Производные гидроксамовой кислоты - RU2196131C2
Код документа: RU2196131C2
Чертежи


Описание
Настоящее изобретение относится к новым соединениям на основе гидроксамовой кислоты, пригодным в качестве фармацевтических препаратов, например, для ингибирования матриксных металлопротеиназ, таких как коллагеназа, и для ингибирования производства TNF, в частности для лечения болезней или состояний, опосредованных сверхпроизводством TNFα или сверхреактивностью к нему.
Фактор некроза опухоли (TNF) представляет собой цитокин, который первоначально продуцируется в качестве предшественника, связанного с мембраной и имеющего молекулярную массу 28 кДа. Затем он расщепляется ферментом (TNF-конвертазой) и высвобождается в виде растворимой активной формы с молекулярной массой 17 кДа. Растворимый TNF существует по крайней мере в двух формах: TNFα и TNFβ, из которых TNFα, по-видимому, более важен в клиническом отношении. Предполагается, что TNFα опосредует воспаление и другие состояния, связанные с септическим шоком или острыми инфекциями. Вероятно, продолжительная сверхстимуляция TNFα играет определенную роль в аутоиммунных и хронических воспалительных состояниях, таких как артрит, рассеянный склероз и т.п.
Было установлено, что определенные ингибиторы матриксных металлопротеиназ из класса гидроксамовой кислоты, в частности (7-N-гидрокси)диамиды 3-имино-4-оксогептан-1,7-диовой кислоты (которые, кроме того, необязательно могут быть замещены в 1-N-, 2-, 5- и 6-положениях), обладают способностью опосредовать производство TNFα, возможно, вследствие ингибирования TNF-конвертазы. Известные представители этого класса соединений обобщены и описаны, например, в WO 94/10990.
Согласно изобретению неожиданно было установлено, что новый класс производных гидроксамовой кислоты (далее называемых "новые соединения") являются сильными супрессорами TNFα и обладают ценными фармацевтическими свойствами, в частности биологической доступностью при оральном введении.
Новые соединения представляют собой (7-N-гидрокси)диамиды 3-имино-4-оксо-6-(оксиметил)гептан-1,7-диовой кислоты. Предпочтительно 6-оксиметильный заместитель представляет собой соединение формулы II, приведенной ниже, например гидроксиметил или моно- или полиалкоксиметил. Кроме того, как известно в данной области, например, как описано в WO 94/10990 или как описано в данной заявке, новые соединения могут иметь другие замещения в 1-N-, 2- и 5-положениях. Например, новые соединения могут быть замещены в l-N-положении метилом, пиридилом или группой формулы X-Y- или X'-Y-, как описано ниже, например, представлять собой (1-N -морфолинокарбонилалкил- 7- N - гидрокси) диамид 3-имино-4-оксо-6-(оксиметил)гептан-1,7-диовой кислоты и находиться в свободной форме или в форме фармацевтически приемлемой соли.
Наиболее предпочтительным классом новых соединений являются (7-N-гидрокси) диамиды 3-имино-4-оксо-5-арил-6- (оксиметил) гептан -1,7 -диовой кислоты. Кроме того, как описано ниже, 5-арильный заместитель, как, например, в случае, когда 5-арильный заместитель представляет собой фенил, необязательно может быть замещен, предпочтительно в 4-положении, например, гидроксигруппой, С1 -С6алкилом, С1-С6алкокси-, аминогруппой, галогеном или цианогруппой. Такие новые соединения, имеющие 5-арильный заместитель, могут находиться в свободной форме или в форме фармацевтически приемлемой соли.
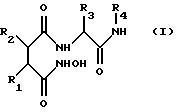
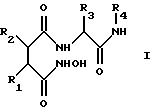
Предпочтительно новые соединения представляют собой соединения формулы I
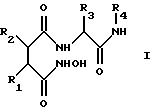
где R1 обозначает заместитель формулы II:
A-(O-(CR5H)n)m-O-CH2- II
где
n равно 1, 2, 3 или 4, предпочтительно 2,
m равно 0, 1, 2 или 3,
R5 в каждом случае независимо обозначает Н, С1-С10алкил (необязательно замещенный гидрокси-, С1-С6алкокси-, амино-, С1-С6алкиламино-, тиольной группой, C1-С6алкилмеркапто- или защищенной гидрокси-, амино- или тиольной группой), С2-С6алкенил, C6-С14арил (необязательно замещенный гидрокси-, C1-С6алкокси-, амино-, С1-С6алкиламиногруппой, галогеном или цианогруппой), или С6-С14(арил)C1-С6алкил; предпочтительно Н, фенил, бензил или C1-С5алкил,
А обозначает водород, С1-С10алкил, С6-С14арил, С6-С14арил(С1-С6алкил), (С6-С14арил)карбонил или (С1-С10алкил)карбонил, предпочтительно водород, С1-С6алкил (например, метил или циклогексил), фенил или бензил,
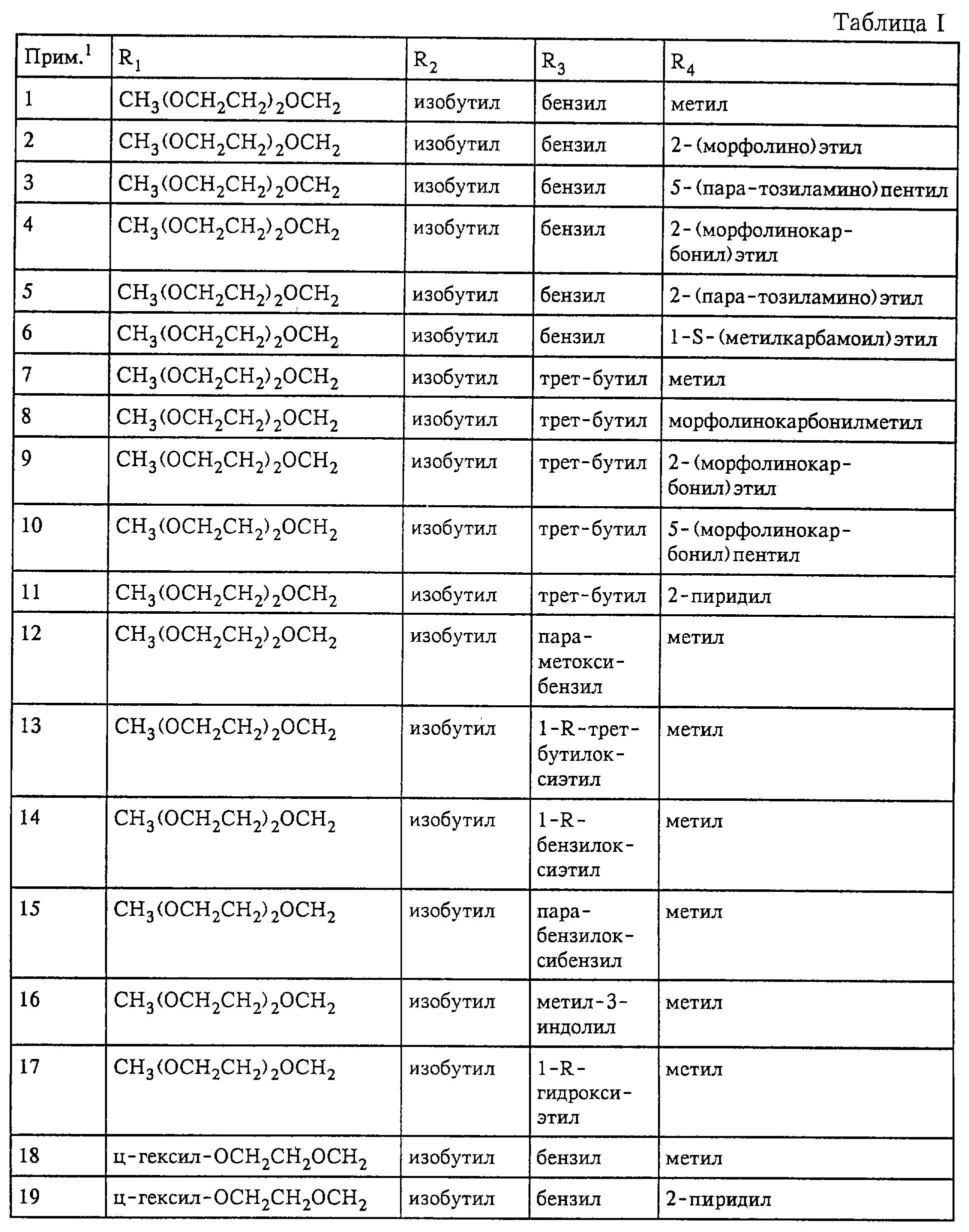
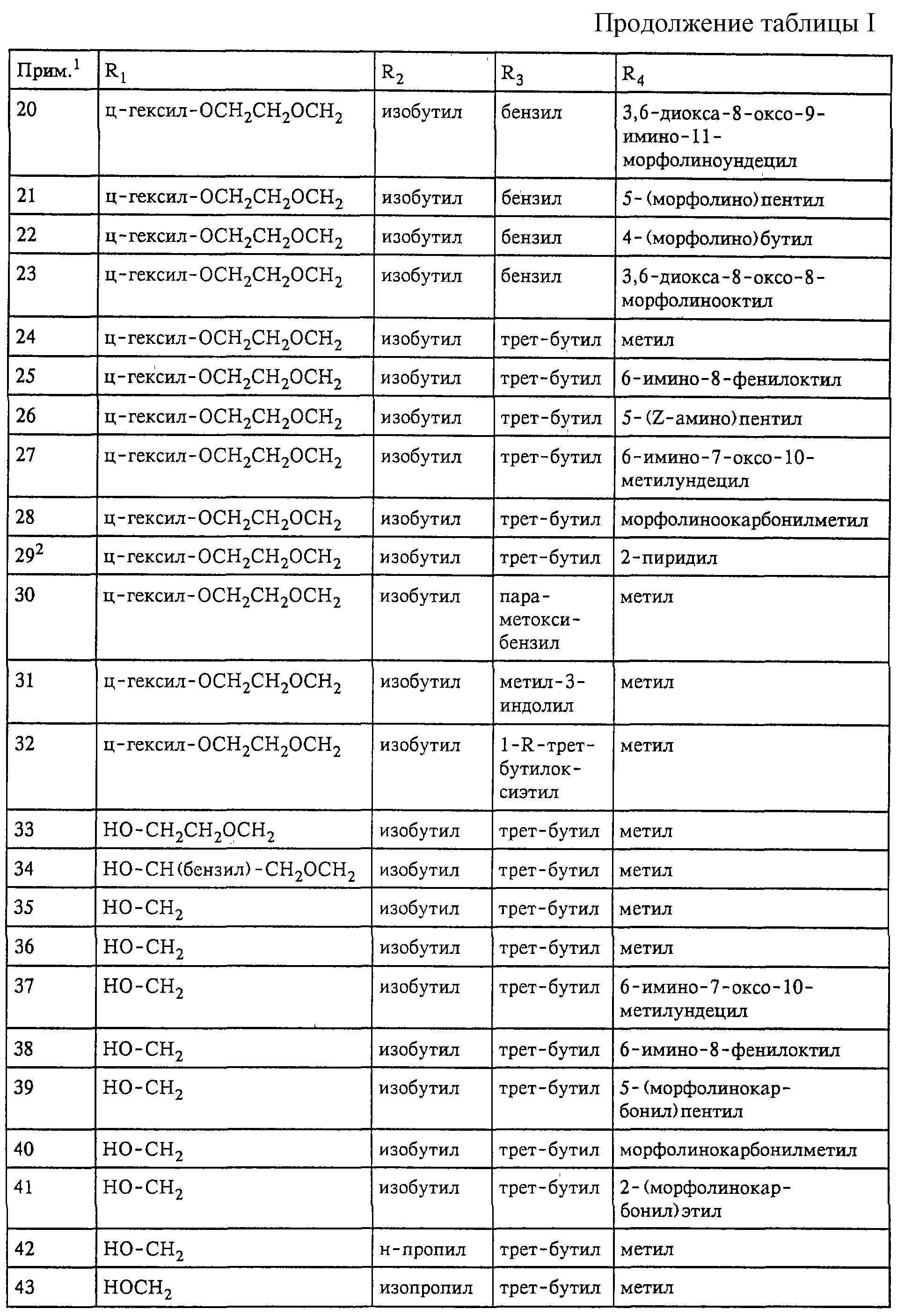
R2 обозначает С2 -С12алкил, С3-С12алкенил, С3-С7циклоалкил (необязательно замещенный гидрокси-, С1-С6алкокси-, амино- или С1 -С6алкиламиногруппой), С5-С14арил или С5-С14арил(C1-С6алкил), где арильные группы необязательно замещены гидроксигруппой, С1-С6алкилом, С1-С6алкокси-, аминогруппой, галогеном или цианогруппой, предпочтительно фенил, 4-метилфенил, 4-метоксифенил, циклогексил или изобутил,
R3 обозначает С1-С10алкил (необязательно замещенный гидрокси- или C1-С6алкокси-, амино-, С1-С6алкиламино-, тиольной группой, С1-С6алкилмеркапто- или защищенной гидрокси-, амино- или тиольной группой), (например, трет-бутил или циклогексилметил), С6-С14арил (необязательно замещенный гидрокси-, С6-С14арилокси- или С1-С6алкокси-, амино-, С1-С6алкиламиногруппой, галогеном или цианогруппой), (например, бензил, пара-метоксибензил, пара-бензилоксибензил) или индолилметил (например, 2-индолилметил), предпочтительно бензил или трет-бутил,
R4 обозначает метил, пиридил или группу формулы X-Y-, где Х обозначает морфолиногруппу, пиридил или арил (предпочтительно морфолиногруппу) и Y обозначает С1-С12алкилен, в котором до четырех метиленовых (-СН2 -) звеньев необязательно замещены -СО-, -NH-, -SO2- или -О-, например, метил, 2-пиридил, морфолинокарбонилметил, 5-(морфолино)пентил или 5-(морфолинокарбонил)пентил.
Понятие "алкил" включает линейный, циклический или разветвленный алкил, а понятие "арил" обозначает одновалентный ароматический радикал, имеющий одно или два ароматических кольца, например фенил, бензил или толил, и включает гетероарил, имеющий один или несколько гетероатомов, например, N, О или S.
Галоид или галоген в контексте настоящего описания обозначает F, Cl, Вr или I, если не указано иное.
Предпочтительно R1 обозначает заместитель формулы II'
А-(О-(СH2)n)m-О-СН2- II'
где А, n и m имеют
указанные выше значения.
В другом варианте R1 обозначает заместитель формулы II''
А-(О-(СHR5-CH2)n)m'-О-СН2
- II''
где А, n и R5 имеют указанные выше значения, а m' равно 0, 1 или 2.
Если R4 в формуле I обозначает группу формулы X-Y-, то предпочтительно он является заместителем формулы X'-Y-, где X' обозначает морфолиногруппу, a Y имеет указанные выше значения.
В еще одном из вариантов изобретение относится к новым соединениям формулы I,
в которых независимо:
n в формуле II равно 3 или 4, или
R5 в формуле II не обозначает Н, или
R2 обозначает С7-С12алкил, С3-С12алкенил, С3-С7циклоалкил (необязательно замещенный гидрокси-, С1-С6алкокси-, амино- или С1-С6алкиламиногруппой),
С5-С14арил или С5-С14арил(C1-С6алкил), где арильные группы необязательно замещены гидроксигруппой, С1-С6алкилом,
C1-С6алкокси-, аминогруппой, галогеном или цианогруппой, предпочтительно фенил, 4-метилфенил, 4-метоксифенил или циклогексил, или
R3 обозначает C1
-С10алкил (замещенный амино-, C1-С6алкиламино-, тиольной группой, С1-С6алкилмеркапто- или защищенной гидрокси-, амино- или тиольной группой),
С6-С14арил (замещенный амино-, C1-С14алкиламиногруппой, галогеном или цианогруппой), или любая арильная группа в этой формуле обозначает гетероарил, имеющий
один или несколько гетероатомов, например, N, О или S.
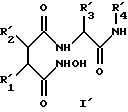
В других вариантах осуществления изобретение относится к новым соединениям формулы I'

в которой R1' обозначает заместитель формулы II'''
А'-(О-(СН2)n')m'-O-СН2- II'''
где n' обозначает целое число, равное 1 или 2,
m' обозначает целое число, равное 0, 1, 2 или 3,
А' обозначает водород, С6-С14арил, С1-С10алкил, (С6-С14арил)карбонил или (С1-С10алкил)карбонил, предпочтительно C1-С6алкил (например, метил или циклогексил),
R2' обозначает С2-С6алкил, предпочтительно изобутил,
R3' обозначает C1-С10алкил (необязательно замещенный гидрокси- или С1-С6алкоксигруппой) (например, трет-бутил или циклогексилметил), С6-С14арил (необязательно замещенный гидрокси-, С6 -С14арилокси или C1-С6алкоксигруппой) (например, бензил, пара-метоксибензил, пара-бензилоксибензил) или индолилметил (например, 2-индолилметил), предпочтительно бензил или трет-бутил,
R4' обозначает метил, пиридил или заместитель формулы X-Y-, где Х обозначает морфолиногруппу, пиридил или арил (предпочтительно морфолиногруппу) и Y обозначает С1-С12алкилен, в котором до четырех метиленовых (-СН2-) звеньев необязательно замещены -СО-, -NH-, -SO2- или -О-, например, метил, 2-пиридил, морфолинокарбонилметил, 5-(морфолино)пентил или 5-(морфолинокарбонил)пентил.
Предпочтительными новыми соединениями, в которых R2 обозначает арил, являются соединения, в которых R1 обозначает радикал формулы II', как он определен выше, и R2 обозначает фенил, 4-метилфенил или 4-метоксифенил.
Особенно предпочтительной группой
соединений формулы I являются соединения, в которых:
(I) R1 обозначает радикал формулы II' или II'' (предпочтительно радикал формулы II') и А в формуле II обозначает водород, С1-С6алкил, например метил или циклогексил (например, R1 в формуле I обозначает, в частности, гидроксиметил, циклогексилоксиэтоксиметил, метоксиэтоксиэтоксиметил или
гидроксиэтилоксиметил) или (С6-С14арил)карбонил, например бензоил (например, R1 в формуле I обозначает, в частности, бензоилоксиметил, бензоилоксиэтоксиэтил или
бензоилоксиэтоксиэтоксиметил),
(II) R2 в формуле I обозначает циклогексил, фенил, 4-метилфенил, 4-метоксифенил или изобутил,
(III) R3 в формуле I обозначает
бензил или трет-бутил и
(IV) R4 в формуле I обозначает метил или морфолинокарбонил(C1-С6)алкил.
Новые соединения могут существовать в свободной форме или в форме соли, и поэтому подразумевается, что соединения в форме соли подпадают под объем изобретения. В частности, некоторые новые соединения могут существовать в форме физиологически приемлемых кислотно-аддитивных солей или солей присоединения оснований, например, в форме гидрохлоридов, оксалатов или фумаратов.
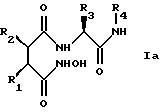
Структура новых соединений
предпочтительно описывается формулой Iа
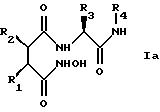
или формулой Ib
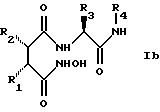
наиболее предпочтительно формулой Iа.
Изобретение включает новые соединения в форме смесей энантиомеров, например в форме рацемических смесей, хотя предпочтительно, чтобы они находились в чистой или практически чистой энантиомерной форме, например в форме, в которой содержание отдельного изомера нового соединения составляет по крайней мере 90%, предпочтительно по крайней мере 95% и особенно предпочтительно по крайней мере 98% (т.е. содержание других изомеров нового соединения составляет менее 10%, предпочтительно менее 5% и особенно предпочтительно менее 2%).
Другие объекты изобретения относятся к новым способам получения соединения формулы I или промежуточных продуктов формул
II, IV или V, приведенных ниже, включающим:
а) для получения соединения формулы I, как определено выше, взаимодействие соединения формулы III

где R1, R2, R3 и R4 имеют указанные выше значения, с гидроксиламином (необязательно в виде соли или в О-замещенной форме, например, в виде гидрохлорида гидроксиламина), выделение продукта формулы I и необязательно удаление защитной группы в полученном продукте или при необходимости выделение требуемого диастереоизомера,
б) для получения соединения формулы III, как определено выше, окисление олефиновой связи в соединении формулы IV
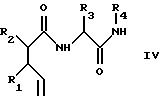
где R1, R2, R3 и R4 имеют указанные выше значения, например, с использованием катализатора окисления, такого как гидрат хлорида рутения (III), с получением кислоты формулы III и необязательно при необходимости выделение требуемого диастереоизомера,
в) для получения соединения формулы IV взаимодействие карбоновой кислоты формулы V
CH2=CH-CH(R1)-CH(R2)-COOH
где R1 и R2 имеют указанные выше значения, с амидом аминокислоты формулы VI
NH2-CH(R3)-CO-NH(R4)
где R3 и R4 имеют указанные выше значения, с получением амида, соответствующего формуле IV, и необязательно при необходимости выделение требуемого диастереоизомера, и
г) для получения соединения формулы V взаимодействие спирта формулы IIIV
А"-(О-(СR5Нn)m-ОН IIIV
где А" имеет значения, указанные для А в формуле II, за исключением случая, когда А обозначает Н, А" обозначает О-защищающую группу (например, группу, способную образовывать легко отщепляемый простой эфир, например бензил) и где R5, n и m имеют значения, указанные выше для формулы II, с дигалогенированным алкеном (транс), например 1,4-дибромбут-2-еном, с получением дизамещенного R1, галоидалкена, например R1-CH=CH-CH2-W (транс), где W обозначает галоген, например бром, который затем подвергают взаимодействию с карбоновой кислотой, соответствующей R2, а именно R2-СН2СООН, с образованием эфира, который затем подвергают перегруппировке, например, в присутствии органического основания, такого как амид диизопропиллития, получая соединение формулы V.
В описанные выше способы необязательно могут быть включены стадии включения и удаления защитных групп, если необходимо сохранить целостность промежуточных продуктов и конечного продукта.
Кроме того, под объем изобретения подпадают новые промежуточные продукты формул III и IV, как они определены выше.
Как описано ниже в примерах по тестированию, новые соединения являются сильными ингибиторами высвобождения TNFα, обладают активностью при оральном введении и не являются цитотоксичными при применении в эффективных дозах. Новые соединения также ингибируют коллагеназу и стромелизин в концентрациях от 0,3 до 10 нМ. Кроме того, испытанные новые соединения проявили активность в опытах in vivo на крысах при оральном введении в дозах, меньших 10 мг/кг, в отношении индуцированного ЛПС (липополисахаридом) высвобождения TNFα и, по-видимому, должны хорошо переноситься в таких дозах. Следовательно, новые соединения обладают фармацевтической пригодностью, заключающейся в следующем.
Новые соединения пригодны для профилактики или лечения болезней и патологических состояний, опосредованых TNF, прежде всего TNFα, например воспалительных состояний, аутоиммунных болезней, серьезных инфекций и отторжения трансплантата органа или ткани, например, при лечении реципиентов, которым трансплантировали сердце, легкое, комбинацию сердце-легкое, печень, почку, панкреатическую железу, кожу или роговицу, и для предотвращения болезни "трансплантат против хозяина", такой как возникающая при трансплантации костного мозга.
Новые соединения особенно пригодны для лечения, предупреждения или облегчения аутоиммунных болезней и воспалительных состояний, в частности воспалительных состояний, этиология которых включает аутоиммунный компонент, таких как артрит (например, ревматоидный артрит, хронический прогрессирующий артрит и деформирующий артрит), и ревматические болезни. Конкретные аутоиммунные болезни, для которых могут применяться новые соединения, включают аутоиммунные гематологические нарушения (включая, например, гемолитическую анемию, гипопластическую анемию, анемию эритроцитов и идиопатическую тромбоцитопению), системную красную волчанку, полихондрит, склеродому), гранулематоз Вегенера, дерматомиозит, активный хронический гепатит, тяжелую псевдопаралитическую миастению, псориаз, синдром Стивена-Джонсона, идиопатическую спру, аутоиммунную воспалительную болезнь кишечника (включая, например, неспецифический язвенный колит и болезнь Крона), эндокринную офтальмопатию, болезнь Грейвса, саркоидоз, рассеянный склероз, первичный билиарный цирроз печени, юношеский диабет (сахарный диабет типа I), увеит (передний и задний), сухой кератоконъюнктивит и вернальный кератоконъюнктивит, интерстициальный фиброз легкого, псориатический артрит и гломерулонефрит (с нефропатическим синдромом и без него, например, включая идиопатический нефротический синдром или минимальное изменение нефропатии).
Новые соединения также пригодны для лечения, предупреждения или ослабления астмы, бронхита, пневмокониоза, эмфиземы легкого и других обструктивных или воспалительных болезней дыхательных путей.
Новые соединения пригодны для лечения нежелательных острых и гиперострых воспалительных реакций, опосредованных TNF, прежде всего TNFα, в частности острых инфекций, например септического шока (в частности, эндотоксинового бактериально-токсического шока и респираторного дистресс-синдрома взрослых), менингита, пневмонии и серьезных ожогов, а также для лечения кахексии или синдрома истощения, связанного с патологическим высвобождением TNF, являющегося следствием инфекции, рака или дисфункции органа, особенно кахексии, связанной со СПИДом, в частности связанной или обусловленной ВИЧ-инфекцией.
Новые соединения, помимо того, что они ингибируют высвобождение TNF, прежде всего TNFα, в результате подавления TNF-конвертазы, также являются ингибиторами матриксных металлопротеиназ, в частности коллагеназы, стромелизина и желатиназ, и, следовательно, пригодны при показаниях, известных для ингибиторов коллагеназы или ингибиторов других матриксных металлопротеиназ, в частности для лечения различных патологических состояний кожи, костей и соединительных тканей, в частности ревматоидного артрита, псориаза, псориатического артрита, остеопороза, остеоартрита, периодонтита, гингивита и изъязвления роговицы, для лечения сердечно-сосудистых болезней, в частности атеросклероза и коронарной ангиопластики, для предупреждения метастаза и инвазии клеток опухоли и для индукции фиброза опухолей, в частности для лечения рака, и для предупреждения нейродегенеративных болезней, в частности болезни Альцгеймера.
Для перечисленных выше показаний соответствующая доза должна, как очевидно, варьироваться в зависимости, например, от конкретно применяемого нового соединения, пациента, подлежащего лечению, формы введения и природы и серьезности состояния, подлежащего лечению. Однако в целом удовлетворительные результаты на животных получают при использовании суточных доз приблизительно от 1 до приблизительно 10 мг/кг/в день перорально. Для крупных млекопитающих, например для человека, суточные дозы составляют приблизительно от 50 до приблизительно 750 мг нового соединения, вводимого орально один раз или более предпочтительно в виде разделенных доз два-четыре раза в день.
Новые соединения могут вводиться любым общепринятым путем, в частности орально, например, в форме растворов для питья, таблеток или капсул, или парентерально, например, в форме растворов или суспензий для инъекции. Как правило, для системного введения предпочтительны оральные дозируемые формы, хотя для определенных показаний новые соединения также могут применяться локально или наноситься на кожу, например, в форме кожного крема или геля или аналогичного препарата, или для применения на глазе - в форме глазного крема, геля или препарата в форме глазных капель, или могут вводиться путем ингаляции, в частности, при лечении астмы. Пригодные стандартные дозируемые формы для орального введения включают, в частности, от 25 до 250 мг нового соединения на стандартную дозу.
В соответствии с вышеизложенным другими объектами изобретения также являются следующие:
А. способ
ингибирования производства растворимого TNF, прежде всего TNFα, или уменьшения воспаления у пациента (например, у млекопитающего, прежде всего у человека), нуждающегося в таком лечении,
включающий введение пациенту эффективного количества нового соединения, или способ лечения любого из вышеуказанных состояний, прежде всего способ лечения воспалительной или аутоиммунной болезни или
состояния, в частности рассеянного склероза или ревматоидного артрита, или облегчения одного или нескольких симптомов любого из вышеуказанных состояний,
Б. новое соединение, предназначенное
для применения в качестве фармацевтического препарата, в частности для применения в качестве иммунодепрессанта или противовоспалительного агента, или для применения с целью предупреждения, ослабления
или лечения любой из вышеуказанных болезней или состояний, в частности аутоиммунной или воспалительной болезней или состояний,
В. Фармацевтическая композиция, содержащая новое соединение
вместе с фармацевтически приемлемым растворителем или носителем, например, предназначенная для применения в качестве иммунодепрессанта или противовоспалительного агента, или для применения с целью
предупреждения, ослабления или лечения любой из вышеуказанных болезней или состояний, в частности аутоиммунной или воспалительной болезней или состояний,
Г. применение нового соединения при
изготовлении лекарственного средства, предназначенного для применения в качестве иммунодепрессанта или противовоспалительного агента или для применения с целью предупреждения, ослабления или лечения
любой из вышеуказанных болезней или состояний, в частности аутоиммунной или воспалительной болезней или состояний.
Пример 1: Получение
{1- [S-фенилаланин-1-метиламид]-4- [N-гидроксил]}диамида R-2-изобутил-S-3-(2,5,8-триоксанонил)янтарной кислоты (соединение формулы I, где R1 обозначает 2,5,8-триоксанонил, R2
обозначает изобутил, R3 обозначает бензил и R4 обозначает метил)
а) Раствор, содержащий транс-1,4-дибром-2-бутен (CAS Reg. 821-06-7) (50,00 г), монометиловый эфир
диэтиленгликоля (CAS Reg. 111-77-3) (30,89 г), бисульфат тетрабутиламмония (CAS Reg. 32503-27-8) (7,94 г) и 50%-ный водный раствор гидроксида натрия (113,70 мл) в метиленхлориде (200 мл), перемешивают
при комнатной температуре (КТ) в течение 16 ч. Реакционную смесь разбавляют водой и простым эфиром, отделяют органическую фазу и продукт в виде олефина очищают хроматографией.
б) Раствор, содержащий продукт в виде транс-олефина и полученный на стадии а) (27,11 г), и ДБУ (1,8-диазабицикло[5.4.0]ундец-7-ен, CAS Reg. 6674-22-2) (17,6 мл) в метиленхлориде (200 мл), обрабатывают изокапроновой кислотой (12,44 г). Через 1 ч добавляют безводный карбонат натрия (18 г). Смесь выдерживают в течение ночи. Органическую фазу отделяют и продукт в виде сложного эфира очищают хроматографией.
в) Из диизопропиламина (22,65 мл) и бутиллития в гексане (1,6 н., 99,89 мл) получают при -70oС раствор ЛДА (амид диизопропиллития) в тетрагидрофуране (400 мл). При этой же температуре добавляют раствор продукта, полученного на стадии б) (43,90 г), в тетрагидрофуране (100 мл). Через 30 мин добавляют хлортриметилсилан (20,22 мл). Температуру сначала повышают до комнатной, а затем смесь выдерживают при температуре дефлегмации в течение ночи. Смесь разбавляют простым эфиром. Некислотные продукты удаляют из органической фазы, получая 35,12 г неочищенной кислоты, которую затем подвергают хроматографии, получая 30,70 г чистого продукта в виде карбоновой кислоты.
г) Раствор, содержащий продукт, полученный на стадии в) (10,50 г), (L)-фенилаланин-1-метиламид (8,60 г) (например, полученный путем взаимодействия поступающего в продажу N-карбобензокси-(L)-фенилаланина с метиламином в стандартных условиях с получением метиламида и гидрирования в присутствии палладия для удаления аминозащитной группы) и 4-диметиламинопиридин (4,89 г) в метиленхлориде (120 мл), обрабатывают ЭДКИ (гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, CAS Reg. 25952-53-8) (7,68 г) и триэтиламином (7,61 мл) и выдерживают в течение ночи. Добавляют простой эфир и органическую фазу сушат и упаривают. Неочищенный продукт, представляющий собой смесь двух изомеров, хроматографируют на силикагеле, разделяя изомеры на основе их относительной полярности.
д) Интенсивно перемешиваемый раствор, содержащий менее полярный продукт, полученный на стадии г) (5,30 г), в четыреххлористом углероде (150 мл), ацетонитриле (150 мл) и воде (20 мл) обрабатывают гидратом хлорида рутения (III) (0, 49 г) и пергидратом натрия (15,16 г). Через 2 ч добавляют простой эфир и значение рН доводят до 4. Органическую фазу отделяют, сушат и упаривают. Остаток хроматографируют на силикагеле, получая чистую кислоту.
е) Раствор, содержащий продукт, полученный на стадии д) (5 г), гидроксибензитриазол (2,00 г) и ЭДКИ (2,51 г) в ДМФ (N,N-диметилформамид) (20 мл), выдерживают при комнатной температуре в течение 2,5 ч. Затем добавляют гидрохлорид гидроксиламина (1,90 г) и N-метилморфолин (4,61 мл) и смесь выдерживают в течение ночи. Растворитель выпаривают в глубоком вакууме при 50oС. Остаток очищают с помощью ЖХВД на колонке типа RP18 на силикагеле, получая чистую гидроксамовую кислоту в виде кристаллического порошка белого цвета.
tпл 195-197oС, вращение плоскости поляризации света [α]D20=-8,5o, с= 0,175 в МеОН.
Примеры 2-17
Соединения, соответствующие примерам 2-17
из таблицы I, получают, работая аналогично способу, описанному в примере 1. Продукт, полученный на стадии в) примера 1, подвергают взаимодействию с соответствующим производным амида аминокислоты, как
описано для стадии г) примера 1. Работая согласно способам стадий д) и е) примера 1, получают чистые гидроксамовые кислоты.
Примеры 18-32
Для взаимодействия с транс-1,
4-дибромбутеном согласно способу стадии а) примера 1 вместо монометилового эфира диэтиленгликоля используют циклогексилгликоль. Работая согласно способам стадий б)-е) примера 1, получают чистые
гидроксамовые кислоты из примеров 18-32 таблицы I.
Примеры 33 и 34
Для взаимодействия с транс-1,4-дибромбутеном согласно способу стадии а) примера 1 вместо монометилового
эфира диэтиленгликоля используют монобензилгликоль или монометиловый эфир (2-бензил) гликоля. Работая согласно способам стадий б)-е) примера 1, получают гидроксамовые кислоты формулы I, имеющие на
R1 защищенную бензилом гидроксигруппу. Бензильную группу удаляют с помощью гидрирования в присутствии каталитического количества палладия или сульфата бария и после очистки с помощью ЖХВД
на колонке типа RP18 на силикагеле получают соответствующие чистые соединения формулы I (см. таблицу I).
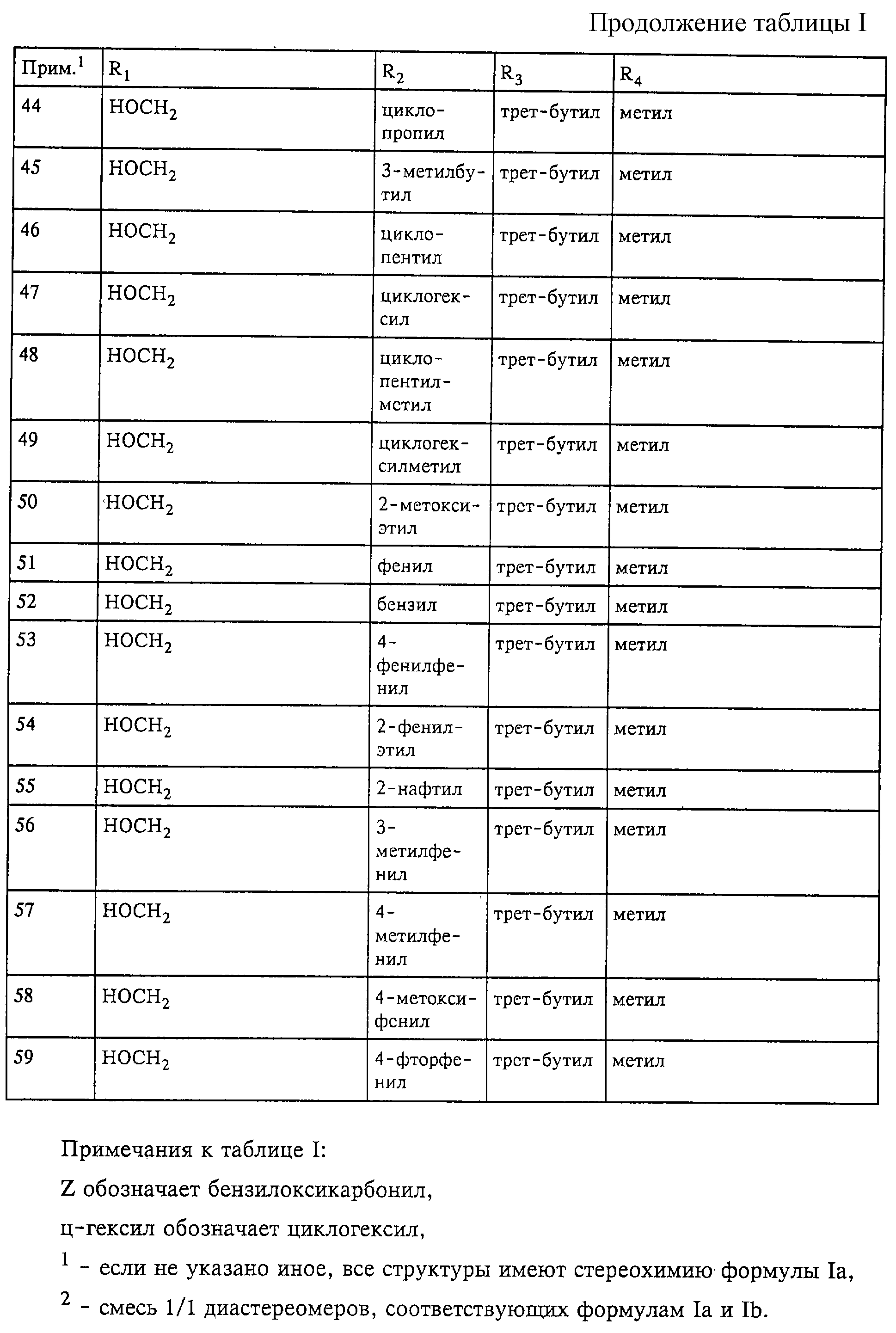
Примеры 35-59
Для взаимодействия с транс-1,4-дибромбутеном согласно
способу стадии а) примера 1 вместо монометилового эфира диэтиленгликоля используют бензиловый спирт. Работая согласно способам стадий б)-е) примера 1 и выбирая соответствующие исходные реагенты,
количества и т.д., получают гидроксамовые кислоты формулы I, имеющие на радикале R1 группу бензилоксиметила. Бензильную группу удаляют с помощью гидрирования в присутствии каталитических
количеств палладия или сульфата бария и после очистки с помощью ЖХВД на колонке типа RP18 на силикагеле получают соответствующие чистые продукты из примеров 35-59 таблицы I.
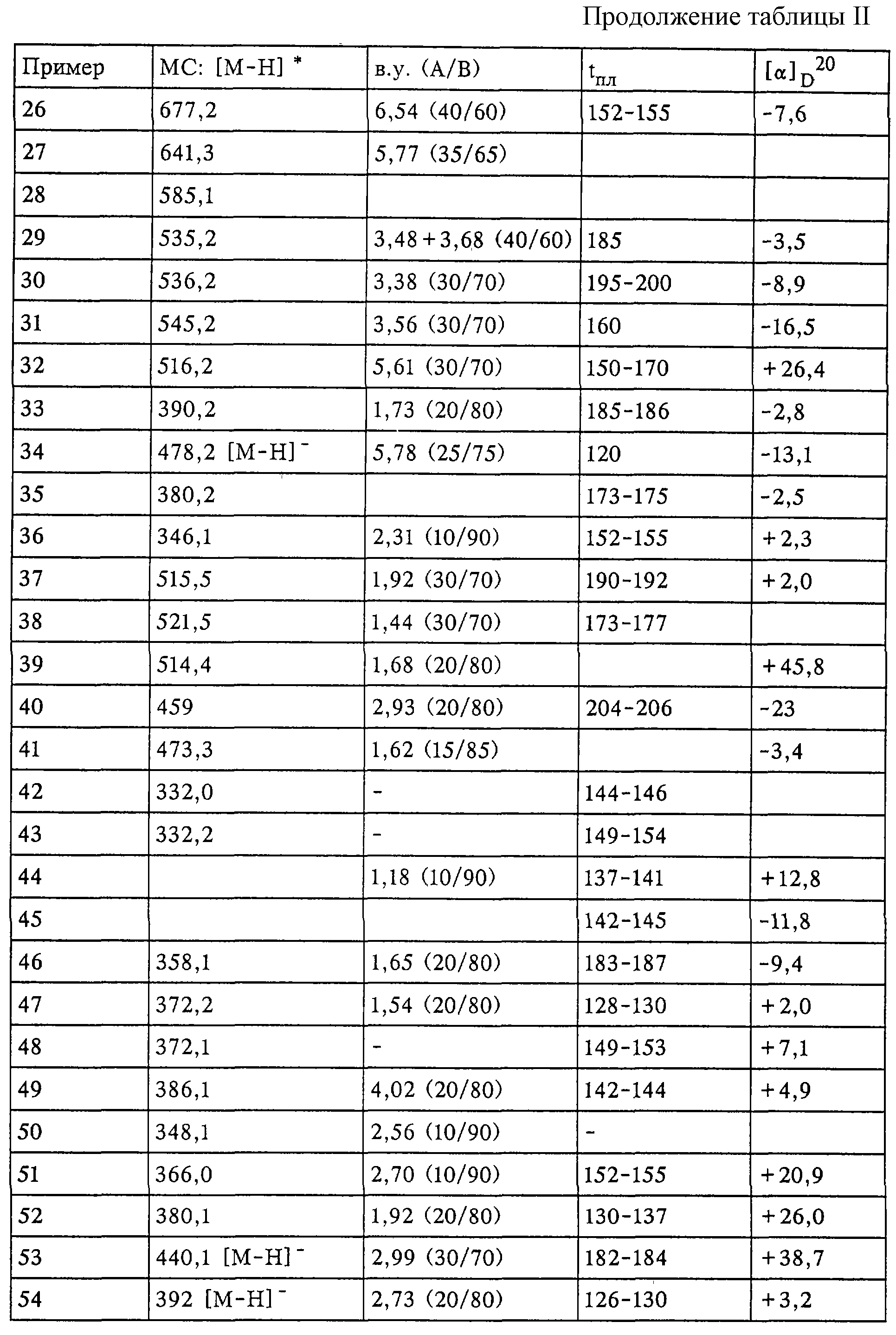
Все соединения охарактеризованы с помощью масс-спектроскопии и1H-ЯМР-спектроскопии. В таблице II обобщены аналитические данные для соединений из примеров 1-59.
Пример по
тестированию 1: Ингибирование высвобождения TNF
Одноядерные клетки получают из периферической крови здоровых добровольцев путем фракционирования на основе плотности с использованием
фиколл-гипак согласно методу Hansell и др., J. Imm. Methods (1991) 145: 105 и используют в концентрации 105 клеток/лунку в среде RPMI 1640, дополненной 10% ФТС (фетальная телячья
сыворотка). Клетки инкубируют с серийными разведениями тестируемых соединений в течение 30 мин при 37oС перед добавлением IFNγ (100 ед. /мл) и ЛПС (5 мкг/мл) и затем дополнительно
инкубируют в течение 3 ч. Инкубацию прекращают путем центрифугирования при 1400 об/мин в течение 10 мин. Содержание TNFα в надосадочной жидкости измеряют с использованием имеющегося в продаже
набора ELISA (Innotest hTNFα, поставляется фирмой Innogenetics N. V. , Zwijnaarde, Бельгия). Новые соединения тестируют в концентрациях от 0 до 10 мкМ. В этом анализе приведенные в примерах
соединения формулы I и прежде всего формулы Iа подавляют высвобождение TNF со значением IC50 приблизительно от 50 нМ до приблизительно 5 мкМ.
Пример по тестированию 2:
Цитотоксичность
Цитотоксичность определяют на клетках ТНР1 (5х104 клеток/лунку), которые инкубируют при 37oС в течение 24 ч в присутствии IFNγ (100 ед./мл) и ЛПС
(5 мкг/мл) и в присутствии тестируемого соединения или без него. Проценты живых и мертвых клеток оценивают с помощью колориметрического метода исключения (ММТ), который позволяет определять
митохондриальные ферменты дегидрогеназы в живых клетках, как описано у Mosman, J. Imm. Methods (1983) 65: 55. Исследованные новые соединения проявляют менее чем 50%-ную цитотоксичность в концентрации
10 мкМ, и это свидетельствует о том, что новые соединения не являются цитотоксичными в концентрациях, достаточных для подавления TNF.
Пример тестирования 3: Ингибирование
коллагеназы
Ингибирование коллагеназы оценивают с использованием активной коллагеназы с ММР-субстратом, содержащим тиопептид, согласно методу, описанному Stein и Izquierdo-Martin, Arch.
Biochem. Biophys. 308 (1994), стр. 274-277. Тестируемое соединение инкубируют с коллагеназой перед добавлением субстрата при рН 6,5, 25oС в буфере из 2-морфолинэтансульфоновой кислоты (50
мМ), дополненном 10 мМ CaCl2. Абсорбцию измеряют при 405 нм с регулярными интервалами в течение 40 мин. Ингибирующую активность тестируемого соединения определяют в виде функции
коллагеназной активности в контроле в присутствии тестируемого соединения и без него. Новые соединения обладают значительной зависящей от дозы способностью ингибировать коллагеназу в низких
наномолярных концентрациях, в частности ниже 10 нМ.
Пример тестирования 4: Биологическая доступность при оральном введении
Метод анализа из предшествующего примера
стандартизуют путем измерения активности конкретного тестируемого соединения в различных известных концентрациях и применяют для измерения концентрации тестируемого соединения в плазме после орального
введения. Тестируемые соединения вводят орально находящимся в сознании крысам в дозе 10 мг/кг. Образцы крови берут из отрезанного кончика хвоста через 30, 60, 120 и 240 мин после орального введения.
Плазму подвергают экстракции с помощью трихлоруксусной кислоты. Экстракт тестируют согласно описанному выше методу анализа ингибирования коллагеназы для получения оценки концентрации лекарства,
присутствующего в плазме. Новые соединения проявляют высокую биологическую доступность при оральном введении, а их концентрации в плазме составляют 300-5000 нМ через 30 мин и 50-500 нМ через 240 мин.
Таким образом, фармацевтически эффективные уровни в плазме (как описано в примерах по тестированию 1 и 3) являются легко достижимыми при оральном введении регулируемых доз, в частности 10 мг/кг. Кроме
того, достигаемые уровни в плазме значительно ниже цитотоксического уровня, и у крыс не наблюдалось каких-либо побочных действий при использовании этой дозы.
Реферат
Настоящее изобретение относится к новым производным гидроксамовой кислоты формулы (I), где R1 обозначает заместитель формулы II, n равно 1-4, m равно 0-3, R5 обозначает водород, алкил или алкенил, А обозначает водород, алкил, арил, арилалкил, R2 обозначает алкил, алкенил, циклоалкил, арил, арилалкил, R3 обозначает алкил, арил или индолилметил, R4 обозначает метил, пиридил или X-Y-, где Х обозначает морфолингруппу, пиридил или арил и Y обозначает алкилен, в котором до четырех метиленовых (-СН2-) звеньев необязательно замещены -СО-, -NH-, -SO2- или О. Эти соединения обладают ингибирующей активностью в отношении высвобождения TNF-α и благодаря этому свойству они могут использоваться в качестве фармацевтического средства. 9 з.п. ф-лы, 2 табл.
А-(О-(СR5Н)n)m-О-СН2- (II)
Формула
где R1 обозначает заместитель формулы II
A-(O-(CR5H)n)m-O-CH2- II
где n равно 1, 2, 3 или 4,
m равно 0, 1, 2 или 3,
R5 в каждом случае независимо обозначает Н, С1-С10алкил, С2-С6алкенил;
А обозначает водород, С1-С10алкил, С6-С14арил, С6-С14арил(С1-С6алкил);
R2 обозначает С3-С12алкил, С3-С12 алкенил, С3-С7циклоалкил, С5-С14арил или С5-С14арил(С1-С6алкил), где арильные группы необязательно замещены гидроксигруппой, С1-С6алкилом, С1-С6алкокси- или галогруппой;
R3 обозначает С1-С10алкил (необязательно замещенный гидрокси- или С1-С6алкоксигруппой), С6-С14арил (необязательно замещенный гидрокси-, С6-С14арилокси- или С1-С6 алкоксигруппой) или индолилметил;
R4 обозначает метил, пиридил или заместитель формулы X-Y-, где Х обозначает морфолиногруппу, пиридил или арил и Y обозначает С1-С14алкилен, в котором до четырех метиленовых (-СН2-) звеньев необязательно замещены -СО-, -NH-, -SO2- или-О-,
в свободной форме или в форме фармацевтически приемлемой соли.
А-(O-(СH2)n)m-О-СН2- II'
где А, n и m имеют значения, указанные в п. 1.
А-(O-(СHR5-CH2 )n)m'-О-СН2- II''
где А и R5 имеют значения, указанные в п. 1, n равно 1 или 2, а m' равно 0, 1 или 2.
n в формуле II равно 3 или 4, или
R5 в формуле II не обозначает Н, или
R2 обозначает С7-С12алкил, С3-С12 алкенил, С3-С7циклоалкил, С5-С14арил или С5-С14арил(С1-С6алкил), где арильные группы необязательно замещены гидроксигруппой, С1-С6алкилом, С1-С6алкокси- или галогруппой, или
R3 обозначает С1-С10алкил, С6-С14арил или любая арильная группа в этой формуле обозначает гетероарил, имеющий один или несколько гетероатомов, например, N, О или S.
где R'1 обозначает заместитель формулы II'''
А'-(O-(СН2)n')m'-O-СН2- II'''
где n' обозначает целое число, равное 1 или 2,
m' обозначает целое число, равное 0, 1, 2 или 3,
А' обозначает водород, С6-С14арил, С1-С10алкил,
R'2 обозначает С2-С6алкил,
R'3 обозначает С1-С10алкил (необязательно замещенный гидрокси- или С1-С6алкоксигруппой), С6-С14арил (необязательно замещенный гидрокси-, С6-С14арилокси или С1-С6алкоксигруппой) или индолилметил,
R'4 обозначает метил, пиридил или заместитель формулы X-Y-, где Х обозначает морфолиногруппу, пиридил или арил и Y обозначает С1-С14алкилен, в котором до четырех метиленовых (-СН2-) звеньев необязательно замещены -СО-, -NH-, -SO2- или -О- в свободной форме или в форме фармацевтически приемлемой соли.
(I) R1 обозначает радикал формулы II' или II'' и А в формуле II обозначает водород или С1-С6алкил;
(II) R2 в формуле I обозначает циклогексил, фенил, 4-метилфенил, 4-метоксифенил или изобутил,
(III) R3 в формуле I обозначает бензил или трет-бутил и
(IV) R4 в формуле I обозначает метил или морфолинокарбонил(С1-С6)алкил.
9. Соединение по любому из пп. 1-8, выбранное из соединений
{ 1-[S-фенилаланин-1-метиламид] -4-[N-гидрокси] } -диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)-янтарной кислоты;
{ 1-[S-фенилаланин-1-(2-(морфолино)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(5-(пара-тозиламино)пентил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5, 8-триоксаноил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(2-(морфолинокарбонил)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-фенилаланин-1-м(2-(пара-тозиламино)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(1-S-(метилкарбамоил)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(морфолинокарбонилметил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(2-(морфолинокарбонил)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(5-(морфолинокарбонил)пентил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(2-пиридил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-(пара-метоксифенил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5, 8-триоксаноил)янтарной кислоты;
{ 1-[R-(трет-бутилоксиметил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[R-(бензилоксиметил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-(пара-бензилоксифенил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-(3-метилиндолил)глицин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[R-(гидроксиметил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(2,5,8-триоксаноил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(метил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(2-пиридил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(3,6-диокса-8-оксо-9-имино-11-морфолиноундецил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(5-(морфолино)пентил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(морфолино)бутил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-(3,6-диокса-8-оксо-8-морфолинооктил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(3-имино-8-фенилоктил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(5-(бензилоксикарбониламино)пентил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(6-имино-7-оксо-10-метилундецил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(морфолинокарбонилметил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(2-пиридил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-(пара-метоксифенил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-(метил-3-индолил)глицин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[R-(трет-бутилоксиметил)аланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(циклогексилоксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиэтоксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидрокси-2-бензилэтоксиметил)янтарной кислоты;
{ 1-[S-фенилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(6-имино-7-oкco-l0-метилундецил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(6-имино-8-фенилоктил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(5-морфолинокарбонил)пентил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(морфолинокарбонилметил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-(2-(морфолинокарбонил)этил)амид] -4-[N-гидрокси] } диамид R-2-изобутил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-н-пропил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-изопропил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-тpимeтилaлaнин-1-мeтилaмид] -4-[N-гидpoкcи] } диамид R-2-циклопропил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-тpимeтилaлaнин-1-мeтилaмид] -4-[N-гидpoкcи] } диамид R-2-(3-метилбутил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-циклопентил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-циклогексил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-циклопентилметил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-циклогексилметил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(2-метоксиэтил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-фенил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-бензил-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(4-фенилфенил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(2-фенилэтил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(2-нафтил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(3-метилфенил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(4-метилфенил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(4-метоксифенил)-S-3-(гидроксиметил)янтарной кислоты;
{ 1-[S-триметилаланин-1-метиламид] -4-[N-гидрокси] } диамид R-2-(4-фторфенил)-S-3-(гидроксиметил)янтарной кислоты.
Приоритет по пунктам:
02.10.1996 - пп. 1-5, 7-10;
02.04.1997 - пп. 6-10.
Комментарии