Способ получения n-замещенных салициламидов - RU2404158C2
Код документа: RU2404158C2
Описание
Настоящее изобретение относится к способу получения N-замещенных салициламидов или их производных и их солей, гидратов и сольватов. Настоящее изобретение прежде всего относится к способу получения N-(5-хлорсалицилоил)-8-аминокаприловой кислоты (5-CNAC) и соответствующего моногидрата динатриевой соли.
N-Замещенные салициламиды, полученные способом по настоящему изобретению, можно использовать в составе композиций для доставки активных агентов пероральным способом или другими способами введения в организм млекопитающих.
Предпосылки создания изобретения
Способы получения N-замещенных салициламидов описаны, например, в WO 00/59863, WO 01/92206 и WO 01/70219, а типичный способ получения 5-CNAC (N-замещенного салициламида), который известен из уровня техники, показан на схеме 1.
Схема 1

Кроме того, общий метод получения соли 5-CNAC в присутствии NaOH в ацетоне показан на схеме 2.
Схема 2

Целью настоящего изобретения является совершенствование способа, описанного в предшествующем уровне техники, например разработка технологичного высокопроизводительного способа, пригодного для получения больших количеств высококачественного продукта.
Краткое изложение сущности изобретения
В общем случае настоящее изобретение относится к способу получения N-замещенных салициламидов или их производных и их солей. Способ включает взаимодействие хлорзамещенного соединения формулы (III) (указанного ниже) с карсаламом (6-хлор-2Н-1,3-бензоксазин-2,4(3Н)-дионом) или его производным при необходимости в присутствии источника ионов брома, например бромида щелочного металла, например NaBr

где n означает целое число от 1 до 8, Q означает группу, которая легко превращается в карбоксигруппу, a R5 и R6 независимо выбирают из группы, включающей водород, -ОН, NR3R4, галоген, С1-С4 алкил, С1-С4 алкокси, С2-С4 алкенил, где R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, С1-С4 алкил, С1-С4 галогеналкил, С1-С4 алкокси, С2-C4 алкенил.
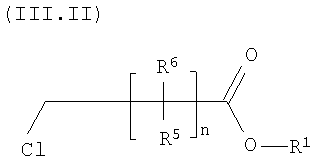
Предпочтительным классом соединений формулы III являются соединения формулы III.II, указанные ниже
Предполагается, что присутствие бромида щелочного металла (предпочтительно NaBr) обеспечит образование бромзамещенного соединения формулы (IIIb)

Кроме того, вышеуказанный способ по настоящему изобретению представляет собой способ получения N-замещенного салициламида формулы (IV) и, например, при обработке кислотой, соответствующей карбоновой кислоте (IV.II)


где t равно 0, 1, 2, 3, 4, 5 или 6, m равно 1, 2, 3 или 4, или, если m>1, то каждый R2 независимо выбирают из группы, включающей -ОН, NR3R4, галоген, C1-С4 алкил, С1-С4 галогеналкил, С1-С4 алкокси, С2-С4 алкенил, а R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, С1-С4 алкил, С1-С4 галогеналкил, С1-С4 алкокси, С2-С4 алкенил.
В другом объекте изобретения соль щелочного металла (V) N-замещенного салициламида получают в присутствии водного раствора катиона щелочного металла Мa+, например Na+, в ацетоне/воде. Другими пригодными растворителями являются любые смеси растворителя с водой, такие, как ацетон/вода, этанол/вода или ацетонитрил/вода.

Более конкретно, в способе по настоящему изобретению используется хлорзамещенное соединение формулы (III.II) (указанное ниже), и прежде всего хлорзамещенный сложный эфир. Более конкретно настоящее изобретение относится к способу получения N-(5-хлорсалицилоил)-8-аминокаприловой кислоты (5-CNAC) и ее соответствующих солей, прежде всего моногидрата динатриевой соли. В предпочтительном варианте способ включает взаимодействие алкилового эфира хлороктановой кислоты, прежде всего этилового эфира хлороктановой кислоты (ЕСО), с 6-хлоркарасаламом в присутствии NaBr с образованием 5-CNAC. Моногидрат натриевой соли получают при взаимодействии 5-CNAC с NaOH в смеси ацетон/вода.
N-Замещенные салициламиды, прежде всего 5-CNAC, полученные способом по настоящему изобретению, можно использовать в составе композиции для доставки активных агентов пероральным или другим способом введения в организм млекопитающего. Более конкретно, соединения, полученные по настоящему изобретению, можно использовать для доставки фармацевтически, физиологически, фармакологически, радиологически активных и других агентов в ткани-мишени в организме теплокровного животного.
Подробное описание предпочтительных вариантов осуществления изобретения
Общая последовательность реакций по изобретению показана на схеме 3. На схеме 3 показан способ получения N-замещенных салициламидов или их производных (IV) и их солей (V) из соответствующего салициламида (I) через промежуточное производное карсалама (II) в присутствии хлорзамещенного соединения (III).
Схема 3

На схеме 3 t равно 0, 1, 2, 3, 4, 5 или 6, предпочтительно t равно 0, р предпочтительно равно 0, 1, 2, 3, 4, 5 или 6, предпочтительно р равно 1. В другом варианте гидраты tH2O и рН2О на схеме 3 могут означать сольват, смешанный гидрат и сольват или смешанный сольват.
Индекс n равен целому числу от 1 до 8, предпочтительно n равно 6, m равно 1, 2, 3 или 4, предпочтительно m равно 1.
M означает щелочной металл, предпочтительно Ма означает Na или К. Наиболее предпочтительно щелочной металл М означает Na (и, следовательно, Мa+ означает Na+). М может присутствовать в форме MaY, где Y означает основный противоион, например карбонат или гидроксид. Более предпочтительно MaY означает NaOH.
Q означает группу, которая легко превращается в карбоновую кислоту формулы (IV.II). Например, Q может означать защищенную карбоксильную группу, причем защитную группу предпочтительно удаляют на конечном этапе стадии В. Таким образом, Q означает группу, которая не принимает участия в реакции соединения формулы (III) на стадии В, но затем легко превращается в свободную карбоксильную группу при обработке в кислотных условиях.
Если m>1, каждый R2 независимо выбирают из группы, включающей -ОН, NR3R4, галоген, С1-С4 алкил, С1-С4 галогеналкил, С1-С4 алкокси, С2-С4 алкенил.
R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, С1-С4 алкил, С1-С4 галогеналкил, С1-С4 алкокси, С2-С4 алкенил.
Соответствующие значения R5 и R6 независимо выбирают из группы, включающей водород, -ОН, NR3R4, галоген, С1-С4 алкил, С1-С4 алкокси, С2-С4 алкенил. R5 могут быть идентичными или различными, аналогичным образом значения R6 могут быть идентичными или различными. Аналогичным образом значения R5 и R6 могут быть идентичными или различными. В предпочтительном варианте каждый R5 и каждый R6 означает водород.
Галоген выбирают из группы, включающей хлор, фтор, бром и йод. Наиболее предпочтительно галоген означает хлор.
Предпочтительно m равно 1, а R2 означает галоген. Предпочтительно R2 означает хлор. Более предпочтительно m равно 1, а R2 находится в положении 5.
Стадия В может включать одну или более промежуточных стадий, на которых получают промежуточные соединения, однако наиболее предпочтительно их не выделяют. Такие промежуточные соединения могут включать представленные ниже соединения формул (IV.I) и (VI)
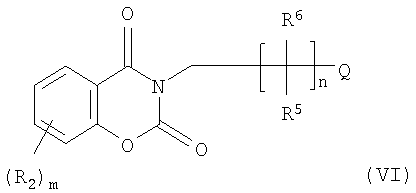

Следовательно, настоящее изобретение может включать дополнительную стадию омыления реакционной смеси, содержащей соединение (IV), с целью последующего получения из промежуточного соединения (VI) целевого соединения. Указанную стадию можно проводить без выделения соединений формулы IV или VI.
Наиболее предпочтительно соединение формулы (III) включает соединения формулы (III.II), указанной ниже

В формуле (III.II) R1 означает карбоксизащитную группу, прежде всего группу, которая практически инертна при проведении реакции по группам NH2-группам на стадии А, но затем ее превращают в карбоксильную группу (-СООН). Более предпочтительно R1 выбирают из группы, включающей C1-С6алкил с прямой или разветвленной цепью, предпочтительно С1-С4 алкил и прежде всего С1-С2алкил, и, следовательно, соединение формулы (III.II) представляет собой сложный эфир. В наиболее предпочтительном варианте R1 означает этил (С2). Таким образом, соединение формулы (III.II) наиболее предпочтительно означает сложный эфир.
Предпочтительно соединение формулы (III) и (III.II), где n равно 6, а каждый CR5R6 означает СН2, т.е. соединения, включающие н-гексил. Предпочтительным соединением является этиловый эфир 8-хлороктановой кислоты (III.III).

Примером предпочтительного класса соединений общей формулы IV, полученных способом по изобретению, являются соединения формулы IV.I
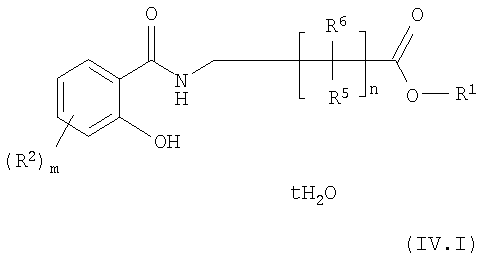
где R1 означает карбоксизащитную группу. Такие соединения предпочтительно являются промежуточными соединениями, которые используют без выделения.
В представленных соединениях, таких, как соединения формул (I) и (IV), которые включают фенольную гидроксигруппу, указанная группа может присутствовать в форме соли, например натриевой соли.
Фенольная гидроксигруппа на стадиях А и В, которые проводят в присутствии основания, может присутствовать в депротонированной форме.
Реагенты
В следующем разделе описаны реагенты, которые предпочтительно используются в настоящем изобретении. Конкретные условия проведения реакций описываются в следующем разделе.
Стадия А
На стадии А салициламид (I) превращают в соответствующее производное карсалама (II) по реакции с избытком хлорформиата, такого, как этилхлорформиат, н-пропилхлорформиат, изопропилхлорформиат, н-бутилхлорформиат или трет-бутилхлорформиат. Более предпочтителен этилхлорформиат. Предпочтительно реакцию проводят в двухфазной системе алкилацетат/слабое органическое основание/вода. Предпочтительно слабое органическое основание практически нерастворимо в воде.
Примерами алкилацетата являются метилацетат, этилацетат, н-пропилацетат, изо-пропилацетат, трет-бутилацетат. Наиболее предпочтительно реакцию проводят в н-бутилацетате.
Примерами слабых органических оснований являются пиридин и производные пиридина, прежде всего алкилзамещенный пиридин, например диалкилпиридин, и более предпочтительно такие производные пиридина, которые практически нерастворимы в воде. Примером предпочтительного производного пиридина является 5-этил-2-метилпиридин.
Более предпочтительно реакцию проводят в двухфазной системе, включающей н-бутилацетат/5-этил-2-метилпиридин/воду и избыток (например, 20% избыток) этилхлорформиата. Наиболее предпочтительно соотношение алкилацетат/вода составляет приблизительно 1:1.
Стадия В
Стадия В предпочтительно включает ряд промежуточных стадий.
Стадия В1
Стадия В1 представляет собой первую реакцию цикла, по которой карсалам или его производное формулы (II) взаимодействует с хлорзамещенным соединением (III). Предпочтительно хлорзамещенное соединение (III) представляет собой сложный эфир, а реакцию проводят в присутствии основания, такого, как карбонат натрия, и апротонного растворителя, такого, как диалкиламид, например диметилацетамид или диметилформамид. Предпочтительно реакцию проводят в присутствии диметилформамида. Кроме того, в реакционной среде присутствует источник ионов брома, например каталитического количества бромида щелочного металла МВr, например КВr или NaBr, прежде всего NaBr.
Апротонные растворители, пригодные для применения по настоящему изобретению, включают, без ограничения перечисленным, натрилы (например, ацетонитрил, бензонитрил, нитрометан), амиды и циклические амиды (например, N,N-диметилформамид, N-метилформамид, N,N-диэтилформамид, N-этилформамид, N,N-диметилацетамид, N-метил-2-пирролидон), сложные эфиры, циклические эфиры и простые эфиры (например, тетрагидрофуран, пропиленкарбонат, этиленкарбонат, γ-бутиролактон, этилацетат, диметиловый эфир), оксиды и сульфосоединения (например, диметилсульфоксид, ацетон, сульфолан, диметилсульфон).
Предпочтительно апротонный растворитель представляет собой амид, выбранный из группы, включающей N,N-диметилформамид, N-метилформамид, N,N-диэтилформамид, N-этилформамид, N,N-диметилацетамид. Наиболее предпочтительно растворитель означает N,N-диметилформамид.
Стадия В2
На второй промежуточной стадии В в реакционную смесь добавляют соль щелочного металла, например гидроксид натрия, в воде.
Стадия В3
На третьей промежуточной стадии В в смесь добавляют кислоту, например серную кислоту, в смеси с алкилацетатом, например метилацетатом, этилацетатом, н-пропилацетатом, изопропилацетатом, трет-бутилацетатом. Наиболее предпочтительно третью промежуточную стадию В проводят в присутствии этилацетата. На стадии В3 получают свободную кислоту формулы (IV.II).
Стадия В4
На предпочтительной четвертой стадии В N-замещенный салициламид (IV.II) кристаллизуют из спирта, например этанола/воды, однако можно также использовать другие растворители, прежде всего смесь этилацетата, этанола, воды или ацетон/вода.
Стадия С
На стадии С N-замещенный салициламид формулы (IV.II) вводят в реакцию с сильным основанием, например гидроксидом натрия, в присутствии ацетона и воды.
При проведении способов по настоящему изобретению в присутствии сильного основания, например MaY, в качестве сильного основания можно использовать гидроксиды, гидриды, амиды, алкоголяты, феноляты, ацетаты, карбонаты, диалкиламиды или алкилсилиламиды щелочных или щелочноземельных металлов, алкиламины, алкилендиамины, необязательно N-алкилированные, необязательно ненасыщенные циклоалкиламины, основные гетероциклы, гидроксиды аммония, а также карбоциклические амины.
Алкилы щелочных металлов выбирают из группы, включающей, например, метиллитий, н-бутиллитий или трет-бутиллитий, необязательно активированные тетраметилэтилендиамином (TMEDA).
Гидриды щелочных металлов выбирают из группы, включающей, например, гидрид натрия и гидрид кальция.
Амиды щелочных металлов выбирают, например, из группы, включающей амид лития или дизопропиламид лития (LDA), диэтиламид лития, изопропилциклогексиламид лития или бис(триметилсилил)амид лития.
Алкоголяты щелочных металлов выбирают из группы, включающей, например, алкоголяты первичных, вторичных или третичных алифатических спиртов, содержащих от 1 до 10 атомов углерода, например метилат натрия, калия или лития, этилат натрия, калия или лития, н-пропилат натрия, калия или лития, изопропилат натрия, калия или лития, н-бутилат натрия, калия или лития, втор-бутилат натрия, калия или лития, трет-бутилат натрия, калия или лития, 2-метил-2-бутилат натрия, калия или лития, 2-метил-2-пентилат натрия, калия или лития, 3-метил-3-пентилат натрия, калия или лития или 3-этил-3-пентилат натрия, калия или лития.
Феноляты щелочных металлов выбирают из группы, включающей, например, орто-алкилфеноляты щелочных металлов, феноляты щелочных металлов, например орто-крезолят натрия или калия.
Кроме того, можно использовать органические амины, которые выбирают из группы, включающей, например, 2,4,6-триметилпиридин, 2-трет-бутил-1,1,3,3-тетраметилгуанидин, 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), 2,3,4,6,7,8,9,10-октагидропиримидо[1,2-а]азепин, 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), диазабициклооктан (DABCO), 1,4-диазабицикло(2.2.2)октан (TED), N,N-дициклогексилметиламин, N,N-диэтиланилин, N,N-диизопропил-2-этилбутиламин, N,N-диизопропилметиламин, N,N-диизопропил-3-пентиламин, N,N-диметиланилин, 2,6-ди-трет-бутил-4-метилпиридин, N,N-диизопропилэтиламин, 2,6-диметилпиридин, 7-метил-1,5,7-триазабицикло(4.4.0)дец-5-ен (MTBD), 3,3,6,9,9-пентаметил-2,10-диазабицикло(4.4.0)дец-1-ен (PMDBD), 1,2,2,6,6-пентаметилпиперидин (РМР), триэтиламин, 1,1,3,3-тетраметилгуанидин (TMG), N,N,N′,N′-тетраметил-1,8-нафталиндиамин, 2,2,6,6-тетраметилпиперидин (ТМР), 1,5,7-триазабицикло(4.4.0)дец-5-ен, 1,3,4,6,7,8-гексагидро-2Н-пиримидо[1,2-а]пиримидин (TBD), трибутиламин, 2,4,6-три-трет-бутилпиридин, трис(триметилсилил)амин и гидроксиды алкиламмония.
Однако можно также использовать смесь вышеуказанных оснований.
Примерами таких оснований являются гидроксид, гидрид, амид, метанолят, ацетат, карбонат натрия, трет-бутанолят, гидроксид, карбонат, гидрид калия, диизопропиламид лития, бис(триметилсилил)амид калия, гидрид кальция, триэтиламин, диизопропилэтиламин, триэтилендиамин, циклогексиламин, N-циклогексил-N,N-диметиламин, N,N-диэтиланилин, пиридин, 4-(N,N-диметиламино)пиридин, хинуклидин, N-метилморфолин, бензилтриметиламмоний, а также 1,5-диазабицикло[5.4.0]ундец-5-ен (DBU).
В способах по настоящему изобретению предпочтительными основаниями являются гидроксиды и карбонаты щелочных металлов, например гидроксид натрия и карбонат натрия.
В предпочтительных условиях реакции на стадии С соотношение ацетон/вода составляет приблизительно 3:1, а также 4:1 или даже приблизительно 5:1, до приблизительно 15:1 при добавлении ацетона в реакционную смесь в процессе проведения реакции.
Подразумевается, что между добавлением реагентов и прежде всего между стадиями А, В или С, или даже между различными промежуточными стадиями В, до начала следующей стадии проводят операции упаривания, фильтрования, экстракции и другие конечные и/или подготовительные операции.
Условия проведения реакций
Если не указано иное, реакции по настоящему изобретению наиболее предпочтительно проводят в атмосфере инертного газа, например в атмосфере азота.
Стадия А
На стадии А реакционную смесь охлаждают до 0-5°С при добавлении хлорформиата, а затем смесь медленно нагревают до температуры кипения растворителя и кипятят с обратным холодильником до завершения реакции. Обычно реакционную смесь кипятят с обратным холодильником в течение приблизительно 3-7 ч, прежде всего приблизительно в течение 5 ч. Температура кипения зависит от свойств используемого алкилацетата. Типичная температура кипения составляет от 80 до 120°С.
Стадия В
На стадии В1 после добавления всех компонентов реакционной смеси кроме карбоната натрия реакционную смесь нагревают до приблизительно 100°С. Затем добавляют карбонат натрия, предпочтительно приблизительно через 2 ч.
На стадии В2 реакционную смесь нагревают при приблизительно 100°С.
На стадии В3 реакционную смесь охлаждают до приблизительно 60°С, а затем добавляют кислоту. Предпочтительно при проведении реакции температуру смеси поддерживают на уровне приблизительно 60°С, причем указанную температуру предпочтительно поддерживают при проведении любых последующих стадий очистки, например экстракции.
На стадия В4 спирт (предпочтительно этанол) добавляют при приблизительно 50-60°С, а затем раствор охлаждают до приблизительно 40-50°С. Наконец, раствор охлаждают до приблизительно 0-5°С.
В предпочтительных вариантах настоящее изобретение относится к способу получения N-(5-хлорсалицилоил)-8-аминокаприловой кислоты (5-CNAC) и ее соответствующих солей, прежде всего моногидрата динатриевой соли. В одном более предпочтительном варианте 5-CNAC получают из 5-хлорсалициловой кислоты через промежуточный 5-хлоркарсалам в присутствии этилового эфира 8-хлороктановой кислоты (ЕСО), как показано ниже на схеме 4.
Схема 4

Реагенты на стадиях А, В и С аналогичны реагентам, показанным на схеме 3.
Наиболее предпочтительные реагенты для осуществления способов по настоящему изобретению, прежде всего для осуществления способа, показанного на схеме 4, приводятся ниже.
На стадии А 5-хлорсалициламид взаимодействует с этилхлорформиатом (присутствующим в избытке) в н-бутилацетате/воде в присутствии 5-этил-2-метилпиридина в качестве необходимого органического основания.
На стадии В1 5-хлоркарсалам взаимодействует с этиловым эфиром хлороктановой кислоты в присутствии карбоната натрия и NaBr (источник ионов брома) в диметилформамиде, при этом NaBr выполняет роль катализатора. На стадии В2 полученный продукт обрабатывают, например, раствором гидроксида натрия. Затем в смесь добавляют кислоту, предпочтительно минеральную кислоту, такую, как серная кислота, а затем этилацетат. Неочищенный продукт перекристаллизовывают из этанола.
На стадии С 5-CNAC обрабатывают раствором гидроксида натрия в смеси ацетон/вода (предпочтительно 3:1).
Вторым объектом настоящего изобретения является применение N-замещенных салициламидов и их производных, прежде всего 5-CNAC, их соответствующих солей, прежде всего моногидрата динатриевой соли, полученных способом по настоящему изобретению, для доставки активных агентов, таких, как биологически или химически активные агенты, в ткани-мишени.
Третьим объектом настоящего изобретения являются фармацевтические композиции N-замещенных салициламидов и их производных и их солей, полученных способом по настоящему изобретению. Настоящее изобретение прежде всего относится к фармацевтическим композициям, включающим 5-CNAC, полученную способом по настоящему изобретению.
В описании заявки описываются преимущества настоящего изобретения главным образом в отношении синтеза 5-CNAC. Однако подразумевается, что описание изобретения в этом отношении не ограничивает объем изобретения, который включает N-замещенные салициламиды общей формулы IV. Таким образом, последующее описание синтеза 5-CNAC главным образом относится к предпочтительному варианту осуществления настоящего изобретения, который позволяет провести сравнение настоящего изобретения с предшествующим уровнем техники. Для специалиста в данной области представляется очевидным, что настоящее изобретение не ограничено только синтезом 5-CNAC.
Предпочтительный синтез по настоящему изобретению представляет собой усовершенствованную методику получения 5-CNAC по сравнению с предшествующим уровнем техники. Следует отметить, что, как показано на схеме 1, предшествующий уровень техники представляет собой способ, согласно которому 5-CNAC получают по реакции с этиловым эфиром 8-бромоктановой кислоты в присутствии карбоната натрия и диметилацетамида.
В синтезе 5-CNAC по настоящему изобретению вместо 5-хлорсалициловой кислоты, как описано в предшествующем уровне техники, в качестве нового исходного материала используют 5- хлорсалициламид, что является преимуществом настоящего изобретения.
В настоящем изобретении также предлагается новый метод синтеза 6-хлоркарсалама, ключевого промежуточного соединения в способе получения 5-CNAC, в котором используется двухфазная смесь (например, н-бутилацетат/вода) с алкилзамещенным пиридином, например диалкилзамещенным пиридином, таким, как 5-этил-2-метилпиридин, в качестве основания. Применение пиридина в качестве слабого органического основания не препятствует осуществлению настоящего изобретения, хотя и не является предпочтительным. Одним конкретным преимуществом применения некоторых производных пиридина, например 5-этил-2-метилпиридина, по сравнению с пиридином из предшествующего уровня техники, является тот факт, что указанный реагент можно регенерировать в указанных условиях. Напротив, в способе из предшествующего уровня техники используется реакционная смесь, содержащая этилхлорформиат в ацетонитриле в присутствии пиридина в качестве основания, который не подлежит регенерации.
Способ по настоящему изобретению также обладает главным преимуществом по сравнению с предшествующим уровнем техники благодаря применению двухфазной системы. В указанной системе происходит гидролиз нежелательных промежуточных соединений, образующихся в ходе побочных реакций, т.е. происходит их удаление из реакционной среды и в результате получают более чистый конечный продукт.
В предпочтительном синтезе 5-CNAC из 6-хлоркарсалама, предварительно полученного способом по настоящему изобретению, этиловый эфир 8-хлороктановой кислоты (ЕСО) вводят в реакцию с 6-хлоркарсаламом в присутствии диметилформамида и карбоната натрия (в качестве основания) и некоторого количества бромида натрия. Заявитель считает, что указанные условия обеспечивают образование более реакционноспособного этилового эфира 8-бромоктановой кислоты (ЕВО) in situ. Бромид натрия можно регенерировать после завершения реакции и по меньшей мере на этом основании принято считать, что бромид натрия выполняет функцию катализатора. Следовательно, способ по настоящему изобретению исключает необходимость применения этилового эфира 8-бромоктановой кислоты (ЕВО) непосредственно в качестве исходного материала. ЕВО является экологически небезопасным соединением из-за его высокой реакционноспособности. Кроме того, ЕСО является более дешевым и более доступным материалом по сравнению с ЕВО.
Промежуточный сложный эфир (или эфиры), полученный по вышеуказанной реакции, наиболее предпочтительно не выделяют, а гидролизуют немедленно после концентрирования конечной реакционной смеси. Указанный гидролиз является преимуществом, так как исключает необходимость выделения продукта, что повышает вероятность достижения более высокого выхода. Эта операция выгодно отличается от соответствующей стадии в способе из предшествующего уровня техники, где необходимо выделение 8-[6-хлор-2Н-1,3-бензоксазин-2,4(3Н)дионил]октаноата. Затем после экстракции и кристаллизации получают высокоочищенную свободную кислоту.
Наконец, получение моногидрата динатриевой соли (стадия С) при добавлении гидроксида натрия в смесь ацетон/вода позволяет проводить кристаллизацию из гомогенного раствора, в отличие от эмульсии в способе из предшествующего уровня техники, где гидроксид натрия добавляют в чистый ацетон. Гомогенный раствор позволяет получать крупные кристаллы, которые можно высушивать в стандартной барабанной лопастной сушилке.
В результате изменений, реализованных в настоящем изобретении (представленных в указанном способе синтеза 5-CNAC), разработан новый способ, который позволяет получать высокоочищенные N-замещенные салициламиды формулы IV, прежде всего 5-CNAC (IVA), рентабельным методом.
Способ по настоящему изобретению с использованием в качестве примера синтеза 5-CNAC, описан ниже более подробно, прежде всего со ссылкой на схемы 5 и 6.
Стадия А
Как показано на схеме 6, температура Т предпочтительно составляет 80-120°С. Наиболее предпочтительно температура Т составляет 85-95°С. Более конкретно температура Т равна 90°С.
Схема 6

Существенным недостатком способа из предшествующего уровня техники является образование продукта, содержащего до 10% исходного амида (IA). Другим недостатком является тот факт, что синтез проводится в ацетонитриле и пиридине (как показано на схеме 1), которые непригодны для регенерации. Указанные недостатки метода делают его более дорогостоящим и менее приемлемым в экологическом отношении.
Соответствующая стадия в способе из предшествующего уровня техники включает две промежуточные стадии:
(1) ацилирование этилхлорформиатом при 0°С и
(2) циклизация при кипячении с обратным холодильником (90°С).
Анализ продуктов в способе из предшествующего уровня техники методом ЖХВР свидетельствует о том, что в ходе ацилирования образуется главным образом одно промежуточное соединение, в то время как исходный материал потребляется полностью. Однако после нагревания быстро образуется второе новое промежуточное соединение, которое медленно превращается в требуемый 6-Сl-карсалам, причем в смеси присутствует некоторое количество исходного материала. Первое промежуточное соединение представляет собой N- ацилированный салициламид (X), а второе соединение является O-ацилированным производным (XX) (по фенольной ОН-группе).
Для решения экологических проблем, связанных со способом из предшествующего уровня техники, заявители настоящего изобретения исследовали возможность применения водной системы с использованием вместо пиридина другого основания, такого, как гидроксид натрия. Однако было установлено, что при проведении реакции в водной среде, например в присутствии гидроксида натрия или карбоната натрия вместо пиридина, после циклизации и образования соединения IIА, в продукте присутствует гораздо больше исходного материала IA по сравнению со способом из предшествующего уровня техники, что свидетельствует о том, что реакция не завершена или происходит разложение материала.
Заявители предполагают, что образуется дополнительный промежуточный продукт, который после нагревания может вновь превращаться в исходный материал. Таким продуктом может являться O-ацилированный амид, который не отделяется при ЖХВР. Образование указанного промежуточного продукта связано с высокой реакционноспособностью атома кислорода амидной группы, что обычно свойственно сильным основаниям, которые способствуют O-ацилированию.
Другой причиной может быть разложение 6-хлоркарсалама в основных условиях аналогично реакции на стадии В (раскрытие цикла под действием карбоната натрия при повышенной температуре в диметилформамиде). Однако это объяснение не применимо к пиридину. Следовательно, очевидно, что более предпочтительным является слабое органическое основание.
С целью исключить вышеуказанную проблему "незавершенной" реакции в настоящем изобретении предлагается двухфазная система. Предпочтительной системой является алкилацетат/органическое основание/вода, например, бутилацетат/замещенный пиридин/вода, и избыток этилхлорформиата. Наиболее предпочтительна система н-бутилацетат/5-этил-2-метилпиридин/вода.
Заявители предполагают, что в присутствии воды происходит быстрый гидролиз любых нежелательных промежуточных соединений (например, O-ацилированного промежуточного соединения), что способствует ацилированию требуемого атома азота (атом азота амидной группы), пока в смеси присутствует достаточное количество хлорформиата. В этих условиях (20% избыток этилхлорформиата и 30% алкилзамещенного пиридина) после циклизации в реакционной смеси найдено только 1-2% исходного материала.
Поскольку пиридин с трудом регенерируется из водных маточных растворов, оказалось предпочтительным заменить его, без потери селективности, на нерастворимое в воде производное пиридина, прежде всего на алкилпиридин, например на нерастворимый в воде 5-этил-2-метил-1-пиридин. Применение нерастворимого в воде алкилпиридина позволяет регенерировать основание и, следовательно, является предпочтительным.
Органический растворитель (алкилацетат) выбирают из группы, включающей метилацетат, этилацетат, н-пропилацетат, изопропилацетат или н-бутилацетат. Предпочтительным растворителем является н-бутилацетат, температура кипения азеотропа которого составляет приблизительно 90°С. Это позволяет проводить реакцию с высокой скоростью при температуре кипения растворителя обычно в течение приблизительно 4-5 ч. Проведение реакции в других растворителях, таких, как этилацетат или изопропилацетат, вполне возможно, но требует больше времени из-за низкой температуры кипения указанных растворителей.
Бутилацетат является более предпочтительным благодаря его низкой растворимости в воде. Поскольку бутилацетат практически нерастворим в воде, он удерживает соединение IA в растворе, обеспечивая получения очень чистого продукта (с чистотой более 98%). По чистоте указанный продукт превосходит продукт из предшествующего уровня техники, представляющего собой смесь IA/IIA. Выход составляет более 90%, аналогичный выход достигается в способе из предшествующего уровня техники, однако благодаря получению более чистого продукта общий выход соединения IIА, полученного способом по настоящему изобретению, выше.
На стадии циклизации смесь предпочтительно медленно (в течение двух часов) нагревают от 0°С до приблизительно 90°С и кипятят с обратным холодильником.
Следует отметить, что в способе по настоящему изобретению не происходит образования сверхалкилированного карсалама (который может образоваться при взаимодействии IIА с этилхлорформиатом), т.е. избыток реагента разрушается до стадии циклизации, что также является преимуществом заявленного способа.
При необходимости соединение IIА очищают обычным кипячением в этилацетате/воде, при котором удаляется до 10% исходного материала без снижения выхода продукта.
Побочные продукты на стадии А
В исходном материале IA может присутствовать соответствующий 3,5-изомер, что приводит к образованию побочного продукта (3,5-дихлоризомера). Предпочтительно, если в исходном материале или в конечном продукте присутствует менее 1%, прежде всего менее 0,5% 3,5-дихлоризомера, более предпочтительно менее 0,07%.
Стадия В
Примечания к схеме 6, приведенной ниже.
Этиловый эфир 8-бромоктановой кислоты (ЕВО), который используется в способе из предшествующего уровня техники, является достаточно дорогостоящим в финансовом и экологическом отношении, не только по причине высокой цены, но из-за его высокой реакционноспособности. Это соединение является эффективным мутагенным алкилирующим агентом и, следовательно, требует соблюдения серьезных мер безопасности как при его применении, так при утилизации. В то же время его хлорсодержащий аналог ЕСО является менее дорогостоящим и благодаря низкой реакционноспособности не столь опасен для окружающей среды и не наносит вред здоровью. Следовательно, указанные преимущества ЕСО, низкая стоимость и безопасность для здоровья и окружающей среды, также являются преимуществом способа по настоящему изобретению.
При разработке способа по изобретению замена ЕВО на ЕСО вначале вызвала большие проблемы на стадии алкилирования/деблокирования, в которой образуется свободная кислотя (IV.II) или (IVA). Однако указанные проблемы были выявлены и преодолены, как указано ниже, в процессе разработки метода по настоящему изобретению.
Схема 6

Установлено, что по сравнению со способом из предшествующего уровня техники реакция алкилирования с использованием ЕСО при 80°С происходит с достаточно низкой скоростью главным образом из-за низкой реакционноспособности хлорсодержащего реагента. Кроме того, при продолжительном времени реакции (необходимом для завершения реакции) возрастает количество побочных продуктов. Например, в аналогичных условиях при использовании ЕВО образуется 0,1% побочного продукта в виде дикислоты, а при использовании ЕСО образуется 7% побочного продукта.
Повышение температуры до 100°С увеличивает скорость реакции, но при этом одовременно возрастает скорость разложения соли "карсалама". Фактически в присутствии влаги карбонат натрия не только образует натриевую соль карсалама, но и частично раскрывает цикл, приводя к образованию соответствующего амида (IA), который реагирует по обоим положениям (по атому О и атому N). Диалкилированный побочный продукт плохо удаляется на конечной стадии и, следовательно, загрязняет конечный продукт. Следовательно, образование диалкилированного продукта является нежелательным.
По результатам экспериментов установлено, что при проведении реакции в присутствии 1 экв. NaBr при 80°С образуется только 2,2% побочного продукта (дикислоты), в то время как в отсутствие NaBr 7%.
В предпочтительных оптимизированных условиях при использовании 1 экв. ЕСО, 0,2 экв. бромида натрия, 0,55 экв. карбоната натрия, при 100°С алкилирование происходит с 95% селективностью, при этом диалкилирование составляет приблизительно только 2%, O-алкилирование составляет менее 1%, и наблюдается полное потребление ЕСО.
Наконец, для подавления деградации (IIА), при синтезе в лабораторном масштабе основание (обычно карбонат натрия) и IIА предпочтительно сначала смешивают в апротонном растворителе (предпочтительно диметилформамиде), нагревают до 100°С и медленно добавляют соединение (III.III). При добавлении 0,98 экв. (III.III), 0,6 экв. основания (обычно карбонат натрия) и 0,1 экв. бромида натрия способ по настоящему изобретению воспроизводимо обеспечивает проведение алкилирования с образованием приблизительно 95% требуемого продукта (смесь циклического и открытого сложного эфира), приблизительно 2% диалкилированнного сложного эфира и приблизительно 3% исходного (IIА).
Установлено, что при использовании в качестве основания карбоната натрия смешивание всех исходных материалов при комнатной температуре и последующее нагревание с данной скоростью приводит к неконтролируемому выделению СО2 и временному загущению суспензии, что является нежелательным.
Следовательно, при проведении синтеза в оптимизированных условиях смешивают все исходные материалы, кроме карбоната натрия. Полученную смесь нагревают до 100°С и лишь после этого медленно добавляют карбонат натрия, предпочтительно порциями, или непрерывно в твердой форме. В отсутствие карбоната натрия реакция не идет. Единственными параметрами, подлежащими контролю, являются количество и скорость добавления основания (карбоната натрия).
В этом альтернативном варианте синтеза по настоящему изобретению предпочтительно используется небольшой избыток карбоната натрия (0,55 экв., что соответствует 1,05-1,1 экв. основания) и предпочтительно карбонат натрия добавляют в течение приблизительно 2 ч. При этом реакция сопровождается незначительными побочными реакциями (менее 1% диалкилирования). Для достижения селективности на уровне более 95% (т.е. 95% требуемой конверсии) предпочтительно используют 1 экв. (III.III) (вместо 0,98) и 0,2 экв. бромида натрия (вместо 0,1 экв.).
Раскрытие цикла происходит при завершении реакции на стадия В1, хотя этот процесс не проходит полностью и предполагают, что это зависит от избытка карбоната натрия. Обычно при завершении стадии раскрытие цикла идет по меньшей мере на 30-50%, что делает нежелательным выделение чистого промежуточного соединения (VIA). Предпочтительно затем проводить омыление и раскрытие цикла непосредственно в том же реакторе, поскольку после омыления оба промежуточных соединения (VIA и VIIA) превращаются в требуемый продукт IVA. В результате образуется только один продукт IVA (5-CNAC, свободная кислота), который выделяют подкислением.
Для оптимизации омыления, чтобы исключить выделение промежуточного соединения, предпочтительно полностью удалить растворитель упариванием. Для этого предпочтительно использовать диметилформамид, а не диметилацетамид, поскольку ДМФА характеризуется более низкой температурой кипения.
Растворитель удаляют упариванием в вакууме при 100°С, при этом получают маслообразный гетерогенный остаток. Остаток смешивают с водой и при 80-100°С обрабатывают избытком гидроксида натрия. Раскрытие цикла и омыление проходят с высокой скоростью. После омыления раствор охлаждают до 60°С, нейтрализуют добавлением серной кислоты до рН 8-9 и разбавляют этилацетатом.
Затем смесь подкисляют до рН 2-3, при этом продукт переходит в органическую фазу. Водную фазу отбрасывают, а органическую фазу промывают водой. Затем добавляют свежую порцию воды и этилацетат упаривают при нормальном давлении, при этом получают суспензию IVA в воде. На этой стадии или даже несколько ранее можно выделить неочищенный продукт.
Суспензию в воде разбавляют этанолом, что позволяет получать раствор кислоты (IVA) при приблизительно 60°С. В указанный раствор добавляют количество гидроксида натрия, которое, как установлено, необходимо для поддержания побочного продукта (дикислоты) в растворе (и таким образом побочный продукт не загрязняет образующиеся кристаллы). После охлаждения из раствора кристаллизуется чистая кислота IVA. Кристаллы отделяют центрифугированием при 0°С и высушивают в вакууме при 60°С. Предпочтительно содержание дикислоты составляет менее 0,6%.
Вышеуказанный процесс кристаллизации IVA оптимизирован с целью удаления побочных продуктов (главным образом оставшегося исходного материала, дикислоты и в некоторых случаях димера). Предпочтительно содержание (чистота) конечного продукта, IVA, составляет более 99% при наличии димера на уровне менее 0,2%.
Побочные продукты на стадии В
(III.III (Этиловый эфир 8-хлороктановой кислоты)
Предпочтительно этиловый эфир 8-хлороктановой кислоты не содержит примесь дихлоргексана. Установлено, что при использовании препарата, содержащего 1% дихлоргексана плюс другой побочный продукт, дихлоргексан взаимодействует с IIА с образованием димерного побочного продукта, который, как указано выше, чрезвычайно трудно отделяется от конечной кислоты (IVA). Предпочтительно использовать этиловый эфир 8-хлороктановой кислоты чистотой >99% с содержанием дихлорметана менее 0,1%.
Карбонат натрия должен представлять собой высокоочищенный препарат, поскольку при использовании неочищенного реагента реакция не завершается, интенсифицируется раскрытие цикла и избыточное образование диалкилированного побочного продукта.
Возможным побочным продуктом является этиловый эфир 5-CNAC, который может образоваться из-за неполного омыления или за счет этерификации свободной кислоты во время конечной кристаллизации из этанола/воды.
Кроме того, как показано выше, если в соединении (III.III) содержится более 0,1% дихлоргексана, в качестве побочного продукта может присутствовать остаточное количество соединения IA, соответствующая 5-хлорсалициловая кислота и так называемый "димер".
Если в соединении (III.III) содержится 0,1% дихлоргексана, то предположительно содержание димера составляет приблизительно 0,3% в соединении IVA и менее 0,1% в соединении (VA).
При необходимости с целью дальнейшей очистки соединения (IVA) можно провести дополнительную экстракцию бутилацетатом после омыления и частичной нейтрализации. На этой стадии при рН 8-9 соединение IVA еще растворимо в воде (в форме мононатриевой соли), и димер, который не образует соли при таком рН (из-за отсутствия карбоксильной группы), можно удалить экстракцией. Основным недостатком такого варианта способа является большой расход бутилацетата, необходимого для разделения фаз.
Другие теоретически возможные примеси, такие, как изомеры, содержащие хлор в ароматическом цикле, и С7, С8 гомологи алкильной цепи могут присутствовать в небольших количествах (менее 0,05%), но предпочтительно их присутствие исключается благодаря контролю на их присутствие в исходных материалах.
Для сведения образования побочных продуктов к минимуму следует прежде всего контролировать следующие параметры:
- качество и количества исходных материалов: (IIА), (III.III), основания (такого, как карбонат натрия), бромида (такого, как бромид натрия),
- температура реакционной смеси и скорость добавления карбоната натрия,
- упаривание апротонного растворителя (такого, как ДМФА), и количество сильного основания (например, гидроксида натрия) (стадия омыления),
- соотношение этанол/вода и количество гидроксида натрия во время кристаллизации.
Кроме того, важными параметрами, от которых зависит качество соединения (IVA), являются качество и относительные количества исходных материалов. Слишком большое количество соединения (III.III) и карбоната натрия (или слишком малое количество соединения IIА), по-видимому, оказывает непосредственное влияние на количество образующегося диалкилированного побочного продукта, который до некоторой степени удаляется при кристаллизации и в принципе подлежит переработке. Однако в образовании побочных продуктов определенную роль может играть также снижение выхода, например, при снижении выхода приблизительно на 20% наблюдается увеличение выхода алкилирования на 10%.
Следовательно, в настоящем изобретении исследован и усовершенствован способ из предшествующего уровня техники и предлагается способ "в одном реакторе", позволяющий увеличить выход на 20% (приблизительно 84% по сравнению с 64%) при равной или более высокой чистоте продукта.
Стадия С
Схема 7

В способе из предшествующего уровня техники метод кристаллизации характеризуется следующими недостатками: кристаллизацию проводят из эмульсии при кипении растворителя (после добавления концентрированного раствора гидроксида натрия к свободной кислоте 5-CNAC в ацетоне) без введения затравки. Следовательно, невозможен контроль кристаллизации, хотя возможно, что в таких условиях кристаллизации достигается определенное термодинамическое равновесие, предотвращающее такой контроль.
В настоящем изобретении предлагается способ кристаллизации, в котором N-замещенный салициламид (IV, IV.II и IVA) кристаллизуют из смеси ацетон/вода в присутствии основания щелочного металла, прежде всего гидроксида натрия. Независимо от условий реакции полигидрат, например гексагидрат соединения IV, IV.II или IVA, кристаллизуется сначала в смеси ацетон/вода, а затем после высушивания образуется моногидрат V или VA. Обычно влажный осадок на фильтре содержит приблизительно 12% воды, что составляет 2-3 молекулы воды на одну молекулу 5-CNAC.
Кристаллизацию с включением только одного эквивалента воды проводят с использованием метилата натрия. При этом образуется сольват, который соответствует другой кристаллической модификации.
Стадия С сама по себе соответствует изобретательскому уровню. На стадиях предпочтительного способа смешивают соединение (IV.II или IVA), ацетон и воду. Соотношение ацетон/вода может составлять от приблизительно 5:1 (об./об.) до приблизительно 15:1 (об./об.), например от приблизительно 10:1 до 11:1. Обычно основание добавляют в смесь при слегка повышенной температуре, например от приблизительно 40°С до 60°С, например от 45°С до 55°С. Затем добавляют еще одну порцию ацетона, например, в виде смеси ацетон/вода (например, от 2:1 об./об. до 4:1 об./об., например, 3:1 об./об.), обычно при слегка повышенной температуре (например, 45°С-55°С). Затем выделяют соль. Одна из методик заключается в следующем: если температура составляет более 50°С, ее уменьшают до 50°С или менее (например, от 40°С до 50°С, до такой, как 45°С-48°С) и добавляют кристаллы-затравку для инициирования кристаллизации. Затем еще раз снижают температуру (например, до 0°С-5°С) для завершения стадии кристаллизации, а затем отделяют кристаллы. Обычно процесс кристаллизации проводят при непрерывном перемешивании. Кристаллы высушивают в вакууме (50-60 мбар) при 50-55°С в течение по меньшей мере 24 ч.
Установлено, что кристаллизация и перемешивание при более высоких температурах (40-50°С) позволяют получать более крупные кристаллы (до 500 мкм). Кроме того, кристаллизация при приблизительно 0°С позволяет получать кристаллические формы другого типа в отличие от кристаллов, полученных при более высоких температурах.
В другом способе кристаллизацию проводят в 2-пентаноне при 80°С.
Предпочтительно кристаллизацию проводят с использованием затравки при 45-50°С, затем при указанной температуре добавляют ацетон и охлаждают до 0°С, при этом образуются требуемая кристаллическая форма. Наиболее предпочтительную кристаллическую форму получают при продолжительном перемешивании в течение 24 ч при 0°С.
Полученная соль V или VA характеризуется чистотой предпочтительно >99% и содержанием любых побочных продуктов не более 0,1%.
Теоретически содержание воды в продукте VA должно составлять 4,8% в расчете на моногидрат. Фактически содержание воды зависит от условий и продолжительности высушивания. Пересушивания не происходит, если процесс проводят при давлении 80-100 мбар и температуре 40-55°С. Однако, чтобы исключить любой риск, рекомендуется контролировать содержание воды в процессе высушивания. Кроме того, после высушивания в некоторых случаях в образцах наблюдается присутствие остаточного количества ацетона, превышающее предельное содержание 0,5%.
Для сведения побочных продуктов к минимуму следует прежде всего контролировать следующие параметры:
- количество и качество основания щелочного металла (гидроксида натрия),
- температуру при внесении затравки и кристаллизации,
- степень насыщения раствора,
- скорость охлаждения,
- время перемешивания при 0°С,
- условия высушивания продукта.
В описании заявки и в пунктах формулы изобретения термины "включает" и "содержит" и их варианты, например "включающий" и "включают", означают "включающий, без ограничения перечисленным", и не означают исключение других фрагментов, добавок, компонентов, целых чисел или стадий.
В способах по настоящему изобретению предполагается, что в соединениях могут присутствовать одна или более защитных групп по одной или более функциональным группам, которые не принимают участие в конкретной реакции, стадии или промежуточной стадии, или которые могли бы принимать участие в такой реакции.
Защита функциональных групп такими защитными группами, пригодные реагенты для введения таких групп, пригодные защитные группы и способы их удаления известны специалисту в данной области. Примеры пригодных защитных групп можно найти в монографиях, таких, как J.F.W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London и New York (1973), T.W. Greene и P.G.M. Wuts, "Protective Groups in Organic Synthesis", 3 edition, Wiley, New York (1999), "The Peptides", т.3 (изд. E. Gross и J. Meienhofer), Academic Press, London end New York (1981), "Methoden der organischen Chemie", Houben-Weyl, 4th edition, т.15/1, Georg Thieme Verlag, Stuttgart (1974), H.-D. Jakubke и H. Jescheit, "Aminosauren, Peptide, Proteine", Verlag Chemie, Weinheim, Deerfield Beach, and Basel (1982) и/или Jochen Lehmann, "Chemie der Kohlenhydrate: Monosaccharide and Derivate", Georg Thieme Verlag, Stuttgart (1974).
Пригодные гидроксизащитные группы прежде всего выбирают из групп типа акрила или сложного эфира, включающих, например, (низш.)алканоил, такой, как формил, ацетил или изобутироил, бензоилформил, хлорацетил, дихлорацетил, трихлорацетил, трифторацетил, метоксиацетил, феноксиацетил, фенилацетил, пара-фенилацетил, дифенилацетил, 2,6-дихлор-4-метилфеноксиацетил, 2,6-дихлор-4-(1,1,3,3-тетраметилбутил)феноксиацетил, 2,4-бис(1,1-диметилпропил)феноксиацетил, хлордифенилацетил, 3-фенилпропионил, 4-азидобутироил, 4-метилтиометоксибутироил, (Е)-2-метил-2-бутеноил, 4-нитро-4-метилпентаноил, 4-пентеноил, 4-оксопентаноил, 4,4-(этилендитио)пентаноил, 5-[3-бис(4-метоксифенил)гидроксиметилфенокси]левулинил, пивалоил, кротоноил, моносукциноил, бензоил, пара-фенилбензоил, 2,4,6-триметилбензоил, 2-(метилтиометоксиметил)бензоил, 2-(хлорацетоксиметил)бензоил, 2-[(2-хлорацетокси)этил]бензоил, 2-[(2-бензилокси)этил]бензоил, 2-[2-(4-метоксибензилокси)этил] бензоил, 2-иодбензоил, орто-(дибромметил)бензоил, орто-(метоксикарбонил)бензоил, 2-хлорбензоил, 4-бромбензоил, 4-нитробензоил, алкоксикарбонил, такой, как метоксикарбонил, этоксикарбонил, изобутоксикарбонил, метоксиметилкарбонил, 9-флуоренилметоксикарбонил, 2,2,2-трихлорэтоксикарбонил, 1,1-диметил-2,2,2-трихлорэтоксикарбонил, 2-(триметилсилил)этоксикарбонил, 2-(фенилсульфонил)этоксикарбонил, 2-(трифенилфосфонио)этоксикарбонил, винилоксикарбонил, аллилоксикарбонил, пара-нитрофеноксикарбонил, бензилоксикарбонил, пара-метоксибензилоксикарбонил, 3,4-диметоксибензилоксикарбонил, орто-нитробензилоксикарбонил, пара-нитробензилоксикарбонил, дансилэтоксикарбонил, 2-(4-нитрофенил)этоксикарбонил, 2-(2,4-динитрофенил)этоксикарбонил, 2-циано-1-фенилэтоксикарбонил, S-бензилтиокарбонил, 4-этокси-1-нафтилоксикарбонил, 3′,5′-диметоксибензоилоксикарбонил, 2-метилтиометоксиэтоксикарбонил, N-фенилкарбамоил, диметилэтилфосфинотиолил, метилдитиокарбонил, N,N,N′,N′-тетраметилфосфордиамидоил, сульфонил, метансульфонил, бензолсульфонил, толуолсульфонил, 2-[(4-нитрофенил)этил]сульфонил, аллилсульфонил, 2-формилбензолсульфонил, нитрокси; или используют защитные группы типа простого эфира, такие, как метил, замещенный метил, предпочтительно (низш.)алкоксиметил, прежде всего метоксиметил (MOM), метилтиометил, (фенилдиметилсилил)метоксиметил, бензилоксиметил, пара-метоксибензилоксиметил, пара-нитробензилоксиметил, гваяколметил, трет-бутоксиметил, 4-пентенилоксиметил, силилоксиметил, (низш.)алкокси(низш.)алкоксиметил, прежде всего 2-метоксиэтоксиметил (MEM), 2,2,2-трихлорэтоксиметил, 2-(триметилсилил)этоксиметил или метоксиметил, тетрагидропиранил, 3-бромтетрагидропиранил, тетрагидротиопиранил, 4-метокситиопиранил, 1-метоксициклогексил, 4-метокситетрагидротиопиранил, S,S-диокси-4-метокситетрагидротиопиранил, 1-[(2-хлор-4-метил)фенил]-4-метоксипиперидин-4-ил, 1-(2-фторфенил)-4-метоксипиперидин-4-ил, 1,4-диоксан-2-ил, тетрагидрофуранил, тетрагидротиофуранил, 2,3,3а,4,5,6,7,7а-октагидро-7,8,8-триметил-4,7-метанобензофуран-2-ил, замещенный этил, такой, как 1-этоксиэтил, 1-(2-хлорэтокси)этил, 1-[2-(триметилсилил)этокси]этил, 1-метилбензилоксиэтил, 1-метилбензилокси-2-фторэтил, 1-метил-1-феноксиэтил, 2,2,2-трихлорэтил, 1,1-дианизил-2,2,2-трихлорэтил, 1,1,1,3,3,3-гексафтор-2-фенилизопропил, 2-триметилсилилэтил, 2-(бензилтио)этил, 2-(фенилселенил)этил, трет-бутил, аллил или пропаргил, замещенные фениловые эфиры, такие как пара-хлорфенил, пара-метоксифенил, пара-нитрофенил, 2,4-динитрофенил или 2,3,5,6-тетрафтор-4-(трифторметил)фенил, бензил, замещенный бензил, такой, как пара-метоксибензил, 3,4-диметоксибензил, орто-нитробензил, пара-нитробензил, пара-галогенбензил, например пара-бромбензил, 2,6-дихлорбензил, пара-цианобензил, пара-фенилбензил, 2,6-дифторбензил, пара-азидобензил, 4-азидо-3- хлорбензил, 2-трифторметилбензил или пара-(метилсульфинил)бензил, 2- или 4-пиколил, 3-метил-2-пиколил, 2-хинолилметил, 1-пиренилметил, дифенилметил, пара, пара′-динитробензгидрил, 5-дибензосуберил, трифенилметил, α-нафтилдифенилметил, пара-метоксифенилдифенилметил, ди(пара-метоксифенил)фенилметил, три(пара-метоксифенил)метил, 4-(4′-бромфенацилокси)фенилдифенилметил, 4,4′,4′-трис(4,5-дихлорфталимидофенил)метил, 4,4′,4′-трис(левулиноилоксифенил)метил, 4,4′,4′-трис(бензоилоксифенил)метил, 4,4′-диметокси-3′′-[N-(имидазолилметил)]тритил, 4,4′-диметокси-3′′-[N-(имидазолилэтил)карбамоил]тритил, 1,1-бис(4-метоксифенил)-1′-пиренилметил, 4-(17-тетрагидробензо[а,с,g,f]флуоренилметил)-4′,4′′-диметокситритил, 9-антрил, 9-(9-фенил)ксантенил, 9-(9-фенил-10-оксо)антрил, 1,3-бензодитиолан-2-ил, или используют S,S-диоксиды типа силиловых эфиров, такие, как три-(низш.)алкилсилил, например триметилсилил, триэтилсилил, триизопропилсилил, диметилизопропилсилил, диэтилизопропилсилил, диметилгексилсилил, трет-бутилдиметилсилил или ди-трет-бутилметилсилил, трет-бутилдифенилсилил, трифенилсилил, дифенилметилсилил, трис(триметилсилил)силил, (2-гидроксистирил)диметилсилил, (2-гидроксистирил)диизопропилсилил, трет-бутилметоксифенилсилил или трет-бутоксидифенилсилил.
Пригодными карбоксизащитными группами прежде всего являются сложноэфирные группы, которые удаляются при ферментативном и/или химическом гидролизе, такие, как гептил, 2-N-(морфолино)этил, холинил, метоксиэтоксиэтил или метоксиэтил; или группы, которые удаляются прежде всего при химическом гидролизе, например алкил, такой, как (низш.)алкил, прежде всего метил, этил, замещенный (низш.)алкил (за исключением бензила и замещенного бензила), такие, как замещенный метил, прежде всего 9-флуоренилметил, метоксиметил, метоксиэтоксиметил, метилтиометил, 2-(триметилсилил)этоксиметил, бензилоксиметил, пивалоилоксиметил, фенилацетоксиметил, триизопропилсилилметил, 1,3-дитианил-2-метил, дициклопропилметил, ацетонил, фенил, пара-бромфенацил, α-метилфенацил, пара-метоксифенацил, дезил, карбамидометил, пара-азобензолкарбоксамидометил, N-фталимидометил или 4-пиколил, 2-замещенный этил, прежде всего 2-иод-, 2-бром- или 2-хлорэтил, 2,2,2-трихлорэтил, 2- (триметилсилил)этил, 2-метилтиоэтил, 2-(пара-нитрофенилсульфенил)этил, 2-(пара-толуолсульфонил)этил, 2-(2'-пиридил)этил, 2-(пара-метоксифенил)этил, 2-(дифенилфосфино)этил, 1-метил-1-фенилэтил, 2-(4-ацетил-2-нитрофенил)этил или 2-цианоэтил, трет-бутил, 3-метил-3-пентил, 2,4-диметил-3- или ω-хлор-(низш.)алкил, прежде всего 4-хлорбутил или 5-хлорпентил, циклопентил, циклогексил, (низш.)алкенил, прежде всего аллил, металил, 2-метилбут-3-ен-2-ил, 3-метилбут-2-енил или 3-бутен-1-ил, замещенный (низш.)алкенил, прежде всего 4-(триметилсилил)-2-бутен-1-ил, циннамил или α-метилциннамил, (низш.)алкинил, такой, как проп-2-инил, фенил, замещенный фенил, прежде всего 2,6-диалкилфенил, такой, как 2,6-диметилфенил, 2,6-диизопропилфенил, 2,6-ди-трет-бутил-4-метилфенил, 2,6-ди-трет-бутил-4-метоксифенил, пара-(метилтио)фенил или пентафторфенил, бензил, замещенный бензил, прежде всего трифенилметил, дифенилметил, бис(орто-нитрофенил)метил, 9-антрилметил, 2-(9,10-диоксо)антрилметил, 5-дибензосуберил, 1-пиренилметил, 2-(трифторметил)-6-хромонилметил, 2,4,6-триметилбензил, пара-бромбензил, орто-нитробензил, пара-нитробензил, пара-метоксибензил, 2,6-диметоксибензил, 4-(метилсульфинил)бензил, 4-сульфобензил, 4-азидометоксибензил, 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино)бензил, пиперонил или пара-полимербензил, тетрагидропиранил, тетрагидрофуранил, или силилрадикалы, такие как три(низш.)алкилсилил, прежде всего триметилсилил, триэтилсилил, трет-бутилдиметилсилил, изопропилдиметилсилил или ди-трет-бутилметилсилил, или фенилди(низш.)алкилсилил, такой, как фенилдиметилсилил; в другом варианте карбоксигруппу можно защитить группами оксазолил, 2-алкил-1,3-оксазолинил, 4-алкил-5-оксо-1,3-оксазолидинил или 2,2-бис-трифтор-4-алкил-5-оксо-1,3-оксазолидинил.
Амидозащитными группами являются прежде всего аллил, трет-бутил, N-метокси, N-бензоилокси, N-метилтио, трифенилметилтио, трет-бутилдиметилсилил, триизопропилсилил, 4-(метоксиметокси)фенил, 2-метокси-1-нафтил, 9-флуоренил, трет-бутоксикарбонил, N-бензилоксикарбонил, N-метокси- или N-этоксикарбонил, толуолсульфонил, N-бутен-1-ил, 2-метоксикарбонилвинил, или прежде всего алкил, такой, как (низш.)алкил, или более предпочтительно замещенный алкил, прежде всего бензил, бензил, замещенный одним или более радикалами, выбранными из группы, включающей (низш.)алкокси, такой, как метокси, (низш.)алканоилокси, такой, как ацетокси, (низш.)алкилсульфинил, такой, как метилсульфицил, дициклопропилметил, метоксиметил, метилтиометил и N-бензоилоксиметил; или бис-(триметилсилил)метил, трихлорэтоксиметил, трет-бутилдиметилсилилоксиметил, пивалоилоксиметил, цианометил, бепзил, 4-метоксибензил, 2,4-диметоксибензил, 3,4-диметоксибензил, 2-ацетокси-4-метоксибензил, орто-нитробензил, бис-(4-метоксифенил)фенилметил, бис-(4-метилсульфинилфенил)метил, пирролидинометил, диэтоксиметил, 1-метокси-2,2-диметилпропил или 2-(4-метилсульфонил)этил.
Защитные группы легко удаляются (т.е. при этом не наблюдается нежелательных побочных реакций), например, сольволизом, восстановлением, фотолизом или в условиях, аналогичных физиологическим условиям, например, ферментативным гидролизом. Обычно защитные и активирующие стадии (реакции) проводятся одновременно.
Способ по настоящему изобретению иллюстрируется следующими примерами синтеза 5-CNAC.
Примеры
Пример 1
Получение 6-хлор-2Н-1,3-бензоксазин-2,4(3Н)-диона

В четырехгорлую круглодонную колбу объемом 3 л в атмосфере азота помещали 5-хлорсалициламид (300 г, 1,75 моля), воду (900 мл) и смесь перемешивали. Затем в смесь добавляли 5-этил-2-метилпиридин (284 г, 2,27 моля) и н-бутилацетат (900 мл), охлаждали до 0-5°С (температура в рубашке -10°С) и добавляли по каплям этилхлорформиат (233 г, 2,10 моля) в течение приблизительно 1 ч. После завершения добавления реагента реакционную смесь медленно нагревали (приблизительно в течение 2 ч) до температуры кипения растворителя и кипячение с обратным холодильником продолжали в течение приблизительно 5 ч (температура в рубашке 110°С, в колбе (IT) 90°C). Полученную суспензию охлаждали до комнатной температуры, добавляли соляную кислоту (28 мл, 37 мас.%, 0,34 моля) и смесь перемешивали в течение приблизительно 30 мин. Полученную суспензию фильтровали в вакууме, осадок па фильтре промывали н-бутилацетатом и водой (600 мл), высушивали досуха в вакууме при 60°С в течение ночи, после высушивания получали 321 г (93%) 6-хлор-2Н-1,3-бензоксазин-2,4(3Н)-диона (6-хлоркарсалама).
Пример 2
Получение N-(5-хлорсалицилоил)-8-аминокаприловой кислоты (5-CNAC)

В четырехгорлую круглодонную колбу объемом 3 л в атмосфере азота помещали 6-хлор-2Н-1,3-бензоксазин-2,4(3Н)-дион (180,5 г, 0,91 моля), бромид натрия (18,7 г, 0,18 моля) и диметилформамид (840 мл) и смесь перемешивали. Затем в смесь одной порцией добавляли этиловый эфир 8-хлороктановой кислоты (188,3 г, 0,911 моля) и добавляли диметилформамид (60 мл). Смесь нагревали до 100-105°С (температура в рубашке 120°С) и порциями в течение 2 ч добавляли безводный карбонат натрия (51,7 г, 0,47 моля). После завершения реакции растворитель выпаривали при пониженном давлении (60-20 мбар, температура в колбе 75-120°С, температура в рубашке 100-130°С), при этом получали маслообразный остаток. Затем при 85-95°С в течение 10-20 мин добавляли воду (700 мл), гидроксид натрия (380 мл, 30 мас.%) и 20 мл воды. Смесь перемешивали при 85-100°С в течение 2 ч, при 60-65°С добавляли серную кислоту (60 мл, 50 мас.%) до рН 8-9, а затем при 60-65°С в течение 30-60 мин добавляли этилацетат (700 мл). Затем при 60-65°С в течение 1 ч добавляли вторую порцию серной кислоты (221 мл, 50 мас.%) до рН 2-3,5. Две фазы разделяли, водную фазу отбрасывали, органическую фазу промывали водой (300-360 мл) при 60-65°С. Затем добавляли воду (600 мл) и гидроксид натрия (10,8 г, 30 мас.%, и 40 мл воды), 20 мл воды (для удаления небольшого количества побочного продукта, N, O-5-(хлорсалилоил)диоктановой кислоты) и органический растворитель выпаривали при атмосферном давлении (температура рубашки 110-120°С, IT 80-100°C). В полученную суспензию при 50-65°С добавляли этанол (1500 мл). Прозрачный раствор охлаждали до 40-45°С, добавляли затравочные кристаллы (0,12 г) и раствор перемешивали в течение 20-30 мин до начала кристаллизации. Смесь охлаждали до 0-5°С в течение 1-2 ч. Твердое вещество отделяли фильтрованием в вакууме, промывали этанолом/водой, 7:3 (540 мл) и высушивали в вакууме (10-50 мбар) при 60°С в течение ночи, при этом получали 237 г N-5-(хлорсалицилоил)-8-аминокаприловой кислоты (84%).
Пример 3
Получение моногидрата динатриевой соли N-(5-хлорсалицилоил)-8-аминокаприловой кислоты
В реактор объемом 50 л в атмосфере азота помещали N-5-(хлорсалицилоил)-8-аминокаприловую кислоту (3,5 кг, 11,15 моля), ацетон (9450 мл) и воду (875 мл, очищенную) и смесь перемешивали при 45-55°С (температура в рубашке 60°С) до образования прозрачного раствора (в течение 20-30 мин). Затем добавляли гидроксид натрия (297 г, 30 мас.%, 22,3 моля), поддерживая температуру 45-55°С, а затем ацетон/воду (3:1, об./об., 1050 мл). Горячий раствор (50°С) пропускали через осветляющий фильтр, фильтрат переносили в другой чистый реактор и нагревали до 45-55°С. Соединительную линию промывали горячим (45-55°С) ацетоном/водой (3:1, об./об., 1050 мл), а затем добавляли ацетон (приблизительно 10,5 л), поддерживая температуру приблизительно 45-55°С (температура в рубашке 55°С). Затем температуру снижали до 45-48°С и добавляли кристаллы-затравку (4 г). Смесь перемешивали в течение приблизительно 20-30 мин до образования тонкой суспензии и начала кристаллизации, затем в течение 1 ч добавляли вторую порцию ацетона (28 л), поддерживая температуру 45-50°С (температура в рубашке 55°С). Смесь медленно перемешивали при 45-50°С в течение 1 ч и температуру снижали до 0-5°С в течение 2 ч. Перемешивание при 0-5°С продолжали в течение 1 ч, кристаллы отделяли центрифугированием, промывали охлажденным ацетоном/водой (95:5, об./об., 7 л) и высушивали в вакууме 50-60 мбар при 50-55°С в течение по меньшей мере 24 ч, при этом получали 4,19 кг моногидрата динатриевой соли 5-CNAC (выход 95%).
Реферат
Изобретение относится к способу получения N-замещенного салициламида, включающему взаимодействие производного карсалама с хлорзамещенным соединением формулы (III) в присутствии каталитического количества источника ионов брома ! ! где n означает целое число от 1 до 8, Q означает защищенную карбоксильную группу, а R5 и R6 независимо выбирают из группы, включающей водород, -ОН, NR3R4, галоген, C1-C4 алкил, C1-C4 алкокси, С2-С4 алкенил, где R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, C1-C4 алкил, С1-С4 галогеналкил, C1-C4 алкокси, C2-C4 алкенил. Также предложен способ получения соединения общей формулы IV. Усовершенствованный способ получения N-замещенного салициламида позволяет получить большие количества высококачественного продукта. 2 н. и 52 з.п. ф-лы.
Формула

где n означает целое число от 1 до 8, Q означает защищенную карбоксильную группу, а R5 и R6 независимо выбирают из группы, включающей водород, -ОН, NR3R4, галоген, C1-C4 алкил, C1-C4 алкокси, C2-C4 алкенил, где R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкокси, С2-С4 алкенил.

где R1 означает карбоксизащитную группу.
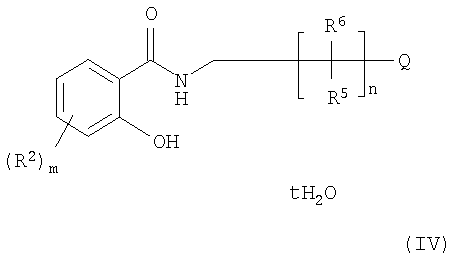
где R5, R6, Q и n имеют значения, указанные в n.1, t равно 0, 1,2, 3, 4, 5 или 6, m равно 1, 2, 3 или 4, или если m>1, то каждый R2 независимо выбирают из группы, включающей -ОН, NR3R4, галоген, C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкокси, С2-С4 алкенил, а R3 и R4 каждый независимо выбирают из группы, включающей водород, -ОН, C1-C4 алкил, C1-C4 галогеналкил, C1-C4 алкокси, С2-С4 алкенил, причем указанный способ включает:
(1) взаимодействие соединения формулы (I)

с хлорформиатом в присутствии слабого основания с образованием соединения формулы (II)

и
(2) взаимодействие соединения формулы (II) с соединением формулы (III) в присутствии каталитического количества источника ионов брома и слабого органического основания

с образованием соединения формулы (IV).

где R2, R5, R6, m, t, n и Q имеют значения, указанные в п.8.

где R1 означает карбоксизащитную группу, а соединением формулы (IV) является соединение формулы (IV.I)



где R2, m, t, n и Q имеют значения, указанные в п.8.
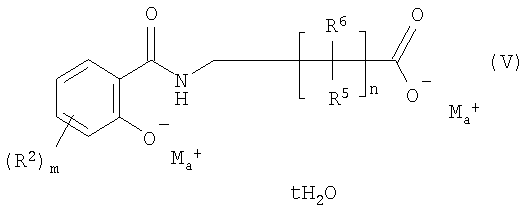
где Ма означает щелочной металл, a Y означает основный противоион.


Комментарии