Производные дезацетилколхицина, способ их получения и противоопухолевая композиция - RU2065863C1
Код документа: RU2065863C1
Чертежи

Описание
Настоящее изобретение относится к новым производным дезацетилколхицина.
Более конкретно, настоящее изобретение относится к производным дезацетилколхицина, представленным
формулой:

где R представляет остаток, получаемый удалением COOH из сахарокарбоновой С3-C7-кислоты, и гидроксил, присутствующий в остатке, может быть соответствующим образом защищен защитной группой для гидроксила.
Давно известно, что колхицин,
представленный формулой:

обладает фармацевтической активностью по отношению к клеткам опухолей, подагре и т. д. (Колхицин в сельском хозяйстве, медицине, биологии и химии (Пресс Амис колледжа штата Айова, Айова, 1955).
Колхицин, однако, характеризуется высокой токсичностью, в связи с чем полностью вытеснен демеколхицином (дезацетил-N-метилколхицином), открытым позднее (Chem. bugng. Huvs, 37, N 41, 67 (1959)/.
Создателями настоящего изобретения в этой связи предприняты многочисленные исследования в поисках производных колхицина, обладающих меньшей токсичностью и лучшей противоопухолевой активностью, в результате которых обнаружено, что производные дезацетилколхицина, представленные формулой (1), оказывают повышенное действие по ингибированию распространения клеток опухоли, и как ожидается, найдут применение в качестве противоопухолевых средств. Указанное открытие и привело к завершению настоящего изобретения.
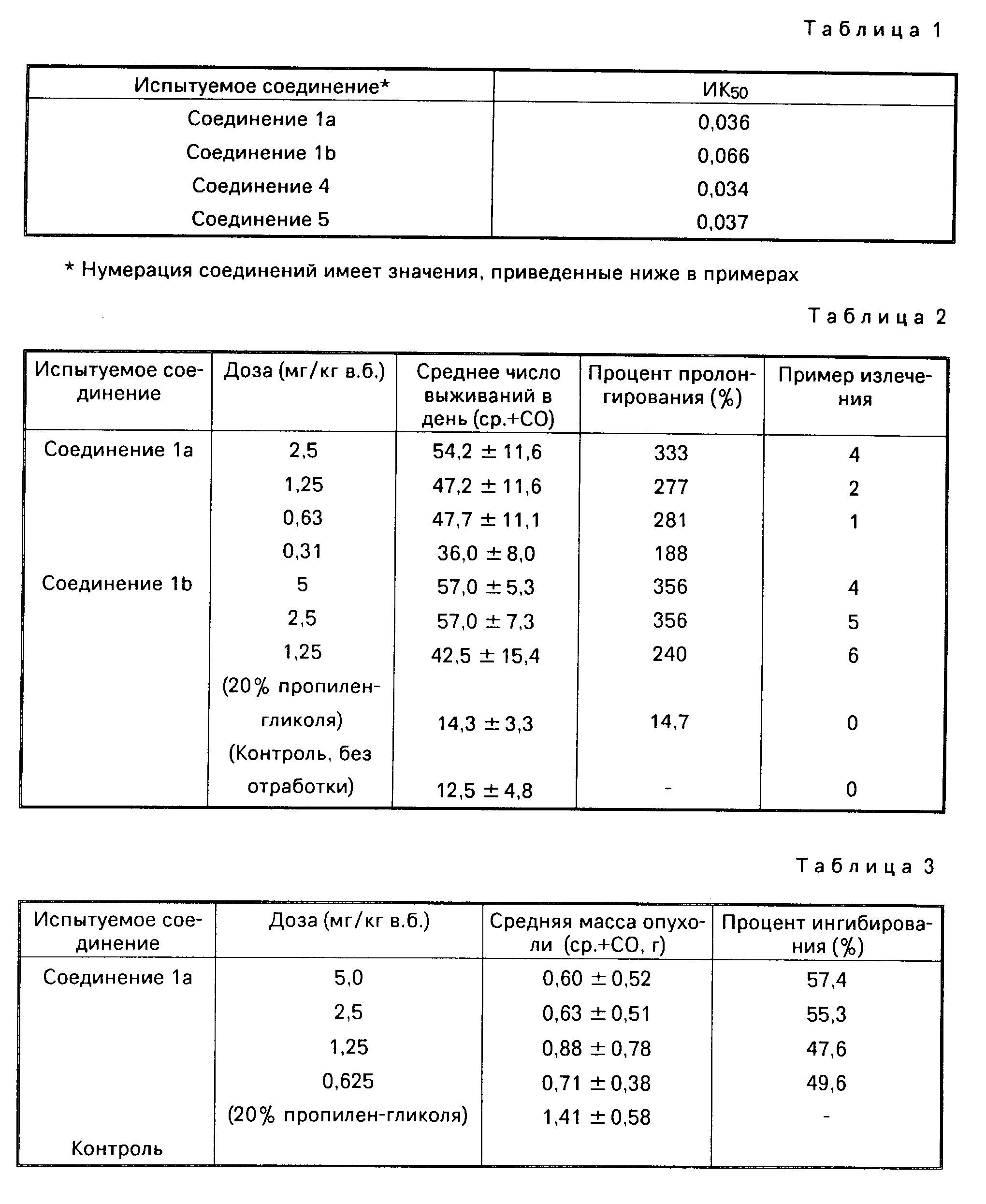
Выражение "остаток, получаемый удалением COOH из сахарокарбоновой С3 -C7-кислоты", представленный R, включает одновалентный остаток (далее "остаток сахара"), получаемый удалением COOH из моносахаридной карбоновой С3-C7-кислоты, такой как: глицериновая кислота, рибоз-карбоновая кислота, глюкуроновая кислота, глуконовая кислота или глюкогептановая кислота. Примеры подобных остатков приведены ниже.




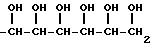
По меньшей мере часть ряда гидроксильных групп, присутствующих в остатке сахара, могут быть соответствующим образом защищены защитной группой для гидроксила. Примеры защитных групп для гидроксила включают: ацильные группы, такие как: ацетил, пропионил, бутурил, пивалоил и бензоил, и ацетальные и кетальные группы, представленные следующими формулами:
Соединения настоящего изобретения могут быть образованы, например, участием дезацетилколхицина, представленного формулой:

в реакции амидирования с использованием сахарокарбоновой кислоты формулы:
R-COOH (III)
где R принимает вышеуказанные значения, или ее реакционноспособных производных.
Амидирование дезацетилколхицина сахарокарбоновой кислотой формулы (III) или ее реакционноспособным производным (например, галоидангидридами и активными эфирами) может быть осуществлено реакцией амидирования, хорошо известной в химии пептидов.
К примеру, соединение настоящего изобретение может быть получено реакцией дезацетилколхицина с галоидангидридом сахарокарбоновой кислоты формулы (III) в присутствии основания. Вышеуказанная реакция может быть осуществлена при температуре 0-30oC, предпочтительно от 0oC до комнатной температуры. Количество галоидангидрида строго не лимитировано, обычно оно составляет 1 1,5 моль, предпочтительно 1 1,2 моль на моль дезацетилколхицина. Примеры оснований включают: третичные амины, такие как триэтиламин и пиридин; гидро(карбонаты) щелочных металлов, такие как: карбонат натрия, гидрокарбонат натрия, карбонат калия и гидрокарбонат калия. Количество основания обычно составляет 1 1, 5 моль, предпочтительно 1 1,2 моль на моль дезацетилколхицина.
Вышеуказанная реакция может быть проведена в инертном растворителе. Примеры растворителей включают: галоидированные углеводороды, такие как: хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтилен и трихлорэтилен; алифатические простые эфиры, такие как: этиловый эфир и метилцеллюлозольв и ароматические углеводороды, такие как: бензол и толуол.
Соединение настоящего изобретения может быть синтезировано прямой реакцией дезацетилколхицина с сахарокарбоновой кислотой в присутствии конденсирующего средства, такого как дициклогексилкарбодиимид или реакцией дезацетилколхицина со сложным эфиром (таким как: метиловый эфир, этиловый эфир или бутиловый эфир) сахарокарбоновой кислоты формулы (III).
Полученное в результате соединение настоящего изобретения может быть выделено и очищено известными методами, такими как: экстракция, хроматография, кристаллизация или их сочетанием.
В случае соединения настоящего изобретения, в котором в остатке сахара, представленного R, присутствует защитная группа для гидроксила, защитная группа может быть удалена в реакции удаления защитной группы, например, при необходимости в реакции гидролиза.
В вышеуказанной реакции
используемый в качестве исходного
соединения дезацетилколхицин является известным соединением ( J. Am. Chem. Soc. 75, 5292 (1953)/ и может быть образован известными методами. Или же дезацетилколхицин
может быть образован реакцией
колхицина формулы:
с триалкилоксонийфторборатом с последующей обработкой реакционной смеси водой по методике, вновь созданной создателями настоящего изобретения.
Реакция колхицина с триалкилоксонийфторборатом может быть проведена при температуре 0-30oC, предпочтительно от 0oC до комнатной температуре в инертном органическом растворителе. Примеры инертного органического растворителя включают: галоидированные углеводороды, такие как: хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтилен и трихлорэтилен; алифатические простые эфиры, такие как: этиловый эфир и метилцеллозольв и ароматические углеводороды, такие как: бензол и толуол.
Триалкилоксонийфторборат, который вводят в реакцию с колхицином, является соединением, отвечающим формуле:
(R′)3
O+•
BF (V)
где R' представляет алкил.
Конкретно рекомендуется триэтилоксонийфторборат /(C2H5)3O+•BF4-/, известный под названием реактива Меервейна.
Количество триалкилоксонийфторбората обычно составляет 1 2 моль, предпочтительно 1 1,5 моль на моль колхицина.
Предполагается, что реакция колхицина с
триалкилоксонийфторборатом дает соединение
формулы:

где R' принимает вышеуказанное значение.
Дезацетилколхицин может быть образован обработкой соединения формулы (VI) как таковое водой. Обработка водой может быть осуществлена путем перемешивания при температуре 0-30oC, особенно при комнатной температуре в течение от 30 мин до трех ч. Количество воды составляет по меньшей мере 1 моль, но обычно применяют избыточное количество на моль колхицина, используемого в качестве исходного продукта.
Полученный в результате дезацетилколхицин находится в водной фазе, из которой может быть выделен и очищен известными методами. К примеру, дезацетилколхицин может быть выделен подщелачиванием водного слоя до рН в интервале 9 10 добавлением щелочи, такой как: гидроокись калия, карбонат натрия и гидрокарбонат натрия, с последующим экстрагированием органическим растворителем, например: галоидированным углеводородом, таким как: хлористый метилен, хлороформ, четыреххлористый углерод, дихлорэтилен и трихлорэтилен, алифатическим простым эфиром, таким как: этиловый эфир и метилцеллозольв или ароматическим углеводородом, таким как: бензол или толуол.
Выделенный в результате дезацетилколхицин может быть очищен, например, его переводом в соль с винной кислотой или яблочной кислотой.
Производные дезацетилколхицина формулы (I), даваемые настоящим изобретением, проявляют прекрасную противоопухолевую активность, о чем свидетельствуют результаты испытаний in vitro и in vivo на клетках опухолей, описанные ниже.
Пример испытаний 1. Испытание на ингибирование in vitro распространения клеток опухоли.
Адриамицин-резистентные клетки лейкоза мышей ПЗ88/ADR в количестве 2 х 105 суспендируют в культурной среде RPMI 1640, содержащей 10% плодной сыворотки теленка, и культивируют 2 дня в присутствии испытуемого соединения (испытуемое соединение растворяют в диметилсульфоксиде до концентрации 1 мг/мл, после чего разбавляют фосфатным буферным раствором). Исследуют влияние на распространение клеток и определяют значения 50% ингибирующей распространение концентрации (ИК50 в мкг/мл). Полученные результаты приведены в табл. 1.
Пример испытаний 2. Испытание in vivo на асцитной опухоли Саркомы 180, трансплантированной мышам.
Клетки Саркомы 180 в количестве 1 х 106 трансплантируют в брюшную полость каждому самцу мышей линии ddy в возрасте 6 недель. Через семь дней после дня трансплантации клеток опухоли каждой группе, состоящей из 6 мышей, в брюшную полость раз в день вводят испытуемое соединение (растворенное в пропиленгликоле и разбавленное фосфатным буферным раствором до конечной концентрации пропиленгликоля 20% или ниже) и определят среднее число выживаний в день и процент пролонгирования

Пример испытаний 3. Испытание in vivo на твердой опухоли Саркомы 180, трансплантированной мышам.
Самкам мышей линии ddy в возрасте 6 недель трансплантируют каждой в спину подкожно клетки Саркомы 180 в количестве 2 х 106 клеток. На 10-ый день от 6-го дня после трансплантации в группах, состоящих из 6 мышей, непрерывно вводят в брюшную полость раз в день испытуемое соединение (растворенное в пропиленгликоле и разбавленное фосфатным буферным раствором до конечной концентрации пропиленгликоля 20% или меньше). Через тридцать дней после трансплантации опухоли ее извлекают. Определяют массу опухоли и подсчитывают среднюю массу опухоли и процент ингибирования

Пример испытаний 4. Испытание на острую токсичность.
Самцам мышей линии ddy в возрасте 5 недель вводят испытуемое соединение и в течение 1 недели определяют смертность. Методом Литчфилда-Уилкоксона подсчитывают 50% летальную дозу (ЛД50). Испытуемое соединение (соединение (а) суспендируют в 10%-ном пропиленгликоле. Готовят градиенты разбавления от максимальной концентрации 80 мг/кг в отношении 1,2 и проводят испытание (табл. 4).
Из результатов испытаний следует, что соединение настоящего изобретения обладает высокой ингибирующей активностью и, как можно ожидать, найдет применение в качестве противоопухолевого средства.
При использовании соединения настоящего изобретения в качестве противоопухолевого средства для обработки и лечения опухоли такое соединение может быть введено либо перорально, либо парантерально (например: внутривенно, внутримышечно, подкожно или внутриректально). Доза соединения может меняться в широком интервале в зависимости от симптомов заболевания, пола и веса больного, пути введения, диагноза доктора и т.п. Обычно доза составляет 1 20 мг/кг. В случае перорального введения приемлемая доза составляет 5 10 мг/кг, в случае внутривенной инъекции 2 4 мг/кг.
Соединение настоящего изобретения может быть введено в состав таблеток, гранул, порошков, капсул, сиропов, инъекций, капель или свеч. Приготовление составов может быть осуществлено известными способами путем смешивания соединения настоящего изобретения с фармацевтически приемлемым носителем или разбавителем. Примеры носителя или разбавителя включают: воду, этанол, крахмал, лактозу, сахарозу, глюкозу, маннит, окись кремния, карбоксиметилцеллюлозу, альгинат, желатин, поливинилпирролидон, глицерин, агар, карбонат кальция, парафин, каолин, тальк, стеарат кальция, стеарат магния и полиэтиленгликоль.
Пример 1. Получение дезацетилколхицина.
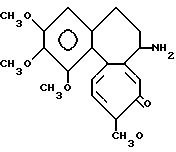
В безводном хлористом метилене растворяют четыре грамма (10 ммоль) колхицина, охлаждают от 0oC и к полученному раствору по каплям прибавляют раствор 15 ммоль триэтилоксонийфторбората (реактив Меервейна) в хлористом метилене. Раствор перемешивают 1 ч при 0oC и еще 5 ч при комнатной температуре. К реакционной смеси добавляют тридцать миллилитров воды и образовавшийся раствор перемешивают 1 ч. Затем водный слой отделяют в делительной воронке, слой хлористого метилена 5 раз экстрагируют 50 мм воды. Слой хлористого метилена сушат над сульфатом магния и используют для извлечения непрореагировавшего колхицина. Водный слой добавлением 1 н. гидроокиси натрия подщелачивают до рН 10 и экстрагируют хлороформом. Слой хлороформа сушат над сульфатом магния и затем концентрируют в испарителе. Остаток растворяют в 30 мл этанола и добавляют 1 г винной кислоты с последующим нагреванием смеси 1 ч. После охлаждения смеси до комнатной температуры осадок отфильтровывают. Полученную соль винной кислоты сушат в эксикаторе (разлагается при температуре плавления 219 220oС).
Соль винной кислоты растворяют в 50 мл воды, добавлением 1 н. гидроокиси натрия устанавливают рН 10 и экстрагируют хлороформом. Экстракт
сушат над сульфатом магния и концентрируют при пониженном давлении в испарителе с получением 1,38 г маслянистого дезацетилколхицина. Выход 39%
Непрореагировавший колхицин может быть выделен
колонной хроматографией на силикагеле начального слоя хлористого метилена из части, элюируемой растворителем бензол-ацетон (1,71 г). Выход дезацетилколхицина с учетом этого количества составляет
61%
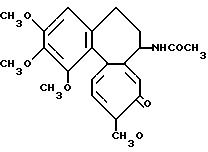
Пример 2. Ацетонидамин дезацетилколхицин-глицериновой кислоты (Соединение I)
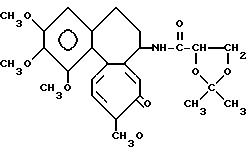
В 30 мл безводного эфира суспендируют ацетонид глицерида калия (3,6 г, 20 ммоль) и к суспензии по каплям прибавляют эфирный (5 мл) раствор 2,4 г (20 ммоль) тионилхлорида. По окончании прикапывания смесь кипятят 3 ч. После охлаждения смеси до комнатной температуры осадок отфильтровывают с отсосом и фильтрат концентрируют при пониженном давлении. Остаток растворяют в добавленном хлористом метилене.
Одновременно в 30 мл хлористого метилена растворяют 2,96 г (8,3 ммоль) дезацетилколхицина и 2,02 г (20 ммоль) триэтиламина. Смесь охлаждают до 0oC и к ней по каплям прибавляют вышеописанный раствор хлорангидрида глицериновой кислоты в хлористом метилене. После перемешивания 3 ч при 0oC раствор хлористого метилена промывают водным раствором гидрокарбоната натрия. Слой хлористого метилена сушат над сульфатом магния и затем концентрируют при пониженном давлении. Остаток разделяют колоночной хроматографией с получением из порции, элюируемой смесью бензол-ацетон (5: 1), 1,11 г продукта (соединение 1а L-изомер). Выход 28% и т.пл. 251-253oC (с разложением).
Кроме того, из фракции, элюируемой смесью
бензол-ацетон (5:2), получают 0,58 г
второго продукта (соединение 1b D-изомер). Выход 14%
Соединение 1а. ИК (KBr): 3250 см-1 (NH), 1670 см-1 (С=0). 1250 см-1
(-0-);
ЯМР (DCD13
): 1,4 (3H, c), 1,6 (3Н, с), 1,69 2,67 (4Н, м), 3,62 (3Н, с), 3,87 (3Н, с), 3,87 (3Н, с), 3,91 (3Н, с), 3,93 (3Н, с), 4-4,5 (4Н, с), 6,49-7,29 (4Н, м).
(0046)
Соединение 1b.
ИК (KBr): 3250 см-1 (NH), 1670 см-1 (С=0), 1250 см-1 (-0-);
ЯМР (DCD13): 1,37 (3Н, с), 1,46 (3Н, с) 1,69
2,67 (4Н, м), 3,62 (3Н, с), 3,86 (3Н, с),
3,91 (3Н, с), 3,97 (3Н, с), 4 4,5 (4Н, с), 6,49 - 7,29 (4Н, м).
Пример 3. Амид дезацетилколхицин-глицериновой кислоты (N-(5,6,7,
9-тетрагидро-1,2,3,
10-тетраметокси-4-оксобензол/a/ гептален-7-ил)глицероамид/ (соединение 2)
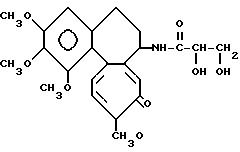
К метанольному раствору (30 мл) ацетонидамида дезацетилколхицин-глицериновой кислоты (0,94 г, 2 ммоль; соединение 1), синтезированного согласно примеру 2, добавляют десять миллилитров 5%-ной соляной кислоты и смесь перемешивают при комнатной температуре 5 ч. По окончании перемешивания добавляют 200 мл хлороформа, смесь промывают водным раствором гидрокарбоната натрия и насыщенным водным раствором NaCl. Слой хлороформа сушат над сульфатом магния и затем концентрируют при пониженном давлении. Остаток разделяют колоночной хроматографией на силикагеле. Элюированием смесью бензол-ацетон (1:2) получают 0,45 г (выход 52%) заглавного соединения (соединение 2.D, L-смесь) с т. пл. 45 59oC.
ИК (KBr): 3350 см-1 (ОН), 1660 см-1 (С=0), 1280 см-1 (-О).
ЯМР (DCDL13): 1,87 2,64 (4Н, м), 3,62 (3Н, с), 3,87 (3Н, с), 3,91 (3Н, с), 3,96 (3Н, с), 3,56 4,84 (6Н, м), 6,51 7,58 (4Н, м), 7,9 (1Н, уш.с.)
Пример 4.
Тетраацетатамид дезацетилколхицин-глюкуроновой кислоты (Соединение 3).
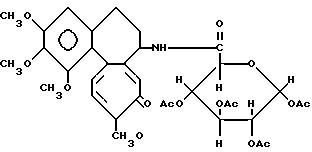
К 30 мл хлороформенного раствора 1,81 г (5 ммоль) тетраацетата глюкуроновой кислоты добавляют тионилхлорид и смесь кипятят 3 ч. После охлаждения смеси до комнатной температуры растворитель и избыток тионилхлорида удаляют при пониженном давлении. Полученный в остатке хлорангидрид кислоты растворяют в 10 мл хлористого метилена. В то же время в 30 мл хлористого метилена растворяют 1,78 г (5 ммоль) дезацетилколхицина и 0,6 г триэтиламина и раствор охлаждают до 0oC. К охлажденной смеси добавляют полученный хлорангидрид кислоты и смесь перемешивают 1,5 ч при 0oC и 15 ч при комнатной температуре. Реакционную смесь промывают водным раствором гидрокарбоната натрия и затем сушат над сульфатом магния. После концентрирования раствора остаток разделяют колоночной хроматографией на силикагеле. Элюированием смесью бензол-ацетон (11:3) получают 1,3 г заглавного соединения (соединение 3). Выход 37% т.пл. 145 147oC (с разложением).
ИК (КВr): 1750 см-1 (ОН), 1680 см-1 (С=0).
ЯМР (DCDL3): 1,91 (3Н, с), 1,96 (3Н, с), 2 (2Н, с), 2,09 (3Н, с), 2,1 2,64 (4Н, м), 3,58 (3Н, с), 3,87 (3Н, с), 3,89 (6Н, с), 4 4,22 (1Н, м), 5 5,38 (4Н, м), 5,8 5,89 (1Н, м), 6,47 7,53) (4Н, м).
Пример 5
Диацетатамид дезацетилколхицин-глюкуроновой кислоты (Соединение 4)
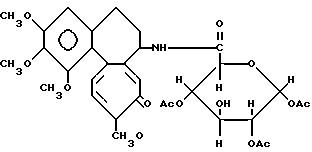
(вероятная формула)
Заглавное соединение получают по методике примера 4 за исключением того, что используют 3,62 г (13 ммоль) диацетата глюкуроновой кислоты, 1,71 г (15 ммоль) тионилхлорида, 2,89 г (8 ммоль) дезацетилколхицина и 1,52 г (15 ммоль) триэтиламина. Получено 1,67 г (выход 35%).
ЯМР (DCDL3): 2,15 (6Н, с), 2,26 2,71 (4Н, м), 3,64 (3Н, с), 3,89 (6Н, с), 3,98 (3Н, с), 3,37 4,48 (8Н, м), 6,53 7,81 (4Н, м).
Пример 6. Амид дезацетилколхицин-глюкуроновой кислоты /N-(5,6,7,9-тетрагидро-1,2,3,10-тетраметокси-9-оксобензо/a/ гептален-7-ил/глюкуронамид/ (соединение 5).

В 30 ммоль метанола растворяют диацетатамид дезацетилколхицин-глюкуроновой кислоты (соединение 4) (1,52 г, 2,5 ммоль), охлаждают до 0oC, по каплям прибавляют пять миллилитров 1 н.гидроокиси натрия и смесь перемешивают 1 ч при 0oC. Добавлением разбавленной соляной кислоты в смеси устанавливают рН 7, после чего концентрируют при пониженном давлении. К остатку добавляют хлороформ и осадок отфильтровывают с последующим разделением фильтрата колоночной хроматографией на силикагеле. В результате их элюируемой смесью бензол-метанол (5: 1) фракции получено 0,93 г (выход 73%) заглавного соединения (соединение 5) с т.пл. 53-57oC.
ЯМР (DCDL3): 2,22 2,67 (4Н, м), 2,96 3,27 (4Н, м), 3,62 (3Н, с), 3,87 (6Н, с), 2,93 (3Н, с), 3,54 4,22 (6Н, м), 6,48 7,78 (4Н, м).
Пример 7. Пентаацетатамид дезацетилколхицин-глюконовой кислоты (Соединение 6).
К 30 мл раствора хлороформа, содержащего 2,57 г (6,3 ммоль) пентаацетата глюконовой кислоты, добавляют тионилхлорид (1,5 г, 13 ммоль) и смесь кипятят 3 ч. После охлаждения смеси до комнатной температуры растворитель и избыток тионилхлорида удаляют при пониженном давлении. Полученный в остатке хлорангидрид кислоты растворяют в 10 мл хлористого метилена. В то же время в 40 мл хлористого метилена растворяют 1,53 г (4,3 ммоль) дезацетилколхицина и 1 г (10 ммоль) триэтиламина и охлаждают до 0oC. К охлажденной смеси добавляют полученный хлорангидрид по каплям и перемешивают 1, 5 ч при 0oC и 1,5 ч при комнатной температуре. Реакционную смесь промывают водным раствором гидрокарбоната натрия и затем сушат над сульфатом магния. После концентрирования раствора остаток разделяют колоночной хроматографией на силикагале. Элюированием смесью бензолацетон (11: 3) получают 1,83 г (выход 57%) заглавного соединения (соединение 6).
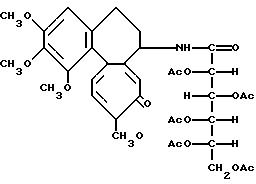
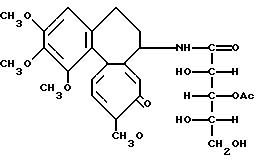
Пример 8. Амид дезацетилколхицин-глюконовой кислоты (Соединение 7).
В 30 мл метанола растворяют пентаацетатамид дезацетилколхицин-глюконовой кислоты (соединение 6) (3,37 г, 5 ммоль), полученного согласно примеру 6, и раствор охлаждают до 0oC. По каплям прибавляют пять миллилитров 1 н.гидроокиси натрия и перемешивают 1 ч при 0oC. Добавлением разбавленной соляной кислоты в смеси устанавливают рН 7 и затем концентрируют при пониженном давлении. К остатку добавляют хлороформ и осадок отфильтровывают. Фильтрат концентрируют и остаток разделяют колоночной хроматографией на силикагеле. В результате из фракции, элюируемой смесью бензол-метанол (5:1), получено 1,12 г (выход 42%) заглавного соединения (соединение 7).
Реферат
Использование: в качестве противоопухолевого соединения. Сущность: продукт - производное
дезацетилколхицина ф-лы I, где R-C(O) - ацильный
остаток сахарокарбоновой С3-C7-кислоты, где гидроксильные группы могут быть защищены ацильной или кетальной группой. Реагент 1:
дезацетилколхицин. Реагент 2: сахарокарбоновая
кислота R-COOH. Условия реакции: в условиях амидирования. 3 с. и 2з. п. ф-лы, 4 табл. Структура соединения ф-лы I:

Формула
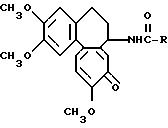
где
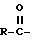

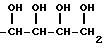

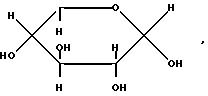

в которых гидроксильные группы могут быть защищены ацильной или кетальной группой.

где

отличающийся тем, что дезацетилколхицин формулы

подвергают амидированию сахарокарбоновой кислотой формулы
R COOH,
где
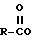

Комментарии