Производные 2-(иминометил)аминофенила, способ их получения, фармацевтическая композиция на их основе и промежуточные вещества - RU2183211C2
Код документа: RU2183211C2
Чертежи
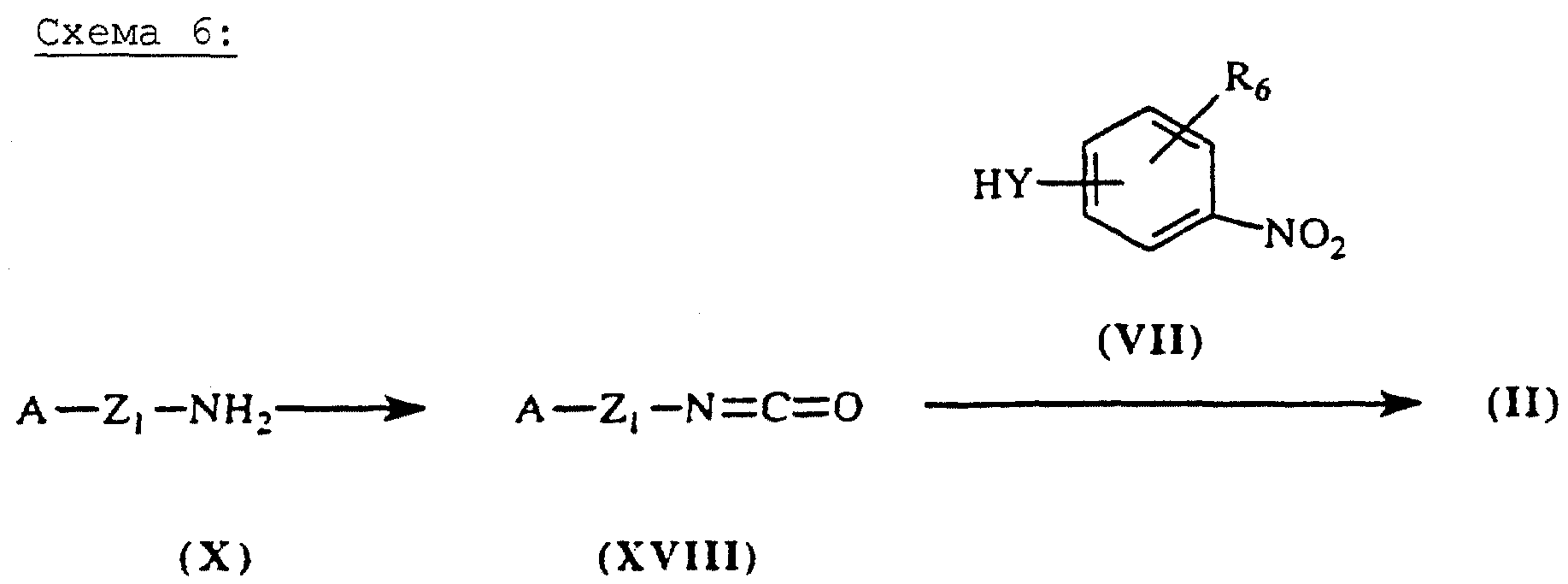
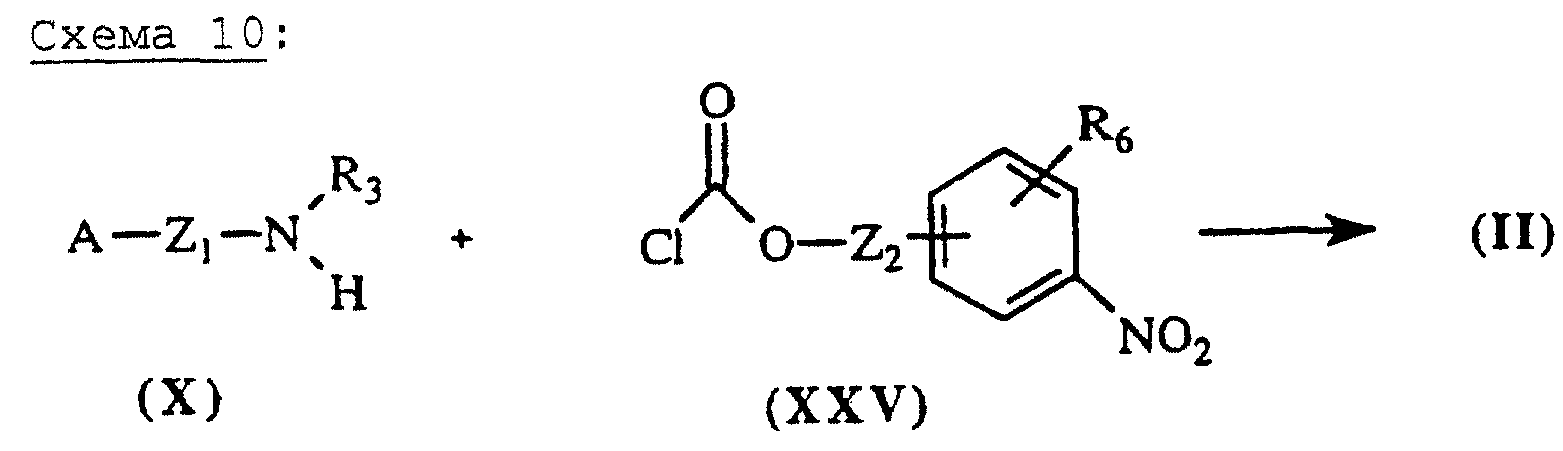
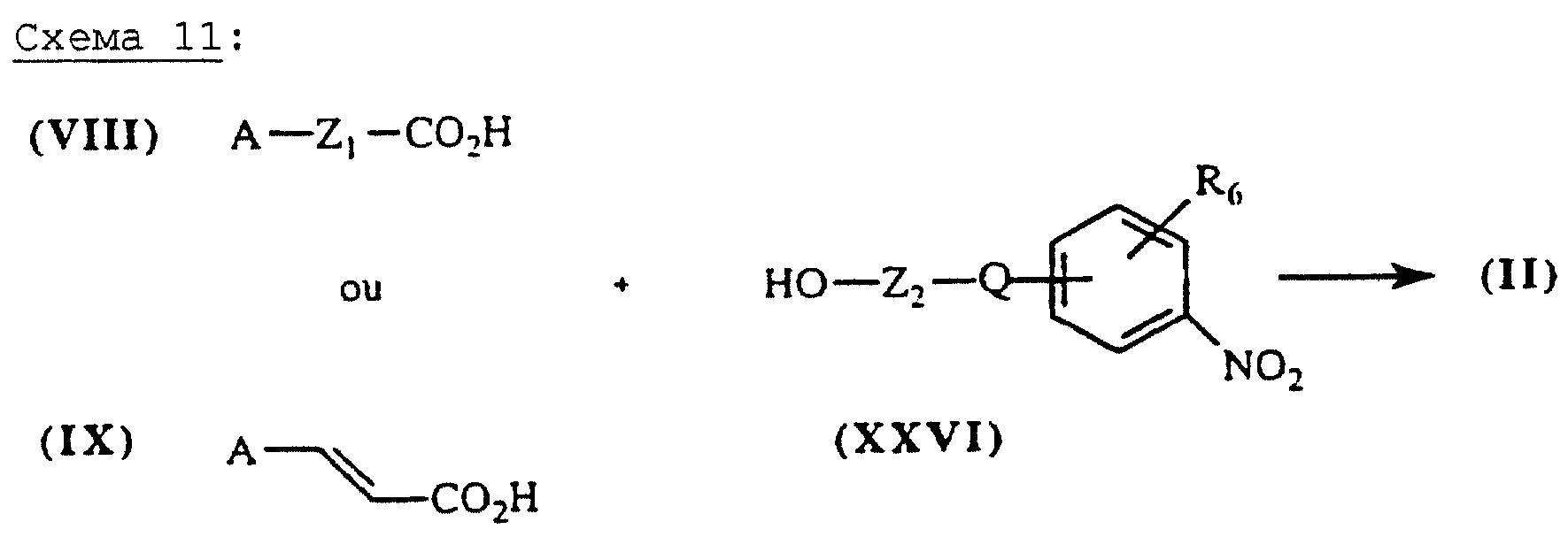
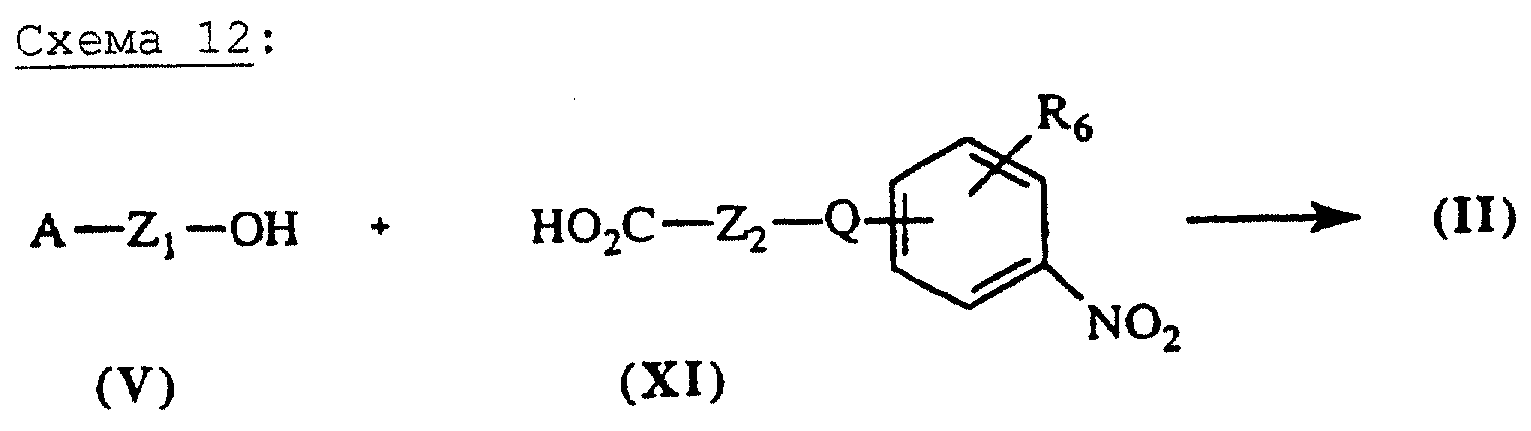
Описание
Объектом настоящего изобретения являются новые производные 2-(иминометил) аминофенила, обладающие ингибирующей активностью в отношении ферментов NO-синтаз, производящие моноксид азота NO, и/или улавливающей активностью по отношению к реактивным формам кислорода (ROS) "reactive охуgene species"). Более конкретно, изобретение касается производных общей формулы (I), определенной ниже, способов их получения, фармацевтических препаратов на их основе и их использования в терапевтических целях, в частности, в качестве ингибитора NO-синтаз и в качестве ловушки, селективной или неселективной, реактивных форм кислорода.
Учитывая потенциальную роль NO и ROS в физиопатологии, новые производные, отвечающие общей формуле (I), могут оказывать полезное или благоприятное действие при лечении
патологий, в которых задействованы эти химические элементы. А именно:
- сердечно-сосудистые и сосудисто-мозговые нарушения, куда входят, например, атеросклероз, мигрень, повышенное
артериальное давление, септический шок, сердечные инфаркты и инфаркты мозга ишемического происхождения или в результате кровоизлияния, ишемия и тромбозы;
- нарушения центральной или
периферической нервной системы, такие как нейродегенеративные заболевания, куда можно отнести, в частности, инфаркты мозга, субарахноидальное кровотечение, старение, старческое слабоумие, включая
болезнь Альцгеймера, хорея Гентингтгона, болезнь Паркинсона, спастический псевдосклероз Крейтцуфельда-Якоба и болезни a prions, боковой амиотрофический склероз, а также боль, травмы головного или
спинного мозга, зависимость от наркотиков; от алкоголя и от веществ, приводящих к чрезмерному привыканию, нарушения эрекции и функции воспроизводимости, расстройство сознания, энцефалопатия,
энцефалопатия вирусного или токсического происхождения;
- нарушения скелетной мышцы и нервномышечных соединений (миопатия, миозит), а также кожные заболевания;
- пролиферативные и
воспалительные нарушения, такие как атеросклероз, легочная гипертония, респираторная недостаточность, гломерулонефрит, портальная гипертензия, псориаз, артроз и ревматоидный артрит, фиброзы,
амилоидозы, воспаления кишечно-пищеварительной системы (колит, болезнь Крона) или легочной системы и дыхательных путей (астма, синуситы, риниты);
- трансплантация органов;
- аутоиммунные и вирусные заболевания, такие как волчанка, спид, вирусные и паразитарные инфекции, диабет, рассеянный склероз;
- рак;
- неврологические заболевания, связанные с
интоксикациями (отравление кадмием, ингаляция н-гексана, пестицида, гербицида), с лечением (рентгенотерапия) или нарушениями генетического происхождения (болезнь Вильсона);
- всех патологий,
характеризующихся избыточным продуцированием или с дисфункцией NO и/или ROS.
Эксперименты подтверждают с очевидностью тот факт, что во всех перечисленных патологиях задействованы NO или ROS (J. Med.Chem. (1995) 38. 4343-4362; Free Radic.Biol.Med. (1996) 20, 675-705; The Neuroscientist (1997) 3, 327-333).
Кроме того, ингибиторы NO-синтаз, их использование и сочетание этих ингибиторов с продуктами, обладающими антиоксидантными или антирадикальными свойствами, описаны в более ранних патентах (патенты США 5,081,148; 5,360,925 и неопубликованная заявка на патент).
Объектом настоящего изобретения являются производные 2-(иминометил) аминофенила, их получение и их применение в терапии.
Соединения согласно изобретению отвечают общей
формуле (I):
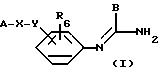
в которой А означает:
либо радикал:
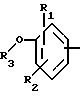
в котором R1 и R2 означают, независимо друг от друга, атом водорода, галоген, группу ОН, линейный или разветвленный алкил или алкокси, имеющий от 1 до 6 атомов углерода,
R3 означает атом водорода, линейный или разветвленный алкил, имеющий от 1 до 6 атомов углерода, или радикал -COR4, где
R4 означает линейный или разветвленный алкил с 1-6 атомами углерода,
либо радикал:
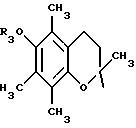
в которой R3 имеет значение, указанное выше, либо радикал:
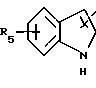
в котором R5 означает атом водорода, группу ОН или линейный или разветвленный алкил или алкокси с 1-6 атомами углерода, В означает линейный или разветвленный алкил с 1-6 атомами углерода, карбоциклический или гетероциклический арил с 5 или 6 звеньями, содержащими от 1 до 4 гетероатомов, выбираемых из О, S, N, и в частности, тиофена, фурана, пирола или тиазола, причем радикал арил при необходимости замещен одной или несколькими группами, выбираемыми из линейных или разветвленных радикалов алкила, алкенила или алкокси, имеющих от 1 до 6 атомов углерода;
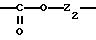
Х означает -Z-, -Z1-CO-, -CH= CH-CO, -Z1-NR3-CO-, -Z1-NR3-CS-, -Z1-NR3-SO2- или простую связь;
Y означает радикал, выбираемый из радикалов -Z2-Q, пиперазина, гомопиперазина, 2-метилпиперазина, 2,5-диметилпиперазина, 4-аминопиперидина, -NR3-Z2-Q-, -NR3-CO-Z2-Q-, -NR3-NH-CO-Z2-, -NH-NH-Z2, -NR3-O-Z2-, -NR3-SQ2-, NR3-Z2-, -O-Z2-Q-, -O-CO2-Z2-Q- или -S-Z2-Q-,
в которых Q означает простую связь, -O-Z3-, R3-N-Z3- или -S-Z3-;
Z1, Z2 и Z3 означают независимо друг от друга простую связь или линейный или разветвленный алкилен с 1-6 атомами углерода, предпочтительно Z1, Z2 и Z3 означают -(CH2)m-, причем m - это целое число, равное от 0 до 6;
R6 означает атом водорода или группу ОН;
или являются их солями.
Соединения общей формулы (I), содержащие асимметрический центр, имеют изомерные формы. Рацематы и энантиомеры этих соединений также составляют часть данного изобретения.
Соединения согласно изобретению могут находиться в виде оснований или аддитивных солей, а именно, в виде солей с органическими или неорганическими кислотами или основаниями, и в частности, в виде гидратов, хлоргидратов, дихлоргидратов, фумаратов или полуфумаратов.
Под линейным или разветвленным алкилом с 1-6 атомами углерода подразумевают, в частности, радикалы метил, этил, пропил, изопропил, бутил, изобутил, втор.-бутил и трет-бутил, пентил, неопентил, изопентил, гексил, изогексил. Под линейным или разветвленным алкокси радикалами с 1-6 атомами углерода подразумевают радикалы, алкил которых имеет вышеуказанное значение.
Под галогеном подразумевают атомы фтора, хлора, брома или иода.
В частности, объектом данного изобретения, являются
описанные в примерах следующие соединения общей формулы (I) (некоторые из них в форме соли):
-3,5-бис-(1,1-диметилэтил) -4-гидрокси-N-{4-[(2-тиенил(имино)метил)амино]фенил}-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{4-[[(2-тиенил(имино)метил)амино]фенил]метил}-бензамид;
-4-ацетокси-3,5-диметокси-N-{ 4-[[(2-тиенил(имино)метил)амино] фенил]метил}-бензамид;
-3,5-диметокси-4-гидрокси-N-{ 4-[[(2-тиенил(имино)метил)амино] фенил]метил}-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{4-[2-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамид;
-4-ацетокси-3,5-диметокси-N-{ 4-[2-[(2-тиенил-(имино)метил)-амино]фенил] этил}-бензамид;
-3,5-диметокси-4-гидрокси-N-{ 4-[2-[(2-тиенил-(имино)метил)амино] фенил] этил}-бензамид;
3,4,
5-тригидрокси-N-{ 4-[2-[(2-тиенил(имино)метил)-амино] фенил] этил}-бензамид;
-N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензоил]-1-пиперазинил]-фенил}-2-тиофенкарбоксимидамид;
-N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензил] -1-пиперазинил]-фенил}-2-тиофенкарбоксимидамид;
-N-{ 4-[4-[3,5-диметокси-4-гидроксибензоил]
-1-пиперазинил]-фенил)-2-тиофенкарбоксимидамид;
-3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{4-[(2-тиенил(имино)метил)амино]фенил}-2Н-1-бензопиран-2-карбоксамид;
-N-{4-[4-[(3,
4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-
бензопиран-2-ил)-карбонил] -1-пиперазинил1-фенил} -2-тиофенкарбоксимидамид;
-N-{
4-[4-[(5-метокси-1Н-индол-3-ил)метилкарбонил]-1- пиперазинил]фенил} -2- тиофенкарбоксимидамид;
-N-[4-[4-[{ 3-[3,5-бис-1,
1-диметилэтил)-4-гидроксифенил]-1-оксо-2-пропенил}-пиперазинил]фенил]]-2-тиофенкарбоксимидамид;
-3,5-бис(1,1-диметилэтил)-4-гидрокси-N{3-[[(2-тиенил-(имино)метил)амино] фенил]метил}-бензамид;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-N'-{{4-[(2-тиенил(имино)метил)амино]фенил}метил}-мочевина;
-N-[5-[{3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил}
амино]-2-гидроксифенил]-2-тиофенкарбоксимидамид;
-N-[3-[{3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино]-4-гидроксифенил]-2- тиофенкарбоксимидамид;
-N-{4-[4-[3,4,5-тригидроксибензоил]-1-пиперазинил]-фенил}-2-тиофенкарбоксимидамид;
-N-[3,5-бис-[1,
1-диметилэтил)-4-гидроксифенил)-N'-{{4-[(2-тиенил(имино)метил)амино]фенил}карбониламино}-мочевина;
-N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил]-N'-{{4-[(2-тиенил(имино)метил)амино]фенил}метил}-тиомочевина;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил} -N'-{ 2-{
4-[(2-тиенил(имино)метил)амино]фенил}этил}-мочевина;
-N'-(4-{ 4-[(3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил}-фенил)-2- тиофенкарбоксимидамид;
-N-[4-{ 4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1Н-1,4-диазепин-1-ил}фенил]-2-тиофенкарбоксимидамид;
-(R)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил)фенил}-2- тиофенкарбоксимидамид;
-(S)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил)фенил}-2-тиофенкарбоксимидамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{2-[3-[(2-тиенил(имино)метил)амино]фенил]этил}-бензамид;
N-{ 4-[4-[2-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-этил]-1-пиперазинил)фенил}-2-тиофенкарбоксимидамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-бензоат
2-{4-[(2-тиенил(имино)метил)амино]фенил]этила;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-бензоат 2-{3-[(2-тиенил(имино)метил)амино]фенил]этила;
-3,5-бис-(1,
1-диметилэтил)-4-гидрокси-бензоат 2-{2-[(2-тиенил(имино)метил)амино]фенил]этила;
а также их соли, в частности, их хлоргидраты, дихлоргидраты, фумараты или полуфумараты.
Предпочтительными соединениями являются соединения общей формулы (I), в которых
- Х означает линейный или разветвленный алкилен с 1-6 атомами углерода, а Y означает пиперазин, гомопиперазин,
2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, NR3-Z2-Q-, -NR3-NH-CO-Z2, -NH-NH-Z2 или -NR3-O-Z2-; или
- Х означает -Z1-CO- или -СН=СН-СО- и Y означает пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, NR3-Z2-Q-, -NR3
-NH-CO-Z2, -NH-NH-Z2, -NR3-O-Z2-, -O-Z2-Q- или -NR3-CO-Q' с Q'=R3-N-Z3; или
- X означает -Z1
-NR3-CO- и Y означает -Z2-Q-, -NH-Z2-Q-,
-NH-CO-Z2-Q"- с Q"= O-Z3-, R3-N-Z3- или S-Z3-, или Y означает
-NR3-SO2-NR3-Z2 или -O-Z2-Q-;
или
- Х означает -Z1-NH-CO- и Y означает пиперазин, гомопиперазин, 2-метилпиперазин, 2,
5-диметилпиперазин, 4 -аминопиперидин,
-NR3-Z2-Q-, -NR3-NH-CO-Z2, -NH-NH-Z2 или -NR3-O-Z2-;
или
- Х означает -Z1-NR3-SO2- и Y означает -Z2-Q"- c Q"=O-Z3, R3-N-Z3- или S-Z3-, или Y означает -NR3
-Z2-Q-;
или
- Х означает -Z1- и Y означает -O-CO-Z2-Q-;
или
- Х означает -Z1-NR3-CS,- и Y означает -NH-Z2-Q- или пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, -NR3-Z2-Q-, -NH-NH-Z2- или -NR3-O-Z2;
или
- X означает связь, a Y означает -O-Z2-NH-, -S-Z2-NH-.
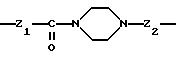
Группу X-Y предпочтительно выбирать из следующих радикалов:
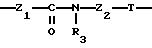
в котором Т означает простую связь, радикал -NR3- или радикал -CO-NR3-,
или
или
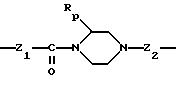
в котором Rp означает атом водорода или метил,
или
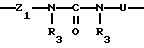
в котором U означает радикал -Z2, -NR3-CO-, -СO-Z2-O-, -СО-, -NR3- или атом кислорода,
или
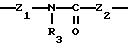
или
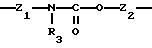
или
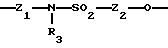
или
или
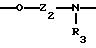
причем радикалы Z1-, Z2 и Z3 имеют значения, указанные выше.
Наиболее предпочтительными из соединений согласно изобретению являются соединения, выбранные из следующих;
-3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-{4-[2-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамид;
-3,4,5-тригидрокси-N-{ 4-[2-[(2-тиенил(имино)метил)-амино] фенил]этил}-бензамид;
-N-{ 4-[4-[3,
5-бис-(1,1-диметилэтил)-4-гидроксибензоил]-1-пиперазинил]-фенил}-2-тиофенкарбоксимидамид;
-N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензил] -1пиперазинил] -фенил}-2-тиофенкарбоксимидамид;
-3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{4-[(2-тиенил(имино)метил)амино]фенил}-2Н-1-бензопиран-2-карбоксамид;
-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил]фенил}-2-тиофенкарбоксимидамид;
-N-{ 4-[4-[(5-метокси-1H-индол-3-ил)метилкарбонил] -1-пиперазинил]фенил} -2-тиофенкарбоксимидамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{3-[[(2-тиенил-(имино)метил)амино]фенил]метил}-бензамид;
-N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил]-N'-{{4-[(2-тиенил(имино)метил)амино)фенил}метил}-мочевина;
-N-[5-[{3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил}
амино]-2-гидроксифенил]-2-тиофенкарбоксимидамид;
-N-[3-[{3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино]-4-гидроксифенил]-2-тиофенкарбоксимидамид;
-N-{4-[4-[(3,4,5-тригидроксибензоил]-1-пиперазинил1фенил}-2-тиофенкарбоксимидамид;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-{ { 4-[(2-тиенил
(имино)метил)амино]фенил}карбониламино}-мочевина;
или соль одного из этих соединений, в частности, хлоргидрат, дихлоргидрат, фумарат или полуфумарат одного из этих соединений.
Другими предпочтительными соединениями согласно изобретению являются следующие:
-4-ацетокси-3,5-диметокси-N-{ 4-[2-[(2-тиенил-(имино)метил)-амино]фенил] этил}-бензамид;
-3,
5-диметокси-4-гидрокси-N-{4-[2-[(2-тиенил-(имино)метил)
-амино]фенил]этил}-бензамид;
или соль одного из них, в частности, хлоргидрат, дихлоргидрат, фумарат или полуфумарат одного из
этих соединений.
Предпочтительными соединениями согласно изобретению являются также следующие:
-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил]фенил}-2- тиофенкарбоксимидамид;
-(R)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил]фенил}-2- тиофенкарбоксимидамид;
- (S)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1-пиперазинил]фенил}-2-тиофенкарбоксимидамид;
или соль одного из этих соединений, в частности, хлоргидрат, дихлоргидрат, фумарат или полуфумарат
одного из этих соединений.
Наконец, для данного изобретения особенно предпочтительными являются соединения общей формулы (I), которые имеют следующие характеристики:
либо:
А означает
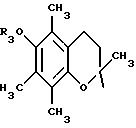
или
Х означает -СО- или -NH-CO-;
a Y означает радикал -NH-Z2-Q- или пиперазин, Q означает простую связь или радикал О-R3, R3-N-Z3 или S-Z3, a Z2 и Z3 означают, независимо друг от друга, связь или линейный или разветвленный алкилен, имеющий от 1 до 6 атомов углерода, а R3 означает атом водорода или линейный или разветвленный алкил с 1-6 атомами углерода.
либо: R6 - группа ОН.
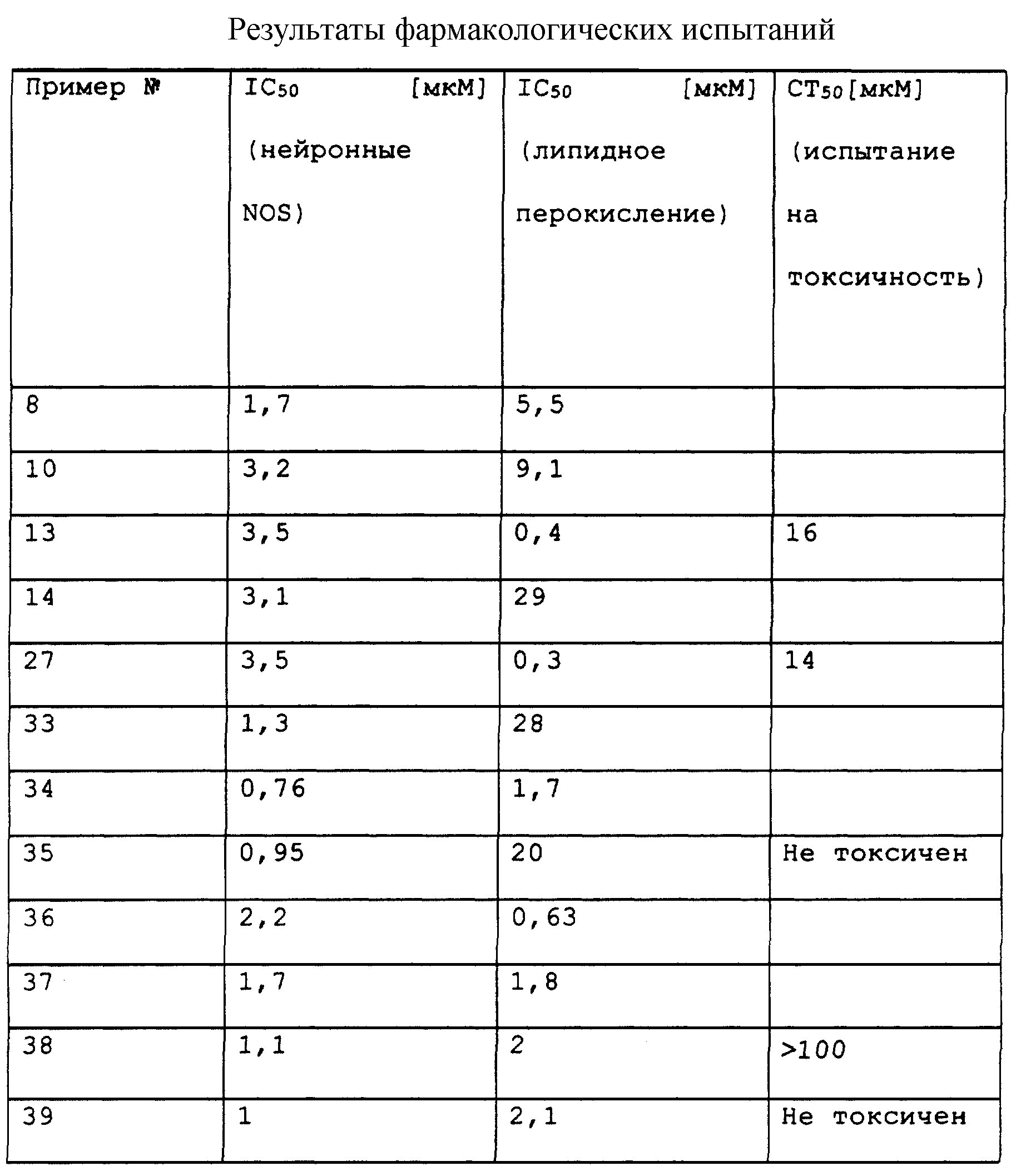
Объектом настоящего изобретения являются также лекарственные средства, в качестве которых используются описанные выше соединения общей формулы (I) и их фармацевтически приемлемые соли. Изобретение относится также к фармацевтическим композициям, содержащим эти соединения в качестве активного начала или их фармацевтически приемлемые соли, и к использованию этих соединений или их фармацевтически приемлемых солей для изготовления лекарственных препаратов, предназначенных для ингибирования нейронной NO-синтазы или индуцируемой NO-синтазы, для ингибирования перекисного окисления липидов или для обеспечения двойной функции ингибирования NO-синтазы и ингибирования перекисного окисления липидов.
Под фармацевтически приемлемой солью подразумевают в частности аддитивные соли неорганических кислот, такие как хлоргидрат, сульфат, дифосфат, бромгидрат и нитрат, или органических кислот, такие как ацетат, малеат, фумарат, тартрат, сукцинат, цитрат, лактат, метансульфонат, паратолуолсульфонат, памоат, оксалат и стеарат. В объем настоящего изобретения входят также соли, образованные основаниями, такими как гидроксид натрия или калия. В качестве других примеров фармацевтически приемлемых солей можно использовать те, которые указаны в документе "Pharmaceutical salts", J.Pharm. Sci. 66:1 (1977).
Фармацевтические композиции, включающие соединения согласно изобретению, могут находиться в твердой форме, например, в виде порошка, гранул, таблеток, желатиновых капсул, липосом или свечей. Соответствующими твердыми носителями могут быть, например, фосфат кальция, стеарат магния, тальк, сахара, лактоза, декстрин, крахмал, желатин, целлюлоза, метилцеллюлоза, карбоксиметилцеллюлоза натрия, поливинилпирролидин и воск.
Фармацевтические композиции, включающие соединения согласно изобретению, могут находиться также в жидкой форме, например, они могут быть в виде растворов, эмульсий, суспензий или сиропов. Соответствующими жидкими носителями могут быть, например, вода, органические растворители, такие как глицерин или гликоли, а также их смеси в воде в разных пропорциях.
Лекарственное средство согласно изобретению может быть введено топическим путем, орально, парентерально, путем внутримышечной инъекции и т.д.
Предлагаемая вводимая доза лекарственного средства согласно изобретению равна от 0,1 мг до 10 г в зависимости от типа используемого активного соединения.
Изобретение относится также к новым промышленным продуктам, представляющим собой соединения, являющиеся промежуточными
продуктами синтеза соединений общей формулы (I), а именно, к соединениям общей формулы (II) А:
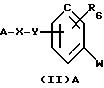
в которой W означает радикал амино или нитро,
А означает:
либо радикал:
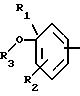
в котором R1 и R2 означают, независимо друг от друга, атом водорода, галоген, группу ОН, линейный или разветвленный алкил или алкокси с 1-6 атомами углерода,
R3 означает атом водорода, линейный или разветвленный алкил с 1-6 атомами углерода или радикал -COR4,
R4 означает линейный или разветвленный алкил с 1-6 атомами углерода,
либо радикал:
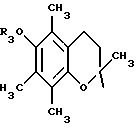
в котором R3 имеет указанное выше значение,
либо радикал:
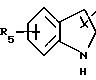
в котором R5 означает атом водорода, группу ОН или линейный или разветвленный алкил или алкокси с 1-6 атомами углерода; Х означает -Z1-, -Z1-CO-, -СН= СН-СО-, -Z1, -NR3-СО-, -Z1-NR3-CS-, -Z1-NR3-SO2- или простую связь;
Y означает радикал, выбираемый из радикалов -Z2-Q-, пиперазина, гомопиперазина, 2-метилпиперазина, 2,5-диметилпиперазина, 4-аминопиперидина, -NR3-Z2-Q-, -NR3-CO-Z2-Q-, -NR3-NH-CO-Z2-, -NH-NH-Z2-, -NR3-O-Z2-, -NR3 -SО2-NR3-Z2-, -O-Z2-Q-, -O-CO2-Z2-Q- или -S-Z2-Q-,
в которых Q означает простую связь, -O-Z3-, R3-N-Z3-, или -S-Z3-;
Z1, Z2 и Z3 означают, независимо друг от друга, простую связь или линейный или разветвленный алкилен с 1-6 атомами углерода, предпочтительно Z1, Z2 и Z3 означают -(СН2)m-, причем m - это целое число, равное от 0 до 6;
R6 означает атом водорода или группу ОН;
за исключением 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-нитрофенил)-бензамида;
или к солям этих соединений.
Изобретение относится к
новым промышленным продуктам, которые представляют собой, в частности, следующие соединения, являющиеся промежуточными в синтезе соединений общей формулы (I):
-3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил]-бензамид;
-3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-[(4-аминофенил)метил]-бензамид;
-4-ацетокси-3,5-диметокси-N-[(4-нитрофенил)метил]-бензамид;
-4-ацетокси-3,5-диметокси-N-[(4-аминофенил)метил]-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-нитрофенил)этил]-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-аминофенил)этил]-бензамид;
-4-ацетокси-3,
5-диметокси-N-[2-(4-нитрофенил)этил]-бензамид;
-4-ацетокси-3,5-диметокси-N-[2-(4-аминофенил)этил]-бензамид;
-3,4,5-тригидрокси-N-[2-(4-нитрофенил)этил]-бензамид;
-3,4,
5-тригидрокси-N-[2-(4-аминофенил)этил]-бензамид;
-2,6-бис-(1,1-диметилэтил)-4-{[4-(4-нитрофенил)-1-пиперазинил]-карбонил} -фенол;
-2,6-бис-(1,1-диметилэтил)-4-{
[4-(4-аминофенил)-1- пиперазинил]-карбонил}-фенол;
-2,6-бис-(1,1-диметилэтил)-4-{ [4-(4-нитрофенил)-1- пиперазинил]-метил} -фенол;
-2,6-бис-(1,1-диметилэтил)-4-{
[4-(4-аминофенил)-1-пиперазинил]-метил}-фенол;
-2,6-диметокси-4-{[4-(4-нитрофенил)-1-пиперазинил]-карбонил}-фенол;
-2,6-диметокси-4-{[4-{4-аминофенил)-1-пиперазинил]-карбонил}-фенол;
-3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-(4-нитрофенил} - 2Н-1-бензопиран-2-карбоксамид;
-3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-(4-аминофенил}
-2Н-1-бензопиран-2-карбоксамид;
-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил} -1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-3,4-дигидро-2,5,7,8-тетраметил-2-{
4-[(4-аминофенил} -1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-1-[(5-метокси-1Н-индол-3-ил)метилкарбонил]-4-(4-нитрофенил)-пиперазин;
-1-[(5-метокси-1Н-индол-3-ил)метилкарбонил]-4-(4-аминофенил)-пиперазин;
-2,6-бис-(1,1-диметилэтил)-4-{ 3-[4-(4-нитрофенил)-1-пиперазинил] -3-оксо-2-пропенил}-фенол;
-2,6-бис-(1,
1-диметилэтил)-4-{ 3-[4-(4-аминофенил)-1-пиперазинил] -3-оксо-2-пропенил}-фенол;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(3-нитрофенил)метил]-бензамид;
-3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-[(3-аминофенил)метил]-бензамид;
-N-3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-[(4-нитрофенил)метил] -мочевина;
-N-[(4-аминофенил)метил] -N'-[3,
5-бис-(1,1-диметилэтил)-4-гидроксифенил] -мочевина;
-3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N-(4-гидрокси-3-нитрофенил)-2-пропенамид;
-3-[(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил] -N-(4-гидрокси-3-аминофенил)-2-пропенамид;
-3-[[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N-(2-гидрокси-5-нитрофенил)-2-пропенамид;
-3-[(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил] -N(2-гидрокси-5-аминофенил)-2-пропенамид;
-5-{[4-(4-нитрофенил)-1-пиперазинил]карбонил}-бензол-1,2,3-триол;
-5-{[4-(4-аминофенил)-1-пиперазинил]карбонил}-бензол-1,2,3-триол;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-N'-[(4-нитрофенил)карбониламино]-мочевина;
-N-[(4-аминофенил]
карбониламино] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-мочевина;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-[(4-нитрофенил)метил] -тиомочевина;
N-[(4-аминофенил)метил] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -тиомочевина;
-N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-[(4-нитрофенил)этил] -мочевина;
N-[2-(4-аминофенил)этил]-N'-[3,5-бис-(1,1-диметилэтил)-4- гидроксифенил] -мочевина;
-1-{ [3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил] карбонил}-4-(4-нитрофенил)пиперазин;
-1-{ [3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил] карбонил}-4-(4-аминофенил)пиперазин;
-гексагидро-4-(4-нитрофенил)-1Н-1,4-диазепин;
-1-[(3,
4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил] карбонил}гексагидро-4-(4-нитрофенил)-1Н-1,4-диазепин;
-1-(4-аминофенил)-4-[(3,4-дигидро-6-гидрокси-2,5,7,
8- тетраметил-2Н-1-бензопиран-2-ил]карбонил}гексагидро-1Н-1,4-диазепин;
-хлоргидрат N-[4-{ 4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)карбонил] -1Н-1,4-диазепин-1-ил}
-фенил]-2-тиофенкарбоксимидамида;
-(R)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-(R)-3,4-дигидро-2,5,7,8-тетраметил-2-{
4-[(4-аминофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-(S)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-(S)-3,
4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-аминофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол;
-3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(3-нитрофенил)этил]-бензамид;
-3,
5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(3-аминофенил)этил]-бензамид;
-3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат 2-(4-нитрофенил) этила;
-3,5-бис-(1,
1-диметилэтил)-4-гидроксибензоат 2-(4-нитрофенил)этила;
или их соли.
Изобретение относится также к способам получения соединений общей формулы (I), которые состоят, например,
во взаимодействии в низшем спирте, таком как метанол, этанол, изопропиловый спирт или трет. бутанол, предпочтительно, в изопропиловом спирте, при температуре от 20 до 90oС, например, при
50oС, и в течение 48 часов, предпочтительно, в течение 15-24 часов, при необходимости, в присутствии ДМФ, соединения общей формулы (III), указанной выше, с соединением общей формулы (IV)
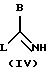
названное соединение формулы (IV) может быть, при необходимости, превращено в соль с помощью минеральной кислоты G, причем В имеет указанное выше значение, a L означает удаляемую группу, в частности, алкокси, тиоалкил, группу сульфоновой кислоты, галогенида, арилового спирта или тозила (другие удаляемые группы, хорошо известные специалисту, и которые, при необходимости, могут быть использованы согласно изобретению, описаны в следующей литературе: Advanced Organic Chemistry, J. March, 3rd Edition (1985), Me Graw-Hill, p.315). Предпочтительно, G означает НСl, HBr или HI.
Другие способы получения рассмотрены, например, в "The Chemistry of amidines and imidates, Vol. 2, Saul PATAL and Zvi RAPPOPORT, John Wiley & Sons, 1991".
Согласно изобретению, соединения общей формулы (I) можно получить способом, описанным ниже.
Получение соединений общей формулы (I):
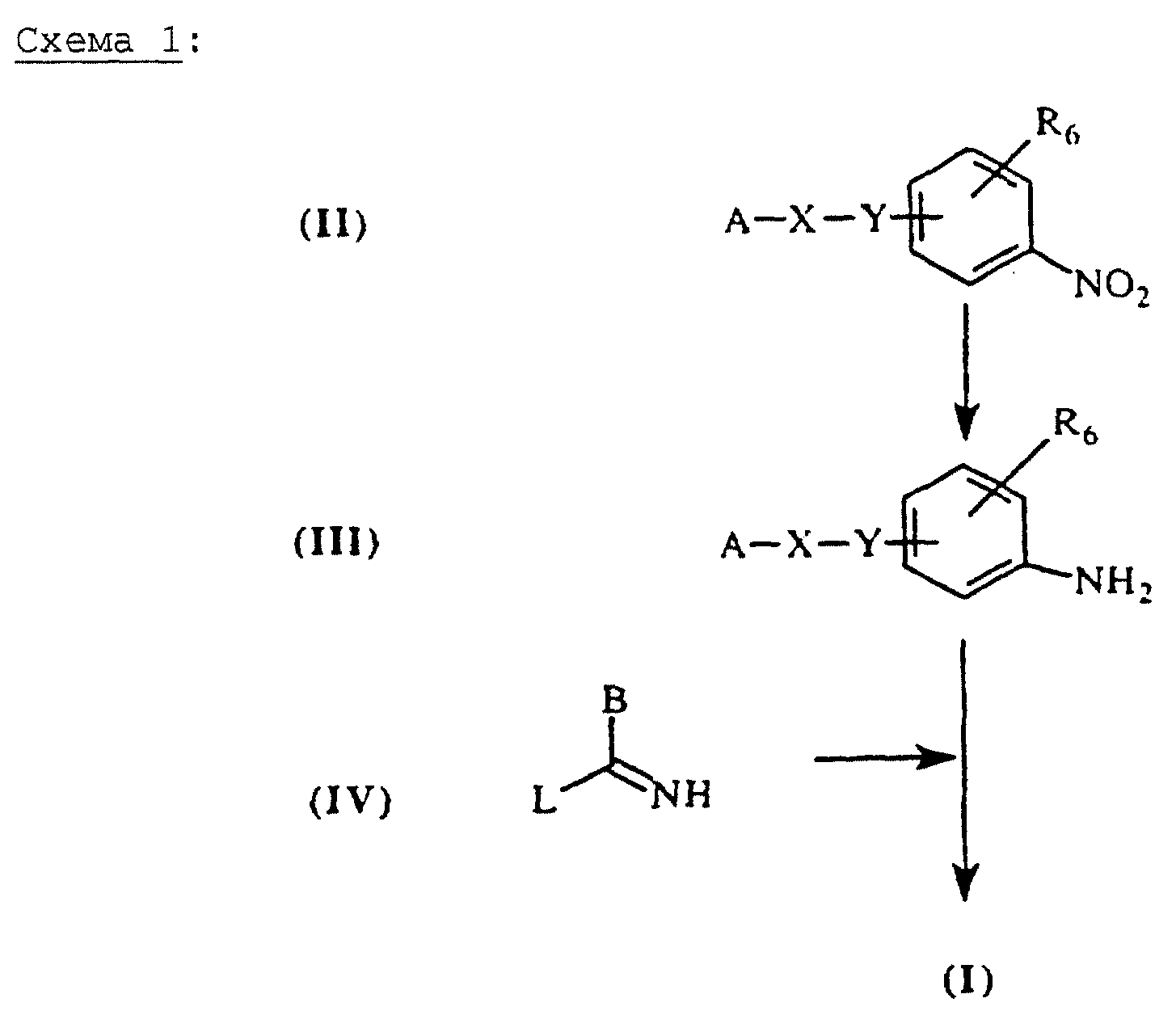
Соединения общей формулы (I) могут быть получены из промежуточных соединений общей формулы (II) согласно схеме 1 (см. в конце описания).
Восстановление нитрогруппы промежуточных соединений общей формулы (II) обычно осуществляют путем каталитического гидрирования в этаноле, в присутствии Pd/C (палладий на угле), за исключением случаев, когда X=-СН=СН-СО- или Y= -O-СН2-, селективно нитрогруппу восстанавливают с помощью, например, SnCl2 (J. Heterocyclic Chem. (1987), 24, 927-930; Tetrahedon Letters (1984), 25, (8), 839-842). Тогда реакцию осуществляют при нагревании смеси при температуре около 70oС в течение, по меньшей мере, трех часов, в этилацетате, к которому, при необходимости, добавляют этанол.
Производные анилина общей формулы (III), полученные таким образом, могут быть конденсированы с производными общей формулы (II), например, производными типа O-алкилтиоимидата или S-алкилтиомидата, чтобы получить конечные соединения общей формулы (I) (см. схема 1). Например, для В=тиофен, можно конденсировать производные общей формулы (III) с иодгидратом 3-метилтиофентиокарбоксамида, полученного по методике, описанной в литературе (Ann. Chim. (1962), 7, 303-337). Конденсацию можно осуществлять путем нагревания в спирте (например, в метаноле или изопропаноле), при необходимости, в присутствии ДМФ при температуре, предпочтительно 50-100oС в течение времени, обычно составляющего от нескольких часов до одной ночи.
Получение промежуточных соединений общей формулы (II):
Промежуточные соединения общей формулы (II) могут быть получены разными способами в зависимости от используемых функциональных
групп; амины, карбоксамиды, мочевины, тиомочевины, сульфонамиды, аминосульфонилмочевины, сульфамиды, карбаматы, эфиры, сложные эфиры, тиоэфиры, ацилмочевины и т.д.:
Если Х = линейный или
разветвленный алкилен с 1-6 атомами углерода, a Y = пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, -NR3-Z2-Q-, -NR3-NH-CO-Z2-, -NH-NH-Z2-, -NR3-O-Z2-.
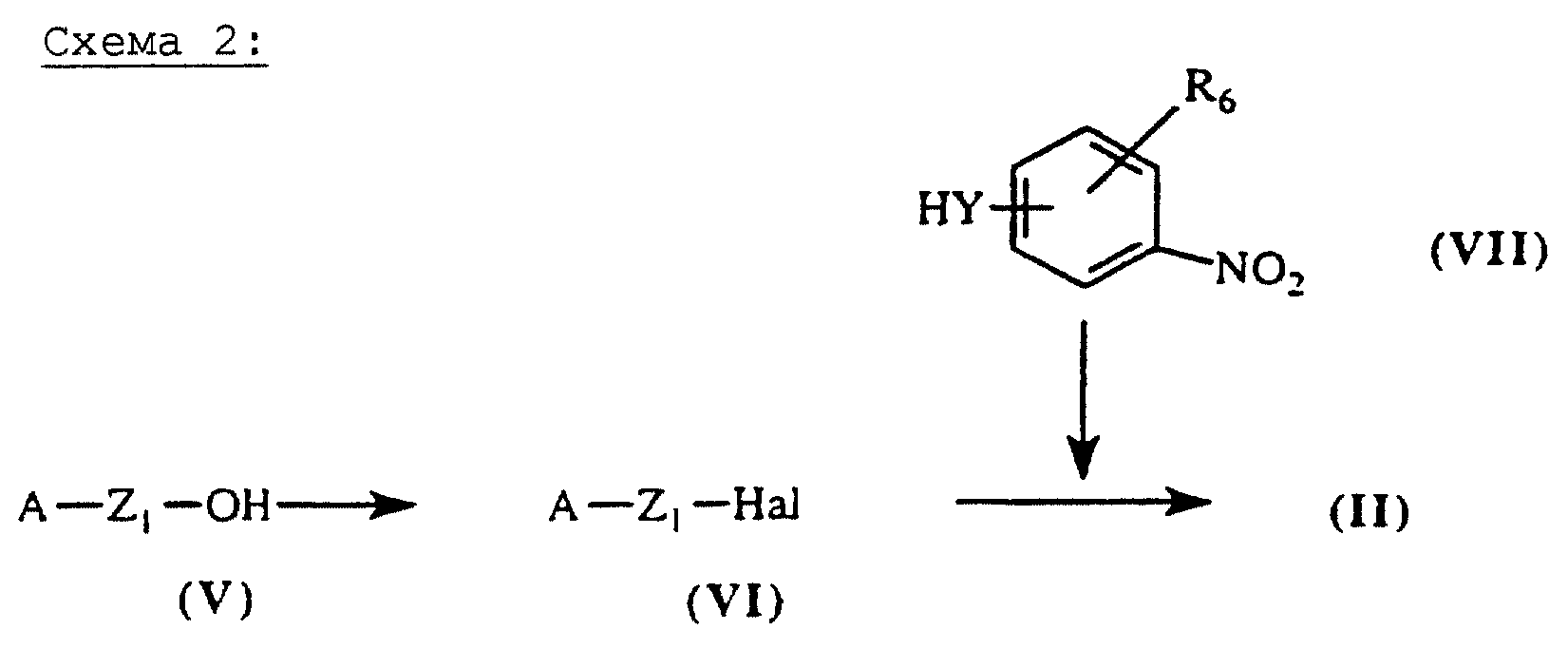
Амины общей формулы (II), схема 2 (см. в конце описания), в которых А, X, Y и R6 определены выше, могут быть получены путем нуклеофильного замещения галогенсодержащих производных общей формулы (VI) амином общей формулы (VII). Реакцию осуществляют, например, в ДМФ в присутствии К2СО3 при 20oС. Галогенсодержащие производные общей формулы (VI) являются доступными, их можно получить, например, в результате бромирования первичных спиртов общей формулы (V) с помощью РВr3 при 0oС в безводном ТГФ. Спирты формулы (V), не являющиеся коммерческими, могут быть получены согласно методикам, описанным в литературе (Tetrahedron Lett. (1983), 24, (24), 2495-2496).
Амины общей формулы (VII), в которых Y означает гомопиперазин, 2,5-диметилпиперазин, 4-аминопиперидин или, как правило, -NR3-Z2-NR3-, синтезируются в три этапа из соответствующих коммерческих диаминов. Диамины селективно моно - защищают в форме карбаматов (Synthesis (1984), (12), 1032-1033; Synth. Commun (1990), 20, (16), 2559-2564) перед взаимодействием путем нуклеофильного замещения на фторнитро-бензоле, в частности, 4-фторнитробензолом. Амины, предварительно защищенные, высвобождаются на последнем этапе, согласно методикам, описанным в литературе (T.W. Greene et P.G.M. Wuts, Protective Groups in Organic Synthesis, Second edition (Wiley-Interscience, 1991), чтобы получить промежуточные соединения общей формулы (VII).
Если Х = -Z1-CO-, -СН=СН-СО-, a Y = пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, -NR3-Z2-Q-, -NR3 -NH-CO-Z2-, -NH-NH-Z2-, -NR3-O-Z2-.
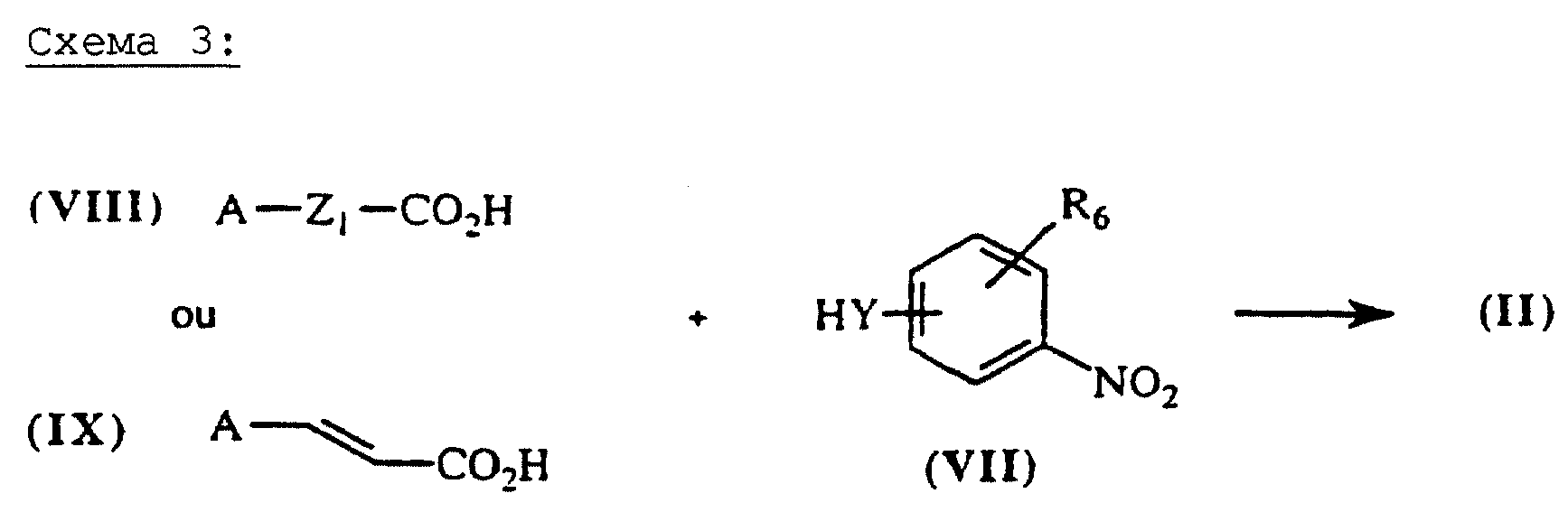
Карбоксамиды общей формулы (II), схема 3 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают путем конденсации коммерческих карбоновых кислот общей формулы ((VIII), если Х = -Z1-CO- и общей формулы (IX), если Х = СН-СН-СО- с аминами общей формулы (VII). Некоммерческие кислоты могут быть синтезированы согласно методикам, аналогичным тем, которые описаны в литературе (J.Org.Chem. (1974), 39(2) 219-222; J.Amer.Chem. Soc. (1957), 79, 5019-5023, et CHIMIA (1991), 45 (4), 121-123, если А означает радикал 6-алкокси-2,5,7,8-тетраметилхроман. Амины общей формулы (VII), в которой Y означает гомопиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, или, как правило, NR3-Z2-NR3-, получают согласно методикам, аналогичным тем, которые описаны в предшествующем абзаце. Карбоксамидные связи образуются в классических условиях пептидного синтеза (М. Bodanszky et A.Bodanszky, The Practice of Peptide Synthesis, 145 (Springer-Verlag, 1984) в ТГФ, дихлорметане или ДМФ в присутствии сочетающего агента, такого как дициклогексилкарбодиимид (ДЦК), 1,1'-карбонилдиимидазол (КДИ) (J.Med.Chem. (1991), 35 (23), 4464-4472) или хлоргидрат 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC или WSCI) (John Jones, The chemical synthesis of peptides, 54 (Clarendon Press, Oxford, 1991)).
Eсли X = -Z1-NR3-CO-, a Y = -Z2-Q-.
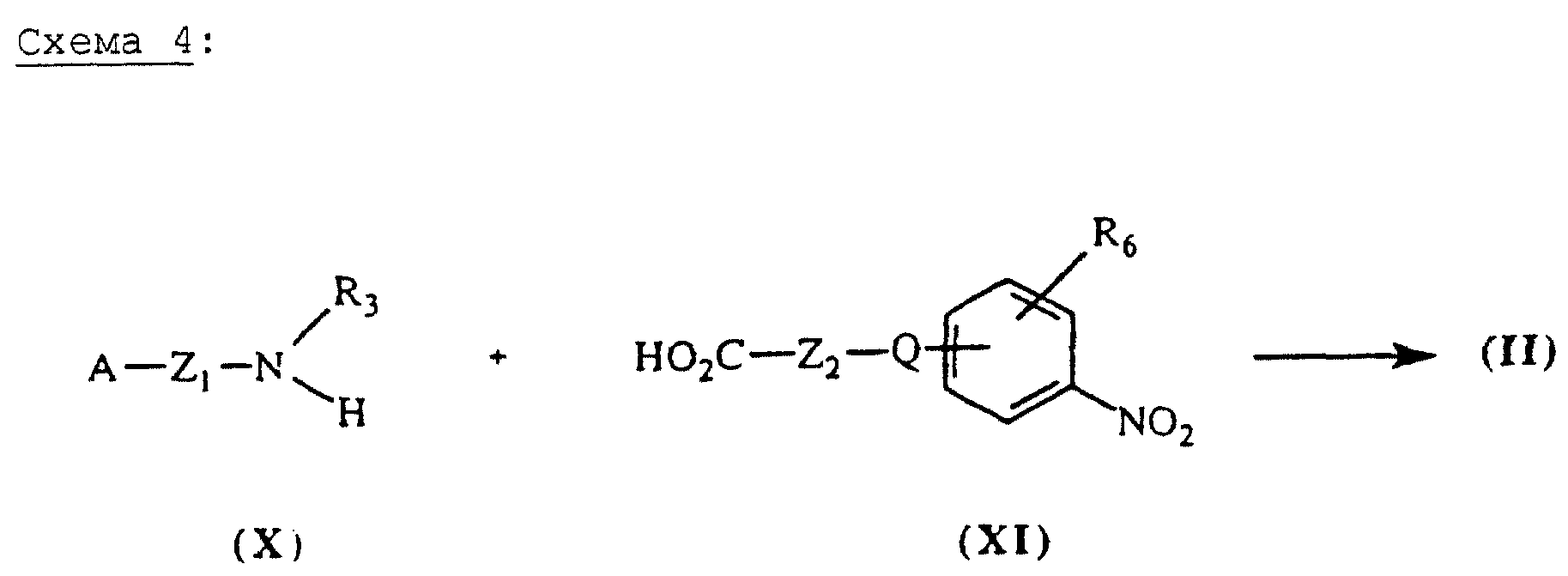
Карбоксамиды общей формулы
(II), в которых А, X, Y и R6 определены выше, могут быть также получены (схема 4, см. в конце описания) путем пептидной конденсации амина общей формулы (X) с коммерческой кислотой общей
формулы (XI). Если Х=-NR3-CO, а R3=Н, соединениями общей формулы (X) являются анилины, которые получают путем гидрирования в присутствии каталитического количества палладия на
угле (Pd/C), соответствующих нитробензольных производных, которые сами синтезируются согласно методике, описанной в литературе (J. Org. Chem. (1968), 33(1), 223-226). Если Х≠-NR3
-CO-, a R3 - линейный или разветвленный алкил с 1-6 атомами углерода, моноалкиламины могут быть получены согласно способу, описанному в американских патентах
3,208,859 и 2,962,531.
Некоммерческие карбоновые кислоты общей формулы (XI) могут быть получены согласно методикам, описанным в литературе: Acta Chem. Scand. (1983), 37, 911-916; Synth.Cononun. (1986), 16 (4), 479-483;
Phophorus, Sulfur Silicon Relat. Elem. (1991), 62, 269-273).
Если Х = -Z1-NR3-CO-, a Y = -NH-Z2-Q-, -NH-CO-Z2-Q-, Q = O-Z3-, R3-N-Z3 или S-Z3-.
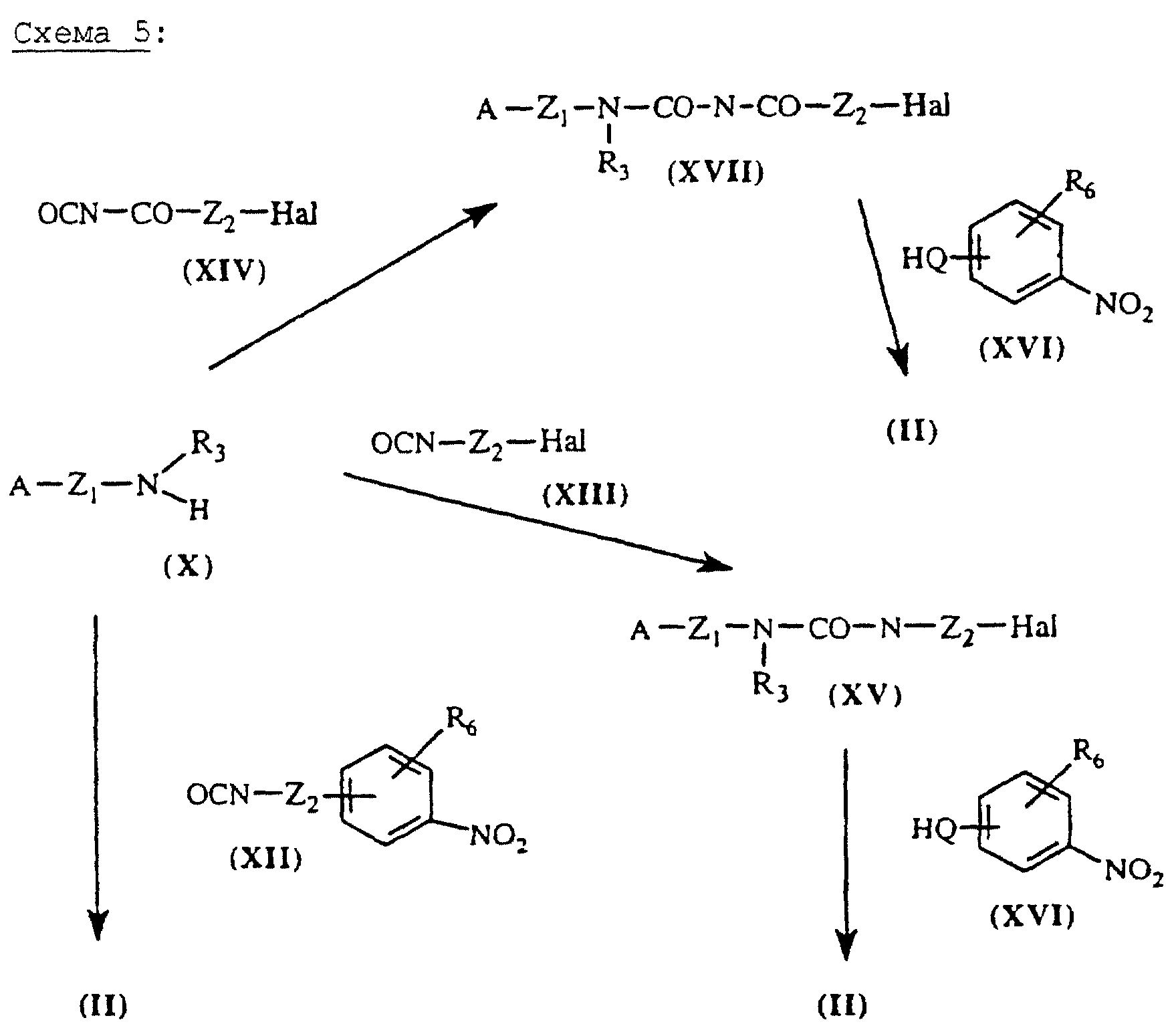
Мочевины общей формулы (II), схема 5 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают путем присоединения амина общей формулы (X) к изоцианату общей формулы (XII), (XIII) или (XIV) в растворителе, таком как хлороформ при 20oС. Синтез некоммерческих изоцианатов общей формулы (XII) описан в литературе (J.Med.Chem. (1992), 35, (21), 3745-3754). Промежуточные галогенсодержащие мочевины (XV) и (XVII) затем замещаются производным общей формулы (XVI), в котором Q означает O-Z3-, R3-N-Z3- или S-Z3-, в присутствии основания, такого как, например, К2СО3 или NaH, в апротонном растворителе, таком как ТГФ или ДМФ, с получением целевого продукта - мочевины общей формулы (II).
Если X = -Z1-NH-CO-, a Y = пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, -NR3-Z2-Q-, -NR3 -NH-CO-Z2-, -NH-NH-Z2-, -NR3-O-Z2-.
Мочевины общей формулы (II), схема 6 (см. в конце описания), в которых А, Х, Y и R6 определены выше, получают путем присоединения амина общей формулы (VII), описанной выше, к изоцианату общей формулы (XVIII) в присутствии основания, такого как диизопропилэтиламин.
Изоцианаты общей формулы (XVIII) синтезируют из первичных аминов общей формулы (X), описанных выше, трифосгена и третичного амина (J.Org.Chem. (1994), 59 (7), 1937-1938).
Амины общей формулы (VII), в которых Y≠-NH-O-, получают согласно методике, описанной в литературе (J.Org.Chem. (1984), 49 (8), 1348-1352).
Если X = -Z1-NR3-CO-, а Y = -NR3-SO2-NR3-Z2-.
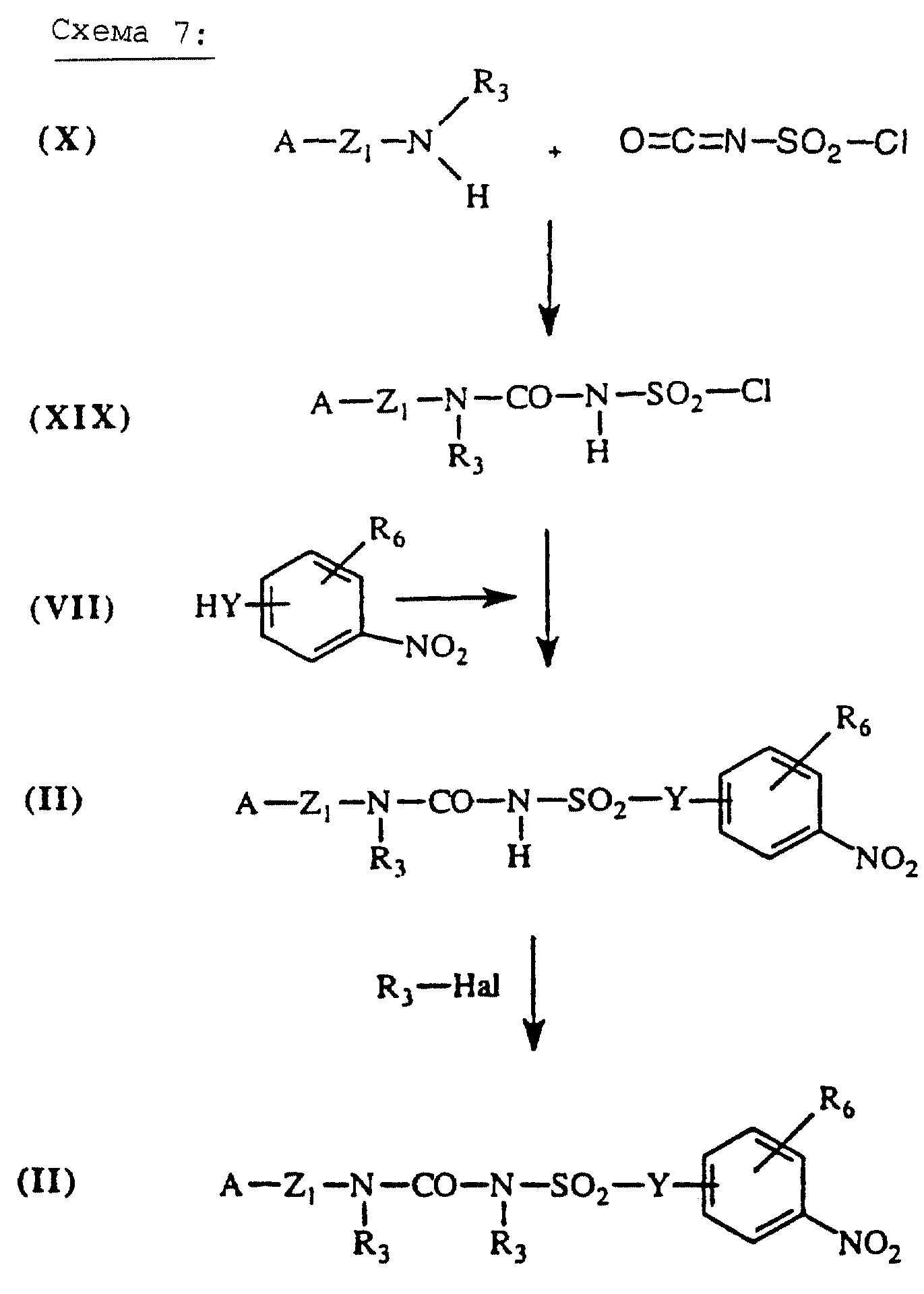
Аминосульфонилмочевины общей формулы (II), схема 7 (см. в конце описания), в которых А, X, Y и Re определены выше, получают путем присоединения аминов общей формулы (X), описанных выше, к хлорсульфонилизоцианату (J.Med. Chem. (1996), 39 (6), 1243-1252). Промежуточную хлорсульфонилмочевину (XIX) затем конденсируют с аминами общей формулы (VII), описанной выше, с получением аминосульфонилмочевин общей формулы (II), которые, при необходимости, могут быть алкилированы с помощью галогенсодержащего производного в присутствии основания, например, такого как NаН, с получением производных общей формулы (II).
Если Х = -Z1-NR3-SO2-, a Y = -Z2-Q-, с Q = O-Z3-, R3-N-Z3- или S-Z3-.
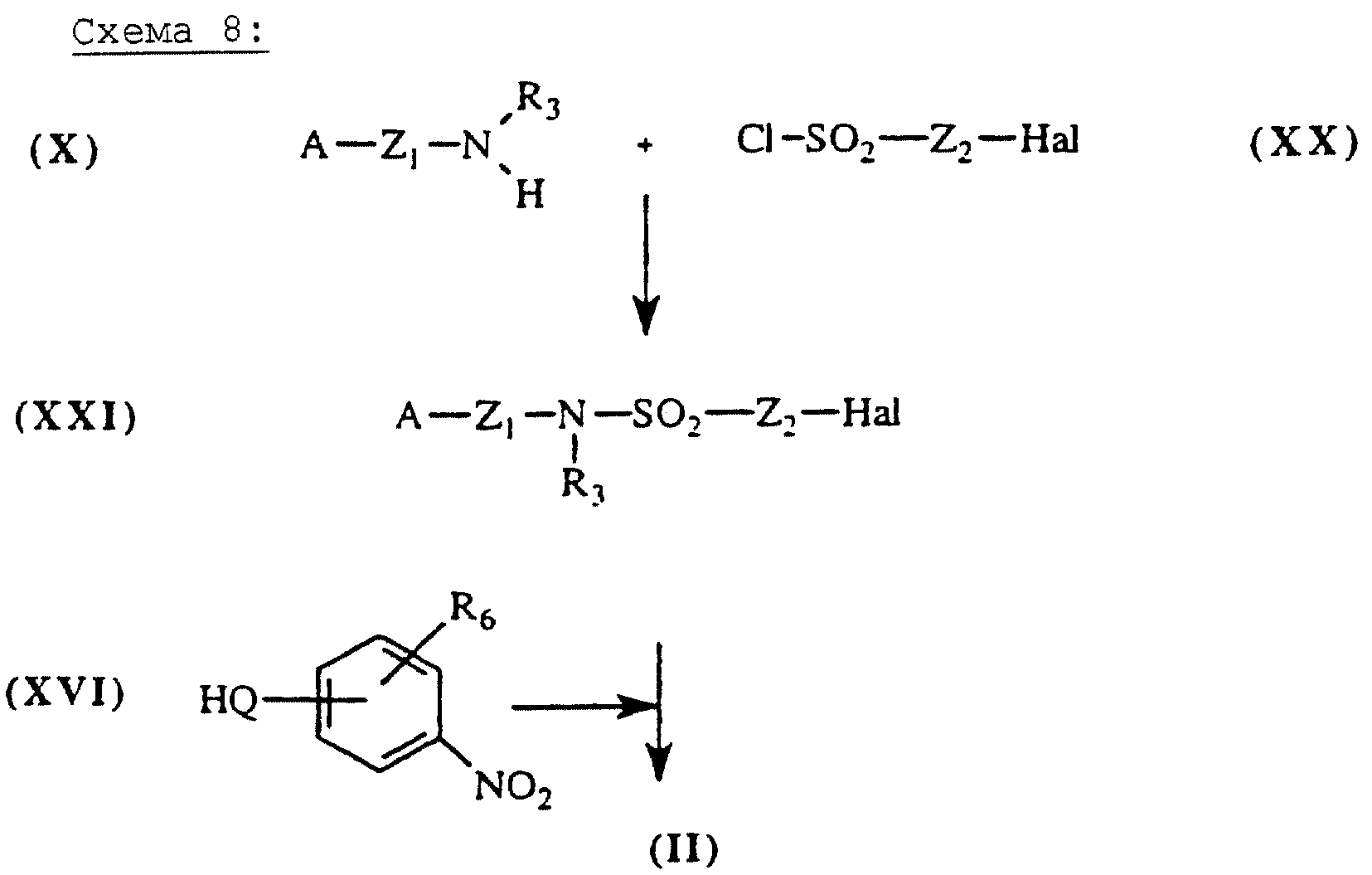
Сульфонамиды общей формулы (II), схема 8 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают путем присоединения аминов общей формулы (X), описанных выше, к хлоридам галогеналкилсульфонила общей формулы (XX). Галогеналкилсульфонамиды общей формулы (XXI), полученные на промежуточной стадии, затем конденсируют со спиртом, амином или тиолом общей формулы (XVI) в присутствии основания, такого например, как К2СО3 или NaH, в полярном растворителе, таком как ацетонитрил или ДМФ.
Если X = -Z1-NR3-SO2-, a Y = -NR3-Z2-Q-.
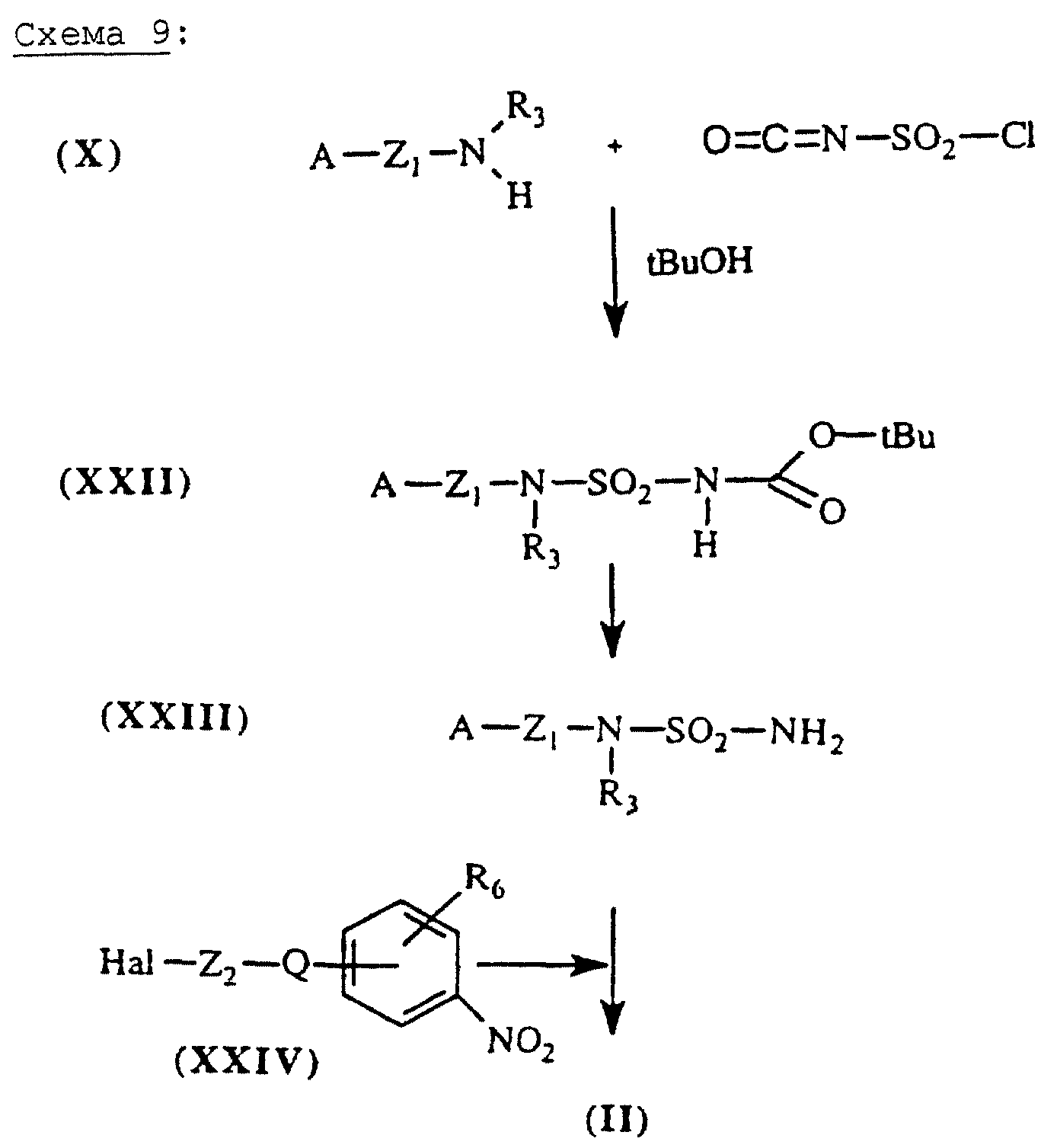
Сульфамиды общей формулы (II), схема 9 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают в три этапа из аминов общей формулы (X) и хлорсульфонилизоцианата. Взаимодействие спирта, такого как tBuOH, с изоцианатной функцией хлорсульфонилизоцианата (Tetrahedron Lett. (1991,), 32 (45), 6545-6546) приводит к промежуточному соединению типа хлорсульфонилкарбамата, которое подвергают взаимодействию с амином общей формулы (X), чтобы получить производное типа карбоксилсульфамида общей формулы (XXII). Обработка этого промежуточного соединения в кислой среде позволяет получить сульфамидное производное общей формулы (XXIII). Алкилирование соединений общей формулы (XXIII) с помощью галогенсодержащих производных общей формулы (XXIV) в присутствии основания, такого как, например NaH, в апротонном полярном растворителе позволяет получить сульфамидные производные общей формулы (II).
Если Х = -Z1-NR3-CO-, a Y = -O-Z2-Q-.
Карбаматы общей формулы (II), схема 10 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают путем взаимодействия аминов общей формулы (X), описанных выше, с хлорформиатными производными общей формулы (XXV), полученными согласно методике, описанной в литературе (Tetrahedron Lett. (1993), 34 (44), 7129-7132).
Если Х = -Z1-CO-, -СН=СН-СО-, а Y = -O-Z2-Q-.
Сложные эфиры общей формулы (II), схема 11 (см. в конце описания), в которых А, X, Y и R6 определены выше, получают путем взаимодействия кислот общей формулы (VIII) или (IX) и спиртов общей формулы (XXVI) в присутствии дициклогексилкарбодиимида и каталитического количества 4-диметиламинопиридина в таком растворителе, как, например, ТГФ или ДМФ при 20oС.
Если X = -Z1-, a Y = -O-CO-Z2-Q-.
Сложные эфиры общей формулы (II), схема 12 (см. в конце описания), в которых А, X, Y и R6 определены выше, могут быть также получены путем взаимодействия кислот общей формулы (XI), описанных выше, со спиртами общей формулы (V) в условиях, описанных выше.
Если Х = -Z1-NR3-CS-, a Y = -NH-Z2-Q-, пиперазин, гомопиперазин, 2-метилпиперазин, 2,5-диметилпиперазин, 4-аминопиперидин, -NR3-Z2-Q-, -NH-NH-Z2, NR3-O-Z2-.
Тиомочевины общей формулы (II), в которых А, Х, Y и R6 определены выше, получают из мочевин, описанных выше, с помощью реагента Lawesson, согласно экспериментальному протоколу, описанному в литературе (J.Med.Chem. (1995), 38 (18), 3558-3565).
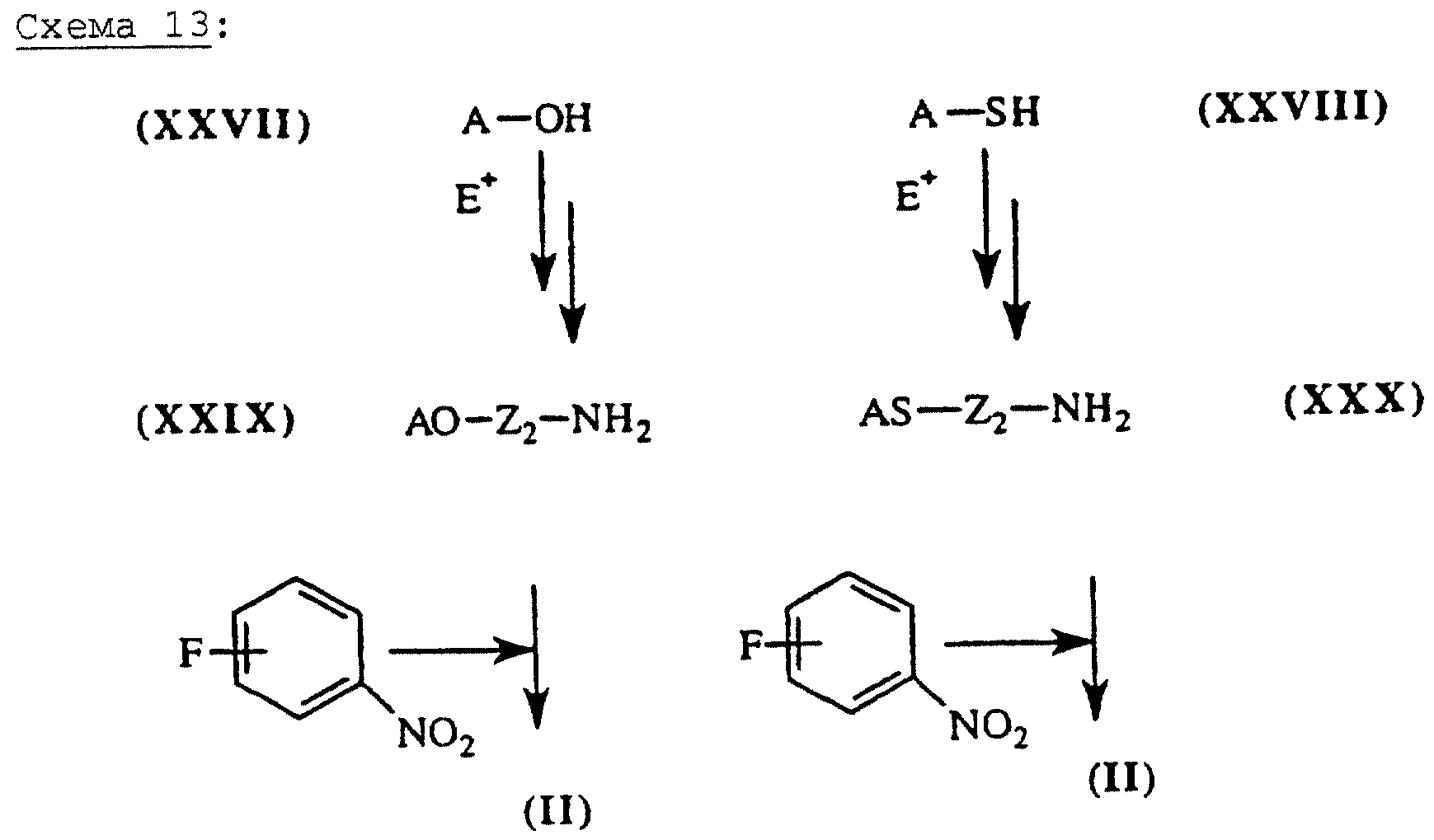
Если Х означает одну связь, a Y = -O-Z2-Q-, -S-Z2-Q-, a Q = -HN-.
Эфироксиды или тиоэфироксиды общей формулы (II), схема 13 (см. в конце описания), в которых А, X, Y и
R6 описаны выше, получают из дигидрохинонов общей формулы (XXVII) (J.Chem. Soc., Perkin Trans, I, (1981), 303-306) или тиофенолов общей формулы (XXVIII) (Bio.Med.Chem. Letters. (1993), 3
(12), 2827-2830) и электрофильного агента (Е+), например такого, как бромацетонитрил или 4-нитрофенилоксазолинон, в присутствии К2СО3 (J.Heterocyclic Chem., (1994), 31,
1439-1443). Нитрилы должны быть восстановлены (гидрид лития или каталитическая гидрогенизация), чтобы получить промежуточные соединения общей формулы (XXIX) или (XXX). Открытие
нитрофенилоксазолинонов, полученных путем взаимодействия соответствующих нитроанилинов с хлорэтилхлорформиатом, как описано в литературе (J. Am. Chem. Soc., (1953), 75, 4596), с помощью фенолов или
тиофенолов приводит непосредственно к соединениям общей формулы (XXIX) или (XXX), которые затем конденсируют с фторнитробензолом, чтобы получить промежуточные соединения общей формулы (II):
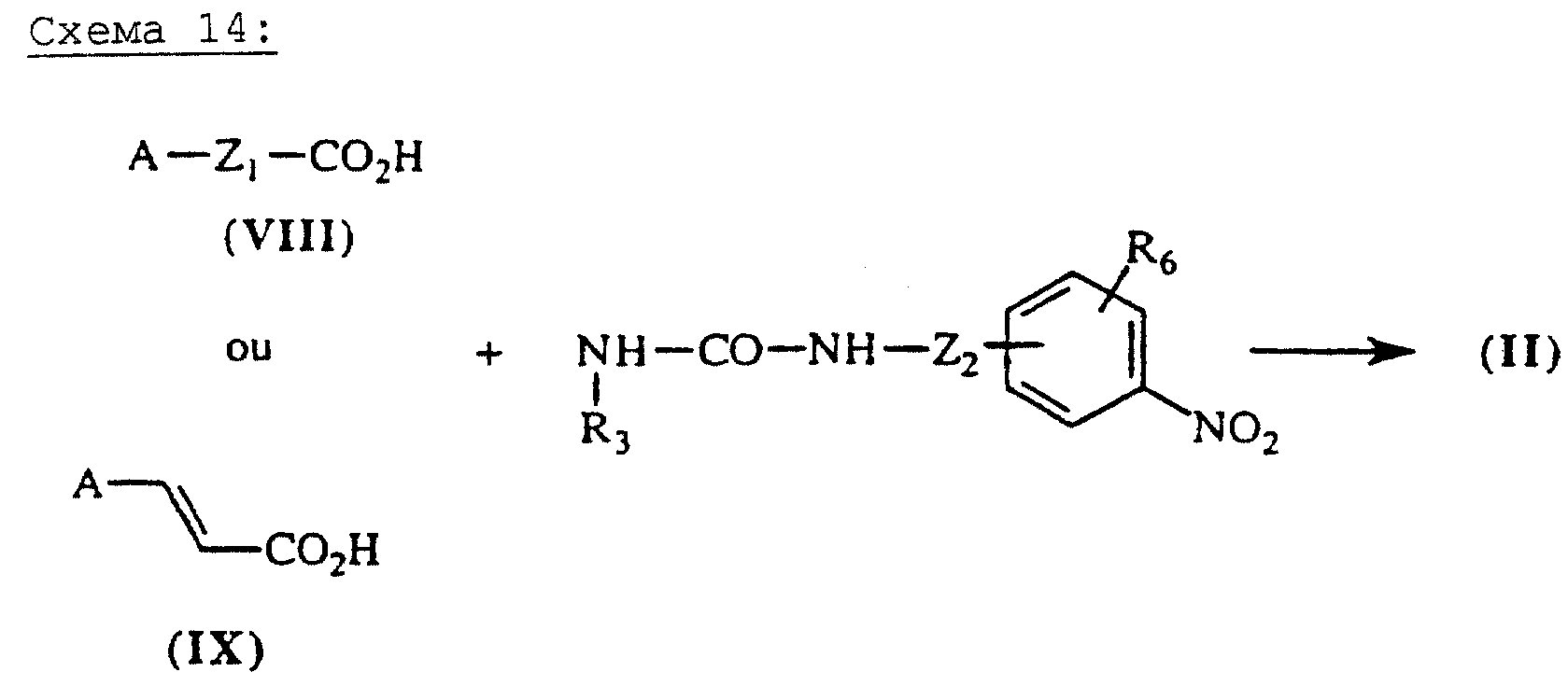
Если X означает -Z1-CO- или -СН=СН-СО-, Y = -NR3-СО-Q-, a Q = R3-N-Z3.
Ацилмочевины общей формулы (II), схема 14 (см. в конце описания), в которых А, X, Y и R6 описаны выше, получают путем конденсации кислот общей формулы (VIII) или (IX), схема 3, и мочевин общей формулы (XXXI) в присутствии агента сочетания, традиционно используемого в синтезе пептидов, как описано выше, в растворителе, таком как дихлорметан или ДМФ. Мочевины общей формулы (XXXI) получают из изоцианатов общей формулы (XII), схема 5, согласно методике, описанной в литературе (J.Chem.Soc., Perkin Trans, I.(1985), (1), 75-79).
Если не дается иных определений, то все технические и научные термины, используемые в данном описании, имеют общепринятые значения, понятные среднему специалисту той области, к которой принадлежит данное изобретение. Все указанные здесь публикации, патентные заявки, патенты и другие ссылочные материалы, включены в качестве справочной литературы.
Ниже представлены примеры, которые иллюстрируют описанные выше процессы и которые не должны рассматриваться как ограничивающие объем изобретения.
Примеры:
Пример 1: хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{4 -[(2-тиенил-(имино)метил)амино]фенил}-бензамида: 1
1.1) 3,5-бис(1,
1-диметилэтил)-4-гидрокси-N-(4-нитрофенил)-бензамид
В колбу емкостью 250 мл, содержащую 20 мл тетрагидрофурана, вводят 1,38 г (10 ммоль) 4-нитроанилина, 2,5 г (10 ммоль) 3,
5-ди-трет-бутил-4-гидроксибензойной кислоты, 2,26 г (11 ммоль) дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при комнатной температуре, появившийся осадок фильтруют и
промывают этилацетатом. После концентрации раствора при пониженном давлении остаток разбавляют в 20 мл этилацетата, а нерастворимое вещество отфильтровывают. Растворитель удаляют под вакуумом, а
остаток осаждают в диэтиловом эфире. Твердое вещество извлекают путем фильтрации, обильно промывают в диэтиловом эфире для получения белого порошка с выходом 65%. Температура плавления: 277-278oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 10,72 (с, 1Н, CONH); 8,30 (м, 4Н,
1.2) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамид:
В сосуде Парра емкостью 250 мл растворяют 2,4 г (6,5
ммоль) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-нитрофенил)-бензамида в 50 мл смеси абсолютный этиловый спирт/дихлорметан (1/1) в присутствии 10%-ного палладия на угле (Pd/C). Смесь перемешивают при
давлении водорода 20 PSI, при 30oС, в течение одного часа. После фильтрации на целите, фильтрат концентрируют в вакууме. Остаток после выпаривания обрабатывают с помощью 25 мл раствора 1М
НСl. Образовавшийся осадок фильтруют и промывают с помощью 50 мл диэтилового эфира, затем с помощью 50 мл этилацетата. Амин высвобождается из своей соли путем перемешивания в смеси 50 мл этилацетата и
50 мл мл NaOH 1M. После декантации органическую фазу промывают с помощью 25 мл NaOH 1M и 25 мл рассола. Органический раствор сушат над сульфатом натрия, фильтруют, промывают и концентрируют досуха при
пониженном давлении, и получают 1,09 г (49%) белого порошка. Температура плавления: 220-221oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 9,80 (с, 1Н, CONH); 7,78 (с, 2Н,
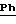
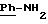
1.3) Хлоргидрат 3,5-бис-(1,1-димвтилэтил)-4-гидрокси-N-{4-[-2-тиенил-(имино)метил)амино]фенил}-бензамида: 1
В колбу емкостью
100 мл, содержащую раствор 1,05 г (3,08 ммоль) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил) -бензамида в 20 мл 2-пропанола, добавляют 880 мг (3,08 ммоль) иодгидрата
S-метил-2-тиофентиокарбоксимида (Ann. Chim. (1962), 7, 303-337). После нагревания при 50oС в течение 15 часов, реакционную смесь концентрируют досуха в вакууме. Остаток обрабатывают с
помощью 50 мл этилацетата и 50 мл насыщенного раствора карбоната натрия. После декантации органическую фазу промывают последовательно с помощью 50 мл насыщенного раствора карбоната натрия, 50 мл воды
и 50 мл рассола. Органический раствор сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Полученные кристаллы обрабатывают диэтиловым эфиром, фильтруют и промывают
последовательно этилацетатом и ацетоном. Получают 0,77 г основания с выходом 58%.
Хлоргидрат получают из 0,77 г (1,71 ммоль) основания, растворенного в 60 мл метанола и превращенного в соль в присутствии 3,42 мл (3,42 ммоль) молярного раствора НСl в безводном диэтиловом эфире. После перемешивания в течение получаса при комнатной температуре растворитель выпаривают в вакууме и остаток осаждается в присутствии диэтилового эфира. Полученные кристаллы фильтруют и промывают большим количеством диэтилового эфира, чтобы получить после сушки 0,65 г (43%) бледно-желтого порошка. Температура плавления: 290-291oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,55 (с, 1Н, NH+); 10,40 (с, 1Н, CONH); 9,83 (с, 1Н, NH+); 8,85 (с, 1Н, NH+); 8,21 (м, 2Н, тиофен); 7,70 (с, 2Н, Ph); 7,67 (м, 4Н,
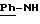
ИК: νOH: 3624 см-1, 3430 см-1; νC=O(амид): 1653 см-1; νC=N (амидин): 1587 см-1.
Пример 2: хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{4-[(2-тиенил-(имино)метил)амино]фенил)метил}-бензамида: 2
2.1) 3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил) метил] -бензамид:
В колбу емкостью 250 мл, содержащую 25 мл тетрагидрофурана, вводят 1,88 г (10 ммоль) хлоргидрата п-нитробензиламина, 2,5 г (10
ммоль) 3,5-ди-трет-бутил-4-гидроксибензойной кислоты, 1,38 г (10 ммоль) триэтиламина и 2,26 г (11 ммоль) дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при комнатной
температуре, появившийся осадок фильтруют и промывают минимальным количеством этилацетата. После концентрации раствора при пониженном давлении остаток осаждают в смеси этилацетат/диэтиловый эфир (1/4)
и фильтруют. Кристаллы обильно промывают в диэтиловом эфире, чтобы получить конечный белый порошок с выходом 74% (2,85 г). Точка плавления: 230-231oС.
ЯМР1H (100 МГц, CDCl3 δ): 7,85 (м, 4Н, Ph-NO2 ); 7,69 (с, 2Н,
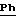
2.2) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N- [(4-аминофенил) метил]-бензамид:
В сосуде Парра
емкостью 250 мл растворяют 2,85 г (7,4 ммоль) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил]-бензамида в 30 мл смеси абсолютного этанолового спирта/дихлорметана (1/1) в присутствии
10%-ного палладия на угле. Смесь перемешивают при давлении водорода 20 PSI, при 30oС, в течение одного часа. После фильтрации на целите фильтрат концентрируют в вакууме. Продукт выпаривания
самопроизвольно кристаллизуется. Его оставляют на одну ночь, фильтруют кристаллы и промывают смесью диэтилового эфира (45 мл) и ацетона (5 мл). Получают 1,63 г (62%) белого порошка. Температура
плавления: 188-189oС.
ЯМР1H (100 МГц, CDC13, δ): 7,62 (с, 2Н, Ph); 6,95 (м, 4Н,
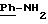
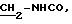
2.3) хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{4-[[(2-тиенил-(имино)метил)амино]фенил]метил}-бензамида: 2
Работают так
же, что описано для соединения 1, причем используют 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-амино-фенил) метил] -бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида.
После солеобразования с помощью молярного раствора НСl в безводном диэтиловом эфире, получают белый порошок с выходом 56%. Температура плавления: 218-219oС.
ЯМР1 H (400 МГц, ДМСО d6, δ): 11,60 (с, 1Н, NH+); 9,83 (с, 1H, NH+); 9,02 (с, 1Н, CONH); 8,90 (c, 1H, NH+); 8,18 (м, 2Н, тиофен); 7,70 (с, 2Н, Ph); 7,42 (м, 6Н, тиофен,
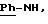
ИК; νOH: 3624 см-1, 3424 см-1; νC=O(амид): 1644 см-1; νC=N(амидин): 1568 см-1.
Пример 3:
4-ацетокси-3,5-диметокси-N-{4-[[(2-тиенил(имино) метил)амино] фенил]метил}-бензамид: 3
3.1) 4-ацетокси-3,5-диметокси-бензойная кислота:
В сосуде емкостью 100 мл, в атмосфере азота,
растворяют 1,50 г (7,57 ммоль) сиреневой кислоты в 15 мл сухого пиридина. Добавляют по капле 0,86 мл (9,08 ммоль) уксусного ангидрида и смесь перемешивают при комнатной температуре в течение 18 часов.
Пиридин испаряют при пониженном давлении, остаток обрабатывают с помощью 25 мл дихлорметана и промывают с помощью 10 мл молярного раствора НСl, затем, с помощью 2 • 10 мл воды. Органическую
фазу сушат над сульфатом натрия, фильтруют и испаряют в вакууме. Получают 1,72 г (95%) бежевого порошка. Температура плавления: 181-183oС.
ЯМР1H (100 МГц, СОСl3, δ): 8,15 (с, 1Н, СО2Н); 7,40 (с, 2Н, Ph); 3,90 (с, 6Н, 2•ОСН3); 2,40 (с, 3Н, СН3).
3.2) 4-ацетокси-3,
5-диметокси-N-[(4-нитрофенил)метил]-бензамид:
Работают так же, как при получении промежуточного продукта 2,1, используют 4-ацетокси-3,5-диметокси-бензойная кислота вместо 3,
5-ди- трет-бутил-4-гидрокси-бензойной кислоты. Получают бесцветное масло с выходом 28%.
ЯМР1H (100 МГц, ДМСО d6, δ): 9,26 (т, 1Н, NHCO, J=6,0 Гц); 7,91 (м, 4Н, Ph-NO2); 7,31 (с, 2Н, Ph); 4,65 (д, 2Н, CH2, J=6,0 Гц); 3,83 (с, 6Н, 2•ОСН3); 2,28 (с, 3Н, СН3).
3.3) 4-ацетокси-3,
5-диметокси-N-[(4-аминофенил)метил]-бензамид
Работают так же, как при получении промежуточного продукта 2.2, причем используют 4 - ацетокси - 3,5 - диметокси
-N-[(4- нитрофенил)метил]-бензамид вместо 3,5-бис-(1,1-диметилэтил)-4- гидрокси-N-[(4-нитрофенил)метил] -бензамида. Получают бесцветное масло с выходом 82%. Продукт используют непосредственно на
следующем этапе без дополнительной очистки.
3.4) 4-ацетокси-3,5-диметокси-Н-{4-[[(2-тиенил(имино)метил)амино]фенил] метил}-бензамид: 3
Работают так же, как и при получении
соединения 1, причем используют 4-ацетокси-3,5-диметокси-N-[(4-аминофенил)метил] -бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Получают основание 3 в виде бежевого
порошка с выходом 65%. Температура: 47-48oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 9,08 (с широкий, 1Н, CONH); 7,75 (м, 1Н, тиофен); 7,62 (м, 1Н, тиофен); 7,30 (с, 2Н, Рh); 7,10 (м, 1Н, тиофен); 7,07 (м, 4Н, Ph-N); 6,48 (с широкий, 2Н, NH2); 4,50 (д, 2Н, CH2, J=4,6 Гц); 3,80 (с, 6Н, 2•ОСН3); 2,30 (с, 3Н, СН3).
ИК: νC=O(эфир): 1760 см-1, νC=O(амид): 1630 см-1; νC=N(амидин): 1540 см-1.
Пример 4: 3,5-диметокси-4-гидрокси-N-(4-[[(2-тиенил(имино) метил)амино]
фенил]метил}-бензамид: 4
В колбу емкостью 50 мл добавляют по капле 1 мл (2 моля) 2N соляной кислоты в
раствор 0,59 г (1 моль) соединения 3 в 5 мл этанола. Реакционную смесь перемешивают в течение 18 часов при 50oС. Растворители выпаривают досуха, остаток обрабатывают дихлорметаном (5 мл) и
промывают молярным раствором щелочи натрия (3•5 мл). После сушки органической фазы, фильтрации и концентрации досуха, полученное масло очищают с помощью хроматографии на силикагеле (элюируя
смесью дихлорметан/метанол: 9/1). Чистые фракции собирают и после выпаривания в вакууме получают бежевый порошок с выходом 60%. Температура плавления: 55-58oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 8,92 (с, 1Н, ОН); 8,84 (м, 1Н, CONH); 7,75 (м, 1Н, тиофен); 7,63 (м, 1Н, тиофен); 7,26 (с, 2Н, Ph); 7,10 (м, 1Н, тиофен), 7,05 (м, 4Н, Ph-N); 6,50 (с, 2Н, NH2); 4,45 (д, 2Н, СН2, J=5,7 Гц); 3,81 (с, 6Н, 2 • ОСН3).
ИК: νOH: 3600 см-1; νC=O(амид): 1630 см-1; νC=N(амидин): 1590 см-1.
Пример 5: иодгидрат 3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-{4-[2-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамида: 5
5.1) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-нитрофенил) этил] -бензамид:
В колбу
емкостью 100 мл, содержащую 20 мл тетрагидрофурана, вводят 2,02 г (10 ммолей) хлоргидрата 4-нитрофенетиламина, 2,5 г (10 ммолей) 3,5-ди-трет-бутил-4-гидрокси-бензойной кислоты, 1,38 г (10 ммолей)
триэтиламина и 2,26 г (11 ммолей) дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при комнатной температуре, появившийся осадок фильтруют и промывают в этилацетате. После
концентрации фильтрата при пониженном давлении остаток осаждают в диэтиловом эфире. Твердое вещество извлекают с помощью фильтрации и промывают в диэтиловом эфире. Получают белый порошок с выходом
73%. Точка плавления: 204-206oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,52 (с, 2Н, Ph); 6,85 (м, 4Н, Ph-NO2); 6,02 (м, 1Н, NHCO); 3,62 (м, 2Н,
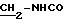
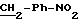
5.2) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-амино-фенил) этил]-бензамид:
Работают так же, как при получении промежуточного соединения 2,2, причем
используют 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-нитрофенил)этил]-бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-нитрофенил)метил] -бензамида. Получают белый порошок с выходом
76%. Температура плавления: 193-195oС.
ЯМР1H (100 МГц, CDC13, δ): 7,80 (м, 4Н, Ph-NH2); 7,55 (с, 2Н, Ph); 6,10 (м, 1Н, NHCO); 5,55 (с, 1Н, ОН); 3,75 (м, 2Н,
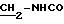
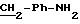
5.3) иодгидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 4-[2-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамида: 5
В
колбу емкостью 50 мл, содержащую 1,01 г (2,74 ммолей) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-аминофенил)этил] -бензамида, растворенного в 20 мл 2-пропанола, добавляют 0,78 г (2,74 ммолей)
иодгидрата 3-метил-2-тиофен-тиокарбоксимида (Ann.Chim. (1962), 7, 303-337). Реакционную смесь нагревают при 40oС в течение 4 часов. Растворитель выпаривают в вакууме, а остаток осаждают в
присутствии 50 мл смеси вода/этилацетат (1/1). Образованные кристаллы фильтруют и промывают последовательно этилацетатом и диэтиловым эфиром. После сушки получают светло-желтый порошок с выходом 68%.
Точка плавления: 185-186oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 9,80 (с, 1Н, NH+); 8,88 (с, 1Н, NH+); 8,40 (с, 1H, CONH); 8,12 (м, 2H, тиофен); 7,60 (с, 2Н, Ph); 7,42 (м, 6Н, тиофен,
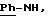
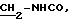
ИК: νOH: 3624 см-1, 3423 см-1; νC=O(амид): 1636 см-1; νC=N(амидин): 1569 см-1.
Пример 6: фумарат 4 -ацетокси-3,5-диметокси-N-{
4-[2-[(2-тиенил- (имино)метил)-амино]фенил]этил}-бензамида: 6
6.1) 4-ацетокси-3,5-диметокси-N-[2-(4-нитрофенил)этил]-бензамид:
Работают так же, как при получении промежуточного
соединения 5.1, причем используют 4-ацетокси-3,5-диметокси-бензойную кислоту (промежуточное соединение 3.1) вместо 3,5-ди-трет-бутил-4-гидрокси-бензойной кислоты. Получают бесцветное масло с выходом
70%. Продукт используют непосредственно на следующем этапе.
6.2) 4-ацетокси-3,5-диметокси-N-[2-(4-аминофенил)этил]-бензамид:
Работают так же, как при получении промежуточного
продукта 2.2, причем используют 4 - ацетокси -3,5-диметокси-N-[2-(4-нитрофенил) этил]-бензамид вместо 3,5-бис-(1,1-диметил-этил)-4-гидрокси-N-[(4-нитрофенил)метил] -бензамида. Получают бесцветное
масло с количественным выходом. Продукт используют непосредственно на следующем этапе без дополнительной очистки.
6.3) фумарат 4-ацетокси-3,
5-диметокси-N-{4-[2-[(2-тиенил- (имино)метил)амино)фенил]этил}-бензамида: 6
Для получения свободного основания работают так же, как при получении соединения 1, причем используют 4-ацетокси-3,
5-диметокси-N-[2-(4-аминофенил)этил] -бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил) -бензамид. Полученный продукт реакции превращают в соль в присутствии эквимолярного
количества фумаровой кислоты в этаноле при нагревании с обратным холодильником. Получают соединение 6 в виде бежевого порошка с выходом 74%. Температура плавления: 178-180oС.
ЯМР1Н (400 МГц, ДМСО d6, δ): 8,60 (м, 1Н, CONH); 7,75 (м, 1Н, тиофен); 7,64 (д, 1Н, тиофен, J=5,0 Гц); 7,20 (с, 2Н, Ph); 7,11 (t, 2Н, тиофен, J=9,0 Гц); 7,02 (м, 4Н, Ph-N); 6,61 (с, 2Н, СН=СН фумарат); 3,81 (с, 6Н, 2 • ОСН3); 3,50 (кв, 2Н, СН2-N, J=6,5 Гц); 2,82 (т, CH2-Ph, J=7,0 Гц); 2,27 (с, Н, СН3).
ИК: νC=O(сложный эфир): 1750 см-1, νC=O(амид): 1640 см-1; νC=N(амидин): 1550 см-1.
Пример 7: хлоргидрат 3,5-дивютокси-4-гидрокси-N-{ 4-[2-[(2-тиенил-(имино)метил)-амино)фенил]этил}-бензамида: 7
В колбу емкостью 50 мл добавляют по капле 1,40 мл (2,80 ммолей) 2N раствора
соляной кислоты в раствор 0,64 г (1,37 ммолей) соединения 6 в виде свободного основания в 5 мл этанола. Реакционную смесь перемешивают в течение 18 часов при 50oС. Растворители выпаривают
досуха и продукт выпаривания осаждают в смеси из 5 мл 2N раствора щелочи едкого натрия и 10 мл дихлорметана. После фильтрации твердый продукт обрабатывают в хлористоводородном этаноле (4N). Удаляют
легкий осадок. Растворитель выпаривают при пониженном давлении, а остаток обрабатывают в ацетоне. Продукт 7 осаждают в виде хлоргидрата с выходом 58%.
Точка плавления: 164-1б7oС.
ЯМР1Н (400 МГц, ДМСО d6, δ): 9,80 (с, 1Н, NH+); 8,90 (с, 2Н, NH+, ОН); 8,54 (м, 1Н, CONH); 8,18 (с, 1Н, тиофен); 8,16 (с, 1Н, тиофен); 7,40 (м, 4Н, Ph-N); 7,21 (с, 2Н, Ph); 7,11 (м, 1Н, тиофен); 3,81 (с, 6Н, 2•ОСН3); 3,51 (кв, 2Н, CH2-N, J=7,0 Гц); 2,92 (т, 3Н,
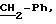
ИК: νOH: 3300 см-1; νC=O(амид): 1620 см-1; νC=N(амидин): 1560 см-1.
Пример 8: полуфумарат 3,4,5-тригидрокси-N-{4-[2-[(2-тиенил- (имино)метил)-амино]фенил]этил}-бензамида: 8
8.1) 3,4,
5-тригидрокси-N-[2-(4-нитрофенил)этил]-бензамид:
В колбу емкостью 100 мл, содержащую 30 мл безводного диметилформамида, вводят 2 г (11,5 ммолей) галловой кислоты, 2,5 г (11,5 ммолей)
хлоргидрата 4-нитрофенетиламина, 1,8 г (11,5 ммолей) гидратированного 1-гидроксибензотриазола, 2,25 г (11,5 ммолей) хлоргидрата 1-(3-диметиламинопропил)-3-этилкарбодиимида и 3,3 мл (23 ммоля)
триэтиламина. Полученный раствор оранжевого цвета перемешивают при 20oС в течение 20 часов и разбавляют в смеси дихлорметана (50 мл) и воды (30 мл). После декантации органическую фазу
промывают молярным раствором соляной кислоты (20 мл) и водой (3 • 20 мл) до нейтрального состояния. После сушки органической фазы над сульфатом магния, фильтрации и концентрации в вакууме,
остаток очищают с помощью хроматографии на силикагеле (элюируя смесью дихлорметан/метанол: 9/1). Получают целевой продукт в виде бесцветного масла с выходом 42% (1,57 г).
ЯМР1H (100 МГц, ДМСО d6, δ): 8,95 (м, 3Н, 3 • ОН); 7,85 (м, 4Н, Ph-NO2); 6,80 (с, 2Н, Ph); 3,36 (м, 2Н, CH2-N); 2,97 (т, 2Н,
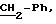
8.2) 3,4,5-тригидрокси-N-[2-(4-аминофенил)этил]-бензамид:
Работают так же, как при получении
промежуточного продукта 2.2, причем используют 3,4,5-тригидрокси-N-[2-(4-нитрофенил)этил]-бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил] -бензамид. Получают бежевый
порошок с выходом 89%. Точка плавления; 167-169oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 8,80 (м, 3Н, ОН); 8,07 (т, 1Н, NHCO, J=5,0 Гц); 6,81 (с, 2Н, Ph); 6,68 (м, 4Н, Ph-NH2); 3,28 (м, 2Н, CH2-N); 2,60 (т, 2Н,
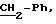
8.3) полуфумарат 3,4,5-тригидрокси-N-{4-[2-[(2-тиенил- (имино)метил)амино)-фенил]этил}-бензамида: 8
Работают так же, как при получении соединения 1, причем используют 3,4,
5-тригидрокси-N-[2-(4-аминофенил)этил] -бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Получают основание 8 в виде порошка, который превращают в соль путем нагревания
с обратным холодильником при температуре кипения этанола, в присутствии эквивалента фумаровой кислоты. Соль самопроизвольно кристаллизуется при 20oС. После фильтрации и промывания этанолом
получают целевой продукт в виде бежевого порошка с выходом 53%. Температура плавления: 245-246oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 8,85 (м, 3Н, 3 • ОН); 8,14 (т, 1Н, NHCO, J= 5,0 Гц); 7,73 (с, 1Н, тиофен); 7,60 (д, 1Н, тиофен, J=5,0 Гц); 7,16 (с, 2Н, Ph): 7,09 (т, 1Н, тиофен, J=4,0 Гц); 6,80 (м, 4Н, Ph-N); 6,59 (с широкий, 2Н, 1/2-СН= СН, NH); 3,41 (м, 3Н, СН2-N+NH); 2,76 (т, 2Н, CH2, J=7,5 Гц).
ИК: νOH: 3300 см-1; νC=O(амид): 1620 см-1; νC=N(амидин): 1590 см-1.
Пример 9: хлоргидрат -N-{ 4-[4-[3,5-бис-(1, 1-диметилэтил)-4-гидроксибензоил]-1-пиперазинил]-фенил}-2- тиофенкарбоксимидамид: 9.
9.1) 2,6-бис-(1,1-диметилэтил)-4-{ [4-(4-нитрофенил)-1-пиперазинил] -карбонил}-фенол:
В
колбу емкостью 100 мл, содержащую 25 мл ДМФ, вводят 2,07 г (10 ммолей) 1-(4-нитрофенил)пиперазина, 2,5 г (10 ммолей) 3,5-ди-трет-бутил-4-гидрокси-бензойной кислоты и 2,26 г (11 ммолей)
дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при комнатной температуре, появившийся осадок фильтруют и промывают в этилацетате. После концентрации фильтрата при
пониженном давлении остаток разводят в 20 мл этилацетата и новый нерастворимый продукт удаляют с помощью фильтрации. Растворитель выпаривают в вакууме, и остаток осаждают в диэтиловом эфире. Твердое
вещество фильтруют, промывают с помощью 2•20 мл этилацетата, чтобы получить порошок желтого цвета с выходом 89%. Температура плавления: 159,5-160,5oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,58 (м, 4Н, Ph-NO2); 7,30 (с, 2Н, Ph); 5,50 (с, 1Н, ОН); 3,85 (м, 4Н, пиперазин); 3,55 (м, 4Н, пиперазин); 1,46 (с, 18Н, 2 х tBu).
9.2) 2,6-бис-(1,1-диметилэтил)-4-{[4-(4-аминофенил)-1-пиперазинил]-карбонил}-фенол:
В сосуде Парра емкостью 250 мл растворяют 2,19 г (5,0 ммолей) промежуточного продукта 9.1 в 50
мл абсолютного этилового спирта в присутствии 10%-ного палладия на угле (Pd/C). Смесь перемешивают при давлении водорода 20 PSI при 30oС, в течение одного часа. После фильтрации на целите
фильтрат концентрируют в вакууме. Продукт выпаривания обрабатывают с помощью 25 мл диэтилового эфира, фильтруют и промывают 2•20 мл диэтилового эфира. Получают бледно-розовый порошок с выходом
82%. Температура плавления: 221-222oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,30 (с, 2Н, Ph); 6,75 (м, 4Н, Ph-NH2); 5,45 (с, 1Н, ОН); 3,80 (м, 4Н, пиперазин); 3,10 (м, 4Н, пиперазин); 1,49 (с, 18Н, 2 х tBu).
9.3) хлоргидрат -N-{4-[4-[(3,5-бис-(1,1-диметилэтил)-4-гидроксибензоил]
-1-пиперазинил]-фенил}-2-тиофен-карбоксимидамида: 9
Работают так же, как и при получении соединения 1, причем используют 2,6-бис-(1,1-диметилэтил)-4-{ [4-(4-аминофенил)-1-пиперазинил]
-карбонил} -фенол вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. После обработки молярным раствором НСl в безводном диэтиловом эфире получают бежевый порошок с выходом 75%.
Температура плавления: 235-236oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,45 (с, 1Н, NH+); 9,78 (с, 1H, NH+); 9,78 (с, 1Н, NH+); 8,75 (с, 1H, NH+); 8,19 (м, 2Н, тиофен); 7,29 (м, 5Н, Ph-N, тиофен); 7,10 (с, 2Н, Ph); 5,60 (с широкий, 1H, ОН); 3,70 (м, 4Н, пиперазин); 3,30 (м, 4Н, пиперазин); 1,40 (с, 18Н, 2 х tBu).
ИК: νOH: 3633 см-1, 3433 см-1; νC=O(амид): 1617 см-1; νC=N(амидин): 1590 см-1.
Пример 10: хлоргидрат -N-{4-[4-[(3,5-бис-(1,1-диметил-этил)-4-гидроксибензил]-1-пиперазинил]-фенил}-2-тиофенкарбоксимидамида: 10
10.1) 2,6-бис-(1,
1-диметилэтил)-4-бромометилфенол:
В трехгорлой колбе емкостью 250 мл в атмосфере азота растворяют 2,36 г (10 ммолей) 3,5-ди-трет-бутил-4-гидроксибензилового спирта в 25 мл безводного
тетрагидрофурана. Раствор охлаждают с помощью ледяной бани, а затем добавляют по каплям 0,95 мл (10 молей) трибромида фосфора, разбавленного 25 мл безводного ТГФ. После перемешивания в течение 15
минут при 0oС раствор разбавляют 100 мл дихлорметана и промывают 3•30 мл воды, затем 30 мл рассола. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют в вакууме,
чтобы получить темно-коричневое масло, которое используют непосредственно на следующем этапе.
10.2) 2,6-бис-(1,1-диметилэтил)-4-{[4-(4-нитрофенил)-1-пиперазинил]-метил)-фенол:
В колбу емкостью 100 мл, содержащую раствор 2,99 г (10 ммолей) 2,6-бис-(1,1-диметилэтил)-4-бромометилфенола в 30 мл ДМФ, добавляют последовательно 1,38 г (10 ммолей) карбоната калия и 2,07 г (10
ммолей) 1-(4-нитрофенил)пиперазина. После двух часов перемешивания при окружающей температуре реакционную смесь разбавляют 150 мл дихлорметана и промывают последовательно в 3х40 мл воды, затем в 40 мл
рассола. Органический раствор сушат над сульфатом натрия, фильтруют и
концентрируют под пониженным давлением. Полученный темно-коричневый остаток очищают хроматографией на силикагеле (элюируя
смесью: петролейный эфир (Еb 40-70o)/этилацетат: 8/2). После концентрации чистых фракций получают 2,31 г (54%) темно-коричневого порошка.
Температура плавления: 177,5-178, 5oС.
ЯМР1H (100 МГц, СDСl3, δ): 7,50 (м, 4Н, Ph-NO2); 7,12 (с, 2Н, Ph); 5,19 (с, 1Н, ОН); 3,50 (с, 2Н, CH2-Ph); 3,40 (м, 4Н, пиперазин); 2,60 (м, 4Н, пиперазин); 1,49 (с, 18Н, 2хtBu).
10.3) 2,6-бис-(1,1-диметилэтил)-4-{[4-(4-аминофенил)-1-пиперазинил]-метил)-фенол:
Работают так же, как и при
получении промежуточного продукта 9,2, причем используют 2,6-бис-(1,1-диметилэтил)-4-{[[4-(4-нитрофенил)-1-пиперазинил]-карбонил] -метил} -фенол вместо 2,6-бис- (1,
1-диметилэтил)-4-{[4-(4-нитрофенил)-1-пиперазинил] -карбонил} -фенол. Получают светло-розовый порошок с выходом 75%. Температура плавления: 152-154oС.
10.4) хлоргидрат
-N-{4-[4-[(3,5-бис-(1,1-диметилэтил)-4-гидроксибензил] -1-пиперазинил]-фенил}-2-тиофенкарбоксимидамида: 10
В колбу емкостью 100 мл, содержащую 0,59 г (1,5 ммоля) промежуточного соединения
10.3 в 20 мл 2-пропанола, добавляют 0,43 г (1,5 ммоля) иодгидрата S-метил-2-тиофен-тиокарбоксимида (Ann. Chim. (1962), 7, 303-337). После нагревания с обратным холодильником в течение 15 часов
реакционную смесь концентрируют досуха в вакууме. Остаток очищают с помощью хроматографии на силикагеле (элюируя смесью: дихлорметан/этанол: 90/10). Чистые фракции концентрируют в вакууме и продукт
выпаривания превращают в соль в присутствии молярного раствора НСl в безводном диэтиловом эфире. Получают порошок светло-желтого цвета с выходом 40%. Температура плавления: 234-236oС.
ЯМР1Н (400 МГц, ДМСО d6, δ): 11,60 (с, 1Н, NH+); 11,40 (с, 1Н, NH+); 9,75 (с, 1H, NH+); 8,70 (с, 1Н, NH+); 8,17 (м, 2Н, тиофен); 7,39 (с, 2Н, Рh); 7,38 (м, 1H, тиофен); 7,24 (м, 5Н, Ph-N, ОН); 4,26 (д, 2Н, CH2-Ph, J= 4,6 Гц), 3,90 (м, 2Н, пиперазин); 3,35 (м, 4Н, пиперазин); 3,15 (м, 2Н, пиперазин); 1,40 (с, 18Н, 2хtBu).
ИК: νOH: 3624 см-1, 3418 см-1; νC=N(амидин): 1610 см-1.
Пример 11: хлоргидрат
-N-{4-[4-[(3,5-диметокси-4-гидроксибензоил]-1-пиперазинил]-фенил}-2-тиофенкарбоксимидамида: 11
11.1) 2,6-диметокси-4-{[4-(4-нитрофенил)-1-пиперазинил]-карбонил}-фенол:
В пробирке
емкостью 100 мл растворяют 0,99 г (5 ммолей) сиреневой кислоты, 0,74 г (5,5 ммолей) гидроксибензотриазола, 1,10 г (5,5 ммолей) дициклогексилкарбодиимида и 1,04 г (5 ммолей) 1-(4-нитрофенил)пиперазина
в 10 мл ДМФ. После 7 часов перемешивания при комнатной температуре смесь фильтруют и осадок промывают 20 мл ДМФ, затем 100 мл хлороформа. Получают 2 г желтого порошка, который содержит приблизительно
20% дициклогексилмочевины. Продукт используют как таковой на следующем этапе.
ЯМР1H (100 МГц, ДМСО d6, δ): 7,69 (м, 4Н, Ph-NO2); 6,88 (с, 2Н, Ph); 5,72 (м, 1Н, ОН); 3,91 (с, 6Н, 2•ОСН3); 3,75 (м, 4Н, пиперазин); 3,49 (м, 4Н, пиперазин).
11.2) 2,
6-диметокси-4-{[4-(4-аминофенил)-1-пиперазинил]-карбонил}-фенол:
В сосуде Парра емкостью 250 мл растворяют 2 г промежуточного продукта 11.1 в 4 мл абсолютного этилового спирта ДМСО (1/3) в
присутствии 10% палладия на угле (Pd/C). Смесь перемешивают под давлением водорода 20 PSI, при 25oС, в течение 15 часов. После фильтрации на целите фильтрат концентрируют в вакууме.
Светло-коричневый продукт выпаривания обрабатывают 50 мл этилацетата, образовавшийся осадок удаляют фильтрацией, промывают 20 мл этилацетата и фильтрат экстрагируют с помощью 2•25 мл молярного
раствора НСl. Водную фазу подщелачивают путем добавления карбоната натрия в порошке и экстрагируют с помощью 2•50 мл этилацетата. Органический раствор сушат над сульфатом натрия, фильтруют и
концентрируют в вакууме. Полученный порошок обрабатывают 20 мл диэтилового эфира, содержащего 3 мл метанола, фильтруют и промывают с помощью диэтилового эфира. Получают 400 мг (22% на двух этапах)
темно-коричневых кристаллов. Температура плавления: 182-183oС.
ЯМР1Н (100 МГц, ДМСО d6, δ): 6,80 (с, 2Н, Ph); 6,74 (м, 4Н, Ph-NH2); 4,80 (м, 2Н, NH2); 3,91 (с, 6Н, 2•ОСН3); 3,77 (м, 4Н, пиперазин); 3,08 (м, 4Н, пиперазин).
11.3) хлоргидрат -N-{ 4-[4-[3,
5-диметoкcи-4-гидpoксибензоил]-1-пиперазинил]-фенил} -2-тиофенкарбоксимидамида: 11
В колбу емкостью 100 мл, содержащую раствор 0,4 г (1,13 ммоля) промежуточного соединения 11,2 в 10 мл
2-пропанола, добавляют 0,32 г (1,13 ммоля) иодгидрата S-метил-2-тиофен-тиокарбоксимида (Ann.Chim. (1962), 7, 303-337). После нагревания при 50oС в течение 15 часов, реакционную смесь
концентрируют досуха в вакууме. Продукт выпаривания обрабатывают 100 мл смеси этилацетат/насыщенный раствор карбоната натрия (1/1). Появившийся осадок фильтруют и промывают последовательно 20 мл воды,
20 мл этилацетата и 50 мл эфира. Полученное основание превращают в соль в присутствии молярного раствора НСl в безводном диэтиловом эфире. После фильтрации, промывания 10 мл ацетона и сушки получают 0,
12 г (20%) светло-желтого порошка. Температура плавления: 184-185oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,47 (с, 1Н, NH+); 9,78 (с, 1Н, NH+); 8,76 (с, 1Н, NH+); 8,18 (м, 2Н, тиофен); 7,37 (м, 1Н, тиофен); 7,28 (м, 4Н, Ph-N); 6,74 (с, 2Н, Ph); 4,27 (с широкий, 1Н, ОН); 3,80 (с, 6Н, 2•ОСН3); 3,70 (м, 4Н, пиперазин); 3,33 (м, 4Н, пиперазин).
ИК: νOH: 3423 см-1, νC=O(амид): 1610 см-1; νC=N(амидин): 1587 см-1.
Пример 12: хлоргидрат 3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{4-[(2-тиенил-(имино)метил)амино]фенил}-2Н-1-бензопиран-2-карбоксамида: 12
12.1) 3,
4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-(4-нитрофенил)-2Н-1-бензопиран-2-карбоксамид:
В колбу емкостью 100 мл добавляют 1,62 г (10 ммолей) 1,1'-карбонилдиимидазола в раствор 2,5 г (10
ммолей) Trolox® в 25 мл ТГФ. После одного часа перемешивания при комнатной температуре добавляют по капле раствор 4-нитроанилина в 20 мл ТГФ. Перемешивание продолжают в течение 15
часов и выпаривают раствор в вакууме. Остаток разводят в 50 мл дихлорметана и промывают последовательно 25 мл молярного раствора соляной кислоты, 25 мл воды и 25 мл рассола. Органическую фазу сушат
над сульфатом натрия, фильтруют и концентрируют под пониженным давлением. Полученное масло очищают с помощью хроматографии на силикагеле (элюируя смесью: петролейный эфир (Еb 40-70o
С/этилацетат: 7/3). После концентрации чистых фракций получают светло-желтый порошок с выходом 77%. Температура плавления: 150-151oС.
ЯМР1Н (100 МГц, CDCl3, δ): 8,68 (с, 1Н, CONH); 7,91 (м, 4Н, Ph); 4,59 (с, 1Н, ОН); 2,95-0,87 (м, 16Н, Trolox®).
12.2) 3,4-дигидро-6-гидрокси-2,5,7,
8-тeтраметил-N-(4-аминофенил)-2Н-1 -бензопиран-2-карбоксамид:
Работают так же, как при получении промежуточного соединения 9.2, причем используют 3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-N-(4-нитрофенил)-2Н-1-бензопиран-2-карбоксамид вместо 2,6-бис-(1,1-диметилэтил)-4-{ [4-(4-нитрофенил)-1-пиперазинил]-карбонил}-фенол. Продукт реакции очищают хроматографией на силикагеле
(элюируя смесью: петролейный эфир (Еb 40-70oС)/этилацетат: 6/4). Чистые фракции собирают, после выпаривания растворителя в вакууме получают бесцветное масло с выходом 45%.
ЯМР1H (100 МГц, CDCl3, δ): 8,19 (с, 1Н, CONH); 7,00 (м, 4Н, Ph); 4,59 (с, 1Н, ОН); 3,65 (с широкий, 2Н, NH2); 2,95-0,87 (м, 16Н, Trolox®).
12.3) хлоргидрат 3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{4-[2-тиенил-(иминометил)амино]фенил}-2Н-1-бензопиран-2-карбоксамида: 12
Работают так же, как при получении
соединения 1, причем используют 3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-(4-аминофенил)-2Н-1-бензопиран-2-карбоксамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида.
Температура плавления: 279-280oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 9,80 (с, 1Н, NH+); 9,50 (с, 1Н, NH+); 8,73 (с, 1H, NHCO); 8,18 (м, 2H, тиофен); 7,60 (м, 1Н, ОН); 7,59 (м, 4Н, Ph); 7,36 (м, 1H, тиофен); 2,60-1,57 (м, 16Н, Trolox®).
ИК: νOH: 3236 см-1; νC=O(амид): 1683 см-1; νC=N(амидин): 1577 см-1.
Пример 13: хлоргидрат N-{4-[4-[(3,4-дигидpo-6-гидpoкси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] -фенил} -2-тиофенкарбоксимидамида: 13
13.1) 3,4-дигидро-2,5,7,8-тетраметил-2-{4-[(4-нитрофенил)-1-пиперазинил]
-карбонил}-2Н-1-бензопиран-6-ол:
В колбу емкостью 100 мл добавляют 1,62 г (10 ммолей) 1,1'-карбонилдиимидазола в раствор 2,5 г (10 ммолей) Trolox® в 25 мл ТГФ. После часа
перемешивания при комнатной температуре добавляют по капле раствор 1-(4-нитрофенил)пиперазина в 10 мл ДМФ. Перемешивание продолжают в течение 15 часов, затем реакционную смесь концентрируют в вакууме.
Продукт выпаривания разводят в 50 мл дихлорметана и промывают последовательно 3•25 мл воды и 25 мл рассола. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при
пониженном давлении. Полученное масло осаждают в 30 мл смеси (95/5) этилацетат/этанол, твердое вещество фильтруют и промывают 2•20 мл этилацетата. Получают светло-желтый порошок с выходом 79%.
Температура плавления 199-200oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,45 (м, 4Н, Ph); 4,41-3,35 (м, 8Н, пиперазин); 2,95-1,25 (м, 16Н, Trolox®).
13.2) 3,4-дигидро-2,5,7,8-тетраметил-2-{4-[(4-аминофенил)-1-пиперизинил] -карбонил)-2Н-1-бензопиран-6-ол:
Работают так же, как при получении промежуточного
соединения 2.2, причем используют 3,4-дигидро-2,5,7,8-тетраметил-2-{4-нитрофенил)-1-пиперазинил]-карбонил} -2Н-1-бензопиран-6-ол вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил]
-бензамид. Продукт реакции очищают хроматографией на силикагеле (элюируя смесью: дихлорметан/метанол: 9/1). Чистые фракции собирают, чтобы после выпаривания растворителя в вакууме получить коричневое
масло с выходом 66%.
ЯМР1H (100 МГц, CDCl3, δ): 6,70 (м, 4Н, Ph); 4,15-2,97 (м, 8Н, пиперазин); 2,80-0,90 (м, 16Н, Trolox®).
13.3) хлоргидрат -N-{4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] -фенил} -2-тиофенкарбоксимидамида: 13
Работают так же, как и
для получения соединения 1, причем используют 3,4-дигидро-2,5,7,8-тетраметил-2-{4-[(4-аминофенил)-1-пиперазинил]-карбонил} -2Н-1-бензопиран-6-ол вместо 3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Однако реакция проходит медленнее и требует 15 часов нагревания. Основание, полученное после экстрагирования, очищают хроматографией на силикагеле
(элюируя смесью: петролейный эфир (Т.кип.40-70oС/этилацетат: 3/7). Чистые фракции концентрируют в вакууме и продукт выпаривания превращают в соль в присутствии молярного раствора НСl в
безводном диэтиловом эфире. Получают бледно-желтый порошок с выходом 40%. Температура плавления: 210-211oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,50 (с, 1Н, NH+); 9,79 (с, 1H, NH+); 8,69 (с, 1Н, NH+); 8,19 (м, 2Н, тиофен); 7,38 (м, 1Н, тиофен); 7,20 (м, 4Н, Ph); 4,58 (с широкий, 1Н, ОН); 4,11 (м, 2Н, пиперазин); 3, 61 (м, 2Н, пиперазин); 3,19 (м, 4Н, пиперазин); 2,62-1,55 (м, 16Н, Trolox®).
ИК: νOH: 3410 см-1; νC=O(амид): 1642 см-1; νC=N(амидин): 1613 см-1.
Пример 14: -N-{4-[4-[(5-метокси-1Н-индол-3-ил)метил-карбонил]-1-пиперазинил]фенил}-2-тиофенкарбоксимидамид:
14
14.1) 1-[(5-метокси-1Н-индол-3-ил)метилкарбонил] -4-(4-нитрофенил)-пиперазин:
В колбу емкостью 100 мл добавляют 1,62 г (10 ммолей) 1,1'- карбонил-диимидазола в раствор 2,05 г (10
ммолей) 5-метоксииндол-3-уксусной кислоты в 10 мл ТГФ. После одного часа перемешивания при комнатной температуре добавляют по капле раствор 1-(4-нитрофенил)-пиперазина в 10 мл ДМФ. Перемешивание
продолжают в течение 15 часов. Затем реакционную смесь концентрируют в вакууме, а продукт выпаривания осаждают в 50 мл смеси этилацетат/вода (1/1). После фильтрации твердое вещество промывают
последовательно в 50 мл воды, 50 мл этилацетата и 50 мл дихлорметана. Получают желтый порошок после сушки в вакууме с выходом 91%. Температура плавления: 239-240oС.
ЯМР1Н (100 МГц, ДМСО d6, δ): 10,90 (м, 1Н, NH); 7,63 (м, 4Н, Ph-NO2); 7,40-7,15 (м, 3Н, индол); 6,87 (дд, 1Н индол, Jopтo=8,7 Гц, Jмета-2,8 Гц); 3,90 (с, 2Н, СН2-СО); 3,88 (с, 3Н, ОСН3); 3,79 (м, 4Н, пиперазин); 3,50 (м, 4Н, пиперазин).
14.2) 1-[(5-метокси-1Н-индол-3-ил)метилкарбонил]
-4-(4-аминофенил)-пиперазин:
В сосуде Парра емкостью 250 мл растворяют 1 г (2,53 ммолей) промежуточного продукта 14.1 в 30 мл ДМСО в присутствии 10%-ного палладия на угле (Pd/C). Смесь
перемешивают под давлением водорода 20 PSI, при 25oС, в течение 7 часов. После фильтрации на целите фильтрат концентрируют в вакууме. Продукт выпаривания разводят в 50 мл этилацетата и
промывают 3•50 мл воды. Органическую фазу затем экстрагируют с помощью 2•25 мл молярного раствора НСl. После промывания кислого раствора с помощью 2•25 мл этилацетата, его
подщелачивают с помощью карбоната натрия в виде порошка. После повторного экстрагирования с помощью 2•50 мл этилацетата, органический раствор сушат над сульфатом натрия, фильтруют, а
растворитель выпаривают в вакууме. Остаток очищают хроматографией на силикагеле (элюируя смесью: дихлорметан/метанол: 98/2). Чистые фракции собирают и после выпаривания растворителя под пониженным
давлением получают 0,39 г бледно-желтого порошка с выходом 46%. Температура плавления: 119-120oС.
ЯМР1H (100 МГц, CDCl3, δ): 8,32 (с, 1Н, индольный); 7,27-6,80 (м, 4Н, индол); 6,69 (м, 4Н, Ph-NH2); 3,82 (с, 3Н, ОСН3); 3,80 (с, 2Н, СН2-СО); 3,80 (с, 2Н, пиперазин); 3,62 (м, 2Н, пиперазин); 3,48 (с, 2Н, NH2); 2,90 (м, 4Н, пиперазин).
14.3) -N-{ 4-[4-[(5 метокси-1Н-индол-3-ил)метилкарбонил]-1-пиперазинил] фенил}-2-тиофенкарбоксимидамид: 14
Работают так же, как при
получении соединения 1, причем используют 1-[(5-метокси-1Н-индол-3-ил)метилкарбонил] -4-(4-аминофенил)-пиперазин вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Целевой продукт
выделяют в виде свободного основания с выходом 20% (бледно-желтый
порошок). Температура плавления: 221-222oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 10, 78 (с, 1Н, NH индольный); 7,72 (м, 1H, тиофен); 7,59 (м, 1Н, тиофен); 7,22 (д, 1Н, индол, J=8,7 Гц); 7,19 (м, 1H, тиофен); 7,09 (м, 2Н, индол); 6,82 (м, 4Н, Ph); 6,72 (м, 1H, индол); 6,35 (с, 2Н, NH2); 3,80 (с, 2Н, CH2); 3,62 (м, 4Н, пиперазин), 2,95 (м, 4Н, пиперазин).
ИК: νOH: 3414 см-1; νC=O(амид): 1628 см-1; νC=N(амидин): 1590 см-1.
Пример 15: фумарат -N-[4-[4-[{3-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил]-1-оксо-2-пропенил}-1-пиперазинил]фенил]]-2-тиофенкарбоксимидамида: 15
15.1) 2,6-бис-(1,
1-диметилэтил)-4-{3-[4-(4-нитрофенил)-1-пиперазинил]-3-оксо-2-пропенил}-фенол:
Работают так же, как при получении промежуточного соединения 11,1, причем используют 3,
5-ди-трет-бутил-гидрокси-коричную кислоту вместо сиреневой кислоты. Получают масло с выходом 60%.
ЯМР1H (100 МГц, CDCl3, δ): 7,71 (д, 1Н, С=СН, J=15,0 Гц); 7,51 (м, 4Н, Ph-NО2); 7,38 (с, 2Н, Ph); 6,69 (д, 1Н, НС=С, J=15,0 Гц); 5,50 (с, 1Н, ОН); 3,88 (м, 4Н, пиперазин); 3,53 (м, 4Н, пиперазин); 1,47 (с, 18Н, 2хtBu).
15.2)
2,6-бис-(1,1-диметилэтил)-4-{3-[4-(4-аминофенил)-1-пиперазинил]-3-оксо-2-пропенил}-фенол:
В колбе емкостью 50 мл, снабженной холодильником, растворяют 0,5 г (1 ммоль) промежуточного
соединения 15.1 в 5 мл концентрированной соляной кислоты и 5 мл абсолютного этилового спирта. Охлаждают смесь до 0oС и добавляют несколькими порциями 1,69 г (7,5 ммолей) дигидратированного
хлорида олова. В конце добавления реакционную среду нагревают с обратным холодильником в течение 30 минут. Затем выпаривают растворители в вакууме, остаток обрабатывают 15 мл воды, нейтрализуют 2N
щелочью натрия и разводят 20 мл дихлорметана. Полученный осадок фильтруют на целите и фильтрат декантируют. Органическую фазу сушат над сульфатом натрия, фильтруют и концентрируют при пониженном
давлении, чтобы получить 0,3 г (67%) желтого масла.
ЯМР1H (100 МГц, CDCl3, δ): 7,66 (д, 1Н, C=СН, J=15,0 Гц); 7,37 (с, 2Н, Ph); 6,75 (м, 4Н, Ph-NH2); 6,30 (Д, 1Н, НС=С, J=15,0 Гц); 5,46 (с, 1H, ОН); 3,80 (м, 4Н, пиперазин); 3,06 (м, 4Н, пиперазин); 1,46 (с, 18Н, 2хtBu).
15.3) фумарат -N-[4-[4-[{3-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил] -1-оксо-2-пропенил}-1-пиперазинил]фенил]]-2- тиофенкарбоксимидамида: 15
Работают так же, как при получении соединения 1, причем используют 2,6-бис(1,
1-диметилэтил)-4-{ 3-[4-(4-аминофенил)-1-пиперазинил] -3-оксо-2-пропенил} -фенол вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида.
Продукт реакции превращают в соль в присутствии эквимолярного количества фумаровой кислоты в этаноле с обратным холодильником. Получают соединение 15 в виде желтого порошка с выходом 22%. Температура плавления: 170,5-173oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 7,77 (с, 1Н, тиофен); 7,67 (д, 1Н, тиофен, J=5,0 Гц); 7,48 (д, 1Н, С=СН, J=15,0 Гц); 7,39 (с, 2Н, Ph); 7,34 (с широкий, 1Н, ОН); 7,13 (т, 1Н, тиофен, J=4,0 Гц); 7,05 (д, 1 Н, НС=С, J=15,0 Гц); 6,92 (м, 4Н, Ph-N); 6,60 (с, 2Н, СН=СН фумарат); 3,78 (м, 4Н, пиперазин); 3,13 (м, 4Н, пиперазин); 1,41 (с, 18Н, 2хtBu).
ИК: νOH: 3619 см-1, 3300 см-1; νC=O(амид): 1640 см-1; νC=C 1600 см-1; νC=N(амидин): 1570 см-1.
Пример 16: хлоргидрат 3,5-бис-(1,1-диметилэтил) -4-гидрокси-N- { 3- [[(2-тиенил-имино)метил) амино]фенил]метил}-бензамида: 16
16.1) 3,5-бис-(1,1-диметилэтил)-4-гидрокcи-N- [(3-нитрофенил) метил]-бензамид:
Работают так же, как и при получении промежуточного соединения 2,1, причем используют хлоргидрат
3-нитробензиламина вместо хлоргидрата 4-нитробензиламина. Получают белый порошок с выходом 63%. Температура плавления: 210-211oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 9,12 (м, 1Н, NH); 8,25 (м, 2Н, Ph-NO2); 7,80 (м, 4Н,
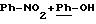
16.2) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N- [(3-аминофенил) метил]-бензамид:
В сосуде Парра емкостью 250 мл
растворяют 2,40 г (6,2 ммолей) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(3-нитрофенил)метил]-бензамида в 45 мл смеси абсолютного этилового спирта ТГФ (1/2) в присутствии 10%-ного палладия на угле
(Pd/C). Смесь перемешивают под давлением водорода 20 PSI, при 30oС, в течение трех часов. После фильтрации на целите фильтрат концентрируют досуха, а остаток очищают хроматографией на
силикагеле (элюируя смесью: гептан/этилацетат; 60/40). Чистые фракции собирают и концентрируют под пониженным давлением, чтобы получить 0,94 г (45%) белого порошка. Точка плавления: 171-172o
С.
ЯМР1H (100 МГц, CDCl3, δ): 7,20 (м, 2Н, Ph-NH2); 6,70 (м, 4Н,
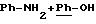
16.3) хлоргидрат
3,5-бис-(1,1-даметилэтил)-4-гидрокси- N-{3-[[(2-тиенил-(имино)метил)амино]фенил]метил}-бензамида: 16
Работают так же, как при получении соединения 1, причем используют 3,5-бис-(1,
1-диметилэтил)-4-гидрокси-N-[(3-амино-фенил)метил] -бензамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. После превращения в соль с помощью молярного раствора НСl в смеси
ацетон/безводный метанол, получают бледно-желтый порошок с выходом 50%. Температура плавления: 226-227oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,71 (с, 1Н, NH+); 9,93 (с, 1Н, NH+); 9,10 (с, 1Н, CONH); 9,00 (с, 1H, NH+); 8,18 (м, 2Н, тиофен); 7,70 (с, 2Н, Ph); 7,42 (м, 6Н, тиофен, Ph-NH, ОН); 4,50 (д, 2Н, CH2-NHCO, J=5,4 Гц); 1,40 (с, 18Н, 2хtBu).
ИК: νOH: 3420 см-1; νC=O(амид): 1639 см-1; νC=N(амидин): 1578 см-1.
Пример 17: хлоргидрат N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-N'-{ {4-[(2-тиенил(имино)метил)амино]фенил}метил}-мочевины: 17
17.1) 4-амино-2,
6-бис-(1,1-диметилэтил)-фенол:
В сосуде Парра емкостью 250 мл растворяют 3,6 г (14 ммолей) 4-нитро-2,6-бис-(1,1-диметилэтил)-фенола (J.Org.Chem. (1968), 33 (1), 223-226) в 60 мл смеси (2/1)
этанола и дихлорметана в присутствии каталитического количества 10%-ного Pd/C. Смесь перемешивают в течение 2 часов при 20oC под давлением водорода 20 PSI. После фильтрации на целите
фильтрат концентрируют досуха при пониженном давлении. Полученный серый порошок суспендируют в гептане (30 мл), фильтруют и промывают тем же объемом гептана. Ожидаемый продукт получают в виде порошка
оранжевато-розового цвета с выходом 50% (1/56 г).
Точка плавления: 123-124oС.
ЯМР1H (100 МГц, CDCl3, δ): 6,60 (с, 2Н, Ph); 4, 65 (с широкий, 1Н, ОН); 3,15 (с широкий, 2Н, NH2); 1,42 (с, 18Н, 2хtBu).
17.2) хлорид 4-нитрофенилуксусной кислоты:
В раствор 0,9 г (5 ммолей) 4-нитрофенилуксусной
кислоты в смеси, состоящей из 10 мл дихлорметана и 0,5 мл ДМФ, добавляют при 20oС 3,75 мл (7,5 ммолей) 2М раствора оксалилхлорида в дихлорметане. После 30 минут перемешивания раствор
концентрируют в вакууме. Полученное желтое масло используют без дополнительной очистки на следующем этапе.
17.3) 4-нитробензилизоцианат:
К водному раствору 0,75 г (11,5
ммолей) азида натрия, охлажденному до 0oС, медленно добавляют хлорид 4-нитрофенилуксусной кислоты, растворенной в сухом ацетоне (7,5 мл). Перемешивание среды поддерживают в течение одного
часа после окончания добавления при 0-5oС. Затем реакционную смесь разбавляют 30 мл хлороформа, декантируют, а органическую фазу промывают водой (20 мл), а затем насыщенным раствором
хлорида натрия (20 мл). После сушки над сульфатом натрия органический раствор фильтруют и концентрируют частично (≈20 мл) в вакууме. Этот раствор ацилазида в хлороформе нагревают с обратным
холодильником в течение одного часа. Полученный изоцианат используют прямо в растворе на следующем этапе.
17.4) -N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]
-N'-[(4-нитрофенил)метил]-мочевина:
К раствору изоцианата (промежуточное соединение 17.3) (теоретические 5 ммолей) в 20 мл хлороформа добавляют за один раз 1,1 г (5 ммолей) 4-амино-2,6-бис-(1,
1-диметилэтил)-фенола. После 2 часов перемешивания при 20oС появившийся осадок фильтруют и промывают с помощью хлороформа (2•20 мл). Получают желтый порошок с выходом 73%.
Температура плавления: 240-241oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 8,60 (с, 1Н,
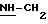
17.5)
N-[(4-нитрофенил)метил] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-мочевина:
В автоклаве емкостью 100 мл растворяют 0,55 г (1,38 ммоля) N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил]-N'-[(4-нитрофенил)метил]-мочевины в смеси (2/1) этанола и этилацетата, в присутствии 10%-ного Pd/C. После 1 часа и 30 минут гидрирования при 20oС
под давлением водорода 20 PSI, смесь фильтруют на целите и концентрируют фильтрат в вакууме. Остаток выпаривания разводят в 20 мл диэтилового эфира и ожидаемый продукт самопроизвольно кристаллизуется.
Кристаллы фильтруют и промывают 20 мл диэтилового эфира. Получают белый порошок с выходом 60% (0,31 г). Температура плавления: 194-195oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,08 (с, 2Н, Ph-OH); 6,87 (м, 4Н,
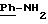
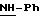
17.6) хлоргидрат N-[3,5-бис-(1,1-диметилэтил)-4- гидроксифенил]-N'-{{ 4-[(2-тиенил(имино)метил)амино) фенил}метил}-мочевины: 17
Работают так же, как при
получении соединения 1, причем используют N-[(4-аминофенил)метил] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -мочевину вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида.
После солеобразования с помощью молярного раствора НСl в безводном диэтиловом эфире получают белый порошок с выходом 45%. Точка плавления: 236-237oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,42 (с широкий, 1Н, NH+); 9,77 (с широкий, 1Н, NH+); 8,54 (с широкий, 1Н, NH+); 8,54 (с, 1Н,
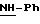
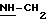
ИК: νOH: 3624 см-1; νC=O (мочевина): 1644 см-1; νC=N(амидин): 1569 см-1.
Пример 18: хлоргидрат N-[5-[{3-(3,5-бис-(1,1-диметилэтил) -4-гидроксифенил)-1-оксо-2-пропенил}
амино] -2-гидроксифенил] -2- тиофенкарбоксимидамида: 18
18.1) 3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N-(4-гидрокси-3-нитрофенид)-2-пропенамид:
В колбу емкостью 50 мл,
содержащую 10 мл ТГФ, вводят 1,78 г (6,4 ммолей) 3,5-ди-трет-бутил-4-гидроксикоричной кислоты, 0,99 г (6,4 ммолей) 4-амино-2-нитрофенола, предварительно разведенного в 10 мл ДМФ, 0,86 г (6,4 ммолей)
гидроксибензотриазола и 1,32 г (6,4 ммолей) дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при комнатной температуре, полученный остаток фильтруют и промывают
этилацетатом. После концентрации раствора при пониженном давлении, остаток разводят в 20 мл этилацетата и нерастворимое вещество снова фильтруют. Фильтрат промывают с помощью 20 мл насыщенного
раствора карбоната натрия. После сушки над сульфатом натрия органический раствор фильтруют и концентрируют досуха при пониженном давлении. Остаток очищают хроматографией на силикагеле (элюант:
гептан/этилацетат: 8/2). Чистые фракции собирают и концентрируют в вакууме, чтобы получить 1,95 г (47%) целевого соединения в виде желто-оранжевого порошка. Температура плавления: 231-232o
С.
ЯМР1H (100 МГц, CDCl3, δ): 10,45 (с, 1Н, NH); 8,45 (д, 1Н, Ph-NO2, J=l, 7 Гц ); 7,98 (дд, 1Н, Ph-NO2, J=1,7 Гц и J=6,8 Гц); 7,78 (д, 1Н, -СН=СН-, J= 10,5 Гц); 7,75 (с, 1Н, ОН); 7,40 (с, 2Н,
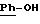
18.2) 3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)]-N-(4-гидрокси-3-аминофенил)-2-пропенамид:
В сосуде емкостью 100 мл с
холодильником растворяют 0,9 г (2,18 ммолей) 3-[(3,5-бис-(1,1 диметилэтил)-гидрокси-фенил]-N-(4-гидрокси-3-нитрофенил)-2-пропенамида в 20 мл этилацетата, добавляют 2,46 г (10,9 ммолей) хлорида олова
(дигидрат) и смесь нагревают при 70oС в течение 3 часов. После возвращения к комнатной температуре реакционную среду выливают в перемешиваемый раствор бикарбоната натрия (0,1 М), образуется
осадок, который удаляют фильтрацией на целите. Фильтрат декантируют и водную фазу экстрагируют с помощью 20 мл этилацетата. Органические фазы группируют и промывают с помощью 20 мл воды, затем 20 мл
насыщенного раствора хлорида натрия. После сушки над сульфатом натрия и фильтрации, органический раствор концентрируют досуха, в частичном вакууме. Остаток выпаривания суспендируют в смеси
гептан/этилацетат (1/1) и фильтруют, чтобы получить желтоватый порошок с выходом 53%. Продукт используют как таковой на следующем этапе.
18.3) хлоргидрат N-[5-[{3-(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил}амино]-2-гидроксифенил]-2-тиофенкарбоксимидамида: 18
Работают так же, как при получении соединения 1, причем используют 3-[(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)] -N-(4-гидрокси-3-аминофенил)-2-пропенамид вместо 3,5-бис-(1,1-диметилэтил) -4-гидрокси-N-(4-аминофенил)-бензамид. Свободное основание очищают хроматографией на
силикагеле (элюируя смесью: гептан/этилацетат: 35/65). Чистые фракции собирают и концентрируют при пониженном давлении. Остаток выпаривания разводят в 10 мл ацетона и превращают в соль в молярном
растворе НСl в безводном эфире, как описано выше. Получают конечный продукт весом 0,35 г в виде желтого порошка с выходом 62%. Температура плавления 199-200oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,11 (с, 1Н, NH+); 10,29 (с, 1Н, NH+); 10,17 (c, 1H, NH+); 9,71 (c, 1H, CONH); 8,61 (с широкий, 1H, ОН), 8,14 (м, 2Н, тиофен); 7,79 (с, 1H, Ph-N); 7,53 (м, 1H, Ph-N); 7,48 (д, 1H, -CH=CH-, J= 14,7 Гц); 7,37 (м, 4Н, Ph-tBu+18Н, Ph-N); 7,05 (м, 1H, тиофен); 6,68 (д, 1H-СН=СН-); 1,41 (с, 18Н, 2хtBu).
ИК: νOH: 3624 см-1, 3415 см-1; νC=O(амид): 1656 см-1; νC=C: 1616 см-1; νC=N(амидин): 1587 см-1.
Пример 19: хлоргидрат N-[3-[{3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил)aмино]-4-гидроксифенил]-2-тиофенкарбоксимидамида: 19
19.1)
3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)]-N-(2-гидрокси-5-нитрофенил)-2-пропенамид:
Работают так же, как при получении промежуточного соединения 18,1, причем используют
2-амино-4-нитрофенол вместо 4-амино-2-нитрофенола. Получают светло-желтый порошок с выходом 25%. Температура плавления: 256-257oС.
ЯМР1H (400 МГц, ДМСО, δ ): 11,79 (с широкий, 1Н, ОН); 9,59 (с, 1Н, NH); 9,21 ( с широкий, 1Н, Ph-NO2); 7,90 (дд mа1 rеsolu), 1Н, Ph-NO2); CONH); 8,61 (с широкий, 1Н, ОН); 8,14 (м, 2Н, тиофен); 7,79 (с, 1Н, Ph-N; J=8,l Гц); 7,52 (д, 1Н, -СН=СН-, J==15,5 Гц); 7,47 (с, 1Н, ОН); 7,42 (с, 2Н, Ph-NO2); 7,15 (д, 1Н, -СН=СН-); 7,04 (д, 1Н, Ph-NO2); 1,42 (с, 18Н, 2хtBu).
19.2) 3-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)] -N-(2-гидрокси-5-аминофенил)-2-пропенамид:
Работают так же, как при получении промежуточного соединения 18,2, причем используют 3-[(3,
5-бис-(1,1-диметилэтил)-4-гидроксифенил)]-N-(2-гидрокси-5-нитрофенил)-2-пропенамид вместо 3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)] -N-(4-гидрокси-2-нитрофенил)-2-пропенамида. Получают желтый
порошок с выходом 74%. Продукт используют без дополнительной очистки на следующем этапе.
19.3) хлоргидрат N-[5-[{3-(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил}амино]-4-гидроксифенил]-2-тиофенкарбоксимидамида: 19
Работают так же, как при получении соединения 1, причем используют 3-[(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)] -N-(2-гидрокси-5-аминофенил)-2-пропенамид вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)бензамида. После образования соли с помощью молярного раствора
НСl в безводном диэтиловом эфире, получают желтый порошок с выходом 54%. Температура плавления: 256-257oС.
ЯМР1Н (400 МГц, ДМСО d6, δ): 11,32 (с, 1Н, NH+); 10,67 (с, NH+); 9,69 (с, 1Н, NH+); 9,55 (с, 1Н, CONH); 8,70 (с широкий, 1Н, ОН); 8,19 (м, 2Н, тиофен); 7,48 (д, 1Н, -СН=СН-, J=15,5 Гц); 7,40 (с, 2Н, Ph-tBu); 7,37 (м, 2Н, Ph-N); 7,34 (с, 1Н, ОН); 7,13 (д, 1Н, -СН=СН-); 7,10 (м, 1Н, Ph-N); 6,99 (м, 1Н, тиофен); 1,41 (с, 18Н, 2хtBu).
ИК: νOH: 3623 см-1, 3410 см-1; νC=O(амид): 1652 см-1; νC=C 1616 см-1; νC=N(амидин): 1587 см-1.
Пример 20:
хлоргидрат N-{4-[4-[3,4,5-тригидроксибензоил)-1-пиперазинил] фенил}-2-тиофенкарбоксимидамид: 20
20.1) 5-{[4-(4-нитрофенил)-1-пиперазинил]карбонил]-бензен-1,2,3-триол:
Работают так же,
как при получении промежуточного соединения 8.1, причем используют 1-(4-нитрофенил)пиперазин вместо 4-нитрофенетиламина. Получают желтый порошок, который содержит еще следы нечистот с выходом 43%.
ЯМР1H (100 МГц, ДМСО, δ): 9,17 (с широкий, 2Н, 2х-ОН); 8,55 (с широкий, 1Н, -ОН); 7,57 (м, 4Н, Ph-NO2); 6,40 (с, 2Н,
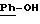
20.2) 5-{[4-(4-аминофенил)-1-пиперазинил]карбонил]-бензен-1,2,3-триол:
Работают так же,
как при получении промежуточного соединения 2.2, причем используют 5-{ [4-(4-нитрофенил)-1-пиперазинил]карбонил]-бензен-1,2,3-триол вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил]
-бензамида. Получают бежевый порошок с выходом 61%. Продукт используют без дополнительной очистки на следующем этапе.
ЯМР1H (100 МГц, ДМСО, δ): 9,12 (с широкий, 2Н, 2х-ОН); 8,55 (с широкий, 1Н, -ОН); 6,61 (м, 4Н, Ph-NH2); 6,34 (с, 2Н,
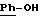
20.3) хлоргидрат N-{4-[4-[3,4,5-тригидроксибензоил]-1-пиперазинил)фенил} -2-тиофенкарбоксимидамида: 20
Работают так же, как при получении соединения 1,
причем используют 5-{ [4-(4-аминофенил)-1-пиперазинил] карбонил]-бензен-1,2,3-триол вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)бензамида. После обработки молярным раствором НСl в
безводном диэтиловом эфире, получают темно-коричневый порошок с выходом 25%. Температура плавления: 198-205oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11, 38 (с, 1Н, NH+); 9,75 (с, 1Н, NH+); 9,00 (с широкий, 1Н, ОН); 8,75 (с, 1H, NH+); 8,15 (м, 2Н, тиофен); 7,39 (м, 1H, тиофен); 7,22 (м, 4Н,
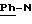
ИК: νOH: 3399 см-1; νC=O(амид): 1696 см-1; νC=N(амидин): 1588 см-1.
Пример 21: хлоргидрат N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-N'{{ 4-[(2-тиенил(имино)метил)амино]фенил} карбониламино}-мочевины: 21
21.1) N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-[{
4-нитрофенил)карбониламино]-мочевина:
В трехгорлую колбу емкостью 50 мл, снабженную капельной воронкой в атмосфере аргона, растворяют 0,22 г (0,73 ммоля) трифосгена при 20oС. К
этой смеси добавляют по капле в течение одного часа раствор из 0,44 г (2 ммоля) 4-амино-2,6-бис-(1,1-диметилэтил)-фенола (промежуточное соединение 17.1) и 0,38 мл (2,2 ммоля) диизопропилэтиламина в 7
мл безводного дихлорметана. Через пять минут после окончания добавления за один раз добавляют раствор из 0,36 г (2 ммоля) 4-нитробензоил и 0,38 мл (2,2 ммоля) диизопропилэтиламина в 4 мл безводного
ДМФ. После 4 часов перемешивания при 20oС реакционную смесь концентрируют досуха при пониженном давлении. Остаток выпаривания разводят в 40 мл этилацетата и органический раствор промывают
последовательно 3 раза с помощью 20 мл воды и 20 мл насыщенного раствора хлорида натрия. После сушки над сульфатом натрия органический раствор фильтруют и фильтрат концентрируют досуха при пониженном
давлении. Полученный остаток суспендируют в гептане, перемешивают и фильтруют, чтобы получить желтый порошок с выходом 86%. Температура плавления: 163-164oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 10,65 (с широкий, 1Н, NH амид); 8,72 (с, 1Н,
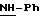
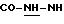
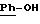
21.2) N-[(4-аминофенил)карбониламино)-N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-мочевина:
В
колбе Парра емкостью 250 мл растворяют 0,72 г (1,68 ммолей) N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-[(4-нитрофенил) карбониламино] -мочевины в 30 мл абсолютного этилового спирта в присутствии
10% Pd/C. Смесь перемешивают под давлением водорода 20 PSI при 30oС в течение двух часов. После фильтрации на целите фильтрат концентрируют в вакууме. Остаток выпаривания суспендируют в
диэтиловом эфире (20 мл), перемешивают и фильтруют, чтобы получить бледно-желтый порошок с выходом 75%. Температура плавления: 245-246oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 9,84 (с широкий, 1H, NH амид); 8,56 (с, 1H,
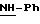
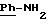
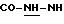
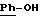
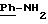
21.3) хлоргидрат N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-N'-{{4-[(2-тиенил(имино)метил)амино]фенил}карбониламино}-мочевины: 21
Работают так же, как при получении соединения 1, причем используют
N-[(4-аминофенил)карбониламино]-N'-[3,5-,bс-(1,1-диметилэтил)-4-гидроксифенил] -мочевину вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Свободное основание очищают
хроматографией на силикагеле (элюируя смесью: гептан/этилацетат: 1/1). Чистые фракции собирают и концентрируют под пониженным давлением. Остаток выпаривания разводят в 15 мл ацетона и превращают в
соль с помощью молярного раствора НСl в безводном эфире, как описано выше. Получают 0,40 г (58%) желтого порошка. Температура плавления: 254-255oС.
ЯМР1H (400 МГц, ДМСО, δ): 1,68 (с широкий, 1Н, NH+); 10,32 (с, 1Н, NH амид); 9,94 (с широкий, 1Н, NH+); 9,13 (с широкий, 1Н, NH+); 9,13 (с широкий, 1Н, NH+); 8, 68 (с, 1Н, NH-CO); 8,18 (м, 2Н, тиофен); 8,07 (м, 3Н,
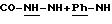
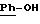
ИК: νOH: 3627 см-1; νC=O(амид),νC=O(мочевина): 1654 см-1; 1602 см-1; νC=N(амидин): 1559 см-1.
Пример 22: хлоргидрат
N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-{ (4-[(2-тиенил(имино)метил)амино]фенил)метил}-тиомочевины: 22
22.1) N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-{ {
4-нитрофенил)метил]-тиомочевина:
Соединение 22.1 получают путем обработки реагентом Lawesson промежуточного соединения 17.4 согласно методике, описанной в литературе (J.Med. Chem. (1995), 38
(18), 3558-3565). Получают светло-желтый порошок с выходом 80%. Температура плавления: 218-220oС.
ЯМР1H (100 МГц, CDCl3, δ); 7,85 (м, 4Н, Ph-NO2); 7,70 (с, 1Н,
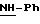
22.2) N-[(4-аминофенил)метил] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил]-тиомочевина:
Работают так же, как при
получении промежуточного соединения 18.2, причем используют промежуточное соединение 22.1 вместо 3-[(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N-(4-гидрокси-3-нитрофенил)-2-пропенамида. Получают
белый порошок с выходом 70%. Температура плавления: 167-169oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,48 (с широкий, 1Н,
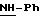
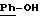
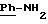
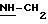
22.3. хлоргидрат N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-N'-{{4-[(2-тиенил(имино)метил)амино]фенил}метил}-тиомочевины: 22
Работают так же, как при получении промежуточного соединения 17,6, причем используют
промежуточное соединение 22.2 вместо N-[(4-аминофенил) метил] -N'-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -мочевины. Получают бледно-желтый порошок с выходом 15%. Точка плавления: 203-205oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,52 (с широкий, 1Н, NH+); 9,86 (с широкий, 1Н, NH+); 8,98 (с широкий, 1Н, NH+); 8,39 (с, 1Н,
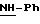
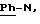
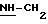
ИК: νOH: 3630 см-1; νC=O(мочевина): 1649 см-1; νC=N(амидин): 1600 см-1 .
Пример 23: хлоргидрат N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-N'-{ 2-{4-[(2-тиенил(имино)метил)амино]фенил}этил}-мочевина: 23
23.1) N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-N'-[2-(4-нитрофенил)этил]-мочевина:
Работают так же, как при получении промежуточного соединения 21.1, причем используют 4-нитрофенетиламин вместо
4-нитробензоил-гидразида. Получают бежевый порошок с выходом 80%. Температура плавления: 185-187oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,75 (м, 4Н, Ph-NO2); 7,00 (с, 2Н, Ph-OH); 6,05 (с, 1Н, ОН); 5,18 (с, 1H, NH); 4,68 (м, 1Н,
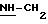
23.2) N-[2-(4-аминофенил)этил] -N'-[3,5-бис-(1,1-диметилэтил)- 4-гидроксифенил]-мочевина:
Работают так
же, как при получении промежуточного соединения 21,2, причем используют промежуточное соединение 23,1 вместо N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-[(4-нитрофенил)карбониламино] -мочевины.
Получают белый порошок с выходом 56%. Температура плавления: 192-194oС.
ЯМР1H (100 МГц, ДМСО d6, δ): 8,25 (с широкий, 1Н, Ph-NH-CO); 7,22 (с, 2Н,
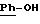
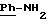
23.3) хлоргидрат N-[3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-N'-{2-{ 4-[(2-тиенил (имино)метил)амино1фенил}этил}-мочевины: 23
Работают так же, как при получении соединения 1, причем используют промежуточное соединение 23,
2 вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамид. Свободное основание очищают хроматографией на силикагеле (элюируя смесью: гептан/этилацетат: 1/1). Чистые фракции собирают и
концентрируют под пониженным давлением. Остаток выпаривания разводят в 15 мл ацетона и превращают в соль с помощью молярного раствора НСl в безводном эфире, как описано выше. Получают 0,25 г (24%)
бледно-желтого порошка. Температура плавления: 207-210oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,48 (с широкий, 1Н, NH+); 9,83 (с широкий, 1Н, NH+); 8,95 (с широкий, 1Н, NН+); 8,50 (с, 1Н, NH-CO); 8,18 (м, 2Н, тиофен); 7,38 (м, 5Н,
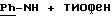
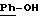
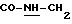
ИК: νOH: 3631 см-1; νC=O(мочевина): 1654 см-1; 1600 см-1; νC=N(амидин): 1560 см-1.
Пример 24: хлоргидрат
N-(4-{4-[(3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил]фенил) -2-тиофенкарбоксимидамида: 24
24.1) 1-{ [3,4-дигидро-6-метокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-4-(4-нитрофенил) пиперазин:
Работают так же, как при получении промежуточного соединения 13.1, причемиспользуют(±)-3,4-дигидро-6-метокси-2,
5,7,8-тетраметил-2Н-бензопиран-2-карбоновую кислоту (полученную согласно CHIMIA (1991), 45 (4), 121-3) вместо (±)-Trolox®. Получают желтый порошок.
ЯМР1H (100 МГц, CDCl3, δ): 7,45 (м, 4Н, Ph); 3,60 (с, 3Р, СН3О); 3,40 (м, 4Н, пиперазин); 3,00 (м, 4Н, пиперазин); 2,50-1,60 (м, 16Н, Trolox®).
24.2) 1-{ [3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-4-(4-аминофенил)пиперазин:
Работают так же, как при получении промежуточного соединения 13,2,
причем используют промежуточное соединение 24.1 вместо 3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ола. Получают масло, которое используют
непосредственно на следующем этапе.
ЯМР1H (100 МГц, CDCl3, δ): 6,70 (м, 4Н, Ph); 3,90 (д широкий, 4Н, пиперазин); 3,60 (с, 3Н, СН3О); 3,45 (с широкий, 2Н, NH2); 2,90 (м, 4Н, пиперазин); 2,60-1,60 (м, 16Н, Trolox®).
24.3) хлоргидрат N-(4-{ 4-[(3,4-дигидро-6-метокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил)-2-тиофенкарбоксимидамида: 24
Работают так же, как при получении соединения 13, причем используют промежуточное соединение
24.2 вместо 3,4-дигидро-2,5,7,8-тетраметил-2-{4-[(4-аминофенил)-1-пиперазинил] -карбонил} -2Н-1-бензопиран-6-ола. Получают бледно-желтый порошок. Температура плавления: 190-195oС.
ЯМР1H (400 МГц, ДМСО, δ): 11,35 (с широкий, 1Н, NH+); 9,70 (с широкий, 1Н, NH+); 8,70 (с широкий, 1Н, NH+); 8,15 (с широкий, 2Н, тиофен); 7,35 (с широкий, 1Н, тиофен); 7,17 (м, 4Н, Ph); 3,90 (д широкий, 4Н, пиперазин); 3,50 (с, 3Н, СН3О); 3,15 (м, 4Н, пиперазин); 2,55-1,55 (м, 16Н, Trolox®).
ИК: νC=O(амид): 1642 см-1; νC=N(амидин): 1618 см-1.
Пример 25: хлоргидрат N-(4-{4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]-1Н-1,4-диазепин-1-ил}фенил]-2-тиофенкарбоксимидамида: 25
25.1) гексагидро-4-(4-нитрофенил)-1Н-1,4-диазепин:
К раствору 2,44 г (12,2
ммолей) гексагидро-lH-l,4-диазепин-1-карбоксилата (1,1-диметил)этила в 50 мл ДМФ добавляют 3,37 г (24,4 ммолей) карбоната калия и 1,89 г (13,4 ммолей) 4-нитрофторбензола. Реакционную смесь нагревают
при 100oС в течение 16 часов. После охлаждения, добавляют 25 мл этилацетата и 50 мл воды. Органический раствор декантируют, а водную фазу экстрагируют с помощью 3•50 мл этилацетата.
Органические фазы объединяют и промывают 50 мл рассола, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Получают 3,7 г твердого вещества ярко-желтого цвета с выходом 95%. Это твердое
вещество растворяют в 100 мл смеси растворителей (дихлорметан/этилацетат 1:1), к которому добавляют по капле, при 0oС, 20 мл водного раствора (6N) соляной кислоты. После сильного
перемешивания при 20oС в течение 1 часа, реакционную смесь декантируют. Водную фазу подщелачивают до рН 11 с помощью 4N щелочи натрия и экстрагируют с помощью 3•50 мл дихлорметана.
Органические фазы объединяют, промывают 50 мл воды, затем 50 мл рассола, сушат над сульфатом натрия и, наконец, фильтруют и концентрируют в вакууме. Получают 1,78 г ярко-желтого порошка с выходом 66%.
Продукт используют непосредственно на следующем этапе без дополнительной очистки.
ЯМР1H (100 МГц, СDСl3, δ): 8,10 (м, 2Н, Рh); 6,65 (м, 2Н, Ph); 3,70 (кв, 4H, CH2N, J=5,2 Гц); 3,10 (т, 2Н, CH2N); 2,85 (т, 2Н, CH2N); 1,95 (кв, 2H, C-CH2-C); 1,65 (с широкий, 1Н NH).
25.2) 1-[(3,
4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]гексагидро-4-(4-нитрофенил)-1Н-1,4-диазепин:
В колбу емкостью 50 мл добавляют 0,71 г (4,4 ммолей) 1,
1'-карбонилдиимидазола в раствор 1,07 г (4.3 ммолей) (±)-Trolox® в 8 мл безводного ТГФ. После перемешивания в течение 1 часа при 20oС добавляют по капле раствор 0,
95 г (4,3 ммолей) промежуточного соединения 25.1 в 4 мл ДМФ. Реакционную смесь перемешивают в течение 16 часов при 20oС. После испарения растворителей в вакууме остаток обрабатывают 30 мл
смеси растворителей (дихлорметан/вода 1:2). После декантации органическую фазу промывают 2х20 мл воды, сушат над сульфатом натрия и концентрируют в вакууме. Получают бледно-желтый порошок с выходом
сырого продукта 97%. Продукт используют непосредственно на следующем этапе без дополнительного очищения.
ЯМР1H (100 МГц, CDCl3, δ): 8,10 (м, 2Н, Ph); 6, 60 (м, 2Н, Рh); 4,40 (с широкий, 1Н, ОН); 3,50 (м, 8Н, CH2N); 2,50-1,50 (м, 18Н, Trolox®+ CH2).
25.3) 1-(4-аминофенил)-4-[(3,
4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]гексагидро-1Н-1,4-диазепин:
Работают так же, как при получении промежуточного соединения 13,2, причем используют
промежуточное соединение 25.2 вместо 3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ола. Полученный продукт очищают хроматографией на силикагеле
(элюируя смесью этилацетатпетролейный эфир 3:2). Получают масло с выходом 57%.
25.4) хлоргидрат N-[4-{4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]
-1Н-1,4-диазепин-1-ил} фенил]-2-тиофенкарбоксимидамида: 25
Смесь из 0,52 г (1,22 ммоля) промежуточного соединения 25.3 и 0,35 г (1,22 ммоля) иодгидрата S-метил-2-тиофен-тиокарбоксимида в 4 мл
изопропанола нагревают при 50oС в течение 40 часов. Реакционную смесь затем фильтруют и полученное твердое вещество обрабатывают в 4 мл насыщенного водного раствора карбоната натрия и 4 мл
этилацетата. Полученную смесь нагревают при 50oС в течение 30 минут, затем декантируют. Органическую фазу промывают 2х10 мл воды, затем 10 мл рассола. Органические фазы объединяют, сушат на
сульфате натрия, фильтруют и концентрируют под пониженным давлением. Полученное твердое вещество очищают хроматографией на силикагеле (элюируя смесью:этилацетат/петролейный эфир 5:1). Получают 0,5 г
продукта с выходом 77%. 0,15 г (0,29 ммоля) этого продукта затем растворяют в 2 мл ацетона. Добавляют по капле 0,84 мл (0,84 ммоля) lN раствора соляной кислоты в безводном этиловом эфире. Содержимое
перемешивают при комнатной температуре в течение 30 минут. Образуется желтый осадок, который фильтруют. Осадок растирают и промывают последовательно 3•5 мл этилового эфира и 5 мл ацетона.
Темно-желтый порошок сушат в вакууме при 70oС в течение 48 часов. Получают продукт с выходом 80%. Температура плавления: 180-185oС.
ЯМР1H (400 МГц, ДМСО, δ): 11,15 (с широкий, 1Н, NH+); 9,60 (с широкий, 1Н, NH+); 8,55 (с широкий, 1Н, NH+); 8,10 (с широкий, 2Н, тиофен); 7,35 (с широкий, 1Н, тиофен); 7,02 (м, 4Н, Ph); 4,80 (с широкий, 1Н, ОН); 3,70 (м, 8Н, CH2N); 2,50-1,40 (м, 18Н, Trolox®+ CH2).
ИК: νOH: 3412 см-1; νC=O(амид): 1613 см-1; νC=N(амидин): 1613 см-1.
Пример 26: хлоргидрат (R) N-{4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-банзопиран-2-ил)-карбонил]-1-пиперазинил]фенил}-2-тиофенкарбоксимидамида: 26
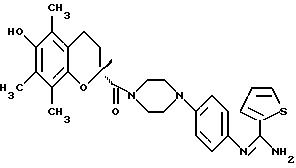
26.1) (R)-3,4-дигидро-2,5,7,8-тераметил-2-{ 4- [(4-нитрофенил)-1-пиперазинил]-карбонил} -2Н-1-бензопиран-6-ол:
Работают так же, как при получении соединения 13.1, причем используют (R)-Trolox® вместо (±)-Trolox®. Получают ярко-желтый порошок с выходом 98%. Точка плавления: 102-105oС.
26.2) (R)-3,4 -дигидро-2,
5,7,8-тетраметил-2-{4-[(4-аминофенил) -1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ол:
Работают так же, как при получении промежуточного соединения 2,2, причем используют промежуточное
соединение 26.1 вместо 3,5-бис- (1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил) метил]-бензамида. Получают розовый порошок с выходом 75%. Продукт используют как таковой на следующем этапе. Температура
плавления: 103-105oС.
26.3) хлоргидрат (R)-N-{4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил]
фенил}-2-тиофенкарбоксимидамида: 26
Работают так же, как при получении соединения 13, используя промежуточное соединение 26.2 вместо 3,4-дигидро-2,5,7,
8-тетраметил-2-{4-[(4-аминофенил)-1-пиперазинил] -карбонил}-2Н-1-бензопиран-6-ола. Продукт получают в виде бледно-желтого порошка, который гидратируется на воздухе. Температура плавления: 195-197oС.
Анализы ЯМР и ИК идентичны анализам для соединения 13.
[α] = -43,5° (с=0,11; DMSO)
Пример 27: дихлоргидрат (S)-N-{4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил]
-1-пиперазинил]фенил}-2-тиофенкарбоксимидамида: 27
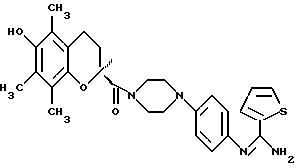
27.1 (S)-3,4-дигидро-2,5,7, 8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил]-карбонил}-2Н-1-бензопиран-6-ол:
Работают так же, как при получении соединения 13,1, при этом используют (S)-Trolox® вместо (±)-Trolox®. Получают желтый порошок с выходом 73%. Температура плавления: 110-111oС.
27.2) (S)-3,4-дигидpo-2,5,7,8-тетраметил-2-{
4-[(4-aминoфенил)-1-пиперазинил]-карбонил}-2Н-1-бензопиран-6-ол:
Работают так же, как при получении промежуточного соединения 2,2, при этом используют промежуточное соединение 27.1 вместо 3,
5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)метил] -бензамида. После очистки хроматографией на силикагеле (элюируя смесью: гептан/этилацетат: 2/8), объединения и выпаривания в вакууме чистых
фракций, получают бежевый порошок с выходом 54%. Температура плавления: 109-111oС.
27.3) дихлоргидрат (S)-N-{4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,
8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил]фенил}-2-тиофенкарбоксимидамида: 27
Работают так же, как при получении соединения 13, при этом используют промежуточное соединение
27.2 вместо 3,4-дигидро-2,5,7, 8-тетраметил-2-{4-[(4-аминофенил)-1-пиперазинил]-карбонил}-2Н-1-бензопиран-6-ола. Продукт получают в виде бледно-желтого порошка, который гидратируется на воздухе.
Температура плавления: 210,6-211,8oС.
Анализы ЯМР и ИК идентичны анализам для соединения 13.
[α] = -43,5° (с=0,11; DMSO)
В качестве варианта соединение 27 может быть получено согласно следующему протоколу:
27.4)
2-тиофен метилкарбоксимидат:
В коническую колбу емкостью 250 мл, продуваемую аргоном, вводят 10,91 г (0,1 моля) 2-тиофенкарбонитрила, 100 мл безводного этилового эфира и 4,5 мл (0,11 моля)
метанола. Раствор охлаждают до 0oС, с помощью ледяной бани, и насыщают безводным газовым потоком НСl в течение 45 минут. Реакционную смесь перемешивают один час при 0oС и одну
ночь при 20oС. Образованный осадок фильтруют, промывают в этиловом эфире и сушат. Полученный хлоргидрат вводят порциями в смесь из 100 мл воды и 150 мл этилового эфира. Среду нейтрализуют
добавлением 8,4 г (0,1 моля) сухого NаНСО3. После декантации и отделения, органическую фазу промывают последовательно 2•30 мл воды и 30 мл рассола. После сушки над сульфатом магния
органический раствор фильтруют и концентрируют в вакууме. Получают бесцветное масло с выходом 66%.
ЯМР1Н (400 МГц, СDСl3, δ): 7,58 (с широкий, 1Н, =N-H); 7,42 (м, 1Н, тиофен); 7,37 (м, 1Н, тиофен); 7,01 (м, 1Н, тиофен); 3,86 (с, 3Н, ОСН3).
ИК: νC=N (карбоксимидат); 1630 см-1.
27.5) дихлоргидрат (S)-N-{4-[4-[(3,4-дигидpo-6-гидpoкcи-2,5,7,8-тетраметил-2Н-1-беназопиран-2-ил)-карбонил]-1- пиперазинил]фенил} -2-тиофенкарбоксимидамида: 27
В конической колбе емкостью 150
мл, под потоком аргона, растворяют 8,2 г (20 ммолей) (S)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-аминофенил)-1-пиперазинил]-карбонил}-2Н-1-бензопиран-6-ола, полученного так же, как промежуточное
соединение 13.2, но из (S)-Trolox® в 60 мл метанола и добавляют 4,2 г (30 ммолей) 2-тиофенметилкарбоксимидата. Реакционную смесь нагревают 18 часов с обратным холодильником. Метанол
выпаривают в вакууме и коричневый маслянистый осадок очищают хроматографией на силикагеле (элюируя смесью: дихлорметан/этанол: 95/5). Чистые фракции собирают и концентрируют в вакууме с получением
коричневого масла с выходом 68%. Это масло обрабатывают с помощью 22 мл 1,3 N этанольного раствора НСl и разбавляют с помощью 180 мл безводного ацетона. Реакционную смесь перемешивают 1 час при 0oС. Образовавшийся осадок фильтруют и промывают последовательно ацетоном и этиловым эфиром. После сушки получают дихлоргидрат в виде бледно-желтого порошка с выходом 53%.
Пример
28: хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 2-[3-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамида: 28
28.1) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 2-(3-нитрофенил)этил}
-бензамид:
Работают так же, как при получении промежуточного соединения 5.1, при этом используют 3-нитрофенетиламин (J. Med. Chem. (1968), 11 (1), 21-26) вместо 4-нитрофенетиламина. Получают
белый порошок с выходом 50%. Температура плавления: 195-197oС.
ЯМР1H (100 МГц, СDСl3 δ): 7,86 (м, 4Н, Ph-NO2); 7,50 (с, 2Н, Ph); 6, 10 (м, 1Н, NHCO); 5,54 (с, 1Н, ОН); 3,75 (м, 2Н,
28.2) 3,5-бис1-(1,1-диметилэтил)-4-гидрокси-N-[2-(3-аминофенил) этил]-бензамид:
Работают так же, как
при получении промежуточного соединения 5.2, при этом используют промежуточное соединение 28.1 вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(4-нитрофенил)этил] -бензамид. Получают белый порошок с
выходом 40%, достаточно чистый, чтобы использовать его на следующем этапе.
28.3) хлоргидрат 3,5 -бис-(1,1
-диметилэтил)-4-гидрокси-N-{2-[3-[(2-тиенил-(имино)метил)амино]фенил]этил}-бензамида: 28
Работают так же, как при получении промежуточного соединения 1,3, при этом используют промежуточное
соединение 28.2 вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Получают бледно-желтый порошок с выходом 35%. Температура плавления: 205-207oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,59 (с широкий, 1Н, NH+); 9,89 (с, 1Н, NH+); 8,95 (с, 1Н, NH+); 8,46 (с, 1Н, CONH); 8,17 (м, 2Н, тиофен); 7,54 (с, 2Н,
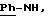
ИК: νOH: 3624 см-1; νC=O(амид): 1631 см-1; νC=N(амидин): 1577 см-1.
Пример 29: хлоргидрат N-{4-(4-[2-(3,5-бис-(1,
1-диметилэтил)-4-гидроксифенил)-1-пиперазинил)фенил} -2-тиофенкарбоксимидамида: 29
Работают так же, как при получении соединения 9, при этом используют 3,
5-ди-трет-бутил-4-гидроксифенилуксусную кислоту вместо 3,5-ди-трет-бутил-4-гидроксибензойной кислоты на первом этапе синтеза. Получают желтый порошок. Температура плавления: 176-180oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,30 (с широкий, 1Н, NH+); 9,70 (с, широкий, 1Н, NH+); 8,65 (с, широкий, 1Н, NH+); 8,10 (с широкий, 21Н, тиофен); 7,35 (с широкий, 1Н, тиофен); 7,12 (м, 4Н, Ph-N); 6,95 (с, 2Н,
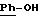
ИК: νOH: 3620 см-1; νC=O((сложный эфир): 1638 см-1; νC=N(амидин): 1612 см-1.
Пример 30: хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат)2-{
4-[(2-тиенил-(имино)метил)амино)фенил}этила: 28
30.1) 3,5-бис-(1,1-диметилэтил)-4-гидрокси-бензоат 2-(4-нитрофенил)этила:
В колбу емкостью 250 мл, содержащую 80 мл ТГФ, в атмосфере
аргона, вводят последовательно, при перемешивании, 2,45 г (9,8 ммолей) 3,5-ди-трет-бутил-4-гидроксибензойной кислоты, 1,8 г (10,8 ммолей) 4-нитробензол-этанола и 2,2 г (10,8 ммолей)
дициклогексилкарбодиимида. Реакционную смесь перемешивают в течение 15 часов при 20oС и появившийся осадок фильтруют. Фильтрат промывают с помощью 2-30 мл насыщенного раствора NaCl,
органическую фазу сушат над сульфатом магния, фильтруют и концентрируют в вакууме. Осадок затем кристаллизуют с помощью диизопропилового эфира. Твердое вещество извлекают фильтрацией и получают 2,4 г
(62%) белых кристаллов после сушки. Температура плавления: 123,5-124,5oС.
ЯМР1H (100 МГц, CDCl3, δ): 7,85 (м, 4Н, Ph-NO2); 7,80 (с, 2Н,
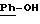
30.2) 3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат 2-(4-аминофенил)этила:
Работают так же, как при получении промежуточного соединения 2,2, при
этом используют промежуточное соединение 30.1 вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[(4-нитрофенил)-метил]-бензамида. Получают белый порошок с выходом 50%. Температура плавления: 135-136oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 7,75 (с, 2Н,
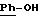
30.3) хлоргидрат 3,
5-бис-(1,1-диметилэтил)-4-гидроксибензоат 2-{4-[(2-тиенил-(имино)метил)амино]фенил}этила: 30
Работают так же, как при получении промежуточного соединения 1.3, при этом используют
промежуточное соединение 30.2 вместо 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-аминофенил)-бензамида. Получают твердое белое вещество с выходом 26%. Температура плавления: 145-150oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,50 (с широкий, 1H, NH+); 9,80 (с широкий, 1H, NH+); 8,90 (с широкий, 1H, NH+); 7,85 (с, 1H, ОН); 7,75 (с, 2Н, Ph-OH); 7,47 (с, 5Н, Ph-N, тиофен); 4,41 (м, 2Н, O-CH2); 3,08 (м, 2Н, CH2); 1,40 (с, 18Н, 2хtBu).
ИК: νC=O((сложный эфир): 1700 см-1; νC=N(амидин): 1592 см-1.
Пример 31: хлоргидрат 3,5-бис(1,1-диметилэтил)- 4-гидроксибензоат 2-{ 3-[(2-тиенил-(имино)метил)
амино]фенил}этила: 31
Работают так же, как при получении соединения 30, при этом используют 3-нитробензол-этанол вместо 4-нитробензол-этанола на первом этапе синтеза. Получают бледно-желтый
порошок. Температура плавления: 145-148oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,50 (с широкий, 1Н, NH+); 9,82 (с, широкий, 1Н, NH+); 8,99 (с, широкий, 1Н, NH+); 8,15 (м, 2Н, тиофен); 7,81 (с, 1Н, ОН); 7,75 (с, 2Н,
ИК: νOH: 3620 см-1; νC=O ((сложный эфир): 1707 см-1; νC=N(амидин): 1654 см-1.
Пример 32: хлоргидрат 3,5-бис-(1,1-диметилэтил)-4-гидрокси-бекзоат 2-{
2-[(2-тиенил-(имино)метил)амино]фенил}этила: 32
Работают так же, как при получении соединения 30, при этом используют 2-нитробензол-этанол вместо 4-нитробензол-этанола на первом этапе
синтеза. Получают бежевый порошок. Температура плавления: 139-145oС.
ЯМР1H (400 МГц, ДМСО d6, δ): 11,50 (с широкий, 1Н, NH+); 9,80 (с широкий, 1Н, NH+); 8,65 (с широкий, 1Н, NH+); 8,15 (м, 2Н, тиофен); 7,80 (с, 1Н, ОН); 7,70 (с, 2Н, Ph-ON); 7,60 (м, 1Н, Ph); 7,45 (м, 3Н, Ph); 7,35 (с, 1Н, тиофен); 4,40 (м, 2Н, O-СН2); 3,00 (м, 2Н, СН2); 1,35 (с, 18Н, 2хtBu).
ИК: νC=O((сложный эфир): 1728 см-1; νC=N(амидин): 1649 см-1.
Пример 33: Гидрохлорид 2-Гидрокси-5-метокси-N-{2-[4-[(2-тиенил(имино)-метил)амино]фенил]этил}бензамида:
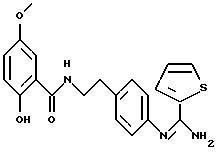
33.1.) 2-гидрокси-5-метокси-N-{2-(4-нитрофенил)этил}бензамид:
Гидрохлорид 4-нитрофениламина (1,81 г; 8,9 ммоль), триэтиламин (2,8 мл; 20 ммоль), гидроксибензотриазол (1,45 г; 10,7 моль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (3,75 г; 19,62 ммоль) добавляют к раствору 1,5 г (8,9 ммоль) 5-метоксисалициловой кислоты в дихлорметане (80 мл). Реакционную среду перемешивают в течение ночи при 25oС. Смесь разбавляют 40 мл воды и перемешивают в течение 10 минут, затем продукт экстрагируют дихлорметаном. Органический раствор сушат над сульфатом натрия, фильтруют и концентрируют в вакууме и полученный после выпаривания остаток очищают на колонке с двуокисью кремния (элюент=этилацетат/гептан; 50/50) для получения белого твердого вещества, выход которого составляет 64%. Температура плавления: 200oС.
ЯМР1H (ДМСО d6, 400 МГц, δ): 3,00 (м, 2Н, СН2); 3,60 (м, 2Н, СН2); 3,70 (С, 3Н, -ОСН3); 6,80 (д, аром., J=8,8 Гц); 7,00 (д, 1Н, аром., J=8,8 Гц); 7,35 (с, 1Н, аром.); 7,50 (д, 2Н, аром., J=7,8 Гц); 8,10 (д, 2Н, аром., J= 7,6 Гц); 8,90 (широкий с, 1Н, CO-NH); 11,90 (широкий с, 1Н, -ОН).
33.2) 2-гидрокси-5-метокси-N-{2-(4-аминофенил)-этил}бензамид:
2,10 г промежуточного продукта 33.1
(6,64 ммоль) растворяют в смеси этанола (40 мл) и дихлорметана (60 мл) и добавляют 0,3 г палладия на углероде (10%). Реакционную среду помещают в атмосферу водорода при давлении 4 бар. Катализатор
отфильтровывают и растворитель выпаривают при пониженном давлении. Полученный после выпаривания остаток очищают на колонке с двуокисью кремния (элюент: этилацетат/гептан; 50/50) с получением
беловато-кремового твердого вещества. Температура плавления: 130oС.
ЯМР1Н (ДМСО d6, 400 МГц, δ): 2,67 (т, 2Н, СН2, J-7,4 Гц); 3,43 (м, 2Н, СН2); 3,73 (С, 3Н, -ОСН3); 4,90 (С, 2Н, NH2), 6,50 (д, аром., J=7,8 Гц); 6,82 (д, 1Н, аром., J=8,8 Гц); 6,90 (д, 2Н, аром., J=8,7 Гц); 7,00 (д, 1Н, аром. , J= 8,8 Гц); 7,39 (С, 1Н, аром.); 8,87 (широкий с, 1Н, CO-NH); 12,09 (широкий с, 1Н, -ОН).
33.3) Гидрохлорид
2-гидрокси-5-метокси-N-{2-[4-[(2-тиенил-(имино)метил)амино]фенил]этил}бензамида:
Промежуточный продукт 33.2 (0,6 г; 2,1 ммоль) растворяют в 2-пропаноле (10 мл), добавляют 0,896 г гидроиодида
S-метил-2-тиофентиокарбоксимида (3,14 ммоль) (Ann.Chim.(1962), 7, 303-337).
После нагревания при 50oС в течение 15 часов реакционную смесь концентрируют в условиях вакуума до сухости. Остаток поглощают в этилацетате и насыщенном растворе карбоната натрия. После декантации органическую фазу последовательно промывают 50 мл насыщенного раствора карбоната натрия, водой и насыщенным солевым раствором. Органический раствор сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Полученный после выпаривания остаток очищают на колонке с двуокисью кремния (элюент: этанол/дихлорметан, 5/95). Получают 0,523 г свободного основания. Гидрохлорид получают из 0,523 г (1,32 ммоль) основания, растворенного в 20 мл ацетона, и образуют соль в присутствии 2,0 мл (2,0 ммоль) молярного раствора НСl в безводном диэтиловом эфире. Полученные кристаллы отфильтровывают и промывают в диэтиловом эфире с целью получения после высушивания 0,58 г (выход 64%) желаемого продукта в форме белого твердого вещества. Температура плавления: 242oС.
ЯМР1H (ДМСО d6, 400 МГц, δ): 2,94 (т, 2Н, CH2, J=7,2 Гц); 3,58 (м, 2Н, СН2); 3,73 (с, 3Н, -ОСНз); 6,85 (д, 1H, аром., J=9,0 Гц); 7,01 (д, 1Н, аром. , J= 8,8 Гц); 7,20-7,70 (м, 6Н, аром.); 8,16 (м, 2Н, тиофен); 8,88 (широкий с, 1Н, NH+); 9,11 (широкий т, 1Н, CO-NH, J=5,15 Гц); 9,83 (широкий с, 1Н, NH+); 11,54 (широкий с, 1Н, NH+), 12,10 (с, 1Н, -ОН).
ИК: νC=N (амидин): 1645 см-1; νC=O(амид): 1645 см-1.
Пример 34: Гидрохлорид N-{4-[2-({2-[3,
5-ди(трет-бутил)-4-гидрроксифенокси]этил}амино)этил]фенил}-2-тиофенкарбоксимидамида:
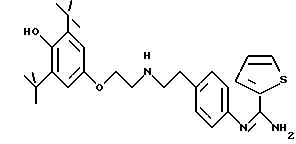
34.1) 2-[3,5-ди(трет-бутил)-4-гидроксифенокси]уксусная кислота:
3,6 мл (46 ммоль) трифторуксусной кислоты добавляют к раствору 1,56 г (4,64 ммоль) трет-бутил 2-[3, 5-ди(трет-бутил)-4-гидроксифенокси] ацетата (полученному в соответствии с J.Heterocycl.Chem. (1994) 31, 1439-1443) в 20 мл дихлорметана. Реакционную смесь перемешивают в течение 1 часа, концентрируют в вакууме и остаток растворяют в 5,0 мл Et2О. Органический раствор дважды экстрагируют 25 мл насыщенного раствора NаНСО3, затем водную фазу промывают 25 мл Et2O. Водный основной раствор затем подкисливают при 0oС насыщенным раствором KHSO4 и затем ожидаемый продукт экстрагируют с использованием 25 мл Et2O дважды. Органический раствор сушат над сульфатом натрия, фильтруют и концентрируют в вакууме с получением белого порошкообразного продукта с выходом 70%. Температура плавления: 172-173oС.
34.2)
2,6-ди(трет-бутил)-4-{2-[(4-нитрофенетил)-амино]-этокси}фенол:
1,92 г (4,5 ммоль) 2-[3,5-ди(трет-бутил)-4-гидроксифенокси]-N-(4-нитрофенетил)ацетамида (полученного в качестве промежуточного
продукта с использованием процедуры примера 1 для промежуточного продукта 34,1) вводят в колбу, содержащую 80 мл безводного ТГФ, в атмосфере аргона. К реакционной смеси добавляют раствор 13,45 мл (13,
45 ммоль) ВН3 в ТГФ и смесь перемешивают при нагревании при температуре кипения с обратным холодильником в течение 4 часов 30 минут. В конце реакции добавляют 10 мл МеОН и нагревание
продолжают еще 30 минут. Затем реакционную смесь концентрируют в вакууме, остаток поглощают в 30 мл 3 N смеси МеОН/НСl (2/1) и смесь кипятят с обратным холодильником еще в течение часа. Реакционной
смеси дают остыть до 22oС, разбавляют 50 мл CH2Cl2 и добавляют 2 М водного раствора едкого натра до получения щелочного рН. После декантации органическую фазу
последовательно промывают 20 мл воды и 20 мл насыщенного солевого раствора, сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Ожидаемый продукт очищают при помощи хроматографии на
колонке с двуокисью кремния (элюент: CH2Cl2/EtOH: 25/1). Получают бледно-желтое твердое вещество.
34.3) трет-Бутил 2-[3,5-ди(трет-бутил)-4-гидроксифенокси]
-этил(4-нитрофенил)карбамат:
1,27 г (3 ммоль) промежуточного продукта 34.2 растворяют в смеси 15 мл CH2Cl2 и 0,53 мл (3 ммоль) N,N-диизопропилэтиламина. Смесь охлаждают
при помощи ледяной бани перед добавлением 0,67 г (3 ммоль) (Вос)2О одной порцией. Реакционную смесь перемешивают в течение 5 часов при 23oС. После концентрации под вакуумом
остаток поглощают в 50 мл этилацетата и выливают в смесь воды со льдом. Органическую фазу декантируют, последовательно промывают 20 мл воды и 20 мл насыщенного солевого раствора. После сушки над
сульфатом натрия, фильтрации и концентрирования в вакууме получаст твердое вещество желтого цвета с количественным выходом.
34.4) трет-Бутил 4-аминофенетил { 2-[3,
5-ди(трет-бутил)-4-гидроксифенокси]этил}карбамат:
1,7 г (3 ммоль) промежуточного продукта 34.3 и 0,8 мл (15 ммоль) гидразингидрата растворяют в 50 мл абсолютного этанола перед добавлением 0,2
г никеля Ренея. Реакционную смесь нагревают до температуры кипения с обратным холодильником до полного исчезновения исходного продукта (4 часа 30 минут). После охлаждения в колбу добавляют небольшое
количество двуокиси кремния и затем растворитель удаляют в условиях вакуума. Полученное порошкообразное вещество помещают непосредственно в верхнюю часть хроматографической колонки. Продукт элюируют с
использованием 1/2 смеси AcOEt/гептана. Ожидаемый продукт получают в форме масла оранжевого цвета с выходом 83%.
34.5) трет-Бутил 4-{ [амино(2-тиенил)метилиден]амино}-фенетил{2-[3,
5-ди(трет-бутил)-4-гидроксифенокси]этил}карбамат:
0,93 г (1,9 ммоль) промежуточного продукта 34.4 и 0,60 г (2,1 ммоль) гидроиодида S-метил-2-тиофентиокарбоксимида (Ann. Chim. (1962) 7,
303-337) растворяют в 50 мл изопропанола. Реакционную смесь перемешивают в течение 15 часов при 60oС. После выпаривания растворителя в вакууме остаток поглощают в 60 мл AcOEt и 40 мл
насыщенного раствора Nа2СО3. Смесь интенсивно перемешивают и затем декантируют. Органическую фазу последовательно промывают в 20 мл воды, 20 мл насыщенного солевого раствора,
сушат над сульфатом натрия, фильтруют и концентрируют в вакууме. Полученный продукт используют непосредственно на следующей стадии без дополнительной очистки.
34.6) N-{ 4-[2-({ 2-[3,
5-ди(трет-бутил)-4-гидроксифенокси] -этил} амино)этил]фенил}-2-тиофенкарбоксимидамид:
0,8 г (1,3 ммоль) промежуточного продукта 34.5 растворяют в 20 мл этанола и добавляют 7 мл 3N раствора
хлористоводородной кислоты. Смесь перемешивают в течение 1 часа при 23oС. После охлаждения при помощи ледяной бани раствор подщелачивают добавлением Nа2СО3 в форме
порошка и смесь затем разбавляют 50 мл AcOEt. После интенсивного перемешивания и декантации органическую фазу промывают 20 мл насыщенного солевого раствора, сушат над сульфатом натрия, фильтруют и
концентрируют до сухости. Остаток очищают на колонке с двуокисью кремния (элюент: АсОЕt /гептан/NН4ОН: 12,5/12,5/0,5).
34.7) Гидрохлорид N-{4-[2-({2-[3,
5-ди(трет-бутил)-4-гидроксифенокси]-этил}амино)этил]фенил}-2-тиофенкарбоксимидамида:
Промежуточный продукт 34.6 (0,29 г, 0,6 ммоль) растворяют в 30 мл абсолютного этанола, смесь охлаждают при
помощи ледяной бани, после чего добавляют 2,4 мл (2,4 ммоль) lN раствора НСl в безводном эфире. После перемешивания в течение 30 минут при 23oС растворитель выпаривают в вакууме с
получением твердого вещества ярко-розового цвета. Температура плавления: 151-153oС.
Пример 35:
(E)-N-(4-{[амино(2-тиенил)метилиден]амино}-фенетил)-3-(2-гидроксифенил)-2-пропенамид:
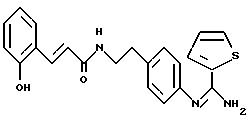
К раствору 8,9 ммоль 2-оксикоричной кислоты в дихлорметане (80 мл) добавляют гидрохлорид 4-нитрофенетиламина (8,9 ммоль), триэтиламин (20 ммоль), гидроксибензотриазол (10,7 ммоль) и гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (19,62 моль). Реакционную среду перемешивают в течение ночи при 25С, смесь разбавляют 40 мл воды и перемешивают в течение 10 минут, затем продукт экстрагируют дихлорметаном. Органический раствор сушат над сульфатом натрия, фильтруют и концентрируют в вакууме и полученный после выпаривания остаток очищают на колонке с двуокисью кремния (элюент: этилацетат/гептан; 50/50) с получением ожидаемого промежуточного продукта.
3,59 г (16 ммоль) SnCl2, 2H2O вводят в раствор промежуточного продукта, полученного на предыдущей стадии (3,18 ммоль) в 50 мл смеси этилацетат/этанол/ацетон (2/1/2). Реакционную смесь нагревают до температуры кипения с обратным холодильником в течение 5 часов и затем, после охлаждения, концентрируют в вакууме до половины объема. Полученный после выпаривания остаток затем выливают в 50 мл насыщенного раствора холодного NаНСО3 и экстрагируют 100 мл этилацетата. Смесь фильтруют на целите и фильтрат декантируют. Органческую фазу последовательно промывают 50 мл воды и 50 мл насыщенного солевого раствора. После высушивания над сульфатом магния и фильтрации органический раствор концентрируют в вакууме. Получают ожидаемый промежуточный продукт, который используют на следующей стадии.
Промежуточный продукт, полученный на предыдущей стадии (2,1 ммоль), растворяют в 2-пропаноле (10 мл) и туда добавляют 3,14 ммоль гидроиодида 5-метил-2-тиофентиокарбоксимида (Ann.Chim. (1962), 7, 303-337). После нагревания в течение 15 часов при 50oС реакционную смесь концентрируют в вакууме до сухости. Остаток поглощают в этилацетат и насыщенный раствор карбоната натрия. После декантации органическую фазу последовательно промывают 50 мл насыщенного раствора карбоната натрия, водой и насыщенным солевым раствором. Органический раствор сушат над сульфатом натрия, фильтруют и выпаривают при пониженном давлении. Полученный после выпаривания остаток очищают на колонке с двуокисью кремния (элюент: этанол/дихлорметан, 5/95). Продукт получают в форме свободного основания, которое преобразуют в гидрохлорид следующим путем: к 1,32 ммоль основания, растворенного в 20 мл ацетона, добавляют 2,0 мл (2,0 ммоль) молярного раствора НСl в безводном диэтиловом эфире. Полученные кристаллы фильтруют и промывают в диэтиловом эфире с целью получения после сушки искомого продукта в форме твердого вещества белого цвета. Температура плавления: 145-155oС.
Пример 36:
(Е)-N-(4-{[амино(2-тиенил)метилиден]амино}-фенетил)-3-(3,4-дигидроксифенил)-2-пропенамид:
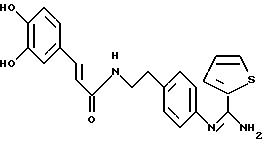
Это соединение получают в соответствии с экспериментальным протоколом, аналогичным описанному для соединения 35. Желтое масло. МН+: 408,08.
Пример 37:
(E)-N-(4-{[амино(2-тиенил)метилиден]амино}-фенетил)-3-(4-гидрокси-3,5-диметоксифенил)-2-пропенамид:
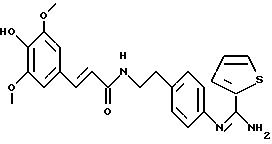
Это соединение получают в соответствии с экспериментальным протоколом, аналогичным описанному для соединения 35. Твердое вещество бледно-желтого цвета. Температура плавления: 196,4-197,6oС.
Пример 38: Гидрохлорид N-(2-гидрокси-5-метокси)-4-{[2-тиенил(имино)метил]амино}бензолбутанамида
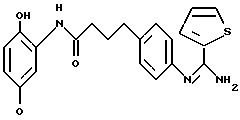
38.1) 2-амино-4-метоксифенол
Используемый экспериментальный протокол такой же, как описано для промежуточного соединения 33.2, при этом вместо 2-гидрокси-5-метокси-N-{2-(4-нитрофенил)этил}бензамида используют 4-метокси-2-нитрофенол.
38.2) N-(2-гидрокси-5-метокси)-4-нитробензолбутанамид
Используемый экспериментальный
протокол такой же, как описано для промежуточного соединения 33.1, при этом вместо 5-метоксисалициловой кислоты используют 4-(4-нитрофенил)бутановую кислоту и вместо гидрохлорида 4-нитрофениламина
используют 2-амино-4-метоксифенол.
38.3) N-(2-гидрокси-5-метокси)-4-аминобензолбутанамид
Используемый экспериментальный протокол такой же, как описано для промежуточного
соединения 33.2, при этом вместо 2-гидрокси-5-метокси-N-{2-(4-нитрофенил)этил} бензамида используют N-(2-гидрокси-5-метокси)-4-нитробензолбутанамид.
38.4) Гидрохлорид N-(2-гидрокси-5-метокси)-4-{ [2-тиенил-(имино)метил] амино}бензолбутанамида.
Используемый экспериментальный протокол такой же, как описано для промежуточного соединения 33, при этом вместо 2-гидрокси-5-метокси-N-{2-(4-аминофенил)этил} бензамида используют N-(2-гидрокси-5-метокси)-4-аминобензолбутанамид. Температура плавления: 199,1-200,7oС.
ЯМР1H (ДМСО d6, 400 МГц, δ): 1,9 (м, 2Н, СН2); 2,45 (м, 2Н, СН2); 2,70 (м, 2Н, СН2); 3,64 (с, 3Н, -ОСН3); 6,51-6,9 (м, 2Н, аром.); 7,36 (м, 6Н, аром. ); 8,17 (м, 2Н, тиофен); 8,88 (широкий с, 1Н, NH+); 9,38 (с, 2Н, -ОН & CONH); 9,81 (широкий с, 1Н, NH+); 11,52 (широкий с, 1Н, NH+).
ИК: νC=N(амидин): 1662 см-1; νC=O(амид): 1693 см-1.
Пример 39: N-(2-гидрокси-5-метокси)-4-{
[2-тиенил(имино)-метил]амино} бензолпропанамид
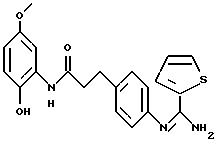
39.1) N-(2-гидрокси-5-метокси)-4-N-Вос-бензолпропанамид.
Используемый экспериментальный протокол такой же, как описано для промежуточного соединения 38.2, при этом вместо 4-(4-нитрофенил)бутановой кислоты используют 3-(4-трет-бутоксикарбонил-амино)фенилпропановую кислоту.
39.2 N-(2-гидрокси-5-метокси)-4-аминобензолпропанамид.
Концентрированную хлористоводородную кислоту (0,6 мл) добавляют к раствору 3,86 ммоль промежуточного продукта 39.1 в метаноле (30 мл). Реакционную среду перемешивают в течение ночи при 25o С. Растворители выпаривают и остаток последовательно промывают 3•40 мл водой и затем 40 мл насыщенного солевого раствора. Органический раствор сушат над сульфатом натрия, фильтруют и концентрируют при пониженном давлении с получением коричневого масла, которое является достаточно чистым для использования непосредственно на следующей стадии.
39.3) Гидрохлорид N-(2-гидрокси-5-метокси)-4-{[2-тиенил-(имино)метиламино}бензолпропанамида.
Используемый экспериментальный протокол такой же, как описано для промежуточного соединения 38, при этом вместо N-(2-гидрокси-5-метокси)-4-аминобензолбутанамида используют N-(2-гидрокси-5-метокси)-4-аминобензолпропанамид.
Температура плавления: 197,6-197,8oС.
ЯМР1H (ДМСО d6, 400 МГц, δ): 2,49 (м, 2Н, СН2); 2,70 (м, 2Н, СН2); 3,61 (с, 3Н, -ОСН3); 6,50-7,0 (м, 2Н, аром.); 7,40 (м, 6Н, аром.); 8, 90 (широкий с, 1Н, NH+); 9,40 (с, 2Н, -ОН & CONH); 9,90 (широкий с, 1Н, NH+); 11,60 (широкий с, 1Н, NН+).
ИК: νC=N(амидин): 1660 см-1; νC=O(амид): 1685 см-1.
Испытание жизнеспособности клеточной линии макрофагов мышей J774-A1
Клеточная линия
Клеточную линию макрофагов мышей J 774 А1 получали от АТСС и поддерживали при 37oС при 5% СO2 в модифицированной Дульбекко минимальной основной среде (DMEM)/10% сыворотки плода
коровы (FBS).
Обработка клеток
Клетки собирали после центрифугирования, промывали и суспендировали в DMEM/10% FBS. Клетки (5•104 клеток на лунку)
культивировали в 96-луночных планшетах в течение 24 часов, а затем в течение 18 часов в присутствии испытываемых соединений при концентрации 10, 30, 50 или 100 мкМ. Испытываемые соединения растворяли
в диметилсульфоксиде (ДМСО) или этаноле в виде исходных растворов 150Х, 500Х или 1000Х. Последующие разведения осуществляли в DMEM. Конечная концентрация ДМСО или этанола не оказывала влияния на
морфологию клеток или жизнеспособность клеток.
Оценка жизнеспособности клеток.
Анализ с использованием синего красителя (Alamar blue) выполняли в соответствии с
инструкциями изготовителя (Interchim). Вкратце, после отсасывания среды, раствор Alamar Blue, разбавленный 1/10 в DMEM без фенольного красного/ 10% FBS, добавляли в течение 3 часов при 37oС
(5% CO2). Колориметрическое определение осуществляли при 570 нм и 620 нм на планшет-ридере. Процент ингибирования метаболической активности в ответ на действие испытываемого соединения в
сравнении с необработанными клетками рассчитывали по формуле:
Величина CI50 является концентрацией, которая снижает % жизнеспособности клеток на 50% и получена экстраполированием из графиков отношений доза-ответ.
Фармакологическое исследование соединений
согласно изобретению
Изучение действия на нейронную конститутивную NO-синтазу мозжечка крысы.
Ингибирующую активность соединений согласно изобретению определяют путем измерения их действия на трансформацию через NO-синтазу [3H]-аргинина в [3H] L-цитрулине в соответствии с модифицированной методикой Bredt и Snyder (Proc. Natl. Acad. Sci. USA, (1990) 87, 682-685). Мозжечки крыс Sprague-Dawley (300 g - Charles River) быстро извлекают, вскрывают при 4oС и гомогенизируют в объеме экстракционного буфера (HEPES 50 мМ, EDTA 1 mM, рН 7, 4, пепсатин А 10 мг/мл, лейкопептин 10 мг/мл). Затем гомогенаты центрифугируют при скорости 21000 g в течение 15 минут при 4oС. Анализ осуществляют в стеклянных пробирках, в которые распределяют 100 мкл инкубационного буфера, содержащего 100 мМ HEPES (рН 7,4), 2 мМ EDTA, 2,5 мМ CaCl2, 2 мМ дитиотреитола, 2 мМ восстановленного NADPH и 10 мкг/мл кальмодулина. Добавляют 25 мкл раствора, содержащего 100 нМ [3H]L-аргинина (удельная активность: 56.4 Ci/ммоль, Amesrsham) и 40 мкМ нерадиоактивного L-аргинина. Реакцию инициируют добавлением 50 мкл гомогената, причем конечный объем равен 200 мкл (недостающие 25 мкл составляет либо вода, либо тестируемое соединение). Через 15 минут реакцию останавливают с 2 мл буферного раствора (20 мМ HEPES, рН 5,5, 2 мМ EDTA). После переноса образцов на 1 мм-ую колонку смолы DOWEX, определяют количество радиоактивности с помощью спектрометра с жидкой сцинциляцией. Соединения примеров 6, 7, 13 и 14, описанных выше, имеют CI50 меньше 3,5 мкМ.
Изучение действия на перекисное окисление липидов коры головного мозга крысы.
Ингибирующую активность соединений согласно изобретению определяют измерением их воздействия на степень перекисного окисления липидов, определяемую концентрацией малондиальдегида (МДА). Малондиальдегид, продуцируемый при перекисном окислении ненасыщенных жирных кислот, является показателем перекисного окисления липидов (Н Esterbauer and KH Cheese-man, Meth. Enzymol. (1990) 186: 407-421). Самцы крыс породы Sprague Dawley весом от 200 до 250 г (Charles River) были умерщвлены путем обезглавливания. Кору головного мозга извлекали, затем гомогенизировали в гомогенизаторе Томаса в буферном растворе Tris-HCl 20 мМ, рН 7,4. Гомогенат центрифугировали в 2 раза при скорости 50000 g в течение 10 минут при 4oС. Осадок хранили при -80oС. В день эксперимента осадок снова суспендировали до концентрации 1 г/15 мл и центрифугировали при скорости 515 g в течение 10 минут при 4oС. Всплывшую часть сразу же использовали для определения липидного перекисного окисления липидов. Гомогенат коры головного мозга крысы (500 мкл) инкубировали при 37oС в течение 15 минут в присутствии тестируемых соединений или растворителя (10 мкл). Реакцию перекисного окисления липидов инициировали добавлением 50 мкл FeCl2 (1 мМ), EDTA (1 мМ) и аскорбиновой кислоты (4 мМ). После 30 минут инкубации при 37oС реакцию останавливали добавлением 50 мкл раствора гидроксилированного трет. бутилтолуола (ВНТ, 0,2%). Количество МДА определяли с помощью колориметрического теста при взаимодействии ауксохромного реагента (R) N-метил-2-фенилиндол (650 мкл) с 200 мкл гомогената в течение 1 часа при 45oС. Конденсация одной молекулы МДА с двумя молекулами реагента R образует стабильный хромофор, у которого длина волны максимальной абсорбции равна 586 нм. (Caldwell et coll. European J.Pharmacol. (1995) 285, 203-206). Соединения примеров 5,8,9,10,12,13,14,16,17,18,19,20,21,26 и 27, описанных выше, все имеют CI50 меньше 30 мкМ. Результаты фармокологических испытаний представлены в таблице.
Реферат
Изобретение относится к новым производным 2- (иминометил) аминобензола общей формулы (I), где А означает: либо радикал, представленный в формуле изобретения, в котором R1 и R2 означают, независимо, атом водорода, группу ОН, линейный или разветвленный алкил или алкоксил, имеющий от 1 до 6 атомов углерода, R3 означает атом водорода, линейный или разветвленный алкил с 1-6 атомами углерода или радикал -COR4, R4 означает линейный или разветвленный алкил с 1-6 атомами углерода, либо радикалы, представленные в формуле изобретения, R5 означает атом водорода, группу ОН или линейный или разветвленный алкил или алкоксил с 1-6 атомами углерода, В означает тиенил, Х означает -Z1-, -Z1-CO-, -Z1-NR3-CO, -СН=СН-СО- или простую связь, Y означает радикал, выбираемый из радикалов -Z2-Q, пиперазинил, гомопиперазинил, -NR3-CO-Z2-Q-, -NR3-O-Z2-, -O-Z2-Q-, в которых Q означает простую связь, -O-Z3 и -N(R3)-Z3-, Z1, Z2 и Z3 означают независимо простую связь или линейный или разветвленный алкилен с 1-6 атомами углерода, предпочтительно Z1, Z2 и Z3 означают -(СН2)m-, причем m - это целое число, равное от 0 до 6, R6 означает атом водорода или группу ОН, или их фармацевтически приемлемые соли. Изобретение может быть использовано в качестве лекарственных средств. Также раскрыты промежуточные соединения, способ получения соединений формулы (I) и фармацевтические композиции. 5 с. и 14 з.п. ф-лы, 1 табл.
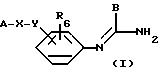
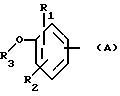
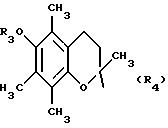
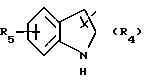
Формула
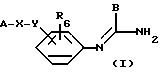
в которой А означает:
либо радикал
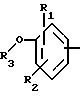
в котором R1 и R2 означают, независимо, атом водорода, группу ОН, линейный или разветвленный алкил или алкоксил, имеющий от 1 до 6 атомов углерода;
R3 означает атом водорода, линейный или разветвленный алкил с 1-6 атомами углерода или радикал -COR4;
R4 означает линейный или разветвленный алкил с 1-6 атомами углерода,
либо радикал
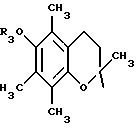
в котором R3 имеет значение, указанное выше,
либо радикал
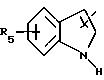
в котором R5 означает атом водорода, группу ОН или линейный, или разветвленный алкил, или алкоксил с 1-6 атомами углерода;
В означает тиенил;
Х означает -Z1-, -Z1-CO-, -Z1 -NR3-CO-, -CН= CH-CO-, или простую связь;
Y означает радикал, выбираемый из радикалов -Z2-Q, пиперазинил, гомопиперазинил, -NR3-CO-Z2-Q-, -NR3-O-Z2-, -O-Z2-Q-, в которых Q означает простую связь, -O-Z3-, -N(R3)-Z3-, Z1, Z2 и Z3 означают независимо простую связь или линейный или разветвленный алкилен с 1-6 атомами углерода, предпочтительно Z1, Z2 и Z3 означают -(СН2)m-, причем m - целое число, равное от 0 до 6;
R6 означает атом водорода или группу ОН;
или фармацевтически приемлемая соль соединения общей формулы (I), за исключением соединения следующей формулы:
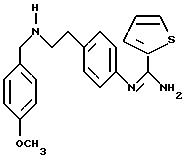
2. Соединение по п. 1, представляющее собой одно из следующих соединений:
3, 5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 4-[(2-тиенил-(имино)метил)амино] фенил} -бензамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 4-{ [(2-тиенил-(имино)метил)амино] фенил] метил} -бензамид;
4-ацетокси-3,5-диметокси-N-{ 4-[[(2-тиенил(имино)метил)амино] фенил] метил} -бензамид;
3,5-диметокси-4-гидрокси-N-{ 4-[[(2-тиенил(имино)метил)амино] фенил] метил} -бензамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 4-[2-[(2-тиенил(имино)метил)амино] фенил] этил} -бензамид;
4-ацетокси-3,5-диметокси-N-{ 4-[2-[(2-тиенил(имино)метил)амино] фенил] этил} -бензамид;
3,5-диметокси-4-гидрокси-N-{ 4-[2-[(2-тиенил(имино)метил)амино] фенил] этил} -бензамид;
3,4,5-тригидрокси-N-{ 4-[2-[(2-тиенил(имино)метил)-амино] фенил-этил} -бензамид;
N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензоил] -1-пиперазинил] -фенил} -2-тиофенкарбоксимидамид;
N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензил] -1-пиперазинил] -фенил} -2-тиофенкарбоксимидамид;
N-{ 4-[4-[3,5-диметокси-4-гидроксибензоил] -1-пиперазинил] -фенил} -2-тиофенкарбоксимидамид;
3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{ 4-[(2-тиенил(имино)метил)амино] фенил} -2Н-1-бензопиран-2-карбоксамид;
N-{ 4-[4-(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-{ 4-[4-[(5-метокси-1Н-индол-3-ил)метилкарбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-[4-[4-[{ 3-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -1-оксо-2-пропенил} -1-пиперазинил] фенил] ] -2-тиофенкарбоксимидамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 3[[2-тиенил-(имино)метил)амино] фенил] метил} -бензамид;
N-[3,5-бис-(1, 1-диметилэтил)-4-гидроксифенил] -N'-{ { 4-[(2-тиенил(имино)метил)амино] фенил} метил} -мочевина;
N-[5-[{ 3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино] -2-гидроксифенил] -2-тиофенкарбоксимидамид;
-N-[3-[{ 3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино] -4-гидроксифенил] -2-тиофенкарбоксимидамид;
N-{ 4-[4-[3,4, 5-тригидроксибензоил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-[3,5-биc-(1,1-димeтилэтил)-4-гидpoкcифeнил] -N'-{ { 4-[(2-тиенил(имино)метил)амино] фенил} карбониламино} -мочевина;
N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-{ { 4-[(2-тиенил(имино)метил)амино] фенил} метил} -тиомочевина;
N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-{ 2-{ 4-[(2-тиенил(имино)метил)амино] фенил} этил} -мочевина;
N(4-{ 4-[(3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил} фенил)-2-тиофенкарбоксимидамид;
N-[4-{ 4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1Н-1,4-диазепин-1-ил} фенил] -2-тиофенкарбоксимидамид;
(R)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7, 8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
(S)-N{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 2-[3-[(2-тиенил(имино)метил)амино] фенил] этил} -бензамид;
N-{ 4-(4-[2-(3,5-бис-(1, 1-диметилэтил)-4-гидроксифенил)-1-оксоэтил] -1-пиперазинил)фенил} -2-тиофенкарбоксимидамид;
3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат2-{ 4-[(2-тиенил(имино)метил)амино] фенил} этила;
3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат2-{ 3-[(2-тиенил(имино)метил)амино] фенил} этила;
3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат2-{ 2-[(2-тиенил(имино)метил)амино] фенил} этила
или одну из солей одного из этих соединений, в частности хлоргидрат, дихлоргидрат, фумарат или полуфумарат одного из этих соединений.
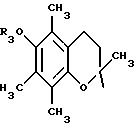
или
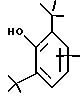
X означает -СО- или -NR3-CO-; Y означает пиперазинил, причем Q означает связь, O-Z3, и Z3 означает связь или линейный или разветвленный алкилен с 1-6 атомами углерода, а R3 означает атом водорода или линейный, или разветвленный алкил с 1-6 атомами углерода.
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 4-[2-[(2-тиенил-(имино)метил)амино] фенил] этил} -бензамид;
3,4,5-тригидрокси-N-{ 4-[2-[(2-тиенил-(имино)метил)-амино] фенил] этил} -бензамид;
N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензоил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-{ 4-[4-[3,5-бис-(1,1-диметилэтил)-4-гидроксибензил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-N-{ 4-[(2-тиенил(имино)метил)-амино] фенил} -2Н-1-бензопиран-2-карбоксамид;
N-{ 4-[4-[(3, 4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-{ 4-[4-[(5-метокси-1Н-индол-3-ил)метилкарбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-{ 3-[[(2-тиенил-(имино)метил)амино] фенил] метил} -бензамид;
N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-{ { 4-[(2-тиенил(имино)метил)амино] фенил} -метил} -мочевина;
N-[5-[{ 3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино] -2-гидроксифенил] -2-тиофенкарбоксимидамид;
N-[3-[{ 3-(3,5-бис-(1,1-диметилэтил)-4-гидроксифенил)-1-оксо-2-пропенил} амино] -2-гидроксифенил] -2-тиофенкарбосимидамид;
N-[4-[4-[3,4,5-тригидроксибензоил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
N-[3,5-бис-(1,1-диметилэтил)-4-гидроксифенил] -N'-{ { 4-[(2-тиенил(имино)метил)амино] фенил} карбониламино} -мочевина;
(R)-N-{ 4-[4-[3, 4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
(S)-N-{ 4-[4-[3,4-дигидро-6-гидрокси-2,5,7, 8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
или соль одного из этих соединений, в частности, хлоргидрат, дихлоргидрат, фумарат или полуфумарат одного из этих соединений.
N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
(R)-N-{ 4-[4[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
(S)-N-{ 4-[4-[(3,4-дигидро-6-гидрокси-2,5,7, 8-тетраметил-2Н-1-бензопиран-2-ил)-карбонил] -1-пиперазинил] фенил} -2-тиофенкарбоксимидамид;
или соль одного из этих соединений, в частности, хлоргидрат, дихлоргидрат, фумарат или полуфумарат одного из этих соединений.
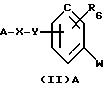
в которой W означает амино или нитро;
А означает:
либо радикал
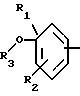
в котором R1 и R2 означают, независимо, атом водорода, группу ОН, линейный или разветвленный алкил или алкокси с 1-6 атомами углерода;
R3 означает атом водорода, линейный или разветвленный алкил с 1-6 атомами углерода или радикал -COR4;
R4 означает линейный или разветвленный алкил с 1-6 атомами углерода,
либо радикал
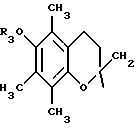
в котором R3 имеет значение, указанное выше,
либо радикал
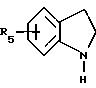
в котором R5 означает алкокси c 1-6 атомами углерода;
Х означает -Z1-, -Z1-CO-, -Z1 -NR3-СО-, -CH= CH-CO-, или простую связь;
Y означает радикал, выбираемый из радикалов -Z2-Q, пиперазинила, гомопиперазинила, -NR3-CO-Z2-Q-, -NR3-O-Z2-, -O-Z2-Q-; в которых Q означает простую связь, -O-Z3-; -N(R3)-Z3-, Z1, Z2 и Z3 означают независимо простую связь или линейный или разветвленный алкилен с 1-6 атомами углерода, причем Z1, Z2 и Z3 означают предпочтительно -(СН2)m-, m - это целое число, равное от 0 до 6;
R6 означает атом водорода или группу ОН;
за исключением 3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-(4-нитрофенил)-бензамида;
или соль соединения общей формулы (II)А, за исключением соединений, соответствующих следующим формулам:
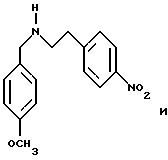
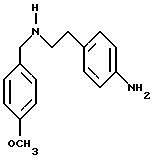
11. Промежуточные соединения общей формулы (II)А, выбранные из группы, включающей
1-{ [3, 4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил] карбонил} -4-(4-нитрофенил)пиперазин;
1-{ [3,4-дигидро-6-метокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил] карбонил} -4-(4-аминофенил)пиперазин;
1-[(3,4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)карбонил] гексагидро-4-(4-нитрофенил)-1Н-1,4-диазепин;
1-(4-аминофенил)-4-[(3, 4-дигидро-6-гидрокси-2,5,7,8-тетраметил-2Н-1-бензопиран-2-ил)карбонил] гексагидро-1Н-1,4-диазепин;
(R)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] -карбонил} -2Н-1-бензопиран-6-ол;
(R)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-аминофенил)-1-пиперазинил] -карбонил} -2Н-1-бензопиран-6-ол;
(S)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-нитрофенил)-1-пиперазинил] карбонил} -2Н-1-бензопиран-6-ол;
(S)-3,4-дигидро-2,5,7,8-тетраметил-2-{ 4-[(4-аминофенил)-1-пиперазинил] -карбонил} -2Н-1-бензопиран-6-ол;
3,5-бис-(1, 1-диметилэтил)-4-гидрокси-N-[2-(3-нитрофенил)-этил] -бензамид;
3,5-бис-(1,1-диметилэтил)-4-гидрокси-N-[2-(3-аминофенил)этил] -бензамид;
3,5-бис-(1, 1-диметилэтил)-4-гидроксибензоат2-(4-нитрофенил)этила;
3,5-бис-(1,1-диметилэтил)-4-гидроксибензоат2-(4-аминофенил)этила;
или соль одного из этих соединений.
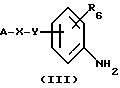
в которой А означает:
либо радикал
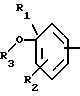
где R1 и R2 означают, независимо, атом водорода, группу ОН, линейный или разветвленный алкил с 1-6 атомами углерода, линейный или разветвленный алкокси с 1-6 атомами углерода;
R3 означает атом водорода, линейный или разветвленный алкил с 1-6 атомами углерода или радикал -COR4;
R4 означает линейный или разветвленный алкил с 1-6 атомами углерода,
либо радикал
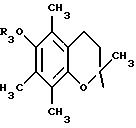
в котором R3 имеет значение, указанное выше,
либо радикал
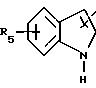
в котором R5 означает алкокси с 1-6 атомами углерода;
Х означает -Z1-, -Z1, -CO-, -Z1, -NR3-CO-, -CH= CH-CO-, или связь;
Y означает -Z2-Q, пиперазинил, гомопиперазинил, -NR3-CO-Z2-Q-, -NR3-O-Z2-, -O-Z2-Q-; в которых Q= связь, -O-Z3-; -N(R3)-Z3-, Z1, Z2 и Z3 означают независимо связь или линейный или разветвленный алкилен с 1-6 атомами углерода;
R6 означает атом водорода или группу ОН,
вводят во взаимодействие в низшем спирте, таком как метанол, этанол, изопропиловый спирт или трет-бутанол, предпочтительно в изопропиловом спирте, с соединением общей формулы (IV)
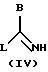
в которой В означает тиенил;
L означает удаляемую группу, в частности тиоалкил, радикал сульфоновой кислоты, трифторметансульфоновой кислоты или тозил, галогенид или фенокси, или солью соединения формулы (IV) с минеральной кислотой G, предпочтительно НСl, НBr или HI.
Комментарии