Способ получения (-)-n-метил-n-[4-(фенил-4-ацетаминопиперидин-1-ил)-2-(3,4-дихлорфенил)бутил]бензамида и его фармацевтически приемлемых солей - RU2097376C1
Код документа: RU2097376C1
Описание
Изобретение относится к способу получения правовращающего 2-(3,4-дихлорфенил)-4-гидроксибутиламина формулы I

Вещество (1) представляет собой ключевое промежуточное вещество при синтезе антагонистов тахикинина. Вещество (1) описано в заявках (европатенты N 0428434, N 0474561, N 0515240 и N 0559538).
В соответствии с указанными заявками, вещество (1) получают посредством разделения рацемата через его соль D -(-)-винной кислоты.
Установлено, что при обработке рацемической 3-циано-3-(3,4-дихлорфенил)пропионовой кислоты D -(-)-N-метилглюкамином происходит асимметрическое превращение второго порядка с образованием (-)-3-циано-3-(3.4-дихлорфенил)пропионовой кислоты, которая при энантиконсервантивном восстановлении бораном дает вещество (1).
Было найдено, что вещество формулы (I) может быть получено из 3,4-дихлорфенилацетонитрила путем взаимодействия с галоидацетатом щелочного металла, предпочтительно хлорацетата натрия, расщепления 3-циано-3-(3,4-дихлорфенил) пропионовой кислоты на месте и энантиконсервативного восстановления, как указано выше.
Таким образом, изобретение относится к способу получения (+)-2-(3,4-дихлорфенил)-4-гидроксибутиламина
формулы
(I),
который включает:
a) обработку 3,4-дихлорфенилацетонитрила формулы (II)

галоидацетатом щелочного металла в жидком аммиаке или в полярном апротонном растворителе, в присутствии сильного основания при температуре от -40 до +25oC;
b) обработку полученной рацемической 3-циано-3-(3,4-дихлорфенил)пропионовой кислоты формулы (III)

-(-)- -метилглюкамино, для того чтобы вся кислота (III) закристаллизовалась в виде -(-)- -метилглюкаминовой соли левовращающей кислоты;
c) обработку указанной соли сильной кислотой; и
d) энантиоселективное восстановление освобожденной 3-циано-3-(3,4-дихлорфенил)пропионовой кислоты формулы (IV)

бораном с получением производной (1).
Более конкретно, стадию (a) проводят с использованием галоидацетата щелочного металла, такого как хлорацетат натрия или калия, или бромацетат натрия или калия, в присутствии сильного основания, такого как амид натрия, трет-бутилат натрия или эталат натрия. Растворители, которые могут быть использованы, включают жидкий аммиак при пониженной температуре (от -40 до -30oC) или полярный апротонный растворитель, который является инертным в условиях этой реакции, такой как диметилсульфоксид или N,N-диметилформамид. Полученную таким образом цианокислоту (III) после 4 5 ч времени реакции выделяют посредством обработки водой или смесями воды с эфиром, например изопропиловым эфиром. Эта цианокислота может быть превращена в одну из солей.
Стадия (b) может быть осуществлена с выделенной цианокислотой (III) или еще без выделения, непосредственно после стадии (а) в растворителе, таком как спирт, предпочтительно этанол. D-(-)-глюкаминовая соль 3-циано-3-(3,4- дихлорфенил) пропионовой кислоты кристаллизуется непосредственно и может быть выделена.
Левовращающаяся кислота получается из ее соли путем обработки сильной кислотой, в соответствии со стадией (c) и выделяется посредством экстракции подходящим растворителем, таким как дихлорметан, дихлорэтан или 1,1,1-трихлорэтан. Кислота (IV) может быть превращена в одну из ее солей.
На стадии (d) энантиоконсервативное восстановление бораном, таким как гидрид бора или гидрид бора или диборан, необязательно в виде комплекса с тетрагидрофураном, или диметилсульфидом, осуществляется при комнатной температуре в растворителе типа эфира, таким как диоксан или тетрагидрофуран. После разложения избытка борана и выпаривания растворителя аминоспирт (I) выделяют путем удаления побочных продуктов с использованием последовательных обработок кислотой и затем основанием с последующей экстракцией подходящим растворителем, таким как дихлорметан, дихлорэтан или 1,1,1-трихлорэтан.
3-циано-3-(3, 4-дихлорфенил)пропионовую кислоту (III) приготавливают посредством взаимодействия хлорацетата натрия с 3,4-дихлорфенилацетонитрилом (II), например в жидком аммиаке в присутствии амида натрия по методике А.Г. Чигарева и Д. В.Иоффе (жур. Органическая химия, 1967, N 3, с. 85 88) или в присутствии другого очень сильного основания, такого как трет-бутилат натрия или калия в жидком аммиаке при -33oC или в безводном диметилсульфоксиде при комнатной температуре. Выход выделенной з-циано-3-(3,4-дихлорфенил)пропионовой кислоты составляет от 74 до 78% однако гораздо лучше, если продукт не выделяется, потому что достаточно, чтобы он взаимодействовал с D-(-)-N-метилглюкамином, недорогим промышленным продуктом, получаемым из D-глюкозы и метиламина (Каррер, Херкенрат, Helv. Chim Acta, 1937, т. 20, с. 37), для того, чтобы вся рацемическая цианокислота (III) закристаллизовалась в виде соли левовращающей кислоты. Выход является очень хорошим, составляя 190% в расчете на содержащийся в рацемате (III) левовращающий энантиомере. Растворителем кристаллизации может быть метанол, этанол, Целлозольв® или любой другой приемлемый растворитель. Температура расщепления находится между температурой кипения растворителя и 0oC. N-метилглюкамин должен присутствовать по меньшей мере в стехиометрическом количестве. Предпочтительно он используется в небольшом избытке.
После выделения 3-циано-3-(3,4-дихлорфенил)пропионовой кислоты из ее D-(-)-N-метилглюкаминовой соли путем обработки сильной кислотой, такой как соляная кислота, щавелевая кислота или ионообменная смесь сульфокислотного типа, ее подвергают энантиоконсервативному восстановлению бораном (энантиомерная чистота 99%).
Выход в этой реакции двойного восстановления составляет по меньшей мере 70% Боран может использоваться в виде димера (диборана B2H6), но предпочтительно он используется в более удобной форме, такой как комплекс с тетрагидрофураном или диметилсульфидом, причем последний комплекс продается как BMS.

Асимметричное превращение второго порядка на стадии (b) в соответствии со способом изобретения является неожиданным и составляет дополнительный признак изобретения, который таким образом относится, согласно другим признакам, к способу получения (-)-3-циано-3-(3,4-дихлорфенил)пропионовой кислоты, который включает обработку рацемической 3-циано-3-(3,4-дихлорфенил)пропионовой кислоты D-(-)-N-метилглюкамином и обработку полученной D-(-)-N-метилглюкаминовой соли (-)-3-(3,4-дихлорфенил)пропионовой кислоты сильной кислотой.
3-Циано-3-(3, 4-дихлорфенил)пропионовая кислота (III) и ее соли, и ее (-) изомер формулы (IV), и его соли являются новыми продуктами и представляют собой дополнительный признак изобретения.
Более конкретно D-(-)-N-метилглюкаминовая соль (-)-3-циано-(3,4-дихлорфенил)пропионовой кислоты представляют собой другой признак изобретения.
Полученные по способу изобретения вещества (I) могут выгодно использоваться для стереоселективного синтеза оптически чистых арилалкиламинов, которые являются антагонистами рецепторов нейрокинина.
В частности вещество (I) может применяться для приготовления арилалкиламинов, описанных в заявках (ЕР N 0428434, N 0474561, N 0515240, N 0559538) в соответствии с общей схемой описанной ниже в cхеме 1, в которой заместители B и D представляют собой все заместители аминированного кольца арилалкиламинов, описанных выше заявках на Европатенты, W, T и Z такие же, как описано в этих патентных заявках и Ar' является дихлорфенильной группой.
Схема 1

Значок "*" означает, что обозначенный этим символом атом углерода имеет определенную конфигурацию (+) или (-).
Предпочтительно вещество (1), полученное по способу изобретения, будет применяться для получения оптически чистых арилалкиламинов формулы (VI).

в которой Y представляет либо группу Cy-N, в которой -Cy представляет собой фенил, незамещенный или замещенный однократно или несколько раз одним из заместителей, выбранных из:
атома водорода, атома галогена, гидроксила,
C1 -C4 -алкокси, C1-C4-алкила, трифторметила, причем эти заместители являются одинаковыми или различными; групп C3-C7-циклоалкила; пиримидинильной группы или пиридильной группы;
либо группу Ar-(CH2)x-C(X), в которой Ar представляет собой фенил, незамещенный или замещенный однократно или несколько раз один из заместителей, выбранных из:
атом водорода, атома галогена, гидроксила,
С1-C4-алкокси-, C1-C4-алкила, трифторметила, причем эти заместители являются одинаковыми или различными; пиридильной группы; тиенильной группы;
x равно 0 или 1;
X представляет собой гидроксил. C1-C4-алкокси-, гидроксиалкил, в котором алкильная группа имеет 1 3 атома углерода; C1-C4-ацилокси; фенацилокси; карбоксил, C1-C4-карбалкокси; циано; аминоалкил, в котором алкилен имеет 1 3 атома углерода; группу -N(X)2, в которой группы X1 независимо представляют собой атом водорода, C1-C4-алкил; группу -NH-CO-Алк, в которой Алк представляет собой алкил C1-C6; группу Алк1-NH-CO-Алк'1 в которой Алк1 является алкиленом C1-C3 и Алк'1 является алкилом C1-C3; ацилом C1-C4; группой S-X2, в которой X2 представляет собой атом водорода или алкильную группу C1-C4; или альтернативно, X образует двойную связь с атомом углерода, с которым он связан и с соседним атомом углерода в гетероцикле;
Ar' представляет собой дихлорфенильную группу;
R является атомом водорода;
T представляет собой группу, выбранную из C-(=O)- и -C(=W)-NH, где W является атомом кислорода или серы и
Z представляет собой или атом водорода, или M или OM, когда T представляет собой группу -CO-, или M, когда T является группой -CW-NH; M представляет собой алкил C1-C6; фенилалкил, в котором алкил является группой C1 -C3, необязательно замещенной в ароматическом кольце галогеном, трифторметилом, алкилом C1-C4, гидроксилом, C1-C4-алкокси; пиридалкилом, в котором алкил является группой C1-C3; нафтилалкильной группой, необязательно замещенной в нафтильном кольце галогеном, трифторметилом, C1-C4 алкилом, гидроксилом, C1-C4-алкокси-; пиридтиоалкилом, в котором алкил является группой C1-C3; стирилом; необязательно замещенной моно-, ди- или трициклической ароматической или гетероароматической группой; или одной из солей с неорганическими или органическими кислотами.
Вещество формулы (VI), которые описаны в Европатенте N 0474561, были приготовлены в соответствии со схемой 1, в которой B-D<представлены в формуле (VI) группой V.
Вещество формулы (I), полученное по способу в соответствии с изобретением, особенно пригодны для получения (-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)-2-(3-, 4-дихлорфенил) бутил]бензамида или его фармацевтически приемлемых солей, таких как гидрохлорид или метансульфонат.
Это вещество обладает выгодными фармакологическими свойствами в качестве антагониста рецепторов нейрокинина A и является полезным, в частности при лечении любой патологии, зависящей от нейрокинина A.
Таким образом, дополнительный предмет изобретения представляет собой способ получения
(-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)-2-(3,
4-дихлорфенил)бутил] бензамида и его
фармацевтически приемлемых солей, отличающийся тем, что он включает:
a) взаимодействие
соединения формулы 1;

с функциональной производной бензойной кислоты;
b) взаимодействие полученного таким образом (-)-N-[2-(3, 4-дихлорфенил-4-гидрокси)бутил]бензамида формулы (VIII):

с дигидопираном,
c) взаимодействие полученного таким образом защищенного по кислороду соединения формулы (IX)

d) удаление защиты по кислороду в полученном таким образом соединения формулы (X)

e) обработку полученного таким образом соединения формулы (XI)

функциональной производной бензолсульфокислоты.
f)
взаимодействие полученного таким образом бензолсульфоната формулы (XII)

с 4-ацетиламино-4-фкенилпиперидином, и
g) выделение (-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)-2-(3,4-дихлорфенил)бутил] бензамида как такового, или по желанию, превращение его в одну из фармацевтически приемлемых солей.
Используемая на стадии (a) функциональная производная бензойной кислоты может быть собственно кислотой, активированной, например, дициклогексилкарбодиимидом, хлоридом кислоты, ангидридом, смешанным ангидридом или активным сложным эфиром кислоты.
Защита по кислороду на стадии (b) может быть проведена в присутствии кислоты, например метансульфокислоты.
Взаимодействие с диметилсульфатом на стадии (c) проводят в присутствии гидрида натрия в полярном апротонном растворителе, таком как диметилформамид.
Удаление защиты по кислороду на стадии (d) может быть проведено в кислой среде, например с использованием хлористоводородной кислоты или метансульфокислоты, а также протонной смолы, такой как Амберлист® 15, в метаноле.
В качестве функциональной производной бензолсульфокислоты, предпочтительно на стадии (e) используется хлорид или ангидрид кислоты.
Взаимодействие соединения формулы (XII) с 4-ацетиламино-4-фенилпиперидином на стадии (f) проводят, предпочтительно в полярном апротонном растворителе, таком как диметилацетамид, диметилформамид или ацетонитрил, в присутствии основания, такого как карбонат натрия или калия, триэтиламин или 4-диметиламинопиридин.
Полученный таким образом (-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)-2-(3, 4-дихлорфенил) бутил] бензамид выделяют в виде свободного основания или в виде солей, например, гидрохлорида, фумарата или сукцината. Также возможно выделять этот продукт, например, в виде соли фумаровой кислоты с превращением ее в другую соль с предварительной ее нейтрализацией и обработкой свободного основания кислотой, например, янтарной кислотой.
Когда (-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)-2-(3,4-дихлорфенил)бутил] бензамид получают в виде свободного основания, образование соли осуществляют путем обработки выбранной кислотой в органическом растворителе. Обработка свободного основания, например, растворенного в спирте, таком как изопропанол, раствором выбранной кислоты в том же самом растворителе дает соответствующую соль, которую выделяют с помощью традиционных методик. Например, таким образом получают гидрохлорид, гидробромид, сульфат, бисульфат, дигидрофосфат, метансульфонат, метилсульфат, оксалат, малеат, фумарат и нафталин-2-сульфонат.
В соответствии с этим способом, возможно объединение двух или более стадий, проводя их в одной емкости. Например, стадии (a) и (b) могут быть объединены, для того чтобы получить соединение формулы (VIII), исходя непосредственно из вещества (1).
Аналогично, N-метилирование и удаление защиты по кислороду, стадии (c) и (d), могут быть объединены, и соединены формулы (X) может быть получено без выделения соединения (IX).
Кроме того, окончательные стадии (e), (f) и (g) могут быть объединены.
Могут быть предусмотрены другие сочетания из более чем двух стадий, и кроме того, возможно осуществление всего процесса в одной емкости, что означает, что две или более стадии объединяются без выделения промежуточных соединений синтеза, таким образом обеспечивается упрощение способа.
Вещества формулы (I), полученные по способу этого изобретения, также
могут применяться для
приготовления оптически чистых арилалкиламинов формулы (VII)

в которой Ar1 представляет собой необязательно замещенную моно- ди- или трициклическую ароматическую или гетероароматическую группу;
T1 является непосредственной связью, гидроксиметиленовой группой, алкоксиметиленовой группой, в которой группа алкокси- представляет собой алкиленовую группу C1-C4 или C1-C5;
Ar' представляет собой дихлорфенильную группу;
R является атомом водорода;
Ams является радикалом

в которой X1, X2 и X3, вместе с атомом азота, с которым они связаны, образующие азабициклическую или азатрициклическую систему, необязательно замещенную фенильной группой; и
A⊖ является фармацевтически приемлемым анионом.
Вещества формулы (VII), которые описаны в заявке (EP N 0591040),
получают
по способу, который практически состоит во взаимодействии:
вещества формулы (I), которое
получают по способу изобретения, с соединением формулы
Ar1-T1
-CO-OH
в которой T1 и Ar1 такие, как определено выше, и
образовавшегося вещества формулы
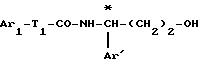
в которой Ar1 такой, как определено выше, с соединением формулы G Cl, в которой G является отщепляемой группой, такой как метил или бензолсульфонил, затем
полученного вещества формулы

с третичным амином формулы

в которой X1, X2 и X3 такие, как определено выше, в органическом растворителе при температуре между комнатной и 120oC и выделении образовавшегося продукта или иначе, если угодно, обменивают метансульфонатный анион образовавшейся четвертичной соли на другой фармацевтически приемлемый анион.
Вещество формулы (I), полученное по способу изобретения является особенно пригодным для приготовления хлористого (+)-1-[2-(3-3, 4-дихлорфенил-1-[(3-изопропоксифенил)-ацетил] пиперидин-3-ил]этил]-4-фенил-1-азоний-бицикло-[2,2,2]октана.
Следующие примеры иллюстрируют изобретение, однако не подразумевается ограничение.
Пример 1. Рацемическая 3-циано-3-(3,4-дихлорфенил)пропионовая кислота (III)
Смесь
18,6 г (0,1 моль) 3,4-дихлорфенилацетонитрила и 12 г (0,103 моль) высушенного
хлорацетата натрия, подвергают взаимодействию в течение 5 ч при комнатной температуре в 150 мл высушенного
диметилсульфоксида в присутствии 10,5 г (0,105 моль) трет-бутилата натрия. После этого
взаимодействия реакционную смесь выливают в 1 л воды со льдом и подкисляют соляной кислотой до значения pH
< 3. Цианокислоту экстрагируют этилацетатом, экстракт промывают до pH > 3,
сушат над сульфатом магния и концентрируют досуха. Остаток затвердевает в 1,2-дихлорэтане, давая 16,1 г
ожидаемого соединения (III), которое было охарактеризовано методом ПМР.
Пример
2. Рацемическая 3-циано-3-(3,4-дихлорфенил)пропионовая кислота (III)
Смесь 93 г (0,5 моль) 3,
4-дихлорфенилацетонитрила и 64 г (0,55 моль) хлорацетата натрия, подвергают взаимодействию в
течение 4 ч при температуре -33oC в 500 мл жидкого аммиака в присутствии 21 г (0,54 моль) амида
натрия. После выпаривания аммиака остаток обрабатывают водой и затем изопропиловым эфиром и
подкисляют соляной кислотой до значения pH < 3. Органическую фазу промывают водой до pH > 3,
отделяют посредством декантации, сушат над сульфатом магния и концентрируют досуха. Остаток
затвердевает в толуоле, он был охарактеризован методом ПМР, т.пл. 106oC.
Пример
3. Рацемическая 3-циано-3-(3,4-дихлорфенил)пропионовая кислота (III)
Смесь 186 г
(1
моль) 3,4-дихлорфенилацетонитрила, 126 г (1,05 моль) третбутилата натрия подвергают взаимодействию в
течение 4 ч при температуре -33oC в 1 л жидкого аммиака. По окончании реакции
выпаривают
аммиак, и остаток обрабатывают 500 мл воды со льдом и затем изопропиловым эфиром и подкисляют
соляной кислотой до значения pH < 3. Водную фазу выливают, а органическую фазу
промывают водой до
pH > 3, отделяют посредством декантации, сушат над сульфатом магния и концентрируют в
вакууме. Остаток затвердевает в 250 мл толуола, и цианокислоту отфильтровывают и сушат
при 50oC
в форвакууме, получая 190 г (выход 78%) ожидаемой 3-циано-3-(3,4-дихлорфенил)пропионовой
кислоты, т.пл. 104oC.
Продукт был охарактеризован методом ПМР
при частоте 200 мГц в
диметилсульфоксиде (ДМСО), ч/млн:
неразрешенные сигналы между 2,85 и 3, 1, 2
протона, -CH2-;
сложный сигнал при 4, 5, 1 протон, -CH-;
ароматические протоны между
7,4 и 7,75, 3 протона;
один кислотный протон при 12,8.
Пример 4. (-)-3-циано-3-(3,4-дихлорфенил)пропионовая кислота (IV)
Смесь 186 г (1
моль) 3,
4-дихлорфенилацетонитрила, 126 г (1,05 моль) хлорацетата натрия и 105 г (1,05 моль) трет-бутилата
натрия подвергают взаимодействию в течение 4 ч при температуре -33oC в 1 л
жидкого
аммиака.
По окончании реакции выпаривают аммиак, и остаток обрабатывают 500 мл воды со льдом и затем изопропиловым эфиром (500 мл) и подкисляют соляной кислотой до значения pH < 3. Водную фазу выливают, а органическую фазу промывают водой до pH > 3, отделяют посредством декантации, сушат над сульфатом магния и концентрируют в вакууме. Концентрат повторно растворяют в 2 л абсолютированного этанола, раствор нагревают и добавляют 292 г D-(-)-N-метилглюкамина. После кристаллизации продукт отфильтровывают, промывают этанолом и сушат в вакууме, получая 396 г N-метилглюкаминовой соли (-)-3-циано-(3,4-дихлорфенил)пропионовой кислоты.
[α] -14,7 (C 1, в метаноле).
Выход составил 91% в расчете на 3,4-дихлорфенилацетонитрил.
Полученную соль растворяют в 900 мл 1-нормальной соляной кислоты и экстрагируют 2 л дихлорметана. Органическую фазу промывают водой, отделяют посредством декантации, сушат над сульфатом магния и концентрируют. Продукт затвердевает в 500 мл циклогексана, давая 187 г ожидаемого продукта. Выход составил 76,5% в расчете на 3,4-дихлорфенилацетонитрил.
Т.пл. 98o C.
[α] -8,6 (C 1, в метаноле).
Энантиомерная чистота равна 99% по данным жидкостной хроматографии высокого разрешения.
Спектр ПМР при 200 мГц в ДМСО был таким же, что и для рацемата.
Пример 5. (+)-2-(3,
4-дихлорфенил)-4-гидроксибутиламин (1)
Добавляют 350 мл 1-молярного раствора
борана в
тетрагидрофуране к раствору 244 г (1 моль) (-)-3-циано-3-(3,4-дихлорфенил)пропионовой кислоты в 500 мл
тетрагидрофурана, охлажденному до 0oC. Когда выделение водорода прекратится,
при
20oC добавляют 650 мл раствора борана и затем 100 мл при 40oC. когда реакция
завершится, избыток борана разлагают добавлением метанола, и реакционную смесь концентрируют
досуха.
Концентрат растворяют в 500 мл воды, подкисляют соляной кислотой и дважды промывают 250 мл
толуола. Водную фазу подщелачивают гидроксидом натрия и экстрагируют дважды 400 мл дихлорметана.
Органическую
фазу промывают водой, отделяют посредством декантации, сушат над сульфатом магния и
концентрируют в вакууме, получая 159 г ожидаемого продукта (Выход 68%).
Хиральная чистота равна 99% по данным жидкостной хроматографии высокого разрешения (ЖХВР).
Продукт был охарактеризован методом ПМР при частоте 200 мГц в дейтерохлороформе, ч/млн:
неразрешенные сигналы
при 1, 8, 2 протона;
один синглет при 2, 4, 3H;
неразрешенные
сигналы между 2,65 и 2, 9, 3 протона;
неразрешенные сигналы между 3,65 и 3, 6, 2
протона;
ароматические протоны между 6,95 и 7,35, 3 протона;
[α] +9,8 (C
1, в метаноле);
т.пл. 80 81oC.
Пример 6. (+)-2-(3,
4-дихлорфенил)-4-гидроксибутиламин (1)
Повторяют методику примера 5, за исключением того, что применяют
комплекс
боран-диметилсульфид и рабочие температуры составляют 20 и затем 50oC. Получают аналогичный продукт с тем же выходом и такими же характеристиками.
Пример 7.
(-)-N-метил-N-[4-(4-фенил-4-ацетиламинопиперидин-1-ил) -2-(3,
4-дихлорфенил)бутил]бензамид
a) К раствору 5,85 кг (+)-2-(3,4-дихлорфенил)-4-гидроксибутиламина (1) и 3,0 кг триэтиламина в 25
мл
дихлорметана добавляют 3,52 кг хлористого бензоила,
растворенного в 15 л дихлорметана, при температуре ниже, чем 15oC. Смесь обрабатывают 20 л воды со льдом, затем водную фазу
декантируют,
экстрагируют 10 л дихлорметана и удаляют. Все
органические фазы объединяют, промывают 5%-ной соляной кислотой, затем 5%-ным водным раствором бикарбоната натрия, водой и сушат над
сульфатом натрия.
b) К полученному таким образом раствору, содержащему вещество (VIII), добавляют 2,5 кг дигидропирана, 10 г метансульфокислоты и смесь перемещают 3 ч при комнатной температуре (примерно 22oC). По завершении взаимодействия реакционную смесь концентрируют, концентрат растворяют в 30 л изопропилового эфира и дают раствору закристаллизироваться. Полученное таким образом вещество (IX) отфильтровывают, промывают изопропиловым эфиром и сушат в вакууме. Выход равен 85% в расчете на (1).
c) Раствор 6,8 г вещества (IX) в 10 л диметилформамида добавляют к суспензии 0,90 кг гидрида натрия (60%) в 5 л диметилформамида. По прекращении выделения водорода в смесь добавляют 3,3 кг диметилсульфата в 5 л толуола. Спустя 1 ч добавляют 45 л воды и затем гидроксид натрия до установления pH более, чем 6. Раствор экстрагируют дважды 25 л хлорметана и водную фазу удаляют. Органический слой промывают водой до pH 7 и концентрируют.
d) Указанный выше концентрат, содержащий вещество (X), растворяют в 25 л метанола и добавляют к раствору 0,5 кг смолы Амберлист® 15. Затем смолу отфильтровывают, промывают 5 л метанола и фильтрат концентрируют. Остаток растворяют в 25 л толуола и дают раствору закристаллизоваться. Полученное таким образом вещество (XI) отфильтровывают, промывают 5 л толуола и сушат в вакууме. Выход равен 85% в расчете на (IX).
e) К раствору 7,06 кг вещества (XI) и 2,7 кг триэтиламина в 25 л толуола при 60oC добавляют 7,06
г хлористого бензолсульфонила. Реакционную
смесь нагревают при 65oC
в течение 15 мин, затем 3 ч при 45oC. Когда реакция завершена, смесь охлаждают до 10oC, затем в
не добавляют 20 л воды. Смесь перемешивают 5
мин, затем декантируют, и водную
фазу удаляют. Органическую фазу перемешивают с 20 л воды, содержащей 6,2 л раствора гидроксида натрия (400 г/л), до тех
пор пока не произойдет полный гидролиз избытка
хлористого бензолсульфонила (3 ч
при комнатной температуре). Затем водную фазы удаляют, органическую фазу сушат над сульфатом натрия и концентрируют в
вакууме. Концентрат содержит вещество формулы
(XII), полученное с выходом 80
100%
f) и g) Часть концентрата, полученного на стадии (e), соответствующая 4,93 кг (± 200 г) вещества по
расчету ЖХВР, и 2,20 кг
4-ацетиламино-4-фенилпиперидина в ацетонитриле кипятят
с обратным холодильником в присутствии 2 кг карбоната калия. По завершении взаимодействия реакционную смесь
концентрируют в вакууме, остаток
растворяют в 50 л воды и 20 л дихлорметана. Смесь
перемешивают 5 мин и декантируют; водную фазу экстрагируют 5 л дихлорметана и удаляют. Все органические фазы
объединяют, промывают 2 раза 50 л
3-нормальной соляной кислоты, затем дважды 25 л воды, 1
раз 25 л 2-нормальным раствором гидроксида натрия и окончательно водой, пока значение pH не станет ниже 10.
После высушивания над сульфатом
натрия растворитель отгоняют в вакууме, остаток растворяют
25 л
ацетона, и раствор добавляют к суспензии 1,275 г фумаровой кислоты в 25 л ацетона при кипячении с
обратным холодильником. Дают смеси
охладиться до комнатной температуры, и отфильтровывают
монофурат (-)-N-метил-N-[4-(4-ацетиламино-4-фенилпиперидин-1-ил)-2-(3,4- дихлорфенил)бутил] бензамида,
промывают его ацетоном и сушат в вакууме.
Выход равен 80%
Смесь 6,68 кг полученного
таким образом продукта в 30 л дихлорметана и 20 л воды подщелачивают, добавляют при перемешивании 2,5 л
водного раствора, содержащего гидроксид
натрия (400 г/л). После 25 мин перемешивания,
реакционную смесь декантируют, водную фазу экстрагируют 10 л дихлорметана и удаляют. Органические фазы
объединяют, промывают водой до pH ниже, чем 8,
сушат над сульфатом натрия и концентрируют в
вакууме. Таким образом получают основание (-)-N-метил-N-[4-(4-ацетиламино-4-фенилпиперидин-1-ил)-2-(3,
4- дихлорфенил)бутил]-бензамид.
Полученное таким образом основание растворяют в 15 л ацетона, и этот раствор выливают в раствор 1,24 кг янтарной кислоты в 25 л ацетона. После охлаждения отфильтровывают полученный таким образом моносукцинат (-)-N-метил-N-[4-(4-ацетиламино-4-фенилпиперидин-1-ил)-2- (3,4-дихлорфенил)(бутил] бензамида, промывают его 10 л ацетоном и сушат в вакууме при 50oC. Выход равен 90%
Реферат
Использование: в медицине в качестве антагонистов рецепторов нейрокинина. Сущность изобретения: продукт: (-)-N-метил-N-/4-фенил-4-ацетоминопиперидин-1-ил)-2-(3,4- дихлорфенил)бутил/бензамид и его фармацевтически приемлемые соли. Реагент 1: (+)HO-CH2CH2-CH(C6H3 Cl2)-CH2-NH2 (I). Реагент 2: функциональное производное бензойной к-ты, продукт их взаимодействия обрабатывают дигидропираном, полученный продукт подвергают взаимодействию с диметилсульфатом, после удаления защиты по кислороду, полученное соединение обрабатывают последовательно производным бензосульфокислоты и 4-ацетиламино-4-фенилпиперидином. 6 з.п.ф-лы.
Формула

подвергают взаимодействию с функциональным производным бензойной кислоты, b) полученный таким образом (-)-N- [2-(3,4-дихлорфенил-4-гидрокси)бутил]бензамид формулы VIII

подвергают взаимодействию с дигидропираном, c) полученное таким образом защищенное по кислороду соединение формулы IX

подвегают взаимодействию с диметилсульфатом, d) удаляют защиту по кислороду в полученном таким образом соединении формулы X

в кислой среде, е) обрабатывают полученное таким образом соединение формулы XI

функциональным производным бензолсульфокислоты, f) полученный таким образом бензолсульфонат формулы XII

подвергают взаимодействию с 4-ацетиламино-4-фенилпиперидином и g) выделяют (-)-N-метил-N-[4-(4-фенил-4-ацетаминопиперидин-1-ил)- 2-(3,4-дихлорфенил)бутил] бензамид как таковой или, по желанию, превращают его в одну из фармацевтически приемлемых солей.
Комментарии