Соединения, обладающие противовоспалительной и иммунодепрессивной активностью и содержащие их фармацевтические композиции - RU2177473C2
Код документа: RU2177473C2
Чертежи

Описание
Изобретение относится к новым соединениям и их применению в качестве противовоспалительных и иммунодепрессивных агентов.
Роль цитокинов (таких как IL-1β и α, TNFα и IL-6) в развитии воспалительных реакций хорошо известна (Dinarello С.А. and Wolff S.M., New Eng. J. Med. 328 (2): 106113, 1993; Tracey K.J. and Cerami A., Crit. Care Med. 21: S415, 1993; Melli and Parente L., Cytokines and lipocortins in inflammation and differentiation. Wiley Liss.New York 1990; Dawson M.M. Lymphokines and Interleukins. CRC Press. Boca Raton, FL 1991). Было проведено большое количество исследований в целях выявления соединений, называемых цитокин-подавляющими противовоспалительными лекарственными средствами (ПЦПВЛС), оказывающих ингибирующее действие на продукцию провоспалительных цитокинов, в частности, IL-1β и TNFα (Lee J.C. et al., Nature 372: 739, 1994; Davidsen S.K. and Summers J. B., Exp. Opin. Ther. Patents. 5(10): 1087, 1995), и недавно были описаны средства широкого спектра действия, называемые нетрадиционными нестероидными противовоспалительными лекарственными средствами (Chiou G.C.Y. and Liu S.X.L., Exp. Opin. Ther. Patents. 6(1): 41, 1996).
Tanaka et al. , Chem. Pharm. Bull., 31(8), 2810-2819 (1983) сообщают о важности гидроксамовой группы для проявления противовоспалительной активности: сообщается, что она играет настолько определяющую роль, что превосходит влияние остальных частей молекулы, где одни группы, такие как метокси, способны увеличивать активность воспаления, тогда как другие группы, такие как ацетамидо, вызывают снижение активности.
Было установлено, что производные гидроксамовой кислоты, содержащие амидобензойный радикал, оказывают, вопреки ранним публикациям, значительное противовоспалительное действие одновременно с иммунодепрессивной активностью.
Таким образом, настоящее изобретение относится к соединениям формулы (I):
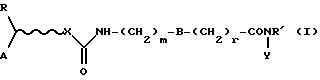
где R' представляет собой водород или (C1-4)алкил;
А представляет собой адамантил или моно-, би- или трициклический остаток, необязательно частично или полностью ненасыщенный, который может содержать один или несколько гетероатомов, выбранных из группы, содержащей N, S или О, и необязательно замещенный гидрокси, алканоилокси, первичным, вторичным или третичным амино, амино (C1-4) алкилом, моно- или ди(С1-4)алкиламино(C1-4)алкилом, галогеном, (С1-4)алкилом, три(С1-4)алкиламмоний (C1-4)алкилом;

R представляет собой водород или фенил;
X представляет собой атом кислорода или группу NR', где R' определен выше, или отсутствует;
r и m независимо равны 0, 1 или 2;
В представляет собой фениленовое или циклогексиленовое кольцо;
Y представляет собой гидрокси или амино(С1-4)алкильную цепь, необязательно прерванную атомом кислорода;
при условии, что трициклическая группа, которая определена в отношении к А, представляет собой флуоренил лишь в том случае, когда X не является О, и Y не является гидрокси, за исключением случая, когда указанный флуоренил замещен три(C1-4)алкиламмоний (C1-4)алкильной группой.
Указанная алкильная группа представляет собой, например, метил, этил, 2-метилэтил, 1,3-пропил, 1,4-бутил, 2-этилэтил, 3-метипропил, 1,5-пентил, 2-этилпропил, 2-метилбутил и аналоги, тогда как моно-, би- или трициклическая группа в том виде, как определена выше, может представлять собой фенил, циклогексил, пиридил, пиперидил, пиримидил, пиридазил, нафтил, инденил, антранил, фенантрил, флуоренил, фуранил, пиранил, бензофуранил, хроменил, ксантил, изотиазолил, изоксазолил, фенотиазил, феноксазил, морфолил, тиофенил, бензотиофенил и им подобные. Атом галогена может представлять собой атом хлора, брома или фтора. Наконец, под алканоилоксигруппой понимают ацетилокси, пропионилокси, изопропионилокси, бутаноилокси и им подобные.
Первой группой предпочтительных соединений формулы 1 являются такие соединения, где R' представляет собой водород; А выбирают из фенила, 1- или 2-нафтила, циклогексила, 1- или 2-1,2,3,4-тетрагидронафтила, адамантила, хинолинила, изохинолинила, 1- или 2-индeнилa, тетрагидрохинолинила, тетрагидроизохинолинила, необязательно замещенных, как указано выше.

Y представляет собой ОН, а R, В, m и r определены выше.
Второй группой предпочтительных соединений являются соединения, где R' представляет собой водород; А представляет собой необязательно замещенный фенил или 1- или 2-нафтил, более предпочтительно, 1- или 2-нафтил; R представляет собой фенил, если А представляет собой фенил, либо водород, если А представляет собой 1- или 2-нафтил; R, B, m и r определены выше; Y представляет собой ОН, а C1-С3 алкиленовая цепь определена выше применительно к первой группе предпочтительных соединений.
Группы А, предпочтительно, замещены (C1-4)алкиламино-(C1-4)алкильной группой.
Другой предмет изобретения относится к применению соединений формулы (I) в качестве противовоспалительных и иммунодепрессивных средств и к их включению в состав фармацевтических композиций, содержащих фармацевтически приемлемые наполнители.
Соединения по данному изобретению получают в
соответствии с методиками, известными специалистам в данной области. Исходные соединения, применяемые для данного получения, имеются в продаже, а также могут быть получены в соответствии с
литературными данными. Соединение формулы (I), в котором содержится X, получают, исходя из соединения формулы (II)
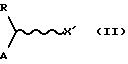
где R и А определены выше, а X' представляет собой атом кислорода или группу NR', где R' определен выше. Данное соединение взаимодействует с реакционноспособным производным карбоновой кислоты, таким как дисукцинимидилкарбонат или карбонилдиимидазол (КДИ), в присутствии третичного амина, такого как триэтиламин, или ароматического амина, такого как пиридин, в инертных растворителях, таких как ацетонитрил, тетрагидрофуран (ТГФ), диоксан, хлорированные растворители, при температуре от комнатной температуры до температуры возгонки растворителя и в течение промежутков времени, варьирующих от приблизительно 1 до приблизительно 48 часов.
Полученное соединение (III)
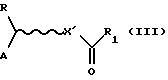
где R, А и X' определены выше, a R' представляет собой имидазолильную или гидроксисукцинимидильную группу, взаимодействует с желаемой аминокислотой в смеси с водой и смешивающимся с водой органическим растворителем, таким как тетрагидрофуран, ацетонитрил или спирты, в присутствии неорганического основания, такого как гидроксид щелочного металла, например, гидроксид натрия, карбонат или бикарбонат щелочного или щелочноземельного металла, например, карбонат натрия, при комнатной температуре в течение промежутков времени, варьирующих от приблизительно 1 до приблизительно 48 часов.
Полученную кислоту (IV)
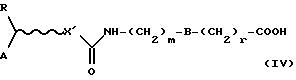
где R, А, X', В, m и r определены выше, активируют как ацилхлорид путем обработки тионилхлоридом в хлорированных растворителях, либо применяя в качестве растворителя тионилхлорид, при температуре от комнатной температуры до температуры возгонки растворителя и в течение времени примерно от 1 до 12 часов.
Ацилхлорид затем взаимодействует с гидроксиламингидрохлоридом в том случае, если желаемым является соединение формулы (I), где Y представляет собой гидрокси, либо с подходящим алкилендиамином в других случаях. Реакция протекает в смеси воды и смешивающегося с водой органического растворителя, такого как тетрагидрофуран, ацетонитрил, в присутствии неорганического основания, описанного на предыдущей стадии, при комнатной температуре в течение промежутков времени, варьирующих от приблизительно 1 до приблизительно 48 часов, с получением желаемого соединения формулы (I).
Альтернативно кислота (IV) взаимодействует с гидроксиламингидрохлоридом или алкилендиамином в присутствии конденсирующего агента, такого как GDI, и третичного амина, такого как триэтиламин, в инертных растворителях, таких как ацетонитрил, тетрагидрофуран, диоксан, хлорированные растворители, при комнатной температуре за время примерно от 1 до 24 часов. Полученные соединения очищают методами обычной хроматографии или кристаллизации.
Соединения формулы (I), в которых X
отсутствует, получают, исходя из соответствующей кислоты формулы (V)
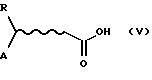
где R и А определены выше, активированной как ацилхлорид путем обработки тионилхлоридом в хлорированных растворителях, либо применяя в качестве растворителя тионилхлорид, при температуре, варьирующей от комнатной температуры до температуры возгонки растворителя в течение приблизительно 1-12 часов. Ацилхлорид взаимодействует с желаемой аминокислотой в смеси с водой и смешивающимся с водой органическим растворителем, таким как ТГФ или ацетонитрил, в присутствии основания, такого как гидроксид щелочного металла, например, гидроксид натрия, карбонат или бикарбонат щелочного или щелочноземельного металла, такой так карбонат натрия, при комнатной температуре в течение приблизительно 1-12 часов.
Полученное соединение формулы (VI)
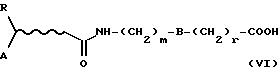
где R, А, В, m и r определены выше, превращают в желаемое соединение формулы (I), следуя методикам, описанным выше.
Ниже приводятся примеры получения некоторых представителей соединений, заявленных в настоящем изобретении.1H-ЯМР спектры снимали в диметилсульфоксиде (ДМСО) на спектрометре Varian Gemini 200, если не указано особо.
Пример 1
4-(5-Фенилпентанамидо)бензогидроксамовая кислота
А. В раствор 5-фенилпентановой кислоты (5 г, 28 ммоль) в хлороформе (100 мл)
добавляли тионилхлорид (2,5 мл, 34 ммоль). Реакционную смесь нагревали с обратным холодильником в течение 4 часов, затем выпаривали досуха. Остаток вновь растворяли в хлороформе и повторно выпаривали
досуха трижды. Полученный 5-фенилпентаноилхлорид растворяли в ТГФ (50 мл) и полученный раствор медленно добавляли к раствору 4-аминометилбензойной кислоты (3,8 г, 28 ммоль) в 1 н. гидроксиде натрия
(56 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем ТГФ выпаривали на холоде и водный раствор подкисляли HCl. Выпавший осадок отфильтровывали, повторно
растворяли в ТГФ, сушили над безводным хлоридом кальция и растворитель выпаривали с получением 6,7 г 4-(5-фенилпентанамидо)бензойной кислоты (выход 81%); т.пл. = 227-230oC.
1H-ЯМР d 12.5 (з, 1H обмен с D2O), 10.3 (s, 1H), 7.92 (d, 2H), 7.75 (d, 2H), 7.25 (m, 5H), 2.63 (t, 2H), 2.41 (t, 2H), 1.65 (m, 4H).
Б. К раствору соединения, полученного в А (6,7 г, 22 ммоль) в хлороформе (100 мл), добавляли тионилхлорид (3,3 мл, 45 ммоль) и 3 капли пиридина. Реакционную смесь перемешивали при комнатной температуре в течение 5 часов, затем выпаривали досуха. Остаток вновь растворяли в хлороформе и повторно выпаривали досуха трижды. Полученный 4-(5-фенилпентанамидо)бензоилхлорид растворяли в ТГФ (50 мл) и полученный раствор медленно добавляли к раствору гидроксиламингидрохлорида (1,9 г, 27 ммоль) и бикарбоната натрия (1,9 г, 22 ммоль) в 1 н. гидроксиде натрия (27 мл, 27 ммоль) и ТГФ (25 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем подкисляли 1 н. HCl и ТГФ выпаривали на холоде. Водную фазу экстрагировали добавлением этилацетата и объединенные органические фазы сушили над безводным сульфатом натрия и растворитель выпаривали. Полученный сырой продукт обрабатывали теплым этиловым эфиром и фильтровали с получением 4,8 г указанного в заголовке соединения (выход 69%); т.пл. = 189-191oC.
1H-ЯМР d 11.12 (s, 1H), 10.13 (s, 1H), 8.97 (s, 1H), 7.691 (m, 4H), 7.33-7.13 (m, 5H), 2.61 (m, 2H), 2.38 (m, 2H), 1.65-1.58 (m, 4H).
Пример 2
4-[2,2-(Дифенил)этоксикарбамоилметил] бензогидроксамовая кислота
А. Смесь 2,2-дифенилэтанола (6 г, 30 ммоль), КДИ (4,9 г, 30 ммоль) в ТГФ (30 мл) перемешивали при комнатной
температуре в течение 2 часов, затем к ним добавляли раствор 4-аминометилбензойной кислоты (4,6 г, 30 ммоль) в 1 н. гидроксиде натрия (30 мл). Реакционную смесь перемешивали при комнатной температуре
в течение 3 часов, затем подкисляли HCl, ТГФ выпаривали на холоде, затем водный раствор экстрагировали добавлением этилацетата. Органическую фазу сушили над безводным сульфатом натрия и растворитель
выпаривали. Полученный сырой продукт обрабатывали теплым н-гексаном и фильтровали с получением 10 г 4-[2,2-(дифенил)этоксикарбамоилметил]бензойной кислоты (выход 90%); т.пл. = 102-105oC.
1H-ЯМР d 12.7 (s, 1H, обмен с D2O), 7.91 (d, 2H), 7.78 (t, 1H), 7.50-7.20 (m, 12H), 4.61 (d, 2H), 4.38 (t, 1H), 4.23 (d, 2H).
Б. К раствору соединения, полученного в стадии А (4,5 г, 12 ммоль) в хлороформе (50 мл), добавляли тионилхлорид (1,3 мл, 18 ммоль) и 3 капли пиридина. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем выпаривали досуха. Остаток вновь растворяли в хлороформе и повторно выпаривали досуха трижды. Полученный 4-[2,2-(дифенил)этоксикарбамоилметил] бензоилхлорид растворяли в ТГФ (50 мл) и раствор медленно добавляли к раствору гидроксиламингидрохлорида (1,0 г, 14,4 ммоль) и бикарбоната натрия (1 г, 12 ммоль) в 1 н. гидроксиде натрия (14,4 мл, 14,4 ммоль) и ТГФ (20 мл). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем подкисляли 1 н. HCl и ТГФ выпаривали на холоде. Водную фазу экстрагировали добавлением этилацетата, объединенные органические фазы сушили над безводным сульфатом натрия и растворитель выпаривали. Полученный сырой продукт очищали по методу колоночной хроматографии на силикагеле (элюент: метиленхлорид/метанол 15;1) и полученный продукт обрабатывали теплым этиловым эфиром и фильтровали, посредством чего получали 1,5 г указанного в заголовке соединения (выход 32%); т.пл. = 164-166oC.
1H-ЯМР d 11.21 (s, 1H), 9.04 (s, 1H), 7.74 (t, 1H), 7.72 (m, 2H), 7.38-7.20 (m, 12H), 4.60 (d, 2H), 4.38 (t, 1H), 4.20 (d, 2H).
Пример 3
4-[2-(Адамант-1-ил)этоксикарбамоил]
бензогидроксамовая кислота
А. Смесь 2-(адамант-1-ил)этанола (2,1 г, 12 ммоль), дисукцинимидилкарбоната (3,3 г, 13 ммоль) и пиридина (0,86 мл, 10 ммоль) в ацетонитриле (50 мл) перемешивали при
комнатной температуре в течение 24 часов, затем растворитель выпаривали на холоде и остаток растворяли в метиленхлориде (100 мл). Раствор промывали водой (50 мл), 1 н. HCl (50 мл), и вновь водой (50
мл). Органическую фазу сушили над безводным сульфатом натрия и растворитель выпаривали с получением 3 г (9 ммоль) сырого 2-(адамант-1-ил)этилсукцинимидилкарбоната, который повторно растворяли в ТГФ
(30 мл). Полученный раствор добавляли к раствору 4-аминобензойной кислоты (1,3 г, 9 ммоль) и карбоната натрия (0,98 г, 9 ммоль) в воде (30 мл), затем перемешивали при комнатной температуре в течение
ночи и подкисляли HCl. ТГФ выпаривали на холоде и водный раствор экстрагировали добавлением этилацетата. Органическую фазу сушили над безводным сульфатом натрия и растворитель выпаривали. Полученный
сырой продукт обрабатывали теплым н- гексаном и фильтровали с получением 3,2 г 4-[2-(адамант-1-ил)этоксикарбамоил] бензойной кислоты (выход 77%); т.пл. = 197-200oC.
1H-ЯМР d 12.6 (s, 1H, обмен с D2O), 10.0 (s, 1H), 7.89 (d, 2H), 7.60 (d, 2H), 4.19 (t, 2H), 1.93 (m, 5H), 1.75-1.40 (m, 12H).
Б. К раствору соединения, полученного в стадии А (3 г, 8,7 ммоль) в хлороформе (50 мл), добавляли тионилхлорид (1,2 мл, 17,4 ммоль) и 3 капли пиридина. Реакционную смесь нагревали с обратным холодильником в течение 5 часов, затем выпаривали досуха. Остаток вновь растворяли в хлороформе и повторно выпаривали досуха трижды. Полученный 4-[2-(адамант-1-ил)этоксикарбамоил]бензоилхлорид растворяли в ТГФ (50 мл) и раствор медленно добавляли к раствору гидроксиламингидрохлорида (0,7 г, 10,4 ммоль) и бикарбоната натрия (0,7 г, 8,7 ммоль), в 1 н. гидроксиде натрия (10,4 мл, 10,4 ммоль) и ТГФ (20 мл), затем перемешивали при комнатной температуре в течение 8 часов, подкисляли 1 н. HCl и ТГФ выпаривали на холоде. Водную фазу экстрагировали добавлением этилацетата, объединенные органические фазы сушили над безводным сульфатом натрия и растворитель выпаривали. Полученный сырой продукт обрабатывали теплым этиловым эфиром и фильтровали с получением 1,5 г указанного в заголовке соединения (выход 48%); т. пл. = 198-200o C.
1H-ЯМР d 11.08 (s, 1H), 9.84 (s, 1H), 8.94 (s, 1H), 7.70 (m, 2H), 7.53 (m, 2H), 4.17 (m, 2H), 1.94 (m, 3H), 1.73-1.37 (m, 14H).
Пример 4
4-[(Нафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота
А. Исходя из нафт-1-илметанола (5 г, 31 ммоль), следуя методике, описанной в Примере 3А, получали 7 г чистой
4-[(нафт-1-ил)метоксикарбамоил]бензойной кислоты (выход 81%).
1H-ЯМР d 12.7 (s, 1H, обмен с D2O), 10.2 (s, 1H), 8.18 (d, 1H), 7.97 (m, 4H), 7.61 (m, 6H), 5.69 (s, 2H).
Б. Исходя из соединения, полученного в стадии А (6 г, 19 ммоль), следуя методике, описанной в Примере 3Б, получали 1,86 г указанного в заголовке соединения (выход 30%); т.пл. = 192-194oC.
1H-ЯМР d 11.12 (s, 1H), 10.06 (s, 1H), 8.98 (s, 1H), 8.14 (m, 1H), 8.03-7.96 (m, 2H), 7.76-7.50 (m, 8H), 5.67 (s, 2H).
Пример 5
4-[(1,2,3,4-Тетрагидронафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота
А. Исходя из (1,2,3,4-тетpaгидpoнaфт-2-ил)мeтaнoлa (4 г, 24 ммоль), следуя методике, описанной в Примере 3А,
получали 5,2 г чистой 4-[(1,2,3,4-тетрагидронафт-2-ил)метоксикарбамоил] бензойной кислоты (выход 67%); т. пл. = 203-206oC.
1H-ЯМР d 12.6 (s, 1H, обмен с D2O), 10.1 (s, 1H), 7.92 (d, 2H), 7.63 (d, 2H), 7.1 (m, 4H), 4.14 (d, 2H), 2.88 (m, 3H), 2.54 (m, 1H), 2.25-1.90 (m, 2H), 1.49 (m, 1H).
Б. Исходя из соединения, полученного в стадии А (5,2 г, 16 ммоль), следуя методике, описанной в Примере 3Б, получали 4,1 г указанного в заголовке соединения (выход 75%); т.пл. = 184-186oC.
1H-ЯМР d 11.10 (s, 1H), 9.97 (s, 1H), 8.96 (s, 1H), 7.72 (m, 2H), 7.54 (m, 2H), 7.09 (s, 4H), 4.11 (d, 2H), 2.92-2.75 (m, 3H), 2.61-2,47 (m, 1H), 2,17-1.93 (m, 2H), 1.56-1,35 (m, 1H).
Пример
6
4-[(3-Фенилпропокси)карбамоил]бензогидроксамовая кислота
А. Исходя из 3-фенилпропанола (5 г, 36 ммоль), следуя методике, описанной в Примере 3А, получали 7,7 г чистой
4-[(3-фенилпропокси)карбамоил] бензойной кислоты (выход 71%); т.пл. = 171-173oC.
1H-ЯМР d 12.6 (s, 1H обмен с D2O), 10.1 (s, 1H), 7.90 (d, 2H), 7.61 (d, 2H), 7.28 (m, 5H), 4.13 (t, 2H), 2.72 (t, 2H), 1.98 (m, 2H).
Б. Исходя из соединения, полученного в стадии А (7 г, 23 ммоль), следуя методике, описанной в Примере 3Б, получали 5,8 г указанного в заголовке соединения (выход 80%); т.пл. = 179-181oC.
1H-ЯМР d 11.09 (s, 1H), 9.94 (s, 1H), 8.95 (s, 1H), 7.71 (m, 2H), 7.54 (m, 2H), 7.36-7.15 (m, 5H), 4.10 (t, 2H), 2.70 (m, 2H), 1.95 (m, 2H).
Пример 7
4-[(Нафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота
А. Исходя из нафт-2-илметанола (5 г, 31 ммоль), следуя
методике, описанной в Примере 3А, получали 6,6 г чистой 4-[(нафт-2-ил)метоксикарбамоил] бензойной кислоты (выход 68%); т.пл. = 241-243oC.
1H-ЯМР d 12.7 (s, 1H, обмен с D2O), 10.2 (s, 1H), 7.95 (m, 6H), 7.60 (m, 5H), 5.39 (s, 2H).
Б. Исходя из соединения, полученного в стадии А (6 г, 18,6 ммоль), следуя методике, описанной в Примере 3Б, получали 4,5 г указанного в заголовке соединения (выход 72%); т.пл. = 220-222oC.
1H-ЯМР d 11,10 (s, 1H), 11.01 (s, 1H), 8.95 (s, 1H), 7.99-7.90 (m, 4H), 7.73 (m, 2H), 7.61-7.50 (m, 5H), 5.36 (s, 2H).
Пример 8
(E)-4-[(3-Фенилпроп-2-енил)карбамоил] бензогидроксамовая кислота
А. 1,75 г (13 ммоль) транс-циннамола растворяли в
130 мл ацетонитрила в атмосфере аргона, затем добавляли 5 г (19 ммоль) N,N'-дисукцинимидилкарбоната, нагревая вплоть до полного растворения, затем смесь охлаждали до 20oC и добавляли
пиридин (0,65 мл, 1 г, 12 ммоль). Реакционную смесь покрывали алюминием и перемешивали при комнатной температуре в течение 30 часов. Растворитель выпаривали на холоде, остаток переносили в этилацетат
и неоднократно промывали 0,1 н. HCl и, наконец, водой. Раствор высушивали, затем выпаривали досуха на холоде с получением сырого продукта, который растворяли в 26 мл диоксана и добавляли к раствору
п-аминобензойной кислоты (1,79 г, 13 ммоль) в 26 мл воды с 1,38 г (13 ммоль) карбоната натрия. Раствор перемешивали при комнатной температуре в течение 60 часов, затем добавляли ТГФ и pacсол.
Органическую фазу отделяли и промывали 0,1 н. HCl (дважды) и вновь рассолом, затем сушили и выпаривали досуха. Полученный сырой продукт переносили в изопропиловый эфир и фильтровали, посредством чего
получали 0,7 г (Е)-4-[(3-фенилпроп-2-енил)карбамоил] бензойной кислоты (выход 18%); т.пл. 176-178oC.
1H-ЯМР d 12.70 (s, 1H, обмен с D2O), 10.20 (s, 1H), 7.93 (d, 2H), 7.61 (d, 2H), 7.55-7.25 (m, 5H), 6.78 (d, 1H), 6.46 (dt, 1H), 4.87 (d, 2H).
Б. Соединение из стадии А (0,7 г, 2,3 ммоль) растворяли в 8 мл безводного ТГФ и добавляли КДИ при 0oC (0,46 г, 2,8 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем к ней добавляли гидроксиламингидрохлорид (0,2 г, 2,3 ммоль) и перемешивание продолжали при комнатной температуре в течение дальнейших 60 часов. Образовавшийся осадок отфильтровывали и суспендировали в 1 н. HCl, затем перемешивали в течение ночи. После фильтрования и сушки в вакууме получали 400 мг указанного в заголовке соединения (выход 55,6%); т.пл. = 194-195oC.
1H-ЯМР d 11.14 (s, 1H, обмен с D2O), 10.07 (s, 1H), 8.97 (s, 1H, обмен с D2O), 7.75 (d, 2H), 7.65-7.25 (m, 7H), 6.79 (d, 1H), 6.48 (dt, 1H), 4.85 (d, 2H).
Пример 9
(Z)-4-[(3-Фенилпроп-2-енил) карбамоил] бензогидроксамовая кислота
А. Исходя из цис-циннамола (2,82 г, 20 ммоль), следуя методике, описанной в Примере 8A, получали 2,4 г (Z)-4- [(3-фенилпроп-2-енил)карбамоил] бензойной кислоты (выход 40%).
1 H-ЯМР d 12.72 (s, 1H, обмен с D2O), 10.20 (s, 1H), 7.92 (d, 2H), 7.60 (d, 2H), 7.55-7.25 (m, 5H), 6.73 (d, 1H), 5,91 (dt, 1H), 4.96 (d, 2H).
Б. Исходя из соединения, полученного в стадии А (0,7 г, 2,3 ммоль), следуя методике, описанной в Примере 8Б, получали 400 мг указанного в заголовке соединения (выход 55,6%); т.пл. = 169-170oC.
1H-ЯМР d 11.13 (s, 1H, обмен с D2O), 10.05 (s, 1H), 8.98 (s, 1H, обмен с D2O), 7,14 (d, 2H), 7,55 (d, 2H), 7,50-7,25 (m, 5H), 6.77 (d, 1H), 5.90 (dt, 1H), 4.94 (d, 2H).
Пример 10
4-[3,3-(Дифенил)пропоксикарбамоил] бензогидроксамовая кислота
А. Исходя из 3,3-дифенилпропанола (3 г, 14 ммоль), следуя методике, описанной в Примере 3А,
получали 4,2 г 4-[3,3-(дифенил)пропоксикарбамоил] бензойной кислоты (выход 80%); т.пл. = 156-159oC.
1H-ЯМР d 12.6 (s, 1H, обмен с D2O), 10.1 (s, 1H), 7.90 (d, 2H), 7.60 (d, 2H), 7.40-7.15 (m, 10H), 4.17 (t, 1H), 4.03 (t, 2H), 2.46 (m, 2H).
Б. Исходя из соединения, полученного в стадии А (4,0 г, 10 ммоль), следуя методике, описанной в Примере 3Б, получали 1 г указанного в заголовке соединения (выход 26%); т.пл. = 196-197oC.
1H-ЯМР d 11.08 (s, 1H), 9.93 (s, 1H), 8.93 (s, 1H), 7.70 (m, 2H), 7.54 (m, 2H), 7.39-7.15 (m, 10H), 4.15 (t, 1H), 3.99 (t, 2H), 2.44 (q, 2H).
Пример 11
N-(2-Аминоэтил-4-[(флуорен-9-ил)метоксикарбамоилметил]бензамид.HCl
А. Раствор
9-флуоренилметилхлорформиата (5 г, 19 ммоль) в ТГФ (50 мл) медленно добавляли к раствору 4-аминометилбензойной кислоты (2,9 г, 19 ммоль). Реакционную смесь перемешивали при комнатной температуре в
течение 3 часов, затем подкисляли 1 н. HCl и ТГФ выпаривали на холоде. Водную фазу экстрагировали добавлением этилацетата, объединенные органические фазы сушили над безводным сульфатом натрия и
растворитель выпаривали. Полученный сырой продукт обрабатывали теплым этиловым эфиром и фильтровали с получением 7 г 4-[(флуорен-9-ил)метоксикарбамоилметил] бензойной кислоты (выход 98%); т. пл. =
232-235oC.
1H-ЯМР d 12.7 (s, 1H, обмен с D2O), 7.94 (m, 5H), 7.77 (d, 2H), 7.58-7.30 (m, 6H), 4.42 (d, 2H), 4.30 (m, 3H).
Б. К раствору соединения, полученного в стадии А (3,7 г, 10 ммоль) в хлороформе (100 мл), добавляли тионилхлорид (1,46 мл, 20 ммоль) и реакционную смесь нагревали с обратным холодильником в течение 2 часов, затем выпаривали досуха. Остаток вновь растворяли в хлороформе и повторно выпаривали трижды. Полученный 4-[(флуорен-9-ил)метоксикарбамоилметил]бензоилхлорид растворяли в ТГФ (30 мл) и раствор медленно добавляли к раствору 2-(трет-бутилоксиимино)этиламина (1,6 г, 10 ммоль) и бикарбоната натрия (0,8 г, 10 ммоль) в воде (20 мл) и ТГФ (30 мл). Реакционную смесь перемешивали при комнатной температуре в течение 12 часов, затем ТГФ выпаривали на холоде, водную фазу подкисляли 1 н. HCl и экстрагировали добавлением этилацетата. Органические фазы сушили над безводным сульфатом натрия и растворитель выпаривали. Полученный сырой продукт обрабатывали теплым этиловым эфиром и фильтровали с получением 3,85 г N-(2- трет-бутилоксикарбамоилметил)-4-[(флуорен-9-ил)метоксикарбамоилметил] бензамида (выход 75%); т.пл. = 167-169oC.
1H-ЯМР d 8.45 (t, 1H), 7.94-7.70 (m, 7H), 7.54-7.25 (m, 6H), 6.96 (t, 1H), 4.42 (d, 2H), 4.27 (m, 3H), 3.32 (q, 2H), 3.13 (q, 2H), 1.41 (s, 9H).
В. Продукт из стадии Б (3,6 г, 7 ммоль) добавляли малыми порциями к трифторуксусной кислоте (25 мл) при 0oC. Смесь перемешивали при комнатной температуре в течение 4 часов, затем кислоту выпаривали, а остаток переносили в теплый этиловый эфир и фильтровали с получением продукта, который растворяли в ТГФ и метаноле, и к нему добавляли раствор HCl в эфире. Растворители выпаривали и процедуру повторяли 4 раза. Сырой продукт переносили в некоторое количество воды при 0oC и фильтровали с получением указанного в заголовке соединения (2 г) (выход 65%); т.пл. = 182-184oC.
1H-ЯМР d 8.79 (t, 1H), 8.17 (s, 3H), 7.96-7.86 (m, 5H), 7.72 (m, 2H), 7.48-7.29 (m, 6H), 4.38 (m, 2H), 4.25 (m, 3H), 3.55 (q, 2H), 3.00 (t, 2H).
Пример 12
Гидрохлорид 4-[6-(диэтиламинометил)нафт-2-илметилоксикарбамоил] бензогидроксамовой кислоты
А. 1-(3-Диметиламинопропил)-3-этилкарбодиимидгидрохлорид
(ЭДКИ) (22,2 г, 115 ммоль) добавляли к раствору 2,6-нафталиндикарбоновой кислоты (25 г, 115 ммоль) и гидроксибензотриазола (15,6 г, 115 ммоль) в диметилформамиде (1800 мл) и смесь перемешивали при
комнатной температуре в течение 2 часов. Добавляли диэтиламин (34,3 мл, 345 ммоль) и раствор перемешивали в течение ночи при комнатной температуре. Растворитель затем выпаривали в условиях пониженного
давления и сырой продукт обрабатывали 1 н. HCl (500 мл) и этилацетатом (500 мл), нерастворимые соединения отфильтровывали и фазы разделяли. Органическую фазу экстрагировали добавлением 5% карбоната
натрия (3 х 200 мл), а объединенные водные растворы подкисляли концентрированной HCl и экстрагировали добавлением этилацетата (3 х 200 мл). Органический раствор затем промывали 1 н. HCl (6 x 100 мл),
сушили над безводным сульфатом натрия и растворитель удаляли в условиях пониженного давления с получением 18,5 г (выход 60%) чистой 6-(диэтиламинокарбонил)-2-нафталинкарбоновой кислоты; т. пл. =
122-124oC.
1H-ЯМР d 8.67 (s, 1H), 8.25-8.00 (m, 4H), 7.56 (d, 1H), 3.60-3.20 (m, 4H), 1.30-1.00 (m, 6H).
Б. Раствор (6-диэтиламинокарбонил)-2-нафталинкарбоновой кислоты (18 г, 66 ммоль) в ТГФ (200 мл) медленно добавляли в возгоняющуюся суспензию литийалюминийгидрида (7,5 г, 199 ммоль) в ТГФ (500 мл). Смесь нагревали с обратным холодильником в течение часа, затем охлаждали до комнатной температуры и обрабатывали смесью ТГФ (25 мл) и воды (3,5 мл), 20% гидроксидом натрия (8,5 мл) и, наконец, водой (33 мл). Белое твердое вещество отфильтровывали и растворитель удаляли в условиях пониженного давления. Сырой продукт растворяли в диэтиловом эфире (200 мл) и экстрагировали добавлением 1 н. HCl (3 х 100 мл). Водный раствор обрабатывали 32% гидроксидом натрия и экстрагировали добавлением диэтилового эфира (3 х 100 мл). Органический раствор затем сушили над безводным сульфатом натрия и растворитель удаляли в условиях пониженного давления с получением 12,7 г (выход 79%) чистого 6-(диэтиламинометил)-2-нафталинметанола в виде густого масла.
1H-ЯМР d 7.90-7.74 (m, 4H), 7.49 (m, 2H), 5.32 (t, 1H, обмен с D2O), 4.68 (d, 2H), 3.69 (s, 2H), 2.52 (q, 4H), 1.01 (t, 6H).
В. Раствор 6-(диэтиламинометил)-2-нафталинметанола (12,5 г, 51 ммоль) и N, N'-дисукцинимидилкарбоната (13,2 г, 51 ммоль) в ацетонитриле (250 мл) перемешивали при комнатной температуре в течение 3 часов, затем растворитель удаляли и сырой продукт растворяли в ТГФ (110 мл). Данный раствор добавляли к раствору 4-аминобензойной кислоты (7,1 г, 51 ммоль) и карбоната натрия (5,5 г, 51 ммоль) в воде (200 мл) и ТГФ (100 мл). Смесь перемешивали в течение ночи при комнатной температуре, затем ТГФ удаляли в условиях пониженного давления и раствор обрабатывали 1 н. HCl (102 мл, 102 ммоль). Осадок отфильтровывали, сушили в условиях пониженного давления, растирали с диэтиловым эфиром и фильтровали с получением 13,2 г (выход 64%) чистой 4-[6-(диэтиламинометил)нафт-2-илметилоксикарбамоил] бензойной кислоты; т. пл. = 201-105oC (с разлож.).
1H-ЯМР d 10.26 (s, 1H), 8.13 (s, 1H), 8.05-7.75 (m, 6H), 7.63 (m, 3H), 5.40 (s, 2H), 4.32 (s, 2H), 2.98 (q, 4H), 1.24 (t, 6H).
Г. Раствор
4-[6-(диэтиламинометил)нафт-2-илметилоксикарбамоил] бензойной кислоты (13,1 г, 32 ммоль) и тионилхлорида (7 мл, 96 ммоль) в хлороформе (300 мл) нагревали с обратным холодильником в течение 4 часов,
затем растворитель и тионилхлорид выпаривали. Сырой продукт растворяли в хлороформе (100 мл) и выпаривали досуха трижды. Сырой продукт в виде твердого вещества добавляли к раствору
гидроксиламингидрохлорида (2,7
г, 39 ммоль) и бикарбоната натрия (5,4 г, 64 ммоль) и 1 н. гидроксида натрия (39 мл, 39 ммоль) в воде (150 мл) и ТГФ (50 мл). Смесь перемешивали в течение ночи
при комнатной температуре, затем ТГФ удаляли в условиях пониженного давления и водную фазу экстрагировали добавлением этилацетата (3 х 100 мл). Объединенные органические фазы сушили над безводным
сульфатом натрия и растворитель удаляли в условиях пониженного давления. Сырой продукт растворяли в ТГФ и обрабатывали 1,5 н. эфирным раствором HCl. Твердый продукт отфильтровывали и сушили с
получением 6 г (выход 41%) чистого гидрохлорида 4-[6-(диэтиламинометил)нафт-2-илметилоксикарбамоил] бензогидроксамовой кислоты в виде белого твердого вещества; т.пл. = 162-165oC (с
разлож.).
1H-ЯМР d 11.24 (s, 1Н, обмен с D2O), 10.88 (s, 1H, обмен с D2O), 10.16 (s, 1H), 8.98 (bs, 1H, обмен с D2O), 8.21 (s, 1H), 8.10-7.97 (m, 3H), 7.89 (d, 1H), 7.80-7.55 (m, 5H), 5.39 (s, 2H), 4.48 (d, 2H), 3.09 (m, 4H), 1.30 (t, 6H).
Пример 13
Гидрохлорид
4-[6-(дипропиламинометил)нафт-2-илметилоксикарбамоил]бензогидроксамовой кислоты
Исходя из 2,6-нафталиндикарбоновой кислоты (5 г) и дипропиламина (9,6 мл), и следуя методике, описанной в
Примере 12, получали 1 г чистого гидрохлорида 4-[6-(дипропиламинометил)нaфт)-2-илмeтилoкcикapбaмoил] бeнзoгидpoкcaмoвoй кислоты в виде белого твердого вещества; т.пл. = 140-142oC (с
разлож.).
1H-ЯМР d 11.15 (s, 1H, обмен с D2O), 10.95 (s, 1H, обмен с D2O), 10.16 (s, 1H), 8.20 (s, 1H), 8.10-7.97 (m, 3H), 7.89 (d, 1H), 7.80-7.55 (m, 5H), 5.40 (s, 2H), 4.50 (d, 2H), 2.98 (m, 4H), 1.79 (m, 4H), 0.88 (t, 6H).
Пример 14
Гидрохлорид 4-[6-(дибутиламинометил)нафт-2- илметилоксикарбамоил] бензогидроксамовой
кислоты
Исходя из 2,6-нафталиндикарбоновой кислоты (5 г) и дибутиламина (11,8 мл), и следуя методике, описанной в Примере 12, получали 1,2 г чистого гидрохлорида
4-[6-(дибутиламинометил)нафт-2-илметилоксикарбамоил] бензогидроксамовой кислоты в виде белого твердого вещества; т.пл. = 137-141oC (с разлож.).
1H-ЯМР d 11.19 (s, 1H, обмен с D2O), 10.91 (s, 1H, обмен с D2O), 10.16 (s, 1H), 8.96 (bs, 1H, обмен с D2O), 8.21 (s, 1H), 8.10-7.98 (m, 3H), 7.87 (d, 1H), 7.80-7.55 (m, 5H), 5.38 (s, 2H), 4.52 (d, 2H), 3.02 (m, 4H), 1.77 (m, 4H), 1.30 (m, 4H), 0.89 (t, 6H).
Пример 15
Гидрохлорид 4-[4-(диэтиламинометил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты
Исходя из 1,4-нафталиндикарбоновой кислоты (5 г) и диэтиламина (7,3 мл), и следуя методике, описанной в Примере 12, получали 1,1 г чистого гидрохлорида
4-[4-(диэтиламинометил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты в виде белого твердого вещества; т.пл. = 162-165oC (с разлож.).
1H-ЯМР d 11.24 (s, 1H, обмен с D2O), 10.48 (s, 1H, обмен с D2O), 10.13 (s, 1H), 8.43 (m, 1H), 8.26 (m, 1H), 8.04 (d, 1H), 7.82-7.70 (m, 5H), 7.58 (d, 2H), 5.73 (s, 2H), 4.84 (d, 2H), 3.17 (m, 4H), 1.32 (t, 6H).
Пример 16
Гидрохлорид 4-[6-(диэтиламинометил)нафт-2-илметиламинокарбамоил] бензогидроксамовой кислоты
А. Раствор
6-(диэтиламинокарбонил)-2-нафталинкарбоновой кислоты (полученной, как описано в Примере 12А) (14 г, 52 ммоль) и тионилхлорида (3,8 мл, 52 ммоль) в хлороформе (200 мл) нагревали с обратным
холодильником в течение 3 часов, затем растворитель и тионилхлорид выпаривали. Сырой продукт растворяли в хлороформе (100 мл) и выпаривали досуха трижды. Сырой продукт растворяли в ТГФ (50 мл) и при
0oC добавляли к раствору 32% гидроксида аммония (10 мл) в воде (50 мл) и ТГФ (50 мл). Смесь перемешивали в течение ночи при комнатной температуре, затем ТГФ удаляли в условиях пониженного
давления и твердое вещество отфильтровывали и сушили с получением 11,6 г (выход 81%) 6-(диэтиламинокарбонил)-2-нафталинкарбоксамида, который применяли на следующей стадии без дальнейшей очистки.
Б. Раствор 6-(диэтиламинокарбонил)-2- нафталинкарбоксамида (11,6 г, 42 ммоль) в ТГФ (100 мл) медленно добавляли в возгоняющуюся суспензию литийалюминийгидрида) 4,9 г, 128 ммоль) в ТГФ (100 мл). Смесь нагревали с обратным холодильником в течение 2 часов, затем охлаждали до комнатной температуры и обрабатывали смесью ТГФ (16 мл) и воды (2,2 мл), 20% гидроксидом натрия (5,5 мл) и, наконец, водой (22 мл). Белое твердое вещество отфильтровывали и растворитель удаляли в условиях пониженного давления. Сырой продукт очищали по методу хроматографии на силикагеле (элюент хлороформ-метанол-гидроксид аммония 15:1:0,1) с получением 8,1 г (выход 80%) чистого 6-(диэтиламинометил)-2-нафтилметиламина в виде восковидного твердого вещества.
1H-ЯМР d 7.78 (m, 4H), 7.49 (m, 2H), 3.89 (s, 2H), 3.67 (s, 2H), 2.50 (q, 4H), 1.00 (t, 6H).
В. Раствор 6-(диэтиламинометил)-2-нафтилметиламина (6 г, 24 ммоль) и N, N'-дисукцинимидилкарбоната (6,3 г, 24 ммоль) в ацетонитриле (200 мл) перемешивали при комнатной температуре в течение 3 часов, затем данный раствор добавляли к раствору 4-аминобензойной кислоты (3,4 г, 24 ммоль) и карбоната натрия (2,6 г, 24 ммоль) в воде (100 мл) и ТГФ (100 мл). Смесь перемешивали при комнатной температуре в течение 48 часов, затем добавляли 1 н. HCl (48 мл, 48 ммоль) и растворители удаляли в условиях пониженного давления. Сырой продукт очищали по методу хроматографии на силикагеле (элюент хлороформ-метанол-гидроксид аммония 7:3:0,5) с получением 3,5 г (выход 36%) чистой 4-[6-(диэтиламинометил)нафт-2-илметиламинокарбамоил] -бензойной кислоты; т.пл. = 179-183oC (с разлож.).
1H-ЯМР d 9.58 (s, 1H), 7.95-7.78 (m, 6H), 7.65-7.45 (m, 4H),
7.29 (t, 1H), 4.51 (d, 2H), 3.81 (s, 2H), 2.62 (q, 4H), 1.07 (t, 6H)
Г. Раствор 4-[6-(диэтиламинометил)нафт-2-илметиламинокарбамоил] бензойной кислоты (3,1 г, 7,6 ммоль) и тионилхлорида (1,1
мл, 15,2 ммоль) в диметилформамиде (30 мл) перемешивали в течение ночи при комнатной температуре, затем суспензию разбавляли диэтиловым эфиром и твердое вещество отфильтровывали и сушили с получением
3,2 г сырого 4-[6-(диэтиламинометил)нафт-2-илметиламинокарбамоил] бензоилхлорида. Данное соединение добавляли в виде твердого вещества к раствору гидроксиламингидрохлорида (0,6 г, 8,5 ммоль) и
бикарбоната натрия (1,2 г, 14 ммоль) и 1 н. гидроксида натрия (8,5 мл, 8,5 ммоль) в воде (30 мл) и ТГФ (40 мл). Смесь перемешивали при комнатной температуре в течение 2 часов, затем смесь насыщали
хлоридом натрия и органическую фазу отделяли, а водную фазу экстрагировали добавлением ТГФ. Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли в условиях
пониженного давления. Сырой продукт растворяли в горячем ТГФ (150 мл) и нерастворимые вещества отфильтровывали. Раствор охлаждали до комнатной температуры и обрабатывали 1,5 н. эфирным раствором HCl.
Твердый продукт отфильтровывали и сушили с получением 1,2 г (выход 38%) чистого гидрохлорида 4-[6-(диэтиламинометил)нафт-2-илметиламинокарбамоил] бензогидроксамовой кислоты в виде белого твердого
вещества; т.пл. = 167-168oC (с разлож.).
1H-ЯМР d 11.07 (s, 1H, обмен с D2O), 10.49 (bs, 1H, обмен с D2O), 9.49 (s, 1H), 8.93 (s, 1H, обмен с
D2O), 8.15 (s, 1H), 8.05-7.87 (m, 3H), 7.81 (d, 1H), 7.72 (d, 2H), 7.65-7.50 (m, 3H), 7.27 (t, 1H), 4.54 (d, 2H), 4.48 (s, 2H), 3.11 (m, 4H), 1.30 (t, 6H)
Пример 17
Гидрохлорид 4-[(N-изопропил-1,2,3,4-тетрагидроизохинол-3-ил)метилоксикарбамоилметил] бензогидроксамовой кислоты
А. Раствор 1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты (9 г, 42 ммоль),
2-бромпропана (8 мл, 84 ммоль) и 1 н. NaOH (168 ммоль) в этаноле (170 мл) нагревали с обратным холодильником в течение 5 часов, затем добавляли 2-бромпропан (8 мл, 84 ммоль) и 1 н. NaOH (168 мл, 168
ммоль) и смесь нагревали с обратным холодильником в течение 5 часов. Этанол удаляли и водный раствор обрабатывали 6 н. соляной кислотой до значения pH 7. Непрореагировавший исходный продукт выделяли
путем фильтрования и растворитель удаляли в условиях пониженного давления. Сырой продукт растворяли в этаноле, неорганические соли отфильтровывали, а органический раствор выпаривали в условиях
пониженного давления. Данную процедуру повторяли трижды с получением 8,1 г (выход 87%) чистой N-изопропил-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты, которую применяли на следующей стадии без
дальнейшей очистки.
1H-ЯМР 87.13 (m, 4H), 4.16 (d, 1H), 3.89 (d, 1H), 3.58 (t, 1H), 3.43 (m, 1H), 3.13 (dd, 1H), 2.94 (dd, 1H), 1.19 (d, 3H), 1.11 (d, 3H).
Б. Суспензию N-изопропил-1,2,3,4-тетрагидро-3-изохинолинкарбоновой кислоты (8,0 г, 36 ммоль) в тетрагидрофуране (100 мл) медленно добавляли в возгоняющуюся суспензию литийалюминийгидрида (2,1 г, 54 ммоль) в тетрагидрофуране (100 мл). Смесь нагревали с обратным холодильником в течение двух часов, затем охлаждали до комнатной температуры и обрабатывали смесью тетрагидрофурана (7 мл) и воды (0,9 мл), 20% гидроксидом натрия (2,3 мл) и, наконец, водой (9,2 мл). Белое твердое вещество отфильтровывали и растворитель удаляли в условиях пониженного давления с получением 5,0 г (выход 68%) чистого N-изопропил-1,2,3,4-тетрагидроизохинол-3-илметанола в виде густого масла.
1H-ЯМР δ 7.12 (m, 4H), 4.54 (bs, 1H, обмен с D2O), 3.68 (s, 2H), 3.55-3.35 (m, 2H), 3.20-2.90 (m, 2H), 2.79 (d, 2H), 1.10 (d, 3H), 1.01 (d, 3H).
В. Раствор N-изопропил-1,2,3,4-тетрагидроизохинол-3-илметанола (4,6 г, 22,4 ммоль) в тетрагидрофуране (50 мл) перемешивали при комнатной температуре в течение 3 часов. Затем данный раствор добавляли к раствору 4-аминометилбензойной кислоты (3,38 г, 22,4 ммоль) и 1 н. гидроксида натрия (22,4 мл, 22,4 ммоль) в воде (20 мл). Раствор перемешивали в течение ночи при комнатной температуре, затем добавляли 1 н. соляную кислоту (22,4 мл, 22,4 ммоль) и растворители удаляли в условиях пониженного давления. Сырой продукт очищали по методу хроматографии на силикагеле (элюент хлороформ-метанол-гидроксид аммония 8: 2: 0,5) с получением 3,1 г (выход 35%) чистой 4-[(N-изопропил-1,2,3, 4-тетрагидроизохинол-3-ил)метилоксикарбамоилметил] бензойной кислоты в виде белого твердого вещества; т.пл. = 93-95oC (с разлож.).
1H-ЯМР δ 7.92 (d, 2H), 7.86 (t, 1H), 7.36 (t, 2H), 7.14 (m, 4H), 4.26 (d, 2H), 4.07 (dd, 1H), 3.73 (s, 2H), 3.68 (dd, 1H), 3.25 (m, 1H), 3.02 (m, 1H), 2.89 (dd, 1H), 2.72 (dd, 1H), 1.10 (d, 3H), 1.04 (d, 3H).
Г. Раствор 4-[(N-изопропил-1,2,3,4-тетрагидроизохинол-3-ил)метилоксикарбамоил] бензойной кислоты (3,0 г, 7,8 ммоль) и тионилхлорида (1,7 мл, 23 ммоль) в хлороформе (100 мл) нагревали с обратным холодильником в течение 3 часов, затем растворитель и тионилхлорид выпаривали. Сырой продукт растворяли в хлороформе (100 мл) и выпаривали досуха трижды. Сырой продукт растворяли в тетрагидрофуране (50 мл) и добавляли к раствору гидроксиламингидрохлорида (0,65 г, 9,4 ммоль) и бикарбоната натрия (1,3 г, 15,7 ммоль) и 1 н. гидроксида натрия (9,4 мл, 9,4 ммоль) в воде (30 мл) и ТГФ (20 мл). Смесь перемешивали при комнатной температуре в течение часа, затем тетрагидрофуран удаляли в условиях пониженного давления и водную фазу экстрагировали добавлением этилацетата (3 х 100 мл). Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли в условиях пониженного давления. Сырой продукт растворяли в тетрагидрофуране и обрабатывали 1,5 н. эфирным раствором соляной кислоты. Твердый продукт отфильтровывали и сушили с получением 2,1 г (выход 62%) чистого гидрохлорида 4-[(N-изопропил-1,2,3,4-тетрагидроизохинол-3-ил)метилоксикарбамоил] бензогидроксамовой кислоты в виде белого твердого вещества; т.пл. = 154-157oC (с разлож. ).
1H-ЯМР δ 11.24 (s, 1H), 10.75 (s, 1H), 9.07 (bs, 1H), 8.01 (t, 1H), 7.70 (d, 2H), 7.34 (d, 2H), 7.32 (m, 4H), 4.38 (d, 2H), 4.26 (m, 4H), 4.02 (m, 1H), 3.78 (m, 1H), 3.15 (d, 2H), 1.42 (d, 3H), 1.30 (d, 3H).
По аналогичной методике могут быть получены следующие
соединения:
4-[(4-диметиламинометилнафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диэтиламиноэтилнафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(димeтилaминoэтилнaфт-2-ил)мeтoкcикapбaмoил] бензогидроксамовая кислота,
4-[(6-димeтилaминoмeтилнaфт-2-ил)мeтoкcикapбaмoил] бензогидроксамовая кислота,
4-[(6-диизопропиламинометилнафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диметиламинометилнафт-2-ил)метоксикарбамоил] метилбензогидроксамовая кислота,
4-[(4-диметиламинометилнафт-2-ил) этоксикарбамоил] бензогидроксамовая кислота,
4-[(5,6,7,8-тетрагидронафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[N-(1,2,3,
4-тетрагидронафт-2-ил)глицинамидо] бензогидроксамовая кислота,
4-[(4-диэтиламинометилнафт-2-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диметиламинометилнафт-2-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диэтиламинометилнафт-2-ил) этоксикарбамоил] бензогидроксамовая кислота,
4-[(1,2,3,
4-тетрагидронафт-2-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диметиламинометилнафт-1-ил) метоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диметиламиноэтилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(5-диметиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(5-диэтиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(5-ди-н-пропиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(5-диизопропиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(5-ди-н-бутиламинометилнафт-1- ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диметиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диэтиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(6-ди-н-пропиламинометилнафт-1- ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диизопропиламинометилнафт-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(6-ди-н-бутиламинометилнафт-1- ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диметиламинометилнафт-1-ил)метоксикарбамоил] метилбензогидроксамовая кислота,
4-[(4-диметиламинометилнафт-1-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(4-диэтиламинометилнафт-1-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(5-диметиламинометилнафт-1-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(5-диэтиламинометилнафт-1-ил) этоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диметиламинометилнафт-1-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[(6-диэтиламинометилнафт-1-ил)этоксикарбамоил] бензогидроксамовая кислота,
4-[N-(нафт-1-илметил)глицинамидо] бензогидроксамовая кислота,
4-[N-(нафт-2-илметил)глицинамидо] бензогидроксамовая кислота,
4-[(N-метил-1,2,3,
4-тетрагидроизохинол-5-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-этил-1,2,3,4-тетрагидроизохинол-5-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(изохинол-5-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-метил-1,2,3,4-тетрагидроизохинол-6-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-этил-1,2,3,
4-тетрагидроизохинол-6-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(изохинол-6-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-метил-1,2,3,
4-тетрагидроизохинол-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-этил-1,2,3,4-тетрагидроизохинол-1- ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(изохинол-1-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-метил-1,2,3,4-тетрагидроизохинол-3-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-этил-1,2,3,
4-тетрагидроизохинол-3-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(изохинол-3-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-метил-1,2,3,
4-тетрагидроизохинол-4-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(N-этил-1,2,3,4-тетрагидроизохинол-4-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(изохинол-4-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[3-(1,2,3,4-тетрагидроизохинол-2-ил)пропионамидо] бензогидроксамовая кислота,
4-[(бензотиофен-4-ил)метоксикарбамоил]
бензогидроксамовая кислота,
4-[(бензотиофен-5-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(бензофуран-4-ил)метоксикарбамоил] бензогидроксамовая кислота,
4-[(бензофуран-5-ил)метоксикарбамоил] бензогидроксамовая кислота,
гидрохлорид 4-[(4-диэтиламинопропил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты,
гидрохлорид
4-[(3-диэтиламинометил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(3-диэтиламиноэтил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты,
гидрохлорид
4-[(3-диэтиламинопропил)нафт-1-илметилоксикарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(4-диэтиламинопропил)нафт-1-илметиламинокарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(3-диэтиламинометил)нафт-1-илметиламинокарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(3-диэтиламиноэтил)нафт-1-илметиламинокарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(3-диэтиламинопропил)нафт-1-илметиламинокарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(6-дипропиламинометил)нафт-2-илметиламинокарбамоил] бензогидроксамовой
кислоты,
гидрохлорид 4-[(6-дибутиламинометил)нафт-2-илметиламинокарбамоил] бензогидроксамовой кислоты,
гидрохлорид 4-[(4-диэтиламинометил)нафт-1-илметиламинокарбамоил]
бензогидроксамовой кислоты,
гидрохлорид 4-[(4-дипропиламинометил)нафт-1-илметил-аминокарбамоил] бензогидроксамовой кислоты,
гидрохлорид
4-[(4-диэтиламиноэтил)нафт-1-илметиламинокарбамоил] бензогидроксамовой кислоты.
Пример 18
Гидрохлорид N-(2-гидроксициклогексил)-4-[6-(N,
N- диэтиламинометил)нафт-2-илметилоксикарбамоил]бензамида
А. 50 г 2,6-нафталиндикарбоновой кислоты (0,23 моль) суспендируют в 350 мл дихлорметана. Добавляют 111 мл трибутиламина (0,46 моль).
Нерастворимый осадок отфильтровывают на бумажном фильтре с получением прозрачного раствора, который перемешивают при комнатной температуре в течение ночи. К реакционной смеси, охлажденной до 0oC, добавляют раствор 45 мл изобутилхлорформиата (0,35 моль) в 180 мл дихлорметана. Получают желтую суспензию, которую перемешивают при 0oC в течение 15 минут. К этой суспензии
добавляют 45 мл диэтиламина (0,46 моль), поддерживая температуру ниже 5oC. После этого добавления реакционную смесь перемешивают в течение 3 часов при комнатной температуре до получения
прозрачного раствора коричневого цвета. Раствор подщелачивают гидроксидом натрия 4 N (200 мл) и разбавляют водой (250 мл). Две фазы разделяют. Водную фазу переносят в большой химический стакан,
охлаждают до 0oC и подкисляют 36-38% хлористоводородной кислотой. Образованное твердое вещество отфильтровывают на пористом мембранном фильтре и промывают этилацетатом для удаления 2,
6-нафталиндикарбоновой кислоты, которая все еще присутствует. Получают 25,25 г 6-(N, N-диэтилкарбамоил)-2-нафтойной кислоты (выход 40,5%).
1H ЯМР: 13,2 (шир.с, 1H), 8,7 (с, 1H), 8,25-8,0 (м, 4H), 7,1 (д, 1H), 3,4 (шир.м, 4H), 1,2 (шир.м, 6H).
В. 7,06 г литийалюминийгидрида (186 ммоль) суспендируют в 200 мл тетрагидрофурана и нагревают при температуре кипения с обратным холодильником. Затем вводят в реакционную смесь по каплям раствор 25,2 г продукта, полученного на стадии А (93,0 ммоль), в тетрагидрофуране. Через 2 часа смесь охлаждают до 0oC и добавляют раствор воды (3,36 мл) и тетрагидрофурана (24,82 мл). Затем добавляют 8,33 мл 5 N гидроксида натрия и в конце добавляют 32,6 мл воды. Смесь переносят в условия комнатной температуры и фильтруют. Растворитель удаляют в вакууме и остаток растворяют в диэтиловом эфире и экстрагируют 1 N хлористоводородной кислотой. Две фазы разделяют. Водную фазу подщелачивают 32% гидроксидом натрия и экстрагируют диэтиловым эфиром. Получают 16,60 г 6-(N,N-диэтиламинометил)-2-нафталинметанола (выход 74,7%).
1H ЯМР: 7,25 (м, 4H), 7,5 (м, 2H), 5,37 (т, 1H), 4,7 (д, 2H), 3,7 (с, 2H), 2,55 (кв, 4H), 1,05 (т, 6H).
С. 6 г продукта, полученного на стадии В (24,69 ммоль), растворяют в ацетонитриле (80 мл). Добавляют 9,5 г N, N'-дисукцинимидил карбоната, смесь перемешивают при комнатной температуре в течение 90 минут. Одновременно 3,39 г 4-аминобензойной кислоты растворяют в водном растворе карбоната натрия (2,62 г в 35 мл воды) и добавляют туда раствор реакционной смеси в ацетонитриле. Затем смесь переносят в условия комнатной температуры на 2 часа. Затем ацетонитрил выпаривают и водный раствор нейтрализуют концентрированной хлористоводородной кислотой (4,11 мл). Образовавшийся ярко-розовый осадок отфильтровывают, суспендируют в воде в течение 2 часов и затем фильтруют. Продукт очищают на силикагеле (элюирование CH2Cl2:CH3ОН=8:2+0,25 NH3). Получают 4,21 г 4-[6-(N, N-диэтиламинометил)нафт-2-илметилоксикарбоксамино] бензойной кислоты (выход: 39,9%).
1H ЯМР: 10,22 (с, 1H), 7,95 (м, 6H), 7,65 (м, 4H), 5,35 (с, 2H), 3,8 (с, 2H), 2,6 (м, 4H), 1,05 (т, 6H).
D. 3,97 г продукта, полученного на стадии С (9,78 ммоль), растворяют в хлороформе (150 мл). К этой смеси добавляют 3,49 г тионилхлорида (29,33 ммоль) и 1 мл N,N-диметилформамида. Смесь нагревают при температуре кипения с обратным холодильником в течение 4 часов и затем переносят в условия комнатной температуры на 2 дня. Растворитель удаляют в вакууме и остаток растворяют в хлороформе, затем растворитель снова удаляют в вакууме. Такую процедуру промывки повторяют дважды. Полученный остаток используют как неочищенный продукт на стадии Е.
Е. Неочищенный продукт, полученный на стадии D (9,78 г), добавляют к охлажденному раствору 2-аминоциклогексанола и карбоната натрия в воде и тетрагидрофуране (0oC). Смесь перемешивают при 0oC в течение 15 минут и затем при комнатной температуре в течение ночи. Растворитель удаляют в вакууме и остаток очищают на силикагеле (элюирование CH2Cl2:CH3ОН=15:1+0,1 NH3). Получают 1,55 г N-(2-гидроксициклогексил)-4-[6-(N, N-диэтиламинометил)нафт-2-илметилоксикарбамоил]бензамида (выход: 31,63%).
1H ЯМР: 10,08 (с, 1H), 7,90 (м, 6H), 7,55 (м, 4H), 5,35 (с, 2H), 4,6 (д, 1H), 3,7 (м, 3H), 2,6 (м, 4H), 1,75 (м, 4H), 1,25 (м, 4H), 1,05 (т, 6H).
F. 1,55 г продукта, полученного на стадии E (3,08 ммоль), растворяют в растворе воды и тетрагидрофурана. Реакционную смесь охлаждают до 0oC и затем добавляют раствор хлористоводородной кислоты в диэтиловом эфире до достижения pH 1. Смесь выдерживают при 0oC в течение 15 минут и при комнатной температуре в течение 2 часов. Растворитель удаляют в вакууме и остаток растворяют в небольшом количестве метанола и выдерживают в течение ночи при +4oC, затем добавляют диэтиловый эфир и осаждают твердый продукт. Продукт фильтруют и сушат в вакууме. Получают 1,59 г N-(2-гидроксициклогексил)-4-[6-(N, N-диэтиламинометил)нафт-2-илметилоксикарбамоил] бензамид гидрохлорида (выход: 95,78%).
1H ЯМР: 10,7 (шир.с, 1H), 10,01 (с, 1H), 8,3-7,5 (м, 10H), 5,38 (с, 2H), 4,67 (шир.с, 1H), 4,47 (д, 2H, J=0,5, 0,4 Гц), 3,1 (м, 4H), 1,9 (м, 2H), 1,6 (м, 2H), 1,29 (т, 6H, J=7,1 Гц).
G. Продукт, полученный на стадии F, содержит неорганическую соль, поэтому его растворяют в насыщенном растворе карбоната натрия и экстрагируют тетрагидрофураном и этилацетатом. Органические растворители объединяют, сушат сульфатом натрия и выпаривают в вакууме. Полученный влажный остаток растворяют в этаноле и выпаривают в вакууме. Высушенный остаток растворяют в растворе тетрагидрофуран:метанол (9:1), охлажденном до 0oC, затем добавляют раствор хлористоводородной кислоты в диэтиловом эфире (2,5 М 1 мл) до достижения pH 1; к смеси добавляют эфир, после чего через несколько часов происходит затвердевание продукта. Продукт фильтруют и сушат в вакууме. Получают 1,14 г продукта.
Пример 19
4-(Флуорен-9-илметилоксикарбамоил)
метилциклогексан гидроксамовая кислота
А. 3 г транс-4-(аминометил)циклогексанкарбоновой кислоты растворяют в водно-щелочном растворе (1,6 г гидрогенкарбоната натрия в 19 мл 1 N гидроксида
натрия и 20 мл тетрагидрофурана), раствор охлаждают до 0oC и добавляют раствор 9-флуоренилметилхлорформиата (5 г, 0,019 моль) в тетрагидрофуране (50 мл). Реакционную смесь перемешивают в
течение 3 часов, затем добавляют хлористоводородную кислоту и отделенную водную фазу экстрагируют этилацетатом. Органический растворитель объединяют, сушат сульфатом натрия и выпаривают в вакууме.
Неочищенный продукт растворяют в диэтиловом эфире и фильтруют. Получают 6,5 г 4-(флуорен-9-илметилоксикарбамоил)-метилциклогексанкарбоновой кислоты (выход: 90,2%).
1H ЯМР: 12,1 (c, 1H), 7,95 (д, 2H), 7,4 (м, 4H), 4,3 (м, 3H), 2,85 (т, 2H), 2,12 (м, 1H), 1,82 (м, 4H), 1,3 (м, 3H), 0,92 (м, 2H).
В. 5,2 г продукта, полученного на стадии А (0,013 моль), растворяют в хлороформе, раствор охлаждают на ледяной бане и затем добавляют 2 мл тионилхлорида (0,027 моль). Реакционную смесь нагревают до температуры кипения с обратным холодильником в течение 4 часов. Растворитель удаляют в вакууме и остаток растворяют в хлороформе и растворитель снова удаляют в вакууме. Такую процедуру промывки повторяют дважды. 5,17 г полученного остатка используют как неочищенный продукт на стадии С.
С. 1,08 г гидрохлорида гидроксиламина растворяют в водно-щелочном растворе (1,09 г гидрогенкарбоната натрия в 15 мл воды и 30 мл тетрагидрофурана) и охлаждают при 0oC. К этой смеси добавляют раствор 5,17 г продукта, полученного на стадии B, в тетрагидрофуране (50 мл). Реакционную смесь перемешивают при комнатной температуре в течение ночи. Смесь охлаждают при помощи ледяной бани и затем добавляют хлористоводородную кислоту, выпаривают органический растворитель и продукт, осажденный в водном растворе, фильтруют. Полученный твердый продукт суспендируют в тетрагидрофуране (100 мл) и эфире (300 мл) при перемешивании в течение 24 часов при комнатной температуре. Продукт фильтруют и сушат в вакууме. Получают 4,5 г 4-(флуорен-9-илметилоксикарбамоил) метилциклогексан гидроксамовой кислоты (выход: 85%).
1H ЯМР: 10,35 (c, 1H), 8,67 (c, 1H), 7,92 (д, 2H), 7,75 (д, 2H), 7,4 (м, 4H), 4,3 (м, 3H), 2,85 (м, 2H), 1,95 (м, 1H), 1,75 (м, 4H), 1,38 (м, 3H), 0,9 (м, 2H).
Фармакологическая активность
Противовоспалительную и иммуносупрессивную активности соединений по
данному изобретению испытывали как in vitro, так и in vivo.
1. Тест на продукцию цитокинов in vitro.
Заявленные соединения испытывают, в частности, на продукцию человеческого IL-1β. Мононуклеарные клетки (РВМС) получали из периферической крови (светлого слоя) здоровых доноров с помощью центрифугирования при градиенте плотности Fi-coll- Hypaque (Biochrom KG, Berlin, Germany). Клетки (2,5 • 106 в мл) культивировали при 37oC во влажной атмосфере, содержащей 5% CO2 в 96-луночных планшетах (Nunc) в конечном объеме 200 мкл. Культуральной средой являлась RPMI 1640 N-ацетил-L-аланил-L-глютамин с низким содержанием эндотоксинов ("low endotoxin"; Biochrom KG) с добавлением 1% околоплодной сыворотки теленка (Hyclone Laboratories Inc., Logan, UT), 100 МЕ/мл пенициллина и 100 мкг/мл стрептомицина. Продукцию цитокинов индуцировали путем стимуляции клеток 10 нг/мл ЛПС (липополисахарида) серотипа 055:B5 E.coli (Sigma Chemical со. , St. Louis, MO). Клетки предварительно обрабатывали соединениями по данному изобретению, растворенными в ДМСО (конечная концентрация: выход 0,05%) в течение 60 минут. Соединения по данному изобретению и ЛПС присутствовали в течение всех 20 часов культивирования. В качестве отрицательного контрольного опыта применяли нестимулированные клетки. По окончании испытания верхние слои отбирают и продукцию IL-1β испытывали с помощью теста ELISA ("sandwich-type antigen capture", R & D System, Minneapolis, MM; все испытания проводили дважды). Для наполнения 96-луночного планшета для микротитрования (Nunc) использовали козью поликлональную сыворотку, специфичную к человеческому IL-1β, очищенную по критерию сродства. После промывки лунки инкубировали в течение 2 часов с 3% бычьего сывороточного альбумина (BSA) в забуференном солевом растворе. Для построения калибровочной кривой для каждого теста применяли рекомбинантный человеческий IL-1β и образцы супернатанта разводили до 1/20 и 1/80 ФБР + 0,1% BSA. Планшеты затем инкубировали при 4oC в течение ночи. После промывки добавляли вторичное антитело, т.е. мышиное моноклональное антитело против человеческого IL-1β. После инкубирования и промывки добавляли козьи конъюгированные с пероксидазой поликлональные антитела против мышиного IgG (Zymed) с последующим добавлением хромогенного субстрата тетраметилбензидиндихлорида (Sigma). Реакцию останавливали 4 н. H2SO4 и с помощью автоматического спектрофотометра (Perkin-Elmer) измеряли поглощение при 450 нм. Ингибирующую активность соединений по данному изобретению выражали в виде ИК50, т.е. концентрации продукта, ингибирующую продукцию IL-1β по сравнению с контрольной культурой на 50%.
Показания ИК50:
Соединение - ИК50 (нмоль/л)
Пример 1 - 305
Пример 2 - 163
Пример 3 - 531
Пример 4 - 28
Пример 5 - 137
Пример 6 - 43
Пример 7 - 12
Пример 8 - 29
Пример 9 - 72
Пример 10 - 101
Пример 12 - 96
Пример
13 - 166
Пример 14 - 382
Пример 15 - 14
Пример 16 - 10
Пример 17 - 21
Дексаметазон - 575
2. ЛПС-индуцированная продукция TNFα in vivo.
Самцов мышей BALB/c (18-20 г) получали из Harlan-Nossan (Correzzana, Italy); липополисахарид (ЛПС) S. enteritidis (код L-6011) получали из Sigma (St. Louis, МО); дексаметазон (Solgesan) получали из Laboratorio Farmacologico Milanese (Corrono P., Italy).
Мышей внутрибрюшинно обрабатывали ЛПС (S. enteritidis, 7,5 мг/кг). Дексаметазон вводили внутрибрюшинно за 30 мин до введения ЛПС. Соединения по данному изобретению вводили перорально за 90 минут до введения ЛПС. Через два часа после введения ЛПС анестезированных животных забивали, кровь собирали путем пункции сердца и давали ей свернуться. Содержание TNFα в сыворотке измеряли по методу ELISA с применением "mouse Reagent Sets" (Genzyme, Cambridge, MA) в соответствии с инструкциями производителя и применяя в качестве стандарта мышиный рекомбинантный TNFα.
Эффект соединения из Примера 12 на ЛПС-индуцированную продукцию сывороточного TNFα приведен в таблице.
Полученные результаты свидетельствуют о том, что тестируемые соединения являются эффективными в ингибировании продукции как IL-1β , так и TNFα, будучи столь же либо более активными, чем дексаметазон, контрольное соединение и хорошо известное противовоспалительное средство.
Изобретение относится также к применению соединений формулы (I) в качестве противовоспалительных и иммуносупрессивных агентов, равно как и ко всем связанным с ними промышленно применимым аспектам, в том числе, к их включению в состав фармацевтических композиций. Примеры таких фармацевтических композиций включают таблетки, пилюли с сахарной оболочкой, кремы, мази и пузырьки, причем последние подходят как для перорального, так и для внутримышечного или внутривенного введения. Они содержат активный ингредиент сам по себе либо в смеси с традиционными фармацевтически приемлемыми носителями и наполнителями.
Дозировки активного ингредиента меняются в широких пределах, в зависимости от типа применяемого соединения, которое назначают один или несколько раз в сутки, соответственно с медицинскими назначениями.
Реферат
Описываются соединения общей формулы (1) где R' представляет собой водород или (C1-4)алкил; А представляет собой адамантил или моно-, би- или трициклический остаток, необязательно частично или полностью ненасыщенный, который может содержать один или несколько гетероатомов, выбранных из группы, содержащей N, S или О, и необязательно замещенный гидрокси, амино(С1-4)алкилом; ди(C1-4)алкил-амино(С1-4)алкилом, (С1-4)алкилом;
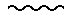
Формула
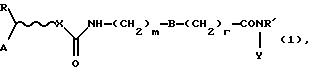
где R' представляет собой водород или (С1-4)алкил;
А представляет собой адамантил или моно-, би- или трициклический остаток, необязательно частично или полностью ненасыщенный, который может содержать один или несколько гетероатомов, выбранных из группы, содержащей N, S и О, и необязательно замещенный гидрокси, амино(С1-4)алкилом, ди(C1-4)алкиламино(C1-4)алкилом, (С1-4)алкилом;
R представляет собой водород или фенил;
Х представляет собой атом кислорода или группу NR', где R' определен выше или отсутствует;
r и m независимо равны 0, 1 или 2;
В представляет собой фениленовое или циклогексиленовое кольцо;
Y представляет собой гидрокси или амино(С1-4)алкильную цепь, необязательно прерываемую атомом кислорода, при условии, что трициклическая группа, которая указана в определении А, представляет собой только флуоренил в том случае, когда одновременно Х не является О и Y не является гидрокси.

Комментарии