Способ получения монозащищенных α,ω-диаминоалканов - RU2747400C2
Код документа: RU2747400C2
Описание
Область техники, к которой относится изобретение
Настоящее изобретение относится к усовершенствованному способу получения монозащищенных α,ω-диаминоалканов.
Уровень техники
α,ω-Диаминоалканы являются важными синтетическими интермедиатами, использующимися в органической химии. Они применяются в синтезе, например, противотуберкулезных препаратов этамбутол и SQ109 (K.A. Sacksteder et al., Future Microbiol., 2012, 7(7), 823-837), в полимерах, например, при сополимеризации 2,2-бис(акриламидо)уксусной кислоты (BAC) и 1,4-бис(акрилоил)пиперазина (BP) с N-трифенилметил-монозамещенными 1,2-диаминами (B. Malgesini et al., Macromol. Biosci., 2003, 3, 59-66), и в дендримерах, таких, как дендример полиамидоамина (D.A. Tomalia et al., Polymer Journal, 1985, 17, 117-132).
Кроме того, α,ω-диаминоалканы применяются в качестве самоотщепляющихся циклизационных спейсерах в пролекарствах (F.M.H. de Groot et al., J. Med. Chem., 2000, 43 (16), 3039-3102, S.C. Jeffrey et al., ACS Med. Chem. Lett., 2010, 1, 277-280). Такие циклизационные спейсеры имеют следующую структуру:

Пролекарства, содержащие спейсерную систему, состоящую из α,ω-диаминоалканового циклизационного спейсера и электронного каскадного спейсера, показали повышенную скорость выделения активного лекарственного соединения после ферментативной активации (F.M.H. de Groot et al., J. Org. Chem., 2001, 66(26), 8815-8830). Быстрое высвобождение активного лекарственного соединения в целевом участке особенно важно в области конъюгатов антитело-лекарственное средство (ADC).
Стабильность в плазме ADC, содержащих α,ω-диаминоалкан в качестве самоотщепляющегося циклизационного спейсера, зависит от замещения на аминах α,ω-диаминоалкана. ADC, в которых замещение на этих аминах является несимметричным, демонстрируют самую высокую стабильность в плазме (R. Elgersma et al., Mol. Pharmaceutics, 2015, 12, 1813-1835).
Для синтеза соединений, содержащих несимметрично замещенные α,ω-диаминоалкановые фрагменты, необходимо использовать α,ω-диаминоалкан, в котором одна аминогруппа является защищенной, т.е., монозащищенное производное, чтобы предотвратить образование двух разных соединений, чего не происходит в случае синтеза соединений, содержащих симметричные α,ω-диаминоалкильные фрагменты. Однако, и для синтеза соединений, содержащих симметричные α,ω-диаминоалкильные фрагменты, тоже выгодно использовать монозащищенные α,ω-диаминоалкановые фрагменты, так как на технологической стадии, приводящей к соединению незащищенных симметричных α,ω-диаминоалканов в молекулу, необходимо использовать избыток незащищенного симметричного α,ω-диаминоалкана, чтобы предотвратить образование побочных продуктов. При использовании монозащищенного α,ω-диаминоалкана такой избыток не требуется.
Однако известные в настоящее время способы синтеза монозащищенных α,ω-диаминоалканов имеют ряд недостатков.
В примере 1 документа WO2011133039 описывается синтез монозащищенных α,ω-диаминоалканов путем восстановительного аминирования (путь А) или путем алкилирования (путь В).
Выход монозащищенных α,ω-диаминоалканов по пути А является низким (27-63%) из-за сложности очистки. Кроме того, реакцию Сверна проводят при температуре -60°C, что невыгодно для промышленного масштаба, например реакция включает использование растворителя ДМСО, который нелегко удалить, и в реакции образуются альдегидные интермедиаты, которые являются нестабильными и поэтому проблематичными в обращении и очистке. В результате процесс восстановительного аминирования требует двух неизбежных и сложных этапов хроматографической очистки.
Путь B также приводит к низкому выходу монозащищенных α,ω-диаминоалканов и имеет ряд дополнительных недостатков. В частности, наблюдается разложение промежуточного мезилата и переалкилирование (двукратное алкилирование амина), что приводит к различным побочным продуктам. Поэтому неизбежна сложная хроматографическая очистка для отделения желаемого продукта от побочных продуктов является неизбежной. Кроме того, последняя стадия процесса проводится в течение ночи при температуре 60°C, а такие условия не являются предпочтительными для промышленного масштаба.
Таким образом, существует потребность в усовершенствованном способе получения монозащищенных α,ω-диаминоалканов. В частности, было бы желательным иметь способ, эффективный с точки зрения выхода и химической чистоты, недорогой в отношении реагентов и реакционных условий, и подходящий для применения на промышленном масштабе.
Краткое описание изобретения
Настоящее изобретение относится к способу синтеза монозащищенных α,ω-диаминоалканов, применению указанного способа в процессе синтеза лекарственного соединения-линкера, содержащего α,ω-диаминоалкановый фрагмент, и к применению способа по настоящему изобретению в процессе получения конъюгата антитело-лекарственное средство, содержащего α,ω-диаминоалкановый фрагмент.
Подробное описание изобретения
α,ω-Диаминоалканы, в частности, монозащищенные α,ω-диаминоалканы, являются важными интермедиатами синтеза, использующимися в органической химии. Наряду с прочим, α,ω-диаминоалканы применяются в качестве самоотщепляющегося циклизационного спейсера в пролекарствах, таких, как ADC.
ADC, содержащие спейсерную систему, состоящую из α,ω-диаминоалкана как циклизационного спейсера и электронного каскадного спейсера, демонстрируют повышенную скорость выделения активного лекарственного соединения после ферментативной активации.
Для синтеза таких пролекарств, содержащих α,ω-диаминоалкан, выгодно использовать α,ω-диаминоалкан, в котором одна аминогруппа является защищенной, т.е., монозащищенный α,ω-диаминоалкан.
В одном варианте осуществления изобретение относится к способу синтеза соединения формулы (III)

включающему стадии: a) превращение соединения формулы (I) в соединение формулы (II) путем реакции соединения формулы (I) с соединением формулы (IV) в присутствии связыващего реагента

и b) обработка соединения формулы (II) борановым реагентом, с последующим кислотным гидролизом;
причем R и R1 независимо выбраны из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8 алкилов, возможно замещенного, C3-15 циклоалкила, возможно замещенного, и C3-15 гетероциклоалкила, возможно замещенного,
P является защитной группой,
m означает целое число от 1 до 3, и
n означает целое число от 1 до 12.
Исходные материалы для настоящего изобретения, т.е., соединения формулы (I) и (IV), либо имеются в продаже, либо могут быть получены способами и по методикам, хорошо известным в уровне техники.
В типичных структурах во всем настоящем описании и в формуле изобретения буквы, кроме R, R1, и R2, используются для определения структурных элементов. Чтобы не принять ошибочно эти буквы за обозначения атомов, они выделены жирным шрифтом, когда не представляют атом.
Разработка пути синтеза позволяет комбинировать стадию образования пептидной связи с высоким выходом и эффективную стадию восстановления бора (относительно такой стадии восстановления бора смотри E.R. Burkhardt et al., Chem. Rev., Am. Chem. Soc., 2006, 106(7), 2617-2650). Со способом по настоящему изобретению соединения формулы (III) можно получить с выходом выше 70%, предпочтительно выше 75%, более предпочтительно выше 80%, и с химической чистотой выше 90% без необходимости каких-либо стадий хроматографической очистки.
Термин "алкил", используемый в настоящем описании, относится к линейной или разветвленной насыщенной углеводородной цепи. Подходящие примеры C1-8 алкильных групп включают метил, этил, изопропил и бутил.
Термин "циклоалкил", используемый в настоящем описании, относится к циклической насыщенной углеводородной цепи. Подходящие примеры циклоалкильных групп включают циклопропан, циклобутан, циклопентан, циклогексан, циклогептан и циклооктан.
Термин "гетероциклоалкил", используемый в настоящем описании, относится к циклической насыщенной углеводородной цепи, в которой один или несколько атомов углерода замещены гетероатомом. Подходящие примеры гетероатомов включают N, O и S.
Термин "циклический насыщенный углеводород", используемый в настоящем описании, относится к циклическим соединениям, в которых все атомы, входящие в состав циклического соединения, соединены одинарной связью с другими атомами, без двойных или тройных связей.
Термин "замещенный", когда он используется в качестве определения к "алкилу", "циклоалкилу" или "гетероциклоалкилу", указывает, что указанный "алкил", "циклоалкил" или "гетероциклоалкил" содержит один или более заместителей, которые замещают водород. Примеры заместителей включают OH, C1-8 алкил и (CH2CH2O)iH, где i означает целое число от 1 до 6.
В одном предпочтительном варианте осуществления настоящего изобретения R и R1 независимо выбраны из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8 алкила, возможно замещенного, C3-15 циклоалкила, возможно замещенного, и C3-15 гетероциклоалкила, возможно замещенного, причем n означает целое число от 1 до 12, предпочтительно от 1 до 6.
Во втором предпочтительном варианте осуществления настоящего изобретения R и R1 независимо выбраны из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, C1-8 алкила, возможно замещенного, где n означает целое число от 1 до 12, предпочтительно от 1 до 6.
В третьем предпочтительном варианте осуществления настоящего изобретения R является C1-8 алкилом, и R1 выбран из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8 алкила, возможно замещенного, C3-15 циклоалкила, возможно замещенного, и C3-15 гетероциклоалкила, возможно замещенного; причем n означает целое число от 1 до 12, предпочтительно от 1 до 6.
В следующем предпочтительном варианте осуществления настоящего изобретения R означает CH3, и R1 выбран из (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3 и C1-8 алкила, возможно замещенного; причем n означает целое число от 1 до 12, предпочтительно от 1 до 6.
В следующем предпочтительном варианте осуществления настоящего изобретения R означает CH3, и R1 выбран из (CH2CH2O)nH, (CH2CH2O)nCH3 и C1-8 алкила, возможно замещенного; причем n означает целое число от 1 до 12, предпочтительно от 1 до 6.
Типично P является аминозащитной группой. Подходящие аминозащитные группы широко описаны в литературе, например, P.G.M. Wuts и T.W. Greene в "Greene's Protective Groups in Organic Synthesis", Fourth Edition, 2006 (ISBN: 978-0-471-69754-1). Специалист в данной области способен выбрать подходящие защитные группы для применения в соответствии со способом по настоящему изобретению.
В одном варианте осуществления настоящего изобретения P выбран из группы, состоящей из 1,1-диметил-2-галоэтилкарбамата, 1,1-диметил-2,2-дибромэтилкарбамата, трет-бутилкарбамата, 1-адамантилкарбамата, N-(2-пивалоиламино)-1,1-диметилэтил карбамата, 2-[(2-нитрофенил)дитио]-1-фенилэтилкарбамата, 9-антрилметилкарбамата, метилкарбамата, этилкарбамата, п-метоксибензилкарбамата, N-гидроксипиперидинил,п-нитробензил карбамата, дифенилметилкарбамата, м-хлор-п-ацилоксибензил карбамата, п-(дигидроксиборил)бензилкарбамата, 5-бензизоксазолилметил карбамата, бензил,N-2,4,6-триметилбензолсульфонила, N-толуолсульфонила, N-бензилсульфонила и N-трифторметилсульфонила.
Предпочтительно, P выбран из группы, состоящей из 1,1-диметил-2-галоэтилкарбамата, 1,1-диметил-2,2-дибромэтил карбамата, трет-бутилкарбамата, 1-адамантилкарбамата, N-(2-пивалоиламино)-1,1-диметилэтилкарбамата, 2-[(2-нитрофенил)дитио]-1-фенилэтилкарбамата и 9-антрилметилкарбамата.
Более предпочтительно, P является трет-бутилкарбаматом (Boc).
В одном варианте осуществления способ по изобретению применяется для синтеза соединения формулы (III), выбранного из

где j означает целое число от 1 до 12, предпочтительно от 1 до 6.
В одном предпочтительном варианте осуществления способ по изобретению применяется для синтеза соединения формулы (III), выбранного из

где j означает целое число от 1 до 12, предпочтительно от 1 до 6.
Более предпочтительно, способ по изобретению применяется для синтеза соединения формулы (III), выбранного из

Еще более предпочтительно, способ по изобретению применяется для синтеза соединения формулы (III), представляющего собой

Термин "связывающий реагент" используемый в настоящем описании, означает реагент для образования пептидной связи, известный специалистам в данной области. Подходящими реагентами для образования пептидной связи являются карбодиимидные реагенты, фосфониевые реагенты и аминиевые реагенты. Подходящие примеры описаны в литературе, например, в статье Ayman El-Faham et al., Chem. Rev., 2011, 111, 6557-6602.
Предпочтительным связывающим реагентом является карбодиимидный реагент. Более предпочтителен карбодиимидный реагент, выбранный из группы, состоящей из DCC (N,N′-дициклогексилкарбодиимид), DIC (N,N'-диизопропилкарбодиимид) и EDC (1-[(3-диметиламинопропил)]-3-этилкарбодиимидгидрохлорид). Наиболее предпочтительным карбодиимидным реагентом является EDC.
Подходящий фосфониевый реагент выбран из группы, состоящей из BOP (бензотриазол-1-илокси-трис-(диметиламино)фосфоний гексафторфосфат), PyBOP (бензотриазол-1-илокси-трипирролидинофосфоний гексафторфосфат), PyBrOP (бром-трипирролидинофосфоний гексафторфосфат), PyAOP (7-аза-бензотриазол-1-илокси-трипирролидинофосфоний гексафторфосфат) и PyOxim (этилциано(гидроксиимино)ацетато-O2)-три-(1-пирролидинил)-фосфоний гексафторфосфат).
Подходящий аминиевый реагент выбран из группы, состоящей из TBTU (O-(бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторборат), HCTU (2-(6-хлор-1H-бензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат), HDMC (N-[(5-хлор-1H-бензотриазол-1-ил)-диметиламиноморфолино]уроний гексафторфосфата N-оксид), HATU (O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилуроний гексафторфосфат), COMU (1-[1-(циано-2-этокси-2-оксоэтилиденаминоокси)-диметиламиноморфолино]уроний гексафторфосфат), TOTT (2-(1-оксипиридин-2-ил)-1,1,3,3-тетраметилизотиоуроний тетрафторборат), HBTU (N,N,N′,N′-тетраметил-O-(1H-бензотриазол-1-ил)уроний гексафторфосфат) и TFFH (фтор-N,N,N′,N′-тетраметилформамидиний гексафторфосфат). В настоящем изобретении аминиевый реагент для использования в реакции сочетания может использоваться с или без основания.
Подходящим растворителем для использования в реакции сочетания является, без ограничений, органический растворитель, предпочтительно полярный апротонный растворитель. Предпочтительно, растворитель выбран из группы, состоящей из DMA (N,N-диметилацетамид), DMF (диметилформамид), DMSO (диметилсульфоксид), NMP (метилпирролидон), HMPA (гексаметилфосфорамид), метилэтилкетона, ацетонитрила, THF (тетрагидрофуран), DCM (дихлорметан), ацетона, EtOAc (этилацетат) и 2-бутанона. Предпочтительным растворителем является DCM.
Можно использовать типичную температуру реакции от 0°C до 40°C. В одном частном варианте осуществления настоящего изобретения реакционную смесь сначала охлаждают до 0°C, добавляют соединение формулы (IV), и затем реакционную смесь нагревают до комнатной температуры.
Термин "комнатная температура" используемый в настоящем описании, означает температуру в интервале от 17°C до 27°C.
Подходящим борановым реагентом для реакции с соединением формулы (II) является боран, диборан или комплекс боран-лиганд. Предпочтительно, комплекс боран-лиганд представляет собой боран-тетрагидрофуран, 9-борабицикло[3.3.1]нонан (9-BBN) или борансульфид. Более предпочтительными боран-лигандными комплексами являются боран-тетрагидрофуран или боран-диметилсульфид. Боран-диметилсульфид является особенно предпочтительным комплексом боран-лиганд. Согласно настоящему изобретению боран, диборан или комплекс боран-лиганд используются в количестве от 1 до 10 эквивалентов. Специалист в данной области сможет выбрать подходящее количество борана, диборана или комплекса боран-лиганд для использования в соответствии со способом по настоящему изобретению.
В предпочтительном варианте осуществления настоящего изобретения реакция проводится с использованием около 5 эквивалентов борандиметилсульфида.
Подходящим растворителем для реакции соединения формулы (II) с борановым реагентом является, без ограничений, органический растворитель, предпочтительно апротонный растворитель. Предпочтительным растворителем является эфирный растворитель, особенно предпочтителен THF.
Типично можно использовать температуру реакции от 0°C до 50°C. В одном частном варианте осуществления настоящего изобретения реакционную смесь сначала охлаждают до 0°C, добавляют борный реагент и затем смесь нагревают до комнатной температуры. Затем смесь перемешивают при комнатной температуре.
Полученный борановый интермедиат гидролизуют путем кислотного гидролиза, получая циклизационное спейсерное соединение формулы (III). Согласно настоящему изобретению, кислота, подходящая для применения в указанном кислотном гидролизе, является сильной неорганической или органической кислотой. Подходящую кислоту предпочтительно выбирать из группы, состоящей из алкил- или (факультативно замещенный алкил)сульфоновой кислоты, арил- или (факультативно замещенный арил)сульфоновой кислоты, гидросульфата калия, серной кислоты, соляной кислоты, бромистоводородной кислоты, фосфорной кислоты, трифторуксусной кислоты, трихлоруксусной кислоты, хлоруксусной кислоты, фосфоновой кислоты, бензилфосфорной кислоты, уксусной кислоты и муравьиной кислоты.
Предпочтительная кислота выбрана из группы, состоящей из алкил- или (факультативно замещенный алкил)сульфоновой кислоты и арил- или (факультативно замещенный арил)сульфоновой кислоты.
В более предпочтительном варианте осуществления настоящего изобретения кислота представляет собой алкил- или (факультативно замещенный алкил)сульфоновую кислоту, выбранную из группы, состоящей из метансульфоновой кислоты, этансульфоновой кислоты, камфорсульфоновой кислоты, нонанфторбутан-1-сульфоновой кислоты и 2-гидроксиэтансульфоновой кислоты.
В еще более предпочтительном варианте осуществления настоящего изобретения кислота представляет собой арил- или (факультативно замещенный арил)сульфоновую кислоту, выбранную из группы, состоящей из бензолсульфоновой кислоты, 4-толуолсульфоновой кислоты и нафталинсульфоновой кислоты.
В наиболее предпочтительном варианте осуществления кислота является 4-толуолсульфоновой кислотой.
Кислотный гидролиз может протекать в органическом растворителе, воде или смеси того и другого. Предпочтительным растворителем является низший спирт (т.е., C1-C4 алифатический спирт), особенно предпочтительным растворителем является MeOH (метанол). Можно использовать температуру реакции от 0°C до 80°C, но в одном предпочтительном варианте осуществления настоящего изобретения реакция проводится при температуре ниже 30°C. В следующем предпочтительном варианте осуществления настоящего изобретения реакция проводится при температуре от 17°C до 27°C. В одном частном варианте осуществления настоящего изобретения реакция проводится при 25°C.
Специалист в данной области способен выбрать подходящую кислоту и подходящий растворитель, чтобы провести гидролиз при температуре ниже 30°C.
В дополнительном варианте осуществления способ синтеза соединения формулы (III) используется в процессе синтеза лекарственного соединения-линкера формулы (VIII)

где RM означает химически активный фрагмент;
L означает связывающую группу, связывающую RM с V1;
V1 означает пептид, моно, ди- или олигосахарид;
Y либо отсутствует, либо означает электронный каскадный спейсер;
Z означает терапевтический или диагностический фрагмент;
R и R1 независимо выбраны из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8 алкила, возможно замещенного,C3-15 циклоалкила, возможно замещенного, и C3-15 гетероциклоалкила, возможно замещенного;
P означает защитную группу;
m означает целое число от 1 до3; и
n означает целое число от 1 до 12.
Предпочтительно, n означает целое число от 1 до 6.
В настоящем изобретении RM является химически активным фрагментом. Термин "химически активный фрагмент" относится здесь к функциональной группе, которая может реагировать со второй функциональной группой в относительно мягких условиях и без необходимости предварительной функционализации химически активного фрагмента. Примеры химически активных фрагментов включают, без ограничений, карбамоилгалогенид, ацилгалогенид, активный сложный эфир, ангидрид, α-галоацетил, α-галоацетамид, малеимид, изоцианат, изотиосианат, дисульфид, тиол, гидразин, гидразид, сульфонилхлорид, альдегид, метилкетон, винилсульфон, галометил и метилсульфонат.
Предпочтительно, RM выбран из группы, состоящей из

где
X выбран из галогенида, гидроксила, OC(O)Ra и OC(O)ORa, или C(O)-X является активным сложным эфиром;
X' выбран из галогенида, мезилокси, трифлилокси и тозилокси; и
Ra выбран из C1-10 алкила, C1-10 гетероалкила, C3-10 циклоалкила, C1-10 гетероциклоалкила, C5-10 арила и C1-10 гетероарила, возможно замещенных.
В более предпочтительном варианте осуществления RM выбран из
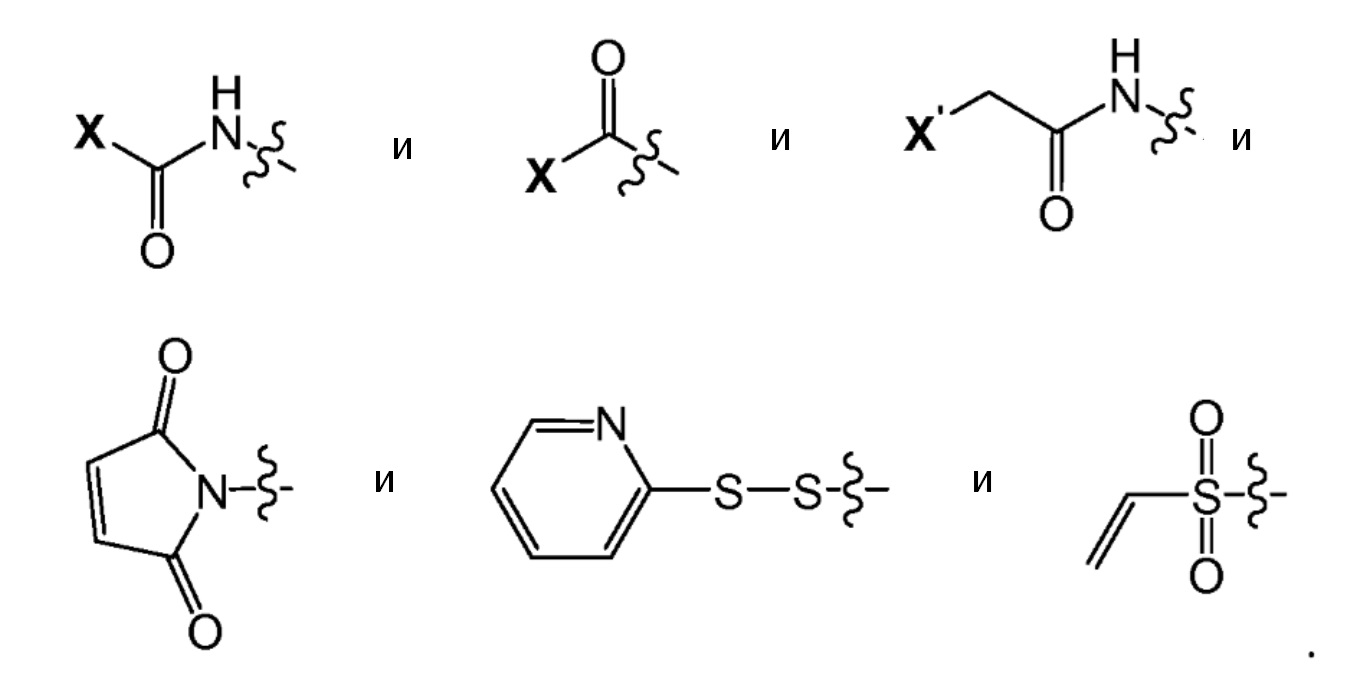
В наиболее предпочтительном варианте осуществления RM представляет собой

В настоящем изобретение L является связывающей группой. Термин "связывающая группа" относится к химической структуре, которая ковалентно связывает структурный элемент RM с V1. Вместе с V1, Y и указанным ниже диамином

L образует спейсерную систему (линкер), которая ковалентно связывает структурный элемент RM с Z. Такая спейсерная система может использоваться в конъюгатах антитело-лекарственное средство (ADC) (L. Ducry et al., Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010, 21(1), 5-13).
Предпочтительно, L выбран из группы, состоящей из

где h и h' независимо друг от друга лежат в интервале от 1 до 8.
Более предпочтительно, L выбран из группы, состоящей из

В наиболее предпочтительном варианте осуществления L представляет собой

V1 означает пептид или моно-, ди- или олигосахарид.
В предпочтительном варианте осуществления изобретения V1 является пептидом. В более предпочтительном варианте осуществления V1 является дипептидом, выбранным из валилцитруллина, валиллизина, фенилаланиллизина, аланилфенилаланиллизина и D-аланилфенилаланиллизина. В наиболее предпочтительном варианте осуществления фрагмент V1 является валилцитруллином.
Y или отсутствует, или является электронным каскадным спейсером. Термин "электронный каскадный спейсер" относится к самоотщепляющемуся спейсеру, разветвленному или неразветвленному, который может сам отщепляться, например, в результате одного или более 1,2+2n электронных каскадных элиминирований (n≥1). Подходящие электронные каскадные спейсеры известны в данной области, смотри, например, F.M.H. de Groot et al., Cascade Elongated Multiple Electronic Cascade and Cyclization Spacer Systems in Activatible Anticancer Prodrugs for Enhanced Drug Release, J. Org. Chem. 2001, 66, 8815-8830; и Drug Delivery in Oncology: From Basic Research to Cancer Therapy, F. Kratz, et al., p. 558-561. Обычно электронный каскадный спейсер содержит фенильное кольцо или гетероциклическое ароматическое кольцо, замещенное сильной электронодонорной группой и связанное в орто- или пара-положении с уходящей группой через CH2, или сопряженную систему с уходящей группой, например, п-аминобензилоксикарбонил.
В предпочтительном варианте осуществления Y выбран из группы, состоящей из

где Rb, Rc, Rd и Re независимо выбраны из H, OH, SH, NH2, N3, NO2, NO, CF3, CN, C(O)NH2, C(O)H, C(O)OH, галогена, Rz, SRz, S(O)Rz, S(O)2Rz, S(O)ORz, S(O)2ORz, OS(O)Rz, OS(O)2Rz, OS(O)ORz, OS(O)2ORz, ORz, NHRz, N(Rz)Rz1,+N(Rz)(Rz1)Rz2, P(O)(ORz)(ORz1), OP(O)(ORz)(ORz1), C(O)Rz, C(O)ORz, C(O)N(Rz1)Rz, OC(O)Rz, OC(O)ORz, OC(O)N(Rz)Rz1, N(Rz1)C(O)Rz, N(Rz1)C(O)ORz, и N(Rz1)C(O)N(Rz2)Rz; причем Rz, Rz1, и Rz2 независимо выбраны из H и C1-6 алкила, C1-6 гетероалкила, C3-20 циклоалкила, C1-20 гетероциклоалкила, C6-20 арила или C1-20 гетероарила, возможно замещенных, причем Rz, Rz1, и Rz2 факультативно соединены одной или более связями, образуя один или более факультативно замещенных углеродных циклов и/или гетероциклов, причем два или более заместителей Rb, Rc, Rd, и Re факультативно соединены одной или более связями, образуя один или более факультативно замещенных углеродных циклов и/или гетероциклов;
Rf и Rh независимо выбраны из H и метила, и
k и p независимо означают целое число от 0 до 10.
В более предпочтительном варианте осуществления Y выбран из группы, состоящей из:

В наиболее предпочтительном варианте осуществления Y представляет собой

В настоящем изобретении Z является терапевтическим или диагностическим фрагментом.
Термин "диагностический фрагмент" относится к веществу, использующемуся для анализа или обнаружения заболевания или другого медицинского состояния.
Термин "терапевтический фрагмент" относится к веществу, использующемуся для лечения заболевания или состояния. Предпочтительно, терапевтический фрагмент Z представляет собой антипролиферативный или противоопухолевый агент, такой как цитотоксический агент, цитостатик, антиангиогенный агент и радиотерапевтический агент. В предпочтительном варианте осуществления Z представляет собой майтансиноид, ауристатин, доластатин, трихотецин, производное дуокармицина, димер дуокармицина, пирролобензодиазепин, α-аманитин, доксорубицин, калихеамицин или другой ендииновый антибиотик, таксан, антрациклин, или их стереоизомеры, изостеры, аналоги или производные.
В одном варианте осуществления Z является производным дуокармицина.
В предпочтительном варианте осуществления изобретения Z представляет собой

где R3 означает CH3, CH2CH3, OCH3, OCH2CH3, CF3, OCF3, Cl или F;
R4, R5, R6 независимо означают H или C1-6 алкил, или
R3 и R4 вместе образуют 5- или 6-звенную (гетеро)циклоалкильную группу;
X1 означает C или N; и
X2 и X3 независимо означают C или N.
В наиболее предпочтительном варианте осуществления изобретения, Z представляет собой

Описанный выше способ синтеза лекарственного соединения-линкера формулы (VIII) дополнительно включает стадию реакции соединения формулы (IX) с соединением формулы (III) с получением соединения формулы (X)

где A означает активированный карбонатный фрагмент;
Z' означает Z, или Z' означает Z, содержащий одну или более спирто-защищающих групп P', когда Z содержит несколько гидроксильных функциональных групп; и
R, R1, P и m таковы, как определено выше.
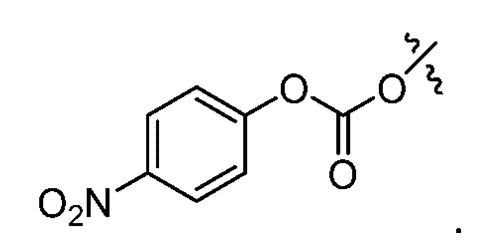
В настоящем изобретении A обозначает активированный карбонатный фрагмент. Термин "активированный карбонатный фрагмент" относится к сложному карбонатному эфиру с повышенной реакционной способностью в реакции с нуклеофилом, содержащему карбонатный фрагмент и уходящую группу.
Активированные карбонатные фрагменты A, подходящие для использования в соответствии с изобретением, специалистам в данной области известны. Примеры подходящих активированных карбонатных фрагментов можно найти, например, в A.K. Ghosh et al. J. Med Chem. 2015, 58, 2895-2940.
Предпочтительно, A выбран из группы, состоящей из

Наиболее предпочтительно, A представляет собой
Z' обычно получают по реакции Z с алкоксикарбонилирующим агентом, например, 4-нитрофенилхлорформиатом, 2-нитрофенилхлорформиатом, пентафорфенилхлорформиатом или бис-(4-нитрофенил)карбонатом, в присутствии основания.
В предпочтительном варианте осуществления настоящего изобретения Z' получен по реакции Z с 4-нитрофенилхлороформиатом.
В предпочтительном варианте осуществления настоящего изобретения Z' представляет собой

Термин "спирто-защищающая группаʺ относится к защитной группе, подходящей для защиты спиртовой группы. Подходящие спирто-защищающие группы широко описаны в литературе, смотри, например, P.G.M. Wuts, T.W. Greene в "Greene's Protective Groups in Organic Synthesis", Fourth Edition, 2006 (ISBN: 978-0-471-69754-1). Специалист в данной области способен выбрать подходящие защитные группы для использования в соответствии со способом по настоящему изобретению.
Спирто-защищающая группа, подходящая для применения в соответствии со способом по настоящему изобретению, является группой, которая стабильна в присутствии слабого основания, такого как триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин или коллидин.
Типично спирто-защищающую группу P' выбирают из группы простых эфиров, сложных эфиров и карбонатов.
В предпочтительном варианте осуществления настоящего изобретения P' является простым эфиром, выбранным из группы, состоящей из MOM (метоксиметиловый эфир), MTM (метилтиометиловый эфир), SMOM [(фенилдиметилсилил)метоксиметиловый эфир], BOM (бензилоксиметиловый эфир), PMBM (п-метоксибензилоксиметиловый эфир), п-нитробензилоксиметиловый эфир, NBOM (о-метоксибензилоксиметиловый эфир), (4-метоксифенокси)метиловый эфир, GUM (гваяколметиловый эфир), трет-бутоксиметиловый эфир, MEM (2-метоксиэтоксиметиловый эфир), 2,2,2-трихлорэтоксиметиловый эфир, бис(2-хлорэтокси)метиловый эфир, SEM [2-(триметилсилил)этоксиметиловый эфир], MM (метоксиметиловый эфир), THP (тетрагидропираниловый эфир), тетрагидротиопираниловый эфир, тетрагидрофураниловый эфир, тетрагидротиофураниловый эфир, EE (1-этоксиэтиловый эфир), CEE [1-(2-хлорэтокси)этиловый эфир], SEE {1-[2-(триметилсилил)этокси]этиловый эфир}, MIP (1-метил-1-метоксиэтиловый эфир), MBE (1-метил-1-бензилоксиэтиловый эфир), 1-метил-1-феноксиэтиловый эфир, 2,2,2-трихлорэтиловый эфир, 2-триметилсилилэтиловый эфир, 2-(бензилтио)этиловый эфир, 2-(фенилселенил)этиловый эфир, трет-бутиловый эфир, п-хлорфениловый эфир, п-метоксифениловый эфир, п-нитрофениловый эфир, бензиловый эфир, MPM (п-метоксибензиловый эфир), DMPM (3,4-диметоксибензиловый эфир), о-нитробензиловый эфир, п-нитробензиловый эфир, 2,6-дихлорбензиловый эфир, п-фенилбензиловый эфир, 2,6-дифторбензиловый эфир, 2-трифторметилбензиловый эфир, дифенилметиловый эфир, трифенилметиловый эфир, TMS (триметилсилиловый эфир), TES (триэтилсилиловый эфир), TIPS (триизопропилсилиловый эфир), IPDMS (диметилизопропилсилиловый эфир), DEIPS (диэтилизопропилсилиловый эфир), TDS (диметилгексилсилиловый эфир), TBDMS (трет-бутилдиметилсилиловый эфир), TBDPS (трет-бутилдифенилсилиловый эфир), трибензилсилиловый эфир, TPS (трифенилсилиловый эфир) и DPMS (дифенилметилсилиловый эфир); или
P' является сложным эфиром, выбранный из группы, состоящей из сложного эфира муравьиной кислоты, уксусной кислоты, никотиновой кислоты, 3-фенилпропионовой кислоты, 4-оксопентановой кислоты, 4,4-(этилендитио)пентановой кислоты, триметилуксусной кислоты, 1-адамантанкарбоновой кислоты и бензойной кислоты; или
P' является карбонатом, выбранным из группы, состоящей из метилкарбоната, метоксиметилкарбоната, алкил-2,2,2-трихлорэтил карбоната, TCBOC (1,1-диметил-2,2,2-трихлорэтилкарбонат), TMSEC [2-(триметилсилил)этилкарбонат], изобутилкарбоната и бензилкарбоната.
В более предпочтительном варианте осуществления настоящего изобретения P' является простым эфиром, выбранным из группы, состоящей из MOM (метоксиметиловый эфир), BOM (бензилоксиметиловый эфир), трет-бутоксиметилового эфира, SEM [2-(триметилсилил)этоксиметиловый эфир], EE (1-этоксиэтиловый эфир), SEE {1-[2-(триметилсилил)этокси]этиловый эфир}, трет-бутилового эфира, п-хлорфенилового эфира, п-метоксифенилового эфира, бензилового эфира, TMS (триметилсилиловый эфир), TES (триэтилсилиловый эфир), TIPS (триизопропилсилиловый эфир) и TBDMS (трет-бутилдиметилсилиловый эфир).
В наиболее предпочтительном варианте осуществления P' является сложным метоксиметиловым эфиром (MOM).
Реакция между соединением формулы (IX) и соединением формулы (III) проводится в подходящем растворителе, предпочтительно в присутствии основания. Основание будет захватывать уходящую группу. Альтернативно, реакция может быть проведена с использованием избытка соединения формулы (III).
Предпочтительно основание присутствует в количестве, эквимолярном количеству соединения (IX). Подходящим основанием является третичный амин, например триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин или коллидин. Предпочтительным основанием является триэтиламин.
Более предпочтительно, реакцию между соединением формулы (IX) и соединением формулы (III) проводят в присутствии добавки, которая дополнительно активирует карбонат соединения (IX). Добавка повышает эффективность синтеза. Подходящие добавки широко описаны в литературе (смотри, например, So-Yeop et al., Tetrahedron, 2004, 60, 2447-2467; и WO2009138985). Специалист в данной области сможет выбрать подходящую добавку для использования в соответствии со способом по настоящему изобретению.
В предпочтительном варианте осуществления добавка выбрана из группы, состоящей из HOBt (гидроксибензотриазол), HOAt (1-гидрокси-7-азабензотриазол), HODhbt (3-гидрокси-1,2,3-бензотриазин-4(3H)-он), N-гидрокситетразола, HOCt (этил-1-гидрокси-5-метил-1Н-1,2,3-триазол-4-карбоксилат), PTF (трифенил(фенилметил)фосфоний дигидротрифторид) и COMU (этил(гидроксиимино)цианоацетат.
В более предпочтительном варианте осуществления добавка представляет собой HOBt.
Подходящим растворителем для реакции между соединением формулы (IX) и соединением формулы (III) является апротонный растворитель. В предпочтительном варианте осуществления растворитель представляет собой DCM, трихлорметан, 1,2-дихлорэтан, толуол, THF, DME (диметоксиэтан), диоксан, EtOAc, ацетонитрил, ацетон, DMF или DMA. Более предпочтительно, растворитель представляет собой THF, диоксан или DMF.
Еще более предпочтительно, растворитель представляет собой DMF.
В одном частном варианте осуществления способа синтеза лекарственного соединения-линкера формулы (VIII) соединение формулы (IX) представляет собой

а соединение формулы (X) представляет собой

где R, R1, R2, R3, R4, R5, X1, X2, X3 и m определены выше.
В следующем частном варианте осуществления изобретения соединение формулы (IX) представляет собой

а соединение формулы (X) представляет собой

В другом варианте осуществления способ синтеза лекарственного соединения-линкера формулы (VIII) дополнительно включает стадию a) удаления защиты с соединения формулы (X), чтобы получить соединение формулы (Xa); и b) реакцию соединения формулы (Xa) с соединением формулы (XI) в присутствии основания:

где RM, L, V1, Y, A, R, R1, Z и m определены выше.
Реакцию удаления защиты проводят путем обработки соединения формулы (X) кислотой в подходящем растворителе. Подходящей кислотой для реакции удаления защиты является сильная кислота. Предпочтительными сильными кислотами являются трифторуксусная кислота и соляная кислота.
Подходящим растворителем для реакции удаления защиты является неводный апротоновый растворитель. Предпочтительными растворителями являются DCM, трихлорметан, 1,2-дихлорэтан, толуол, THF, DME, EtOAc или диоксан. Наиболее предпочтительным растворителем является DCM.
Реакцию между соединением формулы (Xa) и соединением формулы (XI) проводят в присутствии основания в подходящем растворителе. Подходящим основанием для реакции соединения формулы (Xa) с соединением формулы (XI) является третичный амин. Предпочтительными основаниями являются триэтиламин, диизопропилэтиламин, N-метилморфолин, пиридин или коллидин. Наиболее предпочтителен триэтиламин.
Подходящим растворителем для реакции соединения формулы (Xa) с соединением формулы (XI) является неводный апротонный растворитель, предпочтительно DCM, трихлорметан, 1,2-дихлорэтан, толуол, THF, DME, диоксан, EtOAc, ацетонитрил, ацетон, DMF или DMA. Наиболее предпочтителен DMF.
Предпочтительно, способ по изобретению применяется для синтеза лекарственного соединения-линкера формулы (VIII), которое представляет собой

где RM, L, V1, R, R1, Z и m определены выше.
Более предпочтительно, способ по изобретению применяется для синтеза лекарственного соединения-линкера формулы (VIII), которое представляет собой

где RM, L, V1, R, R1, R3, R4, R5, R6, X1, X2, X3 и m определены выше.
Более предпочтительно, способ по изобретению применяется для синтеза лекарственного соединения-линкера формулы (VIII), которое представляет собой

Кроме того, настоящее изобретение относится также к применению способа синтеза лекарственного соединения-линкера формулы (VIII), какое определено выше, в процессе получения ADC. Процесс получения ADC из лекарственного соединения-линкера формулы (VIII) можно реализовать, используя процедуры и оборудование, хорошо известные специалисту в данной области, как показано, например, в примере 15 на странице 218 документа WO2011133039 и на странице 32 документа WO2015177360.
В контексте настоящего изобретения любое антитело, в частности, любое антитело, известное как обладающее терапевтической активностью, или любое антитело, известное в области ADC, или любой его антигенсвязывающий фрагмент, например, фрагмент F(ab')2 или Fab', одноцепочечное (sc) антитело, scFv, однодоменное (sd) антитело, диатело или миниантитело может быть использовано для конъюгации (дикого типа или сайт-специфической) лекарственного соединения-линкера формулы (VIII), какое определено выше. Антитела могут иметь любой изотип, как, например, антитела IgG, IgA или IgM. Предпочтительно, антитело представляет собой антитело IgG, более предпочтительно антитело IgG1 или IgG2.
Антитело может быть моноспецифическим антителом (то есть специфическим для одного антигена, такой антиген может быть общим для видов или иметь родственные антигены между видами) или биспецифическим антителом (то есть специфическим для двух разных антигенов). В одном варианте осуществления настоящего изобретения антитело представляет собой моноспецифическое антитело или его антигенсвязывающий фрагмент.
Эти антитела могут быть получены рекомбинантным, синтетическим или другими подходящими способами, известными в данной области.
Предпочтительно, антитело связывается с антигеном-мишенью, которая экспрессируется в или на клеточной мембране опухолевой клетки (например, на поверхности клетки). Более предпочтительно, антитело поглощается клеткой после связывания с мишенью (антигеном), после чего токсин высвобождается внутриклеточно.
Как правило, антитело, которое может быть использовано в синтезе ADC с применением способа по настоящему изобретению, включает, без ограничений, антитела к аннексину Al, анти-B7H4 антитело, анти-CA6 антитело, анти-CA9 антитело, анти-CA15-3 антитело, анти-CA19-9 антитело, анти-CA125 антитело, анти-CA242 антитело, анти-CCR2 антитело, анти-CCR5 антитело, анти-CD2 антитело, анти-CD19 антитело, анти-CD20 антитело, анти-CD22 антитело, анти-CD30 антитело, анти-CD33 антитело, анти-CD37 антитело, анти-CD38 антитело, анти-CD40 антитело, анти-CD44 антитело, анти-CD47 антитело, анти-CD56 антитело, анти-CD70 антитело, анти-CD74 антитело, анти-CD79 антитело, анти-CD115 антитело, анти-CD123 антитело, анти-CD138 антитело, анти-CD203c антитело, анти-CD303 антитело, анти-CD333 антитело, анти-CEA антитело, анти-CEACAM антитело, анти-CLCA-1 антитело, анти-CLL-1 антитело, анти-c-MET антитело, анти-Cripto антитело, анти-CTLA4 антитело, анти-DLL3 антитело, анти-EGFL антитело, анти-EGFR антитело, анти-EPCAM антитело, антитела против EPh, такие, как анти-EphA2 антитело или анти-EPhB3 антитело, анти-ETBR антитело, анти-FAP антитело, анти-FcRL5 антитело, анти-FGF антитело, анти-FGFR3 антитело, анти-FOLR1 антитело, анти-GCC антитело, анти-GPNMB антитело, анти-HER2 антитело, анти-HMW-MAA антитело, анти-интегрин, анти-IGF1R антитело, анти-L6 антитело, антитело к углеводу подобному Льюису A, анти-Льюис X антитело, анти-Льюис Y антитело, анти-LIV1 антитело, анти-мезотелин антитело, анти-MUC1 антитело, анти-MUC16 антитело, анти-NaPi2b антитело, анти-нектин-4 антитело, анти-PSMA антитело, анти-PTK7 антитело, анти-SLC44A4 антитело, анти-STEAP-1 антитело, анти-5T4 (или анти-TPBG, трофобластический гликопротеин) антитело, анти-TF (тканевый фактор) антитело, анти-TF-Ag антитело, анти-Tag72 антитело, анти-TNF антитело, анти-TROP2 антитело, анти-VEGF антитело и анти-VLA антитело.
В одном предпочтительном варианте осуществления изобретения антитело представляет собой антитело против HER2, предпочтительно трастузумаб.
В следующем предпочтительном варианте осуществления изобретения конъюгат антитело-лекарственное средство представляет собой

В альтернативном варианте осуществления изобретение относится к способу синтеза соединения формулы (III)

включающему стадии:
a) превращение соединения формулы (V) в соединение формулы (VI) путем реакции соединения формулы (V) с соединением формулы (VII) в присутствии связывающего реагента

и
b) обработку соединения формулы (VI) борановым реагентом с последующим кислотным гидролизом;
причем R выбран из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8 алкила, возможно замещенного, C3-15 циклоалкила, возможно замещенного, и C3-15 гетероциклоалкила, возможно замещенного;
R2 выбран из H, (CH2)n-1OCH3,CH2O(CH2CH2O)n-1H, CH2O(CH2CH2O)n-1CH3, (CH2)n-1NHP, (CH2)n-1N(CH3)2, (CH2)n-1NHCONH2, (CH2)n-1NHSO2CH3, (CH2)n-1SO2NH2 и C1-7 алкила, возможно замещенного;
P означает защитную группу, какая определена выше;
m означает целое число от 1 до3, и
n означает целое число от 1 до 12.
Исходные материалы для описанного выше способа, т.е., соединения формулы (V) и (VII), либо доступны для приобретения, либо могут быть получены способами и процедурами, хорошо известными в уровне техники.
Превращение соединения (VI) в соединение (III) включает восстановление карбонильной группы, внешней по отношению к диаминоалкильной цепи, с образованием заместителя CH2R2, который соответствует заместителю R1 соединения формулы (III). Структура альтернативного способа также позволяет комбинировать стадию образования пептидной связи с высоким выходом и эффективную стадию восстановления бора.
В предпочтительном варианте осуществления настоящего изобретения R является C1-8 алкилом, а R2 выбран из H, (CH2)n-1OCH3,CH2O(CH2CH2O)n-1H, CH2O(CH2CH2O)n-1CH3, (CH2)n-1NHP, (CH2)n-1N(CH3)2, (CH2)n-1NHCONH2, (CH2)n-1NHSO2CH3, (CH2)n 1SO2NH2 и C1-7 алкила, возможно замещенного; n означает целое число от 1 до 12, предпочтительно n от 1 до 6.
Во втором предпочтительном варианте осуществления настоящего изобретения R означает CH3, а R2 выбран из (CH2)n-1OCH3, CH2O(CH2CH2O)n-1H, CH2O(CH2CH2O)n-1CH3 и C1-7 алкила, возможно замещенного; n означает целое число от 1 до 12, предпочтительно от 1 до 6.
В третьем предпочтительном варианте осуществления настоящего изобретения R означает CH3, а R2 выбран из CH2O(CH2CH2O)n-1H, CH2O(CH2CH2O)n-1CH3 и C1-7 алкила, возможно замещенного; n означает целое число от 1 до 12, предпочтительно от 1 до 6.
Технологические условия реакции сочетания, реакции с борановым реагентом и кислотного гидролиза аналогичны описанным выше для процесса, исходящего из соединения формулы (I).
Кроме того, в процессе синтеза вышеописанного соединения формулы (VIII) и в процессе синтеза ADC можно использовать альтернативный способ, исходящий из соединения формулы (V).
Примеры
Пример 1
Получение соединения (4) (соответствует соединению формулы (III)), из соединений (1) (соответствует соединению формулы (I)), и (5) (соответствует соединению формулы (IV)) согласно следующей схеме:

Соединение (1) (9,46 г, 50,0 ммоль) растворяли в DCM (100 мл), добавляли соединение (5) (5,52 г, 52,5 ммоль) и смесь охлаждали на льду. Добавляли EDC (9,59 г, 50,0 ммоль), смесь доводили до комнатной температуры (RT) и перемешивали 3 часа (ТСХ указывает на полную конверсию). Смесь промывали 0,5 М раствором KHSO4 (25 мл), сушили над MgSO4 и концентрировали в вакууме, получая 11,81 г соединения (2) в виде бесцветного масла (выход 85%) (химически чистое, согласно ТСХ и ЯМР).
1H-ЯМР (300 МГц, CDCl3): δ=1.47 (с, 9H, Boc), 2.75 (шир.с, 1H, OH), 2.94 (с, 3H, MeN), 3.48 (шир.кв., 2H, NCH2), 3.58 (м, 4H, OCH2), 3.73 (м, 2H, OCH2), 3.86 (с, 2H, NCH2C=O), 6.68 (шир. д, 1H, NH).
Соединение (2) (11,39 г, 41,2 ммоль) растворяли в сухом THF (200 мл) и охлаждали на льду. Затем по каплям добавляли боран диметилсульфид (19 мл, 200 ммоль) и перемешивании при RT в течение ночи. Реакционную смесь осторожно гасили МеОН (50 мл) с последующим добавлением воды (50 мл). Смесь THF/МеОН выпаривали и остаток подщелачивали NaOH (4М, 50 мл) и экстрагировали посредством DCM (4×50 мл). Затем объединенные органические слои сушили с использованием осушающего агента (например, MgSO4), фильтровали и концентрировали в вакууме, получая 11,76 г боранового интермедиата (3) (99%). Борановый интермедиат (3) (11,76 г, 41,2 ммоль) растворяли в МеОН (100 мл), добавляли гидрат 4-толуолсульфоновой кислоты (8,98 г, 47,2 ммоль) и смесь перемешивали в течение 4 часов при RT. Смесь подщелачивали NaOH (4М, 12 мл) и концентрировали в вакууме. Остаток экстрагировали посредством DCM (4×50 мл) и затем объединенные органические слои промывали NaOH (4М, 50 мл), сушили с использованием осушающего агента (например, MgSO4), фильтровали и концентрировали в вакууме, получая 8,95 г соединения (4) (выход 80%, химическая чистота 94%) в виде бесцветного масла.
1H-ЯМР (300 МГц, CDCl3): δ=1.46 (с, 9H, Boc), 2.18 (шир.с, 2H, OH+NH), 2.75-2.87 (м, 4H, 2xCH2), 2.88 (с, 3H, NMe), 3.35 (т, 2H, J=6.6 Гц, CH2), 3,56-3.63 (м, 4H, 2xCH2, CH), 3.70-3.75 (м, 2H, CH2, CH); MS(ESI) m/z=263,5 (M+H+).
Пример 2
Соединения формулы (III), полученные в соответствии с методикой примера 2, превращали в лекарственные соединения-линкеры формулы (XII) в соответствии с методикой примера 10 документа WO2011133039, страница 209.
Реферат
Изобретение относится к способу синтеза монозащищенных α,ω-диаминоалканов, а именно к способу синтеза соединения формулы (III). Способ включает стадии a) и b). На стадии a) осуществляют превращение соединения формулы (I) в соединение формулы (II) путем реакции соединения формулы (I) с соединением формулы (IV) в присутствии связывающего реагента. На стадии b) осуществляют обработку соединения формулы (II) борановым реагентом с последующим кислотным гидролизом с получением соединения формулы (III). В указанных формулах R и R1независимо выбраны из H, (CH2)nOCH3, (CH2CH2O)nH, (CH2CH2O)nCH3, (CH2)nNHP, (CH2)nN(CH3)2, (CH2)nNHCONH2, (CH2)nNHSO2CH3, (CH2)nSO2NH2, C1-8алкила, возможно замещенного, C3-15циклоалкила, возможно замещенного, и C3-15гетероциклоалкила, возможно замещенного, P является защитной группой, m означает целое число от 1 до 3 и n означает целое число от 1 до 12. Предлагаемый способ является эффективным и позволяет получать соединение формулы (III) с хорошим выходом и химической чистотой. Изобретение относится также к применению указанного способа в процессе синтеза лекарственного соединения-линкера, содержащего α,ω-диаминоалкановый фрагмент, а также в процессе получения конъюгатов антитело-лекарственное средство. 3 н. и 12 з.п. ф-лы, 2 пр.
Формула






Комментарии