Способ получения полипептидов - RU2037498C1
Код документа: RU2037498C1
Чертежи








Описание
Изобретение относится к некоторым полиаминам и полипептидам, которые были обнаружены в яде паука Agelenopsis aperta. Полиамины и их приемлемые с фармацевтической точки зрения соли подавляют возбуждающие аминокислотные нейротрансмиттеры, эти нейротрансмиттеры оказывают влияние на клетки, включающие нейтронные клетки самых разнообразных организмов, включая беспозвоночных и позвоночных. Полипептиды и один из упомянутых полиаминов, и их приемлемые с фармацевтической точки зрения соли блокируют кальциевые каналы в клетках, включающих нейронные и мышечные клетки различных организмов, включающих беспозвоночных и позвоночных. Изобретение относится также к использованию таких полиаминов и их солей с целью подавления возбуждающих аминокислотных нейтротрансмиттеров, упомянутые нейротрансмиттеры оказывают влияние на такие клетки, как клетки нервной системы организма как таковые, при лечении заболеваний и симптомов, вызванных возбуждающими аминокислотными нейротрансмиттерами у млекопитающего, и в борьбе с беспозвоночными вредителями, и к композициям, содержащим упомянутые полиамины и их соли. Кроме того, изобретение относится к использованию упомянутых полипептидов и одного из упомянутых полиаминов, и их солей к блокированию кальциевых каналов в таких клетках, как клетки нервной и мышечной системы организма как таковых, при лечении заболеваний и симптомов, вызванных кальциевыми каналами, и для борьбы с беспозвоночными вредителями, и к композициям, содержащим упомянутые полипептиды, полиамин и их соли.
Известно, что яд паука Agelenopsis aperta содержит, по крайней мере два токсина, которые влияют на кальциевые потоки: токсин, именуемый АСТ2, который имеет мол.м. менее 1000 дальтон и который оказывается подавляет кальциевые потоки в самых разнообразных тканях, другой токсин из Agelenopsis aperta, содержащий компоненту в примерно 6000 мол.м. этот токсин устанавливает предсинаптическую блокаду передачи импульса и было выдвинуто предположение, что этот токсин блокирует кальциевые каналы, ассоциированные с высвобождением нейротрансмиттера.
Соединения, которые являются антагонистами возбуждающих аминокислотных нейротрансмиттеров, находят самые разнообразные применения. Антагонисты возбуждающих аминокислотных нейтротрансмиттеров могут найти клиническое применение при лечении таких симптомов, как пароксизм, "удар", церебральная ишемия, расстройства, связанные с нейронными нарушениями, такие как заболевание Алзеймера и эпилепсия, и в качестве психотерапевтических агентов, среди прочих. Кроме того, такие соединения используют при изучении физиологии клеток, таких как нейронные клетки, и в борьбе с беспозвоночными вредителями.
Соединения, которые являются антагонистами кальция, обладают многими полезными свойствами. Антагонисты кальция могут найти клиническое применение при лечении таких симптомов, как ангина, гипертония, кардиомиопатия, надвентрикулярная аритмия, aesophogeal ахалазия, преждевременные роды и заболевание Рейно, среди прочих. Кроме того, такие соединения используют при изучении физиологии клеток, таких как нейронные и мышечные клетки, и для борьбы с беспозвоночными вредителями.
Изобретение касается полиаминов и полипептидов, которые были обнаружены в яде жука Agelenopsis aperta. Полиамины изобретения и фракции, в которых они
присутствуют в соответствии с изобретением, имеют следующую структуру:
(AGEL 452):
Фракция А1












Фракция А1
(AGEL 468):








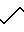



Фракция А2
(AGEL 448):











Фракция В1
(AGEL 505):






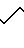





Фракция В2
ФАБ МС: высокое разрешение: 505,3861 рассчитано для С27Н48N6O3.
(AGEL 489):
Фракция Е











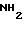
Полипептиды настоящего изобретения и фракции, в которых они присутствуют в соответствии с изобретением, имеют следующий вид.
Фракция G:
Амино-концевая аминокислотная последовательность, содержащая:


мол.м. для полного полипептида в соответствии с ФАБ МС: 7267.
Фракция Н2:
Амино-концевая аминокислотная последовательность, содержащая:

мол.м. полного полипептида в соответствии ион-распылительной МС: 7793.
Фракция I:


Фракция J:

мол.м. полного полипептида в соответствии с ФАБ МС: 5506.
Фракция L1:
Амино-концевая аминокислотная последовательность,
содержащая:


приблизительная мол.м. для полного полипептида составила примерно 20000.
Фракция L2: Полипептид, полученный в соответствии с описанием, приведенным в примере 9.
Фракция М: Амино-концевая аминокислотная последовательность, содержащая:

примерная мол.м. для полного полипептида примерно 80000.
Полиамины изобретения и их приемлемые с фармацевтической точки зрения соли подавляют возбуждающие аминокислотные нейротрансмиттеры, которые воздействуют на клетки. Таким образом, упомянутые полиамины можно использовать для подавления упомянутых нейротрансмиттеров, как таковые. Полиамины изобретения можно также использовать для борьбы с беспозвоночными вредителями и при лечении заболеваний и симптомов у млекопитающих, вызванных возбуждающими аминокислотными нейротрансмиттерами. Упомянутые полиамины полезны также в качестве психотерапевтических агентов для млекопитающих.
Полиамин В1, упомянутый выше, и полипептиды изобретения блокируют кальциевые каналы в клетках. Таким образом, упомянутые полиамин В1 и полипептиды можно использовать для блокирования кальциевых каналов как таковых. Упомянутый полиамин В1 и у помянутые полипептиды можно также использовать для борьбы с беспозвоночными вредителями и при лечении заболеваний и симптомов у млекопитающих, вызванных изменением функционирования кальциевого канала в клетках.
Кроме того, изобретение касается полипептидов, которые обладают по существу той же аминокислотной последовательностью и по существу такой же блокирующей кальциевые каналы активностью, что и указанные полипептиды.
Изобретение касается фармацевтических композиций, содержащих упомянутые полиамины и полипептиды, и способов применения упомянутых полиаминов и полипептидов.
Яд получали от жука Agelenopsis aperta в результате процесса "доения" при помощи стимулирования в соответствии со стандартными приемами, хорошо известными каждому специалисту в этой области техники. В предпочтительном варианте используют такой прием, в соответствии с которым гарантируется отсутствие примесей в яде из-за аномального отрыгивания или наличия гемолимфы. Такие приемы хорошо известны каждому специалисту в этой области техники. Весь яд, полученный таким образом, хранят в замороженном состоянии при температуре примерно -78оС, до очистки, которая описана ниже.
Очистку составляющих полного яда осуществляют при помощи обращенно-фазовой высокоэффективной жидкостной хроматографии (ВЭЖХ) на самых разнообразных препаративных и полупрепаративных колоннах, таких как С-4 и С-18 Видак® ( фирма Райнин Инструмент Ко./Мак Ронд/Уоберн, Массачузетс 01801). Обнаружение пиков осуществляют монохроматически в диапазоне 220-230 нм. Последующий анализ фракций можно осуществить, например, при помощи полихром-УФ данных, собранных при помощи детектора с серией диодов типа Уотерз 990 (фирма Миллипор Корпорэйшн, Отдел Уотерз Хроматографи, ул. Мэпл 34, Милфорд, Массачузетс 01757). Фракции из колонны собирали при помощи известных приемов, например с использованием коллектора фракций ИСКО ("ФОКСИ" и детектора пиков ИСКО 2159 (ИСКО, 4700 Супериор, Линкольн, Небраска 68504). Фракции собирают в соответствующим образом размеченные сосуды, такие как стерильная полиэтиленовая посуда. Концентрирование фракции далее осуществляют при помощи лиофилизации из элюента с последующей лиофилизацией из воды. Чистоту полученных в результате составляющих фракций в последующем можно определить при помощи хроматографического анализа, используя аналитическую колонну с градиентной системой, которая является более изократной, чем система, используемая при финальной очистке фракции.
Структуры, составляемые соответствующими фракциями, определяют в соответствии с известными аналитическими методами, такими как масс-спектроскопия и ядерный магнитный резонанс. Последовательности предлагаемых полипептидов определяют при помощи известных приемов. Например, S-пиридилэтилирование цистиновых остатков полипептида при исследовании можно осуществить в растворе с последующим определением аминокислотных последовательностей этого полипептида. Одна такая процедура для S-пиридилэтилирования состоит в следующем. От примерно 1 до 10 мкг полипептида растворяют или разбавляют в примерно до 50 мкл буфера, полученного в результате смешения 1 части 1М ТрисHCl, рН 8,5, содержащего 4 ммоль ЭДТК и 3 части 8M гуанидина HCl. Затем добавляли 2,5 мкл 10%-ного водного 2-меркаптоэтанола и смесь инкубировали при комнатной температуре в темноте в атмосфере аргона в течение 2 ч. После инкубирования добавляют 2 мкл 4-винилпиридина (свежего реагента, хранимого в аргоне при температуре -20оС) и смесь инкубируют еще в течение 2 ч при комнатной температуре в темноте в атмосфере аргона. Затем смесь подвергают обессоливанию, в предпочтительном варианте при помощи хроматографии на короткой обращенно-фазовой колонне. Извлеченный алкилированный полипептид затем анализируют на состав последовательностей при помощи известных приемов.
При осуществлении изобретения и использовании общей процедуры, кратко описанной выше, было установлено, что соответствующей колонной для первоначального фракционирования яда является колонна С-18 Видак®, 22 х 250 мм, размер пор 300


Фракции A, B, G, H, I/J и L подвергают дальнейшей очистке, используя разнообразные колонны и градиентные программы. Специфические данные для каждой подфракции и другие результаты приведены в примерах 3, 4, 6, 7, 8 и 10 помещенных ниже.
Как это показано в следующих примерах, фракции A', А1, А2, В1, В2 и Е содержат полиаминовые соединения.
После того, как описано преимущество соединений, содержащихся во фракциях яда паука Agelenopsis aperta, теперь имеется возможность получать упомянутые соединения с использованием приемов, отличных от изоляции, очистки из полного яда. Например, полиамины, которые содержатся во фракциях А', А1, А2, В1, В2 и Е, могут быть получены непосредственно синтетическими методами. Полипептиды, которые содержатся во фракциях G, H2, I, J, L1, L2 и М, и для которых описана полная последовательность аминокислот, могут быть получены синтетическим способом при помощи синтеза протеинов ин витро в соответствии с известными приемами. Например, такие полипептиды могут быть синтезированы с использованием синтезатора пептидов в твердой фазе АБИ 430А (фирма Апплаед Биосистемз, Инк. 850 Линкольн Центер Драйв, Фостер Сити, Калифорния 94404), используя известные химические приемы Меррифилда или другие химические приемы синтеза в твердой фазе, известные в этой области техники. Полипептиды могут быть также получены с использованием техники рекомбинантной ДНК, при помощи клонирования последовательностей, кодирующих полипептиды или его отдельные части. Те полипептиды, для которых известны только часть аминокислотной последовательности, например, могут быть клонированы через использование зондов гибридизации, которые имеют преимущество на основании известной в настоящее время информации относительно аминокислотных последовательностей. Для получения предлагаемых полипептидов можно также использовать комбинацию приемов рекомбинантной ДНК и синтеза протеинов ин витро.
В этой области техники хорошо известно, что в полипептидах можно осуществить некоторые аминокислотные замещения, которые не нарушают или по существу не нарушают функции упомянутых полипептидов. Всевозможные замещения варьируются от полипептида к полипептиду. Определение допустимых замещений осуществляют в соответствии с процедурами, которые хорошо известны каждому специалисту в этой области техники, Так, все полипептиды, имеющие по существу ту же аминокислотную последовательность и по существу ту же активность по блокировке кальциевого канала, являются предметом изобретения.
Синтетическая схема для
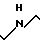
получения некоторых полиаминов изобретения формулы:













в которой R является Н или ОН, приведена в реакционных схемах A-D (см. фиг.1-4).
В соответствии с реакционной схемой А, полиаминовое промежуточное соединение формулы Х получают через последовательность стадий, начиная с диаминобутана. Реакционные условия, соответствующие получению промежуточного соединения формулы Х в соответствии с реакционной схемой А, приведены в примере 11, части А-1. Реакционная схема В служит иллюстрацией способа получения промежуточного соединения формулы XIII. Реакционные условия, соответствующие получению этого промежуточного соединения в соответствии с реакционной схемой В, приведены в примере 11, части I и L. Получение промежуточного соединения формулы XXI показано в реакционной схеме С. Реакционные условия, соответствующие получению соединения формулы XXI в соответствии с реакционной схемой С, приведены в примере 12, части A-F. Получение предлагаемых соединений полиамина формул XVI и XXV указано в реакционной схеме D. Реакционные условия соответствуют соединению промежуточных соединений формул Х и XIII или Х и XXI, и последующему получению соединений формул XVI и XXV, и они приведены в примере 11, части М-О и примере 12, части G-1.
Реакционные схемы Е и F (см. фиг.5-6) иллюстрируют способы получения промежуточных соединений XXVIII и XXXI соответственно. Реакционные условия, соответствующие получению этого промежуточного соединения в соответствии с реакционной схемой Е, приведены в примере 13, части А-С. Реакционные условия, соответствующие получению промежуточного соединения в соответствии с реакционной схемой G (см. фиг.7), приведены в примере 14, части А-С. Получение предлагаемых соединений полиамина формул XXXVI и XXXVII указаны в реакционной схеме G. Реакционные условия, соответствующие соединению промежуточных соединений формул Х и XXXVIII или X и XXXI и последующему получению соединений формул XXXVI и XXXVII, приведены в примере 13, части D-E и примере 14, части D-F соответственно.
Реакционная схема Н (см. фиг.8) иллюстрирует способ получения полиаминовых соединений, являющихся предметом изобретения, имеющих формулу XLI. Реакционные условия, соответствующие получению промежуточного соединения формулы XXXVIII, его соединению с промежуточным соединением формулы XXII и последующему получению полиаминового соединения формулы XLI, указаны в примере 15.
Предлагаемые полиамины обратимо противодействуют возбуждающим аминокислотным нейротрансмиттерам, которые оказывают воздействие на такие клетки, как клетки нервной системы разнообразных микроорганизмов, включая беспозвоночных и позвоночных. Термин позвоночные, как он используется в соответствии с настоящим описанием, включает млекопитающих. Термин беспозвоночные в соответствии с настоящим описанием включает, например, насекомых, эктопаразитов и эндопаразитов. Полиамин фракции В1, описанной выше, также обратимо блокирует кальциевые каналы, содержащиеся в самых разнообразных клетках нервной и мышечной, включая сердечно-сосудистую, систем беспозвоночных и позвоночных.
Способность предлагаемых полиаминов подавлять возбуждающие аминокислотные нейротрансмиттеры подтверждается их способностью блокировать вызванные N-метил-D-аспартоновой кислотой (НМДК) повышения цГМФ в мозжечках новорожденных крыс в соответствии со следующей процедурой. Мозжечки у десяти крыс Вистар в возрасте 8-14 дней быстро вырезают и помещают при температуре 4оС в Креос/бикарбонатный буфер, рН 7,4, а затем разрезают на кусочки размером 0,5 х 0,5 мм, используя ножи для тканей типа МакИлуэин (фирма Де Никл Лэборатори Инжиниеринг Ко. Гомшэлл, Суррей, Англия). Полученные в результате кусочки мозжечка помещают в 100 мл Креос/бикарбонатного буфера при температуре 37оС, который непрерывно уравновешивают смесью 95:5 O2-CO2. Кусочки мозжечка инкубируют таким образом в течение 90 мин с тремя заменами буфера. Затем буфер сливают, ткань центрифугируют (1 мин, 3200 об/мин), а ткань снова суспендирует в 20 мл Креос/бикарбонатного буфера. Затем извлекают порции в 250 мкл (приблизительно 2 мг) и помещают в 1,5 мл микрофуговые пробирки. В эти пробирки добавляют 10 мкл исследуемого соединения из резервуара с раствором, а затем 10 мкл 2,5 ммоль раствора НМДК, чтобы инициировать реакцию. Финальная концентрация НМДК составляет 100 мкмоль. В контрольные пробы не добавляли НМДК. Пробирки инкубируют в течение 1 мин при 37оС в вибрирующей водяной ванне, а затем, чтобы прекратить реакцию добавляют 750 мкл 50 ммоль трис-Cl, 5 ммоль ЭДТК. Немедленно пробирки помещают в ванну с кипящей водой на 5 мин. Содержимое каждой пробирки затем обрабатывают ультразвуком в течение 15 с, используя гомогенизатор проб, установленный на уровне мощности три. Извлекают десять микролитров, а протеин определяют по методу Лоури (Anal. Biochem. т. 100, стр. 201-220, 1979). Далее пробирки центрифугируют (5 мин, 10000 g), извлекают 100 мкл из верхнего слоя, а содержание циклической ГМФ (цГМФ) анализируют, используя РИА-анализ фирмы Нью Инглэнд Нуклеа (Бостон, Массачузетс) на цГМФ по методу фирмы-производителя. Данные приведены в виде пмолей цГМФ, образующейся на мг протеина.
Полипептиды изобретения необратимо блокируют кальциевые каналы, имеющиеся в самых различных клетках, таких как клетки нервной и мышечной системы беспозвоночных и позвоночных.
Способность полиамина фракции В1 и полипептидов фракции G, H1, H2, I, J, K, L1, L2 и М блокировать кальциевые каналы подтверждается при помощи следующей процедуры. Диссоциированные неокортикальные нейроны крысы суспендируют в бикарбонатный буфер Кребса, рН 7,4, содержащий 2 ммоль CaCl2 и 1% альбумина бычьей сыворотки (АБС). Нейроны осаждают центрифугированием и снова суспендируют в тот же буфер, который дополнительно содержит 1 мкмоль фура 2/АМ (фирма Сигма Кем. Ко. Почт. Ящик 14508, Сент-Луис, Миссури 63178). Эти клетки инкубируют в течение 15 мин при 37оС, промывают, а затем инкубируют еще в течение 15 мин в том же буфере без фура 2/AM. Далее клетки промывают в упомянутом буфере, содержащем теперь 1,5 ммоль CaCl2 и уравновешенном при комнатной температуре в форме концентрированной суспензии клеток в течение от примерно 5 до 10 мин. В кварцевую кювету добавляют 1,2 мл предварительно нагретого (37оС), не содержащего АБС, буфера, содержащего 1, 5 ммоль CaCl2 и 10 ммоль глюкозы, затем 0,3 мл указанной концентрированной суспензии клеток. Кювету помещают в термостатированный держатель (37оС), снабженный магнитной мешалкой, а флуоресценцию измеряют при помощи флуоресцентного спектрометра, такого как Перкин Элмер 650-40 (фирма Перкин Элмер, Уилтон, Коннектикут 06897). Флуоресцентным сигналам дают возможность стабилизироваться в течение примерно 1 мин. Затем в кювету добавляют 1-4 мкл раствора соединения, подлежащего изучению, в бикарбонатном буфере Кребса при соответствующих концентрациях. Калибровку флуоресцентных сигналов и коррекцию на утечку фура-2 осуществляют с использованием авторитетных процедур Немата и др. J. Biol. Chem. т. 262, стр. 5188 (1987). При завершении каждого испытания максимальное значение флуоресценции (Fmax) определяли при помощи добавления иономицина, а минимальное значение флуоресценции (Fmin) определяли при помощи последующего добавления 5 ммоль ЭДТК, чтобы кальций превратить в хелат. Используя упомянутую процедуру, тот факт, что кальциевый канал блокируется предлагаемым соединением, устанавливается в результате снижения флуоресценции при добавлении целевого соединения.
Кроме того, предметом изобретения являются полипептиды, содержащие по существу ту же аминокислотную последовательность, что и полипептиды из фракции G, H1, I, J, L1, L2 и М, и которые имеют по существу ту же активность по блокировке кальциевых каналов, что и упомянутые полипептиды. Установлено, что IC50 соединений по изобретению колеблется от 100 нм до 1 мкМ.
Полиамины изобретения можно использовать для противодействия возбуждающим аминокислотным нейротрансмиттерам как таковым. Как таковые, полиамины также эффективны при использовании для борьбы с беспозвоночными вредителями и для лече- ния заболеваний и симптомов у млекопитающих, вызванных возбуждающими аминокислотными нейротрансмиттерами, таких как пароксизм, "удар", церебральная ишемия нейронные дегенеративные расстройства, такие как заболевание Алзеймера и эпилепсия. Такие полиамины эффективны также в качестве психотерапевтических агентов для млекопитающих. Кроме того, такие полиамины можно использовать при исследовании физиологии клеток, включая клетки нервной системы (но не только их).
Полиамины фракции В1 и предлагаемые полипептиды можно использовать в качестве блокаторов кальциевых каналов в клетках. Эти соединения полезны также в борьбе с беспозвоночными вредителями и при лечении заболеваний и симптомов, вызванных нарушением функции кальциевых каналов у млекопитающих, таких как ангина, гипертония, кардиомиопатия, надвентрикулярные аритмии, aesophogeal, ахалазия, преждевременные роды и заболевание Рейно. Кроме того, эти соединения можно использовать при исследовании физиологии клеток, включая клетки нервной и мышечной систем и др.
Кроме того, в области, охватываемой изобретением, содержатся приемлемые с фармацевтической точки зрения соли полиаминов и полипептидов настоящего изобретения. Такие соли получают с использованием известных приемов. Например, присоединенные соли кислот полиаминов и соли оснований полипептидов могут быть получены в соответствии с известными приемами.
Когда предлагаемые полиамин или полипептид должен быть применен к млекопитающему, он может быть применен отдельно или в комбинации с приемлемыми с фармацевтической точки зрения носителями или разбавителями в фармацевтической композиции в соответствии с известной фармацевтической практикой. Полиамины и полипептиды могут быть применены стоматическим способом или парентерально, причем парентеральный способ применения более предпочтителен для полипептидов. Парентеральное применение включает внутривенное, внутримышечное, внутрибрюшинное, подкожное и местное применение.
При стоматическом применении предлагаемых полиамина или полипептида это соединение может быть применено, например, в форме таблеток или капсул, или в форме водных растворов, или суспензии. В случае таблеток для стоматического применения носители, которые в общем случае можно использовать, включают лактозу и кукурузный крахмал, а смазывающие агенты включают стеарат магния. Для стоматического применения в форме капсул эффективными разбавителями являются лактоза и высушенный кукурузный крахмал. Когда для стоматического применения необходимы водные суспензии, активный ингредиент соединяют с эмульгирующими и суспендирующими агентами. Если это необходимо, можно добавлять некоторые ароматизирующие и/или вкусовые агенты.
Для внутримышечного, внутрибрюшинного, подкожного и внутривенного использования в общем случае приготавливают стерильные растворы активного ингредиента, причем рН растворов должно быть соответствующим образом отрегулировано и буферировано. При внутривенном использовании общая концентрация растворенного вещества должна быть отрегулирована, чтобы получить изотонический раствор.
Когда полиамин или полипептид, или его соль изобретения используют для лечения человека, то ежедневную дозу в общем случае определяют по предписанию врача. Кроме того, дозировка будет варьироваться в зависимости от возраста, веса и реакции каждого отдельного пациента, а также от степени серьезности заболевания пациента и эффективности применяемого соединения.
Когда полиамин или полипептид, или его соль используют для борьбы с беспозвоночными вредителями, упомянутое соединение применяют к упомянутым беспозвоночным непосредственно или его применяют к окружающей среде упомянутого беспозвоночного. Например, соединения могут быть разбрызганы в форме раствора на беспозвоночное. Количество соединения, необходимое для борьбы с беспозвоночным, будет зависеть от вида самого беспозвоночного и условий окружающей среды и может быть определено специалистом, применяющим такое соединение.
Когда полиамин или полипептид, или его соль используют для физиологического исследования клеток, соединение применяют к клеткам в соответствии с известными процедурами. Например, соединение может быть применено к клеткам в соответствующем физиологическом буфере. Соответствующая концентрация соединения при использовании в таких исследованиях составляет 100 мкмоль. Однако концентрация соединений в таких исследованиях может быть как большей, так и меньшей 100 мкмоль.
П р и м е р 1. Предварительное фракционирование полного яда паука Agelenopsis aperta.
Полный яд паука Agelenopsis aperta, полученный от фирмы Нэйчурэл Продакт Сайенсиз Инк. Солт Лэйк Сити, Юта 84 108, который хранили в замороженном состоянии при температуре примерно -78оС, оттаивали, и от 10 до 60 мкл его разбавляли до 200 мкл, загружали на колонну С-18 Видак® 22 х 250 мм, 300

Фракция Время элюирования, мин, примерно А 21 В 22,75 Е 27,5 G 38 Н 39 I 40 F 40 L 43 M 48,3
П р и м е р 2. Фракционирование цельного яда паука Agelenopsis aperta с целью получения фракции А'.
Получали цельный яд Agelenopsis aperta и загружали на колонну С-18 Видак®, как это описано в примере 1. Колонну элюировали, используя объемную скорость 15 мл/мин и систему растворителей, используя программу с линейным градиентом 5%__→ 10% В, 95%__→ 90% А [0__→30 мин] где А является 0,1% водным раствором ТФК, а В ацетонитрилом. Обнаружение пиков и сбор фракций осуществляли в соответствии с описанием, приведенным в примере 1. Получали фракцию A', которую элюировали на примерно 12,5 мин. Затем получали фракцию A' для спектрального анализа при помощи лиофилизации из элюента с последующей лиофилизацией из дистиллированной воды в соответствии со стандартными приемами.
Структуру соединения, которое содержится во фракции A', затем определяли при помощи ФАБ МС. Полученные таким образом данные и структура, выведенная из них, имеют следующий
вид:
A':ФАБ МС:M/Z 453 (M+1)
Структура: Бензамид, N-(20-амино-4-окси-4,8,12,17-тетраазейкоз-1-ил)-4-окси.






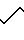





П р и м е р 3. Субфракционирование фракции А и определение в них структур. Фракцию А, полученную в соответствии с примером 1, загружали на колонну типа С-4 Видак (10 х 250 мм, размер пор 300

Фракция Время элюирования, мин,примерно:
А1 7
А2 9
Затем получали фракции А1 и А2 для спектрального анализа при помощи лиофилизации из элюента с последующей лиофилизацией из воды в соответствии с известными приемами.
Структуру соединений, которые содержатся во фракциях А1
и А2, затем определяли при помощи ФАБ МС. Данные, полученные таким образом, и структуры, полученные из них, имеют следующий вид:
A1:
ФАБ МС: M/Z 469 (M+1),
высокое разрешение: 469, 3863, рассчитано для C23H45N6O с погрешностью 2,0 мм. Структура: бензамид, N-(20-амино-4-окси-4,8,12,17-тетраазейкоз-1-ил)-2,5-дио-кси












A2:
ФАБ МС: M/Z 449 (M+1), 433, 376, 260, 203.
Структура:
1Н-индол-3-ацетамид, N-(16-амино-4-оксо-4,8,13- триазагексадец-1-ил)-4-окси










П р и м е р 4. Субфракционирование фракции В и определение в ней структур.
Фракцию В, полученную по примеру 1, загружали на колонну типа С-4 Видак® (22 х 250 мм, размер пор 300

ФАБ МС: M/Z 490 (M+1), 472, 433, 362, 333, 260, 215, 203, 177, 155, 119.
высокое разрешение: 490, 3860, рассчитано для С26Р48N7O2 с погрешностью 0,6 мм
1Н-ЯМР (500 МГц, d6-ДМСО):
10,89 (синглет, 1Н), 8,90-7,85 (мультиплет, 6Н), 7,55 (дублет, J 8,0 Гц, 1Н), 7,34 (дублет, J 8,0 Гц, 1Н), 7,19 (синглет, 1Н, 7,08) двойной дублет, J 8,0 Гц, J 8,0 Гц, 1Н), 6,98 (двойной дублет, J 8,0
Гц, J 8,0 Гц, 1Н), 3,50 (синглет, 2Н), 3,13 (мультиплет, 2Н), 3,03-2,82 (мультиплет, 18Н), 1,88 (мультиплет, 6Н), 1,78 (мультиплет, 2Н), 1,62 (мультиплет, 4Н).
13С-ЯМР (125, 76 МГц, d6-ДМСО): 170,94, 136,10; 127,17; 123,75; 120,92; 118,67; 118,26; 111,33; 108,97; 57,39; 57,07; 46,12; 46,12; 44,06; 43,89; 43,89; 36,52; 36,22; 32,78; 26,71; 23,79; 23,06; 22,65; 22,65; 22, 45.
Структура: 1Н-индол-3-ацетамид, N-(20-амино-4-окси-4,8,12,17-тетраазейкоз-1-ил)











В качестве альтернативы и в предпочтительном варианте цельный яд подвергали фракционированию как это описано в примере 1, за тем исключением, что используемой системой растворителей и программой с линейным градиентом были 0%__→20% В, 100%__→80% А [0__→30 мин] с обнаружением пиков
Фракция: Время элюирования, мин,примерно:
В1 18,5
В2 21,5
Далее получали фракции для спектрального анализа при помощи лиофилизации из воды в соответствии с известными приемами.
Структуру соединения, которое содержится во фракции В1, определяли с использованием ФАБ МС, и получали следующие результаты:
ФАБ МС: M/Z 506 (M+1), 489, 461, 433, 378, 333, 260,
231, 215, 203, 155, 119
высокое разрешение: 506, 3810 рассчитано для C26H48N7O3
Структура: 1Н-индол-3-ацетамид, N-(20-амино-4-окси-4,8,12,
17-тетраазейкоз-1-ил)-4-окси




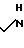



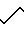

Молекулярную массу соединения, которое содержится во фракции В2, определяли с использованием ФАБ МС, при этом были получены следующие результаты:
ФАБ МС: высокое разрешение 505, 3861, рассчитано для C27H48N6O3.
П р и м е р 5. Структура соединения, содержащегося во фракции Е.
Фракцию Е, полученную по примеру 1, приготавливали для спектрального анализа при помощи лиофилизации из элюента с последующей лиофилизацией из воды в соответствии с известными приемами. Структуру соединения, содержащегося во фракции Е, определяли с использованием в диапазоне 220 нм. Фракцию, которую элюировали на примерно 26,0 мин, загружали на колонну типа Динамакс Фенил (4,6 х 250 мм, 60

П р и м е р 6. Субфракционирование фракции.
Фракцию G, полученную по примеру 1, загружали на колонну типа С-18 Видак (22 х 250 мм, 300

Аминокислотный анализ алкилированного полипептида фракции осуществляли с использованием Пико-Таг-метода Уотерза в соответствии с инструкциями производителя. Данные относительно последовательности собирали при помощи протеин-пептидного анализатора модели 470 А фирмы Апплайед Биосистемз (фирма Апплайед Биосистемз Инк. 850 Линкольн Центер Драйв, Фосетер Сити, Калифорния 94404) с превращением водной ТФК. Анализ полученных в результате фенилтиогидантоиновых аминокислот осуществляли в режиме он лайн при помощи РТН-анализатора модели 120А фирмы Апплайед Биосистемз или в режиме офф лайн на РТН-колонне Дю-Пон Зорбакс (Отдел Биомедицинских Продуктов, Продукты Хроматографии, фирма Е.И.Дюпон де Немюрз и Ко. Инк. 1007 Маркет Стрит, Уилмингтон, Дэлавер 19898).
В результате анализа аминокислот и ФАБ МС полного полипептида было установлено, что амино-концевая
последовательность части полипептида, содержащегося во фракции G, имеет вид:
H2N-glu-lys-gly-leu-pro-glu-gly-ala-glu-cys-asp-
gly-asn-glu-ser-asp-cys-lys-cys-ala-gly-gln-trp-
ile-lys-cys-arg-cys-pro-trp-lys-trp-his-ile-thr-
gly-glu-gly-pro-cys-thr-cys-glu-arg-gly-
leu-lys-lys-thr-cys-ile-ser-lys-leu-ser-
asp-pro-asn-arg-asn-glu-trp-leu-ser;
а ФАБ МС: 7267.
П р и м е р 7. Субфракционирование фракции Н и определение в ней структур.
Фракцию Н, полученную по примеру 1, загружали на колонну типа С-18 Видак® (22 x 250 мм, 300

Фракция: Время элюирования, мин,примерно:
Н2 36
Н3 41
Фракции Н2 и Н3, которые содержат полипептиды, затем подготавливали для исследования последовательности и последовательности определяли в соответствии с процедурами, описанными в примере 6.
Амино-концевая аминокислотная последовательность части полипептида, содержащегося во фракции Н2, имеет следующий вид:
H2
N-ala-lys-ala-leu-pro-pro-gly-ser-
val-cys-asp-gly-asn-glu-ser-asp-cys-lys-
cys-tyr-gly-lys-trp-his-lys-cys-
arg-cys-pro-trp-lys-trp-his-phe-thr-
gly-glu-gly-pro-cys-thr-cys-glu-lys-gly-
met-lys-his-thr-cys-ile-thr-lys-leu-his-
cys-pro-asn-lys-ala-glu-trp-gly-leu-asp-trp-;
а молекулярную массу полного олипептида
определяли при помощи Ион-Струйной МС: 7793.
П р и м е р 8. Субфракционирование фракций I и J, и определение в них структур.
Фракции I и J, полученные по примеру 1, и которые не адекватно разделяются из-за похожих времен элюирования, как это указано в примере 1, загружали вместе на колонну типа С-4 Видак® (22 х 250 мм, размер пор 300

Фракция: Время элюирования, мин,примерно:
I 22
J 27,5
Фракции I и J, которые содержат полипептиды, подготавливали для анализа последовательности, и последовательности определяли в соответствии с процедурами, описанными в примере 6.
Аминокислотная последовательность и ФАБ МС полипептида, содержащегося во фракции I, имели следующий вид:
H2N-asp-cys-val-gly-glu-ser-gln-gln-
cys-ala-asp-trp-ala-gly-pro-his-cys-cys-
asp-gly-tyr-tyr-cys-thr-cys-arg-tyr-
phe-pro-lys-cys-ile-cys-val-asn-asn-CONH2
и ФАБ МС: 4158.
Амино-концевая аминокислотная последовательность части полипептида и ФАБ МС полного полипептида, содержащегося во фракции J имеют следующий вид:
H2
N-asp-glu-pro-cys-ile-pro-leu-gly-lys-
ser-cys-ser-trp-lys-ile-gly-thr-pro-tyr-cys-
cys-pro-his-pro-asp-asp-ala-gly-arg-arg-
thr-trp-cys-leu-val-asp-tyr-ser-arg-phe-
val-thr-ile-cys-ser-gly-arg-lys-tyr-CONH2
а ФАБ МС: 5506.
П р и м е р 9. Субфракционирование фракции L и определение в ней структур.
Фракцию L, полученную по примеру 1, наносили на колонну типа С-18 Видак® (10 х 250 мм, размер пор 300

Фракция Время элюирования, мин, примерно: L1 20,25 L2 22,5
Фракции L1 и L2 , которые содержат полипептид, приготавливали для анализа последовательностей и последовательности определяли в соответствии с приемами, описанными в примере 6.
Амино-концевую
аминокислотную последовательность части полипептида, содержащегося во фракции L1, можно описать следующим образом:
H2N-ile-val-gly-gly-lys-thr-ala-lys-phe-
gly-asp-tyr-pro-trp-met-val-ser-ile-
gln-gln-lys-asn-lys-lys-gly-gly-phe-asp- а молекулярную массу полного полипептида определяли с использованием гелевого электрофореза в соответствии с
известными приемами: примерно 20000.
П р и м е р 10. Определение структуры соединения, содержащегося во фракции М.
Фракцию М, полученную по примеру 1, и которая содержит полипептид, подготавливали для исследования последовательности и последовательность определяли в соответствии с процедурами, описанными в примере 6.
Амино-концевая
аминокислотная последовательность части полипептида, содержащегося во фракции М, имеет вид:
H2N-glu-ala-thr-glu-ala-ala-lys-val-leu-
ser-asn-leu-asp-glu-thr-val-asp-pro- а
молекулярную массу полного полипептида определяли при помощи гелевого электрофореза в соответствии с известными приемами: примерно 80000.
П р и м е р 11. 1Н-индол-3-ацетамид, N-(20-амино-4-окси-4,8,12,17-тетраазейкоз-1-ил.
Синтез соединения из заголовка примера, которое, как было установлено, содержится во фракции Е, в соответствии с описанием, приведенным в примере 4, осуществляли согласно описанию, приведенному ниже.
A. H2N





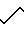


В атмосфере азота 13,22 г (0,15 моль) диаминобутана и 3, 5 мл метанола перемешивали, и через насос шприца в течение 2 ч добавляли 9,94 мл (0,15 моль) акрилонитрила при охлаждении до 0-5оС. Смесь спустя примерно 18 ч подвергали хроматографии на 600 г силикагеля, используя систему растворителей 3:1 CH2Cl2/MeOH (2 л), затем 3:1 CH2Cl2/MeOH, содержащую 5 об. изопропиламина (2 л). Фракции, содержащие соединенный продукт, концентрировали при помощи выпаривания под вакуумом, после чего получали желтое вязкое масло. ЯМР-анализ подтверждал, что продуктом является соединенный продукт формулы I.
B. H2N





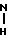
_____→ BOC -





В атмосфере азота 5,78 г (0, 041 моль) соединения формулы I, полученного как это описано в части А, добавляли в 75 мл CH3CN и 11,48 г KF целита, затем перемешивали. В перемешиваемую смесь добавляли раствор 9,75 г (0, 041 моль) соединения формулы II, полученного в соответствии с процедурой, описанной в препарации А, в 25 мл CH3CN.Реакцию нагревали до дефлегмации и анализировали при помощи ТСХ (тонкослойной хроматографии) (3:1 CH2Cl2/MeOH). Спустя 3 ч реакции давали возможность остыть и выдерживали при комнатной температуре. Далее целит отделяли фильтрацией, фильтровальную лепешку хорошо промывали дихлорметаном. Фильтрат выпаривали под вакуумом. Сырой продукт, содержащийся в концентрированном фильтрате, подвергали хроматографии на силикагеле, используя 2 л 3:1 CH2Cl2-MeOH, затем 2 л 3:1 CH2Cl2-MeOH и 10 мл изопропиламина, затем 2 л 3:1 CH2Cl2-MeOH и 30 мл изопропиламина. Продукт, элюированный из колонны после того, как колонна становилась насыщенной изопропиламином, все-таки был с примесями. Все фракции продукта соединяли и концентрировали. Силикагель подготавливали при помощи приготовления шлама 500 г силикагеля в CH2Cl2 и 125 мл изопропиламина, а затем его использовали для снаряжения колонны при помощи известных приемов. Неочищенный продукт наносили на колонну, используя дихлорметан. Затем 500 мл дихлорметана пропускали через колонну, затем 1 л 3:1 CH2Cl2-MeOH. Фракции продукта соединяли и концентрировали под вакуумом, чтобы получить 1,5 г продукта формулы III. Еще 2 г продукта формулы III элюировали из колонны, используя 1 л 3:1 CH2Cl2-MeOH и 30 мл изопропиламина.
C. III + [BOC]2O__→ BOC-




В атмосфере азота 3,5 г (11,8 моль) соединения формулы III, полученного в соответствии с описанием, приведенным в части В, и 150 мл дихлорметана перемешивали, и добавляли 5,2 г (23,6 ммоль) ди-трет-бутилдикарбоната. Смесь перемешивали в течение приблизительно 68 ч при комнатной температуре. Затем смесь концентрировали под вакуумом и подвергали хроматографии, используя 400 г силикагеля и 60:40 гексан-этилацетат в качестве растворителя, чтобы получить 5,62 г соединения формулы IV в виде масла.
D. NC






H2N



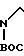


В атмосфере азота 3,0 г соединения формулы IV, полученного в соответствии с описанием, приведенным в части С, растворяли в 30 мл уксусной кислоты в 250 мл колбе Парра®. В этот раствор добавляли 3,0 г Pd(OH2) углерод и смесь подвергали гидрогенизации при давлении Н2 50 фунтов на кв. дюйм (3,5 кг/см2) в течение 2 ч. Катализатор удаляли фильтрацией, а лепешку тщательно промывали уксусной кислотой. Фильтрат концентрировали, переносили в 75 мл дихлорметана, дважды промывали 75 мл 1 н. раствора NaOH и сушили над К2СО3. Раствор концентрировали под вакуумом, чтобы получить 2,86 г продукта формулы V.
Е. V + NC










В атмосфере азота 2,86 г (5,7 ммоль) соединения формулы V, полученного в соответствии с описанием, приведенным в части D, растворяли в 75 мл метанола. При перемешивании 0,41 мл (6,3 ммоль) акрилонитрила добавляли в смесь и реакцию перемешивали в течение ночи. Затем реакционную смесь концентрировали, еще три раза концентрировали из 30 мл дихлорметана, а растворитель отгоняли под вакуумом, чтобы получить 3,18 г соединения формулы VI в виде масла.
F. VI __→NC
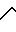






В атмосфере азота 3,18 г (5,7 ммоль) соединения формулы VI, полученного в соответствии с описанием, приведенным в части Е, соединяли с 100 мл дихлорметана и перемешивали в растворе. В этот раствор добавляли 1,37 г (6,3 ммоль) ди-трет-бутилдикарбоната и реакцию перемешивали в течение 90 мин при комнатной температуре. Затем реакционную смесь концентрировали и подвергали хроматографии на 300 г силикагеля, используя 60:40 гексан-этилацетат в качестве элюента. Фракции продукта соединяли, концентрировали, экстрагировали 3 х 20 мл дихлорметана, и растворитель отгоняли под вакуумом, чтобы получить 3,12 г продукта формулы VII.
G. VII ___→H2N



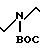


В соответствии с процедурой, описанной в части D, 3,12 г (4,76 ммоль) соединения формулы VII, полученного в соответствии с описанием, приведенным в части F, растворяли в 30 мл уксусной кислоты и подвергали гидрогенизации при давлении 55 фунтов на кв. дюйм (3,867 кг/см2) в течение 2 ч в присутствии 3,0 г Pd(OH2) углерод, чтобы получить 3,02 г продукта формулы VIII в виде масла.








В атмосфере азота 1,0 г (1,5 ммоль) соединения формулы VIII, полученного в соответствии с описанием, приведенным в части G, растворяли в 10 мл метанола. В этот раствор добавляли 0, 1 мл (1,67 ммоль) акрилонитрила и реакцию перемешивали в течение ночи. Далее реакционную смесь концентрировали, еще три раза концентрировали из 20 мл дихлорметана, и растворитель отгоняли под вакуумом, чтобы получить 1,0 г продукта формулы IX.






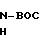
Следуя процедуре, описанной в части D, 1,0 г (1,4 ммоль) соединения формулы IX, полученного в соответствии с описанием, приведенным в части Н, растворяли в 30 мл уксусной кислоты и подвергали гидрогенизации при давлении 50 фунтов на кв. дюйм (3,5 кг/см2) в течение 2,5 ч, чтобы получить 0,85 г продукта формулы Х.






Раствор, содержащий 6,78 г (20 ммоль) кислого сульфата тетрабутиламмония и 75 мл воды, получали при помощи перемешивания, затем добавляли 3,36 г (40 моль) NaHCO3, чтобы получить пену. Затем добавляли 3,5 г (20 ммоль) уксусной кислоты индола, смесь перемешивали 5 мин и добавляли 75 мл CHCl3. Смесь перемешивали еще в течение 5 мин и два нижних слоя разделялись. Водный слой насыщали солью сульфата натрия и экстрагировали при помощи CHCl3. Все CHCl3-экстракты соединяли и сушили над сульфатом натрия. Полученный в результате прозрачный, янтарного цвета, раствор концентрировали, вытесняли 3х75 мл ацетона и, наконец, растворяли в 75 мл ацетона. Затем добавляли 1,9 мл (22 ммоль) аллилбромида и смеси давали возможность отстояться в течение 30 мин. Затем смесь концентрировали и подвергали хроматографии на силикагеле, используя 3:1 гексан-этилацетат, чтобы получить 3,57 г продукта формулы XI в виде ярко-желтого масла.
K XI _________→



Следуя процедуре из Angew. Chem. Int. Ed. Engl. т.23, стр.298 (1984), 2,15 г (10 ммоль) соединения формулы XI, полученного в соответствии с описанием, приведенным в J, соединяли с 20 мл ацетонитрила и в смесь добавляли 2,61 г (12 ммоль) ди-трет-бутилдикарбоната. Затем добавляли 0,122 г (1 ммоль) 4-(N,N-диметиламино)пиридина и реакции давали возможность продолжаться в течение 15 мин. Затем смесь разбавляли до 125 мл этилацетатом, промывали 2 х разбавленной HCl, 3 х 25 мл воды, 1 х соляным раствором, сушили и концентрировали. Продукт подвергали очистке при помощи хроматографии на силикагеле, используя 14,5:1 гексан-этилацетат в качестве элюента, чтобы получить 2,87 г соединения формулы XII, в виде бесцветного масла.
L. XII ______→
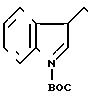

В 3,27 г (10,4 ммоль) соединения формулы XII, полученного в соответствии с частью К, в 20 мл дихлорметана добавляли 7,4 мл 2-этилгексаноата натрия в этилацетате (0,231 г/мл). Затем добавляли 0,5 г трифенилфосфина и 0,5 г трифенилфосфатпалладия, и смесь перемешивали 60 мин. Смесь затем концентрировали, переносили в 150 мл этилацетата и промывали 5 х 25 мл воды. Водные экстракты соединяли и снова экстрагировали 1 х 25 мл этилацетата и 1 х 25 мл диэтилового простого эфира. В водный слой добавляли 75 мл свежего этилацетата, а затем подкисляли до рН 3. Слой этилацетата отделяли, а водный слой экстрагировали при помощи 1 х 50 мл свежего этилацетата. Два этилацетатных экстракта соединяли и промывали 2 х 20 мл воды, затем 1 х 20 мл соляного раствора, сушили, концентрировали, вытесняли с использованием 3 х 30 мл гексана, в котором образуются кристаллы во время второго "вытеснения". Смесь разбавляли до 75 мл гексаном. Твердые частицы отделяли фильтрацией, тщательно промывали гексаном и сушили воздухом, при этом получали 1,37 г белого твердого вещества. При помощи ЯМР подтверждали структуру формулы XIII.





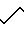
В атмосфере азота 200 мг (0,73 ммоль) соединения формулы XIII соединяли с 25 мл дихлорметана, 84 мг (0, 73 ммоль) N-оксисукцинимида и 150 мг (0,73 ммоль) дициклогексилкарбодиимида, при этом почти сразу выпадал осадок. Смесь перемешивали в течение 3 ч. Далее дициклогексилмочевину отделяли фильтрацией и в фильтрат по каплям добавляли 40 мл раствора соединения формулы Х, полученного в соответствии с описанием, приведенным в примере 11, части А-1, в дихлорметане. Смесь перемешивали 12 ч, затем промывали 0,1% NaOH и сушили над К2СО3, концентрировали и подвергали хроматографии, используя 150 г силикагеля и элюируя при помощи смеси 9:1 дихлорметана-метанола (0,5 л), чтобы удалить примеси, затем смесью 9:1:0,1 дихлорметана-метанола-изопропиламина (2 л). Затем продукт концентрировали, вытесняли дихлорметаном, а затем еще раз концентрировали, чтобы получить 400 мг соединения формулы XIV, что подтверждали при помощи ЯМР 300 МГц.










В атмосфере азота 0,35 г (0,35 ммоль) соединения формулы XIV, полученного в соответствии с приведенным описанием, соединяли с 30 мл ацетона, перемешивали в растворе и охлаждали до примерно 0о С в ванне лед-ацетон. Затем по каплям в течение 8-10 мин добавляли 0,212 г (1,05 ммоль) 85% раствора 3-хлорпероксибензойной кислоты в 20 мл ацетона. Реакцию перемешивали в течение 30 мин, затем разбавляли до 150 мл диэтиловым простым эфиром и промывали 2 х 25 мл 10% К2СО3, 2 х 25 мл воды, 2 х 25 мл 10% К2СО3, 2 х 25 мл воды и сушили над К2СО3. Смесь концентрировали, вытесняли 2 х 30 мл дихлорметана и концентрировали под вакуумом до стабильного веса в 0,33 г. ЯМР-анализ показал, что продукт является смесью целевого продукта формулы XV, исходного материала и побочного продукта. Смесь продуктов добавляли в 3 мл уксусной кислоты, в которую добавляли 20 мг NaCNBH3. Смесь перемешивали в течение ночи, уксусную кислоту отгоняли под вакуумом, растворяли в 50 мл дихлорметана и промывали 1 х 75 мл водного буфера, рН 7. Если необходимо, рН регулировали до рН 7 при помощи 1 н. NaOH, а слой дихлорметана отделяли и промывали 1 х 50 мл водного буфера, рН 7 и 1 х 25 мл соляного раствора, сушили над сульфатом натрия и концентрировали под вакуумом до стабильного веса 0,31 г. Полученный в результате продукт подвергали хроматографии на 100 г силикагеля, используя смесь 95:5 этил ацетат-метанол для элюирования и сбора 25 л фракции. Очищенный продукт формулы XV извлекали из фракции 18-25. Все эти фракции соединяли, концентрировали, вытесняли 2 х 5 мл дихлорметана и концентрировали под вакуумом до белой пены весом 68 мг, которую хранили при температуре -80оС. Фракции 26-40, которые также содержали целевой продукт, содержат также некоторые примеси. Фракции 26-40 соединяли, концентрировали, вытесняли дихлорметаном и концентрировали под вакуумом до стабильного веса в 66 мг. Последний продукт соединяли с 6 мг первого продукта, который ранее использовали для ЯМР-исследований, и со смесью, которую получали в соответствии с описанной процедурой. Полученную в результате смесь подвергали хроматографии на 75 г силикагеля, используя смесь 9:5 этил ацетат-метанол, чтобы элюировать и собрать 25 мл фракции. Очищенный продукт формулы XV содержался во фракциях 16-12. Все эти фракции соединяли, концентрировали, вытесняли 2 х 5 мл дихлорметана и концентрировали под вакуумом до стабильного веса в 46 мг, который хранили при -80оС.










В атмосфере азота 108 мг (0,109 ммоль) соединения формулы XV, полученного в соответствии с приведенным описанием, растворяли в 1,5 мл дихлорметана. Далее добавляли 2 мл трифторуксусной кислоты и смесь перемешивали в течение 1 ч при комнатной температуре. Реакционную смесь затем концентрировали до пленки и добавляли 15 мл диэтилового простого эфира. Пленка превращалась в твердое вещество, а через 30 мин перемешивания давала белый порошок. Порошок отделяли фильтрацией, тщательно промывали диэтиловым эфиром, сушили азотом, затем обрабатывали под вакуумом до стабильного веса в 109 мг названного соединения.
П р и м е р 12. 1Н-индол-3-ацетамид, N-(20-амино-4-окси-4,8,12,17-тетраазеикоз- 1-ил)-4-окси.
Синтез соединения, которое, как было установлено, содержится во фракции В1, как это описано в примере 3, осуществляли в соответствии с приведенным ниже описанием.



В атмосфере аргона, используя высушенную на пламени химическую посуду, 4,0 г (30 ммоль) 4-оксииндола соединяли с 25 мл сухого ДМФ Олдрич, и перемешивали в растворе. Затем добавляли 1,44 г (30 ммоль) NaOH в виде 50% масляной дисперсии. После того, как пена оседала, добавляли 2,5 мл хлорметилметилового простого эфира (Олдрич) и полученный в результате темно-зеленый раствор перемешивали в течение ночи. Далее раствор разбавляли до 125 мл этилацетатом, промывали 5 х 25 мл воды и 1 х 25 мл соляного раствора, сушили над сульфатом натрия и подвергали хроматографии, используя смесь 4:1 гексана-этилацетата. Фракции, содержащие продукт, соединяли и концентрировали, чтобы получить 2,70 г соединения XVII в виде масла.




В 50 мл 3-горлой колбе с круглым дном, снабженной механической мешалкой и атмосферой азота, охлаждали 1,3 г водного формальдегида (37%) и 2,28 г уксусной кислоты. Затем добавляли 3,23 мл охлажденного диметиламина (25% в воде). В полученный в результате охлажденный раствор добавляли 2,7 г (15,2 ммоль) соединения формулы XVII, используя примерно 1,5-2,0 мл тетрагидрофурана в качестве растворителя-носителя. Реакцию нагревали до комнатной температуры и перемешивали в течение ночи. Далее добавляли 40 мл 10% NaOH, после чего выпадал осадок. После перемешивания осадок отделяли фильтрацией, промывали водой и сушили воздухом, чтобы получить 3,26 г соединения формулы XVIII.


В атмосфере азота 4,62 г KCN соединяли с 10 мл воды и перемешивали до получения раствора, затем добавляли 30 мл этанола и 3,26 г (14 ммоль) соединения формулы XVIII, и смесь перемешивали и нагревали до дефлегмации на примерно 65 ч. Реакцуию охлаждали и концентрировали. Полученный в результате осадок, содержащий соединение ХХ, отделяли фильтрацией, промывали водой и сушили воздухом. Водный слой экстрагировали 4 х 15 мл дихлорметана, сохраняя органические экстракты. Водный слой затем дополняли 50 мл этил ацетата и подкисляли до рН 2. Слой этилацетата отделяли, удаляли газ при помощи барботажа азота через слой этилацетата, сушили и концентрировали до твердого вещества, которое растирали с диизопропиловым простым эфиром, чтобы получить 1,03 г соединения XIX в виде не совсем белого твердого вещества. Далее все экстракты из щелочного водного слоя соединяли, сушили, соединяли с фильтратами из описанной выше стадии, и концентрировали, чтобы получить 0,4 г соединения ХХХ в виде твердого вещества.
D. XX _____→ XIX
В атмосфере азота 1,6 г соединения ХХ
соединяли с 7,5 мл этанола, 30 мл воды и 1,3 г КОН. Смесь нагревали до дефлегмации и перемешивали в течение ночи. Затем реакцию охлаждали, экстрагировали 2 х 15 мл этил ацетата, доливали свежий
этилацетат и подкисляли до рН 2. Соединенные слои этилацетата сушили и концентрировали. Полученное в результате твердое вещество растирали с ИПЭ, чтобы получить 1,1 г соединения XIX.
E.




В 10 мл воды растворяли 1,44 г (4,25 ммоль) ТВАН SO4. Далее добавляли 714 мг (8,5 ммоль) NaHCO3. После того как пена оседала, добавляли 1,0 г (4,25 ммоль) соединения формулы XIX и смесь перемешивали в течение 1 мин перед тем, как добавить 40 мл хлороформа. Реакционную смесь перемешивали в течение 5 мин, слой хлороформа отделяли, а затем водную смесь экстрагировали 1 х 15 мл хлороформа. Соединенные экстракты хлороформа сушили, концентрировали и вытесняли 2 х 30 мл ацетона. Далее продукт снова растворяли в 30 мл ацетона в атмосфере азота, добавляли 0,37 мл (4,25 ммоль) аллилбромида и реакцию перемешивали в течение 2 ч. Смесь концентрировали и подвергали хроматографии, используя этилацетат в качестве элюента. Продукт концентрировали, чтобы получить 1,1 г масла. Весь продукт соединяли с 25 мл дихлорметана и добавляли 0,98 г (4,5 ммоль) ди-трет-бутилдикарбоната и 51 мг (0,425 ммоль) 4-(N, N-диметиламино)пиридина. Реакцию перемешивали в течение ночи. Затем реакционную смесь концентрировали и подвергали хроматографии, используя смесь 9: 1 гексана-этилацетата, чтобы получить после концентрирования 1,33 г соединения формулы XXI в виде вязкого масла.
F.




В 2 мл метанола растворяли 64 мг (0,17 ммоль) соединения формулы XXI. В этот раствор добавляли 1,7 мл 0,1 NaOH, а полученную в результате реакционную смесь перемешивали в течение ночи. Затем реакцию подкисляли, сушили, экстрагировали этилацетатом и концентрировали. ЯМР-анализ показывал, что весь аллиловый сложный эфир соединения XXI был удален, но при этом имела место некоторая трансэтерификация и содержалось некоторое количество метилового сложного эфира. Поэтому продукт соединяли с оставшимся количеством соединения XXI (приблизительно 1,26 г), растворенного в 5 мл тетрагидрофурана, и охлаждали до 0оС. Далее добавляли 3,8 мл 1 н. раствора NaOH, реакцию перемешивали в течение 5 мин при 0оС, затем ей давали возможность нагреться до комнатной температуры. Реакция приводила к образованию двух фаз, что требовало добавления 5 мл метанола, чтобы исключить фазы. Реакцию перемешивали в течение ночи. Затем тетрагидрофуран и метанол отгоняли, а полученный в результате водный раствор дополняли 50 мл этилацетата и подкисляли до рН 2. Слой этилацетата отделяли, промывали 1 х 20 мл воды, один раз соляным раствором, сушили над сульфатом натрия, фильтровали и концентрировали в смолу, которая кристаллизовалась, когда ее растирали с гексаном, при этом получали 1,08 г белого твердого вещества. Это твердое вещество подвергали хроматографии, используя смесь 70:30 этилацетата-гексана и чистые фракции продукта соединяли, концентрировали и кристаллизовали из этилацетата-гексана, чтобы получить 0,884 г соединения формулы XXII.




















В атмосфере азота 271 мг (0,81 ммоль) соединения формулы XXII растворяли при перемешивании в 4 мл безводного тетрагидрофурана. Затем добавляли 93 мг (0,81 ммоль) N-оксисукцинимида и 167 мг (0,81 ммоль) дициклогексилкарбодиимида, и смесь перемешивали. Спустя 1 ч и 50 мин добавляли 0,58 г (0,8 ммоль) соединения формулы Х, полученного в соответствии с процедурой, описанной в примере 11, в 30 мл дихлорметана и смесь перемешивали в течение уикенда. Далее дициклогексилмочевину отделяли фильтрацией, и смесь экстрагировали 2 х 3 мл 1 н. раствора NaOH, сушили над К2СО3 и концентрировали. Далее продукт подвергали хроматографии на 100 г силикагеля, используя смесь 9:1 дихлорметана-метанола, чтобы элюировать все вместе с целевым продуктом, затем смесь 90:10:1 дихлорметана-метанола-изопропиламина, чтобы элюировать сам продукт. Фракции, содержащие продукт, концентрировали, вытесняли несколько раз дихлорметаном и отгоняли растворитель, чтобы получить 349 мг соединения формулы XXIII в виде белой пены.
В качестве альтернативы, и в предпочтительном варианте 50 мл раствора, содержащего 0,480 г (1,43 ммоль) соединения формулы XXII в дихлорметане обрабатывали при помощи 0,165 г (1,43 ммоль) N-оксисукцинимида, а затем 0,294 г (1,43 ммоль) дициклогексилкарбодиимида, затем перемешивали в течение 5 ч. Реакционную смесь фильтровали, а фильтрат, содержащий неочищенный оксисукцинимид, добавляли по каплям в течение 20 мин в 100 мл раствора 1,03 г (1,43 ммоль) соединения формулы Х, полученного в соответствии с описанием, приведенным в примере 11, в дихлорметане. Реакцию перемешивали в течение 72 ч. Затем неочищенную реакционную смесь дважды промывали 20 мл 1 н. раствора NaOH, сушили над К2СО3, фильтровали и концентрировали. Далее продукт подвергали хроматографии на силикагеле, используя смесь 4:1 дихлорметана-метанола, затем смесь 9:1:1 дихлорметана-метанола-диизопропиламина, чтобы получить 912 мг соединения формулы XXIII.


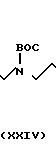





В атмосфере азота индолацетамид XXIII (900 мг, 0,87 ммоль) растворяли в 40 мл дихлорметана. В этот раствор добавляли 2-(фенилсульфонил)-3-фенилоксазиридин (500 мг, 1,92 ммоль). Течение реакции наблюдали при помощи тонкослойной хроматографии. Через 1 ч реакционную смесь концентрировали под вакуумом, чтобы получить главным образом неочищенный нитрон, который растворяли в 35 мл уксусной кислоты. Добавляли большой избыток цианоборогидрида натрия (1,00 г, 15,9 ммоль) и реакцию перемешивали в течение примерно 2 ч. Реакцию концентрировали под вакуумом, переносили в дихлорметан (50 мл), промывали буфером рН 7 (1 х 50 мл), обеспечивая рН 7 при помощи 1 н. раствора NaOH. Органический слой снова промывали буфером рН 7 (1 х 50 мл), сушили над карбонатом калия и концентрировали под вакуумом, чтобы получить сырой продукт, который подвергали хроматографии на силикагеле, используя смесь 95: 5 этилацетата-метанола, чтобы получить 612 мг (67%) соединения формулы XXIV в виде белой пены.






В качестве альтернативы, создавая вакуум и заполняя аргоном три раза, подвергали дегазации 3 мл уксусной кислоты. Далее колбу заворачивали в алюминиевую фольгу и добавляли 98 мг соединения XXIV с использованием примерно 2,5 мл дихлорметана в качестве "несущего" агента. Спустя 45 мин смесь концентрировали, чтобы получить масло, которое после растирания с диэтиловым простым эфиром давало 88 мг соединения XXV в виде белого твердого вещества, которое хранили при температуре -80оС.
В качестве альтернативы и в предпочтительном варианте в насыщенные 100 мл раствора диоксана-HCl, продутые аргоном, добавляли в течение 1,5 мин 0,520 г (0,495 ммоль) соединения формулы XXIV, полученного в соответствии с процедурой, описанной в части Н, в 10 мл диоксана. Сразу же образовывался осадок и реакцию перемешивали 1 ч. Далее добавляли диэтиловый простой эфир, а через 5 мин раствор фильтровали. Полученные в результате твердые частицы промывали диэтиловым простым эфиром, сушили в атмосфере азота, а затем при пониженном давлении, чтобы получить 345 мг кислоты формулы XXIV', которую можно хранить при температуре -78оС. Затем в водный раствор, продутый аргоном, добавляли 150 мг (0,205 ммоль) кислоты формулы XXIV'. Реакцию наблюдали при помощи ВЭЖХ. После завершения реакции примерно спустя 7 ч реакционную смесь сушили вымораживанием, чтобы получить 103 мг соединения формулы XXV в виде хлоргидратной соли.
П р и м е р 13. Бензамид, N-(20-амино-4-окси-4,8, 12,17- тетраазейкоз-1-ил)-4-окси.
Синтез соединения, которое содержится во фракции A', как это описано в примере 2, осуществляли следующим образом.


В 400 мл химическом стакане 6,78 г (20 ммоль) кислого сульфата тетрабутиламмония соединяли с 60 мл воды и этот раствор перемешивали. Далее добавляли 3,36 г (40 ммоль) NaHCO3, а после образования пены добавляли 40 мл раствора; содержащего 2,76 г (20 ммоль) пара-оксибензойной кислоты, 0,8 г NaOH и 40 мл воды. Реакционную смесь перемешивали 5 мин, а затем добавляли 200 мл дихлорметана. Эту смесь перемешивали еще в течение 5 мин и слоями давали возможность разделиться. Водный слой экстрагировали дважды 50 мл дихлорметана. Соединенные экстракты дихлорметана сушили над сульфатом натрия, концентрировали, дважды вытесняли 75 мл ацетона и снова растворяли в 50 мл ацетона. Далее добавляли 1,9 мл (22 ммоль) аллил бромида и смесь перемешивали, затем реакционную смесь концентрировали, переносили в 100 мл этилацетата, дважды промывали 30 мл воды, один раз 50 мл насыщенного бикарбоната, один раз 30 мл воды, один раз соляным раствором, один раз водой и один раз соляным раствором. Полученный в результате продукт сушили и концентрировали, чтобы получить масло. Масло растирали с 50 мл петролейного эфира в течение 1 ч. Далее смесь фильтровали, промывали тщательно петролейным эфиром и сушили воздухом, чтобы получить 1,7 г соединения формулы XXVI.


В атмосфере азота 1,7 г (9,55 ммоль) соединения формулы XXVI, полученного в соответствии с описанием, приведенным в части А, соединяли с 50 мл сухого ДМФ (Олдрич) и перемешивали в растворе. Затем добавляли 0,38 г (9,55 ммоль) NaH в виде 60% масляной дисперсии и реакцию перемешивали в течение ночи. Далее добавляли 0,76 мл хлорметилметилового простого эфира (Олдрич), при этом сразу же выпадал осадок. Реакционную смесь перемешивали в течение 18 ч. Реакцию быстро прекращали при помощи 200 мл этилацетата (100 мл воды). Слои этилацетата отделяли, промывали 3 раза 50 мл воды, один раз 50 мл 1н. раствора NaOH, один раз 50 мл воды и один раз соляным раствором. Полученный в результате раствор сушили, концентрировали и подвергали хроматографии на силикагеле, используя смесь 4:1 гексана-этилацетата, чтобы получить 1,63 г соединения формулы XXVII.


В 75 мл тетрагидрофурана растворяли 1,62 г (7,3 ммоль) соединения формулы XXVII, полученного в соответствии с процедурой, описанной в части В. Далее, добавляли раствор, содержащий 0,4 г (10 ммоль) NaOH в 15 мл воды, что приводило к образованию двух слоев. Добавляли метанол до тех пор, пока не получится однородная смесь. Затем реакцию перемешивали в течение ночи. Тетрагидрофуран и метанол отгоняли под вакуумом, а оставшийся водный раствор экстрагировали дважды при помощи 15 мл этилацетата. Затем водный слой накрывали 50 мл свежего этилацетата и подкисляли до рН 2,5 при помощи 6 н. раствора HCl. Далее слой этилацетата отделяли, промывали один раз 10 мл воды и один раз 25 мл соляного раствора. Полученный в результате раствор концентрировали, чтобы получить твердые частицы, которые растирали с гексаном, фильтровали и сушили воздухом, чтобы получить 1,2 г соединения формулы XXVIII.






20 мл дихлорметанового раствора, содержащего 0,127 г (0,7 ммоль) соединения формулы XXVIII, полученного в соответствии с описанием, приведенным в части С, обрабатывали 0,081 г (0,7 ммоль) N-оксисукцинимида, а затем 0,144 г (0,7 ммоль) дициклогексилкарбодиимида, а затем перемешивали 4 ч. Затем реакционную смесь фильтровали, а фильтрат, содержащий сырой оксисукцинимид, по каплям добавляли в течение 20 мин в 80 мл раствора в дихлорметане, содержащего 0,50 г (0,7 ммоль) соединения формулы Х, полученного в соответствии с описанием, приведенным в примере 11, части A-I. Далее реакцию перемешивали, затем реакционную смесь промывали 1 н. раствором NaOH (2 х 20 мл), сушили над карбонатом калия, фильтровали и концентрировали. Концентрат подвергали хроматографии на силикагеле, используя смесь 9:1 дихлорметана-метанола, затем смесь 9:1:0,25 дихлорметана-метанола-диизопропиламина, чтобы получить 370 мг соединения формулы XXXII.







В атмосфере азота 0,335 г (0,38 ммоль) соединения формулы XXXII, полученного в соответствии с описанием, приведенным в части D, растворяли в 30 мл дихлорметана. В этот раствор добавляли 0,230 г (0,88 ммоль) 2-(сульфонилфенил)-3-фенилоксазиридина. Спустя 0,5 ч реакцию концентрировали под вакуумом, а концентрат растворяли в 15 мл уксусной кислоты. Добавляли большой избыток цианоборогидрида (0,100 г, 1,6 ммоль) и реакцию перемешивали 2 ч. Затем реакцию концентрировали под вакуумом, переносили в 50 мл дихлорметана, промывали один раз 50 мл буфера рН 7, регулируя рН на уровне 7 при помощи 1 н. NaOH. Органический слой снова промывали 50 мл буфера рН 7, сушили над карбонатом калия и концентрировали под вакуумом. Концентрат подвергали хроматографии на силикагеле, используя смесь 50:50 ацетона-гексана, чтобы получить 253 мг соединения формулы XXXIV.






В атмосфере аргона при комнатной температуре соединяли 0,20 г (0,223 ммоль) соединения формулы XXXIV, полученного в соответствии с процедурой, описанной в части Е, и 5 мл трифторуксусной кислоты. Реакцию перемешивали 1,5 ч. Добавляли дополнительно 2 мл трифторуксусной кислоты, чтобы промыть стенки колбы. Реакции давали возможность продолжаться еще в течение 1,5 ч. Далее реакцию концентрировали под вакуумом, растирали этиловым простым эфиром, фильтровали и сушили в атмосфере азота, чтобы получить 218 мг соединения формулы XXXVI.
П р и м е р 14. Бензамид, N-(20-амино-4-окси-4, 8,12,17-тетраазейкоз-1-ил)-2,5-диок- си.
Синтез соединения, которое содержится во фракции А1, как это описано в примере 3, осуществляли следующим образом:
A.




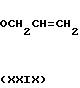
Используя процедуру, описанную в примере 12, часть А, но исходя из 3,08 г (20 ммоль) 2,5-диоксибензойной кислоты, получали 3,0 г соединения формулы XXIX.
B. XXIX ______→

В атмосфере азота 3,0 г (15,5 ммоль) соединения формулы XXIX соединяли с 50 мл сухого ДМФ (Олдрич) и перемешивали в растворе. Затем добавляли 1,24 г (31 ммоль) NaH в виде 60% масляной дисперсии, и смесь перемешивали. Спустя 3 ч добавляли 2,5 мл (33 ммоль) хлорметилметилового простого эфира и реакцию перемешивали в течение 18 ч. Затем эту реакционную смесь добавляли в 300 мл этилацетата и 100 мл воды. Слой этилацетата затем отделяли и промывали 3 раза 75 мл воды, дважды 75 мл 1 н. раствора NaOH, дважды 50 мл воды и один раз соляным раствором. Полученный в результате раствор сушили и концентрировали, чтобы получить 2,9 г масла, которое подвергали хроматографии на силикагеле, используя смесь 4:1 гексана-этилацетата, чтобы получить две фракции, причем вторая фракция содержит 1,16 г соединения формулы ХХХ.
C. XXX ________→

В 10 мл тетрагидрофурана добавляли 1,16 г (4,1 ммоль) соединения формулы ХХХ. Затем добавляли 0,24 г (6 ммоль) NaOH в 10 мл воды, что приводило к образованию двух фаз. В смесь добавляли метанол до тех пор, пока не будет получена одна фаза, а затем смесь перемешивали в течение 18 ч. Далее растворители удаляли, а оставшийся водный раствор дважды экстрагировали 15 мл этилацетата. Затем водный слой наполняли 35 мл свежего этилацетата, а рН обеспечивали на уровне 2,5 при помощи 6 н. раствора HCl. Затем слой этилацетата отделяли, промывали один раз 10 мл воды и один раз соляным раствором, сушили и концентрировали, чтобы получить 1,0 г соединения формулы XXXI в виде масла.






Используя процедуру, описанную в примере 13, часть D, и используя 0,17 г (0,7 ммоль) соединения формулы XXXI, полученного в соответствии с описанием, приведенным в части С, 0,081 г (0,7 ммоль) N-оксисукцинимида, 0,144 г (0,7 ммоль) дициклогексилкарбодиимида и 0,50 г (0,7 ммоль) соединения формулы Х, полученного в соответствии с процедурой, описанной в примере 11, части A-I, получали после хроматографии 370 мг соединения формулы XXXIII.







Используя процедуру, описанную в примере 13, часть Е, и используя 0,308 г (0,33 ммоль) соединения формулы XXXIII, полученного в соответствии с процедурой, описанной в части D, и 0,250 г (0,96 ммоль) 2-(сульфонилфенил)-3-фенилоксазиридина, получали после хроматографии на силикагеле, используя этилацетат, затем смесь 50:50 ацетона-гексана, 210 мг соединения формулы XXXV.









Используя процедуру, описанную в примере 13, часть F, и используя 0,100 г (0,104 ммоль) соединения формулы XXXV, полученного в соответствии с процедурой, описанной в части Е, получали 108 мг соединения формулы XXXVII.
П р и м е р 15. 1Н-индол-3-ацетамид, N-(16-амино-4-окси-4,8, 13-триазагексадец- 1-ил)-4-окси.
Синтез соединения, которое содержится во фракции А2, как это описано в примере 3, осуществляли следующим образом:






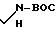
Используя процедуру, описанную в примере 11, часть D, используя 1,15 г (2,07 ммоль) соединения формулы VI, полученного как это описано в примере 11, части А-Е, получали 1,10 г неочищенного соединения формулы XXXVIII.



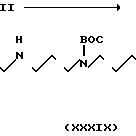



Используя альтернативную процедуру, описанную в примере 12, часть G, и используя дихлорметановый раствор (15 мл), содержащий 0,228 г (6,8 ммоль) соединения формулы XXII, полученного как это описано в примере 12, части А-F, 0,380 г (6,8 ммоль) соединения формулы XXXVIII, полученного как описано в части А, 0,078 г (6,8 ммоль) N-оксисукцинимида и 0,140 г (6,8 ммоль) дициклогексилкарбодиимида, получали 0,407 г соединения формулы XXXIX после хроматографии на силикагеле, используя смесь 9:1 дихлорметана-метанола, затем смесь 9:1:0,5 дихлорметана-метанола- диизопропиламина.







Используя процедуру, описанную в примере 12, часть Н, и используя 0,350 г (0,4 ммоль) соединения формулы XXXIX и 0,230 г (0,88 ммоль) 2-(сульфонилфенил)-3-фенилоксизиридина, получали 0,274 г соединения формулы XL, после хроматографии на силикагеле, используя смесь 50:50 ацетона-гексана. Продукт, содержащий небольшое количество фенилсульфонамида, далее еще раз подвергали очистке на силикагеле, используя этилацетат, затем смесь 50: 50 ацетона-гексана.






Используя альтернативную процедуру из примера 12, часть I, и используя 0,166 г (0,186 ммоль) соединения формулы XL, полученного в соответствии с процедурой, описанной в части С, получали после обработки диоксаном-HCl неочищенную кислоту. Кислоту формулы XL растворяли в воде на 8 ч, затем сушили вымораживанием, чтобы получить 88 мг соединения формулы Х
Препарация А.
Br



В атмосфере азота перемешивали 34,5 г (157,6 ммоль) 3-бромпропиламина. HBr в 600 мл N,N-диметилформамида. В этот раствор добавляли 34,4 г (157,6 ммоль) ди-трет-бутилдикарбоната, затем 32,3 мл (236 ммоль) триэтиламина. Немедленно образовывался осадок. Реакцию перемешивали в течение ночи. Затем реакционную смесь разбавляли до 1,5 л этилацетатом, промывали один раз 500 мл 1 н. раствора HCl, три раза 500 мл воды, один раз соляным раствором и сушили над сульфатом натрия. После концентрирования продукт подвергали хроматографии на 800 г силикагеля, используя смесь 4:1 гексана-этилацетата, а фракции анализировали при помощи тонкослойной хроматографии (KMNO4/12). Фракции, содержащие продукт, соединяли, концентрировали под вакуумом, дважды вытесняли 50 мл дихлорметана и обрабатывали под высоким вакуумом, чтобы получить 25,8 г продукта этой препарации.
Реферат
Использование: в медицинской промышленности для получения полипептидов, которые могут найти применение в биохимии и медицине. Сущность изобретения: по изобретению получают полипептиды из яда паука Agelenopsis aperta колоночной хроматографией на колонке типа с-18 Видак®. 8 ил.
Формула
A Pro B,
где I. A H2N Glu Lys Gly Leu Pro Glu Gly Ala - Glu Gys Asp Gly Asn Glu Ser Asp Cys Lys Cys Ala Gly Gln Trp Jle Lys Cys Ary Cys Pro Trp Lys Trp His Jle Thr Gly Glu Gly Pro Cys Thr Cys Glu Arg Gly Leu [Lus]2 Thr Cys Jle Ser Lys Leu Ser Asp -;
B Asn Arg Asn Glu Trp Leu Ser OH,
или II
A H2N Asp Cys Val Gly Glu Ser Gln Gln Cys Ala Asp Trp Ala Gly Pro His /Gys/2 Asp Gly /Tyr/2 Cys Thr Cys Arg Tyr Phe-;
B Lys Cys Jle Cys Val /Asn/3CONH2,
или
III. A H2N Ala Lys Ala Leu /Pro/2 Gly Ser - Val Cys Asp Gly Asn Glu Ser Asp Cys Lys Cys Tyr Gly Lys Trp His Lys Cys Arg Cys Pro Trp Lys Trp His Phe
Thr Gly Glu Gly Pro Cys Thr Cys Glu Lys Gly Met Lys - His Thr Cys Jle Thr Lys Leu His Cys -;
B Asn Lys Ala Glu Trp Gly Leu Asp TprOH,
или
IV. A H2N Asp Glu Pro Cys Jle Pro Leu Gly Lys - Ser Cys Sep Trp Lys Jle Gly Thr Pro Tyr /Cys/2 Pro His -;
B [Asp]2 Ala Gly [Arg]2 Thr Trp Cys Leu - Val Asp Tyr Ser Arg Phe Val Thr Jle Cys Ser Gly Arg Lys Tyr CONH2,
или
V. A H2N Jle Val [Gly]2 Lys Thr Ala Lys Phe Gly Asp Tyr -;
B Trp Met Val Ser Jle [Glu]2 Lys Asn - [Lys]2 [Gly]2 Phe AspOH,
или
VI. A H2N Glu Ala Thr Glu [Ala]2 Lys Val - Leu Ser Asn Leu Asp Glu Thr Val Asp -;
B OH,
отличающийся тем, что цельный яд паука Agelenopsis Aperta подвергают хроматографическому разделению на колонке типа С-18 Видак® (22 мм · 250 мм, размер пор 300oА, размер частиц 10 μ, элюирование осуществляют при объемной скорости 15 мл/мин, используя при элюировании программу с линейным градиентом 5% __→ 20% ацетонитрила, 95% __→ 80% 0,1%-ной водной трифторуксусной кислоты (0 __→ 30 мин) затем 20% ___→ 70% ацетонитрила, 80% ___→ 30% 0,1% -ной водной трифторуксусной кислоты (30 __→ 55 мин) и индивидуально собирают на 38, 39, 40, 43 и 48,3 мин фракции, соответствующие монохроматическому пику в диапазоне 220 230 нм, причем фракция, элюируемая на 48,3 мин, соответствует пептиду VI, а фракции, полученные на 38, 39, 40 и 43 мин, подвергают повторной хроматографической очистке следующим образом: фракцию, которую собирают на 38 мин, повторно подвергают хроматографированию на той же колонке при объемной скорости элюента 10 мл/мин, используя программу с нелинейным градиентом: 20% __→ 30% ацетонитрила, 80% _ _→ 70% 0,1%-ной водной трифторуксусной кислоты (0 __→ 40 мин) и на 22 мин собирают фракцию, соответствующую пику при 220 230 нм, и пептиду I или фракцию, собранную на 40 мин, очищают на колонке типа С-4 Видак® с теми же характеристиками, что и для Видак® С-18, при объемной скорости 10 мл/мин, используя программу с нелинейным градиентом 20% __→ 30% ацетонитрила и 80% __→ 70% 0,1-ной трифторуксусной кислоты в течение 0 __→ 30 мин затем 30% __ → 50% ацетонитрила, 70% __→ 50% 0,1% -ной трифторуксусной кислоты (30 __→ 45 мин) и на 22 мин собирают фракцию, соответствующую пику 220 230 нм, и пептиду II либо на 27,5 мин собирают фракцию, соответствующую пику при 220 230 нм, и пептиду IV или фракцию, собранную на 39 мин, очищают на указанной колонке Видак® С-18, при объемной скорости 10 мл/мин, используя программу с нелинейным градиентом 20% __→ 25% ацетонитрила, 80% __→ 75% 0,1%-ной водной трифторуксусной кислоты (0 __→ 30 мин) затем 25% __→ 50% ацетонитрила, 75% __→ 50% 0,1%-ной водной трифторуксусной кислоты (30 __→ 45 мин) и на 36 мин собирают фракцию, соответствующую пику при 220 230 нм, и пептиду III или фракцию полученную на 43 мин, повторно подвергают хроматографированию на колонке типа С-18 Видак® (10 мм х 250 мм, размер пор 300oА, размер частиц 5 μ ), элюирование проводят с объемной скоростью 3,5 мл/мин, используя программу с линейным градиентом: 25% ___ → 40% ацетонитрила, 75% ___→ 60% 0,1%-ной водной трифторуксусной кислоты ((0 __→ 30 мин) и на 20,25 мин собирают фракцию, соответствующую пику в диапазоне 220 230 нм, и пептиду V и полученные фракции, в случае необходимости, лиофилизируют, ресуспендируют в воде и лиофилизируют.
Комментарии