Эффективный способ получения производных гидразинофенола - RU2759291C1
Код документа: RU2759291C1
Чертежи



Описание
Область техники, к которой относится изобретение
Данное изобретение относится к области органической, биоорганической и медицинской химии, а именно, к новому и эффективному способу синтеза гидразинофенолов, позволяющему получить новые производные фенола общей формулы (I).
Уровень техники
Фрагмент гидразинофенола играет важную роль в медицинской химии при дизайне кандидатов в препараты. Наиболее известным и изученным применением такого фрагмента является реакция синтеза индола по Фишеру [F. Zhan, G. Liang, Formation of enehydrazine intermediates through coupling of phenylhydrazines with vinyl halides: entry into the Fischer indole synthesis. / Angew. Chem. Int. Ed., 2013, 52 (4): 1266-1269], которая используется при синтезе лекарственных препаратов, содержащих индольный фрагмент, и в полном синтезе природных веществ, среди которых проводится скрининг кандидатов в препараты. К числу таких биологически важных веществ, которые синтетически возможно получить из гидразинофенола по реакции Фишера, относятся гормон-регулятор циркадных ритмов мелатонин и нейромедиатор и фактор роста серотонин, а также производные от них нейромедиаторные гормоны-метаболиты: 6-гидрокси-1-метил-1,2,3,4-тетрагидро-β-карболин (адреногломерулотропин) и 6-гидрокси-1-метил-1,2,3,4-тетрагидро-β-карболин, соответственно. Последний образуется в результате неферментативной реакции Пикте-Шпенглера между серотонином и ацетальдегидом (продуктом окисления этилового спирта в результате алкогольного опьянения) или же присутствует в природе в виде алкалоида, например, в какао-бобах [Т. Herraiz. Tetrahydro-β-carbolines, Potential Neuroactive Alkaloids, in Chocolate and Cocoa / J. Agric. Food Chem., 2000, 48, 10, 4900-4904]. Кроме того, гидразинофенол является промежуточным продуктом в полном синтезе многих алкалоидов: ингибиторов топоизомеразы II типа (9-hydroxyellipticine [J. Bonjoch, N. Casamitjana, J. Quirante, M. Rodriguez, J. Bosch. Functionalized 2-azabicyclo[3,3,1]nonanes. 6. Studies directed to the synthesis of pentacyclic Strychnos indole alkaloids / J. Org. Chem., 1987, 52 (2), 267-275]), кандидатов в препараты для терапии холинергических нарушений и болезни Альцгеймера ((-)-esermethole, (-)-eseroline, (-)-physostigmine (-)-physovenine) [S. Takano, M. Moriya, K. Ogasawara. Enantiocontrolled total syntheses of (-)-physovenine and (-)-physostigmine / J. Org. Chem., 1991, 56 (21): 5982-5984], а также обладающих противораковой активностью (montamine [L.M. Blair, J. Sperry. Studies towards the synthesis of montamine: synthesis of the 1,2-bis(indolyl)ethylhydrazine fragment / Tetrahedron Letters, 2013, 54 (15): 1980-1982]) и противомикробной активностью (koeniginequinone В [H.J. Knolker, K.R. Reddy. Indoloquinones, Part 8. Palladium(II)-catalyzed total synthesis of murrayaquinone A, koeniginequinone A, and koeniginequinone В / Heterocycles, 2003, 60 (5): 1049-1052]). Окисленный фрагмент 4-гидразинофенола также присутствует в стимуляторе белкового фактора роста нервов NG-061, полученного из штамма Penicillium minioluteum F-4627 [R. Bhandari, Т. Eguchi, A. Serine, Y. Ohashi, K. Kakinuma, M. Ito, K. Mizoue. Structure of NG-061, a Novel Potentiator of Nerve Growth Factor (NGF) Isolated from Penicillium minioluteum F-4627 / J. Antibiotics, 1999, 52 (3): 231-234]. 4-Гидразинофенол является промежуточным продуктом в производстве таких важных лекарств, как Индометацин [A.R. Maguire, S.J. Plunkett, S. Papot, M. Clynes, R. O'Connor, S. Touhey. Synthesis of indomethacin analogues for evaluation as modulators of MRP activity / Bioorg. Med. Chem., 2001, 9 (3), 745-762] и Базедоксифен [H.S. Kwan, P.C. Ha, S.R. Seok, C.S. Нее, K.K. Deok, K.D. Hwa, Methods of preparing Bazoedoxifene, Patent KR 10-2018-0095239 (A), date of application: 2017-02-17; H. Lizhi, Preparation method of selective estrogen receptor modulator bazedoxifene and key intermediate thereof, Patent CN 107935906 (A), date of application: 2018-04-20].
Лишь небольшое количество арилгидразинов являются коммерчески доступными или легко синтезируемыми. Для стандартного синтеза арилгидразинов применяется восстановление солей диазония, которые, в свою очередь, получают из соответствующих замещенных производных анилина. Альтернативно, Бухвальд и Хартвиг с коллегами предложили палладий-катализируемое кросс-сочетание для синтеза арилгидразинов [S. Wagaw, В.Н. Yang, S.L. Buchwald. A Palladium-Catalyzed Method for the Preparation of Indoles via the Fischer Indole Synthesis / J. Am. Chem. Soc., 1999, 121 (44): 10251-10263; F. Ma, Z. Peng, W. Li, X. Xie and Z. Zhang, Synlett, 2011, 2011 (17), 2555-2558].
Развитие метода кросс-сочетания привело к тому, что в последние десятилетия гидразинирование ароматических соединений зачастую проводится с помощью металлокомплексного катализа. При этом катализаторами являются либо кислоты Льюиса с переходными металлами, либо соли благородных металлов, либо радикальные инициаторы, а оптимальная температура для реакции составляет 40-70°С. Условия проведения реакции являются слабой стороной кросс-сочетания с технологической точки зрения из-за дороговизны катализатора, необходимости лицензии для работы с ним и связанного с этим учета. Кроме того, для многих функционализированных соединений кислые условия оказываются деструктивными; а, образующиеся продукты гидразинирования должны быть тщательно выделены и очищены от дорогих и сложных катализаторов на основе переходных металлов (из-за необходимости учета расхода катализатора). При получении особо чистых лекарственных субстанций для фармацевтики и медицинской химии продукты гидразинирования должны иметь степени чистоты выше 99,5%.
В настоящее время реакций фенол-содержащих соединений с эфирами азодикарбоксилатов описано крайне мало. Одним из примеров таких реакций является прямое гидразинирование фенола эфирами азодикарбоксилатов в эквимолярных количествах по отношению к фенольной компоненте. Однако оно приводит к образованию смеси продуктов окисления и пара-замещения в соотношении примерно 1:1; использование эфира азодикарбоксилата в избытке приводит к дальнейшему окислению образующегося побочного 3,3',5,5'-тетраалкил-4,4'-дифенилдиола в соответствующий дифенилхинон [S.H. Schroeter. Reaction of phenols with ethyl azodicarboxylate / J. Org. Chem., 1969, 34 (12): 4012-4015]; в ранних работах такие превращения были описаны как радикально протекающий процесс [R. Huisgen, F. Jakob, W. Siegel, A. Cadus. Addition reactions of the N,N-double bond. I. Aromatic side-chain addition of azodicarboxylic ester / Justus Liebigs Ann. Chem., 1954, 590 (1): 1-36; L. Horner and W. Naumann. Azo-diacyl-Verbindungen als Analoga des elementaren Sauerstoffs / Justus Liebigs Ann. Chem., 1954, 587 (2): 81-92].
Другой пример гидразинирования фенола - использование активированного бис(2,2,2-трихлорэтил)азодикарбоксилата (BTCEAD) в трифторметансульфоновой кислоте и/или в среде LiClO4 - был описан И. Лебланом и коллегами, предложившими синхронный механизм превращения [Y. Leblanc, N. Boudreault. Para-Directed Animation of Electron-Rich Arenes with Bis(2,2,2-Trichloroethyl) Azodicarboxylate / J. Org. Chem., 1995, 60 (13): 4268-4271].
Позднее, В. Кинарт предложил сразу два способа гидразинирования фенола и 2-нафтола: через N:→Sn-координированный металлоеновый механизм или по электрофильному замещению через стабилизированный интермедиат Виланда [W. Kinart, С. Kinart. Catalysis of reactions of allyltin compounds and organotin phenoxides by lithium perchlorate / J. Organomet. Chem., 2006, 691 (8): 1441-1451]. Кроме того, описан синтез атропоизомеров - через катализ алкалоидами растений семейства cinchona, при этом авторы упоминают, что сама система 6-гидроксихинолина в структуре катализатора так же взаимодействует с исходным реагентом и, при повышении температуры, аминируется по 5-ому положению. [S. Brandes, М. Bella, A.


Таким образом, ранее в научных публикациях было описано пять различных механизмов и подходов к катализу для родственных описываемому превращению реакций. Каждый из этих способов нуждается в дополнительных стадиях тщательной очистки продукта от катализатора, в большинстве случаев который содержит ион переходного металла.
Карбоксилатный остаток возможно деблокировать с гидразинового остатка при обработке кислотой или эфиратом трифторида бора трет-бутильного эфира продукта [E.F. Evans, N.J. Lewis. N-tert-Butoxycarbonyl (ВОС) Deprotection Using Boron Trifluoride Etherate / Synth. Commun., 1997, 27 (11): 1819-1825], получив незамещенные гидразины с фенольным фрагментом.
Раскрытие сущности изобретения
Сущность изобретения заключается в применении эфиров азодикарбоксилатов в качестве гидразинирующих агентов для получения производных 2- или 4-гидразинофенольного каркаса в молекуле, в реакциях с фенолом, 1-нафтолом, хинолин-8-олом и хинолин-8-ол N-оксидом в присутствии основного катализатора.
Изобретение решает задачу получения гидразиновых производных фенола и родственных ему ароматических соединений с количественными выходами (70-99%) в одну стадию, исходя из доступных эфиров азодикарбоксилатов (эфиров диазендикарбоксилатов) с помощью реакции электрофильного аминирования при основном катализе.
Способ электрофильного гидразинирования производных фенола осуществим в условиях общего основного катализа (Фиг. 1). Для синтеза используются эфиры азодикарбоксилатов, коммерчески доступные как в виде индивидуальных веществ, так и в виде растворов в органических растворителях. Ароматические углеводороды, содержащие фенольный гидроксил в качестве направляющей группы, необратимо подвергаются гидразинолизу в присутствии неорганических и органических оснований с выходом, близким к количественному. Реакция позволяет получать о- или n-замещенные гидразинофенолы при комнатной температуре, при этом следует отметить, что высокий выход продукта реакции не зависит от количества загрузки. В качестве основного катализатора в реакции используются гидриды или амиды щелочных металлов или другие основания (Табл. 1.), как в индивидуальном виде, так и в виде дисперсии в минеральном или силиконовом масле в различных процентных содержаниях. Было показано, что вне зависимости от типа ароматической системы наилучшим образом проявляют себя гидрид натрия и амид лития, как наиболее экономически оправданные (ежегодно производятся в многотоннажном количестве в мире и коммерчески легкодоступны) и требующие каталитических количеств (достаточно 0,05-0,5 мольных эквивалентов на исходные реагенты). При этом роль основания заключается в образовании фенолята и активации ароматической системы, необходимых для нуклеофильной атаки молекулы эфира азодикарбоксилата. В ходе получения фенолята возможно получение газообразных побочных продуктов, например, водорода, поэтому рекомендуется использование хлоркальциевой трубки вместо пробки, или другого приспособления, регулирующего внутреннее давление в реакционном сосуде. Использованные в реакции основания выводятся из реакционной массы экстракцией в водно-органических фазах, с последующим гашением эквимолярным количеством уксусной или соляной кислоты. Следует отметить, что при использовании гидридов и солей щелочных или щелочноземельных металлов или амида лития регенерация катализатора для повторного использования невозможна, в то время как при использовании оснований на основе органических аминов после отделения от продукта реакции возможна регенерация использованного основания.
Контроль за ходом реакций проводится с помощью ТСХ и/или ВЭЖХ, примерное время протекания процесса до количественного выхода составляет от 1 часа (в случае катализа более активным гидридом натрия) до 24 часов (в случае катализа гидридом лития).
Для деблокирования заместителей с гидразинового остатка предлагается проводить гидролиз трет-бутильных эфиров карбоксилатов, которые по своей сути являются защитной группой (трет-бутилоксикарбонил-, или ВОС) в исходных диазенах и/или в конечных продуктах. Проведение деблокирования осуществляется стандартными методами классического органического синтеза в мягких условиях [E.F. Evans, N.J. Lewis. N-tert-Butoxycarbonyl (ВОС) Deprotection Using Boron Trifluoride Etherate / Synth. Commun., 1997, 27 (11): 1819-1825] и приводит к N-незамещенным ароматическим гидразинам или их солям-гидрохлоридам (Фиг. 2.).
Таким образом, предлагаемый нами способ позволяет получать незамещенные гидразины и ди(этилоксикарбонил), ди(изопропилоксикарбонил) и ди(трет-бутилоксикарбонил) эфиры гидразинов фенола, 1-нафтола, 8-гидроксихинолина и N-оксида 8-гидроксихинолина (Фиг. 3) исходя из доступных исходных фенольных субстратов и эфиров азодикарбоксилатов.
Достоинствами данного способа являются: использование легкодоступных реагентов, стереоспецифичность протекающих реакций, обусловленная направляющей фенольной группой в ароматической структуре, полнота превращения исходных соединений и высокий выход целевых соединений.
Ниже приводятся конкретные примеры осуществления заявляемого технического решения, но которые не предназначены для ограничения объема притязаний.
Краткое описание фигур и таблиц
Фигура 1. Схема получения эфиров N,N'-дикарбоксилатов гидразинов фенол-содержащих соединений и условия протекания реакции.
Фигура 2. Схема получения N-незамещенных гидразинов фенол-содержащих соединений и их гидрохлоридов, и условия протекания реакции.
Фигура 3. Примеры полученных эфиров N,N'-дикарбоксилатов и N-незамещенных гидразинов фенол-содержащих соединений на основе предложенного метода.
Таблица 1. Зависимость выхода продуктов 1b и 7b от выбора основания-катализатора при проведении реакции с диизопропиловыми эфирами азодикарбоксилатов (аналогично для этиловых и трет-бутиловых эфиров). Условия: 20±5°С (если иное не указано в таблице), атмосферное давление, тетрагидрофуран в роли растворителя (от 200% по объему по отношению к остальным реагентам), хлоркальциевая трубка для высвобождения образующихся газообразных продуктов.
Осуществление изобретения
Структуры заявленных соединений подтверждали методами ЯМР- и масс-спектроскопии высокого разрешения. ЯМР-спектры регистрировали на приборе Broker АМХ 300 (Германия). Химические сдвиги (δ) в1Н-ЯМР приведены в миллионных долях (м.д.) и измерены относительно остаточного сигнала растворителя: D2O - 4,79 м.д. Величины констант спин - спинового взаимодействия (J) измерены в герцах (Гц). При описании спектров1Н-ЯМР приняты следующие сокращения: с - синглет, уш.с - уширенный синглет, д - дублет, т - триплет, м - мультиплет. Тонкослойную хроматографию проводили на пластинах Merck Kieselgel-60 с УФ-детекцией (λ=254 нм) и термической детекцией (нагревание ТСХ-пластин до 400°С). Колоночную хроматографию продуктов проводили на силикагеле размером 0,035-0,070 мм, 60

Следующие примеры иллюстрируют изобретение.
Пример 1. Общий способ синтеза производных 4-гидразинофенола и 4-гидразино-1-нафтола в граммовых количествах.
Раствор фенол-содержащего исходного вещества (5,31 ммоль) и 8,0 мг NaH (0,27 ммоль, 80% чистоты) в 10 мл тетрагидрофурана в круглодонной колбе объемом 50 мл охлаждали до -5…0°С и продували незанятый жидкостью объем колбы инертным газом или азотом. Порцию азодикарбоксилата (5,57 ммоль) добавляли при перемешивании при комнатной температуре. В ходе протекания процесса реакционная масса меняла цвет с желтого на темно-оранжевый, после чего перемешивание реакции в колбе продолжали еще в течение 1,5 часов. Растворитель упаривали, и реакционную массу экстрагировали в CH2Cl2 / насыщенный раствор NaHCO3,. Органический слой осушали над Na2SO4, раствор фильтровали от образующегося кристаллогидрата, растворитель упаривали, переупаривали с толуолом, образующийся остаток сушили в вакууме. Продукт растворяли в хлороформе и наносили на колонку с силикагелем, элюировали хлороформом. Очищенный продукт получали в виде бесцветных твердых кристаллов.
Пример 2. Диизопропиловый эфир 1-(4-гидроксифенил)гидразинил-1,2-дикарбоксилата (1b)
Продукт получали в виде бесцветных кристаллов согласно общему способу получения (Пример 1.) из 0,500 г фенола и 1,095 мл диизопропилазокдикарбоксилата в количестве 1,416 г (Выход: 90%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,71 & 9,37 (2×уш.с, 1Н, NH (2 конформера)), 9,37-9,71 (уш.с, 1Н, ОН), 7,13 (д, J=8,7 Гц, 2Н, СН-2 & СН-6), 6,72 (д, J=8,8 Гц, 2Н, СН-3 & СН-5), 4,82 (2×септ, J=6,1 Гц, 2Н, СН (i-Pr)), 1,21 & 1,18 (2×д, 12Н, Me (i-Pr)).
13С ЯМР (DMSO-d6, δ, Гц): 155,71 (s, С4), 155,55 (2×c, CO-NH), 154,25 (с, CO-N), 133,9 (с, С1), 125,98 (уш.с, С2 & С6), 114,9 (с, С3 & С5), 69,36 & 68,34 (2×c, С (i-Pr)), 21,87 & 21,77 (2×c, Me (i-Pr)).
Масс-спектр C14H20N2O5 (m/z): рассчитано для [М+Н]+ 297,1445, найдено 297,1442; рассчитано для [M+Na]+ 319,1264, найдено 319,1256; рассчитано для [М+K]+ 335,1004, найдено 335,0998.
Пример 3. Ди-трет-бутиловый эфир 1-(4-гидроксифенил)гидразинил-1,2-дикарбоксилата (1с)
Продукт получали в виде бесцветных кристаллов согласно общему способу получения (Пример 1.) из 0,500 г фенола и 1,284 г ди-трет-бутилазокдикарбоксилата в количестве 1,551 г (Выход: 90%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,47 & 9,37 & 9,06 (3×уш.с, 1H, NH+OH (2 конформера)), 7,09 (д, J=8,7 Гц, 2Н, СН-2 & СН-6), 6,69 (д, J=8,8 Гц, 2Н, СН-3 & СН-5), 1,42 & 1,39 (2×c, 18Н, Me (t-Bu)).
13С ЯМР (DMSO-d6, δ, Гц): 155,60 (с, С4), 155,53 (с, CO-NH), 154,00 (с, CO-N), 134,67 (с, С1), 126,49 (с, С2 & С6), 114,9 (уш.с, С3 & С5), 80,75 & 79,96 (2×c, С (t-Bu)), 28,54 & 28,30 (2×c, Me (t-Bu)).
Масс-спектр C16H24N2O5 (m/z): рассчитано для [М+Н]+ 325,1758, найдено 325,1763; рассчитано для [M+Na]+ 347,1577, найдено 347,1577; рассчитано для [М+K]+ 363,1317, найдено 363,1315.
Пример 4. Ди-трет-бутиловый эфир 1-(4-гидрокси-2,5-диметилфенил)гидразинил-1,2-дикарбоксилата (2с)
Продукт получали в виде бесцветных кристаллов согласно общему способу получения (Пример 1.) из 0,650 г 2,4-диметилфенола и 1,284 г ди-трет-бутилазокдикарбоксилата в количестве 1,797 г (Выход: 93%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,46 & 9,39 (2×уш.c, 1Н, NH (2 конформера)), 9,20 (с, 1H, ОН), 7,00 (с, 1H, СН-5), 6,55 (с, 1Н, СН-2), 2,09 & 2,04 (2×уш.c, 6Н, 2×Ме), 1,42 & 1,31 (2×c, 18H, Me (t-Bu)).
13С ЯМР (DMSO-d6, δ, Гц): 155,07 (с, CO-NH), 154,22 (с, CO-N), 153,64 (с, С4), 133,66 (с, С2), 132,53 (с, С1), 129,27 (с, С6), 128,93 (с, С5), 115,51 (с, С3), 79,85 & 79,24 (2×c, С (t-Bu)), 28,07 & 27,83 (2×c, Me (t-Bu)), 17,13 (а, 2-Ме), 15,53 (а, 5-Ме).
Масс-спектр C16H24N2O5 (m/z): рассчитано для [М+Н]+ 353,2071, найдено 353,2073; рассчитано для [M+Na]+ 375,1890, найдено 375,1891.
Пример 5. Ди-трет-бутиловый эфир 1-(4-гидроксинафт-1-ил)гидразино-1,2-дикарбоксилата (3с)
Продукт получали из 0,775 г 1-нафтола и 1,284 г ди-трет-бутилазокдикарбоксилата согласно общему способу получения (Пример 1.). Реакционную массу после проведения экстракции наносили на колонку с силикагелем и элюировали в системе хлороформ-этанол (97%: 3% по объему). Фракции, содержащие целевой продукт, упаривали в вакууме, в результате чего выделяли 1,646 г 3с в виде вязкой жидкости бежевого цвета (Выход 80%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,90-9,70 (уш.с, 2Н, NH+OH), 8,29 (дд, J=0,8 Гц, J=9,0 Гц, 1Н, СН-8), 8,05 (д, J=8,2 Гц, 1Н, СН-5), 7,62-7,52 (м, 3Н, СН-6+СН-7+СН-8), 7,43 (д, 1Н, J=7,3 Гц, СН-3), 7,34 (д, 1H, J=7,3 Гц, СН-2), 1,47 и 1,26 (2×c, 18Н, Me (t-Bu)).
Масс-спектр C20H26N2O5 (m/z): рассчитано [M+Na]+ 397,1734, найдено 397,1740.
Пример 6. Тетра-трет-бутиловый эфир N,N'-(4-гидроксинафтал-1,3-диил)-бис-(гидразино-1,2-дикарбоксилата) (4с)
Продукт получали из 0,383 г 1-нафтола (2,66 ммоль) согласно общему способу получения (Пример 1.). Реакционную массу после проведения экстракции наносили на колонку с силикагелем и элюировали в хлороформе. Фракции, содержащие целевой продукт под данным ТСХ, упаривали в вакууме, в результате чего выделяли 1,350 г 4 с в виде желтой вязкой жидкости (Выход 84%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,94-9,29 (м, 3Н, 2×NH+ОН), 8,22 (д, J=7,5 Гц, 1H, СН-5), 8,01 (с, 1H, СН-3), 7,54 (м, 3Н, СН-6+СН-7+СН-8), 1,46 и 1,43 и 1,40 и 1,30 и 1,22 (5×c, 36Н, Me (t-Bu) (2 конформера)).
13С ЯМР (DMSO-d6, δ, Гц): 158,25 (с, CO-NH), 155,38 (с, CO-NH) 154,14 (с, СО-N), 153,59 (с, CO-N), 148,35 (с, С1), 130,45 (с, С4), 126,79 (с, С5), 124,91 (с, С6), 123,32 (с, С7), 122,68 (с, С8), 122,08 (с, С10), 121,69 (с, С9), 81,22 и 80,35 и 79,47 и 78,82 (4×с, С (t-Bu)), 28,08 и 27,91 и 27,66 (3×c, Me (t-Bu)).
Масс-спектр C30H44N4O9 (m/z): рассчитано [M+Na]+ 627,3000, найдено 627,3003; рассчитано [М+K]+ 643,2740, найдено 643,2739.
Пример 7. Общий способ синтеза 4-замещенных производных 2-гидразино-1-нафтола и 5-замещенных производных 7-гидразино-8-гидроксихинолина
Раствор 0,100 ди-трет-бутилазодикарбоксилата (0,43 ммоль) и 5,9 мг гидрида натрия (0,20 ммоль, 80% чистоты) смешивали с соответствующим количеством замещенного фенол-содержащего исходного вещества (0,39 ммоль) в 3 мл тетрагидрофурана, перемешивали при комнатной температуре в течение 6 часов. Конверсию превращения отслеживали по данным ТСХ и при необходимости перемешивали еще 12 часов. По окончании реакции растворитель упаривали, реакционную массу экстрагировали в CH2Cl2 / насыщенный раствор NaHCO3, Органический слой осушали над Na2SO4, растворитель упаривали, остаток растворяли в CHCl3 и наносили на колонку с силикагелем, целевые фракции упаривали в вакууме.
Пример 8. Ди-трет-бутиловый эфир 1-(4-хлор-1-гидроксинафт-2-ил)гидразинил-1,2-дикарбоксилата (5с)
Продукт получали из 0,070 г 4-хлор-1-нафтола (0,39 ммоль) согласно общему способу получения (Пример 7.). Реакционную массу после проведения экстракции промывали CCl4, Остаток, содержащий целевой продукт, дополнительно сушили в вакууме, в результате чего получали 0,153 г 6с в виде молочно-белого твердого остатка (Выход 95%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,96 (уш.с, 1Н, NH), 9,70 (уш.с, 1Н, ОН), 8,29 (дд, J=0,8 Гц, J=9,0 Гц, 1H, СН-8), 8,09 (д, J=8,2 Гц, 1H, СН-5), 7,71 (дд, J=6,9 Гц, J=8,2 Гц, J=1,0 Гц, 1Н, СН-6), 7,63 (дд, 1Н, J=6,9 Гц, J=9,0 Гц, J=1,0 Гц, СН-7), 7,43 (с, 1H, СН-3), 1,44 и 1,30 (2×c, 18Н, Me (t-Bu)).
13С ЯМР (DMSO-d6, δ, Гц): 157,57 (с, CO-NH), 153,38 (с, CO-N), 148,65 (с, С8), 130,17 (с, С9), 128,34 (с, С6), 128,10 (с, С10), 126,59 (с, С7), 126,43 (с, С5), 125,83 (с, С4), 123,48 (с, С), 123,27 (с, С8), 119,65 (с, С2), 81,53 и 81,46 (2×c, С (t-Bu)), 27,87 и 27,69 (2×c, Me (t-Bu)).
Масс-спектр C20H25ClN2O5 (m/z): рассчитано [M+NH4]+ 426,1790, найдено 426,1797; рассчитано [M+Na]+ 431,1344, найдено 431,1350; рассчитано [М+K]+ 447,1084, найдено 447,1089.
Пример 9. Ди-трет-бутиловый эфир 1-(5-фтор-8-гидроксихинолин-7-ил)гидразино-1,2-дикарбоксилата (7с)
Продукт получали из 0,064 г 5-фтор-8-гидроксихинолина (0,39 ммоль) согласно общему способу получения (Пример 7.). Реакционную массу после проведения экстракции наносили на колонку с силикагелем и элюировали в системе CCl4: диоксан (9:1) с добавкой 1% уксусной кислоты. Фракции, содержащие целевой продукт, упаривали в вакууме, в результате чего выделяли 0,108 г 8с в виде твердого продукта светло-зеленого цвета (Выход 70%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,95 (уш.с, 2Н, NH+ОН), 8,86 (д, J=4,2 Гц, 1Н, СН-2), 8,25 (д, J=8,4 Гц, 1Н, СН-4), 7,47 (с, 1Н, СН-6), 7,31 (дд, J=8,4 Гц, J=4,2 Гц, 1H, СН-3), 1,42 и 1,40 (2×уш.c, 18Н, Me (t-Bu)).
Масс-спектр C19H24FN3O5 (m/z): рассчитано [М+Н]+ 394,1773, найдено 394,1764.
Пример 10. Ди-трет-бутиловый эфир 1-(5-хлор-8-гидроксихинолин-7-ил)гидразино-1,2-дикарбоксилата (8с)
Продукт получали из 0,071 г 5-хлор-8-гидроксихинолина (0,39 ммоль) согласно общему способу получения (Пример 7.). Реакционную массу после проведения экстракции наносили на колонку с силикагелем и элюировали в системе CCl4: диоксан (9:1) с добавлением 1% (по объему) уксусной кислоты. Фракции, содержащие целевой продукт, упаривали в вакууме, в результате чего выделяли 0,156 г 6с в виде твердого продукта бежевого цвета (Выход 97%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,92-9,38 (уш.с, 2Н, NH+ОН), 8,86 (дд, J=1,0 Гц, J=4,1 Гц, 1H, СН-2), 8,40 (дд, J=8,4 Гц, J=1,5 Гц, 1H, СН-4), 7,64 (дд, J=8,4 Гц, J=4,2 Гц, 1Н, СН-3), 7,56 (с, 1H, СН-6), 1,42 и 1,39 (2×уш.c, 18Н, Me (t-Bu)).
13С ЯМР (DMSO-d6, δ, Гц): 155,74 (с, CO-NH), 153,31 (с, CO-N), 148,65 (с, С2), 140,25 (с, С9), 132,31 (с, С4), 127,03 (уш.с, С5+С7), 125,94 (с, С8), 125,06 (с, С6), 122,75 (с, С3), 115 (с, С10), 80,89 и 80,24 (2×c, С (t-Bu)), 28,50 и 28,28 (2×c, Me (t-Bu)).
Масс-спектр C19H24ClN3O5 (m/z): рассчитано [М+Н]+ 410,1477, найдено 410,1480; рассчитано [M+Na]+ 432,1297, найдено 432,1302.
Пример 11. Общий способ синтеза производных 7-гидразино-8-гидроксихинолина
К раствору, содержащему 2 г 8-гидроксихинолина (13,8 ммоль) и 21 мг гидрида натрия (0,69 ммоль, 80% чистоты) в 40 мл растворителя добавляли порцию азодикарбоксилата (14,47 ммоль) при перемешивании при комнатной температуре. Реакционную массу перемешивали при комнатной температуре от 30 до 180 мин до окончания реакции (прохождение реакции контролировали по данным тонкослойной хроматографии). Растворитель упаривали под вакуумом, реакционную массу экстрагировали в CH2Cl2 / насыщенный водный раствор NaHCO3. Органическую фазу дополнительно осушали Na2SO4, фильтровали от образовавшегося кристаллогидрата и упаривали от растворителя, переупаривали с толуолом.
Пример 12. Диизопропиловый эфир 1-(8-гидроксихинолин-7-ил)гидразинил-1,2-дикарбоксилата (6b)
Продукт получали в виде молочно-белого твердого вещества исходя из общего способа (Пример 11.) из 2,925 г диизопропилазодикарбоксилата в количестве 4,773 г (Выход: 99,7%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,91 (2×уш.c, 1H, NH), 9,47 (2×уш.c, 1Н, ОН), 8,87 (дд, J=1,6 Гц, J=4,2 Гц, 1Н, СН-2), 8,31 (дд, J=1,5 Гц, J=8,3 Гц, 1Н, СН-4), 7,56 (дд, J=4,2 Гц, J=8,3 Гц, 1Н, СН-3), 7,52 (д, J=8,8 Гц, 1H, СН-6), 7,38 (д, J=8,8 Гц, 1Н, СН-5), 4,85 (септ, 2Н, J=6,2, СН (i-Pr)), 1,21 (д, 12Н, Me (i-Pr)).
13С ЯМР (DMSO-d6, δ, Гц): 156,87 и 154,59 (2×c, CO-NH (2 конформера)), 148,89 (с, С2), 148,68 (с, CO-N), 138,1 (с, С9), 136,38 (с, С4), 128,17 (с, С8), 127,48 (2×c, С5 и С10), 126,03 (с, С7), 122,46 (с, С3), 117,37 (с, С6), 69,98 и 69,00 (2×c, С (i-Pr)), 22,32 и 22,25 (2×c, Me (i-Pr)).
15N ЯМР (DMSO-d6, δ, Гц): 297,94 (с, N1), 119,18 (с, NH-CO).
Масс-спектр C17H21N3O5 (m/z): рассчитано [М+Н]+ 348,1554, найдено 348,1562.
Пример 13. Ди-трет-бутиловый эфир 1-(8-гидроксихинолин-7-ил)гидразинил-1,2-дикарбоксилата (6с)
Продукт получали в виде молочно-белого твердого вещества исходя из общего способа (Пример 11.) из 3,173 г ди-трет-бутилазодикарбоксилата в количестве 5,170 г (Выход 99,9%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,73 (2×уш.c, 1H, NH), 9,35 (2×уш.c, 1H, ОН), 8,86 (дд, J=2,5 Гц, J=4,2 Гц, 1H, СН-2), 8,50 (д, J=8,4 Гц, 1Н, СН-4), 7,61 (дд, J=8,6 Гц, J=4,1 Гц, 1Н, СН-3), 7,51 (д, J=8,2 Гц, 1Н, СН-6), 7,08 (д, J=8,2 Гц, 1Н, СН-5), 1,46 и 1,41 и 1,19 (3×уш.с, 18Н, Me (t-Bu) (2 конформера)).
13С ЯМР (DMSO-d6, δ, Гц): 155,27 и 154,00 (с, CO-NH (конформеры)), 153,16 (с, CO-N), 148,11 (с, С2), 138,14 (с, С9), 132,30 (с, С4), 129,30 (с, С10), 126,96 (с, С8), 126,35 (с, С5), 125,87 (с, С7), 121,82 (с, С3), 110,39 (с, С6), 80,55 и 80,12 и 79,63 (3×c, С (t-Bu) (2 конформера), 28,02 и 27,74 (2×c, Me (t-Bu) (2 конформера)).
15N ЯМР (DMSO-d6, δ, Гц): 299,3 (с, N1), 245,5 (с, NH-CO), 111,2 (с, N-CO).
Масс-спектр C19H25N3O5 (m/z): рассчитано [М+Н]+ 376,1867, найдено 376,1867.
Пример 14. 7-(1,2-Бис(трет-бутоксикарбонил)гидразинил)-8-гидроксихинолин N-оксид (9с)
Способ 1: Продукт получали в виде твердого вещества кремового цвете исходя из общего способа синтеза (Пример 11.) из 0,437 г исходного N-оксида 8-гидроксихинолина 0,705 и ди-трет-бутилазодикарбоксилата в количестве 0,948 г (Выход: 83%).
Способ 2: В охлажденный до ~0°С раствор с 0,500 г с 7с (1,33 ммоль) в 5 мл хлористого метилена добавляли порциями раствор 0,300 г мета-хлорпербензойной кислоты (1,73 ммоль) в 2 мл хлористого метилена. Реакцию перемешивали при комнатной температуре в течение 5 ч, протекание процесса контролировали по данным ТСХ. По окончании реакции смесь экстрагировали в CH2Cl2 / насыщенный раствор NaHCO3, Органический слой осушали над Na2SO4, фильтровали раствор от образовавшегося кристаллогидрата, растворитель упаривали, остаток растворяли в CHCl3 и наносили на хроматографическую колонку с силикагелем. Колоночной хроматографией выделяли продукт в градиенте этанола в хлороформе (0→6% спирта по объему), целевые фракции упаривали в вакууме, получая 0,481 г (92%) твердого продукта бежевого цвета.
1H ЯМР (DMSO-d6, δ, Гц): 8,27 (д, J=5,8 Гц, 1H, СН-2), 8,20 (уш.с, 1Н, NH), 8,50 (д, J=5,9 Гц, 1Н, СН-4), 7,60 (д, J=8,3 Гц, 1H, СН-6), 7,29 (дд, J=8,7 Гц, J=6,1 Гц, 1H, СН-3), 7,01 (д, J=8,5 Гц, 1Н, СН-5), 1,46 и 1,45 и 1,35 (3×уш.с, 18Н, Me (t-Bu) (2 конформера)).
13С ЯМР (DMSO-d6, δ, Гц): 155,60 (с, CO-NH), 154,53 (с, CO-N), 154,21 (с, С2), 138,1 (с, С9), 134,71 (с, С4), 130,17 (с, С7), 129,69 (с, С10), 128,59 (с С8), 126,44 (с, С5), 120,88 (с, С3), 114,25 (с, С6), 80,80 и 81,96 (2×c, С (t-Bu)), 28,33 и 28,19 (2×c, Me (t-Bu) (2 конформера)).
Масс-спектр C19H25N3O6 (m/z): рассчитано [М-O+Н]+ 376,1867, найдено 376,1867; рассчитано [М+Н]+ 392,1816, найдено 392,1813; рассчитано [M+Na]+ 414,1636, найдено 414,1628; рассчитано [М+Н]+ 430,1375, найдено 430,1374.
Пример 15. Общий способ получения гидразиновых производных фенолсодержащих ароматических соединений.
Порцию молекулярных сит (размер пор: 4
Пример 16. 4-Гидроксифенилгидразин (1d)
Продукт получали из 0,43 г вещества 1 с (1,33 ммоль) согласно общему способу получения (Пример 15.) в количестве 0,164 г в виде бесцветного твердого осадка (Выход 99%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,69 (уш.с, 3Н, NH+NH2 (один из двух)+ОН), 7,18 (д, J=8,8 Гц, 2Н, СН-3+СН-5), 7,08 (т (равной интенсивности), JH,15N=51,1 Гц, 1Н, NH2), 6,90 (д, J=8,8 Гц, 2Н, СН-2+СН-6).
13С ЯМР (DMSO-d6, δ, Гц): 157,26 (с, С4), 124,31 (с, С3+С5), 122,10 (с, С1), 116,16 (с, С2+С6).
Масс-спектр C6H8N2O (m/z): рассчитано для [М+Н]+ 125,0709, найдено 125,0713.
Пример 17. 7-Гидразино-8-гидроксихинолин (6d)
Продукт получали из 0,5 г вещества 7с (1,33 ммоль) согласно общему способу получения (Пример 15.) в количестве 0,590 г в виде твердого темно-малинового осадка (Выход 98%).
1Н ЯМР (DMSO-d6, δ, Гц): 10,63 (уш.с, 2Н, NH+OH), 8,95 (д, J=4,5 Гц, 1Н, СН-2), 8,80 (д, J=8,4 Гц, 1H, СН-4), 7,76 (дд, J=4,5 Гц, J=8,5 Гц, 1H, СН-3), 7,52 (д, J=8,3 Гц, С5), 7,52 (д, J=8,3 Гц, С6).
Масс-спектр C9H9N3O (m/z): рассчитано для [М+Н]+ 176,0818, найдено 176,0820.
Пример 18. Общий способ получения гидрохлоридов гидразиновых производных фенолсодержащих ароматических соединений.
Навеску Вос-защищенного гидразинового производного (1,33 ммоль) растворяли в 5 мл диоксана в круглодонной колбе при комнатной температуре и добавляли 2 мл водного раствора соляной кислоты (33-37%), закрывали хлоркальциевой трубкой или другим приспособлением, регулирующим давление в реакционном сосуде, и оставляли перемешиваться при температуре 20-40°С. Растворители и избыток кислоты упаривали в вакууме и переупаривали с метанолом, в результате чего получали целевой незамещенный ароматический гидразин в виде его соли гидрохлорида с переменным составом от одной до двух молекул HCl на молекулу ариометического гидразина.
Пример 19. 7-Гидразино-8-гидроксихинолина гидрохлорид (6е)
Продукт получали из 1,000 г вещества 7 с (2,66 ммоль) согласно общему способу получения (Пример 18.) в количестве 1,180 г в виде твердого темно-малинового осадка (Выход 97%).
1Н ЯМР (DMSO-d6, δ, Гц): 9,50 (уш.с, 3Н, NH+OH+HCl), 8,97 (д, J=4,4 Гц, 1Н, СН-2), 8,83 (д, J=8,2 Гц, 1H, СН-4), 7,78 (дд, J=4,4 Гц, J=8,3 Гц, 1H, СН-3), 7,55 (д, J=8,2 Гц, С5), 7,54 (д, J=8,3 Гц, С6).
Масс-спектр C9H10N3OCl (m/z): рассчитано для [М+Н-Cl]+ 176,0818, найдено 176,0822.

Реферат
Изобретение относится к области органической химии, конкретно к способу получения замещенных и незамещенных фенолсодержащих гидразин-N,N'-дикарбоксилатов указанной ниже формулы. Способ заключается в реакции гидразинирования замещенных и незамещенных фенолов с одно- и бициклической конденсированной структурой (1-нафтолы, хинолин-8-олы) эфирами азодикарбоксилатов в присутствии основного катализатора при комнатной температуре в инертных растворителях. При этом возможно последующее деблокирование N-карбоксилатных остатков в частных случаях Boc-замещенных гидразиновых производных для образования N-незамещенных фенолсодержащих гидразинов. В формуле X представляет собой СН, N, N+-O-в фиксированном положении или отсутствует в случае отсутствия цикла; R представляет собой -Н, -COOEt, -COOi-Pr, -COOt-Bu; R' представляет собой -Н, -Ме, -Cl, -F; Z представляет собой nHCl (n=1-2) в случае, когда R=Н. Предлагаемый способ позволяет получать замещенные и незамещенные фенолсодержащие гидразин-N,N'-дикарбоксилаты с выходами 70-99% в одну стадию исходя из доступных эфиров азодикарбоксилатов. 3 ил., 1 табл., 18 пр.
Формула
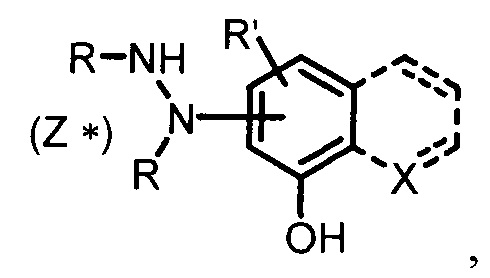
Комментарии