Усовершенствованный способ получения ингибитора дипептидилпептидазы-iv и промежуточного соединения - RU2498976C9
Код документа: RU2498976C9
Описание
ПЕРЕКРЕСТНЫЕ ССЫЛКИ НА РОДСТВЕННУЮ ЗАЯВКУ
Эта заявка на патент испрашивает приоритет заявки на патент Кореи №10-2009-0027106, поданной 30 марта 2009 года, содержание которой включено сюда путем ссылки.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
1. Область изобретения
Настоящее изобретение относится к усовершенствованному способу производства ингибитора дипептидилпептидазы-IV и промежуточного соединения.
2. Описание предшествующего уровня техники
ДПП-IV (дипептидилпептидаза-IV) является ферментом, выполняющим функцию расщепления N-концевого дипептида пептида, имеющего концевую последовательность H-Xaa-Pro-Y (или H-Xaa-Ala-Y, где Хаа представляет собой любую липофильную аминокислоту, Pro представляет собой пролин, и Ala представляет собой аланин) (Heins J era/. Biophys Acta 1988; 161), и также называемым ДП-IV, ДП-4 или ДАП-IV. После открытия факта, что ДПП-IV разрушает глюкагоноподобный пептид-1 (в дальнейшем именуемый ГПП-1), который, как известно, оказывает мощное воздействие на функцию инсулина по контролю содержания глюкозы в крови после приема пищи (Mentlein R et al. EurJBiochem 1993:829-35), появилась возможность получения очень мощного терапевтического агента для лечения диабета II типа, и, следовательно, исследование по разработке ингибитора ДПП-IV ускорилось.
Компания Merck разработала триазолопиперазиновое соединение со структурой бета-аминокислоты, ситаглиптин, при изыскании ингибитора ДПП-IV. Соединение является первым ингибитором ДПП-IV для лечения диабета II типа и в настоящее время имеется в продаже под товарным знаком Januvia™ по всему миру после получения одобрения лекарственного средства от FDA (Управление по контролю за качеством пищевых продуктов и лекарственных препаратов) США в 2006 году. По этому вопросу в публикации патента Кореи №2008-0094604 раскрыто, что когда триазолопиперазиновая часть ситаглиптина замещена пиперазиноном, содержащим гетероатом, он обладает превосходной ингибиторной активностью в отношении ДПП-IV и также значительно улучшенной биодоступностью по сравнению с традиционным ингибитором ДПП-IV; и предложено гетероциклическое соединение, содержащее новую бета-аминогруппу, представленное следующей химической формулой 1, или его фармацевтически приемлемая соль, способ получения этого соединения и фармацевтическая композиция, которая содержит это соединение в качестве эффективного компонента, для предупреждения и лечения диабета или ожирения.
[Химическая формула 1]

(В вышеприведенной химической формуле 1, X представляет собой OR1, SR1 или NR1R2, где R1 и R2 представляют собой низший C1-С5алкил, соответственно; и R1 и R2 в NR1R2 могут представлять собой 5-7-членное кольцо, содержащее гетероатом, O.)
Как показано на следующей схеме реакции A, в публикации патента Кореи №2008-0094604 раскрыт способ получения гетероциклического соединения, представленного химической формулой 1, с бета-аминогруппой, включающий: (I) получение соединения, представленного химической формулой 4, связанного пептидной связью, путем взаимодействия соединения с бета-аминогруппой, представленного химической формулой 2, и замещенного гетероциклического соединения, представленного химической формулой 3, с использованием 1-гидроксибензотриазола (НОВТ), 1-этил-3-(3-диметиламинопропил)карбодиимида (EDC) и третичного амина; и (II) взаимодействие соединения, представленного химической формулой 4, в кислой среде:
[Схема реакции A]
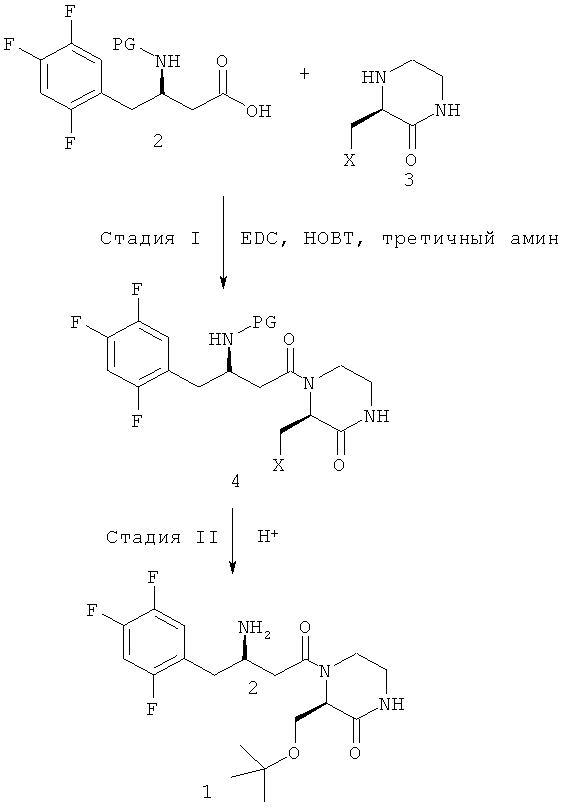
(на вышеприведенной схеме реакции A, X является таким, как определено в вышеприведенной химической формуле 1).
В то же время соединение с бета-аминогруппой, представленное химической формулой 2 в вышеприведенной схеме реакции А, может быть использовано для получения различных ингибиторов ДПП-IV, как раскрыто в бюллетенях с опубликованными международными заявками на патент WO 03/000181, WO 03/004498, WO 03/082817, WO04/007468, WO 04/032836, WO 05/011581, WO 06/097175, WO 07/077508, WO 07/063928, WO 08/028662, WO 08/087560 и им подобным, помимо получения ингибитора ДПП-IV, представленного вышеприведенной химической формулой 1, и может быть получено различными способами.
Например, соединение, представленное вышеприведенной химической формулой 2, может быть получено с использованием способа, раскрытого в J. Med. Chem. 2005; 141 и Synthesis 1997,873, как показано на следующей схеме реакции:
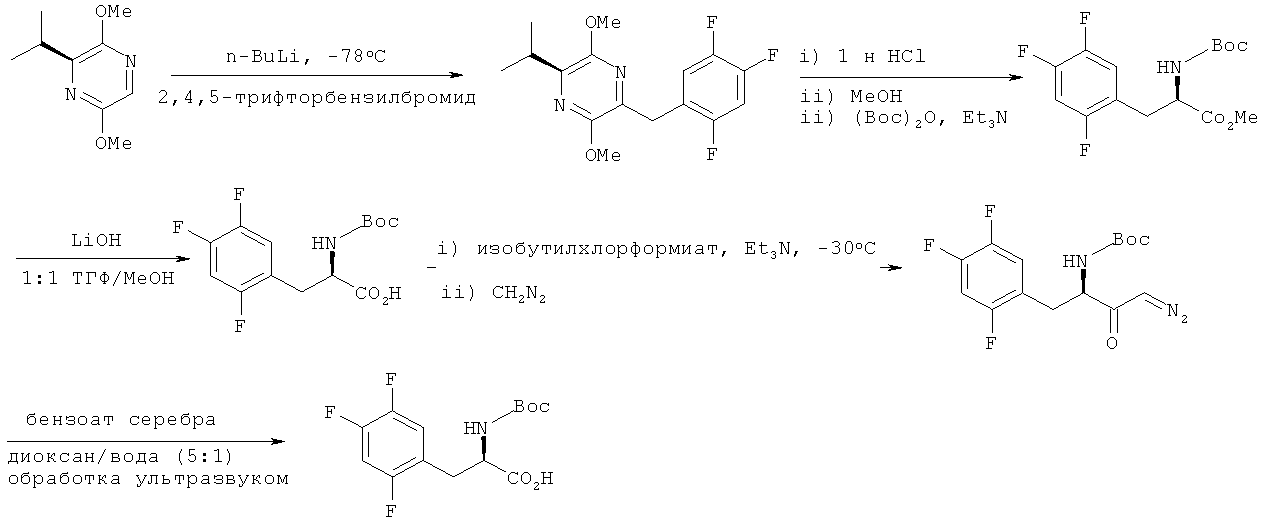
В частности, сложноэфирное соединение получают путем защитной реакции амина после взаимодействия (2S)-(+)-2,5-дигидро-3,6-диметокси-2-изопропилпиразина с 2,4,5-трифторбензилбромидом и обработки кислотой. Сложноэфирное соединение может быть снова гидролизовано с получением 3-(2,4,5-трифторфенил)-2-аминопропионовой кислоты; затем диазокетон может быть получен с использованием изобутилхлорформиата, третичного амина, такого как триэтиламин или диизопропилэтиламин, и диазометана; и соединение, представленное химической формулой 2, может быть получено путем взаимодействия диазокетона с бензоатом серебра. Однако упомянутое выше взаимодействие имеет недостатки, поскольку его следует проводить при низкой температуре (-78°C) или следует использовать дорогостоящую альфа-аминокислоту и крайне опасный диазометан.
Другой способ получения соединения, представленного вышеприведенной химической формулой 2, также известен из Tetrahedron: Asymmetry 2006; 205 или подобным образом Bioorganic & Medicinal Chemistry Letters 2007; 2622, как показано на следующей схеме реакции:

В частности, 2,4,5-трифторфенилуксусную кислоту активируют с использованием 1,1'-карбонилдиимидазола и затем подвергают взаимодействию с монометилмалонатом калия с получением бета-кетоэфирного соединения. Бета-кетоэфирное соединение подвергают взаимодействию с ацетатом аммония и водным раствором аммиака с получением сложного енаминового эфира, и сложноэфирное соединение затем подвергают взаимодействию с димером хлоро(1,5-циклооктадиен)родия(1) и хиральным ферроценовым лигандом (I) путем реакции гидрогенирования при высоком давлении с получением соединения, которое представляет собой бета-аминоэфир, имеющий только хиральный первичный амин. И затем соединение может быть гидролизовано с получением соединения, представленного химической формулой 2. Однако, способ, описанный выше, имеет недостаток в том, что реакцию гидрогенирования при высоком давлении следует проводить с использованием дорогостоящего металлического катализатора.
Кроме того, способ получения соединения, представленного химической формулой 2, также раскрыт в публикации международной заявки на патент №WO 04/87650.
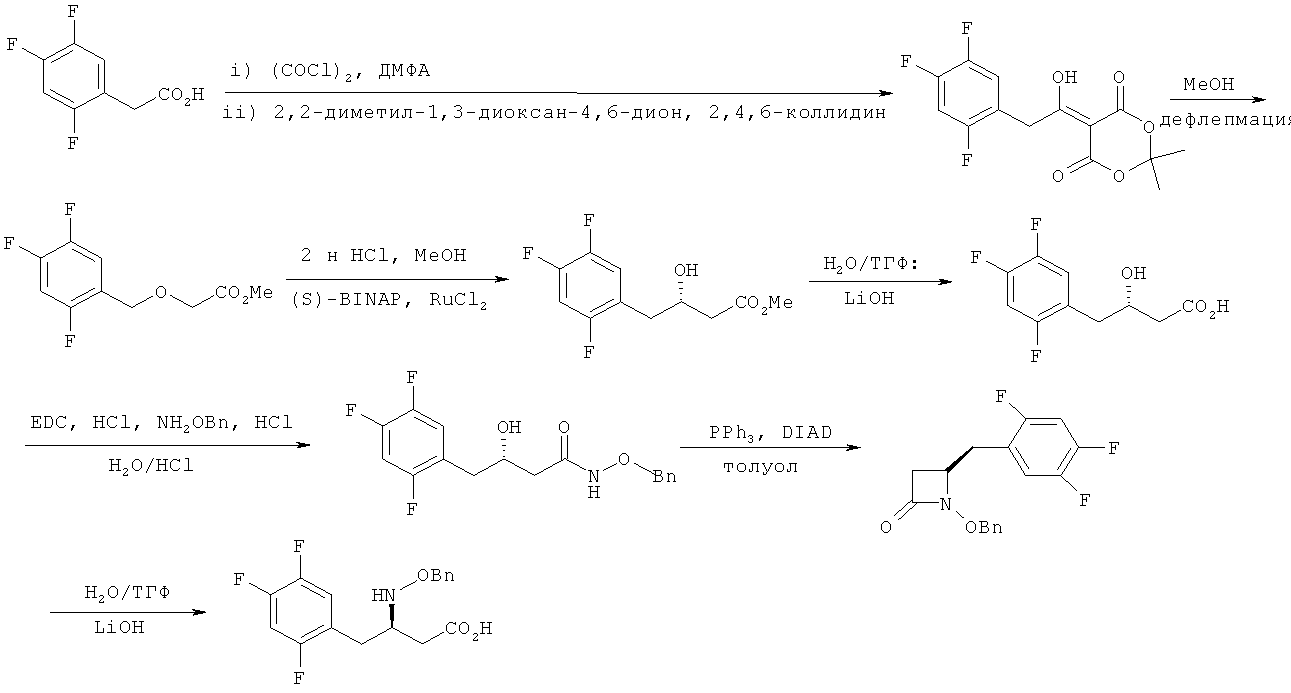
В частности, 2,4,5-трифторфенилуксусную кислоту подвергают взаимодействию с 2,2-диметил-1,3-диоксан-4,6-дионом и оксалилхлоридом, которые являются агентом кислотного активирования, и затем полученный продукт нагревают с обратным холодильником в метаноле с получением соответствующего соединения. Соответствующее соединение подвергают взаимодействию с (s)-BINAP-RuCl2, который является восстановителем, обладающим энантиоселективностью, путем гидрогенирования с получением соединения с (S)-координацией, и затем полученное соединение снова гидролизуют и затем подвергают реакции сочетания с О-бензилгидроксиламином с получением промежуточного соединения. Промежуточное соединение, полученное, как упомянуто выше, может быть подвергнуто реакции конденсации с замыканием кольца в присутствии трифенилфосфина и диизопропилазодикарбоксилата (DIAD) и обработано водным раствором гидроксида лития с получением соединения, представленного химической формулой 2, с (Р)-координацией, в котором аминогруппа также защищена О-бензилом. Однако, способ, описанный выше, имеет недостаток в том, что процесс в целом является длительным и трудоемким, так что выход реакции низкий, и взаимодействие следует проводить в течение длительного времени.
Как упомянуто выше, традиционный известный способ получения соединения, представленного химической формулой 2, имеет некоторые недостатки, такие как использование дорогостоящего реагента, длительное время синтеза и низкий выход и поэтому неприемлем для массового промышленного производства.
Далее, соединение, представленное химической формулой 3, может быть получено с использованием следующей схемы реакции, как раскрыто в публикации патента Кореи №2008-0094604:

(в вышеприведенной схеме реакции, X является таким, как определено в химической формуле 1).
В частности, соединение D-серинметиловый эфир, который является исходным веществом, подвергают замещению тритилхлоридом; затем гидроксильную группу снова замещают мезильной группой, и затем нагревают с обратным холодильником до превращения в азиридиновое соединение.
Тритильную группу удаляют из азиридинового соединения с использованием трифторуксусной кислоты (ТФУК); затем азиридиновое соединение защищают бензилоксикарбонилом (Cbz) и затем подвергают взаимодействию с НХ; и снимают Cbz-защиту с получением метил-2-амино-3-замещенного карбоната. Промежуточное соединение может быть получено с использованием соединения, полученного путем защиты вторичного амина соединения, полученного путем взаимодействия N-бутилоксикарбонил-2-аминоацетальдегида с восстановителем (цианборгидрид натрия, триацетоксиборгидрид натрия, боргидрид натрия и тому подобное), и вторичный амин этого соединения защищают бензилоксикарбонилом (Cbz), и с этого соединения снимают бутилоксикарбонильную (Boc) защиту. Соединение, полученное как упомянуто выше, подвергают циклизации в присутствии триметилалюминия (или диизопропилэтиламина/этанола, гидрокарбоната натрия/метанола и тому подобного) для того, чтобы снять Cbz-защиту, с тем, чтобы можно было получить соединение, представленное химической формулой 3.
Однако способ, описанный выше, имеет недостаток, поскольку в нем используют дорогостоящий реагент, время синтеза длительное, а выход низкий, поэтому данный способ не подходит для массового промышленного производства.
Кроме того, поскольку 1-гидроксибензотриазол (НОВТ) и 1-этил-3-(3-диметиламинопропил)карбодиимид (EDC), используемые для получения обычного соединения, представленного химической формулой 1, являются дорогостоящими реагентами, стоимость реакции будет высокой, поэтому она не подходит для массового промышленного производства.
По этой причине авторы настоящего изобретения разработали настоящее изобретение, подтвердив, что соединение, представленное химической формулой 1, может быть получено экономично с высоким выходом путем использования нового способа производства используемых соединений, представленных химической формулой 2 и химической формулой 3, с более дешевыми реагентами, при изучения способа получения, подходящего для массового промышленного производства, в котором используют более дешевые реагенты; который является экономичным; и который повышает выход.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Одна задача настоящего изобретения состоит в том, чтобы предложить способ производства соединения, полезного в качестве промежуточного соединения при производстве ингибитора дипептидилпептидазы-IV.
Другая задача настоящего изобретения состоит в том, чтобы предложить усовершенствованный способ производства ингибитора дипептидилпептидазы-IV.
Для решения этих задач согласно настоящему изобретению предложен новый способ производства промежуточного соединения ингибитора дипептидилпептидазы-IV.
Согласно настоящему изобретению также предложен усовершенствованный способ производства ингибитора дипептидилпептидазы-IV.
Настоящее изобретение может быть полезно при массовом производстве благодаря снижению стоимости производства путем использования более дешевых реагентов в реакции и повышению выхода.
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВОПЛОЩЕНИЙ
В дальнейшем настоящее изобретение будет описано подробно.
Согласно настоящему изобретению предложен новый способ производства промежуточного соединения ингибитора дипептидилпептидазы-IV, представленного химической формулой 2, как показано на следующей схеме реакции 1, и этот метод включает: (стадия а) получение соединения, представленного химической формулой 6, путем раскрытия азиридинового кольца в соединении, представленном химической формулой 5, с использованием реактива Гриньяра; и (стадия b) получение соединения, представленного химической формулой 2, путем введения защитной группы амина после гидролиза соединения, представленного химической формулой 6,
[Схема реакции 1]

(на вышеприведенной схеме реакции 1, PG представляет собой защитную группу, a R представляет собой низший C1-C5алкил).
В частности, на вышеприведенной стадии (a), азиридиновое соединение, представленное химической формулой 5, подвергают взаимодействию с бромидом 2,4,5-трифторфенилмагния в присутствии комплекса бромид меди(1)-диметилсульфид с получением сложноэфирного соединения, представленного химической формулой 6. В данном случае соединение, представленное химической формулой 5, может быть приобретено у коммерческого поставщика или получено с использованием способа, известного в данной области техники, к которой принадлежит настоящее изобретение. Например, с использованием способов, раскрытых в Tetrahedron Letter 1991; 923, Tetrahedron Letter 1993; 6513, Tetrahedron Letter 1992, 6389, Tetrahedron Letter 2004, 821, Tetrahedron Letter 2006,3509 и им подобным, кислотную функциональную группу трет-бутилового эфира N-Boc-L-аспарагиновой кислоты активируют изобутилхлорформиатом при температуре от -40°C до комнатной температуры и затем подвергают взаимодействию с боргидридом натрия, то есть восстановителем, получая при этом соединение, кислотная функциональная группа в котором замещена на спиртовую группу. И затем полученное соединение может быть подвергнуто взаимодействию с трифенилфосфином и диизопропилазодикарбоксилатом (DIAD) с получением соединения, представленного химической формулой 5.
Затем, на стадии (b), соединение, представленное химической формулой 6, гидролизуют в условиях присутствия кислоты, такой как трифторуксусная кислота, соляная кислота, серная кислота и им подобные, и затем может быть введена защитная группа амина с получением соединения, представленного химической формулой 2. В данном случае бутоксикарбонил (Boc) или бензилоксикарбонил (Cbz) могут быть использованы в качестве защитной группы амина.
Кроме того, согласно настоящему изобретению, предложен новый способ производства промежуточного соединения ингибитора дипептидилпептидазы-IV, представленного химической формулой 3, как показано в следующей схеме реакции 2, включающий: (стадия a') получение соединения, представленного химической формулой 8, путем взаимодействия соединения, представленного химической формулой 7, с аминоальдегидным соединением с защищенной аминогруппой и восстановителем; и (стадия b') получение соединения, представленного химической формулой 3, или его соли путем удаления защитной группы амина с соединения, представленного химической формулой 8, путем инициирования реакции гидрогенирования и индуцирования циклизации,
[Схема реакции 2]
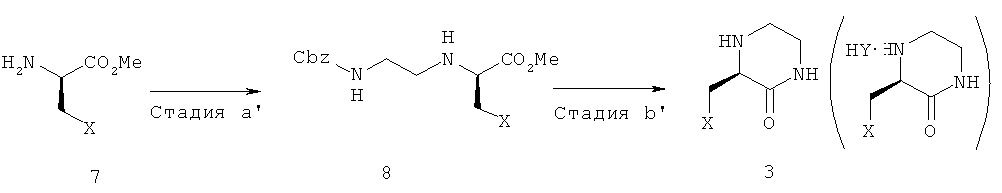
(в вышеприведенной схеме реакции 2, X является таким, как определено в вышеприведенной химической формуле 1, и HY представляет собой свободную кислоту).
В частности, на вышеприведенной стадии (а'), соединение, представленное химической формулой 7, подвергают взаимодействию с аминоальдегидным соединением с защищенной аминогруппой и восстановителем с получением соединения, представленного химической формулой 8. В данном случае соединение, представленное химической формулой 7, может быть приобретено у коммерческого поставщика или получено с использованием способа, известного в данной области техники, к которой принадлежит настоящее изобретение. Например, когда X представляет собой трет-бутокси, гидрохлорид метилового эфира D-cерина подвергают взаимодействию с гидрокарбонатом натрия и бензилоксихлорформиатом в присутствии тетрагидрофурана при температуре от 0°C до комнатной температуры для защиты аминогруппы, затем подвергают взаимодействию с газообразным изобутиленом в присутствии серной кислоты в качестве катализатора при температуре от 0°C до комнатной температуры с получением промежуточного соединения и затем подвергают гидрогенированию в присутствии катализатора палладий на угле с получением соединения, представленного химической формулой 7. В данном случае аминоальдегидное соединение с защищенной аминогруппой может представлять собой аминоальдегидное соединение, которое может быть приобретено у коммерческого поставщика и аминогруппа которого защищена Cbz, и подвергается взаимодействию с соединением, представленным химической формулой 7, в присутствии цианборгидрида натрия и хлорида цинка, которые являются восстановителем, с получением соединения, представленного химической формулой 8.
Затем, на вышеуказанной стадии (b'), защитную группу амина удаляют с соединения, представленного химической формулой 8, путем проведения гидрогенирования и одновременно индуцируют циклизацию с получением соединения, представленного химической формулой 3. В данном случае гидрогенирование предпочтительно проводят в присутствии палладия на угле. Кроме того, соединение, представленное химической формулой 3, может быть использовано в форме приемлемой соли, и соль присоединения кислоты, которую получают с помощью свободной кислоты, полезна в качестве соли. В качестве свободной кислоты может быть использована органическая кислота или неорганическая кислота. В данном случае неорганическая кислота может включать соляную кислоту, бромноватую кислоту, серную кислоту, фосфорную кислоту и им подобные, и органическая кислота может включать ди-пара-толуоил-L-тартрат, лимонную кислоту, уксусную кислоту, молочную кислоту, малеиновую кислоту, фумаровую кислоту, глюконат, метансульфоновую кислоту, гликолевую кислоту, янтарную кислоту, винную кислоту, 4-толуолсульфоновую кислоту, галактуроновую кислоту, эмбоновую кислоту, глутаминовую кислоту, аспарагиновую кислоту и им подобные.
Кроме того, согласно настоящему изобретению предложено соединение, представленное следующей химической формулой 8, полученное в качестве промежуточного соединения при получении соединения, представленного химической формулой 2.
[Химическая формула 8]

(в вышеприведенной химической формуле 8, X является таким, как определено в вышеприведенной химической формуле 1).
Кроме того, согласно настоящему изобретению предложен усовершенствованный способ производства ингибитора дипептидилпептидазы-IV, представленного химической формулой 1, как показано на следующей схеме реакции 3, включающий: (стадия 1) получение соединения, представленного химической формулой 4, путем связывания пептидной связью соединения, представленного химической формулой 2, и соединения, представленного химической формулой 3, путем осуществления их взаимодействия с использованием изохлорформиата и основания в присутствии реакционного растворителя; и (стадия 2) получение соединения, представленного химической формулой 1, удалением защитной группы амина с соединения, представленного химической формулой 4, полученного на вышеприведенной стадии (1),
[Схема реакции 3]

(в вышеприведенной схеме реакции 3, PG представляет собой защитную группу, X является таким, как определено в вышеприведенной химической формуле 1, и HY является таким, как определено в вышеприведенной схеме реакции 2).
Прежде всего, стадия (1) заключается в получении соединения, представленного химической формулой 4, путем связывания пептидной связью соединения, представленного химической формулой 2 и соединения, представленного химической формулой 3, путем осуществления взаимодействия с использованием изохлорформиата и основания.
В настоящем изобретении, реакционный растворитель может включать толуол, тетрагидрофуран, метиленхлорид, ацетонитрил, N,N-диметилформамид и им подобные.
В настоящем изобретении, основание может включать более чем одно соединение, выбранное из группы, состоящей из третичных аминов, таких как N-метилморфолин, изопропилэтиламин, триэтиламин, пиридин и им подобных.
В настоящем изобретении, соединение, представленное химической формулой 2 или 3, может быть приобретено у коммерческого поставщика или получено с использованием известного способа или способа, который раскрыт на вышеприведенной схеме реакции 1 или схеме реакции 2.
В настоящем изобретении, взаимодействие на вышеприведенной стадии (1) предпочтительно проводят при температуре от -20°C до комнатной температуры, а в случае выхода за пределы вышеуказанного диапазона может возникнуть проблема, заключающаяся в том, что взаимодействие будет протекать с затруднениями, так что выход снизится.
Далее, стадия (2) заключается в получении соединения, представленного химической формулой 1, путем удаления защитной группы амина с соединения, представленного химической формулой 4, полученного на вышеприведенной стадии (1).
Удаление защитной группы на вышеприведенной стадии (2) может быть проведено в кислой среде или путем гидрогенирования. В частности, когда защитная группа амина представляет собой бутоксикарбонил (Boc), она может быть удалена путем взаимодействия в условиях присутствия кислоты, такой как трифторуксусная кислота/дихлорметан, этилацетат/хлороводород, диэтиловый эфир/хлороводород, хлороводород/дихлорметан, метанол/хлороводород и им подобных; а когда защитная группа амина представляет собой бензилоксикарбонил (Cbz), она может быть удалена путем гидрогенирования в присутствии палладия на угле.
Ингибитор дипептидилпептидазы-IV по настоящему изобретению, представленный химической формулой 1, может быть использован в форме фармацевтически приемлемой соли, и в качестве соли полезна соль присоединения кислоты, полученная с помощью фармацевтически приемлемой свободной кислоты. В качестве свободной кислоты может быть использована неорганическая кислота и органическая кислота. Неорганическая кислота может включать соляную кислоту, бромноватую кислоту, серную кислоту, фосфорную кислоту и им подобные, и органическая кислота может включать лимонную кислоту, уксусную кислоту, молочную кислоту, малеиновую кислоту, фумаровую кислоту, глюконат, метансульфоновую кислоту, уксусную кислоту, гликолевую кислоту, янтарную кислоту, винную кислоту, 4-толуолсульфоновую кислоту, галактуроновую кислоту, эмбоновую кислоту, глутаминовую кислоту, аспарагиновую кислоту и им подобные. Предпочтительно соляная кислота может быть использована в качестве неорганической кислоты, и винная кислота может быть использована в качестве органической кислоты.
Соль присоединения согласно настоящему изобретению может быть получена обычным способом. Например, соединение, представленное химической формулой 1, растворяют в водорастворимом органическом растворителе, таком как ацетон, метанол, этанол, ацетонитрил, и им подобных; к этому добавляют избыток органической кислоты или раствор неорганической кислоты; и затем осаждают или кристаллизуют с получением соли присоединения. Затем растворитель или избыток кислоты выпаривают из вышеуказанной смеси, и затем смесь может быть высушена или осажденная соль может быть отфильтрована под вакуумом с получением соли присоединения.
После получения промежуточных соединений или соединений, представленных химическими формулами 1-3, согласно настоящему изобретению их молекулярные структуры могут быть установлены с использованием инфракрасной спектроскопии, спектра протонного ядерного магнитного резонанса, масс-спектрометрии, жидкостной хроматографии, рентгеновского метода определения структуры, поляриметра и сравнения между подсчитанным значением и фактическим значением элементного анализа представленных соединений.
Как упомянуто выше, способ производства согласно настоящему изобретению может снижать стоимость производства соединения, представленного химической формулой 1, благодаря использованию более дешевых реагентов и также повышать выход, так что он может быть успешно применен при массовом производстве.
В дальнейшем настоящее изобретение будет описано более подробно со ссылкой на примеры. Однако следующие примеры даны только для иллюстрации, и настоящее изобретение не ограничено ими.
Пример 1. Получение (R)-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая формула 2) из (S)-4-трет-бутокси-2-(трет-бутоксикарбониламино)-4-оксобутановой кислоты
Стадия 1: получение (3)-трет-бутил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноата
2,0 г (3)-4-трет-бутокси-2-(трет-бутоксикарбониламино)-4-оксобутановой кислоты и 14 мл тетрагидрофурана добавляли в колбу на 50 мл, и затем полученный реакционный раствор охлаждали до 0°C. В то время как реакционный раствор перемешивали, по каплям добавляли 1,0 мл 4-метилморфолина и через 10 минут по каплям добавляли 1,2 мл изобутилхлорформиата, и затем перемешивали в течение 1 часа. Полученное твердое вещество отфильтровывали с помощью диатомита, промывали 14 мл тетрагидрофурана, и затем фильтрат охлаждали до 0°C. 523 мг боргидрида натрия добавляли к охлажденному фильтрату и перемешивали в течение 4 часов до тех пор, пока температура реакции самопроизвольно не возрастала до комнатной температуры. После завершения взаимодействия реакционный раствор охлаждали до 0°C и затем по каплям добавляли 10 мл водного раствора хлорида аммония. Добавляли 20 мл этилацетата и 10 мл воды и затем перемешивали в течение 10 минут. Органический слой отделяли, сушили сульфатом магния и затем концентрировали при пониженном давлении. Концентрированный остаток выделяли путем колоночной хроматографии (н-гексан:этилацетат, 2:1) и затем концентрировали при пониженном давлении с получением 1,86 г указанного в заголовке соединения.
1Н ЯМР (CDCl3, 400 МГц) δ 5.19 (s, 1H), 3.94 (уширенный, 1H), 3.67 (s, 2H), 2.46 (m, 3H), 1.43 (s, 9H), 1.42 (s, 9H).
Стадия 2: получение (S)-трет-бутил-2-(2-трет-бутокси-2-оксоэтил)азиридин-1-карбоксилата (химическая формула 5)
2,90 г трифенилфосфина и 15 мл тетрагидрофурана добавляли в колбу на 100 мл, и полученный реакционный раствор охлаждали до 0°C. 2,17 мл диизопропилазодикарбоксилата добавляли по каплям, в то время как реакционный раствор перемешивали. Через 30 минут по каплям добавляли 10 мл тетрагидрофуранового раствора с 1,52 г (S)-трет-бутил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноата и перемешивали в течение 16 часов до тех пор, пока температура реакции самопроизвольно не возрастала до комнатной температуры. После завершения взаимодействия 40 мл этилацетата и 40 мл воды добавляли в реакционный раствор и затем перемешивали в течение 10 минут. Органический слой отделяли, сушили сульфатом магния и затем концентрировали при пониженном давлении. Концентрированный остаток выделяли путем колоночной хроматографии (н-гексан:этилацетат, 15:1) и затем концентрировали при пониженном давлении с получением 1,04 г указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 2.69 (m, 1H), 2.61 (dd, 1H), 2.31 (d, 1H), 2.16 (dd, 1H), 1.97 (d, 1H), 1.44 (d, 18H).
Стадия 3: получение (R)-трет-бутил-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутаноата (химическая Формула 6)
4,2 мл 1-бром-2,4,5-трифторбензола и 10,8 мл тетрагидрофурана добавляли в колбу на 50 мл, и полученный реакционный раствор охлаждали до 0°C. 15 мл хлорида изопропилмагния [2,0 М раствор в тетрагидрофуране] по каплям добавляли в реакционный раствор в атмосфере азота и перемешивали в течение 30 минут с получением реактива Гриньяра. 1,95 г (S)-трет-бутил-2-(2-трет-бутокси-2-оксоэтил)азиридин-1-карбоксилата и 50 мл тетрагидрофурана добавляли в другую колбу на 250 мл, и полученный реакционный раствор охлаждали до 0°C. Затем добавляли 778 мг комплекса бромид меди(1)-диметилсульфид. По каплям добавляли 22,7 мл реактива Гриньяра, полученного в атмосфере азота, и перемешивали в течение 6 часов, при этом температуру реакции поддерживали при 0°C. После завершения взаимодействия 50 мл водного раствора хлорида аммония по каплям добавляли в реакционный раствор; добавляли 100 мл этилацетата и 50 мл воды и затем перемешивали в течение 10 минут. Органический слой отделяли, сушили сульфатом магния и затем концентрировали при пониженном давлении. Концентрированный остаток выделяли путем колоночной хроматографии (н-гексан:этилацетат, 20:1) и затем концентрировали при пониженном давлении с получением 2,62 г указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) 6 7.02 (m, 1H), 6.87 (m, 1H), 5.11 (уширенный, 1H), 4.07 (уширенный, 1H), 2.82 (dd, 1H), 2.77 (dd, 1H), 2.45 (dd, 1H), 2.35 (dd, 1H), 1.44 (s, 9H), 1.35 (s, 9H).
Стадия 4: получение (R)-3-(трет-бутоксикарбониламино)-4-(2.4.5-тоифторфенил)бутановой кислоты (химическая Формула 2)
1,31 г (R)-трет-бутил-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутаноата, 16 мл метиленхлорида и 16 мл трифторуксусной кислоты добавляли в колбу на 100 мл, и полученный реакционный раствор перемешивали в течение 6 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении, и 16 мл метанола добавляли к концентрированному остатку. Реакционный раствор охлаждали до 0°C, добавляли 2,82 г гидрокарбоната натрия и 0,77 мл ди-трет-бутилдикарбоната и затем перемешивали в течение 6 часов до тех пор, пока температура реакции самопроизвольно не возрастала до комнатной температуры. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении; затем добавляли 30 мл этилацетата и 30 мл воды; и затем перемешивали в течение 10 минут. Водный слой отделяли, охлаждали до 0°C и затем по каплям добавляли 2 н. водный раствор соляной кислоты для доведения значения pH до 3-4. Водный слой экстрагировали растворителем метиленхлорид:метанол, 10:1, сушили сульфатом магния и затем концентрировали при пониженном давлении с получением 828 мг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.04 (m, 1H), 6.89 (m, 1H), 6.08 (уширенный, 1H), 5.04 (уширенный, 1H), 4.13 (уширенный, 1H), 2.88 (уширенный, 2H), 2.62 (m, 2H), 1.36 (s, 18H).
Масс-спектр (M+Na): 356.
Пример 2. Получение (PR)-3-(бензилоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая формула 2) из (S)-4-трет-бутокси-2-(трет-бутоксикарбонил)-4-оксобутановой кислоты 64 мг 3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутаноата получали с использованием способа, аналогичного способу для стадий 1-3 примера 1. Для стадии 4 примера 1 использовали смесь тетрагидрофуран/вода и N-(бензилоксикарбонилокси)сукцинимид вместо метанола и ди-трет-бутил-дикарбоната, соответственно, с получением 40 мг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.45-7.18 (m, 5H), 7.05 (m, 1H), 6.83 (m, 1H), 5.37 (d, 1H), 5.10 (s, 2H), 4.52-4.16 (m, 1H), 3.01-2.85 (m, 2H), 2.78-2.42 (m, 2H)
Масс-спектр (М+1): 368.
Пример 3. Получение (R)-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая формула 2) из (S)-4-бензилокси-2-(трет-бутоксикарбониламино)-4-оксобутановой кислоты
Стадия 1: получение (S)-бензил-3-(трет-бутоксикарбониламин)-4-гидроксибутаноата
402 мг указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 1 примера 1, за исключением того, что (S)-4-(бензилокси)-2-(трет-бутоксикарбониламино)-4-оксобутановую кислоту (500 мг) использовали вместо (S)-4-трет-бутокси-2-(трет-бутоксикарбониламино)-4-оксобутановой кислоты на стадии 1 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 7.27 (m, 5H), 5.16 (m, 3H), 4.00 (m, 1H), 3.68 (m, 2H), 2.66 (m, 2H), 2.40 (s, 1H), 1.41 (s, 9H).
Стадия 2: получение (S)-трет-бутил-2-(2-бензилокси-2-оксоэтил)азиридин-1-карбоксилата (химическая формула 5)
239 мг указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 2 примера 1, за исключением того, что (S)-бензил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноат (402 мг) использовали вместо (S)-трет-бутил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноата на стадии 2 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 7.34 (m, 5H), 5.13 (m, 2H), 2.59 (m, 2H) 2.37 (m, 2H), 1.99 (d, 1H), 1.43 (s, 9H).
Стадия 3: получение (R)-бензил-3-(трет-бутоксикарбониламино)-4-(2.4.5-трифторфенил)бутаноата (химическая Формула 6)
58 мг указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 3 примера 1, за исключением того, что (S)-трет-бутил-2-(2-бензилокси-2-оксоэтил)азиридин-1-карбоксилат (100 мг) использовали вместо (S)-трет-бутил-2-(2-трет-бутокси-2-оксоэтил)азиридин-1-карбоксилата на стадии 3 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 7.37 (m, 5H), 6.96 (m, 1H), 6.86 (m, 1H), 5.11 (m, 3H), 4.12 (m, 1H), 2.81 (m, 2H) 2.56 (m, 2H), 1.35 (s, 9H).
Стадия 4: получение (R)-3-(трет-бутоксикарбониламино)-4-(2,4,5-триФторфенил)бутановой кислоты (химическая Формула 2)
58 мг (R)-бензил-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутаноата, 3 мл метанола и 20 мг 10 мас.% палладия на угле добавляли в колбу на 25 мл, и полученный реакционный раствор перемешивали. Газообразный водород барботировали в течение 2 часов при комнатной температуре; реакционный раствор фильтровали путем пропускания через целит, промывали 15 мл этилацетата, и фильтрат концентрировали при пониженном давлении с получением 44 мг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.04 (m, 1H), 6.89 (m, 1H), 6.08 (уширенный, 1H), 5.04 (уширенный, 1H), 4.13 (уширенный, 1H), 2.88 (уширенный, 2H), 2.62 (m, 2H), 1.36 (s, 18H).
Масс-спектр (M+Na): 356.
Пример 4. Получение (R)-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая формула 2) из (S)-2-(трет-бутоксикарбониламино)-4-метокси-4-оксобутановой кислоты
Стадия 1: получение (S)-метил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноата
1,23 г указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 1 примера 1, за исключением того, что (8)-2-(трет-бутоксикарбониламино)-4-метокси-4-оксобутановая кислота (2,0 г) использовали вместо (S)-4-трет-бутокси-2-(трет-бутоксикарбониламино)-4-оксобутановой кислоты на стадии 1 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 5.19 (s, 1H), 3.97 (m, 1H), 3.68 (m, 5H), 2.62 (m, 2H), 2.45 (s, 1H), 1.42 (s, 9H).
Стадия 2: получение (S)-трет-бутил-2-(2-метокси-2-оксоэтил)азиридин-1-карбоксилата (химическая формула 5)
820 мг указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 2 примера 1, за исключением того, что (8)-метил-3-(трет-бутоксикарбониламино)-4-гидроксибутаноат (1,23 г) использовали вместо (S)-трет-бутил-3-трет-бутоксикарбониламино)-4-гидроксибутаноата на стадии 2 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 3.68 (s, 3H), 2.72 (m, 1H), 2.65 (dd, 1H), 2.35 (m, 2H), 1.98 (d, 1H), 1.43 (s, 9H).
Стадия 3: получение (R)-метил-3-(трет-бутоксикарбониламино)-4-(2.4,5-триФторфенил)бутаноата (химическая Формула 6)
53 мг указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 3 примера 1, за исключением того, что (S)-трет-бутил-2-(2-метокси-2-оксоэтил)азиридин-1-карбоксилат (70 мг) использовали вместо (8)-трет-бутил-2-(2-трет-бутокси-2-оксоэтил)азиридин-1-карбоксилата на стадии 3 примера 1.
1H ЯМР (CDCl3, 400 МГц) δ 6.96 (m, 1H), 6.87 (m, 1H), 5.09 (уширенный, 1H), 4.10 (уширенный, 1H), 3.69 (s, 3H), 2.83 (m, 2H), 2.56 (m, 2H), 1.36 (s, 9H).
Стадия 4: получение (R)-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты (химическая Формула 2)
53 мг (R)-метил-3-(трет-бутоксикарбониламино)-4-(2,4,5-трифторфенил)бутаноата, 1,5 мл тетрагидрофурана и 0,5 мл воды добавляли в колбу на 25 мл и полученный реакционный раствор охлаждали до 0°C. 7,32 мг гидроксида лития добавляли в реакционный раствор и перемешивали в течение 6 часов до тех пор, пока температура реакции самопроизвольно не возрастала до комнатной температуры. После завершения взаимодействия 5 мл этилацетата и 5 мл воды добавляли в реакционный раствор и перемешивали в течение 10 минут. Водный слой отделяли, охлаждали до 0°C и по каплям добавляли 2 н. водный раствор соляной кислоты для доведения значения рН до 3-4. Водный слой экстрагировали растворителем метиленхлорид:метанол, 10:1, сушили сульфатом магния и затем концентрировали при пониженном давлении с получением 40,8 мг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.04 (m, 1H), 6.89 (m, 1H), 6.08 (уширенный, 1H), 5.04 (уширенный, 1Н), 4.13 (уширенный, 1H), 2.88 (уширенный, 2H), 2.62 (m, 2H), 1.36 (s, 18H).
Масс-спектр (M+Na): 356.
Пример 5. Получение (R)-3-(трет-бутоксиметил)пиперазин-2-она или его соли (химическая формула 3)
Стадия 1: получение (R)-метил-2-(бензилоксикарбониламино)-3-трет-бутоксипропаноата
Добавляли 130 л метиленхлорида; 20,5 г (R)-метил-2-(бензилоксикарбониламино)-3-гидроксипропаноата в реактор; затем перемешивали в течение 30 минут; и затем добавляли 0,4 кг серной кислоты. Газообразный изобутилен барботировали в течение 24 часов, при этом поддерживали температуру при 20-25°C. После завершения взаимодействия медленно добавляли 18 л насыщенного водного раствора гидрокарбоната натрия, перемешивали в течение 1 часа и затем отделяли органический слой. 5 кг сульфата натрия добавляли к органическому слою, перемешивали в течение 1 часа, фильтровали, промывали, и затем фильтрат концентрировали при пониженном давлении с получением 29,3 кг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.36-7.30 (m, 5H), 5.59 (d, 1H), 5.10 (s, 2H), 4.44 (m, 1H), 3.80 (m, 1H), 3.73 (s, 3H), 3.56 (m, 1H), 1.10 (s, 9H).
Стадия 2: получение (R)-метил-2-амино-3-трет-бутоксипропаноата (химическая формула 7)
Добавляли 330,0 л метанола и добавляли 66,0 кг (R)-метил-2-(бензилоксикарбониламино)-3-трет-бутоксипропаноата в реактор гидрогенирования; и затем продували азотом. Добавляли 4,95 кг палладия на угле (10% водная смесь) и заполняли водородом для поддержания давления 5 бар (0,5 МПа). Перемешивали в течение 60 минут, фильтровали, промывали и затем концентрировали при пониженном давлении. 132,0 л этилацетата и 88 л воды добавляли к концентрированному остатку; перемешивали в течение 10 минут; органический слой отделяли (за 6 раз), сушили и затем концентрировали при пониженном давлении с получением 27,5 кг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 4.21 (m, 1H), 3.82 (s, 3H), 3.74-3.88 (m, 2H), 1.20 (s, 9H).
Стадия 3: получение (Р0-метил-2-(2-(бензилоксикарбониламино)-этиламино)-3-трет-бутоксипропаноата (химическая формула 8)
155 л (122,5 кг) метанола и 5,04 кг цианборгидрида натрия добавляли в первый реактор, охлаждали до температуры меньше чем 0°C и затем добавляли 5,47 кг хлорида цинка. 155 л метанола и 31 кг 2-оксоэтилкарбамата добавляли во второй реактор, охлаждали до 0°C и затем добавляли 28,1 кг (R)-метил-2-амино-3-трет-бутоксипропаноата. Раствор, полученный в первом реакторе, сразу же добавляли по каплям во второй реактор; его температуру повышали до комнатной температуры и затем перемешивали в течение 2 часов. После завершения взаимодействия реакционный раствор концентрировали при пониженном давлении; добавляли 93 л этилацетата и 186 л изопропилового эфира; перемешивали в течение 5 минут; полученное твердое вещество отфильтровывали через набивку из целита и затем промывали смесью изопропиловый эфир:этилацетат, 2:1 (93 л). Фильтрат промывали 310 л насыщенного гидрокарбоната натрия 7 раз и затем промывали 310 л рассола. Органический слой сушили 50,0 кг сульфата натрия, фильтровали, промывали и затем концентрировали при пониженном давлении с получением 35,5 кг указанного в заголовке соединения.
1H ЯМР (CDCl3, 400 МГц) δ 7.36-7.28 (m, 5H), 5.09 (s, 2H), 3.72 (s, 2H), 3.71-3.52 (m, 3H), 3.33 (m, 4H), 1.13 (s, 9H).
Стадия 4: получение (R)-3-(трет-бутоксиметил)пиперазин-2-она (химическая формула 3)
39,5 кг (R)-метил-2-(2-(бензилоксикарбониламино)этиламино)-3-трет-бутоксипропаноата растворяли в 276 л метанола в реакторе; продували азотом; добавляли 5,9 кг палладия на угле (10% водная смесь) и перемешивали в течение 3 часов, поддерживая давление водорода 10 бар (1 МПа). Реакционный раствор фильтровали, концентрировали при пониженном давлении и затем снова подвергали азеотропной перегонке при добавлении 30 л изопропилового эфира. 158 л (115 кг) изопропилового эфира, 39 л (35 кг) этилацетата и 36,4 кг силикагеля добавляли к концентрированному раствору, перемешивали в течение 1 часа, фильтровали при пониженном давлении и затем концентрировали при пониженном давлении. Концентрированный остаток подвергали азеотропной перегонке при добавлении 30 л метанола, и затем концентрированный раствор и 221 л метанола добавляли в реактор. После продувки азотом добавляли 11,85 кг палладия на угле (10% водная смесь), и затем перемешивали в течение 6 часов, поддерживая давление водорода 15 бар (1,5 МПа). Реакционный раствор фильтровали и затем концентрировали при пониженном давлении. Водный слой дважды отделяли путем добавления 80 л изопропилового эфира и 80 л очищенной воды к концентрированному раствору. Органический слой 5 раз отделяли после добавления смеси метиленхлорид/изопропанол, 5:1 (126 л) к водному слою и последующего перемешивания. Органический слой сушили с использованием 50 кг сульфата натрия с получением 9,7 кг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ 6.41 (уширенный s, 1H), 3.76 (m, 3H), 3.63 (m, 1H), 3.52 (m, 1H), 3.42 (m, 1H), 3.28 (m, 1H), 3.16 (m, 1H), 2.95 (m, 1H), 2.45 (уширенный s, 1H), 1.17 (s, 9Н).
Стадия 5: получение ди-пара-толуоил-1-тартрата (R)-3-(трет-бутоксиметил)пиперазин-2-она (химическая Формула 3)
Раствор, который получали путем растворения 100,0 г (R)-3-(трет-бутоксиметил)пиперазин-2-она в 500 мл ацетона и затем путем растворения 207,4 г ди-пара-толуоил-1-винной кислоты в 700 мл ацетона, медленно добавляли в реактор. Полученный реакционный раствор перемешивали в течение 1 часа, и затем полученное твердое вещество фильтровали с получением 251,4 г указанного в заголовке соединения.
1H ЯМР (400 МГц, ДМСО) δ 8.03 (уширенный s, 1H), 7.83 (d, 4H), 7.32 (d, 4H), 5.67 (s, 2H) 3.55-3.66 (m, 3H), 3.18-3.29 (m, 3H), 3.04 (m, 1H), 2.36 (s, 6H), 1.10 (s, 9H).
Пример 6. Получение гидрохлорида (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она (химическая формула 1)
Стадия 1: получение трет-бутил-(R)-4-[(R)-2-(трет-бутоксиметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата
1,0 г (R)-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)бутановой кислоты (химическая формула 2) растворяли в 15 мл метиленхлорида, и затем полученный реакционный раствор охлаждали до 0°C. Пока реакционный раствор перемешивали, по каплям добавляли 0,43 мл 4-метилморфолина; через 10 минут по каплям добавляли 0,47 мл изобутилхлорформиата и затем перемешивали в течение 1 часа. Полученное твердое вещество отфильтровывали с помощью диатомита; промывали 5 мл метиленхлорида; и затем фильтрат охлаждали до 0°C. Раствор, который получали путем растворения 838 мг (R)-(3-трет-бутоксиметил)пиперазин-2-она (химическая формула 3) в 3 мл тетрагидрофурана и 1,1 мл диизопропилэтиламина, добавляли к охлажденному фильтрату и затем перемешивали в течение 1 часа. Затем разбавляли 20 мл этилацетата; дважды промывали рассолом; и затем органический слой осушали сульфатом магния. Остаток очищали путем колоночной хроматографии с получением 838 мг указанного в заголовке соединения.
1H ЯМР (400 МГц, CDCl3) δ 7.03 (m, 1H), 6.88 (m, 1H), 5.97 (m, 1H), 5.48 (m, 1H), 4.16-4.07 (m, 1H), 4.02-3.91 (m, 1H), 3.74 (m, 2H) 3.37 (m, 2H), 3.24 (m, 1H), 2.92 (m, 2H), 2.80 (m, 1H), 2.59 (m, 2H), 1.34 (d, 9H), 1.13 (s, 9H).
Стадия 2: получение гидрохлорида (R)-4-f(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил1-3-(трет-бутоксиметил)пиперазин-2-она (химическая Формула 1)
97 мг трет-бутил-(R)-4-[(R)-2-(трет-бутоксиметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата с вышеприведенной стадии 1 растворяли в 3 мл метанола, добавляли 2 мл смеси 2 н. соляная кислота/диэтиловый эфир и затем перемешивали в течение 3 часов при комнатной температуре. Полученную реакционную смесь концентрировали и сушили при пониженном давлении с получением 64 мг указанного в заголовке соединения в виде пенообразного твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.37 (m, 1H), 7.23 (m, 1H), 4.80 (m, 1H), 4.59-4.40 (m, 1H), 3.93 (m, 1H), 3.90-3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89-2.66 (m, 2H), 1.18 (s, 3H), 1.11 (s, 6H).
Масс-спектр (М+1): 402.
Пример 7. Получение (R)-4-[(R)-3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она (химическая формула 1)
Стадия 1: получение бензил-(R)-4-(R)-2-(трет-бутоксиметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата
65,7 г указанного в заголовке соединения получали с использованием способа, аналогичного способу для стадии 1 примера 6, за исключением того, что 50,0 г (R)-3-(бензилоксикарбониламино)-4-(2,4,5-трифторфенил)бутановой кислоты и 85,7 г ди-пара-толуоил-L-тартрата (R)-(3-трет-бутоксиметил)пиперазин-2-она использовали вместо (R)-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)бутановой кислоты и (R)-(3-трет-бутоксиметил)пиперазин-2-она, соответственно, на стадии 1 примера 6.
1H ЯМР (400 МГц, CDCl3) δ 7.20-7.38 (m, 5H), 7.04 (m, 1H), 6.86 (m, 1H), 6.74 и 6.61 (уширенный s, 1H), 5.79 (m, 1H), 5.00 (m, 2H), 4.91 и 4.69 (m, 1H), 4.41 и 4.25 (m, 1H), 4.16 и 3.99 (m, 1H), 3.68-3.90 (m, 3H), 3.21-3.38 (m, 2H), 2.96-3.12 (m, 2H), 2.59-2.90 (m, 2H), 1.45 и 1.11 (s, 9H).
Стадия 2: получение (R)-4-f(R)-3-амино-4-(2,4,5-триФторфенил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она (химическая Формула 1)
65,7 г бензил-(R)-4-[(R)-2-(трет-бутоксиметил)-3-оксопиперазин-1-ил]-4-оксо-1-(2,4,5-трифторфенил)бутан-2-илкарбамата с вышеприведенной стадии 1 растворяли в 409 мл метанола; добавляли раствор, в который добавлено 13,1 г палладия на угле, смоченного 92 мл этилацетата; и затем перемешивали в течение 2 часов под давлением водорода 15 бар (1,5 МПа). Полученный реакционный раствор фильтровали с помощью диатомита и затем концентрировали при пониженном давлении с получением 34,8 г указанного в заголовке соединения.
1H ЯМР (400 МГц, CD3OD) δ 7.27 (m, 1H), 7.14 (m, 1H), 4.56-4.39 (m, 1H), 3.96-3.81 (m, 3H), 3.70 (m, 1H), 3.46(m, 1H), 3.43-3.32 (m, 1H), 2.83-2.65 (m, 3H), 2.58-2.40 (m, 2H), 1.16 (s, 3H), 1.11 (s, 6H).
Масс-спектр (М+1): 402.
Пример 8. Получение тартрата (R)-4-[(R)-3-амино-4-(2,4,5-трифторметил)бутаноил]-3-(трет-бутоксиметил)пиперазин-2-она (химическая формула 1)
Стадия 1: получение (R)-4-(R)-3-амино-4-(2.4.5-триФторфенил) бутаноил1-3-(трет-бутоксиметил)пиперазин-2-она (химическая Формула 1)
60 мг гидрохлорида соединения, представленного химической формулой 1, полученного по примеру 6, добавляли к 10 мл 5% водного раствора гидрокарбоната натрия; добавляли 10 мл смешанного раствора дихлорметан/2-пропанол [4/1 (об./об.)]; дважды экстрагировали с получением органического слоя; и затем органический слой сушили при пониженном давлении с получением 55 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.27 (m, 1H), 7.14 (m, 1H), 4.56-4.39 (m, 1H), 3.96-3.81 (m, 3H), 3.70 (m, 1H), 3.46 (m, 1H), 3.43-3.32 (m, 1H), 2.83-2.65 (m, 3H), 2.58-2.40 (m, 2H), 1.16 (s, 3H), 1.11 (s, 6H).
Масс-спектр (М+1): 402.
Стадия 2: получение тартрата (R)-4-[(R)-3-амино-4-(2,4,5-триФторфенил)бутаноил1-3-(трет-бутоксиметил)пиперазин-2-она (химическая формула 1)
55 мг соединения с вышеприведенной стадии 1 или по примеру 7 растворяли в 0,56 мл ацетона; медленно добавляли раствор, который приготовлен путем растворения 26 мг L-винной кислоты в 0,35 мл смеси этанол/вода [9/1 (об./об.)], и затем перемешивали в течение 30 минут. Кроме того, снова добавляли 0,56 мл 2-пропанола; перемешивали в течение 10 минут и затем фильтровали с получением 77 мг указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, CD3OD) δ 7.38 (m, 1H), 7.22 (m, 1H), 4.80 (m, 1H), 4.59-4.40 (m, 1H), 4.40 (s, 2H), 3.93 (m, 1H), 3.90-3.83 (m, 2H), 3.70 (m, 1H), 3.38 (m, 2H), 3.27 (m, 1H), 3.07 (m, 2H), 2.89-2.66 (m, 2H), 1.15 (s, 3H), 1.11 (s, 6H).
Масс-спектр (М+1): 402.
Реферат
Настоящее изобретение относится к усовершенствованному способу производства ингибитора дипептидилпептидазы-IV, представленного химической формулой 1. Способ показан на схеме реакцииСпособ включает следующие стадии: (стадия 1) получение соединения, представленного химической формулой 4, путем связывания пептидной связью соединения, представленного химической формулой 2, и соединения, представленного химической формулой 3, путем осуществления их взаимодействия с использованием изобутилхлорформиата и основания в присутствии реакционного растворителя; и (стадия 2) получение соединения, представленного химической формулой 1, удалением защитной группы амина с соединения, представленного химической формулой 4, полученного на вышеприведенной стадии (1). На схеме реакции PG представляет собой защитную группу; X представляет собой OR, где Rпредставляет собой низший C-Cалкил; HY представляет собой свободную кислоту. Предлагаемый способ позволяет повысить выход ингибитора дипептидилпептидазы-IV, представленного химической формулой 1. Изобретение относится также к способу производства соединения, представленного химической формулой 2, и к промежуточному соединению, используемому при получении соединения, представленного химической формулой 3. 3 н. и 9 з.п. ф-лы, 8 пр.
Формула
(стадия a) получение соединения, представленного химической формулой 6, путем раскрытия азиридинового кольца с использованием галогенида 2,4,5-дифторфенилмагния в соединении, представленном химической формулой 5; и
(стадия b) получение соединения, представленного химической формулой 2, путем введения защитной группы амина после гидролиза соединения, представленного химической формулой 6,
[Схема реакции 1]

(на вышеприведенной схеме реакции 1, PG представляет собой защитную группу, a R представляет собой низший С1-С5алкил).
(стадия 1) получение соединения, представленного химической формулой 4, путем связывания пептидной связью соединения, представленного химической формулой 2, и соединения, представленного химической формулой 3, путем осуществления их взаимодействия с использованием изобутилхлорформиата и основания в присутствии реакционного растворителя; и
(стадия 2) получение соединения, представленного химической формулой 1, удалением защитной группы амина с соединения, представленного химической формулой 4, полученного на вышеприведенной стадии (1),
[Схема реакции 3]
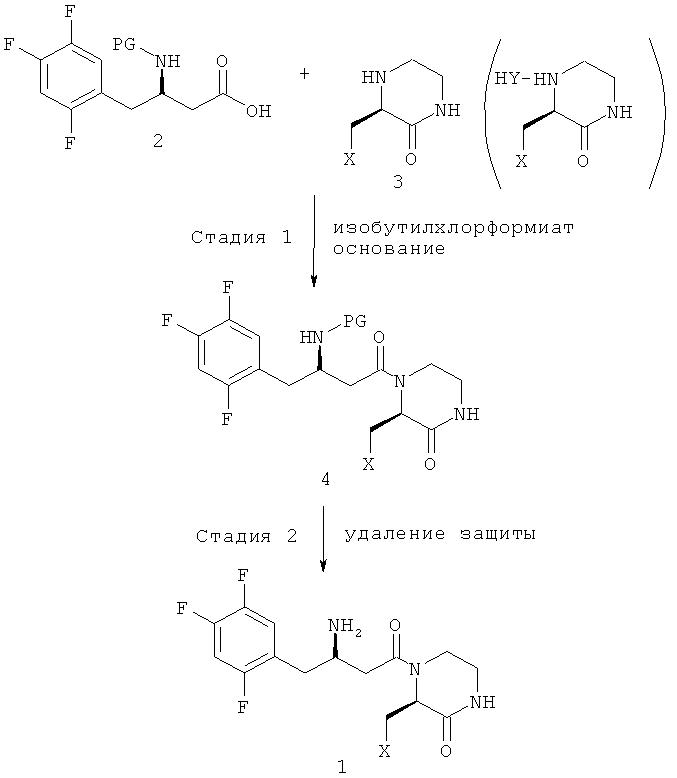
(на вышеприведенной схеме реакции 3 PG представляет собой защитную группу; X представляет собой OR1, где R1 представляет собой низший C1-C5алкил; и HY представляет собой свободную кислоту).
(стадия a') получение соединения, представленного химической формулой 8, путем взаимодействия соединения, представленного химической формулой 7, с бензилоксикарбониламиноацетальдегидом и восстановителем; и
(стадия b') получение соединения, представленного химической формулой 3, или его соли путем удаления бензилоксикарбонильной группы амина с соединения, представленного химической формулой 8, путем инициирования реакции гидрогенирования и индуцирования циклизации,
[Схема реакции 2]
(на вышеприведенной схеме реакции 2, X представляет собой OR1, где R1представляет собой низший C1-C5алкил; и HY представляет собой свободную кислоту).
[Химическая формула 8]
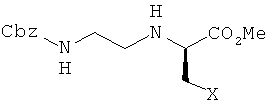
(в вышеприведенной химической формуле 8, X является таким, как определено на схеме реакции 3, приведенной в п.3).
Комментарии