Конъюгаты антагонистов интегрина для нацеленной доставки к клеткам, экспрессирующим альфа-v-бета-3 - RU2623441C2
Код документа: RU2623441C2
Чертежи











Описание
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к синтезу и реакции эффективных и селективных низкомолекулярных антагонистов интегрина, содержащих подходящие линкеры и функциональные группы для химической реакции с другими молекулами, содержащими реакционно-способные нуклеофильные группы, например тиольные группы, для образования ковалентной связи между молекулой, которую нужно конъюгировать, и нацеливающим фрагментом. Низкомолекулярные нацеливающие антагонисты связываются с распознаваемыми ими системами рецепторов, как, например, антагонисты рецептора интегрина типа альфа-V-бета-3 (αVβ3) с димером αVβ3. Ковалентно связываемые молекулы включают низкомолекулярные терапевтические вещества, полимеры, пептиды и олигонуклеотиды. Сюда входят 5ʹ-тиосодержащие олигонуклеотиды для получения производных 5ʹ-тио-миРНК в качестве средств для обеспечения нацеленной доставки указанных миРНК. Такие дериватизированные миРНК в составе конъюгата с подходящими агентами для трансфекции способствуют селективной доставке миРНК в клетки, экспрессирующие такие рецепторы интегрина, таким образом предотвращая экспрессию целевых генов за счет РНК-интерференции (РНКи).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Интегрин αVβ3 типа представляет собой рецептор витронектина [Hermann, P. et al. "The vitronectin receptor and its associated CD47 molecule mediates proinflammatory cytokine synthesis in human monocytes by interaction with soluble CD23" [The Journal of cell biology 144 (1999): 767-75]. Он состоит из двух компонентов, интегрина альфа V и интегрина бета 3 (CD61) и экспрессируется тромбоцитами и клетками других типов. Было показано, что ингибиторы αVβ3, такие как этарацизумаб, можно применять в качестве антиангиогенных средств.
РНК-интерференция - хорошо известный процесс, при котором трансляция информационной РНК (иРНК) в белок затруднена за счет ассоциации или связывания комплементарных или частично комплементарных олигонуклеотидов, таких как малая интерферирующая РНК (миРНК), короткая шпилечная РНК (кшРНК), микро-РНК (миРНК) или антисмысловые олигонуклеотиды. Молекулы миРНК представляют собой двуспиральные молекулы РНК, длиной обычно в интервале 19-25 нуклеотидов, которые образуют ассоциаты с рядом белков в цитоплазме, известных как RISC (индуцированный РНК комплекс сайленсинга). В конечном итоге RISC разделяет двуспиральную миРНК, давая возможность одной цепи связаться или образовать ассоциат с комплементарным или частично комплементарным участком молекулы иРНК, после чего иРНК разрушается под действием RISC, или ее трансляция предупреждается иным путем, вследствие чего подавляется экспрессия кодируемого белка или продукта гена.
Одна из проблем в использовании нуклеиновых кислот, таких как миРНК, в терапевтических целях (в частности, для систематического введения у человека) заключается в доставке нуклеиновых кислот: (1) к конкретной целевой ткани или типам клеток и (2) к цитоплазме указанных клеток (т.е. туда, где указанная иРНК присутствует и транслируется в белок). Проблема доставки частично обусловлена тем фактом, что нуклеиновые кислоты отрицательно заряжены и легко разрушаются (особенно, если они не модифицированы), эффективно фильтруются почками, и их нельзя как таковые легко транспортировать в цитоплазму. Таким образом, основная масса исследований сфокусирована на разрешении проблемы доставки с помощью различных носителей и композиций, включая лизосомы, мицеллы, пептиды, полимеры, конъюгаты и аптамеры. См., например: Ling et al, Advances in Systemic siRNA Delivery, Drugs Future 34(9): 721 (September 2009). Некоторые более перспективные средства доставки включают применение липидных систем, включая липидные наночастицы. См., например: Wu et al., Lipidic Systems for In Vivo siRNA Delivery, AAPS J. 11(4): 639-652 (December 2009); Международная Заявка на патент №WO 2010/042877, Hope et al ("Improved Amino Lipids And Methods For the Delivery of Nucleic Acids"). Тем не менее, сохраняется необходимость в дальнейшем улучшении нацеленной доставки миРНК; а также в таких веществах, как низкомолекулярные соединения, пептиды, другие нуклеиновые кислоты, флуоресцирующие молекулы и полимеры, для конкретных целевых клеток и цитоплазмы таких клеток.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I:

где R1, R2 и n определены в подробном описании и формуле изобретения. В частности, настоящее изобретение относится к соединениям формулы I для улучшенной доставки конъюгированных молекул, таких как низкомолекулярные вещества, пептиды, нуклеиновые кислоты, флуоресцирующие молекулы и полимеры, к клеткам-мишеням, экспрессирующим димер αVβ3, для различных терапевтических целей и других видов применения. Настоящее изобретение относится также к способам изготовления и применения таких соединений.
КРАТКОЕ ОПИСАНИЕ ФИГУР
Фиг. 1а: В Табл. 1. представлен состав конкретных дериватизированных по 5ʹ-концу одинарных и двойных цепей миРНК.
Фиг. 1b: В Табл. 2 представлены данные анализа для низкомолекулярных конъюгатов миРНК.
Фиг. 1c: В Табл. 3 представлены активности низкомолекулярных конъюгатов миРНК в анализах антагонистов интегрина и данные по KD для миРНК.
Фиг. 1d: В Табл. 4 представлены идентичность, характеристика и связывающая активность меченых изомером FITC реагентов.
На Фиг. 1е представлена гистограмма (красный: Дуплекс-27 500 нМ и Пример 140, 10 мкМ; зеленый: Дуплекс-27).
На Фиг. 2 представлен пример отображения захвата миРНК (Дуплекс-27 (500 нМ).
На Фиг. 3 представлены изображения клеток Jurkat с FITC, конъюгированным с соединением из Примера FITC-5 (LFA-1 антагонист-меченный FITC) при концентрации 10 мкМ.
На Фиг. 4 представлены изображения клеток Jurkat с FITC, конъюгированным с соединением из Примера FITC-14 (VLA-4 антагонист-меченный FITC) в концентрации 10 мкМ. На гистограмме показан сдвиг в присутствии дуплекса миРНК с элементом, нацеливающим на VLA-4. В присутствии антагониста VLA-4 из примера 140, указанный сдвиг оказывается сжатым.
На Фиг. 5 показано снижение экспрессии АНА1 в клетках H1299 при их обработке дуплексами миРНК, которые были дериватизированы по 5ʹ-смысловой цепи с помощью низкомолекулярного соединения, нацеливающего к интегрину. Y-ось отображает наблюдаемый уровень экспрессии АНА1. Более низкий столбик отражает более высокую степень выключения (более высокую степень трансфекции миРНК); высокий столбик - более низкую степень выключения (т.е. более низкую степень трансфекции миРНК). Дуплексы голубого цвета включают нацеливающую модификацию на 5ʹ-конце смысловой цепи; дуплексы розового цвета содержат нацеливающую модификацию на 5ʹ-конце смысловой цепи, а также флюорофор Nu547, присоединенный по 5ʹ-концу антисмысловой цепи.
На Фиг. 6 показаны уровни экспрессии иРНК GAPDH, маркера клеточного здоровья. Идентичность уровней экспрессии для тех клеток, которые были обработаны дериватизированной миРНК, по сравнению обработанными холостым образцом и необработанными клетками, говорит об отсутствии токсичности для клеток при используемых концентрациях и длительности обработки.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, следующие специфические термины и выражения, используемые в описании и формуле изобретения, имеют приведенные ниже определения.
Термин "радикал" обозначает атом или группу химически связанных атомов, которые присоединены к другому атому или молекуле посредством одной или более химических связей, составляя таким образом часть молекулы. Например, переменные R1 и R2 формулы I обозначают радикалы, которые присоединены к структуре, изображенной на формуле I, посредством ковалентной связи, где это указано.
Термин "конъюгированный фрагмент" обозначает структуру, представляющую собой терапевтическое средство или полезное соединение, пептид, полимер, низкомолекулярное вещество, флюоресцирующую молекулу, олигонуклеотид или нуклеиновую кислоту. Примерами являются лекарственные вещества, терапевтические пептиды, антисмысловые олигонуклеотиды, миРНК и флюоресцеинизотиоцианат (FITC).
Если не указано иное, термин "водород" или "гидро" обозначает радикал, состоящий из атома водорода (-Н), а не Н2.
Термин "галоген" обозначает радикал фтора, хлора, бром или йода.
Термин "алкил" обозначает моновалентную линейную или разветвленную насыщенную углеводородную группу, включающую от 1 до 12 атомов углерода. В частных воплощениях алкил включает от 1 до 7 атомов углерода, и в частности, от 1 до 4 атомов углерода. Примеры алкила включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил. Термин "TFA" обозначает трифторуксусную кислоту.
Если не указано иное, термин "соединение формулы" или "соединения формулы" обозначает любое соединение, выбранное из рода соединений, охватываемых указанной формулой (включая любые фармацевтически приемлемые соль или эфир любого такого соединения, если не указано иное).
Термин "фармацевтически приемлемые соли" обозначает такие соли, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются нежелательными с биологической или иной точки зрения. Такие соли могут быть образованы неорганическими кислотами, такими как хлороводородная кислота, бромоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., предпочтительно хлороводородная кислота, и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, салициловая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, N-ацетилцистеин и т.п. Кроме того, соли можно получать добавлением неорганического основания или органического основания к свободной кислоте. Соли, образованные неорганическим основанием, включают, без ограничения, соли натрия, калия, лития, аммония, кальция и магния и т.п. Соли, образованные органическими основаниями, включают, без ограничения, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, лизин, аргинин, N-этилпиперидин, пиперидин, полиаминовые смолы и т.п. В зависимости от природы заместителей, соединения по настоящему изобретению могут существовать также в форме цвиттер-ионов.
Соединения по настоящему изобретению могут присутствовать в форме фармацевтически приемлемых солей. Соединения по настоящему изобретению могут также присутствовать в форме фармацевтически приемлемых эфиров (т.е. метиловых и этиловых эфиров кислот формулы I для применения в качестве пролекарств). Соединения по настоящему изобретению можно также сольватировать, т.е. гидратировать. Сольватацию можно осуществлять в ходе процесса получения или она может протекать как следствие гигроскопических свойств изначально безводного соединения формулы I (гидратация).
Соединения, которые имеют одинаковую молекулярную формулу, но различаются по природе или последовательности связей составляющих атомов или расположения атомов в пространстве носят название "изомеры". Изомеры, которые различаются по расположению атомов в пространстве, называются "стереоизомеры". Диастереомеры представляют собой стереоизомеры с противоположной конфигурацией при одном или более хиральных центров, которые не являются энантиомерами. Стереоизомеры, включающие один или более асимметрических центров, представляющие собой несовместимые друг с другом зеркальные отображения друг друга, носят название "энантиомеры". Если соединение содержит асимметрический центр, например, если атом углерода связан с четырьмя различными группами, возможна пара энантиомеров. Энантиомер можно охарактеризовать по абсолютной конфигурации его асимметрического центра или центров, и она описывается правилами для определения R- и S-конфигурации Канна, Ингольда и Прелонга, или же исходя из того, каким образом данная молекула вращает плоскость поляризованного света, и обозначается как правовращающий или левовращающий (т.е. как (+) или (-)-изомеры, соответственно). Хиральное соединение может существовать как в виде индивидуального энантиомера, так и в виде их смеси. Смесь, содержащая равные части энантиомеров, называется "рацемическая смесь".
Термин "терапевтически эффективное количество" обозначает количество соединения, которое эффективно для предотвращения, облегчения или улучшения симптомов заболевания или удлинения срока жизни субъекта, которому проводят лечение. Определение терапевтически эффективного количества находится в компетенции специалиста в данной области техники. Терапевтически эффективное количество или дозировка соединения по настоящему изобретению может изменяться в широких пределах и ее можно определять способом, известным в данной области техники. Такую дозировку следует подбирать в соответствии с индивидуальными требованиями в каждом конкретном случае, включая конкретное вводимое(ые) соединение(я), способ введения, состояние, подлежащее лечению, а также пациента, которому проводят лечение. Суточную дозу можно вводить единственной дозой или разделенными дозами, или, в случае парентерального введения, ее можно вводить путем непрерывной инфузии.
Термин "фармацевтически приемлемый носитель" включает всевозможные материалы, совместимые с фармацевтическим введением, включая растворители, дисперсионные среды, оболочки, антибактериальные и противогрибковые агенты, изотонические и агенты, замедляющие абсорбцию и другие материалы и соединения, совместимые с фармацевтическим введением. За исключением тех случаев, когда любая стандартная среда или агент не совместимы с действующим веществом, их применение в композициях по настоящему изобретению рассматривается. В композиции можно также включать дополнительные действующие вещества.
В частности, настоящее изобретение относится к соединениям формулы I:

или их фармацевтически приемлемым солям, или эфирам; где n равно от 1 до 24 и
где
R1 выбран из группы, состоящей из:
(1) соединения формулы:

где m равно 0 или 1;
(2) соединения формулы:

где X представляет собой N или CH;
(3) соединения формулы:

(4) соединения формулы:

(5) соединения формулы:

R2 выбран из группы, состоящей из:
(1) соединения формулы:

(2) соединения формулы:

(3) соединения формулы:

(4) соединения формулы:

где R3 представляет собой конъюгированный фрагмент, а X представляет собой либо серу, либо соединение формулы:
Используемый в приведенных выше структурах символ
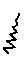

где X представляет собой соединение формулы:

то структура, соответствующая формуле I, будет следующей:

где R1, R3 и n определены в формуле I.
Настоящее изобретение относится также к способам изготовления и применения соединений формулы I, а также фармацевтических композиций, содержащих такие соединения. Соединения формулы I полезны для улучшения доставки низкомолекулярных веществ, белков, нуклеиновых кислот, полимеров, флюоресцирующих маркеров и других веществ к клеткам-мишеням, экспрессирующим рецептор αVβ3. В некоторых воплощениях настоящее изобретение относится к композициям и лекарственным формам, содержащим соединения формулы I, которые полезны для доставки миРНК в цитоплазму клеток-мишеней, экспрессирующих рецептор αVβ3, для ингибирования экспрессии конкретных целевых белков за счет РНК-интерференции.
В более частных воплощениях, настоящее изобретение относится к применению соединений формулы I в композициях для облегчения доставки нуклеиновых кислот, таких как миРНК, к опухолевым клеткам и клеткам других типов, экспрессирующим рецепторы αVβ3. Кроме того, частью настоящего изобретения является применение соединений формулы I в изготовлении композиций для доставки, предназначенных для лечения воспалительных и пролиферативных расстройств, таких как рак.
R1 представляет собой низкомолекулярный антагонист интегрина, который нацеливает соединения формулы I к комплексам рецепторов интегрина, облегчая таким образом их доставку к клеткам, которые экспрессируют такие рецепторы.
В некоторых воплощениях нацеливающие фрагменты структуры R1 низкомолекулярного антагониста интегрина присоединены по такому положению, чтобы аффинность низкомолекулярного вещества к рецептору интегрина существенно не снижалась, по сравнению со свободным низкомолекулярным антагонистом интегрина. Фрагменты R1 формулы I нацелены к димеру интегрина αVβ3.
В частных воплощениях R1 представляет собой нацеливающий к интегрину αVβ3 фрагмент формулы:

или его фармацевтически приемлемую соль, или эфир, где m равно 0 или 1.
В других воплощениях R1 представляет собой нацеливающий к интегрину αVβ3 фрагмент формулы:

или его фармацевтически приемлемую соль, или эфир, где X представляет собой N или CH.
В других воплощениях R1 представляет собой нацеливающий к интегрину αVβ3 фрагмент формулы:

или его фармацевтически приемлемую соль, или эфир.
В других воплощениях R1 представляет собой нацеливающий к интегрину αVβ3 фрагмент формулы:

или его фармацевтически приемлемую соль, или эфир.
В других воплощениях R1 представляет собой нацеливающий к интегрину αVβ3 фрагмент формулы:
или его фармацевтически приемлемую соль, или эфир.
R2 может представлять собой реакционно-способные радикалы, которые могут давать ковалентные связи с терапевтическими или иными полезными соединениями или конъюгированными молекулами, содержащими сильные нуклеофилы, например с тиол-содержащими молекулами. Примеры таких реакционно-способных радикалов включают радикалы, выбранные из группы, состоящей из:



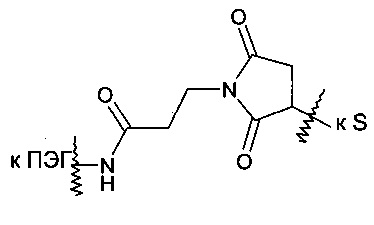
Как вариант, R2 может представлять собой радикал, который уже присоединен к конъюгированному фрагменту, такому как терапевтическое или другое полезное соединение, белок или олигонуклеотид (R3). В частности, R2 может представлять радикал формулы:

где R3 представляет собой конъюгированный фрагмент, а X представляет собой либо серу, либо соединение формулы:

В некоторых воплощениях R3 представляет собой олигонуклеотид. В частных воплощениях R3 представляет собой 5ʹ-конец смысловой цепи молекулы РНК, который может существовать в виде одинарной цепи или в дуплексе, например молекула миРНК. Такие молекулы миРНК, известные также как агенты РНКи, ингибируют экспрессию целевого гена в клетке. В частных воплощениях R3 представляет собой молекулу миРНК, которая состоит по существу из олигорибонуклеотидной цепи длиной от 15 до 30 нуклеотидов, в которой 5ʹ-конец смысловой олигорибонуклеотидной цепи присоединен к R2, как показано в выше приведенных структурах, и комплементарен по меньшей мере одной части иРНК, соответствующей целевому гену. В других воплощениях R3 представляет собой олигонуклеотид ДНК, присоединенный по своему 5ʹ-концу. Такая дериватизированная ДНК может существовать в виде одинарной цепи или одной цепи, гибридизованной с комплементарной цепью другого олигонуклеотида. Олигонуклеотидные цепи могут быть как немодифицированными, так и модифицированными в целях метаболической стабильности. Подобные модификации включают, без ограничения, замещения в конкретных положениях по фосфатной группе (например, фосфоротиоат) и 2ʹ-гидроксигруппе (например, 2ʹ-O-метил и 2ʹ-фтор).
В некоторых воплощениях R2 формулы I представляет собой -X-S-CH2-R3, при этом R3 включает смысловую цепь РНК, как показано ниже в формуле 5 (на основе формулы I):

5
где R1, n и X таковы, как определено в формуле I.
В других частных воплощениях смысловая цепь может быть связана с антисмысловой цепью.
В других частных воплощениях R2 представляет собой -X-S-CH2-R3, при этом R3 представляет собой низкомолекулярное соединение или белок, образуя таким образом ковалентно связанную, специфически нацеленную молекулу формулы I.
В более частных воплощениях R2 представляет собой -X-S-CH2-R3, при этом R3 представляет собой терапевтические низкомолекулярное соединение или белок.
В других частных воплощениях R2 представляет собой -X-S-CH2-R3, при этом R3 представляет собой флюоресцирующий радикал, полезный в целях визуализации указанных связей интегринового рецептора с применением технологий клеточной микроскопии.
В других частных воплощениях R2 представляет собой -X-S-CH2-R3, при этом R3 представляет собой полимер, содержащий первичные реакционно-способные сульфидные группы. Более конкретно, R3 может представлять собой катионный полимер, полезный для комплексирования и доставки миРНК к поверхностям клеток и цитоплазматическим доменам клеток.
В более частных воплощениях настоящее изобретение относится к соединениям формулы I, где R3 представляет собой один из структурных изомеров флуоресцеин изотиоцианата (FITC), показанных ниже:


В других более конкретных воплощениях настоящее изобретение относится к соединениям формулы I, где R3 представляет собой один из структурных изомеров FITC-14, показанный ниже:
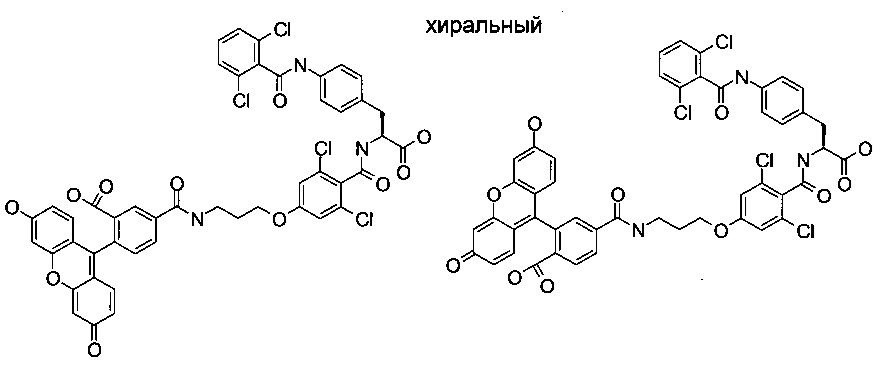
В других воплощениях настоящее изобретение относится к соединению формулы I, где n равно от 9 до 13, предпочтительно 12.
В частных воплощениях настоящее изобретение относится к соединению формулы I, выбранному из группы, состоящей из одного из следующих соединений (или его фармацевтически приемлемой соли или эфира):
αVβ3 Лиганд - Реагент 1: полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)-пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]-пропиониламино]пропокси]-фенил]-3-[2-[3-(гуанидино)-бензоиламино]-ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 2: полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропиониламино]-пропокси]фенил]-3-[2-[3-(гуанидино)бензоиламино]ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 3: полуамид (S)-N-[[[4-[3-[2-[2-[2-[2-[2-[2-[2-(2-ацетил-сульфанилэтокси)-этокси]этокси]этокси]этокси]этокси]этокси]этокси]-1-оксопропил]-амино]пропокси]-фенил]-3-[2-[3-[гуанидино]бензоиламино]ацетиламино]-янтарной кислоты, трифторацетатная соль;
αVβ3 Лиганд - Реагент 4: полуамид (S)-N-[[[4-[3-[2-[2-[2-[2-[2-[2-[2-(2-ацетил-ульфанилтокси)этокси]этокси]этокси]этокси]этокси]этокси]этокси]-1-оксопропил]-амино]пропокси]фенил]3-[2-[3-(тетрагидропиримидин-2-илиденамино)-бензоиламино]ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 5: полуамид (S)-N-[[4-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]пропиониламино]метил]фенил]3-[2-[[2-(3-бензилуреидо)-тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 6: полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]пропиониламино]пропокси]фенил]3-[2-[[2-(3-бензилуреидо)-тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 7: полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]пропиониламино]этокси]-фенил]-3-[2-[[2-(3-бензил-уреидо)тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 8: полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-ацетилсульфанилэтокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропиониламино]пропокси]фенил]3-[2-[[2-(3-бензил-уреидо)тиазол-4-карбонил]-амино]-ацетиламино]-янтарной кислоты;
αVβ3 Лиганд - Реагент 9: (R)-3-[2-{(2-[3-{2-[3-(3-(2-{2-[2-(2-ацетилсульфанил-этокси}-этокси)этокси]этокси}-пропиониламино)пропокси]бензил}-уреидо]-тиазол-4-карбонил)амино}-ацетиламино]фенил-3-ил-пропионовая кислота;
αVβ3 Лиганд - Реагент 10: 3-[2-{(2-[3-{2-[3-(3-{2-[2-(2-{2-[2-(2-{2-[2-(2-{2-[2-(2-Ацетилсульфанилэтокси)этокси]этокси}-этокси)этокси]этокси}-этокси)этокси]-этокси}этокси)этокси]этокси}-пропиониламино)пропокси]бензил}-уреидо]-тиазол-4-карбонил)амино}-ацетиламино]фенил-3-ил-пропионовая кислота;
αVβ3 Лиганд - Реагент 11: (R)-3-[2-{(2-[3-{2-[3-(3-(2-{2-[2-(2-ацетилсульфанил-этокси}-этокси)этокси]этокси}-пропиониламино)пропокси]бензил}-уреидо]-тиазол-4-карбонил)амино}-ацетиламино]-3-пиридин-3-ил-пропионовая кислота;
αVβ3 Лиганд - Реагент 12: (R)-3-[2-{(2-[3-{2-[3-(3-{2-[2-(2-{2-[2-(2-{2-[2-(2-{2-[2-(2-ацетилсульфанилэтокси)этокси]этокси}-этокси)этокси]этокси}-этокси)этокси]-этокси}этокси)этокси]этокси}-пропиониламино)пропокси]бензил}-уреидо]-тиазол-4-карбонил)амино}-ацетиламино]-3-пиридин-3-ил-пропионовая кислота;
Кроме того, настоящее изобретение относится к новым композициям и лекарственным формам, содержащим соединения формулы I, для получения наночастиц путем комбинирования с миРНК, что обеспечивает улучшенную доставку нуклеиновых кислот, таких как миРНК, в цитоплазму клеток-мишеней, экспрессирующих димеры αVβ3. В некоторых воплощениях настоящее изобретение относится к композиции миРНК, включающей: (1) соединение формулы I, в котором R2 включает 5ʹ-миРНК олигонуклеотид; и (2) поликатионный агент для трансфекции.
Настоящее изобретение относится также к способам получения и применения таких соединений и композиций. Соединения формулы I полезны как компоненты композиций или лекарственных форм, улучшающих доставку лекарственных веществ, нуклеиновых кислот или других терапевтических соединений к тканям или клеткам, экспрессирующим димеры αVβ3. В некоторых воплощениях настоящее изобретение относится к композициям, содержащим соединения формулы I, которые полезны для доставки миРНК в цитоплазму клеток-мишеней, экспрессирующих димеры αVβ3, с целью ингибирования экспрессии конкретных белков за счет РНК-интерференции. В более частных воплощениях настоящее изобретение относится к соединениям формулы I и композициям, содержащим такие соединения, которые могут эффективно доставлять миРНК к опухолевым клеткам и клеткам других типов, экспрессирующим димеры αVβ3, для лечения раковых или воспалительных заболеваний. Такие соединения и композиции более эффективны и демонстрируют улучшенную выключающую способность, по сравнению с такими же композициями, не содержащими соединения формулы I.
ОБЩИЙ СПОСОБ СИНТЕЗА СОЕДИНЕНИЙ ПО НАСТОЯЩЕМУ ИЗОБРЕТЕНИЮ
Подходящие способы для синтеза соединений формулы I представлены в примерах. В целом, соединения формулы I можно получить в соответствии со схемами, приведенными ниже. Если не указано иное, переменные n, R1 и R2 в приведенных ниже схемах имеют такое же определение, как и в представленной ранее родовой формулы I.
Общий синтез конъюгирующих агентов малеимид-(ПЭГ)n-антагонистов интегрина
Соединения структуры 26 на схеме 1 с различной длиной ПЭГ коммерчески доступны (например, в Pierce Bioscience). Такие соединения можно также получать ацилированием амино-концов ПЭГ-аминокислот с помощью 3-(2,5-диоксо-2,5-дигидропиррол-1-ил)-пропионовой кислоты в условиях для образования амидной связи, с последующим образованием реакционно-способных N-гидроксисукциновых эфиров в ходе реакции N-гидроксиянтарной кислоты в условиях для образования эфира. Как показано на схеме 1, введение в реакцию соединений 26 с соединениями, содержащими первичную или вторичную аминогруппу, такими как соединения 27, проводят в апротонных или протонных растворителях в присутствии основных аминов, таких как DIEA (диизопропилэтиламин), при комнатной температуре, с образованием ПЭГ-илированных промежуточных продуктов 28.
Соединения структуры 29 на схеме 2, где R4 представляет собой тиоацетил или 2-дитиопиридил, содержащие ПЭГ-группы различной длины, также имеются в продаже (например, в Pierce Bioscience). Реакцию соединений структуры 29 с соединениями, содержащими первичные или вторичные аминогруппы, такими как соединения 27, проводят в апротонных или протонных растворителях в присутствии основных аминов, таких как DIEA (диизопропилэтиламин), при комнатной температуре, с получением ПЭГ-илированных промежуточных продуктов 30.

В качестве частного, не налагающего ограничений примера настоящего изобретения, промежуточное соединение 26 вводят в реакцию с соединением 31 с получением промежуточного малеимида 32, как показано на схеме 3.
Аналогичным образом, промежуточное соединение 26 можно вводить в реакцию с соединением 33 с получением промежуточного малеимида 34, как показано на схеме 4.
Аналогичным образом, промежуточное соединение 29 можно вводить в реакцию с соединением 35 с получением промежуточного соединения 36, как показано на схеме 5, на которой R4 представляет собой либо тиоацетил, либо 2-дитиопиридил.


Аналогичным образом, промежуточное соединение 29 можно вводить в реакцию с соединением 37 с получением промежуточного соединения 38, как показано на схеме 6, где R4 представляет собой либо тиоацетил, либо 2-дитиопиридил.
Для соединений общей структуры 26 или 29 коммерчески доступны ПЭГ различной длины, или их легко может получить специалист в данной области техники; предпочтительно n равно от 8 до 24. По данной теме имеется много статей и обзоров (например: Chemistry for peptide and protein PEGylation, Advanced Drug Delivery Reviews Volume 54, Issue 4, 17 June 2002, Pages 459-476).
Промежуточное соединение 31 можно синтезировать таким же способом, как уже сообщалось в литературе (например, Sidduri, A. et al. Bioorganic & Medicinal Chemistry Letters, 2002, 12, 2475-2478), как показано на схеме 7.


В частности, как показано на схеме 7, промежуточное соединение 41 получали из коммерчески доступной (S)-3-[4-нитрофенил]-2-трет-бутоксикарбонил-аминопропионовой кислоты 40. Нитрогруппу коммерчески доступного исходного вещества 40 в метанольном растворе восстанавливали цинковой пылью в присутствии хлорида аммония при комнатной температуре в течение нескольких часов с получением анилина 41. Другие способы для восстановления нитрогруппы известны специалистам в данной области техники. Анилин 41 ацилировали производными бензоилгалида, такими как 2,6-дихлорбензоилхлорид 42, в апротонном растворителе, таком как дихлорметан, в присутствии основания, такого как диизопропилэтиламин, при комнатной температуре. Таким образом получали амид 43. Трет-бутилкарбонильную (Boc) аминопротекторную группу удаляли стандартными способами, известными специалистам в данной области техники, например путем обработки раствором HCl в диоксане при комнатной температуре; в результате этого получали гидрохлорид 44. Гидрохлорид 44 обрабатывали в условиях образования амидной связи (также хорошо известных специалистам в данной области техники) в присутствии хорошо известной 1-(2-азидоэтил)-циклопентанкарбоновой кислоты 45 с получением диамида 46. Азидную группу в промежуточном соединении 46 восстанавливали путем обработки триалкилфосфином в апротонном растворителе, таком как тетрагидрофуран, при комнатной температуре. Затем полученный метиловый эфир омыляли путем обработки гидроксидом натрия в смеси растворителей, таких как этанол и тетрагидрофуран, при повышенной температуре, например при 50°С, в течение 15 ч. В результате этого получали промежуточное соединение 31, которое может также существовать в виде цвиттер-иона.
Присоединение ПЭГ-фрагмента возможно также с помощью промежуточного соединения 39, которое синтезируют, как показано на схеме 8. В частности, 3,5-дихлорфенол 47 защищают три-изопропилсилилхлоридом в присутствии основания, такого как имидазол, в полярном апротонном растворителе, таком как DMF, после чего проводят реакцию с сильным основанием, таким как бутиллитий, в безводном тетрагидрофуране при низкой температуре, например при -78°С. Получаемый комплекс лития гасят диоксидом углерода, который добавляют в форме сухого льда, с получением промежуточного соединения 48, производного бензойной кислоты. Промежуточное соединение 48 затем хлорируют с образованием ацилхлорида, путем обработки сульфонилхлоридом (SOCl2) в апротонном растворителе, таком как толуол. Полученный ацилхлорид далее вводят в реакцию с гидрохлоридом амина 49 в присутствии основания, такого как диизопропилэтиламин (DIPEA), в апротонном растворителе, таком как дихлорметан (DCM), получая при этом промежуточное соединение 50. Силильную защитную группу в промежуточном соединении 50 удаляют путем обработки фторидом тетрабутиламмония (TBAF) в протонном растворителе, таком как тетрагидрофуран, при комнатной температуре. Это фенольное промежуточное соединение вводят в реакцию в присутствии основания, такого как карбонат калия (K2CO3), в апротонном растворителе, таком как диметилформамид (DMF), с 3-N-трет-бутилкарбомат-1-бромпропаном. Таким образом получают промежуточное соединение 52, которое при снятии защиты с помощью трифторуксусной кислоты (TFA) и последующем гидролизе с помощью основания, такого как гидроксид натрия, в протонном растворителе, таком как этанол, образует промежуточное соединение 39:

Синтез дериватизирующих агентов - антагонистов αVβ3
Промежуточное соединение 117, αVβ3-нацеливающий модуль, можно синтезировать, как показано ниже на Схеме 15. Вкратце, мета-аминобензойную кислоту 110 вводят в реакцию с Ν,Ν-ди-Boc-метилтиомочевиной 111 в DMF, дихлорметаном и пиридином в присутствии ацетата ртути. На данном этапе глицин, защищенный бензиловым эфиром, конденсируют с карбоновой кислотой продукта выше описанной реакции в стандартных условиях для образования пептидной связи. Бензиловый эфир удаляют в условиях гидрогенолиза, получая при этом свободную кислоту 112. В отдельной последовательности реакций пара-нитрофенол 113 конденсируют с трет-бутиловым эфиром (2-гидроксиэтил)-карбаминовой кислоты в присутствии трифенилфосфина и диизопропилазодикарбоксилата в апротонном растворителе, таком как тетрагидрофуран. Нитрогруппу в этом продукте восстанавливают до соответствующего анилина 114, который конденсируют в условиях образования амидной связи с β-трет-бутиловым эфиром N-α-Fmoc-L-аспарагиновой кислоты. После удаления амино-протекторной Fmoc-группы путем обработки пиперидином, амино-конец конденсируют с промежуточным соединением 115, также в стандартных условиях образования амидной связи, с получением промежуточного соединения 116. При обработке сильной кислотой, такой как трифторуксусная кислота, получают промежуточное соединение 117 и выделяют его способами, хорошо известными специалистам в данной области техники (например с помощью препаративной ВЭЖХ).
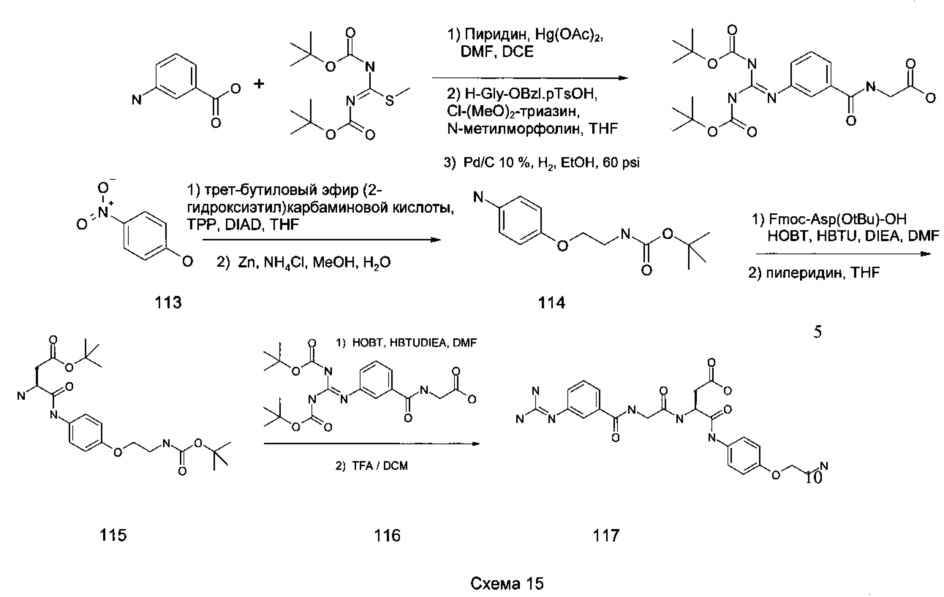
Аналог амина с 3-углеродной цепью 117 можно получить таким же образом, как показано на Схеме 15, с использованием трет-бутилового эфира (3-бром-пропил)карбаминовой кислоты вместо трет-бутилового эфира (2-гидроксиэтил)-карбаминовой кислоты.
Другие αVβ3-нацеливающие модули можно синтезировать, как показано ниже на Схеме 16. Получение метилового эфира 2-аминотиазол-4-карбоновой кислоты 120 описано в литературе (Amide derivatives and their preparation and use as pesticides, By Kobayashi, Yumi; Daido, Hidenori; Katsuta, Hiroyuki; Nomura, Michikazu; Tsukada, Hidetaka; Hirabayashi, Atsushi; Takahashi, Yusuke; Aoki, Yoji; Kawahara, Atsuko; Fukazawa, Yasuaki; et al US Pat. Appl. Publ. (2011), US 20110201687 A1 20110818). Промежуточное соединение 120 вводят в реакцию в стандартных условиях для образования амидной связи с гидрохлоридом метилового эфира аланина 121, при этом получают промежуточный эфир 122. Отдельно получают трет-бутил-3-(2-(аминометил)фенокси)пропилкарбамат 129 в ходе реакции N-(2-гидроксибензил)ацетамида 127 в апротонном растворителе, таком как DMF, с трет-бутил-3-бромпропилкарбаматом 124 в присутствии основания, такого как карбонат калия. Аминогруппу в промежуточном соединении 129 высвобождают обработкой гидразингидратом. Получаемый свободный амин 130 обрабатывают трифосгеном с получением изоцианата 131, который объединяют с метиловым эфиром [(2-аминотиазол-4-карбонил)амино]уксусной кислоты 132 в апротонном растворителе, таком как DMF, с получением промежуточного соединения 133. Превращение метилового эфира промежуточного соединения 133 осуществляют в стандартных условиях омыления, с последующей конденсацией с коммерчески доступным метил-3-амино-3-фенилпропионатом гидрохлоридом 146 или энантиомерно чистыми изомерами метил-3-амино-3-(3-пиридил)пропионата гидрохлорида 147 и 148 в стандартных условиях реакции образования амидной связи, с получением при этом промежуточного соединения 135. Защитную Вос-группу удаляют в стандартных условиях, с получением αVβ3-нацеливающего низкомолекулярного соединения 136.
Схема реакции 16 для следующих примеров: αVβ3 Лиганд - Реагент 9, αVβ3 Лиганд - Реагент 10, αVβ3 Лиганд - Реагент 11 и αVβ3 Лиганд - Реагент 12:


ПОЛЕЗНОСТЬ
Соединения формулы I полезны для доставки конъюгированных молекул, таких как терапевтические вещества, низкомолекулярные вещества, пептиды, нуклеиновые кислоты, флуоресцирующие молекулы и полимеры, к клеткам-мишеням, экспрессирующим комплексы рецептора интегрина αVβ3, в различных терапевтических целях и для других типов применения. Таким образом, соединения формулы I можно применять для лечения различных заболеваний и состояний, связанных с экспрессией или сверхэкспрессией αVβ3. Такие заболевания и состояния могут включать воспалительные, раковые и метаболические заболевания.
В конкретных воплощениях настоящее изобретение включает способ лечения или профилактики рака у млекопитающих (предпочтительно у человека), нуждающихся в таком лечении, включающий введение терапевтически эффективного количества соединения формулы I. Указанные композиции можно вводить в соответствии с надлежащей медицинской практикой. Факторы, которые следует учитывать в данном контексте, включают конкретное расстройство, подлежащее лечению, конкретное млекопитающее, которого нужно лечить, клиническое состояние индивидуального пациента, причину расстройства, сайт доставки агента, способ введения, схему введения и другие факторы, известные лечащим врачам. "Эффективное количество" соединения, которое нужно ввести, определяют с учетом этих факторов как минимальное количество, необходимое для лечения или профилактики данного заболевания или состояния (например, для ингибирования экспрессии целевого белка), не допуская неприемлемой токсичности. Например, такое количество может быть ниже количества, которое токсично для нормальных клеток или для млекопитающего в целом. Такие композиции, содержащие соединение формулы I по настоящему изобретению, можно вводить парентерально, внутрибрюшинно и внутрилегочно. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение.
ПРИМЕРЫ
Понимание настоящего изобретения будет более полным со ссылкой на ниже следующие примеры. Однако их не следует воспринимать как ограничение объема настоящего изобретения.
Реагенты приобретали в компаниях Aldrich, Sigma и Pierce Bioscience или у других поставщиков, указанных ниже, и использовали без дополнительной очистки. Очистку в масштабе от нескольких миллиграмм до нескольких грамм проводили способами, известными специалистам в данной области техники, такими как элюирование с колонки для флэш-хроматографии с силикагелем. В некоторых случаях проводили также очистку с помощью препаративной колоночной флэш-хроматографии с использованием одноразовых предварительно упакованных мультиграммовых колонок с силикагелем (RediSep), элюировали с использованием системы CombiFlash. В целях данного изобретения для очистки промежуточных продуктов можно применять также устройства для колоночной флэш-хроматографии Biotage™ и ISCO™.
Для оценки чистоты и идентичности соединения снимали спектры LC/MS (жидкостная хроматография/масс-спектрометрия) с использованием следующей системы. Система для получения масс-спектров представляла собой спектрометр Micromass Platform II: электроспрэй-ионизация в положительном режиме (диапазон масс: 150-1200 а.е.м.). Одновременное хроматографическое разделение осуществляли с помощью следующей системы для ВЭЖХ: колоночный картридж ES Industries Chromegabond WR С-18 3u 120Å (3,2×30 мм); подвижная фаза А: вода (0,02% TFA) и фаза В: ацетонитрил (0,02% TFA); градиент от 10% В до 90% В за 3 минуты; время уравновешивания 1 минута; скорость потока 2 мл/мин. В некоторых случаях использовали ацетат аммония в 20-миллимолярной концентрации в качестве модификатора эффективной ионизации в ходе препаративной ВЭЖХ. В таких случаях выделяли соль аммония.
В некоторых случаях разделения может быть также полезно применение суперкритической флюидной хроматографии. Разделение суперкритической флюидной хроматографией осуществляли с применением системы Mettler-Toledo Minigram в следующих стандартных условиях: 100 бар; 30°С; 2,0 мл/мин; элюировали с колонки AD 12 мм с 40% MeOH в суперкритическом флюиде CO2. Если аналиты содержали основные аминогруппы, добавляли 0,2% изопропиламин к метанольному модификатору.
Соединения характеризовали либо с помощью1H-ЯМР, с использованием ЯМР-спектрометра Varian Inova 400 МГц, либо или ЯМР-спектрометра Varian Mercury 300 МГц, а с помощью также масс-спектрометрии высокого разрешения с использованием масс-спектрометра высокого разрешения Bruker Apex-II 4.7Т FT. Конечные соединения характеризовали с помощью масс-спектрометрии высокого разрешения с использованием системы LTQ CL Orbitrap, поставляемой фирмой Thermo Electron.
Использована следующая аббревиатура:
СИНТЕЗ НАЦЕЛИВАЮЩИХ СОЕДИНЕНИЙ αVβ3
Пример 1
Полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)-пропиониламино]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропиониламино]пропокси]фенил]-3-[2-[3-(гуанидино)бензоиламино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 1

Стадия 1: Получение 3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензойной кислоты:
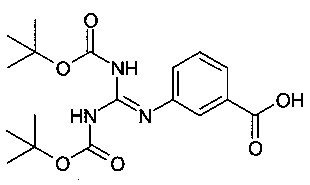
Раствор 3-аминобензойной кислоты (82,3 г; 0,60 моль), Ν,Νʹ-бис(трет-бутоксикарбонил)S-метилизотиомочевины (1,3-ди-boc-2-метилизотиомочевина, CAS # 107819-90-9) (174,2 г; 0,6 моль) и пиридина (94,92 г; 97 мл; 1,20 моль, 2,0 экв.) в смеси безводного диметилформамида (600 мл) и безводного 1,2-дихлорэтана (600 мл) обрабатывали ацетатом ртути (95,6 г; 0,30 моль, 0,5 экв.) и перемешивали с помощью верхнеприводной магнитной мешалки в течение 5 ч при комнатной температуре. Затем твердое вещество отфильтровывали, промывали дихлорметаном, и объединенные фильтрат и смывы выпаривали с получением неочищенного продукта (~307 г). К этому неочищенному веществу добавляли метанол (240 мл) и полученную смесь интенсивно перемешивали в течение 2 ч. Затем медленно вносили 2400 мл воды при интенсивном перемешивании. Фильтровали, тщательно промывали твердое вещество водой и высушивали отсасыванием в течение ночи с получением 3-(N,N-бис-трет-бутокси-карбонилгуанидино)бензойной кислоты с выходом больше теоретического. Высушивали с помощью насоса в высоком вакууме.
Масса полученного вещества превышала теоретическое значение (теоретически = 227,6 г; фактически = 251,2 г; по данным 1H ЯМР присутствовало ~10% DMF).
Стадия 2: Получение бензилового эфира 2-(3-(N,N-бис-трет-бутоксикарбонил-гуанидино)бензоил)аминоуксусной кислоты:

Светло-коричневый раствор 3-(N,N-бис-трет-бутоксикарбонилгуанидино)-бензойной кислоты (171,56 г; 0,4073 моль), 2-хлор-4,6-диметокситриазина (71,52 мг; 0,4073 моль) и N-метилморфолина (41,2 мг; 44,78 мл, 0,4073 моль) в безводном тетрагидрофуране (1600 мл) перемешивали (с помощью верхнеприводной механической мешалки) в течение 2 ч при комнатной температуре и затем добавляли глицин-бензиловый эфир, соль p-TsOH (137,44 г; 0,4073 моль), и еще один эквивалент N-метилморфолина (41,2 г; 44,78 мл; 0,4073 моль). Полученную смесь перемешивали при комнатной температуре в течение 36 ч. Затем удаляли тетрагидрофуран на роторном испарителе и добавляли этилацетат (2000 мл). Полученную смесь промывали последовательно ледяной 0,5 н. HCl (3×1000 мл), водой (1×1000 мл), 5% водным раствором карбоната натрия (1×1000 мл), водой (1×1000 мл), насыщенным водным раствором хлорида натрия (1×1000 мл) и высушивали над сульфатом натрия. Твердый осадок отфильтровывали и растворитель выпаривали с получением неочищенного продукта (228,5 г) в виде масла. Неочищенное вещество очищали хроматографией на приборе Waters Prep500 (10 прогонов), с использованием в качестве элюента смеси дихлорметан: гексан: этилацетат в соотношении 40:45:15, с получением бензилового эфира 2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)-бензоил)-аминоуксусной кислоты (выход 79,3%). (Примечание: на первых прогонах получали 152 г чистого вещества и 33 г незначительно загрязненного вещества, которое повторно хроматографировали двумя прогонами). ES(+)-HRMS m/e расч. для C27H34N4O7 (М+Н)+ 527,2500; фактич. 527,2499.
Стадия 3: Получение 2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензоил)-аминоуксусной кислоты

Раствор бензилового эфира 2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)-бензоил)-аминоуксусной кислоты (170,0 г; 0,323 моль) в абсолютном этаноле (2000 мл) гидрогенировали над 10% Pd на угле (20 г влажного катализатора, содержащего ~50% воды) при давлении 60 psi в течение ночи (18 ч) в режиме Высокого Давления, при комнатной температуре. Катализатор отфильтровывали и растворитель выпаривали с получением продукта. Этот продукт подвергали азеотропной перегонке с толуолом (3 раза) для полного удаления этанола, с получением 2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензоил)-аминоуксусной кислоты (выход 97,88%) в виде белого твердого вещества. ES(+)-HRMS m/e расч. для C20H28N4O7 (М+Н)+ 437,2031; фактич. 437,2030.
Стадия 4: Получение трет-бутилового эфира [3-(4-нитрофенокси)-пропил]-карбаминовой кислоты:

К раствору трет-бутилового эфира (3-гидроксипропил)-карбаминовой кислоты (7,03 г; 40,1 ммоль) в безводном THF (40 мл) добавляли 4-нитрофенол (5,07 г; 36,5 ммоль) и трифенилфософин (10,5 г; 40,1 ммоль) при комнатной температуре, в атмосфере азота. Полученный раствор охлаждали до ~0°С на водно-ледяной бане и затем в течение 15-20 минут добавляли по каплям диизопропил азодикарбоксилат (DIAD, 8,1 г; 40,1 ммоль). По окончании внесения полученный раствор нагревали до комнатной температуры и перемешивали в течение 15 ч, при этом анализ с помощью LCMS показал присутствие 16% исходного вещества. После этого добавляли еще по 0,1 экв. каждого из указанных выше реагентов и реакционную смесь перемешивали еще в течение 15 ч. Полученный твердый осадок отфильтровывали и промывали этилацетатом, затем фильтрат промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом магния. В результате фильтрации и концентрирования получали неочищенный остаток, который очищали хроматографией на колонке ISCO (340 г) с получением трет-бутилового эфира [3-(4-нитрофенокси)-пропил]-карбаминовой кислоты (выход 64%) в виде белого твердого вещества. ES(+)-HRMS m/e расч. для C14H20N2O5 (M+Na)+ 319,1264; фактич. 319,1266.
Стадия 5: Получение трет-бутилового эфира [3-(4-аминофенокси)пропил]-карбаминовой кислоты:

К раствору трет-бутилового эфира [3-(4-нитрофенокси)пропил]карбаминовой кислоты (7,7 г; 26 ммоль) в метаноле (200 мл; нагретом для растворения исходного вещества) добавляли при комнатной температуре воду (10 мл), хлорид аммония (20,9 г; 390 ммоль; 15 экв.) и цинковую пыль (16,4 г; 260 ммоль; 10 экв., тремя порциями). По окончании внесения цинковой пыли (экзотермическая реакция), реакционную смесь перемешивали в течение 1-2 ч, при этом анализ с помощью ТСХ полученной смеси показал отсутствие исходного вещества. Затем твердое вещество отфильтровывали и промывали водой и этилацетатом, а органическое соединение из фильтрата экстрагировали этилацетатом (3×100 мл). Объединенные экстракты промывали солевым раствором и высушивали над безводным сульфатом магния. В результате фильтрации и концентрирования получали неочищенный остаток, который очищали хроматографией на колонке ISCO (330 г) с выделением трет-бутилового эфира [3-(4-аминофенокси)пропил]-карбаминовой кислоты (выход 79%) в виде белого твердого вещества. ES(+)-HRMS m/e расч. для C14H22N2O3 (M+Na)+ 289,1522; фактич. 289,1523.
Стадия 6: Получение трет-бутилового эфира (S)-N-[4-(3-трет-бутоксикарбонил-аминопропокси)фенил]-3-(9Н-флюорен-9-илметоксикарбониламино)янтарной кислоты:

К раствору трет-бутилового эфира [3-(4-аминофенокси)пропил]-карбаминовой кислоты (5,41 г; 20,2 ммоль) и трет-бутилового эфира (S)-2-(9H-флюорен-9-илметоксикарбониламино)янтарной кислоты в DMF (40 мл) добавляли при комнатной температуре НОВТ (3 г; 22,2 ммоль) и DIPEA (8,52 г; 66,6 ммоль). Полученный раствор охлаждали до 0°С на ледяной бане и добавляли твердый HBTU (8,43 г; 22,2 ммоль) тремя порциями в течение 5-10 минут. По окончании внесения охлаждающую баню убирали, реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2,5 ч, при этом анализ с помощью LCMS показал отсутствие исходного вещества. Затем реакционную смесь разводили этилацетатом (400 мл) и промывали водой (400 мл), насыщенным раствором бикарбоната натрия (400 мл) и солевым раствором (400 мл). Высушивали над безводным сульфатом магния, фильтрат концентрировали и неочищенный остаток очищали хроматографией на колонке ISCO (330 г) с выделением трет-бутилового эфира полуамида (S)-N-[4-(3-трет-бутоксикарбонил-аминопропокси)фенил]-3-(9Н-флюорен-9-илметоксикарбониламино)янтарной кислоты (выход 95%) в виде белого твердого вещества. ES(+)-HRMS m/e расч. для C37H45N3O8 (M+Na)+ 682,3099; фактич. 682,3105.
Стадия 7: Получение трет-бутилового эфира полуамида (S)-3-амино-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты:

К раствору трет-бутилового эфира полуамида (S)-N-[4-(3-трет-бутокси-карбониламинопропокси)фенил]-3-(9Н-флюорен-9-илметоксикарбониламино)-янтарной кислоты (11 г; 16,67 ммоль) в THF (95 мл) добавляли при комнатной температуре пиперидин (4,26 г; 50 ммоль). Полученный раствор перемешивали 4 ч, при этом анализ с помощью LCMS показал отсутствие исходного вещества. Затем растворитель удаляли в вакууме и остаток подвергали азеотропной перегонке с толуолом с получением белого твердого вещества, которое растворяли в минимальном объеме этилацетата (25-30 мл) в горячем состоянии, а затем его разводили гексаном (250-300 мл) для образования осадка. Полученный твердый осадок собирали фильтрацией и промывали гексаном с получением, после высушивания на воздухе, трет-бутилового эфира полуамида (S)-3-амино-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (выход 81%) в виде белого твердого вещества. ES(+)-HRMS m/e расч. для C22H35N3O6 (M+Na)+ 460,2418; фактич. 460,2416.
Стадия 8: Получение трет-бутилового эфира полуамида (S)-3-(2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензоиламино)ацетиламино)N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты:
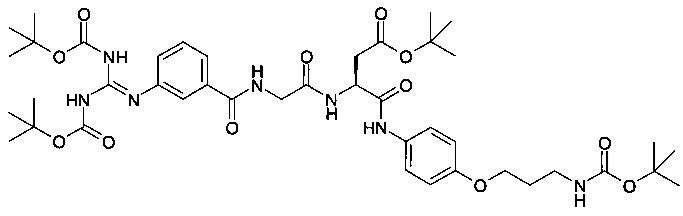
К смеси трет-бутилового эфира полуамида (S)-3-амино-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (2,0 г; 4,58 ммоль), 2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензоил)аминоуксусной кислоты (2,0 г; 4,58 ммоль), HBTU (1,91 г; 5,04 ммоль) и НОВТ (681 мг; 5,04 ммоль) добавляли DMF (15 мл), а затем DIPEA (1,95 г; 15,12 ммоль) при комнатной температуре, в атмосфере азота. Полученный светло-коричневый раствор перемешивали в течение 2 суток, при этом образовывалось большое количество гелеобразного твердого вещества. После этого добавляли воду (~50 мл) и полученную светло-коричневую пасту растворяли в этилацетате (~200 мл) в горячем состоянии. Затем эти два слоя разделяли, и водный слой еще раз экстрагировали этилацетатом (100 мл). Объединенные этилацетатные экстракты промывали насыщенным раствором бикарбоната натрия, водой и солевым раствором и затем органический слой высушивали над безводным сульфатом магния. В результате фильтрации и концентрирования получали неочищенное светло-коричневое твердое вещество, которое очищали хроматографией на колонке ISCO (120 г) с выделением трет-бутилового эфира полуамида (S)-3-(2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)-бензоиламино)ацетиламино)N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (выход 94%) в виде белого твердого вещества. ES(+)HRMS m/e расч. для C42H61N7O12 (М+Н)+ 856,4450; фактич. 856,4451.
Стадия 9: Получение полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-(2-(3-(гуанидино)-бензоиламино)ацетиламино)янтарной кислоты, трифторацетатной соли αVβ3 Лиганд - 1:

К раствору трет-бутилового эфира полуамида (S)-3-(2-(3-(N,N-бис-трет-бутоксикарбонилгуанидино)бензоиламино)ацетиламино)N-[4-(3-трет-бутокси-карбониламинопропокси)фенил]-янтарной кислоты (3,7 г; 4,32 ммоль) в дихлорметане (80 мл) добавляли избыток трифторуксусной кислоты (40 мл) при 0°С (на ледяной бане) в атмосфере азота. Полученный бесцветный раствор перемешивали в течение 1-2 ч при этой температуре и затем нагревали смесь до комнатной температуры, убрав охлаждающую баню. Перемешивали в течение 15 ч, после чего растворитель удаляли в вакууме и остаток подвергали азеотропной перегонке с толуолом. Полученную темно-синюю пасту обрабатывали трет-бутил-метиловым эфиром, но это не привело к образованию хорошего твердого осадка. Затем растворитель удаляли в вакууме и остаток обрабатывали дихлорметаном и диэтиловым эфиром. Полученное светло-коричневое твердое вещество собирали фильтрацией и промывали диэтиловым эфиром. После высушивания на воздухе выделяли 2,7 г (S)-N-[4-(3-аминопропокси)фенил]-3-(2-(3-(гуанидино)бензоил-амино)ацетиламино)янтарной кислоты в виде трифторацетатной соли (выход 85%). ES(+)HRMS m/e расч. для C23H29N7O6 (М+Н)+ 500,2252; фактич. 500,2252.
Стадия 10: Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]пропиониламино]пропокси]фенил]3-[2-[3-(гуанидино)-бензоиламино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 1:

К раствору 2,5-диоксопирролидин-1-илового эфира полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-(2-(3-(гуанидино)бензоиламино)ацетиламино)янтарной кислоты (245 мг; 0,289 ммоль) и 3-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]-пропионовой кислоты (200 мг; 0,289 ммоль) в DMSO (5 мл) добавляли избыток DIPEA (186 мг; 252 мкл; 1,44 ммоль) при комнатной температуре в атмосфере азота. Полученный светло-желтый раствор перемешивали в течение 2 ч, при этом анализ с помощью LCMS показал отсутствие исходного вещества. Затем избыток DIPEA удаляли в вакууме и желаемый продукт выделяли в ходе очистке с помощью ВЭЖХ с получением 212 мг (выход 68%) полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]пропиониламино]пропокси]фенил]3-[2-[3-(гуанидино)бензоиламино]-ацетиламино]-янтарной кислоты в виде светло-желтого твердого. ES(+)HRMS m/e расч. для C49H71N9O18 (М+Н)+ 1074,4990, фактич. 1074,4984.
Пример 2
Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]пропиониламино]пропокси]-фенил]-3-[2-[3-(гуанидино)бензоиламино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 2:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)фенил]3-(2-(3-(гуанидино)бензоиламино)ацетиламино)-янтарной кислоты (245 мг; 0,289 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]-этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (250 мг; 0,289 ммоль) и DIPEA (373 мг; 503 мкл; 2,89 ммоль), после очистки с помощью ВЭЖХ, с получением светло-коричневого масла (312 мг; 86%). ES(+)HRMS m/e расч. для C57H87N9O22 (М+Н)+ 1250,6039, фактич. 1250,6032.
Пример 3
Получение полуамида (S)-N-[[[4-[3-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)-этокси]этокси]этокси]этокси]этокси]этокси]этокси]-1-оксопропил]амино]пропокси]-фенил]-3-[2-[3-[гуанидино]бензоиламино]ацетиламино]-янтарной кислоты трифторацетатной соли; αVβ3 Лиганд - Реагент 3

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)-фенил]-3-(2-(3-(гуанидино)бензоиламино)ацетиламино)янтарной кислоты (245 мг; 0,289 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (172 мг; 0,289 ммоль) и DIPEA (503 мкл; 2,89 ммоль), после очистки с помощью ВЭЖХ, с получением светло-желтого вязкого масла (172 мг; 73%). ES(+)HRMS m/e расч. для C44H67N7O16S (М+Н)+ 982,4438, фактич. 982,4432.
Пример 4
Полуамид (S)-N-[[[4-[3-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]-этокси]этокси]этокси]этокси]этокси]этокси]1-оксопропил]амино]пропокси]фенил]-3-[2-[3-(тетрагидропиримидин-2-илиденамино)бензоиламино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 4:

Стадия 1: Получение ди-трет-бутилового эфира 2-[3-(бензилоксикарбонил-метилкарбамоил)фенилимино]дигидропиримидин-1,3-дикарбоновой кислоты:
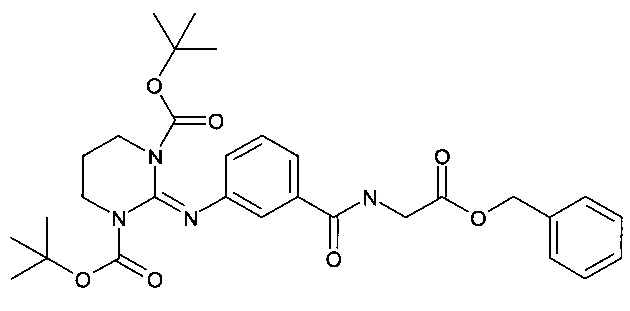
Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 2, исходя из ди-трет-бутилового эфира 2-[3-карбоксифенилимино]дигидропиримидин-1,3-дикарбоновой кислоты (4,85 г, 11,56 ммоль), 2-хлор-4,6-диметокситриазина (2,03 г; 11,56 ммоль), N-метилморфолина (1,17 г; 1,27 мл; 11,56 ммоль), глицин-бензилового эфира, соли p-TsOH (3,9 г; 11,56 ммоль), и второго эквивалента N-метилморфолина (1,17 г; 1,27 мл; 11,56 ммоль) в безводном тетрагидрофуране (90 мл), после очистки хроматографией на колонке ISCO, с получением бесцветного вязкого масла (3,19 г; 49%). ES(+)HRMS m/e расч. для C30H38N4O7 (М+Н)+ 567,2813, фактич. 567,2810.
Стадия 2: Получение ди-трет-бутилового эфира 2-[3-(карбоксиметил-карбамоил)фенилимино]дигидро-пиримидин-1,3-дикарбоновой кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 3, исходя из ди-трет-бутилового эфира 2-[3-(бензилоксикарбонилметилкарбамоил)фенилимино]дигидропиримидин-1,3-дикарбоновой кислоты (475 мг; 0,84 ммоль) и 10% Pd на угле (250 мг) в абсолютном этаноле (20 мл), с получением аморфного белого твердого вещества (355 мг; 89%). ES(+)HRMS m/e расч. для C23H32N4O7 (М+Н)+ 477,2344, фактич. 477,2344.
Стадия 3: Получение ди-трет-бутилового эфира 2-[3-[[[(S)2-трет-бутоксикарбонил-1-[4-(3-трет-бутоксикарбониламинопропокси)фенилкарбамоил]этилкарбамоил]-метил]карбамоил]фенилимино]дигидропиримидин-1,3-дикарбоновой кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 8, исходя из ди-трет-бутилового эфира 2-[3-(карбоксиметил-карбамоил)фенилимино]дигидропиримидин-1,3-дикарбоновой кислоты (332 мг; 0,69 ммоль), трет-бутилового эфира полуамида (S)-3-амино-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (305 мг; 0,69 ммоль), HBTU (290 мг; 0,76 ммоль), НОВТ (104 мг; 0,76 ммоль) и DIPEA (297 мг; 400 мкл; 2,3 ммоль) в DMF (5 мл), после очистки хроматографией на колонке ISCO, с получением аморфного белого твердого вещества (602 мг; 97%). ES(+)HRMS m/e расч. для C45H65N7O12 (М+Н)+ 896,4764, фактич. 896,4764.
Стадия 4: Получение полуамида (S)-N-[4-(3-аминопропокси)фенил]3-[2-[3-(тетрагидропиримидин-2-илиденамино)бензоиламино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - 2:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 9, исходя из ди-трет-бутилового эфира 2-[3-[[[(S)2-трет-бутоксикарбонил-1-[4-(3-трет-бутоксикарбониламинопропокси)фенил-карбамоил]этилкарбамоил]метил]карбамоил]фенилимино]дигидропиримидин-1,3-дикарбоновой кислоты (595 мг; 0,66 ммоль) и трифторуксусной кислоты (10 мл) в дихлорметане (20 мл), с получением светло-коричневого твердого вещества в виде трифторацетатной соли (555 мг; 96%). ES(+)HRMS m/e расч. для C23H29N7O6 (М+Н)+500,2252, фактич. 500,2252.
Стадия 5: Получение полуамида (S)-N-[[[4-[3-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]этокси]этокси]этокси]этокси]этокси]этокси]-1-оксопропил]амино]пропокси]фенил]3-[2-[3-(тетрагидропиримидин-2-илиденамино)-бензоиламино]ацетиламино]-янтарной кислоты (αVβ3 Лиганд - Реагент 4):

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-[2-[3-(тетрагидропиримидин-2-илиденамино)бензоил-амино]ацетиламино]-янтарной кислоты (265 мг; 0,3 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]этокси]-этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (179 мг; 0,3 ммоль) и DIPEA (387 мг; 526 мкл; 3,0 ммоль), после очистки с помощью ВЭЖХ, с получением светло-желтого вязкого масла (208 мг; 68%). ES(+)HRMS m/e расч. для C47H71N7O16S (М+Н)+1022,4751, фактич. 1022,4742.
Пример 5
Получение полуамида (S)-N-[[4-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанил-этокси)этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-этокси]пропиониламино]метил]фенил]-3-[2-[[2-(3-бензилуреидо)тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 5:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-аминометилфенил]3-[2-[[2-(3-бензилуреидо)тиазол-4-карбонил]амино]ацетил-амино]-янтарной кислоты (166 мг; 0,3 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-(2-ацетилсульфанилэтокси)этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (232 мг; 0,3 ммоль) и DIPEA (387 мг; 522 мкл; 3,0 ммоль), после очистки с помощью ВЭЖХ, с получением светло-коричневого масла (362 мг; 99%). ES(+)HRMS m/e расч. для C54H81N7O20S2 (М+Н)+ 1212,5051, фактич. 1212,5058.
Пример 6
Полуамид (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропиониламино]пропокси]фенил]-3-[2-[[2-(3-бензил-уреидо)тиазол-4-карбонил]-амино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 6:

Стадия 1: Получение гидробромида этилового эфира 2-амино-тиазол-4-карбоновой кислоты:
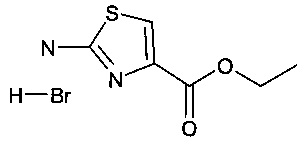
Трехгорловую колбу на 3 л с обратным холодильником, термометром и механической мешалкой помещали на масляную баню и вносили в нее тиомочевину (48,2 г; 634 ммоль) и этилбромпируват (137 г; 88,3 мл; 634 ммоль). Реакционную смесь медленно и осторожно нагревали до момента образования прозрачного раствора (60°С), при котором реакция становилась экзотермической (температура реакционной смеси достигала 110°С), и перемешивали по мере отвердевания реакционной смеси. К смеси добавляли ЕА (500 мл), убирали нагрев реакционной смеси и остужали ее до комнатной температуры. Твердое вещество отфильтровывали и промывали с помощью ЕА и Et2O с получением этилового эфира 2-аминотиазол-4-карбоновой кислоты, гидробромидной соли, в виде белого твердого вещества (157 г; 98%).
Стадия 2: Получение этилового эфира 2-(3-бензилуреидо)тиазол-4-карбоновой кислоты:

В реакционный сосуд, содержащий гидробромидную соль этилового эфира 2-аминотиазол-4-карбоновой кислоты (66,6 г; 263 ммоль), 4-этилморфолин (60,7 г; 67,1 мл; 527 ммоль) и безводный DMF (660 мл), добавляли бензилизоцианат (42,1 г; 316 ммоль), реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение 7 ч, вносили дополнительное количество бензилизоцианата (42,1 г; 316 ммоль) и реакционную смесь перемешивали при комнатной температуре в атмосфере аргона в течение ночи. На следующий день реакционную смесь концентрировали на 2/3, разбавляли водой (1,6 л), и полученный осадок фильтровали и высушивали отсасыванием в течение ночи с получением этилового эфира 2-(3-бензилуреидо)-тиазол-4-карбоновой кислоты в виде грязно-белого твердого вещества (146 г; 182%). Анализ ЯМР показал, что влажное вещество содержало воду и DMF, и его использовали в таком виде.
Стадия 3: Получение 2-(3-бензилуреидо)тиазол-4-карбоновой кислоты:

В реакционный сосуд, содержащий влажный этиловый эфир 2-(3-бензил-уреидо)тиазол-4-карбоновой кислоты (146 г; теоретически приняли за 263 ммоль) добавляли этанол (1,2 л) и 1 н. NaOH (1,31 л). Реакционную смесь нагревали (60°С) и перемешивали в атмосфере аргона в течение 4 ч. Реакционную смесь фильтровали и твердое вещество отбрасывали. Фильтрат охлаждали на ледяной бане и подкисляли с помощью 1 н. HCl (1,31 л), полученный осадок фильтровали, промывали водой и высушивали отсасыванием в течение двух суток с получением 2-(3-бензилуреидо)-тиазол-4-карбоновой кислоты (72 г; 99%).
Стадия 4: Получение этилового эфира {[2-(3-бензил-уреидо)тиазол-4-карбонил]амино}-уксусной кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 2, исходя из 2-(3-бензилуреидо)тиазол-4-карбоновой кислоты (72 г; 259 ммоль), 2-хлор-4,6-диметокситриазина (45,5 г; 259 ммоль), N-метилморфолина (26,2 г; 28,5 мл, 259 ммоль), глицин-этилового эфира гидрохлорида (36,2 г; 259 ммоль) и второго эквивалента N-метилморфолина (26,2 г; 259 ммоль), после проведения кристаллизации и хроматографии, с получением белого кристаллического твердого вещества (72,6 г; 76%).
Стадия 5: Получение {[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-уксусной кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 3, исходя из этилового эфира {[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-уксусной кислоты (72,3 г; 199 ммоль), метанола (200 мл), THF (1 л), 1 н. NaOH (0,2 л) и 1 н. HCl (0,2 л), после нейтрализации, выпаривания и кристаллизации, с получением белых кристаллов (73,1 г; 109%). По данным анализа ЯМР, продукт содержал растворитель, и его использовали в таком виде.
Стадия 6: Получение трет-бутилового эфира полуамида (S)-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)N-[4-(3-трет-бутокси-карбониламинопропокси)фенил]-янтарной кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 8, исходя из {[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-уксусной кислоты, трет-бутилового эфира полуамида (S)-3-амино-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (1,5 г; 3,43 ммоль), HBTU (1,43 г; 3,77 ммоль), НОВТ (0,51 г; 3,77 ммоль) и DIPEA (1,46 г; 1,97 мл; 11,3 ммоль) с получением белого твердого вещества (1,83 г; 70%). ES(+)HRMS m/e расч. для C36H47N7O9S (M+Na)+ 776,3048, фактич. 776,3050.
Стадия 7: Получение полуамида (S)-N-[4-(3-аминопропокси)-фенил]-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)янтарной кислоты, αVβ3 Лиганд - Реагент 8, 40389-052) αVβ3 Лиганд - 3:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 9, исходя из трет-бутилового эфира полуамида (S)-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)-N-[4-(3-трет-бутоксикарбониламинопропокси)фенил]-янтарной кислоты (1,83 г; 2,48 ммоль), с получением белого твердого вещества (1,29 г; 88%). ES(+)HRMS m/e расч. для C17H31N7O7S (М+Н)+598,2079, фактич. 598,2077.
Стадия 8: Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]пропиониламино]пропокси]фенил]-3-[2-[[2-(3-бензилуреидо)-тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)янтарной кислоты, αVβ3 Лиганд - Реагент 8, 40389-052) (242 мг; 0,405 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (280 мг; 0,405 ммоль) и DIPEA (262,2 мг; 353 мкл; 2,03 ммоль), после очистки с помощью ВЭЖХ, с получением светло-коричневой смолы (206 мг; 43%) ES(+)HRMS m/e расч. для C53H73N9O19S (М+Н)+ 1172,4816, фактич. 1172,4806.
Пример 7
Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси] этокси]этокси]этокси]этркси]этокси]пропиониламино]этокси]фенил]-3-[2-[[2-(3-бензилуреидо)тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 7, 40389-058):

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)янтарной кислоты αVβ3 Лиганд - Реагент 8, 40389-052) (155 мг; 0,260 ммоль), 2,5-диоксопирролидин-1-илового эфира 3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (225 мг; 0,260 ммоль) и DIPEA (167 мг; 226 мкл; 1,29 ммоль), после очистки с помощью ВЭЖХ, с получением светло-желтой смолы (149 мг; 42%) ES(+)HRMS m/e расч. для C61H89N9O23S (М+Н)+ 1348,5865, фактич. 1348,5864.
Пример 8
Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-ацетилсульфанилэтокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]пропиониламино]пропокси]-фенил]3-[2-[[2-(3-бензилуреидо)тиазол-4-карбонил]амино]ацетиламино]-янтарной кислоты; αVβ3 Лиганд - Реагент 8:

Указанное в заголовке соединение получали таким же способом, как описано в Примере 1, Стадия 10, исходя из полуамида (S)-N-[4-(3-аминопропокси)фенил]-3-(2-{[2-(3-бензилуреидо)тиазол-4-карбонил]амино}-ацетиламино)-янтарной кислоты, 40389-052) (120 мг; 0,201 ммоль), 2,5-диоксопирролидин-1 -илового эфира 3-[2-[2-[2-[2-[2-[2-[2-[2-ацетилсул ьфанил-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропионовой кислоты (120 мг; 0,201 ммоль) и DIPEA (129,2 мг; 174 мкл; 1,01 ммоль), после очистки с помощью ВЭЖХ, с получением светло-коричневой смолы (206 мг; 43%) ES(+)HRMS m/e расч. для C48H69Ν7Ο17S2 (М+Н)+ 1080,4264, фактич. 1080,4257.
Получение N-(2-метоксибензил)ацетамида

К чистому 2-метоксибензиламину (40 г; 0,29 моль) добавляли по каплям Ac2O (80 мл; 0,85 моль) на водно-ледяной бане. Затем реакционную смесь перемешивали при комнатной температуре в течение еще 2 ч. Полученную смесь вливали в 50 мл воды и экстрагировали этилацетатом (3×300 мл). Объединенные органические слои промывали водой (3×150 мл), солевым раствором (100 мл) и высушивали над безводным сульфатом натрия. После фильтрации и концентрирования остаток суспендировали в петролейном эфире (200 мл) и перемешивали в течение 10 мин, затем фильтровали и высушивали с получением указанного в заголовке соединения (36,5 г; 69,9%) в виде чистого белого твердого вещества. 1Н ЯМР (300 МГц, CDCl3): δ 7,31-7,25 (m, 2Н), 6,95-6,87 (m, 2Н), 6,01 (brs, 1Н), 4,44 (d, 2Н, J=6,0 Гц), 3,87(s, 3H), 1,98 (s, 3H).
Получение N-(2-гидроксибензил)ацетамида

N-(2-метоксибензил)ацетамид (6,5 г; 0,036 моль) растворяли в DCM (100 мл), и полученный раствор охлаждали до -10°С в атмосфере азота. К этому раствору добавляли по каплям BBr3 (15 мл; 0,15 моль). По окончании внесения ледяную баню убирали и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Реакционную смесь повторно охлаждали до -10°С и гасили водой (20 мл). Смесь экстрагировали с помощью DCM (3×100 мл), и органический слой промывали водой (3×100 мл), солевым раствором (100 мл) и высушивали с получением указанного в заголовке соединения (4,3 г; 71,7%) в виде твердого вещества. Его использовали непосредственно на следующей стадии без дополнительной очистки. 1Н ЯМР (300 МГц, DMSO): δ 9,56 (brs, 1Н), 8,27 (brs, 1Н), 7,09-7,03 (m, 2Н), 6,79-6,72 (m, 2Н), 4,16 (d, 2Н, J=6,0 Гц), 1,87 (s, 3H). LC-MS: 166,2 [М+Н]+, tR=1,15 мин.
Получение трет-бутил 3-бромпропилкарбамата

К суспензии 3-бромпропиламина гидробромида (50 г; 0,228 моль) в DCM (1000 мл) добавляли (Вос)2O (52 г; 0,238 моль) и триэтиламин (100 мл; 0,722 моль). Затем реакционную смесь перемешивали при комнатной температуре еще в течение 3 ч. Растворитель удаляли в вакууме и остаток промывали петролейным эфиром (500 мл). Полученную смесь фильтровали и полученный фильтрат выпаривали с получением указанного в заголовке соединения (54 г; 99,2%) в виде бесцветного масла. 1Н ЯМР (300 МГц, CDCl3): δ 4,69 (brs, 1Н), 3,43 (t, 2Н, J=6,6 Гц), 3,27-3,23 (m, 2Н), 2,10-2,03 (m, 2Н), 1,27 (s, 9Н).
Получение трет-бутилового эфира {3-[2-(ацетиламино-метил)фенокси]пропил}-карбаминовой кислоты

К раствору N-(2-гидроксибензил)ацетамида (18,8 г; 0,11 моль) в DMF (100 мл) добавляли при перемешивании трет-бутил-3-бромпропилкарбамат (32 г; 0,135 моль) и K2CO3 (47 г; 0,34 моль) при комнатной температуре. Затем реакционную смесь перемешивали при комнатной температуре в течение 16 ч. Полученную смесь фильтровали и растворитель удаляли в вакууме с получением неочищенного продукта, который очищали хроматографией на силикагеле (петролейный эфир / этилацетат = 1:3-1:4) с получением указанного в заголовке соединения (23 г; 62,8%) в виде твердого вещества. 1Н ЯМР (300 МГц, CDCl3): δ 8,08 (t, 1Н, J=5,8 Гц), 7,20-7,13 (m, 2Н), 6,94-6,86 (m, 3H), 4,21 (d, 2Н, J=5,4 Гц), 3,97 (t, 2H, J=5,9 Гц), 3,14-3,08 (m, 2H), 1,87 (s, 3H), 1,82-1,86 (m, 2H), 1,36 (s, 9H). LC-MS: 323,1 [M+H]+, tR=2,98 мин.
Получение трет-бутил 3-(2-(аминометил)фенокси)пропилкарбамата

трет-Бутиловый эфир {3-[2-(ацетиламинометил)фенокси]пропил}-карбаминовой кислоты (10,4 г; 0,032 моль) суспендировали в гидразине гидрате (150 мл; 85%) и эту реакционную смесь кипятили с обратным холодильником при перемешивании в течение 20 ч. Полученную смесь охлаждали до комнатной температуры и экстрагировали диэтиловым эфиром (3×150 мл). Объединенные органические фазы промывали солевым раствором (150 мл) и высушивали над безводным сульфатом натрия. После фильтрации и концентрирования остаток очищали хроматографией на силикагеле (DCM/МеОН=20:1~1:1) с получением указанного в заголовке соединения (4 г; 44,6%). 1Н ЯМР (300 МГц, CDCl3): δ 7,33-7,28 (m, 2Н), 7,01-6,90 (m, 2Н), 5,41-5,36 (m, 1Н), 4,15-4,11(m, 2Н), 3,91 (s, 2Н), 3,44-3,40(m, 2Н), 2,09-2,05 (m, 2Н), 1,90 (brs, 2Н), 1,50 (s, 9Н). LC-MS: 281,1 [М+Н]+, tR=2,33 мин.
Синтез 2-аминотиазол-4-карбоновой кислоты 120

К суспензии соединения 150 (125 г; 0,525 моль) в THF (2500 мл) добавляли по каплям раствор NaOH (63 г в 790 мл воды) в течение одного часа. Полученную смесь перемешивали при комнатной температуре в течение 2 ч. Растворитель выпаривали и остаток обрабатывали с помощью 2 н. HCl (770 мл), фильтровали и высушивали с получением соединения 120 (75 г; 99,1%) в виде желтого твердого вещества. 1Н ЯМР (300 МГц, DMSO): δ 7,38 (s, 1Н), 7,13 (brs, 2Н).
Синтез метилового эфира [(2-аминотиазол-4-карбонил)амино]уксусной кислоты 8

К раствору соединения 120 (1 г; 6,93 ммоль) в DMF (50 мл) добавляли соединение 121 (0,97 г; 7,73 ммоль), DIPEA (3,5 мл; 21,1 ммоль) и HATU (2,93 г; 7,7 ммоль). Полученный раствор перемешивали при комнатной температуре в течение ночи. Этот реакционный раствор выпаривали в вакууме при 85°С досуха, обрабатывали с помощью 30 мл THF и перемешивали при комнатной температуре в течение около 0,5 ч, затем фильтровали, промывали небольшим количеством этанола и высушивали с получением соединения 122 (0,7 г; 46,9%) в виде желтого твердого вещества. 1Н ЯМР (300 МГц, DMSO): δ 8,07 (t, 1Н, J=6,0 Гц), 7,22 (s, 1Н), 7,12 (brs, 1Н), 3,97(d, 2Н, J=6,0 Гц), 3,64 (s, 3H).
Получение метилового эфира [(2-{3-[2-(3-трет-бутоксикарбониламинопропокси)-бензил]уреидо}-тиазол-4-карбонил)амино]уксусной кислоты

К раствору трет-бутил 3-(2-(аминометил)фенокси)пропилкарбамата (489 мг; 1,74 ммоль) в DCM (50 мл) добавляли при перемешивании раствор DIPEA (449 мг; 3,48 ммоль) в DCM (5,0 мл). Этот раствор добавляли по каплям к другому раствору трифосгена (180 мг; 0,61 ммоль) в DCM (5 мл) при 0°С в атмосфере азота. По окончании внесения полученную смесь перемешивали при 0°С еще в течение 15 мин. Этот раствор выпаривали с получением трет-бутилового эфира [3-(2-изоцианатометилфенокси)пропил]карбаминовой кислоты в виде белого твердого вещества. Его растворяли в 2,5 мл DMF и использовали непосредственно на следующей стадии.
К раствору метилового эфира [(2-аминотиазол-4-карбонил)амино]уксусной кислоты (374 мг; 1,74 ммоль) и DIPEA (449 мг; 3,48 ммоль) в DMF (2,5 мл) добавляли раствор трет-бутилового эфира [3-(2-изоцианатометилфенокси)пропил]-карбаминовой кислоты в DMF (2,5 мл приготовленного выше) при комнатной температуре. Эту реакционную смесь перемешивали при 80°С в течение 2 ч. Затем полученную смесь охлаждали и вливали в 50 мл воды, экстрагировали этилацетатом (3×100 мл). Органическую фазу промывали солевым раствором (20 мл) и высушивали над безводным сульфатом натрия. После фильтрации и концентрирования остаток очищали хроматографией (петролейный эфир / этилацетат = 1:2~1:1) с получением указанного в заголовке соединения (165 мг; 18,2% в две стадии) в виде желтого твердого вещества.
1Н ЯМР (300 МГц, CD3OD): δ 7,55 (s, 1Н), 7,17-7,14 (m, 2Н), 6,87-6,77 (m, 2Н), 4,34 (s, 2Н), 4,01-3,96 (m, 4Н), 3,64 (s, 3H), 3,22-3,19 (m, 2Н), 1,91-1,86 (m, 2Н), 1,31 (s, 9Н). LC-MS: 522,2 [М+Н]+, tR=2,73 мин.
Получение [(2-{3-[2-(3-трет-бутоксикарбониламинопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]уксусной кислоты

К раствору метилового эфира [(2-{3-[2-(3-трет-бутоксикарбониламино-пропокси)бензил]уреидо}-тиазол-4-карбонил)амино]уксусной кислоты (0,165 г; 0,31 ммоль) в THF (5 мл) добавляли по каплям раствор LiOH.H2O (0,132 г; 3,1 ммоль) в воде (1 мл) при комнатной температуре. Затем этот раствор перемешивали при данной температуре в течение 16 ч. Растворитель выпаривали и остаток разбавляли с помощью 5 мл воды. Водный раствор экстрагировали этилацетатом (10 мл) и водную фазу подкисляли 1 н. раствором лимонной кислоты до рН~5. Водный раствор экстрагировали этилацетатом (3×15 мл), и объединенные органические слои промывали солевым раствором (10 мл) и высушивали над Na2SO4. После фильтрации и концентрирования, указанное в заголовке соединение (0,080 г; 50,9%) получали в виде твердого вещества.
1Н ЯМР (300 МГц, DMSO): δ 10,57 (brs, 1Н), 8,06 (t, 1Н, J=5,8 Гц), 7,63 (s, 1Н), 7,26-7,20 (m, 2Н), 6,98-6,88 (m, 4Н), 4,32 (d, 2Н, J=5,7 Гц), 4,03-3,91 (m, 4Н), 3,16-3,10 (m, 2Н), 1,90-1,83 (m, 2Н), 1,36 (s, 9Н). LC-MS: 508,2 [М+Н]+, tR=2,58 мин
Получение метил-3-(2-(2-(3-(2-(3-(трет-бутоксикарбониламино)пропокси)бензил)-уреидо)тиазол-4-карбоксамидо)ацетамидо)3-фенилпропионата

К раствору [(2-{3-[2-(3-трет-бутоксикарбониламинопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]уксусной кислоты (0,7 г; 1,38 ммоль) в THF (25 мл) добавляли при комнатной температуре DIPEA (1,4 г; 10,8 ммоль) и HATU (0,525 г; 1,38 ммоль). Затем эту реакционную смесь перемешивали в течение 20 мин. Добавляли метил-3-амино-3-фенилпропионат гидрохлорид (0,29 г; 1,34 ммоль) одной порцией, и эту реакционную смесь перемешивали в течение 16 ч. Растворитель удаляли при пониженном давлении и остаток растворяли в 50 мл этилацетата, промывали 1 н. раствором NaOH (3×15 мл), затем 1 н. HCL (3×15 мл) и водой (3×15 мл), после этого солевым раствором (15 мл) и высушивали над сульфатом натрия. После фильтрации и концентрирования остаток очищали хроматографией на силикагеле (элюировали смесью этилацетат / петролейный эфир / метанол = 25:25:2) с получением указанного в заголовке соединения (0,54 г; 58,6%) в виде твердого вещества.
1Н ЯМР (300 МГц, CD3OD): δ 7,67 (s, 1Н), 7,37-7,24 (m, 7Н), 6,99-6,92 (m, 2Н), 5,40 (t, 1Н, J=7,3 Гц), 4,46 (s, 2Н), 4,13-4,04 (m, 4Н), 3,67 (s, 3H), 3,34-3,32 (m, 2Н), 2,90-2,86 (m, 2Н), 2,03-1,98 (m, 2Н), 1,43 (s, 9Н). LC-MS: 669,2 [М+Н]+, tR=3,11 мин
Получение метил-3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)3-фенилпропионата гидрохлорида

К насыщенному раствору HCl в этилацетате (250 мл) добавляли по каплям при перемешивании раствор метил-3-(2-(2-(3-(2-(3-(трет-бутоксикарбониламино)-пропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)3-фенилпропионата (5,5 г; 8,23 ммоль) в этилацетате (30 мл) при комнатной температуре, затем полученную смесь перемешивали при комнатной температуре в течение 16 ч. Органический растворитель отгоняли при пониженном давлении, и полученное твердое вещество обрабатывали диэтиловым эфиром (50 мл), и затем фильтровали с получением указанного в заголовке соединения (4,7 г; 94,5%) в виде белого твердого вещества.
1Н ЯМР (300 МГц, CD3OD): δ 7,69 (s, 1Н), 7,36-7,27 (m, 7Н), 7,02-6,95 (m, 2Н), 5,40 (t, 1Н, J=7,4 Гц), 4,48 (s, 2Н), 4,19 (t, 2Н, J=5,6 Гц), 4,06 (d, 2Н, J=2,1 Гц), 3,63 (s, 3H) 3,24 (t, 2Н, J=7,2 Гц), 2,90-2,86 (m, 2Н), 2,24-2,15 (m, 2Н). LC-MS: 569 [М+Н]+, tR=3,00 мин. ВЭЖХ: 99,85% при 214 нм, 99,14% при 254 нм, tR=4,05 мин
Получение 3-{2-[(2-{3-[2-(3-аминопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]ацетиламино}-3-фенил-пропионовой кислоты, αVβ3 Лиганд-4:

К раствору гидрохлорида метилового эфира 3-{2-[(2-{3-[2-(3-аминопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]ацетиламино}-3-фенил-пропионовой кислоты (2,0 г; 3,31 ммоль, экв.: 1,00) в метаноле (10 мл) добавляли 2 M гидроксид натрия (33,1 ммоль, экв.: 10,00). Эту реакционную смесь перемешивали при 55°С в течение ночи. В ходе нейтрализации с помощью 1 н. HCl неочищенный продукт выпадал в осадок и его собирали фильтрацией (2,19 г). Неочищенный продукт очищали с помощью обращенно-фазной ВЭЖХ с получением 1041 мг указанного в заголовке соединения в виде соли TFA.
HRMS m/e 555,2006 (М+Н)+
Получение (R)-этил 3-(2-(2-(3-(2-(3-(трет-бутоксикарбониламино)пропокси)бензил) уреидо)тиазол-4-карбоксамидо)ацетамидо)-3-(пиридин-3-ил)пропионата

К раствору [(2-{3-[2-(3-трет-бутоксикарбониламинопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]уксусной кислоты (7,08 г; 13,9 ммоль) и HATU (5,8 г; 15,2 ммоль) в THF (150 мл) при комнатной температуре в атмосфере азота добавляли DIPEA (13,9 г; 107,7 ммоль). Затем эту реакционную смесь перемешивали при комнатной температуре еще в течение 20 мин. Добавляли одной порцией (R)-этил-3-амино-3-(пиридин-3-ил)пропионат гидрохлорид (4,45 г; 16,8 ммоль) и эту реакционную смесь перемешивали при данной температуре в течение 16 ч. Растворитель выпаривали и остаток растворяли в 500 мл этилацетата, промывали с помощью 0,3 н. HCL (2×100 мл), солевым раствором (100 мл) и высушивали с получением неочищенного продукта, который очищали хроматографией на силикагеле (элюировали 10% метанолом в этилацетате) с получением указанного в заголовке соединения (7,2 г; 77,3%) в виде твердого вещества. LC-MS: 684,2 [М+Н]+, tR=2,60 мин.
Получение (R)-этил 3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)-3-(пиридин-3-ил)пропионата гидрохлорида

К раствору (R)-этил-3-(2-(2-(3-(2-(3-(трет-бутоксикарбониламино)пропокси) бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)3-(пиридин-3-ил)пропионата (2,3 г; 3,36 ммоль) в этаноле (50 мл) добавляли при перемешивании ацетилхлорид (5 мл) при 0°С. Затем этот раствор перемешивали при комнатной температуре в течение 1 ч. Растворитель удаляли при пониженном давлении и остаток промывали этилацетатом (100 мл) с получением указанного в заголовке соединения (1,1 г; 52,6%) в виде белого твердого вещества. 1Н ЯМР (300 МГц, CD3OD): δ 8,97 (s, 1Н), 8,83-8,81 (m, 1Н), 8,73 (d, 1Н, J=8,1 Гц), 8,13 (d, 1Н, J=7,0 Гц), 7,73-7,75 (m, 1Н), 7,34-7,28 (m, 2Н), 7,04-6,96 (m, 2Н), 5,56-5,51 (m, 1Н), 4,49 (s, 2Н), 4,23-4,07 (m, 6Н), 3,26 (t, 2Н, J=6,9 Гц), 3,11 (d, 2Н, J=6,9 Гц), 2,26-2,22 (m, 2Н), 1,24 (t, 3H, J=7,0 Гц). LC-MS: 584,0 [М+Н]+, tR=2,17 мин. ВЭЖХ: 100% при 214 нм, 100% при 254 нм, tR=5,91 мин.
Получение (R)3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)-3-(пиридин-3-ил)пропионовой кислоты, αVβ3 Лиганд-5

К раствору (R)-этил 3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)3-(пиридин-3-ил)пропионата дигидрохлорида (1,99 г; 3,1 ммоль) в МеОН (10 мл) добавляли 2 н. гидроксид натрия (15,5 мл; 31,0 ммоль) и полученную реакционную смесь перемешивали при 55°С в течение ночи. Затем ее нейтрализовали 1 н. хлороводородной кислотой и очищали обращенно-фазной ВЭЖХ с получением 1,10 г указанного в заголовке соединения.
Пример 11
Получение αVβ3 Лиганда - Реагента 9

Указанное в заголовке соединение получали аналогичным образом из 3-{2-[(2-{3-[2-(3-аминопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]ацетиламино}-3-фенил-пропионовой кислоты и 2,5-диоксопирролидин-1-илового эфира 3-(2-{2-[2-(2-ацетилсульфанилэтокси)этокси]этокси}-этокси)-пропионовой кислоты, как показано в Примере 7. HRMS m/e 883,2978 (M+Na)+
Пример 12
Получение αVβ3 Лиганда - Реагента 10

Указанное в заголовке соединение получали аналогичным образом из 3-{2-[(2-{3-[2-(3-аминопропокси)бензил]уреидо}-тиазол-4-карбонил)амино]ацетиламино}-3-фенил-пропионовой кислоты и N-гидроксисукцинимидилового эфира 1-(S-ацетил)меркапто-3,6,9,12,15,18,21,24,27,30,33,3б-додекаоксанонатриаконтан-39-овой кислоты, как показано в Примере 7. HRMS m/e 1213,5248 (М+Н)+
Пример 13
Получение αVβ3 Лиганда - Реагента 11

Указанное в заголовке соединение получали аналогичным образом из (R)-3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)-3-(пиридин-3-ил)-пропионовой кислоты трифторацетата и 2,5-диоксопирролидин-1-илового эфира 3-(2-{2-[2-(2-ацетилсульфанилэтокси)этокси]этокси}-этокси)пропионовой кислоты, как показано в Примере 7. HRMS m/e 884,2924 (М+Н)+
Пример 14
Получение αVβ3 Лиганда - Реагента 12

Указанное в заголовке соединение получали аналогичным образом из (R)-3-(2-(2-(3-(2-(3-аминопропокси)бензил)уреидо)тиазол-4-карбоксамидо)ацетамидо)3-(пиридин-3-ил)пропионовой кислоты трифторацетата и N-гидроксисукцинимидилового эфира 1-(8-ацетил)меркапто-3,6,9,12,15,18,21,24,27,30,33,36-додекаоксанонатриаконтан-39-овой кислоты, как показано в Примере 7. HRMS m/e 1214,5205 (М+Н)+.
ПОЛУЧЕНИЕ МЕЧЕННЫХ ФЛЮОРЕСЦЕИНОМ (FITC) НАЦЕЛИВАЮЩИХ РЕАГЕНТОВ
Специфические реагенты можно дериватизировать флюорофорами, которые могут быть полезны в исследованиях по отслеживанию их связывания с клетками, которые экспрессируют рецепторы к специфическим низкомолекулярным соединениям. Такие молекулы можно изготавливать любым из двух способов. Во-первых, можно проводить реакцию нацеленных малеимидов с 2-[(5-флюоресцеинил)аминокарбонил]этилмеркаптаном. Или же можно проводить реакцию в однореакторном сосуде нацеливающих низкомолекулярных лигандов - антагонистов интегрина с 2-[(5-флюоресцеинил)аминокарбонил]этилмеркаптаном и бифункциональным ПЭГ-реагентом, как показано на схемах 17 и 18.
Пример по Способу а)
Получение полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)-пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]-пропиониламино]пропокси]-фенил]-3-[2-[3-(гуанидино)-бензоиламино]-ацетиламино]-янтарной кислоты-FITC
К желтой суспензии полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5- -диоксо-2,5-дигидропиррол-1-ил)-пропиониламино]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]этокси]пропиониламино]-пропокси]фенил]-3-[2-[3-(гуанидино)-бензоиламино]-ацетиламино]-янтарной кислоты (37,5 мг; 0,03 ммоль) и 2-[(5-флюоресцеинил)аминокарбонил]этил-меркаптана (FITC-реагент) (15,6 мг; 0,036 мл) в метаноле (5 мл) добавляли при комнатной температуре, в атмосфере азота избыток DIPEA (38,7 мг; 52 мкл; 0,3 ммоль). Полученную светло-желтую суспензию перемешивали в течение 2 ч, при этом анализ с помощью LCMS показал отсутствие исходного вещества. Затем избыток DIPEA удаляли в вакууме и выделяли желаемый продукт после проведения очистки с помощью ВЭЖХ, с получением 25 мг (выход 50%) FITC-производного полуамида (S)-N-[4-[3-[3-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-диоксо-2,5-дигидропиррол-1-ил)пропиониламино]этокси]этокси]этокси]этокси]этокси]-этокси]этокси]этокси]этокси]этокси]этокси]этокси]пропиониламино]пропокси]-фенил]-3-[2-[3-(гуанидино)-бензоиламино]-ацетиламино]-янтарной кислоты в виде коричневого твердого вещества.
ES(+)-HRMS m/e расч. для C80H104N10O28S (М+2Н)2+ 843,3444; фактич. 843,3437. Данные LCMS=М+Н, 1687,6

Стадия 1. Цистамин дигидрохлорид (68 мг; 0,301 ммоль) и DIEA (110 мкл; 2,1 экв.) растворяли в DMF (10 мл), после чего добавляли NHS-флуоресцеин, смесь 5- и 6-карбоксифлуоресцеина (300 мг; 0,634 ммоль) и полученную реакционную смесь перемешивали в течение ночи при комнатной температуре. Затем ее разводили этилацетатом и промывали три раза водой и один раз солевым раствором. Экстракт высушивали над безводным сульфатом натрия, концентрировали при пониженном давлении, перерастворяли в небольшом количестве метанола и этилацетата и затем обрабатывали диэтиловым эфиром с получением 140 мг аддукта флуоресцеина и цистамина в виде ярко-оранжевого твердого вещества.
Стадия 2. Аддукт флуоресцеина и цистамина (80 мг; 0,092 ммоль) растворяли в смеси 3:1 метанола и воды (4 мл) и добавляли ТСЕР гидрохлорид (80 мг; 3 экв.). Полученную реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Продукт очищали путем ВЭЖХ с получением 78 мг вещества. LRMS (ESI) 435,0.
ПОЛУЧЕНИЕ КОНЪЮГАТОВ, МЕЧЕННЫХ ФЛУОРЕСЦЕИНОМ НИЗКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ С ПЭГ

Общий способ. К раствору лиганда (1 экв.) в DMSO добавляли DIEA (2 экв.) и SM(PEG)4n (1 экв.). Полученную реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Затем добавляли флуоресцеин с тиольной ножкой (1 экв.) и реакционную смесь перемешивали еще в течение 10 мин. Продукт очищали путем ВЭЖХ.
Способы ковалентного присоединения низкомолекулярных интегрин-нацеливающих лигандов к 5ʹ-тиол-миРНК олигонуклеотидам
Получение миРНК
Синтез олигорибонуклеотидов
Олигорибонуклеотиды синтезировали по фосфоамидитной (амидофосфитной) технологии на твердой фазе с использованием установки для синтеза ABI 394 (Applied Biosystems), в масштабе 10 мкмоль. Информацию о последовательности РНК см. в табл.1 и 2. Соответствующие миРНК направлены к конститутивному гену АНА1. Синтез проводили на твердой подложке из стекла с контролируемым размером пор (CPG,


Расщепление и снятие защиты олигомера, связанного с подложкой
После завершения твердофазного синтеза высушенную твердую подложку переносили в пробирку на 15 мл и обрабатывали метиламином в метаноле (2 М, Aldrich) в течение 180 мин при 45°С. После центрифугирования супернатант переносили в новую пробирку на 15 мл, и стекло CPG промывали с помощью 1200 мкл N-метилпирролидин-2-она (NMP, Fluka, Бухс, Швейцария). Смыв объединяли с метанольным раствором метиламина и добавляли 450 мкл триэтиламин-тригидрофторида (TEA⋅3HF, Alfa Aesar, Карлсруэ, Германия). Эту смесь доводили до 65°С в течение 150 мин. После охлаждения до комнатной температуры добавляли 0,75 мл NMP и 1,5 мл этокситриметилсилана (Fluka, Бухс, Швейцария). Через 10 мин выпавший в осадок олигорибонуклеотид собирали центрифугированием, супернатант отбрасывали и твердое вещество растворяли в 1 мл буфера А (см. ниже).
Очистка олигорибонуклеотидов
Неочищенные олигорибонуклеотиды очищали с помощью ВЭЖХ на сильном катионообменнике (SAX) с использованием препаративной колонки 22×250 мм DNA Рас 100 (Dionex, Идштайн, Германия) на установке АКТА Explorer (GE Healthcare). Буфер А содержал 10 мМ NaClO4, 1 мМ EDTA, 10 мМ Tris, рН 7,4, 6 M мочевину и 20% ацетонитрил. Буфер В содержал 500 мМ NaClO4 в Буфере А. Скорость потока составляла 4,5 мл/мин. Вели запись УФ-сигнала при 260 и 280 нм. Использовали градиент от 20%В до 45%В за 55 мин. Нужные фракции объединяли и осаждали с помощью 3 M NaOAc, рН=5,2 и 70% этанола.
Неочищенные, меченные с помощью Nu547 олигомеры очищали в ходе препаративной ВЭЖХ с использованием колонки XTerra Prep MS С8 10×50 мм (Waters, Эшборн, Германия) на установке АКТА Explorer (GE Healthcare). Буфер А содержал 100 мМ ацетат триэтиламмония (Biosolve, Валкенсвард, Нидерланды), а буфер В содержал 50% ацетонитрил в буфере А. Скорость потока составляла 5 мл/мин. Записывали УФ-сигнал при 260, 280 и 547 нм (в случае меченного с помощью Nu547 олигорибонуклеотида). Использовали градиент от 5%В до 60%В за 58 объемов колонки (CV). Нужные фракции объединяли и осаждали с помощью 3 M NaOAc, рН=5,2 и 70% этанола.
В конце очищенный олигомер обессаливали с помощью эксклюзионной хроматографии на колонке, содержащей Sephadex G-25 (GE Healthcare). Концентрацию этого раствора определяли по измерению абсорбции при 260 нм на УФ-фотомере (Beckman Coulter, Крефельд, Германия). До ренатурации отдельные цепи хранили в виде замороженных растворов при -20°С.
Получение низкомолекулярных конъюгатов РНК
Низкомолекулярные соединения, содержащие малеимидную функциональную группу, ковалентно конъюгировали с РНК через тиоэфирную связь. Для осуществления этого химического процесса ~60 мг РНК, содержащей 5'-дисульфидный линкер, восстанавливали в воде (5 мл) до соответствующего тиола, с помощью 1 мл ТСЕР (0,5 М в воде, производства Sigma Aldrich). Как только анализ с помощью аналитической анионнообменной ВЭЖХ показывал завершение реакции (~2 ч при комнатной температуре), РНК осаждали с помощью 30 мл смеси этанол/3 M NaOAc (рН 5,4) 32:1 (об.%) в течение ночи при -20°С. Осадок собирали центрифугированием и использовали для последующей конъюгации низкомолекулярного соединения.
В стандартной реакции конъюгации 10 мг РНК растворяли в 2 мл натрий-фосфатном буфере (0,1 М; рН 7,0). К этому раствору добавляли низкомолекулярное соединение (0,12 мМ) в ACN/NMP 1:1 (об.%) в течение 5 минут. Когда, по данным анализа RPLC-ESI MS, внесенная РНК была израсходована, смесь разбавляли водой (~10 мл) и добавляли ~40 мл смеси этанол/3 M NaOAc (рН 5,4) 32:1 (об.%) для осаждения конъюгированной РНК в течение ночи при -20°С. Осадок собирали центрифугированием, растворяли в воде и при необходимости очищали с помощью анионообменной ВЭЖХ, следуя описанной выше процедуре. Если конъюгат был достаточно чистым, реакционную смесь фильтровали через колонку для гель-фильтрации (Sephadex G-25, GE Healthcare).
Гибридизация олигорибонуклеотидов для получения миРНК
Комплементарные цепи гибридизировали путем соединения эквимолярных растворов РНК. Полученную смесь лиофилизовали и вновь растворяли в подходящем объеме буфера для гибридизации (100 мМ NaCl, 20 мМ фосфат натрия, рН 6,8) до достижения желаемой концентрации. Этот раствор помещали на водяную баню при 95°С, которую охлаждали до КТ в течение 3 ч.
Проводили ниже следующий анализ для оценки αVβ3-связывающей активности нацеленных к αVβ3 соединения по настоящему изобретению.
Твердофазный анализ αVβ3 человека:
Иммунологические 96-луночные планшеты (NUNC, Партия №439454) сенсибилизировали с помощью αvβ3 (R&D, кат.№3050-AV), для чего в каждую лунку вносили по 100 мкл αvβ3 (1х) и инкубировали планшеты в течение ночи при 4°С. Используемый буфер представлял собой Буфер А: 20 мМ Tris, 150 мМ NaCl, 1 мМ CaCl2, 1 мМ MgCl2, рН 7,4. После удаления сенсибилизирующего реагента в каждую лунку вносили 150 мкл 3,5% BSA в Буфере А для блокирования планшетов в течение 105 минут при 37°С. После блокирования планшеты промывали 5 раз по 200 мкл Буфера В (Буфер А+1 мМ MnCl2). Затем в каждую лунку вносили по 50 мкл раствора тестируемого соединения (2-кратного) в 5% DMSO и 50 мкл фибриногена (2-кратного) (Innovative Research, кат. № IFIB). Планшеты встряхивали в течение 2 минут и затем инкубировали в течение 2 ч при 37°С. После того как планшеты промывали 5 раз по 200 мкл Буфера В, на планшеты вносили первые антитела - антитела кролика против фибриногена человека (Innovative Research, Cat IASHFBGN-GF) в количестве 100 мкл/лунку, и вторые антитела - козьи антитела против IgG кролика, конъюгированные с пероксидазой хрена (ПХ) (Invitrogen, кат. №G21234) в количестве 50 мкл/лунку, соответственно. После внесения первых антител и после внесения вторых антител планшеты встряхивали 2 мин и инкубировали в течение 60 минут при 37°С, затем промывали 5 раз по 200 мкл Буфера В, соответственно. Конечные условия твердофазного анализа: [αvβ3]=1,25 мкг, [фибриноген] = 0,75 мкг/мл, [Anti-FG] = 1/2400 (разведенный), [антитела против IgG кролика-ПХ] = 1/1000 (разведенные). По окончании анализа связывания на планшеты вносили по 100 мкл/лунку детектирующего реагента ABTS (смесь реагента А и реагента В) (KPL, кат.№50-62-00). Планшеты шейкировали при КТ в течение 5-8 мин, при этом наблюдалось постепенное развитие зеленого окрашивания. После добавления 100 мкл/лунку Стоп-Буфера (1,0 М Фосфорная кислота (H3PO4)), планшеты считывали на приборе Envision в режиме Абсорбции при 450 нм.
Для контрольного соединения (141) была определена величина IC50 равная приблизительно 2 нМ (т.е. 50% клеток не связалось с αVβ3 на поверхности лунок, поскольку αVβ3 рецепторы клеток предположительно связались или дали ассоциаты с контрольным соединением). Эти результаты показаны ниже в Таблицах 3 и 4.

Доказательство клеточной проницаемости и локализации производных низкомолекулярных соединений для ковалентно связанных антагонистов интегрина с FITC-флюорофорами и миРНК для нацеленной доставки.
Процедура
AML MV4-11 клетки в питательной среде (RPMI 1640 с 10% FBS) инкубировали с Дуплексом-27 (500 нМ) в течение 1 ч при 37°С. Для определения VLA-4-независимого связывания, включали 140 (10 мкМ) в одних условиях для блокировки VLA-4-зависимого связывания. Затем по окончании инкубации клетки дважды промывали с помощью D-PBS и фиксировали в 1% параформальдегиде в течение 10 минут. Захват миРНК анализировали с помощью проточной цитофлуориметрии с визуализацией, с использованием ImageStreamx (Aminis Corporation, Сиэтл). Результаты представлены в Таблице А и на Фигурах 1-4.

Анализ миРНК с модифицированной 5ʹ-смысловой цепью на выключение иРНК АНА1 в клеточных системах

Обратная трансфекция: клетки Н1299, трансфицированные с помощью указанной миРНК при конечной концентрации 50 нМ, с использованием реагента для трансфекции DharmaFect-1 в количестве 0,15 мкл/лунку. Затем клетки высевали в 96-луночный планшет в количестве 5000 клеток/лунку и инкубировали при 37°С в течение 48 ч.
Эффективность выключения миРНК измеряли с помощью анализа разветвленной ДНК, как рекомендовано производителем; результаты такого выключения показаны на Фиг. 5. Относительную жизнеспособность клеток оценивали по абсолютной экспрессии GAPDH в той же лунке (на Фиг. 6).
Если не указано обратное, все соединения в примерах получали и характеризовали, как описано выше. Все патенты и публикации, на которые даны ссылки в настоящем описании, полностью включены посредством ссылки.
Реферат
Настоящее изобретение относится к соединениям формулы I для применения в изготовлении и доставке конъюгированных молекул, таких как низкомолекулярные соединения, пептиды, нуклеиновые кислоты, флюоресцирующие молекулы и полимеры, которые соединены с антагонистами интегрина альфа-V-бета-3, для нацеливания к клеткам, экспрессирующим альфа-V-бета-3. 5 н. и 12 з.п. ф-лы, 10 ил., 1 табл., 14 пр.
Формула
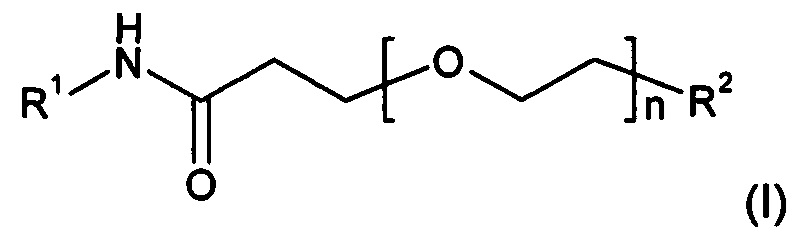














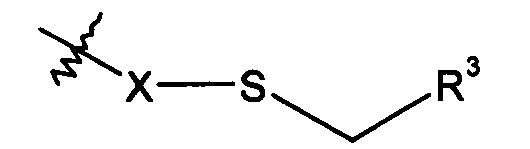

Комментарии