Способ получения 5-алкилсалицилалдоксимов и их применение - RU2721407C2
Код документа: RU2721407C2
Описание
Изобретение относится к способу получения 5-алкилсалицилальдоксимов (5-алкил-2-гидроксибензальдоксимов) и применению продукта реакции в качестве антикоррозионного средства.
5-алкилсалицилальдоксимы образуют стабильные комплексы с железом, нерастворимы в воде и тем самым эффективно ингибируют процесс коррозии металла и делают его поверхность гидрофобной. Например, в патенте ЕР 0178850 описан антикоррозионный препарат, содержащий альдоксим с двумя оксимовыми группами в положении о/то к гидроксильной группе в качестве активного вещества. Оксимы могут быть использованы для непосредственного нанесения на поверхность металла, а также в виде раствора в соответствующем органическом растворителе или в виде водной эмульсии. В качестве растворителя в упомянутом препарате могут быть использованы спирты, простые эфиры, кетоны, ароматические и алифатические углеводороды, например этанол, изопропанол, толуол, ксилол, хлороформ или 1,1,1-трихлорэтан. Учитывая распространенность коррозии, важно, чтобы препараты, используемые для предотвращения этого явления, были простыми в получении и недорогими.
Известные способы получения альдоксимов заключаются в реакции соответствующего альдегида с гидроксиламином. Самый старый из известных способов заключается в добавлении водного или метанольного [1] раствора гидрохлорида гидроксиламина [2], или сульфата гидроксиламина [6] в концентрированный раствор альдегида в спирте, например метаноле [2] или этаноле, преимущественно при температуре кипения реакционной смеси, при регулировании рН в пределах 5-8. [1] [


Для получения соответствующих 5-алкилсалицилальдоксимов в целом их производители должны иметь в своем распоряжении методику получения исходных альдегидов. В большинстве случаев чистые 5-алкилсалицилальальдегиды являются дорогостоящими или коммерчески недоступными - как правило, они не являются коммерческими продуктами (кроме лабораторных реагентов).
Одним из основных методов синтеза 5-алкилсалицилальальдегидов является реакция Раймера-Тимана, в которой чаще всего в качестве реагентов используют гидроксид натрия, алкилфенол и хлороформ и в качестве растворителя - смесь вода-метанол. Согласно стандартной методике раствор соответствующего фенола в метаноле вводят в водный раствор NaOH или суспензию, а затем хлороформ медленно добавляют с обратным холодильником при температуре кипения метанола [1]. Как вариант, соответствующий фенол и всю порцию хлороформа вводят в водный раствор NaOH или суспензию и реакцию проводят под давлением при температуре 80-88°С [7] [W.Е. Smith, The Dow Chemical Company, US 4324922, 1982]. В обоих вариантах, когда реакция завершена, всю систему охлаждают до комнатной температуры, непрореагировавшие NaOH и NaCl, образующиеся во время реакции, растворяют в воде, и затем послереакционную смесь подкисляют серной (VI) или соляной кислотой, получая два слоя: водную фазу и органическую фазу. Отделенную органическую фазу промывают водой, и образовавшийся альдегид в зависимости от применяемого фенола очищают фракционной перегонкой под вакуумом или кристаллизацией, например, из гексана [1]. На второй стадии полученный таким образом альдегид подвергают реакции конденсации с гидроксиламином, чтобы получить 5-алкилсалицилальдоксим, используя способ, описанный выше.
Описанный выше двухэтапный синтез имеет недостаток - относительно низкий выход первой стадии или получения 5-алкилсалицилальдегида - обычно он не превышает 35% по отношению к алкилфенолу. Низкие выходы и низкая степень превращения алкилфенолов, несомненно, вызваны ингибированием протекания синтеза после достижения равновесия между субстратами и продуктами реакционной системы. Легко подсчитать, что даже если бы выход конденсации полученного таким образом альдегида с гидроксиламином был близок к 100%, конечный выход синтеза оксима в двухэтапном процессе: алкилфенол → альдегид → оксим (являющийся продуктом выхода отдельных этапов) не превысит 35%, а на самом деле он может быть еще ниже. Более того, синтез альдегидов с использованием этого метода приводит к образованию многих побочных продуктов, включая продукты поликонденсации [4]. Кроме того, существенным неудобством является необходимость выделения и очистки исходного альдегида.
В 1994 году был описан способ получения оксимов в результате взаимодействия магниевых солей соответствующих 5-алкилсалицилальдегидов с гидроксиламином, непосредственно в смеси после синтеза этих солей, исключая трудоемкие выделение и очистку альдегида [8] [D. Levin, Zeneca Limited, ЕР 0584988 А1, 1994]. Сначала получают соль соответствующего п-алкилфенола с магнием. К ее смеси в толуоле по частям добавляют параформ(альдегид), и одновременно метанол, образующийся в качестве побочного продукта в реакции этих субстратов, отгоняют в форме азеотропа с толуолом, достигая формилирования orto-положения исходной п-алкилфенольной соли и образования магниевой соли соответствующего 5-алкилсалицилальдегида, суспендированного или растворенного в толуоле. Непосредственно после формилирования при температуре 40-45°С в послереакционную смесь вводят водный раствор гидроксиламина (или сульфата гидроксиламина(VI)) для получения магниевой соли оксима. В конце смесь подкисляют разбавленной серной кислотой(VI), а толуол отгоняют из выделенной органической фазы и промывают водой, получая неочищенный продукт - соответствующий 5-алкилсалицилальдоксим, который, необязательно, очищают фракционной перегонкой в вакууме или кристаллизацией. Этот способ дает возможность получать необходимые 5-алкилсалицилальдоксимы из исходных алкилфенолов в системе «один горшок» с высокими выходами, но весь процесс занимает много времени и требует больших затрат.
Целью изобретения является создание способа, позволяющего решить вышеупомянутые проблемы.
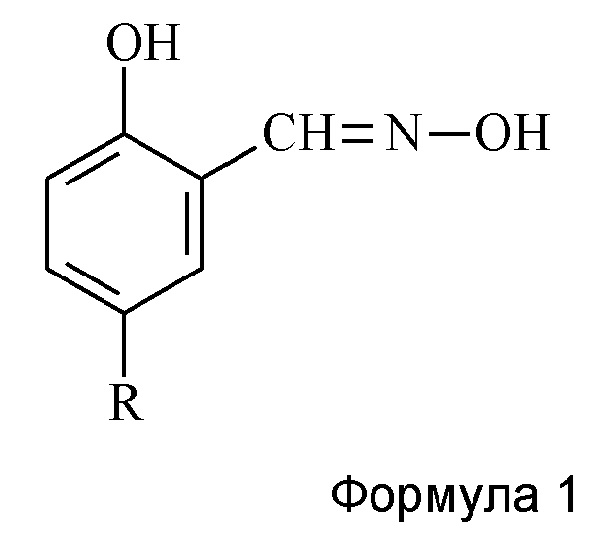
Способ получения продукта, содержащего 60-75 мас. % 5-алкилсалицилальдоксимов с формулой 1,

где R представляет собой С6-С16-алкильную группу, заключающийся в том, что в растворяющую смесь вода-спирт С2-С4 с массовым соотношением 2:1 - 3:1, вводят п-алкилфенол, гидроксид натрия, хлороформ и гидроксиламин, причем относительно используемого алкилфенола гидроксид натрия и хлороформ используют в количествах до 100% избытка, предпочтительно в избытке 50-100%, и гидроксиламин используют в количествах до 60% избытка, и реакцию проводят при температуре 60-75°С, предпочтительно при 63-71°С, в течение 1,5-4 часов, а затем при температуре 20-30°С послереакционную смесь подкисляют до достижения рН водной фазы <7,0, а затем азеотроп спирт-вода отгоняют с примесью непрореагировавшего хлороформа, остаток смешивают с нейтральным углеводородным растворителем С5-С10, слои разделяют, и растворитель отгоняют из органической фазы.
Гидроксиламин вводят в виде водного раствора, предпочтительно приблизительно 50%; необязательно, он может быть синтезирован в реакционной системе в реакции сульфата(VI) или гидрохлорида гидроксиламина с концентрированным раствором гидроксида натрия (вводится в реакционную систему в количестве 2 молей NaOH/моль сульфата(VI) гидроксиламина или 1 моль щелочи/моль гидрохлорида гидроксиламина).
В качестве спирта предпочтительно используют изопропанол.
Изобретение также включает применение продукта, полученного способом согласно изобретению, в качестве ингибитора коррозии, также повышающего пластичность и адгезию лакокрасочных покрытий. Неочищенный оксим в растворителе, в концентрациях до 30%, может быть использован после удаления минеральных примесей для покрытия поверхности металла без дополнительной очистки. В качестве растворителя используют низшие спирты С2-С4, простые эфиры, кетоны и ароматические и алифатические углеводороды, например этанол, изопропанол, толуол, ксилол, хлороформ, гексан или 1,1,1-трихлорэтан; необязательно - смеси этих растворителей, для пропитки стальных поверхностей перед нанесением покрывающих антикоррозионных систем.
Способ получения 5-алкилсалицилальдоксимов в соответствии с изобретением представляет собой одноэтапный процесс.Натриевая соль подходящего 5-алкилсалицилальдегида, образующегося в реакции типа Раймера-Тимана, конденсируется в состоянии выделения, или in statu nascendi в водно-спиртовой среде с гидроксиламином, вводимым в виде водного раствора, необязательно синтезированного в реакционной системе в реакции сульфата(VI)/гидрохлорида гидроксиламина с гидроксидом натрия, образуя натриевую соль оксима с фенолятным характером, и последняя превращается в соответствующий оксим после подкисления послереакционной смеси (схема 1). Содержание оксимов в продукте обычно составляет 60-75%, а выход образования чистого продукта - 50-85%.
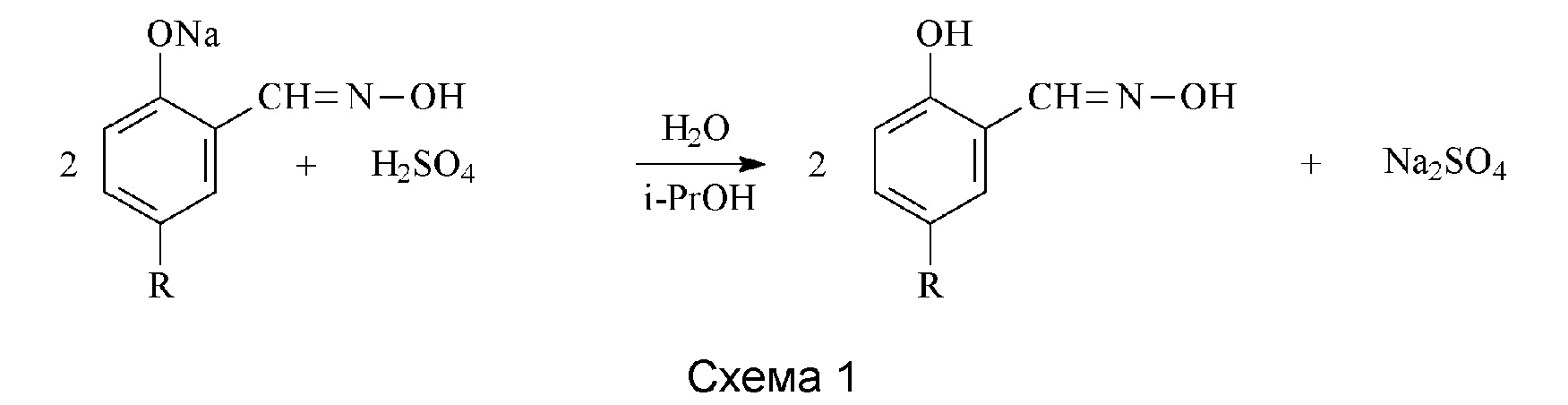
Такое немедленное превращение соли образующегося альдегида в соль оксима оказалось возможным с использованием применяемой процедуры и одновременно и неожиданно дает ряд положительных эффектов: ограничивает побочные реакции, имеющие характеристики конденсации или полимеризации альдегидов, ускоряет синтез альдегидов, повышает степень превращения исходного алкилфенола. В результате весь процесс синтеза оксима происходит быстро, с удовлетворительной селективностью и выходом. Продукт реакции представляет собой готовый ингибитор коррозии, и нет необходимости в его отделении от реакционной смеси.
Слой, образующийся после нанесения раствора продукта, полученного в соответствии с изобретением, на металлическую поверхность, обеспечивает хорошую адгезию типичных лаковых покрытий к корродированной стальной подложке после удаления рыхлой ржавчины, а также с мелкими покрытиями после затирки стальной щеткой. Применение продукта дает возможность исключить дорогостоящие, обычно технически сложные и экологически вредные процедуры тщательной очистки стальных поверхностей, например пескоструйную обработку, измельчение, химическое травление, которые требуются при использовании большинства красок.
Способ получения и применения оксимов в соответствии с изобретением иллюстрируется следующими примерами.
Пример 1. Получение 2-гидрокси-5-нонилбензальдоксима
В реакционной колбе, имеющей емкость 0,25 л, 0,85 моль (34,0 г) гидроксида натрия растворяли в 54,0 г воды при перемешивании и охлаждении. Затем, при температуре приблизительно 40°С, в колбу вводили 0,1 моль (22,0 г) л-нонилфенола (С15Н24О) в 23,0 г изопропанола. Одновременно готовили раствор 0,075 моль (12,5 г) сульфата(VI) гидроксиламина (SHA) в 12,5 г воды при 60°С. К содержимому реакционной колбы, нагретой до 65°С, при интенсивном перемешивании по каплям добавляли 0,175 моль (20,8 г) хлороформа в течение 0,5 часа и в то же время добавляли приготовленный раствор SHA, поддерживая при этом температуру в пределах 65-68°C (реакция экзотермическая). После добавления по каплям этих реагентов перемешивание реакции продолжали приблизительно 1,5 часа при вышеуказанной температуре. Протекание реакции контролировали хроматографически, методом тонкослойной хроматографии (TLC) (хлороформ/гексан 4:1), наблюдая распад исходного пятна алкилфенола (общее время реакции: приблизительно 2 часа). Послереакционную смесь затем охлаждали до комнатной температуры и затем, при перемешивании и охлаждении, 35% серную(VI) кислоту добавляли по частям до достижения рН водной фазы <7,0. При 80-95°С из колбы отгоняли азеотроп изопропанола с водой с примесью непрореагировавшего хлороформа.
Остаток после перегонки охлаждали до температуры приблизительно 35°С и затем перемешивали в течение 0,25 часа вместе с 30 г (приблизительно 44,0 мл) гексана. После разделения фаз верхнюю (органическую) фазу промывали водой. Гексан отгоняли из органического раствора. Остаток после дистилляции представляет собой неочищенный продукт - 2-гидрокси-5-нонилбензальдоксим (m=26,5 г) в виде маслянистой оливково-коричневой вязкой жидкости.
Исходя из содержания азота, определенного с помощью элементарного анализа, рассчитали приблизительное количество основного компонента - оксима, которое составило 64,2%. Выход по отношению к исходному п-нонилфенолу составлял 64,8%.
Пример 2. Получение 2-гидрокси-5-нонилбензальдоксима
Процедура была такой же, как в примере 1, но использовали следующие реагенты: 0,7 моль (28,0 г) гидроксида натрия в 45,0 г воды, 0,1 моль (22,0 г) л-нонилфенола (С15Н24О) в 23,0 г изопропанола и 0,15 моль (4,95 г) гидроксиламина в виде 50%-ного водного раствора при комнатной температуре, а также 0,175 моль (20,8 г) хлороформа. Расход серной(VI) кислоты и гексана - как в примере 1. Получали 31,7 г неочищенного продукта - 2-гидрокси-5-нонилбензальдоксима - с приблизительным содержанием основного компонента - оксима, равным 68,9%. Выход по отношению к исходному п-нонилфенолу составлял 82,9%.
Пример 3. Получение 2-гидрокси-5-додецилбензальдоксима
Процедура была такой же, как в примере 2, но использовали следующие реагенты: 0,7 моль (28,0 г) гидроксида натрия в 45,0 г деминерализованной воды, 0,1 моль (26,3 г) п-додецилфенола (C18H30O) и 0,15 моль (4,95 г) гидроксиламина в виде 50%-ного водного раствора при комнатной температуре, а также 0,175 моль (20,8 г) хлороформа. Общее время реакции: 2,5 часа. Расход серной(VI) кислоты и гексана - как в примере 1.
Получали 31,1 г неочищенного продукта
2-гидрокси-5-додецилбензальдоксима - в виде маслянистой оливково-коричневой вязкой жидкости, при этом приблизительное содержание основного компонента - оксима - составляло 62,5%. Выход по отношению к исходному л-додецилфенолу составлял 63,6%.
Пример 4.
На поверхность 6 стальных пластин, очищенных до степени St3 в соответствии со стандартом PN-EN ISO 8501 (очистка щеткой, приблизительно 50% поверхности с остатками ржавчины), наносили препарат А - неочищенный оксим, полученный в соответствии с примером 1, разбавляли изопропанолом (приблизительно 10% раствор), получая после 4 часов сушки при комнатной температуре слой, содержащий приблизительно 5,5 г неочищенного оксима на метр стальной поверхности. Затем наносили систему покрытия, состоящую из эпоксидного грунтовочного слоя толщиной 50 мкм, межслойного эпоксидного покрытия толщиной 100 мкм и полиуретанового верхнего покрытия толщиной 60 мкм. Такую же систему покрытия наносили на следующие 6 стальных пластин, подготовленных таким же образом, но без препарата А. После естественного высыхания покрытий в центре пластины делали поперечный разрез с углом пересечения 30° на 3 пластинах каждой серии.
Все пластины помещали в камеру для испытания в солевом тумане и тестировали в соответствии со стандартом ISO 9227, наблюдая за повреждениями в покрытиях (распыляли 3%-ный раствор хлорида натрия при температуре 30±2°С). В таблицах 1 и 2 можно видеть повреждения покрытий после 1440-часового испытания в камере солевого тумана, оцененные в соответствии со стандартом PN-EN ISO 4628 (I - пластины без препарата А; II - пластины с препаратом А; X - пластины с поперечным разрезом).


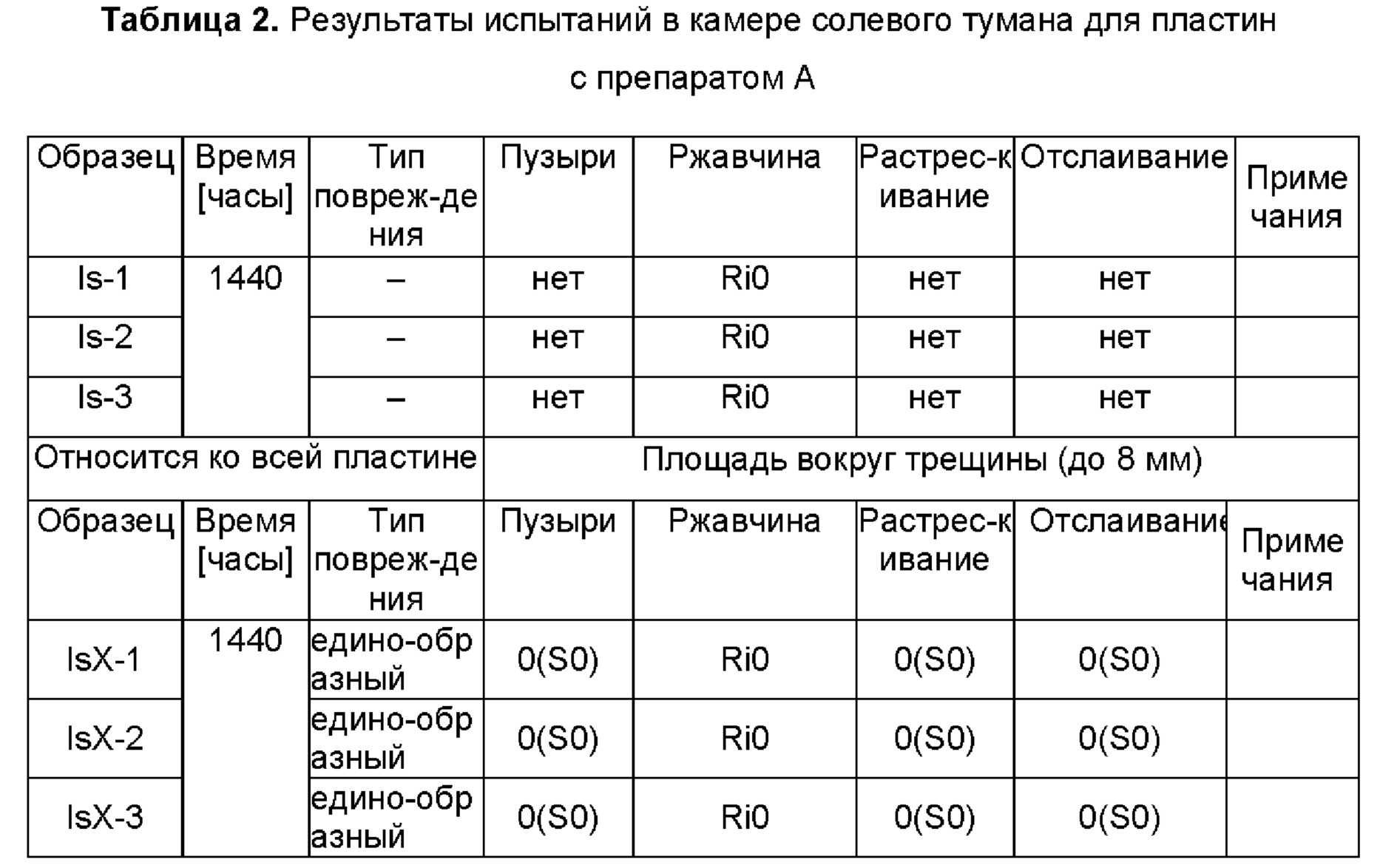
где:
Ri1 - степень ржавления, обозначающая ржавчину на 0,05% поверхности. Степени ржавления отмечаются как возрастающие от Ri0 до Ri5.
Образование пузырей, растрескивание и отслаивание определяются их плотностями и размерами (значение в скобках) по шкале от 0 до 5 (0 - самая низкая плотность и наименьший размер). Как можно видеть, наблюдается небольшая коррозия необработанных образцов и образование пузырей вблизи разреза на пластинах без препарата А и коррозия на пластинах с разрезом. На обоих типах пластин с препаратом А никаких повреждений не было.
Пример 5.
На поверхность 3 пластин из стали S215 (ранее St3), очищенной до степени St3 в соответствии со стандартом PN-EN ISO 8501 (очистка щеткой, около 50% поверхности с остатками ржавчины), наносили препарат А - неочищенный оксим, полученный в соответствии с примером 1, разбавляли изопропанолом (10% раствор), получая после 4 часов сушки при комнатной температуре слой, содержащий приблизительно 5,5 г неочищенного оксима на метр стальной поверхности. Затем наносили систему покрытия, состоящую из эпоксидного грунтовочного слоя толщиной 50 мкм и межслойного эпоксидного покрытия толщиной 100 мкм. Такую же систему покрытия наносили на следующие 3 стальные пластины, подготовленные таким же образом, но без препарата А. После естественного высыхания покрытий проводили исследования методом импедансной спектроскопии и испытания на адгезию покрытия методом вытягивания в соответствии со стандартом ISO 16276. Пластины помещали в камеру влажности и в течение 1440 часов проводили испытание на коррозионную стойкость в соответствии со стандартом ISO. После извлечения пластин из камеры их внешний вид оценивали, и повторяли тест методом импедансной спектроскопии. Значение логарифма модуля импеданса при частоте 0,1 Гц показывает барьерные свойства покрытий. Значения, превышающие 6, подтверждают наличие барьерных свойств. Чем выше значение, тем лучше барьерные свойства. Никакого повреждения, видимого невооруженным глазом, не было обнаружено ни на одной из испытуемых пластин. Для спектров импеданса в системе тел были найдены следующие значения логарифма модуля импеданса при частоте 0,1 Гц:
- перед испытаниями в камере влажности: 8-10 для всех пластин;
- после испытаний в камере влажности:

Значения адгезии покрытия, измеренные методом вытягивания, составили:
- перед испытаниями в камере влажности: 7-9 МПа для всех пластин;
- после испытаний в камере влажности: 7-9 МПа для пластин с препаратом А и 4-5 МПа для пластин без препарата А.
Полученные результаты подтверждают лучшие барьерные свойства систем с препаратом А после воздействия в камере влажности, а также их лучшую адгезию после того же воздействия.
Реферат
Изобретение относится к способу получения продукта, содержащего 60-75 мас. % 5-алкилсалицилальдоксимов с формулой 1, где R представляет собой С-С-алкильную группу, который заключается в том, что в водно-спиртовую систему растворителей вводят п-алкилфенол, гидроксид натрия, хлороформ и гидроксиламин, причем относительно используемого алкилфенола гидроксид натрия и хлороформ используют в количествах от стехиометрического количества до 100% избытка, и гидроксиламин используют в количествах от стехиометрического количества до 60% избытка, и реакцию проводят при температуре 60-75°С в течение 1,5-4 часов, а затем при температуре 20-30°С послереакционную смесь подкисляют до достижения рН водной фазы <7,0, а затем азеотроп спирт-вода отгоняют с примесью непрореагировавшего хлороформа, остаток смешивают с нейтральным углеводородным растворителем С-С, слои разделяют, и растворитель отгоняют из органической фазы. 3 з.п. ф-лы, 2 табл., 5 пр.
Формула
Патенты аналоги
METHOD FOR PREPARATION OF 5-ALKYLSALICYLALDOXIMES AND APPLICATION THEREOF
СПОСОБ ПОЛУЧЕНИЯ 5-АЛКИЛСАЛИЦИЛАЛДОКСИМОВ И ИХ ПРИМЕНЕНИЕ
METHOD FOR PREPARATION OF 5-ALKYLSALICYLALDOXIMES AND APPLICATION THEREOF
Method for preparation of 5-alkylsalicylaldoximes and application thereof
METHOD FOR PREPARATION OF 5-ALKYLSALICYLALDOXIMES AND APPLICATION THEREOF

Комментарии