Способ получения солей разветвлённого олигогексаметиленгуанидина для их применения в качестве фармацевтических субстанций (варианты) - RU2750869C1
Код документа: RU2750869C1
Описание
Область техники, к которой относится изобретение
Изобретение относится к химической технологии полимеров медицинского назначения. Более конкретно, оно обеспечивает способ получения солей разветвлённого олигогексаметиленгуанидина (ОГМГ), которые могут найти применение в фармацевтической промышленности для изготовления антимикробных, антипаразитарных, противовирусных и противогрибковых препаратов. Предлагаемый способ может служить альтернативой способу получения солей ОГМГ, особенно гидрохлорида ОГМГ, в емкостных реакторах смешения.
Уровень техники
В описании изобретения к авторскому свидетельству SU 1616898 (опубл. 30.12.1990) раскрыт способ получения дезинфицирующего средства взаимодействием гексаметилендиамина (ГМДА) и расплава гуанидингидрохлорида (ГГХ) при молярном соотношении 1:(0,85-0,95), для осуществления которого гексаметилендиамин предварительно расплавляют и при 180 °С расплав в течение 2,5 часов равномерно добавляют к расплаву гуанидингидрохлорида с последующим повышением температуры до 240 °С и выдержкой при этой температуре в течение 5 часов. В результате с выходом 98 % получают гидрохлорид полигексаметиленгуанидина (ПГМГ-ГХ), имеющий молекулярную массу от 0,8 до 30 кДа. Длительные выдержки реакционной массы при высоких температурах требуют значительного расхода энергии. Кроме того, величина LD50, определённая для мышей, составляет 1000-1500 мг/кг, тогда как значение LD50, характеризующее соединения, получаемые в соответствии с данным изобретением, в аналогичных исследованиях находится на уровне 3500-4000 мг/кг, что свидетельствует о присутствии значительных количеств исходных мономеров и непригодности известного продукта для использования в фармацевтике.
Описание изобретения к авторскому свидетельству SU 1808832 (опубл. 15.04.1993) раскрывает способ получения полимера гексаметиленгуанидина (ПГМГ) взаимодействием ГМДА и ГГХ при нагревании, которое осуществляют при молярном соотношении гуанидингидрохлорида и гексаметилендиамина 1:(1,2-1,5) в течение 10 часов при 150-160 °С с последующим охлаждением до получения пенообразной твёрдой массы, измельчением и отмывкой водой. Целью изобретения являлось создание полимера с ионообменными свойствами, проявляющего также бактерицидную активность, что было продемонстрировано на примере E. сoli: 5 г полимера после набухания уничтожают 4×108 кол/л бактерий в течение 15 минут. Из описания изобретения следует, что целевой полимер характеризуется трёхмерной структурой и имеет на порядок большую молекулярную массу. Образование таких полимеров при осуществлении поликонденсации ГМДА и ГГХ в проточных реакторах малого поперечного сечения нежелательно, поскольку значительное увеличение вязкости реакционной массы повышает энергозатраты на её перекачивание через реактор и даже способно привести к остановке процесса.
В описании изобретения к патенту RU 2318803 (опубл. 10.03.2008) описан способ получения соли полигуанидина, включающий предварительное перемешивание в реакторе диамина и соли гуанидина, постепенное нагревание полученной смеси до достижения оптимальной температуры и проведение реакции поликонденсации в расплаве, подачу расплава в охлаждаемую форму, измельчение конечного продукта, в котором поликонденсацию проводят при достижении оптимальной температуры 180-220 °С с одновременным вакуумированием расплава при Рост=0,05-0,2 атм в течение 4-6 часов до полного удаления аммиака и остатков диамина, измельчение конечного продукта проводят без доступа влаги. В качестве соли гуанидина могут быть использованы гидрохлорид или цитрат гуанидина. При этом достигаются низкие уровни содержания остаточных гексаметилендиамина (0,02 масс.%) и гуанидингидрохлорида (ниже 0,1 масс.%), а минимальная концентрация, задерживающая рост Ps. aeruginosa, для обеих солей равна 0,006 мг/мл. Однако существенным недостатком предлагаемого способа является необходимость вакуумирования расплава, что усложняет технологическое оборудование.
В описании изобретения к патенту RU 2487118 (опубл. 10.07.2013) раскрыт способ получения полигуанидинов путем поликонденсации соли гуанидина с диамином, взятых в эквимольных соотношениях, при нагревании, при осуществлении которого поликонденсацию проводят в присутствии органической кислоты или смеси органических кислот и ступенчатом нагревании: на первой ступени – до 120-130 С; на второй – до 150-160°С и на третьей – до 170-180 °С, а времена выдержки реакционной смеси на каждой ступени нагревания составляют соответственно 0,5-1 ч; 3,5-4 ч; 0,5-1,5 ч. Предпочтительно, в качестве диамина применяют гексаметилендиамин, солью гуанидина является гидрохлорид, а органическую кислоту выбирают из монокарбоновых кислот, таких как лимонная или салициловая, которую вводят в количестве 0,01-16 мас.%, преимущественно 0,05-1,0 мас.% от реакционной смеси. В соответствии с приведёнными примерами, минимальное содержание остаточного мономера составляет 0,05 мас.%, что соответствует требованиям, изложенным в XIV издании Государственной Фармакопеи РФ XIV издание (ГФ XIV), но при этом в половине синтезов оно находится на уровне 0,15-0,30 мас.%, что существенно превышает допустимую норму. Время синтеза составляет 5,5-6 часов, что при проведении реакции в объёме связано с повышенным расходом энергии.
Из публикации RU 2489452 (опубл. 10.08.2013) известен способ получения дезинфицирующего средства, включающий поликонденсацию ГМДА и ГГХ и последующий ступенчатый нагрев реакционной массы, при осуществлении которого процесс поликонденсации начинают с приготовления реакционной массы в виде суспензии кристаллического ГГХ в расплаве ГМДА, взятых в соотношении 1:(1-1,5), при этом суспензию получают плавным введением в расплав ГМДА кристаллического ГГХ, предварительно разогретого до температуры 90-120 °С, и последующим их перемешиванием, после чего полученную реакционную массу выдерживают 4 ч при температуре 120 °С, затем 8 ч при температуре 160 °С, 3 ч при температуре 180 °С и затем температуру постепенно поднимают до 210 °С со скоростью 3-4 °С/ч, затем реакционную массу подвергают вакуумированию и охлаждению. При соотношении ГГХ:ГМДА равном 1:1,1, с выходом 97,2 % получают ПГМГ-ГХ с молекулярной массой ≈28 000 Да и содержанием остаточного ГГХ равным 0,025 масс.%. При этом содержание ГМДА равно 0, 054 %, а значение рН 1 % водного раствора равно 9,2. Основным недостатком способа является его длительность (более суток) и связанная с этим энергоёмкость. Кроме того, получаемый продукт не удовлетворяет требованиям ГФ XIV.
Способ получения продукта, а именно – разветвленных олигомеров ГМДА и ГГХ формулы (I):

где R представляет


а n1, n2 и n3 равны 1-3, z равно 0,15-1,10 с молекулярно-массовым распределением Mw/Mn от 5,4 до 9,3 при среднемассовой молекулярной массе Mw в интервале от приблизительно 3800 до 6300 и среднечисловой молекулярной массе Mn в интервале от приблизительно 600 до 1100, раскрыт в примерах 1-6 описания изобретения к патенту RU 2443684 (опубл. 27.02.2012). Так, реакцию поликонденсации ведут при мольном соотношении ГМДА и ГГХ 1:(1,00-1,20) и начальной температуре (температуре прибавления ГМДА в расплав ГГХ) равной 160 °С, которую быстро повышают до 180-230 °С и выдерживают реакционную массу при конечной температуре в течение 5-12 часов. Получаемые таким способом олигогексаметиленгуанидины (ОГМГ) обладают высокой бактерицидной активностью. В частности, в эксперименте контактирование E. coli с 1 % раствором таких олигомеров на поверхности из линолеума в течение 2 минут приводит к гибели 80-97 % колоний, что доказывает их высокую бактерицидную активность. Величина LD50, определённая для мышей, составляет 3500-4000 мг/кг, что свидетельствует об их низкой токсичности. В соответствии с описанием, получаемые таким способом олигомеры предназначены для изготовления дезинфицирующего средства для удаления бактериальных, вирусных и грибковых загрязнений, а также загрязнений спорами с различных объектов. Для данных целей содержание остаточных мономеров не столь критично, как при производстве фармацевтических препаратов. Поэтом описание изобретения таких данных не содержит.
Интерес к солям разветвлённых ОГМГ как к перспективным фармацевтическим субстанциям обусловлен их высокой бактерицидной активностью в отношении различных патогенных микроорганизмов.
Так в источнике Ха, Кам Ань. Разработка технологии получения субстанции гидросукцината олигогексаметиленгуанидина и глазных капель на ее основе: дисс. на соискание уч. ст. канд. фарм. наук: 14.04.01. – Всеросс. научно-иссл. ин-т лекарств. и ароматич. растений, Москва, 2013 – URL: https://search.rsl.ru/ru/record/01005096248 предложен трёхстадийный способ получения гидросукцината ОГМГ формулы (I) из гидрохлорида ОГМГ, полученного в соответствии с примерами, приведёнными в RU 2443684.
На первой стадии одну часть 50 % водного раствора ОГМГ-ГХ вводят в реакцию с 10 частями раствора, содержащего 2-4 эквивалента КОН в 96 % этиловом спирте, что позволяет снижать содержание хлора в получаемом основании ОГМГ до 0,2-0,5 %. Однако, высокая растворимость КОН в водно-спиртовой среде неизбежно приводит к росту содержания золы в конечном продукте.
Для уменьшения зольности на второй стадии в полученный щелочной раствор основания ОГМГ барботируют углекислый газ, в результате чего в растворе образуется гидрокарбонат ОГМГ, а малорастворимый в водно-спиртовой среде КНСО3 отделяют фильтрованием. Получение целевого гидросукцината ОГМГ (ОГМГ-ГС) сводилось к добавлению эквивалентного количества янтарной кислоты и упариванию раствора. В диссертации отмечено, что предложенный подход является общим для получения солей ОГМГ с кислотами более сильными, чем угольная, т.е. позволяет получить любую соль органической кислоты.
К количествам примесей были предъявлены следующие требования:
которым удовлетворяла получаемая субстанция. В частности, удалось снизить содержание ГМДА до 0,09 %. Тем не менее, более жёсткие требования действующей в настоящее время ГФ XIV не позволяют использовать раскрытый в диссертации способ изготовления ОГМГ-ГС, предназначенного для применения в качестве фармацевтической субстанции на территории РФ.
Дальнейшие исследования в области офтальмологических препаратов, содержащих соли ОГМГ показали их высокую эффективность. Так пример 4 в описании изобретения к патенту RU 2513997 (опубл. 27.04.2014) содержит следующие данные о минимальных бактериостатических концентрациях (МПК) ОГМГ-ГХ:
В примере 4 описания изобретения к патенту RU 2699377 (опубл. 05.09.2019) препарат, содержащий сукцинат ОГМГ в качестве единственного действующего вещества, охарактеризован следующими значениями МПК:
В описании изобретения к патенту RU 2052453 (опубл. 20.01.1996) раскрыто получение очищенного ПГМГ-ГХ с молекулярной массой 10 тыс. у.е. (пример 2), через промежуточное образование основания ПГМГ (ПГМГ-О) под действием концентрированного водного раствора едкого натра, взятого в количестве 1,125 моль/моль ПГМГ-ГХ в виде расслаивающейся суспензии. После осторожного сливания раствора NaCl слой, содержащий ПГМГ-О, трижды промывают водой с температурой 50-80 °С и высушивают до постоянной массы. Получают очищенное ПГМГ-О, содержащее по результатам элементного анализа приблизительно 25 % воды и около 10 % незамещённых гидрохлоридных звеньев исходного полимера. К полученному основанию добавляют при перемешивании по каплям концентрированную соляную кислоту до достижения раствором рН 5 и получают вязкий прозрачный раствор, содержащий ≈ 75 % ГМГ-ГХ и 5 % NaCl. Содержание токсичного ГМДА в таком растворе по данным газожидкостной сверхкритической хроматографии (ГЖСХ) не превышает 0,1 %. Достигаемая данным способом степень очистки ПГМГ-ГХ с учётом требований ГФ XIV не является достаточной.
Описание изобретения к патенту RU 2052453 содержит также пример 1.2 получения гидрокарбоната ПГМГ (ПГМГ-ГК), в соответствии с которым смесь 1 моль карбоната гуанидина и 1 моль ГМДА в конической колбе емкостью 0,5 л, снабженной воздушным обратным холодильником (стеклянная трубка длиной 0,5 м и диаметром 1 см) нагревают до 135-140 °С и при этом вначале начинается интенсивное «кипение». Реакционную массу выдерживают при указанной температуре 10-15часов, в продолжение которых она загустевает и превращается в светло-желтую пенообразную смолу, заполняющую почти всю колбу. Получают 170 г ограниченно растворимого в воде ПГМГ-ГК, по данным элементного анализа, соответствующего брутто-формуле С7,5Н16N3O1,5. Молекулярная масса М или степень полимеризации ПГМГ не указана и не может быть вычислена по приведённой в описании изобретения характеристической вязкости [η] равной 0,06 дл/г, поскольку значения констант К и а в степенном уравнении модели [η] = К·Ма не даны.
Таким образом, существует потребность в способе получения солей разветвлённого ОГМГ с воспроизводимыми молекулярными характеристиками и физико-химическими свойствами и пониженным содержанием балластных и токсичных веществ и степенью чистоты, достаточной для их применения в качестве фармацевтической субстанции в соответствии с действующей ГФ XIV.
Раскрытие сущности изобретения
Согласно ГФ XIV (ОФС.1.1.0006.15 «Фармацевтические субстанции») соли разветвлённого ОГМГ должны отвечать, в частности, следующим критериям качества:
* в фармацевтической субстанции ОГМГ-ГХ контроль не вводят
Вследствие ужесточения требований, предъявляемых к качеству фармацевтических субстанций, возрастает значение решения двух важных проблем, возникающих при получении солей разветвлённого ОГМГ, а именно – одновременного соответствия продукта нормам содержания хлоридов и сульфатной золы и существенного снижения содержания токсичного гексаметилендиамина и гуанидина гидрохлорида.
Авторами было установлено, что при действии 10 объёмами 15-30 масс./об.% раствора КОН в 96 об.% этиловом спирте на 1 объём 50-60 масс./об.% водного раствора ОГМГ-ГХ при мольном соотношении КОН: ОГМГ-ГХ от 2:1 до 4:1 не удаётся достичь содержания хлоридов в получаемом ОГМГ-О менее 0,2-0,5 масс.%. В то же время, увеличение мольного избытка щелочи на порядок неизбежно приводит к росту содержания сульфатной золы в конечном продукте вследствие высокой растворимости КОН в водно-спиртовой среде. Его замена на приблизительно вдвое менее растворимый NaOH также не даёт приемлемо низкого содержания золы.
В случае применения большого мольного избытка щёлочи значительно понизить зольность продукта возможно удалением из раствора избытка ионов щелочного металла в виде гидрокарбоната, поскольку при этом ОГМГ-О превращается в гидрокарбонат (ОГМГ-ГК), хорошо растворимый в водно-спиртовых средах. В то же время, из уровня техники известно, что гидрокарбонаты натрия и калия практически нерастворимы в спиртах и малорастворимы (на уровне 0,1 мол.%) в водно-спиртовых средах с преобладанием спирта (G. H. Kim et al. Antisolvent Precipitation of Potassium Bicarbonate from KHCO3 + H2O + Ethanol/2-Propanol Systems in the CO2 Capture Process // Ind. Eng. Chem. Res. 2015, 54, 8287−8294). Для осаждения избытка ионов щелочного металла в виде гидрокарбоната в раствор барботируют углекислый газ. Данная реакция нейтрализации происходит в гетерофазной системе «жидкость-газ», характеризуемой малыми значениями растворимости углекислого газа в воде (2,1 г/л) и константы диссоциации угольной кислоты в воде по первой ступени (4,3·10–7). В связи с этим, для завершения реакции и практически полного выделения гидрокарбоната щелочного металла в твёрдом виде требуется барботирование большого избытка углекислого газа 200-450 моль/моль щёлочи) с небольшим расходом в течение длительного времени. Для присутствия в растворе 1 моль щёлочи оптимальный расход углекислого газа оценен величиной 3-6 н.л/мин, при этом время барботирования углекислого газа в раствор составит от 12 до 24 часов.
Авторы изобретения установили, что для выделения твёрдого ОГМГ-ГК из раствора после удаления гидрокарбоната щелочного металла фильтрацией или центрифугированием эффективно введение ацетона в тройную систему «вода-спирт- ОГМГ-ГК» в качестве антирастворителя. Одновременно с этим достигается содержание гексаметилендиамина в получаемом ОГМГ-ГК ниже уровня 0,05 %, установленного ГФ XIV, поскольку практически всё количество этой примеси остаётся в растворе. Для целей настоящего изобретения ацетон обладает преимуществом высокой летучести, что обеспечивает его простое и полное удаление из получаемого твёрдого продукта, и низким классом опасности. Кроме того, при необходимости значительная доля использованного ацетона может быть регенерирована и возвращена в технологический процесс, что улучшает экологические показатели предлагаемого способа.
После выпаривания ацетона требуемую соль ОГМГ, выбранную из гидросукцината, гидрохлорида, гидроцитрата, гидросалицилата или гидросульфосалицилата, получают добавлением к водному раствору ОГМГ-ГК соответствующей кислоты в количестве, близком к эквивалентному. Хлористоводородную кислоту добавляют в виде концентрированного, например, 35 % водного раствора. Лимонную и сульфосалициловую кислоты также можно вводить в реакцию с ОГМГ-ГК в виде концентрированных или насыщенных водных растворов, а также в твёрдом виде. Салициловую и янтарную кислоты вводят в твёрдом виде.
В контексте описания настоящего изобретения понятие «количество кислоты, близкое к эквивалентному» определяет количество выбранной кислоты, взаимодействующее с основными группами ОГМГ, способными к солеобразованию в водной среде, таким образом, чтобы 1 масс./об.% водный раствор полученной в результате обменной реакции фармацевтической субстанции соли ОГМГ формулы (I) имел величину рН в интервале от 6,5 до 8,5. Специалисту в данной области очевидно, что 1 масс./об.% раствор соли ОГМГ со слабыми кислотами, такими как янтарная, вследствие её гидролиза по аниону будет иметь рН более 7. Настоящее изобретение допускает присутствие в продукте обменной реакции небольшого избытка твёрдой кислоты при условии соответствия фармацевтической субстанции требованиям, предъявляемым к значению рН её 1 масс./об.% водного раствора.
Установление подлинности, а также определение среднечисловой молекулярной массы Mn и степени разветвления, характеризуемой значением z, и суммы индексов n1 + n2 + z·n3, характеризующей степень олигомеризации, выполняют с применением спектроскопии ЯМР13С (ГФ XIV, ОФС 1.2.1.1.0007.15) как раскрыто в описании изобретения к патенту RU 2443684. Получаемые данные позволяют также вычислить приблизительную молярную массу эквивалента ОГМГ-ГК для оценки нижней границы количества выбранной кислоты, близкого к эквивалентному, при получении фармацевтической субстанции в соответствии с изобретением. Необходимый для этого математический аппарат полностью находится в компетенции специалиста в данной области. Определение остаточных мономеров осуществляют обращеннофазной ВЭЖХ в ион-парном варианте.
В результате обширных исследований авторы настоящего изобретения установили, что недостатки, присущие известным техническим решениям, могут быть в значительной мере преодолены проведением поликонденсации гуанидина гидрохлорида (Г-ГХ) с гексаметилендиамином (ГМДА) или, альтернативно, гуанидина гидрокарбоната (Г-ГК) с гексаметилендиамином (ГМДА) в проточном микрореакторе.
Существенным недостатком проведения поликонденсации Г-ГХ с ГМДА в емкостных реакторах смешения является анизотропия температурного поля в объёме реактора, что приводит к анизотропии физико-химических свойств реакционной массы. Вследствие большой вязкости реакционной массы, анизотропию невозможно в достаточной мере устранить перемешиванием, поэтому эффективные константы скоростей поликонденсации могут существенно различаться по объёму реакционной массы и по-разному изменяться во времени при её нагревании. Это приводит к ухудшению воспроизводимости выхода и разбросу молекулярных характеристик и биологической активности продукта в широком интервале. Кроме того, масштабирование процесса поликонденсации требует, в частности, выявления и учёта сложных взаимосвязей геометрии реактора и мешалки и интенсивности перемешивания с гидродинамическим режимом и теплопередачей при нагревании.
Применение изотермического микрореактора позволяет решить проблему анизотропии, поскольку градиенты температуры и концентраций реагентов и продуктов в направлении, перпендикулярном их потоку, пренебрежимо малы вследствие малого диаметра микрореактора, а при малой линейной скорости эти градиенты также незначительны в направлении потока. Масштабирование процесса вместо замены реакторного узла сводится к увеличению числа микрореакторов, параллельно питаемых реагентами по раздельности или в виде смеси.
Однако осуществление поликонденсации ГМДА и ГГХ в расплаве, известное из уровня техники, встречает существенные трудности при проведении процесса в микрореакторе. Длительность реакции обусловливает необходимость применения микрореакторов с длинной канала, оцениваемой метрами, что, вместе с высокой вязкостью расплава реакционной массы требует приложения давления в десятки атмосфер. Кроме того, образование олигомеров со значительно более высокой среднечисловой молекулярной массой приведёт к существенному увеличению вязкости расплава. Принимая применимость закона Гагена-Пуазейля, давление, необходимое для перекачивания реакционной массы с постоянным расходом, возрастёт прямо пропорционально увеличившейся вязкости, превысит максимальное значение рабочего давления подающего насоса, и подача реагентов будет автоматически остановлена. Это потребует прекращения ведения процесса, что приведёт к потере части продукта и затратам времени на восстановление работоспособности технологической установки.
Авторами изобретения было неожиданно установлено, что загрузка реагентов или их смесей в микрореактор в виде концентрированных водных растворов позволяет вести процесс поликонденсации стабильно, т. е. с малым числом технологических сбоев и с хорошей воспроизводимостью качества продукта. Не ограничиваясь данным объяснением, авторы полагают, что образующийся при нагревании реакционной массы водяной пар уменьшает сопротивление движению реакционной массы в канале микрореактора и снижает вероятность агломерации реакционной массы на стенках канала, которая может привести к остановке процесса. Подача реагентов или их заранее приготовленной смеси в микрореактор также облегчается, поскольку не требуется на протяжении всего времени проведения поликонденсации поддерживать в узле подачи реагентов температуру не ниже 100 °С для предотвращения конденсации расплава.
Кроме того, было установлено, что поликонденсацию с образованием разветвлённого ОГМГ-ГХ можно эффективно вести при более низкой оптимальной температуре на уровне температуры смешения реагентов в соответствии с примерами, приведёнными в RU 2443684 (160 °С), далее не повышая её до 180-230 °С и получать на выходе микрореактора продукт, соответствующий требованиям ГФ XIV.
Таким образом, в соответствии с первым вариантом осуществления настоящего изобретения предложен способ получения соли разветвлённого олигогексаметиленгуанидина (ОГМГ), представленного общей формулой (I):

где R представляет

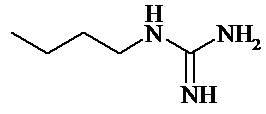
а n1, n2 и n3 равны 1-3, z равно 0,15-1,10 при среднечисловой молекулярной массе Mn в интервале от 850 до 1500 Да, выбранной из гидрохлорида, гидросукцината, гидроцитрата, гидросалицилата, гидросульфосалицилата, имеющей чистоту, достаточную для её применения в качестве фармацевтической субстанции, для осуществления которого:
а) гуанидина гидрохлорид (Г-ГХ) растворяют в воде и к раствору Г-ГХ прибавляют гексаметилендиамин (ГМДА) в мольном соотношении Г-ГХ:ГМДА от 1,0:0,5 до 1,0:1,0 с получением водного раствора, содержащего 60-70 масс.% реагентов,
б) водный раствор реагентов подают в проточный микрореактор с постоянной по его длине температурой 150-160 °С, обеспечивающий время пребывания в нём реакционной смеси от 2 до 3 часов, в котором происходит реакция конденсации Г-ГХ и ГМДА с образованием технического ОГМГ гидрохлорида (т-ОГМГ-ГХ) в виде водного раствора, который на выходе из микрореактора охлаждают до температуры окружающей среды,
в) из охлаждённого водного раствора т-ОГМГ-ГХ полностью выпаривают воду с получением твёрдого т-ОГМГ-ГХ,
г) твёрдый т-ОГМГ-ГХ растворяют в воде с получением 10 масс./об.% раствора, небольшими порциями добавляют в него ацетон до прекращения помутнения и выдерживают систему до появления чётко различимой границы раздела прозрачного и мутного слоёв,
д) удаляют верхний прозрачный слой, а из нижнего мутного слоя выделяют твёрдый солевой продукт ОГМГ с пониженным содержанием примесей (ОГМГ-ПСП), содержащий ОГМГ-ГХ в качестве основного компонента,
е) ОГМГ-ПСП, полученный на стадии д), растворяют в воде с образованием 60 масс./об.% водного раствора, добавляют спиртовой раствор щёлочи с концентрацией от 10 масс./об.% до концентрации раствора, насыщенного при температуре окружающей среды, и получают смесь щелочного водно-спиртового раствора, содержащего ОГМГ основание (ОГМГ-О), насыщенного хлоридом щелочного металла, и твёрдого хлорида щелочного металла, при этом спирт выбран из группы, состоящей из метанола, этанола, пропанола и изопропанола, а щёлочь выбрана из группы, состоящей из гидроксида натрия и гидроксида калия и взята в 20-30-кратном мольном избытке по отношению к количеству ОГМГ-ПСП,
ж) из смеси, полученной на стадии е), удаляют твёрдый хлорид щелочного металла и получают щелочной водно-спиртовой раствор, содержащий ОГМГ-О,
з) через раствор, полученный на стадии ж), барботируют углекислый газ в течение времени, превышающего время прекращения выделения тепла, и затем кипятят реакционную массу с обратным холодильником с получением водно-спиртового раствора ОГМГ гидрокарбоната (ОГМГ-ГК) и твёрдых солей щелочного металла,
и) из смеси, полученной на стадии з), удаляют твёрдые соли щелочного металла и получают водно-спиртовой раствор ОГМГ-ГК,
к) из водно-спиртового раствора осаждают ОГМГ-ГК добавлением ацетона до прекращения помутнения и выдерживают систему до появления чётко различимой границы раздела прозрачного и мутного слоёв,
л) удаляют верхний прозрачный слой, а из нижнего мутного слоя выделяют ОГМГ-ПСП, содержащий ОГМГ-ГК в качестве основного компонента,
м) ОГМГ-ПСП, полученный на стадии л), растворяют в воде с образованием 10-25 масс./об.% водного раствора,
н) в полученный раствор ОГМГ-ГК добавляют кислоту, выбранную из группы, состоящей из хлористоводородной кислоты, янтарной кислоты, лимонной кислоты, салициловой кислоты или сульфосалициловой кислоты, в количестве, близком к эквивалентному,
о) полученную реакционную массу перемешивают до растворения кислоты, отбирают аликвоту реакционной массы, из аликвоты готовят 1 масс./об.% водный раствор, определяют величину его рН и, при условии, что эта величина находится в пределах от 6,5 до 8,5, осуществляют стадию р), а если величина рН более 8,5, то осуществляют стадию п),
п) оценочно рассчитывают требуемое количество добавляемой кислоты, вносят соответствующий объём раствора или навеску кислоты в реакционную массу и повторяют стадию е),
р) получают водный раствор соли ОГМГ с выбранной кислотой и лиофилизируют её сублимационной сушкой.
Микрореактор для осуществления первого варианта изобретения предпочтительно имеет внутренний диаметр канала от 3,18 мм до 1,59 мм (от 1/8 дюйма до 1/16 дюйма) и длину канала 2 м.
На стадиях д) и л) верхний прозрачный слой предпочтительно удаляют декантацией или откачкой, а ОГМГ-ПСП выделяют из нижнего мутного слоя выпариванием растворителя при пониженном давлении.
Более предпочтительно, перед выпариванием растворителя нижний мутный слой концентрируют центрифугированием.
На стадии е) спиртом предпочтительно является 96 об.% этанол, а щёлочь предпочтительно является гидроксидом натрия, взятым в 20-кратном мольном избытке, или гидроксидом калия, взятым в 30-кратном мольном избытке.
Предпочтительно в течение времени, определённого для осуществления стадии з), барботируют 200-450-кратный мольный избыток углекислого газа по отношению к количеству щёлочи.
При осуществлении стадий н) и п) выбранную кислоту добавляют предпочтительно в виде водного раствора, насыщенного при температуре окружающей среды.
Число технологических стадий процесса получения соли ОГМГ, представленного общей формулой (I), можно уменьшить, применяя на стадии поликонденсации гуанидина гидрокарбонат (Г-ГК) вместо Г-ГХ.
Таким образом, в соответствии со вторым вариантом осуществления изобретения предложен способ получения соли разветвлённого олигогексаметиленгуанидина (ОГМГ), представленного общей формулой (I):
где R представляет
а n1, n2 и n3 равны 1-3, z равно 0,15-1,10 при среднечисловой молекулярной массе Mn в интервале от 850 до 1500 Да, выбранной из гидрохлорида, гидросукцината, гидроцитрата, гидросалицилата, гидросульфосалицилата, имеющей чистоту, достаточную для её применения в качестве фармацевтической субстанции, для осуществления которого:
а) гуанидина гидрокарбонат (Г-ГК) растворяют в воде и к раствору Г-ГК прибавляют гексаметилендиамин (ГМДА) в мольном соотношении Г-ГК:ГМДА от 1,0:0,5 до 1,0:1,0 с получением водного раствора реагентов, содержащего 30-40 масс./об.% Г-ГК,
б) водный раствор реагентов подают в проточный микрореактор с постоянной по длине реактора температурой 135-160 °С, обеспечивающий время пребывания в нём реакционной смеси от 2 до 5 часов, в котором происходит реакция конденсации Г-ГК и ГМДА с образованием ОГМГ гидрокарбоната (ОГМГ-ГК) в виде водного раствора, который охлаждают до температуры окружающей среды,
в) из охлаждённого водного раствора ОГМГ-ГК полностью выпаривают воду с получением твёрдого ОГМГ-ГК,
г) твёрдый ОГМГ-ГК растворяют в воде с получением 10-25 масс./об.% водного раствора ОГМГ-ГК,
д) в полученный раствор ОГМГ-ГК добавляют кислоту, выбранную из группы, состоящей из хлористоводородной кислоты, янтарной кислоты, лимонной кислоты, салициловой кислоты или сульфосалициловой кислоты, в количестве, близком к эквивалентному,
е) полученную реакционную массу перемешивают до растворения кислоты, отбирают аликвоту реакционной массы, из аликвоты готовят 1 масс./об.% водный раствор, определяют величину рН и, при условии, что эта величина находится в пределах от 6,5 до 8,5, осуществляют стадию з), а если величина рН более 8,5, то осуществляют стадию ж),
ж) оценочно рассчитывают требуемое количество добавляемой кислоты, вносят соответствующий объём раствора или навеску кислоты в реакционную массу и повторяют стадию е),
з) получают водный раствор соли ОГМГ с выбранной кислотой и лиофилизируют её сублимационной сушкой.
Микрореактор для осуществления второго варианта изобретения предпочтительно имеет внутренний диаметр канала от 3,18 мм до 1,59 мм (от 1/8 дюйма до 1/16 дюйма) и длину канала 2 м.
При осуществлении стадий д) и ж) выбранную кислоту добавляют предпочтительно в виде водного раствора, насыщенного при температуре окружающей среды.
В контексте описания настоящего изобретения термин «температура окружающей среды» относится к температуре в объёме пространства, к котором размещено технологическое оборудование, в частности, ёмкости для приготовления и/или хранения растворов щелочей и кислот. Температура окружающей среды предпочтительно находится в пределах от 15 ºС до 25 С, и более предпочтительно она составляет 20 ºС, что определено термином «комнатная температура».
Техническим результатом осуществления предложенного способа получения солей разветвлённого ОГМГ в любом из его вариантов является снижение содержания балластных (хлоридов и сульфатной золы) и токсичных (остаточных ГМДА и ГГХ) веществ ниже уровней, позволяющих применять такие соли в качестве субстанций для изготовления препаратов медицинского и/или ветеринарного назначения в соответствии с действующей ГФ XIV.
Другим техническим результатом применения заявленного способа является существенное улучшение воспроизводимости свойств и выхода получаемого продукта по сравнению с известным способом, осуществляемым в емкостном реакторе смешения.
Осуществление изобретения
Настоящее изобретение далее будет проиллюстрировано примерами его осуществления, подтверждающими достижение указанных в описании технических результатов.
Примеры 1-8. Получение ОГМГ-ГХ
Стадия 1. Получение т-ОГМГ-ГХ в микрореакторе
Операция 1. В примерах 1-4 примеряют микрореактор с внутренним диаметром 1,59 мм (1/16 дюйма) и R составляет 1,0:0,50, а в примерах 5-8 применяемый микрореактор имеет внутренний диаметр 3,18 мм (1/8 дюйма) и R равно 1,0:1,5.
Исходя из заданного значения мольного соотношения R реагентов Г-ГХ и ГМДА, вычисляют массы навесок Г-ГХ (m1, г) и ГМДА (m2, г), а также объём V (мл) воды очищенной, необходимый для приготовления раствора реагентов с заданной концентрацией (с1 , масс.%) в интервале от 60 до 70 для осуществления серии из трёх синтезов для каждой комбинации условий «R – с1 – температура реакции t (°С) – время пребывания τ (ч)».
В стеклянный стакан загружают воду очищенную, при перемешивании вносят навеску Г-ГХ, и после её растворения прибавляют навеску ГМДА и перемешивают до получения прозрачного бледно-жёлтого раствора. Растворение реагентов ведут при температуре от комнатной до приблизительно 40ºС.
Раствор реагентов с помощью прецизионного шприцевого насоса (neMESYS Syringe Pump; производитель: Wingflow AG, Швейцария) с рабочим объёмом 10 мл подают в автоматическую микрореакторную систему (Qmix Pro Ext; производитель: Wingflow AG, Швейцария) со стальным змеевиковым микрореактором, который имеет внутренний диаметр 1,59 мм (1/16 дюйма) или 3,18 (1/8 дюйма) и рабочую длину 200 см, при этом рабочий объём равен 4,0 мл или 6,0 мл соответственно.
По всей длине микрореактора с точностью ±1°С поддерживают температуру t (°С). Расход раствора реагентов устанавливают постоянным в соответствии с заданным временем пребывания реакционной массы τ (ч) в микрореакторе выбранной модели. На выходе реакционную массу собирают во взвешенную круглодонную колбу. Условия получения т-ОГМГ-ГХ в микрореакторе приведены в таблице 1.
Таблица 1
Операция 2. Выделение т-ОГМГ-ГХ, полученного в микрореакторе
Из собранной реакционной массы на роторном испарителе при пониженном давлении (10-15 мм. рт. ст., 90-95ºС) выпаривают воду, определяют массу остатка и готовят из него 10 масс./об.% водный раствор, к которому небольшими порциями добавляют ацетон (приблизительно 200 мл). Помутневшую смесь выдерживают до появления чётко различимой границы раздела верхнего прозрачного и нижнего мутного слоёв. Добавляют небольшую порцию ацетона и, если не происходит помутнения верхнего слоя, то верхний слой декантируют.
Нижний слой помещают в стаканы низкоскоростной лабораторной центрифуги (Sigma 2-6, производитель: Sigma Laborzentrifugen GmbH, Германия) и центрифугируют при 3600 об/мин в течение 1 часа. Пасту из центрифужных стаканов переносят во взвешенную круглодонную колбу и выпаривают при пониженном давлении (10-15 мм. рт. ст., 90-95ºС) на роторном испарителе с получением сухого технического ОГМГ-ГХ (т-ОГМГ-ГХ).
Операция 3. Анализ т-ОГМГ-ГХ, полученного в микрореакторе
В соответствии с описанием изобретения к патенту RU 2443684 методом13С ЯМР определяют величину среднечисловой молекулярной массы Mn (с точностью до 30 Да) и степень разветвления z в общей формуле (I).
Содержание исходных мономеров ГМДА и Г-ГХ определяют методом ВЭЖХ в условиях, приведённых а примерах выполнения общих анализов (примеры А1 и А2).
Стадия 2. Получение ОГМГ-ПСП из т-ОГМГ-ГХ
Значения Mn и z, определённые при выполнении операции 3 на стадии 1 для серии из трёх синтезов в каждом из примеров 1-8, позволяют объединить продукты для последующей очистки.
Операция 1. Очистка т-ОГМГ-ГХ
В каждом из примеров 1-8 объединённые продукты трёх серий синтезов растворяют в воде с получением 10 масс./об.% раствора, небольшими порциями добавляют в него ацетон (приблизительно 500 мл) до прекращения помутнения и выдерживают систему до появления чётко различимой границы раздела прозрачного и мутного слоёв. Добавляют небольшую порцию ацетона и, если не происходит помутнения верхнего слоя, то верхний слой декантируют, а нижний слой обрабатывают, как указано при описании операции 2 на стадии 1, с получением ОГМГ-ГХ с пониженным содержанием примесей (ОГМГ-ПСП).
Операция 2. Анализ ОГМГ-ПСП, полученного из т-ОГМГ-ГХ
Данную операцию выполняют аналогично операции 3 на стадии 1. Характеристики полученных продуктов по стадиям приведены в таблице 2.
Таблица 2
Вспомогательные примеры
Пример В1. Вариант 1 получения гидрокарбоната ОГМГ (ОГМГ-ГК)
из ОГМГ-ГХ с пониженным содержанием примесей
взаимодействием с КОН
Стадия 1. Получение основания ОГМГ (ОГМГ-О)
Навеску (m3, г) (10 ммоль) гидрохлорида ОГМГ-ПСП (объединённого продукта из примеров, указанных в таблице 3) с определёнными значениями Mn (Да) и z растворяют в V0 (мл) воды очищенной (ФС.2.2.0020.18). Полученный раствор при комнатной температуре в течение 5 минут добавляют при перемешивании в 60 мл 5 М раствора КОН в 96 об.% спирте этиловом (ФС.2.1.0036.15), при этом мольное соотношение КОН:ОГМГ-ГХ составляет 30:1, и реакционную массу перемешивают 15 минут. Перемешивание прекращают, и смесь выдерживают до выпадения осадка калия хлорида, который удаляют фильтрацией через стеклянный фильтр при пониженном давлении.
Стадия 2. Получение ОГМГ-ГК
Фильтрат, содержащий ОГМГ-О, переносят в колбу, снабжённую мешалкой, термометром, барботёром и обратным холодильником с водяной рубашкой, и при перемешивании в течение 15 часов барботируют углекислый газ с расходом 5 л/мин. После прекращения выделения тепла смесь кипятят до прекращения образования осадка карбонатов калия, который затем отделяют фильтрацией через стеклянный фильтр при пониженном давлении.
К полученному водно-спиртовому раствору добавляют ацетон (приблизительно 400 мл) до прекращения помутнения и разделяют жидкую и твёрдую фазы центрифугированием на низкоскоростной лабораторной центрифуге (Sigma 2-6, производитель: Sigma Laborzentrifugen GmbH, Германия) при 3600 об/мин в течение 1 часа. Пасту из центрифужных стаканов переносят во взвешенную круглодонную колбу и выпаривают при пониженном давлении (10-15 мм. рт. ст., 55-60ºС) на роторном испарителе с получением сухого ОГМГ-ГК массой (m4, г) с выходом (ω0, %), приведенными в таблице 3.
Таблица 3
Пример В2. Вариант 2 получения гидрокарбоната ОГМГ (ОГМГ-ГК)
из ОГМГ-ГХ с пониженным содержанием примесей
взаимодействием с NaOH
Стадия 1. Получение основания ОГМГ (ОГМГ-О)
Навеску (m5, г) (10,5 ммоль) гидрохлорида ОГМГ-ПСП (объединённого продукта из примеров, указанных в таблице 4) с определёнными значениями Mn (Да) и z растворяют в V0 (мл) воды очищенной (ФС.2.2.0020.18). Полученный раствор при комнатной температуре в течение 5 минут добавляют при перемешивании в 70 мл 3 М раствора NaOH в 96 об.% спирте этиловом (ФС.2.1.0036.15), при этом мольное соотношение КОН:ОГМГ-ГХ составляет 20:1, и реакционную массу перемешивают 20 минут. Перемешивание прекращают, и смесь выдерживают до выпадения осадка натрия хлорида, который удаляют фильтрацией через стеклянный фильтр при пониженном давлении.
Стадия 2. Получение ОГМГ-ГК
Выполняют все операции, приведённые в описании примера В1 для стадии 1, в тех же условиях. Получают сухого ОГМГ-ГК массой (m6, г) с выходом (ω0, %), приведенными в таблице 4.
Таблица 4
Получение солей ОГМГ, пригодных для применения в качестве фармацевтических субстанций
В примерах получения солей применяют оборудование производства НПП «Эконикс Эксперт» (РФ): магнитную мешалку «Ритм-01» и рН-метр «Эксперт-рН» с электродом комбинированным ЭСК-10303/7 (производитель: ООО «Измерительная техника», РФ).
Примеры 9-11. Вариант 1 получения солей ОГМГ (с раствором кислоты)
Разбавлением кислоты хлористоводородной (37 %, ФС.2.2.0034.18) водой очищенной (ФС.2.2.0020.18) готовят 5 М раствор, применяемый в примере 9.
Навеску 24,01±0,01 г кислоты лимонной (ФС.2.1.0024.15) при комнатной температуре растворяют в 20 мл 96 об.% этанола (ФС.2.1.0036.15), доводят объём раствора до 25,0 мл и получают 5 М этанольный раствор лимонной кислоты, применяемый в примере 10.
Навеску 2,952±0,001 г кислоты сульфосалициловой дигидрата (ГОСТ 4478-78) при комнатной температуре растворяют в 20 мл воды очищенной (ФС.2.2.0020.18) и доводят объём раствора до 25,0 мл и получают 0,5 М раствор сульфосалициловой кислоты, применяемый в примере 11.
Берут навески 10,0 г±0,05 г продуктов ОГМГ-ГК, полученных в соответствии со вспомогательными примерами (В. П.), указанными в таблице 5, и растворяют их в воде очищенной с получением растворов, имеющих концентрацию (с, масс./об.%) и содержащих (n, ммоль) ОГМГ-ГК. В раствор каждого продукта ОГМГ-ГК при перемешивании добавляют объём раствора выбранной кислоты (V, мл) и выдерживают реакционную массу 5 минут.
Из каждого раствора, содержащего солевой продукт ОГМГ, отбирают аликвоту объёмом (Vа, мл), вносят её в 20 мл воды и при перемешивании магнитной мешалкой добавляют отмеряемый объём десятикратно разбавленного применяемого раствора соответствующий кислоты до достижения заданного значения рН, определяемого потенциометрически. На основании определённого объёма добавленного разбавленного раствора кислоты рассчитывают добавляемый объём применяемого раствора кислоты, вносят его в реакционную массу и выдерживают её 5 минут при перемешивании.
Полученный раствор охлаждают и помещают в лиофильную сушилку на 48 часов. Получают лиофилизированный солевой продукт ОГМГ со значениями массы (m, г), общего выхода (ω, %), содержания хлоридов (Хл, масс.%) и сульфатной золы (СЗ, масс.%), и рН 1 % водного раствора, указанными в таблице 5.
Таблица 5
Примеры 12-13. Вариант 2 получения солей ОГМГ (с твёрдой кислотой)
Берут навески 10,0 г±0,05 г продуктов ОГМГ-ГК, полученных в соответствии со вспомогательными примерами (В. П.), указанными в таблице 6, и растворяют их в воде очищенной (ФС.2.2.0020.18) с получением растворов, имеющих концентрацию (с, масс./об.%) и содержащих (n, ммоль) ОГМГ-ГК.
В раствор каждого продукта ОГМГ-ГК при перемешивании порциями по 0,3-0,4 г добавляют тонкоизмельчённую навеску (mк, г) выбранной кислоты и выдерживают реакционную массу по 20 минут после каждого прибавления.
В бюксе с точностью до 0,001 г взвешивают около 0,5 г тонкоизменьчённой выбранной кислоты, определяя общую массу. Из каждого раствора, содержащего солевой продукт ОГМГ, отбирают аликвоту объёмом (Vа, мл), вносят её в 20 мл воды и при перемешивании магнитной мешалкой с выдержкой 5 минут небольшими порциями добавляют соответствующую кислоту до достижения заданного значения рН, определяемого потенциометрически. После этого бюкс с кислотой повторно взвешивают и на основании массы добавленной кислоты рассчитывают навеску этой кислоты, которую вносят в реакционную массу и выдерживают её 20 минут при перемешивании.
Полученный раствор охлаждают и помещают в лиофильную сушилку на 48 часов. Получают лиофилизированный солевой продукт ОГМГ со значениями массы (m, г), общего выхода (ω, %), содержания хлоридов (Хл, масс.%) и сульфатной золы (СЗ, масс.%), и рН 1 % водного раствора, указанными в таблице 6.
Таблица 6
Общие анализы
Все реагенты поставлены ООО «Мерк» (РФ) с сертификатами соответствия требованиям Европейской Фармакопеи (Ph. Eur. 9) к реагентам для ВЭЖХ.
Определения выполняют в условиях внутрилабораторной воспроизводимости в соответствии с руководством по эксплуатации высокоэффективного жидкостного хроматографа, снабжённого спектрофотометрическим детектором и пригодного для данного применения с учётом требований, предъявляемых к правильности результатов.
Пример А1. Условия определения содержания ГМДА в образцах солей ОГМГ
Градуировочные растворы: растворы ГМДА (98 % (ГЖХ) с концентрацией 0,25 мкг/мл, 0,50 мкг/мл, 2,50 мкг/мл, 5,0 мкг/мл в воде для хроматографии.
Буферный раствор:0,1 М боратный буферный раствор, полученный добавлением свежеприготовленного 1 М раствора NaOH (98,5 % (титр.) к раствору 6,20 г борной кислоты (99,5 % (титр.) в воде для хроматографии.
Раствор 9-флуоренилметилхлорформиата (FMOC): свежеприготовленный раствор 10,0 мг FMOC (98 % (ВЭЖХ) в 10,0 мл ацетонитрила для хроматографии (поставщик: ООО «Мерк», РФ; 99,9 % (ГЖХ).
Раствор образца: навеску исследуемого образца соли ОГМГ массой приблизительно 150 мг, взятую с точностью 0,1 мг, растворяют в воде для хроматографии и доводят объём раствора до 250,0 мл. К аликвоте 100 мкл приготовленного раствора добавляют 100 мкл боратного буферного раствора, 1000 мкл ацетонитрила и 100 мкл раствора FMOC, тщательно перемешивают и выдерживают при комнатной температуре 5 минут.
Колонка: обращено-фазовая колонка C18, 250×4,6 мм, 5 мкм (Luna C18(2), производитель: Phenomenex International или аналогичная).
Подвижная фаза А: вода для хроматографии.
Подвижная фаза В: ацетонитрил для хроматографии.
Пример А2. Условия определения содержания Г-ГХ в образцах ОГМГ-ГХ
Градуировочные растворы: растворы Г-ГХ (98 % (ГЖХ) с концентрацией 10 мкг/мл, 25 мкг/мл, 75 мкг/мл, 100 мкг/мл в подвижной фазе А.
Раствор образца: навеску исследуемого образца соли ОГМГ массой приблизительно 1 г, взятую с точностью до 1 мг, при ультразвуковом воздействии растворяют в 50-70 мл подвижной фазы А и доводят объём раствора до 100,0 мл.
Колонка: обращено-фазовая колонка C18, 150×4,6 мм, 3 мкм (Gemini C18 110Å, производитель: Phenomenex International или аналогичная или аналогичная;
Подвижная фаза А:навеску 870±1 мг пентан-1сульфоновой кислоты натриевой соли растворяют приблизительно в 300 мл воды для хроматографии, приливают 10,0 мл ортофосфорной кислоты концентрированной (99 % (титр.) и доводят объём раствора до 1000 мл.
Подвижная фаза В: ацетонитрил для хроматографии.
Реферат
Настоящее изобретение относится к вариантам способов получения соли разветвлённого олигогексаметиленгуанидина (ОГМГ). Соль разветвлённого олигогексаметиленгуанидина имеет общую формулу (I):(I),где R представляетили, а n1, n2и n3равны 1-3, z равно 0,15-1,10 при среднечисловой молекулярной массе Mnв интервале от 850 до 1500 Да, выбранной из гидрохлорида, гидросукцината, гидроцитрата, гидросалицилата, гидросульфосалицилата, имеющей чистоту, достаточную для её применения в качестве фармацевтической субстанции. Технический результат – получение солей разветвлённого олигогексаметиленгуанидина с воспроизводимыми молекулярными характеристиками и физико-химическими свойствами и пониженным содержанием балластных и токсичных веществ и степенью чистоты, достаточной для их применения в качестве фармацевтической субстанции. 2 н. и 8 з.п. ф-лы, 6 табл., 13 пр.
Формула


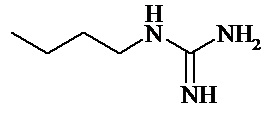
Комментарии