Способы получения замещенных арилконденсированныхазаполициклических соединений, промежуточные продукты и способы их получения - RU2282619C2
Код документа: RU2282619C2
Описание
Данное изобретение относится к способу получения промежуточных 1,3-замещенных инденов формул (Ia), (Ib) и (Ic) или их смесей:
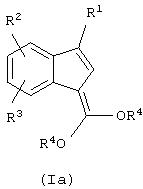


где R1, R2, R3, R4 и R5 определены ниже.
Соединения формул (Ia), (Ib) и (Ic) и их смеси применимы для получения соединений формулы (II):

где R2, R3 и R6 также определены ниже. Способы получения соединений формулы (II) изложены в WO 99/35131, соответствующей заявкам США № 09/402010, поданной 28 сентября 1999, и США № 09/514002, поданной 25 февраля 2000, обе включены в качестве ссылок. В данном изобретении предлагается более эффективный альтернативный путь получения соединений формулы (II) посредством использования катализируемого палладием процесса циклизации с получением промежуточных соединений формул (Ia), (Ib) и (Ic), которые затем далее преобразуют в соединения формулы (II).
Соединения формулы (II) связываются с нейронными никотиновыми ацетилхолиновыми специфическими рецепторными сайтами и могут использоваться для модулирования холинергической функции. Такие соединения могут использоваться для лечения воспалительной болезни кишечника (включая, но без ограничения указанным, язвенный колит, гангренозную пиодермию и болезнь Крона), синдром раздраженной толстой кишки, спастическую дистонию, хроническую боль, острую боль, нарушение всасывания в кишечнике, pouchitis, вазоконстрикцию, тревожное состояние, паническиое расстройство, депрессию, биполярное расстройство, аутизм, нарушения сна, расстройство нормального циркадного ритма, боковой амиотрофический склероз (ALS), нарушение познавательной способности, гипертензию, булимию, анорексию, ожирение, сердечные аритмии, повышенную секрецию желудочной кислоты, язвы, феохромацитому, прогрессивный супрануклеарный паралич, привыкания и зависимости от химических веществ (например, привыкания или зависимости от никотина (и/или табачных изделий), спирта, бензодиазепинов, барбитуратов, опиоидов или кокаина), головную боль, мигрень, удар, травматическое повреждение мозга (TBI), обсессивно-компульсивное расстройство (OCD), психоз, хорею Нантингтона, позднюю дискинезию, гиперкинезию, дислексию, шизофрению, постинсультное слабоумие, возрастное ухудшение познавательной способности, эпилепсию, включая малые эпилептические припадки, сенильную деменцию типа Альцгеймера (AD), болезнь Паркинсона (PD), гиперактивность с дефицитом внимания (ADHD) и болезнь Туретта.
Соединения по данному изобретению могут быть также использованы в сочетании с антидепрессантом, таким как, например, трициклический антидепрессант или антидепрессант, ингибирующий повторное усвоение серотонина (SRI), для того, чтобы лечить и ухудшение познавательной способности, и депрессию, связанную с AD, PD, ударом, хореей Хантингтона или травматическим повреждением мозга (TBI); в сочетании с мускариновыми агонистами для того, чтобы стимулировать как центральные мускариновые, так и никотиновые рецепторы для лечения, например, ALS, нарушения познавательной способности, возрастного ухудшения познавательной способности, AD, PD, удара, хореи Хантингтона и TBI; в сочетании с нейротрофическими факторами, такими как NGF, для того, чтобы максимально стимулировать холинергическое усовершенствование для лечения, например, ALS, нарушения познавательной способности, возрастного ухудшения познавательной способности, AD, PD, удара, хореи Хантингтона и TBI, или в сочетании с агентами, которые замедляют или прекращают AD, такими как средства, улучшающие познавательную способность, ингибиторы агрегации амилоида, ингибиторы секретазы, ингибиторы тау-киназы, нейронные противовоспалительные агенты и лечебные средства, подобные эстрогену.
Краткое описание изобретения
Данное изобретение относится к способу получения любого из промежуточных 1,3-замещенных инденов формул (Ia), (Ib) и (Ic) или их смесей:
где R2 и R3 выбраны независимо из водорода, фтора, хлора, -SOq(C1-C6)алкила, где q равно нулю, единице или двум, ((C1-C6)алкил)2амино-, -CO2R7, -CONR8R9, -SO2NR10 R11, арил-(C0 -C3)алкил- или арил-(C0-C3)алкил-O-, где указанный арил выбран из фенила и нафтила, гетероарил-(C0-C3 )алкила- или гетероарил-(C0-C3)алкил-O-, где указанный гетероарил выбран из пяти-семичленных ароматических колец, содержащих от одного до четырех гетероатомов, выбранных из кислорода, азота и серы; X2(C0-C6)алкила- и X2(C1-C6)алкокси-(C0-C6)алкила-, где X2 отсутствует или X2 означает (C1-C6)алкиламино- или ((C1-C6)алкил)2амино- и где группа (C0-C6)алкил- или (C1-C6)алкокси-(C0-C6 )алкил- указанного X2(C0-C6)алкила- или X2(C1-C6)алкокси-(C0-C6 )алкила- содержит по меньшей мере один атом углерода, и где от одного до трех атомов углерода указанных групп (C0-C6)алкила- или (C1-C6)алкокси-(C0-C6)алкила- необязательно могут быть замещены атомом кислорода, азота или серы при условии, что любые два таких гетероатома должны быть отделены друг от друга по меньшей мере двумя атомами углерода, и где любая из алкильных групп указанных групп (C0-C6)алкила или (C1-C6)алкокси-(C0-C6)алкила необязательно может быть замещена двумя-семью атомами фтора, и где один из атомов углерода каждой алкильной группы указанного арил-(C0-C3)алкила- и указанного гетероарил-(C0-C3 )алкила- необязательно может быть замещен атомом кислорода, азота или серы, и где каждая из указанных арил- и гетероарильной групп необязательно может быть замещена одним или несколькими заместителями, предпочтительно, от ноля до двух заместителей, независимо выбранными из (C1-C6)алкила, необязательно замещенного одним-семью атомами фтора, (C1-C6)алкокси, необязательно замещенного двумя-семью атомами фтора, хлора, фтора, ((C1-C6)алкил)2амино-, -CO2R7, -CONR8R9и -SO2NR10R11;
или R2 и R3, вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленное моноциклическое или десяти-четырнадцатичленное бициклическое карбоциклическое кольцо, которое может быть насыщенным или ненасыщенным, где от одного до трех неконденсированных атомов углерода указанных моноциклических колец и от одного до пяти атомов углерода указанных бициклических колец, которые не являются частью бензольного кольца, показанного в формуле (II), необязательно и независимо могут быть замещены азотом, кислородом или серой, и где указанные моноциклические и бициклические кольца необязательно могут быть замещены одним или несколькими заместителями, предпочтительно, от ноля до двух заместителей для моноциклических колец и от ноля до трех заместителей для бициклических колец, которые выбраны независимо из (C0-C6)алкила- и (C1-C6)алкокси-(C0-C6)алкила-, где суммарное число атомов углерода не превышает шести и где любая из алкильных групп необязательно может быть замещена одним-семью атомами фтора; оксо, фтор, хлор, ((C1-C6)алкил)2 амино-,-CO2R7, -CONR8R9 и -SO2NR10R11;
каждый R7, R8, R9, R10 и R11 независимо выбран из водорода и (C1-С6) алкила, или R8 и R9, или R10 и R11 вместе с атомом азота, к которому они присоединены, образуют кольцо пирролидина, пиперидина, морфолина, азетидина, пиперазина, -N-(C1-С6)алкилпиперазина или тиоморфолина или кольцо тиоморфолина, где атом серы кольца замещен сульфоксидом или сульфоном; и
каждый Х независимо означает (C1-C6)алкилен;
R5 означает C1-С6 алкил или Si(R12)3
R1 означает электроноакцепторную группу, выбранную из циано, алкоксикарбонила, алкилкарбонила, арилкарбонила, арила, нитро, трифторметила и сульфонила;
каждый R4 независимо означает Н,


каждый L независимо означает Н, C1-C6 алкил, фенил или бензил; и
каждый R12 независимо означает C1 -С6 алкил или фенил; включающему стадию
взаимодействия соединения формулы (III)

где R1, R2, R3 и R5 - такие, как указано выше; и X' означает йод, бром, хлор или OSO2R13, где R13 означает (C1-С6) алкил, Si((C1 -С6)алкил)3 или Si(фенил)3, в присутствии палладиевого(0) катализатора и основания.
Предпочтительным вариантом этого способа получения инденов является такой, где палладиевый катализатор получают из сочетания соединения палладия, выбранного из группы, включающей ацетат палладия(II), хлорид палладия(II), хлорид бис-ацетонитрил палладия(II), хлорид бис-бензонитрил палладия(II), бромид палладия(II), трис(дибензилиденацетон)дипалладий(0), более предпочтительно, ацетат палладия(II), и фосфинового лиганда, выбранного из дициклогексилфенилфосфина, трициклогексилфосфина, три-трет-бутилфосфина, три-изопропилфосфина, три-н-пропилфосфина, три-изобутилфосфина, три-н-бутилфосфина, три-о-толилфосфина, трифенилфосфина, 2-(дициклогексилфосфино)бифенила или 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенила, более предпочтительно, трициклогексилфосфина или три-трет-бутилфосфина. Следующим предпочтительным вариантом является такой, где указанную стадию осуществляют в присутствии основания, выбранного из трет-бутоксида натрия, трет-бутоксида калия, трет-пентоксида натрия, этоксида натрия, карбоната натрия, карбоната калия, карбоната цезия, гидрида натрия, гидрида лития и гидрида калия, более предпочтительно, выбранного из трет-бутоксида натрия или трет-пентоксида натрия. Далее, предпочтительным аспектом изобретения является такой, где указанный процесс проводят в растворителе, выбранном из 1,2-диметоксиэтана, тетрагидрофурана, 2-метилтетрагидрофурана, 1,2-диэтоксиэтана, 1,4-диоксана, толуола, N-метилпирролидинона, диметилацетамида и диметилформамида, более предпочтительно, выбранном из 1,2-диметоксиэтана или тетрагидрофурана. Кроме того, предпочтительно, когда указанную стадию способа проводят при температуре между комнатной температурой и 120°С, более предпочтительно, между 40 и 90°С, в течение периода времени от 1 до 48 часов и, более предпочтительно, 8 часов.
Другим вариантом способа получения любого из промежуточных соединений формул (Ia), (Ib) и (IC) и их смесей является способ, дополнительно включающий более важную стадию взаимодействия соединения формулы (IV):

где X' означает йод, бром, хлор или OSO2R13, где R13 означает (C1-C6)алкил, Si((C1-C6)алкил)3 или Si(фенил)3, с соединением формулы (V):

где R5 определен выше, в присутствии основания.
Предпочтительным вариантом этой стадии конденсации является такой, где эту стадию проводят в растворителе, выбранном из тетрагидрофурана, 2-метилтетрагидрофурана, 1,2-диметоксиэтана, 1,2-диэтоксиэтана, 1,4-диоксана, диметилсульфоксида, диметилформамида, диметилацетамида, N-метилпирролидинона, метиленхлорида, 1,2-дихлорметана, метанола, этанола, изопропанола и пропанола, более предпочтительно, когда растворитель выбран из тетрагидрофурана, 2-метилтетрагидрофурана и 1,2-диметоксиэтана. Другим предпочтительным вариантом осуществления изобретения является такой, когда основание на этой стадии выбрано из трет-бутоксида натрия, трет-бутоксида калия, метоксида натрия, этоксида натрия, трет-пентоксида натрия, карбоната натрия, карбоната калия, карбоната цезия, диизопропиламида лития, бис(триметилсилил)амида лития, бис(триметилсилил)амида натрия, бис(триметилсилил)амида калия, гидрида натрия и гидрида лития, более предпочтительно, когда основание выбрано из трет-бутоксида натрия, этоксида натрия и метоксида натрия. Далее, предпочтительным способом является такой, где эту стадию проводят при температуре между -78°С и 50°С; более предпочтительно, между 0°С и 30°С, предпочтительно, в течение периода от 1 до 24 часов, более предпочтительно, 6 часов.
Более предпочтительным вариантом является такой, где способ получения какого-либо из промежуточных соединений формул (Ia), (Ib) и (Ic) и их смесей является тандемом конденсации и катализируемой палладием циклизации, включающим единственную стадию взаимодействия соединения формулы (IV)
где X' означает йод, бром, хлор или OSO2R13, где R13 означает (C1-C6)алкил, Si((C1-C6)алкил)3 или Si(фенил)3, с соединением формулы (V)

где R5 указано выше, в присутствии основания и катализатора палладия(0).
Предпочтительно, конденсацию и катализируемую палладием циклизацию проводят в одну стадию, сначала растворением соединений формулы (IV) и (V) в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 1,4-диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, N-метилпирролидинон, метиленхлорид, 1,2-дихлорметан, предпочтительно, тетрагидрофуран, 2-метилтетрагидрофуран или 1, 2-диметоксиэтан, и затем добавлением раствора к смеси, содержащей и основание, и катализатор палладий(0) также в одном из указанных выше растворителей. Предпочтительно, основание выбрано из группы, включающей трет-бутоксид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, трет-пентоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, гидрид натрия и гидрид лития, более предпочтительно, выбрано из трет-бутоксида натрия, этоксида натрия или метоксида натрия. Предпочтительно, палладиевый катализатор получают объединением соединения палладия, выбранного из ацетата палладия(II), хлорида палладия(II), хлорида бис-ацетонитрил палладия(II), хлорида бис-бензонитрил палладия(II), бромида палладия(II), трис(дибензилиденацетон)дипалладия(0), более предпочтительно ацетата палладия(II), и фосфинового лиганда, такого как дициклогексилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, три-изопропилфосфин, три-н-пропилфосфин, три-изобутилфосфин, три-н-бутилфосфин, три-о-толилфосфин, трифенилфосфин, 2-(дициклогексилфосфино)бифенил или 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, более предпочтительно, трициклогексилфосфин или три-трет-бутилфосфин. Предпочтительно, эту единственную стадию образования инденов осуществляют при температуре между -78°С и 50°С; более предпочтительно, между 0°С и 30°С, в течение периода от 1 до 24 часов, более предпочтительно, от 1 до 5 часов, и с последующим нагреванием при температуре между 25°С и 120°С, более предпочтительно, между 40°С и 90°С, в течение периода от 1 до 24 часов, предпочтительно, 8 часов.
Предпочтительный вариант изобретения относится к способам получения соединений формул (Ia), (Ib) и (Ic) и их смесей, где один из R2 и R3 или оба означают Н, (C1-C6)алкокси, -CF3, фтор или C2F5 и R1 означает CN. Более предпочтителен вариант, где R2 и R3 означают Н, R1 означает CN и X' означает Br. Предпочтительно, R6 в соединении формулы (V) означает метил или этил.
Данное изобретение также относится к соединениями 1,3-замещенного индена формул (Ia), (Ib) и (Ic). К предпочтительным соединениям индена по изобретению относятся следующие:
натриевая соль 3-(гидроксиметоксиметилен)-5-трифторметил-3H-инден-1-карбонитрила,
натриевая соль 3-(гидроксиметоксиметилен)-3H-инден-1-карбонитрила,
натриевая соль 3-(этоксигидроксиметилен)-3H-инден-1-карбонитрила,
натриевая соль 3-(этоксигидроксиметилен)-5,6-диметокси-3H-инден-1-карбонитрила,
натриевая соль 3-(гидроксиэтоксиметилен)-5-трифторметил-3H-инден-1-карбонитрила,
натриевая соль 3-(этоксигидроксиметилен)-7-фтор-3H-инден-1-карбонитрила,
натриевая соль 3-(этоксигидроксиметилен)-5-фтор-3H-инден-1-карбонитрила,
3-[1,3]диоксолан-2-илиден-5-трифторметил-3H-инден-1-карбонитрил;
3-[1, 3]диоксолан-2-илиден-3H-инден-1-карбонитрил и
этиловый эфир 3-бензолсульфонил-3H-инден-1-карбоновой кислоты.
Данное изобретение относится также к способу получения соединений формулы (VII)

где R2, R3 и R5 являются такими, как определено выше; включающему способ получения соединений любой из формул (Ia), (Ib) и (Ic), где R1 означает CN, и их смесей и дополнительно включающему стадию (А) гидрирования любого из соединений формул (Ia)', (Ib)' и (Ic)' или их смеси:



Предпочтительным вариантом осуществления изобретения является такой, где условия гидрирования стадии (А) являются, предпочтительно, такими, которые подходят для осуществления желаемого восстановления, где катализатор и условия гидрирования известны специалисту в этой области, как отраженные в ссылке, такой как Comprehensive Organic Synthesis, Vol.8, "Reduction", Eds.-in-Chief, B.M.Trost and Ian Fleming (Pergamon Press NY 1991). Однако предпочтительным вариантом осуществления изобретения является такой, где гидрирование на стадии (А) проводят, используя катализатор гидрирования, выбранный из палладия на углероде, гидроксида палладия на углероде, платины на углероде, оксида платины и оксида платины на углероде; более предпочтительно, выбранный из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, в присутствии кислоты, выбранной из серной кислоты, хлористоводородной кислоты, метансульфоновой кислоты, толуолсульфоновой кислоты, трифторуксусной кислоты, уксусной кислоты, муравьиной кислоты, фосфорной кислоты и перхлорной кислоты, более предпочтительно, выбранной из серной кислоты, метансульфоновой кислоты и смеси любых из указанных выше кислот. Предпочтительно, растворитель для гидрирования выбирают из метанола, этанола, изопропанола, бутанола, пропанола, этилацетата, изопропилацетата, тетрагидрофурана, толуола, этиленгликоля и смеси любых из указанных растворителей; более предпочтительно, растворитель выбирают из этилацетата, метанола, этанола и пропанола. Гидрирование (А), предпочтительно, проводят в атмосфере водорода вплоть до 7 атмосфер (приблизительно 7,03 кг/см2), более предпочтительно, от 3 до 4 атмосфер (приблизительно 3,52 кг/см2); предпочтительно, в течение периода времени от 1 до 24 часов; более предпочтительно, 6 часов. Далее, стадию (A), предпочтительно, проводят при температуре между 0 и 60°С; более предпочтительно, между 20 и 40°С.
Данное изобретение относится также к способу получения соединения формулы (VII), как определено выше, где стадию (А) заменяют двумя отдельными стадиями
(а) гидрированием одного из соединений формул (Ia)', (Ib)' и (Ic)' или их смеси с получением соединения формулы (IX):

где R2, R3 и R9 являются такими, как определено выше;
(b) и далее восстановлением соединения формулы (IX) восстановительными условиями, с получением соединения формулы (VII):

где R2, R3 и R9 являются такими, как определено выше.
Условия гидрирования любой из стадий (а) и (b) предпочтительно такие, которые подходят для осуществления желаемого восстановления, где катализаторы и условия гидрирования известны специалисту в этой области, как отраженные в ссылке, такой как Comprehensive Organic Synthesis, Vol.8, "Reduction", Eds.-in-Chief, B.M.Trost and Ian Fleming (Pergamon Press NY 1991). Однако предпочтительным вариантом осуществления изобретения является такой, где условия гидрирования на стадии (А) для восстановления какого-либо из соединений формул (Ia)', (Ib)' и (Ic)' или их смесей до соединения формулы (IX) включает обработку катализатором гидрирования, выбранным из группы, включающей палладий на углероде, гидроксид палладия на углероде, платину на углероде, оксид платины и оксид платины на углероде; более предпочтительно, выбранным из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, необязательно, в присутствии кислоты, такой как уксусная кислота, муравьиная кислота, бензойная кислота или салициловая кислота; более предпочтительно, муравьиная кислота или уксусная кислота; в растворителе, таком как метанол, этанол, изопропанол, бутанол, пропанол, этилацетат, изопропилацетат, тетрагидрофуран, толуол или любая смесь указанных растворителей; более предпочтительны, метанол или этанол; в атмосфере водорода вплоть до 7 атмосфер (приблизительно 7,03 кг/см2), более предпочтительно, от 3 до 4 атмосфер (приблизительно, 3,52 кг/см2); в течение периода времени от 1 до 48 часов; предпочтительно, 12 часов. Далее, эту, стадию предпочтительно, проводят при температуре между 0 и 60°С; более предпочтительно, между 20 и 30°С.
Дополнительно, предпочтительным вариантом осуществления изобретения является такой, где условия восстановления на стадии (b) для восстановления нитрила формулы (IX) до соединения формулы (VII) включает обработку катализатором гидрирования, выбранным из группы, включающей палладий на углероде, гидроксид палладия на углероде, платину на углероде, оксид платины и оксид платины на углероде; более предпочтительно, выбранным из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, в присутствии кислоты, такой как серная кислота, хлористоводородная кислота, метансульфоновая кислота, толуолсульфоновая кислота, трифторуксусная кислота, уксусная кислота, муравьиная кислота, фосфорная кислота или перхлорная кислота, предпочтительны серная кислота, метансульфоновая кислота или любое сочетание указанных выше кислот; в растворителе, таком как метанол, этанол, изопропанол, бутанол, пропанол, этилацетат, изопропилацетат, тетрагидрофуран, толуол или любая смесь указанных растворителей; предпочтительны метанол или этанол; в атмосфере водорода, вплоть до 7 атмосфер (приблизительно, 7,03 кг/см2), более предпочтительно, от 3 до 4 атмосфер (приблизительно, 3,52 кг/см2); в течение периода времени от 1 до 48 часов; предпочтительно, 6 часов. Далее, эту стадию, предпочтительно, проводят при температуре между 0 и 60°С; более предпочтительно, между 20 и 40°С.
Данное изобретение также относится к соединениям формулы (IX):

где R2, R3 и R9 являются такими, как определено выше. Особенно предпочтительными соединениями формулы (IX) являются:
этиловый эфир 3-циано-индан-1-карбоновой кислоты,
этиловый эфир 6-трифторметил-3-циано-индан-1-карбоновой кислоты,
этиловый эфир 5,6-диметокси-3-циано-индан-1-карбоновой кислоты,
этиловый эфир 4-фтор-3-циано-индан-1-карбоновой кислоты,
этиловый эфир 6-фтор-3-циано-индан-1-карбоновой кислоты, и
этиловый эфир 3-бензолсульфонил-3-циано-индан-1-карбоновой кислоты.
Данное изобретение также относится к способу получения соединения формулы (VIII):

включающему указанные выше стадии, относящиеся к получению соединений формулы (VII) непосредственно выше, и дополнительно включающему стадию
(В) воздействия на соединение формулы (VII) основания для циклизации амида.
Предпочтительным способом по изобретению является такой, где воздействие основания на стадии (В) включает обработку основанием, выбранным из группы, включающей из трет-бутоксид натрия, метоксид натрия, этоксид натрия, гидроксид натрия, трет-бутоксид калия, метоксид калия, этоксид калия, гидроксид калия, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия, триэтиламин, метилимидазол, лютидин, пиридин, метилморфолин, этилморфолин или диизопропилэтиламин; более предпочтительным является трет-бутоксид натрия или метоксид натрия; в растворителе, выбранном из группы, включающей метанол, этанол, изопропанол, пропанол, бутанол, этиленгликоль, этилацетат, ацетонитрил, толуол, воду и смеси любых из указанных растворителей; более предпочтительно, растворителем является метанол, этанол, пропанол или любая смесь метанола, этанола или пропанола. Предпочтительно, стадию (B) проводят при температуре между 0 и 120°С; более предпочтительно, при комнатной температуре, предпочтительно, в течение периода времени между 30 минутами и 72 часами, более предпочтительно, от 6 до 12 часов.
Данное изобретение также относится к получению соединений формулы (II):

где R6 означает водород и R2 и R3 являются такими, как определено выше; включающему указанные выше стадии, относящиеся к получению соединений формулы (VIII), и дополнительно включающему стадию
(С) восстановления карбонильной группы соединения формулы (VIII) восстановителем с получением соединения формулы (II).
Предпочтительным вариантом осуществления изобретения является такой, где стадию восстановления (С) проводят с восстановителем, выбранным из группы, включающей комплекс борана и тетрагидрофурана, диборан, комплекс борана и диметилсульфида, смешанный гидрид лития и алюминия и сочетание боргидрида натрия и трифторида бора, более предпочтительно, восстановителем является либо комплекс борана и тетрагидрофурана, либо сочетание боргидрида натрия и трифторида бора. Предпочтительно, стадию восстановления проводят в растворителе, выбранном из группы, включающей тетрагидрофуран, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 2-метилтетрагидрофуран, 1,4-диоксан и метил-трет-бутиловый эфир, более предпочтительно, растворителем является тетрагидрофуран. Предпочтительно, восстановление проводят при температуре между 0 и 80°С; более предпочтительно, при 50°С, в течение периода времени между 1 и 24 часами, предпочтительно, 5 часов.
В предпочтительном варианте изобретения продукт формулы (II) может быть затем выделен удалением растворителя. Более предпочтительно, продукт выделяют кристаллизацией в виде соли, наиболее предпочтительно, в виде соли с кислотой, такой как п-толуолкарбоновая кислота, метансульфоновая кислота, хлористоводородная кислота, щавелевая кислота, лимонная кислота или уксусная кислота, более предпочтительно, используют п-толуолкарбоновую кислоту, в растворителе, таком как изопропанол, гексан, ацетон, этилацетат, метилэтиловый кетон или толуол, более предпочтительны изопропанол или этилацетат.
Данное изобретение также относится к способу получения соединения формулы (II), указано выше, где R6 означает R6' и R6' означает (C1-C6)алкил, неконъюгированный (C3-C6)алкенил, бензил, -(C1-C6)алкил-СНО, -(C1-C6)алкил-(С=О)-(C1-C6)алкил или -СН2 СН2-О-(C1-C4)алкил, включающему стадии, которые установлены здесь выше для получения соединения формулы (II), где R6 означает Н; и дополнительно включающему стадию (D) взаимодействия продукта стадии (C) с соединением формулы R6'-Y, где Y означает удаляемую группу, такую как хлор, бром, йод или мезилат.
Данное изобретение также относится к способу получения соединения формулы (II), которое определено выше, где R6 означает (СН2)-R6'' и R6'' означает (C1-C5 )алкил, неконъюгированный (C3-C5)алкенил, фенил, -(C1-C5)алкил-СНО, -(C1-C5 )алкил-(С=О)-(C1-C6)алкил или -СН2-О-(C1-C4)алкил, включающему стадию взаимодействия в условиях восстановительного аминирования соединения формулы (II) (где R6 означает Н) с соединением формулы R6''-СНО. Предпочтительно, условиями восстановительного аминирования данного способа являются либо каталитическое гидрирование, либо обработка некоторыми гидридными реагентами в инертном в условиях реакции растворителе. Более предпочтительно, каталитическое гидрирование проводят в присутствии металлического катализатора, такого как палладий или никель Ренея. Наиболее предпочтительно, условием восстановительного аминирования является обработка гидридным реагентом, выбранным из боргидридов, таких как боргидрид натрия (NaBH4), цианборгидрид натрия (NaBH3CN) и триацетоксиборгидрид натрия (NaB(ОАс)3Н), бораны, реагентов на основе алюминия и триалкилсиланов, в подходящем растворителе, выбранном из полярных растворителей, таких как метанол, этанол, метиленхлорид, тетрагидрофуран (ТГФ), диоксан и этилацетат. Предпочтительно, это восстановительное аминирование проводят при температуре от -78°С до температуры кипения растворителя с обратным холодильником, более предпочтительно, от 0°С до 25°С, в течение от 5 минут до 48 часов, более предпочтительно, от 0,5 до 12 часов.
Примерами предпочтительных гетероарильных групп в пределах определения R2 и R3 являются следующие: тиенил, оксазолил, изоксазолил, пиридил, пиримидил, тиазолил, тетразолил, изотиазолил, триазолил, имидазолил, тетразолил, пирролил и следующие группы:





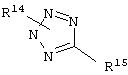
где один из R14 и R15 означает водород или (C1-C6)алкил, а другой означает связь с бензольным кольцом формулы (II).
Примеры бициклических кольцевых систем, которые R2 и R3 могут образовывать вместе с бензольным кольцом формулы (II), могут быть выбраны из следующих:



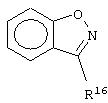
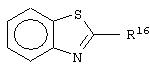
где R16 и R17 выбраны независимо из Н, (C1-C6)алкила-, (C1-C6)алкокси-(C0-C6)алкила-, где общее число атомов углерода не превышает шести и где любая из алкильных групп необязательно может быть замещена одним-семью атомами фтора; оксо, фтора, хлора, ((C1-C6)алкил)2амино-, -СО2 R4, -CONR5R6 и -SO2NR7R8, где R4, R5, R6, R7 и R8 определены выше.
Другие варианты осуществления данного изобретения относятся к способам получения соединений формулы (II) и их фармацевтически приемлемых солей, где R2 и R3 вместе с бензольным кольцом формулы (II) образуют бициклическую или трициклическую кольцевую систему, выбранную из следующих:











где R16 и R17 определены выше и m равно нулю, единице или двум, и где один из атомов углерода кольца А необязательно может быть замещен кислородом или N(C1-C6)алкилом.
Другие предпочтительные варианты осуществления данного изобретения относятся к способам получения соединений формулы (II) и их фармацевтически приемлемых солей, где R2 и R3 не образуют вместе с бензольным кольцом формулы (II) бициклическую или трициклическую кольцевую систему.
Другие предпочтительные варианты осуществления данного изобретения относятся к способам получения соединений формулы (II), где один из R2 и R3 или оба означают (C1 -C6)алкокси, -CF3, фтор или C2F5.
Хотя соединения формулы (II) могут быть получены с группами R2 и R3 уже имеющимися, в качестве альтернативы, соединения формулы (II) могут быть получены из соединения формулы (II), где обе группы R2 и R3 означают Н, способами по данному изобретению с дополнительной обработкой соединения формулы (II), где R2, R3 и R6 все означают Н, следующими способами.
Данное изобретение также относится к способам получения соединений формулы (II), включающим дополнительные стадии
(i) введения защитной группы, Q, на атом азота соединения формулы (II), где R2, R3 и R6 означают Н:

полученного в соответствии со способом, указанным выше;
(ii) взаимодействия соединения с защитной Q-группой со стадии (i) c трифторметансульфоновой кислотой (CF3SO2OH) и эквивалентами азотной кислоты с получением соединения формулы (XIII-Q):

и (iii) восстановления нитрогрупп соединения формулы (XIII-Q) с получением соединения формулы (XIV-Q):

Защищающая азот группа Q может быть выбрана из подходящих групп, известных специалисту в этой области, включая -COCF3 , -COCCl3, -COOCH2CCl3, -COO(C1-C6)алкил и -COOCH2C6H5. Эти группы могут быть присоединены или удалены способами, описанными для каждой из них, в T.W.Greene and G.M.Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons, New York, 1991). Предпочтительно, защищающей азот группой Q является группа трифторацетил или трет-бутоксикарбонил.
Предпочтительно, стадию (ii) проводят в смеси 4 или более эквивалентов трифторметансульфоновой кислоты (CF3SO2OH) и 2-3 эквивалентов азотной кислоты в хлорированном углеводородном растворителе, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид. Предпочтительно, полученной смеси дают взаимодействовать от около 5 до 24 часов, более предпочтительно, при температуре в пределах от около -78°С до около 0°С в течение около 2 часов, и затем дают нагреться до комнатной температуры в течение остального времени. Предпочтительно, стадию (iii) осуществляют с использованием газообразного водорода и палладиевого катализатора, такого как гидроксид палладия, 5% палладий на углероде или 10% палладий на углероде.
Изобретение также относится к способу получения соединения формулы (IA):

где R16 определен выше, включающему стадию взаимодействия соединения формулы (XIV-Q), полученного в соответствии со способом, установленным выше:

где Q означает защищающую азот группу, с соединением формулы (XXVIII):

где R20 и R21, каждый, независимо, означают (C1-C6 )алкил и где R16 определен выше; и
(ii) удаления защитной группы Q.
Защищающая азот группа Q может быть выбрана из подходящих групп, известных специалисту в этой области, включая -COCF3, -COCCl3, -COOCH2CCl3, -COO(C1-C6)алкил и -COOCH2C6H5. Эти группы могут быть присоединены или удалены способами, описанными для каждой из них, в T.W.Greene and G.M.Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons, New York, 1991). Предпочтительно, защищающей азот группой Q является группа трифторацетил или трет-бутоксикарбонил.
Изобретение также относится к способу получения соединения формулы (IB):

где R16 и R17определены выше, включающему стадии
(i) взаимодействия соединения формулы (XIV-Q), полученного в соответствии со способом, определенным выше:

где Q означает защищающую азот группу, с соединением формулы (XXVIII):
где R20 и R21, каждый независимо, означают (C1-C6)алкил, и где R16 определен выше; и
(ii) взаимодействия продукта со стадии (i) с соединением формулы R17Z, где R17 определен выше и Z означает удаляемую группу, в присутствии основания;
(iii) удаления защитной группы Q.
Предпочтительно, в способе получения IB удаляемую группу выбирают из группы, включающей галоген, галогенсульфонат, мезилат и тозилат, и основанием является гидрид, гидроксид или карбонат щелочного металла. Предпочтительно, защитной группой Q является группа трифторацетил или трет-бутоксикарбонил.
Изобретение также относится к другому способу получения соединения формулы (IB):
где R16 и R17 определены выше, включающему стадии
(i) взаимодействия соединения формулы (XIV-Q), полученного в соответствии со способами, описанными выше, с одним эквивалентом соединения формулы R17Z, где R17 определен выше и Z означает удаляемую группу, в присутствии основания с образованием соединения формулы (XX-Q);

где Q означает защищающую азот группу,
(iii) взаимодействия соединения формулы XX-Q с соединением формулы (XXVIII):
где R20 и R21, каждый независимо, означают (C1 -C6)алкил и где R16 определен выше; и
(iii) удаления защитной группы Q.
Предпочтительно, в этом способе получения IB защитной группой Q является группа трифторацетил или трет-бутоксикарбонил.
Изобретение также относится к способу получения соединения формулы (IC):

где R16 и R17определены выше, включающему стадии
(i) взаимодействия соединения формулы (XIV-Q), полученного в соответствии со способами, описанными выше,
где Q означает защищающую азот группу, с соединением формулы
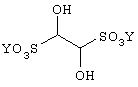
где Y означает катион щелочного металла или щелочно-земельного металла, или с соединением формулы

где R16 и R17 определены выше; и
(iii) удаления защитной группы Q.
Защитной группой Q, предпочтительно, является группа трифторацетат или трет-бутоксикарбонил. Предпочтительным способом по изобретению является такой, где стадию (i) проводят с соединением формулы
где R16 и R17 оба означают Н. Предпочтительно, стадию (i) проводят в полярном растворителе, более предпочтительно, в воде, ТГФ, ДМФ, ДМСО, в смеси воды и любого из ТГФ, ДМФ или ДМСО.
Если не указано иначе, используемый здесь термин "галоген" включает фтор, хлор, бром и йод.
Если не указано иначе, используемый здесь термин "алкил" включает группы с прямой цепью с достаточным числом атомов углерода, разветвленные и циклические группы.
Используемый здесь термин "алкокси" означает "-О-алкил" или "алкил-О-", где "алкил" определен выше.
Используемый здесь термин "алкилен" означает радикал алкил, имеющий два доступных центра связывания (т.е. -алкил-), где "алкил" определен выше.
Если не указано иначе, используемый здесь термин "один или несколько заместителей" относится к числу заместителей от одного до максимального числа заместителей, возможного на основе числа доступных центров связывания.
Если не указано иначе, используемый здесь термин "лечение" относится к обратному развитию, облегчению, ингибированию развития или предотвращению болезни или состояния, по отношению к которому указанный термин применяется, или одного или нескольких симптомов такого состояния или болезни. Используемый здесь термин "лечение" относится к лечебному воздействию, где лечебное воздействие определено выше.
Соединения формулы (II), которые получают согласно изобретению, могут иметь оптические центры и поэтому могут иметь различные энантиомерные конфигурации. Изобретение включает все энантиомеры, диастереомеры и другие стереоизомеры таких соединений формулы (II), а также их рацемические смеси. Данное изобретение также относится ко всем радиоактивно меченным формам соединений формулы (II), полученным способами по изобретению. Предпочтительными мечеными соединениями формулы (II) являются такие, где радиоактивные метки выбраны из3Н,11С,14С,18F,123I и125I. Такие меченые соединения могут использоваться в качестве исследовательских и диагностических инструментов в исследованиях метаболизма, таких как фармакокинетические исследования и т.п., и в анализах связывания в организме животных и человека.
Это изобретение также относится к способам получения фармацевтически приемлемых аддитивных солей кислот и соединений формулы (II) с использованием способов по изобретению. Примерами фармацевтически приемлемых аддитивных солей кислот и соединений формулы (II) являются соли хлористоводородной кислоты, п-толуолсульфоновой кислоты, фумаровой кислоты, лимонной кислоты, янтарной кислоты, салициловой кислоты, щавелевой кислоты, бромистоводородной кислоты, фосфорной кислоты, метансульфоновой кислоты, винной кислоты, яблочной кислоты, ди-п-толуоилвинной кислоты и миндальной кислоты, а также соли, полученные из других кислот, известные специалистам в этой области, образующие фармацевтически приемлемые аддитивные соли кислот с соединениями основного характера. Другими возможными аддитивными солями кислот являются, например, соли, содержащие фармацевтически приемлемые анионы, такие как гидройодид, нитрат, сульфат или бисульфат, фосфат или кислый фосфат, ацетат, лактат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат и памоат (например, 1,1'-метилен-бис-(2-гидрокси-3-нафтоат) соли).
Данное изобретение также относится к способам получения меченных изотопами соединений, идентичных тем, которые соответствуют формуле (II), или их фармацевтически приемлемых солей, но в которых фактически один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличающиеся от атомной массы или атомного числа, обычно встречающихся в природе. Примеры изотопов, которые могут быть введены в соединения по данному изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, фтора и хлора, такие как2Н,3Н,13С,14С,15 N,18О,17O,31P,32P,35S,18F и36Cl, соответственно.
Изобретение также относится к способам получения соединений по данному изобретению, их пролекарств и фармацевтически приемлемых солей указанных соединений или указанных пролекарств, которые содержат указанные изотопы и/или другие изотопы других атомов, они находятся в сфере действия данного изобретения. Конкретные меченные изотопами соединения по данному изобретению, например те, в которые введены радиоактивные изотопы, такие как3Н и14С, могут использоваться, например, в анализах распределения в тканях лекарства и/или субстрата. Изотопы трития, т.е.3Н, и углерода-14, т.е.14С, особенно предпочтительны, благодаря легкости их получения и обнаружения. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е.2Н, позволяет достигать конкретных терапевтических преимуществ, являющихся результатом большей метаболической стабильности, например увеличенного in vivo периода полураспада или требований уменьшенной дозировки, и, следовательно, может быть предпочтительным в некоторых случаях. Меченные изотопами соединения формулы I по изобретению и их пролекарства, как правило, могут быть получены осуществлением способов, описанных здесь, путем замещения легко доступным меченным изотопом реагентом не меченного изотопом реагента.
Подробное описание изобретения
Соединения формул (Ia), (Ib) и (Ic) или их смеси могут быть получены в соответствии с новым способом, показанном на схеме реакции 1. Схема 2 относится к дополнительному альтернативному пути получения этих соединений. Соединения формул (VII), (VIII), (II), (II') и (II'') могут быть получены согласно схемам 3 и 4. Если не указано иначе, значения R1, R2, R3, R4, R5, R6, R6', R6'', R7, R8, R9, R10, R11, R12, R13, R14, R15, R16, R17, R18, R20, X, X', L, Q и Z являются такими, как описано выше.

Данное изобретение относится к новым средствам построения структуры соединений формул (Ia), (Ib) и (Ic) посредством конденсации арил-производного с алкоксиакрилатом с получением промежуточного соединения для циклизации в объединенных или в раздельных стадиях. Палладиевый катализатор способствует циклизации до соответствующих производных индена формул (Ia), (Ib) и (Ic) или их смесей. Схема 1 изображает общий процесс получения инденов по изобретению.
Стадия 1 схемы 1 является реакцией конденсации. Арил-производное формулы (IV) подвергают взаимодействию с алкоксиакрилатом формулы (V) в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 1,4-диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, N-метилпирролидинон, метиленхлорид, 1,2-дихлорметан, метанол, этанол, изопропанол или пропанол, предпочтительно, тетрагидрофуран, 2-метилтетрагидрофуран или 1,2-диметоксиэтан, и добавляют к основанию, такому как трет-бутоксид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, трет-пентоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, гидрид натрия или гидрид лития, предпочтительно, трет-бутоксид натрия или метоксид натрия, в одном из указанных выше растворителей при температуре между -78°С и 50°С, предпочтительно, между 0°С и 30°С, в течение периода от 1 до 24 часов, предпочтительно, 6 часов, с получением соединение формулы (III), которое может быть смесью олефиновых изомеров.
Стадия 2 схемы 1 является катализируемой палладием реакцией циклизации. Соединение формулы (III) обрабатывают источником палладия, таким как ацетат палладия(II), хлорид палладия(II), хлорид бис-ацетонитрил палладия(II), хлорид бис-бензонитрил палладия(II), бромид палладия(II), трис(дибензилиденацетон) дипалладий(0), предпочтительно, ацетат палладия(II), и фосфиновым лигандом, таким как дициклогексилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, три-изопропилфосфин, три-н-пропилфосфин, три-изобутилфосфин, три-н-бутилфосфин, три-о-толилфосфин, трифенилфосфин, 2-(дициклогексилфосфино)бифенил или 2-дициклогексилфосфино-2'-(N, N-диметиламино)бифенил, предпочтительно, трициклогексилфосфин или три-трет-бутилфосфин, и основанием, таким как трет-бутоксид натрия, трет-бутоксид калия, трет-пентоксид натрия, этоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия, гидрид лития или гидрид калия, предпочтительно, трет-бутоксид натрия или трет-пентоксид натрия, в растворителе, таком как 1, 2-диметоксиэтан, тетрагидрофуран, 2-метилтетрагидрофуран, 1,2-диэтоксиэтан, 1, 4-диоксан, толуол, N-метилпирролидинон, диметилацетамид, диметилформамид, предпочтительно, 1,2-диметоксиэтан или тетрагидрофуран, при температуре между комнатной температурой и 120°С, предпочтительно, между 50 и 90°С, в течение периода времени от 1 до 48 часов, предпочтительно, 8 часов, с получением соединения формулы (Ia), (Ib) или (Ic) (или их смесь), которое может быть смесью олефиновых региоизомеров (Ib) и (Ic). Катализируемые палладием альфа-арилирования карбонильных соединений обсуждаются во многих литературных источниках. Kawatsura, M., Hartwig, J.F., J. Am. Chem. Soc, 1999, 121, 1473-1478; патент США №6057456; Hamann, B.C.; Hartwig, J.F., J. Am. Chem. Soc, 1997, 119, 12382-12383; Fox, J.M., Huang, X., Chieffi, A., Buchwald, S.L. J. Am. Chem. Soc., 2000, 122, 1360-1370; Palucki M., Buchwald, S.L. J. Am. Chem. Soc., 1997, 119, 11108-11109; WO 00/02887.

В качестве альтернативы, вместо двух стадий 1 и 2 на схеме 1 может быть осуществлена единственная стадия схемы 2. Единственная стадия схемы 2 является тандемом конденсации и катализируемой палладием циклизации. Производное арена формулы (IV) подвергают взаимодействию с алкосиакрилатом формулы (V) в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, 1, 2-диметоксиэтан, 1,2-диэтоксиэтан, 1,4-диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, N-метилпирролидинон, метиленхлорид, 1,2-дихлорметан, предпочтительно, тетрагидрофуран, 2-метилтетрагидрофуран или 1,2-диэтоксиэтан, и добавляют к реакционной смеси, содержащей основание, такое как трет-бутоксид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, трет-пентоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, гидрид натрия или гидрид лития, предпочтительно, трет-бутоксид натрия или метоксид натрия, источник палладия (здесь может быть вставлена кипятильная плитка с палладиевым катализатором), таким как ацетат палладия(II), хлорид палладия(II), хлорид бис-ацетонитрил палладия(II), хлорид бис-бензонитрил палладия(II), бромид палладия(II), трис(дибензилиденацетон)дипалладий(0), предпочтительно, ацетат палладия(II), и фосфиновый лиганд, такой как дициклогексилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, три-изопропилфосфин, три-н-пропилфосфин, три-изобутилфосфин, три-н-бутилфосфин, три-о-толилфосфин, трифенилфосфин, 2-(дициклогексилфосфино)бифенил или 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, предпочтительно, трициклогексилфосфин или три-трет-бутилфосфин, в одном из указанных выше растворителей при температуре между -78°С и 50°С; предпочтительно, между 0°С и 30°С, в течение периода от 1 до 24 часов, предпочтительно, от 1 до 5 часов, и затем нагревают между 25°С и 120°С, предпочтительно, между 50°С и 90°С, в течение периода от 1 до 24 часов, предпочтительно, 8 часов, чтобы получить одно из соединений формул (Ia), (Ib) или (Ic) (или смесь всех их), которое может быть также только смесью олефиновых региоизомеров формул (Ib) и (Ic).
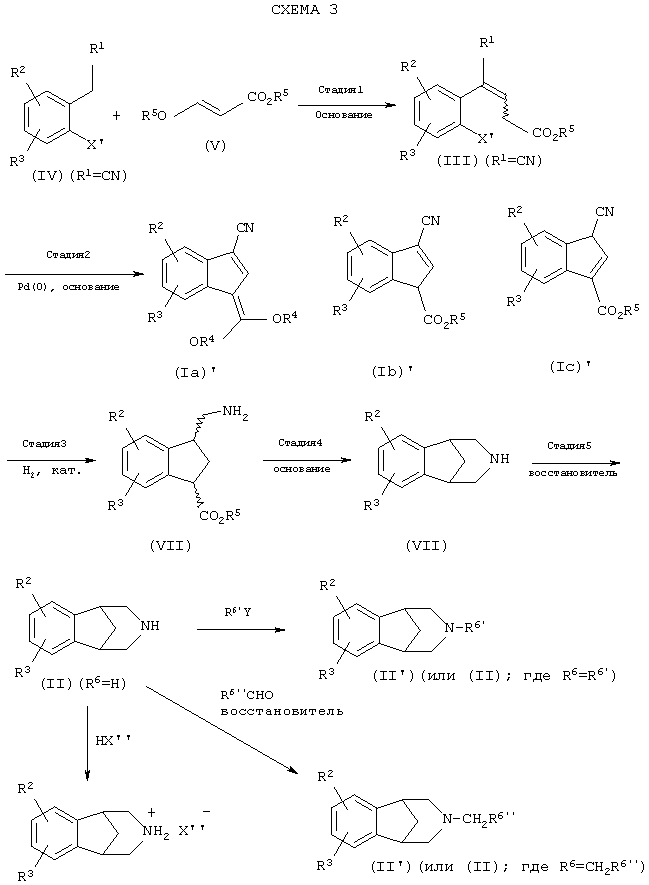
Данное изобретение также относится к новым средствам для образования структуры соединений формулы (II) с использованием производных индена формул (Ia)', (Ib)' и/или (Ic)' или их смесей (через соединения формул (Ia), (Ib) и/или (Ic), где R1 означает CN), как показано на схемах 1 и 2. Схема 3 относится к этому новому пути получения соединений формулы (II) с использованием метода катализируемой палладием циклизации. Циклизация по схеме 2 в одном реакторе может быть использована вместо стадий 1 и 2 схемы 3. После катализируемой палладием циклизации до производного (производных) индена гидрирование (схема 4 обеспечивает альтернативное двухстадийное восстановление) и обработка основанием приводят к эффективной конструкции бициклического ядра. Восстановление промежуточного циклического амида приводит к структуре ядра соединений формулы (II).

Стадия 1 схемы 3 является реакцией конденсации. Производное арилацетонитрила формулы (IV) подвергают взаимодействию с алкоксиакрилатом формулы (V) в растворителе, таком как тетрагидрофуран, 2-метилтетрагидрофуран, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 1,4-диоксан, диметилсульфоксид, диметилформамид, диметилацетамид, N-метилпирролидинон, метиленхлорид, 1,2-дихлорметан, метанол, этанол, изопропанол или пропанол, предпочтительно, тетрагидрофуран, 2-метилтетрагидрофуран или 1,2-диметоксиэтан, и добавляют к основанию, такому как трет-бутоксид натрия, трет-бутоксид калия, метоксид натрия, этоксид натрия, трет-пентоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, диизопропиламид лития, бис(триметилсилил)амид лития, бис(триметилсилил)амид натрия, бис(триметилсилил)амид калия, гидрид натрия или гидрид лития, предпочтительно, трет-бутоксид натрия или метоксид натрия, в одном из указанных выше растворителей при температуре между -78°С и 50°С, предпочтительно, между 0°С и 30°С, в течение периода от 1 до 24 часов, предпочтительно, 6 часов, с получением соединения формулы (III), которое может быть смесью олефиновых изомеров.
Стадия 2 схемы 3 является катализируемой палладием реакцией циклизации. Соединение формулы (III) обрабатывают источником палладия, таким как ацетат палладия(II), хлорид палладия(II), хлорид бис-ацетонитрил палладия(II), хлорид бис-бензонитрил палладия(II), бромид палладия(II), трис(дибензилиденацетон)дипалладий(0), предпочтительно, ацетат палладия(II), и фосфиновым лигандом, таким как дициклогексилфенилфосфин, трициклогексилфосфин, три-трет-бутилфосфин, три-изопропилфосфин, три-н-пропилфосфин, три-изобутилфосфин, три-н-бутилфосфин, три-о-толилфосфин, трифенилфосфин, 2-(дициклогексилфосфино)бифенил или 2-дициклогексилфосфино-2'-(N,N-диметиламино)бифенил, предпочтительно, трициклогексилфосфин или три-трет-бутилфосфин, и основанием, таким как трет-бутоксид натрия, трет-бутоксид калия, трет-пентоксид натрия, этоксид натрия, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия, гидрид лития или гидрид калия, предпочтительно, трет-бутоксид натрия или трет-пентоксид натрия, в растворителе, таком как 1,2-диметоксиэтан, тетрагидрофуран, 2-метилтетрагидрофуран, 1,2-диэтоксиэтан, 1, 4-диоксан, толуол, N-метилпирролидинон, диметилацетамид, диметилформамид, предпочтительно, 1,2-диметоксиэтан или тетрагидрофуран, при температуре между комнатной температурой и 120°С, предпочтительно, между 50 и 90°С, в течение периода времени от 1 до 48 часов, предпочтительно, 8 часов, с получением соединения формул (Ia)', (Ib)' и/или (Ic)' (или их смесь), которое может быть смесью олефиновых региоизомеров (Ib)' и (Ic)'. Катализируемые палладием альфа-арилирования карбонильных соединений обсуждаются во многих литературных источниках. Kawatsura, M., Hartwig, J.F., J. Am. Chem. Soc., 1999, 121, 1473-1478; патент США №6057456; Hamann, B.C.; Hartwig, J.F., J. Am. Chem. Soc., 1997, 119, 12382-12383; Fox, J.M., Huang, X., Chieffi, A., Buchwald, S.L. J. Am. Chem. Soc., 2000, 122, 1360-1370; Palucki M., Buchwald, S.L. J. Am. Chem. Soc., 1997, 119, 11108-11109; WO 00/02887. В качестве альтернативы, вместо стадий 1 и 2 схемы 3 может быть проведена единственная стадия схемы 2 (например, где R1 означает CN).
Стадия 3 схемы 3 является реакцией гидрирования нитрила и олефина. Производное индена формул (Ia)', (Ib)' или (Ic)' (или смесь любых из трех) обрабатывают катализатором гидрирования, подходящим для осуществления желаемого восстановления. Такой катализатор и условия должны быть известны специалисту в этой области, как отраженные в ссылке, такой как Comprehensive Organic Synthesis, Vol.8, "Reduction", Eds.-in-Chief, B.M.Trost and Ian Fleming (Pergamon Press NY 1991). Предпочтительно, условия гидрирования такие, где катализатором является один из следующих: палладий на углероде, гидроксид палладия на углероде, платина на углероде, оксид платины, оксид платины на углероде, более предпочтительно, выбранный из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, с кислотой, такой как серная кислота, хлористоводородная кислота, метансульфоновая кислота, толуолсульфоновая кислота, трифторуксусная кислота, уксусная кислота, муравьиная кислота, фосфорная кислота или перхлорная кислота, предпочтительно, серная кислота, метансульфоновая кислота или смесь каких-либо двух из указанных выше кислот, в растворителе, таком как метанол, этанол, изопропанол, бутанол, пропанол, этилацетат, изопропилацетат, тетрагидрофуран, толуол или любая смесь указанных растворителей, предпочтительно, метанол или этанол, в атмосфере водорода вплоть до 7 атмосфер (приблизительно 7,03 кг/см2), более предпочтительно, от 3 до 4 атмосфер (приблизительно 3,52 кг/см2), в течение периода от 1 до 24 часов, предпочтительно, 6 часов, с получением соединения формулы (VII), которое может быть смесью диастереомеров.
В качестве альтернативы стадии 3 схемы 3, вместо нее могут быть осуществлены стадии 1 и 2 схемы 4. Стадия 1 схемы 4 является гидрированием олефина. Производное индена формул (Ia)', (Ib)' или (Ic)' (или смесь любых из трех) обрабатывают катализатором гидрирования, подходящим для осуществления желаемого восстановления. Такой катализатор и условия известны специалисту в этой области, как отраженные в ссылке, такой как Comprehensive Organic Synthesis, Vol.8, "Reduction", Eds.-in-Chief, B.M.Trost and Ian Fleming (Pergamon Press NY 1991). Предпочтительно, в этом случае условия гидрирования такие, где катализатором является один из следующих: палладий на углероде, гидроксид палладия на углероде, платина на углероде, оксид платины, оксид платины на углероде, более предпочтительно, выбранный из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, с кислотой, такой как уксусная кислота, муравьиная кислота, бензойная кислота или салициловая кислота, предпочтительно, муравьиная кислота или уксусная кислота, в растворителе, таком как метанол, этанол, изопропанол, бутанол, пропанол, этилацетат, изопропилацетат, тетрагидрофуран, толуол или любая смесь указанных растворителей; предпочтительно, метанол или этанол; в атмосфере водорода вплоть до 7 атмосфер (приблизительно 7,03 кг/см2), более предпочтительно, от 3 до 4 атмосфер (приблизительно 3,52 кг/см2), в течение периода времени от 1 до 48 часов, предпочтительно, 12 часов, с получением соединения формулы (IX), которое может быть смесью диастереомеров. Условия (т.е. выбор кислоты) гидрирования на стадии 1 схемы 4, как правило, более мягкие и более селективные, чем на следующей стадии 2.
Стадия 2 схемы 4 является гидрированием нитрила. Производное индена формулы (VIII) обрабатывают катализатором гидрирования, подходящим для осуществления желаемого восстановления. Такой катализатор и условия должны быть известны специалисту в этой области, как отраженные в ссылке, такой как Comprehensive Organic Synthesis, Vol.8, "Reduction", Eds.-in-Chief, B.M.Trost and Ian Fleming (Pergamon Press NY 1991). Предпочтительно, условия гидрирования такие, где катализатором является один из следующих: палладий на углероде, гидроксид палладия на углероде, платина на углероде, оксид платины, оксид платины на углероде, более предпочтительно, выбранный из 5% палладия на углероде, 10% палладия на углероде и 5% платины на углероде, с кислотой, такой как серная кислота, хлористоводородная кислота, метансульфоновая кислота, толуолсульфоновая кислота, трифторуксусная кислота, уксусная кислота, муравьиная кислота, фосфорная кислота или перхлорная кислота, предпочтительны серная кислота, метансульфоновая кислота или любое сочетание указанных выше кислот, в растворителе, таком как метанол, этанол, изопропанол, бутанол, пропанол, этилацетат, изопропилацетат, тетрагидрофуран, толуол или любая смесь указанных растворителей, предпочтительно, метанол или этанол, в атмосфере водорода вплоть до 7 атмосфер (приблизительно 7,03 кг/см2 ), более предпочтительно, от 3 до 4 атмосфер (приблизительно, 3,52 кг/см2), в течение периода от 1 до 48 часов, предпочтительно, 6 часов, с получением соединения формулы (VII), которое может быть смесью диастереомеров.
Стадия 4 схемы 3 является образованием амида. Амин формулы (VII) обрабатывают основанием, таким как трет-бутоксид натрия, метоксид натрия, этоксид натрия, гидроксид натрия, трет-бутоксид калия, метоксид калия, этоксид калия, гидроксид калия, карбонат натрия, карбонат калия, карбонат цезия, гидрид натрия, триэтиламин, метилимидазол, лютидин, пиридин, метилморфолин, этилморфолин или диизопропилэтиламин, предпочтительно, трет-бутоксид натрия или метоксид натрия, в растворителе, таком как метанол, этанол, изопропанол, этилацетат, ацетонитрил, толуол, вода или смесь указанных растворителей, предпочтительно, метанол или смесь метанола и этилацетата, при температуре между 0 и 120°С, предпочтительно, при комнатной температуре, в течение периода времени между 30 минутами и 72 часами, предпочтительно, 6 часов, с получением соединения формулы (VIII).
Стадией 5 схемы 3 является восстановление амида. Амид формулы (VIII) обрабатывают восстановителем, таким как комплекс борана и тетрагидрофурана, диборан, комплекс борана и диметилсульфида, смешанный гидрид лития и алюминия или сочетание боргидрида натрия и трифторида бора, предпочтительно, сочетание боргидрида натрия и трифторида бора, в растворителе, таком как тетрагидрофуран, 1,2-диметоксиэтан, 1,2-диэтоксиэтан, 2-метилтетрагидрофуран, 1,4-диоксан или метил-трет-бутиловый эфир, предпочтительно, тетрагидрофуран, при температуре между 0 и 80°С, предпочтительно, при 50°С, в течение периода между 1 и 24 часами, предпочтительно, 5 часов. Продукт выделяют кристаллизацией в виде соли, такой как соль кислоты, такой как п-толуолкарбоновая кислота, метансульфоновая кислота, хлористоводородная кислота, щавелевая кислота, лимонная кислота или уксусная кислота, предпочтительна п-толуолкарбоновая кислота, в растворителе, таком как изопропанол, гексан, ацетон, этилацетат, метилэтиловый кетон или толуол, предпочтительно, изопропанол, чтобы получить соль соединения формулы (II).
Также, как показано на схеме 3, продукт формулы (II) (где R6 означает теперь Н) может быть затем выделен удалением растворителя или выделен кристаллизацией в виде соли, предпочтительно, такой как соль кислоты, такой как п-толуолкарбоновая кислота, метансульфоновая кислота, хлористоводородная кислота, щавелевая кислота, лимонная кислота или уксусная кислота, в растворителе, таком как изопропанол, гексан, ацетон, этилацетат, метилэтиловый кетон или толуол. Применение п-толуолкарбоновой кислоты с изопропанолом дает очень благоприятные результаты.
Далее, в соответствии со схемой 3, продукт формулы (II), где R6 означает R6', и R6' (или формулы (II')) означает (C1-C6)алкил, неконъюгированный (C3-C6)алкенил, бензил, -(C1-C6)алкил-СНО, -(C1 -C6 )алкил-(С=О)-(C1-C6)алкил или -СН2СН2-О-(C1-C4)алкил, может быть выделен путем взаимодействия соединения формулы (II) (где R6 означает Н) с соединением формулы R6'-Y, где Y означает удаляемую группу, такую как хлор, бром, йод или мезилат.
В дополнение, продукт формулы (II), где R6 означает (СН2)-R6''и R6'' (или формулы (II'')) означает (C1-C5)алкил, неконъюгированный (C3-C5)алкенил, фенил, -(C1-C5)алкил-СНО, -(C1-C5)алкил-(С=О)-(C1-C6)алкил или -СН2-О-(C1-C4)алкил, может быть получен взаимодействием в условиях восстановительного аминирования соединения формулы (II) (где R6 означает Н) с соединением формулы R6''-СНО. Предпочтительно, такое восстановительное аминирование может быть проведено каталитическим гидрированием или с некоторыми гидридными реагентами в инертном растворителе. Каталитическое гидрирование может быть проведено в присутствии металлического катализатора, такого как палладий или никель Ренея. Подходящие гидридные реагенты включают боргидриды, такие как боргидрид натрия (NaBH4), цианборгидрид натрия (NaBH3CN) и триацетоксиборгидрид натрия (NaB(ОАс)3Н), бораны, реагенты на основе алюминия и триалкилсиланы. Подходящие растворители включают полярные растворители, такие как метанол, этанол, метиленхлорид, тетрагидрофуран (ТГФ), диоксан и этилацетат. Эту реакцию обычно проводят при температуре от -78°С до температуры кипения растворителя с обратным холодильником, более предпочтительно, от 0°С до 25°С, в течение от 5 минут до 48 часов, предпочтительно, от 0,5 до 12 часов.
Способ по изобретению может быть осуществлен, когда группы R2 и R3, которые определены выше, уже присоединены к соединению формулы (II). В качестве альтернативы, получение соединения формулы (II) может быть осуществлено вначале получением соединения формулы (II), где одна или обе группы R2 и R3 означают Н, и затем присоединением дополнительных циклических структур (т.е. где R2 и R3 вместе с атомами углерода, к которым они присоединены, образуют четырех-семичленное моноциклическое или десяти-четырнадцатичленное бициклическое карбоциклическое кольцо, которое может быть насыщенным или ненасыщенным, где от одного до трех неконденсированных атомов углерода указанных моноциклических колец и от одного до пяти атомов углерода указанных бициклических колец, которые не являются частью бензольного кольца, показанного в формуле (II), могут быть необязательно и независимо замещены азотом, кислородом или серой) после осуществления способа по схеме 3, где одна или обе группы R2 и R3 означают водород.
Главные соединения формулы (II) могут быть получены из соединений формулы (II), где R2 и R3оба означают Н, способами, описанными в WO 99/35131, соответствующей заявке США № 09/402010, поданной 28 сентября 1999, и заявке США № 09/514002, поданной 25 февраля 2000, обе включены здесь в качестве ссылок.
Соединения формулы (II) в объеме всех значений R2 и R3, которые определены, могут быть получены соответственно или аналогично схемам 5-12 ниже.

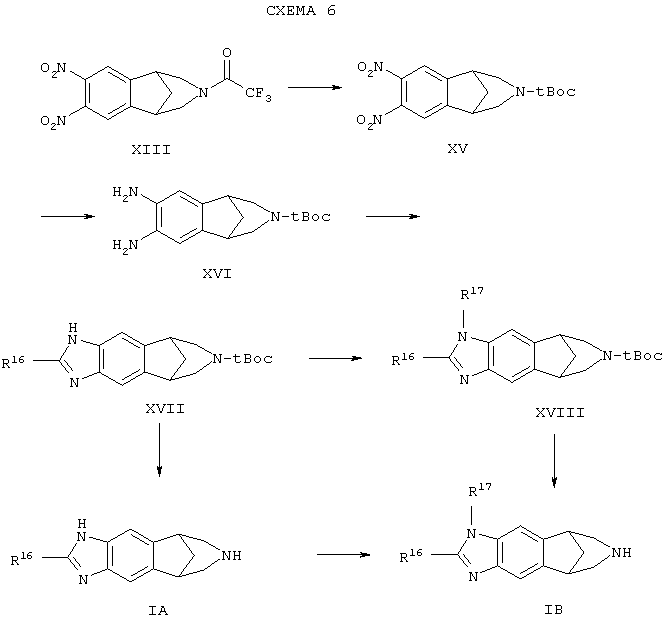






Относительно схемы 5, исходный материал формулы (XI) подвергают взаимодействию с трифторуксусным ангидридом в присутствии пиридина, с получением соединения формулы (XII). Эту реакцию обычно проводят в метиленхлориде при температуре от около 0°С до около комнатной температуры. Другие способы введения трифторуксусной защитной группы, которые могут быть использованы, известны специалистам в этой области.
Соединение формулы (XII) затем преобразуют в динитропроизводное формулы (XIII) следующим способом. Соединение формулы (XII) добавляют к смеси 4-х или более эквивалентов трифторметансульфоновой кислоты (CF3SO2OH) и 2-3-х эквивалентов азотной кислоты в хлорированном углеводородном растворителе, таком как хлороформ, дихлорэтан (ДХЭ) или метиленхлорид. Полученной смеси дают взаимодействовать от около 5 до 24 часов. Обе указанные выше реакции обычно проводят при температуре в пределах от около -78°С до около 0°С в течение 2 часов и затем дают нагреться до комнатной температуры в течение остального времени.
Восстановление соединения формулы (XIII) с использованием методов, хорошо известных специалистам, дает соединение формулы (XIV). Это восстановление может быть осуществлено, например, с использованием водорода и палладиевого катализатора, такого как гидроксид палладия или палладий на углероде, и проведением реакции в метаноле при температуре около комнатной. Стадии схемы 5 могут быть также осуществлены с защищающей азот группой, иной, чем трифторацетильная группа, которая будет специалистами признана подходящей. Другие подходящие защищающие азот группы, которые могут быть использованы в процедурах, описанных в данном документе, включают -COCF3, -COCCl3, -COOCH2CCl3, -COO(C1-C6)алкил и -COOCH2C6 H5. Эти группы могут быть присоединены или удалены способами, описанными для каждой из них, в T.W.Greene and G.M.Wuts, Protective Groups in Organic Synthesis (John Wiley & Sons, New York, 1991).
Что касается схемы 6, соединение формулы (XIII) преобразуют в соответствующее соединение, где защитную трифторацетильную группу заменяют защитной группой трет-Boc (формула (XV) путем его взаимодействия вначале с гидроксидом или карбонатом щелочного металла или щелочно-земельного металла (или аммония) и затем взаимодействия выделенного продукта предшествующей реакции с ди-трет-бутилдикарбонатом. Хотя в данном случае используют трет-Boc, могут быть использованы и другие подходящие защищающие азот группы, известные специалисту. Взаимодействие с гидроксидом или карбонатом щелочного или щелочно-земельного металла (или аммония) обычно проводят в водном спирте, диоксане или тетрагидрофуране (ТГФ) при температуре от около комнатной температуры до около 70° С, предпочтительно, при около 70°С, от около одного до около 24 часов. Взаимодействие выделенного незащищенного амина или соли такого амина с присоединенной кислотой по указанной выше реакции с ди-трет-бутилдикарбонатом, предпочтительно, проводят в растворителе, таком как ТГФ, диоксан или метиленхлорид, при температуре от около 0°С до около комнатной температуры. Эта реакция может быть проведена в присутствии основания или без него. Когда реагент является солью амина, предпочтительно, применение основания. Полученное соединение формулы (XV) может быть преобразовано в соответствующее диаминопроизводное формулы (XVI) с использованием процедуры, описанной выше для преобразования динитросоединения формулы (XIII) в соответствующее диаминосоединение формулы (XIV), или других общепринятых методов восстановления нитрогрупп, которые известны специалистам, например восстановлений, опосредуемых цинком, оловом или железом, и т.п.
Преобразование соединения формулы (XVI) в желаемое соединение формулы (XVII) может быть осуществлено взаимодействием соединения формулы (XVI) с соединением формулы (XVIII)

где R16 означает водород, (C1-C6)алкил, необязательно замещенный одним-семью атомами фтора, арил-(C0-C3)алкил, где указанный арил выбран из фенила и нафтила, или гетероарил-(C0-C3)алкила, где указанный гетероарил выбран из пяти-семичленных ароматических колец, содержащих от одного до четырех гетероатомов, выбранных из кислорода, азота и серы, и где каждая из указанных групп арила и гетероарила необязательно может быть замещена одним или несколькими заместителями, предпочтительно, от ноля до двух заместителей, независимо выбранными из (C1-C6)алкила, необязательно замещенного одним-семью атомами фтора, (C1-C6)алкокси, необязательно замещенного одним-семью атомами фтора. Предпочтительным растворителем для этой реакции является смесь 10:1 этанол/уксусная кислота. Температура реакции может находиться в пределах от около 40°С до около 100°С. Предпочтительно, она составляет около 60°С. Другие подходящие растворители включают уксусную кислоту, этанол и изопропанол.
Альтернативные способы получения соединений формулы (XVII) и формулы (XVI) описаны в Selegelstein et al., Tetrhedron Lett., 1993, 34, 1897.
Удаление трет-Boc защитной группы из соединения формулы (XVII) дает соответствующее соединение формулы (IA). Защитная группа может быть удалена с использованием методов, хорошо известных специалистам. Например, соединение формулы (XVII) может быть обработано безводной кислотой, такой как хлористоводородная кислота, бромистоводородная кислота, метансульфоновая кислота или трифторуксусная кислота, предпочтительно, хлористоводородной кислотой в этилацетате, при температуре от около 0°С до около 100°С, предпочтительно, от около комнатной температуры до около 70°С, в течение от около одного до 24 часов.
Соединение формулы (XVII) может быть преобразовано в соответствующее соединение формулы (IB) взаимодействием его с соединением формулы R17Z, где R17 определен как R16, и R16 определен выше и Z означает удаляемую группу, такую как галоген или сульфонат (например, хлор, бром, мезилат или тозилат), в присутствии основания, такого как гидрид, гидроксид или карбонат щелочного металла, предпочтительно, гидроксид калия, в полярном растворителе, таком как вода, диметилсульфоксид (ДМСО), ТГФ или ДМФ, предпочтительно, смесь ДМСО и воды, и затем удалением защитной группы, как описано выше. Реакцию с R17Z обычно проводят при температуре от около комнатной температуры до около 100°С, предпочтительно, при около 50°С, в течение около пяти часов.
Схема 7 поясняет альтернативный способ получения соединений формулы (IB) из соединения формулы (XV). Этот способ является предпочтительным способом получения соединений формулы (IB), где R17 означает объемную группу, такую как группа, содержащая арил или гетероарил, или когда группа R17 не может быть присоединена, как показано на схеме 6, методами алкилирования или замещения арила. Что касается схемы 7, соединение формулы (XV) подвергают взаимодействию с подходящим соединением формулы R17NH2 в полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно ТГФ, при температуре от около комнатной температуры до около 100°С, предпочтительно, при температуре кипения с обратным холодильником, в течение от около четырех до восемнадцати часов. Полученное соединение формулы (XIX) затем преобразуют в соответствующее соединение формулы (ХХ) восстановлением нитрогруппы в аминогруппу с использованием методов, хорошо известных специалистам в этой области. Такие методы указаны выше для преобразования соединений формулы (XIII) в соединение формулы (XIV) на схеме 1 и представлены в экспериментальных примерах 22В и 28В. Закрытие кольца имидазола с образованием соответствующего соединения формулы (XXI) может быть осуществлено затем взаимодействием соединения формулы (ХХ) из указанной выше реакции с соединением формулы (XXVIII):
где R16 имеет значения, указанные выше, как описано выше, для преобразования соединений формулы (XVI) в соединения формулы (XVII).
Удаление защитной группы из соединения формулы (XXI) дает соответствующее соединение формулы (IB). Это может быть осуществлено с использованием методов, хорошо известных в технике, например, как описано выше для получения соединений формулы (IA) из соответствующих соединений формулы (XVII).
Схема 8 поясняет способ получения соединений формулы (IC), где R16 и R17 имеют значения, указанные выше. Что касается схемы 8, соединение формулы (XVI) или аналогично формулы (XIV) на схеме 5 подвергают взаимодействию с соединением формулы

(продукт присоединения этандиона и бисульфита натрия) в воде или другом полярном растворителе, таком как ТГФ, ДМФ или ДМСО, предпочтительно, в смеси воды и смешивающегося с водой растворителя, такого как ТГФ, в течение от около одного до четырех часов. Температура реакции может находиться в пределах от около 40°С до около 100°С и, предпочтительно, около температуры кипения с обратным холодильником.
Альтернативно соединение формулы (XVI) может быть подвергнуто взаимодействию с соединением формулы

(реакция двойной конденсации) в полярном растворителе, таком как ТГФ, вода или уксусная кислота, предпочтительно, в смеси воды и ТГФ. Эту реакцию обычно проводят при температуре от около 40°С до около 100°С, предпочтительно, при температуре кипения с обратным холодильником, в течение от около одного до четырех часов. Желаемый хиноксалин формулы (IC) может быть затем образован удалением защитной группы из соединения, полученного в одной из предшествующих реакций, с использованием методов, описанных выше для преобразования соединения формулы (XVII) в соединение формулы (IA). В качестве варианта, вместо соединения (XVI) в схеме 8 в этом способе аналогично может быть использовано соединение (XIV) схемы 5 с удалением/повторным введением защитной группы, как обрисовано на схеме 6 (т.е. процесс превращения (XIII) в (XV)), для того чтобы вплотную приблизиться к соединению (IC). Как правило, альтернативные защищающие азот группы также подходят для процедуры схемы 8.
Схема 9 поясняет способ получения соединений формулы (II), где R2 и R3 вместе с бензольным кольцом, к которому они присоединены, образуют кольцевую систему бензоксазола. Так соединение, где R1 означает водород, изображено на схеме 9 в виде химической формулы (IE). Что касается схемы 9, соединение формулы (XXII), где Y' означает нитро, галоген, трифторметансульфонат или соль диазония, подвергают взаимодействию с ацетатом калия или карбоксилатом другого щелочного или щелочно-земельного металла в растворителе, таком как диметилсульфоксид (ДМСО), ДМФ или ацетонитрил, предпочтительно, ДМСО. Этому взаимодействию позволяют осуществляться в течение около 12-24 часов. Подходящий диапазон температур реакции от около 70°С до около 140°С. Предпочтительно, приблизительно 100°С.
Указанная реакция дает соединение формулы (XXIII), которое затем может быть преобразовано в желаемое соединение, имеющее формулу (IE), следующим способом. Во-первых, соединение формулы (XXIII) восстанавливают взаимодействием с водородом и палладиевым или платиновым катализатором, таким как гидроксид палладия, в метаноле при температуре от около 0° С до около 70°С, предпочтительно, при комнатной температуре, с образованием соответствующего аминопроизводного. Продукт этой реакции затем подвергают взаимодействию с хлорангидридом кислоты формулы R16COCl или ангидридом кислоты формулы (R16CO)2O, где R16 означает (C1-C6)алкил, или соединением формулы R16 С(ОС2Н5)3 в подходящем инертном растворителе, таком как декалин, хлорбензол или ксилолы. Предпочтительна смесь ксилолов. Эту реакцию обычно проводят при температуре от около 120-150°С, предпочтительно, при около 140°С. Когда в качестве реагента используют R16COCl, предпочтительно добавлять к реакционной смеси стехиометрическое количество триэтиламина (TEA) или другого органического основания третичного амина и каталитическое количество пиридиний-п-толуолсульфоновой кислоты или пиридиний-п-толуолсульфоната (PPTs). Когда в качестве реагента используют R16С(ОС2Н5)3, к реакционной смеси предпочтительно добавлять каталитическое количество PPTs.
Удаление защищающей азот трифторацетильной группы дает желаемое соединение формулы (IE). Это может быть осуществлено с использованием методов, хорошо известных специалистам, например взаимодействием защищенного соединения с низшим алканолом и водным гидроксидом или карбонатом щелочного или щелочно-земельного металла (или аммония), водным карбонатом натрия при температуре от около 50°С до около 100°С, предпочтительно, при около 70°С, в течение от около двух до шести часов.
Схема 10 иллюстрирует получение соединений формулы (II), где R1 означает водород и R2 и R3 вместе с бензольным кольцом, к которому они присоединены, образуют кольцевую систему бензотиазола. Что касается схемы 10, соединение формулы (XI) подвергают взаимодействию с трифторуксусным ангидридом, чтобы получить соответствующее соединение, где атом азота кольца защищен трифторацетильной группой, и полученное соединение с защищенным азотом затем подвергают взаимодействию с двумя эквивалентами трифторметансульфонового ангидрида и одним эквивалентом азотной кислоты, чтобы получить соответствующее соединение формулы (XXIV), где имеется единственный нитрозаместитель на бензольном кольце. Взаимодействие с трифторуксусной кислотой обычно проводят в присутствии пиридина. Обе указанные реакции обычно проводят в инертном в условиях реакции растворителе, таком как хлорированный углеводородный растворитель, предпочтительно, метиленхлорид, при температуре от около 0°С до около комнатной температуры.
Указанное преобразование может быть также осуществлено с использованием других методов нитрования, известных специалистам. Восстановление нитрогруппы до аминогруппы может быть осуществлено, как описано выше, с получением соединения формулы (XXV).
Соединение формулы (XXV) затем подвергают взаимодействию с галогенангидридом или ангидридом карбоновой кислоты формулы R16COX''' или (R16CO)2O, где X''' означает галоген и R16 означает водород или (C1-C6)алкил, и пиридином, ТЭА или другим основанием третичного амина, с получением соединения формулы (XXVI), которое затем может быть преобразовано в желаемое соединение, имеющее формулу (XXVII) при взаимодействии с реагентом Лавессона:

Реакцию с R16COX''', где X''' означает галоген, или с (R16 CO)2O обычно проводят при температуре от около 0°С до около комнатной температуры, предпочтительно, при комнатной температуре. Реакцию с реагентом Лавессона обычно проводят в инертном в условиях реакции растворителе, таком как бензол или толуол, предпочтительно, в толуоле, при температуре от около комнатной температуры до около температуры кипения реакционной смеси с обратным холодильником, предпочтительно, около температуры кипения с обратным холодильником.
Замыкание кольца бензотиазола и удаление защиты азота, чтобы получить желаемое соединение формулы (IF), может быть осуществлено при взаимодействии соединения формулы (XXVII) с феррицанидом калия и гидроксидом натрия в смеси воды и метанола (NaOH/H2O/CH3OH) при температуре от около 50°С до около 70°С, предпочтительно, при около 60°С, в течение около 1,5 часа.
Схемы 11 и 12 поясняют способы получения соединений формулы (II), где R1 означает водород и R2 и R3 представляют варианты различных заместителей, которые указаны выше, но не образуют кольца.
Схема 11 поясняет способы получения соединений формулы (II), где (а) R1 означает водород и R2 означает R7R8NO2S-, (b) R1 и R2 оба означают хлор, и (с) R1 означает водород и R2 означает R13С(=О)-. Эти соединения представлены на схеме 11, соответственно, как соединения формул IJ, IK и IL.
Что касается схемы 11, соединения формулы (IJ) могут быть получены при взаимодействии соединения формулы (XII) с двумя или более эквивалентами галогенсульфоновой кислоты, предпочтительно, хлорсульфоновой кислоты, при температуре от около 0°С до около комнатной температуры. Взаимодействие полученного таким образом производного хлорсульфоновой кислоты с амином, имеющим формулу R7R8NH, где R7 м R8 определены выше, с последующим удалением защищающей азот группы дают желаемое соединение, имеющее формулу (IJ).
Соединения формулы (IK) могут быть получены при взаимодействии соединения формулы (XII) с трихлоридом йода в хлорированном углеводородном растворителе с последующим удалением защищающей азот группы. Реакцию с трихлоридом йода обычно проводят при температуре от около 0°С до около комнатной температуры. Подобным образом, моно- или дибромированные или моно- или дииодированные соединения могут быть получены при взаимодействии соединения XII с N-иодсукцинамидом или N-бромсукцинамидом в растворителе трифторметансульфоновой кислоте с последующим удалением защищающей азот группы, как описано выше.
Взаимодействие соединения формулы XII с галогенангидридом кислоты R18COCl или ангидридом кислоты формулы (R18CO)2O, где R18 означает водород или (C1-C6)алкил, с инертным в условиях реакции растворителем, таким как хлорированный углеводородный растворитель, предпочтительно, метиленхлорид, или без него в присутствии кислоты Льюиса, такой как хлорид алюминия, при температуре от около 0°С до около 100°С с последующим удалением защиты азота дает соединение формулы (IL). Реакция с галогенангидридом или ангидридом кислоты может быть проведена с использованием других известных кислот Льюиса или других методов ацилирования Фриделя-Крафта, которые известны в технике.
Реакции, описанные здесь, когда -NO2, -SO2NR7R8, -COR1, I, Br или Cl вводят в соединение формулы (XII), как отображено на схеме 11 и описано выше, могут быть осуществлены с другим аналогичным соединением, где R2 означает водород, (C1-C6)алкил, галоген, (C1-C6)алкокси или -NHCONR7R8, с получением соединений формулы (II), где R2 и R3 имеют значения, указанные в определении соединений формулы (II) выше.
Соединения, которые идентичны соединениям формулы (IL), но которые сохраняют защищающую азот группу, могут быть преобразованы в соответствующие О-ацил-замещенные соединения, т.е. такие, где группа -С(=О)R18 в формуле (IL) заменена группой -О-С(=О)R18 с использованием процессов Байера-Виллиджера, известных специалистам. Полученные соединения могут быть частично гидролизованы, чтобы получить соответствующие гидрокси-замещенные соединения, и затем алкилированы, чтобы получить соответствующие алкокси-замещенные соединения. Кроме того, О-ацил-замещенные соединения могут быть использованы для получения разнообразно замещенных бензизоксазолов.
Схема 12 иллюстрирует способы получения соединений формулы (II), где: (а) R1 означает водород и R2 означает хлор; (b) R1 означает водород, и бензольное кольцо замещено циано; (с) R1 означает водород, и бензольное кольцо замещено амино, и (d) R1 означает водород, и бензольное кольцо замещено R18 С(=O)N(H)-. Эти соединения представлены на схеме 12, соответственно, как соединения формул (IM), (IN), (IP) и (IQ).
Соединения формулы (IM) могут быть получены из соединений формулы (XXV) путем образования соли диазиния, например, с нитритом щелочного металла и сильной минеральной кислотой (например, хлористоводородной кислотой, серной кислотой и бромистоводородной кислотой) в воде с последующим взаимодействием с солью галогенида меди, такой как хлорид меди (II). Удаление защиты азота методами, описанными выше, дает желаемое соединение формулы (IM). Альтернативные методы образования солей диазония, как известные и практикуемые специалистами в этой области, также могут быть использованы. Указанную выше реакцию обычно проводят при температурах в пределах от около 0°С до около 60°С, предпочтительно, около 60°С, в течение от около 15 минут до одного часа.
Взаимодействие соли диазония, полученной, как описано выше, с йодидом калия в водной среде обеспечивает аналогичное производное йодида. Эту реакцию обычно проводят при температуре от около 0°С до около комнатной температуры, предпочтительно, около комнатной температуры. Полученное соединение или его аналогичная N-трет-бутилкарбонатная защищенная форма могут быть использованы для получения соответствующего цианопроизводного взаимодействием с цианидом меди (II) и цианидом натрия в ДМФ, N,N-диметилпропилмочевине (DMPU) или ДМСО, предпочтительно ДМФ, при температуре от около 50°С до около 180°С, предпочтительно, около 150°С. Удаление защиты азота, как было описано выше, обеспечивает желаемое соединение формулы (IM).
Описанное выше производное йодида также может быть использовано для введения разнообразных других заместителей, таких как арил, ацетилен и винил заметители, а также соответствующих карбонильных сложных эфиров и амидов путем известных специалистам процессов, катализируемых палладием и никелем, таких как реакции сочетания Heck, Suzuki и Stille и карбонилирования Heck. Указанные соединения и другие, где R2 означает галоген, алкил, алкокси и т.д., могут быть подобным образом функционализованы, чтобы получить соединения, где R2 и R3 являются такими, как указано выше.
Удаление защиты азота соединения формулы (XXV) обеспечивает соединение формулы (IP). Соединение формулы (XXV) подвергают взаимодействию с ацильной группой, имеющей формулу R18 COCl или (R18СО)2О, с использованием методов, описанных выше, с последующим удалением защиты азота, чтобы получить соединения формулы (IQ). Подобным образом, обработка защищенного амина соединением, имеющим формулу R18SO2 X'''', где X'''' означает хлор или бром, с последующим удалением защиты азота обеспечивает соответствующее производное сульфонамида.
Как отмечалось выше, подходящие защищающие амин группы, которые могут быть использованы альтернативно в процедурах, описанных в данном документе, включают -COCF3, -COCCl3, -COOCH2CCl3, -COO(C1 -C6)алкил и -COOCH2C6H5. Эти группы могут быть удалены способами, описанными для каждой из них, в Greene et al., Protective Groups in Organic Chemistry, как упоминалось выше. Случаи, когда защитные группы могли быть модифицированы в условиях реакции, такие как, например, группа -COOCH2C6H5 в процессе нитрования, также позволяют задействовать такие способы, как описано с указанной модифицированной защитной группой. Модифицирование порядка введения защитной группы и/или методы введения или модифицирования функциональной группы также могут быть применены, где это возможно.
В каждой из реакций, обсуждавшихся выше или иллюстрированных на схемах 5-12 выше, давление не является критическим, если не указано иначе. Как правило, приемлемы велечины давления от около 0,5 атмосферы до около 5 атмосфер, по соображениям удобства предпочтительно давление окружающей среды, т.е. около 1 атмосферы.
Биологический анализ
Эффективность активных соединений, полученных способами по данному изобретению, в подавлении связывания никотина со специфическими рецепторными сайтами определяют по следующему методу, который является модификацией методов Lippiello, P.M. и Fernandes, K.G. (в The Binding of L-[3H]Nicotine To a Single Class of High-Affinity Sites in Rat Brain Membranes, Molecular Pharm., 29, 448-54, (1986)) и Anderson, D.J. и Arneric, S.P. (в Nicotinic Receptor Binding of3H-Cystisine,3H-Nicotine and3H-Methylcarbamylcholine in Rat Brain, European J.Pharm., 253, 261-67 (1994)).
Соединения формулы (II) и их фармацевтически приемлемые соли (здесь далее "активные соединения"), полученные способами по данному изобретению, могут быть введены пероральным, трансдермальным (например, с использованием пластыря), интраназальным, сублингвальным, ректальным, парентеральным или местным путем. Предпочтительны трансдермальное и пероральное введения. Эти соединения вводят, наиболее желательно, в дозировках в пределах от около 0,01 мг до около 1500 мг в сутки, предпочтительно, от около 0,1 до около 300 мг в сутки, единственной или раздельными дозами, хотя изменения обязательно будут в зависимости от массы и состояния субъекта, подвергаемого лечению, и конкретного выбранного пути введения. Однако наиболее желательно использовать уровень дозирования в пределах от около 0,001 мг до около 10 мг на кг массы тела в сутки. Изменения, тем не менее, могут происходить в зависимости от массы и состояния персон, подвергаемых лечению, и их индивидуальных реакций на указанный медикамент, а также от типа выбранного фармацевтического состава и периода времени и интервалов в то время, когда проводят такое введение. В некоторых случаях уровни дозирования ниже нижнего предела указанного диапазона могут быть более чем адекватны, тогда как в других случаях еще более высокие дозы могут быть использованы без каких-либо вредных побочных эффектов при условии, что такие повышенные дозы вначале делят на несколько малых доз для введения в течение суток.
Активные соединения по данному изобретению могут быть введены одни или в сочетании с фармацевтически приемлемыми носителями или разбавителями каким-либо из различных указанных ранее путем. Более конкретно, активные соединения могут быть введены в широком разнообразии различных лекарственных форм, например, они могут быть объединены с различными фармацевтически приемлемыми инертными носителями в виде таблеток, капсул, трансдермальных пластырей, пастилок, лепешек, леденцов, порошков, спреев, кремов, мазей, суппозиториев, желе, гелей, паст, лосьонов, мазей-суспензий, водных суспезий, растворов для инъекций, эликсиров, сиропов и тому подобное. Такие носители включают твердые разбавители или наполнители, стерильную водную среду и различные нетоксичные органические растворители. В дополнение, пероральные фармацевтические составы могут быть подходящим образом подслащены и/или ароматизированы. Как правило, активные соединения присутствуют в таких лекарственных формах при уровнях концентрации в пределах от около 5,0% до около 70% по массе.
Для перорального введения могут быть использованы таблетки, содержащие различные разбавители, такие как микрокристаллическая целлюлоза, цитрат натрия, карбонат кальция, фосфат дикальция и глицин, наряду с различными разрыхляющими веществами, такими как крахмал (предпочтительно, кукурузный, картофельный или крахмал тапиоки), альгиновая кислота и некоторые комплексные силикаты, вместе со свзующими грануляции, такими как поливинилпирролидон, сахароза, желатин и камедь акации. Дополнительно для целей таблетирования могут быть использованы смазывающие агенты, такие как стеарат магния, лаурилсульфат натрия и тальк. Твердые составы подобного типа также могут быть использованы в качестве наполнителей в желатиновых капсулах; предпочтительные материалы в этой связи также включают лактозу или молочный сахар, а также высокомолекулярные полиэтиленгликоли. Когда для орального введения желательны водные суспензии и/или эликсиры, активный ингредиент может быть объединен с различными подслащивающими или ароматизирующими агентами, окрашивающим материалом и, если желательно, эмульгирующими и/или суспендирующими агентами вместе с такими разбавителями, как вода, этанол, пропиленгликоль, глицерин и различные их сочетания.
Для парентерального введения может быть использован раствор активного соединения в кунжутном или арахисовом масле или в водном пропиленгликоле. Водные растворы могут быть подходяще забуферены (предпочтительно, рН более чем 8), если необходимо, и жидкому разбавителю придают изотоничность. Такие водные растворы являются подходящими для целей внутривенного вливания. Маслянистые растворы являются подходящими для целей внутрисуставных, внутримышечных и подкожных инъекций. Приготовление всех этих растворов в стерильных условиях легко осуществимо стандартными фармацевтическими методиками, которые хорошо известны специалистам в этой области.
Возможно также вводить активные соединения, полученные способами по данному изобретению, местно, и это может быть осуществлено посредством кремов, пластырей, желе, гелей, паст, мазей-суспензий и тому подобное в соответствии со стандартной фармацевтической практикой.
Получение других соединений по данному изобретению, не описанное конкретно в последующей экспериментальной части, может быть осуществлено с использованием сочетаний реакций, описанных выше, что очевидно специалисту. Промежуточные соединения по изобретению, указанные выше, могут содержать хиральные центры и поэтому могут существовать в различных энантиомерных и диастереомерных формах; данное изобретение относится ко всем таким оптическим и стереоизомерам указанных промежуточных соединений, а также к их смесям.
ПРИМЕРЫ
Данное изобретение поясняется следующими примерами. Следует учесть, однако, что изобретение не ограничивается конкретными деталями этих примеров.
ПРИМЕР 1

МЕТИЛОВЫЙ ЭФИР 4-(2-ХЛОР-4-ТРИФТОРМЕТИЛ-ФЕНИЛ)-4-ЦИАНО-БУТ-3-ЕНОВОЙ КИСЛОТЫ
К раствору трет-бутоксида натрия (3,28 г, 34,2 ммоль) в тетрагидрофуране (20 мл) при 0°C добавляют раствор метил-3-метоксиакрилата (2,65 г, 22,8 ммоль) и (2-хлор-4-трифторметил-фенил)ацетонитрила (5,00 г, 22,8 ммоль) в тетрагидрофуране (20 мл) по каплям в течение 45 минут. Раствор красного цвета перемешивают при 0°C в течение 3 часов и затем дают нагреться до 10°C в течение еще 2 часов. Реакционную смесь затем обрабатывают 0,2 M водной лимонной кислотой (50 мл) и экстрагируют смесью 2:1 метил-трет-бутилового эфира и гексана (75 мл). Органический слой промывают водным насыщенным раствором хлорида натрия (50 мл). Органический раствор пропускают через короткую колонку силикагеля, промывая остаточный материал через пад дополнительным метил-трет-бутиловым эфиром. Фильтрат концентрируют в вакууме и получают метиловый эфир 4-(2-хлор-4-трифторметилфенил)-4-циано-бут-3-еновой кислоты в виде масла красного цвета (6,74 г, 97%). Основной олефиновый изомер:1H ЯМР (400 МГц, CDCl3) δ 7,71 (с, 1H), 7,58 (д, 1H, J=8,3), 7,49 (д, 1H, J=8,3), 6,89 (т, 1H, J=7,1), 3,78 (с, 3H), 3,69 (д, 2H, J=7,1);13C ЯМР (100 МГц, CDCl3) δ 169,4, 145,9, 136,1, 133,7, 133,0 (кв., J=33, 5), 131,5, 127,6 (кв., J=3,8), 124,5 (кв., J=3,8), 123,2 (кв., J=292), 115,4, 114,7, 52,7, 36,7; IR (ATR, neat) 2957, 2224, 1740, 1438, 1395, 1319, 1268, 1172, 1128, 1080, 1010, 963, 892, 831, 719, 655 см-1, Побочный олефиновый изомер:1H-ЯМР (400 МГц, CDCl3) δ 7,76 (с, 1H), 7,61 (д, 1H, J=7,8), 7,43 (д, 1H, J=7,8), 7,02 (т, 1H, J=7,8), 3,71 (с, 3H), 3,11 (д, 2H, J=7,8).
ПРИМЕР 2

3-(ГИДРОКСИМЕТОКСИМЕТИЛЕН)-5-ТРИФТОРМЕТИЛ-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
Раствор метилового эфира 4-(2-хлор-4-трифторметилфенил)-4-циано-бут-3-еновой кислоты (1,00 г, 3,29 ммоль), ацетата палладия(II) (37,0 мг, 0,165 ммоль) и трициклогексилфосфина (55 мг, 0,197 ммоль) в диметиловом эфире этиленгликоля (8,0 мл) перемешивают в течение 10 минут при комнатной температуре в атмосфере азота и затем добавляют трет-бутоксид натрия (791 мг, 8,23 ммоль). Реакционную смесь нагревают с обратным холодильником в течение 48 часов. Реакционную смесь охлаждают до комнатной температуры и распределяют между метил-трет-бутиловым эфиром (25 мл) и 0,25 M водным дигидрофосфатом калия (25 мл). Водный слой отделяют и обрабатывают этилацетатом (30 мл) и 4,0 M водной соляной кислотой (3 мл). Экстракт этилацетата промывают насыщенным водным раствором хлорида натрия (20 мл), разбавляют метанолом (10 мл) и перемешивают с карбонатом калия (25 мг, 0,181 ммоль) в течение 1 часа. Органический раствор концентрируют в вакууме и получают 3-(гидроксиметоксиметилен)-5-трифторметил-3H-инден-1-карбонитрил, натриевую соль в виде вспенного твердого вещества красного цвета (727 мг, 83%).1H ЯМР (400 МГц, CD3OD) δ 8,29 (с, 1H), 7,73 (с, 1H), 7,50 (д, 1H, J=8,3), 7,12 (д, 1H, J=8,3), 3,82 (с, 3H);13C ЯМР (100 МГц, CD3OD) δ 169,1, 138,7, 134,7, 132,2, 127,9 (кв., J=270), 123,4, 121,8 (кв., J=30,5), 118,5 (кв., J=4,3), 118,3, 115,7 (кв., J=3,8), 105,2, 81,0, 50,9; IR (ATR, neat) 2953, 2179, 1612, 1466, 1325, 1389, 1325, 1283, 1194, 1153, 1101, 1073, 1014, 900, 838, 814, 777, 753, 708, 644 см-1.
ПРИМЕР 3
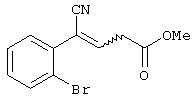
МЕТИЛОВЫЙ ЭФИР
4-(2-БРОМФЕНИЛ)-4-ЦИАНО-БУТ-3-ЕНОВОЙ КИСЛОТЫ
Раствор 2-бром-фенилацетонитрила (6,62 мл, 51,0 ммоль) и метил-3-метоксиакрилата (5,32 мл, 49,5 ммоль) в тетрагидрофуране (10 мл) добавляют к суспензии трет-бутоксида натрия (5,05 г, 51,0 ммоль) в тетрагидрофуране (70 мл) при 0°C в течение 10 минут в токе азота. Реакционную смесь нагревают до комнатной температуры и через 15 минут разбавляют метил-трет-бутиловым эфиром (240 мл). К смеси добавляют 0,2 н водную лимонную кислоту до тех пор, пока водный слой не будет иметь pH 2 (80 мл). Органический слой отделяют и промывают насыщенным водным раствором хлорида натрия (2×80 мл), сушат над безводным сульфатом натрия, фильтруют через пад из целита и диоксида кремния (4:1) и концентрируют в вакууме и получают метиловый эфир 4-(2-бромфенил)-4-циано-бут-3-еновой кислоты в виде масла бледно-желтого цвета (13,8 г, 96%). Главный изомер1H ЯМР (400 МГц, CDCl3) δ 7,59 (дд, 1H, J=7,6, 1,2), 7,57-7,19 (м, 3H), 6,71 (т, 1H, J=7,1), 3,71 (с, 3H), 3,61 (д, 2H, J=7,1);13C ЯМР (100 МГц, CDCl3) δ 169,7, 144,4, 133,7, 131,4, 131,1, 128,3, 122,5, 118,1, 117,3, 115,3, 52,7, 36,6; IR (ATR, neat) 1737, 1434, 1319, 1199, 1171, 1026, 755 см-1. Побочный изомер1H ЯМР (400 МГц, CDCl3) δ 7,61 (дд, 1H, J=7,6, 1,0), 7,57-7,19 (м, 3H), 6,9 (т, 1H, J=7,5), 3,64 (с, 3H), 3,06 (д, 2H, J=7,5).
ПРИМЕР 4

3-(ГИДРОКСИМЕТОКСИМЕТИЛЕН)-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
Раствор метилового эфира 4-(2-бромфенил)-4-циано-бут-3-еновой кислоты (12,4 г, 44,4 ммоль) в диметиловом эфире этиленгликоля (100 мл) продувают азотом в течение 10 минут и помещают в него трициклогексилфосфин (311 мг, 1,11 ммоль) и затем ацетат палладия(II) (199 мг, 0,890 ммоль). Смесь перемешивают при комнатной температуре до тех пор, пока реакционная смесь не станет гомогенным раствором, и помещают трет-бутоксид натрия (11,0 г, 111 ммоль). Реакционную смесь нагревают до 85°C и перемешивают в течение 5 часов, затем охлаждают до комнатной температуры и разбавляют метил-трет-бутиловым эфиром (300 мл). К мутной смеси добавляют 0,25 M водный раствор дигидрофосфата калия (300 мл) и водный слой отделяют и подкисляют 4,0 M водной соляной кислотой (50 мл), затем экстрагируют этилацетатом (300 мл). Органический слой отделяют и промывают насыщенным водным раствором хлорида натрия (2× 200 мл), разбавляют метанолом (150 мл) и сушат над карбонатом калия (2,00 г). Через 2 часа гетерогенную суспензию фильтруют через пад из целита и сырой продукт концентрируют в вакууме с получением натриевой соли 3-(гидроксиметоксиметилен)-3H-инден-1-карбонитрила в виде твердого вещества темнокоричневого цвета (6,80 г, 77%). Сырое твердое вещество растворяют в безводном MeOH (30 мл) и хранят в виде раствора.1H ЯМР (400 МГц, d4-MeOH) δ 7,82-7,30 (м, 5H), 3,66 (с, 3H);13C ЯМР (100 МГц, d4-MeOH) δ 168,3, 135,7, 132,3, 131,9, 123,2, 120,2, 119,3, 118,6, 117,2, 102,7, 79,4, 49,6; IR (ATR, neat) 2175, 1608, 1453, 1385, 1322, 1256, 1189, 1065, 1013, 751, 632 см-1.
ПРИМЕР 5

3-(ЭТОКСИГИДРОКСИМЕТИЛЕН)-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
В раствор трициклогексилфосфина (21,5 мг, 0,0770 ммоль) в диметиловом эфире этиленгликоля (10 мл) в атмосфере азота помещают ацетат палладия(II) (11,5 мг, 0,0510 ммоль). Реакционную смесь перемешивают при комнатной температуре до образования гомогенного раствора (приблизительно 15 минут), охлаждают до 0°C и добавляют трет-бутоксид натрия (2,53 г, 25,5 ммоль). Через 5 минут добавляют по каплям в течение 10 минут раствор 2-бром-фенилацетонитрила (1,32 мл, 10,2 ммоль) и этил-3-этоксиакрилата (1,47 мл, 10,2 ммоль) в диметиловом эфире этиленгликоля (10 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры, затем нагревают до 85°C в течение 1 часа. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют этилацетатом (50 мл) и выливают в водный дигидрофосфат калия (0,25 M, 50 мл), pH=7. Водный слой насыщают добавлением хлорида натрия в виде твердого вещества и органический слой отделяют и промывают водным насыщенным хлоридом натрия (1×50 мл), сушат над безводным сульфатом натрия, фильтруют и концентрируют в вакууме, получая натриевую соль 3-(этоксигидроксиметилен)-3H-инден-1-карбонитрила в виде масла темно-коричневого цвета (1,74 г, 84%), которое затвердевает при стоянии.1H ЯМР (400 МГц, CD3CN) δ 8,04 (д, 1H, J=6,0), 7,58 (с, 1H), 7,43 (д, 1H, J=6,0), 6,98-6,91 (м, 2H), 4,25 (кв., 2H, J=7,2), 1,35 (т, 3H, J=7,2);13C ЯМР (100 МГц, CD3CN) δ 166,7, 135,5, 132,3, 131,3, 122,8, 120,5, 119,0, 118,4, 117,7, 103,3, 79,2, 58,2, 14,6; IR (ATR, neat) 2176, 1597, 1465, 1257, 1195, 1068, 1029, 754 см-1.
ПРИМЕР 6

3-(ЭТОКСИГИДРОКСИМЕТИЛЕН)-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
В раствор трициклогексилфосфина (204 мг, 0,720 ммоль) в диметиловом эфире этиленгликоля (10 мл) в атмосфере азота помещают ацетат палладия(II) (148 мг, 0,660 ммоль). Реакционную смесь перемешивают при комнатной температуре до образования гомогенного раствора (приблизительно 25 минут), охлаждают до 0°C и добавляют трет-бутоксид натрия (1,63 г, 16,6 ммоль). Через 10 минут добавляют по каплям в течение 5 минут раствор 2-хлорфенилацетонитрила (1,00 г, 6,60 ммоль) и этил-3-транс-этоксиакрилата (953 мкл, 6,60 ммоль) в диметиловом эфире этиленгликоля (10 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры и затем нагревают до 85°C в течение 22 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют метил-трет-бутиловым эфиром (30 мл) и выливают в водный дигидрофосфат калия (0,25 M, 40 мл), pH=7. Водный слой отделяют, затем насыщают добавлением твердого хлорида натрия и экстрагируют этилацетатом (1×50 мл). Органический слой отделяют и промывают водным насыщенным хлоридом натрия (2×30 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая натриевую соль 3-(этоксигидроксиметилен)-3H-инден-1-карбонитрила в виде масла светло-оранжевого цвета (1,06 г, 5,0 ммоль, 75%). Физические данные смотри в примере 5 выше.
ПРИМЕР 7

3-(ЭТОКСИГИДРОКСИМЕТИЛЕН)-5,6-ДИМЕТОКСИ-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
В раствор трициклогексилфосфина (82,0 мг, 0,293 ммоль) в диметиловом эфире этиленгликоля (10 мл) в атмосфере азота помещают ацетат палладия(II) (43,7 мг, 0,195 ммоль). Реакционную смесь перемешивают при комнатной температуре, пока раствор не станет гомогенным, (приблизительно 15 минут) и перемешивают дополнительные 5 минут перед охлаждением до 0°C и добавляют трет-бутоксид натрия (996 мг, 9,75 ммоль). Через 5 минут добавляют по каплям в течение 10 минут раствор 2-бром-4, 5-диметоксифенилацетонитрила (1,00 г, 3,90 ммоль) и этил-3-этоксиакрилата (0,564 мл, 3,90 ммоль) в диметиловом эфире этиленгликоля (5 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры и затем нагревают до 85°C в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют метил-трет-бутиловым эфиром (50 мл) и выливают в водный дигидрофосфат калия (0,25 M, 100 мл). Водный слой отделяют и твердый хлорид натрия добавляют к водному слою до насыщения. Водный слой экстрагируют этилацетатом (1×125 мл) и этот органический слой промывают водным насыщенным хлоридом натрия (2×35 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая натриевую соль 3-(этокси-гидроксиметилен)-5, 6-диметокси-3H-инден-1-карбонитрила в виде темно-коричневого масла (906 мг, 3,3 ммоль, 85%), которое кристаллизуется при стоянии.1H ЯМР (400 МГц, d4-MeOH) δ 7,64 (с, 1H), 7,46 (с, 1H), 6,99 (с, 1H), 4,56 (кв., 2H, J=7,1), 3,86 (с, 6H), 1,38 (т, 3H, J=7,05);13C ЯМР (100 МГц, d4-MeOH) δ 167,8, 145,0, 144,5, 130,2, 129,4, 126,4, 123,3, 112, 5, 104,0, 102,6, 100,7, 79,0, 58,4, 55,6, 14,1; IR (ATR, neat) 3499, 2164, 1629, 1482, 1449, 1282, 1207, 1157, 1124, 1076, 845, 769 см-1 .
ПРИМЕР 8
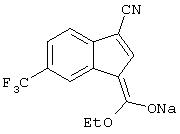
3-(ГИДРОКСИЭТОКСИМЕТИЛЕН)-5-ТРИФТОРМЕТИЛ-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
Раствор ацетата палладия(II) (102 мг, 0,454 ммоль) и трициклогексилфосфина (153 мг, 0,546 ммоль) в диметиловом эфире этиленгликоля (10 мл) перемешивают в течение 15 минут при комнатной температуре в атмосфере азота и затем добавляют трет-бутоксид натрия (2,19 г, 22,8 ммоль). Реакционную смесь охлаждают до 0°C и добавляют по каплям раствор (2-хлор-4-трифторметилфенил)ацетонитрила (2,00 г, 9,11 ммоль) и этил-3-этоксиакрилата (1,45 мл, 10,0 ммоль) в диметиловом эфире этиленгликоля (10 мл). Сразу после завершения добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 4 часов. Реакционную смесь затем нагревают до 60°C в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры и распределяют между метил-трет-бутиловым эфиром (45 мл), гексаном (20 мл) и 0,25 M водным дигидрофосфатом калия (60 мл). Водный слой отделяют, насыщают хлоридом натрия и экстрагируют этилацетатом (60 мл). Экстракт этилацетата сушат над безводным сульфатом натрия и концентрируют в вакууме с получением натриевой соли 3-(гидроксиэтоксиметилен)-5-трифторметил-3H-инден-1-карбонитрила в виде вспененного твердого вещества красного цвета (1,86 г,67%).
1H ЯМР (400 МГц, CD3OD) δ 8,31 (с, 1H), 7,70 (с, 1H), 7,49 (д, 1H, J=7,9), 7,11 (д, 1H, J=7,9), 4,28 (кв., 2H, J=7,1), 1,39 (т, 3H, J=7,1);13C ЯМР (100 МГц, CD3OD) δ 167,6, 137,4, 133,4, 131,0, 126,7 (кв., J=201), 122,1, 120,5 (кв., J=22,7), 117,3 (кв., J=3,3), 117,1, 114,3 (кв., J=2,5), 104,2, 79,6, 58,7, 14,0; IR (ATR, neat) 2986, 2943, 2180, 1606, 1465, 1326, 1284, 1197, 1156, 1105, 1076, 1027, 900, 853, 814, 778, 753, 708, 645 см-1.
ПРИМЕР 9
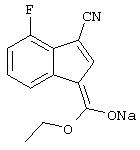
3-(ЭТОКСИГИДРОКСИМЕТИЛЕН)-7-ФТОР-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
В раствор трициклогексилфосфина (248 мг, 0,880 ммоль) в диметиловом эфире этиленгликоля (5 мл) в атмосфере азота помещают ацетат палладия(II) (132 мг, 0,590 ммоль). Реакционную смесь перемешивают при комнатной температуре до образования гомогенного раствора (приблизительно 15 минут), охлаждают до 0°C и добавляют трет-бутоксид натрия (1,46 г, 14,7 ммоль). Через 5 минут добавляют по каплям в течение 5 минут раствор 2-хлор-6-фторфенилацетонитрила (1,00 г, 5,90 ммоль) и этил-3-этоксиакрилата (877 мкл, 5,90 ммоль) в диметиловом эфире этиленгликоля (5 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры и затем нагревают до 60°C в течение 17 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют метил-трет-бутиловым эфиром (30 мл) и выливают в водный дигидрофосфат калия (0,25 M, 40 мл), pH=7. Водный слой отделяют, затем насыщают добавлением твердого хлорида натрия и экстрагируют этилацетатом (1×40 мл). Органический слой отделяют и промывают водным насыщенным хлоридом натрия (2×25 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая натриевую соль 3-(этоксигидроксиметилен)-7-фтор-3H-инден-1-карбонитрила в виде масла светло-оранжевого цвета (1,21 г, 4,8 ммоль, 81%), которое затвердевает при стоянии. Растиранием в CH2Cl2 (10 мл) в течение 2 часов и фильтрованием получают натриевую соль 3-(этокси-гидроксиметилен)-7-фтор-3H-инден-1-карбонитрила в виде твердого вещества белого цвета (1,09 г, 4,4 ммоль, 74%).1 H ЯМР (400 МГц, CD3CN) δ 7,81 (д, 1H, J=7,9), 7, 46 (с, 1H), 6,82 (кв., 1H, J=2,5), 6,52 (дд, 1H, J=7,5, 11,2), 4,20 (кв., 2H, J=7,5), 1,32 (т, 3H, J=7,1);13C ЯМР (100 МГц, d4-MeOH) 167,8, 156,3, 135,8, 132,8, 123,5, 122,3, 119,3, 116,2, 104,0, 102,8, 75,5, 58,6, 14,0; IR (ATR, neat) 2194, 1628, 1546, 1484, 1462, 1238, 1219, 1095, 1019, 925, 788, 737 см-1.
ПРИМЕР 10

3-(ЭТОКСИГИДРОКСИМЕТИЛЕН)-5-ФТОР-3H-ИНДЕН-1-КАРБОНИТРИЛ, НАТРИЕВАЯ СОЛЬ
В раствор трициклогексилфосфина (248 мг, 0,880 ммоль) в диметиловом эфире этиленгликоля (5 мл) в атмосфере азота помещают ацетат палладия(II) (132 мг, 0,590 ммоль). Реакционную смесь перемешивают при комнатной температуре до образования гомогенного раствора (приблизительно 15 минут), охлаждают до 0° C и добавляют трет-бутоксид натрия (1,46 г, 14,7 ммоль). Через 5 минут добавляют по каплям в течение 5 минут раствор 2-хлор-6-фторфенилацетонитрила (1,00 г, 5,90 ммоль) и этил-3-этоксиакрилата (877 мкл, 5,90 ммоль) в диметиловом эфире этиленгликоля (5 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры и затем нагревают до 60°C в течение 16 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют метил-трет-бутиловым эфиром (30 мл) и выливают в водный дигидрофосфат калия (0,25 M, 40 мл), pH=7. Водный слой отделяют, затем насыщают добавлением твердого хлорида натрия и экстрагируют этилацетатом (1×40 мл). Органический слой отделяют и промывают водным насыщенным хлоридом натрия (2×25 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая натриевую соль 3-(этоксигидроксиметилен)-5-фтор-3H-инден-1-карбонитрила в виде масла светло-оранжевого цвета (1,29 г, 5,1 ммоль, 86%), которое затвердевает при стоянии. Растиранием в CH2Cl2 (10 мл) в течение 3 часов и фильтрованием получают натриевую соль 3-(этоксигидроксиметилен)-5-фтор-3H-инден-1-карбонитрила в виде твердого вещества желтовато-белого цвета (1,21 г, 4,8 ммоль, 82%).1H ЯМР (400 МГц, d4-MeOH) δ 7,63 (дд, 1H, J=2,5, 11,4), 7,60 (с, 1H), 7,32 (dd, 1H, J=5,2, 8,5), 6,67 (дт, 1H, J=2,5, 7,1), 4,26 (кв., 2H, J=7,1), 1,38 (т, 3H, J=7,1);13C ЯМР (100 МГц, d4-MeOH) 168,1, 159,2, 132,65, 131,92, 122,92, 117,7, 112,5, 106,4, 105,2, 102,8, 79,8, 58,7, 14,1, IR (ATR, neat) 2179, 1602, 1558, 1465,1250, 1196, 1108, 1061, 1026, 775 см-1; т.пл. 250-260°C.
ПРИМЕР 11
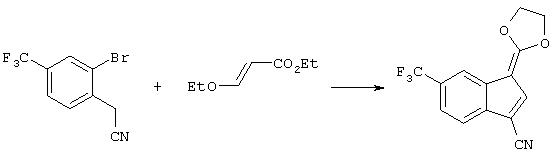
3-[1,3]ДИОКСОЛАН-2-ИЛИДЕН-5-ТРИФТОРМЕТИЛ-3H-ИНДЕН-1-КАРБОНИТРИЛ
Раствор ацетата палладия(II) (79,2 мг, 0,353 ммоль) трициклогексилфосфина (148 мг, 0,527 ммоль) в тетрагидрофуране (5 мл) перемешивают в течение 30 минут при комнатной температуре в атмосфере азота и затем добавляют трет-бутоксид натрия (1,69 г, 17,5 ммоль). Реакционную смесь охлаждают до 0°C и добавляют по каплям раствор (2-бром-4-трифторметил-фенил)-ацетонитрила (2,00 г, 7,04 ммоль) и этил 3-этоксиакрилата (1,12 г, 7,77 ммоль) в тетрагидрофуране (5 мл). После завершения добавления реакционной смеси дают нагреться до комнатной температуры и перемешивают в течение 4 1/2 часов. Реакционную смесь затем нагревают до 60°C в течение 18 часов. Реакционную смесь охлаждают до комнатной температуры и добавляют этиленгликоль (20 мл) с последующим добавлением по каплям концентрированной серной кислоты (3 мл). После перемешивания реакционной смеси в течение ночи продукт осаждается из раствора. Реакционную смесь фильтруют, чтобы изолировать твердое вещество, и твердое вещество растирают с 15 мл толуола (чтобы удалить окраску). Получают 3-[1, 3]диоксолан-2-илиден-5-трифторметил-3H-инден-1-карбонитрил в виде твердого вещества бледно-желтого цвета (1,22 г, 62%).1H ЯМР (400 МГц, CD3CN) δ 8,15 (д, 1H, J=0,9), 7,90 (с, 1H), 7,75 (д, 1H, J=8,1), 7,57 (дд, 1H, J=8,1, 0,9), 5,06-5,00 (м, 2H), 4,94-4,88 (м, 2H);13C ЯМР (100 МГц, d8-ТГФ) 169,5, 141,4, 138,5, 134,6, 127,5 (кв., J=271), 127,4 (кв., J=31,6), 121,6 (кв., J=3,8), 121,3, 119,1 (кв., J=4,1), 117,9, 100,8, 94,6, 72,6, 71,4; IR (ATR, neat) 2205, 1605, 1330, 1268, 1235, 1216, 1161, 1101, 1065, 1015, 985, 914, 859, 828,704, 639, 545, 531 см-1.
ПРИМЕР 12

3-[1, 3]ДИОКСОЛАН-2-ИЛИДЕН-3H-ИНДЕН-1-КАРБОНИТРИЛ
В раствор трициклогексилфосфина (536 мг, 1,91 ммоль) в тетрагидрофуране (25 мл) помещают ацетат палладия(II) (287 мг, 1,27 ммоль) в атмосфере азота. Через 1 час реакционную смесь охлаждают до 0°C и добавляют трет-бутоксид натрия (31,6 г, 319 ммоль). Через 5 минут добавляют по каплям в течение 15 минут раствор 2-бром-фенилацетонитрила (25,0 г, 128 ммоль) и этилового эфира β-этоксиакриловой кислоты (18,4 мл, 128 ммоль) в тетрагидрофуране (75 мл). Реакционную смесь нагревают 60°C. Через 2 часа 30 минут реакционную смесь охлаждают до комнатной температуры и добавляют этиленгликоль (200 мл) в течение 5 минут и затем добавляют по каплям в течение 15 минут серную кислоту (18,8 M, 36 мл). Через 15 часов реакционную смесь разбавляют водой (90 мл) и твердый продукт фильтруют через стеклянную фритту. Твердое вещество сушат в вакууме, получая 3-[1,3]диоксолан-2-илиден-3H-инден-1-карбонитрил (21, 6 г, 102 ммоль, 80%) в виде твердого вещества желтоватого цвета. Сырой материал суспендируют в изопропаноле (50 мл) в течение 2 часов, фильтруют и сушат в вакууме, получая 3-[1, 3]диоксолан-2-илиден-3H-инден-1-карбонитрил (20,8 г, 98,5 ммоль, 77%) в виде твердого вещества желтоватого цвета.1H ЯМР (400 МГц, d6-DMSO) δ 7,75 (с, 1H), 7,74 (д, 1H, J=7,9), 7,50 (д, 1H, J=7,1), 7,22 (м, 2H), 4,97 (т, 2H, J=7,8), 4,85 (т, 2H, J=7,8);13C ЯМР (100 МГц, d6-DMSO) 167,4, 136,7, 135,7, 133,1, 124,7, 123,9, 121,1, 119,4, 118,1, 97, 8, 92,7, 71,1, 69,9; т.пл. (разложение) 228°C.
ПРИМЕР 13

ЭТИЛОВЫЙ ЭФИР
3-БЕНЗОЛСУЛЬФОНИЛ-3H-ИНДЕН-1-КАРБОНОВОЙ КИСЛОТЫ
В раствор трициклогексилфосфина (46 мг, 0,164 ммоль) в тетрагидрофуране (2,5 мл) в атмосфере азота помещают ацетат палладия(II) (24,5 мг, 0,109 ммоль). Реакционную смесь перемешивают в течение 45 минут, затем охлаждают до 0° C и добавляют трет-бутоксид натрия (270 мг, 2,73 ммоль). Через 5 минут добавляют по каплям в течение 2 минут раствор этилового эфира β-этоксиакриловой кислоты (158 мкл, 1,09 ммоль) и 2-бромбензилфенилсульфона (340 мг, 1,09 ммоль) в тетрагидрофуране (2,5 мл). После завершения добавления реакционную смесь нагревают до комнатной температуры и затем нагревают до 60°C в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры, затем разбавляют метил-трет-бутиловым эфиром (25 мл) и выливают в водный дигидрофосфат калия (0,25 M, 25 мл), pH=7. Водный слой отделяют и насыщают добавлением твердого хлорида натрия и экстрагируют этилацетатом (2×50 мл). Органический слой отделяют и промывают дважды водным насыщенным хлоридом натрия (2×30 мл), сушат над сульфатом натрия, фильтруют и концентрируют в вакууме, получая этиловый эфир 3-бензолсульфонил-3H-инден-1-карбоновой кислоты в виде масла светло-оранжевого цвета (304 мг, 0,925 ммоль, 85%).1H ЯМР (400 МГц, CDCl3) δ 7,88-7,86 (м, 1H), 7,75-7,72 (м, 1H), 7,49-7,43 (м, 3H), 7,36-7,32 (м, 2H), 7,27-7,25 (м, 3H), 5,15 (д, 1H, J=2,1), 4,27 (кв., 2H, J=7,1), 1,34 (т, 3H, J=7,1); IR(ATR, neat) 1709, 1649, 1463, 1444, 1314, 1271, 1247, 1188, 1133, 1081, 1044, 773, 752, 722, 686, 575, 528.
ПРИМЕР 14

2,3,4,5-ТЕТРАГИДРО-1,5-МЕТАНО-2-ОКСО-1H-3-БЕНЗАЗЕПИН
Раствор метилового эфира 3-циано-1H-инден-1-карбоновой кислоты (6,40 г, 32,2 ммоль) в MeOH (30 мл) в реакторе, работающем под давлением, продувают азотом в течение 5 минут и добавляют 5% палладий на углероде (6,84 г, 1,61 ммоль) и концентрированную серную кислоту (3,57 мл, 64,3 ммоль). Реактор, работающий под давлением, продувают азотом и откачивают его три раза, затем продувают водородом и откачивают его три раза перед наполнением сосуда высокого давления водородом до давления 50 фунт/кв.дюйм (3,4 атмосферы) и механически встряхивают. Через 5 минут давление водорода снижают до 15 фунт/кв.дюйм (приблизительно 1 атмосфера) и реактор заполняют водородом, чтобы повысить давление до 50 фунт/кв.дюйм (3,4 атмосферы). Через 16 часов реактор откачивают и продувают азотом три раза перед фильтрованием реакционной смеси через найлоновый диск. Метанол вытесняют изопропанолом и упаривают досуха с изопропанолом, чтобы удалить воду. Концентрированный раствор растворяют в безводном метаноле и добавляют трет-бутоксид натрия (15,9 г, 161 ммоль) при 0°C и суспензию перемешивают в атмосфере азота при постепенном нагревании до комнатной температуры. После 24 часов при комнатной температуре реакционную смесь концентрируют в вакууме, и полученное твердое вещество растворяют а этилацетате (60 мл) и гасят 0,5 M водным дигидрофосфатом калия (100 мл). Органический слой промывают насыщенным водным раствором хлорида натрия (2×125 мл) и сушат над безводным сульфатом натрия. Органический раствор концентрируют в вакууме, получая 2,3,4,5-тетрагидро-1,5-метано-2-оксо-1H-3-бензазепин в виде твердого вещества коричневого цвета (4,02 г, 72%).1H ЯМР (400 МГц, CDCl3): δ 7,33 (д, 1H, J=7,6), 7,31 (д, 1H, J=7,6), 7,22 (т, 1H, J=7,6), 7,18 (т, 1H, J=7,6), 5,62 (с, 1H), 3,68 (дд, 1H, J=11,2, 4,1), 3,55 (д, 1H, J=3,7), 3,43-3,37 (м, 1H), 3,18 (д, 1H, J=11,2), 2,52-2,45 (м, 1H), 2,32 (д, 1H, J=11,2);13C ЯМР (100 МГц, CDCl3): δ 173,6, 144,7, 144,6, 128, 0, 127,7, 123,2, 122,9, 49,3, 47,9, 39,1, 38,4; IR (чистый) 3218, 2949, 2872, 1666, 1485, 1459, 1400, 1328, 1303, 1288, 1250, 1215, 1122, 1104, 1045, 1004, 946, 910, 756, 730, 643, 613 см-1 .
ПРИМЕР 15

ЭТИЛОВЫЙ ЭФИР 3-ЦИАНОИНДАН-1-КАРБОНОВОЙ КИСЛОТЫ
Раствор натриевой соли 3-(этоксигидроксиметилен)-3Н-инден-1-карбонитрила (1,00 г, 4,70 ммоль) в этаноле (40 мл) в реакторе, работающем под давлением, продувают азотом в течение 5 минут и добавляют 5% палладий на углероде (1, 00 г, 0,230 ммоль) и муравьиную кислоту (0,180 мл, 4,70 ммоль), рН=5 при начале реакции. Реактор, работающий под давлением, продувают азотом и откачивают его три раза, затем продувают водородом и откачивают его три раза перед загрузкой сосуда высокого давления водородом при давлении 50 фунт/кв.дюйм (3,4 атмосферы) и механически встряхивают. Через 2 часа реактор откачивают и продувают азотом три раза перед фильтрованием реакционной смеси через найлоновый диск. Азеотропно удаляют муравьиную кислоту в вакууме этанолом и после концентрирования в вакууме получают этиловый эфир 3-цианоиндан-1-карбоновой кислоты в виде твердого вещества светло-желтого цвета (880 мг, 87%).1H ЯМР (400 МГц, CD3CN) δ 7,47-7,45 (м, 2H), 7,37-7, 35 (м, 2H), 4,28 (т, 1H, J=8,0), 4,20 (м, 2H), 4,16 (т, 1H, J=3), 2,80 (м, 1H), 2,60 (м, 1H), 1,27 (т, 3H, J=7,2);13C ЯМР (100 МГц, CD3CN) δ 172,3, 140,8, 138,5, 129,0, 128,7, 125,4, 124,7, 117,6, 61,3, 48,9, 33,4, 33,3, 13,8;13C ЯМР (100 МГц, d4-MeOH) δ 172,6, 140,4, 138,1, 129,0, 128,8, 125,4, 124,7, 120,9, 61,4, 48,9, 33,2, 33,1, 13, 4; IR (ATR, neat) 1732, 1478, 1457, 1370, 1324, 1263, 1208, 1180, 1035, 747.
ПРИМЕР 16
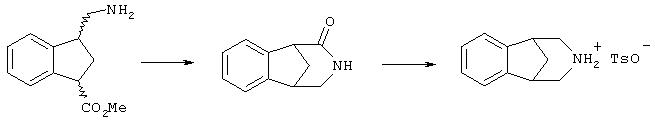
10-АЗА-ТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-ТОЗИЛАТ
А) 9-Оксо-10-аза-трицикло[6.3.1.02,7]додека-2(7),3, 5-триен
К раствору метилового эфира 3-аминометилиндан-1-карбоновой кислоты (предположительно 20,0 ммоль, 1 эквивалент) в 50 мл метанола добавляют 3,84 г трет-бутоксида натрия (40,0 ммоль, 2,0 эквивалента). Реакционную смесь нагревают до температуры кипения с обратным холодильником в течение 2 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют в вакууме. Остаток распределяют между 60 мл этилацетата и 40 мл 5% водного раствора бикарбоната натрия. Водный слой экстрагируют дважды еще 50 мл этилацетата. Объединенные органические слои сушат над безводным сульфатом натрия и концентрируют с получением твердый материал. Перекристаллизация твердого вещества из 10 мл толуола дает белые кристаллы указанного в заголовке соединения (1,78 г, 51%). Т.пл.=172-173°С;1H ЯМР (400 МГц, CDCl3): δ 7,33 (д, J=7,6 Гц, 1H), 7,31 (д, J=7,6Гц, 1H), 7,22 (т, J=7,6 Гц, 1H), 7,18 (т, J=7,6 Гц, 1H), 5,62 (с, 1H), 3,68 (дд, J=11,2, 4,1 Гц, 1H), 3,55 (д, J=3,7 Гц, 1H), 3,43-3,37 (м, 1H), 3,18 (д, J=11,2 Гц, 1H), 2,52-2,45 (м, 1H), 2,32 (д, J=11,2 Гц, 1H);13C ЯМР (100 МГц, CDCl3): δ 173,6, 144,7, 144,6, 128,0, 127,7, 123,2, 122,9, 49,3, 47,9, 39,1, 38,4; IR (neat, см-1): 3218, 2949, 2872, 1666, 1485, 1459, 1400, 1328, 1303, 1288, 1250, 1215, 1122, 1104, 1045, 1004, 946, 910, 756, 730, 643, 613; Анал. рассч. для С11Н21 NO: С, 76,28; H, 6,40; N, 8,09; Найдено: С, 75,94; H, 6,27; N, 7,99.
B) 10-Аза-трицикло[6.3.1.02,7 ]додека-2(7),3,5-триен тозилат
К раствору 1,38 г 9-оксо-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триена (8,00 ммоль, 1 эквивалент) в 8 мл тетрагидрофурана добавляют 603 мг боргидрида натрия (16,0 ммоль, 2,0 эквивалента) с последующим медленным добавлением 2,77 мл диэтил-эфирата трифторида бора (21,6 ммоль, 2,7 эквивалента). После завершения выделения газа реакционную смесь нагревают до 50°C в течение 5 часов. Реакционную смесь затем охлаждают до комнатной температуры для добавления 10 мл метанола (сначала добавляют по каплям) и 0,125 мл концентрированной хлористоводородной кислоты. Нагревание продолжают при кипении с обратным холодильником в течение 12 часов. Реакционную смесь затем охлаждают до комнатной температуры и концентрируют в вакууме. Остаток разбавляют 20 мл 20% водного гидроксида натрия, а затем 30 мл метил-трет-бутилового эфира. Смесь перемешивают в течение 30 минут и затем водный слой экстрагируют еще 30 мл метил-трет-бутилового эфира. Объединенные органические слои промывают 40 мл насыщенного водного раствора хлорида натрия и сушат над безводным сульфатом натрия. После концентрирования в вакууме добавляют 1,67 г моногидрата п-толуолсульфоновой кислоты (8,80 ммоль, 1,1 эквивалента) с 20 мл изопропанола. Раствор нагревают, пока он не станет гомогенным, и затем дают постепенно охладиться до комнатной температуры при перемешивании. Белые кристаллы указанного в заголовке соединения получают и собирают фильтрованием (2,17 г, 81%), т.пл.: 207-208°C;1H ЯМР (400 МГц, CD3OD): δ 7,69 (д, J=7,9 Гц, 2H), 7,43-7,32 (м, 4H), 7,23 (д, J=7,9 Гц, 2H), 3,37 (д, J=11,2 Гц, 4H), 3,30 (ушир.с, 2H), 3,15 (д, J=12,4 Гц, 2H), 2,36 (с, 3H), 2,40-2,35 (м, 1H), 2,08 (д, J=11,2 Гц, 1H);13C ЯМР (100 МГц, CD3OD): δ 140,8, 140,5, 139,1, 127, 2, 127,2, 124,3, 122,3, 45,1, 39,7, 37,3, 18,7; IR (KBr, см-1): 3438, 3021, 2958, 2822, 2758, 2719, 2683, 2611, 2424, 1925, 1606, 1497, 1473, 1428, 1339, 1302, 1259, 1228, 1219, 1176, 1160, 1137, 1122, 1087, 1078, 945, 914, 876, 847, 829, 818, 801, 710, 492; Анал. рассч. для C18H21NO3S: C, 65,23; H, 6,39; N, 4,23; Найдено: C, 65,05; H, 6,48; N, 4,26.
ПРИМЕР 17
ГИДРОХЛОРИД 4-НИТРО-10-АЗАТРИЦИКЛО[6.3.1.02, 7]ДОДЕКА-2(7),3,5-ТРИЕНА
А) 1-(10-Аза-трицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон
Соль гидрохлорид 10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триена (12,4 г, 63,9 ммоль) перемешивают в CH2Cl2 (200 мл). Это охлаждают (на на бане со льдом) и обрабатывают пиридином (12,65 г, 160 ммоль), а затем трифторуксусным ангидридом (TFAA) (16,8 г, 11,3 мл, 80 ммоль) из капельной воронки в течение 10 минут. Через ˜3 часа раствор выливают в 0,5 н водную HCl (200 мл) и слои разделяют. Водный слой экстрагируют CH2Cl2 (3×50 мл) и объединенный органический слой промывают 0, 5 н водной HCl (50 мл), Н2О (2×50 мл) и насыщенным водным раствором NaHCO3 (50 мл). Этот раствор сушат через хлопковую пробку, затем разбавляют ˜3% этилацетата и фильтруют через пад из диоксида кремния, элюируют смесью ˜3% этилацетат/CH2Cl2. Концентрирование дает прозрачное масло, которое кристаллизуется с образованием белых игольчатых кристаллов (15,35 г, 60,2 ммоль, 94%). (ТСХ 30% этилацетат/гексан Rf0,53).1H ЯМР (400 МГц, CDCl3) δ 7,18 (м, 4H), 4,29 (ушир.д, J=12,6 Гц, 1H), 3, 84 (ушир.д, J=12,6 Гц, 1H), 3,51 (дд, J=12,6,1,5 Гц, 1H), 3, 21 (ушир.с, 1H), 3,10 (ушир.с, 1H), 3,10 (ушир.д, J=12,6 Гц, 1H), 2,37 (м, 1H), 1,92 (д, J=10,8 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 255 (M+), т.пл. 67-68°C.
В) 1-(4-Нитро-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон
(На основе метода, описанного Coon, C.L.; Blucher, W.G.; Hill, M.E. J. Org. Chem. 1973, 25, 4243). К раствору трифторметансульфоновой кислоты (2,4 мл, 13,7 ммоль) в CH2Cl2 (10 мл), перемешиваемому при 0°С, медленно добавляют азотную кислоту (0,58 мл, 27,4 ммоль), получая белый осадок. Через 10 минут полученную смесь охлаждают до -78°С и обрабатывают 1-(10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтаноном (3,5 г, 13,7 ммоль) в CH2Cl2 (15 мл) по каплям из капельной воронки в течение 5 минут. Реакционную смесь перемешивают при -78°С в течение 30 минут, затем нагревают до 0°С в течение 1 часа. Реакционную смесь выливают на энергично перемешиваемый лед (100 г). Слои разделяют и водный слой экстрагируют CH2Cl2 (3×30 мл). Органический слой объединяют и промывают Н2О (3×30 мл). Объединенный органический слой промывают насыщенным водным раствором NaHCO3 (20 мл) и Н2О (20 мл), затем сушат через хлопковую пробку и концентрируют с получением масла оранжевого цвета, которое затвердевает при стоянии (4,2 г). Хроматография дает чистый продукт в виде кристаллического твердого вещества (3,2 г, 78%) (ТСХ 30% этилацетат/гексан Rf0,23).1H ЯМР (400 МГц, CDCl3) δ 8,12 (ушир.д, J=8,0 Гц, 1H), 8,08 (ушир.с, 1H), 7,37 (ушир.д, J=8,0 Гц, 1H), 4,38 (ушир.д, J=12,6 Гц, 1H), 3,94 (ушир.д, J=12,6 Гц, 1H), 3,59 (ушир.д, J=12,6 Гц, 1H), 3,43-3,35 (м, 2H), 3,18 (ушир.д, J=12, 6 Гц, 1H), 2,48 (м, 1H), 2,07 (д, J=10,8 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 300 (M+).
C) Гидрохлорид 4-нитро-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триена
1-(4-Нитро-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (182 мг, 0,61 ммоль) перемешивают с Na2CO3 (160 мг, 1,21 ммоль) в метаноле (3 мл) и H2O (1 мл) при 70°C в течение 18 часов. Смесь концентрируют, добавляют воду и продукт экстрагируют CH2Cl2. Органический слой экстрагируют 1 н. водной HCl (3 x 20 мл) и кислотный слой промывают CH2Cl2 (2×20 мл). Водный слой подщелачивают Na2CO3 (тв.) до pH ˜10 и продукт экстрагируют CH2Cl2 (3×30 мл). Органический слой сушат через хлопковую пробку и концентрируют до масла. Его растворяют в метаноле и обрабатывают 1 н. HCl в метаноле, концентрируют до твердых веществ, которые подвергают перекристаллизации из смеси метанол/Et2O с получением продукта в виде твердого вещества белого цвета (73 мг, 50%), (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,38).1H ЯМР (400 МГц, DMSO-d6) δ 8,21 (с, 1H), 8,18 (дд, J=8,0,2,0 Гц, 1H), 7,59 (д, J=8,0 Гц, 1H), 3,43 (ушир.с, 2H), 3,28 (м, 2H), 3,07 (дд, J=13,0,13,0 Гц, 2H), 2,24 (м, 1H), 2,08 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 205,1 [(M+1)+], т.пл. 265-270°C.
ПРИМЕР 18
ГИДРОХЛОРИД 4-АМИНО-10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА
4-Нитро-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен (500 мг, 2,08 ммоль) перемешивают в 1,4-диоксане (40 мл) и обрабатывают насыщенным водным раствором Na2CO3 (15 мл). К этому добавляют ди-трет-бутил-дикарбонат (1,8 г, 8,31 ммоль), После перемешивания 18 часов реакционную смесь обрабатывают H2O (50 мл), экстрагируют CH2Cl2 (4×30 мл), сушат через хлопковую пробку и концентрируют с получением масла (500 мг, 91%).
Это масло (500 мг, 1,64 ммоль) растворяют в метаноле (30 мл), обрабатывают 10% Pd/C (˜50 мг) и гидрируют в атмосфере водорода (45 фунт/кв.дюйм или приблизительно 3 атмосферы) в течение 1 часа. Смесь фильтруют через пад из целита и концентрируют до прозрачного масла (397 мг, 88%).
Это масло (50 мг, 0,18 ммоль) перемешивают в 3 н. HCl в этилацетате (3 мл) в течение 2 часов, затем концентрируют до твердого вещества белого цвета (25 мг, 56%).1H ЯМР (400 МГц, DMSO-d6) δ 7,38-7,10 (3H), 3,60 (ушир.с, 2H), 3,25 (м, 2H), 2,98 (м, 2H), 2,18 (м, 1H), 1,98 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 175,1 [(M+1)+], т.пл. 189-192°C.
ПРИМЕР 19
ГИДРОХЛОРИД N1-[10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-4-ИЛ]АЦЕТАМИДА
A) 1-(4-Амино-10-аза-трицикло[6.3.1.02,7]додека-2(7), 3,5-триен-10-ил)-2,2,2-трифторэтанон
Гидрирование 1-(4-нитро-10-аза-трицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанона (2,0 г, 6,66 ммоль) в атмосфере водорода (40 фунт/кв.дюйм или приблизительно 2,7 атмосферы) и в присутствии 10% Pd/C (200 мг) в метаноле в течение 1,5 часов, фильтрование через целит и концентрирование дают масло желтого цвета (1,7 г). (ТСХ 50% этилацетат/гексан Rf 0,27).1H ЯМР (400 МГц, CDCI3) δ 6,99 (м, 1H), 6,64 (ушир.с, 1H), 6,57 (м, 1H), 4,25 (м, 1H), 3,82 (м, 1H), 3,50 (м, 1H), 3, 17-3,07 (м, 3H), 2,35 (м, 1H), 1,90 (д, J=10,8 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 270 (M+).
B) N-(10-Трифторацетил-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-4-ил)ацетамид
1-(4-Амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон (850 мг, 3,14 ммоль) перемешивают в CH2 Cl2 (5 мл) и обрабатывают триэтиламином (0,53 мл, 3,76 ммоль) и ацетилхлоридом (0,23 мл, 3,2 ммоль), затем перемешивают 18 часов. Стандартная обработка NaHCO3 дает масло, которое затем подвергают хроматографии и получают прозрачное масло (850 мг, 87%), (50% этилацетат/гексан Rf 0,28).
C) Гидрохлорид N1-[10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ил]ацетамида
N-(10-Трифторацетил-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-4-ил)ацетамид (100 мг, 0,32 ммоль) перемешивают с Na2CO3 (70 мг, 0,64 ммоль) в метаноле (10 мл) и H2O (2 мл) при 70°C в течение 18 часов. Смесь концентрируют, добавляют воду и продукт экстрагируют этилацетатом. Органический слой экстрагируют 1 н. водной HCl (3×20 мл) и кислотный слой промывают этилацетатом (2×20 мл). Водный слой подщелачивают Na2CO3 (тв.) до pH ˜10 и продукт экстрагируют этилацетатом (3×20 мл). Органический слой сушат (сульфат натрия (Na2SO4 )) и концентрируют до масла. Этот материал растворяют в метаноле и обрабатывают 3 н. HCl в этилацетате (3 мл), концентрируют и подвергают перекристаллизации из смеси метанол/Et2O с получением твердого вещества (40 мг, 50%).1H ЯМР (400 МГц, DMSO-d6) δ 9,98 (с, 1H), 9,02 (ушир.м, NH), 7,65 (с, 1H), 7,55 (ушир.с, NH), 7,38 (д, J=8,0 Гц, 1H), 7,20 (д, J=8,0 Гц, 1H), 3,33 (м, 4H), 2,96 (м, 2H), 2,13 (м, 1H), 2,00 (с, 3H), 1,96 (д, J=10,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 217,2 [(M+1)+], т.пл. 225-230° C.
ПРИМЕР 20
ГИДРОХЛОРИД 6-МЕТИЛ-5-ТИА-7, 13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
A) N-(10-Трифтортиоацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-4-ил)тиоацетамид
N-(10-Трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-4-ил)ацетамид (850 мг, 2,72 ммоль) и 2,4-бис(4-метоксифенил)-1,3-дитиа-2, 4-дифосфетан-2,4-дисульфид (реагент Лавессона) (1,1 г, 2,72 ммоль) объединяют в толуоле (10 мл) и доводят до кипения с обратным холодильником в течение 1,5 часов. После охлаждения реакционную смесь обрабатывают этилацетатом/насыщенным водным раствором NaHCO3. Органический слой сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии на силикагеле с получением продукта (410 мг, 44%). (50% этилацетат/гексан Rf 0,38).
B) Гидрохлорид 6-метил-5-тиа-7,13-диазатетрацикло [9.3.1. 02,10,04,8 ]пентадека-2(10),3,6,8-тетраена
Указанное масло, а именно 2,2, 2-трифтор-N-(10-трифтортиоацетил-10-азатрицикло[6.3.1.04,8]додека-2(7),3,5-триен-4-ил)тиоацетамид, (360 мг, 1, 05 ммоль) растворяют в метаноле (10 мл) и 1 н. NaOH (5 мл) и добавляют к феррицианиду калия (K3Fe(CN)6) (1,72 г, 5,23 ммоль) в H2O (10 мл). Эту смесь нагревают до 60°C в течение 1,5 часов, охлаждают, концентрируют и обрабатывают этилацетатом/H2O. Этот материал перемешивают в диоксане (20 мл) и обрабатывают H2O (50 мл) и Na2CO3 до достижения pH 10. К этому добавляют ди-трет-бутилдикарбонат (436 мг, 2,0 ммоль) и смесь перемешивают в течение 18 часов. Реакционную смесь концентрируют, обрабатывают H2O и экстрагируют CH2Cl2. Продукт подвергают хроматографии (Диоксид кремния 30% этилацетат/гексан Rf 0,41) с получением масла (100 мг).
Указанный продукт обрабатывают 3 н. HCl/этилацетатом (3 мл) и нагревают до кипения с обратным холодильником в течение ˜15 минут, затем концентрируют до твердого вещества, которое подвергают азеотропной обработке CH2Cl2 (два раза). Эти твердые вещества растворяют в минимальном количестве метанола, затем насыщают Et2O и перемешивают. Полученный белый кристаллический порошок собирают фильтрованием (40 мг, 14%).1H ЯМР (400 МГц, DMSO-d6) δ 9,46 (с, NH), 7,65 (с, 1H), 7,82 (с, 1H), 7,65 (ушир.м, NH), 3,36 (м, 2H), 3,24 (м, 2H), 3,02 (м, 2H), 2,76 (с, 3H), 2,23 (м, 1H), 2,06 (д, J=10,8 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 231,1 [(M+1)+], т.пл. 183-184°C.
ПРИМЕР 21
4,5-ДИНИТРО-10-АЗАТРИЦИКЛО[6.3.1.02,7 ]ДОДЕКА-2(7),3,5-ТРИЕН
A)1-(4,5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон
(На основе метода, описанного в Coon, C. L.; Blucher, W. G.; Hill, M. E. J.Org. Chem. 1973, 25, 4243. Дополнительный относящийся к делу пример динитрования смотри: Tanida, H.; Ishitobi, H.; Irie, T.; Tsushima, T. J. Am. Chem. Soc. 1969, 91, 4512.)
К раствору трифторметансульфоновой кислоты (79,8 мл, 902,1 ммоль) в CH2Cl2 (550 мл), перемешиваемому при 0°C, медленно добавляют азотную кислоты (19, 1 мл, 450,9 ммоль), получая белый осадок. Через 10 минут добавляют по каплям 1-(10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (50 г, 196 ммоль) в CH2 Cl2 (300 мл) из капельной воронки в течение 30 минут. Реакционную смесь перемешивают при 0°C в течение 2,5 часов и затем перемешивают при комнатной температуре в течение 24 часов. Реакционную смесь выливают в энергично перемешиваемую смесь H2O (500 мл) и льда (400 г). Слои разделяют и водный слой снова экстрагируют CH2Cl2 (3×300 мл). Органический слой объединяют и промывают H2O (3×300 мл). Объединенные водные слои повторно экстрагируют CH2Cl2 (2×100 мл). Органический слой объединяют и промывают насыщенным водным раствором NaHCO3 (200 мл) и H2O (200 мл), затем сушат через хлопковую пробку и концентрируют до твердых веществ. Растирание с этилацетатом/гексанами дает белые твердые вещества, которые фильтруют и сушат (52 г, 151 ммоль, 77%). Маточную жидкость подвергают хроматографии и получают дополнительные 4,0 г для общего выхода 56,0 г (82,8%). (ТСХ 50% этилацетат/гексан Rf 0,29).1H ЯМР (400 МГц, CDCl3) δ 7,77 (с, 1H), 7,75 (с, 1H), 4,39 (ушир.д, J=13,0 Гц, 1H), 3,98 (ушир.д, J=13,0 Гц, 1H), 3,65 (д, J=13,0 Гц, 1H), 3,49 (ушир.с, 1H), 3,44 (ушир.с, 1H), 3,24 (ушир.д, J=12,6 Гц, 1H), 2,53 (м, 1H), 2,14(д, J=11,5Гц, 1 H), газовая хроматография и масс-спектроскопия m/e 345 (M+).
B) 4, 5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен
1-(4,5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон (3,7 г, 10,7 ммоль) и Na2CO3 (2,3 г, 21,4 ммоль) объединяют в метаноле (50 мл) и H2O (20 мл), затем нагревают до кипения с обратным холодильником в течение 18 часов. Реакционную смесь охлаждают, концентрируют, обрабатывают H2O и экстрагируют CH2Cl2 (3×50 мл), затем сушат через хлопковую пробку. После концентрирования остаток подвергают хроматографии и получают твердое вещество коричневого цвета (1,9 г, 71%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,36).1H ЯМР (400 МГц, CDCl3) δ 7,69 (с, 2H), 3,17 (ушир.с, 2H), 3,11 (д, J=12, 6 Гц, 2H), 2,53 (м, 1H), 2,07 (д, J=11,0 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 249 (M+).
ПРИМЕР 22
ГИДРОХЛОРИД 6-МЕТИЛ-7-ПРОПИЛ-5,7, 13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]-ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
A) Трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-карбоновой кислоты
4,5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен (1,9 г, 7,6 ммоль) перемешивают в 1,4-диоксане (75 мл) и обрабатывают насыщенным водным раствором Na2CO3 (10 мл). К этому добавляют ди-трет-бутил-дикарбонат (3,31 г, 15,2 ммоль). После перемешивания в течение 6 часов реакционную смесь обрабатывают H2O (50 мл) и экстрагируют этилацетатом (4×25 мл), сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии с получением продукта (1,9 г, 71%). (ТСХ 30% этилацетат/гексан (NH3) Rf 0,58).1H ЯМР (400 МГц, CDCl3) δ 7, 77 (ушир.с, 1H), 7,72 (ушир.с, 1H), 4,08 (м, 1H), 3,92 (м, 1H), 3,39 (ушир.с, 1H), 3, 27 (ушир.с, 1H), 3,25 (м, 1H), 3,18 (м, 1 H), 2,46 (м, 1 H), 2,02 (д, J=11,0 Гц, 1 H).
B) Трет-бутиловый эфир 4,5-диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-карбоновой кислоты
Трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-карбоновой кислоты (1,9 г, 5,44 ммоль) гидрируют в метаноле в атмосфере H2 (45 фунт/кв.дюйм или приблизительно 3 атмосферы) над 10%Pd/C (100 мг) в течение 1,5 часов, затем фильтруют через пад из целита и концентрируют до твердого вещества белого цвета (1,57 г, 100%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf0,14).
C) Трет-бутиловый эфир 6-метил-5,7,13-триазатетрацикло[9.3.1.02,10 .04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты (Условия смотри: Segelstein, B.E.; Chenard, B.L.; Macor, J.E.; Post, R.J. Tetrahedron Lett, 1993, 34, 1897.)
Трет-бутиловый эфир 4,5-диамино-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (700 мг, 2,42 ммоль) растворяют в этаноле (10 мл) и уксусной кислоте (HOAc) (1 мл) и обрабатывают 1-этоксиэтиленмалононитрилом (329 мг, 2,42 ммоль). Полученную смесь нагревают до 60°C и перемешивают 18 часов. Реакционную смесь охлаждают, концентрируют, обрабатывают H2O и насыщенным водным раствором Na2CO3 и экстрагируют этилацетатом (3×50 мл), затем сушат (Na2SO4). После фильтрования и концентрирования остаток подвергают хроматографии и получают твердое вещество коричневого цвета (247 мг, 36%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf0,28).
D) Трет-бутиловый эфир 6-метил-7-пропил-5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5, 8-тетраен-13-карбоновой кислоты
(Условия смотри: Pilarski, B. Liebigs Ann. Chem. 1983, 1078.) Трет-бутиловый эфир 6-метил-5,7,13-триазатетрацикло[9.3.1.02,10,04,8 ]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты (80 мг, 0,267 ммоль) перемешивают в 50% водном растворе NaOH (3 мл) и ДМСО (1 мл), затем обрабатывают 1-йодпропаном (0,03 мл, 0,321 ммоль). Эту смесь нагревают до 40°C в течение 2 часов, затем охлаждают, обрабатывают H2O и экстрагируют этилацетатом. Органический слой промывают H2O (3 раза), затем сушат (Na2SO4), фильтруют и концентрируют до масла (90 мг, 0, 253 ммоль). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,15).
E) Гидрохлорид 6-метил-7-пропил-5,7,13-триазатетрацикло [9.3.1.02,10.04, 8]пентадека-2(10),3,5,8-тетраена
Трет-бутиловый эфир 6-метил-7-пропил-5,7, 13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты (90 мг, 0,253 ммоль) растворяют в 3 н. HCl в этилацетате (5 мл) и нагревают до 100° C в течение 1/2 часа. Смесь охлаждают, концентрируют, суспендируют в этилацетате и фильтруют с получением твердого вещества белого цвета (25 мг, 34%). ЯМР (400 МГц, DMSO-d6) δ 9,56 (с, NH), 7,91 (с, 1H), 7,83 (ушир.м, NH), 7,74 (с, 1H), 4,38 (м, 2H), 3,48 (м, 2H), 3,32 (м, 2H), 3,10 (м, 2H), 2,87 (с, 3H), 2,28 (м, 1H), 2,15 (д, J=11,0 Гц, 1H) 1,85 (м, 2H), 0,97 (м, 3H), т.пл. 147-150°C.
ПРИМЕР 23
ГИДРОХЛОРИД 5,7, 13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]-ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
A) Трет-бутиловый эфир 5,7,13-триазатетрацикло[9.3.1.02,10.04,8 ]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты (Условия смотри: Segelstein, B.E.; Chenard, B.L.; Macor, J.E.; Post, R.J. Tetrahedron Lett, 1993, 34, 1897.)
Трет-бутиловый эфир 4, 5-диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (1,0 г, 3,45 ммоль) растворяют в этаноле (10 мл) и HOAc (1 мл) и обрабатывают этоксиметилмалононитрилом (421 мг, 3,45 ммоль). Полученную смесь нагревают до 60°C и перемешивают 18 часов. Реакционную смесь охлаждают, концентрируют, обрабатывают H2O и насыщенным водным раствором Na2 CO3 и экстрагируют этилацетатом (3×50 мл), затем сушат (Na2SO4). После фильтрования и концентрирования остаток подвергают хроматографии и получают коричневое твердое вещество (580 мг, 56%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0, 28).
B) Гидрохлорид 5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраена
Трет-бутиловый эфир 5,7,13-триазатетрацикло [9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение методами, описанными в примере 22E.1H ЯМР (400 МГц, D2O) δ 8,95 (с, 1H), 7,67 (с, 2H), 3,45 (ушир.с, 2H), 3,31 (д, J=12,5 Гц, 2H), 3,13 (д, J=12,5 Гц, 2H), 2,30 (м, 1H), 1,99 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 200,1 [(M+1)+], т.пл. >250°C.
ПРИМЕР 24
ГИДРОХЛОРИД 7-МЕТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04, 8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 22D, трет-бутиловый эфир 5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5, 8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение путем реакции с йодметаном с последующим удалением защитной группы, как описано в примере 12E.1H ЯМР (400 МГц, D2O) δ 8,97 (с, 1H), 7,71 (с, 1H), 7,67 (с, 1H), 3,94 (с, 3H), 3,48 (м, 2H), 3,33 (д, J=12,2 Гц, 2H), 3,14 (д, J=12,2 Гц, 2H), 2,34 (м, 1H), 2,03 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 214,2 [(M+1)+].
ПРИМЕР 25
ГИДРОХЛОРИД 6-МЕТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8 ]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Трет-бутиловый эфир 6-метил-5,7, 13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение методами, описанными в примере 22E.1H ЯМР (400 МГц, DMSO-d6) δ 9,40 (ушир.м, NH), 7,77 (ушир.м, NH), 7,70 (с, 1H), 3,44 (м, 2H), 3,30 (м, 2H), 3,05 (ушир.д, J=11,0 Гц, 2H), 2,79 (с, 3H), 2,23 (м, 1H), 2,10 (д, J=10,8 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 213,5 (M+).
ПРИМЕР 26
ГИДРОХЛОРИД 6,7-ДИМЕТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10 .04,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 22D, трет-бутиловый эфир 6-метил-5,7,13-триазатетрацикло[9.3.1.02,10,04,8 ]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение путем реакции с йодметаном с последующим удалением защитной группы, как описано в примере 22E.1H ЯМР (400 МГц, DMSO-d6) δ 9,52 (с, NH), 7,84 (с, 1H), 7,82 (ушир.м, NH), 7,72 (с, 1H), 3,90 (с, 3H), 3,45 (м, 2H), 3,28 (м, 2H), 3,04 (м, 2Н), 2,82 (с, 3Н), 2,23 (м, 1Н), 2, 12 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении MS т/в 228,2 [(M+1)+]. Т.пл. 225-230°C.
ПРИМЕР 27
ГИДРОХЛОРИД 7-ПРОПИЛ-5,7, 13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]-ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 22D, трет-бутиловый эфир 5,7, 13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение путем реакции с йодпропаном с последующим удалением защитной группы, как описано в примере 22E.1H ЯМР (400 МГц, DMSO-d6) δ 9,52 (с, 1H), 9,45 (ушир.с, NH), 7,97 (с, 1H), 7,85 (с, 1H), 7,83 (ушир.м, NH), 4,43 (м, 2H), 3,49 (м, 2H), 3,33 (м, 2H), 3,08 (м, 2H), 2,28 (м, 1H), 2,15 (д, J=11,0 Гц, 1H), 1,92 (м, 2H), 0,93 (м, 3H), химическая ионизация при атмосферном давлении МС m/e 242,2 [(M+1)+], т.пл. 170-171°C (субл.).
ПРИМЕР 28
ГИДРОХЛОРИД 7-БУТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02, 10.04,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
A) Трет-бутиловый эфир 4-бутиламино-5-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (Условия смотри: Senskey, M.D.; Bradshaw, J.D.; Tessier, C.A.; Youngs, W.J.Tetrahedron Lett, 1995, 36, 6217.)
Трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-карбоновой кислоты (500 мг, 1,43 ммоль) и 1-бутиламин (1,42 мл, 14,3 ммоль) объединяют в ТГФ (5 мл) и перемешивают 4 часа. Смесь разбавляют этилацетатом (50 мл) и промывают H2O (3×30 мл), затем сушат (Na2SO4), фильтруют и концентрируют до масла. Это масло пропускают через фильтр-колонку с гелем диоксида кремния, чтобы удалить примеси нулевой линии, элюируя 30% этилацетатом/гексаном (510 мг, 1,41 ммоль, 99%).
B) Трет-бутиловый эфир 4-бутиламино-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-карбоновой кислоты
Трет-бутиловый эфир 4-бутиламино-5-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (460 мг, 1,27 ммоль) обрабатывают формиатом аммония (850 мг, 12,7 ммоль) и 10%Pd(OH)2 /C (50 мг) в метаноле (20 мл) и доводят до кипения с обратным холодильником в течение 1 часа, затем фильтруют через пад из целита и концентрируют. Твердое вещество обрабатывают насыщенным водным раствором Na2CO3, экстрагируют CH2Cl2 (3×30 мл) и сушат фильтрованием через хлопковую пробку с получением масла (440 мг, 100%).
C) Трет-бутиловый эфир 7-бутил-5,7,13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5, 8-тетраен-13-карбоновой кислоты
Трет-бутиловый эфир 4-бутиламино-5-амино-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (440 мг, 1,27 ммоль) растворяют в этаноле (20 мл) и HOAc (2 мл) и обрабатывают этоксиметиленмалононитрилом (186 мг, 1,52 ммоль). Полученную смесь нагревают до 60°C и перемешивают 18 часов. Реакционную смесь охлаждают, концентрируют, обрабатывают H2O и насыщенным водным раствором Na2CO3, затем экстрагируют этилацетатом (3×50 мл) и сушат (Na2SO4 ). После фильтрования и концентрирования остаток подвергают хроматографии и получают желтое масло (400 мг, 89%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,70).
D) Гидрохлорид 7-бутил-5,7, 13-триазатетрацикло [9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраена
Трет-бутиловый эфир 7-бутил-5,7, 13-триазатетрацикло[9.3.1.02,10.04,8 ]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение методами, описанными в примере 22E.1H ЯМР (400 МГц, DMSO-d6) δ 9,93 (ушир.с, NH), 9,68 (с, 1H), 7,99 (с, 1H), 7,92 (ушир.м, NH), 7,87 (с, 1H), 4,50 (м, 2H), 3,49 (м, 2H), 3,30 (м, 2H), 3,08 (м, 2H), 2, 26 (м, 1H), 2,15 (д, J=11,0 Гц, 1H), 1,88 (м, 2H), 1, 32 (м, 2H), 0,82 (т, J=7,0 Гц, 3H), химическая ионизация при атмосферном давлении МС m/e 256,2 [(M+1)+], т.пл. 204-208°C.
ПРИМЕР 29
ГИДРОХЛОРИД 7-ИЗОБУТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Трет-бутиловый эфир 4, 5-динитро-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-карбоновой кислоты и изобутиламин преобразуют в указанное в заголовке соединение используя методы, описанные в примере 28A-D.1H ЯМР (400 МГц, CDCl3) δ 7,74 (с, 1H), 7,52 (с, 1H), 7,14 (с, 1H), 3,90 (дд, J=7,5,2,0 Гц, 2H), 3,04-2,97 (м, 4H), 2,70 (дд, J=12,8,2,3 Гц, 2H), 2,42 (м, 1H), 2,19 (м, 1H), 1,98 (д, J=10,5 Гц, 1H), 0,93 (м, 6H), химическая ионизация при атмосферном давлении МС m/e 256,2 [(M+1)+], т.пл. 147-150°C (субл.).
ПРИМЕР 30
ГИДРОХЛОРИД 6-МЕТИЛ-7-ИЗОБУТИЛ-5,7, 13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,1004,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
A) Трет-бутиловый эфир 6-метил-7-изобутил-5,7, 13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты
Трет-бутиловый эфир 4-амино-5-изобутиламино-10-азатрицикло[6.3.1.02, 7]додека-2(7),3,5-триен-10-карбоновой кислоты (250 мг, 0,74 ммоль) из примера 29B растворяют в этаноле (10 мл) и HOAc (2 мл) и обрабатывают 1-этоксиэтиленмалононитрилом (118 мг, 0,87 ммоль). Реакция происходит, как в примере 28C (18 ч), и обработку проводят подобным образом с получением продукта (ТСХ 3% метанол/CH2Cl2 (NH3) Rf 0,57).
B) Гидрохлорид 6-метил-7-изобутил-5,7, 13-триазатетрацикло [9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраена
Трет-бутиловый эфир 6-метил-7-изобутил-5,7, 13-триазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,5,8-тетраен-13-карбоновой кислоты преобразуют в указанное в заголовке соединение методами, описанными в примере 22E. APCI MS m/e 270,3 [(М+1)+]. Т.пл. 129-130°С (субл.).
ПРИМЕР 31
ГИДРОХЛОРИД 7-ФЕНИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8 ]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 28A, трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты и анилин преобразуют в трет-бутил-производное 4-фениламино-5-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты при 75°C в течение 4 часов на стадии сочетания. Затем его преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 28B,C,D.1H ЯМР (400 МГц, DMSO-d6) δ 9,08 (1H), 7,78-7, 57 (м, 7H), 3,47-3,00 (м, 6H), 2,23 (м, 1H), 2,09 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении MS т/в 276,2 [(M+1)+], т.пл. 210-213°C.
ПРИМЕР 32
ГИДРОХЛОРИД 6-МЕТИЛ-7-ФЕНИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 31 и примере 30, трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты и анилин преобразуют в указанное в заголовке соединение.1H ЯМР (400 МГц, DMSO-d6) δ 7,79 (с, 1H), 7,73-7,56 (м, 5H), 7,32 (с, 1H), 3,46-2,99 (м, 6H), 2,66 (с, 3H), 2,23 (м, 1H), 2,08 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 290,2 [(M+1)+], т.пл. >250°C.
ПРИМЕР 33
ГИДРОХЛОРИД 7-НЕОПЕНТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1. 02,10.04,8 ]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примере 28A-D, трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты и неопентиламин преобразуют в указанное в заголовке соединение. трет-Boc предшественник GCMS m/e 369 (M+), (HCl соль). Т.пл.>250°C.
ПРИМЕР 34
ГИДРОХЛОРИД 6-МЕТИЛ-7-НЕОПЕНТИЛ-5,7,13-ТРИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,5,8-ТЕТРАЕНА
Используя методы, описанные в примерах 21 и 20, трет-бутиловый эфир 4,5-динитро-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-карбоновой кислоты и неопентиламин преобразуют в указанное в заголовке соединение.1H ЯМР (400 МГц, DMSO-d6) δ 7,31 (с, 1H), 7,27 (s,1H), 7,02 (ушир.с, NH), 4,41 (т, J=13,0 Гц, 2H), 3,90 (с, 3H), 3,47-3,26 (м, 6H), 2,20 (м, 1H), 2,00 (д, J=11,5 Гц, 1H), 0,90 (с, 9H), химическая ионизация при атмосферном давлении МС m/e 384,2 [(М+1)+]. Т.пл. >250°C.
ПРИМЕР 35
ГИДРОХЛОРИД 6,7-ДИМЕТИЛ-5,8, 14-ТРИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
(На основе следующей процедуры: Jones, R.G.; Mclaughlin, K.C. Org. Syn. 1963, 4, 824, b) Ehrlich, J., Bobert, M.T. J.Org.Chem, 1947, 522.) Трет-бутиловый эфир 4,5-диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (100 мг, 0,35 ммоль) нагревают до 80°C в H2O (5 мл). К нему добавляют в атмосфере N2 в течение 2 часов бутан-2,3-дион (0,034 мл, 0,38 ммоль). Реакционную смесь охлаждают до комнатной температуры и экстрагируют этилацетатом (3×40 мл). Объединенный органический слой промывают H2O (2×30 мл), сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии на силикагеле с получением масла (120 мг, 100%). Масло растворяют в 2 н. HCl в метаноле (5 мл) и нагревают до кипения с обратным холодильником в течение 30 минут, затем концентрируют. Перекристаллизация из смеси метанол/Et2O обеспечивает белый порошок (50 мг, 43%). (ТСХ этилацетат Rf 0,14).1H ЯМР (400 МГц, DMSO-d6) δ 7,85 (с, 2H), 3,50 (ушир.с, 2H), 3,32 (д, J=12,5 Гц, 2H), 3,10 (д, J=12,5 Гц, 2H), 2,64 (с, 6H), 2,24 (м, 1H), 2,13 (д, J=11,0 Гц, 1H), t-Boc предшественник химической ионизации при атмосферном давлении МС m/e 340, 3 [(M+1)+].
ПРИМЕР 36
ГИДРОХЛОРИД 5,8,14-ТРИАЗАТЕТРАЦИКЛО[10.3.1.02,1104,9] ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1-(4, 5-Диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон
1-(4,5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2, 2, 2-трифторэтанон (3,0 г, 8,70 ммоль) гидрируют в метаноле (30 мл) в атмосфере водорода (45 фунт/кв.дюйм, или приблизительно 3 атмосферы) над Pd(OH)2 (300 мг 20 мас.%/C, 10 мас.%). Через 2, 5 часа реакционную смесь фильтруют через пад из целита и промывают метанолом (30 мл). Раствор концентрируют до светло-коричневого масла, которое кристаллизуется (2,42 г, 96%). (ТСХ 10% метанол/CH2Cl2 Rf 0,56). Химическая ионизация при атмосферном давлении MS m/e 286,2 [(М+1)+]. Т.пл. 129-131°C.
B) 1-(5,8, 14-Триазатетрацикло[10.3.1.02,l1.04,9]гексадека-2(11),3,5,7,9-пентаен)-2,2,2-трифторэтанон
1-(4,5-Диамино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон (500 мг, 1,75 ммоль) перемешивают в ТГФ (2 мл). Эту смесь обрабатывают H2O (2 мл) и гидратом продукта присоединения глиоксаля и бисульфита натрия (931 мг, 3,50 ммоль), затем перемешивают при 55°C в течение 2,5 часов. Реакционную смесь охлаждают до комнатной температуры и экстрагируют этилацетатом (3×40 мл). Объединенный органический слой промывают H2O (2×30 мл), сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии на силикагеле с получением не совсем белого порошок (329 мг, 60%), (ТСХ 25% этилацетат/гексан Rf 0,40). Т.пл. 164-166°C.
C) Гидрохлорид 5,8,14-триазатетрацикло[10.3.1.02,11.04,9] гексадека-2(11),3,5,7, 9-пентаена
1-(5,8,14-Триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаен)-2,2,2-трифторэтанон (320 мг, 1,04 ммоль) суспендируют в метаноле (2,0 мл) и обрабатывают Na2CO3 (221 мг, 2,08 ммоль) в H2O (2,0 мл). Смесь нагревают до 70°C в течение 2 часов, затем концентрируют, обрабатывают H2O (20 мл) и экстрагируют CH2Cl2 (3×10 мл). Органический слой сушат через хлопковую пробку и концентрируют с получением масла светло-желтого цвета (183 мг, 83%), которое затвердевает при стоянии (Т.пл. 138-140°C). Этот материал растворяют в метаноле (10 мл), обрабатывают 3M HCl/этилацетатом (3 мл), концентрируют и подвергают азеотропной обработке метанолом (2×20 мл) с получением твердого вещества, которое подвергают перекристаллизации из смеси метанол/Et2O, и получают продукт в виде белого твердого вещества (208 мг, 97%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,26).1H ЯМР (400 МГц, CD3OD) δ 8,94 (с, 2H), 8,12 (с, 2H), 3,70 (м, 2H), 3,54 (д, J=12,5 Гц, 2H), 3,35 (д, J=12,5 Гц, 2H), 2,49 (м, 1H), 2,08 (д, J=11,0 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 211 (M+), т.пл. 225-230°C.
ПРИМЕР 37
ГИДРОХЛОРИД 14-МЕТИЛ-5,8,14-ТРИАЗАТЕТРАЦИКЛО [10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
5,8,14-Триазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,5,7,9-пентаен (207 мг, 0,98 ммоль) обрабатывают 37% водным раствором формалина (1 мл) и муравьиной кислотой (1 мл), затем нагревают до 80°C в течение 1 часа. Реакционную смесь выливают в воду, подщелачивают (NaOH, pH ˜11) и экстрагируют этилацетатом. Органический слой сушат (Na2SO4), концентрируют и подвергают хроматографии на силикагеле с получением твердого вещества желтого цвета. Его перемешивают в метаноле (2 мл) и обрабатывают 3 н. HCl в этилацетате (2 мл). После концентрирования твердое вещество подвергают перекристаллизации из смеси метанол/Et2O и получают продукт в виде белого твердого вещества (70 мг, 27%). (2% метанол/CH2Cl2 (NH3) Rf 0,47).1H ЯМР (400 МГц, CDCl3) δ 8,71 (с, 2H), 7,80 (с, 2H), 3,37 (ушир.с, 2H), 3,03 (м, 2H), 2,47 (м, 2H), 2,32 (м, 1H), 2,18 (ушир.с, 3H), 1,84 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении MS т/в 226,2 [(М+1)+], т.пл. >250°C.
ПРИМЕР 38
ГИДРОХЛОРИД 5-OXA-7, 13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
A) 2,2,2-Трифтор-1-(4-гидрокси-5-нитро-10-азатрицикло [6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)этанон
1-(4,5-Динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (900 мг, 2,61 ммоль) и ацетат калия (KOAc) (2,6 г, 26,1 ммоль) растворяют в ДМСО (10 мл) и нагревают с перемешиванием до 100°C в течение 16 часов. Смесь охлаждают и разбавляют H2O (50 мл), затем экстрагируют смесью 80% этилацетат/гексан (6×25 мл). Органический слой промывают H2O (3×20 мл), сушат (Na2SO4), фильтруют и концентрируют и очищают хроматографией с получением масла (575 мг, 70%). (ТСХ 50% этилацетат/гексан (NH3) Rf 0,56).
B) 2,2,2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)этанон
2,2, 2-Трифтор-1-(4-гидрокси-5-нитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон (575 мг, 1,82 ммоль) гидрируют в метаноле в атмосфере H2 (45 фунт/кв.дюйм или приблизительно 3 атмосферы) над 10%Pd/C (80 мг) в течение 1,5 часов, затем фильтруют через пад из целита и концентрируют до белого твердого вещества (450 мг, 86%). (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,6).1H ЯМР (400 МГц, CD3OD) δ 6,67-6,59 (м, 2H), 4,12 (м, 1H), 3,73 (м, 1H), 3,73 (м, 1H), 3, 51 (м, 1H), 3,07 (м, 2H), 2,24 (м, 1H), 1,94 (д, J=10,5 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 286 (M+).
C) 2,2,2-Трифтор-1-(5-окса-7,13-диазатетрацикло [9.3.1.02,10.04,8 ]пентадека-2(10),3,6,8-тетраен)этанон (Goldstein, S.W.; Dambek, P.J. J. Het. Chem. 1990, 27, 335)
2,2, 2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)этанон (150 мг, 0,524 ммоль), триметилортоформиат (0,19 мл, 1,73 ммоль), пиридиний-п-толуолсульфоновую кислоту (PPTS, 18 мг, 0,07 ммоль) и ксилолы (10 мл) объединяют в атмосфере азота и перемешивают при 135°C в течение 18 часов. Смесь охлаждают, обрабатывают H2O и экстрагируют этилацетатом. Экстракты сушат (Na2SO4), фильтруют, концентрируют и очищают хроматографией с получением масла (110 мг, 71%). (ТСХ 20% этилацетат/гексан Rf 0,40).
D) Гидрохлорид 5-окса-7,13-диазатетрацикло[9.3.1.02,10.04, 8] пентадека-2(10),3,6,8-тетраена
2,2,2-Трифтор-1-(5-окса-7,13-диазатетрацикло [9.3.1.02, 10.04,8]пентадека-2(10),3,6,8-тетраен)этанон (110 мг, 0,37 ммоль) перемешивают в метаноле (5 мл) и обрабатывают Na2CO3 (78 мг, 0,74 ммоль) в H2O (2 мл). Перемешиваемую смесь нагревают до 80°C в течение 2 часов, концентрируют до твердого вещества, разбавляют H2O и экстрагируют этилацетатом (3×40 мл). Продукт экстрагируют в 1 н. водный раствор HCl (2×40 мл), который промывают этилацетатом, затем нейтрализуют насыщенным водным раствором Na2CO3 до pH˜10. Продукт экстрагируют этилацетатом (3×40 мл), сушат (Na2SO4), концентрируют и подвергают хроматографии на силикагеле с получением масла (ТСХ 5% метанол/CH2Cl2 (NH3) Rf 0,19).
Масло растворяют в метаноле и обрабатывают 3 н. HCl в этилацетате (4 мл), затем концентрируют, перемешивают в минимальном количестве CH2Cl2 и пропитывают гексанами. Через 18 часов продукт собирают фильтрованием (55 мг, 63%).1H ЯМР (400 МГц, CD3OD) δ 8,47 (с, 1H), 7,70 (с, 1H), 7,65 (с, 1H), 3,41 (м, 2H), 3,30 (м, 2H), 3,10 (д, J=12,5 Гц, 2H), 2,47 (м, 1H), 2,15 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 201,03 [(M+1)+].
ПРИМЕР 39
ГИДРОХЛОРИД 6-МЕТИЛ-5-ОКСА-7,13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
A) 2,2,2-Трифтор-1-(6-метил-5-окса-7,13-диазатетрацикло [9.3.1.02,10.04,8]пентадека-2(10),3,6,8-тетраен)этанон
2,2,2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон (150 мг, 0,524 ммоль), триэтил-ортоацетат (0,34 мл, 1,83 ммоль), пиридиний-п-толуолсульфоновую кислоту (PPTS, 20 мг, 0,08 ммоль) и ксилолы (10 мл) объединяют в атмосфере азота и перемешивают при 135°C в течение 18 часов. Переработка, изоляция и очистка, как в примере 38C, обеспечивают указанное в заголовке соединение (90 мг, 55%).
B) Гидрохлорид 6-метил-5-окса-7, 13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2(10),3,6, 8-тетраена
2,2,2-Трифтор-1-(6-метил-5-окса-7,13-диазатетрацикло[9.3.1.02,10.04,8 ]пентадека-2(10),3,6,8-тетраен)этанон (90 мг, 0,30 ммоль) перемешивают в метаноле (5 мл) и обрабатывают Na2CO3 (61 мг, 0,58 ммоль) в H2O (2 мл). Перемешиваемую смесь нагревают до 80°C в течение 2 часов, концентрируют до твердого вещества, разбавляют H2O и экстрагируют этилацетатом (3×40 мл). Раствор сушат (Na2SO4), концентрируют и подвергают хроматографии на силикагеле, чтобы получить масло (ТСХ 10% метанол/CH2Cl2 (NH3) Rf 0,18).1H ЯМР (свободное основание) (400 МГц, CDCl3) (7,40 (с, 1H), 7,26 (с, 1H), 3,05-2,98 (м, 4H), 2,72 (д, J=12,8 Гц, 2H), 2,59 (с, 3H), 2,46 (м, 1H), 1,98 (д, J=10,5 Гц, 1H).
Масло растворяют в метаноле и обрабатывают 3 н. HCl в этилацетате (4 мл), затем концентрируют, перемешивают в минимуме CH2Cl2 и насыщают гексанами. Через 18 часов продукт собирают фильтрованием (10 мг, 13%). Химическая ионизация при атмосферном давлении MS m/e 215,2 [(М+1)+]. Т.пл. >250°C.
ПРИМЕР 40
ГИДРОХЛОРИД 2-ФТОР-N-(4-ГИДРОКСИ-10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-5-ИЛ)БЕНЗАМИДА
2,2, 2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло [6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-этанон (150 мг, 0,524 ммоль), 2-фторбензоилхлорид (0,07 мл, 0,576 ммоль), пиридиний-п-толуолсульфоновую кислоту (PPTS, 20 мг, 0,08 моль), пиридин (0,046 мл, 0,576 ммоль) и ксилолы (5 мл) объединяют в атмосфере азота и перемешивают при 135°C в течение 18 часов. Спустя 24 часа, добавляют дополнительную PPTS (50 мг) и материал перемешивают при 135°C в течение следующих 24 часов. Указанная переработка обеспечивает сырой подукт (145 мг, 0,375 ммоль), который объединяют с Na2CO3 (тв.) (80 мг, 0,75 ммоль) в метаноле (5 мл) и H2O (2 мл) и нагревают до кипения с обратным холодильником. Спустя 3 часа, реакционную смесь охлаждают и разбавляют водой, затем экстрагируют CH2Cl2 (4×40 мл), сушат через хлопковую пробку, затем подвергают хроматографии, чтобы удалить примесь нулевой линии (5% метанол/CH2Cl2 (NH3)). Сырой материал обрабатывают избытком 3 н. HCl в этилацетате и концентрируют, затем растворяют в минимуме метанола и раствор насыщают Et2O и перемешивают. После перемешивания в течение 4 часов продукт собирают фильтрованием (85 мг, 68%).1H ЯМР (400 МГц, CD3OD) (7,99 (м, 2H), 7,59 (м, 1H), 7,36-7,23 (м, 2H), 6,82 (с, 1H), 2,99 (м, 4H), 2,78 (м, 2H), 2,35 (м, 1H), 1,96 (д, J=10,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 313,1 [(M+1)+], т.пл. 125-130°C (субл.).
ПРИМЕР 41
ГИДРОХЛОРИД 4-ХЛОР-10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА
A) 1-(4-Хлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон
Хлорид меди (I)(CuCl) получают следующим образом: CuSO4 (4,3 г) и NaCl (1,2 г) растворяют в горячей H2O (14 мл). Гидросульфит натрия (NaHSO3) (1 г) и гидроксид натрия (NaOH) (690 мг) растворяют в H2O (7 мл) и добавляют к горячему кислотному раствору в течение 5 минут. Осажденное белое твердое вещество фильтруют и промывают водой.
1-(4-Амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон (460 мг, 1,7 ммоль) растворяют в H2O (2 мл) и концентрированном растворе HCl (1 мл), затем охлаждают до 0°C и обрабатывают раствором нитрита натрия (NaNO2) (275 мг) в H2O (1 мл) по каплям. К полученному раствору добавляют CuCl (202 мг, полученный, как описано выше, 2,04 ммоль) в концентрированном растворе HCl (2 мл) в течение 10 минут (наблюдается газовыделение). Полученный раствор нагревают до 60°C в течение 15 минут, затем охлаждают до комнатной температуры и экстрагируют этилацетатом (4×30 мл). После сушки над Na2SO4 раствор фильтруют и концентрируют до масла, которое фильтруют через пад диоксида кремния, чтобы удалить материал нулевой линии, элюируя смесью 50% этилацетат/гексан, и получают масло (470 мг, 95%).
B) Гидрохлорид 4-хлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена
1-(4-Хлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифтор-этанон (470 мг, 1,62 ммоль) и Na2CO3 (344 мг, 3,24 ммоль) в метаноле (30 мл) и H2O (10 мл) нагревают до кипения с обратным холодильником. Спустя 2 часа реакционную смесь охлаждают и разбавляют водой, затем экстрагируют этилацетатом (4×40 мл), сушат (Na2 SO4), фильтруют и концентрируют до желтого масла. Сырой материал обрабатывают избытком 3 н. HCl в этилацетате и концентрируют, затем растворяют в минимуме CH2Cl2 и раствор насыщают гексанами и перемешивают. После перемешивания в течение 4 часов продукт собирают фильтрованием (155 мг, 42%).
1H ЯМР (свободное основание) (400 МГц, CDCl3) δ 7,15 (м, 2H), 7,09 (д, J=8,0 Гц, 1H), 3,00-2,94 (м, 4H), 2,68, (м, 2H), 2,38 (м, 1H), 1,92 (д, J=10,5 Гц, 1H),1H ЯМР (HCl соль) (400 МГц, DMSO-d6) δ 7,30-7,20 (м, 3H), 3,30-3,15 (м, 6H), 2,37 (м, 1H), 1,89 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 194,1 [(M+1)+].
ПРИМЕР 42
ГИДРОХЛОРИД 10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-4-ИЛ ЦИАНИДА
A) 1-(4-иод-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон
1-(4-Амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон (500 мг, 1,85 ммоль) растворяют в H2O (5 мл) и концентрированном растворе H2SO4 (0,5 мл), затем охлаждают до 0°C и обрабатывают раствором нитрита натрия (NaNO2) (140 мг, 2,04 ммоль) в H2O (2 мл) по каплям. Йодид калия (460 мг, 2,78 ммоль) в 1 н. растворе H2SO4(0,5 мл) добавляют в течение 10 минут (реакционная смесь становится темно-красной). Полученный раствор нагревают до комнатной температуры и перемешивают 18 часов. Реакцию гасят NaHSO3 и водой (pH 2,5), затем экстрагируют этилацетатом (4×30 мл). После сушки (Na2SO4) раствор фильтруют и концентрируют до желтого масла, которое подвергают хроматографии на силикагеле, и получают желтое масло (260 мг, 37%). (ТСХ 30% этилацетат/гексан Rf 0,70). (переработка, как указано выше, в масштабе 5,4 г дает 5 г, 67%).
B) Трет-бутиловый эфир 4-йод-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты
1-(4-йод-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (5 г, 13,1 ммоль) и 37% насыщенный водный раствор NH4OH (50 мл) перемешивают в метаноле (250 мл) в течение 2 часов, затем концентрируют и подвергают азеотропной обработке с метанолом (2×50 мл). Полученный продукт перемешивают в 1,4-диоксане (75 мл) и обрабатывают насыщенным раствором Na2CO3 (15 мл). К этому добавляют ди-трет-бутил-дикарбонат (5,71 г, 26,2 ммоль). После перемешивания в течение 18 часов реакционную смесь обрабатывают H2O (50 мл) и экстрагируют CH2Cl2 (4×30 мл), сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии на силикагеле (ТСХ 20% этилацетат/гексан) с получением продукта в виде масла (4,9 г, 98%).
C) Трет-бутиловый эфир 4-циано-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-карбоновой кислоты
(Используют методы, описанные в: House, H.O.; Fischer, W.F. J. Org. Chem. 1969, 3626.)
CuCN (108 мг, 1,21 ммоль) и NaCN (59 мг, 1,21 ммоль) объединяют в сухом ДМФ (6 мл) и нагревают до 150°C в атмосфере N2. Раствор образуется за 20 минут. К нему добавляют трет-бутиловый эфир 4-йод-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-карбоновой кислоты (232 мг, 0,6 ммоль) в ДМФ (3,5 мл) и смесь перемешивают в течение 18 часов при 150°C. Реакционную смесь охлаждают и разбавляют 50% насыщенным водным раствором NaCl и экстрагируют смесью 50% этилацетат/гексан (3×30 мл). После сушки (Na2SO4), фильтрования и концентрирования продукт изолируют хроматографией (86 мг, 50%). (ТСХ 20% этилацетат/гексан Rf 0,28).
D) Гидрохлорид 10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ил цианида
Трет-бутиловый эфир 4-циано-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты обрабатывают 3 н. HCl в этилацетате (6 мл) и нагревают до кипения с обратным холодильником в течение 2 часов, затем концентрируют, растворяют в минимуме метанола, который насыщают Et2O и перемешивают в течение 18 часов. Продукт собирают фильтрованием (49 мг, 73%).1H ЯМР (400 МГц, DMSO-d6) δ 9,66 (ушир.с, NH), 7,86 (ушир.с, NH), 7,74-7,70 (м, 2H), 7,49 (д, J=7,5 Гц, 1H), 3,33-2,97 (м, 6H), 2,17 (м, 1H), 2,01 (д, J=11,0 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 184 (M+). Т.пл. 268-273°C.
ПРИМЕР 43
ГИДРОХЛОРИД 3-(10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3, 5-ТРИЕН-4-ИЛ)-5-МЕТИЛ-1, 2,4-ОКСАДИАЗОЛА
Трет-бутиловый эфир 4-циано-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (300 мг, 1,1 ммоль) перемешивают в этаноле (10 мл). К этому добавляют гидрохлорид гидроксиламина (382 мг, 5,5 ммоль) и NaOH (242 мг, 6,05 ммоль) и смесь нагревают до кипения с обратным холодильником. Спустя 45 минут реакционную смесь охлаждают, разбавляют H2O и экстрагируют этилацетатом. Органический слой сушат (Na2SO4) и концентрируют с получением твердого вещества желтого цвета (110 мг, 0,35 ммоль). Это твердое вещество растворяют в пиридине (1 мл) и обрабатывают ацетилхлоридом (0,03 мл, 0,415 ммоль) и нагревают до 100°C в течение 18 часов. Реакционную смесь охлаждают, обрабатывают H2O и экстрагируют этилацетатом. Органические экстракты промывают водой и насыщенным водным раствором NaCl, сушат (Na2SO4) и концентрируют. Хроматография на силикагеле дает продукт (50 мг, 0,15 ммоль). (25% этилацетат/гексан Rf 0,18). Этот продукт обрабатывают 2 н. HCl в метаноле (10 мл), нагревают до 70°C в течение 1 часа, охлаждают, концентрируют и подвергают перекристаллизации из смеси метанол/Et2O с получением продукта (15 мг), химическая ионизация при атмосферном давлении MS m/e 242,2 [(М+1)+].
ПРИМЕР 44
ГИДРОХЛОРИД 1-(10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-4-ИЛ)-1-ЭТАНОНА
A) 1-(4-Ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон
1-(10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (253 мг, 1,0 ммоль) и AcCl (0,68 мл, 10 ммоль) растворяют в ДХЭ (3 мл) и обрабатывают хлоридом алюминия (AlCl3) (667 мг, 5,0 ммоль). Полученную желтую смесь перемешивают в течение 30 минут, затем выливают на лед и насыщенный водный раствор NaHCO3. После перемешивания в течение 20 минут смесь экстрагируют CH2Cl2 (3×30 мл). Органический слой сушат через хлопковую пробку, затем концентрируют до оранжево-желтого масла (255 мг, 86%).
B) Трет-бутиловый эфир 4-ацетил-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты
1-(4-Ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (1,3 г, 4,37 ммоль) и 37% водный раствор NH4OH (10 мл) перемешивают в метаноле (30 мл) в течение 3 часов, затем концентрируют и подвергают азеотропной обработке с метанолом (2×50 мл). (Этот продукт может быть превращен в соль с HCl непосредственно: смотри следующий пример.) Полученный продукт перемешивают в 1,4-диоксане (20 мл) и обрабатывают насыщенным водным раствором Na2CO3(5 мл). К этому добавляют ди-трет-бутил-дикарбонат (1,91 г, 8,74 ммоль). После перемешивания в течение 2 часов реакционную смесь обрабатывают H2O (50 мл), экстрагируют CH2Cl2 (4×30 мл), сушат (Na2SO4), фильтруют, концентрируют и подвергают хроматографии с получением масла (1,3 г, 100%). (ТСХ 40% этилацетат/гексан Rf 0,56).
C) Гидрохлорид 1-(10-азатрицикло[6.3.1.02, 7 ]додека-2(7),3,5-триен-4-ил)-1-этанона
Трет-бутиловый эфир 4-ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты (190 мг, 0,63 ммоль) обрабатывают избытком 3 н. HCl в этилацетате и нагревают до 70°C в течение 1 часа, затем концентрируют и растворяют в минимуме метанола. Полученный раствор насыщают Et2O и перемешивают. Спустя 18 часов, белый кристаллический продукт собирают фильтрованием (81 мг, 54%).1H ЯМР (400 МГц, DMSO-d6) δ 9,75 (ушир.с, NH), 7,89 (с, 1H), 7,88 (д, J=8,0 Гц, 1H), 7,74 (ушир.с, NH), 7,44 (д, J=8,0 Гц, 1H), 3,33 (ушир.с, 2H), 3,22 (ушир.с, 2H), 3,00 (ушир.м, 2H), 2,54 (с, 3H), 2,17 (м, 1H), 2,02 (д, J=11,0 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 201 (M+). Т.пл. 198-202°C.
ПРИМЕР 45
ГИДРОХЛОРИД 10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕН-4-ОЛА
A) 10-трифторацетил-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-иловый эфир уксусной кислоты
1-(4-Ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон (2,5 г, 8,41 ммоль) и 3-хлорпероксибензойную кислоту (m-CPBA) (7,5 г, 42 ммоль) перемешивают в CH2Cl2 (20 мл) и нагревают до 40°C в течение 18 часов. Смесь охлаждают до комнатной температуры, затем обрабатывают диметилсульфидом (Me2S) (3 мл, 40,8 ммоль) и перемешивают в течение 24 часов. Полученную смесь выливают в лед и насыщенный водный раствор Na2CO3 (100 мл), затем экстрагируют Et2O (4×40 мл). Органический слой промывают насыщенным водным раствором Na2CO3 (3×40 мл), затем сушат (Na2SO4), фильтруют и концентрируют с получением масла (1,83 г, 69%). (ТСХ этилацетат Rf 0,80).
B) 2,2, 2-Трифтор-1-(4-гидрокси-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил)этанон
10-трифторацетил-10-аза-трицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-иловый эфир уксусной кислоты (900 мг, 2,87 ммоль) перемешивают в метаноле (20 мл) и насыщенном водном растворе NaHCO3(15 мл) в течение 48 часов. Смесь концентрируют, разбавляют H2O и экстрагируют CH2Cl2 (3×20 мл), затем сушат через хлопковую пробку. Хроматография на силикагеле дает чистый продукт (420 мг, 54%). (ТСХ 5% метанол/CH2Cl2 Rf 0,44).1H ЯМР (400МГц, CDCl3) δ 7,05 (м, 1H), 6,70 (м, 1H), 6,62 (м, 1H), 4,32 (м, 1H), 3,84 (м, 1H), 3,48 (м, 1H), 3,21 (ушир.с, 1H), 3,16 (ушир.с, 1H), 3,09 (м, 1H), 2,38 (м, 1H), 1,97 (д, J=11,0 Гц, 1H).
C) Гидрохлорид 10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ола
2,2, 2-Трифтор-1-(4-гидрокси-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон (50 мг, 0,184 ммоль) растворяют в метаноле/H2O (3/1, 5 мл), обрабатывают Na2CO3 (тв.) (40 мг, 0,369 ммоль) и нагревают до 65°C в течение 2 часов. Смесь концентрируют, разбавляют H2O и экстрагируют CH2Cl2 (3×20 мл), затем сушат через хлопковую побку. Фильтрование через пад из силикагеля дает масло (10% метанол/CH2Cl2), которое обрабатывают 3 н. HCl в этилацетате (3 мл), затем концентрируют, растворяют в минимуме метанола, который насыщают Et2O, и перемешивают. Через 18 часов собирают фильтрованием белый кристаллический продукт (10 мг, 26%).1H ЯМР (400 МГц, CDOD3) δ 7,16 (д, J=8,0 Гц, 1H), 6,80 (д, J=2,0 Гц, 1H), 6,72 (дд, J=8,0,2,0 Гц, 1H), 3,32-3,28 (4H), 3,09 (дд, J=14,5,12,0 Гц, 2H), 2,32 (м, 1H), 2,03 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 176,2 [(М+1)+], т.пл. 308°C (разл.).
ПРИМЕР 46
ГИДРОХЛОРИД 7-МЕТИЛ-5-ОКСА-6, 13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2,4(8),6,9-ТЕТРАЕНА
A) 1-(4-Ацетил-5-гидрокси-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон
10-трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-иловый эфир уксусной кислоты (800 мг, 2,55 ммоль) объединяют с AlCl3 (1,0 г, 7, 65 ммоль) и нагревают до 170°C в течение 2 часов. Смесь охлаждают и обрабатывают 1 н. водным раствором HCl (20 мл), экстрагируют этилацетатом и сушат (Na2SO4). После хроматографии получают масло (190 мг, 24%). (ТСХ этилацетат Rf 0,75).1H ЯМР (400 МГц, CDCl3) (12,58 (с, 0,5H), 12,52 (с, 0,5H), 7,53 (с, 1H), 6,86 (с, 1H), 4,33 (м, 1H), 3,91 (м, 1H), 3,56 (м, 1H), 3,28 (ушир.с, 1H), 3,24 (ушир.с, 1H), 3,14 (м, 1H), 2,35 (м, 1H), 1,97 (ушир.д, J=11,2 Гц, 1H).
B) 2,2, 2-Трифтор-1-[4-гидрокси-5-(1-гидроксииминоэтил)-10-азатрицикло[6.3.1.02,7]-додека-2(7),3,5-триен-10-ил]этанон
1-(4-Ацетил-5-гидрокси-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (190 мг, 0,605 ммоль), гидроксиламин HCl (99 мг, 1,21 ммоль) и ацетат натрия (118 мг, 1,21 ммоль) объединяют в метаноле (4 мл) и H2O (1 мл) и нагревают до 65°C в течение 18 часов. Смесь охлаждают, разбавляют H2O и экстрагируют этилацетатом, который сушат (Na2SO4), фильтруют и концентрируют с получением масла желтого цвета (177 мг, 93%).
C) 2,2,2-Трифтор-7-метил-5-окса-6,13-диазатетрацикло [9.3.1.02,10.04,8]пентадека-2,4(8),6,9-тетраенэтанон
Указанное выше масло, а именно 2,2,2-трифтор-1-[4-гидрокси-5-(1-гидроксииминоэтил)-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-10-ил]этанон (177 мг, 0,54 ммоль), перемешивают в ДХЭ (3 мл), обрабатывают триэтиламином (0,4 мл, 2,8 ммоль) и уксусным ангидридом (Ac2O) (0,3 мл, 2,8 ммоль), затем перемешивают в течение 18 часов. Реакционную смесь обрабатывают H2O и экстрагируют этилацетатом. Экстракты сушат (Na2SO4), фильтруют и концентрируют до желтого масла, которое растворяют в безводном ДМФ (3 мл) и обрабатывают 60% NaH в масле (32 мг, 1,08 ммоль). После перемешивания в течение 18 часов вводят дополнительный 60% NaH в масле (33 мг) и смесь перемешивают 2 часа. Реакцию гасят H2O (5 мл) и экстрагируют смесью 80% этилацетат/гексан (3×30 мл). Органический слой промывают H2O (3×20 мл), сушат (Na2SO4), фильтруют и концентрируют и подвергают хроматографии с получением масла (40% этилацетат/гексан Rf 0,56).
D) Гидрохлорид 7-метил-5-окса-6,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2,4(8),6,9-тетраена
Используя методы, описанные в примере 19C, 2,2,2-трифтор-7-метил-5-окса-6,13-диазатетрацикло[9.3.1.02,10.04,8]пентадека-2,4(8),6,9-тетраенэтанон преобразуют в указанное в заголовке соединение. Его обрабатывают 3 н. HCl в этилацетате (3 мл), концентрируют и растворяют в минимуме CH2Cl2, который насыщают гексанами и перемешивают. Спустя 18 часов белый кристаллический продукт собирают фильтрованием (18 мг, общий выход 13%).1H ЯМР (400 МГц, DMSO-d6) δ 7,72 (с, 1H), 7,63 (с, 1H), 3,42-2,98 (м, 6H), 2,50 (с, 3H), 2,23 (м, 1H), 2,08 (д, J=10,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 215,2 [(M+1)+].
ПРИМЕР 47
ГИДРОХЛОРИД 4-(2-МЕТИЛ-2H-ПИРАЗОЛ-3-ИЛ)-10-АЗАТРИЦИКЛО [6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА И ГИДРОХЛОРИД 4-(1-МЕТИЛ-1H-ПИРАЗОЛ-3-ИЛ)-10-АЗА-ТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА
1-(4-Ацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (1,0 г, 3,3 ммоль) и диметилформамид-диметилацеталь (ДМФ-DMA) (4,0 г, 33,6 ммоль) нагревают до 140°C в течение 18 часов. После охлаждения кристаллический осадок отфильтровывают и промывают этилацетатом (690 мг, 58%).
Указанное твердое вещество, а именно 3-диметиламино-1-(10-трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-ил)пропенон (200 мг, 0,56 ммоль), растворяют в этаноле (2 мл) и обрабатывают 5 н. HCl в этаноле (0,1 мл), а затем метилгидразином (0,6 ммоль). Полученную смесь нагревают до 70°C в течение 4 часов. Смесь охлаждают, разбавляют водой и экстрагируют этилацетатом, сушат (Na2SO4) и концентрируют. Хроматография на силикагеле обеспечивает 3/1 смесь региоизомерных продуктов (130 мг, 68%). (ТСХ 50% этилацетат/гексан Rf 0,40).
Указанное масло (130 мг, 0,388 ммоль) и Na2CO3(тв.) (82 мг, 0,775 ммоль) перемешивают в метаноле (10 мл) и H2O (5 мл) в течение 18 часов. После охлаждения реакционную смесь разбавляют водой, экстрагируют CH2Cl2, сушат через хлопковую пробку и концентрируют. Продукт очищают хроматографией на силикагеле и концентрируют до масла. Соль получают с 2 н. HCl в метаноле, концентрируют и подвергают перекристаллизации из смеси метанол/этилацетат с получением 3/1 смеси региоизомерных пиразолов (85 мг, 58%). (5% метанол/CH2Cl2 (NH3) Rf 0,25). TFA-предшественник. Химическая ионизация при атмосферном давлении MS m/e 336,2 [(М+1)+].
ПРИМЕР 48
ГИДРОХЛОРИД 4, 5-ДИХЛОР-10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА
A) 1-(4,5-Дихлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (На основе Campaigne, E.; Thompson, W. J. Org. Chem. 1950, 72, 629.)
1-(10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (539 мг, 2,1 ммоль) перемешивают в CH2Cl2 (5 мл) и обрабатывают ICl3(тв.) (982 мг, 4,21 ммоль). Полученный оранжевый раствор перемешивают в течение 0,5 часа, выливают в насыщенный водный раствор NaHSO3(25 мл), экстрагируют CH2Cl2 (3×25 мл), сушат через хлопковую пробку и концентрируют до масла (570 мг, 84%). (ТСХ 50% этилацетат/гексан Rf 0, 62).
B) Гидрохлорид 4,5-дихлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триена
1-(4,5-Дихлор-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-10-ил)-2,2,2-трифторэтанон (570 мг, 1,75 ммоль) перемешивают в метаноле (25 мл) и обрабатывают Na2CO3 (тв.) (5 г, 47 ммоль) в H2O (5 мл). Перемешиваемую смесь нагревают до 70°C в течение 4 часов, концентрируют до твердого вещества, разбавляют H2O и экстрагируют этилацетатом (3×40 мл). Продукт экстрагируют в 1 н. водном растворе HCl (2×40 мл), который промывают этилацетатом, затем нейтрализуют насыщенным водным раствором Na2CO3 до рН ˜10. Продукт экстрагируют CH2Cl2 (3×40 мл), фильтруют через хлопковую пробку и концентрируют до масла (400 мг, 100%).
Масло растворяют в метаноле и обрабатывают 3 н. HCl в этилацетате (4 мл) и концентрируют, затем растворяют в минимуме метанола и насыщают Et2O и перемешивают 18 часов. Продукт собирают фильтрованием (210 мг, 45%). (ТСХ 50% этилацетат/гексан (NH3) Rf 0,08).1H ЯМР (400 МГц, DMSO-d6) δ 7,58 (с, 2H), 3,33-2,97 (м, 6H), 2,18 (м, 1H), 1,99(д, J=10,5 Гц, 1H),13C ЯМР (100 МГц, DMSO-d6) δ 141,02, 130, 60, 126,58, 45,54, 40,55, 38,30, газовая хроматография и масс-спектроскопия m/e 227, 229 (M+), т.пл. 283-291°C.
ПРИМЕР 49
ГИДРОХЛОРИД N4, N4-ДИМЕТИЛ-10-АЗАТРИЦИКЛО[6.3.1.02,7]
ДОДЕКА-2(7),3,5-ТРИЕН-4-СУЛЬФОНАМИДА
A) 10-Трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3, 5-триен-4-сульфонилхлорид
1-{10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (530 мг, 2,1 ммоль) добавляют к хлорсульфоновой кислоте (2 мл, 30 ммоль) и перемешивают в течение 5 минут. Смесь резко охлаждают льдом, экстрагируют этилацетатом, сушат (Na2SO4), фильтруют и концентрируют с получением масла (640 мг, 87%). (ТСХ 30% этилацетат/гексан Rf 0,15).
B) Гидрохлорид N4,N4-диметил-10-азатрицикло[6.3.1.02,7] додека-2(7),3,5-триен-4-сульфонамида
10-Трифторацетил-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-4-сульфонилхлорид (320 мг, 0,9 ммоль) перемешивают в ТГФ (10 мл) и обрабатывают 40% Me2 NH/H2 O (1,5 мл). Через 10 минут смесь концентрируют и подвергают хроматографии на силикагеле (ТСХ 30% этилацетат/гексан Rf 0,31) с получением масла (256 мг, 78%). Этот материал растворяют в метаноле (6 мл) и NH4OH (2 мл) и перемешивают 18 часов. Смесь концентрируют и подвергают азеотропной обработке с метанолом (3 раза). Полученное масло растворяют в метаноле и обрабатывают 3 н. HCl в этилацетате (4 мл), концентрируют, растворяют в минимуме метанола и насыщают Et2O и перемешивают 18 часов. Продукт собирают фильтрованием в виде белого порошка (163 мг, 59%). (ТСХ 10% метанол/CH2Cl2 (NH3) Rf 0,54).1H ЯМР (data, свободное основание) (400 МГц, CDCl3) δ 7,64 (м, 2H), 7,41 (д, J=8,0 Гц, 1H), 3,30 (м, 2H), 3,20 (д, J=12,5 Гц, 2H), 3,07 (дд, J=12,5,2,2 Гц, 2H), 2,69 (с, 6H), 2,45, (м, 1H), 2,00 (д, J=11,0 Гц, 1H),13C ЯМР (100 МГц, CDCl3) δ 128, 43, 124,16, 122, 75, 46,67, 46,55, 42,11, 39,44, 37,81, газовая хроматография и масс-спектроскопия m/e 266 (M+), (data HCl соль)1H ЯМР (400 МГц, DMSO-d6) δ 7, 68-7,52 (3H), 3,38 (м, 2H), 3,24 (м, 2H), 3,04 (м, 2H), 2,58 (с, 6H), 2,22 (м, 1H), 2,04 (д, J=11,0 Гц, 1H), газовая хроматография и масс-спектроскопия m/e 266 (M+). Анал. рассч. для C13H18N2O2HCl: C, 51,56; H, 6,32; N, 9,25. Найдено C, 51,36; H,6,09; N,9,09.
ПРИМЕР 50
ГИДРОХЛОРИД 4-(1-ПИРРОЛИДИНИЛСУЛЬФОНИЛ)-10-АЗАТРИЦИКЛО[6.3.1.02,7]ДОДЕКА-2(7),3,5-ТРИЕНА
Аналог пирролидина получают из 10-трифторацетил-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-4-сульфонилхлорида (320 мг, 0,9 ммоль) как замещением пирролина на стадии сочетания, как описано в примере 49B. TFA продукт изолируют в виде масла (314 мг, 89%). Удаление защитной группы и превращение в соль, как в примере 49B, дает белый порошок (189 мг, 63%). (ТСХ 10% метанол/CH2Cl2 (NH3) Rf 0,60). (ТСХ 50% этилацетат/гексан Rf 0,65).1H ЯМР (400 МГц, CDCl3) δ 7,66 (д, J=8,0 Гц, 1H), 7,64 (с, 1 H), 7,37 (д, J=8,0 Гц, 1 H), 3,30-3,15 (м, 8H), 3,00 (м,2H), 2,39 (м, 1 H), 1,98 (д, J=11,5 Гц, 1H), 1,72 (м, 4H),13C ЯМР (100 МГц, CDCl3) δ 146,91, 144,08, 136,65, 127,90, 124,18, 122,36, 50,43, 47,87, 46,80, 46,63, 42,11, 39,63, 25,10, химическая ионизация при атмосферном давлении МС m/e 293 [(M+1)+], (data HCI соль)1H ЯМР (400 МГц, DMSO-d6) δ 9,78 (ушир.с, NH), 8,1 (ушир.с, NH), 7,73 (д, J=1, 5 Гц,1H), 7,66 (дд, J=8,0,1,5 Гц, 1H), 7,53 (д, J=8,0 Гц, 1H), 3,39-3,01 (10H), 2,21 (м, 1H), 2,04 (д, J=11,0 Гц, 1H), 1,66 (м, 4H), газовая хроматография и масс-спектроскопия m/e 292 (M+). Анал. рассч. для C13H18N2O2HCI-1/2метанол: C, 54,07; H, 6,47; N, 8,51. Найдено C, 53,98; H,6,72; N, 8,12.
ПРИМЕР 51
ГИДРОХЛОРИД 5,13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2,4(8),9-ТРИЕН-6-ОНА
(Указанное в заголовке соединение получают, следуя процедурам, описанным в Quallich, G.J.; Morrissey, P.M.Synthesis 1993, 51-53, обработкой трет-бутилового эфира 4,5-динитро-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-карбоновой кислоты в качестве эквивалента ортофторфенильной группе.)1H ЯМР (400 МГц, DMSO-d6) δ 10,42 (с, NH), 9,88 (ушир.с, NH), 7,52 (ушир.с, 1H), 7,15 (с, 1H), 6,79 (с, 1H), 3,41 (д, J=5, 0 Гц, 2H), 3,35-3,13 (м, 4H), 2,93 (м, 2H), 2,12 (м, 1H), 1,95 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 215,2 [(M+1)+].
ПРИМЕР 52
ГИДРОХЛОРИД 6-ОКСО-5-ОКСА-7,13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
(Смотри ссылки: Nachman, R.J. J. Het. Chem. 1982, 1545.) 2,2, 2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон (317 мг, 1,11 ммоль) перемешивают в ТГФ (10 мл), обрабатывают карбонилдиимидазол (269 мг, 1, 66 ммоль) и нагревают до 60°C в течение 18 часов. Смесь концентрируют, разбавляют CH2Cl2 (50 мл) и промывают 1 н. водным раствором HCl (3×10 мл). Органический слой сушат через хлопковую пробку, концентрируют и подвергают хроматографии на силикагеле (50% этилацетат/гексан) с получением масла (130 мг). Этот материал преобразуют в указанное в заголовке соединение методами, описанными в примере 19C.1H ЯМР (400 МГц, DMSO-d6) (11,78 (с, NH), 9,56 (ушир.с, NH), 7,63 (ушир.с, NH), 7,24 (с, 1H), 7,07 (с,1H), 3,26 (ушир.с, 2H), 3,16 (ушир.т, J=9,5 Гц, 1H), 2,93 (ушир.с, 1H), 2,18 (м, 1H), 1,97 (д, J=11,0 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 217,2 [(M+1)+].
ПРИМЕР 53
ГИДРОХЛОРИД 6-БЕНЗИЛ-5-ОКСА-7,13-ДИАЗАТЕТРАЦИКЛО [9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
2,2,2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло [6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон и фенил-ацетил хлорид преобразуют в указанное в заголовке соединение следуя процедурам, описанным в примере 54.1H ЯМР (400 МГц, CD3 OD) δ 7,63 (с, 1H), 7,58 (с, 1Н), 7,36-7,24 (5Н), 4,29 (с, 2Н), 3,46 (д, J=2,5 Гц, 2H), 3,39 (д, J=12,0 Гц, 2H), 3,18 (2H), 2,42 (м, 1H), 2,15 (д, J=11,5 Гц, 1H), химическая ионизация при атмосферном давлении MS m/e 291,2 [(M+1)+].
ПРИМЕР 54
ГИДРОХЛОРИД 6-ЭТИЛ-5-ОКСА-7,13-ДИАЗАТЕТРАЦИКЛО[9.3.1.02,10.04,8]ПЕНТАДЕКА-2(10), 3, 6,8-ТЕТРАЕНА
2,2,2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон и пропионилхлорид преобразуют в указанное в заголовке соединение, следуя процедурам, описанным в примере 40 и Goldstein, S.W.; Dambek, P.J. J. Het. Chem. 1990, 27, 335.1H ЯМР (400 МГц, CD3OD) δ 7,64 (с, 1H), 7,62 (с, 1H), 3, 48 (д, J=2,5 Гц, 2H), 3,41 (д, J=12,0 Гц, 2H), 3,20 (2H), 3,01 (q, J=7,5 Гц, 2H), 2,45 (м, 1H), 2,17 (д, J=11,5 Гц, 1H), 1,42 (т, J=7,5 Гц, 3H), химическая ионизация при атмосферном давлении MS m/e 229,2 [(M+1)+].
ПРИМЕР 55
ГИДРОХЛОРИД 6-ИЗОПРОПИЛ-5-ОКСА-7,13-ДИАЗАТЕТРАЦИКЛО[9.3.1. 02,10.04,8]ПЕНТАДЕКА-2(10),3,6,8-ТЕТРАЕНА
2,2,2-Трифтор-1-(4-гидрокси-5-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)этанон изобутирилхлорид преобразуют в указанное в заголовке соединение, следуя процедурам, описанным в примере 54 (ТСХ 25% этилацетат/гексан Rf 0,14).1H ЯМР (400 МГц, CD3OD) δ 7,65 (2H), 3,49 (ушир.с, 2H), 3,41 (д, J=12,0 Гц, 2H), 3,33-3,19 (3H), 2, 45 (м, 1H), 2,18 (д, J=11,5 Гц, 1H), 1,45 (д, J=7,0 Гц, 6H), химическая ионизация при атмосферном давлении МС m/e 243,2 [(M+1)+], (HCl соль) т.пл. 249-251°C.
ПРИМЕР 56
ГИДРОХЛОРИД 5,14-ДИАЗАТЕТРАЦИКЛО [10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1-(5,14-Диазатетрацикло[10.3.1.02,11.04, 9]гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон (На основе метода Campbell, K.N.; Schaffner, I.J. J. Am. Chem. Soc. 1945, 67, 86.)
1-(4-Амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (607 мг, 1,98 ммоль) растворяют в смеси 95% этанол/H2O (5 мл) и обрабатывают FeCl3 .6H2O (800 мг, 2,97 ммоль), ZnCl2 (27 мг, 0,20 ммоль) в этаноле (2 мл). Смесь нагревают до 65°C в течение 15 мин, обрабатывают акролеином (0,2 мл, 2,97 ммоль) и нагревают до кипения с обратным холодильником в течение 2,5 часов. Смесь полностью анализируют ТСХ, охлаждают и гасят в насыщенном водном растворе NaHCO3 (40 мл). Смесь (pH 8,5) экстрагируют CH2Cl2(8 x 30 мл). Органический слой промывают H2O и насыщенным водным раствором NaCl, затем сушат через хлопковую пробку. Концентрирование дает темное масло, которое подвергают хроматографии на силикагеле с получением масла желтого цвета (105 мг, 17%). (ТСХ 50% этилацетат/гексан Rf 0,08).
B) Гидрохлорид 5, 14-диазатетрацикло[10.3.1.02,11.04,9] гексадека-2(11),3,5,7,9-пентаена
1-(5,14-Диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7, 9-пентаен-10-ил)-2,2,2-трифторэтанон (94,7 мг, 0,31 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17 и получают кристаллическое твердое вещество (36,9 мг).1H ЯМР (400 МГц, CD3OD) δ 9,19 (м, 2H), 8,33 (с, 1H), 8,27 (с, 1H), 8,10 (дд, J=8,3, 5,6 Гц, 1H), 3,78 (ушир.с, 1H), 3,74 (ушир.с, 1H), 3,58 (ушир.д, J=11,4 Гц, 2H), 3,40 (м, 2H), 2,50 (м, 1H), 2,34 (д, J=11,6 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 210,9 [(М+1)+]. Т.пл. 260°C (разл.). Анал. рассч. для С14 H14N2.2HCl: C, 59,38; H, 5,69; N, 9,89. Найдено C, 59,69; H, 5,82; N, 9,79.
ПРИМЕР 57
ГИДРОХЛОРИД 6-МЕТИЛ-5, 14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9 ]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1((6-Метил-5,14-диазатетрацикло[10.3.1.02,11.04,9] гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон
Следуя методу, описанному в примере 56A, 1-(4-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2, 2-трифторэтанон (686 мг, 2,00 ммоль) подвергают взаимодействию с (E)-2-бутеналем (0,2 мл, 2,97 ммоль) и получают желтое масло (335,6 мг, 52%). (ТСХ 75% этилацетат/гексан Rf 0,25).
B) Гидрохлорид 6-метил-5,14-диазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,5,7,9-пентаена
1-(6-Метил-5,14-диазатетрацикло[10.3.1.02,11 .04,9]гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2, 2-трифторэтанон (308 мг, 0,96 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают кристаллическое твердое вещество (186 мг).1H ЯМР (400 МГц, CD3OD) δ 9,00 (д, J=8,5 Гц, 1H), 8,25 (с, 1H), 8,17 (с, 1H), 7,94 (д, J=8,5 Гц, 1H), 3,76 (ушир.с, 1H), 3,71 (ушир.с, 1H), 3,57 (ушир.д, J=11,8 Гц, 2H), 3,38 (м, 2H), 3,01 (с, 3H), 2,49 (м, 1H), 2,32 (д, J=11,6 Гц, 1H), химическая ионизация при атмосферном давлении МС m/e 225,2 [(М+1)+]. Т.пл. >300°C (разл.). Анал. рассч. для C15H16N2·2HCl,1/2H2O: C, 58,83; H, 6,25; N, 9,15. Найдено C, 58,49; H, 6,22; N, 9,02.
ПРИМЕР 58
ГИДРОХЛОРИД 7-МЕТИЛ-5, 14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1-(7-Метил-5, 14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3, 5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон
Следуя методу, описанному в примере 56A, 1-(4-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (686 мг, 2,00 ммоль) подвергают взаимодействию с 2-метилпропеналем (0,25 мл, 3,00 ммоль) и получают желтое масло (94 мг, 15%). (ТСХ 10% метанол/CH2Cl2 Rf 0,16).
B) Гидрохлорид 7-метил-5,14-диазатетрацикло[10.3.1.02,11.04,9 ]гексадека-2(11),3,5,7,9-пентаена
1-(7-Метил-5,14-диазатетрацикло[10.3.1.02,11 .04,9]гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон (86 мг, 0,27 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают кристаллическое твердое вещество (12,6 мг).1H ЯМР (400 МГц, CD3OD) δ 9,10 (с, 1H), 9,00 (с, 1H), 8,22 (с, 1H), 8,20 (с, 1H), 3,76 (ушир.с, 1H), 3,72 (ушир.с, 1H), 3,57 (ушир.д, J=11,5 Гц, 2H), 3,39 (м, 2H), 2,71 (с, 3H), 2,48 (м, 1H), 2,32 (д, J=11,6 Гц, 1 H), химическая ионизация при атмосферном давлении МС m/e 225,0 [(M+1)+].
ПРИМЕР 59
ГИДРОХЛОРИД 7-ЭТИЛ-5,14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04, 9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1-(7-Этил-5,14-диазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон
Следуя методу, описанному в примере 56A, 1-(4-амино-10-азатрицикло[6.3.1.02,7]додека-2(7), 3,5-триен-10-ил)-2,2,2-трифторэтанон (686 мг, 2,00 ммоль) подвергают взаимодействию с 2-этилпропеналем (0,35 мл, 3,60 ммоль) и получают желтое масло (110 мг, 16%). (ТСХ 75% этилацетат/гексан Rf 0,32).
B) Гидрохлорид 7-этил-5,14-диазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,5,7,9-пентаена
1-(7-Этил-5, 14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаен-10-ил)-2,2, 2-трифторэтанон (94 мг, 0,28 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают кристаллическое твердое вещество (33 мг).1H ЯМР (400 МГц, CD3OD) δ 9,12(с, 1H), 9,00 (с, 1H), 8,23 (с, 1H), 8,18 (с, 1H), 3,76 (ушир.с, 1H), 3,72 (ушир.с, 1Н), 3,56 (ушир.д, J=11,5 Гц, 2Н), 3,37 (м, 2Н), 3,05 (кв., J=7,5 Гц, 2Н), 2,48 (м, 1Н), 2,32 (д, J=11,6 Гц, 1Н), 1,44 (т, J=7,5 Гц, 3Н), химическая ионизация при атмосферном давлении МС m/е 239,1 [(М+1)+]. Т.пл. 288-291°С (разл.). Анал. рассч. для С16Н18N2·2НСl·Н2O: С, 58,36; Н, 6,73; N, 8,51. Найдено С, 57,98; Н, 5,99; N, 8,41.
ПРИМЕР 60
ГИДРОХЛОРИД 8-МЕТИЛ-5, 14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7, 9-ПЕНТАЕНА
A) 1-(8-Метил-5,14-диазатетрацикло[10.3.1.02,11,.04,9]гексадека-2(11), 3,5,7, 9-пентаен-10-ил)-2,2,2-трифторэтанон
Следуя методу, описанному в примере 56А, 1-(4-амино-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (775 мг, 2,52 ммоль) подвергают взаимодействию с 1-бутен-3-оном (0,32 мл, 3, 79 ммоль) и получают желтое масло (424 мг, 52%). (ТСХ 50% этилацетат/гексан Rf 0,08).
B) Гидрохлорид 8-метил-5,14-диазатетрацикло[10.3.1.02,11.04,9 ]гексадека-2(11),3,5,7,9-пентаена
1-(8-метил-5,14-диазатетрацикло[10.3.1.02,11.04, 9]гексадека-2 (11),3,5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон (403 мг, 1,26 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают кристаллическое твердое вещество (266 мг).1Н ЯМР (400 МГц, CD3OD) δ 9,01 (д, J=5,6 Гц, 1Н), 8,49 (с, 1Н), 8,22 (с, 1Н), 7,97 (д, J=5,6 Гц, 1Н), 3,76 (ушир.м, 2Н), 3,58 (ушир.д, J=11,5 Гц, 2Н), 3,40 (м, 2Н), 3,06 (с, 3Н), 2,48 (м, 1 Н), 2,33 (д, J=11,6 Гц, 1Н). Анал. рассч. для C15H16N2·HCl·H2O: С, 57,15; Н, 6,39; N, 8,89. Найдено С, 57,43; Н, 6,44; N, 8,82.
ПРИМЕР 61
ГИДРОХЛОРИД 5,14-ДИАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,7, 9-ТЕТРАЕН-6-ОНА
A) Соль лития 3,3-диметоксипропановой кислоты
(Имеет отношение к способам, описанным в Alabaster, С.Т. et. al., J. Med. Chem. 1988, 31, 2048-2056.)
Метиловый эфир 3,3-диметоксипропановой кислоты (14,25 г, 96,2 ммоль) в ТГФ (100 мл) обрабатывают LiOH·H2O (2,5 г, 106 ммоль) и Н2О (2 мл). Смесь поддерживают при кипении с обратным холодильником в течение 4 часов, охлаждают до комнатной температуры и азеотропно сушат из ТГФ (4 раза) и получают белое твердое вещество (13,3 г).
B) 1-(4-(N-3', 3'-Диметоксипропионамид)-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2, 2,2-трифторэтанон
Соль лития 3,3-диметоксипропановой кислоты (840 мг, 6,0 ммоль) в ТГФ (15 мл) обрабатывают трифторуксусным ангидридом (0,85 мл, 6,0 ммоль) по каплям и перемешивают в течение 15 минут. Полученный желтый раствор добавляют по каплям к энергично перемешиваемой смеси 1-(4-амино-10-азатрицикло [6.3.1.02.77]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанона (540 мг, 2 ммоль) в ТГФ (5 мл) и насыщенного водного раствора NaHCO3 (2 мл). Через 3 часа реакционную смесь разбавляют Н2О и экстрагируют этилацетатом (3 раза). Органический слой промывают насыщенным раствором соли, сушат над Na2SO4, фильтруют и концентрируют до масла, которое очищают хроматографией на силикагеле, и получают белое твердое вещество (477 мг, 62%). (ТСХ 50% этилацетат/гексан Rf 0,37).
C) 1-(5, 14-Диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,1,9-тетраен-6-он-10-ил)-2,2, 2-трифторэтанон
1-(4-(N-3',3'-Диметоксипропионамид)-10-азатрицикло[6.3.1.02,7]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (460 мг, 1,19 ммоль) обрабатывают трифторуксусной кислотой (4 мл) и перемешивают в течение 18 часов, концентрируют, разбавляют СН2 Cl2 и Н2O. Водный слой экстрагируют СН2Cl2 (4 раза) и органический слой промывают насыщенным водным раствором NaHCO3 (40 мл) и насыщенным водным раствором NaCl, затем сушат через хлопковую пробку. Концентрирование дает желтое твердое вещество (320 мг, 83%).
D) Гидрохлорид 5,14-диазатетрацикло[10.3.1.02,11.04, 9]гексадека-2(11),3,7,9-тетраен-6-она
1-(5, 14-Диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,7,9-тетраен-6-он-10-ил)-2,2,2-трифторэтанон (540 мг, 2 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают указанное в заголовке соединение в виде розового кристаллического твердого вещества (72 мг, 71%).1Н ЯМР (400 МГц, CD3OD) δ 8,42 (д, J=8,8 Гц, 1Н), 7,90 (с, 1H), 7,66 (с, 1Н), 6,98 (д, J=8,8 Гц, 1Н), 3,59 (ушир.с, 1H), 3,56 (ушир.с, 1H), 3,49 (дд, J=12,4, 5,8 Гц, 2Н), 3,29 (м, 2Н), 2,42 (м, 1H), 2,23 (д, J=11,6 Гц, 1H), химическая ионизация при атмосферном давлении МС m/е 227 [(М+1)+]; т.пл. 300°С (разл.). Анал. рассч. для C14 H14N2O·2HCl: С, 56,20; Н, 5,39; N, 9,36. Найдено С, 56,40; Н, 5,63; N, 9,25.
ПРИМЕР 62
ГИДРОХЛОРИД 6-ХЛОР-5, 14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) 1-(6-Хлор-5,14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3, 5,7,9-пентаен-10-ил)-2,2,2-трифторэтанон
1-(5,14-Диазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,7,9-тетраен-6-он-10-ил)-2,2,2-трифторэтанон (156 мг, 0,49 ммоль) обрабатывают POCl3 (5 мл) и нагревают до 100°С с перемешиванием в течение 3 часов. После концентрирования в вакууме остаток разбавляют СН2Cl2 (15 мл) и осторожно обрабатывают насыщенным раствором NaHCO3 (10 мл) с перемешиванием. Как только замедляется выделение СО2, смесь разделяют и водный слой экстрагируют СН2 Cl2 (3 раза). Органический слой промывают Н2O и насыщенным раствором NaCl, фильтруют через хлопок и концентрируют до коричневого масла (217 мг, 93%). (ТСХ этилацетат, Rf 0,3).1H ЯМР (400 МГц,2HCCl3) δ 8,03 (д, J=8,5 Гц, 1Н), 7,83 (с, 1Н), 7,62 (с, 1Н), 7,35 (д, J=8,5 Гц, 1Н), 4,43 (м, 1Н), 4,01 (м, 1Н), 3,62 (м, 1Н), 3,29 (м, 2Н), 3,23 (м, 1Н), 2,45 (м, 1Н), 2,10 (д, J=11,6 Гц, 1 Н), химическая ионизация при атмосферном давлении МС m/е 341,1 [(М+1)+].
B) Гидрохлорид 6-хлор-5, 14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена
1-(6-Хлор-5,14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7, 9-пентаен-10-ил)-2,2,2-трифторэтанон (26 мг, 0,076 ммоль) преобразуют в указанное в заголовке соединение, используя методы, описанные в примере 17, и получают указанное в заголовке соединение в виде твердого вещества (5,8 мг, 24%).1H ЯМР (свободное основание, 400 МГц,2 HCCl3) δ 8,01 (д, J=8,5 Гц, 1Н), 7,77 (с, 1Н), 7,57 (с, 1Н), 7,30 (д, J=8,5 Гц, 1H), 3, 28 (ушир.с, 1Н), 3,24 (ушир.с, 1H), 3,12 (ушир.д, J=12,5 Гц, 2Н), 2,96 (ушир.д, J=12,5 Гц, 2Н), 2,41 (м, 1H), 2,02 (д, J=11,6 Гц, 1H), химическая ионизация при атмосферном давлении МС m/е 245,1 [(М+1)+].
ПРИМЕР 63
ГИДРОХЛОРИД 6-МЕТОКСИ-5, 14-ДИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,5,7,9-ПЕНТАЕНА
A) Трет-бутиловый эфир 6-хлор-5,14-диазатетрацикло[10.3.1.02,11.04,9 ]гексадека-2(11),3,5,7,9-пентаен-10-карбоновой кислоты
6-Хлор-5,14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаен (2,82 г, 11,53 ммоль) преобразуют указанное в заголовке соединение, как описано в примере 22А, и получают коричневое масло (3,55 г, 89%). (ТСХ: 5% метанол/СН2Cl2, Rf 0,37).
B) Трет-бутиловый эфир 6-метокси-5,14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7, 9-пентаен-10-карбоновой кислоты
Металлический натрий (-12 мг) растворяют в метаноле (1 мл) в атмосфере азота с перемешиванием и обрабатывают раствором трет-бутилового эфира 6-хлор-5, 14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7, 9-пентаен-10-карбоновой кислоты (118 мг, 0,33 ммоль) в метаноле (3 мл) и поддерживают при кипении с обратным холодильником в течение 18 часов. Смесь охлаждают, концентрируют, обрабатывают Н2 O и экстрагируют СН2Cl2. Органический слой промывают насыщенным раствором NaCl и фильтруют через хлопковую пробку, затем концентрируют до масла (165 мг). (ТСХ: 5% метанол/СН2Cl2 Rf 0,55).
С) Гидрохлорид 6-метокси-5, 14-диазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,5,7,9-пентаена
Трет-бутиловый эфир 6-метокси-5,14-диазатетрацикло[10.3.1.02,11.04,9 ]гексадека-2(11),3,5,7,9-пентаен-10-карбоновой кислоты (138 мг, 0,41 ммоль) растворяют в трифторуксусной кислоте (4 мл), поддерживают при кипении с обратным холодильником в течение 4 часов. Смесь охлаждают и концентрируют до масла, которое растворяют в этилацетате и обрабатывают 3 н. HCl/этилацетатом (1 мл). После концентрирования остаток подвергают перекристаллизации из смеси метанол/диэтиловый эфир и получают бежевое твердое вещество (51 мг, 26%).1H ЯМР (400 МГц, CD3OD) δ 8,77 (д, J=9,5 Гц, 1Н), 8,01 (с, 1Н), 7,90 (с, 1Н), 7,54 (д, J=9,5 Гц, 1H), 4,30 (с, 3Н), 3,65 (ушир.с, 1Н), 3,61 (ушир.с, 1H), 3,50 (дд, J=12,4, 3,8 Гц, 2Н), 3,29 (м, 2Н), 2,44 (м, 1H), 2,24 (д, J=11,6 Гц, 1H), химическая ионизация при атмосферном давлении МС m/е 241,2 [(M+1)+]; Т.пл. 240, (темнеет), 275°С (разл.); (ТСХ: 10% метанол (NH3)/CH2Cl2, Rf 0,38).
ПРИМЕР 64
ГИДРОХЛОРИД 5,8,14-ТРИАЗАТЕТРАЦИКЛО[10.3.1.02,11.04,9]ГЕКСАДЕКА-2(11),3,7,9-ТЕТРАЕН-6-ОНА
А) 1-(5,8,14-Триазатетрацикло[10.3.1.02,11.04,9 ]гексадека-2(11),3,7,9-тетраен-6-он-10-ил)-2,2,2-трифторэтанон
1-(4,5-Диамино-10-азатрицикло[6.3.1.02,7 ]додека-2(7),3,5-триен-10-ил)-2,2,2-трифторэтанон (536 мг, 1,88 ммоль) перемешивают в этаноле (4 мл). Эту смесь обрабатывают метил-2-гидроксил-2-метоксиацетатом (0,203 мл, 2,07 ммоль) и перемешивают при 70°С в течение 2,5 часов. Реакционную смесь охлаждают до комнатной температуры и концентрируют. Растирание с метанолом и фильтрование обеспечивают светло-желтое твердое вещество (337 мг, 55%). (ТСХ 10% метанол/СН2Cl2 Rf 0,57).
В) Гидрохлорид 5,8,14-триазатетрацикло[10.3.1.02,11.04,9]гексадека-2(11),3,7, 9-тетраен-6-она
1-(5,8,14-Триазатетрацикло[10.3.1.02, 11.04,9]гексадека-2(11),3,7,9-тетраен-6-он-10-ил)-2,2,2-трифторэтанон (145 мг, 0,45 ммоль) преобразуют в указанное в заголовке соединение методами, описанными в примере 17С, и получают коричневое твердое вещество (26 мг, 46%).1H ЯМР (400 МГц, D2O) δ 7,94 (с, 1Н), 7,58 (с, 1Н), 7,18 (с, 1Н), 3,39 (ушир.с, 2Н), 3,28 (ушир.д, J=12,5 Гц, 1Н), 3,12 (ушир.д, J=12,5 Гц, 1Н), 2,29 (м, 1Н), 1,99(д, J=12,0 Гц, 1Н), химическая ионизация при атмосферном давлении МС m/е 228,2 [(М+1)+]; Т.пл. 296, (темнеет), 310°С (разл.); (ТСХ: 10% СН2Cl2/метанол(NH3), Rf 0,10).
Реферат
Изобретение относится к новому способу получения замещенных арилконденсированных азаполициклических соединений формул (II) и (VIII), новым промежуточным продуктам и способам их получения. Замещенные арилконденсированные азаполициклические соединения формулы (II) обладают свойствами связываться с нейроновыми никотиновыми ацетилхолиновыми специфическими рецепторными сайтами и могут быть использованы при модулировании холинергической функции для лечения различных заболеваний. Способ получения соединения формулы (II)

где R6 представляет собой водород, R2 и R3 выбраны независимо из водорода, галогена, (С1-С6)алкила, необязательно замещенного от 1 до 3 атомами фтора и (С1-С6)алкокси, необязательно замещенного от 1 до 3 атомами фтора, включает гидрирование соединения формулы (1а)':


полученного из соединения формулы (III), с получением промежуточно образующихся соединений формулы (IX) и (VII):

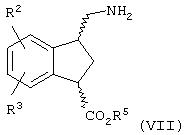
Соединение (VII) при обработке основанием циклизуется, с получением соединения формулы (VIII),

которое восстанавливают. Соединение формулы (III) может быть получено взаимодействием соединения (IV) с соединением формулы (V)


7 н. и 7 з.п. ф-лы.
Формула













Комментарии