Защищенные 3,5-дигидрокси-2,2-диметилвалеронитрилы для синтеза эпотилонов и их производных, способ их получения и их применение - RU2303590C2
Код документа: RU2303590C2
Описание
Настоящее изобретение относится к объектам, охарактеризованным в формуле изобретения, а именно к новым промежуточным продуктам и способу их получения, а также к их применению. Предлагаемый в изобретении способ получения новых промежуточных продуктов, основанный на использовании недорогих исходных материалов, позволяет получать промежуточные продукты в виде чистых энантиомеров с высокой химической чистотой и с хорошим выходом и может быть реализован в промышленном масштабе.
Изобретение предлагается для применения при синтезе структурного звена А природных и синтетически модифицированных эпотилонов или их производных. Эпотилоны представляют собой 16-членные макролидные кольца, выделенные из культур миксобактерии Sorangium Cellosum, и являются представителями класса перспективных противоопухолевых средств, эффективность которых против целого ряда раковых заболеваний была подтверждена в ходе их тестирования. Различные методы синтеза описаны в обзорной статье J.Mulzer и других авторов, опубликованной в Journ. Org. Chem. 65, 2000, cc.7456-7467

В литературе наряду с природными эпотилонами описаны и многие синтетические производные эпотилонов, варьирующиеся преимущественно по остаткам М и Т. М представляет собой в этих случаях большей частью гетероциклический остаток. При осуществлении большинства вариантов синтеза природных эпотилонов, равно как и синтетических производных эпотилонов, используют основное структурное звено А, которое образовано атомами углерода C5-C10 в макролиде. В этом структурном звене А (см. далее) C1 является атомом C5 в макролиде, а С6 - атомом С10 в макролиде и т.д.

В этом структурном звене Т представляет собой С1-С4алкил или алкенильный остаток, Sg1 и Sg2 обозначают обычные известные специалистам защитные группы, такие, например, как ТБДМС-группа (ТБДМС обозначает трет-бутилдиметилсилил).
Один из возможных вариантов получения структурного звена А описан, например, в заявке WO 00/58254 (на имя University of Wisconsin). В этой заявке предлагается синтез, осуществляемый исходя из β-кетоэфиров, которые по многостадийному механизму можно переводить в структурное звено А. Хиральность достигается асимметрическим гидрированием соответствующего β-кетоэфира по методу Нойори (Noyori):

Трансформацию эфирной группы в кетон можно реализовать в этих случаях только в несколько последовательно проводимых стадий. При этом после защиты 1- и 3-гидроксигруппы эфирную группу (атом С5) восстанавливают до спирта, далее осуществляют окисление до альдегида, затем присоединением алкильного остатка к алкилмагнию, соответственно алкиллитиевому соединению по реакции Гриньяра получают вторичный спирт, который затем окисляют. Для получения из эфира кетона требуется в общей сложности 8 стадий. Непосредственное химическое превращение эфира протекает не избирательно, поскольку получаемый промежуточный продукт участвует в дальнейшей реакции. На приведенной ниже схеме показан весь описанный синтез в его последовательности:

Еще один метод построения основного структурного звена А описан у В.Paniker и др. в Tetrahedron 56, 2000, сс.7859-7868. В этой публикации указывается, что реакция альдольного типа с хиральным структурным элементом протекает с малой степенью избирательности. В публикации описывается синтез с образованием хирального центра при атоме С3, осуществляемый в отсутствие необходимости применять N-метилтиоацетилоксазолидинон по многостадийному механизму с повышенной диастереомерной избирательностью с помощью енолята бора. Для достижения приемлемой диастереомерной избирательности требуется замещение метилтиогруппы, при этом после реакции альдольного типа тиоэфир отщепляют.
Из уровня техники известен далее синтез, при котором для реакции Гриньяра используют сложный фениловый эфир (см. R.E.Taylor, Y.Chen, Org. Lett. 3 (14), 2001, сс.2221-2224). Достигаемый при этом выход составляет, как указано в публикации, 77%. В примере, описанном у A.Fürstner в Chem. Comm., 2001, сс.1057-1059, продукт получают с выходом 67%. Таким образом, выход, обеспечиваемый реакцией Гриньяра из уровня техники, заметно ниже по сравнению с выходом, достигаемым согласно настоящему изобретению.
В Journ. Org. Chem. 65, 2000, сс.7456-7467, описан далее асимметрический синтез β -кетоэфира, при этом один из вариантов проводят по асимметрическому механизму по реакции альдольного типа. В качестве катализатора в этом методе используют D-Ts-валин, который можно получить из дорогостоящей аминокислоты D-валин. Данный метод обеспечивает ее-показатель (энантиомерный избыток), равный 90%. В качестве еще одного примера, иллюстрирующего этот метод, у R.E.Taylor и Y.Chen в Org. Lett. 3 (14), 2001, сс.2221-2224, описана асимметрическая реакция альдольного типа, дающая выход в 71%.
В завершение обзора уровня техники можно назвать еще один метод получения двузащищенного ТБДМС-группой структурного звена А-этилкетона, описанного у Nicolaou в Chem. Eur. Journ. 6, 2000, сс.2783-2800.
Исходя из вышеизложенного, в основу настоящего изобретения была положена задача получить пригодный для универсального применения исходный промежуточный продукт общей формулы I, а также оптически чистые антиподы общих формул Ia и Ib

В этих формулах
R1 и R2 могут иметь идентичные либо разные значения и независимо друг от друга представляют собой спиртовую защитную группу, например бензил, 4-метоксибензил, 3,4-диметоксибензил, ТГП, ТБДМС, ТМС, ТЭС, ТИП, ТБДФС, МЭМ, MOM, аллил, тритил,
или в случае, когда R1 и R2 соединены мостиковой связью, представляют собой кетальную защитную группу, такую, например, как

Соединения представленных выше формул I, Ia и Ib предусматриваются для получения основного структурного звена А, предназначенного для общего синтеза эпотилонов. В этих целях соединения общей формулы I подвергают химическому превращению согласно следующей схеме и более подробно описанному ниже.

Реакции по превращению соединений общей формулы I, равно как и их антиподов формул Ia и Ib, в кетоны АК осуществляют взаимодействием с метиллитием или метальными соединениями Гриньяра по стандартным, известным специалистам методам, получая в результате последующей водной переработки требуемый кетон. Путем последующего алкилирования соответствующим алкилом или алкенилгалогенидом формулы Т-гал (гал обозначает Cl, Br, I или тозилат, мезилат, трифлат и т.д.) при добавлении соответствующего основания образуют указанное структурное звено А.
Вместе с тем существует также возможность получать структурное звено А прямым путем, подвергая амиды общей формулы I непосредственно взаимодействию с металлоорганическими соединениями, такими, например, как соединение лития Li-СН2-Т, и проводя затем соответствующую водную переработку.
Описанные реакции протекают, как правило, без каких-либо затруднений и позволяют получать структурные звенья А с высоким выходом. С учетом этого возросла потребность в разработке предусмотренного для реализации в промышленном масштабе способа, который позволял бы получать в качестве промежуточного продукта основное структурное звено А, пригодное для универсального применения при общем синтезе эпотилонов.
Наряду с высоким выходом получаемых структурных звеньев А следует отметить и такое преимущество, как относительно легкая доступность соединений общей формулы I, т.е. возможность их беспроблемного получения из относительно недорогих исходных материалов. Кроме того, в отличие от известных из литературы эфиров и кетонов предлагаемые в изобретении соединения отличаются устойчивостью при хранении и по мере необходимости их можно подвергать химическому превращению на различных этапах осуществляемого синтеза. Соединения общей формулы I представляют собой в основном кристаллические твердые вещества, и их можно очищать путем кристаллизации. Благодаря этому обеспечивается возможность получать продукт с высоким химическим и оптическим выходом (ее>98%).
Положенная в основу настоящего изобретения задача решается благодаря предлагаемым в нем новым соединениям общих формул I, Ia и Ib

где R1 и R2 могут иметь идентичные либо разные значения и независимо друг от друга представляют собой спиртовую защитную группу, такую, например, как бензил, 4-метоксибензил, 3,4-диметоксибензил, ТГП, ТБДМС, ТМС, ТЭС, ТИП, ТБДФС, МЭМ, MOM, аллил или тритил,
или в случае, когда R1 и R2 соединены мостиковой связью, представляют собой кетальную защитную группу, такую, например, как
Для получения предлагаемых в изобретении соединений можно предложить в общей сложности 4 варианта:
Вариант I (общая методика получения с помощью реакций альдольного типа)
а) В том случае, когда R1 и R2 представляют собой кетальную защитную группу или когда R1 идентичен R2, соединения общей формулы I можно получать из соединений общей формулы II, 2,2-диметил-3,5-дигидроксивалеронитрила,

по известным специалистам методам из химии защитных групп; так, например, получение и отщепление подобных групп описано у P.J.Kocienski в "Protecting Groups", изд-во Georg Thieme Verlag, Stuttgart, New York, 1994, и в Houben Weyl, 4-е изд., том VI/1b, с.737, изд-во Thieme, Stuttgart, 1984.
б) В том случае, когда R1 и R2 не представляют собой кетальную защитную группу, но вместе с тем могут иметь идентичные либо разные значения, соединения общей формулы I можно непосредственно получать из соединений общей формулы III за счет введения защитной группы R2 по известным из литературы методам (см. J.Mulzer и др., Journ. Org. Chem. 65, 2000, cc.7456-7467).
Соединения общей формулы II можно получать из соединений общей формулы III

где R1 представляет собой одну из указанных выше защитных групп, за счет отщепления этой защитной группы R1 по методу, известному специалистам из практики отщепления защитных групп от спиртов (см. P.J.Kocienski в "Protecting Groups", изд-во Georg Thieme Verlag, Stuttgart, New York (1994) и в Houben Weyl, 4-е издание, том VI/1b, с.737, изд-во Thieme, Stuttgart (1984)).
Соединения общей формулы III можно в свою очередь получать из соединений общей формулы IV

взаимодействием с соединением общей формулы V - 2-метилпропионитрилом
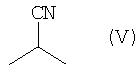
в которой R1 имеет указанные выше значения, по известной специалистам методике проведения альдольной конденсации.
Что касается соединений общей формулы IV, то их получение специалистам известно, при этом R1 представляет собой ТГП (см. JOC 49, 1984, cc.2301-2309), бензил (см. Journ. Chem. Soc. Perk. Trans 1, 2000, cc.2429-2454) или ТБДМС (см. JOC 65, 2000, cc.7456-7467).
Соединение формулы V, т.е. 2-метилпропионитрил, является коммерчески доступным продуктом.
Вариант II (получение оптически активных промежуточных продуктов общей формулы Ia)

При получении оптически активных соединений общей формулы Ia работают аналогично тому, как описано в варианте I. Исходя из оптически активного промежуточного продукта общей формулы IIa и IIIa

получают соединения общей формулы Ia.
Соединения общей формулы IIa получают аналогичным путем из оптически активного соединения-предшественника общей формулы IIIa

Оптически активные соединения общей формулы IIIa получают следующим образом:
1. Рацемическое соединение общей формулы III разделяют на хиральной фазе (литература: G.Roussel, P.Piras, Chirabase, Pure and Applied Chemistry 65, 1993, cc.235-244) прежде всего по SMB-технологии: (A.Seidel-Morgenstern и др., Chromat. A 827/2, 1998, cc.175-191).
2. Исходя из рацемического спирта общей формулы III по известным специалистам методам этерификации получают сложные эфиры общей формулы VI

в которой R3 представляет собой C1-С6алкильную группу или аллильную, фенильную либо бензильную группу, и затем указанные эфиры ферментативными или микробиологическими методами омыляют с энантиоселективностью. Образующийся спирт по своему Rf-показателю заметно отличается от используемого сложного эфира, что позволяет без проблем оба эти соединения разделять, например, посредством колоночной хроматографии.
3. Под действием хиральных катализаторов осуществляют альдольную конденсацию, заключающуюся в том, что соединения общей формулы IV и V подвергают химическому превращению с использованием в этих целях каталитического или стехиометрического количества соответствующего хирального катализатора альдольной конденсации:

Литература: см., например, Journ. Org. Chem. 65, 2000, cc.7456-7467.
4. По известным специалистам методам осуществляют хиральное восстановление кетона общей формулы VII

Литература: Noyori и др., Journ. Am. Chem. Soc. 109, 1987, с.5850; Noyori и др., Journ. Am. Chem. Soc. 110, 1988, с.629; R.C.Larock, "Comprehensive Organic Transformations", изд-во VCH Publishers New York, 1989, ISBN 0-89573-710-8, cc.540-548.
Соединения общей формулы VII, где R1 имеет указанные выше значения, можно получать взаимодействием соединения общей формулы V с соединениями общей формулы VIII

в которой Nu обозначает уходящую группу, такую как Cl, Br, имидазол, -OPh, -O-C6H4NO2, -O-С1-С4алкил и т.д. Указанное взаимодействие осуществляют по известной специалистам методике.
Получение соединений общей формулы VIII описано в литературе: см. Journ. Med. Chem., 1999, cc.706-721.
В некоторых случаях оказалось целесообразным получать соединения общей формулы VII путем окисления из рацемических спиртов общей формулы II по известным специалистам методам окисления (например, окислением по Сверну, PDC, РСС и т.п.).
В некоторых случаях оказалось целесообразным подвергать соединение общей формулы V взаимодействию с пропиолактоном с получением в результате соединений общей формулы IX

Соединения общей формулы IX за счет введения защитных групп по известным специалистам методам можно очень легко переводить в соединения общей формулы VII (см. P.J.Kocienski, "Protecting Groups", изд-во Georg Thieme Verlag, Stuttgart, New York, 1994, и Houben Weyl, 4-е изд., том VI/1b, изд-во Thieme, Stuttgart, 1984, с.737).
Вместе с тем существует также возможность исходя из соединений общей формулы IX получать соединения общей формулы IIa

за счет хирального восстановления кетогруппы с помощью химических или микробиологических методов (см., например, Journ. Org. Chem. 50, 1985, с.127, или Journ. Chem. Soc., Chem. Commun., 1987, с.1368).
Вариант III
Соединения общей формулы Ia
можно получать также за счет введения защитных групп по известным из литературы методам, используемым при введении спиртовых защитных групп, из соединений общей формулы Х

(см. указанную выше литературу касательно введения защитных групп).
Соединения общей формулы Х можно в свою очередь получать из соединений общей формулы XI

в которой R4 представляет собой метильную, этильную или бензильную группу, путем восстановления сложных эфиров по известным специалистам методам.
Соединения же общей формулы XI можно получать из соединений общей формулы XII

в которой R4 представляет собой С1-С6алкильную, метильную, этильную, трет-бутильную, фенильную или бензильную группу, за счет введения защитной группы R2 по известным специалистам методам (см. выше).
Соединения общей формулы XII можно получать в свою очередь из β-кетоэфиров общей формулы XIII

по методам хирального восстановления (химическим или ферментативным путем).
Соединения общей формулы XIII получают исходя из соединений общей формулы XIV взаимодействием с соединениями общей формулы V

Соединения общей формулы XIV известны из литературы или их можно получать также взаимодействием соединений общей формулы XIIIa с соединениями общей формулы XIIIb

При этом Nu обозначает указанную выше уходящую группу, Q обозначает атом водорода или СООН-группу. Если Q представляет собой атом водорода, то соединение XIIIa депротонируют с помощью органического основания, такого, например, как ДАЛ (диизопропиламид лития), после чего по известным специалистам методам подвергают взаимодействию с активированным производным кислоты. Если же Q представляет собой СООН-группу, то в этом случае используют методы конденсации полуэфиров малоновой кислоты, как это описано, например, в Journ. Am. Chem. Soc. 121, 1999, cc.7050-7062; Synth. Commun. 27, 1997. cc.3227-3234.
Соединения общей формулы XIIIa являются коммерчески доступными продуктами и поставляются, в частности, фирмой Aldrich.
Соединения общей формулы XIIIb получают по методике, описанной у R.C. Larock в "Comprehensive Organic Transformations", изд-во VCH Publishers New York, 1989, ISBN 0-89573-710-8, cc.963-964.
В некоторых случаях оказалось целесообразным диолы общей формулы IIa

получать непосредственно из соединений общей формулы XII

путем восстановления сложноэфирной группы по указанным выше методам.
При получении рацемического диола общей формулы II можно исходить также из β-кетоэфиров общей формулы XIII
используя при этом обычные методы восстановления сложных эфиров и кетонов.
Вариант IV
При создании изобретения было установлено, что для получения оптически активных диолов общей формулы IIa в некоторых случаях целесообразно осуществлять хроматографическое разделение или кристаллизацию диастереомерных кеталей общих формул XIVa и XIVb

в которых А обозначает остаток соответствующего оптически активного кетона, например (-)-ментон, (-)-камфора и т.п., и затем по известным специалистам методам, используемым в химии защитных групп, отщеплять кетальную группу.
Диастереомерные кетали 1,3-диолов общих формул XIVa и XIVb получают из рацемического диола общей формулы II взаимодействием с хиральными кетонами по известным из литературы методам. Литература: Т.Harada и др., Journ. Org. Chem. 57, 1992, cc.1412-1421.
Очевидной является также возможность получения соответствующих энантиомерных соединений общей формулы Ib при использовании в этих целях катализаторов изомеризации или каких-либо иных ферментативных систем

В равной степени существует возможность при использовании промежуточных продуктов общей формулы IIIb

путем инверсии гидроксильной группы, например, по методике Мицунобу (литература: О.Mitsunobu, Synthesis, 1981, cc.1-28) получать соответствующие энантиомеры.
Из используемых в синтезе защитных групп R1 и R2 особенно предпочтительными являются бензильная группа и ТБДМС-группа. В тех же случаях, когда R1 и R2 представляют собой кетальную защитную группу, особенно предпочтительна группа -(С(СН3)2)-.
Из числа представленных различных вариантов синтеза особенно предпочтительными являются таковые, которые предназначены для образования ахиральных соединений-предшественников. К ним относятся следующие:
1. Получение соединений общей формулы VII из промежуточных продуктов общих формул V и VIII, где R1 представляет собой бензил, a Nu обозначает Cl

2. Получение соединений общей формулы XIII из соединений общих формул V и XIV, где R4 представляет собой этил, a Nu обозначает Cl

3. Получение соединений общей формулы VII путем альдольной конденсации и последующим окислением. При этом R1 представляет собой бензил, а Nu обозначает Cl

4. Получение соединений общей формулы IX (Y обозначает диметиламиногруппу)

Для получения хиральных соединений-предшественников особенно предпочтительными являются следующие операции:
1. Хиральная альдольная конденсация с использованием хирального катализатора

2. Энантиоселективное омыление ацетата с помощью соответствующего фермента

3. Хиральное восстановление β-кетонитрила (по типу реакции Нойори)

4. Хиральное восстановление β-кетоэфира с последующим восстановлением

Предлагаемые в изобретении соединения предпочтительно получать в описанной ниже последовательности.
1. Получение ацетонкеталей

2. Получение соединения, защищенного диТБДМС-группой

Ниже получение предлагаемых в изобретении соединений и используемые в этих целях способы более подробно поясняются на примерах осуществления изобретения, которые не ограничивают его объем.
Пример 1
Пример 1а
5-бензилокси-2,2-диметил-3(R, S)-гидроксипентаннитрил
К раствору ДАЛ (диизопропиламида лития) (полученного из 33,64 г, (79,17 ммоля) н-бутиллития, 15%-ного в гексане (1,6-молярный) и 80,1 г (79,17 ммоля) диизопропиламина) по каплям добавляют при -65°С 5,47 г (79,17 ммоля) нитрила изомасляной кислоты и в течение 20 мин перемешивают при -65°С. Затем по каплям (в течение 60 мин) добавляют раствор из 10 г (60,9 ммоля) 3-бензилокси-1-пропанальдегида в 20 мл ТГФ. Температуру при этом поддерживают на уровне -65°С. После 1-часового перемешивания смесь нагревают до -20°С, а затем по каплям добавляют раствор 20%-ной серной кислоты и температуре дают повыситься до +10°С. Далее добавляют 50 мл МТБ-эфира (метил-трет-бутилового эфира) и органическую фазу отделяют. Сначала органическую фазу промывают водой, а затем насыщенным раствором гидрокарбоната натрия. В завершение повторно промывают водой и концентрируют досуха в вакууме.
Выход: 13,1 г (92% от теории), бесцветное масло.
Пример 1б
5-бензилокси-2,2-диметил-3(R,S)-ацетоксипентаннитрил
К раствору из 25,6 г (109,7 ммоля) 5-бензилокси-2,2-диметил-3-гидроксипентаннитрила (соединения, указанного в заголовке примера 1а), 14,43 г (142,64 ммоля) триэтиламина и 200 мг 4-диметиламинопиридина (ДМАП), растворенного в 128 мл МТБ-эфира, добавляют при 0°С 14,56 г (42,64 ммоля) ангидрида уксусной кислоты и перемешивают в течение 5 ч при комнатной температуре. Затем реакционную смесь сливают в 2 л смеси льда и воды и дважды экстрагируют МТБ-эфиром его порциями по 300 мл. Объединенные МТБ-фазы сначала один раз промывают 300-ми мл 5%-ной соляной кислоты, а затем водой. В завершение упаривают досуха в вакууме.
Выход: 28,82 г (95% от теории), бесцветное масло.
Пример 1в
5-бензилокси-2,2-диметил-3(S)-гидроксипентаннитрил
10 г (36,31 ммоля) 5-бензилокси-2,2-диметил-3(R,S)-ацетоксипентаннитрила (соединения, указанного в заголовке примера 1б) вводят в буферный раствор, полученный из 0,88 г дигидрофосфата калия и 1,82 г динатрийгидрофосфата в 250 мл воды. Затем добавляют 5 г фермента Lipase AYS "Amano" (поставщик фирма Amano) и перемешивают в течение 24 ч при 40°С. Добавлением 2,062 г динатрийгидрофосфата значение рН устанавливают на 7, после чего через 12 ч, контролируя при этом ход реакции посредством ЖХВР (жидкостная хроматография высокого разрешения), продолжают перемешивание, пока на пик (R)-ацетата не придется менее 1% от всей площади под кривой. Последующая переработка состоит в следующем. Сначала дважды экстрагируют 200-ми мл этилацетата. Затем органические фазы объединяют и упаривают досуха в вакууме. Очистку проводят хроматографией на силикагеле (элюент: гексан/этилацетат с градиентом соотношения). В результате получают одну фракцию, содержащую 4,2 г (45% от теории) 5-бензилокси-2,2-диметил-3(R)-гидроксипентаннитрила, и вторую фракцию, содержащую 4,8 г (48% от теории) 5-бензилокси-2,2-диметил-3(S)-ацетоксипентаннитрила.
4,8 г (17,5 ммоля) 5-бензилокси-2,2-диметил-3(S)-ацетоксипентаннитрила из второй фракции растворяют в 50 мл метанола и смешивают с 1,4 г (35 ммолей) NaOH. Затем перемешивают в течение 3 ч при 25°С, сливают в 200 мл воды, дважды экстрагируют МТБ-эфиром его порциями по 200 мл, сушат над сульфатом натрия и концентрируют.
Выход: 4 г (47% от теории) 5-бензилокси-2,2-диметил-3(S)-гидроксипентаннитрила в виде бесцветного масла.
Пример 1г
5-гидрокси-2,2-диметил-3(S)-гидроксипентаннитрил
К 11, 13 г (47,70 ммоля) 5-бензилокси-2,2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1в), растворенных в 100 мл тетрагидрофурана, добавляют 16 г катализатора Перлмана (Pearlman) (Pd(OH)2 на угле, 20%-ный). Затем гидрируют в течение 7,5 ч при давлении 10 бар и комнатной температуре. Далее отфильтровывают от катализатора и фильтрат досуха концентрируют в вакууме.
Выход: 6,73 г (98% от теории) бесцветного вязкого масла.
Пример 1д
3(S)-(3,5)-ацетондиметилкеталь-2,2-диметилпентаннитрил
6,73 г (47 ммолей) 5-гидрокси-2, 2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1 г) растворяют в 27 мл ацетондиметилкеталя и затем добавляют 546 мг камфор-10-сульфоновой кислоты. Смесь нагревают до 50°С и поддерживают на этом уровне в течение 15 ч. Далее концентрируют досуха в вакууме, остаток растворяют в 200 мл метиленхлорида и промывают сначала насыщенным раствором гидрокарбоната натрия, а затем насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом натрия и концентрируют досуха в вакууме. При стоянии полученное масло кристаллизуется.
Выход: 5,55 г (77% от теории), бесцветное кристаллическое твердое вещество.
Пример 2
3(S)-3,5-ди-трет-бутилдиметилсилилокси-2,2-диметилпентаннитрил
К раствору из 3 г (20,95 ммоля) 5-гидрокси-2, 2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1 г) в 20 мл диметилформамида добавляют 7,13 г (104,75 ммоля) имидазола и 7,9 г (52,37 ммоля) трет-бутилдиметилсилилхлорида и перемешивают в течение 16 ч при комнатной температуре. Затем раствор сливают в 200 мл воды и дважды экстрагируют циклогексаном его порциями по 50 мл. Органические фазы объединяют и упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 7,39 г (95% от теории) бесцветного вязкого масла.
Пример 3
3(S)-3, 5-циклогексанондиметилкеталь-2,2-диметилпентаннитрил
К раствору из 3 г (20,95 ммоля) 5-гидрокси-2,2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1 г) в 30,21 г (0,2095 моля) циклогексанондиметилкеталя добавляют 10 мг n-толуолсульфоновой кислоты и перемешивают в течение 6 ч при 100°С. Затем раствор сливают в 200 мл воды и дважды экстрагируют этилацетатом его порциями по 50 мл. Органические фазы объединяют и упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 4,21 г (90% от теории) бесцветного вязкого масла.
Пример 4
3(S)-3,5-бензальдегиддиметилацеталь-2,2-диметилпентаннитрил
К раствору из 3 г (20,95 ммоля) 5-гидрокси-2, 2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1 г) в 20 мл диметилформамида добавляют 31,9 г (0,2095 моля) бензальдегиддиметилацеталя и 50 мг n-толуолсульфоновой кислоты и перемешивают в течение 16 ч при 100°С. Затем раствор сливают в 200 мл воды и дважды экстрагируют этилацетатом его порциями по 50 мл. Органические фазы объединяют и упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 4,26 г (88% от теории) бесцветного вязкого масла.
Пример 5
3(S)-3, 5-дихлордифенилсилан-2,2-диметилпентаннитрил
К раствору из 3 г (20,95 ммоля) 5-гидрокси-2,2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 1 г) в 20 мл диметилформамида добавляют 3,14 г (46,09 ммоля) имидазола и 5,83 г (23,05 ммоля) дихлордифенилсилана и перемешивают в течение 16 ч при комнатной температуре. Затем раствор сливают в 200 мл воды и дважды экстрагируют метиленхлоридом его порциями по 50 мл. Органические фазы объединяют и упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 5,76 г (85% от теории) бесцветного вязкого масла.
Пример 6а
5-трет-бутилдиметилсилил-2,2-диметил-3(R,S)-гидроксипентаннитрил
К раствору ДАЛ (полученному из 28,6 г (66, 99 ммоля) н-бутиллития (15%-ного, 1,6-молярного) и 6,82 г (66,99 ммоля) диизопропиламина) по каплям добавляют при -65°С 4,62 г (66,99 ммоля) нитрила изомасляной кислоты и перемешивают в течение 20 мин при -65°С. Затем в течение 60 мин по каплям добавляют раствор из 11,47 г (60,9 ммоля) 5-трет-бутилдиметилсилил-1-пропанальдегида в 20 мл ТГФ, поддерживая при этом температуру на том же уровне -65°С. После 1-часового перемешивания смеси дают нагреться до -20°С. Далее по каплям добавляют раствор 130 мл 1 н. соляной кислоты и дают температуре повыситься до +10°С. После этого добавляют 50 мл МТБ-эфира и органическую фазу отделяют. Сначала органическую фазу промывают водой, а затем насыщенным раствором гидрокарбоната натрия. В завершение повторно промывают водой и затем концентрируют в вакууме.
Выход: 13,65 г (87% от теории).
Пример 6б
5-гидрокси-2,2-диметил-3(R,S)-гидроксипентаннитрил
К раствору из 3 г (11,65 ммоля) 5-трет-бутилдиметилсилил-2,2-диметил-3(R,S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 6а) в 40 мл тетрагидрофурана добавляют 12,18 г (46,61 ммоля) гидрата тетрабутиламмонийфторида и перемешивают в течение 16 ч при комнатной температуре. Затем смесь упаривают досуха в вакууме. Остаток очищают RP-18-хроматографией (элюент: ацетонитрил/вода с градиентом соотношения).
Выход: 1,41 г (85% от теории) бесцветного вязкого масла.
Пример 6в, (-)-камфоркеталь
3(S)-(3,5)-камфордиметилкеталь-2,2-диметилпентаннитрил
6,73 г (47 ммолей) 5-гидрокси-2,2-диметил-3(R,S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 6б) растворяют в 27 мл метиленхлорида с 93 г (1S)-(-)-камфоркеталя (полученного из (1S)-(-)-камфоры, метанола и n-толуолсульфоновой кислоты) и добавляют 546 мг камфор-10-сульфоновой кислоты. Смесь нагревают в течение 15 ч с обратным холодильником. Далее эту смесь разбавляют 200-ми мл метиленхлорида и сначала промывают насыщенным раствором гидрокарбоната натрия, а затем насыщенным раствором хлорида натрия. Органическую фазу сушат над сульфатом натрия и концентрируют досуха в вакууме. Остаток хроматографируют на хиральной фазе (элюент: ацетонитрил/вода с градиентом соотношения). Таким путем получают масло, которое при стоянии кристаллизуется.
Выход: 10 г (77% от теории) бесцветного кристаллического твердого вещества.
Пример 6г
5-гидрокси-2,2-диметил-3(S)-гидроксипентаннитрил
Расщепление камфоркеталя
13 г (47 ммоля) 3(S)-(3, 5)-камфордиметилкеталь-2,2-диметилпентаннитрила (соединения из примера 6в) растворяют в 40 мл тетрагидрофурана, добавляют 12,18 г (46,61 ммоля) гидрата тетрабутиламмонийфторида и перемешивают в течение 16 ч при комнатной температуре, затем упаривают досуха в вакууме. Остаток очищают RP-18-хроматографией (элюент: ацетонитрил/вода с градиентом соотношения).
Выход: 5,72 г (85% от теории) бесцветного вязкого масла.
Пример 7
5-бензилокси-2,2-диметил-3(S)-гидроксипентаннитрил и 5-бензилокси-2,2-диметил-3(R)-гидроксипентаннитрил
Соединение, указанное в заголовке примера 1а, 5-бензилокси-2,2-диметил-3(R,S)-гидроксипентаннитрил, хроматографируют на хиральной фазе (10 г колонка Chiralpak AD с размером частиц 20 мкм (элюент: гексан/этанол в соотношении 98:2, длина волны 208 нм).
Таким путем получают R-изомер, выход: 3,8 г (38% от теории) бесцветного вязкого масла.
Помимо этого получают также S-изомер, выход: 4,1 г (41% от теории) бесцветного вязкого масла.
Пример 8а
5-трет-бутилдиметилсилил-2,2-диметил-3(R,S)-ацетоксипентаннитрил
К 28,24 г (109,7 ммоля) 5-трет-бутилдиметилсилил-2,2-диметил-3(R, S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 6а) добавляют при 0°С 14,43 г (142,64 ммоля) триэтиламина и 200 мг 4-диметиламинопиридина (ДМАП), растворенных в 128 мл МТБ-эфира, и 14,56 г (142,64 ммоля) ангидрида уксусной кислоты и перемешивают в течение 5 ч при комнатной температуре. Затем сливают в 2 л смеси воды со льдом и дважды экстрагируют МТБ-эфиром его порциями по 300 мл. Объединенные МТБ-фазы промывают один раз сначала 300-ми мл 5%-ной соляной кислоты, а затем водой. После этого упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 34,21 г (95% от теории) бесцветного масла.
Пример 8б
5-трет-бутилдиметилсилил-2,2-диметил-3(S)-гидроксипентаннитрил
10 г (33,39 ммоля) 5-трет-бутилдиметилсилил-2,2-диметил-3(R,S)-ацетоксипентаннитрила (соединения, указанного в заголовке примера 8а) вводят в буферный раствор, полученный из 0,88 г дигидрофосфата калия и 1,82 г динатрийгидрофосфата в 250 мл воды. Затем добавляют 5 г фермента Lipase AYS "Amano" (поставщик фирма Amano) и перемешивают в течение 42,5 ч при комнатной температуре. Добавлением 2,062 г динатрийгидрофосфата значение рН устанавливают на 7 и продолжают перемешивание в течение последующих 44,5 ч. Затем трижды экстрагируют 200-ми мл этилацетата. Органические фазы объединяют и упаривают досуха в вакууме. Очистку продукта осуществляют хроматографией на силикагеле (элюент: гексан/этилацетат с градиентом соотношения). Таким путем получают 3,8 г (45% от теории) 5-трет-бутилдиметилсилил-2, 2-диметил-3(R)-гидроксипентаннитрила и 4,8 г (48% от теории) 5-трет-бутилдиметилсилил-2,2-диметил-3(S)-ацетоксипентаннитрила.
4,8 г (16 ммолей) 5-трет-бутилдиметилсилил-2, 2-диметил-3(S)-ацетоксипентаннитрила растворяют в 50 мл этанола и смешивают с 1,28 г (32 ммоля) NaOH. Далее перемешивают в течение 3 ч при 25°С, сливают в 200 мл воды, дважды экстрагируют МТБ-эфиром его порциями по 200 мл, сушат над сульфатом натрия и концентрируют.
Выход: 3,43 г (40% от теории).
Пример 8в
3(S)-3, 5-ди-трет-бутилдиметилсилилокси-2, 2-диметилпентаннитрил
К раствору из 3 г (11,65 ммоля) 5-трет-бутилдиметилсилил-2,2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 8б) в 10 мл диметилформамида добавляют 2,37 г (34,95 ммоля) имидазола и 2,63 г (17,47 ммоля) трет-бутилдиметилсилилхлорида и перемешивают в течение 16 ч при комнатной температуре. Затем раствор сливают в 100 мл воды и дважды экстрагируют МТБ-эфиром его порциями по 50 мл. Органические фазы объединяют и упаривают досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 4,11 г (95% от теории) бесцветного вязкого масла.
Пример 9
5-гидрокси-2,2-диметил-3(S)-гидроксипентаннитрил
К раствору из 3 г (11,65 ммоля) 5-трет-бутилдиметилсилил-2, 2-диметил-3(S)-гидроксипентаннитрила (соединения, указанного в заголовке примера 8б) в 40 мл тетрагидрофурана добавляют 12,18 г (46,61 ммоля) гидрата тетрабутиламмонийфторида и перемешивают в течение 16 ч при комнатной температуре. Затем упаривают досуха в вакууме. Остаток очищают RP-18-хроматографией (элюент: ацетонитрил/вода с градиентом соотношения).
Выход: 1,41 г (85% от теории) бесцветного вязкого масла.
Пример 10а
5-бензилокси-2,2-диметил-3-кетопентаннитрил
К раствору ДАЛ (полученному из 33,64 г (79,17 ммоля) н-бутиллития (15%-ного, 1, 6-молярного) и 80,1 г (79,17 ммоля) диизопропиламина) по каплям добавляют при -65°С 5,47 г (79,17 ммоля) нитрила изомасляной кислоты и перемешивают в течение 20 мин при -65°С. Затем в течение 60 мин по каплям добавляют раствор из 14,29 г (71,97 ммоля) хлорангидрида 3-бензилокси-1-пропионовой кислоты в 20 мл ТГФ, поддерживая при этом температуру на том же уровне -65°С. После 1-часового перемешивания смеси дают нагреться до -20°С и по каплям добавляют раствор 20%-ной серной кислоты, температуре при этом дают повыситься до +10°С. Далее добавляют 50 мл МТБ-эфира и органическую фазу отделяют. Сначала органическую фазу промывают водой, а затем насыщенным раствором гидрокарбоната натрия. В завершение повторно промывают водой, после чего концентрируют досуха в вакууме. Остаток очищают экспресс-хроматографией на силикагеле (гексан/МТБ-эфир).
Выход: 14,15 г (85% от теории) бесцветного вязкого масла.
Пример 10б
5-гидрокси-2, 2-диметил-3-кетопентаннитрил
К 10 г (43,23 ммоля) 5-бензилокси-2,2-диметил-3-кетопентаннитрила (соединения, указанного в заголовке примера 10а), растворенных в 100 мл метанола, добавляют 3 г катализатора Перлмана (Pd(OH)2 на угле, 20%-ный).
Затем гидрируют в течение 7,5 ч при давлении 10 бар и комнатной температуре. Далее отфильтровывают от катализатора и фильтрат концентрируют досуха в вакууме.
Выход: 5,98 г (98% от теории) бесцветного вязкого масла.
Пример 10в
3(S)-5-дигидрокси-2, 2-диметилпентаннитрил
5 г (35,41 ммоля) 5-гидрокси-2,2-диметил-3-кетопентаннитрила (соединения, указанного в заголовке примера 10б) гидрируют в присутствии катализатора (приготовленного из 233 мг RuCl2(Ph)2 и 626 мг (R)-бинаф согласно R.Selke, Angew. Chem. 110, 1998, cc.1927-1930) при 40°С и давлении 100 бар. Затем отфильтровывают от катализатора и фильтрат концентрируют досуха в вакууме.
Выход: 4,96 г (98% от теории) бесцветного вязкого масла.
Пример 11
S-3-(2,2-диметил-[1,3]диоксан-4-ил)-3-метилбутан-2-он
К 3,26 г (17,79 ммоля) 3(S)-(3, 5)-ацетондиметилкеталь-2,2-диметилпентаннитрила (соединения, указанного в заголовке примера 1д), растворенных в 5 мл диэтилового эфира, по каплям добавляют при -20°С 35,6 мл комплекса метиллитий-бромид лития (1,5-молярный в диэтиловом эфире). Затем в течение 30 мин перемешивают при -20°С, после чего нагревают до комнатной температуры. Далее перемешивают при комнатной температуре в течение ночи. После этого добавляют 10 мл насыщенного раствора хлорида аммония и перемешивают в течение 6 ч при комнатной температуре. Затем органическую фазу отделяют и дважды промывают водой. В завершение органическую фазу концентрируют досуха в вакууме. Очистку проводят хроматографией на силикагеле (элюент:гексан/этилацетат с градиентом соотношения).
Выход: 2,77 г (78% от теории) масла.
Элементный анализ:
Пример 12
(S)-1, 3-бис(трет-бутилдиметилсиланилокси)-4,4-диметилпентан-5-он
К 5 г (13,45 ммоля) 3(S)-3, 5-ди-трет-бутилдиметилсилилокси-2,2-диметилпентаннитрила (соединения, указанного в заголовке примера 2), растворенных в 5 мл диэтилового эфира, по каплям добавляют при -20°С 40,35 мл этилата лития (1-молярный раствор в ТГФ). Затем перемешивают в течение 30 мин при -20°С и после этого нагревают до комнатной температуры. Перемешивание при комнатной температуре продолжают в течение ночи. Далее добавляют 10 мл насыщенного раствора хлорида аммония и перемешивают в течение 6 ч при комнатной температуре. Органическую фазу отделяют и дважды промывают водой. В завершение органическую фазу концентрируют досуха в вакууме. Очистку проводят хроматографией на силикагеле (элюент: гексан/этилацетат с градиентом соотношения).
Выход: 4,06 г (75% от теории) масла.
Пример 13
(S)-2-(2,2-диметил-[1,3]диоксан-4-ил)-2-метилгептан-3-он
К 3,26 г (17,79 ммоля) 3(S)-(3,5)-ацетондиметилкеталь-2,2-диметилпентаннитрила (соединения, указанного в заголовке примера 1д), растворенных в 5 мл ТГФ, по каплям добавляют при -65°С 34 мл н-бутиллития (15%-ного, 1,6-молярного в гексане). Затем в течение 5 ч перемешивают при -65°С и после этого нагревают до комнатной температуры. Перемешивание при комнатной температуре продолжают в течение ночи. Далее добавляют 10 мл насыщенного раствора хлорида аммония и перемешивают в течение 6 ч при комнатной температуре. Органическую фазу отделяют и дважды промывают водой. В завершение органическую фазу концентрируют досуха в вакууме. Очистку проводят хроматографией на силикагеле (элюент: гексан/этилацетат с градиентом соотношения).
Выход: 4,13 г (96% от теории) масла.
Пример 14
(4S)-4-(2-метил-3-оксогепт-6-ен-2-ил)-2,2-диметил-[1,3]-диоксан
К 3,26 г (17,79 ммоля) 3(S)-(3,5)-ацетондиметилкеталь-2, 2-диметилпентаннитрила (соединения, указанного в заголовке примера 1д), растворенных в 5 мл ТГФ, по каплям добавляют при -90°С 50 мл раствора 3-бутениллития (полученного из 4-бром-1-бутена и литиевой проволоки либо трет-бутиллития (см. Journ. Org. Chem. том 56, №21, 1991, cc.6094-6103, или Journ. Chem. Soc. Perkin Trans. I, 1988, с.2937). Затем перемешивают в течение 17 ч при -90° С и после этого нагревают до комнатной температуры.
Перемешивание при комнатной температуре продолжают в течение 17 ч. Далее добавляют 10 мл насыщенного раствора хлорида аммония и перемешивают в течение 6 ч при комнатной температуре. Органическую фазу отделяют и дважды промывают водой. В завершение органическую фазу концентрируют досуха в вакууме. Очистку продукта проводят хроматографией на силикагеле (элюент: гексан/этилацетат с градиентом соотношения).
Выход: 2,74 г (70% от теории) бесцветного масла.
Элементный анализ:
Пример 15
Метиловый эфир 4-циано-4-метил-3-оксопентановой кислоты
а) К 50,6 г (0,5 моля) диизопропиламина в 200 мл тетрагидрофурана при -30°С добавляли 312,5 мл (0,5 моля) 1,6 М раствора бутиллития в гексане. Смесь перемешивали в течение 30 мин. Затем смесь охлаждали до -60°С, в течение 30 мин добавляли по каплям 34,5 г (0,5 моля) изобутиронитрила и перемешивали в течение еще 30 мин.
б) В 200 мл ТГФ помещали 20 г гидрида натрия (60% раствор в белом парафине), добавляли 61 г (0,5 моля) диметилового эфира малоновой кислоты в 100 мл ТГФ и перемешивали при 20°С в течение 1 ч. Затем раствор охлаждали до -50°С и при -50°С добавляли приготовленный ранее раствор (см. раздел а) и перемешивали в течение 8 ч при нагревании до 20°С. В смесь добавляли насыщенный раствор хлорида аммония и продукт экстрагировали этилацетатом. После хроматографии на силикагеле получали 43 г (74% от теории) метилового эфира 4-циано-4-метил-3-оксопентановой кислоты.
C8H11NO3: MM 169,18; МС (Cl-NH3, 70 эВ): m/z 170 (М+Н)+
1Н-ЯМР (400 МГц, CDCl3): δ 1,55 (s, 6H), 2,9 (s, 6H), 3,72 (s, 2H), 3,95 (s, 3H).
Для обозначения эфирных защитных групп в описании используются следующие сокращения:
Реферат
Изобретение относится к соединениям, которые применяются для получения эпотилонов или их производных, а именно соединениям формулы I, соединениям общей формулы III, к соединениям общей формулы XII, где R4 представляет собой C1-С6алкильную группу, где R1 и R2 могут иметь идентичные либо разные значения и независимо друг от друга представляют собой спиртовую защитную группу, такую, например, как бензил, трет-бутилдиметилсилил, триметилсилил, триэтилсилил, трет-бутилдифенилсилил, или в случае, когда R1 и R2 соединены мостиковой связью, представляют собой кетальную защитную группу, такую, например,
как

Изобретение также относится к способу получения соединений формулы (I), который заключается в том, что исходное соединение общей формулы (II) подвергают обработке с целью защиты спиртовых групп защитными группами R1 и R2. Также к способу получения оптически активных соединений общей формулы IIIa, который заключается в том, что сложный рацемический эфир общей формулы VI, где R3 представляет собой C1-С6алкильную группу или аллильную, фенильную либо бензильную группу, энантиоселективно омыляют с помощью ферментативной реакции с использованием фермента Lipase Amano AY, или заключающийся в том, что при использовании исходных соединений формулы VII осуществляют хиральное восстановление кетогруппы. Способ получения соединений общей формулы XIII заключается в том, что соединения общей формулы XIV, где R4 представляет собой C1-С6алкильную группу, a Nu обозначает уходящую группу, такую как Cl, Br, имидазол, -OPh, -О-С6Н4NO2 или -O-С1-С4алкил, подвергают взаимодействию с соединением формулы V. Способ получения кетонов общей формулы А, где V обозначает С1-С5алкил или алкенил, заключается в том, что соединения общей формулы Ia подвергают взаимодействию с соединениями общей формулы Т-V, где М обозначает Li или TgCl, TgBr либо TgI, и затем перерабатывают путем гидролиза. 19 н. и 11 з.п. ф-лы.

Формула

























Комментарии