Симметричные и несимметричные производные дифенилмочевины (варианты), фармацевтическая композиция, способ подавления роста опухолевых клеток, опосредованного киназой raf - RU2247109C9
Код документа: RU2247109C9
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к применению производных группы арилмочевины для лечения заболеваний, опосредованных киназой raf, и фармацевтических композиций для проведения такой терапии.
Уровень техники
Онкоген p21ras вносит основной вклад в развитие и прогрессию солидных опухолей человека и мутирован в 30% всех опухолей в организме человека (см. статьи Bollton с соавт. Ann. Rep. Med. Chem., 1994, т.29, стр.165-74; Bos. Cancer Res., 1989, т.49, стр.4682-9). В нормальной, немутированной форме белок ras является ключевым элементом каскада трансдукции сигнала, управляемого рецепторами ростового фактора практически во всех тканях (см. статью Avruch с соавт. Trends Biochem. Sci., 1994, т.19, стр.279-83). В биохимическом отношении ras представляет собой гуаниннуклеотид-связывающий белок, а взаимопревращение между ГТФ-связанной активированной формой и ГДФ-связанной инактивированной формой строго контролируется эндогенной ГТФазной активностью ras и другими регуляторными белками. Мутантные формы ras, присутствующие в опухолевых клетках, характеризуются сниженной эндогенной ГТФазной активностью, и, следовательно, этот белок осуществляет передачу неконтролируемых ростовых сингналов опосредованным с ним эффекторам, например, такому ферменту, как киназа raf. Это приводит к опухолевой трансформации клеток, несущих мутантные формы ras (см. статью Magnuson с соавт. Semin. Cancer Biol., 1994, т.5, стр.247-53). Показано, что ингибирующее действие активной формы ras путем ингибирования сигнального пути с участием киназы raf введением инактивирующих антител против киназы raf или за счет совместной экспрессии доминантной негативной киназы raf или доминантной негативной формы МЕК, субстрата киназы raf, приводит к реверсии трансформированных клеток в нормально растущий фенотип (см. статьи Daum с соавт. Trends Biochem. Sci., 1994, т.19, стр.474-80; Fridman с соавт. J. Biol. Chem., 1994, т.269, стр.30105-8). Кроме того, в статье Kolch с соавт. (Nature, 1991, т.349, стр.426-28) показано, что ингибирование экспрессии киназы raf с помощью антисмысловой ДНК блокирует пролиферацию клеток, вызванную мембранно-ассоциированными онкогенами. Аналогично, ингибирование киназы raf (антисмысловыми олигонуклеотидами) коррелирует in vitro и in vivo с ингибированием роста различных типов опухолей человека (см. статью Monia с соавт. Nat. Med., 1996, т.2, стр.668-75).
Сущность изобретения
Настоящее изобретение относится к соединениям, которые являются ингибиторами фермента киназы raf. Поскольку этот фермент является эффектором p21ras, ингибиторы быстрого действия могут найти применение в фармацевтических композициях для лечения человека или для применения в ветеринарии, если показано ингибирование raf киназного пути, например, при лечении опухолей и/или опухолевого роста клеток, опосредованного киназой raf. Прежде всего, такие соединения применимы при лечении рака человека или животных, например, мышей, солидных опухолей, поскольку прогрессия таких опухолей зависит от каскада трансдукции сигнала белком ras и, следовательно, чувствительна к воздействиям, прерывающим этот каскад, например, путем ингибирования киназы raf. Соответственно, соединения по изобретению могут найти применение при лечении солидных опухолей, например таких, как карциномы (например, легких, поджелудочной железы, щитовидной железы, мочевого пузыря или толстой кишки, заболеваний спинного или костного мозга, например, лейкозе, или аденомы, например, ворсинчатой опухоли (полип толстой кишки).
Настоящее изобретение представляет соединения, которые обычно носят название производные арилмочевины, включая как арил-, так и гетероарилзамещенные аналоги, которые ингибируют путь передачи сигнала с участием киназы raf. Изобретение также предлагает способ лечения заболеваний человека или млекопитающих, опосредованных киназой raf. Таким образом, изобретение относится к соединениям и способам подавления роста опухолевых клеток, опосредованного киназой raf, включающим введение соединения формулы I:
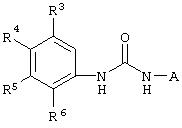
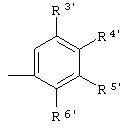
где А представляет собой
R3 означает Н, галоген, NO2, С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения; С1-10aлкокси, по выбору замещенный галогеном вплоть до полного замещения;
R4, R5 и R6 каждый независимо означает Н, галоген, NO2, С1-4алкил, по выбору замещенный галогеном вплоть до полного замещения, С1-4алкокси, по выбору замещенный галогеном вплоть до полного замещения; и либо один из заместителей R4, R5 и R6 означает -X-Y, либо два соседних заместителя из R4, R5 и R6 объединены с основным фенильным кольцом с образованием нафтила или индольной группы, замещенной пиридинильной группой;
R3', R4', R5' и R6' независимо означают Н, галоген, NO2, С1-4aлкил, по выбору замещенный галогеном вплоть до полного замещения, С1-4алкокси, по выбору замещенный галогеном вплоть до полного замещения, С5-гетероарил, содержащий атом азота или серы и возможно замещенный С1-4алкилом,
или 2 соседних заместителя из R3', R4', R5' и R6' объединены с основным фенильным кольцом с образованием нафтильной группы;
Х означает -СН2-, -S-, -N(СН3)-, -NHC(O)-, -CH2-S-, -S-CH2, -С(O)- или -О-;
Y означает фенил, замещенный следующими группами: С1-10алкокси, ОН, -SСН3,
пиридил, по выбору замещенный следующими группами: С1-10алкил, С1-10-алкокси, галоген, ОН, -SСН3 или NO2;
нафтил, по выбору замещенный следующими группами: С1-10алкил, С1-10-алкокси, галоген, ОН, -SСН3 или NО2;
пиридон, по выбору замещенный следующими группами: С1-10алкил, C1-10-алкокси, галоген, ОН, -SСН3 или NO2;
пиразин, по выбору замещенный следующими группами: С1-10алкил, С1-10-алкокси, галоген, ОН, -SСН3 или NО2;
пиримидин, по выбору замещенный следующими группами: С1-10алкил, С1-10-алкокси, галоген, ОН, -SСН3 или NO2;
бензодиоксан, по выбору замещенный следующими группами: С1-10алкил, С1-10алкокси, галоген, ОН, -SСН3 или NO2;
бензопиридин, по выбору замещенный следующими группами: С1-10алкил, одним С1-10алкокси, галоген, ОН, -SСН3 или NO2; или
бензотиазол, по выбору замещенный следующими группами: С1-10алкил, одним С1-10алкокси, галоген, ОН, -SСН3 или NО2;
или его фармацевтически приемлемой соли,
при условии, что, если Х означает -О-, -CH2-, -С(O)- или -S-, R3 и R6 означают
Н, a Y означает фенил, незамещенный ОН, то R6' означает алкокси.
R3 предпочтительно означает Н, галоген, СН3 или СF3; R4 означает Н, галоген, СН3 или СF3; R5 означает Н, галоген или и R6 означает Н, галоген или С1-4-алкил. Более предпочтительно R3' означает С1-4алкил, Cl, F или СF3; R4'означает Н, Cl, F; R5' означает Н, Cl, F или С4-10алкил и R6' означает Н или ОСН3. Наиболее предпочтительно R3' и R5' означают трет-бутил. Х предпочтительно означает -СН2- или -S-, a Y означает фенил или пиридил, или Х означает -О-, а Y препочтительно означает фенил, пиридил и бензотиазол.
Кроме того, изобретение относится к способу подавления роста опухолевых клеток, опосредованного киназой raf и включающему введение соединения формулы I
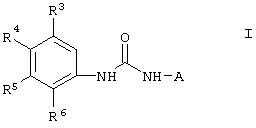
где А означает
R3 означает Н, галоген, NО2, С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения; С1-10 алкокси, по выбору замещенный галогеном вплоть до полного замещения;
R4, R5 и R6 каждый независимо означает Н, галоген, NO2, С1-4 алкил, по выбору замещенный галогеном вплоть до полного замещения, С1-4алкокси, по выбору замещенный галогеном вплоть до полного замещения; и либо один из заместителей R4, R5 и R6 означает -X-Y, либо два соседних заместителя из R4, R5 и R6 объединены с основным фенильным кольцом с образованием нафтила или индольной группы, замещенной пиридинильной группой;
R3', R4', R5' и R6' независимо означают Н, галоген, NO2, С1-C4алкил, по выбору замещенный галогеном вплоть до полного замещения,
С1-С4алкокси, по выбору замещенный галогеном вплоть до полного замещения, C5-гетероарил, содержащий атом азота или серы и возможно замещенный С1-4алкилом,
или 2 соседних заместителя из R3', R4', R5' и R6' объединены с основным фенильным кольцом с образованием нафтильной группы;
Х означает -СН2-, -S-, -N(СН3)-, -NHC(O)-, -CH2-S-, -S-CH2, -C(O)-, или -О;
Y означает фенил, по выбору замещенный следующими группами: С1-10алкокси, ОН, -SСН3,
а также пиридил, нафтил, пиридон, пиразин, пиримидин, бензодиоксан, бензопиридин, бензотиазол, каждый из по выбору замещен следующими группами: С1-10алкил, С1-10алкокси, галоген, ОН, -SCH3 или NO2;
или его фармацевтически приемлемой соли,
при условии, что, если Х означает -О-, -СН2-, -С(O)- или -S-, R3 и R6 означают Н, а Y означает фенил, незамещенный ОН, то R6' означает алкокси.
Предпочтительными соединениями являются соединения формулы IIа:
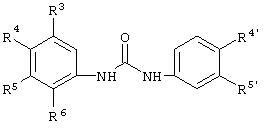
где каждый из R3, R4, R5 и R6 независимо означает Н, галоген, NO2, С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения, или С1-10-алкокси, по выбору замещенный галогеном вплоть до полного замещения; и один из R3-R6 может означать -X-Y; или 2 соседних R3-R6 могут быть объединены с образованием арильного или гетероарильного цикла, содержащего 5-12 атомов, по выбору замещенного следующими группами: С1-10 алкил, С1-10алкокси, С3-10циклоалкил, С2-10алкенил, С1-10алканоил, С6-12арил, С5-12гетероарил; С6-12алкарил, галоген; NR1; -NО2; -СF3; -COOR1; -NHCOR1; -CN; -CONR1R1; -SO2R2; -SOR2; -SR2; в которых R1 означает Н или С1-10алкил, по выбору замещенный на галоген вплоть до полного замещения, и R2 oзначает С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения.
В формуле I примеры подходящих гетероциклических групп включают, не ограничиваясь перечисленным, ароматические кольца, содержащие 5-12 атомов углерода, или циклические системы, содержащие 1-3 кольца, из которых по крайней мере одно является ароматическим, в которых один или более, например, 1-4 углеродных атома в одном или более колец могут быть заменены на атомы кислорода, азота или серы. Обычно каждое кольцо содержит 3-7 атомов. Например, кольцо может означать 2- или 3-фурил, 2- или 3-тиенил, 2- или 4-триазинил, 1-, 2- или 3-пирролил, 1-, 2-, 4- или 5-имидазолил, 1-, 3-, 4- или 5-пиразолил, 2-, 4- или 5-оксазолил, 3-, 4- или 5-изоксазолил, 2-, 4- или 5-тиазолил, 3-, 4- или 5-изотиазолил, 2-, 3- или 4-пиридил, 2-, 4-, 5- или 6-пиримидинил, 1,2, 3-триазол-1-, -4- или -5-ил, 1,2,4-триазол-1-, -3- или -5-ил, 1- или 5-тетразолил, 1,2,3-оксадиазол-4- или -5-ил, 1,2,4-оксадиазол-3- или -5-ил, 1,3,4-тиадиазол-2- или -5-ил, 1,2,4-оксадиазол-3- или -5-ил, 1,3,4-тиадиазол-2- или -5-ил, 1,3,4-тиадиазол-3- или -5-ил, 1,2,3-тиадиазол-4- или -5-ил, 2-, 3-, 4-, 5- или 6-2Н-тиопиранил, 2-, 3- или 4-4Н-тиопиранил, 3- или 4-пиридазинил, пиразинил, 2-, 3-, 4-, 5-, 6- или 7-бензофурил, 2-, 3-, 4-, 5-, 6- или 7-бензотиенил, 1-, 2-, 3-, 4-, 5-, 6- или 7-индолил, 1-, 2-, 4- или 5-бензимидазолил, 1-, 3-, 4-, 5-, 6- или 7-бензопиразолил, 2-, 4-, 5-, 6- или 7-бензоксазолил, 3-, 4-, 5-, 6- или 7-бензизоксазолил, 1-, 3-, 4-, 5-, 6- или 7-бензотиазолил, 2-, 4-, 5-, 6- или 7-бензизотиазолил, 2-, 4-, 5-, 6- или 7-бенз-1,3-оксадиазолил, 2-, 3-, 4-, 5-, 6-, 7- или 8-хинолинил, 1-, 3-, 4-, 5-, 6-, 7- или 8-изохинолинил, 1-, 2-, 3-, 4- или 9-карбазолил, 1-, 2-, 3-, 4-, 5-, 6-, 7-, 8- или 9-акридинил или 2-, 4-, 5-, 6-, 7- или 8-хиназолинил, или дополнительно по выбору замещенные фенил, 2-или 3-тиенил, 1,3,4-тиадиазолил, 3-пиррил, 3-пиразолил, 2-тиазолил или 5-тиазолил и т.п. Например, В может означать 4-метилфенил, 5-метил-2-тиенил, 4-метил-2-тиенил, 1-метил-3-пиррил, 1-метил-3-пиразолил, 5-метил-2-тиазолил или 5-метил-1,2,4-тиадиазол-2-ил.
Подходящие алкильные группы и алкильная часть в заместителях, например, в алкоксигруппе и т.п., в ключают метил, этил, пропил, бутил и т.п., включая все линейные и разветвленные изомеры, такие как изопропил, изобутил, втор-бутил, трет-бутил и т.п.
Подходящие арильные группы включают, например, фенил и 1- и 2-нафтил.
Подходящие циклоалкильные группы включают циклопропил, циклобутил, циклогексил и т.п. Термин "циклоалкил", используемый в тексте заявки, означает циклические структуры, имеющие или не имеющие алкильных заместителей, причем, например, ″циклоалкил" включает как циклопропильные группы, замещенные метильной группой, так и циклобутильные группы. Кроме того, термин "циклоалкил" включает насыщенные гетероциклические группы.
Подходящие галогены включают F, Cl, Вr и/или I, причем возможны группы, содержащие от одного заместителя до полностью замещенных (когда все атомы водорода в группе замещены на атом галогена), кроме того возможны остатки со смешанным замещением различными галогенами.
Ряд соединений формулы I имеют асимметрические атомы углерода и, следовательно, могут существовать в виде рацематов и оптически активных форм. Способы разделения смесей энантиомеров и диастереомеров хорошо известны специалистам в данной области техники. Настоящее изобретение включает любую выделенную рацемическую или оптически активную форму соединений формулы I, которая обладает ингибирующей активностью в отношении киназы raf.
Соединения формулы I можно получить с использованием известных химических реакций и методов. Тем не менее, далее приводятся следующие общие препаративные методы, которые помогут специалисту в данной области синтезировать ингибиторы, а в экспериментальной части даны примеры синтеза конкретных соединений.
Сведения, подтверждающие возможность осуществления изобретения
Общие препаративные методы
Соединения формулы I получают с использованием известных химических реакций и методик, некоторые из исходных материалов, которые являются коммерчески доступными. Тем не менее, ниже здесь предложены общие препаративные методы, чтобы специалисты в данной области техники могли использовать их для синтеза этих соединений, причем более подробные примеры приводятся в экспериментальной части, которая следует далее.
Замещенные анилины получают по стандартным методам [см. в книгах March, Advanced Organic Chemistry (Органическая химия), 3ье изд., John Wiley:New York (1985); Larock, Comprehensive Organic Transformations (Превращения органических соединений), VCH Publishers: New York (1989)]. Как показано на схеме I, ариламины обычно синтезируют восстановлением нитроарилов с использованием металлических катализаторов, таких как Ni, Pd или Pt, и H2 или переносчиков гидрида, таких как формиат, циклогексадиен или боргидрид [см. в книге Rylander, Hydrogenation Methods (Методы гидрирования), Academic Press: London, UK(1985)]. Нитроарилы можно также восстанавливать непосредственно с использованием эффективных источников гидрида, такого как LiAlH4 [см. в книге Seyden-Penne, Reduction by the Alumino- and Borohydrides in Organic Synthesis (Восстановление алюмо- и боргидридами в органическом синтезе), VCH Publishers: New York (1991)], или при использовании металлов с нулевой валентностью, таких как Fe, Sn или Са, в большинстве случаев в кислой среде. Существует множество методов синтеза нитроарилов [см. в книгах March, Advanced Organic Chemistry (Органическая химия), 3ье изд., John Wiley:New York (1985); Larock, Comprehensive Organic Transformations (Превращения органических соединений), VCH Publishers: New York (1989)].
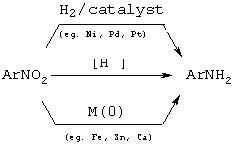
Схема I Восстановление нитроарилов в ариламины
Нитроарилы обычно получают электрофильным ароматическим нитрованием с использованием НNО3 или альтернативного источника NO2+. Перед восстановлением нитроарилы могут быть модифицированы.
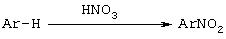
Таким образом нитроарилы, замещенные потенциально удаляемой группой (F, Cl, Вr и т.п.), могут вступать в реакции замещения при взаимодействии с нуклеофилами, такими как тиолат (как показано на схеме II) или феноксиды. Нитроарилы можно также вводить в реакцию конденсации типа реакции Ульмана (Ullman) (см. схема II)
Схема II
Некоторые реакции нуклеофильного ароматического замещения с использованием нитроарилов
Нитроарилы могут также вступать в реакцию перекрестного сочетания, инициированную переходным металлом. Например, электрофильные нитроарилы, такие как нитроарил бромиды, иодиды или трифлаты, вступают в реакцию перекрестного сочетания, иницированную палладием, с нуклеофильными арилами, такими как алкилборными кислотами (реакция Сузуки, пример которой приведен ниже), арилами олова (реакции Штилле) или арилами цинка (реакция Негиши), при этом получают биарилы (5).
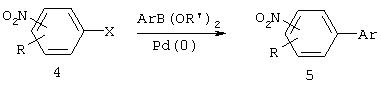
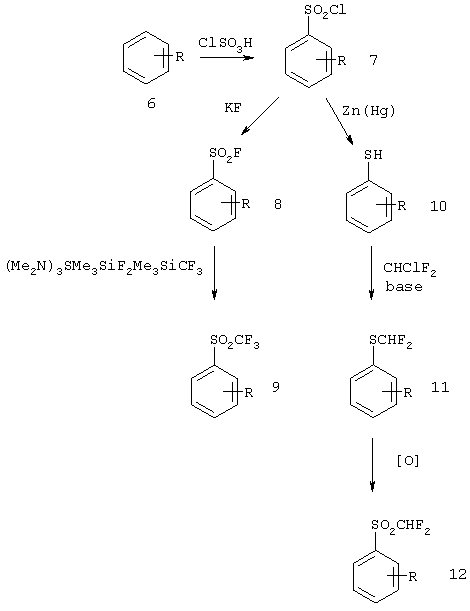
Нитроарилы или анилины можно преобразовать в соответствующий аренсульфонилхлорид (7) обработкой хлорсульфоновой кислотой. Взаимодействие сульфонилхлорида с источником фтора, таким как KF, приводит к образованию сульфонилфторида (8). Взаимодействие сульфонилфторида 8 с триметилсилилтрифторметаном в присутствии источника фтора, такого как дифтортриметилсиликонат трис(диметиламино)сульфония (TASF), приводит к образованию соответствующего трифторметилсульфона (9). По альтернативному способу сульфонилхлорид (7) может быть восстановлен до арентиола (10), например, амальгамой цинка. Взаимодействие тиола 10 с CHClF2 в присутствии основания приводит к образованию дифторметилмеркаптана (11), который может быть окислен до сульфона (12) любым оксидантом, включая СrO3 - уксусный ангидрид (Sedova и соавт. Zh. Org. Khim. 1970, т.6, стр.568).
Схема III Отдельные методы синтеза фторированных алкилсульфонов
Как показано на схеме IV, синтез несимметричных производных мочевин может включать взаимодействие арилизоцианата (14) с ариламином (13). Гетероарилизоцианаты могут быть синтезированы из гетероариламинов обработкой фосгеном или аналогами фосгена, такими как
трихлорметилхлорформиат (дифосген), бис(трихлорметил)карбонат (трифосген) или N,N'-карбонилдиимидазол (CDI). Изоцианат также можно получить из производного гетероциклической карбоновой кислоты, такого как сложный эфир, галогенид кислоты или ангидрид, с использованием перегруппировки Куртиуса. Таким образом, реакция кислотного производного (16) с источником азида с последующей перегруппировкой приводит к образованию изоцианата. Соответствующая карбоновая кислота (17) может также вступать в реакцию типа перегруппировки Куртиуса при использовании дифенилфосфорилазида (DPPA) или аналогичного реагента.
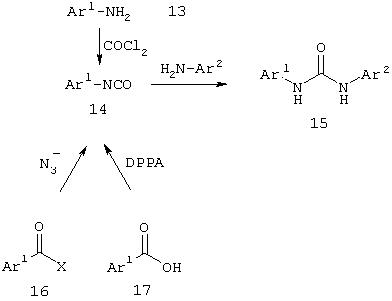
Схема IV Отдельные методы синтеза несимметричных производных мочевины
В конечном итоге, производные мочевины в дальнейшем могут быть модифицированы с использованием способов, хорошо известных специалистам в данной области техники.
Настоящее изобретение включает также фармацевтические композиции, включающие соединения формулы I, и физиологически приемлемый носитель.
Соединения можно вводить перорально, местно, парентерально, ингаляцией или в виде аэрозоля или под язык, ректально или вагинально с использованием композиций, содержащих унифицированные дозы. Термин "введение путем инъекции" включает внутривенные, внутримышечные, подкожные и парентеральные инъекции, а также использование метода вливания. Кожное введение может включать местную аппликацию или чрескожное введение. Одно или несколько соединений можно использовать в сочетании с одним или несколькими нетоксичными фармацевтически приемлемыми носителями и, если необходимо, с другими активными комопонентами.
Композиции, предназначенные для перорального применения, получают по любому известному специалистам способу получения фармацевтических композиций. Для придания приятного вкуса такие композиции могут включать один или несколько агентов, которые выбирают из группы, включающей разбавители, подсластители, ароматизаторы, красители и консерванты. Таблетки содержат активный компонент в смеси с нетоксичными фармацевтически приемлемыми наполнителями, пригодными для изготовления таблеток. Например, такими наполнителями могут быть инертные разбавители, такие как карбонат кальция, карбонат натрия, лактоза, фосфат кальция или фосфат натрия; гранулирующие или дезинтегрирующие агенты, например, кукурузный крахмал или альгиновая кислота; и связующие агенты, например, стеарат магния, стеариновая кислота или тальк. Таблетки могут быть без оболочки или могут быть покрыты известными методами с целью замедлить дезинтеграцию и всасывание в желудочно-кишечном тракте и тем самым обеспечить пролонгированное действие в течение определенного времени. Например, может использоваться материал, обеспечивающий замедленное действие, такой как глицерилмоностеарат или глицерилдистеарат. Эти соединения также можно получить в твердой, быстро рассасывающейся форме.
Композиции для перорального применения могут быть получены в виде твердых желатиновых капсул, где активный компонент смешан с инертным твердым разбавителем, например, карбонатом кальция, фосфатом кальция или каолином, или в виде мягких желатиновых капсул, где активный компонент смешан с водой или масляной средой, например, арахисовым маслом, жидким парафином или оливковым маслом.
Могут также использоваться водные суспензии, содержащие активные компоненты в смеси с наполнителями, пригодными для получения водных суспензий. Такими наполнителями являются суспендирующие агенты, например, натриевая соль карбоксиметилцеллюлозы, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, камедь трагаканта и аравийская камедь; диспергирующими и смачивающими агентами могут быть природные фосфатиды, например, лецитин или продукты конденсации алкиленоксида с жирными кислотами, например, полиоксиэтиленстеарат, или продукты конденсации этиленоксида с высшими алифатическими спиртами, например, гептадекаэтиленоксиэтанол, или продукты конденсации этиленоксида с неполными эфирами сорбита с жирными кислотами, такие как полиоксиэтиленсорбитмоноолеат, или продукты конденсации этиленоксида с неполными эфирами ангидросорбита с жирными кислотами, например, полиэтиленангидросорбитмоноолеат. Водные суспензии могут также содержать один или несколько консервантов, например, этил- или н-пропил-пара-гидроксибензоат, один или несколько красителей, один или несколько ароматизаторов и один или несколько подсластителей, такик как сахароза или сахарин.
Диспергируемые порошки и гранулы, пригодные для приготовления водной суспензии путем добавления воды, содержат активный компонент в смеси с диспергирующим или смачивающим агентом, суспендирующим агентом и одним или несколькими консервантами. Примером пригодных диспергирующих или смачивающих агентов являются описанные выше компоненты. Кроме них в композиции могут присутствовать дополнительные наполнители, например, подсластители, ароматизаторы и красители.
Соединения можно также использовать для получения неводных композиций, например, масляных суспензий, которые можно получить суспендированием активных компонентов в растительном масле, например, арахисовом масле, оливковом масле, кунжутном масле или масле ореха арахис, или в минеральном масле, таком как жидкий парафин. Масляные суспензии могут содержать загустители, например, пчелиный воск, твердый парафин или цетиловый спирт. Для придания приятного вкуса в композиции для перорального применения можно добавить подсластители, упоминавшиеся выше, и ароматизаторы. Эти композиции могут быть защищены добавлением антиоксиданта, такого как аскорбиновая кислота.
Фармацевтические композиции по изобретению могут быть изготовлены в форме водно-масляных эмульсий. В качестве масляной фазы может использоваться растительное масло, например, оливковое масло или арахисовое масло, или минеральное масло, например, вазелиновое масло, или их смеси. Подходящими эмульгирующими агентами могут быть природные камеди, например, аравийская камедь или камедь трагаканта, природные фосфатиды, например, соевые бобы, лецитин, и эфиры или неполные эфиры ангидросорбита с жирными кислотами, например, ангидросорбит моноолеат, и продукты конденсации указанных неполных эфиров с этиленоксидом, например, полиоксиэтиленсорбитанмоноолеат. Эмульсии также могут содержать подсластители и ароматизаторы.
Сиропы и эликсиры могут быть приготовлены с подсластителями, например, глицерином, пропиленгликолем, сорбитом и сахарозой. Такие композиции могут также содержать средство, снижающее раздражение, консервант, ароматизатор и краситель.
Соединения по изобретению можно также использовать в форме суппозиториев для ректального или вагинального применения. Эти композиции получают путем смешивания лекарственного средства с подходящим, не вызывающим раздражения наполнителем, который является твердым при обычной температуре, но становится жидким при температуре тела, т.е. будет плавиться при ректальном или вагинальном введении с высвобождением лекарственного средства. Такие материалы включают кокосовое масло и полиэтиленгликоли.
Соединения по изобретению можно вводить чрескожно с использованием способов, известных специалистам в данной области [см., например, в книге Chien, Transdermal Controlled Systemic Medications (Трансдермально-регулируемое системное медикаментозное лечение), Marcel Dekker, Inc., (1987); Lipp с соавт., публикация международной патентной заявки WO94/04157, 3.03.94]. Например, раствор или суспензия соединения формулы I в подходящем летучем растворителе, по выбору содержащем агенты, способствующие проникновению, объединяют с другими добавками, известными специалистам в данной области, такими как материалы матрицы и бактерицидные средства. После стерилизации полученную смесь можно переработать по известной технологии в лекарственные формы. Кроме того, после обработки эмульгирующими агентами и водой из раствора или суспензии соединения формулы I можно получить лосьон или мазь.
Подходящие растворители для приготовления систем чрескожной доставки известны специалистам в данной области, они включают низшие спирты, такие как этанол или изопропиловый спирт, низшие кетоны, такие как ацетон, эфиры низших карбоновых кислот, такие как этилацетат, полярные простые эфиры, такие как тетрагидрофуран, низшие углеводороды, такие как гексан, циклогексан или бензол, или галогенированные углеводороды, такие как дихлорметан, хлороформ, трихлортрифторэтан или трихлорфторэтан. Подходящие растворители могут также включать смеси одного или нескольких соединений, которые выбирают из низших спиртов, низших кетонов, эфиров низших карбоновых кислот, полярных простых эфиров, низших углеводородов, галогенированных углеводородов.
Подходящие способствующие проникновению агенты для систем чрескожной доставки известны специалистам в данной области, они включают, например, моногидрокси- или полигидроксиспирты, такие как этанол, пропиленгликоль или бензиловый спирт, насыщенные или ненасыщенные жирные C8-С18спирты, такие как лауриловый спирт или цетиловый спирт, насыщенные или ненасыщенные жирные C8-С18кислоты, такие как стеариновая кислота, насыщенные или ненасыщенные жирные сложные эфиры, содержащие до 24 атомов углерода, такие как метиловый, этиловый, пропиловый, изопропиловый, н-бутиловый, втор-бутиловый, изобутиловый, трет-бутиловый или моноглицериновый эфиры уксусной кислоты, капроновой кислоты, лауриновой кислоты, миристиновой кислоты, стеариновой кислоты и пальмитиновой кислоты, или диэфиры насыщенных или ненасыщенных дикарбоновых кислот, содержащие до 24 углеродных атомов, такие как диизопропиладипат, диизобутиладипат, диизопропилсебацат, диизопропилмалеат или диизопропилфумарат. Дополнительные материалы, способствующие проникновению, включают фосфатидилпроизводные, такие как лецитин или кефалин, терпены, амиды, кетоны, мочевину и их производные, а также простые эфиры, такие как диметилизосорбит и моноэтиловый эфир диэтиленгликоля. Подходящие композиции, способствующие проникновению, могут также включать смеси одного или нескольких агентов, которые выбирают из группы: моногидрокси- или полигидроксиспирты, насыщенные или ненасыщенные жирные C8-С18спирты, насыщенные или ненасыщенные жирные С8-С18кислоты, насыщенные или ненасыщенные жирные сложные эфиры, содержащие до 24 атомов углерода, диэфиры насыщенных или ненасыщенных дикарбоновых кислот, содержащие до 24 углеродных атомов, фосфатидилпроизводные, такие как лецитин или кефалин, терпены, амиды, кетоны, мочевину и их производные, и простые эфиры.
Подходящие связующие материалы для систем чрескожной доставки известны специалистам в данной области, они включают, например, полиакрилаты, силиконы, полиуретаны, блокполимеры, сополимеры стирола и бутадиена, природные и синтетические смолы. В качестве компонентов матрицы могут также использоваться эфиры целлюлозы, производные полиэтилена и силикаты. Для повышения вязкости матрицы могут быть добавлены дополнительные материалы, такие как вязкие смолы или масла.
Для всех схем применения, заявленных в описании для соединений формулы I, суточная пероральная доза предпочтительно составляет от 0,01 до 200 мг/кг веса тела. Суточная доза для введения путем инъекции, включая внутривенные, внутримышечные, подкожные и парентеральные инъекции, и использование метода вливания, предпочтительно составляет от 0,01 до 200 мг/кг веса тела. Суточная ректальная доза предпочтительно составляет от 0,01 до 200 мг/кг веса тела. Суточная вагинальная доза применения лекарственного средства предпочтительно составляет от 0,01 до 200 мг/кг веса тела. Суточная местная доза предпочтительно составляет от 0,1 до 200 мг при введении от одного до четырех раз в день. При чрескожном введении концентрация предпочтительно должна обеспечивать суточную дозу от 0,01 до 200 мг/кг. При ингаляции суточная доза предпочтительно составляет от 0,01 до 10 мг/кг веса тела.
Для специалиста в данной области техники представляется очевидным, что конкретный способ введения зависит от ряда факторов, каждый из которых при назначении лекарственного средства рассматривается в установленном порядке. Однако следует также полагать, что индивидуальный уровень доз для данного пациента зависит от множества факторов, включающих активность конкретного используемого соединения, возраст пациента, вес тела пациента, общее состояние здоровья пациента, пол пациента, диету пациента, время введения, схемы введения, скорости выведения лекарственного средства, сочетания лекарственных средств, тяжести заболевания, подлежащего терапевтическому лечению. Для специалиста в данной области представляется также очевидным, что оптимальный курс лечения, т.е. схема лечения и суточное число доз соединения формулы I или его фармацевтически приемлемой соли в течение определенного времени, может быть установлено специалистом в данной области на основании обычных медицинских анализов.
Соединения формулы I получают из известных соединений (или из исходных материалов, которые, в свою очередь, получают из известных соединений) с использованием общих препаративных методов, приведенных ниже. Активность конкретного соединения в отношении ингибирования киназы raf определяют общепринятым способом по методике, описанной ниже. Последующие примеры даны для иллюстрации сущности изобретения и ни в коей мере не ограничивают объем изобретения.
Все заявки, патенты и публикации, цитированные выше и далее по тексту, включены в качестве ссылки в текст заявки, включая предварительную заявку на выдачу патента Attorney Docket BAYER 8 V1, поданную 22 декабря, 1997 г. под серийным №08/996343, преобразованную 22 декабря, 1998 г.
Примеры
Все реакции проводят в стеклянной посуде, высушенной в пламени или в сушильном шкафу, при избыточном давлении сухого аргона или сухого азота и, если не указано иное, при перемешивании на магнитной мешалке. Чувствительные (к кислороду или влаге) жидкости и растворы переносят с помощью шприца или канюли и вносят в реакционные сосуды через резиновые прокладки. Если не указано иное, термин "концентрирование при пониженном давлении" относится к использованию роторного испарителя Бюхи при давлении приблизительно 1,99 кН/м2 (15 мм рт.ст.).
Все значения температур приводятся в градусах Цельсия (°С) без поправки. Если не указано иное, все доли и проценты указаны по массе.
Все реагенты и растворители товарного качества используют без дополнительной очистки. Тонкослойную хроматографию (ТСХ) проводят на стеклянных пластинках (фирмы Whatman®), покрытых слоем силикагеля 60А F-254 толщиной 250 мкм. Проявление хроматограмм проводят по одному или нескольким следующим способам: (а) при освещении УФ-светом, (б) экспозицией в парах иода, (в) погружением пластинок в 10%-ный раствор фосфорномолибденовой кислоты в этаноле с последующим нагреванием, (г) погружением пластинок в раствор сульфата церия с последующим нагреванием, и/или (д) погружением пластинок в раствор 2,4-динитрофенилгидразина в подкисленном этаноле с последующим нагреванием. Хроматографию на колонке (экспресс-хроматографию) проводят на силикагеле с размерами частиц 230-400 меш (фирмы ЕМ Science®).
Температуру плавления (т.пл.) определяют на приборе Thomas-Hoover или на приборе Mettler FP66 и приводят без поправки. Инфракрасные спектры с Фурье-преобразованием снимают на спектрофотометре Mattson 4020 Galaxy Series. Протонные спектры (1Н) ядерного магнитного резонанса (ЯМР) снимают на спектрометре General Electric GN-Omega 300 (300 МГц) с использованием в качестве стандартов Me4Si (δ 0,00) или остаточно протонированный растворитель (СНСl3 δ 7,26; МеОН δ 3,30; ДМСО δ 2,49).
Спектр углерода13С снимают на спектрометре General Electric GN-Omega 300 (75 МГц) с использованием в качестве стандарта растворителя (СDСl3 δ 77,0; MeOD-d3 δ 49,0; ДМСО-d6 δ 39, 5). Масс-спектры низкого разрешения (MS) и масс-спектры высокого разрешения (HRMS) получают как в виде спектров ионизации электронным ударом (ЕI), так и в виде спектров ионизации быстрыми атомами (FAB). Спектры ионизации электронным ударом (EI-MS) получают на масс-спектрометре Hewlett Packard 5989A, снабженном блоком для ввода образца с десорбцией химической ионизацией (фирмы Vacumetrics). Источник ионов имеет температуру 250°С. Ионизацию электронным ударом проводят при энергии электронов 70 эВ и токе в ловушке 300 мкА. Масс-спектры вторичных ионов с захватом цезия (FAB-MS), современный вариант метода с ионизацией быстрыми атомами, получают на спектрометре Kratos Concept 1-H. Масс-спектры с химической ионизацией (CI-MS) получают на приборе Hewlett Packard MS-Engine 5989A с метаном или аммиаком в качестве газа-реагента (133,32×10-4-333,3×10-4 (1×10-4-2,5×10-4 торр). Блок прямого ввода образца десорбцией с химической ионизацией (фирмы Vaccumetrics, Inc.) (DCI) функционирует при перепаде давления от 0-147,1 кН/м2 (0-1,5 ат.), а затем в течение 10 с при 980,6 кН/м2 (10 ат.), пока не исчезают все следы образца (˜ 1-2 мин). Спектр сканируют в диапазоне от 50-800 единиц атомной массы (одно сканирование в течение 2 с). ВЭЖХ-масс-спектр с электроспреем (HPLC ES-MS) получают на приборе Hewlett Packard 1100 HPLC, снабженном четырехканальным насосом, детектором с переменной длиной волны, колонкой С-18 и масс-спектрометром с ионной ловушкой фирмы Finnigan LCQ с ионизацией электроспреем. Спектр сканируют в диапазоне от 120-800 единиц атомной массы с использованием переменного времени счета числа ионов в источнике. Газово-хроматографический ион селективный масс-спектр (GC-MS) получают на газовом хроматографе Hewlett Packard 5890, снабженном HP-1 метилсиликоновой колонкой (25 м × 0,2 мм; пропитка 0,33 мМ) и масс-селективным детектором Hewlett Packard 5971 (с энергией ионизации 70 эВ).
Элементный анализ выполняют в лаборатории Robertson Microlit Labs, Madison NJ.
Все соединения охарактеризованы спектрами ЯМР, масс-спектрами низкого разрешения (MS) и элементными анализами или масс-спектрами высокого разрешения (HRMS), которые соответствуют приведенным структурам.
Список сокращений
АсОН - уксусная кислота,
ВОС - трет-бутоксикарбонил,
DMPU - 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон,
ДМФ - N,N-диметилформамид,
ДМСО - диметилсульфоксид,
DPPA - дифенилфосфорилазид,
EtOAc - этилацетат,
ЕtOН - этанол (100%),
Et2O - диэтиловый эфир,
Еt3N - триэтиламин,
m-СРВА - 3-хлорпероксибензойная кислота,
МеОН - метанол,
пет. - эфир,
(pet ether) - петролейный эфир (температура кипения 30-60°С),
ТГФ - тетрагидрофуран,
ТФУ - трифторуксусная кислота,
Tf - трифторметансульфонил,
acetone - ацетон,
hexane - гексан,
СН2Сl2 - хлористый метилен,
СНСl3 - хлороформ,
t-Bu - трет-бутил.
А. Общие методы синтеза замещенных анилинов
А1. Синтез 2,5-диоксопирролидиниланилинов
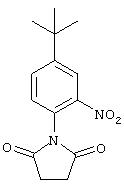
Стадия 1. 4-трет-Бутил-1-(2,5-диоксо-1-пирролидинил)-2-нитробензол:
К раствору 4-трет-бутил-2-нитроанилина (1,04 г, 5, 35 ммоль) в ксилоле (25 мл) добавляют янтарный ангидрид (0,0535 г, 5,35 ммоль) и триэтиламин (0,75 мл, 5,35 ммоль). Реакционную смесь кипятят с обратным холодильником в течение 24 ч, охлаждают до комнатной температуры и разводят Et2O (25 мл).
Полученную смесь последовательно промывают 10%-ным раствором HCl (50 мл), насыщенным раствором NH4Cl (50 мл) и насыщенным раствором NaCl (50 мл), сушат (МgSO4) и концентрируют при пониженном давлении. Остаток очищают экспресс-хроматографией (60% ЕtOАс/40% гексан), при этом получают сукцинимид в виде твердого вещества желтого цвета (1,2 г, 86%): т.пл. 135-138°С;1H ЯМР (СНСl3) δ 1,38 (s, 9H), 2,94-2,96 (m, 4H), 7,29-7,31 (m, 1H), 7,74-7,78 (m, 1Н), 8,18-8,19 (m, 1H).
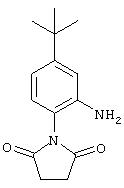
Стадия 2. 5-трет-Бутил-2-(2,5-диоксо-1-пирролидинил)анилин: К раствору 4-трет-бутил-1-(2,5-диоксо-1-пирролидинил)-2-нитробензола (1,1 г, 4,2 ммоль) в ЕtOАс (25 мл) добавляют 10% Pd/C (0,1 г). Полученную суспензию помещают в атмосферу Н2, производя три цикла процедуры откачки-продувки, и перемешивают в атмосфере Н2 в течение 8 ч. Реакционную смесь фильтруют через слой целита и остаток промывают СНСl3. Объединенный фильтрат концентрируют при низком давлении, при этом получают требуемый анилин в виде твердого вещества серо-белого цвета (0,75 г, 78%): т.пл. 208-211°С;1H ЯМР (DMSO-d6) δ 1,23 (s, 9H), 2,62-2,76 (m, 4H), 5,10 (ушир. s, 2H), 6,52-6,56 (m, 1H), 6,67-6,70 (m, 2H).
А2. Общий метод синтеза тетрагидрофуранилоксианилинов
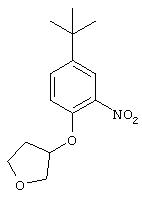
Стадия 1. 4-трет-Бутил-1-(3-тетрагидрофуранилокси)-2-нитробензол:
К раствору 4-трет-бутил-2-нитрофенола (1,05 г, 5,4 ммоль) в безводном ТГФ (25 мл) добавляют 3-гидрокситетрагидрофуран (0,47 г, 5,4 ммоль) и трифенилфосфин (1,55 г, 5,9 ммоль), а затем диэтилазодикарбоксилат (0, 93 мл, 5,9 ммоль), и реакционную смесь перемешивают при комнатной температуре в течение 4 ч. Полученную смесь разводят Et2O (50 мл) и промывают насыщенным раствором NH4Cl (50 мл) и насыщенным раствором NaCl (50 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток очищают экспресс-хроматографией (30% ЕtOАс/70% гексан), при этом получают требуемый эфир в виде твердого вещества желтого цвета (1,3 г, 91%):1H ЯМР (СНСl3) δ 1,30 (s, 9Н), 2,18-2,24 (m, 2H), 3,91-4,09 (m, 4H), 5,00-5,02 (m, 1H), 6,93 (d, J=8,8 Гц, 1Н), 7,52 (dd, J=2,6; 8,8 Гц, 1Н), 7,81 (d, J=2,6 Гц, 1Н).
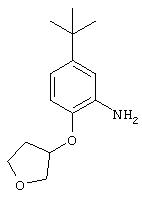
Стадия 2. 5-трет-Бутил-2-(3-тетрагидрофуранилокси)анилин: К раствору 4-трет-бутил-1-(3-тетрагидрофуранилокси)-2-нитробензола (1,17 г, 4,4 ммоль) в EtOAc (25 мл) добавляют 10% Pd/C (0,1). Полученную суспензию помещают в атмосферу H2, проводя три цикла процедуры откачки-продувки, и перемешивают в атмосфере Н2 в течение 8 ч. Реакционную смесь фильтруют через слой целита и остаток промывают СНСl3. Объединенный фильтрат концентрируют при низком давлении, при этом получают требуемый анилин в виде твердого вещества желтого цвета (0,89 г, 86%): т.пл. 79-82°С;1H ЯМР (СНСl3) δ 1,30 (s, 9H), 2,16-2,20 (m, 2H), 3,78 (ушир. s, 2H), 3,85-4,10 (m, 4H), 4,90 (m, 1H), 6,65-6,82 (m, 3Н).
A3. Общий метод синтеза трифторметансульфониланилинов
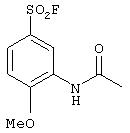
Стадия 1. 2-Метокси-5-(фторсульфонил)ацетанилид: К раствору 4-метоксиметанилилфторида (1,0 г, 4,8 ммоль) в пиридине (15 мл) добавляют уксусный ангидрид (0,90 мл, 9,6 ммоль). После перемешивания при комнатной температуре в течение 4 ч реакционную смесь концентрируют при пониженном давлении. Полученный остаток растворяют в CH2Cl2 (25 мл), промывают насыщенным раствором NаНСО3 (25 мл), сушат (Nа2SО4) и концентрируют при пониженном давлении, при этом получают пену, которую растирают с раствором Еt2О/гексан и получают конечный продукт (0,85 г):1H ЯМР (CDCl3 ) δ 2,13 (s, 3H), 3,98 (s, 3Н), 7,36 (d, J=8,5 Гц, 1Н), 7,82 (dd, J=2,6; 8,8 Гц, 1Н), 8,79 (d, J=2,2 Гц, 1Н), 9,62 (ушир. s, 1H).
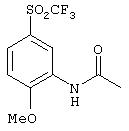
Стадия 2. 2-Метокси-5-(трифторметансульфонил)ацетанилид: К охлажденной до 0°С суспензии трис(диметиламино)сульфоний дифтортриметилсиликоната (0,094 г, 0,34 ммоль в ТГФ (4 мл), добавляют раствор (трифторметил)триметилсилана (1,0 мл, 6,88 ммоль) в ТГФ (3 мл), а затем раствор 2-метокси-5-(фторсульфонил)ацетанилида (0, 85 г, 3,44 ммоль) в ТГФ (3 мл). Реакционную смесь перемешивают на ледяной бане в течение 2 ч, затем нагревают до комнатной температуры, после чего концентрируют при пониженном давлении. Полученный остаток растворяют в CH2Cl2 (25 мл), промывают водой (25 мл), сушат (Nа2SO4) и концентрируют при пониженном давлении. Полученный материал очищают экспресс-хроматографией (3% МеОН/97% CH2Cl2), при этом получают конечный продукт в виде твердого вещества белого цвета (0,62 г):1H ЯМР (CDCl3) δ 2, 13 (s, 3Н), 4,00 (s, 3H), 7,42 (d, J=8,8 Гц, 1H), 7,81 (dd, J=2,6; 8,8 Гц, 1H), 8,80 (d, J=2,2 Гц, 1H), 9,64 (ушир. s, 1Н); МС-ББА (бомбардировка быстрыми атомами) m/z 298 ((М+1)+).
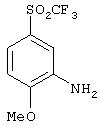
Стадия 3. 2-Метокси-5-(трифторметансульфонил)анилин: Раствор 2-метокси-5-(трифторметансульфонил)ацетанилида (0,517 г, 1,74 ммоль) в ЕtOН (5 мл) и 1Н. растворе HCl (5 мл) кипятят с обратным холодильником в течение 4 ч и полученную смесь концентрируют при пониженном давлении. Остаток растворяют в CH2Cl2 (30 мл), промывают водой (30 мл), сушат (Na2SO4) и концентрируют при пониженном давлении, при этом получают конечный продукт в виде смолы (0,33 г):1H ЯМР (CDCl3) δ 3,90 (s, 3H), 5,57 (ушир. s, 2H), 7,11-7,27 (m, 3H); FAB-MS m/z 256 ((M+1)+). Этот материал используют для получения мочевины без дальнейшей очистки.
А4. Общий метод получения ариламина путем нитрования фенольного кольца с последующим получением эфира и восстановлением
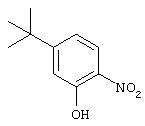
Стадия 1. 2-Нитро-5-трет-бутилфенол: Смесь дымящей азотной кислоты (3,24 г, 77,1 ммоль) и ледяной уксусной кислоты (10 мл) при 0°С прикапывают к раствору м-трет-бутилфенола (11,58 г, 77,1 ммоль) в ледяной уксусной кислоте (15 мл). Смесь перемешивают при 0°С в течение 15 мин и нагревают до комнатной температуры. Через 1 ч смесь вливают в ледяную воду (100 мл) и экстрагируют Et2O (2×50 мл). Органический слой промывают насыщенным раствором NaCl (100 мл), сушат (MgSO4) и концентрируют в вакууме. Остаток очищают экспресс-хроматографией (30% ЕtOс/70% гексан), при этом получают требуемый фенол (4,60 г, 31%):1Н ЯМР (DMSO-d6 ) δ 1,23 (s, 9H), 7,00 (dd, J=1,84; 8,83 Гц, 1Н), 7,07 (d, J=1,84 Гц, 1Н), 7,82 (d, J=8,83 Гц, 1Н), 10,74 (s, 1Н).
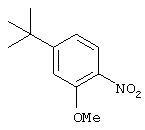
Стадия 2. 2-Нитро-5-трет-бутиланизол: Суспензию 2-нитро-5-трет-бутилфенола (3,68 г, 18,9 ммоль) и К2СО3 (3,26 г, 23,6 ммоль) в безводном ДМФ (100 мл) перемешивают при комнатной температуре в течение 15 мин, затем с помощью шприца добавляют иодметан (2,80 г, 19,8 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, затем добавляют воду (100 мл) и экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои промывают насыщенным раствором NaCl (50 мл), сушат (MgSO4) и концентрируют в вакууме, при этом получают требуемый эфир (3,95 г, 100%):1H ЯМР (DMSO-d6) δ 1,29 (s, 9H), 3,92 (s, 3Н), 7,10 (dd, J=1,84; 8,46 Гц, 1Н), 7,22 (d, J=1,84 Гц, 1Н), 7,79 (d, J=8,46 Гц, 1Н). Этот материал используют на следующей стадии без дальнейшей очистки.
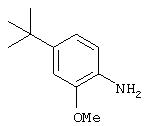
Стадия 3. 4-трет-Бутил-2-метоксианилин: Раствор 2-нитро-6-трет-бутиланизола (3,95 г, 18,9 ммоль) в МеОН (65 мл) добавяют во флакон, содержащий 10% Pd/C в МеОН (0,400 г), затем помещают в атмосферу Н2 (баллон). Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, затем фильтруют через слой целита и концентрируют в вакууме, при этом получают конечный продукт в виде липкого твердого вещества темного цвета (3,40 г, 99%).1H ЯМР (DMSO-d6) δ 1,20 (s, 9H), 3,72 (s, 3Н), 4,43 (ушир. s, 2H), 6,51 (d, J=8,09 Гц, 1Н), 6,64 (dd, J=2,21; 8,09 Гц, 1Н), 6,76 (d, J=2,21 Гц, 1Н).
А5. Общий метод получения ариламинов путем этерификации карбоновой кислоты с последующим восстановлением
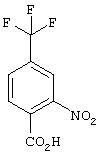
Стадия 1. Метил 2-Нитро-4-(трифторметил)бензоат: К раствору 2-нитро-4-(трифторметил)бензойной кислоты (4,0 г, 17,0 ммоль) в МеОН (150 мл) при комнатной температуре добавляют концентрированную H2SO4 (2,5 мл). Смесь кипятят с обратным холодильником в течение 24 ч, охлаждают до комнатной температуры и концентрируют в вакууме. Остаток разводят водой (100 мл) и экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют в вакууме. Остаток очищают экспресс-хроматографией (14% ЕtOАс/86% гексан), при этом получают требуемый эфир в виде масла светло-желтого цвета (4,17 г, 98%):1H ЯМР (DMSO-d6) δ 3,87 (s, 3H), 8,09 (d, J=7,72 Гц, 1Н), 8,25 (dd, J=1,11; 8,09 Гц, 1Н), 8,48 (d, J=1,11 Гц, 1Н).
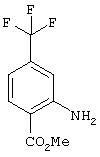
Стадия 2. Метил 2-Амино-4-(трифторметил)бензоат: Раствор метил 2-нитро-4-(трифторметил)бензоата (3,90 г, 15,7 ммоль) в ЕtOАс (100 мл) добавяют во флакон, содержащий 10% Pd/C (0,400 мг) в ЕtOАс (10 мл), затем помещают в атмосферу H2 (баллон). Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, затем фильтруют через слой целита и концентрируют в вакууме, при этом получают требуемый продукт в виде кристаллического твердого вещества белого цвета (3,20 г, 93%).1H ЯМР (DMSO-d6) δ 3,79 (s, 3H), 6,75 (dd, J=1,84; 8,46 Гц, 1Н), 6,96 (ушир. s, 2H), 7,11 (d, J=0,73 Гц, 1Н), 7,83 (d, J=8,09 Гц, 1Н).
А6. Общий метод получения ариламинов путем образования эфира с последующим омылением эфира, с помощью перегруппировки Курциуса и снятия защитных групп с карбамата
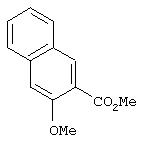
Стадия 1. Метил 3-метокси-2-нафтоат: Суспензию метил 3-гидрокси-2-нафтоата (10,1 г, 50,1 ммоль) и К2СО3 (7,96 г, 57,6 ммоль) в ДМФ (200 мл) перемешивают при комнатной температуре в течение 15 мин, затем добавляют иодметан (3,43 мл, 55,1 ммоль). Смесь перемешивают при комнатной температуре в течение ночи, затем добавляют воду (200 мл). Полученную смесь экстрагируют ЕtOАс (2×200 мл). Объединенные органические слои промывают насыщенным раствором NaCl (100 мл), сушат (MgSO4) и концентрируют в вакууме (около 53,32 н/м2 (0,4 мм рт.ст.) в течение ночи), при этом получают требуемый эфир в виде масла янтарного цвета (10,30 г):1H ЯМР (DMSO-d6) δ 2,70 (s, 3H), 2,85 (s, 3Н), 7, 38 (арр t, J=8,09 Гц, 1Н), 7,44 (s, 1Н), 7,53 (арр t, J=8,09 Гц, 1Н), 7,84 (d, J=8,09 Гц, 1Н), 7,90 (s, 1Н), 8,21 (s, 1H).
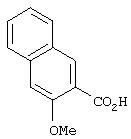
Стадия 2. 3-Метокси-2-нафтойная кислота: К раствору метил 3-метокси-2-нафтоата (6,28 г, 29,10 ммоль) и воды (10 мл) в МеОН (100 мл) при комнатной температуре добавляют 1н. раствор NaOH (33,4 мл, 33,4 ммоль). Смесь кипятят с обратным холодильником в течение 3 ч, охлаждают до комнатной температуры и подкисляют 10%-ным раствором лимонной кислоты. Полученный раствор экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют в вакууме. Остаток растирают с гексаном и несколько раз промывают гексаном, при этом получают требуемую карбоновую кислоту в виде кристаллического твердого вещества белого цвета (5,40 г, 92%).1H ЯМР (DMSO-d6) δ 3,88 (s, 3Н), 7,34-7,41 (m, 2Н), 7,49-7,54 (m, 1H), 7,83 (d, J=8,09 Гц, 1H), 7,91 (d, J=8,09 Гц, 1H), 8,19 (s, 1H), 12,83 (ушир. s, 1H).
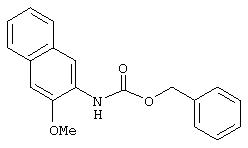
Стадия 3. 2-(N-Карбобензилокси)амино-3-метоксинафталин: Раствор 3-метокси-2-нафтойной кислоты (3,36 г, 16,6 ммоль) и Et3N (2,59 мл, 18,6 ммоль) в безводном толуоле (70 мл) перемешивают при комнатной температуре в течение 15 мин, затем пипеткой добавляют раствор дифенилфосфорилазида (5,12 г, 18,6 ммоль) в толуоле (10 мл). Полученную смесь нагревают при температуре 80°С в течение 2 ч. После охлаждения смеси до комнатной температуры через шприц добавляют бензиловый спирт (2,06 мл, 20 ммоль). Затем смесь в течение ночи выдерживают при температуре 80°С. Полученную смесь охлаждают до комнатной температуры, реакцию останавливают добавлением 10%-ного раствора лимонной кислоты и экстрагируют EtOAc (2×100 мл). Объединенные органические слои промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют в вакууме. Остаток очищают экспресс-хроматографией (14% ЕtOАс/86% гексана), при этом получают бензил карбамат в виде масла светло-желтого цвета (5,1 г, 100%):1H ЯМР (DMSO-d6) δ 3,89 (s, 3H), 5,17 (s, 2H), 7,27-7,44 (m, 8H), 7,72-7,75 (m, 2H), 8,20 (s, 1Н), 8,76 (s, 1H).
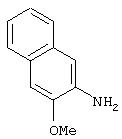
Стадия 4. 2-Амино-3-метоксинафталин: Суспензию 2-(N-карбобензилокси)амино-3-метоксинафталина (5,0 г, 16,3 ммоль) и 10% Pd/C (0,5 г) в EtOAc (70 мл) выдерживают в атмосфере H2 (баллон) в течение ночи при комнатной температуре. Полученную смесь фильтруют через целит и концентрируют в вакууме, при этом получают требуемый амин в виде порошка светло-розового цвета (2,40 г, 85%).1H ЯМР (DMSO-d6) δ 3, 86 (s, 3H), 6,86 (s, 2H), 7,04-7,16 (m, 2H), 7,43 (d, J=8,0 Гц, 1Н), 7,56 (d, J=8,0 Гц, 1H); EI-MS m/z 173 (M+).
А7. Общий метод синтеза ариламинов путем перекрестного сочетания, инициированного металлом, с последующим восстановлением
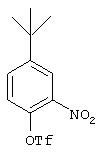
Стадия 1. 5-трет-Бутил-2-(трифторметансульфонил)окси-1-нитробензол: К раствору 4-трет-бутил-2-нитрофенола (6,14 г, 31,5 ммоль) и пиридина (10 мл, 125 ммоль) в СН2Сl2 (50 мл), охлажденному льдом, с помощью шприца медленно добавляют трифторметансульфоновый ангидрид (10 г, 35,5 ммоль). Реакционную смесь перемешивают в течение 15 мин, затем нагревают до комнатной температуры и разбавляют CH2Cl2 (100 мл). Полученную смесь последовательно промывают 1М раствором NaOH (3×100 мл) и 1М раствором HCl (3×100 мл), сушат (МgSO4) и концентрируют при пониженном давлении, при этом получают конечный продукт (8,68 г, 84%):1H ЯМР (CDCl3) δ 1,39 (s, 9H), 7,30-8,20 (m, 3Н).
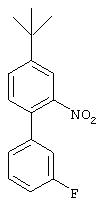
Стадия 2. 5-трет-Бутил-2-(3-фторфенил)-1-нитробензол: К раствору 5-трет-бутил-2-(трифторметансульфонил)окси-1-нитробензола (6 г, 18,4 ммоль) в диоксане (100 мл) добавляют смесь 3-фторбензолбороновой кислоты (3,80 г, 27,5 ммоль), КВr (2,43 г, 20,4 ммоль), К3РO4 (6,1 г, 28,8 ммоль) и Pd(PPh3 )4 (1,0 г, 0,9 ммоль). Реакционную смесь нагревают при 80°С в течение 24 ч, после чего данные ТСХ показывают завершение реакции. Реакционную смесь обрабатывают насыщенным раствором NH4Cl (50 мл) и экстрагируют ЕtOАс (3×100 мл). Объединенные органические слои сушат (MgSO4) и концентрируют при пониженном давлении. Остаток очищают экспресс-хроматографией (3% ЕtOАс/97% гексан), при этом получают указанное соединение (4,07 г, 81%):1H ЯМР (CDCl3) δ 1,40 (s, 9H), 6,90-7,90 (m, 7H).
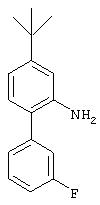
Стадия 3. 5-трет-Бутил-2-(3-фторфенил)анилин: К раствору 5-трет-бутил-2-(3-фторфенил)-1-нитробензола (3,5 г, 12,8 ммоль) и ЕtOН (24 мл) в ЕtOАс (96 мл) добавляют 5% Pd/C (0,350 г) и полученную суспензию перемешивают в течение 24 ч в атмосфере Н2, после чего данные ТСХ показывают, что произошло полное превращение исходного материала. Реакционную смесь отфильтровывают через слой целита, при этом получают конечный продукт (2,2 г, 72%):1H ЯМР (СDСl3) δ 1,35 (s, 9H), 3,80 (ушир. s, 2Н), 6,90-7,50 (m, 7H).
A8. Общий метод синтеза нитроанилинов
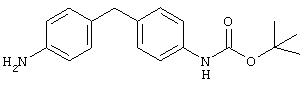
Стадия 1. 4-(4-(2-Пропоксикарбониламино)фенил)метиланилин: Раствор ди-трет-бутилдикарбоната (2,0 г, 9,2 ммоль) и 4, 4'-метилендианилина (1,8 г, 9,2 ммоль) в ДМФ (100 мл) кипятят с обратным холодильником в течение 2 ч, затем охлаждают до комнатной температуры. Эту смесь разбавляют ЕtOАс (200 мл), последовательно промывают насыщенным раствором NH4Cl (200 мл) и насыщенным раствором NaCl (100 мл) и сушат (MgSO4). Остаток очищают экспресс-хроматографией (30% ЕtOАс/70% гексан), при этом получают требуемый карбамат (1,3 г, 48%):1H ЯМР (CDCl3) δ 1,51 (s, 9H), 3,82 (s, 2H), 6,60-7,20 (m, 8H).
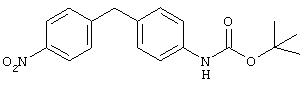
Стадия 2. 4-(4-(2-Пропоксикарбониламино)фенил)метил-1-нитробензол: К раствору 4-(4-(2-пропоксикарбониламино)фенил)метиланилина (1,05 г, 3,5 ммоль) в CH2Cl2 (15 мл), охлажденному льдом, добавляют m-СРВА (1,2 г, 7,0 ммоль). Реакционную смесь медленно нагревают до комнатной температуры и перемешивают в течение 45 мин, после чего данные ТСХ показывают исчезновение исходного материала. Полученную смесь разбавляют ЕtOАс (50 мл), последовательно промывают 1 М раствором NaOH (50 мл) и насыщенным раствором NaCl (50 мл) и сушат (MgSO4). Остаток очищают экспресс-хроматографией (20% ЕtOАс/80% гексан), при этом получают требуемый нитробензол (0,920 г): FAB-MS m/z 328 (М+).
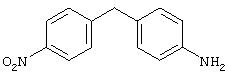
Стадия 3. 4-(4-Нитрофенил)метиланилин: К раствору 4-(4-(2-пропоксикарбониламино)фенил)метил-1-нитробензола (0,920 г, 2,8 ммоль) в диоксане (10 мл) добавляют концентрированную HCI (4,0 мл) и полученную смесь нагревают до 80°С в течение 1 ч, после чего данные ТСХ показывают исчезновение исходного материала. Реакционную смесь охлаждают до комнатной температуры. Полученную смесь разводят ЕtOАс (50 мл), промывают 1М раствором NaOH (3× 50 мл) и сушат (MgSO4), при этом получают требуемый анилин (0,570 мг, 89%):1H ЯМР (CDCl3) δ 3,70 (ушир. s, 2Н), 3,97 (s, 2Н), 6,65 (d, J=8,5 Гц, 2Н), 6,95 (d, J=8,5 Гц, 2Н), 7,32 (d, J=8,8 Гц, 2H), 8,10 (d, J=8,8 Гц, 2Н).
A9. Общий метод синтеза ариланилинов путем алкилирования нитрофенола с последующим восстановлением
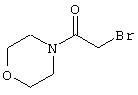
Стадия 1. 4-(α-Бромацетил)морфолин: К охлажденному до 0°С раствору морфолина (2,17 г, 24,9 ммоль) и диизопропилэтиламина (3,21 г, 24,9 ммоль) в СН2Сl2 (70 мл) с помощью шприца добавляют раствор бромацетилбромида (5,05 г, 25 ммоль) в CH2Cl2 (8 мл). Полученный раствор выдерживают при 0°С в течение 45 мин, затем нагревают до комнатной температуры. Реакционную смесь разбавляют ЕtOАс (500 мл), промывают последовательно 1 М раствором HCl (250 мл) и насыщенным раствором NaCl (250 мл), сушат (MgSO4), при этом получают требуемый продукт (3,2 г, 62%):1H ЯМР (DMSO-d6) δ 3,40-3,50 (m, 4Н), 3,50-3,60 (m, 4H), 4,11 (s, 2H).
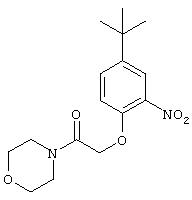
Стадия 2. 2-(N-Морфолинилкарбонил)метокси-5-трет-бутил-1-нитробензол: Суспензию 4-трет-бутил-2-нитрофенола (3,9 г, 20 ммоль) и K2СО3 (3,31 г, 24 ммоль) в ДМФ (75 мл) перемешивают при комнатной температуре в течение 15 мин, затем добавляют раствор 4-(α-бромацетил)морфолина (4,16 г, 20 ммоль) в ДМФ (10 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи, затем разбавляют ЕtOАс (500 мл), промывают последовательно насыщеннным раствором NaCl (4×200 мл) и 1М раствором NaOH (400 мл). Остаток очищают экспресс-хроматографией (75% ЕtOАс/25% гексан), при этом получают нитробензол (2,13 г, 33%):1H ЯМР (DMSO-d6) δ 1,25 (s, 9H), 3,35-3,45 (m, 4H), 3,50-3,58 (m, 4H), 5,00 (s, 2H), 7,12 (d, J=8,8 Гц, 1Н), 7,50-7, 80 (m, 2H).
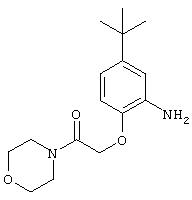
Стадия 3. 2-(N-Морфолинилкарбонил)метокси-5-трет-бутиланилин: К раствору 2-(N-морфолинилкарбонил)метокси-5-трет-бутил-1-нитробензола (2,13 г, 6,6 ммоль) и ЕtOН (10 мл) в ЕtOАс (40 мл) добавляют 5% Pd/C (0,215 г). Полученную суспензию перемешивают в атмосфере H2 в течение 6 ч, после чего данные ТСХ показывают полное превращение исходного материала. Реакционную смесь фильтруют через слой целита, при этом получают требуемый продукт (1,9 г, 98%):1H ЯМР (DMSO-d6) δ 1,18 (s, 9H), 3,40-3,50 (m, 4Н), 3,50-3,60 (m, 4H), 4,67 (ушир. s, 2Н), 4,69 (s, 2H), 6,40-6,70 (m, 3H).
А 10. Общий метод получения ариламина путем алкилирования нитрофенола с последующим восстановлением
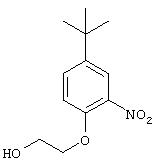
Стадия 1. 5-трет-Бутил-2-(2-гидроксиэтокси)-1-нитробензол: Раствор 4-трет-бутил-2-нитрофенола (30 г, 0,15 моль) и тетра-н-бутиламмоний фторида (0,771 г, 3,0 ммоль) в этиленкарбонате (10,24 мл, 0,15 моль) нагревают при 150°С в течение 18 ч, затем охлаждают до комнатной температуры и распределяют между водой (50 мл) и CH2Cl2 (50 мл). Органический слой сушат (МgSO4 ) и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (20% ЕtOАс/80% гексан), при этом получают требуемый продукт в виде масла коричневого цвета (35,1 г, 90%):1H ЯМР (DMSO-d6) δ 1,25 (s, 9H), 3,66-3,69 (m, 2H), 4,10-4,14 (t, J=5,0 Гц, 2Н), 4,85 (t, J=5,0 Гц, 1Н), 7,27 (d, J=8,8 Гц, 1H), 7,60-7,64 (m, 1H), 7,75 (d, J=2,6 Гц, 1Н).
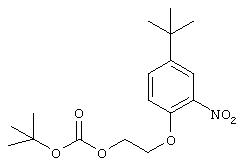
Стадия 2. 5-трет-Бутил-2-(2-трет-бутоксикарбонилокси)этокси-1-нитробензол: Раствор 5-трет-бутил-2-(2-гидроксиэтокси)-1-нитробензола (0,401 г, 1,68 ммоль), ди-трет-бутилдикарбоната (0,46 мл, 2,0 ммоль) и диметиламинопиридина (0,006 г, 0,05 ммоль) в СН2Сl2 (15 мл) перемешивают при комнатной температуре в течение 30 мин, после чего данные ТСХ показывают полное превращение исходного материала. Полученную смесь промывают водой (20 мл), сушат (МgSO4) и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (3% МеОН/97% CH2Cl2), при этом получают конечный продукт в виде масла желтого цвета (0, 291 г, 51%);1H ЯМР (DMSO-d6) δ 1,25 (s, 9H), 1,38 (s, 9H), 4,31 (ушир. s, 4Н), 7,27 (d, J=9,2 Гц, 1H), 7,64 (dd, J=2,6; 8,8 Гц, 1Н), 7,77 (d, J=2,6 Гц, 1Н).
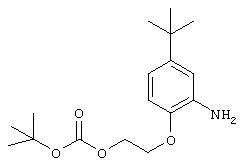
Стадия 3. 5-трет-Бутил-2-(2-трет-бутоксикарбонилокси)этокси)анилин:
К смеси 5-трет-бутил-2-(2-трет-бутоксикарбонилокси)этокси-1-нитробензола (0,290 г, 0,86 ммоль) и 5% Pd/C в МеОН (2 мл) добавляют формиат аммония (0,216 г, 3,42 ммоль) и полученную суспензию перемешивают при комнатной температуре в течение 12 ч, затем фильтруют через слой целита с использованием ЕtOН. Фильтрат концентрируют при пониженном давлении и очищают колоночной хроматографией (2% МеОН/98% CH2Cl2), при этом получают требуемый продукт в виде масла светло-желтого цвета (0,232 г, 87%): ТСХ (20% ЕtOАс/80% гексан), Rf 0,63;1H ЯМР (DMSO-d6) δ 1,17 (s, 9H), 1,39 (s, 9H), 4,03-4,06 (m, 2Н), 4,30-4,31 (m, 2H), 4,54 (ушир. s, 2H), 6,47 (dd, J=2,2; 8,1 Гц, 1H), 6,64-6,67 (m, 2H).
А11. Общий метод получения замещенного анилина путем гидрирования нитроарена
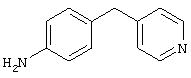
4-(4-Пиридинилметил)анилин: К раствору 4-(4-нитробензил)пиридина (7,0 г, 32,68 ммоль) в ЕtOН (200 мл) добавляют 10% Pd/C (0,7 г) и полученную суспензию встряхивают с помощью шейкера Parr в атмосфере H2 при 50 psi (344,79 кН/м2). Через 1 ч данные ТСХ и1H-ЯМР показывают завершение реакции. Смесь фильтруют через тонкий слой целита. Фильтрат концентрируют в вакууме, при этом получают твердое вещество белого цвета (5,4 г, 90%):1H-ЯМР (ДМСО-d6) δ 3,74 (s, 2H), 4,91 (ушир. s, 2H), 6,48 (d, J=8,46 Гц, 2H), 6,86 (d, J=8,09 Гц, 2H), 7,16 (d, J=5,88 Гц, 2H), 8,40 (d, J=5,88 Гц, 2H); EI-MS m/z 184 (M+). Полученный материал используют для синтеза мочевины без дополнительной очистки.
А12. Общий метод синтеза замещенных анилинов с использованием восстановления в растворе нитроароматических соединений металлами
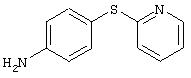
4-(2-Пиридинилтио)анилин: К раствору 4-(2-пиридинилтио)-1-нитробензола (Menai ST 3355A; 0,220 г, 0,95 ммоль) и H2O (0,5 мл) в АсОН (5 мл) добавляют порошкообразное железо (0,317 г, 5,68 ммоль) и полученную суспензию перемешивают в течение 16 ч при комнатной температуре. Реакционную смесь разбавляют ЕtOАс (75 мл) и H2O (50 мл), подщелачивают порционным добавлением К2СО3 до рН 10 (Внимание: пенообразование). Органические слои промывают насыщенным раствором NaCl, сушат (MgSO4) и концентрируют в вакууме. Твердый остаток очищают MPLC (30% ЕtOАс/70% гексан), при этом получают требуемый продукт в виде вязкого масла (0,135 г, 70%): ТСХ (30% ЕtOАс/70% гексан); Rf 0,20.
А13а. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
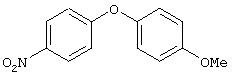
Стадия 1. 1-Метокси-4-(4-нитрофенокси)бензол: К суспензии NaH (95%, 1,50 г, 59 ммоль) в ДМФ (100 мл) при комнатной температуре добавляют по каплям раствор 4-метоксифенола (7,39 г, 59 ммоль) в ДМФ (50 мл). Реакционную смесь перемешивают в течение 1 ч, затем по каплям добавляют раствор 1-фтор-4-нитробензола (7,0 г, 49 ммоль) в ДМФ (50 мл), при этом получают раствор темно-зеленого цвета. Реакционную смесь нагревают при 95°С в течение ночи, затем охлаждают до комнатной температуры, реакцию останавливают Н2О и концентрируют в вакууме. Остаток распределяют между ЕtOАс (200 мл) и Н2О (200 мл). Органический слой последовательно промывают Н2О (2×200 мл), насыщенным раствором NаНСО3 (200 мл) и насыщенным раствором NaCl (200 мл), сушат (Na2SO4) и концентрируют в вакууме. Остаток растирают (Et2О/гексан), при этом получают 1-метокси-4-(4-нитрофенокси)бензол (12,2 г, 100%):1H-ЯМР (CDCl3) δ 3,83 (s, 3H), 6,93-7,04 (m, 6Н), 8,18 (d, J=9,2 Гц, 2H); EI-MS m/z 245 (M+).
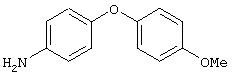
Стадия 2. 4-(4-Метоксифенокси)анилин: К раствору 1-метокси-4-(4-нитрофенокси)бензола (12,0 г, 49 ммоль) в ЕtOАс (250 мл) добавляют 5% Pt/C (1,5 г) и полученную смесь встряхивают в атмосфере H2 при 50 psi (344,79 кН/м2) в течение 18 ч. Реакционную смесь фильтруют через слой целита с помощью ЕtOАс и концентрируют в вакууме, при этом получают масло, которое медленно затвердевает (10,6 г, 100%):1H-ЯМР (CDCl3) δ 3,54 (ушир. s, 2H), 3,78 (s, 3H), 6,65 (d, J=8,8 Гц, 2H), 6,79-6,92 (m, 6H); EI-MS m/z 215 (M+).
A13b. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
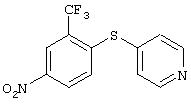
Стадия 1. 3-(трифторметил)-4-(4-пиридинилтио)нитробензол: Раствор 4-меркаптопиридина (2,8 г, 24 ммоль), 2-фтор-5-нитробензотрифторида (5 г, 23,5 ммоль) и карбоната калия (6,1 г, 44,3 ммоль) в безводном ДМФ (80 мл) перемешивают при комнатной температуре в атмосфере аргона в течение ночи. Данные ТСХ показывают завершение реакции. Смесь разбавляют Et2O (100 мл) и водой (100 мл) и водный слой снова экстрагируют Et2O (2×100 мл). Органические слои промывают насыщенным раствором NaCl (100 мл), сушат (МgSO4) и концентрируют при пониженном давлении. Твердый остаток растирают с Et2O, при этом получают требуемый продукт в виде твердого вещества желто-коричневого цвета (3,8 г, 54%): ТСХ (30% EtOAc/70% гексан); Rf 0,06;1H-ЯМР (ДМСО-d6) δ 7,33 (dd, J=1,2, 4,2 Гц, 2Н), 7,78 (d, J=8,7 Гц, 1Н), 8,46 (dd, J=2,4, 8,7 Гц, 1Н), 8,54-8,56 (m, 3H).
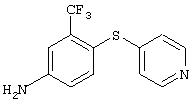
Стадия 2. 3-(Трифторметил)-4-(4-пиридинилтио)анилин: Суспенизию 3-(трифторметил)-4-(4-пиридинилтио)нитробензола (3,8 г, 12,7 ммоль), порошкобразного железа (4,0 г, 71,6 ммоль), уксусной кислоты (100 мл) и воды (1 мл) перемешивают при комнатной температуре в течение 4 ч. Смесь разбавляют Et2O (100 мл) и водой (100 мл). Водную фазу доводят до рН 44 н. раствором NaOH. Объединенные органические слои промывают насыщенным раствором NaCl (100 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток фильтруют через слой силикагеля (градиент от 50% EtOAc/50% гексан до 60% ЕtOАс/40% гексан), при этом получают требуемый продукт (3,3 г): ТСХ (50% EtOAc/50% гексан); Rf 0,101H-ЯМР (ДМСО-d6) δ 6,21 (s, 2H), 6,84-6,87 (m, 3H), 7,10 (d, J=2,4 Гц, 1Н), 7,39 (d, J=8,4 Гц, 1Н), 8,29 (d, J=6,3 Гц, 2Н).
А13с. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
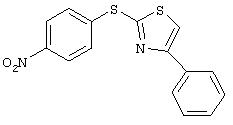
Стадия 1. 4-(2-(4-Фенил)тиазолил)тио-1-нитробензол: Раствор 2-меркапто-4-фенилтиазола (4,0 г, 20,7 ммоль) в ДМФ (40 мл) обрабатывают 1-фтор-4-нитробензолом (2,3 мл, 21,7 ммоль), затем K2CO3 (3,18 г, 23 ммоль) и полученную смесь нагревают приблизительно при 65°С в течение ночи. Затем реакционную смесь разбавляют ЕtOАс (100 мл), последовательно промывают водой (100 мл) и насыщенным раствором NaCl (100 мл), сушат (МgSO4 ) и концентрируют при пониженном давлении. Твердый остаток растирают с раствором Et2O/гексан, при этом получают требуемый продукт (6,1 г): ТСХ (25% ЕtOАс/75% гексан); Rf 0,49;1H-ЯМР (CDCl3) δ 7,35-7,47 (m, 3Н), 7,58-7,63 (m, 3Н), 7,90 (d, J=6,9 Гц, 2Н), 8,19 (d, J=9,0 Гц, 2Н).
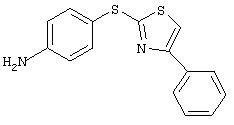
Стадия 2. 4-(2-(4-Фенил)тиазолил)тиоанилин: 4-(2-(4-фенил)тиазолил)тио-1-нитробензол восстанавливают с использованием методики, указанной для получения 3-(3-фторметил)-4-(4-пиридинилтио)анилина: ТСХ (25% ЕtOАс/75% гексан); Rf 0,181H-ЯМР (CDCl3) δ 3,89 (ушир. s, 2H), 6,72-6,77 (m, 2H), 7, 26-7,53 (m, 6H), 7,85-7,89 (m, 2H).
A13d. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
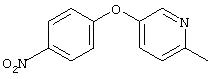
Стадия 1. 4-(6-Метил-3-пиридинилокси)-1-нитробензол: К раствору 5-гидрокси-2-метилпиридина (5,0 г, 45,8 ммоль) и 1-фтор-4-нитробензола (6,5 г, 45,8 ммоль) в безводном ДМФ (50 мл) добавляют К2СО3 (13,0 г, 91,6 ммоль) одной порцией. Смесь кипятят с обратным холодильником при перемешивании в течение 18 ч, затем охлаждают до комнатной температуры. Полученную смесь выливают в воду (200 мл) и экстрагируют ЕtOАс (3×150 мл). Объединенные органические слои последовательно промывают водой (3×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (Nа2SO4) и концентрируют в вакууме, при этом получают требуемый продукт (8,7 г, 83%). Полученный материал используют на следующей стадии без дополнительной очистки.
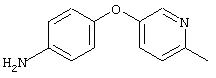
Стадия 2. 4-(6-Метил-3-пиридинилокси)анилин: Раствор 4-(6-метил-3-пиридинилокси)-1-нитробензола (4, 0 г, 17,3 ммоль) в ЕtOАс (150 мл) добавляют к 10% Pd/C (0,500 г, 0,47 ммоль) и полученную смесь помещают в атмосферу Н2 (баллон) и перемешивают в течение 18 ч при комнатной температуре. Затем смесь фильтруют через слой целита (Celite®) и концентрируют в вакууме, при этом получают требуемый продукт в виде твердого вещества желто-коричневого цвета (3,2 г, 92%). EI-MS m/z 200 (М+).
А13е. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
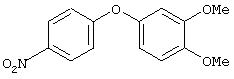
Стадия 1. 4-(3,4-Диметоксифенокси)-1-нитробензол: К раствору 3,4-диметоксифенола (1,0 г, 6,4 ммоль) и 1-фтор-4-нитробензола (700 мкл, 6,4 ммоль) в безводном ДМФ (50 мл) добавляют К2СО3 (1,8 г, 12,9 ммоль) одной порцией. Полученную смесь кипятят с обратным холодильником при перемешивании в течение 18 ч и затем охлаждают до комнатной температуры. Затем полученную смесь выливают в воду (100 мл) и экстрагируют ЕtOАс (3×100 мл). Объединенные органические слои последовательно промывают водой (3×50 мл) и насыщенным раствором NaCl (2×50 мл), сушат (Na2SO4) и концентрируют в вакууме, при этом получают требуемый продукт (0,8 г, 54%). Неочищенный продукт используют на следующей стадии без дополнительной очистки.
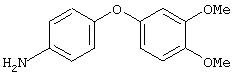
Стадия 2. 4-(3,4-Диметоксифенокси)анилин: Раствор 4-(3,4-диметоксифенокси)-1-нитробензола (0,8 г, 3,2 ммоль) в ЕtOАс (50 мл) добавляют к 10% Pd/C (0,100 г) и полученную смесь помещают в атмосферу H2 (баллон) и перемешивают в течение 18 ч при комнатной температуре. Затем смесь фильтруют через слой целита и концентрируют в вакууме, при этом получают требуемый продукт в виде твердого вещества белого цвета (0,6 г, 75%). EI-MS m/z 245 (M+).
A13f. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
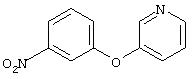
Стадия 1. 3-(3-Пиридинилокси)-1-нитробензол: К раствору 3-гидроксипиридина (2,8 г, 29,0 ммоль), 1-бром-3-нитробензола (5,9 г, 29,0 ммоль) и бромида меди (I) (5,0 г, 34,8 ммоль) в безводном ДМФ (50 мл) добавляют К2СО3 (8,0 г, 58,1 ммоль) одной порцией. Полученную смесь кипятят с обратным холодильником при перемешивании в течение 18 ч и затем охлаждают до комнатной температуры. Полученную смесь выливают в воду (200 мл) и экстрагируют ЕtOАс (3×150 мл). Объединенные органические слои последовательно промывают водой (3×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (Na2SO4) и концентрируют в вакууме. Полученное масло очищают экспресс-хроматографией (30% EtOAc/70% гексан), при этом получают требуемый продукт (2,0 г, 32%). Полученное вещество используют на следующей стадии без дополнительной очистки.
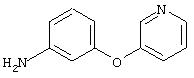
Стадия 2. 3-(3-Пиридинилокси)анилин: Раствор 3-(3-пиридинилокси)-1-нитробензола (2,0 г, 9,2 ммоль) в ЕtOАс (100 мл) добавляют к 10% Pd/C (0,200 г) и полученную смесь помещают в атмосферу H2 (баллон) и перемешивают в течение 18 ч при комнатной температуре. Затем смесь фильтруют через слой целита и концентрируют в вакууме, при этом получают требуемый продукт в виде масла красного цвета (1,6 г, 94%). EI-MS m/z 186 (М+).
А13g. Общий метод синтеза замещенных анилинов через образование нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
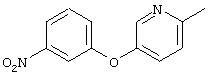
Стадия 1. 3-(5-Метил-3-пиридинилокси)-1-нитробензол: К раствору 3-гидрокси-5-метилпиридина (5,0 г, 45,8 ммоль), 1-бром-3-нитробензола (12,0 г, 59,6 ммоль) и иодида меди (I) (10,0 г, 73,3 ммоль) в безводном ДМФ (50 мл) добавляют К2СО3 (13,0 г, 91,6 ммоль) одной порцией. Полученную смесь кипятят с обратным холодильником при перемешивании в течение 18 ч и затем охлаждают до комнатной температуры. Полученную смесь выливают в воду (200 мл) и экстрагируют ЕtOАс (3×150 мл). Объединенные органические слои последовательно промывают водой (3×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (Na2SO4) и концентрируют в вакууме. Полученное масло очищают экспресс-хроматографией (30% ЕtOАс/70% гексан), при этом получают требуемый продукт (1,2 г, 13%).
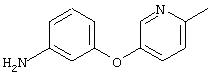
Стадия 2. 3-(5-Метил-3-пиридинилокси)анилин: Раствор 3-(5-метил-3-пиридинилокси)-1-нитробензола (1,2 г, 5,2 ммоль) в ЕtOАс (50 мл) добавляют к 10% Pd/C (0,100 г) и полученную смесь помещают в атмосферу Н2 (баллон) и перемешивают в течение 18 ч при комнатной температуре. Затем смесь фильтруют через слой целита и концентрируют в вакууме, при этом получают требуемый продукт в виде масла красного цвета (0,9 г, 86%). CI-MS (CI-химическая ионизация) m/z 201 ((М+Н)+).
А13h. Общий метод синтеза замещенных анилинов через образование нитроароматических соединений нуклеофильным ароматическим замещением с последующим восстановлением
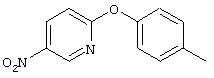
Стадия 1. 5-нитро-2-(4-метилфенокси)пиридин: К раствору 2-хлор-5-нитропиридина (6,34 г, 40 ммоль) в ДМФ (200 мл) добавляют 4-метилфенол (5,4 г, 50 ммоль, 1,25 эквивалент) и К2СО3 (8,28 г, 60 ммоль, 1,5 эквивалент).
Смесь перемешивают в течение ночи при комнатной температуре. Полученную смесь обрабатывают водой (600 мл) для получения осадка. Полученную смесь перемешивают в течение 1 ч, твердое вещество отделяют и последовательно промывают 1н. раствором NaOH (25 мл), водой (25 мл) и пет. эфиром (25 мл), при этом получают требуемый продукт (7,05 г, 76%). т.пл. 80-82°С; ТСХ (30% ЕtOАс/70% пет. эфира) Rf 0,79;1H-ЯМР (ДМСО-d6) δ 2,31 (s, 3H), 7,08 (d, J=8,46 Гц, 2Н), 7,19 (d, J=9,20 Гц, 1Н), 7,24 (d, J=8,09 Гц, 2Н), 8,58 (dd, J=2,94, 8,82 Гц, 1Н), 8,99 (d, J=2,95 Гц, 1Н); FAB-MS m/z (отн. масса) 231 ((М+Н)+; 100%).
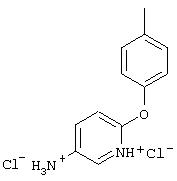
Стадия 2. Дигидрохлорид 5-амино-2-(4-метилфенокси)пиридина:
Раствор 5-нитро-2-(4-метилфенокси)пиридина (6,94 г, 30 ммоль, 1 эквивалент) и ЕtOН (10 мл) в ЕtOАс (190 мл) продувают аргоном, затем обрабатывают 10% Pd/C (0,60 г). Затем полученную смесь помещают в атмосферу Н2 (баллон) и интенсивно перемешивают в течение 2,5 ч. Затем смесь фильтруют через слой целита. К фильтрату по каплям добавляют раствор HCl в Et2O. Полученный осадок отделяют и промывают ЕtOАс, при этом получают требуемый продукт (7,56 г, 92 %): т.пл. 208-210°С (разл.); ТСХ (50% ЕtOАс/50% пет.), Rf 0,42;1H-ЯМР (ДМСО-d6) δ 2,25 (s, 3H), 6,98 (d, J=8,45 Гц, 2Н), 7, 04 (d, J=8,82 Гц, 1Н), 7,19 (d, J=8,09 Гц, 2Н), 8,46 (dd, J=2,57, 8,46 Гц, 1Н), 8,63 (d, J=2,57 Гц, 1Н); EI-MS m/z (отн. масса) ((M+; 100%).
А13i. Общий метод синтеза замещенных анилинов путем образования нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
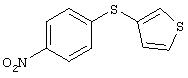
Стадия 1. 4-(3-Тиенилтио)-1-нитробензол: К раствору 4-нитротиофенола (чистота 80%; 1,2 г, 6,1 ммоль), 3-бромтиофена (1, 0 г, 6,1 ммоль) и оксида меди (II) (0,5 г, 3,7 ммоль) в безводном ДМФ (20 мл) добавляют КОН (0,3 г, 6,1 ммоль) и полученную смесь нагревают при 130°С при перемешивании в течение 42 ч и затем охлаждают до комнатной температуры. Затем реакционную смесь выливают в смесь льда и 6 н. раствора HCl (200 мл), полученную водную смесь экстрагируют ЕtOАс (3×100 мл). Объединенные органические слои последовательно промывают 1 М раствором NaOH (2×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (MgSO4) и концентрируют в вакууме. Полученное масло очищают MPLC (силикагель; градиент от 10% ЕtOАс/90% гексан до 5% ЕtOАс/95% гексан), при этом получают требуемый продукт (0,5 г, 34%). GC-MS m/z 237 (M+).
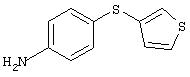
Стадия 2. 4-(3-Тиенилтио)анилин: 4-(3-тиенилтио)-1-нитробензол
восстанавливают до анилина аналогичным способом, как описано в методе В1.
A13j. Общий метод синтеза замещенных анилинов путем получения нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
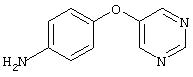
4-(5-Пиримидинилокси)анилин: 4-Аминофенол (1,0 г, 9,2 ммоль) растворяют в ДМФ (20 мл), затем добавляют 5-бромпиримидин (1,46 г, 9,2 ммоль) и К2СО3 (1,9 г, 13,7 ммоль). Полученную смесь нагревают при 100°С в течение 18 ч и при 130°С в течение 48 ч, после чего по данным анализа GC-MS в реакционной смеси обнаруживают некоторое количество исходного вещества. Реакционную смесь охлаждают до комнатной температуры и разбавляют водой (50 мл). Полученный раствор экстрагируют ЕtOАс (100 мл). Органические слои промывают насыщенным раствором NaCl (2×50 мл), сушат (MgSO4) и концентрируют в вакууме. Твердый остаток очищают MPLC (50% EtOAc/50% гексан), при этом получают требуемый амин (0,650 г, 38%).
А13k. Общий метод синтеза замещенных анилинов путем получения нитроароматических соединений с использованием нуклеофильного ароматического замещения с последующим восстановлением
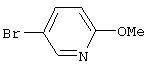
Стадия 1. 5-Бром-2-метоксипиридин: Смесь 2,5-дибромпиридина (5,5 г, 23,2 ммоль) и NaOMe (3,76 г, 69,6 ммоль) в МеОН (60 мл) нагревают при 70°С в герметически закрытом реакционном сосуде в течение 42 ч, затем охлаждают до комнатной температуры. Реакционную смесь обрабатывают водой (50 мл) и экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои сушат (Na2SO4) и концентрируют при пониженном давлении, при этом получают летучее масло светло-желтого цвета (4,1 г, 95%). ТСХ (10% ЕtOАс/90% гексан) Rf 0,57.
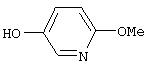
Стадия 2. 5-Гидрокси-2-метоксипиридин: К раствору 5-бром-2-метоксипиридина (8,9 г, 47,9 ммоль) в ТГФ (175 мл) при -78°С при перемешивании по каплям добавляют раствор N-бутиллития (2,5 М в гексане; 28,7 мл, 71,8 ммоль) и полученную смесь перемешивают при -78°С в течение 45 мин. Триметилборат (7,06 мл, 62,2 ммоль) добавляют с помощью шприца и полученную смесь дополнительно перемешивают в течение 2 ч. Реакционную смесь ярко-оранжевого цвета нагревают до 0°С и обрабатывают смесью 3 н. раствора NaOH (25 мл, 71,77 ммоль) и раствора пероксида водорода (30%; прибл. 50 мл). Полученную слегка мутную реакционную смесь желтого цвета нагревают до комнатной температуры в течение 30 мин и затем кипятят с обратным холодильником в течение 1 ч. Реакционную смесь охлаждают до комнатной температуры. Водный слой нейтрализуют 1н. раствором HCl, затем экстрагируют Et2O (2×100 мл). Объединенные органические слои сушат (Na2SO4) и концентрируют при пониженном давлении, при этом получают вязкое масло желтого цвета (3,5 г, 60%).
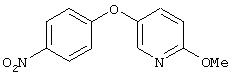
Стадия 3. 4-(5-(2-Метокси)пиридил)окси-1-нитробензол: К суспензии NaH (97%, 1,0 г, 42 ммоль) в безводном ДМФ (100 мл) при перемешивании добавляют раствор 5-гидрокси-2-метоксипиридина (3,5 г, 28 ммоль) в ДМФ (100 мл). Полученную смесь перемешивают при комнатной температуре в течение 1 ч, затем шприцом добавляют 4-фторнитробензол (3 мл, 28 ммоль). Реакционную смесь нагревают при 95°С в течение ночи, затем обрабатывают водой (25 мл) и экстрагируют ЕtOАс (2×75 мл). Органический слой сушат (MgSO4) и концентрируют при пониженном давлении. Маслянистый остаток коричневого цвета кристаллизуют (ЕtOАс/гексан), при этом получают кристаллы желтого цвета (5,23 г, 75%)
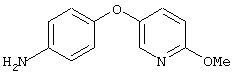
Стадия 4. 4-(5-(2-Метокси)пиридил)оксианилин: 4-(5-(2-метокси)-пиридил)окси-1-нитробензол восстанавливают до анилина аналогичным способом, как описано в методе B3d, стадия 2.
А14а. Общий метод синтеза замещенных анилинов путем нуклеофильного ароматического замещения с использованием галогенпиридина
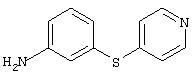
3-(4-пиридинилтио)анилин: К раствору 3-аминотиофенола (3,8 мл, 34 ммоль) в безводном ДМФ (90 мл) добавляют гидрохлорид 4-хлорпиридина (5,4 г, 35,6 ммоль), затем добавляют К2СО3 (16,7 г, 121 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 1,5 ч, затем разбавляют ЕtOАс (100 мл) и водой (100 мл). Водный слой экстрагируют ЕtOАс (2×100 мл). Объединенные органические слои промывают насыщенным раствором NaCl (100 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Остаток фильтруют через слой силикагеля (градиент от 50% ЕtOАс/50% гексан до 70% ЕtOАс/30% гексан) и полученное вещество растирают с раствором Еt2О/гексан, при этом получают требуемый продукт (4,6 г, 66%): ТСХ (100% этилацетат) Rf 0,29;1H-ЯМР (ДМСО-d6) δ 5,41 (s, 2H), 6,64-6,74 (m, 3Н), 7,01 (d, J=4,8 Гц, 2H), 7,14 (t, J=7,8 Гц, 1Н), 8,32 (d, J=4,8 Гц, 2Н).
A14b. Общий метод синтеза замещенных анилинов путем нуклеофильного ароматического замещения с использованием галогенпиридина
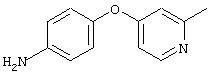
4-(2-Метил-4-пиридинилокси)анилин: К раствору 4-аминофенола (3,6 г, 32,8 ммоль) и 4-хлорпиколина (5,0 г, 39,3 ммоль) в безводном DMPU (50 мл) добавляют трет-бутоксид калия (7,4 г, 65,6 ммоль) одной порцией. Реакционную смесь нагревают при 100°С при перемешивании в течение 18 ч, затем охлаждают до комнатной температуры. Полученную смесь выливают в воду (200 мл) и экстрагируют ЕtOАс (3×150 мл). Объединенные экстракты последовательно промывают водой (3×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (Nа2SO4) и концентрируют в вакууме. Полученное масло очищают экспресс-хроматографией (50% ЕtOАс/50% гексан), при этом получают требуемый продукт в виде масла желтого цвета (0,7 г, 9%): CI-MS m/z 201 ((М+Н)+).
А14с. Общий метод синтеза замещенных анилинов путем нуклеофильного ароматического замещения с использованием галогенпиридина
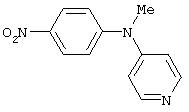
Стадия 1. Метил(4-нитрофенил)-4-пиридиламин: К суспензии N-метил-4-нитроанилина (2,0 г, 13,2 ммоль) и К2СО3 (7,2 г, 52,2 ммоль) в DMPU (30 мл) добавляют гидрохлорид 4-хлорпиридина (2,36 г, 15,77 ммоль). Реакционную смесь нагревают при 90°С в течение 20 ч, затем охлаждают до комнатной температуры. Полученную смесь разбавляют водой (100 мл), экстрагируют ЕtOАс (100 мл). Органический слой промывают водой (100 мл), сушат (Nа2SO4) и концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (силикагель; градиент от 80% ЕtOАс/20% гексан до 100% ЕtOАс), при этом получают метил(4-нитрофенил)-4-пиридиламин (0,42 г).
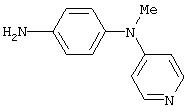
Стадия 2. Метил(4-аминофенил)-4-пиридиламин: Метил(4-нитрофенил)-4-пиридиламин восстанавливают аналогичным способом, как описано в методе В1.
А15. Общий метод синтеза замещенных анилинов путем алкилирования фенолов с последующим восстановлением нитроароматических соединений.
Стадия 1: 4-(4-Бутоксифенил)тио-1-нитробензол: К раствору 4-(4-нитрофенилтио)фенола (1,50 г, 6,07 ммоль) в безводном ДМФ (75 мл) добавляют при 0°С NaH (60% в минеральном масле, 0,267 г, 6,67 ммоль). Полученную суспензию коричневого цвета перемешивают при 0°С до прекращения выделения газа (15 мин), затем по каплям добавляют раствор иодбутана (1,12 г, 0,690 мл, 6,07 ммоль) в безводном ДМФ (20 мл) в течение 15 мин при 0°С. Реакционную смесь перемешивают при комнатной температуре в течение 18 ч, после чего данные ТСХ показывают присутствие непрореагировавшего фенола. Затем добавляют дополнительное количество изобутана (56 мг, 0,035 мл, 0,303 ммоль, 0,05 эквивалента) и NaH (13 мг, 0,334 ммоль). Реакционную смесь дополнительно перемешивают в течение 6 ч при комнатной температуре, затем реакцию останавливают добавлением воды (400 мл). Полученную смесь экстрагируют Et2O (2×500 мл). Объединенные органические экстракты промывают водой (2×400 мл), сушат (MgSO4) и концентрируют при пониженном давлении, при этом получают прозрачное масло желтого цвета, которое очищают хроматографически на колонке с силикагелем (градиент от 20% EtOAc/80% гексан до 50% EtOAc/50% гексан), при этом получают требуемый продукт в виде твердого вещества желтого цвета (1,24 г, 67%): ТСХ (20% EtOAc/80% гексан) Rf 0,75;1H-ЯМР (ДМСО-d6) δ 0,92 (t, J=7,5 Гц, 3Н), 1,42 (арр. hex, J=7,5 Гц, 2Н), 1,70 (m, 2H), 4,01 (t, J=6,6 Гц, 2H), 7,08 (d, J=8,7 Гц, 2H), 7,17 (d, J=9 Гц, 2Н), 7,51 (d, J=8, 7 Гц, 2H), 8,09 (d, J=9 Гц, 2Н).
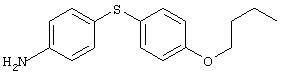
Стадия 2. 4-(4-Бутоксифенил)тиоанилин: 4-(4-Бутоксифенил)тио-1-нитробензол восстанавливают до анилина аналогичным способом, как описано для синтеза 3-(трифторметил)-4-(4-пиридинилтио)анилина (Метод В3b, стадия 2): ТСХ (33% ЕtOАс/77% гексан) Rf 0,38.
A16. Общий метод синтеза замещенных анилинов путем ацилирования диаминоароматических соединений
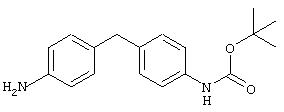
4-(4-трет-Бутоксикарбамоилбензил)анилин: К раствору 4,4'-метилендианилина (3,00 г, 15,1 ммоль) в безводном ТГФ (50 мл) добавляют при комнатной температуре раствор ди-трет-бутилдикарбоната (3,30 г, 15,1 ммоль) в безводном ТГФ (10 мл). Реакционную смесь кипятят с обратным холодильником в течение 3 ч, после чего данные ТСХ показывают присутствие непрореагировавшего метилендианилина. Затем добавляют дополнительное количество ди-трет-бутилдикарбоната (0,664 г, 3,03 ммоль, 0,02 эквивалента) и реакционную смесь дополнительно кипятят с обратным холодильником в течение 16 ч. Полученную смесь разбавляют Et2O (200 мл), последовательно промывают насыщенным раствором NаНСО3 (100 мл), водой (100 мл) и насыщенным раствором NaCl (50 мл), сушат (МgSO4) и концентрируют при пониженном давлении. Полученное твердое вещество белого цвета очищают хроматографически на колонке с силикагелем (градиент от 33% ЕtOАс/67% гексан до 50% ЕtOАс/50% гексан), при этом получают требуемый продукт в виде твердого вещества белого цвета (2,09 г, 46%): ТСХ (50% ЕtOАс/50% гексан) Rf 0,45;1H-ЯМР (ДМСО-d6) δ 1,43 (s, 9Н), 3,63 (s, 2H), 4,85 (ушир. s, 2Н), 6,44 (d, J=8,4 Гц, 2H), 6,80 (d, J=8,1 Гц, 2Н), 7,00 (d, J=8,4 Гц, 2Н), 7,28 (d, J=8,1 Гц, 2H), 9,18 (ушир. s, 1H). FAB-MS m/z 298 (M+).
A17. Общий метод синтеза ариламинов путем электрофильного нитрования с последующим восстановлением
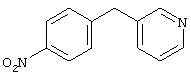
Стадия 1. 3-(4-нитробензил)пиридин: Раствор 3-бензилпиридина (4,0 г, 23,6 ммоль) и 70% азотной кислоты (30 мл) нагревают в течение ночи при 50°С. Полученную смесь охлаждают до комнатной температуры, затем выливают в ледяную воду (350 мл). Водную смесь подщелачивают 1н. раствором NaOH, затем экстрагируют Еt2О (4×100 мл). Объединенные экстракты последовательно промывают водой (3×100 мл) и насыщенным раствором NaCl (2×100 мл), сушат (Nа2SO4) и концентрируют в вакууме. Полученное масло очищают MPLC (силикагель; 50% ЕtOАс/50% гексан), затем перекристаллизовывают (ЕtOАс/гексан), при этом получают требуемый продукт (1,0 г, 22%). GC-MS m/z 214 (М+ ).
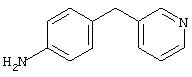
Стадия 2. 3-(4-пиридинил)метиланилин: 3-(4-нитробензил)пиридин восстанавливают до анилина аналогичным способом, как описано в методе В1.
A18. Общий метод синтеза ариламинов путем замещения с использованием нитробензилгалогенидов с последующим восстановлением
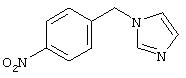
Стадия 1. 4-(1-имидазолилметил)-1-нитробензол: К раствору имидазола (0,5 г, 7,3 ммоль) и бромида 4-нитробензила (1,6 г, 7,3 ммоль) в безводном ацетонитриле (30 мл) добавляют К2СО3 (1,0 г, 7,3 ммоль). Полученную смесь перемешивают при комнатной температуре в течение 18 ч и затем выливают в воду (200 мл). Полученный водный раствор экстрагируют ЕtOАс (3×50 мл). Объединенные органические слои последовательно промывают водой (3×50 мл) и насыщенным раствором NaCl (2×50 мл), сушат (MgSO4) и концентрируют в вакууме. Полученное масло очищают MPLC (силикагель; 25% ЕtOАс/75% гексан), при этом получают требуемый продукт (1,0 г, 91%): EI-MS m/z 203 (M+).
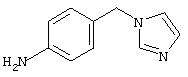
Стадия 2. 4-(1-имидазолилметил)анилин: 4-(1-имидазолилметил)-1-нитробензол восстанавливают до анилина аналогичным способом, как описано в методе В2.
A19. Синтез замещенных гидроксиметиланилинов путем окисления соединений нитробензила с последующим восстановлением
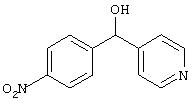
Стадия 1. 4-(1-Гидрокси-1-(4-пиридил)метил)-1-нитробензол: К раствору 3-(4-нитробензил)пиридина (6,0 г, 28 ммоль) в CH2Cl2 (90 мл) при перемешивании при 10°С добавляют m-СРВА (5, 80 г, 33,6 ммоль) и полученную смесь перемешивают при комнатной температуре в течение ночи. Реакционную смесь последовательно промывают 10% раствором NаНSО3 (50 мл), насыщенным раствором К2СО3 (50 мл) и насыщенным раствором NaCl (50 мл), сушат (MgSO4) и концентрируют при пониженном давлении. Полученное твердое вещество желтого цвета (2,68 г) растворяют в безводном уксусном ангидриде (30 мл) и кипятят с обратным холодильником в течение ночи. Полученную смесь концентрируют при пониженном давлении. Остаток растворяют в МеОН (25 мл) и обрабатывают 20% водным раствором NН3 (30 мл). Смесь перемешивают при комнатной температуре в течение 1 ч, затем концентрируют при пониженном давлении. Остаток выливают в смесь воды (50 мл) и CH2 Cl2 (50 мл). Органический слой сушат (MgSO4), концентрируют при пониженном давлении и очищают колочной хроматографией (80% ЕtOАс/20% гексан), при этом получают требуемый продукт в виде твердого вещества белого цвета (0,53 г, 8%): т.пл. 110-118°С; ТСХ (80% этилацетат/20% гексан) Rf 0,12; FAB-MS m/z 367 ((М+Н)+; 100%).
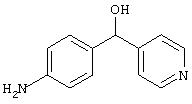
Стадия 2. 4-(1-Гидрокси-1-(4-пиридил)метил)анилин: 4-(1-гидрокси-1-(4-пиридил)метил)-1-нитробензол восстанавливают до анилина аналогичным способом, как описано в методе B3d, стадия 2.
А20. Синтез 2-(N-метилкарбамоил)пиридинов с использованием реакции Менисци (Menisci)
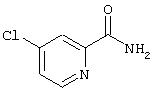
Стадия 1. 2-(N-метилкарбамоил)-4-хлорпиридин. (Внимание: взрывоопасная реакция, возможен взрыв). К раствору 4-хлорпиридина (10 г) в N-метилформамиде (250 мл) в атмосфере аргона при температуре окружающей среды добавляют конц. H2SO4 (3,55 мл) (экзотермичная реакция). К полученному раствору добавляют Н2О2 (17 мл), 30 мас.% в Н2О, затем FеSO4·7Н2О (0,55 г), при этом происходит экзотермическая реакция. Реакционную смесь перемешивают в темноте при температуре окружающей среды в течение 1 ч, затем медленно нагревают в течение 4 ч при 45°С. После окончания выделения пузырьков газа реакционную смесь нагревают при 60°С в течение 16 ч. Непрозрачный раствор коричневого цвета разбавляют Н2О (700 мл), затем 10% раствором NaOH (250 мл). Водную смесь экстрагируют ЕtOАс (3×500 мл) и органические слои промывают отдельно насыщенным раствором NaCl (3×150 мл). Объединенные органические экстракты сушат (MgSO4) и фильтруют через слой силикагеля, с использованием ЕtOАс в качестве элюента. Растворитель удаляют в вакууме и остаток коричневого цвета очищают хроматографически на колонке с силикагелем (градиент от 50% ЕtOАс/50% гексан до 80% EtOAc/20% гексан). Полученное масло желтого цвета кристаллизуют при 0°С в течение 72 ч, при этом получают 2-(N-метилкарбамоил)-4-хлорпиридин (0,61 г, 5,3%): ТСХ (50% ЕtOАс/50% гексан) Rf 0,50;1H-ЯМР (CDCl3) δ 8,44 (d, J=5,1 Гц, 1Н, CHN), 8,21 (s, 1H, СНССО), 7,96 (ушир. s, 1H, NH), 7,43 (dd, J=2,4, 5,4 Гц, 1Н, CICHCN), 3,04 (d, J=5,1 Гц, 3Н, метил); CI-MS m/z 171 ((М+Н)+).
A21. Общий метод синтеза ω-сульфонилфениланилинов
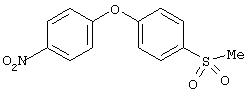
Стадия 1. 4-(4-Метилсульфонилфенокси)-1-нитробензол: К раствору 4-(4-метилтиофенокси)-1-нитробензола (2 г, 7,66 ммоль) в CH2Cl2 (75 мл) медленно добавляют при 0°С m-СРВА (57-86%, 4 г) и реакционную смесь перемешивают при комнатной температуре в течение 5 ч. Реакционную смесь обрабатывают 1н. раствором NaOH (25 мл). Органический слой последовательно промывают 1н. раствором NaOH (25 мл), водой (25 мл) и насыщенным раствором NaCl (25 мл), сушат (MgSO4) и концентрируют при пониженном давлении, при этом получают 4-(4-метилфенил-сульфонилфенокси)-1-нитробензол в виде твердого вещества (2,1 г).
Стадия 2. 4-(4-Метилсульфонилфенокси)-1-анилин:
4-(4-Метилфенилсульфонилфенокси)-1-нитробензол восстанавливают до анилина аналогичным способом, как описано в методе B3d, стадия 2.
А22. Общий метод синтеза ω-алкокси-ω -карбоксифениланилинов
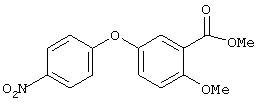
Стадия 1. 4-(3-Метоксикарбонил-4-метоксифенокси)-1-нитробензол:
К раствору 4-(3-карбокси-4-гидроксифенокси)-1-нитробензола (приготовленного аналогичным способом, как описано в методе В3а, стадия 1, 12 ммоль) в ацетоне (50 мл) добавляют К2СО3 (5 г) и диметилсульфат (3,5 мл). Полученную смесь кипятят с обратным холодильником в течение ночи, затем охлаждают до комнатной температуры и фильтруют через слой целита. Полученный раствор концентрируют при пониженном давлении, адсорбируют на силикагеле и очищают колоночной хроматографией (50% ЕtOАс/50% гексан), при этом получают 4-(3-метоксикарбонил-4-метоксифенокси)-1-нитробензол в виде порошка желтого цвета (3 г): т.пл. 115-118°С.
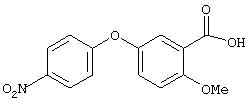
Стадия 2. 4-(3-Карбокси-4-метоксифенокси)-1-нитробензол: Смесь 4-(3-метоксикарбонил-4-метоксифенокси)-1-нитробензола (1,2 г), КОН (0,33 г) и воды (5 мл) в МеОН (45 мл) перемешивают при комнатной температуре в течение ночи и затем кипятят с обратным холодильником в течение 4 ч.
Полученную смесь охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток растворяют в воде (50 мл) и водную смесь подкисляют 1н. раствором HCl. Полученную смесь экстрагируют ЕtOАс (50 мл). Органический слой сушат (МgSO4) и концентрируют при пониженном давлении, при этом получают 4-(3-карбокси-4-метоксифенокси)-1-нитробензол (1,04 г).
В. Основные методы получения производных мочевины
В1а. Основной метод проведения реакции ариламина с арилизоцианатом
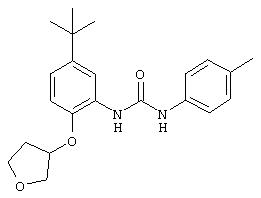
N-(5-трет-Бутил-2-(3-тетрагидрофуранилокси)фенил)-N'-(4-метилфенил) мочевина: К раствору 5-трет-бутил-2-(3-тетрагидрофуранилокси)анилина (0,078 г, 0,33 ммоль) в толуоле (2,0 мл) добавляют п-толилизоцианат (0,048 г, 0,36 ммоль), полученную смесь перемешивают при коматной температуре в течение 8 ч и получают осадок. Реакционную смесь отфильтровывают и остаток последовательно промывают толуолом и гексаном, при этом получают требуемую мочевину в виде твердого вещества белого цвета (0,091 г, 75%): т.пл. 229-231°С;1H ЯМР (DMSO-d6) δ 1,30 (s, 9H), 1, 99-2,03 (m, 1H), 2,19-2,23 (m, 4H), 3,69-3,76 (m, 1Н), 3,86-3,93 (m, 3H), 4,98-5,01 (m, 1H), 6,81-6,90 (m, 2H), 7,06 (d, J=8,09 Гц, 2Н), 7,32 (d, J=8,09 Гц, 2H), 7,84 (s, 1H), 8,22 (d, J=2,21 Гц, 1H), 9,26 (s, 1H).
B1b. Общий метод проведения реакции ариламина с арилизоцианатом
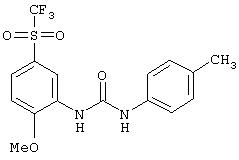
N-(2-Метокси-5(трифторметансульфонил)фенил)-N'-(4-метилфенил)мочевина: К раствору 2-метокси-5-(трифторметансульфонил)анилина (0,330 г, 1,29 ммоль) в ЕtOАс (5 мл) добавляют п-толилизоцианат (0,19 мл, 1,55 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 18 ч. Полученный осадок собирают фильтрованием и промывают Et2O, при этом получают твердое вещество белого цвета (0,28 г). Затем этот материал очищают ВЭЖХ (колонка С-18, 50% СН3СN/50% H2O) и полученное твердое вещество растирают с Et2O, при этом получают конечный продукт (0,198 г):1H ЯМР (СDСl3) δ 7,08 (d, J=8,5 Гц, 2H), 7,33 (d, J=8,5 Гц, 2H), 7,40 (d, J=8,8 Гц, 1H), 7,71 (dd, J=2,6; 8,8 Гц, 1H), 8,66 (s, 1H), 8,90 (d, J=2,6 Гц, 1H), 9,36 (s, 1H); FAB-MS m/z 389 ((M+1)+).
В1с. Общий метод проведения реакции ариламина с арилизоцианатом
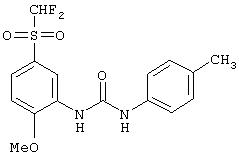
N-(2-Метокси-5-(дифторметансульфонил)фенил)-N'-(4-метилфенил)мочевина: п-Толил изоцианат (0,058 мл, 0,46 ммоль) добавляют к раствору 2-метокси-5-(дифторметансульфонил)анилина (0,100 г, 0,42 ммоль) в ЕtOАс (0,5 мл) и реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Полученный осадок отфильтровывают и промывают Et2O, при этом получают конечный продукт в виде твердого вещества белого цвета (0,092 г):1H ЯМР (CDCl3) δ 2,22 (s, 3H), 4,01 (s, 3H), 7,02-7,36 (m, 6H), 7,54 (dd, J=2,4; 8,6 Гц, 1Н), 8,57 (s, 1H), 8,79 (d, J=2,6 Гц, 1H), 9,33 (s, 1H); EI-MS m/z 370 (M+).
B1d. Общий метод проведения реакции ариламина с арилизоцианатом
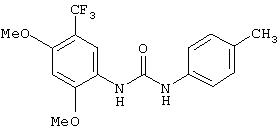
N-(2, 4-Диметокси-5-(трифторметил)фенил)-N'-(4-метилфенил)мочевина: п-Толилизоцианат (0,16 мл, 1,24 ммоль) добавляют к раствору 2,4-диметокси-5-(трифторметил)анилина (0,25 г, 1,13 ммоль) в EtOAc (3 мл) и полученную смесь перемешивают при комнатной температуре в течение 18 ч. Полученный осадок промывают Et2O, при этом получают конечный продукт в виде твердого вещества белого цвета (0,36 г):1H ЯМР (CDCl3) δ 2,21 (s, 3H), 3,97 (s, 3Н), 3,86 (s, 3H), 6,88 (s, 1H), 7,05 (d, J=8,5 Гц, 2H), 7,29 (d, J=8,5 Гц, 2Н), 8,13 (s, 1H), 8,33 (s, 1H), 9,09 (s, 1H); FAB-MS m/z 355 ((M+1)+).
B1e. Общий метод проведения реакции ариламина с арилизоцианатом
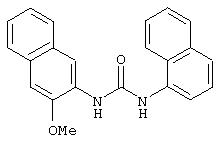
N-(3-Метокси-2-нафтил)-N'-(1-нафтил)мочевина: К раствору 2-амино-3-метоксинафталина (0,253 г, 1,50 ммоль) в СН2Сl2 (3 мл) при комнатной температуре добавляют раствор 1-нафтилизоцианата (0,247 г, 1,50 ммоль) в CH2Cl2 (2 мл) и полученную смесь перемешивают в течение ночи. Полученный осадок отделяют и промывают CH2Cl2, при этом получают требуемую мочевину в виде порошка белого цвета (0,450 г, 90%): т.пл. 235-236°С;1H ЯМР (DMSO-d6) δ 4,04 (s, 3H), 7,28-7,32 (m, 2H), 7,38 (s, 1H), 7,44-7,72 (m, 6H), 7,90-7,93 (m, 1H), 8,05-8,08 (m, 1H), 8,21-8,24 (m, 1H), 8,64 (s, 1H), 9,03 (s, 1H), 9,44 (s, 1H); FAB-MS m/z 343 ((M+H)+).
B1f. Общий метод проведения реакции ариламина с арилизоцианатом
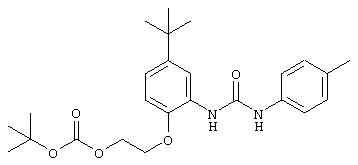
N-(5-трет-Бутил-2(2-трет-бутоксикарбонилокси)этокси)фенил)-N'-(4-метилфенил)мочевина: Смесь 5-трет-бутил-2-(2-трет-бутоксикарбонилокси)этокси)анилина (Метод А10, 0,232 г, 0,75 ммоль) и п-толилизоцианата (0,099 мл, 0,79 ммоль) в ЕtOАс (1 мл) перемешивают при комнатной температуре в течение 3 дней, при этом получают твердое вещество, которое отделяют. Фильтрат очищают колоночной хроматографией (100% CH2Cl2) и осадок растирают (Et2O/гексан), при этом получают требуемый продукт (0,262 г, 79%): т.пл. 155-156°С; ТСХ (20% ЕtOАс/80% гексан), Rf 0,49;1H ЯМР (DMSO-d6) δ 1,22 (s, 9H), 1,37 (s, 9H), 2,21 (s, 3H), 4,22-4,23 (m, 2Н), 4,33-4,35 (m, 2H), 6,89-7,00 (m, 4Н), 7,06 (d, J=8,5 Гц, 2Н), 7,32 (d, J=8,1 Гц, 2H), 7,96 (s, 1Н), 8,22 (d, J=1,5 Гц, 1Н), 9,22 (s, 1H); FAB-MS m/z (отн. масса) 443 ((М+Н)+, 6%).
В2а. Общая методика (проведения) реакции ариламина с фосгеном с последующим добавлением второго арил амина
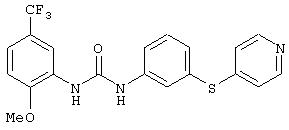
N-(2-Метокси-5-(трифторметил)фенил)-N'-(3-(4-пиридинилтио)фенил)мочевина: К раствору пиридина (0,61 мл, 7,5 ммоль, 3,0 экв) и фосгена (20% в толуоле; 2,65 мл, 5,0 ммоль, 2 экв) в СН2Сl2 (20 мл) при 0°С добавляют 2-метокси-5-(трифторметил)анилин (0,48 г, 2,5 ммоль). Полученую смесь нагревают до комнатной температуры, перемешивают в течение 3 ч, затем добавляют безводный толуол (100 мл) и концентрируют при пониженном давлении. Остаток суспендируют в смеси CH2Cl2 (10 мл) и безводного пиридина (10 мл), добавляют 3-(4-пиридинилтио)анилин (0,61 г, 2, 5 ммоль, 1,0 экв). Смесь перемешивают при комнатной температуре в течение ночи, затем выливают в воду и экстрагируют CH2Cl2 (3×25 мл). Объединенные органические слои сушат (МgSO4) и концентрируют при пониженном давлении. Остаток растворяют в минимальном количестве CH2Cl2 и обрабатывают петролейным эфиром, при этом получают требуемый продукт в виде твердого вещества белого цвета (0,74 г, 70%): т.пл. 202°С; ТСХ (5% ацетон/95% СН2Сl2) Rf 0,09;1H ЯМР (DMSO-d6) δ 7, 06 (d, J=5,5 Гц, 2Н), 7,18 (dd, J=2,4; 4,6 Гц, 2Н), 7,31 (dd, J=2,2; 9,2 Гц, 1Н), 7,44 (d, J=5,7 Гц, 1Н), 7,45 (s, 1Н), 7,79 (d, J=2,2 Гц, 1H), 8,37 (s, 2Н), 8,50 (dd, J=2,2; 9,2 Гц, 2Н), 9,63 (s, 1H), 9,84 (s, 1H); FAB-MS m/z 420 ((M+H)+, 70%).
В2b. Общий метод проведения реакции ариламина с фосгеном с последующим добавлением второго ариламина
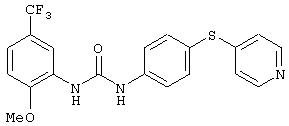
N-(2-Метокси-5-(трифторметил)фенил)-N'-(4-(4-пиридинилтио)фенил)мочевина: К раствору пиридина (0,61 мл, 7,5 ммоль, 3,0 экв) и фосгена (20% в толуоле; 2,65 мл, 5,0 ммоль, 2,0 экв) в CH2Cl2 (20 мл) при 0°С добавляют 4-(4-пиридинилтио)анилин (0,506 г, 2,5 ммоль). После перемешивания в течение 3 ч при комнатной температуре к смеси добавляют безводный толуол (100 мл), затем концентрируют при пониженном давлении. Остаток суспендируют в смеси CH2Cl2 (10 мл) и безводного пиридина (10 мл) и добавляют 2-метокси-5-(трифторметил)анилин (0,50 г, 2,5 ммоль, 1,0 экв). Смесь перемешивают при комнатной температуре в течение ночи, затем выливают в 1н. раствор NaOH (50 мл) и экстрагируют CH2Cl2 (3×25 мл). Объединенные органические слои сушат (MgSO4) и концентрируют при пониженном давлении, при этом получают требуемую мочевину (0,74 г, 71%): т.пл. 215°С; ТСХ (5% ацетон/95% CH2Cl2), Rf 0,08;1H ЯМР (DMSO-d6) δ 3,96 (s, 3Н), 6,94 (dd, J=1, 1; 4,8 Гц, 2Н), 7,19 (d, J=8,4 Гц, 1Н), 7,32 (dd, J=2,2; 9,3 Гц, 1Н), 7,50 (d, J=8,8 Гц, 2Н), 7,62 (d, J=8,8 Гц, 2Н), 8,32 (d, J=5,1 Гц, 2Н), 8,53 (d, J=0,7 Гц, 1Н), 8,58 (s, 1Н), 9,70 (s, 1Н); FAB-MS m/z 420 ((М+Н)+).
В3а. Общий метод проведения реакции ариламина с фосгеном с выделением изоцианата и последующей реакцией со вторым ариламином
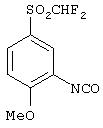
Стадия 1. 5-(Дифторметансульфонил)-2-метоксифенилизоцианат: К раствору фосгена (1,95 М в толуоле; 3, 0 мл, 5,9 ммоль) в CH2Cl2 (40 мл) при 0°С по каплям добавляют раствор 5-(дифторметансульфонил)-2-метоксианилина (0,70 г, 2,95 ммоль) и пиридина (0,44 мл, 8,85 ммоль) в CH2Cl2 (10 мл). После перемешивания при 0°С в течение 30 мин и при комнатной температуре в течение 3 ч реакционную смесь концентрируют при пониженном давлении, а затем добавляют толуол. Полученную смесь концентрируют при пониженном давлении, затем добавляют Еt2O (50 мл), при этом образуется осадок (гидрохлорид пиридиния). Полученный фильтрат концентрируют при пониженном давлении, при этом получают конечный продукт в виде твердого вещества белого цвета (0,33 г). Этот материал используют на следующей стадии без дальнейшей очистки.
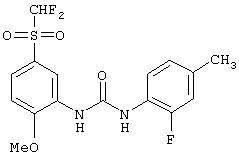
Стадия 2. N-(2-Метокси-5-(дифторметансульфонил)фенил)-N'-(2-фтор-4-метилфенил)мочевина: К раствору 5-(дифторметансульфонил)-2-метоксифенилизоцианата (0,046 г, 0,17 ммоль) в ЕtOАс (1 мл) добавляют 2-фтор-4-метиланилин (0,022 мл, 0,19 ммоль). Реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Полученный осадок промывают Et2O, при этом получают конечный продукт в виде твердого вещества белого цвета (0,055 г).1H ЯМР (CDCl3) δ 2,24 (s, 3H), 4,01 (s, 3Н), 6,93 (d, J=8,5 Гц, 1Н), 7,01-7,36 (m, 3Н), 7,56 (dd, J=2,4; 8,6 Гц, 1Н), 7,98 (арр t, J=8,6 Гц, 1H), 8,79 (d, J=2,2 Гц, 1Н), 9,07 (s, 1Н), 9,26 (s, 1H); FAB-MS m/z 389 ((M+1)+).
В3b. Общий метод проведения реакции ариламина с фосгеном с выделением изоцианата и последующей реакцией со вторым ариламином
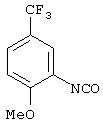
Стадия 1. 2-Метокси-5-трифторметилфенилизоцианат: К раствору фосгена (1,93 М в толуоле; 16 мл, 31,4 ммоль) в CH2 Cl2 (120 мл) при 0°С по каплям добавляют раствор 2-метокси-5-(трифторметил)анилина (3,0 г, 15,7 ммоль) и пиридина (2,3 мл, 47,1 ммоль) в CH2Cl2 (30 мл). Полученную смесь перемешивают при 0°С в течение 30 мин и при комнатной температуре в течение 3 ч, затем концентрируют при пониженном давлении. Остаток разбавляют в толуоле (30 мл), концентрируют при пониженном давлении и добавляют Et2O. Полученный осадок (гидрохлорид пиридиния) удаляют, а фильтрат концентрируют при пониженном давлении, при этом получают конечный продукт в виде масла желтого цвета (3,0 г), которое кристаллизуется при комнатной температуре в течение нескольких дней.
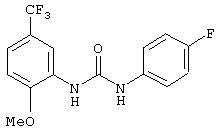
Стадия 2. N-(2-Метокси-5-(трифторметил)фенил)-N'-(4-фторфенил)мочевина: К раствору 2-метокси-5-(трифторметил)фенил-изоцианата (0,50 г, 2,30 ммоль) в ЕtOАс (6 мл) добавляют 4-фторанилин (0,24 мл, 2,53 ммоль) и реакционную смесь перемешивают при комнатной температуре в течение 3 дней. Полученный осадок промывают Et2O, при этом получают конечный продукт в виде твердого вещества белого цвета (0,60 г): ЯМР: 3,94 (s, 3H), 7,13-7,18 (m, 3Н), 7,30 (dd, J=1,5; 8,4Гц, 1H), 7,44 (m, 2H), 8,45 (s, 1H), 8,52 (d, J=2,2 Гц, 1H), 9,42 (s, 1H); FAB-MS m/z 329 ((M+1)+).
B4. Общий метод синтеза производных мочевины с использованием перегруппировки Курциуса с последующим захватом с амином
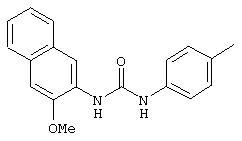
N-(3-Метокси-2-нафтил)-N'-(4-метилфенил)мочевина: К раствору 3-метокси-2-нафтойной кислоты (метод А6, стадия 2; 0,762 г, 3,80 ммоль) и Et3N (0,588 мл, 4,2 ммоль) в безводном толуоле (20 мл) при комнатной температуре добавляют раствор дифенилфосфорилазида (1,16 г, 4,2 ммоль) в толуоле (5 мл). Полученную смесь нагревают до 80°С в течение 2 ч, охлаждают до комнатной температуры и добавляют п-толуидин (0,455 г, 4,1 ммоль). Смесь нагревают при температуре 80°С, охлаждают до комнатной температуры, реакцию останавливают добавлением 10%-ного раствора лимонной кислоты, экстрагируют ЕtOАс (2×25 мл). Объединенные органические слои промывают насыщенным раствором NaCl (25 мл), сушат (МgSO4) и концентрируют в вакууме. Остаток растирают с CH2Cl2, при этом получают требуемую мочевину в виде порошка белого цвета (0,700 г, 61%): т.пл. 171-172°С;1H ЯМР (DMSO-d6) δ 2,22 (s, ЗН), 3,99 (s, 3H), 7,07 (d, J=8,49 Гц, 2Н), 7,27-7,36 (m, 5H), 7,67-7,72 (m, 2Н), 8,43 (s, 1H), 8,57 (s, 1H), 9,33 (s, 1H); FAB-MS m/z 307 ((M+H)+).
B5. Общий метод проведения реакции замещенного анилина с N,N'-карбонилдиимидазолом с последующей реакцией со вторым амином
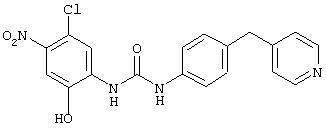
N-(5-Хлор-2-гидрокси-4-нитрофенил)-N'-(4-(4-
пиридинилметил)фенил)мочевина: Раствор 4-(4-пиридинилметил)анилина (0,300 г, 1,63 ммоль) и N,N'-карбонилдиимидазола (0,268 г, 1,65 ммоль) в CH2Cl2 (10 мл) перемешивают при комнатной температуре в течение 1 ч, после чего данные ТСХ показывают отсутствие исходного анилина. Затем в реакционную смесь добавляют 2-амино-4-хлор-5-нитрофенол (0,318 г, 1,65 ммоль) и перемешивают при температуре 40-45°С в течение 48 ч. Полученную смесь охлаждают до комнатной температуры и разбавляют ЕtOАс (25 мл). Полученный осадок отделяют, при этом получают требуемый продукт (0,416 г, 64%): ТСХ (50% ацетон/50% CH2 Cl2), Rf 0,40;1H ЯМР (DMSO-d6) δ 3,90 (s, 2Н), 7,18 (d, J=8,4 Гц, 2H), 7,21 (d, J=6 Гц, 2Н), 7,38 (d, J=8,4 Гц, 2H), 7,54 (s, 1Н), 8,43-8,45 (m, 3Н), 8,78 (s, 1H), 9,56 (s, 1H), 11,8 (ушир. s, 1H); FAB-MS m/z (отн. масса) 399 ((М+Н)+, 10%).
В6. Общий метод синтеза симметричных дифенил-производных мочевины как побочных продуктов реакций образования мочевины
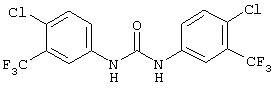
Бис(4-хлоро-3-(трифторметил)фенил)мочевина: К раствору 5-амино-3-трет-бутилизоксазола (0,100 г) в безводном толуоле (5 мл) добавляют 4-хлор-3-(трифторметил)фенилизоцианат (0,395 г). Реакционный сосуд герметично закрывают, нагревают при 85°С в течение 24 ч и охлаждают до комнатной температуры. Реакционную смесь добавляют к суспензии смолы Dowex® 50WX2-100 (0,5 г) в CH2Cl2 (40 мл) и полученную смесь интенсивно перемешивают в течение 72 ч. Смесь фильтруют и фильтрат концентрируют при пониженном давлении. Остаток очищают колоночной хроматографией (градиент от 100% СН2Сl2до 5% МеОН/95% CH2Cl2), при этом получают бис(4-хлор-3-(трифторметил)фенил)мочевину, а затем элюируют N-(3-трет-бутил-5-изоксазолил)-N'-(4-хлор-3-(трифторметил)фенил)мочевину. Остаток, полученный из фракций симметричных производных мочевины, растирают (Еt2О/гексан), при этом получают мочевину в виде твердого вещества белого цвета (0,110 г): ТСХ (3% МеОН/97% CH2Cl2), Rf 0,55; FAB-MS m/z 417 ((M+H)+).
В. Комбинаторный метод синтеза дифенил-производных мочевины с использовнием трифосгена
Один из анилинов, предназначенный для конденсации, растворяют в дихлорэтане (0,10 М). Этот раствор добавляют в сосуд емкостью 8 мл (0,5 мл), содержащий дихлорэтан (1 мл). Затем туда же добавляют раствор трифосгена (0,12 М в дихлорэтане, 0,2 мл, 0,4 экв), а затем диизопропилэтиламин (0,35 М в дихлорэтане, 0,2 мл, 1,2 экв). Сосуд закрывают и нагревают при температуре 80°С в течение 5 ч, затем охлаждают до комнатной температуры в течение приблизительно 10 ч. Добавляют второй анилин (0,10 М в дихлорэтане, 0,5 мл, 1,0 экв), затем диизопропилэтиламин (0,35 М в дихлорэтане, 0,2 мл, 1,2 экв). Полученный раствор нагревают при температуре 80°С в течение 4 ч, охлаждают до комнатной температуры и добавляют МеОН (0,5 мл). Полученную смесь концентрируют при пониженном давлении и полученные продукты очищают обращенно-фазовой ВЭЖХ.
С. Взаимопревращения производных мочевины и прочие реакции
С1. Общий метод алкилирования гидроксифенил-производных мочевины
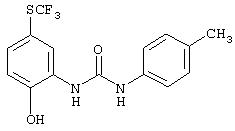
Стадия 1. N-(2-Гидрокси-5-(трифторметилтио)фенил)-N'-(4-метилфенил)мочевина: п-Толилизоцианат (0,066 мл, 0,52 ммоль) добавляют к раствору 2-гидрокси-5-(трифторметилтио)анилина (0,100 г, 0,48 ммоль) в ЕtOАс (2 мл) и реакционную смесь перемешивают при комнатной температуре в течение 2 дней. Полученный остаток промывают ЕtOАс, при этом получают конечный продукт (0,13 г):1H ЯМР (СDСl3) δ 2,24 (s, 3H), 7,44-7,03 (m, 6H), 8,46 (s, 1H), 8,60 (d, J=1,8 Гц, 1Н), 9,16 (s, 1Н), 10,41 (s, 1H); FAB-MS m/z 343 ((M+1)+). Этот материал используют на следующей стадии без очистки.
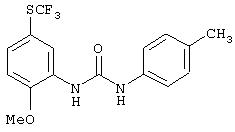
Стадия 2. N-(2-Метокси-5-(трифторметилтио)фенил)-N'-(4-метилфенил)мочевина: Раствор N-(2-гидрокси-5-(трифторметилтио)фенил)-N'-(4-метилфенил)мочевины (0,125 г, 0,36 ммоль), иодметана (0,045 мл, 0,73 ммоль) и К2СО3 (100 мг, 0,73 ммоль) в ацетоне (2 мл) кипятят с обратным холодильником в течение 6 ч, затем охлаждают до комнатной температуры и концентрируют при пониженном давлении. Остаток растворяют в минимальном количестве МеОН, наносят на силикагель и очищают экспресс-хроматографией (3% Еt2O/97% CH2Cl2), при этом получают конечный продукт в виде твердого вещества белого цвета (68 мг):1H ЯМР (CDCl3) δ 2,22 (s, 3Н), 3,92 (s, 3Н), 7,05-7,32 (m, 6H), 8,37 (s, 1Н), 8,52 (d, J=2,2 Гц, 1Н), 9,27 (s, 1Н); FAB-MS m/z 357 ((M+1)+).
С2. Общий метод восстановления производных мочевины, содержащих нитрогруппу
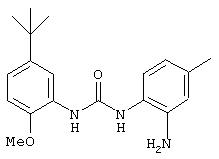
N-(5-трет-Бутил-2-метоксифенил)-N'-(2-амино-4-метилфенил)мочевина:
Раствор N-(5-трет-бутил-2-метоксифенил)-N'-(2-нитро-4-метилфенил)мочевины (полученной способом, аналогичным описанному в методе В1а; 4,0 г, 11,2 ммоль) в ЕtOН (100 мл) добавляют к суспензии 10% Pd/C (0,40 г) в ЕtOН (10 мл) и полученный раствор перемешивают в атмосфере H2 (баллон) при комнатной температуре в течение 18 ч. Смесь отфильтровывают через слой целита и концентрируют в вакууме, при этом получают требуемый продукт (3,42 г, 94%) в виде порошка: т.пл. 165-166°С;1H ЯМР (DMSO-d6) δ 1,30 (s, 9H), 2,26 (s, 3Н), 3,50 (ушир. s, 2H), 3,71 (s, 3Н), 6,39 (ушир. s, 1Н), 6,62 (s, 1Н), 6,73 (d, J=8,46 Гц, 1Н), 6,99 (dd, J=2,21; 8,46 Гц, 1Н), 7,05 (d, J=8,46 Гц, 1Н), 7,29 (s, 1H), 8,22 (d, J=2,57 Гц, 1Н); FAB-MS m/z 328 ((М+Н)+).
С3. Общий метод получения тиомочевины путем реакции с тиоизоцианатом
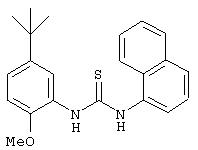
N-(5-трет-Бутил-2-метоксифенил)-N'-(1-нафтил)тиомочевина: К раствору 5-трет-бутил-2-метоксианилина (0,372 г, 2,07 ммоль) в толуоле (5 мл) добавляют 1-нафтилтиоизоцианат (0,384 г, 2,07 ммоль) и полученную смесь перемешивают при комнатной температуре в течение 8 ч, при этом получают осадок. Твердое вещество отделяют и последовательно промывают толуолом и гексаном, при этом получают требуемый продукт в виде порошка серо-белого цвета (0,364 г, 48%): т.пл. 158-160°С;1H ЯМР (DMSO-d6) δ 1,31 (s, 9Н), 3,59 (s, 3H), 6,74 (d, J=8,46 Гц, 1H), 7,13 (dd, J=2,21; 8,46 Гц, 1H), 7,53-7,62 (m, 4H), 7,88-7,95 (m, 4H), 8,06-8,08 (m, 1H), 8,09 (ушир. s, 1H); FAB-MS m/z 365 ((M+H)+).
С4. Общий метод удаления защитных групп с производных мочевины, содержащих трет-бутилкарбонатные группы
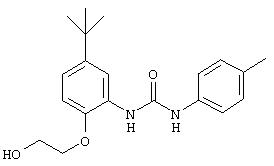
N-(5-трет-бутил-2-(2-гидроксиэтокси)фенил)-N'-(4-метилфенил)мочевина: Раствор N-(5-трет-бутил-2-(2-трет-бутоксикарбонилокси)этокси)фенил)-N'-(4-метилфенил)мочевины (метод В1f; 0,237 г, 0,54 ммоль) и ТФУ (0,21 мл, 2,7 ммоль) в CH2Cl2 (2 мл) перемешивают при комнатной температуре в течение 18 ч, затем промывают насыщенным раствором NaHCO3 (2 мл). Органический слой сушат пропусканием через слой фильтровальной бумаги 1PS (фирма Whatman®) и концентрируют при пониженном давлении. Полученную белую пену растирают (Еt2О/гексан), затем перекристаллизовывают Еt2О), при этом получают требуемый продукт (3,7 мг): ТСХ (50% ЕtOАс/50% гексан), Rf 0,62;1H ЯМР (DMSO-d6) δ 1,22 (s, 9H), 3, 75-3,76 (m, 2Н), 4,00-4,03 (m, 2H), 4,80 (t, J=5,0 Гц, 1Н), 6,88-6,89 (m, 4H), 7,06 (d, J=8,5 Гц, 2H), 7,33 (d, J=8,1 Гц, 2H), 7,97 (s, 1H), 8,20 (ушир. s, 1Н), 9,14 (s, 1H); FAB-MS m/z (отн. масса) 343 ((М+Н)+, 100%).
Следующие соединения синтезируют с использованием приведенных выше общих методов (см. табл.1-5).
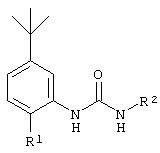
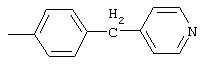
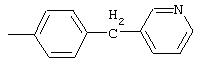
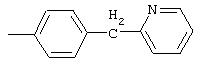
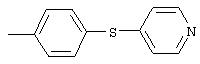
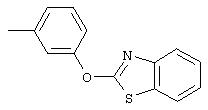
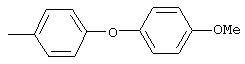
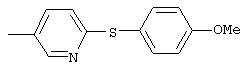
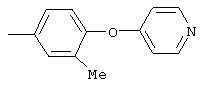
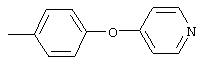
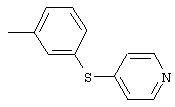
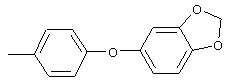
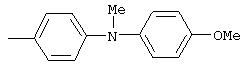
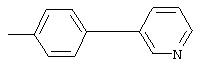
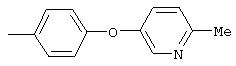
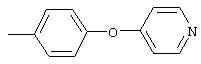
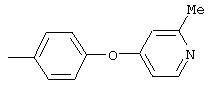
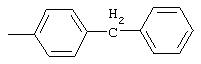
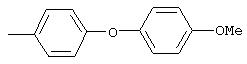
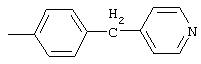
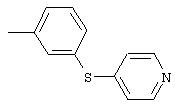
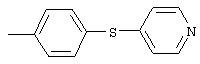
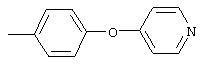
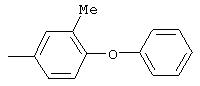
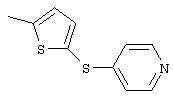
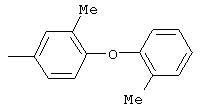
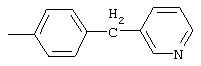
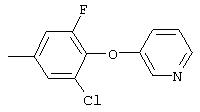
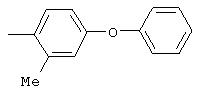
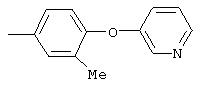
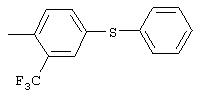
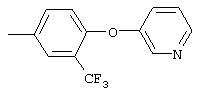
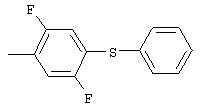
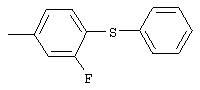
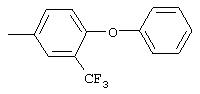
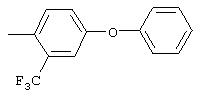
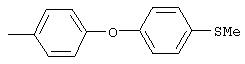
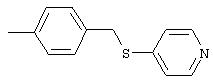
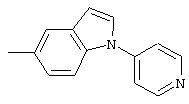
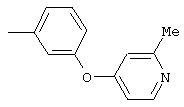
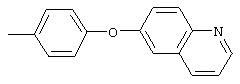
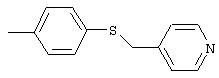
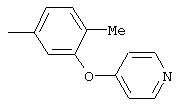
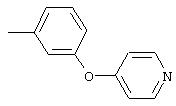
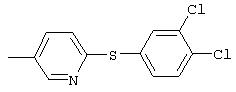
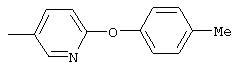
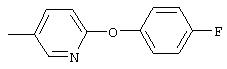
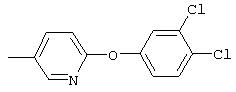
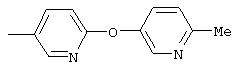
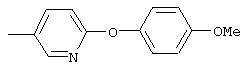
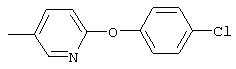
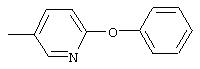
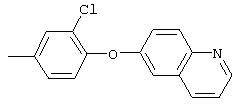
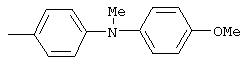
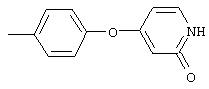
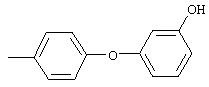
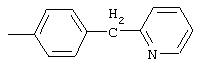
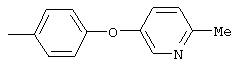
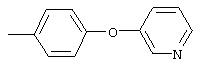
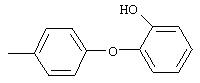
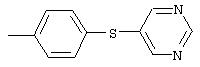
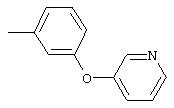
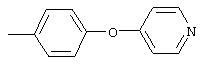
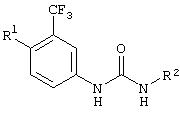
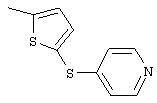
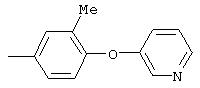
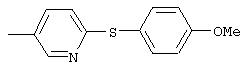
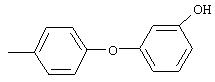
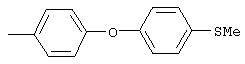
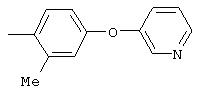
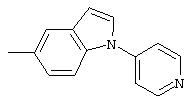
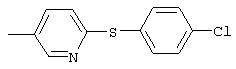
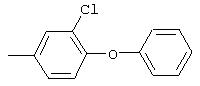
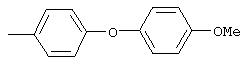
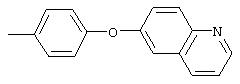
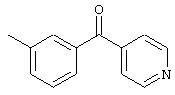
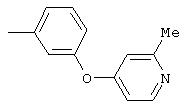
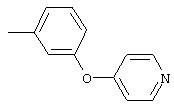
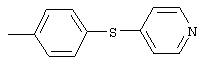
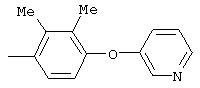
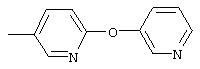
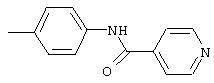
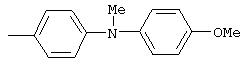
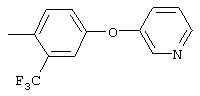
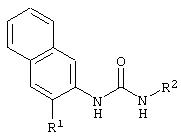
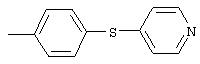
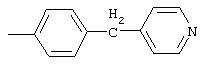
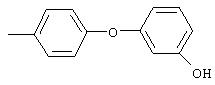
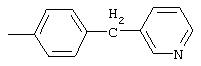
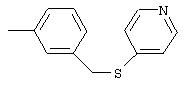
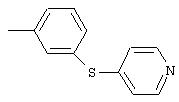
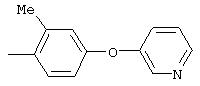
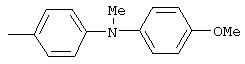
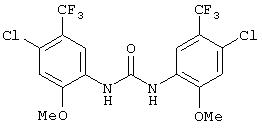
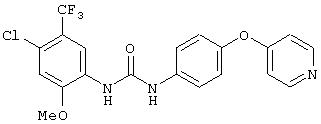
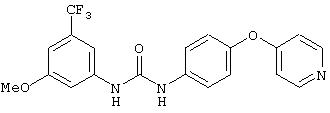
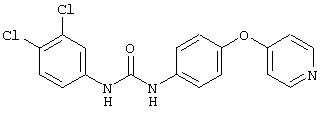
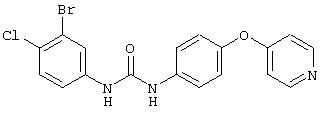
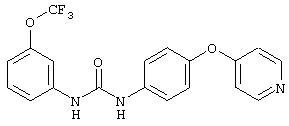
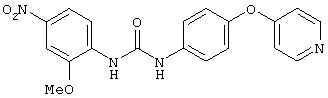
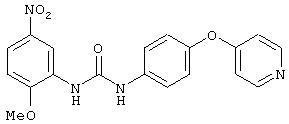
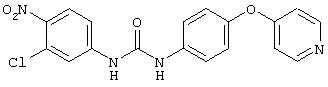
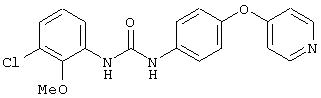
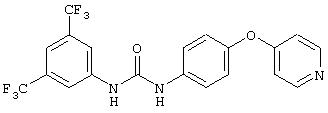
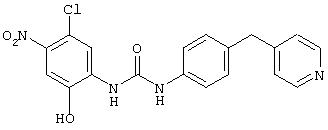
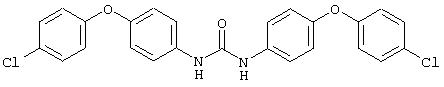
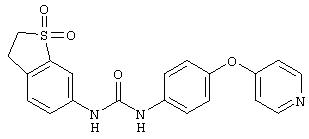
Примеры биологических испытаний
Анализ киназы raf in vitro
При анализе киназы in vitro киназу raf инкубируют с белком МЕК в 20 мМ трис-НСl буферном растворе, рН 8,2, содержащем 2 мМ 2-меркаптоэтанола и 100 мМ NaCl. Затем готовят концентрированный 10 мМ раствор (исходный раствор) соединений в ДМСО. К 20 мкл раствора белков raf+MEK добавляют 5 мкл воды или разбавленный водой исходный раствор соединения. Киназную реакцию инициируют добавлением 25 мкл [γ-33P]ATФ (1000-3000 расп. в мин./пмоль) в 80 мМ трис-НСl буферном растворе, рН 7,5, содержащем 120 мМ NaCl, 1,6 мМ ДТТ, 16 мМ MgCl2. Реакционную смесь инкубируют при 32°С обычно в течение 22 мин. Затем реакционную смесь наносят на фосфоцеллюлозные фильтры, отмывают свободную метку 1%-ным раствором фосфорной кислоты и включение33P в белок определяют количественно в жидком сцинтилляторе. Для высокоэффективного анализа большого числа образцов используют 10 мкМ раствор АТФ и 0,4 мкМ раствор МЕК. В некоторых экспериментах киназную реакцию останавливают добавлением равного количества буфера Лэммли. Образцы кипятят в течение 3 мин и белки разделяют электрофорезом в 7,5%-ном геле по методу Лэммли. Гели фиксируют, сушат и помещают на фотопластинку (фирмы Fuji). Степень фосфорилирования определяют, используя аналитическую систему Fujix Bio-lmaging.
Все приведенные в качестве примеров соединения имеют значения IC50 в диапазоне от 1 нМ до 10 мкМ.
Анализ роста клеток in vitro
Для анализа роста in vitro линии опухолевых клеток человека, включая НСТ116 и DLD-1, но не ограничиваясь перечисленным, содержащие мутантные гены K-ras, используют в стандартных анализах пролиферации при росте на якорной подложке на пластике или при безъякорном росте на мягком агаре. Линии опухолевых клеток человека получают на фирме АТСС (Rockville MD) и поддерживают в среде RPMI с 10%-ной инактивированной нагреванием эмбриональной телячьей сывороткой и 200 мМ глутамином. Культуральную среду и добавки получают на фирме Gibco/BRL (Gaitherburg, MD) за исключением эмбриональной телячьей сыворотки (фирма JRH Biosciences, Lenexa, KS). В стандартном анализе пролиферации при росте на якорной подложке клетки (3×103) высевают в 96-луночные планшеты для клеточных культур и для закрепления в лунках оставляют на ночь при 37°С в термостате с 5%-ным CO2. Соединения раститровывают в среде методом разведения и добавляют в лунки 96-луночного планшета с клеточными культурами. Обычно клетки оставляют расти в течение 5 суток с подачей свежего раствора соединения на третий день. Пролиферацию контролируют измерением метаболической активности стандартным колориметрическим анализом ХТТ (фирма Boehringer Mannheim) с использованием стандартного ридера для планшетов для ИФА (ELISA plate reader) при OD 490/560 или определением включения в ДНК3H-тимидина у 8 ч культуры с использованием3H-тимидина (1 мкКи), например, перенесением клеток сборщиком клеток на стекловолоконные фильтры с последующим измерением включения3H-тимидина по числу импульсов в жидком сцинтилляторе.
Для безъякорного роста клетки в 0,4%-ной агарозе (Seaplaque) в полной среде RPMI в количестве от 1×103 до 3×103 помещают в 24-луночные планшеты, наслаивая на донный слой, содержащий только 0,64%-ный агар в полной среде RPMI. Затем добавляют растворы анализируемых соединений, полученные методом серийных разведений на полной среде, и инкубируют при 37°С в термостате с 5%-ным CO2 в течение 10-14 суток с подачей свежей среды, содержащей анализируемое соединение с интервалом 3-4 дня. Количество и средний размер колоний регистрируют с использованием компьютерного анализа изображения и специальных программ (Image Pro Plus, media Cybernetics).
Данные анализа показывают, что соединения формулы I обладают активностью, как ингибиторы киназы raf и ингибиторы роста опухолевых клеток.
Анализ in vivo
Анализ in vivo ингибирующего воздействия соединений на опухоли (например, солидные опухоли), опосредованные raf киназой, проводят по следующей методике.
Мышам линии CDI nu/nu (возрастом 6-8 недель) вводят подкожно в бок 1×106 клеток аденокарциномы толстой кишки человека. Начиная примерно с 10 дня, когда размер опухоли составляет 50-100 мг, животным вводят внутрибрюшинно, внутривенно или перорально определенными дозами по 10, 30, 100 или 300 мг/кг соединения по изобретению. В течение 14 дней вводят определенные дозы один раз в день; размер опухоли замеряют кронциркулем дважды в неделю.
Кроме того, ингибирующее действие соединений на киназу raf и, следовательно, на опухоли (например, солидные опухоли), опосредованные киназой raf, можно продемонстрировать in vivo по методу, описанному в статье Monia с соавт. (Nat. Med., 1996, т.2, стр.668-75).
Вышеприведенные примеры можно повторить с тем же результатом, заменяя описанные в общем виде или более конкретно реагенты и/или условия (реакции) по настоящему изобретению на соединения или условия, описанные в предшествующих примерах.
Из вышеприведенного описания специалист в данной области техники может легко установить характерные особенности настоящего изобретения и, не выходя за пределы объема и сущности изобретения, может создать различные варианты и модификации изобретения для того, чтобы адаптировать его к разнообразным задачам и условиям.
Реферат
Изобретение относится к новым производным дифенилмочевины формулы (I) или их фармацевтически приемлемым солям, которые могут быть использованы при лечении опухолей, опосредованных киназой raf. В общей формуле (I) соединений настоящего изобретения:
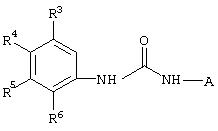
А означает
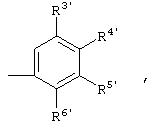
R3 означает Н, галоген, NO2, С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения;
С1-10алкокси, по выбору замещенный галогеном вплоть до полного замещения; R4, R5 и R6 каждый независимо означает Н, галоген, NO2, С1-10алкил, по выбору замещенный галогеном вплоть до полного замещения; С1-10алкокси, по выбору замещенный галогеном вплоть до полного замещения; и либо один из R4, R5 и R6 по выбору означает -X-Y; либо два соседних R4, R5 и R6 могут быть объединены с основным фенильным кольцом с образованием нафтила или индольной группы, замещенной пиридинильной группой, Х означает -CH2-, -S-, -N(СН3)-, -NHC(O)-, -CH2-S-, -S-CH2, -С(O)-, или -О-. Значения других радикалов указаны в формуле изобретения. Изобретение также относится к фармацевтической композиции, включающей соединения настоящего изобретения. 5 н. и 12 з.п. ф-лы, 5 табл.
Формула
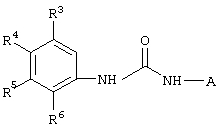
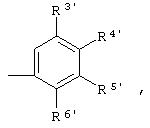
Документы, цитированные в отчёте о поиске
Способ получения диароматических соединений
Комментарии