Производные арилоксиалкилкарбаматов, их получение и их применение в терапии - RU2392269C2
Код документа: RU2392269C2
Описание
Объектом изобретения являются производные арилоксиалкилкарбаматов, их получение и их применение в терапии.
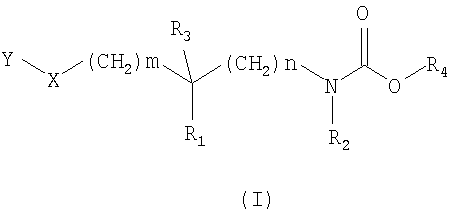
Соединения согласно изобретению отвечают общей формуле (I):
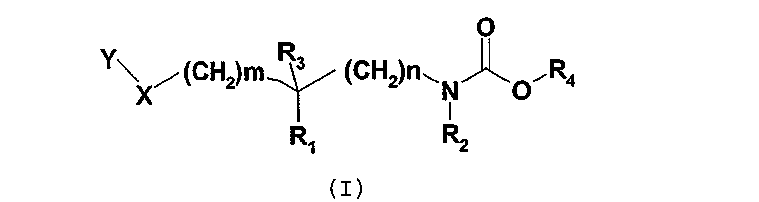
в которой
m означает 0, 1, 2 или 3;
n означает 0, 1, 2 или 3;
X означает атом кислорода или серы или группу SO или SO2;
R1 и R2 независимо друг от друга означают атом водорода или C1-3-алкильную группу, или R1 и R2 вместе образуют группу -(CH2)p-, где p означает целое число от 1 до 5, такое что n + p является целым числом от 2 до 5;
R3 означает атом водорода или фтора или гидроксильную или метильную группу;
R4 означает группу общей формулы CHR5CONHR6, в которой
R5 означает атом водорода или C1-6-алкильную группу, и
R6 означает атом водорода или C1-6-алкильную, C3-7-циклоалкильную, C3-7-циклоалкил-C1-6-алкиленовую группу;
Y означает группу Y1, выбранную, в частности, из фенила, пиридинила, пиридазинила, пиримидинила, пиразинила, триазинила, тиазолила, нафтила, хинолинила, изохинолинила, фталазинила, хиназолинила, хиноксалинила, нафтиридинила, циннолинила, бензофуранила, дигидробензофуранила, бензотиенила, дигидробензотиенила, индолила, изоиндолила, индолинила, бензимидазолила, бензоксазолила, бензизоксазолила, бензотиазолила, бензизотиазолила, бензотриазолила, бензоксадиазолила, бензотиадиазолила; причем группа Y1 при необходимости замещена одним или несколькими заместителями Y2, одинаковыми или отличающимися друг от друга, или группой Y3;
Y2 означает атом галогена, группу циано, нитро, C1-8-алкил, C1-8-алкокси, C1-8-тиоалкил, C1-8-фторалкил, C1-8-фторалкокси, C1-8-фтортиоалкил, C3-7-циклоалкил, C3-7-циклоалкилокси, C3-7-циклоалкил-C1-8-алкилен, C3-7-циклоалкил-C1-8-алкилокси, гидрокси, NR7R8, NHCOR7, NHSO2R7, COR7, CO2R7, CONR7R8, SO2R7, SO2NR7R8, -O-(С1-3-алкилен)-O-, фенилокси, фенилтио, фенил-C1-C8-алкилен, фенил-C1-C8-алкилокси или фенил-C1-C8-алкилтио;
Y3 означает группу, выбранную, в частности, из фенила, пиридинила, пиримидинила, пиразинила или пиридазинила; причем группа или группы Y3 могут быть замещены одной или несколькими группами Y2, одинаковыми или отличающимися друг от друга;
R7 и R8 независимо друг от друга означают атом водорода или С1-6-алкильную группу, или вместе с атомом азота, с которым они связаны, образуют азетидиновый, пирролидиновый, пиперидиновый, морфолиновый, тиоморфолиновый, азепиновый, пиперазиновый цикл, при необходимости замещенный C1-3-алкильной или бензильной группой.
Из соединений общей формулы (I), первую группу соединений составляет группа, для которой:
Y означает группу Y1, выбранную, в частности, из фенила, пиридинила, пиримидинила, тиазолила, нафтила, хинолинила, изохинолинила, бензоксазолила; причем группа Y1 при необходимости замещена одним или несколькими заместителями, в частности, одним или двумя заместителями Y2, одинаковыми или отличающимися друг от друга, или группой Y3;
Y2 означает атом галогена, в частности, хлор, фтор или бром, группу циано, C1-8-алкил, в частности, метил, изопропил, бутил, трет-бутил или тетраметилбутил, C1-8-алкокси, в частности, метокси, этокси или пропокси, C1-8-фторалкил, в частности, трифторметил, C1-8-фторалкокси, в частности, трифторметокси, фенилокси, фенил-C1-C8-алкилен, в частности, фенил-(1,1-диметилметилен);
Y3 означает фенильную группу; причем Y3 может быть замещена одной или несколькими группами, в частности, одной или двумя группами Y2, одинаковыми или отличающимися друг от друга.
Из соединений первой группы, какая определена выше, вторую группу соединений составляет группа, для которой:
Y означает
группу Y1, выбранную, в частности, из фенила или нафтила; причем группа Y1 при необходимости замещена одним или несколькими заместителями, в частности, одним или двумя заместителями Y2, одинаковыми или отличающимися друг от друга, или группой Y3;
Y2 означает атом галогена, в частности, хлор, фтор или бром, группу циано, C1-8-алкил, в частности, метил, изопропил, бутил, трет-бутил или тетраметилбутил, C1-8-алкокси, в частности, метокси, этокси или пропокси, C1-8-фторалкил, в частности, трифторметил, C1-8-фторалкокси, в частности, трифторметокси, фенилокси, фенил-C1-C8-алкилен, в частности, фенил-(1,1-диметилметилен);
Y3 означает фенильную группу; причем Y3 может быть замещена одной или несколькими группами, в частности, одной или двумя группами Y2, одинаковыми или отличающимися друг от друга.
Из соединений общей формулы (I) третью группу соединений составляет группа, для которой:
m означает 0, 1, 2 или 3; и/или
n означает 0, 1, 2 или 3; и/или
R1 и R2 независимо друг от друга означают атом водорода или C1-3-алкильную группу, или R1 и R2 вместе образуют группу -(CH2)p-, где p означает целое число от 1 до 5, такое, что n + p является целым числом от 2 до 5;
при условии, что когда R1 и R2 независимо друг от друга означают атом водорода или C1-3-алкильную группу, то m + n > 1.
Из соединений третьей группы, какая определена выше, четвертую группу соединений составляет группа, для которой:
m означает 0, 1, 2 или 3; и/или
n означает 0, 1, 2 или 3; и/или
R1 и R2 вместе образуют группу -(CH2)p-, где p означает целое число от 1 до 4, такое, что n + p равно 4.
Из соединений общей формулы (I) пятую группу соединений составляет группа, для которой X означает атом кислорода.
Из соединения общей формулы (I) шестую группу соединений составляет группа, для которой R3 означает атом водорода.
Седьмая группа образована соединениями, для которых одновременно R1, R2, R3, R4, R5, R6, R7, R8, X, Y, Y1, Y2, Y3, n и m такие, как определено выше в подгруппах соединений.
Соединения общей формулы (I) могут содержать один или несколько несимметричных углеродов. Они могут существовать в форме энантиомеров или диастереомеров. Эти энантиомеры или диастереомеры, а также их смеси, в том числе рацемические смеси, являются частью изобретения.
Соединения формулы (I) могут существовать в виде оснований или кислотно-аддитивных солей. Такие аддитивные соли являются частью изобретения.
Эти соли предпочтительно получены с фармацевтически приемлемыми кислотами, но и соли других кислот, пригодные, например, для очистки или выделения соединений формулы (I), также составляют часть изобретения. Соединения общей формулы (I) могут находиться в виде гидратов или сольватов, а именно в виде ассоциатов или комбинаций с одной или несколькими молекулами воды или с растворителем. Такие гидраты и сольваты также составляют часть изобретения.
В контексте изобретения подразумевается, что:
- Ct-z, где t и z могут принимать значения от 1 до 8, означает углеродную цепь, которая может иметь от t до z атомов углерода, например, C1-3 - углеродная цепь, которая может иметь от 1 до 3 атомов углерода;
- алкил означает насыщенную алифатическую группу, линейную или разветвленную; например, С1-3-алкильная группа означает углеродную цепь с 1-3 атомами углерода, линейную или разветвленную, в частности, метил, этил, пропил, 1-метилэтил;
- алкилен означает двухвалентную насыщенную алкильную группу, линейную или разветвленную, например, С1-3-алкиленовая группа означает двухвалентную углеродную цепь с 1-3 атомами углерода, линейную или разветвленную, в частности, метилен, этилен, 1-метилэтилен, пропилен, 1,1-диметилметилен;
- циклоалкил означает циклическую алкильную группу, например, C3-5-циклоалкильная группа означает циклическую углеродную группу с 3-5 атомами углерода, в частности, циклопропил, циклобутил, циклопентил;
- алкенилен означает двухвалентную ненасыщенную алифатическую группу с 2 атомами углерода, в частности, этилен;
- алкокси означает -О-алкильную группу с насыщенной алифатической цепью, линейной или разветвленной;
- тиоалкил означает -S-алкильную группу с насыщенной алифатической цепью, линейной или разветвленной;
- фторалкил означает алкильную группу, у которой один или несколько атомов водорода замещены атомом фтора;
- фторалкокси означает алкокси-группу, у которой один или несколько атомов водорода замещены атомом фтора;
- фтортиоалкил означает тиоалкильную группу, у которой один или несколько атомов водорода замещены атомом фтора;
- атом галогена означает фтор, хлор, бром или йод.
Соединения согласно изобретению могут быть получены различными способами, показанными на следующих схемах.
Так, один способ получения (схема 1) состоит в том, чтобы провести реакцию амина общей формулы (II), в которой Y, X, R1, R2, R3, m и n таковы, как определено в общей формуле (I), с карбонатом общей формулы (III), в которой Z означает атом водорода или нитрогруппу, R5 определено в общей формуле (I), и R означает метильную или этильную группу, в растворителе, таком как толуол или дихлорэтан, при температуре, составляющей от 0 до 80°C. Карбаматные эфиры общей формулы (IV), полученные таким путем, затем преобразуют в соединения общей формулы (I) аминолизом с помощью амина общей формулы R6NH2, где R6 определено в общей формуле (I). Реакция аминолиза может быть проведена в таком растворителе, как метанол или этанол, или в смеси растворителей, таких как метанол и тетрагидрофуран.
Схема 1

Другой способ (схема 2) получения соединения общей формулы (I), в которой R2 означает, в частности, атом водорода, состоит в том, чтобы провести реакцию производного общей формулы (IIa), в которой W означает гидроксильную группу, мезилат, тозилат или атом хлора, брома или йода, и в которой Y, X, R1, R3, m, и n определены в общей формуле (I), с оксазолидиндионом общей структуры (V), где R5 определено в общей формуле (I), чтобы получить производное оксазолидиндиона общей структуры (VI). В случае, когда W означает гидроксильную группу, реакция может быть проведена как реакция Мицунобу (Synthesis, 1981, 1-28), например, под действием диэтил- или диизопропилазодикарбоксилата в присутствии трифенилфосфина. В случае, когда W означает атом хлора, брома или йода, или мезилатную или тозилатную группу, реакция может быть проведена в присутствии основания, такого как 1,1,3,3-тетраметилгуанидин, гидрид натрия или трет-бутилат натрия, в растворителе, таком как тетрагидрофуран, ацетонитрил или диметилформамид, при температуре, составляющей от 0°C до температуры кипения растворителя. Производное оксазолидиндиона общей формулы (VI), полученное таким путем, затем преобразуют в соединение общей формулы (I), путем аминолиза с помощью амина общей формулы R6NH2, где R6 определено в общей формуле (I).
Схема 2
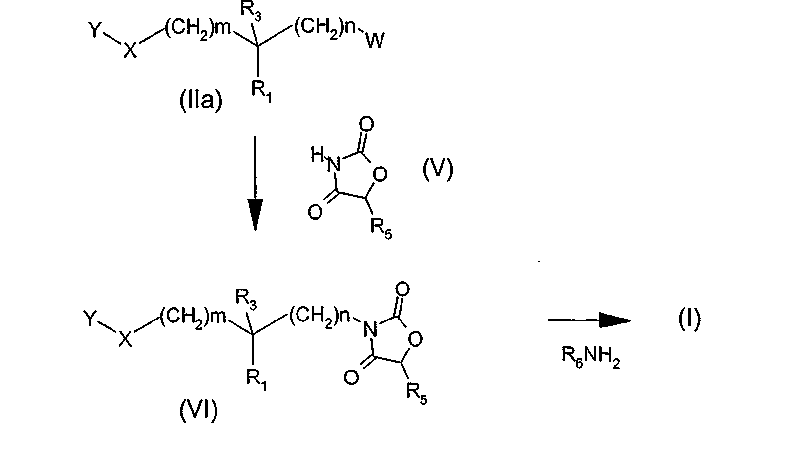
Другой вариант (схема 3) получения соединения общей формулы (I), в которой X означает, в частности, атом кислорода, состоит в проведении реакции производного спирта общей формулы (VIIa), (VIIb) или (VIIc) с производным фенола общей структуры YOH, в которой Y определен в общей формуле (I), например, в условиях реакции Мицунобу (Synthesis, 1981, 1-28) или модифицированным условиям (Tetrahedron Letters 1993, 34, 1639-1642), причем затем производные карбаматных эфиров (IVa) и оксазолидиндиона (VIa) преобразуют в соединения общей формулы (I) путем реакции аминолиза с помощью амина общей структуры R6NH2, где R6 определено в общей формуле (I).
В общих формулах (VIIa), (VIIb) и (VIIc) группы R1, R2, R3, R5, R6, m, n, и R определены выше.
Схема 3
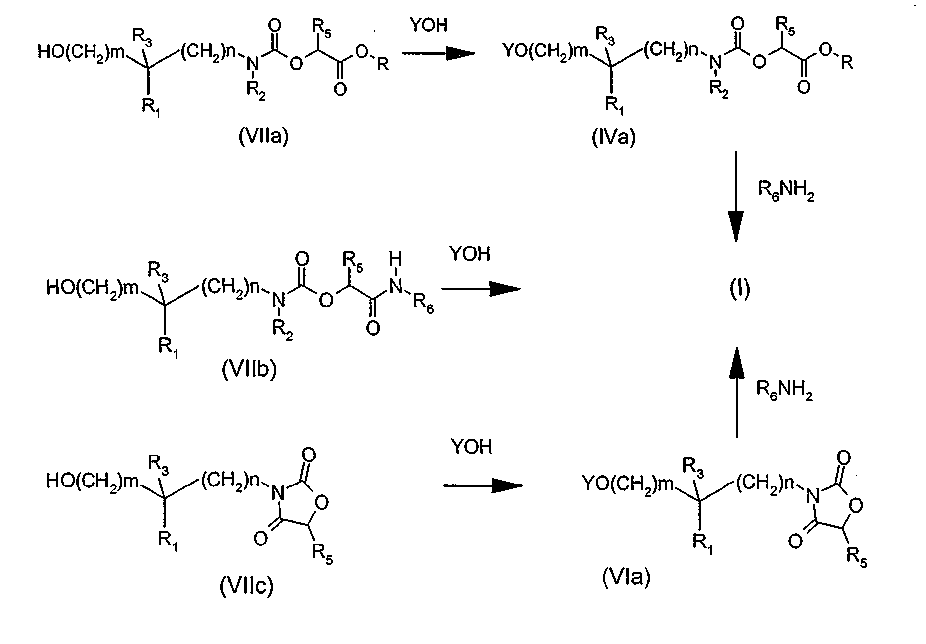
Другой вариант (схема 4) получения соединений общей формулы (I), в которой Y означает, в частности, группу Y1-Y3 арил-арил, арил-гетероарил, гетероарил-арил или гетероарил-гетероарил, состоит в проведении реакции производного арилгалогенида общей структуры (VIII), в которой U является атомом брома или йода, а Y1, X, R1, R2, R3, R5, R6, n и m определены в общей формуле (I), с производным арил- или гетероарилборной кислоты формулы Y3B(OH)2, где Y3 определено в общей формуле (I), следуя условиям реакции Сузуки (Chem. Rev. 1995, 95, 2457-2483), или с производным арил- или гетероарилтриалкилстанната формулы Y3Sn(R')3, где Y3 определено в общей формуле (I), и R' означает C1-4-алкил, следуя условиям реакции Штилле (Angew. Chem. Int. Ed. 1986, 25, 504-524).
Схема 4
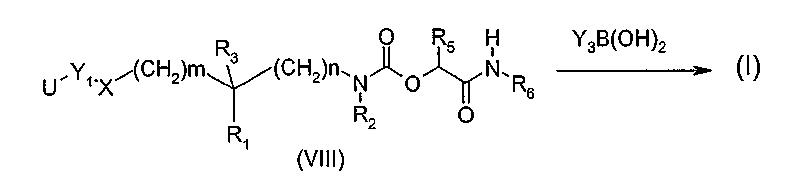
Соединения общих формул (II), (IIa), (III), (V), (VIIa), (VIIb), (VIIc), (VIII) и производные фенола общей структуры YOH, если способ их получения не описан, имеются в продаже или описаны в литературе, или также могут быть получены по способам, которые там описаны или которые известны специалисту.
Амины общей формулы R6NH2 имеются в продаже.
Следующие примеры иллюстрируют получение некоторых соединений согласно изобретению. Эти примеры не являются ограничивающими и только иллюстрируют изобретение. Микроанализ, ИК- и ЯМР-спектры и/или ЖХ-МС (жидкостная хроматография в сочетании с масс-спектроскопией) подтверждают структуры и чистоту полученных соединений.
Т.пл. (°C) означает температуру плавления в градусах Цельсия.
Номера, указанные в скобках в названиях примеров, соответствуют числам в первой колонке нижеследующей таблицы.
Для обозначения соединений в следующих примерах была использована номенклатура ЮПАК (Международный Союз Чистой и Прикладной Химии, IUPAC). Например, для бифенильной группы соблюдалась следующая нумерация:

Пример 1 (соединение 1)
[2-(метиламино)-2-оксоэтил]-{2-[(4-хлорфенил)окси]этил}карбамат
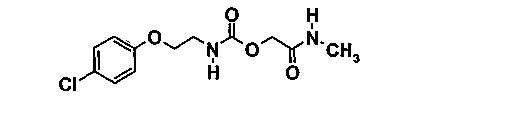
1.1. этил-[(фенилоксикарбонил)окси]ацетат
К раствору 25 г (240 ммоль) этилгликолята и 55 мл (315 ммоль) диизопропилэтиламина в 500 мл толуола медленно добавляют при комнатной температуре 32 мл (256 ммоль) фенилхлорформиата. Продолжают перемешивать 2 часа при комнатной температуре. Образовавшуюся соль отделяют и фильтрат концентрируют при пониженном давлении. Получают 53,7 г маслянистого продукта, используемого на следующем этапе в том виде, как он был получен.
1.2. этил-{[({2-[(4-хлорфенил)окси]этил}амино)карбонил]окси}ацетат
Нагревают до 60°C в течение ночи раствор 0,6 г (3,5 ммоль) [(4-хлорфенил)окси]этиламина (Chim. Ther. 1973, 8, 259-270) и 1,3 г (5,8 ммоль) этил [(фенилоксикарбонил)окси]ацетата, полученного на этапе 1.1, в 30 мл толуола. Выпаривают досуха и продукт очищают хроматографией на силикагеле, элюируя смесью 30/70 этилацетата и циклогексана. Получают 0,7 г маслянистого продукта, содержащего 10% циклизованного продукта оксазолидиндиона, используемого на следующем этапе в том виде, как он был получен.
1.3. [2-(метиламино)-2-оксоэтил]-{2-[(4-хлорфенил)окси]этил}карбамат
3,5 мл (7 ммоль) 2М раствора метиламина в тетрагидрофуране добавляют к раствору 0,7 г (2,3 ммоль) этил{[({2-[(4-хлорфенил)окси]этил}амино)карбонил]окси}ацетата, полученного на этапе 1.2, в 5 мл метанола. Оставляют реагировать в течение ночи при комнатной температуре. Выпаривают досуха и оставшуюся твердую фазу промывают гексаном, затем диизопропиловым эфиром, чтобы получить 0,59 г продукта в виде порошка.
Температура плавления(°C): 147-149
ЖХ-МС: M+H = 287
1Н-ЯМР (ДМСО) δ(м.д.): 7,75 (м, 1H), 7,40 (м, 1H), 7,25 (д, 2H), 6,95 (д, 2H), 4,35 (с, 2H), 3,95 (т, 2H), 3,35 (м, 2H), 2,60 (д, 3H)
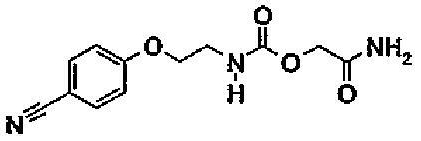
Пример 2 (соединение 11)
[2-амино-2-оксоэтил]-(2-[(4-цианофенил)окси]этил)карбамат
2.1. 3-(2-гидроксиэтил)-1,3-оксазолидин-2,4-дион
В течение 2 часов по каплям добавляют раствор 3 мл (39,6 ммоль) метилгликолята в 25 мл тетрагидрофурана к раствору 49 мл (95 ммоль) 1,9M фосгена в толуоле, разбавленному в 50 мл тетрагидрофурана, и охлаждают на ледяной бане. Затем перемешивают при комнатной температуре в течение 16 часов и выпаривают досуха. Выпаривают 4 раза вместе с 30 мл дихлорметана. Остаток объединяют с 40 мл ацетонитрила и по каплям в течение 1 часа добавляют к раствору 3,4 мл (59,4 ммоль) этаноламина и 30 мл (178 ммоль) диизопропилэтиламина в 50/10 смеси ацетонитрила и дихлорметана, охлажденному на ледяной бане. Затем перемешивают 16 часов при комнатной температуре. Фильтруют через целит, выпаривают досуха и продукт очищают хроматографией на силикагеле, элюируя смесью 70/30, затем 80/20 этилацетата и н-гексана, чтобы получить 4,9 г продукта в виде белой твердой субстанции.
2.2. [2-амино-2-оксоэтил]-(2-[(4-цианофенил)окси]этил)карбамат
0,61 мл (1,35 ммоль) 2,2M раствора диэтилазодикарбоксилата в толуоле по каплям добавляют к раствору 0,13 г (0,88 ммоль) 3-(2-гидроксиэтил)-1,3-оксазолидин-2,4-диона, полученного на этапе 2.1, 0,35 г (1,35 ммоль) трифенилфосфина и 0,10 г (0,89 ммоль) 4-гидроксибензонитрила в 2 мл бензола, охлажденного на ледяной бане. Затем реакционную смесь перемешивают при комнатной температуре в течение 16 часов. Выпаривают досуха и продукт очищают хроматографией на силикагеле, элюируя смесью 99/1, затем 98/2 дихлорметана и этилацетата. Продукт вводят в 1,5 мл 7M раствора аммиака (10,5 ммоль) в метаноле. Перемешивают в течение одного часа. Осадок отфильтровывают и промывают его этилацетатом, чтобы получить 0,035 г белой твердой субстанции.
Температура плавления(°C): 204-206
ЖХ-МС: M+H = 264
1Н-ЯМР (ДМСО) δ(м.д.): 7,55 (д, 2H), 7,05 (м, 1H), 6,90-6,80 (м+д, 4H), 4,35 (с, 2H), 4,05 (т, 2H), 3,45 (м, 2H)
Пример 3 (соединение 58)
[2-амино-2-оксоэтил]-[4-(1-нафталенилокси]бутил]карбамат
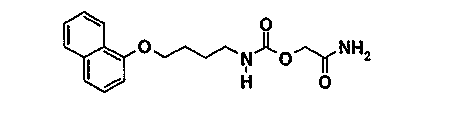
3.1. 3-[4-(1-нафталенилокси]бутил]-1,3-оксазолидин-2,4-дион
К раствору 3,1 г (11,1 ммоль) 1-[(4-бромбутил)окси]нафталина (Eur. J. Med. Chem. 1997, 32, 175-179) и 1,35 г (13,3 ммоль) 1,3-оксазолидин-2,4-диона (J. Med. Chem. 1991, 34, 1542-1543) в 30 мл тетрагидрофурана по каплям добавляют раствор 2,55 г (22,2 ммоль) 1,1,3,3-тетраметилгуанидина в 15 мл тетрагидрофурана. Кипятят с обратным холодильником в течение 8 часов. Добавляют 0,28 г (2,7 ммоль) 1,3-оксазолидин-2,4-диона и 0,32 г (2,7 ммоль) 1,1,3,3-тетраметилгуанидина и кипятят с обратным холодильником в течение дополнительных 4 часов. Реакционную смесь охлаждают на ледяной бане и добавляют 100 мл этилацетата, затем 50 мл 1М водного раствора соляной кислоты. Отстаивают, водную фазу экстрагируют 2 x 80 мл этилацетата. Затем органические фазы промывают 80 мл воды, затем 80 мл насыщенного водного раствора хлорида натрия. Сушат над сульфатом натрия, затем выпаривают досуха. Продукт очищают хроматографией на силикагеле, элюируя смесью 80/20 циклогексана и этилацетата, чтобы получить 2,0 г продукта, используемого на следующем этапе как есть.
3.2. [2-амино-2-оксоэтил]-[4-(1-нафталенилокси]бутил]карбамат
Растворяют 1,50 г (5,0 ммоль) 3-[4-(1-нафталенилокси]бутил]-1,3-оксазолидин-2,4-диона, полученного на этапе 3.1, в смеси 10 мл тетрагидрофурана и 28 мл 7н. раствора аммиака (200 ммоль) в метаноле. Оставляют реагировать на ночь при комнатной температуре, затем выпаривают досуха. Продукт очищают хроматографией на силикагеле, элюируя смесью 97/3 дихлорметана и метанола. Кристаллизуют в этилацетате, затем промывают диэтиловым эфиром, чтобы получить 0,73 г продукта в виде белой твердой субстанции.
Температура плавления (°C): 80-82
ЖХ-МС: M+H =317
1Н-ЯМР (CDCl3) δ(м.д.): 8,25 (дд, 1H), 7,80 (дд, 1H), 7,55-7,30 (м, 4H), 6,80 (д, 1H), 6,00 (м, 1H), 5,65 (м, 1H), 5,05 (м, 1H), 4,65 (с, 2H), 4,20 (т, 2H), 3,35 (м, 2H), 2,00 (м, 2H), 1,90 (м, 2H)
Пример 4 (соединение 85)
[2-(метиламино)-2-оксоэтил]-4-[(4'-фтор-4-бифенил)окси]-1-пиперидинкарбоксилат

4.1. 1,1-диметилэтил-4-[(4-бромфенил)окси]-1-пиперидинкарбоксилат
К раствору 2,01 г (10 ммоль) 1,1-диметилэтил-4-гидрокси-1-пиперидинкарбоксилата в 20 мл диметилформамида добавляют 7 г (40 ммоль) 1-бром-4-фторбензола и 2,5 г (50 ммоль) 50%-ного гидрида натрия в минеральном масле. Смесь перемешивают при 100°C в течение 3 часов, затем выпаривают досуха. Остаток объединяют с 50 мл ледяной воды и экстрагируют дихлорметаном. Органические экстракты выпаривают досуха, чтобы получить 3,5 г маслянистого продукта, используемого на следующем этапе как есть.
4.2. 4-[(4-бромфенил)окси]пиперидин
К раствору 3,5 г (9,83 ммоль) 1,1-диметилэтил-[4-[(4-бромфенил)окси]-1-пиперидинкарбоксилата], полученного на этапе 4.1, в 20 мл дихлорметана, добавляют 10 мл трифторуксусной кислоты и раствор перемешивают при комнатной температуре в течение 1 часа. Выпаривают досуха, затем остаток объединяют с 30 мл толуола, который снова выпаривают досуха. Затем остаток промывают пентаном, затем объединяют со смесью 60 мл дихлорметана и 20 мл 4н. водного раствора аммиака. Интенсивно перемешивают в течение 15 минут, затем органическую фазу сцеживают, сушат над сульфатом натрия и выпаривают досуха, чтобы получить 2,7 г продукта в виде масла, используемого на следующем этапе в том виде, как он был получен.
4.2. [2-(этилокси)-2-оксоэтил]-4-[(4-бромфенил)окси]-1-пиперидинкарбоксилат
Смешивают 2,7 г (7,58 ммоль) 4-[(4-бромфенил)окси]пиперидина, полученного на этапе 4.2, и 1,70 г (7,6 ммоль) этил{[(фенилокси)карбонил]окси}ацетата, полученного по примеру 1.1, в 40 мл толуола, и нагревают раствор до 50°C в течение 20 часов. После охлаждения выпаривают досуха и продукт очищают хроматографией на силикагеле, элюируя смесью 40/60 этилацетата и циклогексана. Затем растирают в диизопропиловом эфире, чтобы получить 2,9 г продукта в виде порошка.
Температура плавления(°C): 87-88
4.3. [2-(метиламино)-2-оксоэтил]-4-[(4-бромфенил)окси]-1-пиперидинкарбоксилат
Перемешивают при комнатной температуре в течение 20 часов 2,9 г (7,5 ммоль) [2-(этилокси)-2-оксоэтил]-4-[(4-бромфенил)окси]-1-пиперидинкарбоксилата, полученного на этапе 4.3, растворенного в 10 мл этанольного раствора, с 33% метиламина. После выпаривания продукт очищают хроматографией на силикагеле, элюируя этилацетатом, чтобы получить 0,8 г продукта в виде резины, используемой на следующем этапе как есть.
4.5. [2-(метиламино)-2-оксоэтил]-4-[(4'-фтор-4-бифенил)окси]-1-пиперидинкарбоксилат
В стеклянную трубку с пробкой помещают 0,1 г (0,27 ммоль) [2-(метиламино)-2-оксоэтил]-4-[(4-бромфенил)окси]-1-пиперидинкарбоксилата, полученного на этапе 4.4, 0,01 г тетракис(трифенилфосфин)палладия(0) и 0,057 г (0,4 ммоль) 4-фторфенилборной кислоты. Добавляют 4 мл толуола, 2 мл 2н. водного раствора карбоната натрия и 0,5 мл этанола. Нагревают до 80°C при перемешивании в течение 2 часов. После охлаждения добавляют 1 мл воды и 2 мл толуола. Органическую фазу выделяют и продукт очищают хроматографией на силикагеле, элюируя смесью 95/5 дихлорметана и метанола. Продукт снова растворяют в 1 мл этанола, затем его осаждают путем добавления 2 мл воды, чтобы получить 0,031 г продукта в виде порошка.
Температура плавления(°C): 117-119
ЖХ-МС: M+H 387
1Н-ЯМР (CDCl3) δ(м.д.): 7,70 (дд, 2H); 7,65 (д, 2H); 7,30 (дд, 2H); 7,20 (д, 2H); 6,25 (шир. с, 1H), 4,80 (с+м, 3H); 4,00-3,70 (м, 4H); 3,05 (д, 3H); 2,25-2,00 (м, 4H)
Пример 5 (соединение 120)
[2-(метиламино)-2-оксоэтил]-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилат

5.1. 1,1-диметилэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.1. Из 2,5 г (11,6 ммоль) 1,1-диметилэтил-4-(гидроксиметил)-1-пиперидинкарбоксилата и 8,13 г (46,4 ммоль) 1-бром-4-фторбензола получают 5,75 г неочищенного продукта в виде масла.
5.2. 4-{[(4-бромфенил)окси]метил}пиперидин
Действуют, как описано в примере 4.2. Из 5,75 г 1,1-диметилэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидкарбоксилата, полученного на этапе 5.1, получают 3 г продукта в виде масла.
5.3. 2-(этилокси)-2-оксоэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.3. Из 1,6 г (5,9 ммоль) 4-{[(4-бромфенил)окси]метил}пиперидина, полученного на этапе 5.2, и 1,32 г (5,9 ммоль) этил-{[(фенилокси)карбонил]окси]ацетата, полученного согласно примеру 1.1, получают продукт в виде масла.
5.4. 2-(метиламино)-2-оксоэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.4. Из 2-(этилокси)-2-оксоэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилата, полученного на этапе 5.3, получают 1,1 г продукта в виде порошка.
Температура плавления(°C): 163-165
ЖХ-МС: M+H = 386
1H-ЯМР (CDCl3) δ(м.д.): 7,35 (д, 2H); 6,75 (д, 2H); 6,05 (шир. с, 1H); 4,70-4,50 (м, 2H); 4,30-4,10 (м, 2H); 3,80 (д, 2H); 3,00-2,75 (м, 2H); 2,85 (д, 3H); 2,10-1,80 (м, 3H); 1,45-1,20 (м, 2H)
Пример 6 (соединение 154)
[2-(метиламино)-2-оксоэтил]-4-{[(4'-(трифторметил)-4-бифенил)окси]метил}-1-пиперидинкарбоксилат

Действуют, как описано в примере 4.5. Из 0,1 г (0,26 ммоль) 2-(метиламино)-2-оксоэтил-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилата, полученного по примеру 5, и 0,074 г (0,389 ммоль) 4-трифторметилфенилборной кислоты получают 0,049 г продукта в виде порошка.
Температура плавления(°C): 197-199
ЖХ-МС: M+H = 451
1Н-ЯМР (ДМСО) δ(м.д): 7,85-7,65 (м, 7H), 7,05 (д, 2H), 4,35 (с, 2H), 4,05 (шир. д, 2H), 3,90 (д, 2H), 2,85 (м, 2H), 2,60 (д, 3H), 2,00 (м, 1H), 1,80 (шир. д, 2H), 1,35-1,10 (м, 2H).
Пример 7 (соединение 137)
[2-амино-2-оксоэтил]-4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилат
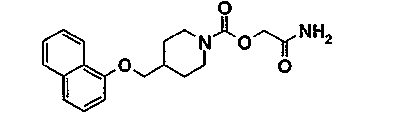
7.1 1,1-диметилэтил-4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилат
К раствору 5,0 г (23,2 ммоль) 1,1-диметилэтил-4-(гидроксиметил)-1-пиперидинкарбоксилата, 4,3 г (29,8 ммоль) 1-нафталенона и 7,82 г (29,8 ммоль) трифенилфосфина в 120 мл тетрагидрофурана, охлажденному в атмосфере азота на ледяной бане, по каплям добавляют раствор 6,03 г (29,8 ммоль) диизопропилазодикарбоксилата. Реакционную смесь доводят до комнатной температуры и продолжают перемешивать в течение ночи. Добавляют 2 мл метанола, затем выпаривают досуха. Остаток объединяют с 200 мл дихлорметана и последовательно промывают 10%-ным водным раствором гидросульфата калия, водой и 1М водным раствором гидроксида натрия. Сушат над сульфатом натрия и выпаривают досуха. Продукт очищают хроматографией на силикагеле, элюируя смесью 80/20, затем 70/30 и 50/50 циклогексана и дихлорметана, чтобы получить 7,96 г продукта в виде масла, которое отверждается.
Температура плавления(°C): 97-100
7.2. 4-[(1-нафталенилокси)метил]пиперидин
Раствор 7,96 г (29,1 ммоль) 1,1-диметилэтил-4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилата, полученного на этапе 7.1, в 120 мл метанола и 28 мл 35%-ного водного раствора соляной кислоты нагревают до 60°C в течение 6 часов. Охлаждают до комнатной температуры и выпаривают досуха, затем 2 раза выпаривают вместе с этанолом. Твердый остаток промывают диэтиловым эфиром, затем сушат в вакууме в присутствии пентоксида фосфора, чтобы получить 3,1 г белого твердого вещества.
Твердую фазу вводят в 80 мл воды и добавляют 30%-ный водный раствор гидроксида натрия до основного pH, затем 2 раза экстрагируют 150 мл диэтилового эфира. Экстракты сушат над сульфатом натрия и концентрируют досуха, чтобы получить 2,75 г маслянистого продукта, используемого на следующем этапе как есть.
7.3. [2-(этилокси)-2-оксоэтил]-4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилат
Раствор 2,75 г (11,4 ммоль) 4-[(1-нафталенилокси)метил]пиперидина, полученного на этапе 7.2, и 2,56 г (11,4 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного по примеру 1.1, в 80 мл толуола нагревают до 50°C в течение ночи. Выпаривают досуха и остаток объединяют со смесью воды, дихлорметана и насыщенного водного раствора гидрокарбоната натрия. Органическую фазу декантируют, сушат над сульфатом натрия и выпаривают досуха. Продукт очищают хроматографией на силикагеле, элюируя смесью 50/50 циклогексана и дихлорметана, затем дихлорметаном и смесью 95/5 дихлорметана и этилацетата. Получают 2,05 г продукта в виде масла, используемого на следующем этапе как есть.
7.4. 2-амино-2-oкcoэтил-[4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилат]
Растворяют 1,0 г (2,69 ммоль) 2-(этилокси)-2-оксоэтил-[4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилата], полученного на этапе 7.3, в 12 мл 7 н. раствора аммиака (84 ммоль) в метаноле. Оставляют реагировать при комнатной температуре в течение 3 дней. Выпаривают досуха и очищают хроматографией на силикагеле, элюируя смесью 90/10, затем 80/20, 70/30 и 50/50 дихлорметана и этилацетата, затем смесью 95/5 этилацетата и метанола. Затем кристаллизуют в этилацетате, чтобы получить 0,77 г продукта.
Температура плавления(°C): 135-136
ЖХ-МС: M+H = 343
1Н-ЯМР (ДМСО) δ(м.д.): 8,15 (дд, 1H), 7,80 (дд, 1H), 7,50-7,30 (м, 4H), 7,30 (м, 1H), 7,15 (м, 1H), 6,95 (д, 1H), 4,35 (с, 2H), 4,15-4,00 (м+д, 4H), 4,90 (м, 2H), 2,10 (м, 1H), 1,90 (д, 2H), 1,45-1,25 (м, 2H)
Пример 8 (соединение 148)
[2-амино-2-оксоэтил]-4-[(7-хинолинилокси)метил]-1-пиперидинкарбоксилат

8.1. [2-(метилокси)-2-оксоэтил]-4-(гидроксиметил)-1-пиперидинкарбоксилат
Действуют, как описано в примере 2.1, используя вместо этаноламина 6,84 г (59,4 ммоль) 4-(гидроксиметил)пиперидина, чтобы получить 7,85 г продукта в виде бесцветного масла.
8.2. [2-амино-2-оксоэтил]-4-[(7-хинолинилокси)метил]-1-пиперидинкарбоксилат
0,26 г (1,03 ммоль) 1,1'-(азодикарбонил)ди-пиперидина (ADDP) добавляют к раствору 0,16 г (0,69 ммоль) [2-(метилокси)-2-оксоэтил]-4-(гидроксиметил)-1-пиперидинкарбоксилата, полученного на этапе 8.1, 0,26 мл (1,03 ммоль) три-н-бутилфосфина и 0,13 г (0,90 ммоль) 7-гидроксихинолина в 2,5 мл бензола, охлажденного на ледяной бане. Смесь перемешивают в течение 15 минут при 0°C, затем 16 часов при комнатной температуре. Фильтруют через целит и промывают диэтиловым эфиром. Фильтраты выпаривают досуха и очищают хроматографией на силикагеле, элюируя смесью 70/30 этилацетата и н-гексана. Полученный продукт растворяют в 3 мл (21 ммоль) 7М раствора аммиака в метаноле. Перемешивают в течение 3 часов, затем выпаривают досуха. Продукт очищают хроматографией на силикагеле, элюируя смесью 90/10 этилацетата и этанола, и кристаллизуют в этилацетате, чтобы получить 0,115 г продукта в виде белого твердого вещества.
Температура плавления(°C): 137-139
ЖХ-МС: M+H = 344
1Н-ЯМР (CDCl3) δ(м.д.): 7,80 (дд, 1H), 8,05 (дд, 1H), 7,70 (д, 1H), 7,40 (д, 1H), 7,30-7,15 (м, 2H), 6,05 (м, 1H), 5,65 (м, 1H), 4,60 (с, 2H), 4,25 (м, 2H), 4,00 (д, 2H), 2,90 (м, 2H), 2,10 (м, 1H), 1,95 (д, 2H), 1,50-1,30 (м, 2H)
Пример 9 (соединение 168)
[2-(метиламино)-2-оксоэтил]-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилат
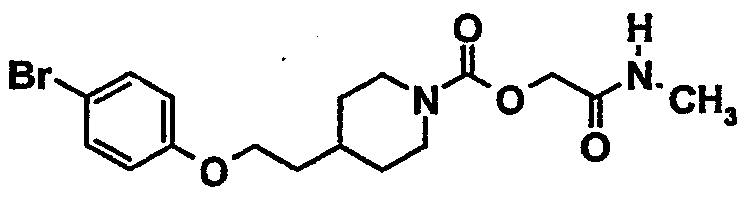
9.1. 1,1-диметилэтил-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.1. Из 1,93 г (8,4 ммоль) 1,1-диметилэтил-4-(2-гидроксиэтил)-1-пиперидинкарбоксилата и 5,88 г (33,6 ммоль) 1-бром-4-фторбензола получают 4,1 г неочищенного продукта в виде масла.
9.2. 4-{2-[(4-бромфенил)окси]этил}пиперидин
Действуют, как описано в примере 4.2. Из 1,1-диметилэтил-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилата, полученного на этапе 9.1, получают 1,79 г продукта в виде порошка.
Температура плавления(°C): 100-102
9.3. [2-(этилокси)-2-оксоэтил]-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.3. Из 1,76 г (6,19 ммоль) 4-{2-[(4-бромфенил)окси]этил}пиперидина, полученного на этапе 9.2, и 1,39 г (6,19 ммоль) этил{[(фенилокси)карбонил]окси}ацетата, полученного по примеру 1.1, получают 1,4 г продукта в виде масла.
9.4. [2-(метиламино)-2-оксоэтил]-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилат
Действуют, как описано в примере 4.4. Из 1,3 г (3,14 ммоль) [2-(этилокси)-2-оксоэтил]-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилата, полученного на этапе 9.3, получают 0,95 г продукта в виде порошка.
Температура плавления(°C): 101-103
ЖХ-МС: M+H = 400
1Н-ЯМР (CDCl3) δ(м.д.): 7,55 (д, 2H); 7,00 (д, 2H); 6,25 (шир. с, NH); 4,90-4,70 (м, 2H); 4,50-4,25 (м, 2H); 4,20 (т, 2H); 3,20-2,90 (м, 2H); 3,10 (д, 3H); 2,05-1,90 (м, 5H); 1,55-1,30 (м, 2H)
Пример 10 (соединение 186)
[2-(метиламино)-2-оксоэтил]-4-{2-[(4'-хлор-4-бифенил)окси]этил}-1-пиперидинкарбоксилат

Действуют, как описано в примере 4.5. Из 0,1 г (0,25 ммоль) 2-(метиламино)-2-оксоэтил-[4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилата], полученного по примеру 9, и 0,117 г (0,75 ммоль) 4-хлорфенилборной кислоты, получают 0,087 г продукта в виде порошка.
Температура плавления(°C): 104-106
ЖХ-МС: M+H =431
1Н-ЯМР (CDCl3) δ(м.д.): 7,70-7,50 (м, 6H); 7,10 (д, 2H); 6,20 (шир. с, NH); 4,85-4,60 (м, 2H); 4,45-4,15 (м, 2H); 4,20 (т, 2H); 3,15-2,95 (м, 2H); 3,05 (д, 3H); 2,10-1,85 (м, 5H), 1,50-1,25 (м, 2H)
Пример 11 (соединение 183)
[2-амино-2-оксоэтил]-4-[2-(7-изохинолинилокси)этил]-1-пиперидинкарбамат
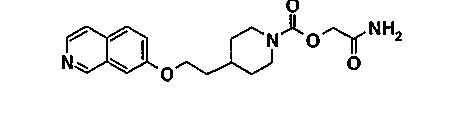
11.1. [2-(метилокси)-2-оксоэтил]-4-(2-гидроксиэтил)-1-пиперидинкарбоксилат
Действуют, как описано в примере 2.1, используя вместо этаноламина 7,6 г (59,4 ммоль) 4-(2-гидроксиэтил)пиперидина, чтобы получить 7,1 г продукта в виде бесцветного масла.
11.2. [2-амино-2-оксоэтил]-4-[2-(7-изохинолинилокси)этил]-1-пиперидинкарбамат
Действуют, как описано в примере 8.2, исходя из 0,46 г (1,84 ммоль) ADDP, 0,30 г (1,24 ммоль) [2-(метилокси)-2-оксоэтил]-4-(2-гидроксиэтил)-1-пиперидинкарбоксилата, полученного на этапе 11.1, 0,46 мл три-н-бутилфосфина и 0,26 г (1,84 ммоль) 7-гидроксиизохинолина в 4 мл бензола. Продукт очищают хроматографией на силикагеле, элюируя этилацетатом, затем смесью 95/5 этилацетата и этанола, чтобы получить 0,25 г продукта в виде белого твердого вещества.
Температура плавления(°C): 179-181
ЖХ-МС: M+H = 358
1Н-ЯМР (CDCl3) δ(м.д.): 9,15 (с, 1H), 8,45 (д, 1H), 7,60 (д, 1H), 7,35 (дд, 1H), 7,20 (д, 1H), 6,05 (м, 1H), 5,75 (м, 1H), 4,60 (с, 2H), 4,20 (т, 4H), 2,90 (м, 2H), 1,90-1,70 (м, 5H), 1,40-1,20 (м, 2H)
Пример 12 (соединение 83)
[2-амино-2-оксоэтил]-3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилат
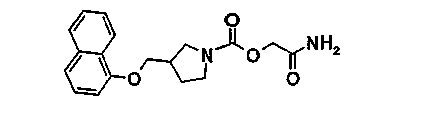
12.1. 1,1-диметилэтил-3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилат
К раствору 1,0 г (4,9 ммоль) 1,1-диметилэтил-3-(гидроксиметил)-1-пирролидинкарбоксилата (описан в документе WO 0066557), 0,95 г (6,4 ммоль) 1-нафталенона и 1,4 г (6,9 ммоль) три-н-бутилфосфина в 40 мл толуола и 20 мл тетрагидрофурана, охлажденному в атмосфере азота на ледяной бане, по каплям добавляют раствор 1,74 г (6,9 ммоль) ADDP. Реакционную смесь оставляют доходить до комнатной температуры и продолжают перемешивать в течение 24 часов. Смесь фильтруют и осадок промывают толуолом. Выпаривают досуха. Остаток объединяют с дихлорметаном и промывают 1М водным раствором гидроксида натрия. Сушат над сульфатом натрия и выпаривают досуха. Остаток очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном, затем смесью 98/2 дихлорметана и метанола, чтобы получить 0,80 г продукта в виде бесцветного масла.
12.2. 3-[(1-нафталенилокси)метил]пирролидин
Раствор 0,42 г (1,28 ммоль) 1,1-диметилэтил-[3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилата], полученного на этапе 12.1, в 10 мл 1,4-диоксана и 6 мл 2н. водного раствора соляной кислоты перемешивают в течение 6 часов. Выпаривают досуха, затем 2 раза выпаривают вместе с толуолом. Твердый остаток промывают диэтиловым эфиром. Твердую фазу вводят в дихлорметан и добавляют концентрированный раствор аммиака до основного pH. Фильтруют через ПТФЭ картридж Whatman и концентрируют органическую фазу, чтобы получить 0,21 г маслянистого продукта, используемого на следующем этапе как есть.
12.3. [2-(этилокси)-2-оксоэтил]-3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилат
Раствор 0,20 г (0,88 ммоль) 3-[(1-нафталенилокси)метил]пирролидина, полученного на этапе 12.2, и 0,35 г (1,5 ммоль) этил[(фенилоксикарбонил)окси]ацетата, полученного по примеру 1.1, в 6 мл толуола нагревают при 60°C в течение ночи. Выпаривают досуха и остаток объединяют со смесью воды, дихлорметана и насыщенного водного раствора гидрокарбоната натрия. Органическую фазу декантируют, сушат над сульфатом натрия и выпаривают досуха. Остаток очищают хроматографией на колонке с силикагелем, элюируя дихлорметаном, затем смесью 99/1 дихлорметана и метанола. Получают 0,24 г продукта в виде масла, используемого на следующем этапе в том виде, как он выделен.
12.4. [2-амино-2-оксоэтил]-3-[(1-нафталенилокси) метил]-1-пирролидинкарбоксилат
Растворяют 0,24 г (0,67 ммоль) [2-(этилокси)-2-оксоэтил]-3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилата, полученного на этапе 12.3, в 15 мл 7н. раствора аммиака (105 ммоль) в метаноле. Перемешивают в запаянной трубке при комнатной температуре в течение 3 дней. Выпаривают досуха и остаток очищают хроматографией на колонке с силикагелем, элюируя смесью 97/3, затем 94/6 дихлорметана и метанола. Полученную твердую фазу растирают в диэтиловом эфире и фильтруют, чтобы получить 0,15 г продукта.
Температура плавления(°C): 161-163
ЖХ-МС: M+H = 329
1Н-ЯМР (ДМСО) δ(м.д.): 8,15 (м, 1H), 7,75 (м, 1H), 7,50-7,30 (м, 4H), 7,10-6,90 (с, 2H), 6,80 (м, 1H), 4,40 (с, 2H), 4,20-4,05 (м, 2H), 3,90-3,30 (м, 4H), 2,90-2,70 (м, 1H), 2,30-2,10 (м, 1H), 2,05-1,85 (м, 1H).
Следующая таблица иллюстрирует химические структуры и физические свойства некоторых соединений согласно изобретению.
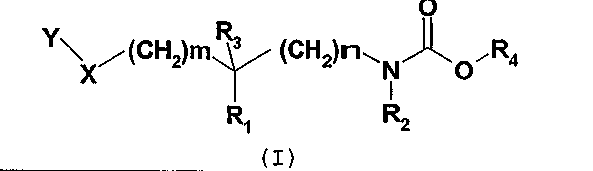
Соединения согласно изобретению были объектом фармакологических тестов, позволяющих определить их ингибирующее действие на ферменты FAAH (амидо-гидролаза жирной кислоты).
Ингибирующая активность была подтверждена в радиоферментом тесте, основанном на измерении продукта гидролиза (этаноламин [1-3H]) анандамида [этаноламин 1-3H] посредством FAAH (Life Sciences (1.995), 56, 1999-2005 и Journal of Pharmacology and Experimental Therapeutics (1997), 283, 729-734). Так, мозги мышей (без мозжечка) были извлечены и содержались при -80°C. Мембранные гомогенаты были приготовлены непосредственно перед экспериментом путем гомогенизирования ткани (Polytron) в буфере Tris-HCl 10 мМ (pH 8,0), содержащем 150 мМ NaCl и 1 мМ ЭДТА. Затем проводилась ферментативная реакция в 70 мкл буфера, содержащего альбумин бычьей сыворотки без жирной кислоты (1 мг/мл). Последовательно добавлялись тестируемые соединения в различных концентрациях: анандамид [этаноламин 1-3H] (удельная активность 15-20 Ci/ммоль), разбавленный до 10 мкМ холодным анандамидом и мембранным препаратом (по 400 мкг замороженной ткани на опыт). Через 15 минут при 25°C ферментативную реакцию останавливали добавлением 140 мкл смеси хлороформ/метанол (2:1). Смесь перемешивали 10 минут, затем центрифугировали в течение 15 минут при 3500 об/мин. Радиоактивность аликвоты (30 мкл) водной фазы, содержащей этаноламин [1-3H] подсчитывали по сцинтилляции жидкости.
В этих условиях самые активные соединения по изобретению имеют CI50 (концентрация, ингибирующая 50% ферментатавной активности контрольной FAAH) составляет от 0,001 до 1 мкМ. Например, соединение 58 таблицы имеет CI50 0,047 мкМ.
Таким образом, видно, что соединения согласно изобретению обладают ингибирующей активностью на фермент FAAH.
Активность in vivo соединений по изобретению была оценена в тесте на анальгезию.
Так, внутрибрюшинное (i.p.) введение PBQ (фенилбензохинон, 2 мг/кг в 0,9%-ном растворе хлорида натрия, содержащем 5% этанола) самцам мышей линии OF1 весом 25-30 г, вызывает растяжение брюшины, в среднем 30 растяжений, или сжатий в течение периода от 5 до 15 минут после инъекции. Испытываемые соединения вводились перорально в суспензии Tween 80 в концентрации 0,5%, за 60 или 120 минут до введения PBQ. В этих условиях самые сильнодействующие соединения по изобретению на 35-70% снижают число растяжений, вызванных PBQ, в диапазоне доз, составляющем от 1 до 30 мг/кг. Например, соединение таблицы снижает через 2 часа число растяжений, вызванных PBQ, на 51% при дозе 1 мг/кг.
Фермент FAAH (Chemistry and Physics of Lipids, (2000), 108, 107-121) катализирует гидролиз эндогенных производных амидов и сложных эфиров разных жирных кислот, таких как N-арахидоноилэтаноламин (анандамид), N-пальмитоилэтаноламин, N-олеоилэтаноламин, олеамид или 2-арахидоноилглицерин. Кроме того, эти производные проявляют разную фармакологическую активность во взаимодействии с каннабиоидными и ваниллоидными рецепторами.
Соединения по изобретению блокируют этот путь деградации и увеличивают тканевую долю этих эндогенных веществ. Они могут применяться для этой цели при профилактике и лечении патологий, в которые вовлечены эндогенные каннабиоиды и/или любые другие субстраты, метаболизованные ферментом FAAH.
Можно назвать, например, следующие заболевания и расстройства:
боли, в частности, острые или хронические боли нейрогенного типа: мигрень, невропатические боли, включая формы, связанные с вирусом герпеса и диабетом; острые и хронические боли, связанные с воспалительными заболеваниями: артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулярит, болезнь Крона, синдром раздраженной кишки;
острые и хронические периферические боли;
головокружения, рвота, тошнота, в частности, являющиеся результатом химиотерапии;
расстройства пищевого поведения, в частности, анорексия и кахексия различной природы;
неврологические и психиатрические патологии: дрожание, дискинезия, дистония, спастичность, обсессивно-компульсивные расстройства, синдром Туретта, все формы депрессии и тревоги любой природы и происхождения, расстройства настроения, психозы; острые и хронические нейродегенеративные заболевания: болезнь Паркинсона, болезнь Альцгеймера, сенильная деменция, хорея Хантингтона, поражения, связанные с церебральной ишемией и черепно-мозговыми травмами;
эпилепсия;
нарушения сна, включая остановку дыхания во сне;
сердечно-сосудистые заболевания, в частности гипертензия, сердечная аритмия, артериосклероз, сердечные приступы, ишемия сердца;
почечная ишемия;
рак: доброкачественные опухоли кожи, папилломы и опухоли мозга, опухоли простаты, опухоли мозга (глиобластомы, медулло-эпителиомы, медуллобластомы, нейробластомы, опухоли эмбрионального происхождения, астроцитомы/астробластомы, эпендиомы, олигодендроглиомы, опухоли в сплетениях, нейроэпителиомы, опухоли эпифиза, эпендимобластомы, злокачественные менингиомы, саркоматозы, злокачественные меланомы, шванномы);
расстройства иммунной системы, в частности аутоиммунные заболевания: псориаз, красная волчанка, заболевания соединительной или связующей ткани, синдром Шегрена, анкилостомный спондилоартрит, недифференцированный спондилоартрит, болезнь Бехчета, аутоиммунные гемолитические анемии, рассеянный склероз, боковой амиотрофический склероз, амилозы, отторжение пересадок, заболевания, действующие на плазмоцетарный ряд;
аллергические заболевания: мгновенная или замедленная гиперчувствительность, риниты или аллергические конъюнктивиты, контактные дерматиты;
инфекционные паразитарные заболевания, вирусные или бактериальные заболевания: СПИД, менингит; воспалительные заболевания, в частности заболевания суставов: артрит, ревматоидный артрит, остеоартрит, спондилит, подагра, васкулярит, болезнь Крона, синдром раздраженной кишки; остеопороз; глазные болезни: глазная гипертензия, глаукома;
болезни легких: заболевания дыхательных путей, бронхоспазмы, кашель, астма, хронический бронхит, хронические непроходимость дыхательных путей, эмфизема; желудочно-кишечные заболевания: синдром раздраженного кишечника, воспалительные кишечные расстройства, язвы, диарея;
недержание мочи и воспаление мочевого пузыря.
Применение соединений формулы (I), в состоянии фармацевтически приемлемого основания, соли, гидрата или сольвата, для получения медикамента, предназначенного для лечения вышеназванных патологий, является составной частью изобретения.
Объектом изобретения являются также лекарственные средства, которые содержат соединение формулы (I) или соль, или также фармацевтически приемлемый гидрат или сольват соединения формулы (I). Эти медикаменты находят свое применение в терапии, в частности при лечении вышеназванных патологий.
Согласно одному из других аспектов, настоящее изобретение относится к фармацевтическим композициям, содержащим в качестве действующего вещества по меньшей мере одно соединение формулы (I). Эти фармацевтические композиции содержат эффективную дозу соединения согласно изобретению, или фармацевтически приемлемую соль, или гидрат, или сольват указанного соединения, и при необходимости один или несколько фармацевтически приемлемых эксципиентов.
Указанные эксципиенты выбирают, в соответствии с желаемой фармацевтической формой и способом введения, из обычных эксципиентов, которые специалисту известны.
В фармацевтических композициях по настоящему изобретению для перорального, сублингвального, подкожного, внутримышечного, внутривенного, топического, местного, интратрахеального, интраназального, трансдермального введения, введения в легкие, глаза или ректально, действующее вещество вышеуказанной формулы (I), или его соль, сольват или возможный гидрат, может быть введено в виде единой формы введения, в смеси с классическими фармацевтическими эксципиентами, животным и человеку для профилактики или лечения указанных выше расстройств или заболеваний.
Подходящие единые формы введения содержат формы для перорального приема, такие как таблетки, мягкие или твердые желатинозные капсулы, порошки, гранулы, жевательные резинки и растворы или суспензии для полости рта, сублингвальные формы приема, через рот, интратрахеальные, внутриглазные, интраназальные, ингаляционные, формы подкожного, внутримышечного или внутривенного введения, формы ректального или вагинального введения. Для топического применения соединения согласно изобретению можно применять в кремах, помадах или лосьонах.
К примеру, одна единая форма введения соединения согласно изобретению в виде таблетки может содержать следующие компоненты:
Соединение согласно изобретению 50,0 мг
Маннитол 223,75 мг
Кроскармелоза натрия 6,0 мг
Кукурузный крахмал 15,0 мг
Гидроксипропил-этилцеллюлоза 2,25 мг
Стеарат магния 3,0 мг
Указанные единые формы могут быть дозированы так, чтобы позволить суточный прием от 0,01 до 20 мг действующего вещества на кг веса тела, согласно галеновой форме.
Могут быть особые случаи, когда подходят более высокие или более низкие дозировки; такие дозировки также не выходят за рамки изобретения. Согласно обычной практике, дозы, подходящие для каждого пациента, определяются врачом в соответствии со способом приема, весом и восприимчивостью указанного пациента.
Настоящее изобретение, согласно другому аспекту, относится также к способу лечения указанных выше патологий, который включает прием пациентом эффективной дозы соединения согласно изобретению, или одной из его фармацевтически приемлемых солей, или гидратов, или сольватов.
Реферат
Изобретение относится к новому соединению, обладающему ингибирующим действием в отношении фермента FAAH, отвечающему общей формуле (I), в которой переменные m, n, X, R1 и R2, R3, R4 и Y принимают значения, приведенные в описании; к способу его получения, а также к применению этого соединения в терапии и к фармацевтической композиции, содержащей соединение формулы (I). 5 н. и 7 з.п.ф-лы.
Формула
в которой m означает 0, 1, 2 или 3;
n означает 0, 1, 2 или 3;
X означает атом кислорода, или серы, или SO2;
R1 и R2 независимо друг от друга означают атом водорода или R1 и R2 вместе образуют группу -(СН2)р-, где р означает целое число от 1 до 5 такое, что n+p является целым числом от 2 до 5;
R3 означает атом водорода или метильную группу;
R4 означает группу общей формулы CHR5CONHR6, в которой
R5 означает атом водорода или C1-6-алкильную группу, и
R6 означает атом водорода или С1-6-алкильную группу;
Y означает группу Y1, выбранную, в частности, из фенила, пиридинила, пиримидинила, тиазолила, нафтила, хинолинила, изохинолинила, бензоксазолила; причем группа Y1 при необходимости замещена одним или несколькими заместителями Y2, одинаковыми или отличающимися друг от друга, или группой Y3;
Y2 означает атом галогена, группу циано, C1-8-алкил, C1-8-алкокси, C1-8-фторалкил, C1-8-фторалкокси, фенилокси, фенил-С1-8-алкилен;
Y3 означает фенильную группу; причем группа или группы Y3 могут быть замещены одной или несколькими группами Y2, одинаковыми или отличающимися друг от друга;
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
причем группа Y1 при необходимости замещена одним или несколькими заместителями Y2, одинаковыми или отличающимися друг от друга, или группой Y3;
Y2 означает атом галогена, группу циано, С1-8-алкил, C1-8-алкокси, C1-8-фторалкил, C1-8-фторалкокси, фенилокси, фенил-С1-С8-алкилен;,
Y3 означает фенильную группу; причем Y3 может быть замещена одной или несколькими группами Y2, одинаковыми или отличающимися друг от друга;
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
n означает 0, 1, 2 или 3;
R1 и R2 независимо друг от друга означают атом водорода или R1 и R2 вместе образуют группу -(СH2)р-, где р означает целое число от 1 до 5 такое, что n+р является целым числом от 2 до 5;
при условии, что когда R1 и R2 независимо друг от друга означают атом водорода или C1-3-алкильную группу, то m+n≥1;
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
m означает 0, 1, 2 или 3; и/или
n означает 0, 1, 2 или 3; и/или
R1 и R2 вместе образуют группу -(СН2)р-, где р означает целое число от 1 до 4 такое, что n+р равно 4;
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
в форме фармацевтически приемлемого основания, фармацевтически приемлемой кислотно-аддитивной соли.
[2-(метиламино)-2-оксоэтил]-{2-[(4-хлорфенил)окси]этил}карбамата;
[2-амино-2-оксоэтил]-(2-[(4-цианофенил)окси]этил)карбамата;
[2-амино-2-оксоэтил]-[4-(1-нафталенилокси]бутил]карбамата;
[2-(метиламино)-2-оксоэтил]-4-[(4'-фтор-4-бифенил)окси]-1-пиперидинкарбоксилата;
[2-(метиламино)-2-оксоэтил]-4-{[(4-бромфенил)окси]метил}-1-пиперидинкарбоксилата;
[2-(метиламино)-2-оксоэтил]-4-{[(4'-(трифторметил)-4-бифенил)окси]метил}-1-пиперидинкарбоксилата;
[2-амино-2-оксоэтил]-4-[(1-нафталенилокси)метил]-1-пиперидинкарбоксилата;
[2-амино-2-оксоэтил]-4-[(7-хинолинилокси)метил]-1-пиперидинкарбоксилата;
[2-(метиламино)-2-оксоэтил]-4-{2-[(4-бромфенил)окси]этил}-1-пиперидинкарбоксилата;
[2-(метиламино)-2-оксоэтил]-4-{2-[(4'-хлор-4-бифенил)окси]этил}-1-пиперидинкарбоксилата;
[2-амино-2-оксоэтил]-4-[2-(7-изохинолинилокси)этил]-1-пиперидинкарбамата;
[2-амино-2-оксоэтил]-3-[(1-нафталенилокси)метил]-1-пирролидинкарбоксилата.
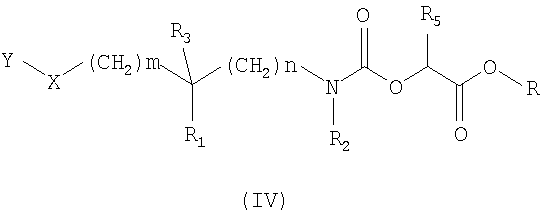
в которой Y, X, R1, R2, R3, R5, m и n определены в любом из пп.1-7 и R означает метильную или этильную группу,
амином формулы R6NH2, где R6 определен в формуле (I) по п.1.
Документы, цитированные в отчёте о поиске
Способ получения замещенных (1-бензилпирролидинил-2-алкил)бензамидов или их кислых или аммонийных солей или их правовращающих или левовращающих изомеров
Комментарии