17β-n-(2,5-бис(трифторметил)фенилкарбамоил-4-аза-5α-андрост-1-ен-3-он, способы его получения, фармацевтический состав, способ лечения - RU2140926C1
Код документа: RU2140926C1
Чертежи

Описание
Предметом настоящего изобретения является конкретное производное 17 β -анилид-4-аза- 5α -андрост-1-ен-3-она как необычайно сильный и избирательный двойственный ингибитор человеческой 5 α -редуктазы типа 1 и 2.
Андрогены ответственны за многие физиологические функции как у мужчин, так и у женщин. Действие андрогена опосредовано специфическими внутриклеточными гормональными рецепторами, экспрессируемыми в чувствительных к андрогену клетках. Тестостерон, основной циркулирующий андроген, секретируется клетками Leydig в тестах при стимуляции продуцируемого гипофизом лютенизирующего гормона (ЛГ). Однако для действия андрогена в некоторых тканях-мишенях, таких как предстательная железа и кожа, необходимо восстановление 4,5 двойной связи тестостерона с образованием дигидротестостерона (ДГТ). Стероид-5 α -редуктазы в тканях-мишенях катализирует превращение тестостерона в ДГТ по НАДФ-зависимому пути, как показано на Схеме А.
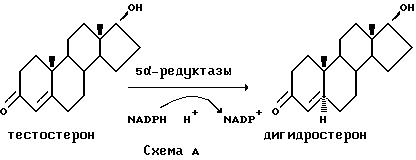
Способность ДГТ действовать в качестве агониста в таких тканях-мишенях было ярко освещено исследованиями дефицитных по стероид-5 α -редуктазе индивидов, у которых имеется рудиментарная предстательная железа и которые не страдают от юношеских угрей или мужского облысения [1]. Таким образом, ожидается, что ингибирование превращения тестостерона в ДГТ в таких тканях-мишенях будет полезным при лечении различных андрогензависимых заболеваний, например доброкачественной гиперплазии предстательной железы, рака предстательной железы, угрей, мужского облысения и гирсутизма.
Кроме того, недавно было установлено, что у людей существуют два изозима 5 α -редуктазы, которые различаются по их тканевой локализации, сродству к тестостерону, профилю pH и чувствительности к ингибиторам [2,3]. Индивиды, дефицитные по стероид5 α -редуктазе, изучавшиеся Imperato-McGinley, являются дефицитными по ферменту 5 α -редуктазе типа 2 [4,5], который является доминирующим изозимом, локализованным в предстательной железе, в то время как изозим типа 1 доминирует в коже. Относительное количество изозимоспецифических и двойственных ингибиторов обоих изозимов 5 α -редуктазы зависит от типа излечиваемого заболевания (доброкачественная гиперплазия предстательной железы, рак предстательной железы, угри, мужское облысение или гирсутизм), а также от стадии заболевания (профилактика в сравнении с лечением) и возможных побочных эффектов у отобранных пациентов (например лечение юношеских угрей у половозрелых мужчин).
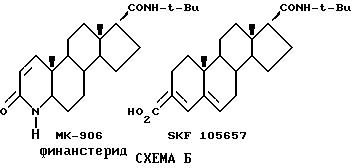
В силу их значительного терапевтического потенциала, ингибиторы тестостерон-5 α -редуктазы [именуемые ниже как "ингибиторы 5α -редуктазы"] являлись объектом активных исследований во всем мире. Например [6-13]. Двумя наиболее многообещающими ингибиторами 5α -редуктазы являются МК-906 (Merck), известный под общим названием финастерид и выпускаемый под торговой маркой Проскар и SKF-105657 (Smithkilne Beecham), представленные на Схеме Б.
Сильное ингибирование 3 β гидрокси- Δ5 - стероид-дегидрогеназа/3-кето- Δ5 -стероид-изомеразы клеток бычьих надпочечников и свиной гранулемы (ЗБГСД) 4-азастероидным производным, 4-МА, представленным на Схеме В, и отсутствие ингибирования лекарственным фенастеридом [14,15] совместно с существенным значением ЗБГСД в стероидном биосинтезе [16] подтверждают тот факт, что оптимальные ингибиторы 5 α -редуктазы типа 1 и 2 также должны быть селективными в отношении ЗБГСД надпочечников человека.
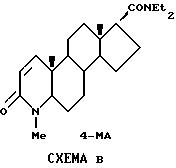
Значение селективности у ингибиторов 5 α -редуктазы особенно подчеркивается сообщениями о гепатотоксичности у определенных 4- азастероидов, таких как 4-МА [17,18].
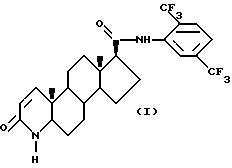
Одним из аспектов настоящего изобретения является соединение формулы (1),
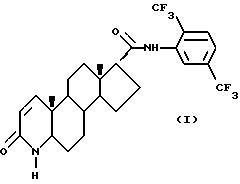
известное также как 17 β -N-(2,5-бис(трифторметил))фенилкарбамоил-4- аза-5 α -андрост-1-ен-3-он и его фармацевтические приемлемые сольваты.
Другими аспектами изобретения являются:
1. Способ ингибирования тестостерон-5 α -редуктаз, включающий взаимодействие тестостерон-5 α
-редуктаз с соединением формулы (1).
2. Способ лечения чувствительного к андрогену или опосредованного им заболевания, включающий введение эффективного количества соединения формулы (1) пациенту, нуждающемуся в таком лечении.
3. Фармацевтичесие препараты, содержащие соединение формулы (1) в качестве активного ингредиента.
4. Способ лечения чувствительного к андрогену или опосредованного им заболевания, включающий введение эффективного количества соединения формулы (1) пациенту, нуждающемуся в таком лечении, в сочетании с таким антиандрогеном, как флутамид.
5. Способ лечения доброкачественной гиперплазии предстательной железы, включающий введение эффективного количества соединения формулы (1) пациенту, нуждающемуся в таком лечении, в сочетании с альфа 1 андренергическим рецепторным блокатором (например теразоцином).
6. Способ лечения доброкачественной гиперплазии предстательной железы, включающий введение эффективного количества соединения формулы (1) пациенту, нуждающемуся в таком лечении, в сочетании с антиэстрогеном.
Специалистам в области органической химии известно, что многие органические соединения могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они выпадают в осадок либо кристаллизуются. Такие комплексы известны как "сольваты". Например комплекс с водой известен как "гидрат". Сольваты соединения (1) находятся в рамках изобретения.
Специалистам в области органической химии следует иметь в виду, что многие органические соединения могут существовать в более чем одной кристаллической форме. Например, кристаллическая форма может изменяться от сольвата к сольвату. Поэтому все кристаллические формы соединений формулы (I) или все их фармацевтически приемлемые сольваты находятся в рамках настоящего изобретения.
Получение соединений
Соединение по
настоящему изобретению может быть получено способами [11, 12] , включенными по сноске. Например, соединение формулы (1) может быть получено способом, представленным на Схеме I и II.
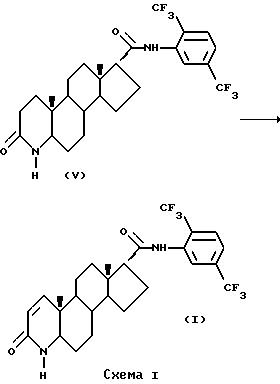
На Схеме I соединение формулы (V) дегидрогенизируют с образованием соединения формулы (1) путем обработки дегидрогенизирующей системой, например 2,3-дихлор-5,6-дициано-1,4-бензохиноном (ДДХ) и бис(триметил-силил)трифторацетамидом в сухом диоксане при комнатной температуре в течение 2-5 часов с последующим прогреванием при дефлегмации в течение 10-20 часов [19].
Соединение формулы (V) может быть получено согласно Схеме 1А
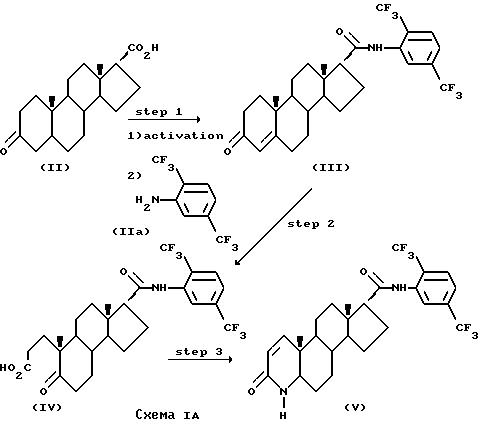
На стадии I Схемы IA 3-оксо-4-андростен-17 β карбоновую кислоту (II) превращают в соответствующий амид формулы (III). Это может быть осуществлено путем активации кислоты и реакции с анилином формулы (IIа). В частности, реакция может представлять собой превращение соединения формулы (II) в соответствующий галогенангидрид посредством обработки галогенизирующим агентом, таким как тионилхлорид, в апротонном растворителе, например толуоле, метиленхлориде или тетрагидрофуране, при температуре от - 5oC до 10oC в присутствии основания, например пиридина.
Промежуточный галогенангидрид, например хлорангидрид, может быть подвергнут взаимодействию с замещенным анилином формулы (IIа) при температуре от 25oC до 70oC в таком апротонном растворителе, как тетрагидрофуран, с образованием амида формулы (III). Соединение формулы (IIa) является коммерчески доступным препаратом (Aldrich Chemical Company, Milwaukee, Wl 53201).
На Стадии 2 соединение формулы (III) превращают посредством окисления, например посредством обработки водным перманганатом калия и периодатом калия в щелочных условиях при дефлегмации в t-бутаноле, в производное 5-оксо-А-нор-3,5-секоандростан-3-оевой кислоты формулы (IV).
На Стадии 3 соединение формулы (IV) превращают в 4-аза-5 α -андростан-3-он формулы (V) посредством обработки аммонием при дефлегмации в этиленгликоле с последующей гидрогенизацией промежуточного 4-аза-андрост-5-ен-3-она в уксусной кислоте при температуре от 60oC до 70oC и давлении водорода, равном 40-60 пси, в присутствии оксида платины в качестве катализатора.
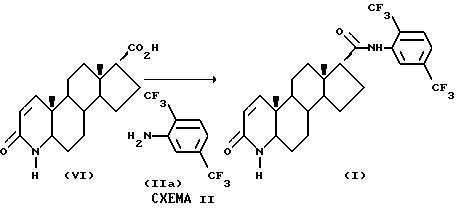
По схеме же II соединение формулы (I) может быть получено из 3-оксо-4-аза-5 α -андрост-1-ен-17 β -карбоновой кислоты формулы (VI) [20] способом по Схеме IА, стадия 1.
Специалисту следует учитывать, что на более ранней стадии получения соединения формулы (I) или его сольвата может оказаться необходимым и/или желательным защитить одну или более чувствительных групп в молекуле для предотвращения нежелательных побочных реакций.
Защитные группы, применяемые при получении соединения формулы (I), могут быть использованы обычным способом [21,22].
Удаление любой имеющейся защитной группы может быть осуществлено обычными способами. Арилалкильная группа, например бензил, может быть отщеплена путем гидролиза в присутствии катализатора, например палладия или древесного угля; ацильная группа, например N- бензилоксикарбонил, может быть удалена посредством гидролиза, например с бромоводородом в уксусной кислоте, или путем восстановления, например каталитической гидрогенизации.
Следует иметь в виду, что в любом из
описанных выше способов может оказаться желательным или даже необходимым защитить любые чувствительные группы в молекуле, как уже отмечалось ранее. Поэтому реакционная стадия, включающая удаление
защитных групп из защищенного производного общей формулы (I) или его соли, может быть осуществлена в результате реализации любого из следующих существующих способов:
(I) удаление любых
защитных групп; и
(II) превращение соединения формулы (I) или его сольвата в его фармацевтически приемлемый сольват.
Будучи используемыми в качестве последней основной стадии в последовательности получения, основные методы, описанные выше для получения соединений по изобретению, могут также быть применены и для введения желательных групп на промежуточной стадии при получении искомого соединения. Поэтому следует иметь в виду, что в таких многостадийных процессах последовательность реакций следует выбирать таким образом, чтобы реакционные условия не оказывали воздействия на те группы, присутствующие в молекуле, наличие которых желательно в конечном продукте.
Соединение формулы (I) и промежуточные соединения (II) - (VI), представленные на Схемах 1 и II, могут быть очищены обычными способами, известными из уровня техники, например хроматографией или кристаллизацией.
In vitrо анализы
Стероид-5 α
-редуктаза
Ферментативные активности можно определять, используя микросомы, полученные из: 1) ткани предстательной железы пациетов с доброкачественной гиперплазией предстательной железы (ДПГ);
2) SF9 клеток, инфицированных рекомбинантным бакуловирусом, который экспрессирует человеческую 5 α -редуктазу типа 2. Микросомы получали путем гомогенизации ткани или клеток с последующим
дифференциальным центрифугированием гомогената. Микросомные экстракты инкубировали с различными концентрациями [1,2,6,7-3H]-тестостерона, 1 мМ НАДФ и различными количествами соединений формулы 1, то
есть тестируемым соединением, в буфере, содержавшем НАДФ-регенерирующую систему, способную поддерживать концентрацию НАДФ в течение периода времени в пределах интервала, равного 0,5-240 минутам. В
качестве контрольного исследования проводили соответствующие инкубации при отсутствии тест-соединения. Для измерений ИК50 для клона 1 все компоненты, задействованные в анализе, за
исключением тестостерона, подвергали предварительной инкубации в течение 10 минут при pH 7,0, а после добавления 100 нМ тестостерона пробы выдерживали в течение 10-120 минут для прохождения анализа.
Для измерения ИК50 для клона 2 все компоненты, задействованные в анализе, за исключением тестостерона, подвергали предварительной инкубации в течение 20 минут при pH 6,0, а после добавления
8 нМ тестостерона пробы выдерживали в течение 20-40 минут для прохождения анализа. Процент превращения тестостерона в ДГТ в присутствии тест-соединений по сравнению с соответствующим превращением в
контрольном исследовании устанавливали с помощью жидкостной хроматографии высокого разрешения (ЖХВР) с радиохимическим обнаружением. Результаты этих анализа, выраженные в виде ИК50,
представлены в Таблице 1.
3 β -гидрокси- Δ5 -стероид-дегидрогеназа/3-кето- Δ5 -стероид- изомераза
Ферментативные активности измеряют,
используя микросомы, полученные из тканей человеческих надпочечников. Микросомы получали посредством гомогенизации этой ткани с последующим дифференциальным центрифугированием гомогената. Микросомные
экстракты инкубировали с различными концентрациями дигидроэпиандростерона (ДГЭА), 1мМ НАД+ и переменными количествами соединения формулы (I), то есть тест-соединения, в буфере с рН 7,5 в
течение периода времени в интервале от 1 до 60 минут. В качестве контрольного исследования проводили соответствующие инкубации при отсутствии тест-соединения. Процент превращения ДГЭА в андростендион
в присутствии тест-соединений по сравнению с соответствующим превращением в контрольном исследовании устанавливали с помощью ЖХВР с радиохимическим обнаружением. Результаты этих анализов, выраженные в
виде Ki, представлены в Таблице 1.
In vivo определение ингибиторов стероид-5 α -редуктазы
In vivo активность ингибиторов стероид-5 α -редуктазы может
быть определена в хронической крысиной модели [23]. В хронической модели используют кастрированных самцов крыс, которым ежедневно подкожно вводят тестостерон (20 мкг/крысу) и тест- соединение (0,01-10
мг/кг) или носитель перорально в течение 7 дней. Затем животных умерщвляют и взвешивают их предстательные железы. Понижение величины веса простимулированных тестостероном предстательных желез
демонстрировало активность тест-соединения. Известные ингибиторы стероид-5 α редуктазы тестировали параллельно для того, чтобы подтвердить согласованность метода анализа.
Практическое применение
Ингибитор стероид-5 α -редуктазы по настоящему изобретению перспективен для лечения андрогеночувствительных заболевании, например доброкачественных и
злокачественных заболеваний предстательной железы, особенно доброкачественной гиперплазии предстательной железы, способом, сходным со способами, разработанными для других ингибиторов 5 α
-редуктазы, таких как финастерид и SKF105657. Однако соединение по настоящему изобретению имеет неожиданно продолжительный период полураспада и эффективность по сравнению с финастеридом и SKF105657.
Данные о корреляции in vitro, in vivo на крысах и клинические результаты на людях в отношении ингибитора 5 α -редуктазы приводят [20,23,24].
Соединение по данному изобретению перспективно для лечения простатитов, рака предстательной железы, опосредованных андрогеном заболеваний кожи, например угрей, гирсутизма и мужского облысения. Другие гормональнозависимые заболевания, в частности поликистоз яичников, также могут излечиваться с помощью этого соединения.
Количество соединения формулы (I), необходимое для того, чтобы оно было эффективно в качестве ингибитора 5 α - редуктазы, будет конечно варьировать в зависимости от конкретного млекопитающего, подлежащего лечению, и оно в конечном счете должно определяться по усмотрению практикующего медика или ветеринара. Факторы, которые должны при этом учитываться, включают состояние, подлежащее излечению, способ введения, природу препарата, вес тела млекопитающего и площадь поверхности, возраст и общее состояние млекопитающего. Однако для пациентов - людей пригодная доза ингибитора 5 α -редуктазы находится в интервале от приблизительно 0,001 до приблизительно 2 мг/кг веса тела в день, предпочтительно в интервале от приблизительно 0,005 до приблизительно 1 мг/кг в день.
Общая дневная доза может быть введена в виде разовой дозы, множественных доз, например от двух до шести раз в день, либо путем внутривенного вливания определенной продолжительности. Дозировки выше или ниже указанного выше интервала находятся в рамках настоящего изобретения и могут быть введены отдельному пациенту в случае, если это желательно или необходимо. Например, для млекопитающего весом 75 кг интервал доз должен составлять от приблизительно 0,4 мг до приблизительно 75 мг в день, типичная доза должна составлять приблизительно 10 мг в день. Из-за продолжительного периода полураспада соединения по настоящему изобретению для многих пациентов лечение может требоваться только через день или даже каждый третий день. Если показаны дискретные множественные дозы, то лечение может обычно заключаться во введении 2,5 мг соединения формулы (I) четырежды в день.
Препараты
Препараты по настоящему изобретению для медицинского применения содержат активное соединение, например соединение формулы (I), совместно с приемлемым носителем и возможно другими
терапевтически активными ингредиентами. Носитель должен быть фармацевтически приемлемым в том смысле, что он должен быть совместимым с другими ингредиентами препарата и не должен быть вредным для его
реципиента.
Настоящее изобретение, кроме того, предлагает фармацевтический препарат, содержащий соединение формулы (I) совместно с его фармацевтически приемлемым носителем.
Препараты включают препараты, пригодные для перорального, локального, ректального или парентерального (включая подкожное, внутримышечное или внутривенное) введение. Предпочтительными являются препараты, пригодные для перорального или парентерального введения.
Допустимо применение препаратов в виде унифицированной дозировочной формы, которая может быть изготовлена любым из известных в фармации способов. Все способы включают стадию ассоциирования активного соединения с носителем, который содержит один или более совместимых ингредиентов. В общем препараты изготовляют путем единообразного и однородного ассоциирования активного соединения с жидким носителем или тщательно измельченным твердым носителем, а затем, если необходимо, формования продукта в виде желаемой унифицированной дозировочной формы.
Препараты по настоящему изобретению, пригодные для перорального введения, могут быть изготовлены в виде дискретных единиц, таких как капсулы, крахмальные капсулы, таблетки или лепешки, каждая из которых содержит предварительно определенное количество активного соединения, таких как порошок или гранулы, или суспензия либо раствор в водном растворе или неводном растворе, например сироп, эликсир, эмульсия или доза жидкого лекарства.
Таблетка может быть изготовлена путем прессования или формования, возможно с одним или более приемлемых ингредиентов. Спрессованные таблетки могут быть изготовлены путем прессования в соответствующей установке активного соединения в свободно- текучей форме, например порошка или гранул, возможно смешанной с приемлемыми ингредиентами, например связующим веществом, смазками, инертными разбавителями, поверхностно-активными или диспергирующими агентами. Формованные таблетки могут быть изготовлены путем формования в соответствующей установке смеси порошкообразного активного соединения с любым приемлемым носителем.
Сироп или суспензия могут быть изготовлены путем добавления активного соединения к концентрированному водному раствору сахара, например сахарозы, к которому также могут быть добавлены любые приемлемые ингредиенты. Такие приемлемые ингредиент(ы) могут включать корригент, агент для замедления кристаллизации сахара или агент для увеличения растворимости любого другого ингредиента, в частности спирта, содержащего несколько атомов водорода, например глицерола или сорбитола.
Препараты для ректального введения могут быть изготовлены в виде суппозиториев с приемлемым носителем, например маслом какао или Witepsol S55 (торговая марка Dynamite Nobel Chemical, Germany) в качестве основы суппозитория.
Препараты, пригодные для парентерального введения, обычно содержат стерильный водный раствор активного соединения, являющийся предпочтительно изотоническим с кровью реципиента. Такие препараты могут как правило содержать дистиллированную воду, 5%-ную декстрозу в дистиллированной воде или рассоле и соединение формулы (I), которое имеет приемлемую растворимость в этих растворителях. Используемые на практике препараты содержат также концентрированные растворы или твердые вещества, содержащие соединение формулы (I), которое при растворении в соответствующем растворителе образует раствор, пригодный для вышеописанного парентерального введения.
Локальные препараты включают мази, кремы, гели и лосьоны, которые могут быть изготовлены соответствующими способами, известными в фармации. В дополнение к основе из мази, крема, геля или лосьона и активному ингредиенту такие локальные препараты могут содержать также консерванты, отдушки и дополнительные активные фармацевтические агенты.
В дополнение к вышеперечисленным ингредиентам препараты по данному изобретению могут включать также один или более дополнительных приемлемых ингредиентов, применяемых в выпускаемых фармацевтических препаратах, например разбавители, буферы, корригенты, связывающие вещества, поверхностно-активные агенты, уплотнители, смазывающие вещества, суспендирующие агенты, консерванты (включая антиоксиданты) и тому подобное.
ПРИМЕРЫ
Следующие
примеры иллюстрируют аспекты этого изобретения, но не рассматриваются как ограничения. Символы и договоренности, используемые в этих примерах, согласуются с символами и договоренностями, которые
используются в современной химической литературе, например [25].
Пример 1
17 β -N-(2,5-бис(трифторметил)фенилкарбамоил-4-аза-5 α - андрост-1-ен-33он
(Синтез по схеме 1)
А. 17 β -N-(2,5-бис(трифторметил))фенилкарбамоил- андрост-4-ен-3-он
К раствору 3-оксо-4-андростен-17 β -карбоновой кислоты [18]
(17,2 г, 54,
4 ммоль), сухого ТГФ (180 мЛ) и сухого пиридина (7 мл) при 2oC добавляют тионилхлорид (5,1 мЛ, 70,8 ммоль). Реакционную смесь перемешивают при 2oC в течение 20 минут, а затем
перемешивают при комнатной температуре в течение 40 минут. Затем реакционную смесь фильтруют, а твердую фазу промывают толуолом. Фильтрат концентрируют под вакуумом до масла, которое разводят сухим
ТГФ (150 мл) и сухим пиридином (7 мЛ). К полученному темному раствору добавляют 2,5-бис-(трифторметил)анилина (9,4 мЛ, 59,8 ммоль), а реакционную смесь подвергают дефлегмации в течение 5 часов,
разбавляют метиленхлоридом, экстрагируют последовательно 1 N HCl и рассолом, сушат над сульфатом натрия и фильтруют. Фильтрат концентрируют и наносят на колонку с 500 г силикагеля, колонку элюируют
15-30% градиентом этилацетат-гексан, получая после концентрирования 17 β -N-(2,5- бис(трифторметил)фенилкарбамоил-андрост-4-ен-3-она в виде не совсем белой пены; выход: 18,3 г (64%).
Б. 17 β -N-(2, 5-бис(трифторметил))фенилкарбамоил-5- оксо-А-нор-3, 5-секоандростан-3-оевая кислота
К кипящему в колбе с обратным холодильником раствору 17 β - N-(2,
5-бис (трифторметил)фенилкарбамоил-андрост-4-ен-3-ону (18, 3 г, 34,9 ммоль), полученному согласно пункту А, добавляют t-бутанол (275 мЛ), карбонат натрия (6,3 г, 50,8 ммоль) и воду (36 мЛ), спустя 45
минут, 75oC-ный раствор перманганата калия (0,38 г, 2,4 ммоль), периодата натрия (52,2 г, 245 ммоль) и воды (311 мЛ). После дополнительной дефлегмации в течение 15 минут гетерогенную смесь
охлаждают до комнатной температуры и добавляют броунмиллерит (50 г). Реакционную смесь фильтруют через слой броунмиллерита (50 г), твердую фазу промывают водой, а фильтрат концентрируют под вакуумом,
удаляя t-бутанол (прибл. 175 мл). Полученный водный раствор подкисляют до рН 2 добавлением 36%- ной HCl и четыре раза экстрагируют хлороформом. Хлороформные слои объединяют и промывают водой, рассолом,
сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с образованием 17 β -N-(2, 5- бис(трифторметил))фенилкарбамоил-5-оксо-А-нор-3, 5-cекоандростан- 3-оевой кислоты в виде не совсем
белого вещества; выход: 20,5 г (100% сырого). Этот продукт прямо передают на стадию В.
В. 17 β -N-(2, 5-бис(трифторметил))фенилкарбамоил-4-аза- андрост-5-ен-3-он
К
суспензии 17 β -N-(2,5- бис(трифторметил))фенилкарбамоил- 5-оксо-А-нор-3,5-секоандростан- 3-оевой кислоты (20,5 г, 34,8 ммоль), полученной на стадии Б, в сухом этиленгликоле (100 мЛ) при
комнатной температуре добавляют в течение 5-минутного периода времени аммоний (прибл. 8 мЛ, 0,32 моль). Полученный раствор нагревают до 180oC в течение 45 минут, а через 12 минут - до
180oC, реакционную смесь охлаждают до 70oC и в течение периода времени, равного 5 мин, добавляют воду (116 мЛ). Полученную суспензию охлаждают до 7oC и перемешивают в
течение 10 минут, а затем фильтруют под вакуумом. Твердую фазу промывают водой (60 мЛ), а затем растворяют в хлороформе и промывают водой, рассолом, сушат над сульфатом натрия, фильтруют и
концентрируют. Остаток растворяют в хлороформе и наносят на колонку со 110 г силикагеля, колонку элюируют 2-5%-ным градиентом изопропанол-хлороформ, получая в результате 17 β -N-(2,
5-бис(трифторметил))фенил-карбамоил-4-аза-андрост-5-ен-3-он в виде не совсем белого вещества; выход: 16,5 г (90%).
Г. 17 β -N-(2,5-6ис(трифторметил))фенилкарбамоил-4-аза- 5
α -андроcтан-3-он
К раствору 17 β -N-(2,5-бис(трифторметил))фенилкарбамоил- 4-аза-андрост-5-ен-3-она (8,9 г, 16,7 ммоль) в уксусной кислоте (120 мЛ) добавляют оксид платины (0,9
г). Полученную смесь насыщают до 50 пси водородом и прогревают при 60-70oC в течение 6 часов. После замещения водородной атмосферы азотом реакционную смесь фильтруют через броунмиллерит,
слой броунмиллерита промывают уксусной кислотой (30 мЛ), хлороформом (60 мЛ) и толуолом (200 мЛ). Фильтрат концентрируют в вакууме до масла, добавляют толуол (200 мЛ), а раствор концентрируют до пены
в вакууме. Пену кристаллизуют из смеси этилацетат-гептан с получением, после сушки в вакууме при 85oC в течение 1 часа, 17 β -N-(2,5- бис(трифторметил)фенилкарбамоил-4-аза-5 α
-андростан-3-она; выход: 4,78 г, (54%); т. пл. 245-247oC. Аналит. расчетный состав для
C27H32F6N2O2: С, 61,12; H, 6,08; N, 5,
28. Обнаружено: С, 61,13; H, 6,12; N, 5,21.
Д. 17 β -N-(2,5-бис(трифторметил))фенилкарбамоил-4- аза-5 α -андростан-1-ен-3-он
К суспензии 17 β N-(2,
5-бис(трифторметил))фенилкарбамоил- -4-аза-5 α -андростан-3-она (7,24 r, 13,7 ммоль) и 2,3-дихлор- 5,6-дициано-1,4-бензохинона (3,41 г, 15 ммоль) в сухом диоксане (168 мЛ) при комнатной
температуре добавляют бис(триметилсилил) трифторацетамид (14,5 мЛ, 54,6 ммоль). После перемешивания при комнатной температуре в течение 7 часов реакционную смесь подвергают дефлегмации в течение 18
часов. Полученный темный раствор охлаждают до комнатной температуры и концентрируют под вакуумом до темного масла. К маслу добавляют метиленхлорид (100 мЛ) и 1%-ный раствор бисульфита натрия (40 мЛ),
двухфазную смесь быстро перемешивают в течение 15 минут и фильтруют. Два профильтрованных слоя разделяют, слой метиленхлорида промывают последовательно 2N HCl и рассолом, сушат над сульфатом натрия,
фильтруют и концентрируют до коричневого масла. Масло разводят толуолом и наносят на колонку с 300 г силикагеля, элюируют градиентом от 12:3:1 до 9:3:1 толуол: ацетон:этилацетат с получением 17
β -N-(2, 5-бис(трифторметил))фенилкарбамоил-4-аза- 5 α -андрост -1-ен-3-она в виде пены; выход: 3,38 r (47%). Этот продукт кристаллизуют из смеси этилацетат-гептан (1:1) с образованием
белого вещества; т. пл. 244-245oC.
13C ЯМР (100 МГц, CHCl3) δ 171.31, 166.77, 151.04, 136.35 (q, J = 1.4 Гц), 135.01 (q, J = 33.1 Гц), 126.73 (q, J = 5.4 Гц), 123.44 (q, J = 273.5 Гц), 123.03 (q, J = 273.2 Гц), 122.84, 121.58 (qq, J = 30.4, 1.0 Гц), 120.37 (q, J = 3.6 Гц), 120.29 (q, J = 3.9 Гц), 59.58, 58.33, 55.69, 47.46, 44.78, 39.30, 37.81, 35.29, 29.34, 25.70, 24.17, 23.59, 21.15, 13.40, 11.91.
Аналит. расчетн. состав для C27H30F6N2O2: С, 61.36; H, 5.72; N, 5.30. Обнаружено: С, 61.36; H, 5.73; N, 5.23.
Пример 2
17 β -N-(2.5-бис(трифторметил))фенилкарбамоил-4- аза-5 α андрост-1-ен-3-он
(Синтез по схеме II)
Суспензию 3-оксо-4-аза-5 α -андрост-1-ен- 17 β карбоновой кислоты (300 г, 0,95 моль) в 9 Л толуола перемешивают механически и прогревают при дефлегмации до тех пор, пока 1 Л толуола не
удаляется посредством дистилляции. Смесь охлаждают до - 2 ± 2oC и разбавляют толуолом (1 Л), диметилформамидом (10 мл) и пиридином (191 мл, 2,37 моль). К
перемешиваемой суспензии
добавляют тионилхлорид (135 г, 1,14 моль) при поддержании температуры ниже 0oC. После перемешивания при 0-20oC в течение 2-х часов добавляют 2,5-бис(трифторметил)-анилин (238 г,
1,04 моль) и диметиламинопиридин (2,0 г, 0,016 моль), смесь прогревают при 100oC в течение 15-16 часов. После охлаждения до 20oC раствор в толуоле промывают 1 N гидроксидом
натрия (2 х 1 Л), рассолом (1 Л), сушат над сульфатом магния и фильтруют. После промывания твердых фаз толуолом (100 мл) раствор концентрируют при 40-50oC (50-100 мм) до объема
приблизительно 2 Л и добавляют ацетонитрил. Смесь снова концентрируют, как и ранее до тех пор, пока не произойдет кристаллизация. Полученную суспензию перемешивают при комнатной температуре в течение
15-16 часов, охлаждают до 0-10oC, продукт собирают фильтрованием, получая в результате 17 β -N-(2,5-бис(трифторметил)фенил)-карбамоил-4-аза-5 α - андрост-1-ен-3-она в виде
белого кристаллического вещества, выход: 243 г (48%); т.пл. 245-245,5oC, которое идентично веществу, получаемому посредством процесса из примера 1.
Пример 3
Фармацевтические препараты "Активным соединением" является соединение формулы (I)
(А) Трансдермальная система - для 1000 фрагментов
Ингредиенты - Количество
Активное
соединение - 40 г
Силиконовая жидкость - 450 г
Коллоидный диоксид кремния - 25 г
Смешивают вместе силиконовую жидкость и активное соединение, а для увеличения вязкости
добавляют диоксид кремния. Затем материал дозируют в полимерный слоистый пластик, запаиваемый затем посредством нагревания, состоящий из следующих компонентов: полиэфирной высвобождающей обертки,
кожного контактного адгезива, состоящего из силиконовых и акрильных полимеров, контрольной мембраны, являющейся полиолефином (например полиэтиленом, поливинилацетатом или полиуретаном), и
непроницаемой основной мембраны, изготовленной из полиэфирного мультиламината. Полученную слоистую пленку затем разрезают на фрагменты площадью 10 см2.
(Б) Пероральная
таблетка - для 1000 таблеток
Ингредиенты - Количество
Активное соединение - 20 г
Крахмал - 20 г
Стеарат магния - 1 г
Активное соединение и крахмал
гранулируют с водой и сушат. К высушенным гранулам добавляют стеарат магния, а смесь тщательно перемешивали. Перемешанную смесь формовали в таблетки
(В) Суппозиторий - для 1000
суппозиториев
Ингредиенты - Количество
Активное соединение - 25 г
Салицилат теобромина натрия - 250 г
Витепсол S55 - 1725 г
Неактивные ингредиенты
смешивают и расплавляют. Затем в расплавленной смеси распределяют активное соединение, разливают в формы и оставляют для остывания.
(Г) Инъекция - для 1000 ампул
Ингредиенты
- Количество
Активное соединение - 5 г
Забуферивающие агенты - q.s.
Пропиленгликоль - 400 мг
Вода для инъекций - 600 мЛ
Активное соединение и
забуферивающие агенты растворяют в пропиленгликоле при температуре приблизительно 50oC. Затем при перемешивании добавляют воду для инъекций, полученный раствор фильтруют, заполняют им
ампулы, запаивают и стерилизуют автоклавированием.
(Д) Капсула - для 1000 капсул
Ингредиенты - Количество
Активное соединение - 20 г
Лактоза - 450 г
Стеарат магния - 5 г
Хорошо измельченное активное соединение смешивают с лактозой и стеаратом и запаковывают в желатиновые капсулы.
Пример 4
17 β -N-(2,
5-бис(трифторметил))фенилкарбамоил-андрост-4-ен-3- он, полученный в результате реализации стадии (А) Примера 1, подвергают взаимодействию с ацетангидридом с получением 17 β N- ацетил-N- (2,
5-бис(трифторметил))фенилкарбамоил-андрост- 4-ен-3-она. Затем это соединение подвергают последующим взаимодействиям согласно стадиям (Б), (В) и (Г). В условиях стадии (Д) полученное таким образом
соединение подвергают взаимодействию с бис(триметилсилил) трифторацетамидом с получением 17 β -N-ацетил-N-(2,5-бис (трифторметил))фенилкарбамоил-4-аза-5 α - андрост-1-ен -3-она, при
гидролизе которого в кислой среде получают 17 β -N-(2, 5-бис(трифторметил))фенилкарбамоил-4-аза- 5 α -андрост-1-ен-3-он.
3-оксо-4-аза-5 α андрост- 1-ен-17 β
карбоновую кислоту подвергают взаимодействию с ацетангидридом с получением 3-оксо-N-ацетил-4-аза-5 α -андрост-1-ен-17 β карбоновой кислоты, которую в условиях примера 2 подвергают
взаимодействию с 2,5- бис(трифторметил)-анилином с получением 17 β -N- (2,5- бис(трифторметил))фенилкарбамоил-N-ацетил-4-аза-5 α -андрост-1- ен-3-она. Это соединение подвергают гидролизу
в кислой среде с получением 17 β-N -(2, 5-бис(трифторметил))фенилкарбамоил- 4-аза-5 α -андрост-1-ен-3-она
ЛИТЕРАТУРА
1. McGinley, J. et a1. , The New England J. of
Medicine, 300, 1233 (1979).
2. Russell, D.W. et a1., J.Clin.Invest., 89, 293 (1992).
3. Russell, D.W. et a1., Nature, 354, 159 (1991).
4. Russell, D.W. et a1., J.Clin.Invest., 90, 799 (1992).
5. Russell, D.W. et a1., New England J. Med., 327, 1216 (1992).
6. Hsia, S. & Voight, W., J.Invest.Derm., 62, 224 (1973).
7. Robaire, В. et a1., J.Steroid Biochem., 8, 307 (1977).
8. Petrow, V. et a1., Steroids, 38, 121 (1981).
9. Liang, Т. et a1., J. Sterold Biochem., 19, 385 (1983).
10. Holt, D. et a1., J. Med. Chem., 33, 937 (1990).
11. U.S. Patent No. 4,377,584.
12. U.S. Patent No. 4,760,071.
13. U.S. Patent No. 5,017,568.
14. Tan, C.H.; Fong, C.Y.; Chan, W.K. Biochem. Biophys. Res. Comm., 144, 166 (1987).
15. Brandt, M.; Levy, M.A. Biochemistry, 28, 140 (1989).
16. Potts, G.O. et a1., Steroids, 32, 257 (1978).
17. McConnell, J.D. The Prostate Suppi., 3, 49 (1990).
18. Rasmusson, G.H. et a1., J.Med.Chem., 27, 1690 (1984).
19. Bhattacharya, A. et a1., J. Am. Chem. Soc., 110, 3318 (1988).
20. Rasmusson, G.H. et a1., J.Med.Chem., 29, 2298 (1986).
21. Protective Groups in Organic Chemistry, Ed. J. F. W. McOmie. Plenum Press, London (1973).
22. Protective Groups in Organic Synthesis. Theodora Gre- en, John Wiley and Sons, New York (1981).
23. Brooks, J.R. et a1., Steroids, 47, 1 (1986).
24. Stoner, E. et a1., J. Steroid Biochem. Molec. Bio1.,37, 375 (1990),
25. Journal of the
American Chemical Society.
Реферат
Описывается новое соединение формулы I: 17β-N-(2,5-бис(трифторметил)фенилкарбамоил-4-аза-5α-андрост-1-ен-3-он как необычайно сильный и избирательный двойственный ингибитор человеческой 5α-редуктазы типа 1 и 2. Описывается также способ его получения, фармацевтический состав на основе указанного соединения и способ лечения. 5 с. и 2 з.п.ф-лы. 1 табл.
Формула
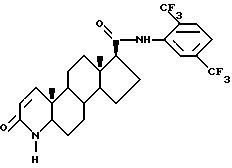
2. Фармацевтический состав для ингибирования фермента тестостерон-5α-редуктазы, отличающийся тем, что он содержит соединение по п. 1 и его фармацевтически приемлемый носитель.
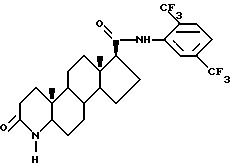
в котором функциональные группы возможно могут быть защищены,
и, если это необходимо и/или желательно, подвергают полученное таким образом соединение одной или более чем одной дополнительной реакции, включающей удаление любой защитной группы или групп.
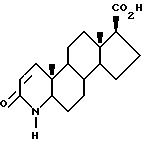
в котором функциональные группы возможно могут быть защищены,
подвергают взаимодействию с соединением формулы IIa
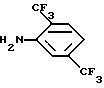
в котором функциональные группы возможно могут быть защищены,
и, если это необходимо и/или желательно, подвергают полученное таким образом соединение одной или более чем одной дополнительной реакции, включающей удаление любой защитной группы или групп.
Комментарии