Производные бифениламидина - RU2197478C2
Код документа: RU2197478C2
Чертежи






























Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым селективным ингибиторам активированного фактора Ха свертывающей системы крови (ниже называемого "Fxa"), с общей формулой (I).
УРОВЕНЬ ТЕХНИКИ
Терапия, имеющая целью антикоагуляцию, играет важную роль в лечении и профилактике тромбоэмболий, таких, как инфаркт миокарда, церебральный тромбоз, тромбоз
периферических артерий и тромбоз глубоко расположенных вен.
В частности, для профилактики хронического тромбоза желательны безвредные и пригодные для перорального введения антикоагулянты, которые можно вводить в течение длительного периода времени. Однако в настоящее время единственным из вышеупомянутых антикоагулянтов являются калий-варфариновые агенты, для которых трудно контролировать степень антикоагуляции, и поэтому сохраняется необходимость в антикоагулянтах, которые были бы более удобными в использовании.
Хотя в прошлом были разработаны антитромбиновые агенты, предназначенные для использования в качестве антикоагулянтов, но известно, что применение этих агентов, например, гирудина, связано с риском возникновения кровотечения, являющегося возможным побочным действием этих лекарств. Сейчас начинают понимать, что ингибирование FXa, расположенного выше тромбина в каскаде свертывания крови, является системно более эффективным, чем ингибирование тромбина, и что ингибиторы FXa не вызывают вышеописанного существенного побочного действия и являются клинически предпочтительными.
Соединения бифениламидина, проявляющие активность ингибирования FXa, описаны в публикации The 17th Symposium of Medicinal Chemistry, The 6th Annual Meeting of Division of Medicinal Chemistry, Abstracts, 184-185, 1997. Однако соединения по настоящему изобретению представляют собой новые соединения, существенно отличающиеся использованием гетероатома в связи между бифениламидиновой структурой, которая может взаимодействовать с карманом S1, и циклической структурой, способной взаимодействовать с сайтом связывания арила, а также присутствием заместителя, такого, как карбоксильная группа, на линкерном бензольном конце.
Кроме того, в японской нерассмотренной патентной публикации (Kokai) 4-264068 описываются производные бифениламидина, как циклические имино-производные. Однако соединения по настоящему изобретению отличаются присутствием связи через гетероатом в бензильном положении.
Таким образен, целью настоящего изобретения является создание нового соединения, которое может являться ингибитором FXa, имеющим возможность клинического применения.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Авторы настоящего изобретения приложили максимальные
усилия для достижения поставленной цели и в результате осуществили следующие изобретения 1-10.
1. Производное бифениламидина общей формулы (1):

в которой R1 представляет собой атом водорода, атом фтора, атом хлора, атом брома, гидроксильную группу, аминогруппу, нитрогруппу, C1-8-алкильную группу или C1-8-алкоксильную группу;
L представляет собой прямую связь или C1-4-алкиленовую группу;
R2 представляет собой атом фтора; атом хлора; атом брома; гидроксильную группу; аминогруппу; C1-8-алкоксильную группу; карбоксильную группу; C1-8-алкоксикарбонильную группу; арилоксикарбонильную группу; аралкоксикарбонильную группу; карбамоильную группу, в которой атом азота, входящий в состав карбамоила, может быть замещен моно- или ди-C1-8-алкильной группой или может представлять собой атом азота в аминокислоте; C1-8-алкилкарбонильную группу; C1-8-алкилсульфенильную группу; C1-8-алкилсульфинильную группу; C1-8 -алкилсульфонильную группу; моно- или ди-C1-8-алкиламиногруппу; моно- или ди-C1-8-алкиламиносулфонильную группу; сульфогруппу; фосфоногруппу; бис(гидроксикарбонил)метильную группу; бис(алкоксикарбонил)-метильную группу); или 5-тетразолильную группу;
R3 представляет собой атом водорода, атом фтора, атом хлора, атом брома, гидроксильную группу, аминогруппу, нитрогруппу, C1-8-алкильную группу, C1-8-алкоксильную группу, карбоксильную группу или C1-8-алкоксикарбонильную группу;
Х представляет собой любую из групп:
-О-, -S-, -SO-, SO2-, -NH-CO-NH-, -N(R4)-, -CO-N(R5)-, -N(R5)-СО-, -N(R5)-SO2-, -SO2-N(R5)-,
в которых R4 представляет собой атом водорода, C1-10-алкильную группу, C1-10-алкилкарбонильную группу, C1-10-алкилсульфонильную группу, С3-8-циклоалкильную группу или арильную группу,
R5 представляет собой атом водорода, C1-10-алкильную группу, С3-8-циклоалкильную группу или арильную группу,
причем алкильная группа в R4 и R5 может быть замещена арильной группой, гидроксильной группой, аминогруппой, атомом фтора, атомом хлора, атомом брома, С1-8-алкоксильной группой, карбоксильной группой, С1-8-алкоксикарбонильной группой, арилоксикарбонильной группой, аралкоксикарбонильной группой, или 5-тетразолильной группой;
Y представляет собой С4-8-циклоалкильную группу, в которой метиленовая группа в С4-8-циклоалкиле может быть замещена карбонильной группой, или может быть замещена атомом фтора, атомом хлора, атомом брома, гидроксильной группой, аминогруппой, С1-8-алкильной группой, С1-8-алкоксильной группой, карбамоильной группой, С1-8 -алкоксикарбонильной группой; карбоксильной группой, аминоалкильной группой, моно- или ди-алкиламиногруппой, моно- или ди-алкиламиноалкильной группой; или следующее 5-8-членное кольцо формул I-1 или I-2:


в которых, в формулах I-1 или I-2, в каждой циклической системе метиленовая группа может быть замещена карбонильной группой, а цикл может иметь ненасыщенные связи,
R6 представляет собой атом водорода, атом фтора, атом хлора, атом брома, гидроксильную группу, аминогруппу, нитрогруппу, С1-8-алкильную группу или С1-8-алкоксильную группу,
W представляет собой С-Н или атом азота, при условии, что W не является атомом азота в случае, когда цикл представляет собой 5-членное кольцо,
Z представляет собой атом водорода; С1-10-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой, за исключением случая, когда Z представляет собой С1-алкил, аминогруппу, C1-8 -алкоксильнуто группу, за исключением случая, когда Z представляет собой C1-алкил, карбоксильную группу, C1-8-алкоксикарбонильную группу, арилоксикарбонильную группу или аралкоксикарбонильную группу; C1-8-алкилкарбонильную группу; арилкарбонильную группу; аралкарбонильную группу; амидиногруппу; или следующую группу формулы I-3:

в которой R7 представляет собой C1-8-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой или C1-8-алкоксильной группой; аралкильную группу; или арильную группу;
m представляет собой целое число от 1 до 3;
n представляет собой целое число от 0 до 3, при условии, что W не является атомом азота, когда n представляет собой 0 или 1;
или его фармацевтически приемлемая соль.
2. Производное
бифениламидина, в котором, в указанной формуле (1),
R1 представляет собой атом водорода, атом фтора, атом хлора, атом брома, гидроксильную группу, аминогруппу, C1-4
-алкильную группу или C1-4-алкоксильную группу;
L представляет собой прямую связь или C1-4-алкиленовую группу;
R2 представляет собой атом фтора; атом
хлора; атом брома; гидроксильную группу; аминогруппу; C1-8-алкоксильную группу; карбоксильную группу; C1-8-алкоксикарбонильную группу; арилоксикарбонильную группу;
аралкоксикарбонильную группу, карбамоильную группу, причем атом азота в карбамоильной группе может быть замещен моно- или ди-C1-8-алкильной группой или может представлять собой атом азота в
аминокислоте; C1-8-алкилкарбонильную группу; C1-8-алкилсульфенильную группу; C1-8-алкилсульфинильную группу; C1-8-алкилсульфонильную группу; моно- или
ди-C1-8-алкиламиногруппу; моно- или ди-C1-8-алкиламиносулфонильную группу; сульфогруппу; фосфоногруппу; бис(гидроксикарбонил)метильную группу; бис(алкоксикарбонил)-метильную
группу; или 5-тетразолильную группу;
R3 представляет собой атом водорода;
Х представляет собой любую из групп:
-О-, -S-, -N(R4)-, -CO-N(R5),
-N(R5)-CO-, -N(R5)-SO2- или -SO2-N(R5)-;
в которых R4 представляет собой атом водорода, C1-10-алкильную группу,
C1-10-алкилкарбонильную группу или C1-10-алкилсульфонильную группу,
R5 представляет собой атом водорода или C1-10-алкильную группу,
причем
алкильная группа в R4 и R5 может быть замещена арильной группой, гидроксильной группой, аминогруппой, атомом фтора, атомом хлора, атомом брома, C1-8-алкоксильной
группой, карбоксильной группой, C1-8-алкоксикарбонильной группой, арилоксикарбонильной группой, аралкоксикарбонильной группой, карбамоильной группой, или 5-тетразоильной группой;
Y
представляет собой С4-8-циклоалкильную группу, в которой метиленовая группа, являющаяся частью С4-8-циклоалкила, может быть замещена карбонильной группой, или может быть замещена
атомом фтора, атомом хлора, атомом брома, гидроксильной группой, аминогруппой, C1-8-алкильной группой, C1-8-алкоксильной группой, карбамоильной группой, C1-8
-алкоксикарбонильной группой; карбоксильной группой, аминоалкильной группой, моно- или ди-алкиламиногруппой, или моно- или ди-алкиламиноалкильной группой; или следующее 5-8-членное кольцо формулы
II-1:

в которой, в формуле II-1, в циклической системе метилен может быть замещен карбонильной группой,
R6 представляет собой атом водорода, атом фтора, атом хлора, атом брома, гидроксильную группу, аминогруппу, С1-4-алкильную группу, или C1-8-алкоксильную группу;
W представляет собой С-Н или атом азота, при условии, что W не является атомом азота в случае, когда цикл представляет собой 5-членное кольцо;
Z представляет собой атом водорода; C1-10-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой, за исключением случая, когда Z представляет собой C1 -алкил, аминогруппу, C1-8-алкоксильную группу, за исключением случая, когда Z представляет собой C1-алкил, карбоксильную группу, C1-8-алкоксикарбонильную группу, арилоксикарбонильную группу или аралкоксикарбонильную группу; C1-8-алкилкарбонильную группу; арилкарбонильную группу; аралкилкарбонильную группу; амидиногруппу; или следующую группу формулы II-2:

в которой R7 представляет собой C1-8-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой или C1-4-алкоксильной группой; аралкильную группу; или арильную группу;
m представляет собой целое число от 1 до 3;
n представляет собой целое число от 0 до 3, при условии, что W не является атомом азота, когда n представляет собой 0 или 1;
или его фармацевтически приемлемая соль.
3.
Производное бифениламидина общей формулы (2):

в которой L представляет собой связь или C1-4-алкиленовую группу;
R2 представляет собой карбоксильную группу; C1-4-алкоксикарбонильную группу; аралкоксикарбонильную группу; карбамоильную группу, в которой входящий в ее состав атом азота может быть замещен моно- или ди-C1-4-алкильной группой или может представлять собой атом азота в аминокислоте; или C1-4-алкилкарбонильную группу;
Х представляет собой -О-, -N(R4)- или NH-CO-,
в которых R4 представляет собой атом водорода, C1-10-алкильную группу, C1-10 -алкилкарбонильную группу или C1-10-алкилсульфонильную группу, причем алкильная группа необязательно может быть замещена гидроксильной группой, аминогруппой, группой фтора, карбоксильной группой или С1-8-алкоксикарбонильной группой;
Y представляет собой С5-6-циклоалкильную группу, причем метиленовая группа, входящая в состав С5-6 -циклоалкильной группы, может быть замещена карбамоильной группой, С1-4-алкоксильной группой или карбоксильной группой; или следующее 5-6-членное кольцо формулы III-1:

в которой W представляет собой С-Н или атом азота, при условии, что W не является атомом азота в случае, когда цикл представляет собой 5-членное кольцо;
Z представляет собой атом водорода; С1-4-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой, за исключением случая, когда Z представляет собой C1-алкил, аминогруппу, карбоксильную группу или C1-4-алкоксикарбонильную группу; C1-4-алкилкарбонильную группу; амидиногруппу; или следующую группу формулы III-2:

в которой R7 представляет собой C1-4-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой;
n представляет собой целое число от 0 до 2, при условии, что W не является атомом азота, когда n равно 0 или 1;
или его фармацевтически приемлемая соль.
4. Производное бифениламидина, в котором, в указанной формуле (2),
Х представляет собой -О- или
-N(R4)-,
в которых R4 представляет собой атом водорода, C1-10-алкильную группу, C1-10-алкилкарбонильную группу или C1-10
-алкилсульфонильную группу, причем алкильная группа необязательно может быть замещена гидроксильной группой, аминогруппой, группой фтора, карбоксильной группой или C1-8-алкоксикарбонильной
группой;
или его фармацевтически приемлемая соль.
5. Производное бифениламидина, в котором, в указанной формуле (2),
Х представляет собой -NH-CO-,
или его
фармацевтически приемлемая соль.
6. Производное бифениламидина, в котором, в общей формуле (2),
L представляет собой связь;
R2 представляет собой
карбоксильную группу или метоксикарбонильную группу;
Х представляет собой -О- или -N(R4)-,
в которых R4 представляет собой атом водорода, метильную группу или
2-гидроксиэтильную группу;
Y представляет собой любую из формул:



n представляет собой 1;
или его фармацевтически приемлемая соль.
7. Пролекарство, которое вырабатывает производное бифениламидина или его фармацевтически приемлемую соль в соответствии с любыми из указанных п. 1-6, in vivo.
8. Ингибитор коагуляции крови, включающий по меньшей мере производное бифениламидина или его фармацевтически приемлемую соль в соответствии с любыми из указанных п. 1-7, а также фармацевтически приемлемый носитель.
9. Агент для профилактики тромбоза или эмболии, включающий по меньшей мере производное бифениламидина или его фармацевтически приемлемую соль в соответствии с любыми из указанных пунктов 1-7, а также фармацевтически приемлемый носитель.
10. Агент для терапии тромбоза или эмболии, включающий по меньшей мере производное бифениламидина или его фармацевтически приемлемую соль в соответствии с любыми из указанных пунктов 1-7, а также фармацевтически приемлемый носитель.
НАИЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Настоящее изобретение подробно описывается ниже.
При описании заместителей соединений формулы (1) по настоящему
изобретению используются следующие дефиниции (определения):
Термин "C1-8-алкил" обозначает разветвленную или прямую углеродную цепь, имеющую от 1 до 8 атомов углерода, и включает,
например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, неопентил, изопентил, 1,2-диметилпропил, гексил, изогексил, 1,1-диметилбутил, 2,2-диметилбутил, 1-этилбутил, 2-этилбутил,
изогептил, октил или изооктил и т.п. Среди них предпочтительны имеющие от 1 до 4 атомов углерода, а особо предпочтительны метил или этил.
Термин "C1-8-алкокси" обозначает алкоксильную группу, имеющую от 1 до 8 атомов углерода, и включает, например, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси, трет-бутокси, пентилокси, неопентилокси, трет-пентилокси, 2-метилбутокси, гексилокси, изогексилокси, гептилокси, изогептилокси, октилокси или изооктилокси и т.п. Среди них предпочтительны имеющие от 1 до 4 атомов углерода, а особо предпочтительны метокси или этокси.
Термин "C1-4-алкилен" обозначает линейный алкилен, имеющий от 1 до 4 атомов углерода, и включает метилен, этилен, пропилен или бутилен.
Термин "C1-8-алкоксикарбонил" обозначает метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил, трет-бутоксикарбонил, пентилоксикарбонил, изопентилоксикарбонил, неопентилоксикарбонил, гексилоксикарбонил, гептилоксикарбонил, или октилоксикарбонил и т.п.; предпочтительно, это метоксикарбонил, этоксикарбонил или трет-бутоксикарбонил, а более предпочтительно - метоксикарбонил.
Термин "арилоксикарбонил" обозначает феноксикарбонил, нафтилоксикарбонил, 4-метилфеноксикарбонил, 3-хлорофеноксикарбонил или 4-метоксифеноксикарбонил и т.п.; и предпочтительно, это феноксикарбонил.
Термин "аралкоксикарбонил" обозначает бензилоксикарбонил, 4-метоксибензилоксикарбонил или 3-трифторметилбензилоксикарбонил и т.п.; и предпочтительно, это бензилоксикарбонил.
Термин "аминокислота" обозначает натуральную или искусственно синтезированную коммерчески доступную аминокислоту; предпочтительно, это глицин, аланин или β-аланин; и более предпочтительно, это глицин.
Термин "C1-8-алкилкарбонил" обозначает карбонильную группу с прямой или разветвленной углеродную цепью, имеющую от 1 до 8 атомов углерода, и включает, например, гормил, ацетил, пропионил, бутирил, изобутирил, валерил, изовалерил, пивалоил, гексаноил, гептаноил или октаноил и т.п.; среди них предпочтительны имеющие от 1 до 4 атомов углерода, и более предпочтительны ацетил или пропионил.
Термин "C1-8 -алкилсульфенил" обозначает алкилсульфенильную группу, имеющую от 1 до 8 атомов углерода, и включает, например, метилтио, этилтио, бутилтио, изобутилтио, пентилтио, гексилтио, гептилтио или октилтио и т.п., и предпочтительно, это метилтио.
Термин "C1-8-алкокилсульфинил" обозначает алкилсульфинильную группу, имеющую от 1 до 8 атомов углерода, и включает, например, метилсульфинил, этилсульфинил, бутилсульфинил, гексилсульфинил или октилсульфинил и т.п., и предпочтительно, это метилсульфинил.
Термин "C1-8-алкокилсульфонил" обозначает алкилсульфонильную группу, имеющую от 1 до 8 атомов углерода, и включает, например, метилсульфонил, этилсульфонил, бутилсульфонил, гексилсульфонил или октилсульфонил и т.п., и предпочтительно, это метилсульфонил.
Термин "моно- или ди-C1-8-алкиламино" обозначает метиламино, диметиламино, этиламино, пропиламино, диэтиламино, изопропиламино, диизопропиламино, дибутиламино, бутиламино, изобутиламино, втор-бутиламино, трет-бутиламино, пентиламино, гексиламино, гептиламино или октиламино и т.п.; предпочтительно, это метиламино, диметиламино, этиламино, диэтиламино или пропиламино; и более предпочтительно, это метиламино или диметиламино.
Термин "моно- или ди-C1-8-алкиламиносульфонил" обозначает, например, метиламиносульфонил, диметиламиносульфонил, этиламиносульфонил, пропиламиносульфонил, диэтиламиносульфонил, изопропиламиносульфонил, диизопропиламиносульфонил, дибутиламиносульфонил, бутиламиносульфонил, изобутиламиносульфонил, втор-бутиламиносульфонил, трет-бутиламиносульфонил, пентиламино-сульфонил, гексиламиносульфонил, гептиламиносульфонил или октиламиносульфонил и т.п.; предпочтительно, это метиламиносульфонил, диметиламиносульфонил, этиламиносульфонил, диэтиламиносульфонил или пропиламиносульфонил; и более предпочтительно, это метиламиносульфонил или диметиламиносульфонил.
Термин "бис(алкоксикарбонил)метил" обозначает, в частности, бис(метоксикарбонил)метил или бис(этоксикарбонил)метил и т.п.; предпочтительно, это бис(метоксикарбонил)метил.
Термин "C1-10-алкил" обозначает прямую или разветвленную углеродную цепь, имеющую от 1 до 10 атомов углерода, и включает, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, неопентил, изопентил, 1,2-диметилпропил, гексил, изогексил, 1,1-диметилбутил, 2,2-диметилбутил, 1-этилбутил, 2-этилбутил, гептил, изогептил, 1-метилгексил, 2-метилгексил, октил, 2-этилгексил, нонил, децил, или метилнонил и т.п. Среди них предпочтительны имеющие от 1 до 4 атомов углерода, а особо предпочтительны метил или этил.
Термин "C1-10-алкилкарбонил" обозначает карбонильную группу с прямой или разветвленной углеродную цепью, имеющую от 1 до 10 атомов углерода, и включает, например, гормил, ацетил, пропионил, бутирил, изобутирил, валерил, изовалерил, пивалоил, гексаноил, гептаноил, октаноил, нонаноил или деканоил и т. п. ; среди них предпочтительны имеющие от 1 до 4 атомов углерода; и более предпочтительны ацетил или пропионил.
Термин "C1-10-алкилсульфонил" обозначает алкилсульфонильную группу, имеющую от 1 до 10 атомов углерода, и включает, например, метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, бутилсульфонил, изобутилсульфонил, пентилсульфонил, изопентилсульфонил, неопентилсульфонил, гексилсульфонил, гептилсульфонил, октилсульфонил, нонилсульфонил или децилсульфонил и т. п.; предпочтительны имеющие от 1 до 4 атомов углерода, и более предпочтительны метилсульфонил или этилсульфонил.
Термин "С3-8-циклоалкил" обозначает циклоалкильную группу, имеющую от 3 до 8 атомов углерода, и включает, в частности, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, или циклооктил; и предпочтительно, циклопропил.
Термин "арил" обозначает, в частности, соединения карбоциклической арильной группы, такие, как фенил или нафтил, или гетероарил, такой, как пиридил или фурил, и предпочтительно, это фенил.
Термин "С4-8-циклоалкил" обозначает циклоалкильную группу, имеющую от 4 до 8 атомов углерода, и включает, в частности, циклобутил, циклопентил, циклогексил, циклогептил, или циклооктил и т.п.; и предпочтительно, это циклопентил или циклогексил.
Термин "аминоалкил" обозначает линейный алкил, имеющий аминогруппу и от 1 до 8 атомов углерода, и включает, в частности, 8-аминооктил, 6-аминогексил, 4-аминобутил, 2-аминоэтил или аминометил; предпочтительно, это 2-аминоэтил или аминометил.
Термин "моно- или ди-С1-8-алкиламино" обозначает, например, метиламино, диметиламино, этиламино, пропиламино, диэтиламино, изопропиламино, диизопропиламино, дибутиламино, бутиламино, изобутиламино, втор-бутиламино, трет-бутиламино и т.п.; предпочтительно, это метиламино, диметиламино, этиламино, диэтиламино, изопропиламино или диизопропиламино; и более предпочтительно, это этиламино, диэтиламино или изопропиламино.
Термин "моно- или ди-С1-8-алкиламиноалкил" обозначает, в частности, метиламиноэтил, диметиламиноэтил, этиламиноэтил, метиламинопропил, диметиламинопропил, этиламинопропил, диэтиламинопропил, метиламинобутил или диметиламинобутил и т.п.; предпочтительно, это метиламиноэтил, диметиламиноэтил, или этиламиноэтил.
"C1-10-алкил", который связывается с атомом азота в качестве элемента Z, обозначает прямую или разветвленную углеродную цепь, имеющую от 1 до 10 атомов углерода, и представляет собой, например, метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, пентил, неопентил, изопентил, 1,2-диметилпропил, гексил, изогексил, 1,1-диметилбутил, 2,2-диметилбутил, 1-этилбутил, 2-этилбутил, гептил, изогептил, 1-метилгексил, 2-метилгексил, октил, 2-этилгексил, нонил, децил или 1-метилнонил и т.п. Среди них предпочтительны имеющие от 1 до 4 атомов углерода, а особо предпочтительны изопропил или пропил.
Термин "арилкарбонил" обозначает бензоил, 4-метоксибензоил, или 3-трифторметилбензоил и т.п., и предпочтительно, это бензоил.
Термин "аралкилкарбонил" включает, в частности, бензилкарбонил, фенэтилкарбонил, фенилпропилкарбонил, 1-нафтилметилкарбонил или 2-нафтилметилкарбонил и т.п.; и предпочтительно, это бензилкарбонил.
Термин "аралкил" включает, в частности, бензил, фенэтил, фенилпропил, 1-нафтилметил или 2-нафтилметил и т.п.; и предпочтительно, это бензил.
Кроме того, в дефинициях, касающихся заместителей в соединении формулы (2) по настоящему изобретению.
Термин "C1-4-алкоксикарбонил" обозначает метоксикарбонил, этоксикарбонил, пропоксикарбонил, изопропоксикарбонил, бутоксикарбонил, изобутоксикарбонил, втор-бутоксикарбонил или трет-бутоксикарбонил; предпочтительно, это метоксикарбонил, этоксикарбонил или трет-бутоксикарбонил, и более предпочтительно - метоксикарбонил.
Термин "C1-4-алкил" обозначает прямую или разветвленную углеродную цепь, имеющую от 1 до 4 атомов углерода, и включает, например, метил, этил, пропил, изопропил, бутил, изобутил или трет-бутил; и предпочтительно, он представляет собой метил или этил.
Термин "C1-4-алкилкарбонил" обозначает карбонильную группу с прямой или разветвленной углеродной цепью, имеющую от 1 до 4 атомов углерода, и включает, например, гормил, ацетил, пропионил, бутирил или изобутирил и т.п.; и предпочтительно, он представляет собой ацетил или пропионил.
Термин "С5-6-циклоалкил" обозначает циклоалкильную группу, имеющую от 5 до 6 атомов углерода, и включает циклопентил или циклогексил; и предпочтительно, он представляет собой циклогексил.
Термин "C1-4-алкокси" обозначает алкоксильную группу, имеющую от 1 до 4 атомов углерода, и включает, в частности, метокси, этокси, пропокси, изопропокси, бутокси, изобутокси, втор-бутокси или трет-бутокси и т.п. Среди них предпочтительны метокси или этокси.
Соединение (1) по настоящему изобретению может образовывать соли присоединения (аддитивные соли). Кроме того, оно может образовывать соли с основаниями, в зависимости от видов заместителей. Применение этих солей не ограничиваются, если только они являются фармацевтически приемлемыми, и они включают, в частности, минеральные соли, такие, как гидрохлорид, гидробромид, гидроиодид, фосфат, нитрат или сульфат, и т.п.; органические сульфонаты, такие, как метансульфонат, 2-гидроксиэтансульфонат или р-толуолсульфонат и т.п.; и органические карбонаты, такие, как ацетат, трифторацетат, пропионат, оксалат, цитрат, малонат, сукцинат, глютарат, адипат, тартрат, малеат, малат или манделат, и т.п. В качестве солей с основаниями, сюда относятся соли с неорганическими основаниями, такие, как соли натрия, соли калия, соли магния, соли кальция или соли алюминия, а также соли с органическими основаниями, такие, как соли метиламина, соли этиламина, соли лизина или соли орнитина и т.п.
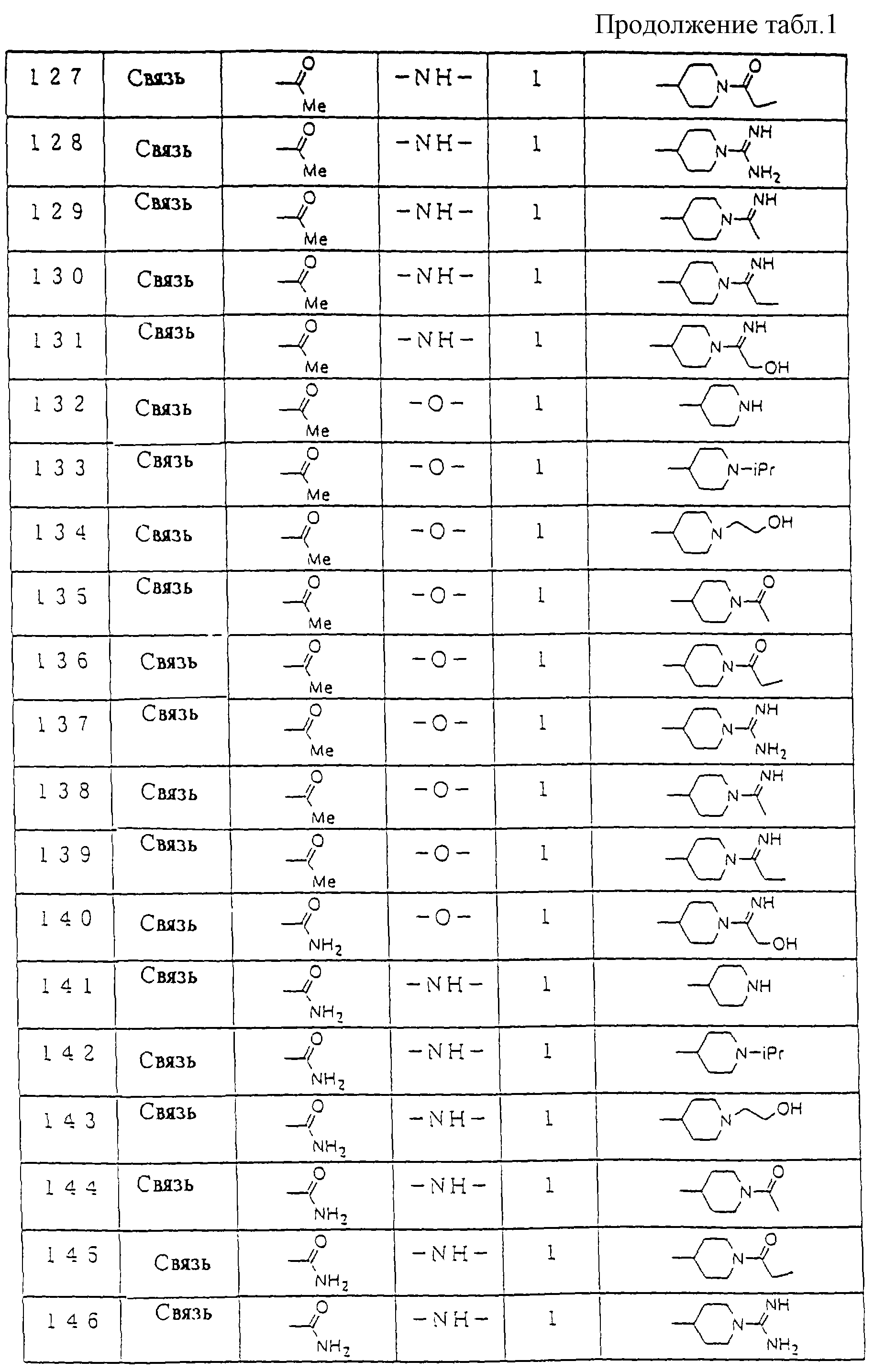
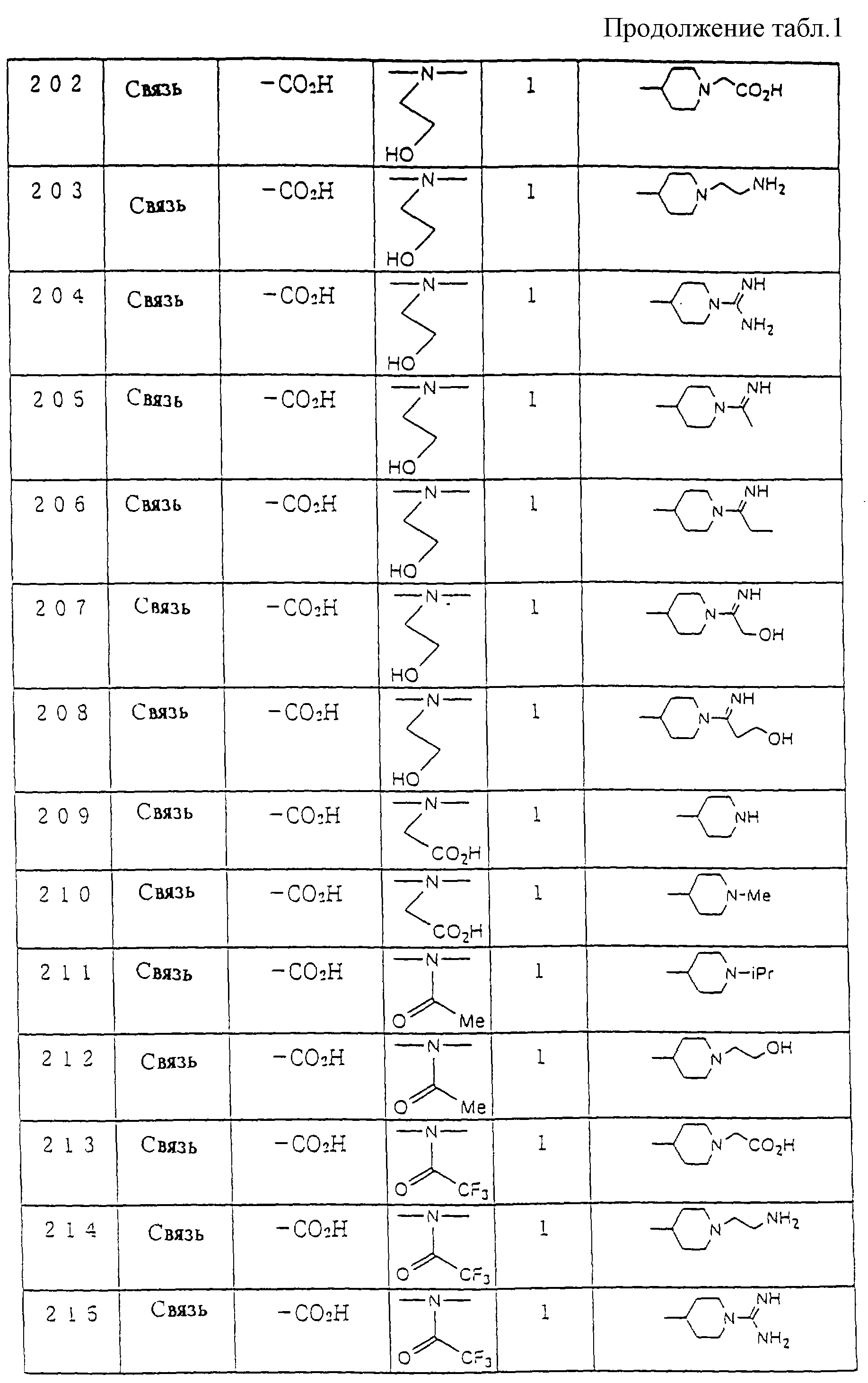
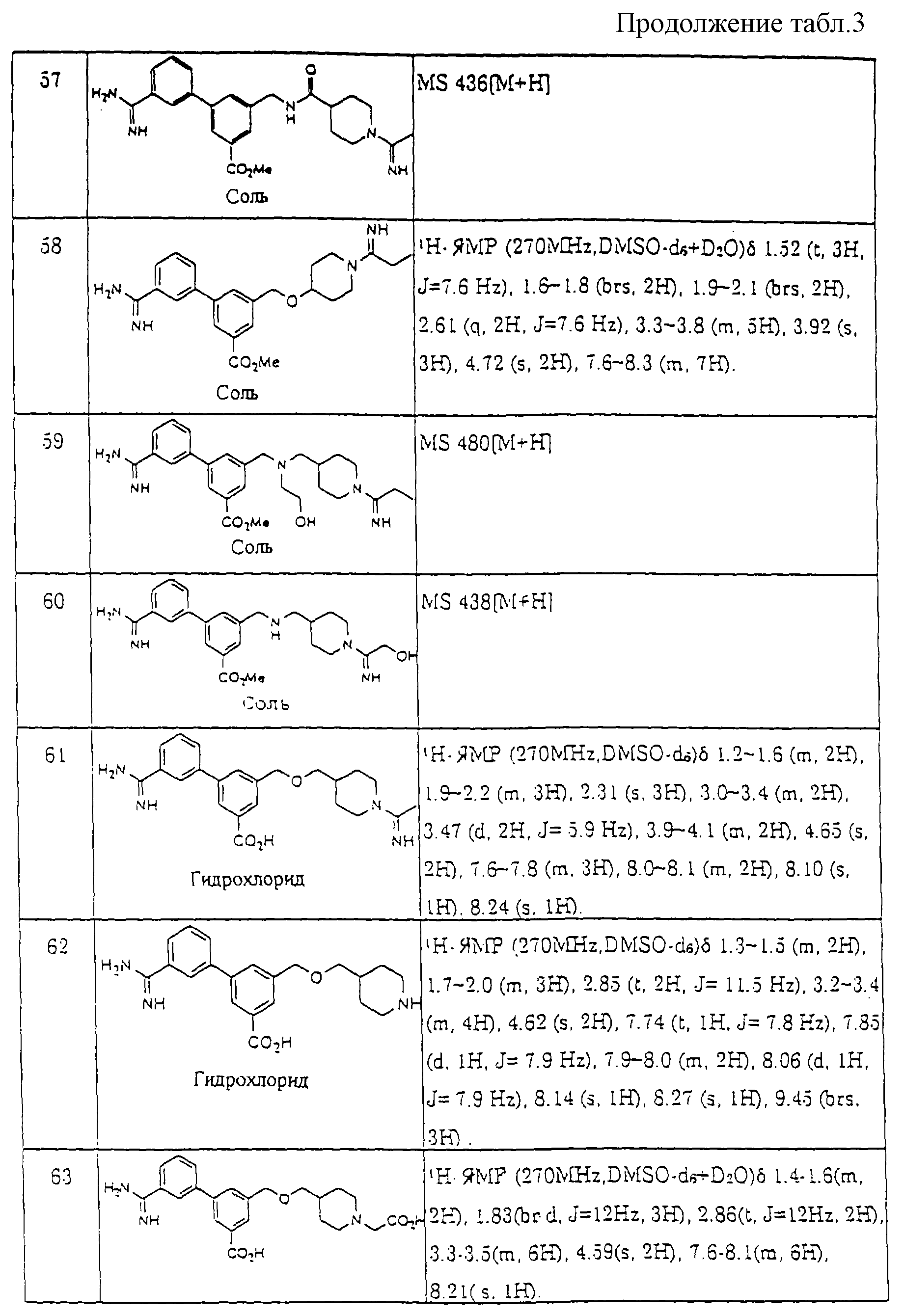
Предпочтительные соединения по настоящему изобретению представлены в таблице 1.
Более предпочтительные соединения по настоящему изобретению те, что обозначены следующими номерами, из соединений, перечисленных в таблице 1.
Соединения : 23, 29, 30, 31, 53, 54, 57, 58, 59, 60, 91, 92, 93, 115, 119, 120, 121, 156, 166, 168, 201, 205, 206, 207, 244, 245 и 246.
Репрезентативные варианты стратегии синтеза соединений формулы (1) по настоящему изобретению подробно описываются ниже.
В соответствии с настоящим изобретением, в случае, если исходные или промежуточные соединения имеют заместители, которые влияют на ход реакции, такие, как гидроксил, амино или карбоксил и т.п., то предпочтительно соответствующим образом защитить такие функциональные группы, чтобы провести реакцию, а затем отщепить защитную группу. Для защитной группы не имеется никаких ограничений, если только она представляет собой обычно применяющуюся защитную группу для соответствующих заместителей и не оказывает отрицательных побочных действий на другие элементы во время процессов защиты и снятия защиты, и включает, например, триалкилсилил, C1-4-алкоксиметил, тетрагидропиранил, ацил или C1-4-алкоксикарбонил в качестве защитных групп для гидроксила; C1-4-алкоксикарбонил, бензилоксикарбонил или ацил в качестве защитных групп для амино; и C1-4-алкил в качестве защитной группы для карбоксила. Реакция снятия защиты (отщепления защитной группы) может проводиться в соответствии со способами, которые обычно практикуются для соответствующих защитных групп.
Среди нитрилов, которые являются предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х кислород, можно синтезировать,
например, в соответствии со следующей реакцией:

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле 1-3 как заместитель Y для элемента Z; R3 обозначает водород, фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, или С1-8-алкокси.
Таким образом, как видно из вышеприведенной реакции (а-1), нитрилы, являющиеся предшественниками соединений по настоящему изобретению, можно получить путем смешивания спирта, представленного формулой: Y1-(СН2)n-ОН, с сырьевым материалом, бифенилалкилбромидом, в присутствии оснований.
Кроме того, из нитрилов, которые являются предшественниками соединений
формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х кислород, можно синтезировать, например, в соответствии со следующей реакцией:

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3, как заместитель Y для элемента Z.
Таким образом, нитрилы, являющиеся предшественниками соединений по настоящему изобретению, можно получить путем смешивания спирта, представленного формулой: Y1-(СН2)n-OH, с исходным материалом, 3-бром-3-иодфенилалкилбромидом, в присутствии оснований, для получения 3-бром-5-иодфенилалкилового эфира, с последующим введением заместителя -L-COOMe в полученный эфир, путем монокарбонилирования или моноалкилирования, с получением 3-бромфенилалкилэфира, и с проведением после этого реакции сочетания с производным цианофенилбороновой кислоты.
Эфиризацию первой стадии в реакциях (а-1) и (а-2) осуществляют, используя в качестве растворителя алифатический эфир, такой, как тетрагидрофуран или диэтилэфир, апротонные углеводороды, такие, как бензол или толуол, апротонные полярные растворители, такие, как DMF (диметилформамид) или НМРА (гексаметилфосфорная кислота), или их смесь, и т.п., а в качестве оснований используют оксид металла, такой, как оксид бария или оксид цинка, гидроксид металла, такой, как гидроксид натрия или гидроксид калия, или гидрид металла, такой, как гидрид натрия и т.п. Реакцию проводят при температуре 0-100oС в течение 3-72 часов при перемешивании. Предпочтительно, ее проводят при 20-80oС в течение 8-36 часов, используя гидрид натрия, в абсолютных алифатических эфирах, таких, как THF (тетрагидрофуран) или простой эфир.
Реакцию для ввода заместителя -L-COOMe к эфирам, являющуюся второй стадией реакции (а-2), можно провести с помощью следующих реакций (i) или (ii).
(i) Монокарбонилирование путем введения моноокиси углерода (в случае, когда L представляет собой связь). Иод можно заместить метоксикарбонильной группой, путем растворения эфиров, полученных на первой стадии реакции (а-1) в метаноле, с добавлением бивалентного палладиевого катализатора и оснований, таких, как третичный амин, такой, как триэтиламин, и необязательно - фосфинового лиганда, такого, как трифенилфосфин, с перемешиванием в течение 3-48 часов при комнатной температуре или при нагревании в атмосфере моноокиси углерода. Предпочтительно, реакцию проводят с использованием в качестве катализатора палладия бистрифенилфосфина или ацетата палладия, а в качестве основания - диизопропилэтиламина или трибутил-амин, при температуре 60-80oС в течение 12-36 часов.
(ii) Моноалкилирование с использованием в качестве реагента органического цинка (в случае, когда L представляет собой C1-8-алкилен). Иод можно заместить алкилом, путем растворения эфиров, полученных на первой стадии реакции (а-1) и 0-валентного палладиевого катализатора, такого, как палладий тетракистрифенилфосфин, в растворителе, таком, как THF или DMF, бензол или толуол, или в их смеси, с добавлением к этому раствору раствора THF, содержащего алкилцинковый реагент формулы: 1-Zn-L-COOMe, с перемешиванием в течение 3-48 часов при комнатной температуре или при нагревании. Предпочтительно, реакцию проводят с использованием в качестве катализатора палладия тетракистрифенилфосфина, а в качестве растворителя - THF, при температуре 20-80oС в течение 6-36 часов.
(iii) Бифенилирование, являющееся третьей стадией реакции (а-2), можно осуществить посредством проведения реакции моногалогенида с цианофенилбороновой кислотой в присутствии палладиевого катализатора. Эту реакцию обычно осуществляют путем нагревания, с перемешиванием в DMF, моногалогенида, полученного на второй стадии реакции (а-2), в присутствии в качестве катализатора бивалентного палладия, такого, как ацетата палладия, и кроме того, оснований, таких, как триэтиламин и триарилфосфины, с получением требуемого соединения - цианобифенила. Предпочтительно, реакцию проводят при температуре 60-100oС в течение 2-24 часов.
Кроме того, среди нитрилов, которые являются предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х азот, можно синтезировать, например, в соответствии со следующими реакциями (b-1) и (b-2).

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле 1-3 как заместитель Y для элемента Z; R10 обозначает заместитель R4 , за исключением применения его для водорода и арила; Е представляет собой отщепляемую группу, такую, как хлор, бром, иод, ацилокси или сульфонилокси.

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R6 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле 1-3 как заместитель Y для элемента Z; Аr обозначает арил; Е представляет собой отщепляемую группу, такую, как хлор, бром, иод, ацилокси или сульфонилокси.
N-алкилирование реакций (b-1) и (b-2) можно проводить с помощью условий алкилирования, которые известны. Таким образом, можно провести реакцию исходного материала, бифенилалкилбромида с аминами формулы: Y1-(CH2)n-NH2 в присутствии минеральных солей, таких, как карбонат калия, или аминов, таких, как третичные амины, которые действуют как основания, получив в результате вторичный амин, который является соединением по настоящему изобретению. Можно провести реакцию этого соединения с алкилирующим агентом формулы: R4-E, получив в результате третичный амин, являющийся соединением по настоящему изобретению. Вышеуказанные реакции обычно проводят посредством смешивания аминов с алкилирующими агентами в различных соотношениях в подходящих растворителях и перемешивания в течение 1-96 часов при охлаждении, при комнатной температуре или при нагревании. Обычно эти реакции проводят с использованием в качестве основания минеральных солей, таких, как карбонат калия или карбонат натрия, или органических третичных аминов, таких, как триэтиламин или пиридин, и с использованием в качестве растворителя спиртов, таких, метанол или этанол, углеводородов, таких, как бензол или толуол, или растворителей, которые не влияют на реакцию, таких как THF, диоксан, ацетонитрил, DMF или DMSO, или их смесей, при соотношении алкилирующих агентов к аминам, составляющем 1:10-10:1. Предпочтительно, реакцию проводят при соотношении алкилирующих агентов к аминам, составляющем 1:5-1:1, при комнатной температуре или при нагревании, в течение 2-24 часов.
Из нитрилов, которые являются предшественниками соединений формулы (1) по
настоящему изобретению, соединения, имеющие в качестве элемента Х серу, можно синтезировать, например, в соответствии со следующими реакциями (с-1) и (с-2):

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R6 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3, как заместитель Y для элемента Z; и Е обозначает отщепляемую группу, такую, как хлор, бром, иод, сульфонат.

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z; и Е представляет собой отщепляемую группу, такую, как хлор, бром, иод или сульфонат.
Тиоэфиризацию реакций (с-1) и (с-2) можно проводить с помощью условий тиоэфиризации, которые известны. Обычно это осуществляется путем смешивания галогенидов с тиолами, в различных соотношениях, в подходящих растворителях, в присутствии оснований, таких, как гидроксид натрия или аммиак, с перемешиванием их при охлаждении, при комнатной температуре или при нагревании, в течение от 30 минут до 96 часов. В качестве растворителей используют соединения, которые не влияют на реакцию, такие, как вода, этанол, DMF или толуол, а в качестве основания применяют гидроксид натрия, аммиак или карбонат цезия и т. п. Предпочтительно, реакции проводят путем смешивания при соотношении алкилгалогенидов к тиолам, составляющем 1:5-5:1, при перемешивании, при комнатной температуре или при нагревании, в течение от 30 минут до 24 часов.
Кроме того, полученный сульфид можно подвергнуть окислению, как показано в нижеследующей реакции (d), получив в результате соединение, имеющее сульфоксид или сульфон в качестве элемента Х соединения формулы (1).

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); и Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z.
Окисление реакции (d) можно проводить в соответствии со способом, описанным в публикации Jikken Kagaku Kohza (The 4thEdition), 24, Organic Synthesis Vi - heteroelement.metallic compounds-, p.350-373, под редакцией Японской Химической Ассоциации. Обычно реакцию проводят с использованием сульфидов или сульфоксидов, используя в качестве растворителей воду или спирты, такие, как этанол, а в качестве окисляющего агента - перекись водорода, надуксусную кислоту, метанадиодную кислоту или m-хлорнадбензойную кислоту и т. п., при охлаждении, при комнатной температуре или при нагревании, с перемешиванием в течение от 30 минут до 24 часов. Предпочтительно, сульфоксид получают в течение от 30 минут до 12 часов при 0-20oС, а сульфон получают в течение 1-12 часов при 0-80oС.
Кроме того, среди нитрилов, которые являются предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х
амидо-связь, можно синтезировать, например, в соответствии со следующими реакциями (е-1) и (е-2):

в которой R1, R3, R5, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z; и G представляет собой галоген, ацилокси, р-нитрофенокси или гидроксил и т.п.

в которой R1, R3, R5, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z; и G представляет собой галоген, ацилокси, р-нитрофенокси или гидроксил и т.п.
Реакции (е-1) и (е-2) можно проводить с помощью условий для амидирования, которые известны. Обычно амиды можно получать путем смешивания активных производных карбоновых кислот с соединениями амина в подходящих растворителях, в присутствии оснований, для ацилирования. В качестве активных производных карбоновых кислот используют активные сложные эфиры, такие, как галогениды кислот, смесь ангидридов карбоновой кислоты, или р-нитрофенол, и т. п. , при охлаждении или при комнатной температуре, в течение от 30 минут до 24 часов. Предпочтительно, реакцию проводят в галогенированных углеводородах, таких, как дихлорметан, алифатические эфиры, такие, как THF или диэтилэфир, или в растворителях, таких, как ацетонитрил или DMF, или используя в качестве растворителя их смесь, с применением в качестве оснований третичных аминов, таких, как триэтиламин, при 0-20oС в течение 1-18 часов.
Кроме того, эти амиды можно получить путем конденсации между аминами и карбоновыми кислотами, в присутствии конденсирующих агентов, таких, как карбодиимиды. В этом случае, галогенированные углеводороды, такие, как DMF или хлороформ, пригодны в качестве растворителей, а N,N-дициклогексилкарбодиимид, 1-этил-(3-(N,N-диметиламино)пропил)карбодиимид, карбонилдиимидазол, дифенилфосфорилазид или диэтилфосфорилцианид пригодны в качестве конденсирующих агентов. Реакцию обычно проводят при охлаждении или при комнатной температуре, в течение 2-48 часов.
Кроме того, среди
нитрилов, которые являются предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х сульфонамидную структуру, можно синтезировать, например, в
соответствии со следующими реакциями (f-1) и (f-2):

в которой R1, R3, R5, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, С1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); и Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3, как заместитель Y для элемента Z.

в которой R1, R3, R5, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); и Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3, как заместитель Y для элемента Z.
Реакции (f-1) и (f-2) можно осуществлять путем проведения реакции амина с активными производными сульфоновых кислот в подходящих растворителях, в присутствии оснований, для получения требуемых сульфонамидов. В качестве активных производных сульфоновых кислот предпочтителен сульфонилгалогенид, и реакцию проводят в галогенированных углеводородах, таких, как дихлорметан, в алифатических эфирах, таких, как THF или диэтилэфир, в растворителях, таких, как ацетонитрил или DMF, или в смеси этих растворителей, при 0-20oС, в течение 1-24 часов, с использованием в качестве основания третичных аминов, таких, как триэтиламин.
Кроме того, среди нитрилов, которые являются
предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве элемента Х мочевинную структуру, можно синтезировать, например, в соответствии со следующей реакцией
(g):

в которой R1, R3, L, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); и Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z.
Таким образом, соединения, имеющие в качестве элемента Х структуру мочевины, можно получить путем проведения реакции амина, как исходного материала, с производными изоцианата, в подходящих растворителях, в температурном режиме от охлаждения до нагревания. Растворитель, используемый в этой реакции, может представлять собой DMF, THF, диоксан, дихлорэтан, хлороформ, ацетонитрил, DMSO, бензол или толуол и т.п.
Нитрилы, являющиеся предшественниками соединения по настоящему изобретению, получаемого с помощью вышеприведенных реакций (а-1), (а-2), (b-1), (b-2), (с-1),
(с-2), (d), (е-1), (е-2), (f-1), (f-2) и (g), можно преобразовать в производные бифениламидина, которые представляют собой соединения по настоящему изобретению, с помощью нижеследующей реакции
амидинирования:

в которой R1, R3, L, X, m и n таковы, как они определены в формуле (1); Y1 обозначает заместитель Y, определенный в формуле (1), за исключением того, который имеет структуры, определенные в формуле I-3 как заместитель Y для элемента Z; R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1); и R11 обозначает C1-4-алкил. Это амидинирование осуществляют в соответствии с условиями реакции, подробно описанными в нижеследующих пунктах (iii) или (iv):
(iii) Амидинирование через имидирование, с помощью галогенида водорода в спиртовом растворе. Реакцию, с помощью которой получают имидаты из нитрилов и спиртов, проводят, например, путем растворения алкоксиметилфенилбензонитрилов в спиртах, имеющих от 1 до 4 атомов углерода (R11OH), содержащих галогениды водорода, такие, как, хлорид водорода или бромид водорода и т.п., при перемешивании. Реакцию обычно проводят при температуре -20 - +30oС, в течение 12-96 часов. Предпочтительно, ее проводят в растворе соляной кислоты в метаноле или в этаноле, при -10 - +30oС, в течение 24-72 часов. Реакцию между имидатом и аммиаком проводят путем размешивания имидата в спирте, имеющем от 1 до 4 атомов углерода, таком, как метанол или этанол, содержащем аммиак или амины, такие, как гидроксиламин, гидразин или сложный эфир карбамата, или в алифатических эфирах, таких, как диэтилэфир, или в галогенированных углеводородах, таких, как дихлорметан или хлороформ, или в их смеси, получая в результате производное бифениламидина, являющееся соединением по настоящему изобретению. Реакцию обычно проводят при температуре -10 - +50oС, в течение 1-48 часов. Предпочтительно, ее проводят при температуре 0-30oС, в течение 2-12 часов.
(iv) Амидинирование через имидат, полученный путем прямого барботирования галогенида водорода. Реакцию между нитрилами и спиртами проводят, например, путем растворения нитрилов в алифатических эфирах, таких, как диэтилэфир, или в галогенированных углеводородах, таких, как, хлороформ, или в апротонных растворителях, таких, как бензол, с добавлением эквивалентного или избыточного количества спирта, имеющего от 1 до 4 атомов углерода (R11OH), и осуществления барботирования галогенидов водорода, таких, как соляная кислота или бромид водорода, при температуре от -30 до 0oС, в течение от 30 минут до 6 часов, при перемешивании, после чего барботирование прекращают, а перемешивание продолжают при температуре 0-50oС, в течение 3-96 часов. Предпочтительно, реакцию проводят путем барботирования соляной кислоты в течение 1-3 часов при температуре от -10 до 0oС, при размешивании в галогенированных углеводородах, содержащих эквивалентное или избыточное количество метанола или этанола, после чего барботирование прекращают и перемешивают при температуре 10-40oС в течение 8-24 часов. Полученные имидаты можно преобразовать в производные бифениламидина (1), которые представляют собой соединения по настоящему изобретению, путем размешивания их в спиртовых растворителях, имеющих от 1 до 4 атомов углерода, таких, как метанол или этанол, содержащих аммиак или амины, такие, как гидроксиламин, гидразин или сложный эфир карбамата, или в растворителях, представляющих собой алифатические эфиры, таких, как диэтилэфир, или в растворителях, представляющих собой галогенированные углеводороды, таких, как хлороформ, или в их смеси. Реакцию обычно проводят при температуре -20 - +50oС, в течение 1-4 часов. Предпочтительно, ее проводят в насыщенном растворе аммиака в этаноле, при температуре 0-30oС, в течение 2-12 часов.
Среди нитрилов, которые являются предшественниками соединений формулы (1) по настоящему изобретению, соединения, имеющие заместитель Y, в
котором заместитель Z имеет структуры, определенные в формуле I-3, можно получить путем осуществления имидоилирования нижеследующих реакций (j-1) и (j-2), после получения соединений бифениламидина,
имеющих вторичную аминогруппу в заместителе Y, с помощью вышеприведенной реакции (h):

в которой R1, R3, R6, L, W, X, Z, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1).

в которой R1, R3, R6, L, W, X, Z, m и n таковы, как они определены в формуле (1); R9 обозначает фтор, хлор, бром, гидроксил или защищенный гидроксил, амино или защищенный амино, C1-8-алкокси или метоксикарбонил, из заместителей R2, определенных в формуле (1).
Это имидоилирование проводят путем смешивания соединений бифениламидина, имеющих вторичную аминогруппу в заместителе Y, с эквивалентным или избыточным количеством имидатов, в воде или в спиртах, имеющих от 1 до 4 атомов углерода, таких, как метанол или этанол, или в алифатических эфирах, таких, как диэтилэфир, или в галогенированных углеводородах, таких, как хлороформ, или в полярных растворителях, таких, как DMF или DMSO, или в их смеси, в присутствии оснований, при перемешивании. Реакцию обычно проводят при комнатной температуре в течение 1-24 часов. В качестве основания можно использовать N-метилморфолин, триэтиламин, диизопропилэтиламин, гидроксид натрия или гидроксид калия и т.п.
Среди соединений формулы (1) по настоящему изобретению, соединения, имеющие в качестве R2 карбоксил, получают путем сложноэфирного гидролиза соединений, имеющих в качестве R9 метоксикарбонил, являющихся соединениями бифениламидина, получаемыми с помощью вышеприведенных реакций (h), (j-1) и (j-2). Этот гидролиз можно проводить в условиях основной среды, в условиях кислой среды или в условиях нейтральной среды, если это необходимо. В реакциях, проводимых в условиях основной среды, в качестве основания используют гидроксид натрия, гидроксид калия, гидроксид лития или гидроксид бария и т.п., а в реакциях, проводимых в условиях кислой среды, используют соляную кислоту, серную кислоту или льюисовы кислоты, такие, как трихлорид бора, трифторуксусная кислота или р-толуолсульфоновая кислота и т.п., и в реакциях, проводимых в условиях нейтральной среды, используют ион галогена, такой, как иодид лития или бромид лития, соли щелочных металлов с тиолом или селенолом, иодтриметилсилан, а также ферменты, такие, как эстераза. Используемые растворители включают полярные растворители, такие, как вода, спирты, ацетон, диоксан, THF, DMF, DMSO и т.п., или их смесь. Реакцию обычно проводят при комнатной температуре или при нагревании в течение 2-96 часов. Подходящие условия температуры или времени реакции и т.п. различны, в зависимости от используемых условий реакции, и подходящие условия можно выбрать обычным способом.
В соединениях, имеющих карбоксил в качестве заместителя R2, полученных с помощью вышеописанного процесса, карбоксил можно преобразовать в другие сложные эфиры, с помощью нижеследующих процессов (v), (vi) или (vii).
(v) Превращение карбоксила в алкоксикарбонил. Карбоксил можно преобразовать в алкоксикарбонил путем проведения реакции соединений, имеющих карбоксил в качестве заместителя R2, из соединений формулы (1), с эквивалентным или избыточным количеством алкилирующих агентов (например, метилацилоксихлоридов, таких, как метилацетоксихлорид или метилпивалоилоксихлорид, или аллилхлоридов, или бензилхлоридов) в галогенированных углеводородах, таких, как дихлорметан, или в алифатических эфирах, таких, как THF, или в апротонных полярных растворителях, таких, как DMF, или в их смеси, в присутствии третичных аминов, таких, как триэтиламин или диизопропилэтиламин, при -10 - +80oС в течение 1-48 часов. Предпочтительно, реакцию проводят с использованием от эквивалентного до слегка избыточного количества алкилирующего агента, в присутствии диизопропилэтиламина, при 20-60oС, в течение 2-24 часов.
(vi) Превращение карбоксила в аралкоксикарбонил. Карбоксил можно преобразовать в аралкоксикарбонил путем проведения реакции соединений, имеющих карбоксил в качестве заместителя R2, из соединений формулы (1), с эквивалентным или избыточным количеством спиртов, таких, как бензиловый спирт, с использованием в качестве растворителей галогенированных углеводородов, таких, как дихлорметан, в присутствии кислых катализаторов, таких, как соляная кислота, серная кислота или сульфоновая кислота. Реакцию обычно проводят при комнатной температуре или при нагревании в течение 1-72 часов. Предпочтительно, реакцию проводят с использованием от эквивалентного до слегка избыточного количества спиртов, в присутствии диизопропилэтиламина, при 20-60oС, в течение 2-24 часов.
(vii) Превращение карбоксила в арилоксикарбонил. Карбоксил можно преобразовать в арилоксикарбонил путем проведения реакции соединений, имеющих карбоксил в качестве заместителя R2, из соединений формулы (1), с эквивалентным или избыточным количеством ароматического соединения, имеющего гидроксил, таких, как фенол, с использованием в качестве растворителей алифатических эфиров, таких, как диэтилэфир, в присутствии конденсирующих агентов, таких, как дициклогексилкарбодиимид. Реакцию обычно проводят при температуре 0-50oС, в течение 1-48 часов. Предпочтительно, реакцию проводят при комнатной температуре, в течение 3-24 часов.
Кроме того, соединения, имеющие карбоксил в качестве R2, можно преобразовать в соединения, имеющие карбамоил, с помощью известных методов, например, путем обработки карбоксила оксалилхлоридом, и т.п., получив в результате галогенангидриды, и проведя реакцию с раствором аммиака. Подобным же образом, их можно преобразовать в N-метил-N-метоксикарбамоил, с помощью галогенангидридов с N-метил-N-метоксиамином, и затем полученное вещество можно преобразовать в алкилкарбонил, путем проведения реакции с различными алкилмагниевыми реагентами.
Из соединений по настоящему изобретению, синтезированных с помощью вышеописанных технологий, соединения, имеющие в качестве заместителя А амидиногруппу, можно ввести через один из атомов азота, входящий в состав амидиногруппы, с различными карбонилами, с помощью нижеследующих способов (ix), (х) или (xi).
(ix) Арилоксикарбонилирование амидино. Арилоксикарбонил можно ввести через один из атомов азота, входящий в состав амидино, путем перемешивания соединений, имеющих амидино в качестве заместителя А, из соединений формулы (1), с эквивалентным или избыточным количеством арилхлороформатов, таких, как фенилхлороформат, в смешанном растворителе, состоящем из воды и галогенированных углеводородов, таких, как дихлорметан, в присутствии оснований, таких, как гидроксид натрия или гидроксид калия. Реакцию обычно проводят при температуре при от -10 до +40oС, в течение 3-48 часов. Предпочтительно, реакцию проводят с применением эквивалентного или слегка избыточного количества арилхлороформата, при температуре от 0 до 30oС, в течение 6-24 часов.
(х) Алкоксикарбонилирование амидино. Алкоксикарбонил можно ввести через один из атомов азота, входящий в состав амидино, путем проведения реакции соединений, имеющих амидино в качестве заместителя А, из соединений формулы (1), с эквивалентным до избыточного количеством сложного эфира алкилкарбоновой кислоты и р-нитрофенила, в абсолютном растворителе, таком, как THF или DMF, в присутствии оснований, таких, как гидриды металлов, таких, как гидрид натрия или третичные амины, при температуре от -10 до +30oС, в течение 3-48 часов. Предпочтительно, реакцию проводят с применением от эквивалентного до слегка избыточного количества р-нитрофенилового сложного эфира алкилкарбонатов, в присутствии третичных аминов, таких, как триэтиламин или диизопропилэтиламин, при температуре от -10 до +40oС, в течение 6-24 часов.
(xi) Арилкарбонилирование амидино: Арилкарбонил можно ввести через один из электронов в азоте, входящем в состав амидино, путем проведения реакции соединений, имеющих амидино в качестве заместителя А, из соединений формулы (1), с эквивалентным до избыточного количеством ароматического хлорида карбоновой кислоты, такого, как бензоилхлорид, с использованием в качестве растворителя галогенированных углеводородов, таких, как метиленхлорид, или таких растворителей, как THF, DMF или пиридин, или их смеси, в присутствии оснований, таких, как амины, при температуре от -10 до +30oС, в течение 1-48 часов. Предпочтительно, реакцию проводят с применением от эквивалентного до слегка избыточного количества ароматического хлорида карбоновой кислоты, в присутствии аминов, таких, как триэтиламин, диизопропилэтиламин или пиридин, при температуре от -10 до +40oС, в течение 2-24 часов.
Кроме того, соединения формулы (1) можно получить путем необязательной комбинации других хорошо известных способов эфиризации, амидинирования, гидролиза, алкилимидоилирования, амидирования или эстерификации, или с помощью способа, обычно используемого специалистами в данной области техники.
Производные алкоксиметилфенилбифениламидина (1), полученные, как описано выше, можно выделить и очистить с помощью известных способов, например, путем экстракции, осаждения, фракционной хроматографии, фракционной кристаллизации или перекристаллизации и т.п. Кроме того, фармацевтически приемлемую соль соединения по настоящему изобретению можно получить, подвергнув его обычной реакции образования соли.
Производные бифениламидина и их фармацевтически приемлемые соли по настоящему изобретению обладают эффектом ингибирования активности FXa, и их можно использовать в качестве профилактического агента и/или терапевтического агента, которые клинически применимы против тромбоэмболии, такой, как инфаркт миокарда, церебральный тромбоз, тромбоз периферических артерий или тромбоз глубоко расположенных вен, в качестве ингибитора FXa.
Кроме того, на основе производных бифениламидина по настоящему изобретению можно составить фармацевтические композиции с фармацевтически приемлемыми носителями, и их можно вводить перорально или парентерально в виде различных лекарственных форм. Парентеральное введение включает, например, введение с помощью внутривенного, подкожного, внутримышечного, трансдермального, интраректального, трансназального и инстилляционного способов.
Лекарственная форма фармацевтической композиции включает следующее: например, в случае перорального введения, можно использовать таблетки, пилюли, гранулы, порошок, раствор, суспензию, сироп или капсулы и т.п.
Что касается способов получения таблеток, их можно штамповать обычными способами, с помощью фармацевтически приемлемого носителя, такого, как наполнитель, связывающее вещество или дезинтегратор, и т.п. Кроме того, лекарственную форму, представляющую собой пилюли, гранулы или порошок, можно получать обычными способами, с помощью наполнителя и т.п., так же, как и таблетки. Лекарственную форму, представляющую собой раствор, суспензию или сироп, можно получать обычными способами, с помощью сложных эфиров глицерина, спиртов, воды или растительных масел и т.п. Лекарственную форму, представляющую собой капсулы, можно получать путем наполнения капсулы, изготовленной из желатина и т.п., гранулами, порошком или раствором и т.п.
Что касается агентов для парентерального введения, то в случае внутривенного, подкожного или внутримышечного введения, их можно вводить с помощью инъекций. Растворы для инъекций можно получать путем растворения производных бифениламидина в водорастворимых растворах, таких, как например, физиологический раствор, или в водонерастворимых растворах, состоящих из органических сложных эфиров, таких, например, как пропиленгликоль, полиэтиленгликоль или растительные масла и т.п.
В случае трансдермального введения, например, может использоваться лекарственная форма в виде мази или крема. Мазь можно получить путем использования производного бифениламидина в смеси жиров и масел или вазелинов, и т. п., а крем можно получить путем смешивания производного бифениламидина с эмульгаторами.
В случае ректального введения, лекарственная форма может применяться в виде суппозитория, с использованием мягкой желатиновой капсулы, и т.п.
В случае трансназального введения (введения через нос), это соединение можно использовать в виде лекарственной формы, состоящей из жидкой или порошкообразной композиции. В качестве основы для жидкой лекарственной формы используют воду, солевой раствор, фосфатный буфер или ацетатный буфер и т.п. , и кроме того, она может содержать поверхностно-активные вещества, антиоксиданты, стабилизаторы, презерванты или вещества, придающие липкость. Основа для порошковой лекарственной формы может включать водопоглощающие материалы, такие, например, как высокорастворимые в воде полиакрилаты, низшие алкилэфиры целлюлозы, полиэтиленгликольполивинилпирролидон, амилозу или пуллулан и т. п. , или не адсорбирующие воду материалы, такие, например, как целлюлозы, крахмалы, белки, смолы или сшитые винильные полимеры. Водопоглощающие материалы предпочтительны. Эти материалы при использовании можно смешивать. Кроме того, к порошковой лекарственной форме можно добавлять антиоксиданты, красители, консерванты, презерванты или антисептики и т.п. Жидкую или порошковую лекарственную форму можно вводить, например, с помощью аэрозольного устройства.
В случае введения в виде глазных капель, можно использовать водные или неводные глазные капли. В водных глазных каплях в качестве растворителя можно использовать стерилизованную и очищенную воду или физиологический раствор и т. п. Если в качестве растворителя используют только стерилизованную и очищенную воду, то глазные капли в виде водной суспензии можно получить путем добавления суспензии таких веществ, как поверхностно-активные вещества или высокомолекулярные вещества, придающие липкость (клейкость), а растворимые глазные капли можно получить путем добавления солюбилизаторов, таких, как неионные поверхностно-активные вещества. В случае неводных глазных капель, глазные капли в виде неводной суспензии можно получить с помощью использования в качестве растворителей, применяющихся для инъекций неводных растворителей.
В случае введения через глаза иным способом, чем глазные капли, можно использовать такие лекарственные формы, как глазные мази, растворы для аппликации, диффундирующие агенты или агенты для вкладок.
Кроме того, в случае ингаляции через нос или через рот, раствор или суспензию, содержащие производное бифениламидина и обычно используемый фармацевтический носитель, вдувают, например, через аэрозольное устройство для ингаляции и т.п. Кроме того, производное бифениламидина, находящееся в форме сухого порошка, можно вводить через ингалятор и т.п., который контактирует непосредственно с легкими.
К этим лекарственным формам при необходимости можно добавлять фармацевтически приемлемые носители, такие, как изотонические агенты, презерваторы, консерваторы, увлажняющие агенты, буферы, эмульгаторы, диспергаторы или стабилизаторы и т.п.
Кроме того, при необходимости эти лекарственные формы можно стерилизовать путем добавления стерилизующего вещества, фильтрации с помощью задерживающего бактерии фильтра или путем обработки теплом или облучения и т.п. В качестве альтернативы, можно получать асептическую твердую лекарственную форму, которую можно использовать для растворения или суспендирования в подходящем асептическом растворе непосредственно перед использованием.
Доза бифениламидина по настоящему изобретению зависит от вида заболевания, способа введения, состояния, возраста, пола и веса пациента, но как правило, она составляет около 1-500 мг/день/вес человека, предпочтительно 10-300 мг/день/вес человека в случае перорального введения, и около 0,1-100 мг/день/вес человека, предпочтительно 0,3-30 мг/день/вес человека в случае внутривенного, подкожного, внутримышечного, трансдермального, интраректального, трансназального, инстилляционного введения или ингаляции.
Если бифениламидин по настоящему изобретению используют в качестве профилактического агента, его можно вводить в соответствии с хорошо известными способами, в зависимости от соответствующих условий.
ВАРИАНТЫ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Ниже настоящее изобретение иллюстрируется с помощью примеров получения, вариантов осуществления и экспериментов. Однако объем изобретения этими
примерами ни в коей мере не ограничивается.
ПРИМЕР ПОЛУЧЕНИЯ 1.
Метил-3-амино-5-гидроксиметилбензоат:

85 г 3-нитро-5-метоксикарбонилбензойной кислоты растворяли в 200 мл ТНF в потоке азота, а затем добавляли 43,4 мл борандиметилсульфидного комплекса, при перемешивании и охлаждении на льду. После продолжавшегося в течение 18 часов перемешивания добавляли 200 мл воды, а затем 96 г карбоната калия. Его экстрагировали этилацетатом, а органический слой промывали cолевым раствором. После сушки с помощью сульфата магния, полученное твердое вещество растворяли в 800 мл этилацетата, добавляли 750 мг 10%-го палладия на угле и продолжали перемешивание в потоке водорода. После окончания реакции произвели фильтрование, а затем фильтрат сконцентрировали, получив 64 г вещества по настоящему изобретению.
1Н-ЯМР (270 МНz, СDСl3): δ 2,30 (s, 1H), 3,89 (s, 3H), 4,64 (s, 1Н), 6,89 (s, 1H), 7,26 (s, 1Н), 7,39 (s, 1Н).
ПРИМЕР ПОЛУЧЕНИЯ 2.
Метил-5-гидроксиметил-3-иодбензоат:

34,3 г соединения, полученного в примере получения 1, растворяли в 200 мл THF, а затем добавляли 75 иодистоводородной кислоты, при перемешивании и охлаждении на льду. Добавляли 100 мл раствора, содержащего 13,73 г нитрита натрия. После перемешивания при 0oС в течение 40 минут, добавляли 150 мл раствора, содержащего 34,6 г иодида калия. После перемешивания при 40oС в течение 2 часов, добавляли 300 мл воды и полученный раствор концентрировали. Проводили экстрагирование этилацетатом, а органический слой промывали солевым раствором. После сушки сульфатом натрия провели очистку с помощью хроматографии на колонке с силикагелем, получив в результате 23,1 г (42%) соединения по настоящему изобретению.
1Н-ЯМР (270 MHz, СDСl3): δ 1,81 (t, 1Н, J=5,6 Hz), 3,92 (s, 3Н), 4,72 (d, 1H, J=5,6 Нz), 7,93 (s, 1Н), 7,98 (s, 1Н), 8,29 (s, 1 Н).
ПРИМЕР ПОЛУЧЕНИЯ 3.
Дигидрокси-(3-цианофенил)боран:

20 г 3-бромбензонитрила растворяли в 100 мл сухого THF, а затем, в атмосфере азота, добавляли 37,6 мл триизопропоксиборана. Этот раствор охлаждали до -78oС и по каплям добавляли 98,3 мл 1,6М раствора n-бутиллитиевого гексана, в течение 30 минут, при перемешивании. После продолжавшегося 30 минут перемешивания при комнатной температуре, смесь охлаждали до 0oС и добавляли 220 мл 4 М серной кислоты. Этот раствор нагревали в колбе с обратным холодильником до следующего дня, а затем снова охлаждали до 0oС. Добавляли 340 мл 5 М гидроксида натрия и экстрагировали с помощью 200 мл диэтилового эфира. Водный слой удаляли, затем добавляли 6 М соляную кислоту, до рН 2. Дважды провели экстракцию с помощью 300 мл этилацетата, высушили сульфатом магния, а затем растворитель удалили. Полученный сухой продукт перекристаллизовали из DMF-воды, получив 11,6 г (72%) соединения по настоящему изобретению, в виде игольчатых светло-желтых кристаллов.
1Н-ЯМР (270 MHz, DMSO-d6): δ 7,6~8,3 (m, 4H), 8,5 (brs, 2H).
ПРИМЕР ПОЛУЧЕНИЯ 4.
Метил-3-(3-цианофенил)-5-(гидроксиметил)бензоат:
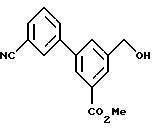
3,08 г соединения, полученного в вышеописанном примере получения 2, растворяли в 500 мл сухого THF в потоке азота, а затем к этому раствору добавляли 2,32 г соединения, полученного в вышеописанном примере получения 3, а также 2,18 г карбоната калия и 456 мг палладия тетракис(трифенолфосфина), и перемешивали при нагревании при 90oС до следующего дня. Реакцию гасили путем добавления воды, экстрагировали этилацетатом и высушивали на сульфате магния, а затем удаляли растворитель. После очистки с помощью хроматографии на колонке с силикагелем получали 2,05 г (73%) соединения по настоящему изобретению, в виде бесцветных кристаллов.
1Н-ЯМР (270 MHz, СDСl3):

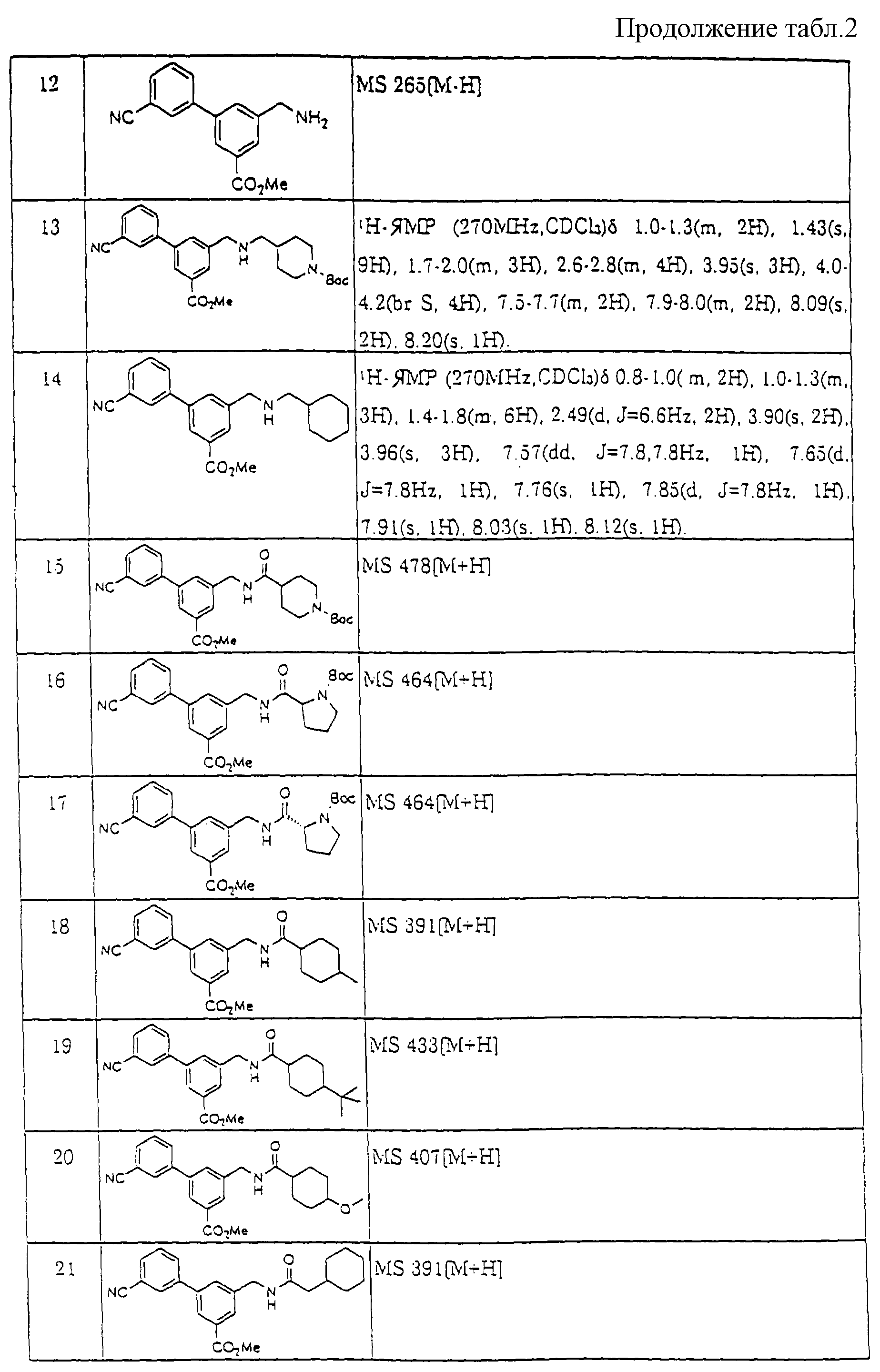
В соответствии с тем же способом, который описан в примере получения 4, синтезировали соединения примеров получения 5-10, перечисленные в таблице 2.
ПРИМЕР ПОЛУЧЕНИЯ 11.
Метил-3-(3-цианофенил)-5-(бромметил)бензоат:

К 1,0 г соединения, полученного в вышеописанном примере получения 4, добавляли 20 мл диэтилового эфира, получив суспензию, а затем медленно, по каплям добавляли 0,5 мл трибромида фосфора. Реакционный раствор перемешивали при комнатной температуре в течение 19 часов, а затем подвергали экстрагированию. Органический слой промывали насыщенным солевым раствором, высушивали сульфатом натрия, а затем растворитель удаляли под вакуумом и получали соединение по настоящему изобретению в форме светло-желтого твердого вещества (1,2 г, 98%).
1Н-ЯМР(270 MHz, СDСl3): δ 3,97 (s, 3H), 4,58 (s, 2H), 7,5~7,9 (m, 5H), 8,l~8,2 (m, 2H).
ПРИМЕР ПОЛУЧЕНИЯ 12.
Метил-3-(3-цианофенил)-5-(аминометил)бензоат:

1,1 г соединения, полученного в вышеописанном примере получения 11, растворяли в 33 мл DMF, а затем добавляли 325 мг азида натрия. Реакционный раствор перемешивали при комнатной температуре в течение 2 часов, после чего добавляли 80 мл воды и 120 мл этилацетата, чтобы экстрагировать органические вещества, и водный слой дважды экстрагировали 100 мл этилацетата. Экстракт промывали насыщенным солевым раствором, высушивали безводным раствором сульфата натрия, а затем растворитель удаляли под вакуумом, получив в результате светло-желтый маслянистый метил-3-(3-цианофенил)-5-(азидометил)бензоат в виде сырого продукта. GC - MS(M-N2) = 264.
Метил-3-(3-цианофенил)-5-(азидометил)бензоат, полученный, как описано выше, помещали в колбу, растворяли в 66 мл этанола, и после добавления 1,1 г карбоната палладия-бария воздух в колбе заменяли на водород. Перемешивание продолжали при комнатной температуре в течение 6 часов, затем катализатор подвергали фильтрации через броунмиллерит, полученный фильтрат концентрировали и очищали с помощью хроматографии на колонке с силикагелем, получив в результате 794 мг соединения по настоящему изобретению (выход из двух стадий: 90%). GC - MS(M-H) = 265.
ПРИМЕР ПОЛУЧЕНИЯ 13.
Метил-3-(3-цианофенил)-5-[((N-t-бутоксикарбонил)-4-бутоксикарбонил) пиперидин-4-илметил]аминометил) бензоат:

5,5 г соединения, полученного в вышеописанном примере получения 11, растворяли в 150 мл сухого THF. К этому раствору добавляли 7,92 мг 4-аминометил-(N-t-бутоксикарбонил)пиперидина и перемешивали при комнатной температуре до следующего дня. Реакцию гасили путем выливания этого раствора в 0,5 М раствор бисульфата калия и экстрагировали этилацетатом. После сушки сульфатом натрия растворитель удаляли, получив в результате 10 г соединения по настоящему изобретению (соль бисульфат калия, количественно).
1Н-ЯМР(270 MHz, СDСl3): δ, 1,0~1,3 (m, 2Н), 1,43 (s, 9H), l,7~2,0 (m, 3Н), 2,6~2,8 (m, 4H), 3,95 (s, 3H), 4,0~4,2 (brs, 4H), 7,5~7,7 (m, 2H), 7,9~ 8,0 (m, 2H), 8,20 (s, 2H).
В соответствии с таким же способом, который описан в примере получения 13, синтезировали соединения примера получения 14, перечисленные в таблице 2.
ПРИМЕР ПОЛУЧЕНИЯ 15.
Метил-3-(3-цианофенил)-5-[((N-t-бутоксикарбонил)пиперидин-4-карбонил) аминометил]бензоат:

53 мг соединения, полученного в вышеописанном примере получения 12, растворяли в 2,0 мл хлороформа. К этому раствору добавляли 57 мг (N-t-бутоксикарбонил)изонипекотиновой кислоты, 27 мг HOBt и 48 мг гидрохлорида EDCI, после чего перемешивали при комнатной температуре до следующего дня. Эту реакцию подвергали воздействию колонки SCX с катионообменной смолой для твердофазной экстракции и колонки SAX с анионообменной смолой для твердофазной экстракции, изготовленных компанией Barian, и экстрагировали метанолом, удаляя примеси. Экстракт концентрировали, получив в результате 100 мг соединения по настоящему изобретению, количественно. MS(M+1)-478.
В соответствии с такой же процедурой, которая описана в примере получения 15, синтезировали соединения примеров получения 16-22, перечисленные в таблице 2.
ПРИМЕР ПОЛУЧЕНИЯ 23.
Метил-3-(3-цианофенил)-5-[N-[(N-t-бутоксикарбонилпиперидин)-4-илметил)] -N-метиламинометил]бензоат:

464 мг соединения, полученного в вышеописанном примере получения 13, растворяли в 13 мл диметилформамида, добавляли 276 мг карбоната калия и 94 мкл метилиодида, и после продолжавшегося в течение 6 часов перемешивания осуществляли экстракцию. Органический слой промывали солевым раствором, высушивали сульфатом натрия, растворитель удаляли под вакуумом, а затем проводили очистку с помощью хроматографии на колонке с силикагелем, получив в результате 289 мг соединения по настоящему изобретению (выход: 61%).
1Н-ЯМР (270 MHz, СDСl3): δ 1,0~1,9 (m, 5H), 1,49 (s, 9H), 2,22 (s, 3H), 2,2~ 2,3 (m, 2H), 2,5~2,8 (m, 2H), 2,70 (t, 2H, J=12,0 Hz), 3,57 (s, 2H), 3,96 (s, 3H), 4,0~4,2 (m, 2H), 4,64 (s, 1Н), 4,72 (s, 1Н), 7,5~7,7 (m, 2Н), 7,72 (s, 1H), 7,85 (d, 1H, J=7,6 Hz), 8,01 (s, 1Н), 8,12 (s, 1Н).
В соответствии с такой же процедурой, которая описана в примере получения 23, синтезировали соединения примеров получения 24-27, перечисленные в таблице 2.
ПРИМЕР ПОЛУЧЕНИЯ 28.
Метил-3-(3-цианофенил)-5-[N-[(N-t-бутоксикарбонил)пиперидин-4-илметил)] -N-ацетиламинометил]бензоат:
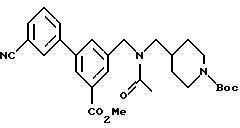
464 мг соединения, полученного в вышеописанном примере получения 13, растворяли в 10 мл диметилформамида и добавляли 277 мкл триэтиламина. Затем добавляли 92 мкл ацетилхлорида и перемешивание продолжали в течение 2 часов. Полученный раствор выливали на раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой промывали солевым раствором, высушивали сульфатом натрия, растворитель удаляли под вакуумом, а затем проводили очистку с помощью хроматографии на колонке с силикагелем, получив в результате 349 мг соединения по настоящему изобретению (выход: 69%).
1H-ЯMP (270 MHz, СDСl3): δ 1,0~2,0 (m, 5Н), 1,45 & 1,46 (s, 9H), 2,15 & 2,21 (s, 3Н), 2,5~2,8 (m, 2Н), 3,2~3,3 (m, 2Н), 3,96 & 3, 97 (s, 3Н), 4,0~4,3 (m, 2H), 4,64 & 4,72 (s, 2H), 7,4~8,0 (m, 6H), 8,1~8,2 (m, 1H).
В соответствии с такой же процедурой, которая описана в примере получения 28, синтезировали соединения примеров получения 29-32, перечисленные в таблице 2.
ПРИМЕР ПОЛУЧЕНИЯ 33.
Метил-3-(3-цианофенил)-5-[N-[(N-t-бутоксикарбонилпиперидин-4-ил)метил]
-N-трифторацетиламинометил]бензоат:

В атмосфере азота 2 г соединения, полученного в вышеописанном примере получения 13, растворяли в 20 мл сухого DMF и этот раствор охлаждали до 0oС. При помешивании добавляли 1,38 мл триэтиламина, а затем добавляли 0,70 мл трифторуксусного ангидрида. После перемешивания при комнатной температуре, продолжавшегося в течение 5 часов, добавляли воду и этилацетат. Осуществляли экстракцию этилацетатом, органический слой промывали разбавленной соляной кислотой и раствором гидрокарбоната натрия, высушивали сульфатом магния и удаляли растворитель. Очистка с помощью хроматографии на колонке с силикагелем привела к получению 1,46 г (78%) соединения по настоящему изобретению. MS(М+1)=560.
ПРИМЕР ПОЛУЧЕНИЯ 34.
Метил-3-(3-цианофенил)-5-[[2-[(4-t-бутоксикарбонил)пиперазин-1-ил] -этокси]метил]бензоат:

После того, как 24 г гидрида натрия (60% в масле) суспендировали в 2,0 мл диметиформамида, добавляли 2,0 мл раствора диметилформамида, содержащего 154 мг 1-t-бутоксикарбонил-4-(2-гидроксиэтил)пиперазина и перемешивали в течение 10 минут. После охлаждения до -30oС добавляли 143 мг соединения, полученного в вышеописанном примере получения 11, растворенного в 2,0 мл диметилформамида, и перемешивали, начиная с -30oС до комнатной температуры в течение 4 часов. Полученный раствор выливали на насыщенный водный раствор хлорида аммония и экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным солевым раствором и высушивали сульфатом магния. После удаления растворителя под вакуумом, проводили очистку с помощью хроматографии на колонке с силикагелем, что привело к получению 21 мг (выход: 10%) соединения по настоящему изобретению.
1Н-ЯМР (270 MHz, СDСl3): δ 1,45 (s, 9Н), 2,4~2,5 (m, 4Н), 2,66 (t, 2H, J= 5,9 Hz), 3,4~3,5 (m, 4H), 3,66 (t, 2H, J=5,8 Hz), 3,97 (s, 3H), 4,65 (s, 2H), 7,5~8,2 (m, 7H).
ПРИМЕР ПОЛУЧЕНИЯ 35.
Метил-3-(3-цианофенил)-5-[(1-ацетилпиперидин-4-ил)-метоксиметил]бензоат:

400 мг соединения, полученного в примере получения 9, растворяли в 20 мл метанола, а затем добавляли 20 мл 2 N соляной кислоты, при перемешивании и охлаждении на льду. После продолжавшегося в течение 7 часов перемешивания при температуре 0oС, в результате концентрации получали сырой продукт метил-3-(3-цианофенил)-5-(1-пиперидин-4-илметоксиметил)бензоат. Этот продукт растворяли в 20 мл дихлорметана, после чего добавляли 3,0 мл триэтиламина. Затем добавляли 460 мкл ацетилхлорида, при перемешивании и охлаждении на льду, и перемешивание продолжали при температуре от 0oС до комнатной температуры в течение 18 часов, после чего полученный раствор выливали на насыщенный раствор гидросульфата калия и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором гидрокарбоната натрия, а затем насыщенным водным раствором соли, и высушивали сульфатом магния. После удаления растворителя под вакуумом, проводили очистку с помощью хроматографии на колонке с силикагелем, что привело к получению 260 мг (выход: 74%) соединения по настоящему изобретению.
1Н-ЯMР (270 MHz, СDСl3): δ 1,0~1,3 (m, 2Н), 1,7~2,0 (m, 3H), 2,09 (s, 3Н), 2,56 (td, 1H, J=12,8, 2,9 Hz), 3,06 (td, 1H, J=13,2, 2,0 Hz), 3,2~3,5 (m, 2H),
3,83 (brd, 1H, J=l3,5 Hz), 3,97 (s, 3H), 4,65 (s, 2H), 4,5~4,8 (m, 1H), 7,58 (t, 1Н, J=7,8 Hz), 7,6~7,8 (m, 1H), 7,72 (s, 1H), 7,85 (d, 1H, J= 7,9 Hz), 7,90 (s, 1H), 8,03 (s, 1H), 8,17 (s, 1Н)
ПРИМЕР ПОЛУЧЕНИЯ 36.
Метил-3-(3-цианофенил)-5-((1-t-бутоксикарбонилметил)-4-пиперидил)метоксиметил)бензоат:

В атмосфере азота 100 мг метил-3-(3-цианофенил)-5-(пиперидин-4-ил-метоксиметил)бензоата, полученного в примере получения 35, растворяли в 5 мл сухого этанола, добавляли 56 мг карбоната калия и 69 мкл t-бутилбромацетата и перемешивали при 60oС до следующего дня. Удаляли растворитель и проводили очистку с помощью хроматографии на колонке с силикагелем, и в результате получили 10 мг (7,6%) соединения по настоящему изобретению.
1Н-ЯМР (270 MHz, CDCl3): δ 1,2~1,4 (m, 2H), 1,46 (s, 9H), 1,6~1,8 (m, 3Н), 2, 17, (d, J=11Hz, 2Н), 2,96 (d, J=9 Hz, 2H), 3,11 (s, 2H), 3,38 (d, J= 6,3 Hz), 3,96 (s, 3H), 4,60 (s, 2H), 7,5~7,9 (m, 5Н), 8,03 (s, 1Н), 8,15 (s, 1Н).
ПРИМЕР ПОЛУЧЕНИЯ 37.
Метил 3-(3-цианофенил)-5-[[N-[N-(2-гидроксиэтил)пиперидин 4-ил-метил]-N-трифторацетит]аминометил]бензоат:

К соединению, полученному в примере получения 35, добавляли 5 мл 2 N трифторуксусной кислоты при температуре 0oС и перемешивали 30 минут. Удаляли растворитель. В атмосфере азота к этому раствору добавляли 20 мл сухого метанола, 300 мг карбоната калия и 250 мкл 2-бромэтанола и перемешивали при температуре 60oС до следующего дня. Удаляли растворитель и проводили очистку с помощью хроматографии на колонке с силикагелем, в результате чего получили 220 мг (выход: 71%) соединения по настоящему изобретению. MS(M = Н) = 504.
ПРИМЕР ПОЛУЧЕНИЯ 38.
3-(3-цианофенил)-5-[2-(N-t-бутоксикарбонилпиперидин-4-ил)-метоксиметил] бензойная кислота:

1,43 г соединения, полученного в примере получения 9, растворяли в 20 мл метанола и добавляли 2 мл воды. Затем добавляли 1,54 мл 4 N раствора гидроксида лития и перемешивали при комнатной температуре в течение 3 часов. После подкисления путем добавления насыщенного водного раствора хлорида аммония осуществляли экстракцию этилацетатом. Органический слой промывали насыщенным водным раствором соли, высушивали сульфатом магния, растворитель удаляли, а затем проводили очистку с помощью хроматографии на колонке с силикагелем, получив в результате 1,03 г (выход: 74%) соединения по настоящему изобретению.
1Н-ЯМР (270 MHz, СDСl3): δ 1,0~1,3 (m, 2Н), 1,46 (s, 9Н), 1,7~2,0 (m, 3H), 2,56 (td, 1H, J=12,8, 2,9 Hz), 3,05 (td, 1H, J=13,2, 2,0 Hz), 3,2~3,5 (m, 2H), 3,83 (brd, 1H, J=13,5 Hz), 4,65 (s, 2H), 4,6~4,8 (m, 1H), 7,60 (t, 1H, J= 7,8 Hz), 7,6~7,8 (m, 1Н), 7,74 (s, 1H), 7,85 (d, 1H, J=7,9 Hz), 7,90 (s, 1H), 8,03 (s, 1H), 8,16 (s, 1H).
ПРИМЕР ПОЛУЧЕНИЯ 39.
Диметиламид 3-(3-цианофенил)-5-[(1-t-бутоксикарбонилпиперидин-4-ил)метоксиметил]бензойной кислоты:

300 мг соединения, полученного в примере получения 38, растворяли в 10 мл дихлорметана, добавляли 116 мкл оксалилхлорида, а затем 135 мкл пиридина при 0oС, и перемешивали при 0oС в течение 1 часа. К этому реакционному раствору добавляли по каплям 40% раствор диметиламина и перемешивали при комнатной температуре в течение 1 часа. Затем добавляли насыщенный раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором соли, высушивали сульфатом магния и удаляли растворитель. Полученный сырой продукт очищали с помощью хроматографии на колонке с силикагелем, получив 268 мг (выход: 84%) соединения по настоящему изобретению.
1H-ЯMP (270 MHz, СDС13):

ПРИМЕР ПОЛУЧЕНИЯ 40.
1-ацетил-3-(3-цианофенил)-5-[(N-t-бутоксикарбонилпиперидин-4-ил)-метоксиметил]бензол:

291 мг соединения, полученного в примере получения 38, растворяли в 10 мл дихлорметана, добавляли 116 мкл оксалилхлорида, а затем 135 мкл пиридина при 0oС, и перемешивали при 0oС в течение 1 часа. Затем добавляли 76 мг гидрохлорида N, О-диметилгидроксиламина и перемешивали при комнатной температуре в течение 1 часа. Затем добавляли насыщенный водный раствор гидрокарбоната натрия и экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли, высушивали сульфатом магния и удаляли растворитель. Полученный сырой продукт растворяли в 10 мл тетрагидрофурана, после чего в атмосфере азота при 0oС добавляли 2,29 мл бромида метилмагния. После перемешивания при 0oС, проводившегося в течение 40 минут, добавляли разбавленный раствор соляной кислоты и экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором соли и высушивали сульфатом магния. Удаляли растворитель под вакуумом, затем осуществляли очистку с помощью хроматографии на колонке с силикагелем, в результате получили 190 мг (выход: 65%) соединения по настоящему изобретению.
1Н-ЯМР (270 MHz, СDСl3): δ 1,0~1,3 (m, 2H), 1,46 (s, 9H), 1,7~2,0 (m, 3Н), 2,09 (s, 3Н), 2,56 (td, 1H, J=12,8, 2,9 Hz), 3,06 (td, 1H, J=13,2, 2,0 Hz), 3,2~ 3,5 (m, 2H), 3,83 (brd, 1H, J=13,5 Hz), 4,65 (s, 2H), 4,5~4,7 (m, 1H), 7,60 (t, 1H, J=7,9 Hz), 7,6~7,8 (m, 1H), 7,70 (s, 1H), 7,85 (d, 1H, J= 7,9 Hz), 7,90 (s, 1H), 8,02 (s, 1H), 8,16 (s, 1Н).
ПРИМЕР 1.
Соль
метил-3-(3-амидинофенил)-5-[(4-пиперидинил)метоксиметил]-бензоат:

6,0 г соединения, полученного в примере получения 9, растворяли в 60 мл дихлорметана и добавляли 3,0 мл метанола. Газообразную соляную кислоту барботировали в полученный раствор при перемешивании, с охлаждением на льду. После перемешивания, проводившегося при 0oС в течение 30 минут, а затем при комнатной температуре в течение 20 часов, раствор сконцентрировали до состояния сухого твердого вещества. Затем добавляли 30 мл насыщенного раствора аммиака в этаноле, перемешивали при комнатной температуре в течение 5 часов и концентрировали. Затем полученный сырой продукт очищали с помощью хроматографии на колонке с HP-20 (30 г, элюент: вода-метанол), получив в результате соединение по настоящему изобретению (4,89 г, выход: 99%).
1Н-ЯМР (270 MHz, DMSО-d6): δ 1,3-1,5 (m, 2H), 1,7-2,0 (m, 3H), 2,7-2,9 (m, 2Н), 3,2-3,3 (m, 2H), 3,38 (d, 2H, J=6,3 Hz), 3,91 (s, 3Н), 4,64 (s, 2Н), 7,69 (t, 1H, J=7,9 Hz), 7,86 (d, 1H, J=7,9 Hz), 7,99 (s, 1H), 8,02 (s, 1H), 8,07 (d, 1H, J=7,6 Hz), 8,15 (s, 1Н), 8,28 (s, 1H), 8,55 & 8,85 (brs, 1H), 9,19 & 9,52 (s, 2H).
В соответствии с той же самой реакцией, что и в вышеописанном примере 1, за исключением того, что для выделения и очистки использовали ВЭЖХ (ODS, элюент: вода-метанол) вместо хроматографии на колонках с НР-2, синтезировали соединения примеров 2-40, перечисленные в таблице 3.
ПРИМЕР 41.
Соль метил-3-(3-амидинофенил)-5-[(1-ацетоимидоил-4-пиперидинил)метоксиметил]бензоат:
К 4,79 г соединения примера 1 и 3,10 г этилацетоимидатмоногидрохлорида добавляли 50 мл этанола. Затем по каплям добавляли 5,25 мл триэтиламина, при помешивании, с охлаждением на льду. После того, как температура поднялась от 0oС до комнатной температуры, перемешивание продолжали в течение 36 часов, а затем полученный продукт сконцентрировали до состояния сухого твердого вещества. После очистки методом ВЭЖХ (ODS, элюент: вода-метанол) получили соединение по настоящему изобретению (4,37 г, выход: 82%).
1Н-ЯМР (270 MHz, DMSO-d6): δ 1,1~1,4 (m, 2H), 1,7~2,1 (m, 3H), 2,50 (s, 3H), 3,0~ 3,5 (m, 4H), 3,8~4,0 (m, 1H), 3,91 (s, 3H), 4,0~4,2 (m, 1H), 4,64 (s, 2H), 7,74 (t, 1H, J=7,8 Hz), 7,87 (d, 1H, J=7,6 Hz), 8,00 (s, 1H), 8,03 (s, 1H), 8,07 (d, 1H, J=7,6 Hz), 8,16 (s, 1H) 8,28 (s, 1H), 8,64 & 9,20 (brs, 1Н), 9,25 & 9,54 (brs, 2H).
В соответствии с той же самой реакцией, что и в вышеописанном примере 41, синтезировали соединения примеров 42-57, перечисленные в таблице 3.
Кроме того, используя ту же самую реакцию, которая описана выше, за исключением того, что вместо этилацетоимидат-моногидрохлорида использовали пропионимидатмоногидрохлорид, синтезировали соединения примеров 58-59, перечисленные в таблице 3.
Кроме того, используя ту же самую реакцию, которая описана выше, за исключением того, что вместо этилацетоимидат-моногидрохлорида использовали гидроксиацетоимидатмоногидрохлорид, синтезировали соединения примера 60, перечисленные в таблице 3.
ПРИМЕР 61.
Гидрохлорил-3-(3-амидинофенил)-5-[(1-ацетоимидоил-4-пиперидинил)метоксиметил]бензойной кислоты:

2,71 г соединения примера 41 растворяли в 27 мл 2 N соляной кислоты, перемешивали при 70oС в течение 24 часов, концентрировали до состояния сухого твердого вещества, а затем осуществляли выделение и очистку методом ВЭЖХ (ODS, элюент: вода-метанол), получив в результате соединение по настоящему изобретению (2, 00 г, выход: 76%).
1Н-ЯМР (270 MHz, DMSО-d6): δ 1,2~1,6 (m, 2Н), l,9~2,2 (m, 3H), 2,31 (s, 3Н), 3,0~3,4 (m, 2H), 3,47 (d, 2Н, J=5,9 Hz), 3,9~4,1 (m, 2H), 4,65 (s, 2H), 7,6~7,8 (m, 3H), 8,0~8,1 (m, 2H), 8,10 (s, 1H), 8,24 (s, 1H).
В соответствии с той же самой реакцией, что и в вышеописанном примере 61, синтезировали соединения примеров 62-68,70 и 72-83, перечисленные в таблице 3.
ПРИМЕР 69.
Соль 3-(3-амидинофенил)-5-[[(4-пиперидил)метил] аминометил] фенилкарбониламиноуксусной кислоты:

73 г соединения примера 68 растворяли в 5 мл DMF, к этому раствору добавляли 38 мг 1-(3-диметиламинопропил)-3-этилкарбодиимидгидрохлорида, 20 мг глицина и 50 мг триэтиламина и перемешивании при комнатной температуре до следующего дня. Растворитель удаляли, а затем осуществляли выделение и очистку методом ВЭЖХ (ODS, элюент: вода-метанол), получив в результате соединение по настоящему изобретению (25 мг, выход: 30%).
В соответствии с той же самой реакцией, что и в вышеописанном примере 69, синтезировали соединения примера 71, перечисленные в таблице 3.
ЭКСПЕРИМЕНТ 1.
(1) Определение ингибирующей активности
антивированного фактора свертывания крови Х (FXa):
Вещество для анализа растворяли в воде или в воде, в которую были добавлены подходящие концентрации органических растворителей (DMSO,
этанола или метанола), и использовали полученные растворы в качестве образцов. К 70 мкл образцов, серийно разбавленных водой, добавляли по 90 мкл 100 мМ трис-буфера (рН 8,4), 20 мкм 50 мМ трис-буфера
(рН 8,4), содержащего 50 мЕ/мл человеческого FXa, и 2 мМ субстрата (Daiichi Chemical S-2765), инкубировали в течение 30 минут, затем добавляли 50 мкл 50% уксусной кислоты и определяли поглощение
(А405). В качестве "холостой пробы", вместо FXa добавляли трис-буфер, а в контрольных вариантах вместо образца добавляли воду. Определяли 50%-ную ингибирующую активность (IC50), в качестве
показателя активности ингибирования FXa. Активность ингибирования человеческого FXa соединением по настоящему изобретению показана в таблице 4.
(2) Определение активности ингибирования тромбина.
К 70 мкл образцов, серийно разбавленных водой, добавляли по 90 мкл 100 мМ трис-буфера (рН 8,4), 20 мкм 50 мМ трис-буфера (рН 8,4), содержащего 1 мЕ/мл человеческого тромбина, и 2 мМ субстрата (Daiichi Chemical S-2238), инкубировали в течение 30 минут, затем добавляли 50 мкл 50% уксусной кислоты и определяли поглощение (А405). В качестве "пустой пробы", вместо тромбина добавляли трис-буфер, а в контрольных вариантах вместо образца добавляли воду. Определяли 50%-ную ингибирующую активность (IC50), в качестве показателя ингибирования активности тромбина. Активность ингибирования человеческого тромбина соединением по настоящему изобретению представлена в таблице 4.
(3) Определение противосвертывающей активности
(АРТТ):
К 100 мкл нормальной человеческой плазмы

(4) Определение активности ингибирования ацетилхолинэстеразы
(AchE):
Подлежащее анализу вещество растворяли в дистиллированной воде, получая таким образом испытуемый образец. К 50 мкл серийно разбавленных испытуемых образцов добавляли по 50 мкл
раствора фермента, в котором человеческая ацетилхолинэстераза (изготовленная компанией Sigma, C-5400) была растворена в дистиллированной воде в концентрации 0,1 мЕ/мл. К этому раствору добавляли 50
мкл раствора, приготовленного путем растворения 5,5'-дитиобис (изготовленного компанией Nacarai Tesque, 141-01) в фосфатном буфере (0,1 М NaH2PO4-Na2HPO4,
pH 7,0) в концентрации 0,5 мМ, перемешивали и проводили реакцию с 50 мкл раствора, в котором ацетилтиохолиниодид (Wako Company, 017-09313) был растворен в фосфатном буфере в концентрации 3 мМ, при
комнатной температуре. В качестве контроля, вместо предназначенного для анализа вещества добавляли воду, и через некоторый промежуток времени определяли поглощение (А450). В качестве "пустой пробы"
вместо раствора фермента добавляли фосфатный буфер и определяли 50% активность ингибирования (IC50).
Активность ингибирования человеческой AChe соединением по настоящему изобретению представлена в таблице 4.
(5) Определение биологической доступности (ВА):
Подлежащее анализу вещество растворяли в дистиллированной воде (для перорального
введения; 10 мг/кг) или в физиологическом растворе (для внутривенного введения; 3 мг/кг), чтобы приготовить раствор для введения. Этот раствор вводили голодавшим мышам линии ICR (самцы, возраст 6
недель), брали образцы цельной крови из сердца под эфирной анестезией, через 5 минут (только для группы с внутривенным введением), 15 мин, 30 мин, 1 час, 2 часа и 4 часа после введения, и отделяли
плазму с помощью центрифугирования (3500 оборотов/мин, 30 мин, 4oС), получая таким образом испытуемые образцы (n=4). Используя вышеописанный метод определения активности ингибирования FXa,
сначала построили калибровочную кривую для подлежащих анализу веществ, а затем определяли концентрацию анализируемых веществ в образцах. Рассчитывали нижнюю область кривой концентрации в плазме
- времени (AUC - область под кривой), а затем рассчитывали биологическую доступность (ВА) для мышей, в соответствии со следующей формулой:
ВА% = (AUC, перорально) /(AUC, внутривенно) •
(Доза/внутривенно) /(Доза, перорально) • 100
Биологическая доступность для мышей соединений по настоящему изобретению представлена в таблице 4.
ПРАКТИЧЕСКАЯ
ПРИМЕНИМОСТЬ
Производное бифениламидина и его фармацевтически приемлемая соль по настоящему изобретению обладают эффектом ингибирования активности FXa и могут использоваться в качестве
профилактического агента и/или терапевтического агента, которые клинически применимы против тромбоэмболии, таких, как инфаркт миокарда, церебральный тромбоз, тромбоз периферических артерий или тромбоз
глубоко расположенных вен, в качестве ингибитора FXa.
Реферат
Изобретение относится к производным бифениламидина общей формулы (1), где R1 представляет собой атом водорода; L представляет собой прямую связь или С1-4-алкиленовую группу; R2 представляет собой карбоксильную группу; С1-8 -алкоксикарбонильную группу; карбамоильную группу, причем атом азота, входящий в состав карбамоильной группы, может быть замещен моно- или ди-С1-8-алкильной группой или может представлять собой атом азота в аминокислоте; С1-8-алкилкарбонильную группу; R3 представляет собой атом водорода; Х представляет любую из групп: -O-, -NH-CO-NH-, -N(R4)-, -CO-N(R5)-, -N(R5)-CO-, в которых R4 представляет собой атом водорода, С1-10-алкильную группу, С1-10-алкилкарбонильную группу, С1-10 -алкилсульфонильную группу, R5 представляет собой атом водорода, С1-10-алкильную группу, Y представляет собой С4-8-циклоалкильную группу, в которой метиленовая группа в С4-8-циклоалкиле может быть замещена С1-8-алкильной группой, С1-8-алкоксильной группой, карбамоильной группой, С1-8-алкоксикарбонильной группой, карбоксильной группой, или следующее 5-8-членное кольцо формулы I-1. Эти соединения обладают ингибирующей активностью активированного фактора свертывания крови Х и благодаря этому свойству могут использоваться в качестве агентов для профилактики или терапии тромбоза или эмболии. 4 с. и 5 з.п. ф-лы, 4 табл.


Формула

в которой R1 представляет собой атом водорода;
L представляет собой прямую связь или С1-4-алкиленовую группу;
R2 представляет собой карбоксильную группу; С1-8- алкоксикарбонильную группу; карбамоильную группу, причем атом азота, входящий в состав карбамоильной группы, может быть замещен моно- или ди С1-8-алкильной группой или может представлять собой атом азота в аминокислоте; С1-8-алкилкарбонильную группу;
R3 представляет собой атом водорода;
Х представляют любую из групп:
-O-, -NH-CO-NH-, -N(R4)-, -CO-N(R5)-, -N(R5)-CO-,
в которых R4 представляет собой атом водорода, С1-10-алкильную группу, С1-10-алкилкарбонильную группу, С1-10-алкилсульфонильную группу;
R5 представляет собой атом водорода, С1-10-алкильную группу, причем алкильная группа в R4 и R5 может быть замещена гидроксильной группой, аминогруппой, атомом фтора, атомом хлора, атомом брома, карбоксильной группой, С1-8-алкоксикарбонильной группой, карбамоильной группой;
Y представляет собой С4-8-циклоалкильную группу, в которой метиленовая группа в С4-8-циклоалкиле может быть замещена С1-8-алкильной группой, С1-8-алкоксильной группой, карбамоильной группой, С1-8-алкоксикарбонильной группой, карбоксильной группой, или следующее 5-8-членное кольцо формулы I-1:

в которой в циклической системе метиленовая группа может быть замещена карбонильной группой, а цикл может иметь ненасыщенные связи;
R6 представляет собой атом водорода;
W представляет собой С-Н или атом азота, при условии, что W не является атомом азота в том случае, когда цикл представляет собой 5-членное кольцо;
Z представляет собой атом водорода; C1-10 -алкильную группу, причем алкильная группа может быть замещена гидроксильной группой, за исключением случая, когда Z представляет собой С1-алкильную группу, карбоксильную группу, С1-8-алкоксикарбонильную группу, С1-8-алкилкарбонильную группу; или следующую группу формулы I-3:

в которой R7 представляет собой С1-8-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой;
m представляет собой целое число от 1 до 3;
n представляет собой целое число от 0 до 3, при условии, что W не является атомом азота, когда n представляет собой 0 или 1;
или его фармацевтически приемлемая соль.

где Y представляет собой С4-8-циклоалкильную группу, в которой метиленовая группа в С4-8-циклоалкиле может быть замещена С1-8 -алкильной группой, С1-8-алкоксильной группой, карбамоильной группой, С1-8-алкоксикарбонильной группой, карбоксильной группой, или следующее 5-8-членное кольцо формулы II-1:

в которой в циклической системе метиленовая группа может быть замещена карбонильной группой.

в которой R2 представляет собой карбоксильную группу; С1-4 - алкоксикарбонильную группу; карбамоильную группу, причем атом азота, входящий в состав карбамоильной группы, может быть замещен моно- или ди С1-4-алкильной группой или может представлять собой атом азота в аминокислоте; С1-4-алкилкарбонильную группу;
Х представляет любую из групп:
-О-, -N(R4)-, NH-CO-,
в которых R4 представляет собой атом водорода, С1-10-алкильную группу, С1-10-алкилкарбонильную группу, С1-10-алкилсульфонильную группу,
Y представляет собой С5-6-циклоалкильную группу, в которой метиленовая группа в С5-6-циклоалкиле может быть замещена С1-4 -алкоксильной группой, карбамоильной группой, С1-8-алкоксикарбонильной группой, карбоксильной группой, или следующее 5-6-членное кольцо формулы III-1:

в которой W представляет собой С-Н или атом азота, при условии, что W не является атомом азота в том случае, когда цикл представляет собой 5-членное кольцо;
Z представляет собой атом водорода; C1-4-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой, за исключением случая, когда Z представляет собой С1-алкильную группу, карбоксильную группу, С1-4-алкоксикарбонильную группу, С1-4-алкилкарбонильную группу; или следующую группу формулы III-2:

в которой R7 представляет собой С1-4-алкильную группу, причем алкильная группа может быть замещена гидроксильной группой;
n представляет собой целое число от 0 до 2, при условии, что W не является атомом азота, когда n представляет собой 0 или 1;
или его фармацевтически приемлемая соль.



n представляет собой 1; или его фармацевтически приемлемая соль.
20.11.1997, 11.05.1998 по пп. 1-3, 7-10;
20.11.1997, 11.05.1998 по пп. 4 и 6.
Документы, цитированные в отчёте о поиске
Способ получения фармацевтически приемлемых солей производных бензамидина
Комментарии