, как, например, аминоалкил-индолы, а также эйкозаноиды. К последним относится эндогенный агонист СВ1-рецепторов анандамид.
В заявках на международные патенты №А-98/37061, №А-00/10967 и №А-00/10968 описываются определенные арилоксифенилалкансульфонаты в качестве агонистов кан-набиноидных рецепторов для лечения нейродегенеративных заболеваний.
В патенте США №3462473, в статьях Biochem. Pharmacol. 1972, 21, 1127-1134, Fed. Proc. Fed. Amer. Soc. Exp. Biol. 1971, 30, 841-847, и J. Pharm. Sci. 1973, 62, 1780-1784, раскрываются определенные n-феноксифенилалкансульфонаты и их гипохолестеринемическое или, соответственно, гиполипидемическое действие.
Кроме того, известны определенные феноксифенилалкансульфонаты и их применение в качестве гербицидов (1), антимикробных средств (2), средств для снижения адгезии (3), сенсибилизаторов для термобумаги (4) и синтетических промежуточных продуктов (5): (1) заявка на европейский патент №0023725; патент США №3929903; патент США №4415354; Chem. Abstr. 1979, 91, 175034 (патент Японии №А-54066631); Chem. Abstr. 1981, 94, 156552 (патент Японии №А-55154953); Chem. Abstr. 1981, 95, 168773 (патент Японии №A-56046859); Chem. Abstr. 1981, 95, 168789 (патент Японии №A-56079665); Chem. Abstr. 1989, 111, 2678 (патент Японии №А-63104903); (2) заявка на патент ФРГ №А-1493 776; заявка на патент ФРГ №А-2131754; Патент США №3629477; патент США №3772445; патент США №3850972; заявка на патент Швейцарии №В-450347; заявка на патент Швейцарии №В-459656; Chem. Abstr. 1975, 83, 72725 (патент Японии №В-50003375); (3) патент США №3346612; (4) патент США №4837197; (5) Chem. Abstr. 1997, 727, 26629 (патент Японии №A-09087210); Tetrahedr. 1990, 46, 4161-4164; J. Am. Chem. Soc. 1998, 39, 435-436.
Задача настоящего изобретения состояла в получении агонистов каннабиноидных рецепторов с улучшенным действием.
Данная задача решается соответствующими изобретению новыми соединениями.
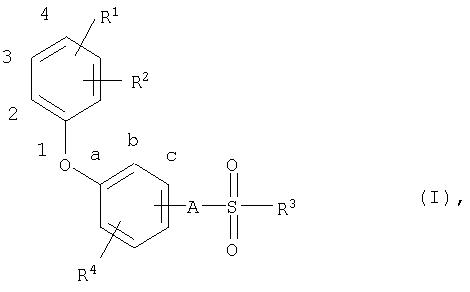
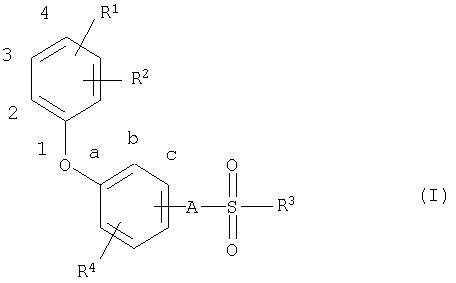
В соответствии с этим настоящее изобретение относится к новым соединениям общей формулы (I)
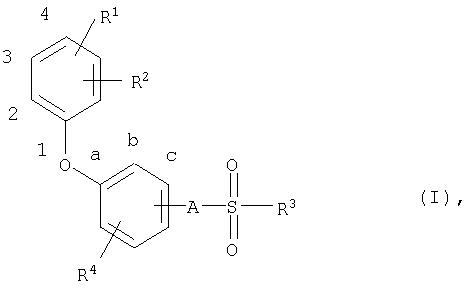
где
R1 означает атом водорода, алкильную группу с числом атомов углерода от одного до четырех, атом галогена, трифторметильную группу, трифторметоксигруппу, пианогруппу или нитрогруппу,
R2 означает атом галогена, трифторметильную группу, трифторметоксигруппу, цианогруппу или нитрогруппу,
R3 означает алкильную группу с числом атомов углерода от четырех до семи, которая может быть от одного до нескольких раз замещена атомами фтора или хлора,
R4 означает атом водорода или галогена и
А означает атом кислорода или NH-группу.
Соответствующие изобретению соединения могут существовать в стереоизомерных формах, которые отличаются как изображение и его зеркальное отражение (энантиомеры) или же не так, как изображение и его зеркальное отражение (диастереомеры). Изобретение относится как к энантиомерам или диастереомерам, так и к любым их смесям. Эти смеси энантиомеров или диастереомеров могут быть разделены известными способами на стереоизомерно однородные составляющие.
Соответствующие изобретению соединения могут также находиться в виде их солей. В общем случае здесь следует назвать соли с органическими или неорганическими основаниями или кислотами.
В рамках настоящего изобретения предпочтение отдается не вызывающим опасений с физиологической точки зрения солям. Приемлемые с физиологической точки зрения соли соответствующих изобретению соединений могут быть представлены солями соответствующих изобретению веществ с минеральными кислотами, с карбоновыми кислотами или с сульфокислотами. Особенно предпочтительны, например, соли с хлористоводородной кислотой, бромистоводородной кислотой, серной кислотой, фосфорной кислотой, метансульфокислотой, этансульфокислотой, толуолсульфокислотой, бензолсульфокислотой, нафталиндисульфокислотой, уксусной кислотой, пропионовой кислотой, молочной кислотой, винной кислотой, лимонной кислотой, фумаровой кислотой, малеиновой кислотой или с бензойной кислотой.
Приемлемыми с физиологической точки зрения солями могут быть также соли соответствующих изобретению соединений с металлами или их аммонийные соли. Особенно предпочтительны, например, натриевые, калиевые, магниевые или кальциевые соли, а также соли аммония, являющиеся производньми аммиака или таких органических аминов, как, например, этиламин, ди- или, соответственно, триэтиламин, ди- или, соответственно, триэтаноламин, дициклогексиламин, диметиламиноэтанол, аргинин, лизин, этилендиамин или 2-фенилэтиламин.
К настоящему изобретению относятся также аммонийные соединения, которые могут быть получены в результате превращения свободных аминов с помощью алкилирования.
В рамках настоящего изобретения заместители в общем случае имеют приведенное далее значение.
Алкильная группа с числом атомов углерода от одного до четырех в рамках изобретения означает линейный или разветвленный алкильный остаток с числом атомов углерода от одного до четырех. В качестве примера следует назвать метильную, этильную, н-пропильную, изопропильную, н-бутильную, изобутильную, втор.-бутильную и трет.-бутильную группу.
Алкильная группа с числом атомов углерода от четырех до семи в рамках изобретения означает линейный или разветвленный алкильный остаток с числом атомов углерода от четырех до семи. В качестве примера следует назвать н-бутильную, изобутильную, втор.-бутильную, трет.-бутильную, изопентильную, н-пентильную, гексильную или гептильную группу. Предпочтение отдается н-бутильной, н-пентильной и н-гексильной группам.
Понятие галогена в рамках изобретения включает атомы фтора, хлора, брома и иода. Предпочтение отдается атомам хлора или фтора.
Предпочтение отдается соединениям общей формулы (I), где
R1 означает атом водорода, фтора, хлора, метильную группу, трифторметильную группу, трифторметоксигруппу, цианогруппу или нитрогруппу,
R2 означает атом фтора, трифторметильную группу, трифторметоксигруппу, цианогруппу или нитрогруппу,
R3 означает н-бутильную, н-пентильную группу, 4,4, 4-трифторбут-1-ильную группу или 5,5,5-трифторпент-1-ильную группу,
R4 означает атом водорода и
А означает атом кислорода.
Особое предпочтение отдается соединениям формулы (I),
где
R1, R2, R3, R4 и А имеют приведенное выше значение, а в фенильном ядре с заместителями R1 и R2 атом водорода находится в положении 4.
Это может быть отражено в следующей формуле:

Наиболее предпочтительны соединения общей формулы (I),
где
R1, R2, R3, R4 и А имеют приведенное выше значение, тогда как R1 и R2 занимают в фенильном ядре положения 2 и 3.
Положения R1 и R2 в фенильном ядре могут быть представлены следующими формулами
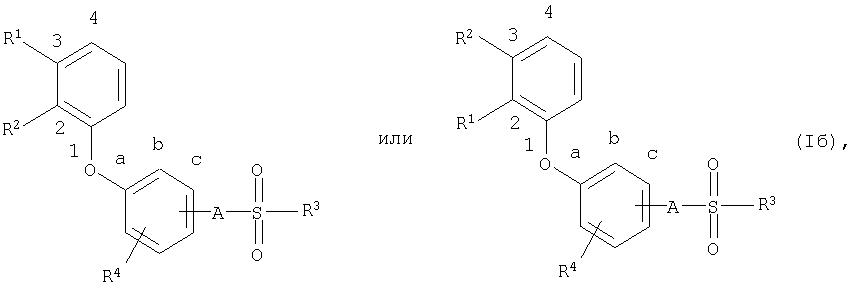
Точно также наиболее предпочтительны соединения общей формулы (I),
где
R1, R2, R3, R4 и А имеют приведенное выше значение, тогда как
А находится в положении с бензольного остатка.
Положение А в бензольном остатке может быть отражено следующей формулой:

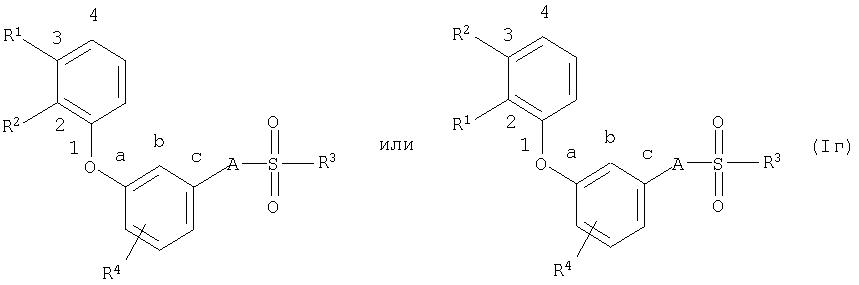
Наиболее предпочтительны соединения общей формулы (I),
где
R1, R2, R3, R4 А имеют приведенное выше значение, тогда как
R1 и R2 занимают в фенильном кольце положения 2 и 3, а
А находится в положении с бензольного остатка.
Положения R1, R2и А могут быть представлены следующими формулами:
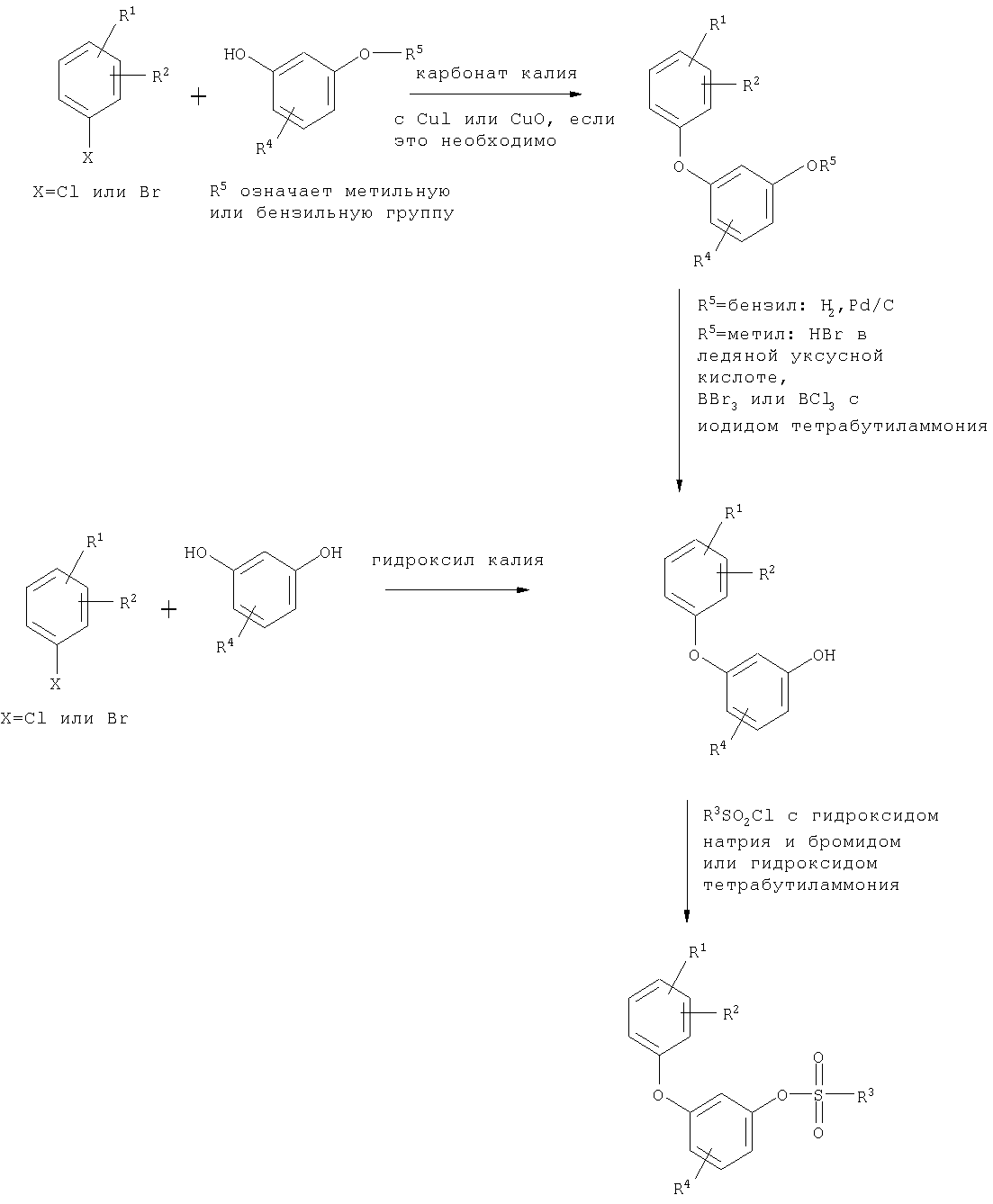
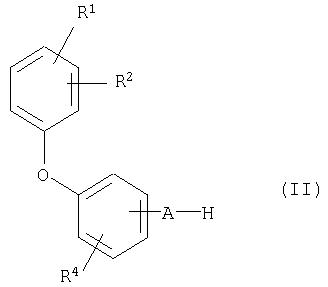
Кроме того, разработан способ получения соединений общей формулы (I), отличающийся, тем, что соединение общей формулы (II)

где R1, R2, R4 и А имеют приведенное выше значение, в инертном растворителе в присутствии подходящего основания и, если это необходимо, в присутствии катализатора межфазного переноса вводят в реакцию с соединением общей формулы (III),

где
X1 означает подходящую уходящую группу и
R3 имеет указанное выше значение.
Соединения общей формулы (II) можно получить по аналогии с общеизвестными способами в результате взаимодействия соединения общей формулы (IV) с соединением общей формулы (V)
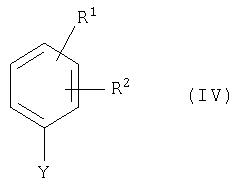
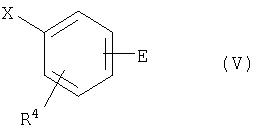
где
R1, R2 и R4 имеют приведенное выше значение,
a) Y означает гидроксильную группу и Х означает подходящую уходящую группу,
или наоборот
б) Y означает подходящую уходящую группу и Х означает гидроксильную группу,
и
Е означает нитрогруппу или группу формулы -О-R5,
где
R5 означает подходящую защитную группу для гидроксильной группы,
при этом взаимодействие проводят в инертном растворителе в присутствии подходящего основания и, если это необходимо, в присутствии соединения одновалентной меди или двухвалентной меди, получая сначала соединение общей формулы (VI)

где R1, R2, R4 и Е имеют приведенное выше значение,
и тогда
[А] это вещество в случае, когда Е означает нитрогруппу, в подходящих условиях обычными способами восстанавливают с образованием соединения общей формулы (IIa)
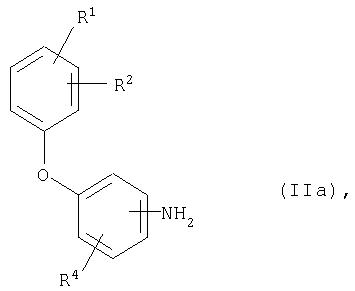
где R1, R2 и R4 имеют приведенное выше значение,
или
[Б] в случае, когда Е означает группу формулы -O-R5, в подходящих условиях обычными способами удаляют защитную группу R5 с образованием соединения общей формулы (IIб)
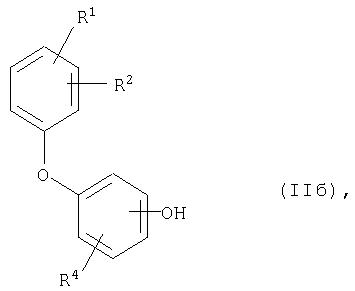
где
R1, R2 и R4 имеют приведенное выше значение.
Соединения общей формулы (II) могут быть также получены в результате взаимодействия соединения общей формулы (IV) с соединением общей формулы (VII)
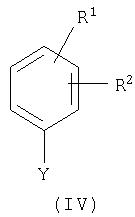
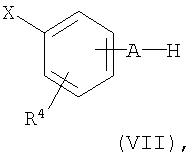
где
R1, R2, R4, А, Х и Y имеют приведенное выше значение,
при этом взаимодействие проводят в инертном растворителе в присутствии подходящего основания и, если это необходимо, в присутствии соединения одновалентной меди или двухвалентной меди.
Соответствующие изобретению способы могут быть иллюстрированы, например, следующей схемой реакций:
Инертные растворители в контексте изобретения представляют собой такие растворители, которые не подвергаются превращениям в выбранных условиях протекания реакций или изменяются незначительно.
Подходящими для способа (II)+(III)→(I) инертные растворители представлены, например, такими простыми эфирами, как, например, диэтиловый эфир, моно- или диметиловый эфир гликоля, диоксан или тетрагидрофуран, или такими углеводородами, как бензол, толуол, ксилол, циклогексан или продукты перегонки нефти, или такими галогензамещенными углеводородами, как дихлорметан, хлороформ, четыреххлористый углерод, а также диметилсульфоксидом, диметилформамидом, гексаметил-триамидом фосфорной кислоты, этилацетатом, пиридином, триэтиламином или пиколином. Можно также использовать смеси названных растворителей, в том числе и с водой. Особое предпочтение отдается дихлорметану, дихлорметану с водой, тетрагидрофурану, диоксану и диоксану с водой.
В качестве оснований для реакции (II)+(III)→(I) подходят органические амины, в частности такие триалкиламины с числом атомов углерода в алкильных группах от одного до шести, как, например, триэтиламин или диизопропилэтиламин, или такие гетероциклы, как пиридин, метилпиперидин, пиперидин или N-метилморфолин, такие гидроксиды или карбонаты щелочных или, соответственно, щелочноземельных металлов, как, например, гидроксид натрия, гидроксид калия, карбонат натрия, карбонат калия, или такие алкоголяты, как, например, метилат натрия или этилат натрия. Особое предпочтение отдается триэтиламину, пиридину и гидроксиду натрия.
В общем случае основания вводят в реакцию в количестве от 0,1 моля до 5 молей, предпочтительно от 1 моля до 3 молей, в каждом отдельном случае из расчета на 1 моль соединений общей формулы (II).
Если это необходимо, способ (II)+(III)→(I) может быть реализован и в присутствии катализатора межфазного переноса. В качестве катализатора межфазного переноса подходят, например, соли четвертичного аммония, предпочтение отдается бромиду тетра-бутиламмония.
В качестве уходящей группы X1 подходит, например, галоген, предпочтительно это хлор.
Взаимодействия могут быть проведены при нормальном давлении, но также и при повышенном или при пониженном давлении (например, в пределах от 0,5 до 3 бар). В общем случае работу проводят при нормальном давлении.
Способ (II)+(III)→(I) реализуют в температурном интервале от 0 до 100°С, предпочтительно от 0 до 30°С.
В роли подходящих инертных растворителей для способов (IV)+(V)→(VI) и (IV)+(VII)→(II) оправдало себя использование, например, таких соединений: такие органические растворители, как, например, диэтиловый эфир, моно- или диметиловый эфир гликоля, диоксан или тетрагидрофуран, или такие углеводороды, как бензол, толуол, ксилол, циклогексан или продукты перегонки нефти, или такие галогензамещенные углеводороды, как дихлорметан, хлороформ, четыреххлористый углерод, а также диметилсульфоксид, диметилформамид, N-метилпирролидон, гексаметилтриамид фосфорной кислоты, этилапетат, пиридин, триэтиламин или пиколин. Можно также использовать смеси названных растворителей, в том числе и с водой. Особое предпочтение отдается пиридину, N-метилпирролидону, диметилформамиду и диметилсульфоксиду.
При необходимости, способы (IV)+(V)→(VI) и (IV)+(VII)→(II) могут быть реализованы также в присутствии соединения одновалентной или двухвалентной меди. Предпочтение отдается иодиду одновалентной меди и оксиду двухвалентной меди.
На роль оснований для способов (IV)+(V)→(VI) и (IV)+(VII)→(II) подходят карбонаты и бикарбонаты щелочных металлов, в частности карбонат натрия и калия, гидроксиды щелочных металлов, в частности гидроксид натрия, или органические амины, в частности такие триалкиламины с числом атомов углерода в алкильных группах от одного до шести, как, например, триэтиламин. Особое предпочтение отдается гидроксиду калия, гидроксиду натрия и карбонату калия.
В общем случае основания вводят в реакцию в количестве от 0,1 моля до 5 молей, предпочтительно от 1 моля до 3 молей, в каждом отдельном случае из расчета на 1 моль соединений общей формулы (IV) или, соответственно, (V).
Для роли уходящей группы Х в способе (IV)+(V)→(VI) в варианте а) и, соответственно, Y в способе (IV)+(V)→(VI) в варианте б) подходит, например, галоген или такая сульфонатная группа, как, например, трифлатная. Предпочтение отдается фтору, хлору или брому.
Взаимодействия могут быть проведены при нормальном давлении, но также и при повышенном или при пониженном давлении (например, в пределах от 0,5 до 5 бар). В общем случае работу проводят при нормальном давлении.
Реакции проводят в температурном интервале от 20 до 200°С, предпочтительно от 100 до 160°С.
Способы восстановления ароматической нитрогруппы для стадии процесса (IV)→(IIa) известны (например, это R.C.Larock, "Comprehensive Organic Transformations", Нью Йорк, 1989, с.411-415, и цитированная в этом источнике литература).
Введение защитных групп для гидроксильных функций, а также способы их отщепления известны (например, T.W.Green, P.G.M.Wuts, "Protective Groups in Organic Synthesis", 2-е изд. Нью Йорк. 1991, и цитированная в этом источнике литература; J. Org. Chem. 1999, 64, 9719-9721).
В качестве защитной группы R5 для реакционной последовательности (IV)+(V)→(VI)→(11б) подходят, например, метильная, бензильная, аллильная, метоксиметильная, 2-триметилсилил-этоксиметильная или триметилсилильная группы. Предпочтение отдается метильной и бензильной группам.
Соединения общей формулы (III) могут быть приобретены коммерческим путем, они известны из литературы или могут быть синтезированы известными из литературных источников способами (ср., например, J. Chem. Soc. С 1968, 1265; Chem. Ber. 1967, 100, 1696; фторированные хлориды алкансульфокислот могут быть получены, например, в соответствии с заявкой на международный патент №А-98/37061 или с заявкой на патент ФРГ №А-1942264).
Соединения общих формул (IV), (V) и (VII) известны или же они могут быть получены известными способами.
Соединения общих формул (IV) и (V) могут быть приобретены коммерческим путем, они известны из литературных источников или они могут быть получены по аналогии с известными из литературы способами (ср., например, J.March, "Advanced Organic Chemistry", 4-е изд., Уайли, 1992, с.с. 531-534 и 1295, и, соответственно, цитированная в этом источнике литература; Synthesis 1990. 1145-1147).
Неожиданным образом соответствующие изобретению соединения показали ценный спектр фармакологической активности, что нельзя было предвидеть заранее.
Они выделяются как высокоэффективные агонисты каннабиноидных рецепторов с высокой метаболической стабильностью и с высокой биодоступностью при приеме их через рот. В соответствии с этим они особенно хорошо подходят для оральной терапии.
В качестве единственных лекарственных средств или в сочетании с другими лекарственными средствами они могут быть использованы для профилактики и лечения острых и/или хронических болевых синдромов (их классификация представлена в "Classification of Chronic Pain, Descriptions of Pain Syndromes and Definitions of Pain Terms", 2-е изд., изд. Мески и Бегдак, lASP-Press, Сиэтл, 1994), а также нейродегенеративных заболеваний, в частности для лечения болей, вызванных раковыми заболеваниями, и таких хронических нейропатических болей, как, например, при диабетической нейропатии, постгерпесной невралгии, повреждениях периферических нервов, при центральных болях (например, как следствие церебральной ишемии) и тригеминальной невралгии, и при таких других хронических болях, как, например, люмбаго, боли в спине (low back pain) или ревматические боли. Наряду с этим, данные вещества подходят также для терапии первичных острых болей любого происхождения и происходящих от них вторичных болевых синдромов, а также для терапии острых болевых синдромов, перешедших в хронические.
Для комбинирования с соответствующими изобретению соединениями с целью лечения острых и/или хронических болей подходят, например, опиаты, например трамадол, морфин, дигидрокодеин, декстропропоксифен, трициклические антидепрессанты, например амитриптилин, антиконвульсанты, например карбамазепин, габапентин, нестероидные противовоспалительные средства, например аспирин, ибупрофен, напроксен, включая СОХ-2-ингибиторы, например рофекоксиб, целекоксиб.
Точно также подходят соответствующие изобретению соединения для терапии первичных и/или вторичных болезненных состояний мозга, например, во время или после церебральных сосудистых спазмов, мигрени, спастичности, гипоксии и/или аноксии, которые не имеют названного выше генеза, перинатальной асфиксии, аутоиммунных заболеваний, заболеваний, связанных с обменом веществ, или заболеваний органов, которые могут иметь происхождение от повреждений мозга, а также повреждений мозга вследствие первичных заболеваний мозга, например судорожных состояний и атеро-и/или артериосклеротических изменений. Соответствующие изобретению соединения подходят также для лечения таких хронических или психиатрических недугов, как, например, депрессия, язва желудка, таких нейродегенеративных заболеваний, как, например, болезни Альцгеймера, Паркинсона или Хантингтона, рассеянный склероз, амиотрофический латеральный склероз, дегенерация нервов в результате острых и/или хронических вирусных или бактериальных инфекций и мультиинфарктной деменции.
Кроме того, они могут быть использованы в лекарственных средствах для лечения рвоты, тошноты, глаукомы, астмы, анорексии, конвульсий, ревматизма, заторможенности и нарушений двигательных функций.
Соответствующие изобретению вещества подходят также для лечения заболеваний, вызываемых бактериальной и/или вирусной инфекцией, основанных на прямых и/или опосредованных изменениях иммунной системы или, соответственно на нарушениях регуляции с участием иммунной системы, например, при локальных или системных аутоиммунных заболеваниях (например, красной волчанки Lupus erythematodes во всех ее вариантах), при воспалительных и/или имеющих аутоиммунологическое происхождение заболеваниях суставов (например, при первичном хроническом полиартрите, при обусловленных травмами воспалениях), при воспалительных и/или имеющих аутоиммунологическое происхождение заболеваниях костного и мышечного аппарата, при воспалительных и/или имеющих аутоиммунологическое происхождение болезненных процессах во внутренних органах (например, при болезни Крона, язвенном колите, гломерулонефрите), в наружных органах (например, при аллергических реакциях на поступающие с воздухом антигены) и в центральной нервной системе (например, при рассеянном склерозе, болезни Альцгеймера, психиатрических заболеваниях), а также в органах чувств, при первичных и/или вторичных и/или аутоиммунологических заболеваниях кроветворной системы и самой иммунной системы (например, при реакциях отторжения, при СПИДе), а также при заболеваниях кожи воспалительного и/или иммунологического происхождения у человека и животных. Кроме того, эти вещества оказывают действие при косвенных симптомах этих заболеваний, например боли.
Предпочтительно их используют для лечения боли, спастичности, церебральных ишемий и травм черепа/мозга.
Действие соответствующих соединений in vitro на каннабиноидные рецепторы может быть иллюстрировано следующими далее биологическими опытами.
1. Опыт на CB1-люциферазе репортерного гена крыс
Основные культуры Репортер линии клеток крыс - СНОСВ1 получают по описанной в заявке на международный патент №А-98/37061, с.55 и сл. методике.
Для скрининга веществ используют следующую далее последовательность экспериментальных операций. Основные культуры выращивают в 50% модифицированной Дальбекко среде Игл / 50% F-12 (DMEM/F12) с 10% FCS при 37°С в атмосфере с 10% диоксида углерода и в каждом отдельном случае по истечении срока от двух до трех дней делят их как 1:10. Подопытные культуры высевают по 5000 клеток на ячейку, используя пластинки с 96 ячейками, и доращивают их в течение 70 часов при 37°С. После этого культуры осторожно промывают солевым раствором с фосфатным буфером и приводят в исходное состояние с помощью свободной от сыворотки среды Ultra-СНО (Bio-Whittaker). Растворенные в диметилсульфоксиде вещества однократно разбавляют в среде и с помощью пипетки прибавляют к подопытным культурам (максимальная конечная концентрация диметилсульфоксида в изучаемой смеси равна 0,5%). Через 20 минут прибавляют форсколин и после этого инкубируют культуры в течение трех часов в камере для выращивания при 37°С. Затем удаляют супернатанты, а клетки лизируют с помощью 25 мкл лизирующего реагента (25 ммоль/л трифосфата, рН 7,8 с 2 ммоль/л дитиотреитола, 10% глицерина, 3% Тритона Х100). Сразу после этого прибавляют раствор субстрата для люциферазы (2,5 ммоль/л аденозинтрифосфата, 0,5 ммоль/л люциферина, 0,1 ммоль/л кофермента А, 10 ммоль/л трицина, 1,35 ммоль/л сульфата магния, 15 ммоль/л дитиотреитола, рН 7,8), быстро встряхивают и определяют активность люциферазы в системе камер Хамаматцу.
Для инактивации Gi-белков подопытные культуры перед проведением опыта обрабатывают в течение 16 часов действием 5 нг/мл (конечная концентрация) токсина пертуссиса.
Значения концентраций ингибирования IC50 рассчитывают с помощью программы GraphPadPrism (Уравнение Хилла, версия: one-site competition).
Вещество по примеру 17 показывает в этом опыте значение IC50, равное 0,81 нмоль/л.
2. Опыт на hСВ2-люциферазе репортерного гена крыс
Проводят стабильную трансфекцию клеток СНOlис9 СВ2-рецептором человека. Транс-фекцию, селекцию клонов и выращивание опытного материала проводят по аналогии с работами на СВ1-рецепторе крыс. Для фармакологической характеризации клеток и для испытания веществ используют следующую далее последовательность экспериментальных операций.
Основные культуры выращивают в 50% модифицированной Дальбекко среде Игл / 50% F-12 (DMEM/F12) с 10% FCS при 37°С в атмосфере с 10% диоксида углерода и в каждом отдельном случае по истечении срока от двух до трех дней делят их как 1:10. Подопытные культуры высевают по 5000 клеток на ячейку, используя пластинки с 96 ячейками, в среду DMEM/F12 с 5% FCS и доращивают их в течение 70 часов при 37°С. После этого культуры отделяют от среды и заменяют ее свободной от сыворотки средой Ultra-CHO (Bio-Whittaker). Растворенные в диметилсульфоксиде вещества (конечная концентрация 200х) с помощью пипетки прибавляют к подопытным культурам (максимальная конечная концентрация диметилсульфоксида в изучаемой смеси равна 0,5%) и через 20 минут прибавляют форсколин. После этого инкубируют культуры в течение трех с половиной часов в камере для выращивания при 37°С. Затем удаляют супернатанты, а клетки лизируют с помощью 25 мкл лизирующего реагента (25 ммоль/л трифосфата, рН 7,8 с 2 ммоль/л дитиотреитола, 10% глицерина, 3% Тритона X100). Сразу после этого прибавляют 50 мкл раствора субстрата для люциферазы с удвоенной концентрацией (5 ммоль/л аденозинтрифосфата, 1 ммоль/л люциферина, 0,2 ммоль/л кофермента А, 10 ммоль/л трицина, 1,35 ммоль/л сульфата магния, 15 ммоль/л дитиотреитола, рН 7,8), быстро встряхивают и определяют активность люциферазы в измерительной системе камер с фотоумножителем (Хамаматцу).
Значения концентраций ингибирования IC50 рассчитывают с помощью программы GraphPadPrism™ (Уравнение Хилла, версия: one-site competition).
3. Связывание с кортикальными мембранами крыс
По стандартным методикам приготавливают мембранный белок из различных тканей или, соответственно, клеток. С помощью пипетки получают смесь буфера, меченого лиганда, диметилсульфоксида или исследуемого вещества, затем прибавляют 100 мкг белка, хорошо перемешивают смесь и инкубируют в водяной бане в течение 60 минут при 30°С. По истечении времени инкубации реакцию останавливают добавлением в каждую трубочку охлажденного во льду инкубационного буфера. После отфильтровывания промывают, используя 3/4 мл инкубационного буфера. Фильтры переносят в минифлаконы, радиоактивность определяют в жидкостном сцинтилляционном счетчике.
Метаболическая стабильность соответствующих изобретению соединений может быть показана с помощью следующих далее опытов in vitro.
4. Изучение микросомальной стабильности
Метаболическая стабильность соответствующих изобретению соединений может быть определена с помощью микросом печени крысы (по аналогии с J. Pharmacol. Exp. Ther. 1997, 283, 46-58).
Для определения микросомальной стабильности и расчета на основе эффекта первичного превращения в печени (реакции фазы I) максимально возможной биодоступности (Fmax) инкубируют вещество в небольшой концентрации с микросомальным белком в течение 15 минут с добавлением кофакторов при 37°С.
Инкубацию и отбор проб проводят на модифицированном пипетирующем автомате фирмы Канберра Пакард.
Как показывает сравнение с примером из заявки на международный патент №А-98/37061, соответствующие изобретению соединения в этом опыте более метаболически стабильны.

Биодоступность соответствующих изобретению соединений, а также другие фармако-кинетические параметры могут быть определены in vivo следующим далее образом.
5. Фармакокинетика в крысе
а) Внутривенное вливание
Вещество вливают с помощью бранулы непосредственно в кровоток через латеральную вену хвоста в течение 15 минут. Для точного введения выбранной дозы и объема используют калиброванный шприц на 20 мл. Для вливания используют насос производства Браун Мельзунген №152440/1.
б) Пероральное введение
Дозу вещества вводят в виде болюса через желудочный зонд.
в) Отбор проб и исследование крови и плазмы
Пробы крови отбирают у животных с катетером (яремная вена) в гепаринизированные трубочки. Кровь центрифугируют и соответствующим образом готовят плазму для аналитического исследования (жидкостная хроматография с масс-спектроскопией, масс-спектроскопия). До проведения анализа плазму хранят при температуре менее -15°С.
г) Результаты фармакокинетических исследований
Микросомальные данные (микросомы печени крысы) позволяют предположить максимально возможную биодоступность до 100%.
Полученные в опытах in vivo (на крысах) фармакокинетические параметры для примера 22:
Пероральные данные (доза 3 мг/кг): AUCnorm - 0,102 кг·час/л, Cmax,norm - 0,0198 кг/л, tmax - 2.29 ч, t1/2 - 2,36 ч, F - 33%.
Внутривенные данные (доза 0,3 мг/кг): AUCnom - 0,307 кг·час/л, Сmax,norm - 0,5978 кг/л, Vss - 4,12/кг, t1/2 - 1,6 ч.
Принятые обозначения:
AUCnorm - приведенная к дозе 1 мг/кг поверхность под кривой зависимости концентрации в плазме от времени;
Сmax,norm - приведенная к дозе 1 мг/кг максимальная концентрация в плазме после однократного введения;
tmax - время, в течение которого достигается максимальная концентрация в плазме после однократного введения;
t1/2 - конечное время полупревращения;
F - биодоступность, в данном случае это доля от дозировки в процентах, которая оказывает системное действие по сравнению с внутривенным введением;
Vss - эффективный объем распределения в стационарном состоянии (steady state).
Действие in vivo соответствующих изобретению соединений может быть представлено, например, в следующих далее опытах на животных.
6. Гипотермия (на крысах)
Эффект агонизма in vivo на СВ1-рецептор демонстрируется в эксперименте на гипо-термию на крысе.
Через пять минут после определения ректальной температуры тела с помощью температурного зонда Oesophagus вводят (перорально) исследуемое вещество. Контрольная группа получает также перорально только растворитель для исследуемых веществ (Cremophore EL 1-10% + дистиллированная вода). Температуру тела измеряют через 120 и 240 минут после перорального введения. Численность группы для каждой дозировки составляет 5-7 животных (крысы).
Гипотермия на крысах - эффект агонизма
a)Эффективная доза для снижения температуры тела на 1°С
Возможность использования соответствующих изобретению соединений для лечения болевых синдромов может быть продемонстрирована следующими далее опытами на животных.
7. Аксотомия разветвлений седалищного нерва на крысах (модель хронических болей)
При анестезии с помощью пентабарбитала препарируют трифуркацию седалищного нерва, после перевязывания нервов рядом с аксотомированным участком аксотомируют ответвление малоберцового нерва и большеберцового нерва. На контрольных животных проводят симуляцию операции. После операции у аксотомированных животных развивается хроническая механическая гиперальгезия. Эту гиперальгезию определяют с помощью датчика давления (электронный анестезиометр Фрея производства ПТС Inc.-Life Science Instruments, Woodland Hills, CA, США) в сравнении с животными, на которых операция только симулировалась.
Введение веществ осуществляют в разные точки времени перед измерением болевого эффекта при различных способах введения (внутривенно, внутрибрюшинно, перорально, i.t., i.c.v., через кожу).
Вещество по примеру 22 снижает гиперальгезию в модельном опыте при минимальной эффективной дозировке 1 мг/кг, перорально (акутное введение, за 60 минут перед опытом).
Возможность использования соответствующих изобретению соединений, например, для лечения нейродегенеративных заболеваний может быть показана в модельном опыте перманентной фокальной церебральной ишемии на крысах (МСА-O) или в модельном опыте субдуральной гематомы на крысах (SDH) (заявка на международный патент №А-98/37061, с.60 и сл.).
Новые активные вещества могут быть известными способами переведены в такие обычные лекарственные формы, как таблетки, драже, пилюли, грануляты, аэрозоли, сиропы, эмульсии, суспензии и растворы, с использованием инертных, нетоксичных, подходящих с фармацевтической точки зрения веществ носителей или растворителей. Для этого терапевтически эффективное соединение в каждом отдельном случае должно находиться в концентрации от примерно 0,5 до 90 мас.% от общей смеси, то есть в количествах, которые достаточны для достижения указанных пределов дозирования.
Лекарственные формы получают, например, разбавлением активных веществ растворителями и/или веществами носителями с применением, если это необходимо, эмульгирующих и/или диспергирующих средств, причем в случае использования, например, в качестве разбавителя воды могут быть также использованы органические растворители в качестве солюбилизаторов.
Введение проводят обычными способами, предпочтительно через рот, через кожу или парентерально, в частности через язык или внутривенно. Возможно также ингаляционное введение через рот или нос, например с помощью аэрозоля, или наружно через кожу.
В общем случае оказалось, что для достижения эффективных результатов лучше всего вводить вещества в количествах от примерно 0,001 до 10 мг/кг, при введении через рот предпочтительно от примерно 0,005 до 1 мг/кг массы тела.
Тем не менее в отдельных случаях может появиться необходимость в отклонении от названных количеств, а именно в зависимости от массы тела или соответственно от способа введения, от индивидуального отношения к медикаменту, от вида его лекарственной формы и от времени или соответственно от интервала времени, в которые происходит его введение. Так, например, в некоторых случаях можно брать меньше чем указанные минимальные количества, тогда как в других случаях приходится превышать названный выше верхний предел. В случае использования повышенных количеств можно рекомендовать распределение их на несколько отдельных доз в течение дня.
Определение времени удерживания исходных соединений и получаемых веществ с помощью ВЭЖХ проводят в следующих условиях:
Колонка: Kromasil С 18 60*2; объем вводимой пробы 1,00 мкл; скорость подачи 0,75 мл/мин; элюент: А=0,01 моль/л фосфорной кислоты в воде, Б = ацетонитрил; градиент [t(мин) - А/Б]: 0-90/10; 0,5-90/10; 4,5-10/90, 6,5-10/90, 7,5-90/10.
Используемые сокращения
Исходные соединения Пример I
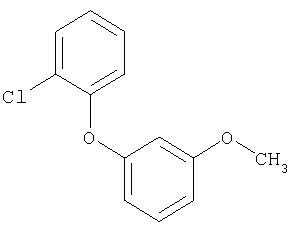
3-Метокси-1-(3-метил-2-нитрофенокси)бензол
(Синтез дифениловых эфиров, способ А)
К примерно 600 мл пиридина прибавляют 14,7 г (96,0 ммолей) 3-метил-2-нитрофенола, 53,9 г (288 ммолей) 3-броманизола и 18,3 г (96,0 ммолей) карбоната калия и нагревают до температуры около 140°С. Смеси дают немного снова остыть и прибавляют 18,3 г (96 ммолей) иодида одновалентной меди. Реакционную массу около 60 часов перемешивают при температуре около 140°С. После удаления растворителя в вакууме остаток растворяют в толуоле и снова упаривают. Остаток растворяют в дихлорметане и фильтруют через инфузорную землю. После промывки небольшим количеством дихлорметана последовательно промывают 5 н. соляной кислотой, 2 н. раствором гидроксида натрия, 5 н. соляной кислотой, водой и раствором хлорида натрия. После сушки над сульфатом магния упаривают в вакууме и очищают полученный сырой продукт перегонкой с шариковым дефлегматором.
Выход 3,50 г (13%; чистота по данным ВЭЖХ 94%).
Значение Rf 0,28 (циклогексан/этилацетат 5:1).
Масс-спектр (ионизация при электронном ударе): 259 (100%, [М]+).
ВЭЖХ: время удерживания 4,94 мин.
1H-ЯМР (300 МГц, дейтерохлороформ): δ (миллионных долей) = 2,37 (с., 3Н), 3,78 (с., 3Н), 6,1-6,65 (м, 2Н), 6,67-6,74 (м, 1Н), 6.83 (д., J=8 Гц, 1Н), 6,99 (д., J=8 Гц, 1Н), 7,14-7,32 (м., 2Н).
Пример II
3-Метокси-1-[2-(трифторметил)фенокси]бензол
(Синтез дифениловых эфиров, способ Б)
К примерно 450 мл пиридина прибавляют 50,0 г (222 ммоля) 2-бромбензотрифторида, 27,6 г (222 ммоля) 3-метоксифенола и 30,7 г (222 ммоля) карбоната калия и непродолжительное время нагревают при температуре около 100°С. Дают немного снова остыть и прибавляют 17,7 г (222 ммоля) оксида двухвалентной меди. Реакционную массу около 48 часов перемешивают при нагревании с обратным холодильником (температура бани около 140°С). После удаления растворителя в вакууме остаток растворяют в дихлорметане и встряхивают с 2 н. соляной кислотой. После этого органическую фазу промывают 1 н. раствором гидроксида натрия и водой. После сушки над сульфатом магния упаривают в вакууме и очищают полученный сырой продукт перегонкой с шариковым дефлегматором.
Выход 37,2 г (62%; чистота по данным ВЭЖХ 98%).
Значение Rf 0,47 (циклогексан/этилацетат 5:1).
Масс-спектр (ионизация при электронном ударе): 268 (100%, [М]+).
ВЭЖХ: время удерживания 5,14 мин.
1 H-ЯМР (200 МГц, дейтерохлороформ): δ (миллионных долей) = 3,79 (с., 3Н), 6,56-6,65 (м, 2Н), 6,71 (д.д.д, J=8 Гц. 2 Гц, 1 Гц, 1Н), 6,97 (д., J=8 Гц, 1Н), 7,17 (т., J=8 Гц, 1Н), 7.25 (т., J=8 Гц, 1Н), 7,46 (т.д., J=8 Гц. 1 Гц, 1Н), 7,66 (д., J=8 Гц, 1Н).
Пример III
1-[2-Циано-3-(трифторметил)фенокси]-3-метоксибензол
(Синтез дифениловых эфиров, способ В)
В атмосфере аргона к безводному диметилформамиду прибавляют 10,7 г (52,1 ммоля) 2-хлор-6-(трифторметил)бензонитрила [пример 5 в заявке на патент ФРГ №А-3836159; 2-хлор-6-(трихлорметил)бензонитрил может быть получен из 2,6-диметилбензонитрила по примеру 3 в заявке на патент ФРГ №А-2214058], прибавляют 7,19 г (52,1 ммоля) карбоната калия и 6,46 г (52,1 ммоля) 3-метоксифенола и 5 часов перемешивают при 100°С. После этого прибавляют 500 мл 2 н. гидроксида натрия и 200 мл насыщенного раствора хлорида натрия. Два раза экстрагируют эфиром примерно по 300 мл, сушат объединенные органические фазы над сульфатом магния, упаривают в вакууме и проводят флэш-хроматографию на 450 г силикагеля, используя в качестве подвижной фазы толуол. Фракции с продуктом упаривают досуха и прибавляют к остающемуся маслу немного эфира, оставляют для кристаллизации, отсасывают и промывают пентаном.
Выход 9,36 г (57%; чистота по данным ВЭЖХ 96%).
Значение Rf 0,39 (толуол).
Температура плавления 68°С
ВЭЖХ: время удерживания 4,89 мин.
1H-ЯМР (200 МГц, дейтерохлороформ): δ (миллионных долей) = 3,72 (с., 3Н), 6,62-6,87 (м, 3Н), 7,08 (д., J=8 Гц, 1Н), 7,33 (т., J=8 Гц, 1Н), 7,44 (д., J=8 Гц, 1Н), 7,57 (т., J=8 Гц, 1Н).
Пример IV
1-[2-Хлор-3-(трифторметил)фенокси]-3-нитробензол
(Синтез дифениловых эфиров, способ Г)
В атмосфере аргона к 10 мл диметилформамида прибавляют 1,00 г (5,09 ммоля) 2-хлор-3-(трифторметил)фенола, 0,72 г (5,09 ммоля) трифторнитробензола и 0,70 г (5,09 ммоля) карбоната калия. Смесь примерно 16 часов кипятят с обратным холодильником. После охлаждения прибавляют к реакционной массе 50 мл 2 н. гидроксида натрия и перемешивают еще один час, затем прибавляют 20 мл раствора хлорида натрия и перемешивают еще 30 минут. Затем экстрагируют дихлорметаном, сушат органическую фазу над сульфатом магния и упаривают в вакууме. Очистку проводят хрома-тографированием на силикагеле, используя в качестве подвижной фазы циклогексан/этилацетат 20:1, получают 0,69 г (42%; чистота по данным ВЭЖХ 100%) целевого соединения.
Значение Rf 0,39 (циклогексан/этилацетат 2:1).
Масс-спектр (ионизация при электронном ударе): 317 (100%, [М]+).
ВЭЖХ: время удерживания 5,22 мин.
1Н-ЯМР (300 МГц, d6-диметилсульфоксид): δ (миллионных долей) = 7,51 (д.д., J=8 Гц, 2 Гц, 1Н), 7,56-7,83 (м, 5Н), 8,05 (д.д, J=8 Гц, 2 Гц, 1Н).
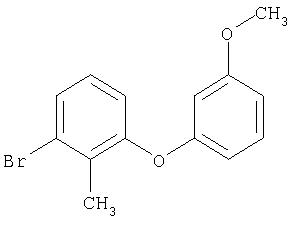
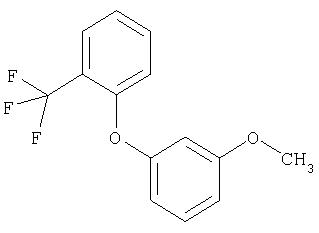
Следующие далее примеры V-XIII получают по аналогичным схемам в соответствии со способами получения исходных соединений А или Б из соответствующих исходных продуктов.



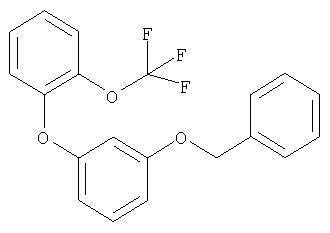


Пример XIV
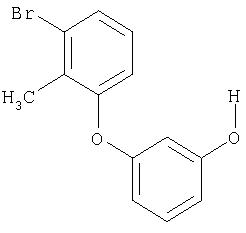
3-(3-Метил-2-нитрофенокси)фенол
(Отщепление метильной группы в простом эфире, способ А)
В атмосфере аргона к 2 мл безводного дихлорметана прибавляют 500 мг (1,93 ммоля) 1-метокси-3-(3-метил-2-нитрофенокси)бензола и охлаждают раствор до -20°С. При этой температуре прибавляют 5,8 мл раствора трибромида бора в дихлорметане с концентрацией 1 моль/л. Температуре дают подняться до 0°С и перемешивают 1 час. После добавления воды три раза экстрагируют дихлорметаном. Объединенные органические фазы промывают раствором бикарбоната натрия, сушат над сульфатом магния и упаривают в вакууме. После очистки хроматографированием на силикагеле со смесью циклогексан/этилацетат 30:1 в качестве подвижной фазы получают 424 мг (89%) целевого соединения.
Значение Rf 0,18 (циклогексан/этилацетат 2:1).
Масс-спектр (ионизация при электронном ударе): 245 ([М]+).
ВЭЖХ: время удерживания 4,40 мин.
1Н-ЯМР (300 МГц, дейтерохлороформ): δ (миллионных долей) = 2,37 (с., 3Н), 4,88 (ш.с, 1Н), 6,54 (т., J=2 Гц, 1Н), 6,57-6,66 (м, 2Н). 6,85 (д., J=8 Гц, 1Н), 7,01 (д., J=8 Гц, 1Н), 7,19 (т., J=8 Гц, 1Н), 7,27 (т., J=8 Гц, 1Н).
Пример XV
3-[2-Циано-3-(трифторметил)фенокси]-фенол
(Отщепление метальной группы в простом эфире, способ Б)
В атмосфере аргона в безводном дихлорметане растворяют 10,0 г (34,1 ммоля) 1-(2-ци-ано-3-трифторметилфенокси)-3-метоксибензола и прибавляют 13,9 г (37,5 ммоля) иодида н-тетрабутиламмония. Охлаждают до -78°С и медленно прибавляют по каплям 120 мл раствора трихлорида бора в дихлорметане с концентрацией 1 моль/л, поддерживая температуру не выше -70°С. В течение двух часов дают нагреться до комнатной температуры. Реакционную смесь выливают в 300 мл воды со льдом, три раза экстрагируют смесь дихлорметаном, органическую фазу два раза промывают насыщенным раствором бикарбоната натрия и один раз раствором хлорида натрия. Сушат над сульфатом магния и проводят очистку с помощью флэш-хроматографии на примерно 400 г силикагеля с дихлорметаном. К полученному маслянистому продукту прибавляют пентан и оставляют для кристаллизации.
Выход 7,75 г (96%, чистота по данным ВЭЖХ 96%).
Значение Rf 0,16 (дихлорметан).
Температура плавления 108°С.
ВЭЖХ: время удерживания 4,41 мин.
1H-ЯМР (300 МГц, дейтерохлороформ): δ (миллионных долей) = 5,13 (с., 1Н), 6,59-6,78 (м, 3Н), 7,11 (д., J=8 Гц, 1Н), 7,28 (т., J=8 Гц, 1Н), 7,45 (д., J=8 Гц, 1Н), 7,58 (т., J=8 Гц, 1Н).
Пример XVI
3-[2-Хлор-3-(трифторметил)фенокси]фенол
(Отщепление метильной группы в простом эфире, способ В)
В 6 мл ледяной уксусной кислоты растворяют 600 мг (1, 98 ммоля) 3-метокси-1-[2-хлор-3-(трифторметил)фенокси] бензола, прибавляют 3,60 мл 48%-ной водной бромистоводородной кислоты и 4 часа кипятят с обратным холодильником. После охлаждения разбавляют водой и экстрагируют этилацетатом. Органические фазы три раза промывают водой, сушат над сульфатом магния и упаривают в вакууме. Хроматографированием на силикагеле со смесью дихлорметан/циклогексан 2:1 в качестве подвижной фазы получают 484 мг (81%, чистота по данным ВЭЖХ 96%) целевого соединения.
Значение Rf 0,39 (циклогексан/этилацетат 2:1).
Масс-спектр (ионизация при электронном ударе): 288 ([М]+).
ВЭЖХ: время удерживания 4,80 мин.
1H-ЯМР (300 МГц, d6-диметилсульфоксид): δ (миллионных долей) = 6,37 (т., J=2 Гц), 6,44 (д.д.д, J=8 Гц, 2 Гц, 1 Гц, 1Н), 6,59 (д.д.д, J=8 Гц, 2 Гц, 1 Гц, 1Н), 7,20 (т., J=8 Гц, 1Н), 7,39 (д.д, J=8 Гц, 1 Гц, 1Н), 7,56 (т., J=8 Гц, 1Н), 7,68 (д.д, J=8 Гц, 1 Гц, 1Н), 9,69 (с., 1Н).
Пример XVII
3-[3-(Трифторметил)фенокси]фенол
(Отщепление бензильной группы в простом эфире, способ Г)
В аппаратуре для гидрирования в 135 мл тетрагидрофурана и 15 мл этанола суспендируют 1,70 г (4,72 ммоля) 3-бензилокси-1-[3-(трифторметил)фенокси]бензола и после прибавления 170 мг 10%-ного палладия на угле в течение ночи гидрируют при нормальной температуре и давлении водорода 1 атм. Для выделения продукта отфильтровывают катализатор через инфузорную землю, фильтрат упаривают и флэш-хроматографируют на 130 г силикагеля с градиентом циклогексан/этилацетат от 10:1 до 1:1.
При удалении растворителя получают 1,27 г (99%, чистота по данным ВЭЖХ 95%) целевого соединения.
Значение Rf 0,28 (циклогексан/этилацетат 5:1).
Масс-спектр (ионизация при электронном ударе): 270 ([М]+).
ВЭЖХ: время удерживания 4,78 мин.
1Н-ЯМР (200 МГц, дейтерохлороформ): δ (миллионных долей) = 4,97 (с., 1Н), 6,53 (т., J=2 Гц, 1Н), 6,56-6,67 (м, 2Н), 6,85-7,01 (м, 3Н), 7,22 (т., J=8 Гц, 1Н), 7,34 (т., J=8 Гц, 1Н).
Следующие далее примеры XVIII-XXV получают аналогично в соответствии с методиками А, В или Г.
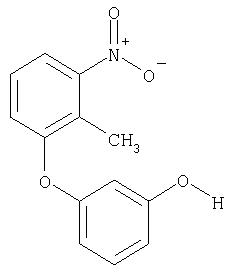

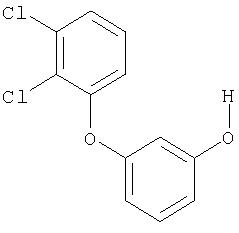
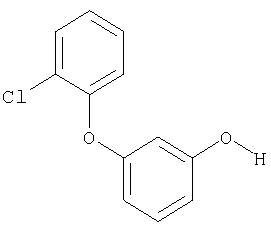

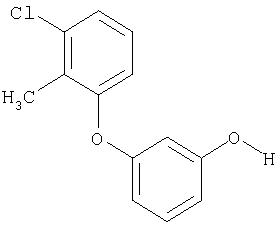
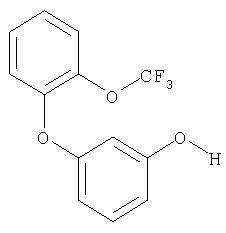
Пример XXVI
3-[2-Хлор-3-(трифторметил)фенокси]-анилин
В атмосфере аргона к 7 мл метанола прибавляют 630 мг (1,98 ммоля) 1-[2-хлор-3-(три-фторметил)фенокси]-3-нитробензола, 625 мг (9,09 ммоля) формиата аммония и 31,5 мг 10-ного палладия на угле в качестве катализатора. Смесь два часа нагревают с обратным холодильником. После охлаждения фильтруют через инфузорную землю, промывают метанолом и упаривают фильтрат. Снова растворяют в дихлорметане, три раза экстрагируют водой, сушат органические фазы над сульфатом магния и снова упаривают. Хроматографической очисткой на силикагеле со смесью циклогексан/этил-ацетат 6:1 в качестве подвижной фазы получают 458 мг (72%, по данным ВЭЖХ чистота 90%) целевого соединения.
Значение Rf 0,48 (циклогексан/этилацетат 1:1).
Масс-спектр (ионизация при электрораспылении): 288 (22%, [М+Н]+).
ВЭЖХ: время удерживания 4,41 мин.
1H-ЯМР (300 МГц, d6-диметилсульфоксид): δ (миллионных долей) = 5,28 (с., 2Н), 6,11-6,19 (м., 2Н). 6,38 (д.д.д, J=8 Гц, 2 Гц, 1 Гц, 1Н), 7,03 (т., J=8 Гц. 1Н), 7,33 (д.д., J=8 Гц, 1 Гц, 1Н), 7.54 (т., J=8 Гц, 1Н), 7,63 (д.д., J=8 Гц, 1 Гц).
Пример XXVII
1-[2-Циано-3-(трифторметил)фенокси]-3-гидроксибензол
(Синтез дифенилового эфира по методике Д)
В 170 мл N-метилпирролидона частично растворяют 44,0 г (0,4 моля) резорцина, прибавляют гидроксид калия [не менее чем 85%-ный, 34,5 г (0,52 моля)] и после этого прибавляют 2-хлор-6-(трифторметил)бензонитрил [20,5 г (0,1 моля)]. Эту смесь 2,5 часа перемешивают при 60-65°С. После прибавления 300 мл толуола и 400 мл воды отделяют водную фазу и еще раз экстрагируют 300 мл толуола. Объединенные органические фазы сушат над сульфатом магния и после фильтрации упаривают. После экстракции маслянистого остатка 150 мл воды, фильтрации и высушивания получают слегка коричневатые кристаллы.
Выход 22 г (79% от теории; сравнить с примером XV).
Примеры получения
Пример 1
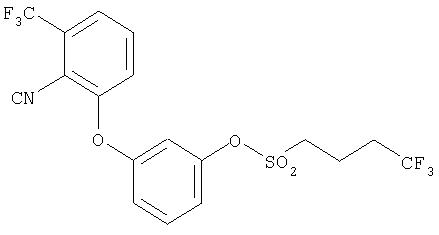
3-[2-Циано-3-(трифторметилфенокси]фенил-4,4,4-трифтор-1-бутансульфонат
(Синтез эфиров сульфокислот, способ А)
В атмосфере аргона в 60 мл дихлорметана растворяют 7,70 г (27,6 ммоля) 3-[2-циано-3-(трифторметил)фенокси] фенола, затем к этому раствору прибавляют 4,32 г (13,1 ммоля) бромида тетрабутиламмония и 3,95 мл 45%-ного раствора гидроксида натрия. При температуре 0°С сразу приливают раствор 6,64 г (31,5 ммоля) 4,4,4-трифторбутан-1-сульфохлорида в 20 мл дихлорметана. Раствор, окрашивающийся в цвета от желтого до оранжевого, перемешивают в течение одного часа. После этого разбавляют водой и три раза экстрагируют дихлорметаном. Объединенные органические фазы промывают раствором хлорида натрия и сушат над сульфатом магния. Очистку проводят с помощью флэш-хроматографии на 360 г силикагеля, используя ступенчатый градиент концентраций подвижной фазы циклогексан/дихлорметан от 1:1 до 1:4. После упаривания на роторном испарителе остается маслянистый остаток, который кристаллизуется после добавления пентана.
Выход: первая фракция 9, 21 г (74%, чистота по данным ВЭЖХ 100%),
вторая фракция 2,29 г (18%, чистота по данным ВЭЖХ 97%).
Значение Rf 0,56 (дихлорметан).
Температура плавления 60-61°С.
Масс-спектр (ионизация электрораспылением): 454 ([М+Н]+).
ВЭЖХ: время удерживания 5,08 мин.
1Н-ЯМР (300 МГц, дейтерохлороформ): δ (миллионных долей) = 2,2-2,5 (м., 4Н), 3,39 (т, J=7 Гц, 2Н), 7,0-7,3 (м, 4Н), 7,50 (т, J=8 Гц, 1Н), 7,53 (д, J=8 Гц, 1Н), 7,64 (д, 1=8 Гц).
Пример 2
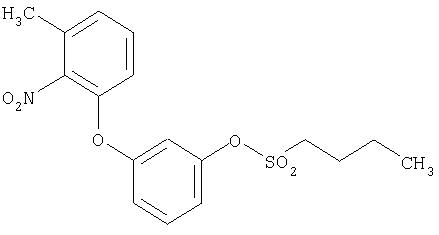
3-(3-Метил-2-нитро-фенокси)фенил-н-пентансульфонат
(Синтез эфиров сульфокислот, способ Б)
К 200 мг (0,82 ммоля) 3-(3-Метил-2-нитро-фенокси)фенола в 5 мл дихлорметана при комнатной температуре сначала прибавляют 1 мл 40%-ного раствора гидроксида тет-рабутиламмония, затем после перемешивания в течение пяти минут прибавляют 153 мг (0,90 ммоля) н-пентансульфохлорида. После перемешивания в течение 1,5 часа прибавляют 0,5 мл 10%-ного раствора бикарбоната натрия, фильтруют смесь через картридж Extrelut (3 г) (Мерк, Дармштадт, номер для заказа 115095) и несколько раз промывают картридж дихлорметаном. При хроматографической очистке на силикагеле со смесью циклогексан/этилацетат 30:1 в качестве подвижной фазы получают 255 мг (82%, по данным ВЭЖХ чистота равна 99%) целевого соединения.
Значение Rf 0,35 (циклогексан/этилацетат 2:1).
Масс-спектр (ионизация электрораспылением): 380 (100%, [M+H]+).
ВЭЖХ: время удерживания 5,29 мин.
1 H-ЯМР (300 МГц, дейтерохлороформ): δ (миллионных долей) = 0,93 (т., J=7 Гц, 3Н), 1,30-1,51 (м, 4Н), 1,89-2,02 (м., 2Н), 2,39 (с., 3Н), 3.18-3,28 (м, 2Н), 6,85-7,42 (м., 7Н).
Пример 3
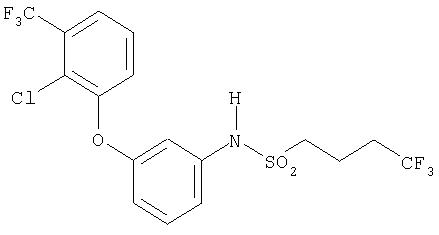
N-{3-[2-Хлор-3-(трифторметил)фенокси]фенил}-4,4, 4-трифторбутан-1-сульфонамид (Синтез амида сульфокислоты, способ В)
В атмосфере аргона в 1 мл дихлорметана растворяют 100 мг (0,35 ммоля) 3-[2-хлор-3-(трифторметил)фенокси]анилина. Прибавляют 106 мг (1,04 ммоля) триэтиламина и раствор 77 мг (0,37 ммоля) 4,4,4-трифторбутан-1-сульфохлорида в 1 мл дихлорметана и перемешивают при комнатной температуре. Через 4 дня прибавляют еще 0, 3 эквивалента 4,4,4-трифторбутансульфохлорида и перемешивают еще три дня. После этого реакционную массу три раза экстрагируют 2 н. соляной кислотой и один раз насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом магния и упаривают в вакууме. При хроматографической очистке на силикагеле со смесью дихлорметан/циклогексан 7:2 в качестве подвижной фазы получают 96 мг (54%, по данным ВЭЖХ чистота равна 90%) целевого соединения.
Значение Rf 0,33 (циклогексан/этилацетат 2:1).
Масс-спектр (прямая химическая ионизация/NH3): 479 (100%, [M+NH4]+).
ВЭЖХ: время удерживания 8,34 мин.
1H-ЯМР (300 МГц, d6-диметилсульфоксид): δ (миллионных долей) = 1,80-1,93 (м., 2Н), 2,31-2,50 (м, 2Н), 3,25 (т., J=8 Гц, 2Н), 6,75 (д.д., J=8 Гц, 2 Гц, 1Н), 6,85 (т, J=2 Гц, 1Н), 7,03 (д.д, J=8 Гц, 1 Гц), 7,38 (т., J=8 Гц, 1Н), 7,43 (д.д., J=8 Гц, 1 Гц. 1Н), 7, 58 (т., J=8 Гц, 1Н), 7,72 (д.д., J=8 Гц, 1 Гц, 1Н), 10,04 (с., 1Н).
Следующие далее примеры от 4 до 24 получают аналогично в соответствии с примерами получения по методикам А, Б или В из соответствующих исходных соединений.





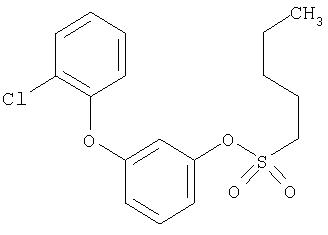
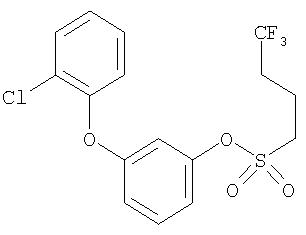
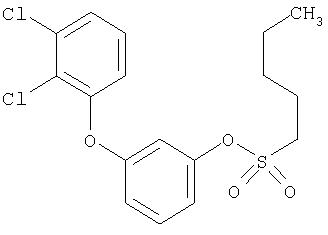
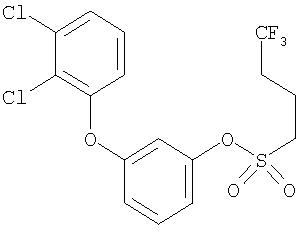
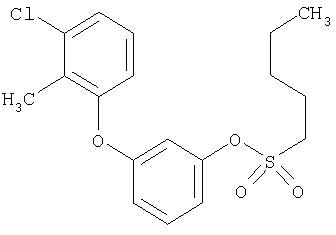
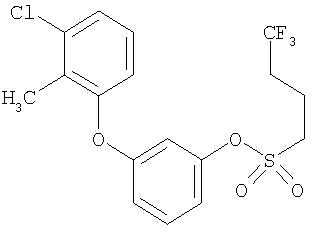

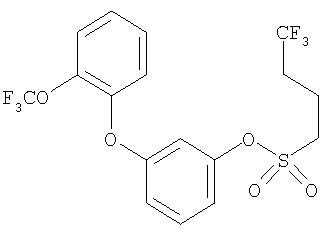
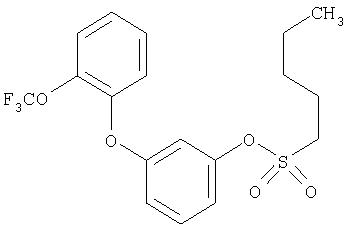
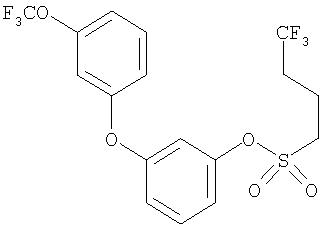
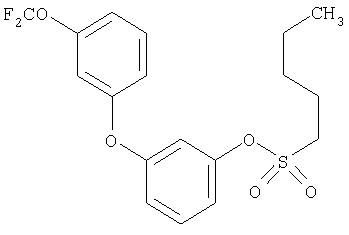
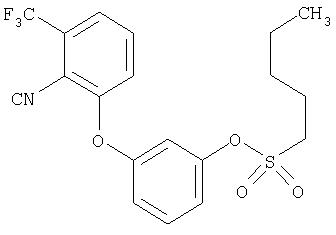



Приведенные выше примеры характеризуются следующими далее данными спектров1H ЯМР.
Реферат
Настоящее изобретение относится к феноксифенилалкансульфонатам общей формулы I
где R3 означает алкильную группу с числом атомов углерода от четырех до семи и эта группа может быть от одного до нескольких раз замещена атомами фтора или хлора, А означает атом кислорода или NH-группу. Описаны также способ получения соединений I и лекарственное средство на их основе. Соединения могут быть использованы для лечения болевых синдромов и нейродегенеративных заболеваний. 3 н. и 4 з.п. ф-лы, 3 табл.
Формула
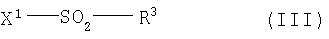
Комментарии