Амиды аминокарбоновых кислот, способ их получения и фармацевтическая композиция - RU2134683C1
Код документа: RU2134683C1
Чертежи


Описание
Изобретение относится к новым амидам аминокарбоновых кислот общей формулы:
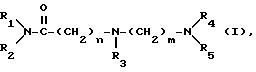
где R1, R2, R3, R4, R5, а также "n" и "m" имеют следующие значения:
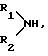
R1, R2 обозначают водород, линейный или разветвленный (C1-C8)-алкил;
(С3-С8)-циклоалкил, как циклогексил; фенил, который может быть одно- или двукратно замещен линейным (C1 -C4)-алкилом, (C1-C2)-алкоксилом, галогеном, циано-группой, нитро-группой, трифторметилом или ациламино-группой; фенилалкил, где алкильная цепь может содержать 1-3 C-атома и фенильное кольцо, которое может быть одно- или двукратно замещено метилом, метокси-группой, галогеном, нитро-группой, циано-группой или ациламидо-группой;
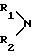
обозначает морфолин или пиперидин, который может быть замещен одно- или двукратно (C1-C2)-алкильной группой или группу
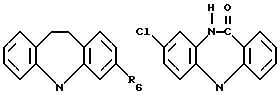
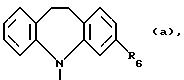
где R6 может обозначать H, NHCО2CH2CH3;
R3 обозначает
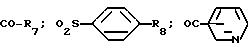
где R7 обозначает (C3-C7)-циклоалкил такой, как циклогексил; фенил, который может быть одно или двукратно замещен линейным (C1-C4)-алкилом, (C1-C2)-алкоксилом, галогеном, циано-группой, нитро-группой, трифторметилом, сульфонамидо-группой, метансульфонамидо-группой или ациламино-группой;
фенилалкил, где алкильная цепь может содержать 1-3 C-атом и фенильное кольцо может быть одно- или двукратно замещено метилом, метокси-группой, галогеном, нитро-группой, циано-группой или ациламидогруппой; и
R8 может обозначать водород или ациламиногруппу;
R4, R5 - обозначает линейный или разветвленный (C1-C6)-алкил; (С3-C6)-циклоалкил, как циклогексил; фенил; фенилалкил, где алкильная группа может содержать 1-3 C-атома или
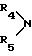
обозначает пиперидин или морфолин
"n" = 1-5;
"m" = 2-4;
их физиологически приемлемым солям присоединения кислот, способу их получения, а также к фармацевтическим композициям и их применению в качестве лекарственных средств, в особенности для лечения нарушений сердечного ритма.
Лекарственные средства для лечения нарушений сердечного ритма на основании их механизма действия по
Vaughan Williams (J.Glin. Pharmacol. 1984, 24, 129-147) подразделяют на 4 класса:
класс I: антагонисты натрия, которые далее подразделяют на классы IA, IB и IC
;
класс II: блокаторы - рецепторов;
класс III: блокаторы калиевых каналов (ингибиторы реполяризации);
класс IV: антагонисты кальция.
Используемые до сих пор антиаритмические средства, которые в подавляющем большинстве относятся к классу I, обладают только незначительной терапевтической широтой. В частности антиаритмические средства класса IC, как показало исследование CAST (N. Engl. J. Med. 1991, 324, 781-788), не в состоянии снижать смертность с течением времени пациентов с инфарктом миокарда. Смертность в группе, в которой вводят лекарство (Verum - Gruppe) даже выше, чем в группе, в которой вводят плацебо. В качестве причины этого указывают на проаритмогенные свойства антиаритмических средств класса I, которые сводятся к замедлению скорости проведения возбуждения, благодаря чему способствуют возникновению повторных аритмий.
Антиаритмические средства класса III, которые называют как ингибиторы реполяризации, напротив, особенно эффективны при повторных аритмиях; так как они удлиняют защитную зону для дополнительно вызываемых некоординированных раздражений в конце фазы реполяризации (так называемая эффективная рефракторная фаза), не влияя на скорость проведения возбуждения.
Вводимые антиаритмические средства со свойствами класса III Амиодарон и Соталол поэтому лучше пригодны для терапии нарушений сердечного ритма, чем антиаритмические средства класса I.
На основании отчасти серьезных побочных действий, в особенности: в случае Амиодарона, эти лекарственные средства также только ограниченно применимы.
Терапия угрожающих жизни аритмий, которыми только в США ежегодно заболевают свыше 1 миллиона людей, а также профилактика скоропостижной смерти от остановки сердца, далее, представляет собой нерешенную проблему, так что существует большая потребность в антиаритмических средствах с улучшенными свойствами.
Поэтому в основу изобретения положена задача получения новых соединений с улучшенной антиаритмической эффективностью и большей терапевтической широтой действия.
Соответственно настоящему изобретению, этими новыми соединениями являются амиды аминокарбоновых кислот общей формулы (I), где R1-R5, "n" и "m" имеют вышеуказанное значение, и их физиологически приемлемые соли присоединения кислот.
Соединения общей формулы (I) в специальной и патентной литературе до сих пор еще не описаны.
В качестве примеров соединений
формулы (I) настоящего изобретения следует назвать:
/(4-ацетиламино-бензолсульфонил)-(2-диэтиламино-этил)- амино/-N,N-дицикло-гексил-ацетамид;
N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-3-метокси-4-нитро-бензамид;
4-циано-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-бензамид,
4-хлор-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-3-нитро-бензамид;
N-(2-дициклогексиламино-карбамоилметил)-N-(3-диэтиламино-пропил)-4-трифторметил-бензамид;
4-ацетиламино-N-(3-дибутиламино-пропил)-N-(дициклогексил-карбамоил-метил)-бензамид;
N-/5-(дициклогексил-карбамоил)-пентил/-N-(2- диэтиламино-этил)-4-нитро-бензамид;
4-амино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино- пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-2-нитро-бензамид;
4-карбэтоксиамино-N-(дициклогексил-карбамоилметил)-N-(3- диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-морфолин-4-ил-пропил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-3-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диметиламино-пропил)-4-нитро-бензамид;
4-ацетиламино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид;
4-ацетиламино-N-(дициклогексил-карбамоилметил)-N-(2- диэтиламино-этил)-бензамид;
N-(дициклогексил-карбамоилметил)-4-нитро-N-(2-пиперидин-1-ил-этил)-бензамид;
4-ацетиламино-N-(бензил-метил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид-иодметилат;
N-(дициклогексил-карбамоилметил)-N-(3-морфолин-4-ил-пропил)-4-нитро-бензамид-иодметилат;
N-(3-диэтиламино-пропил)-N-/2-(10,11-дигидро-дибензо/b,
f//азепин-5-ил)-2-оксо-этил/-4-нитро-бензамид;
N-(2-диэтиламино-этил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)-бензамид;
N-(бензил-метил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-метансульфонил-амино-бензамид;
N-(3-диэтиламино-пропил)-N-/2-(3-фторфенил-1-ил-амино)-2-оксоэтил/-4-нитро-бензамид;
N-(3-диэтиламино-пропил)-N-(2-морфолин-4-ил-2-оксо-этил)-4-нитро-бензамид;
N-/2-(3-карбэтокси-амино-10,11-дигидро-дибензо/b, f/азепин-5-ил)-2-оксо-этил/-N-(2-диэтиламино-этил)-4-нитро-бензамид;
8-хлор-5-{
/N-/3-(диэтиламино)пропан-1-ил/-N-(п-метилсульфониламинобензоил)/-аминоацетил}-5,10-дигидро-11H-дибензо/b,e/ /1,4/диазепин-11-он;
8-хлор-5-{
/6-/N-(3-диэтиламино)пропан-1-ил/-N- бензоил/-амино-1-оксо-гексил}-5,10-дигидро-11Н-дибензо/b,e/ /1,4/диазепин-11-он.
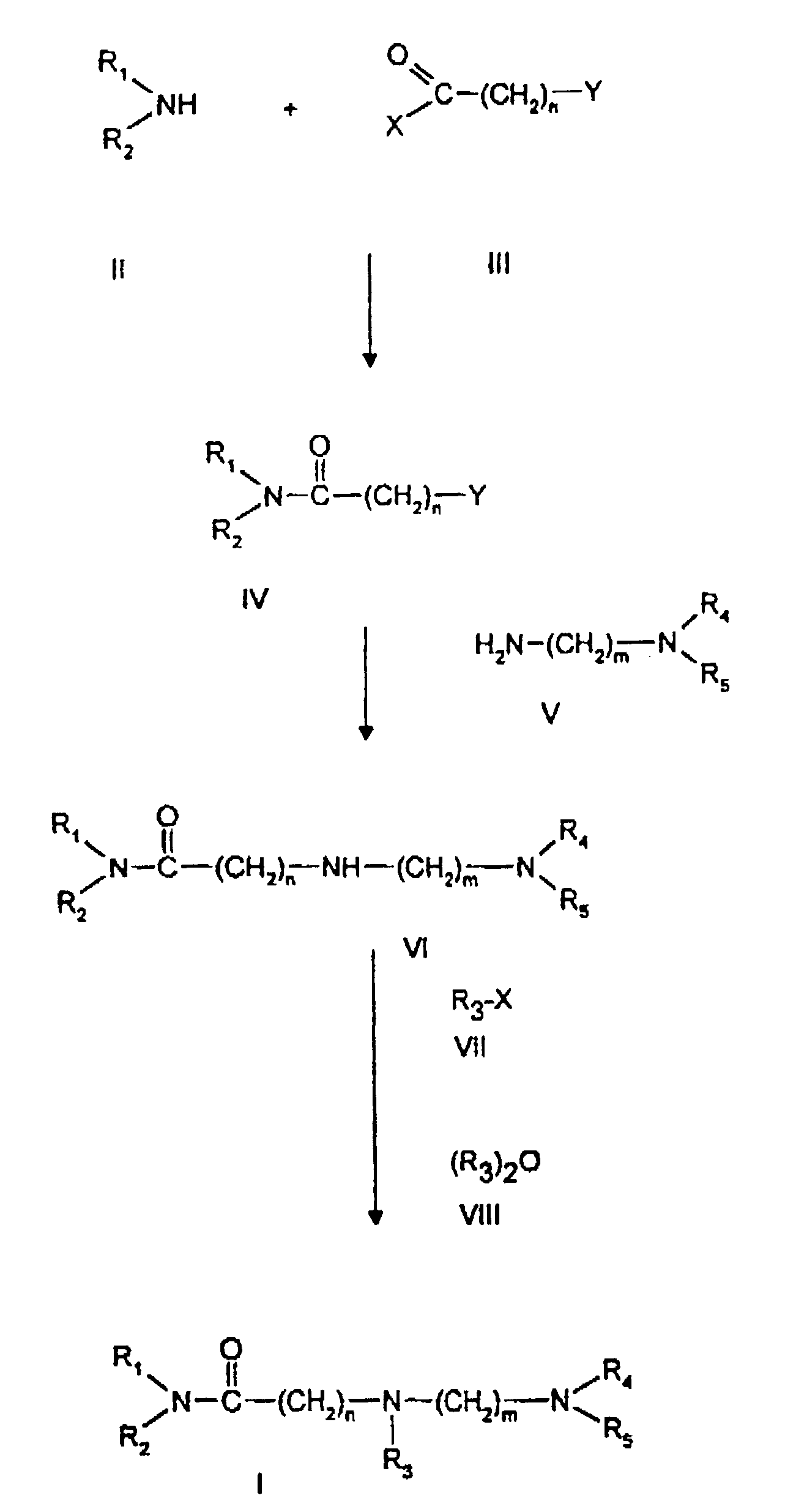
Соответственно настоящему изобретению, соединения общей формулы (I) можно получить способом, приведенным на схеме в конце описания.
Первичные или вторичные амины общей формулы (II), где R1 и R2 имеют вышеуказанное значение, вводят во взаимодействие с галогенидами ω - галогеналканкарбоновых кислот общей формулы (III), где X и Y, смотря по обстоятельствам, обозначают атом галогена, предпочтительно атом хлора или брома, и "n" имеет вышеуказанное значение, при температурах от 0oC вплоть до температуры кипения обратного холодильника используемого растворителя, в инертных органических растворителях или в двухфазных смесях растворителей, как вода/толуол, вода/циклогексан или вода/диэтиловый эфир в присутствии акцепторов кислот, как, например, карбонат калия или третичный амин, с получением амидов общей формулы (IV), причем R1, R2 и Y имеют вышеуказанное значение.
В качестве инертных органических растворителей пригодны, например, простые эфиры, как диоксан или тетрагидрофуран; ароматические углеводороды, как бензол, толуол или ксилол; галогенированные алифатические или ароматические углеводороды, как хлороформ, 1,2-дихлорэтан, четыреххлористый углерод или хлорбензол; алифатические углеводороды, как циклогексан или гептан; или диполярные апротонные растворители, как диметилформамид, ацетонитрил или диметилсульфоксид.
Соединения общей формулы (IV) вводят во взаимодействие с диаминами общей формулы (V), причем R4, R5 и "m" имеют вышеуказанное значение, в инертных растворителях, в присутствии акцепторов кислот, как, например, карбонат калия или третичные амины, или предпочтительно в избытке диамин общей формулы (V) без улавливателя НУ.
В качестве инертных растворителей пригодны, например, спирты, как этанол, н-пропанол или изопропанол; простые эфиры, как диоксан или тетрагидрофуран; ароматические углеводороды, как бензол, толуол или ксилол; галогенированные алифатические или ароматические углеводороды, как хлороформ, 1,2-дихлорэтан, четыреххлористый углерод или хлорбензол; алифатические углеводороды, как циклогексан или гептан; или диполярные апротонные растворители, как диметилформамид, ацетонитрил или диметилсульфоксид.
Особый вариант осуществления настоящего изобретения состоит в том, что в качестве растворителя применяют избыток соединения формулы (V).
При получении соединений формулы (VI) температуру можно изменять в широких пределах от комнатной до 150oC.
Соединения общей формулы (VI) вводят во взаимодействие с галогенидами карбоновых кислот или галогенидами сульфокислот общей формулы (VII) или (VIII), где R3 имеет вышеуказанное значение и X обозначает атом галогена, предпочтительно атом хлора или брома, в инертных растворителях без добавки или в присутствии акцептора кислоты, с получением целевых соединений общей формулы (I), где R1, R2, R3, R4, R5, а также "n" и "m" имеют вышеуказанное значение.
В качестве акцепторов кислот пригодны, например, карбонаты щелочных металлов, гидрокарбонаты щелочных металлов или третичные органические амины, как триэтиламин, пиридин или N,N-диметиланилин.
В качестве инертных растворителей можно применять, например, простые эфиры, как диоксан или тетрагидрофуран; ароматические углеводороды, как бензол, толуол или ксилол; галогенированные алифатические или ароматические углеводороды, как хлороформ, 1,2-дихлорэтан, четыреххлористый углерод или хлорбензол; алифатические углеводороды, как циклогексан или гептан; или диполярные апротонные растворители, как диметилформамид, ацетонитрил или диметилсульфоксид.
При получении соединений формулы (I) температура может изменяться в пределах от -5oC до +120oC.
Для получения физиологически приемлемых солей присоединения кислот, амиды аминокарбоновых кислот общей формулы (I) вводят во взаимодействие с неорганическими или органическими кислотами, как соляная кислота, бромистоводородная кислота, серная кислота, фосфорная кислота, уксусная кислота, щавелевая кислота, винная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, молочная кислота или янтарная кислота.
Изобретение относится также к фармацевтическим композициям, содержащим в качестве активного ингредиента по меньшей мере одно соединение общей формулы I или его физиологически приемлемую соль кислотного присоединения и инертные фармацевтически приемлемые носители и вспомогательные вещества, как например: сахароза, лактоза, поливинилпирролидон, метилцеллюлоза, МКЦ (микрокристаллическая целлюлоза), желатин, стеараты кальция или магния, кальций фосфат двузамещенный, тальк и т.д., которые составляют остальную часть композиции. Способы получения указанных композиций являются традиционными и общеизвестными для фармацевтической химии.
Соединения общей формулы (I) в свободной форме или в форме соли с физиологически приемлемой кислотой можно вводить перорально, парентерально, внутривенно или чрескожно.
В качестве вводимых форм поэтому пригодны в особенности таблетки, драже, капсулы, пластыри, растворы, ампулы или свечи. Их можно приготовлять общеизвестными и обычными в фармацевтической практике способами.
Дозировка фармацевтических композиций зависит от возраста, состояния и веса пациента, а также от вводимой формы. Как правило, суточная доза биологически активного вещества при внутривенном применении составляет 0,01-10 мг/кг, а при оральном применении - 0,1-25 мг/кг веса тела.
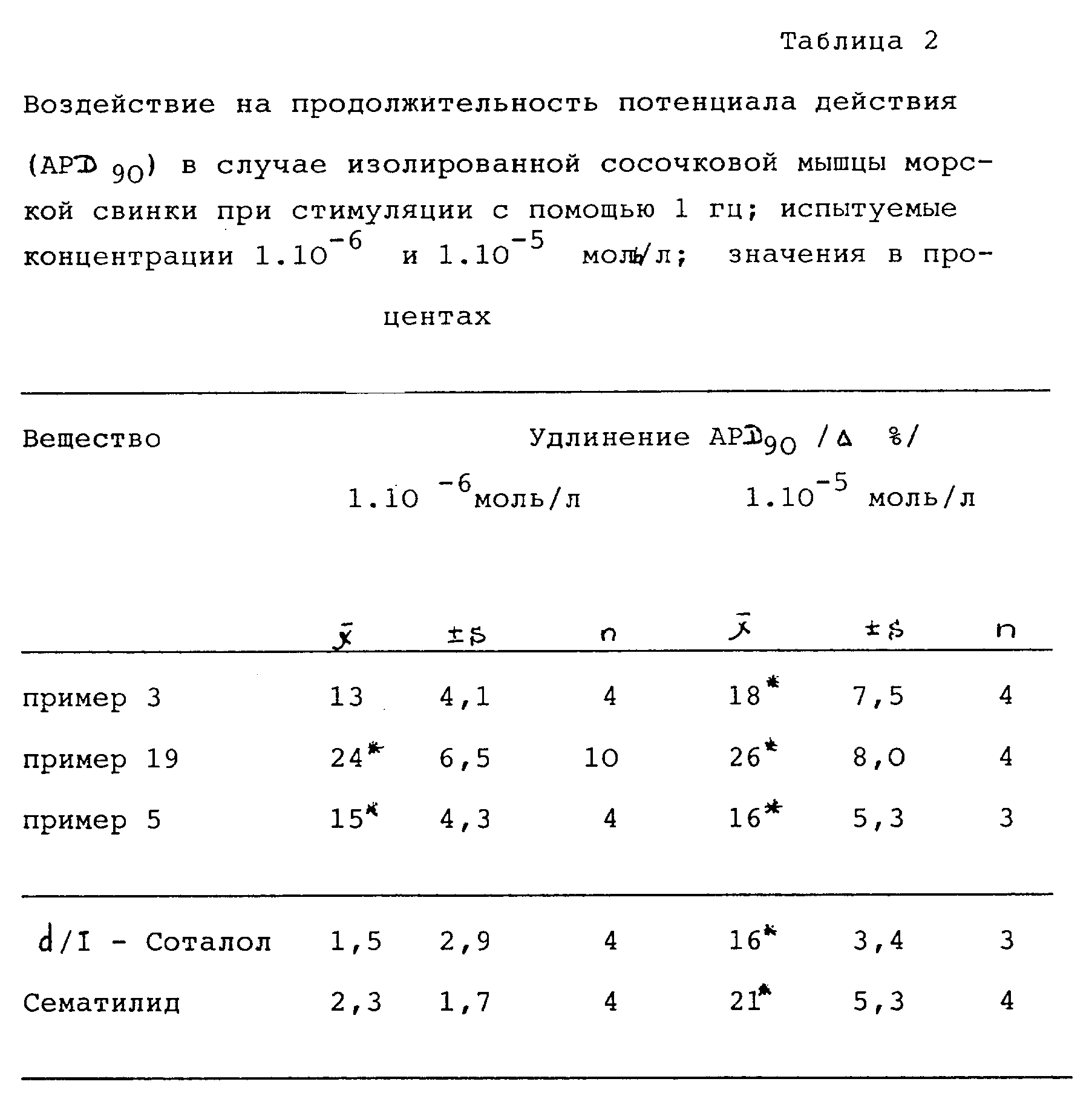
Предлагаемые согласно изобретению соединения общей формулы (I) представляют собой новые антиаритмические средства, в особенности класса III по Vaughan-Williams (J. Clin. Pharmacol. 24,129-147 (1984). Они в значительной степени и в зависимости от дозы удлиняют рефракторную фазу (см. табл. 1) и параллельно с этим удлиняют продолжительность потенциала действия (см. табл. 2) в случае изолированных стимулированных сосочковых мышц (сердца) морских свинок (табл. 1-3 см. в конце описания).
Соединения общей формулы (I) при стимуляции с помощью 1 Гц удлиняют эффективную рефракторную фазу (ЭРФ) в более сильной мере, чем сравнительные вещества Сематилид, проявляющее вещество, и d/I-Соталол (см. табл. 1). Особое преимущество новых соединений состоит в том, что при быстром возбуждении (стимуляция с помощью 3 Гц) увеличивается воздействие на эффективную рефракторную фазу при одинаковой испытуемой концентрации. Вместе с этим новые соединения обладают полезными (use dependence) свойствами, в то время как в случае d/I-Соталола и Сематилида имеются противоположные полезные свойства. При терапии нарушений ритма типа тахикардии новые соединения особенно эффективны, в то время как сравнительные вещества - Сематилид и d/I-Соталол - при аритмиях типа тахикардии проявляют только слабое действие.
Воздействие предлагаемых согласно изобретению соединений общей формулы (I) на продолжительность потенциала действия (APD90) приводится в табл. 2.
Новые соединения вызывают зависящее от дозы и значительное удлинение APD90 без изменения в значительной степени времени излияния крови из одной камеры сердца в другую и максимальной скорости покрытия.
Воздействие при стимуляции с помощью 1 Гц больше, чем воздействие сравнительных препаратов Соталола и Сематилида.
Исследования ин виво на бодрствующих кошках показывают, что после орального введения QT - время в ЭКГ сильно и на длительный период удлиняется. Метод испытания на воздействие при оральном введении в случае бодрствующих кошек описывается Poppe Н. и др., PHARMAZIE, 40(12), 857-61 (1985).
Новые соединения поэтому пригодны в особенности для лечения аритмий, которые имеют место вместе с нарушениями фазы реполяризации сердечного ритма.
Соединения общей формулы (I) являются сильно антифибрилляционно эффективно действующими. Они повышают порог мерцания желудочков сердца кошек отчетливо сильнее, чем сравнительное вещество Лидокаин, антиаритмическое средство класса I. Соединение согласно примеру 8 в дозе 2,4 мг/кг внутривенно вызывает 6,7 - 10- кратное повышение порога мерцания желудочков сердца, в то время как Лидокаин в дозе 5,0 мг/кг внутривенно повышает порог мерцания желудочков сердца в 2-2,5 раза. Метод определения порога мерцания желудочков сердца в случае находящихся под наркозом кошек описывается Кавериной Н.В. и др., PHARMAZIE, 40 (12), 840-844 (1985).
Дальнейшее существенное преимущество предлагаемых в изобретении новых соединений заключается в их отчетливо большей терапевтической широте действия по сравнению со сравнительными веществами класса I, как Лидокаин Хинидин. Как показано в табл. 3, терапевтический индекс соединения согласно примеру 8 составляет 167, в то время как Хинидин имеет индекс 11,3, а Лидокаин имеет индекс 5,0. Терапевтические индексы (ТИ) рассчитывают из частного от деления 50%-ной летальной дозы (ЛД50) в случае крысы при внутривенном введении на 50%-ную эффективную дозу (ЭД50) при аритмии за счет конитина у бодрствующей крысы при внутривенном введении.
Исследования в отношении антиаритмического воздействия на аритмию за счет Аконитина в случае бодрствующей крысы и определение острой токсичности в случае крысы описываются Сеновой 3.П. и др., КАРДИОЛОГИЯ, 26(8), 24-28 (1986).
Удлинение эффективной рефракторной фазы и продолжительности потенциала действия определяют следующим образом.
Определение эффективной рефракторной фазы на изолированной сосочковой мышце морской свинки (правый желудочек сердца)
Самцов или самок
морских свинок с массой тела примерно 400 г умерщвляют ударом в затылок и обескровливают. Сердце быстро извлекают и фиксируют в препаровальной ванне, через которую барботируют карбоген. После
раскрытия правого желудочка сердца препарируют сосочковую мышцу (диаметр 1 мм) и немедленно вносят в корпус ванны (объем ванны 39 мл), в котором находится раствор Krebs-Henseleit следующего состава
(данные в ммоль/л): NaCl = 113,8; CaCl2 = 3,2; NaHCO3 = 25,0; MgSO4 = 1,2; KCl = 4,7; KH2PO4 = 1,2; глюкоза = 10,0. Раствор имеет температуру
35oC и непрерывно продувается газовой смесью из 95% кислорода и 5% CO2. После подвешивания в корпусе ванны сосочковые мышцы нагружают нагрузкой 2 мН (=0,2 г). Сосочковые мышцы
раздражают через биполярные платиновые электроды с помощью электростимулятора "TUR RS 21". Учет измеряемых величин осуществляют с помощью линейного измерителя (Linearcorder) марки VII. Порог
раздражения (в миллиамперах) определяют после 10-минутной паузы при частоте 1 Гц, соответственно, 3 Гц, с широтой раздражения 0,5 мс, удваивают и поддерживают постоянным в течение всей
продолжительности испытания. После периода адаптации 60-90 минут осуществляют определение рефракторной фазы по стандартизированной программе стимуляции (отдельные раздражения). Интервал между базисным
раздражением и экстрастимуляцией после каждого удара (толчка) автоматически увеличивается каждый на величину 2 мс вплоть до достижения эффективной рефракторной фазы. Эффективную рефрактерную фазу
определяют как самый маленький интервал между базисным раздражением и экстрастимуляцией (в мс), который имеет следствием измеримое сокращение. После определения базисного значения осуществляют
введение испытуемых веществ в жидкость ванны. После времени воздействия 1 час снова определяют эффективную рефракторную фазу.
Оценку осуществляют в процентах, причем базисное значение принимают равным 100%. Из 3-6 отдельных значений при одной испытуемой концентрации рассчитывают средние значения со стандартным отклонением. Средние значения из 3-4 испытуемых концентраций используют для расчета эффективной концентрации, которая вызывает 20%-ное удлинение эффективной рефракторной фазы (EC20 моль/л). Расчет осуществляют посредством регрессионного анализа со степенью достоверности p<0,05.
Электрофизиологические исследования на изолированной сосочковой мышце морской свинки
Самцов или самок морских свинок с массой тела примерно 400 г
умерщвляют ударом в затылок и обескровливают. Сердце быстро извлекают и препарируют тонкую правую сосочковую мышцу. Затем сосочковую мышцу вносят в камеру аппаратуры Steiert (Hugo-Sachs Electronik KG),
через которую непрерывно протекает раствор Krebs-Henseleit. Скорость протекающего раствора составляет 18 мл/мин. Состав раствора Krebs-Henseleit в ммоль/л: NaCl = 113,9; CaCl2 = 3,2;
KH2PO4 = 1,2; KCl = 4,7; MgSO4 = 1,2; глюкоза = 10,0. pH-Значение устанавливают равным 7,4 с помощью NaHCO3. Раствор имеет температуру 35oC и
непрерывно продувается газовой смесью из 95% кислорода и 5% CO2.
Сосочковую мышцу помещают на биполярный электрод Ag-AgCl. Порог раздражения (в милливольтах) определяют при частоте 1 Гц. При непрерывной стимуляции при 1 Гц, соответственно, 3 Гц, и удвоенном пороге раздражения время адаптации составляет 1 час. Ширина прямоугольного импульса составляет 1 мс (миллисекунд). Потенциалы действия устанавливают с помощью стеклянных микроэлектродов (Kwik - Fil TM, K-серии капилляров из боросиликатного стекла, Sarasota, Florida 34240), которые заполнены с помощью 3 моль/л KCl и имеют максимальное сопротивление 10-25 мегаом. Сигналы усиливаются с помощью электродного усилителя (Hugo-Sachs Electronik KG, тип 309), становятся видимыми на осциллографе и регистрируются на оперативном анализаторе, соединенном с матричным печатающим устройством. Определяют параметры: RP (электрический потенциал покоя/милливольт/); OV (потенциал отклонения от установленного значения /милливольт/); Vmax (максимальная скорость покрытия [объем/сек.]); tUe (время излияния крови из одной камеры сердца в другую /миллисекунды/); и APD90 (длительность потенциала действия при 90%-ной реполяризации /миллисекунды/).
Оценку действия вещества осуществляют после времени воздействия 1 час в суперфузионной среде по сравнению с базисным значением. Расчет отклонения в процентах (Δ%) осуществляют тем, что из процентных изменений отдельных опытов (п) рассчитывают средние значения и стандартное отклонение (
Примеры осуществления
Пример 1: /(4-Ацетиламино-бензенсульфонил)-(2-диэтиламино-этил)амино/-N, N-дициклогексил-ацетамид-оксалат
10,2 г (0.03 моль) N,N-Дициклогексил-2-(2-диэтиламино-этиламино)-ацетамида растворяют
в 200 мл толуола. В полученном растворе при перемешивании суспендируют 5,5 г (0,04 моль) карбоната калия (марки ч.д.а.). При охлаждении, вносят при t=0-5oC, в течение 10 минут порциями 12,0
г (0,05 моль) N-ацетил-сульфанилил-хлорида. Затем перемешивают в течение 6 часов при комнатной температуре, примешивают 200 мл воды и перемешивают следующие 4 часа. Жидкие фазы декантируют, прилипшее
к стенкам колбы масло перемешивают с 200 мл толуола и 100 мл воды примерно в течение 2 часов при 50oC до тех пор, пока все не перейдет в раствор. Объединенные толуольные фазы дополнительно
экстрагируют путем встряхивания с 300 мл воды, затем экстрагируют путем встряхивания с 150 мл 10%-ной соляной кислоты, и толуольную фазу дополнительно экстрагируют путем встряхивания с 200 мл воды.
Солянокислую фазу и последнюю водную фазу вместе с прилипшим ко дну колбы маслом смешивают с 300 мл толуола и с помощью 10%-ного раствора гидроксида натрия устанавливают pH 13-14. После интенсивных
встряхиваний фазы разделяют, толуольную фазу сушат над сульфатом натрия и перегоняют в вакууме досуха. Остаток доводят до постоянного веса и растворяют в небольшом количестве абсолютного этанола при
нагревании. К этому раствору при 50-60oC добавляют насыщенный раствор эквимолярного количества щавелевой кислоты в абсолютном этаноле. После охлаждения и по окончании кристаллизации
отсасывают полученные кристаллы и перекристаллизовывают их из этанола при добавке примерно 0,2 г активного угля. Выход: 8,7 г (44,5% от теории). Т.пл. 103,5-106oC.
C28H46N4O4S•C2H2O4•1,5 H2O (масса моля 651,81)
рассчитано, %: С 55,27; H 7,88; N 8,59; S 4,91;
найдено, %: C 55,19; H 7,52; N 8,18; S 4,91.
Получение исходного продукта:
N,N-Дициклогексил-2-(2-диэтиламино-этиламино)ацетамид
78 г (0,3 моль) 2-Хлор-N,
N-дициклогексилацетамида и 174,3 г (1,5 моль) N, N-диэтилэтилендиамина в 400 мл циклогексана в течение 10 часов кипятят с обратным холодильником, затем циклогексан и избыточный амин отгоняют в
вакууме. Остаток обрабатывают с помощью 1 л воды, устанавливают в полученном растворе pH 12 с помощью раствора гидроксида натрия, экстрагируют дважды по 500 мл толуолом, объединенные толуольные фазы
дважды экстрагируют путем встряхивания с водой, беря каждый раз по 250 мл воды, сушат над сульфатом натрия, отфильтровывают и перегоняют в вакууме досуха.
Выход: 100,5 г (99% от теории).
C20H39N3O (масса моля 337,55)
рассчитано, %: С 71,16; H 11,64; N 12,45;
найдено, %: C 71,47; H 11,65; N 12,14.
Оксалат: C20H39N3O•1,5 C2H2O4 (масса моля 472,60)
Т.пл. = 205-215oC
рассчитано, %: С 58,
45; H 8,95; N 8,89;
найдено: %: C 57,21; H 9,00; N 8,90.
N,N-Дициклогексилхлорацетамид
К смеси 108,8 г (0,6 моль) дициклогексиламина в 600 мл дихлорметана и 122 г (0,
88 моль) карбоната калия в 160 мл воды, при перемешивании и при температуре 15-20oC, в течение 30 минут добавляют 95 г (0,84 моль) хлорацетилхлорида. Перемешивают еще 2 часа дополнительно
при комнатной температуре. Дихлорметановый раствор дважды экстрагируют путем встряхивания с 300 мл воды. Дихлорметан перегоняют и
остаток перекристаллизуют из изопропанола. Выход: 120,5 г (78% от
теории).
Т.пл. = 111-112oC.
C14H24ClNO:
рассчитано, %: С 62,35; H 8,07; N 9,86;
найдено, %: C 62,21; H 8,09; N 10,
16.
Пример 2: N-(Дициклогексил-карбамоилметил)-N-(2-диэтил-амино-этил)-3-метокси-4-нитро-бензамид-оксалат
6,76 г (0,02 моль) N,
N-Дициклогексил-2-(2-диэтиламино-этиламино)-ацетамида растворяют в 50 мл N,N-диметилформамида. При перемешивании и охлаждении на водяной бане в полученный раствор порциями вносят 4,33 г (0,02 моль)
3-метокси-4-нитро-бензоилхлорида и перемешивают при комнатной температуре в течение 14 часов. После этого добавляют 130 мл циклогексана и 70 мл воды, фазы разделяют и водную фазу концентрируют в
вакууме досуха. Остаток распределяют между 100 мл воды и 150 мл этилацетата, устанавливают pH 13-14, фазы разделяют и органическую фазу дважды дополнительно экстрагируют путем встряхивания с водой,
беря каждый раз по 50 мл воды. После высушивания над сульфатом натрия и перемешивания с 0.2 г активного угля отфильтровывают, этилацетат отгоняют в вакууме, остаток растворяют в смеси толуола с
метанолом в соотношении 6/4 и очищают путем колоночной хроматографии (силикагель 60-100; толуол/метанол = 6/4). Собранные фракции концентрируют досуха, остаток доводят до постоянного веса и растворяют
в 50 мл абсолютного этанола. К полученному последним раствору при нагревании добавляют раствор эквимолярного количества щавелевой кислоты в 10 мл абсолютного этанола, концентрируют досуха, остаток
перемешивают со смесью из 30 мл безводного ацетона и 30 мл циклогексана, отсасывают и промывают ацетоном. Продукт высушивают в течение 4 часов при 100oC. Выход: 2,6 г (21,4% от теории).
Т.пл. = 183-187oC.
C28H46N4O6•C2H2O4 (масса моля 606,708)
рассчитано, %: С 59,39;
H 7,64; N 9,23;
найдено, %: C 59,45; H 7,64; N 9,07.
Пример 3: Циано-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-бензамид-оксалат
7,4 г (0,02 моль) N,
N-Дициклогексил-2-(2-диэтиламино-этиламино)-ацетамида растворяют при нагревании в 200 мл ацетонитрила. При перемешивании и при комнатной температуре в полученный раствор сначала порциями вносят 2,8 г
(0,02 моль) карбоната калия (марки ч.д.а.), затем 3,31 г (0,02 моль) 4-цианобензоилхлорида и в течение 12 часов при перемешивании кипятят с обратным холодильником. Ацетонитрил отгоняют, остаток
распределяют между 300 мл воды и 400 мл толуола, с помощью аммиака устанавливают pH 9-10, фазы разделяют и толуольную фазу еще раз экстрагируют путем встряхивания с 200 мл воды. После высушивания над
сульфатом натрия и перемешивания с 0,2 г активного угля толуол отгоняют досуха, остаток растворяют при нагревании в 50 мл абсолютного этанола, отфильтровывают и добавляют раствор эквимолярного
количества щавелевой кислоты в 10 мл абсолютного этанола. Затем концентрируют досуха и остаток "вываривают" в 50 мл циклогексана, перемешивают 2 часа, отсасывают и промывают циклогексаном,
Выход: 2,2 г (20% от теории). Т.пл. = 173-181oC.
C28H42N4O2•C2H2O4 (масса моля 556,
69)
рассчитано, %: С 64,72; H 7,97; N 10,06;
найдено, %: C 64,75; H 8,29; N 9,75.
Пример 4:
4-Хлор-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-3-нитро-бензамид-оксалат
6.75 г (0,02 моль) N,N-Дициклогексил-2-(2-диэтиламино-этиламино)-ацетамида растворяют в 50 мл N,
N-диметилформамида. При перемешивании и охлаждении в полученный раствор вносят порциями 4,84 г (0,022 моль) 4-хлор-3-нитро-бензоилхлорида и перемешивают в течение 6 часов при комнатной температуре.
Затем добавляют 150 мл циклогексана и 150 мл воды, фазы разделяют и водную фазу концентрируют в вакууме досуха. Остаток распределяют между 100 мл воды и 150 мл этилацетата, устанавливают pH 13-14 с
помощью раствора гидроксида натрия, фазы разделяют и органическую фазу дополнительно экстрагируют путем встряхивания дважды с водой, беря каждый раз по 50 мл воды. После высушивания над сульфатом
натрия и перемешивания с активным углем этилацетат отгоняют в вакууме, остаток растворяют в 10 мл абсолютного этанола и при нагревании добавляют раствор эквимолярного количества щавелевой кислоты в
абсолютном этаноле. По окончании кристаллизации отсасывают образовавшиеся кристаллы и промывают их с помощью 10 мл абсолютного этанола. Выход: 5,0 г (41% от теории). Т.пл. = 174-178oC.
C27H41ClN4O4•C2H2O4 (масса моля 611,124)
рассчитано, %: С 56,99; H 6,93; N 9,17; Cl 5,80;
найдено, %: C 57,04; H 7,09; N 9,03; CI 6,18.
Пример 5: N-(2-Дициклогексиламино-карбамоилметил)-N-(3-диэтиламино-пропил)-4-трифторметил-бензамид
4,23 г (0,012 моль) N,
N-Дициклогексил-2-(3-диэтиламино-пропиламино)-ацетамида и 20 мл триэтиламина растворяют в 200 мл толуола. При перемешивании и при комнатной температуре в полученный раствор порциями вносят 5 г (0,02
моль) 4-(трифторметил)-бензоилхлорида, перемешивают 4 часа при комнатной температуре и 7 часов кипятят с обратным холодильником при перемешивании. После этого добавляют 250 мл воды, фазы разделяют,
толуольную фазу смешивают со 100 мл воды и 10%-ной соляной кислотой вплоть до pH 1. К солянокислой фазе добавляют 100 мл толуола, с помощью раствора гидроксида натрия устанавливают pH 10, встряхивают,
толуольную фазу сушат над сульфатом натрия, отфильтровывают и фильтрат концентрируют досуха. Остаток (3,9 г кристаллического продукта) очищают с помощью препаративной ВЭЖХ (высокоэффективной
жидкостной хроматографии) (RP - 18; колонка размерами 250 х 50 мм; ацетонитрил/вода = 9/1; 40 мл/мин) и очищенную фракцию концентрируют досуха. Выход: 0,8 г (13% от теории); Т.пл. = 110-126o
C.
C29H44F3N3O2 (масса моля 523,666)
рассчитано, %: С 66,51; H 8,47; N 8,02;
найдено, %: C 66,67; H 8,76; N 7,
76.
Пример 6: 4-Ацетиламино-N-(3-дибутиламино-пропил)-N-(дициклогексил-карбамоилметил)-бензамид-оксалат
8,2 г (0,02 моль) N,
N-Дициклогексил-2-(3-дибутиламино-пропиламино)-ацетамида, 3,45 г карбоната калия (0,025 моль) и 6,0 г (0,03 моль) 4-ацетиламино-бензоилхлорида в 200 мл толуола вводят во взаимодействие аналогично
примеру 2. Выход: 4,6 г (33,5% от теории)
Т.пл.: = 85-97oC
C34H56N4)3•C2H2O4•1,5
H2O (масса моля 685,888)
рассчитано, %: С 63,04; H 8,96; N 8,17;
найдено, %: C 63,13; H 9,16; N 8,17.
Получение исходного продукта:
N,
N-Дициклогексил-2-(3-дибутиламино-пропиламино)-ацетамид
25,3 г (0,1 моль) 2-Хлор-N,N-дициклогексил-ацетамида и 90,3 г (0,5 моль) 3-дибутиламино-1-пропиламина в 200 мл циклогексана кипятят с
обратным холодильником в течение 10 часов, затем циклогексан и избыточный амин отгоняют в вакууме. Остаток смешивают с 200 мл воды и 150 мл толуола, устанавливают pH 11-12 с помощью аммиака или
раствора гидроксида натрия и перемешивают в течение 30 минут, до тех пор, пока масляные фазы не растворяются. Фазы разделяют, водную фазу дополнительно экстрагируют путем встряхивания с толуолом, беря
каждый раз по 70 мл толуола, объединенные толуольные фазы экстрагируют путем встряхивания с 80 мл воды, толуольную фазу сушат над сульфатом натрия, отфильтровывают и концентрируют досуха. Выход: 38,5
г (94,5% от теории)
C25H49N3O (масса моля 407,69)
рассчитано, %: С 73,65; H 12,11; N 10,31;
найдено, %: C 73,73; H 12,09; N 10,33.
Пример 7: N-/5-(Дициклогексил-карбамоил)пентил/-N-(2-диэтиламино-этил)-4-нитро-бензамид-оксалат
9,85 г (0,025 моль) Дициклогексиламида 6-(2-диэтиламино-этиламин)-гексановой кислоты
и 5,56 г (0,03 моль) 4-нитробензоилхлорида в 200 мл толуола вводят во взаимодействие аналогично примеру 2.
Выход: 14,5 г (91% от теории). Т.пл. = 125-129oC.
C31H50N4O4•C2H2O4•2H2O (масса моля 638,818)
рассчитано, %: С 62,04; H 8,83; N 8,77;
найдено, %: C 62,43; H 8,22; N 8,73.
Получение исходных продуктов:
Дициклогексиламид 6-бром-гексановой кислоты
К смеси из 275 мл дихлорэтана и 38,1 г (0,275
моль) карбоната калия, растворенных в 220 мл воды, добавляют 50 г (0,275 моль) дициклогексиламина и затем, при перемешивании и охлаждении, в течение 15 минут прикапывают 71,4 г (0,33 моль)
хлорангидрида 6-бром-гексановой кислоты. После перемешивания в течение 7 часов при комнатной температуре фазы разделяют, органическую фазу экстрагируют дважды путем встряхивания со 110 мл воды, сушат
над сульфатом натрия, в течение 30 минут перемешивают примерно с 2 г активного угля, отфильтровывают, дихлорэтан отгоняют и с помощью 50 мл циклогексана удаляют оставшийся дихлорэтан. Остаток при
перемешивании вносят в 250 мл циклогексана. После перемешивания в течение 3 часов выпавший осадок отсасывают и высушивают. Выход: 48,8 г (50% от теории). Т.пл. = 70,5-73,5oC.
C13H32BrNO (масса моля 358,36)
рассчитано, %: С 60,32; H 9,00; N 3,91; Br 22,30;
найдено, %: C 60,23; H 9,11; N 4,02; Br 22,39.
Дициклогексиламид 6-(2-диэтиламино-этиламин)-гексановой кислоты
35,8 г (0,1 моль) Дициклогексиламида 6-бром-гексановой кислоты и 58,1 г (0,5 моль) N,N-диэтилэтилендиамина в 200 мл толуола
перемешивают в течение 72 часов при комнатной температуре. После этого добавляют 100 мл воды и 4,4 г гидроксида натрия. Перемешивают в течение 1 часа, фазы разделяют и органическую фазу экстрагируют
дважды путем встряхивания со 100 мл воды. Органическую фазу сушат над сульфатом натрия и перемешивают примерно с 2 г активного угля, отфильтровывают, толуол и избыточный амин отгоняют в вакууме.
Выход: 38,2 г (97% от теории)
C24H47N3O (масса моля 393,64)
рассчитано, %: С 73,22; H 12,03; N 10,67;
найдено, %: C 73,07; H 12,21; N 10,
47.
Пример 8: N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-нитро-бензамид
12,8 г (0,05 моль) 2-Хлор-N,N-дициклогексил-ацетамида и 32,6 г (0,25 моль)
3-диэтиламино-1-пропиламина в 100 мл бензола кипятят с обратным холодильником в течение 8 часов. Затем бензол и 3-диэтиламино-1-пропиламин отгоняют. К остатку добавляют воду и концентрированный
раствор аммиака, и N,N-дициклогексил-2-(3-диэтиламино-пропиламино)-ацетамид экстрагируют бензолом. Бензольный раствор встряхивают с водой и затем бензол отгоняют. Выход маслянистого N,
N-дициклогексил-2-(3-диэтиламино-пропиламино)ацетамида составляет 16,2 г (92% от теории).
7,03 г (0,02 моль) N,N-Дициклогексил-2-(3-диэтиламино-пропиламино)-ацетамида растворяют в 70 мл бензола, смешивают с 4,2 г (0,04 моль) карбоната натрия и при перемешивании и при комнатной температуре прикапывают раствор 5,6 г (0,03 моль) п-нитро-бензоилхлорида в 30 мл бензола. Реакционную смесь перемешивают в течение 8 часов при комнатной температуре, смешивают со 100 мл воды и перемешивают еще 4 часа при комнатной температуре. Затем бензольный раствор отделяют, экстрагируют его путем встряхивания с водой, целевой продукт экстрагируют из бензольной фазы с помощью разбавленной 1:5 соляной кислоты, подщелачивают с помощью разбавленного 1: 5 раствора аммиака и экстрагируют бензолом. После промывки бензольной фазы водой бензол отгоняют. Остаток перекристаллизуют из 70%-ного этанола. Выход: 6,21 г (62% от теории). Т.пл.: = 121-122oC.
C28H44N4O4
рассчитано, %: С 67,17; H 8,86; N 11,19;
найдено, %: C 69,94; H 8,91; N 11,47.
К раствору 5 г (0,01 моль) N-(дициклогексил-карбамоил-метил)- N-(3-диэтиламино-пропил)-4-нитро-бензамида в 10 мл этанола добавляют раствор 1,26 г (0,01 моль) дигидрата щавелевой кислоты в 5 мл этанола и смесь оставляют стоять в холодильнике. Осадок отфильтровывают и перекристаллизуют из этанола.
Выход: 4,9 г (86,3% от теории).
Т.пл. = 183,5-185oC.
C28H44N4O4•0,75 (COOH)2:
рассчитано, %: С 62,35; H 8,07; N 9,86;
найдено, %: C 62,21; H 8,09; N 10,16.
Пример 9:
N-(Дициклогексил-карбамоилметил)-N-(3-диметиламино-пропил)-4-нитро-бензамид-гидрохлорид-гидрат
К раствору из 7,2 г (0,022 моль) N,N-дициклогексил-2-(3-диметиламино-пропиламино)ацетамида в 60
мл бензола добавляют 4,8 г (0,045 моль) карбоната натрия и при перемешивании и при комнатной температуре прикапывают раствор 6,1 г (0,033 моль) 4-нитро-бензоилхлорида в 30 мл бензола. Реакционную
смесь перемешивают 6 часов при комнатной температуре, смешивают со 100 мл воды и перемешивают при комнатной температуре следующие 4 часа. Бензольный раствор отделяют, экстрагируют путем встряхивания с
водой и целевой продукт экстрагируют из бензольной фазы с помощью разбавленной 1:5 соляной кислоты. Солянокислую фазу подщелачивают с помощью разбавленного раствора аммиака и экстрагируют бензолом.
После промывки бензольной фазы водой, бензол отгоняют, остаток растворяют в безводном эфире и добавляют раствор газообразного HCl в этилацетате. Выпавший осадок отфильтровывают и перекристаллизуют из
изопропанола. Выход: 6,0 г (51,6% от теории). Т.пл. = 203-204oC.
C26H40N4O4•HCI•H2O
рассчитано,
%: С 59,24; H 8,22; N 10,63; Cl 6,73; H2O 3,4;
найдено, %: C 59,52; H 8,35; N 10,91; CI 7,01; H2O 3,7.
Пример 10:
N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-2-нитро-бензамид-оксалат
3,51 г (0,01 моль) N,N-Дициклогексил-2-(3-диэтиламино-пропиламино)-ацетамида растворяют в 40 мл бензола,
смешивают с 2,12 г (0,02 моль) карбоната натрия и при перемешивании и при комнатной температуре прикапывают раствор 2,8 г (0,015 моль) 2-нитро- бензоилхлорида в 10 мл бензола. Реакционную смесь
перемешивают в течение 6 часов при комнатной температуре, смешивают с 60 мл воды и перемешивают еще 4 часа при комнатной температуре. Бензольный раствор отделяют, встряхивают с водой и целевой продукт
экстрагируют из бензольной фазы с помощью разбавленной 1:5 соляной кислоты. Солянокислую фазу подщелачивают с помощью разбавленного 1:5 раствора аммиака и экстрагируют бензолом. После промывки
бензольной фазы водой, бензол отгоняют. Остаток (4,2 г) растворяют в 8 мл изопропанола, затем добавляют раствор 0,82 г (0,009 моль) щавелевой кислоты в 7 мл изопропанола и небольшое количество эфира и
выдерживают в холодильнике вплоть до кристаллизации. Выпавший осадок отфильтровывают и высушивают при 80-90oC.
Выход: 3,3 г (55,9% от теории). Т.пл. = 153-154o C.
C28H44N4O4•(COOH)2:
рассчитано, %: C 61,00; H 7,85; N 9,48;
найдено, %: C 61,35; H 7,86; N 9,74.
Пример 11: N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-3-нитро-бензамид-оксалат
7,03 г (0,02 моль) N,N-Дициклогексил-2-(3-диэтиламино-пропиламино)-ацетамида, 4,2 г
(0,04 моль) карбоната натрия и 5,6 г (0,03 моль) 3-нитро-бензоилхлорида в бензоле вводят во взаимодействие аналогично примеру 10.
Остаток (8,5 г) растворяют в 15 мл изопропанола, смешивают с 2,4 г (0,019 моль) дигидрата щавелевой кислоты, растворенными в 8 мл изопропанола, добавляют 3 мл эфира и раствор оставляют стоять в холодильнике. Выпавший осадок отфильтровывают и перекристаллизуют из изопропанола и эфира.
Выход: 7,5 г (63,5% от теории). Т.пл. = 145-146oC.
C28H44N4O4•
(COOH)2:
рассчитано, %: С 61,00; H 7,85; N 9,48;
найдено, %: C 61,06; H 7,87; N 9,67.
Пример 12:
N-(Дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид
13,5 г (0,04 моль) N,N-Дициклогексил-2-(2-диэтиламино-этиламино)ацетамида, 8,4 г (0,08 моль) карбоната натрия и 11,1 г
(0,06 моль) 4-нитро-бензоилхлорида вводят во взаимодействие аналогично примеру 10. Остаток перекристаллизуют из 70%-ного этанола. Выход: 10,2 г (52,3% от теории).
Т.пл. = 117-119oC.
C27H42N4O4:
рассчитано, %: С 66,64; H 8,7; N 11,51;
найдено, %: C 67,02; H 8,6; 11,77.
Пример
13: N-(Дициклогексил-карбамоилметил)-N-(3-морфолин-4-ил-пропил)-4-нитро-бензамид
N,N-Дициклогексил-2-(3-морфолин-4-ил-пропил)-ацетамид получают аналогично примеру 8 из 2-хлор-N,
N-дициклогексилаце тамида и 3-морфолино-1-пропиламина.
9,1 г (0,025 моль) N,N-Дициклогексил-2-(3-морфолин-4-ил-пропил)ацетамида, 5,3 г (0,05 моль) карбоната натрия и 7,5 г (0,04 моль) 4-нитро-бензоилхлорида вводят во взаимодействие аналогично примеру 10. Остаток (11,5 г) растворяют при нагревании в 25 мл этанола и оставляют стоять в течение нескольких дней. Выпавший осадок отсасывают и перекристаллизуют из этанола. Выход: 7,8 г (60,6% от теории).
Т.пл. = 153-154oC.
C28H42N4O5:
рассчитано, %: С 65,34; H 8,23; N 10,87;
найдено, %: C 62,28; H 8,26; N 11,14.
Пример 14:
N-(Дициклогексил-карбамоилметил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)-бензамид-оксалат
N, N-Дициклогексил-2-(2-пиперидин-1-ил-этиламино)ацетамид получают из 2-хлор-N,
N-дициклогексил-ацетамида и 2-пиперидиноэтиламина аналогично примеру 8.
13,96 г (0,04 моль) N,N-дициклогексил-2-(2-пиперидин-1-ил-этиламино)-ацетамида, 7,5 г (0,07 моль) карбоната натрия и 11,1 г (0,06 моль) 4-нитро-бензоилхлорида вводят во взаимодействие аналогично примеру 10. Остаток перекристаллизуют из разбавленного этанола (80 мл этанола и 30 мл воды). Выход: 11,4 г (55,6% от теории). Т.пл. = 164-165oC.
C28H42N4O4:
рассчитано, %: C 67,44; H 8,49; N 11,27;
найдено, %: C 67,63; H 8,42;
N 11,65.
Пример 15: N-(Бензил-метил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид-гидрохлорид
19,8 г (0,1 моль) N-Метил-бензил-хлорацетамида и 32,5 г (0,25 моль)
3-диэтиламино-1-пропиламина в 100 мл бензола кипятят с обратным холодильником в течение 8 часов. Бензольный раствор экстрагируют путем встряхивания с водой и целевой продукт экстрагируют из бензольной
фазы с помощью 3%-ной соляной кислоты. Солянокислую фазу подщелачивают с помощью разбавленного 1:3 раствора аммиака и экстрагируют бензолом. После промывки бензольного раствора водой, бензол отгоняют
и остаток перегоняют в вакууме. Получают прежде всего 21,5 г (74% от теории) метилбензиламида N-(3-диэтиламинопропил)аминоуксусной кислоты с т. кип. 300-305oC при 3 мм рт.ст.
C17H29N3O
рассчитано, %: С 70,13; H 9,97; N 14,45;
найдено, %: C 70,07; H 10,24; N 14,40.
21,5 г (0,074 моль) Метилбензиламида
N-(3-диэтиламино-пропил)-аминоуксусной кислоты, 16,3 г (0,154 моль) карбоната натрия и 18,6 г (0,1 моль) 4-нитро-бенэоилхлорида вводят во взаимодействие аналогично примеру 10. Остаток растворяют в
изопропаноле, смешивают с небольшим количеством эфира и оставляют стоять в холодильнике. Осадок отсасывают и перекристаллизуют из изопропанола. Выход: 17,8 г (50,4% от теории)
Т.пл. =
160-162oC.
C24H32N4O4•HCl
рассчитано, %: C 60,43; H 6,97; N 11,75; Cl 7.43;
найдено, %: C 60,62; H 7,23;
N 11,73; CI 7,50.
Получение исходного продукта:
N-Метил-N-бензил-хлорацетамид
К смеси из 30,2 г (0,25 моль) N-метилбензиламина в 300 мл хлороформа и 54,4 г (0,4 моль)
карбоната калия в 80 мл воды, при перемешивании и при температуре 20-25oC, в течение 30 минут прикапывают 40,6 г (0,36 моль) хлорацетилхлорида. После перемешивания в течение 2 часов при
комнатной температуре хлороформный раствор экстрагируют путем встряхивания сначала с водой, затем с 3%-ной соляной кислотой и с водой. Хлороформ отгоняют и остаток перегоняют в вакууме.
Выход: 46,7 г (94,5% от теории). Т. кип.: 155-158oC при 5 мм рт.ст.
C10H12ClNO:
рассчитано, %: С 60,91; H 6,06; N 7,10; Cl 17,97;
найдено, %: C 60,87; H 5,97; N 6,81; CI 17,94.
Пример 16: N-(3-Диэтиламино-пропил)-N-/2-(10,11-дигидро-дибензо/b,f/-азепин-5-ил)-2-оксо-этил/-4-нитро-бензамид-оксалат
27,2 г (0,1 моль) 2-Хлор-N-(10,11-дигидро-дибензо/b,f/азепин-5-ил)-ацетамида и 39 г (0,3 моль) 3-диэтиламино-1-пропиламина перемешивают в течение 3 часов при 100oC, затем отгоняют
избыточный 3-диэтиламино-1-пропиламин. Остаток смешивают с водой и концентрированным раствором аммиака и продукт встряхивают с бензолом. Бензольную фазу экстрагируют путем встряхивания с водой и
целевой продукт экстрагируют из бензольной фазы с помощью разбавленной 1: 5 соляной кислоты. Солянокислую фазу подщелачивают с помощью разбавленного 1:5 раствора аммиака и экстрагируют бензолом. После
промывки бензольной фазы водой бензол отгоняют. Выход 2-(3-диэтиламино-пропиламино)-N-(10,11-дигидро-дибензо-/b, f/азепин-5-ил)-ацетамида составляет 33 г (90% от теории).
14,61 г (0, 04 моль) 2-(3-диэтиламино-пропиламино)-N-(10,11-дигидро-дибензо/b, f/азепин-5-ил)-ацетамида растворяют в 200 мл бензола, в полученный раствор вносят 8,4 г (0,08 моль) карбоната натрия и при перемешивании и при комнатной температуре прикапывают раствор 11,1 г (0,06 моль) 4-нитро-бензоил-хлорида в 50 мл бензола. Реакционную смесь перемешивают в течение 5 часов при комнатной температуре, затем смешивают со 150 мл воды и перемешивают еще 4 часа при комнатной температуре. Бензольную фазу отделяют, встряхивают с водой и бензол отгоняют. Выход маслянистого N-(3- диэтиламино-пропил)-N-/2-(10,11-дигидро-дибензо/b, f/азепин-5-ил)-2-оксо-этил/-4-нитро-бензамида составляет 19,75 г (95% от теории).
К раствору 10,3 г (0,02 моль) N-(3-диэтиламино-пропил)-N-/2-(10,11-дигидро-дибензо/b, f/азепин-5-ил/-2-оксо-этил/-4-нитро-бензамида в 100 мл изопропанола добавляют раствор 2,8 г (0,022 моль) дигидрата щавелевой кислоты в 100 мл 50%-ного изопропанола и оставляют стоять в холодильнике вплоть до кристаллизации. Осадок отсасывают и высушивают. Выход: 7,8 г (64,5% от теории). Температура разложения: 209-210oC.
C30H34N4O4•(COOH)2:
рассчитано, %: C 63,56; H 6,02; N 9,26;
найдено, %: C 63,57; H 6,23; N 9,52.
Пример 17: 4-Амино-N-(дициклогексил-карбамоилметил)-N-(3- диэтиламино-пропил) бензамид- оксалат-гидрат
5 г (0,01 моль)
N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-нитро-бензамида в 50 мл метанола при добавке 0,3 г палладия-на-угле (10% Pd) гидрируют при перемешивании и при комнатной температуре.
Теоретическое количество водорода поглощается в течение 2 часов. Реакционную смесь перемешивают в течение 3 часов в атмосфере водорода, катализатор отфильтровывают и метанол отгоняют.
Маслянистый 4-амино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид (4,7 г) растворяют в 30 мл абсолютного этанола и смешивают с раствором 2,5 г (0,02 моль) щавелевой кислоты в 15 мл абсолютного этанола. Этанол отгоняют, остаток растворяют в 40 мл изопропанола, добавляют небольшое количество эфира и оставляют стоять в холодильнике вплоть до кристаллизации. Осадок отсасывают и высушивают при 50-60oC в вакууме. Выход; 4,1 г (66% от теории). Т.пл.=90,5-92oC.
C28H46N4O2•1,5 (COOH)2•H2O
рассчитано, %: С 59,64; H 8,24; N 9,72; H2O 2,88;
найдено, %: C 59,82; H 8,17; N 9,85; H2O 2,18.
Пример 18:
4-Ацетиламино-N-(дициклогексил-карбамоилметил)-N-(3-диэтил-амино-пропил)бензамид-полугидрат
К раствору 4,7 г (0,01 моль)
4-амино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламинопропил)бензамида в 40 мл безводного бензола добавляют раствор 5,1 г (0,05 моль) уксусного ангидрида в 10 мл безводного бензола. При этом
температура повышается до 45oC. Реакционную смесь в течение 6 часов кипятят с обратным холодильником. Бензол и уксусный ангидрид отгоняют. К остатку (5 г) добавляют воду и разбавленную
соляную кислоту и раствор выливают в разбавленный раствор аммиака. Осадок отфильтровывают, промывают разбавленным раствором аммиака и водой и высушивают в вакууме. Выход: 3,8 г (74% от теории.). Т.пл.
= 92-93oC.
C30H48O3N4•0,25 H2O
рассчитано, %: C 69,66; H 9,45; N 10,85; H2O 0,87;
найдено, %: C 69,28; H 9,4; N 10,81; H2O 0,73.
Пример 19: 4-Ацетиламино-N-(дициклогексил-карбамоилметил)-N-(2-диэтил-амино-этил)-бензамид-оксалат
4-Амино-N-(дициклогексил-карбамоиламино)-N-(2-диэтиламино-этил)-бензамид получают из N-(дициклогексил-карбамоилметил)-N-(2- диэтиламино-этил)-4-нитро-бензамида, согласно методике примера 17.
К раствору 4,57 г (0,01 моль) 4-амино-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-бензамида в 40 мл бензола добавляют раствор 5,1 г (0,05 моль) уксусного ангидрида в 10 мл безводного бензола. Реакционную смесь кипятят с обратным холодильником в течение 6 часов и затем концентрируют досуха. Остаток растворяют в воде, подщелачивают с помощью разбавленного 1:5 раствора аммиака и экстрагируют бензолом. После промывки бензольной фазы водой бензол отгоняют.
Остаток (4,9 г) растворяют в 10 мл изопропанола, добавляют раствор 1,26 г (0,01 моль) дигидрата щавелевой кислоты в 15 мл изопропанола и оставляют стоять вплоть до кристаллизации. Осадок отсасывают и перекристаллизуют из изопропанола.
Выход: 4,04 г (68,6% от теории)
Т.пл. = 195-197oC.
C29H46N4O3•(COOH)2:
рассчитано, %: C 63,24; H 8,22; N 9,52;
найдено, %: C
63,32; H 8,44; N 9,66.
Пример 20: 4-Ацетиламино-N-(бензил-метил-карбамоилметил)-N-(3- диэтил-амино-пропил)-бензамид-гидрат
4-Амино-N-(бензил-метил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид получают из N-(бензилметил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-нитро-бензамида, согласно методике примера 17.
5,9 г (0,0143 моль) 4-Амино-N-(бензил-метил-карбамоилметил)-N-(3-диэтиламино-пропил)бензамида и 7,45 г (0,073 моль) уксусного ангидрида в 50 мл бензола кипятят с обратным холодильником в течение 6 часов и затем перегоняют досуха. Остаток смешивают с водой и 10%-ным раствором аммиака и экстрагируют бензолом. Бензольный раствор при нагревании встряхивают с водой и концентрируют на треть. Остаток смешивают с небольшим количеством воды и оставляют стоять вплоть до кристаллизации. Осадок отсасывают и высушивают в вакууме в течение 3 часов при температуре 30-40oC. Выход: 5,22 г (78% от теории). Т.пл. = 55-56oC.
C26H36N4O3•H2O
рассчитано, %: C 66,33; H 8,14; N 11,90;
H2O 3,79;
найдено, %: C 66,19; H 8,11; N 12,05; H2 4.20.
Пример 21:
4-Карбэтоксиамино-N-(дициклогексил-карбамоил-метил)-N-(3-диэтиламино-пропил)-бензамид-оксалат-гидрат
4,7 г (0,01 моль)
4-Амино-N-(дициклогексил-карбамоилметил)- (3-диэтиламино-пропил)бензамида растворяют в 60 мл бензола, затем смешивают с 2,12 г (0,02 моль) карбоната натрия и при перемешивании и при температуре
15-20oC прикапывают раствор 2,16 г (0,02 моль) этилового эфира хлормуравьиной кислоты в 10 мл бензола. Реакционную смесь перемешивают 3 часа при комнатной температуре, добавляют 50 мл воды
и перемешивают следующие 30 минут при комнатной температуре. Бензольную фазу отделяют, встряхивают с водой и перегоняют досуха. Остаток (5,2 г) растворяют в 20 мл изопропанола. Добавляют 1 г (0,011
моль) щавелевой кислоты, растворенной в 5 мл изопропанола, и небольшое количество воды, и оставляют стоять в холодильнике вплоть до кристаллизации. Осадок отсасывают и перекристаллизуют из
изопропанола и эфира.
Выход: 4,8 г (75% от теории). Т.пл. = 160-161oC (разложение).
C31H50N4O4•
(COOH)2•H2O
рассчитано, %: C 61,76; H 8,32; N 8,73; H2O 1,4;
найдено, %: C 61,60; H 8,40; N 9,01; H2O 2,0.
Пример
22: N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид-оксалат
К раствору 7,1 г (0,015 моль)
4-амино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамида и 3,03 г (0,03 моль) триэтиламина при перемешивании и при температуре 15-20oC добавляют раствор 2,06 г (0,018
моль) метансульфонил-хлорида в 10 мл хлороформа. Реакционную смесь перемешивают в течение 6 часов при комнатной температуре, затем кипятят с обратным холодильником в течение 6 часов, добавляют
следующие 1,72 г (0,015 моль) метансульфонилхлорида и кипятят с обратным холодильником в течение 6 часов. После охлаждения добавляют 50 мл воды и перемешивают в течение 2 часов. Затем добавляют
раствор 4,8 г (0,035 моль) карбоната калия в 20 мл воды и перемешивают в течение 3 часов. Хлороформную фазу отделяют и перегоняют досуха. Остаток смешивают с разбавленным раствором аммиака и
экстрагируют бензолом. После промывки бензольной фазы водой бензол отгоняют. Остаток растворяют в 70 мл метанола и 60 мл триэтиламина и кипятят с обратным холодильником в течение 6 часов. После этого
перегоняют досуха, остаток смешивают с водой и экстрагируют бензолом. После промывки бензольной фазы водой бензол отгоняют, остаток (6 г) растворяют в 15 мл этанола и добавляют раствор 1,4 г (0,011
моль) дигидрата щавелевой кислоты в 5 мл этанола. По окончании кристаллизации (холодильник) осадок отсасывают и перекристаллизуют из этанола. Выход: 4,9 г (51% от теории). Т.пл. = 175-176o
C.
C29H48N4O4S•(COOH)2:
рассчитано, %: C 58,27; H 7,88; N 8,77; S 5,02;
найдено, %: C 58,26; H 7,81; N
9,06; S 4,97.
Пример 23: N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид-иодметилат
2,7 г (0,005 моль)
N-(Дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамида и 2,13 г (0,015 моль) иодметана в 30 мл ацетона кипятят с обратным холодильником в течение 3 часов. После
охлаждения осадок отсасывают и промывают ацетоном. Осадок в 30 мл ацетона кипятят с обратным холодильником в течение 20 минут, отсасывают
и перекристаллизуют из изопропанола.
Выход: 2,6 г (76% от теории). Т.пл. = 139-140,5oC.
C30H51N4SO4
рассчитано, %: C 52,16; H 7,44; N 8,11; S 4,64;
найдено, %: C 52,49; H 7,48; N 7,84; S 4,47.
Пример 24: N-(Дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-метансульфониламино-бензамид-оксалат
7,0 г (0,0155 моль)
4-Амино-N-(дициклогексил-карбамоил-метил)-N-(2-диэтиламино-этил)бензамида и 2,7 г (0,0235 моль) метансульфонилхлорида вводят во взаимодействие согласно методике примера 22.
Маслянистый N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-метансульфониламино-бензамид (6,9 г) растворяют в 9 мл изопропанола, добавляют раствор 0,4 г (0,003 моль) дигидрата щавелевой кислоты в 2 мл изопропанола, выпавший осадок отсасывают и перекристаллизуют из этанола. Выход: 2,2 г (73,1% от теории).
C28H46N4SO4•
0,5 (COOH)2:
рассчитано, %: C 60,08; H 8,17; N 9,66; S 5,53;
найдено, %: C 59,79; H 8,14; N 9,58; S 5,56.
Пример 25:
N-(3-Диэтиламино-пропил)-N-/2-(3-фторфенил-1-ил-амино)-2-оксо-этил/-4-нитро-бензамид
3,7 г (0,01 моль) 2-(3-Диэтиламино-пропиламино)-N-(3-фторфенил)-ацетамида•2HCl•H2
O, 50 мл хлороформа, 2,8 г (0,015 моль) п-нитро-бензоилхлорида и 2,8 мл (0,02 моль) триэтиламина кипятят с обратным холодильником в течение 2 часов. За протеканием реакции следят с помощью ТСХ
(тонкослойной хроматографии) (толуол: метанол: концентрированный раствор NH3 = 60:40:1). По окончании взаимодействия реакционную смесь экстрагируют трижды с помощью 20 мл насыщенного
раствора карбоната калия и затем дважды с помощью 10 мл концентрированной HCl. Объединенные солянокислые фазы экстрагируют путем встряхивания один раз с 20 мл хлороформа, подщелачивают с помощью
концентрированного раствора аммиака, экстрагируют дважды 20 мл хлороформа и объединенные хлороформные фазы экстрагируют один раз с помощью 20 мл воды. Хлороформные фазы сушат над сульфатом натрия и,
после концентрирования, получают кристаллический остаток массой 2,4 г, который перекристаллизуют из 5 мл изопропанола. Выход: 1,9 г (44,2% от теории).
Т.пл. = 106 - 109o
C
C22H27N4O4F (масса моля 430,48)
рассчитано, %: С 61,38; H 6,32; N 13,02;
найдено, %: C 61,49; H 6,35; N 13,45.
Исходный продукт для получения вышеуказанного соединения синтезируют следующим образом:
2-(3-Диэтиламино-пропиламино)-N-(3-фторфенил)-ацетамид-дигидрохлорид-гидрат
3,5 г (0,02 моль)
Хлорацетил-3-фторанилида и 13,0 г (0,1 моль) 3-диэтиламино-1-пропиламина объединяют вместе, причем внутренняя температура повышается примерно до 50oC. После этого в течение 1 часа
перемешивают при комнатной температуре и избыток амина отгоняют в вакууме. Кристаллический остаток смешивают с ацетонитрилом, нагревают вплоть до полного растворения и осторожно охлаждают. В этот
раствор пропускают газообразный HCl вплоть до достижения кислой реакции. Гидрохлорид частично выкристаллизовывается. Реакционную смесь концентрируют и остаток перекристаллизуют из изопропанола.
Выход: 3,4 г (45,8% от теории). Т.пл. = 174-178oC.
C15H24N3OF•2HCl•H2O (масса моля 372,32):
рассчитано, %: С 48,51; H 7,50; N 11,31; Cl- 19,09;
найдено, %: C 48,13; H 7,62; N 11,33; CI- 19,10.
Пример 26:
N-(3-Диэтиламино-пропил)-N-(2-морфолин-4-ил-3-оксо-этил)-4-нитро-бензамид
2-(3-Диэтиламино-пропиламино)-ацетилморфолид (из 0,022 моль хлорацетилморфолида) растворяют в 30 мл хлороформа,
смешивают с 3,9 г (0,02 моль) 4-нитро-бензоилхлорида и при кипячении с обратным холодильником перемешивают в течение 5 часов. Затем реакционную смесь экстрагируют путем встряхивания один раз с 10 мл
разбавленного 1:1 раствора аммиака, один раз с 10 мл воды и дважды с разбавленной 1:1 HCl. Объединенные солянокислые фазы экстрагируют один раз с помощью 10 мл воды, подщелачивают с помощью
концентрированного раствора NH3 дважды экстрагируют хлороформом, беря каждый раз по 20 мл хлороформа, и объединенные хлороформные фазы экстрагируют путем встряхивания с 10 мл воды. После
этого хлороформные фазы сушат и концентрируют. Кристаллический остаток (4,7 г) перекристаллизуют из 5 мл изопропанола и высушивают при 70oC. Выход: 2,2 г (39,5% от теории, в расчете на
морфолид хлоруксусной кислоты).
C20H30N4O5 (масса моля 406,49)
рассчитано, %: С 59,10; H 7,44; N 13,78;
найдено, %: C 58,
80; H 7,38; N 13,86.
Получение исходного продукта:
2-(3-Диэтиламино-пропиламино)ацетил-морфолид
13 г (0,1 моль) 3-Диэтиламино-1-пропиламина при охлаждении смешивают
с 3,3 г (0,022 моль) морфолида хлоруксусной кислоты. Реакционную смесь перемешивают в течение 1 часа при комнатной температуре, затем отгоняют избыточный амин, дважды смешивают с 50 мл воды и каждый
раз воду отгоняют (30-40 мбар). Остаток еще раз высушивают при 140oC и давлении 0,8-1 торр. Получают маслянистый остаток, который без дальнейшей очистки используют для последующей реакции
ацилирования.
Пример 27: N-/2-(3-Карбэтокси-амино-10,11-дигидро-дибензо/b,f/азепин-5-ил)-2-оксо-этил/-N-(2-диэтиламино-этил)-4-нитро-бензамид
8,8 г (0,02 моль)
2-(Диэтиламино-этиламино)-N-/(3-карбэтокси- амино-10,11-дигидро-дибензo/b, f/азепин)-5-ил/-ацетамида. 100 мл толуола и 9,25 г (0,05 моль) 4-нитро-бензоил- хлорида перемешивают в течение 1 часа при
кипячении с обратным холодильником. После охлаждения толуол сливают от выделившегося (осадившегося) продукта и остаток обрабатывают хлороформом. Хлороформную фазу экстрагируют путем встряхивания
трижды с 20 мл насыщенного раствора карбоната калия и один раз с 20 мл воды. Затем сушат над сульфатом натрия и концентрируют. Остаток (11,6 г) обрабатывают 20 мл изопропанола. Спустя некоторое время
продукт кристаллизуется. Выход: 10,7 г (90,9% от теории).
C32H31N5O6 (масса моля 588,69):
рассчитано, %: C 65,40; H 6,35; N 11,92;
найдено, %: C 65,36; H 6,34; N 12,18.
Пример 28: 8-Хлор-5-{/N-[3-(диэтиламино)пропан-1-ил]-N-(п-метилсульфониламинобензоил)/аминоацетил} -5,10-дигидро-11H-дибензо-/b,e//1,
4/диазепин-11-он-0,5 гидрохлорид-гидрат
2,55 г (0,005 моль) 8-Хлор-5-/(3-диэтиламинопропан-1-ил)аминоацетил/-5,10-дигидро-11Н-дибензо /b,e//1,4/диазепин-11-он-гидрохлорида растворяют в 50 мл
воды и с помощью концентрированного раствора аммиака высвобождают основание. Затем экстрагируют путем встряхивания дважды с 25 мл хлороформа, и объединенные хлороформные фазы экстрагируют путем
встряхивания с 20 мл воды. Объединенные хлороформные фазы, затем сушат над сульфатом натрия, смешивают с 0,4 мл (0,005 моль) пиридина, нагревают до температуры бани 80oC (кипение с обратным
холодильником), добавляют 1,8 г (0,0075 моль) 4-метил-сульфониламинобензоилхлорида и перемешивают в течение 3 часов при кипении с обратным холодильником. При этом выделяется мазеобразный продукт,
который осаждается на краю колбы. Реакционную смесь затем смешивают с 20 мл разбавленного 1: 1 раствора аммиака, хорошо перемешивают, разделяют в делительной воронке, хлороформную фазу еще раз
экстрагируют путем встряхивания с 20 мл воды и экстрагируют разбавленной 2:1 соляной кислотой (при большем разбавлении соляной кислоты также приходят к выделению мазеобразного продукта между фазами).
Солянокислую фазу еще раз экстрагируют путем встряхивания с 20 мл хлороформа, подщелачивают с помощью концентрированного раствора аммиака и затем экстрагируют дважды хлороформом, беря каждый раз по 25
мл хлороформа. Объединенные хлороформные фазы (50 мл) встряхивают теперь с 30 мл воды, сушат над сульфатом натрия и концентрируют досуха (остаток: 2,9 г). Остаток перекристаллизуют из метанола
(добавка активного угля). Выход: 1,3 г (40%).
C30H34N5O5SCl•0,5 HCl•H2O (масса моля 666,63):
рассчитано,
%: C 55,57; H 5,67; N 10,80; Cl 8,20; Cl- 2,73;
найдено, %: C 55,81; H 5,62; N10,90; CI 8,30; CI- 2,81;
рассчитано, %: S 4,95; H2O 2,8.
найдено, % S 4,96; H2O 2,4.
Пример 29: 8-Хлор-5-{ /6-[N-(3-диэтиламино)пропан-1- ил]-N-бензоил/-амино-1-оксогексил} -5,10-дигидро-11Н-дибензо/b, e//1,
4/диазепин-11-он-гидрохлорид-полугидрат
5,44 г (0,01 моль) 8-Хлор-5-{6-/[3-(диэтиламино)пропан-1-ил]амино/-1-оксогексил} -5,10-дигидро-11Н-дибензо/b, e/ /1,
4/диазепин-11-он-дигидрохлорид-полугидрата растворяют в 50 мл воды, смешивают с концетрированным раствором аммиака вплоть до сильно щелочной реакции и экстрагируют с помощью 50 мл и 20 мл толуола.
После встряхивания с 20 мл воды и высушивания над сульфатом натрия к объединенным толуольным фазам, при температуре бани 70-80oC и при перемешивании, добавляют 3,5 г (0,025 моль)
бензоилхлорида. Время реакции составляет 2 часа. После охлаждения реакционной смеси толуол сливают от выделившегося масла, остаток обрабатывают при нагревании 30 мл изопропанола, изопропанол снова
отгоняют и остаток высушивают при давлении 80 мбар (4,2 г аморфного продукта). 3,7 г вещества растворяют в 35 мл метанола и очищают с помощью ВЭЖХ. При этом получают 1,8 г целевого соединения, после
высушивания при давлении 2 мбар и 60oC (3 часа) в виде моногидрохлорида с 0,5 моля воды.
C33H39N4O3Cl•HCl•0,5
H2O (масса моля 620,63):
рассчитано, %: C 63,87; H 6,66; N 9,03; Cl 11,43;
найдено, %: C 64,20; H 6,56; N 9,20; CI 11,27.
Пример 30:
N-(2-Диэтиламино-этил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)бензамид•(COOH)2
41,32 г (0,256 моль) N-Хлор ацетил-пиперидина и 66,1 г (0,57 моль) N, N-диэтилэтилендиамина в
300 мл бензола кипятят с обратным холодильником в течение 12 часов, бензол и N,N-диэтилэтилендиамин отгоняют и остаток растворяют в 5%-ной соляной кислоте. Затем с помощью концентрированного раствора
аммиака подщелачивают и экстрагируют хлороформом. После промывки хлороформной фазы водой хлороформ отгоняют. Выход маслянистого N-/2-диэтиламино-этиламино) ацетил/пиперидина составляет 40,6 г (65% от
теории). 40,6 г (0,168 моль) N-/2-(2-диэтиламино-этиламино)ацетил/пиперидана растворяют в 200 мл бензола и смешивают с 35,6 г (0,33 моль) Na2CO3. При перемешивании и при
температуре 15-20oC прикапывают раствор 46,9 г (0,253 моль)-4-нитробензоилхлорида в 200 мл бензола. Реакционную смесь перемешивают в течение 6 часов при комнатной температуре, смешивают с
200 мл воды и перемешивают следующие 4 часа при комнатной температуре. Затем бензольный раствор отделяют, встряхивают его с водой, целевой продукт экстрагируют с помощью разбавленной 1: 5 соляной
кислотой, кислую фазу после отделения подщелачивают с помощью концентрированного раствора аммиака и основную (щелочную) фазу экстрагируют путем встряхивания с хлороформом. После промывки хлороформной
фазы водой эту фазу концентрируют. Выход маслянистого N-(2-диэтиламино-этил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)бензамида составляет 34,3 г (52% от теории).
К раствору 11,15 г (0, 0286 моль) N-(2-диэтиламино-этил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)бензамида в 10 мл подогретого изопропанола добавляют раствор 2,6 г (0,0288 моль) щавелевой кислоты в 10 мл изопропанола и раствор оставляют стоять в холодильнике. Осадок отсасывают и перекристаллизуют из изопропанола. Выход: 7,62 г (55,5% от теории).
Т.пл. = 132-134oC.
C20H30N4O4•(COOH)2 (масса моля 480,52)
рассчитано, %: С 54,99; H 6,71; N 11,66;
найдено, %: C 55,27; H 6,77; N 11,73.
Реферат
Амиды аминокарбоновых кислот формулы I, где R1, R2 - Н, C1-8-алкил, С3-8-циклоалкил; фенил или фенилалкил, возможно замещенный алкилом, алкоксилом, галогеном, цианогруппой, нитро, СF3 или ациламиногруппой; или R1 -N-R2 обозначает морфолин, пиперидин или группа (а) или (б), R3-CO-R7 или O2S-C6H4-R8 (значения других радикалов см. п.1 формулы изобретения), их физиологически приемлемые соли и фармацевтические композиции на их основе представляют собой новые антиаритмические средства. 3 с. и 2 з.п. ф-лы, 3 табл.
R1(R2)N-CO-(CH2)n-N(R3)-(CH2)m-N(R4)R5 (I)
Формула
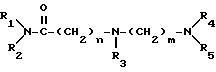
где R1, R2 обозначают водород; линейный или разветвленный (C1-C8)-алкил, (C3-C8)-циклоалкил, такой, как циклогексил; фенил, который может быть одно- или двукратно замещен линейным (C1-C4)-алкилом, (C1-C2)-алкоксилом, галогеном, цианогруппой, нитрогруппой, трифторметилом или ациламиногруппой; фенилалкил, где алкильная цепь может содержать 1-3 C-атома и фенильное кольцо, которое может быть одно- или двукратно замещено метилом, метоксигруппой, галогеном, нитро-, цианогруппой или ациламидогруппой; или
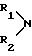
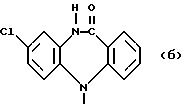
обозначает морфолин или пиперидин, причем пиперидин может быть замещен одно- или двукратно (C1-C2)-алкилом; или группу
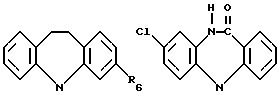
где R6 может обозначать H, NHCO2CH2CH3;
R3 обозначает CO - R2,
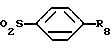
R7 обозначает (C3-C7)-циклоалкил, такой, как циклогексил; фенил, который может быть одно- или двукратно замещен линейным (C1-C4)-алкилом, (C1-C2)-алкоксилом, галогеном, циано-, нитрогруппой, трифторметилом, сульфонамидо-, метансульфонамидогруппой или ациламиногруппой; фенилалкил, где алкильная цепь может содержать 1-3 С-атома и фенильное кольцо может быть одно- или двукратно замещено метилом, метоксигруппой, галогеном, нитро-, цианогруппой или ациламидогруппой;
R8 обозначает водород, ациламиногруппу;
R4, R5 обозначает линейный или разветвленный (C1-C6)-алкил; (C3-C6)-циклоалкил, такой как циклогексил; фенил; фенил-алкил, где алкильная группа может содержать 1-3 С-атома; или
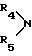
обозначает пиперидин или морфолин,
n = 1-5;
m = 2-4,
или их физиологически приемлемые соли присоединения кислот.
/4-ацетиламино-бензолсульфонил/-/2-диэтиламино-этил/-амино/-N, N-дициклогексил-ацетамид;
N-/дициклогексил-карбамоилметил/-N-/2-диэтиламино-этил/-3-метокси-4-нитро-бензамид;
4-циано-N-/дициклогексил-карбамоилметил/-N-диэтиламиноэтил/бензамид;
4-хлор-N-/дициклогексил-карбамоилметил/-N-/2-диэтиламиноэтил/-3-нитро-бензамид;
N-/2-дициклогексиламино-карбамоилметил/-N-/3-диэтиламинопропил/-4-трифторметил-бензамид;
4-ацетиламино-N-/3-дибутиламино-пропил/-N-/дициклогексилкарбамоилметил/-бензамид;
N-/5-(дициклогексил-карбамоил)пентил/-N-(2-диэтиламино-этил)-4-нитро-бензамид;
4-амино- N-(дициклогексил-карбамоилметил)- N-(3-диэтиламино-пропил)бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-2-нитро-бензамид;
4-карбэтоксиамино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-морфолин-4-ил-пропил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-3-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диметиламино-пропил)-4-нитро-бензамид;
4-ацетиламино-N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид;
4-ацетиламино-N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-бензамид;
N-(дициклогексил-карбамоилметил)-4-нитро-N-(2-пиперидин-1-ил-этил)бензамид;
4-ацетиламино-N-(бензил-метил-карбамоилметил)-N-(3-диэтиламино-пропил)-бензамид;
N-(дициклогексил-карбамоил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид;
N-(дициклогексил-карбамоилметил)-N-(3-диэтиламино-пропил)-4-метансульфониламино-бензамид-иодметилат;
N-(дициклогексил-карбамоилметил)-N-(3-морфолин-4-ил-пропил)-4-нитро-бензамид-иодметилат;
N-(3-диэтиламино-пропил)-N-/2-(10,11-дигидро-дибензо/b, f/азепин-5-ил)-2-оксо-этил/-4-нитро-бензамид;
N-(2-диэтиламино-этил)-4-нитро-N-(2-оксо-2-пиперидин-1-ил-этил)-бензамид;
N-(бензил-метил-карбамоилметил)-N-(2-диэтиламино-этил)-4-нитро-бензамид;
N-(дициклогексил-карбамоилметил)-N-(2-диэтиламино-этил)-4-метансульфониламино-бензамид;
N-(3-диэтиламино-пропил)-N-/2-(3-фторфенил-1-ил-амино)-2-оксоэтил/-4-нитро-бензамид;
N-(3-диэтиламино-пропил)-N-(2-морфолин-4-ил-2-оксо-этил)-4-нитро-бензамид;
N-/2-(3-карбэтокси-амино-10,11-дигидро-дибензо/b, f/азепин-5-ил)-2-оксо-этил/-N-(2-диэтиламино-этил)-4-нитро-бензамид;
8-хлор-5-{ /N-[3-(диэтиламино)пропан-1-ил] -N-(п-метилсульфониламино-бензоил)/-аминоацетил}-5, 10-дигидро-11H-дибензо/ b, e//1, 4/диазепин-11-он;
8-хлор-5-{ /6-[N-(3-диэтиламино)пропан-1-ил] -N-бензоил/-амино-1-оксо-гексил}-5, 10-дигидро-11Н-дибензо/b, e//1, 4/диазепин-11-он.
где R1 и R2 имеют вышеуказанное значение,
вводят во взаимодействие с галогенидами w-галогенкарбоновых кислот общей формулы III
XC(O)-(CH2)n -Y,
где X и Y обозначают атом галогена,
в инертных органических растворителях или двухфазных смесях растворителей, в присутствии акцептора кислоты с получением амидов общей формулы IV
R1R2N-C(O)-(CH2)n-Y,
причем R1, R2 и Y имеют вышеуказанное значение,
и амиды далее вводят во взаимодействие с диаминами общей формулы V
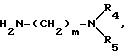
причем R4 и R5 имеют вышеуказанное значение,
в инертных растворителях, в присутствии акцептора кислоты с получением соединений общей формулы VI
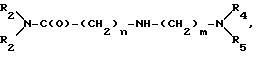
где R1, R2, R4 и R5, а также n и m имеют вышеуказанное значение,
и переводят их с помощью галогенидов карбоновых кислот или галогенидов сульфокислот общей формулы VII
R3-X
или VIII
(R3)2O,
где R3 имеет вышеуказанное значение,
в инертных растворителях, без добавки или в присутствии акцептора кислоты, в соединения общей формулы I.
Комментарии