Амидины и их производные и содержащие их фармацевтические композиции - RU2375346C2
Код документа: RU2375346C2
Описание
Настоящее изобретение относится к амидинам и их производным и содержащим их фармацевтическим композициям, которые применяют для предупреждения и лечения поврежденной ткани вследствие усиленного рекрутинга полиморфно-ядерных (PMN) нейтрофилов (PMN лейкоцитов) в местах воспаления.
Отдельные клетки крови (макрофаги, гранулоциты, нейтрофилы, полиморфно-ядерные клетки) отвечают на химический стимул (когда происходит стимуляция веществами, называемыми хемокинами) путем миграции по градиенту концентрации стимулирующего агента вследствие процесса, называемого хемотаксисом. Основные известные стимулирующие агенты или хемокины представляют собой продукты расщепления C5a-комплемента, некоторые N-формилпептиды, генерируемые за счет лизиса бактериальной поверхности, или пептиды синтетического происхождения, такие как формил-метионил-лейцил-фенилаланин (f-MLP), и, главным образом, различные цитокины, включая интерлейкин-8 (IL-8, также называемый CXCL8). Интерлейкин-8 является эндогенным хемотаксическим фактором, вырабатываемым большинством ядросодержащих клеток, таких как фибробласты и макрофаги.
При некоторых патологических состояниях, характеризующихся усиленным рекрутингом нейтрофилов, более тяжелое повреждение ткани на участке связано с инфильтрацией нейтрофилов. В последнее время продемонстрирована роль активации нейтрофилов при определении повреждения, связанного с постишемической реперфузией и легочной гипероксией.
Биологическая активность IL-8 опосредована взаимодействием интерлейкина с CXCR1- и CXCR2-мембранными рецепторами, относящимися к семейству семи трансмембранных рецепторов, экспрессируемых на поверхности человеческих нейтрофилов и некоторых типов Т-клеток (L. Xu et al., J. Leukocyte Biol., 57, 335, 1995). Известны селективные лиганды, которые могут распознавать CXCR1 и CXCR2: GRO-α, например, является селективным хемотаксическим фактором CXCR2.
Хотя известно, что CXCR1-активация играет ключевую роль в опосредованном IL-8 хемотаксисе, недавно было предположено, что CXCR2-активация может играть патофизиологическую роль в хронических воспалительных заболеваниях, таких как псориаз. В действительности, патофизиологическая роль IL-8 в псориазе также подтверждается воздействием IL-8 на кератиноцитные функции.
В самом деле, показано, что IL-8 является эффективным стимулятором пролиферации эпидермальных клеток, а также ангиогенеза - двух важных аспектов псориатического патогенеза (A. Tuschil et al., J. Invest Dermatol, 99, 294, 1992; Koch A.E. et al., Science, 258, 1798, 1992).
Кроме того, известно, что патофизиологическая роль IL-8 в прогрессировании и метастазе меланомы может быть опосредована CXCR2-активацией (L.R. Bryan et al., Am. J. Surg, 174, 507, 1997).
Потенциальная патогенная роль IL-8 в легочных заболеваниях (повреждение легких, синдром острого респираторного дистресса, астма, хроническое воспаление легких и кистозный фиброз) и, в особенности, в патогенезе COPD (хроническое обструктивное заболевание легких) посредством метаболизма CXCR2-рецептора описана (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology 2001, 1:242-247).
Исследования в отношении вклада отдельных (S)- и (R)-энантиомеров кетопрофена в противовоспалительную активность рацемата и их роли в модуляции хемокина (P. Ghezzi et al., J. Exp. Pharm. Ther., 287, 969, 1998) показывают, что оба энантиомера и их соли с хиральными и нехиральными органическими основаниями могут ингибировать зависимым от дозы образом хемотаксис и увеличение внутриклеточной концентрации Са2+-ионов, индуцируемых IL-8 в человеческих полиморфно-ядерных лейкоцитах (заявка на патент США 6069172). Позже было показано (C. Bizzarri et al., Biochem. Pharmacol. 61, 1429, 2001), что кетопрофен обладает свойством ингибирования биологической активности IL-8 вместе с другими молекулами, принадлежащими к классу нестероидных противовоспалительных средств (NSAID), таких как флурбипрофен, ибупрофен и индометацин. Типичная активность типичных NSAID в отношении ингибирования циклооксигеназ (COX) ограничивает терапевтическое применение этих соединений при лечении нейтрофильнозависимых патологических состояний и воспалительных состояний, таких как псориаз, идиопатический легочный фиброз, острая дыхательная недостаточность, повреждения от реперфузии и гломерулонефрит. Ингибирование синтеза простагландина происходит вследствие воздействия на циклооксигеназы, вызывающие увеличение продуцирования цитокинов, которые, подобно TNF-α, играют роль в усилении нежелательных противовоспалительных эффектов нейтрофилов.
Новые классы эффективных и селективных ингибиторов биологической активности IL-8 пригодны для «in vivo» введения. Амиды R-2-арилпропионовой кислоты и N-ацилсульфонамиды описаны как эффективные ингибиторы IL-8, индуцирующего хемотаксис и дегрануляцию нейтрофилов (WO 01/58852; WO 00/24710). Более того, новые R- и S-2-фенилпропионовые кислоты недавно описаны как эффективные ингибиторы IL-8, у которых полностью отсутствует нежелательный эффект в отношении ингибирования СОХ, и раскрыты в WO 03/043625.
Подробное описание данного изобретения
Найден новый класс амидинов и их производных, проявляющих способность эффективно ингибировать хемотаксис и дегрануляцию нейтрофилов, индуцируемые IL-8.
Настоящее изобретение относится к амидинам формулы (I) и их производным:

и их фармацевтически приемлемым солям, где
Ar выбирают из группы, состоящей из 3'-бензоилфенила, 3'-(4-хлорбензоил)фенила, 3'-(4-метилбензоил)фенила, 3'-ацетилфенила, 3'-пропионилфенила, 3'-изобутаноилфенила, 4'-трифторметансульфонилоксифенила, 4'-бензолсульфонилоксифенила, 4'-трифторметансульфониламинофенила, 4'-бензолсульфониламинофенила, 4'-бензолсульфонилметилфенила, 4'-ацетоксифенила, 4'-пропионилоксифенила, 4'-бензоилоксифенила, 4'-ацетиламинофенила, 4'-пропиониламинофенила, 4'-бензоиламинофенила, 4'-изобутилфенила, (3'-фтор-4'-фенил)-фенила, 5'-бензоил-2'-тиофена;
R представляет Н; R' означает Н, С1-5алкил, фенил, C1-5фенилалкил; или - остаток формулы - (СН2)n-NRaRb, где n означает целое число от 1 до 3 и каждый из Ra и Rb могут быть одинаковыми или различными и означать C1-С6-алкил или, альтернативно, Ra и Rb вместе с атомом азота, с которым они связаны, образуют 6-членный гетероцикл формулы (II):

где
W представляет собой одинарную связь;
R и R', альтернативно, могут образовывать 6-членный гетероцикл формулы (III):
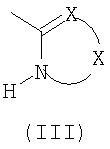
где
Х представляет собой остаток -O(СН2)n-, где n означает 2, или остаток -(СН2)n-, где n означает 2,
при условии, что исключается соединение 2-(4-изобутилфенил)пропионамидин(α-метил-4-(2-метилпропил)-бензолэтанимидамид).
Соединения формулы (I) являются хиральными соединениями, и данное изобретение относится как к рацемическим, так и к индивидуальным (R)- и (S)-энантиомерам.
Другим объектом согласно настоящему изобретению являются соединения формулы (I), как описано выше, пригодные в качестве лекарственных средств. В особенности, данное изобретение относится к соединениям формулы (I) в качестве ингибиторов индуцируемого IL-8 хемотаксиса человеческих полиморфно-ядерных нейтрофилов.
Предпочтительными R'-группами являются Н, С1С5-алкил, C1-C5-фенилалкил; или - остатоком формулы - (СН2)n-NRaRb, где n означает целое число от 2 до 3, более предпочтительно 3, и группа NRaRb означает N,N-диметиламин, N,N-диэтиламин, 1-пиперидил, 1-пирролидил, 1-пиперазинил, 1-(4-метил)пиперазинил.
Более предпочтительной группой NRaRb является N,N-диметиламин или 1-пиперидил.
Предпочтительной R'-группой является Н.
Когда R и R' образуют гетероцикл формулы (III), X предпочтительно представляет собой остаток -O(СН2)n- где n означает целое число 1 или 2, или остаток -(СН3)2.
Особенно предпочтительными соединениями согласно данному изобретению являются
(R,S)-(2-(4-изобутилфенил)пропионамидингидрохлорид;
(+)-(2-(4-изобутилфенил)пропионамидингидрохлорид;
(R,S)-(2-(4-изобутилфенил)пропионамидингидрохлорид;
(+)-(2-(4-изобутилфенил)пропионамидингидрохлорид;
(-)-(2-(4-изобутилфенил)пропионамидингидрохлорид;
(R,S)-2-(3-бензоилфенил)пропионамидингидрохлорид;
(R,S)-2-[(3-фтор-4-фенил)фенил]пропионамидингидрохлорид;
(R,S)-2-(4-трифторметансульфонилоксифенил)пропионамидингидрохлорид;
(R,S)-2-(5-бензоил-2-тиофен)пропионамидингидрохлорид;
(R,S)-2-(4-изобутилфенил)-N-[3"-(N'-пиперидино)пропил]пропионамидиндигидрохлорид;
(R,S)-2-(4-изобутилфенил)-N-метилпропионамидингидрохлорид;
(R,S)-2-(3-бензоилфенил)-N-[3-(N,N-диметиламино)пропил]пропионамидингидрохлорид;
(R,S)-2-(4-изобутилфенил)пропионамидинацетат;
(R,S)-2-(4-изобутилфенил)-N-[3-(N,N-диметиламино)пропил]пропионамидин;
(R,S)-2-(4-изобутилфенил)-N-бензилпропионамидин;
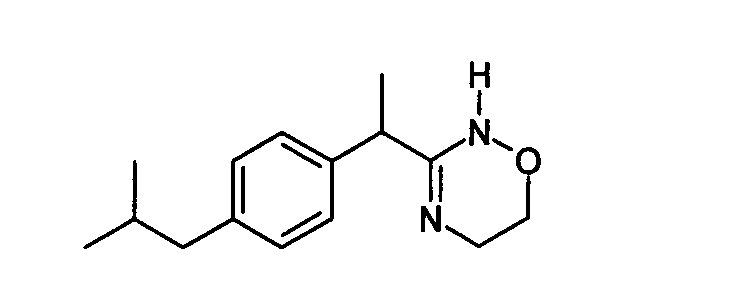
(R,S)-3-[1-(4-изобутилфенил)этил]-5,6-дигидро-2H-1,2,4-оксадиазин;
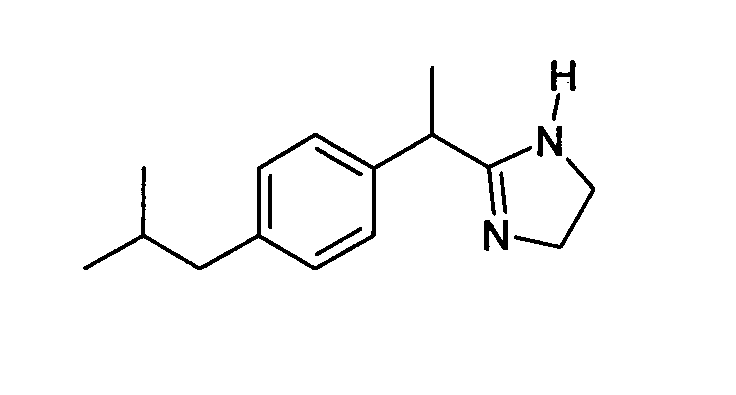
(R,S)-2-[1-(4-изобутилфенил)этил]-4,5-дигидро-2H-1,3-имидазол.
Соединения согласно данному изобретению являются эффективными и селективными ингибиторами индуцируемого IL-8 хемотаксиса человеческих полиморфно-ядерных клеток.
Соединения формулы (I) согласно данному изобретению, как правило, выделяют в форме их аддитивных солей как с органическими, так и с неорганическими фармацевтически приемлемыми кислотами.
Примерами таких кислот являются кислоты, выбираемые из группы, состоящей из соляной кислоты, серной кислоты, фосфорной кислоты, метансульфокислоты, фумаровой кислоты и лимонной кислоты.
Соединения формулы (I) получают путем обработки соответствующих нитрильных производных формулы (IV):

где Ar имеет то же самое значение, как описано выше,
в растворе MeOH/HCl и с последующим взаимодействием промежуточных имидатов с аминами формулы NHR, где R имеет то же самое значение, как описано выше, в безводном органическом растворителе, таком как дихлорметан.
Соединения формулы (I), где группы R и R' образуют гетероцикл формулы (III), получают путем прямой циклизации амидов формулы (V):
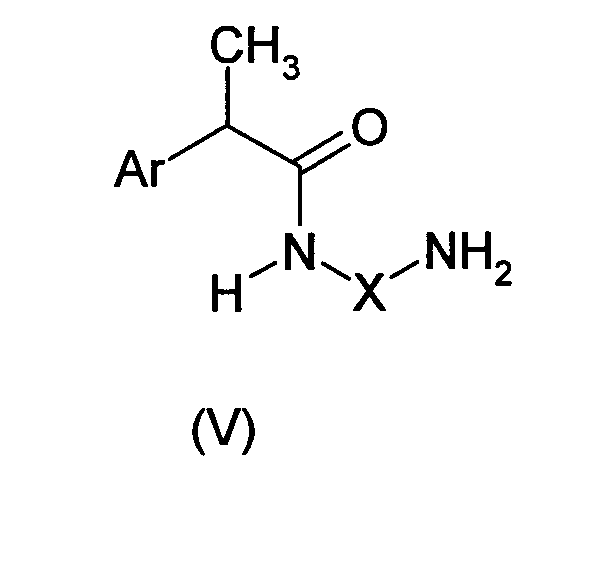
где Х имеет то же самое значение, как описано выше,
в присутствии подходящего катализатора, такого как Al(CH3)3.
Альтернативно, соединения формулы (I), где группы R и R' образуют гетероцикл формулы (III), получают путем прямого взаимодействия амидинов формулы (I), где R' означает Н и R означает Н или ОН, с реагентом формулы L-K-L', в присутствии основания, где L и L' являются обычными удаляемыми группами, такими как галогены, мезилат и т.д., и, когда группы R и R', обе, означают Н, К представляет собой остаток -(CH2)n-, где n означает целое число от 2 до 4; когда R означает ОН и R' означает Н, К представляет собой остаток -(CH2)n-, где n означает целое число от 1 до 3.
Соединения формулы (I) согласно данному изобретению оценивали in vitro в отношении их способности ингибировать хемотаксис полиморфно-ядерных лейкоцитов (в дальнейшем называемых PMN) и моноцитов, индуцируемый фракциями IL-8 и GRO-α. С этой целью, для выделения PMN из гепаринизированной человеческой крови, взятой у здоровых взрослых добровольцев, мононуклеарные клетки удаляли посредством седиментации на декстране (в соответствии с методикой, описанной W.J. Ming et al., J. Immunol., 138, 1469, 1987), а эритроциты удаляли с помощью гипотонического раствора. Жизнеспособность клеток выявляли путем исключения при окраске трипановым синим, в то время как соотношение циркулирующих полиморфно-ядерных клеток оценивали в цитоцентрифугате после окрашивания с помощью Diff Quick.
Человеческий рекомбинантный IL-8 (Pepro Tech) использовали в качестве стимулирующего агента в экспериментах по хемотаксису, получая практически идентичные результаты: лиофилизированный белок растворяли в объеме HBSS (сбалансированный солевой раствор Хенкса), содержащем 0,2% бычьего сывороточного альбумина (BSA), получая таким образом исходный раствор с концентрацией 10-5 М, разбавляемый в HBSS до концентрации 10-9 М для анализов на хемотаксис.
Во время анализа на хемотаксис (согласно W. Falket и др., J. Immunol. Methods, 33, 239, 1980) использовали не содержащие поливинилпирролидона (PVP) фильтры с пористостью 5 мкм и микрокамеры, подходящие для репликации.
Соединения формулы (I) согласно данному изобретению оценивали при концентрации в диапазоне от 10-6 М до 10-10 М; для этой цели их добавляли, в одной и той же концентрации, как в поры нижнего отсека, так и в поры верхнего отсека микрокамеры. Оценку способности соединений формулы (I) согласно данному изобретению к ингибированию индуцируемого IL-8 хемотаксиса человеческих моноцитов осуществляли в соответствии с методикой, описанной в Van Damme J. et al. (Eur. J. Immunol., 19, 2367, 1989).
Особенно предпочтительными соединениями согласно данному изобретению являются соединения формулы (I), где группы Ar означают 3'-бензоилфенил, 3'-(4-хлорбензоил)фенил, 3'-(4-метилбензоил)фенил, 3'-ацетилфенил, 3'-пропионилфенил, 3'-изобутаноилфенил, 4'-трифторметансульфонилоксифенил, 4'-бензолсульфонилоксифенил, 4'-трифторметансульфониламинофенил, 4'-бензолсульфониламинофенил, 4'-бензолсульфонилметилфенил, 4'-ацетоксифенил, 4'-пропионилоксифенил, 4'-бензоилоксифенил, 4'-ацетиламинофенил, 4'-пропиониламинофенил, 4'-бензоиламинофенил, которые проявляют дополнительное свойство эффективного ингибирования индуцируемого GRO-α хемотаксиса PMN; эта активность делает возможным терапевтическое использование этих соединений при патологиях, связанных с IL-8, где особенно затрагивается путь метаболизма CXCR2 или вместе с передачей сигнала CXCR1.
Двойственные ингибиторы индуцируемых IL-8 и GRO-α биологических активностей являются наиболее предпочтительными для терапевтических применений, однако, описанные соединения с селективным воздействием на IL-8-рецептор CXCR1 или GRO-α/IL-8-рецептор CXCR2 могут найти полезные терапевтические применения при лечении специфических патологий, как описано ниже.
Найдено, что соединения формулы (I), оцениваемые ex vivo в крови, in toto, в соответствии с методикой, описанной Patrignani и др., в J. Pharmacol. Exper. Ther., 271, 1705, 1994, полностью неэффективны в качестве ингибиторов циклооксигеназ (COX).
В большинстве случаев соединения формулы (I) не препятствуют продуцированию PGE2, индуцируемого в мышиных макрофагах за счет стимуляции липополисахаридами (LPS, 1 мкг/мл) при концентрации в диапазоне от 10-5 до 10-7 М. Ингибирование продуцирования PGE2, которое может быть зарегистрировано, обычно находится на пределе статистического значения и чаще составляет ниже, чем 15-20% от основного значения. Пониженная эффективность в отношении ингибирования СО представляет собой преимущество для терапевтического применения соединений согласно данному изобретению, так как ингибирование синтеза простагландина представляет собой стимул для макрофагов в отношении усиленного синтеза TNF-α (индуцируемого LPS или пероксидом водорода), который является важным медиатором активации нейтрофилов и стимулом для продуцирования цитокина интерлейкина-8.
Принимая во внимание экспериментальные данные, обсужденные выше, и роль, которую выполняет интерлейкин-8 (IL-8) и имеющие с ним общее происхождение лимфокины в процессах, которые включают активацию и инфильтрацию нейтрофилов, соединения согласно данному изобретению особенно пригодны при лечении заболеваний, таких как псориаз (R. J. Nicholoff et al., Am. J. Pathol., 138, 129, 1991). Дальнейшими заболеваниями, которые можно лечить с помощью соединений согласно настоящему изобретению, являются кишечные хронические воспалительные патологии, такие как язвенный колит (Y. R. Mahida и др., Clin. Sci., 82, 273, 1992) и меланома, хроническое обструктивное заболевание легких (СОРD), буллезный пемфиго, ревматоидный артрит (M. Selz et al., J. Clin. Invest., 87, 463, 1981), идиопатический фиброз (E. J. Miller, цитировано выше, и P. C. Carré et al., J. Clin. Invest., 88, 1882, 1991), гломерулонефрит (T. Wada et al., J. Exp. Med., 180, 1135, 1994), и их можно использовать для предупреждения и лечения повреждений, вызываемых ишемией и реперфузией.
Ингибиторы активации CXCR1 и CXCR2 находят полезные применения, как подробно описано выше, особенно при лечении хронических воспалительных патологий (например, псориаз), в случае которых предполагают, что активация обоих IL-8-рецепторов играет ключевую патофизиологическую роль в развитии заболевания.
В действительности, известно, что активация CXCR1 является необходимой в опосредуемом IL-8 хемотаксисе PMN (Hammond M. et al., J. Immunol, 155, 1428, 1995). С другой стороны, предполагают, что активация CXCR2 является необходимой в опосредуемой IL-8 эпидермальной клеточной пролиферации и ангиогенезе у псориатических больных (Kulke R. et al., J. Invest Dermatol, 110, 90, 1998).
Кроме того, селективные антагонисты CXCR2 находят особенно полезные терапевтические применения при лечении существенных легочных заболеваний, подобно хроническому обструктивному заболеванию легких (СОРD) (D. WP Hay and H.M. Sarau., Current Opinion in Pharmacology, 2001, 1:242-247).
Следовательно, другой объект согласно настоящему изобретению относится к соединениям, предназначенным для лечения псориаза, язвенного колита, меланомы, хронического обструктивного заболевания легких (СОРD), буллезного пемфиго, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и для предупреждения и лечения повреждений, вызываемых ишемией и реперфузией, а также к применению таких соединений для получения лекарственного средства для лечения заболеваний, которые описаны выше. Фармацевтические композиции, содержащие соединение согласно данному изобретению и подходящий для этого носитель, также входят в объем настоящего изобретения.
Соединения согласно данному изобретению вместе с обычно применяемым адъювантом, носителем, разбавителем или эксципиентом, фактически могут находиться в форме фармацевтических композиций и их унифицированных (стандартных) доз и в такой форме могут быть использованы в виде твердых форм, таких как таблетки или наполненные лекарством капсулы, или в виде жидких форм, таких как растворы, суспензии, эмульсии, эликсиры или наполненные тем же самым капсулы, причем все для перорального применения, или в форме стерильных инъецируемых растворов для парентерального (включая подкожное) применения. Такие фармацевтические композиции и их унифицированные дозированные формы могут содержать ингредиенты в стандартных пропорциях, вместе или без дополнительных активных соединений или действующих начал, и такие унифицированные дозированные формы могут содержать любое подходящее эффективное количество активного ингредиента в соответствии с имеющимся в виду используемым диапазоном суточных доз.
Амидины согласно данному изобретению, используемые в качестве фармацевтических средств, обычно вводят в форме фармацевтической композиции. Такие композиции можно получать способом, хорошо известным в фармацевтической области, и они включают, по меньшей мере, одно активное соединение. Как правило, соединения согласно данному изобретению вводят в фармацевтически эффективном количестве. Фактически вводимое количество соединения обычно определяют в зависимости от состояния, которое нужно лечить, выбранного пути введения, реально вводимого соединения, возраста, массы тела и ответной реакции отдельного пациента, серьезности симптомов пациента, и т.п.
Фармацевтические композиции согласно данному изобретению можно вводить различными путями, включая пероральный, ректальный, трансдермальный, подкожный, внутривенный, внутримышечный и интраназальный. В зависимости от предназначенного пути введения соединения предпочтительно используют для получения готовой формы в виде или инъецируемых или пероральных композиций. Композиции для перорального введения могут быть в форме, в основной массе, жидких растворов или суспензий, или, в основной массе, порошков. Обычно, однако, композиции находятся в виде унифицированных дозированных форм, что обеспечивает точное дозирование. Термин «унифицированные дозированные формы» относится к физически дискретным единицам, пригодным в качестве стандартных доз для человека и других млекопитающих, причем каждая унифицированная дозированная форма содержит предопределенное количество активного вещества, рассчитанное для достижения желательного терапевтического эффекта, в сочетании с подходящим фармацевтическим эксципиентом. Традиционные унифицированные дозированные формы включают предварительно заполненные, заранее соразмеренные ампулы или шприцы с жидкой композицией или пилюли, таблетки, капсулы, или т.п., в случае твердых композиций. В таких композициях соединение кислотного характера обычно находится в незначительном количестве (от примерно 0,1 до примерно 50 мас.% или, предпочтительно, от примерно 1 до примерно 40 мас.%), а остальное включает различные разбавители или носители и технологически вспомогательные средства, пригодные для получения желательной дозированной формы.
Жидкие формы, подходящие для перорального введения, могут включать пригодный водный или неводный разбавитель с буферами, суспендирующими и диспергирующими агентами, красителями, ароматизаторами и т.п. Жидкие формы, включая описанные в данном контексте ниже инъецируемые композиции, всегда хранятся в отсутствие света, так как нужно избегать любого каталитического воздействия света, такого как образование гидропероксида или пероксида. Твердые формы могут включать, например, любой из следующих ингредиентов или соединений подобной природы: связующее вещество, такое как микрокристаллическая целлюлоза, трагакантовая камедь или желатин; эксципиент, такой как крахмал или лактоза; дезинтегрирующий агент, такой как альгиновая кислота, примогель или кукурузный крахмал; смазочный материал, такой как стеарат магния; придающий скользкость агент, такой как гель кремниевой кислоты; подсластитель, такой как сахароза или сахарин; или ароматизатор, такой как мята перечная, метилсалицилат или апельсиновый ароматизатор.
Инъецируемые композиции обычно базируются на инъецируемом стерильном физиологическом растворе или забуференном фосфатом физиологическом растворе или других инъецируемых носителях, известных в уровне техники. Как указано выше, производное кислотного характера формулы (I) в таких композициях обычно является используемым в незначительном количестве компонентом, часто присутствующим в пределах от 0,05 до 10 мас.%, а остальное представляет носитель для инъекции и т.п. Средняя суточная доза зависит от различных факторов, таких как тяжесть заболевания и состояния пациента (возраст, пол и масса тела). Доза, как правило, может изменяться от 1 мг или нескольких мг до 1500 мг соединений формулы (I) в сутки, необязательно разделенная для многократного введения. Также можно вводить более высокие дозировки, благодаря низкой токсичности соединений согласно данному изобретению, в течение длительных периодов времени.
Вышеописанные компоненты для перорального введения или инъецируемые композиции являются только показательными. Дальнейшие материалы, а также технологии производства и т.п. изложены в части 8 “Remington's Pharmaceutical Sciences Handbook”, 18-е издание, 1990, Mack Publishing Company, Истон, штат Пенсильвания, которая включена в данный контекст путем ссылки.
Соединения согласно данному изобретению также можно вводить в виде форм с замедленным высвобождением или в виде систем доставки лекарственного средства с замедленным высвобождением. Описание показательных материалов с замедленным высвобождением также можно найти во включенных в данный контекст вышеуказанных материалах Remington's Handbook, как указано выше.
Настоящее изобретение проиллюстрировано с помощью следующих примеров, которые нельзя рассматривать как ограничивающие объем охраны изобретения.
Аббревиатура:
THF: тетрагидрофуран;
DMF: диметилформамид;
AcOEt: этилацетат.
Экспериментальные методики
Пример 1
Следуя методике, описанной Granik, Russ. Chem. Rev., 52, 377-393 (1983), могут быть получены следующие незамещенные амидины:
1a (R,S)-(2-(4-Изобутилфенил)пропионамидингидрохлорид
2-(4-Изобутилфенил)пропионитрил
4-Изобутил-α-метилфенилацетамид (2 г, 9,7 ммоль), полученный в соответствии с методикой, описанной в WO 00/24710, растворяют в смеси толуола и трихлорметана (2:1; 30 мл). Добавляют 20%-ный раствор фосгена в толуоле (15,5 мл, 30 ммоль) и полученную смесь оставляют перемешиваться 12 часов в инертной атмосфере до полного растворения исходного реагента. После выпаривания растворителей при пониженном давлении сырой продукт растворяют в этилацетате (20 мл), органическую фазу промывают насыщенным раствором NaHCO3 (2×20 мл) и насыщенным раствором NaCl (2×15 мл), сушат над Na2SO4 и выпаривают в вакууме с получением 2-(4-изобутилфенил)пропионитрила в виде бесцветного масла (1,45 г, 7,76 ммоль). Выход 80%.
1H-ЯМР (CDCl3): δ 7,42 (д, 2H, J=7 Гц); 7,28 (д, 2H, J=7 Гц); 4,05 (кв., 1H, J=8 Гц); 2,65 (д, 2H, J=8 Гц); l,95 (м, 1H); 1,80 (д, 3H, J=8 Гц); 1,05 (д, 6H, J=8 Гц).
Раствор 2-(4-изобутилфенил)пропионитрила (0,2 г, 1,07 ммоль) в смеси диэтилового эфира и метанола (1:1; 20 мл) охлаждают до температуры 0-5°С и через раствор барботируют газообразный HCl в течение 1 часа. Затем температуру доводят до комнатной температуры и смесь перемешивают в течение ночи. После выпаривания растворителя при пониженном давлении сырой продукт растворяют в метаноле (10 мл) и охлаждают до температуры 0-5°С. Через раствор барботируют аммиак в течение 1 часа и полученную смесь оставляют перемешиваться в течение ночи при комнатной температуре. После выпаривания растворителя при пониженном давлении сырой продукт суспендируют в диэтиловом эфире (15 мл) и оставляют перемешиваться при комнатной температуре в течение 2 часов. 2-(4-Изобутилфенил)пропионамидингидрохлорид формулы (I) выделяют путем отфильтровывания под вакуумом в виде белого твердого вещества (0,193 г, 0,80 ммоль). Выход 75%.
1H-ЯМР (ДМСО-d6): δ 8,80-8,50 (уш.с, NH3+Cl-); 7,40 (д, 2H, J=7 Гц); 7,15 (д, 2H, J=7 Гц); 3,98 (кв., 1H, J=8 Гц); 2,42 (д, 2H, J=8 Гц); l,90 (м, 1H); 1,57 (д, 3H, J=8 Гц); 0,88 (д, 6H, J=8 Гц).
В соответствии с вышеописанной методикой и при использовании подходящей карбоновой кислоты могут быть получены следующие соединения:
1b (R,S)-2-(3-бензоилфенил)пропионамидингидрохлорид
Получают из 2-(3'-бензоилфенил)пропионитрила, полученного по вышеописанной методике, и соответствующего α-метилфенилацетамида. Общая методика получения описана в WO/0158852. Выход 70%. Температура плавления: 110-113оС.
1H-ЯМР (ДМСО-d6): δ 7,86 (с, 1H); 7,80-7,50 (м, 8H + NH2++ NH2); 4,13 (кв., 1H, J=7 Гц); 1,60 (д, 3H, J=7 Гц).
1c (R,S)-2-[(3-фтор-4-фенил)фенил]пропионамидингидрохлорид
Получают из 2-(3-фтор-4-фенил)пропионитрила, полученного по вышеописанной методике, и соответствующего α-метилфенилацетамида. Общая методика получения описана в WO/0158852. Выход 53%. Температура плавления: 143-145оС.
1H-ЯМР (ДМСО-d6): δ 9,18 (уш.с, NH2+Cl-); 8,85 (уш.с, NH2); 7,67-7,30 (м, 8H); 4,15 (кв., 1H, J=7 Гц); 1,62 (д, 3H, J=7 Гц).
1d (R,S)-2-(4-трифторметансульфонилоксифенил)пропионамидингидрохлорид
Получают из 2-(4'-трифторметансульфонилоксифенил)пропионитрила, полученного по вышеописанной методике, и соответствующего α-метилфенилацетамида. Выход 68%.
1H-ЯМР (ДМСО-d6): δ 7,47 (д, 2H, J=8 Гц); 7,25 (д, 2H, J=8 Гц); 6,55 (уш.с, NH2+NH2+Cl-); 3,92 (кв., 1H, J=7 Гц); 1,56 (д, 3H, J=7 Гц).
1e (R,S)-2-(5-бензоил-2-тиофен)пропионамидингидрохлорид
Получают из 2-(5-бензоил-2-тиофен)пропионитрила, полученного по вышеописанной методике, и соответствующего пропионамида. Выход 60%.
1H-ЯМР (ДМСО-d6): δ 7,9 (д, 2H, J=8 Гц); 7,7-7,4 (м, 4H); 7,0 (д, 1H, J=8 Гц); 6,55 (уш.с, NH2+ NH2+Cl-); 3,9 (кв., 1H, J=7 Гц); 1,56 (д, 3H, J=7 Гц).
Разделение на оптические антиподы (R,S)-2-(4-изобутилфенил)пропионамидина
Индивидуальные (+)- и (-)-энантиомеры 2-(4-изобутилфенил)пропионамидина получают путем разделения на оптические антиподы, исходя из (R,S)-2-(4-изобутилфенил)пропионамидингидрохлорида. Свободное основание получают путем обработки гидрохлорида с помощью сильно основной смолы AMBERLITE IRA-910.
Соответствующие (L)- и (D)-тартраты получают путем обработки (R,S)-2-(4-изобутилфенил)пропионамидина с помощью (L)- и (D)-тартрата в метаноле. Оптически чистые изомеры (+)- и (-)-2-(4-изобутилфенил)пропионамидина получают посредством последовательных стадий кристаллизации тартратов из раствора в изопропаноле (или ацетоне).
Свободные основания получают путем обработки тартратов с помощью сильно основной смолы AMBERLITE IRA-910.
1f (+)-(2-(4-изобутилфенил)пропионамидин
[α]D=+28,1 (c=0,5, MeOH)
1g (-)-(2-(4-изобутилфенил)пропионамидин
[α]D=-28,0 (c=0,5, MeOH).
Пример 2
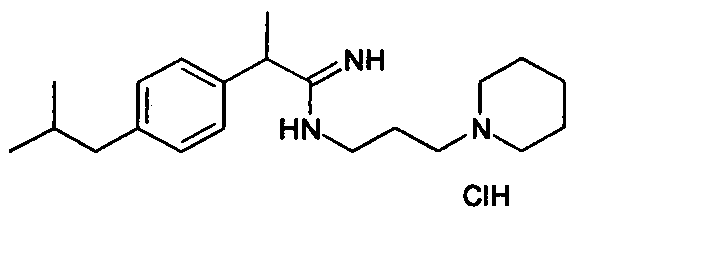
2a (R,S)-2-(4-Изобутилфенил)-N-[3-(N-пиперидино)пропил]пропионамидиндигидрохлорид
Раствор 2-(4-изобутилфенил)пропионитрила (0,15 г, 0,80 ммоль) в смеси диэтилового эфира и метанола (1:1; 10 мл) охлаждают до температуры 0-5°С и через раствор барботируют газообразный HCl в течение 1 часа. Затем температуру доводят до комнатной температуры и смесь перемешивают в течение ночи. После выпаривания растворителя при пониженном давлении сырой продукт растворяют в метаноле (10 мл) и охлаждают до температуры 0-5°С. Добавляют по каплям раствор 3-пиперидинпропиламина (0,15 г, 0,96 ммоль) в метаноле (5 мл) и полученную смесь оставляют перемешиваться в течение ночи при комнатной температуре. После выпаривания растворителя при пониженном давлении сырое масло суспендируют 2 н. HCl (рН раствора равно 2) и продукт экстрагируют дихлорметаном (3×15 мл). Объединенные органические экстракты затем промывают насыщенным раствором NaCl (2×15 мл), сушат над Na2SO4 и выпаривают в вакууме с получением 2-(4'-изобутилфенил)-N-[3-(N-пиперидино)пропил]пропионамидиндигидрохлорида в виде стекловидного твердого вещества (0,193 г, 0,48 ммоль). Выход 60%.
1H-ЯМР (CDCl3): δ 10,88 (уш.с, NH+Cl-); 10,22 (уш.с, NH+Cl-); 9,82 (уш.с, NH+Cl-); 7,64 (уш.с, NH); 7,41 (д, 2H, J=8 Гц); 7,15 (д, 2H, J=8 Гц); 4,39 (кв., 1H, J=8 Гц); 3,78 (м, 2H); 3,45 (м, 2H); 3,10 (м, 2H); 2,75 (м, 2H); 2,46 (д, 2H, J=8 Гц); 2,32-2,05 (м, 3H); 2,00-1,68 (м, 9H); 0,90 (д, 6H, J=8 Гц).
В соответствии с вышеописанной методикой и при использовании подходящего амина в качестве свободного основания получают следующие соединения:
2b (R,S)-2-(4-изобутилфенил)-N-метилпропионамидингидрохлорид
Получают из 2-(4-изобутилфенил)пропионитрила, полученного по методике, описанной в примере 1, и соответствующего α-метилфенилацетамида. Выход 75%.
1H-ЯМР (ДМСО-d6): δ 10,15 (уш.с, NH+Cl-); 7,12 (м, 4H); 4,25 (уш.с, NH2); 3,71 (м, 1H); 2,90 (с, 3H); 2,48 (д, 2H, J=8 Гц); 1,91 (м, 1H); 1,55 (д, 3H, J=8 Гц); 0,93 (д, 6H, J=8 Гц).
2с (R,S)-2-(3-бензоилфенил)-N-[3-(N,N-диметиламино)пропил]пропионамидингидрохлорид
Получают из 2-(3-бензоилфенил)пропионитрила, полученного по методике, описанной в примере 1, и соответствующего α-метилфенилацетамида. Выход 48%.
1H-ЯМР (ДМСО-d6): δ 7,81 (д, 2H, J=8 Гц); 7,74 (с, 1H); 7,67 (д, 1H, J=8 Гц); 7,59 (д, 1H, J=8 Гц); 7,52-7,27 (м, 4H+NH); 3,65 (кв., 1H, J=7 Гц); 3,25 (т, 2H, J=6 Гц); 2,27 (т, 2H, J=6 Гц); 2,09 (с, 6H); 1,66 (м, 2H); 1,46 (д, 6H, J=7 Гц).
Пример 3
(R,S)-2-(4-Изобутилфенил)пропионамидинацетат
В качестве альтернативной методики получения 2-(4-изобутилфенил)пропионамидинов следует использовать методику, описанную Judkins B.D., Allen D.G., Cook T.A., Evans B. and Sardharwala T.E., Synth. Comm., 26(23), 4315-4367 (1996).
(R,S)-2-(4-Изобутилфенил)-N-гидроксипропионамидин
Смесь гидроксиламингидрохлорида (0,38 г, 5,32 ммоль) и трет-бутилата натрия (0,5 г, 5,28 ммоль) в этаноле (10 мл) перемешивают при комнатной температуре в течение 15 минут; осадок отфильтровывают и по каплям добавляют маточный раствор к раствору 2-(4-изобутилфенил)пропионитрила (0,11 г, 0,49 ммоль) в абсолютном этаноле (3 мл). Полученный раствор кипятят с обратным холодильником в течение 18 часов. После охлаждения до комнатной температуры растворители выпаривают при пониженном давлении и сырой остаток разбавляют трихлорметаном (25 мл), промывают 5%-ным раствором лимонной кислоты (2×15 мл), затем насыщенным раствором NaCl (2×15 мл), сушат над Na2SO4 и выпаривают в вакууме с получением 2-(4-изобутилфенил)-N-гидроксипропионамидина, выделенного после кристаллизации из н-гексана в виде белого твердого вещества (0,075 г, 0,34 ммоль). Выход 70%. Температура плавления: 75-78оС.
1H-ЯМР (CDCl3): δ 7,25 (д, 2H, J=7 Гц); 7,12 (д, 2H, J=7 Гц); 5,030 (уш.с, 1H, NH); 4,35 (уш.с, 2H, NH-OH); 3,58 (кв., 1H, J=8 Гц); 2,48 (д, 2H, J=8 Гц); l,87 (м, 1H); 1,50 (д, 3H, J=8 Гц); 0,92 (д, 6H, J=8 Гц).
2-(4-Изобутилфенил)-N-гидроксипропионамидин (0,097 г, 0,44 ммоль) растворяют в уксусной кислоте (3 мл) и обрабатывают при комнатной температуре уксусным ангидридом (0,06 мл, 0,66 ммоль). Добавляют 10%-ный Pd на активированном угле (0,03 г) и через раствор в колбе барботируют водород до полного растворения исходного реагента. Добавляют метанол (5 мл), катализатор отфильтровывают на слое целита и растворители выпаривают при пониженном давлении с получением маслянистого остатка. После кристаллизации сырого остатка из н-гексана получают 2-(4-изобутилфенил)пропионамидинацетат в виде белого твердого вещества (0,106 г, 0,4 ммоль). Выход 91%. Температура плавления >220°С.
1H-ЯМР (ДМСО-d6): δ 8,70-8,50 (уш.с, NH3++ NH); 7,42 (д, 2H, J=7 Гц); 7,23 (д, 2H, J=7 Гц); 3,85 (кв., 1H, J=8 Гц); 2,52 (д, 2H, J=8 Гц); l,97 (м, 1H); 1,75 (с, 3H); 1,60 (д, 3H, J=8 Гц); 0,95 (д, 6H, J=8 Гц).
Пример 4
Для получения 2-(4-изобутилфенил)-N-алкилпропионамидинов следуют методике, описанной Weintraub L., Oles S.R. и Kalish N., J. Org. Chem., 33(4), 1679-1681 (1968).
4a (R,S)-2-(4-Изобутилфенил)-N-[3-(N,N-диметиламино)пропил]пропионамидин
4-Изобутил-α-метилфенилацетамид (1 г, 4,9 ммоль), полученный в соответствии с методикой, описанной в WO 00/24710, растворяют в безводном дихлорметане (10 мл) в инертной атмосфере при комнатной температуре и обрабатывают триэтилоксонийтетрафторборатом (1,0 М в CH2Cl2, 5 мл, 5 ммоль). Полученный раствор оставляют перемешиваться в течение ночи при комнатной температуре. После выпаривания растворителя при пониженном давлении сырой промежуточный продукт разбавляют диэтиловым эфиром (5 мл) при комнатной температуре и в инертной атмосфере и обрабатывают 3-(диметиламино)пропиламином (0,61 мл, 4,9 ммоль). Полученный раствор кипятят с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры растворители выпаривают при пониженном давлении и сырой продукт очищают флэш-хроматографией (элюент: CHCl3/циклогексан/CH3OH/NH4OH: 60:24:17:2). Получают чистый 2-(4-изобутилфенил)-N-[3-(N,N-диметиламино)пропил]пропионамидин в виде бледно-желтого масла (0,82 г, 2,84 ммоль). Выход 58%.
1H-ЯМР (ДМСО-d6): δ 7,39 (д, 2H, J=8 Гц); 7,14 (д, 2H, J=8 Гц); 4,15 (кв., 1H, J=7 Гц); 3,25 (т, 2H, J=7 Гц); 2,42 (д, 2H, J=7 Гц); 2,16 (т, 2H, J=7 Гц); 2,06 (с, 3H); 1,80 (м, 1H); 1,65 (м, 2H); 1,53 (д, 3H, J=7 Гц); 0,84 (д, 6H, J=7 Гц).
В соответствии с вышеописанной методикой и при использовании N-бензиламина получают следующее соединение:
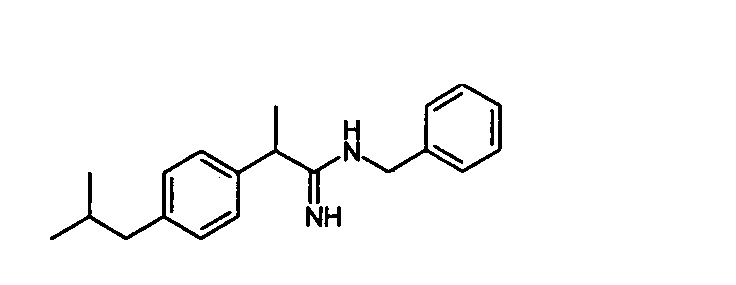
4b (R,S)-2-(4-изобутилфенил)-N-бензилпропионамидин
Выход 65%.
1H-ЯМР (CDCl3): δ 7,35-7,18 (м, 5H); 7,15 (д, 2H, J=8 Гц); 7,05 (д, 2H, J=8 Гц); 5,05 (уш.с, 2H, NH); 4,30 (с, 2H); 3,65 (кв., 1H, J=7 Гц); 2,45 (д, 2H, J=7 Гц); 1,91 (м, 1H); 1,55 (д, 3H, J=7 Гц); 0,95 (д, 6H, J=7 Гц).
Пример 5
(R,S)-3-[1-(4-Изобутилфенил)этил]-5,6-дигидро-2H-1,2,4-оксадиазин
(R,S)-2-(4-Изобутилфенил)-N-гидроксипропионамидин (50 мг, 0,23 ммоль, получен, как описано в примере 3) растворяют в 10 мл хлороформа при комнатной температуре. К этому раствору при комнатной температуре добавляют избыток карбоната натрия и 0,28 ммоль 1,2-дихлорэтана (28 мг, 20%-ный избыток). Суспензию кипятят с обратным холодильником в течение 5 часов. После охлаждения неорганические соли отфильтровывают и раствор промывают рассолом (2×10 мл). Растворитель удаляют при пониженном давлении и указанное в заголовке соединение очищают колоночной хроматографией на силикагеле (н-гексан/этилацетат: 9:1) с получением 29 мг бледно-желтого масла (выход 51%).
1H-ЯМР (CDCl3): δ 7,35 (д, 2H, J=7 Гц); 7,15 (д, 2H, J=7 Гц); 3,70 (кв., 1H, J=8 Гц); 3,6-3,4 (м, 4H); 2,42 (д, 2H, J=8 Гц); 2,3-2,1 (м, 2H); l,90 (м, 1H); 1,57 (д, 3H, J=8 Гц); 0,88 (д, 6H, J=8 Гц).
Пример 6
(R,S)-2-[1-(4-Изобутилфенил)этил]-4,5-дигидро-2H-1,3-имидазол
(R,S)-2-[(4-Изобутил)фенил]пропионамидингидрохлорид (100 мг, 0,49 ммоль, получен, как описано в примере 1а) суспендируют в 25 мл безводного хлороформа при комнатной температуре в инертной атмосфере, затем обрабатывают большим избытком (10-50 экв.) трет-бутилата калия. К суспензии (0,59 ммоль) добавляют 1,2-дихлорэтан (58 мг, 20%-ный избыток). Суспензию затем кипятят с обратным холодильником в течение 24 часов. При комнатной температуре суспендированное твердое вещество отфильтровывают и раствор промывают 5% фосфатным буфером с рН 5 и рассолом. Раствор, высушенный над сульфатом натрия, выпаривают; остаточное масло хроматографируют на колонке с силикагелем, получая указанное в заголовке чистое соединение (73 мг, выход 65%).
1H-ЯМР (CDCl3): δ 7,40 (д, 2H, J=7 Гц); 7,15 (д, 2H, J=7 Гц); 3,75 (кв., 1H, J=8 Гц); 3,5-3,6 (м, 4H); 2,42 (д, 2H, J=8 Гц); l,90 (м, 1H); 1,57 (д, 3H, J=8 Гц); 0,88 (д, 6H, J=8 Гц).
Химическая структура соединений примеров 1-6 представлена в таблице.









Реферат
Изобретение относится к амидинам формулы (I) и их производным, способам их получения и включающим амидины формулы (I) фармацевтическим композициям. Согласно данному изобретению, амидины пригодны для ингибирования хемотаксиса нейтрофилов, индуцируемого IL-8, и могут применяться для получения лекарственных средств для лечения псориаза, язвенного колита, меланомы, хронического обструктивного заболевания легких (COPD), буллезного пемфиго, ревматоидного артрита, идиопатического фиброза, гломерулонефрита и для предупреждения и лечения повреждений, вызываемых ишемией и реперфузией. 6 н. и 1 з.п.ф-лы, 1 табл.
Формула
и их фармацевтически приемлемые соли,
где Ar выбирают из группы, состоящей из
-3'-бензоилфенила, 3'-(4-хлорбензоил)фенила, 3'-(4-метилбензоил)фенила, 3'-ацетилфенила, 3'-пропионилфенила, 3'-изобутаноилфенила, 4'-трифторметансульфонилоксифенила, 4'-бензолсульфонилоксифенила, 4'-трифторметансульфониламинофенила, 4'-бензолсульфониламинофенила, 4'-бензолсульфонилметилфенила, 4'-ацетоксифенила, 4'-пропионилоксифенила, 4'-бензоилоксифенила, 4'-ацетиламинофенила, 4'-пропиониламинофенила, 4'-бензоиламинофенила, 4'-изобутилфенила, (3'-фтор-4'-фенил)фенила, 5'-бензоил-2'-тиофена;
R представляет Н;
R' означает Н, С1-5алкил, фенил, С1-5фенилалкил;
- остаток формулы -(CH2)n-NRaRb, где n означает целое число от 1 до 3 и каждый из
Ra и Rb, могут быть одинаковыми или различными и означать С1-С6-алкил или, альтернативно, Ra и Rb вместе с атомом азота, с которым они связаны, образуют 6-членный гетероцикл формулы (II):
где W представляет собой одинарную связь;
R и R', альтернативно, могут образовывать 6-членный гетероцикл формулы (III):
где Х представляет собой остаток -O(СН2)n-, где n означает 2, или остаток -(СН2)n-, где n означает 2,
при условии, что исключается соединение 2-(4-изобутилфенил) пропионамидин(α-метил-4-(2-метилпропил)-бензолэтанимидамид).
(R,S)-(2-(4-изобутилфенил)пропионамидин гидрохлорида;
(+)-(2-(4-изобутилфенил)пропионамидин гидрохлорида;
(-)-(2-(4-изобутилфенил)пропионамидин гидрохлорида;
(R,S)-2-(3-бензоилфенил)пропионамидин гидрохлорида;
(R,S)-2-[(3-фтор-4-фенил)фенил]пропионамидин гидрохлорида;
(R,S)-2-(4-трифторметансульфонилоксифенил)пропионамидин гидрохлорида;
(R,S)-2-(5-бензоил-2-тиофен)пропионамидин гидрохлорида;
(R,S)-2-(4-изобутилфенил)-N-[3''-(N'-пиперидино)пропил]пропионамидин дигидрохлорида;
(R,S)-2-(4-изобутилфенил)-N-метилпропионамидин гидрохлорида;
(R,S)-2-(3-бензоилфенил)-N-[3-(N,N-диметиламино)пропил]пропион амидин гидрохлорида;
(R,S)-2-(4-изобутилфенил)пропионамидин ацетата;
(R,S)-2-(4-изобутилфенил)-N-[3-(N,N-диметиламино)пропил]пропион амидина;
(R,S)-2-(4-изобутилфенил)-N-бензилпропионамидина;
(R,S)-3-[1-(4-изобутилфенил)этил]-5,6-дигидро-2Н-1,2,4-оксадиазина;
(R,S)-2-[1-(4-изобутилфенил)этил]-4,5-дигидро-2Н-1,3-имидазола и их фармацевтически приемлемые соли.

где Ar имеет то же самое значение, как описано в п.1, с амином формулы NHR, где R имеет то же самое значение, как описано в п.1.
Документы, цитированные в отчёте о поиске
Соли производных амидина и ингибитора циклооксигеназы, способ их получения и фармацевтические композиции на их основе
Комментарии