Производные n-метилдеацетилколхицинамида, способы их получения и противоопухолевая композиция - RU2073006C1
Код документа: RU2073006C1
Чертежи

Описание
Изобретение относится к новым производным
N-метилдеацетилколхицеинамида и, более детально, к производным N-метилдеацетилколхицеинамида, представленным следующей формулой, и их солям:

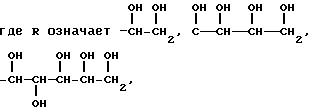
где R обозначает остаток, полученный отщеплением СООН от С3-C7 сахарных карбоксильных кислот, и присутствующие в остатке гидроксильные группы могут соответственно быть защищены с помощью существующих для них защитных групп.
Уже известно, что колхицин, представленный следующей формулой

обладает фармакологической активностью по отношению к раковым клеткам, сгусткам и т.д. (E. E. Van. Tamelen, T. A. Spencer, Ir. D. S. Allen, Ir. и R. L. Orvis, Tetrahedron, 14, 8 (1961)).
Однако колхицин обладает сильной токсичностью и может уже совершенно не рассматриваться после появления
найденного позднее димеколхицина) [Деацетил-N-метилколхицин; Chem. Engng. News. 37, Nos. 41 и 67 (1959)]
Авторы изобретения, изучив производные колхицина, имеющие исключительно низкую
токсичность и, кроме того, предельное противораковое действие, обнаружили ранее, что производные деацетилколхицина, представленные следующей формулой
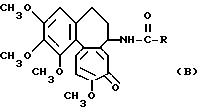
где R такой, как определено выше,
оказывают сильный ингибирующий эффект на рост раковых клеток и предложили их (см. ЕР-А-493, 064).
С другой стороны, также уже известно, что колхицеинамид, представленный следующей формулой
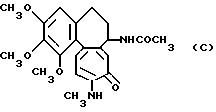
оказывает фармакологическое воздействие на раковые клетки [Journal of the National Cancer Institute,

Авторы провели интенсивное изучение производных колхицина и родственных им соединений, обладающих еще более пониженной токсичностью и, кроме того, еще более высоким противораковым действием, и в результате установили, что производные N-метилдеацетилколхицеинамида, представленные приведенной выше формулой (1) и полученные замещением на метиламиногруппу метокси группу в положении 10 соединений приведенной выше формулы (В) также обладают пониженной токсичностью и сильным ингибирующим эффектом на рост раковых клеток и могут быть предложены для использовании в качестве канцеростатических противораковых агентов.
Примерами "остатка, полученного отщеплением СООН от С3-C7 сахарных карбоновых кислот", представленного с помощью R в приведенной выше формуле (I), служат моновалентные остатки, полученные отщеплением СООН от 3-7 карбоновых кислот моносахаридов, таких, как глицериновая кислота, рибозокарбоновая кислота, глюкуроновая кислота, глюконовая кислота и глюкогептановая кислота (далее здесь ссылаются как на остатки сахаров), и конкретные примеры, указаны ниже.

По крайней мере часть множественных гидроксильных групп, находящихся в таким образом описанных остатках сахаров, может соответственно быть защищена с помощью существующих для них защитных групп. Примерами защитных групп являются ацильные группы, такие, как C1-C10 (преимущественно С2-C6) алканоильные группы, например ацетил, пропионил, бутирил и пивалоил и арильная группа, например бензол, а также ацетильные и кетальные группы, представленные формулой
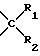
где R1 обозначает атом водорода или алкильную группу и R2 обозначает C1-C6 (преимущественно С1-C4) алкильную или фенильную группу, например

Здесь, также, как и для R, в качестве предпочтительных групп могут быть упомянуты группы, представленные следующей формулой
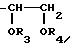
где R3 и R4 каждый обозначают атом водорода или защитную группу для гидроксильной группы (например, ацильная группа, как упомянуто выше), или Р3 и P4 объединяются для обозначения защитной группы для гидроксильных групп (например, ацетальная или кетальная группа, как упомянуто выше).
Кроме того, в качестве солей производных N-метилдиацетилколхицеинамида формулы (1) здесь могут, например, быть упомянуты соли неорганических кислот, такие, как хлоргидрат или сульфат, и соли органических кислот, такие, как ацетат, пропионат, бутират, лактат, тартрат, малат, цитрат, глюконат, сукцинат, малеат, фумарат, глицитиллитинат бензоат и т.д.
К примеру, соединение,
отвечающее приведенной выше формуле (I) изобретения, может быть приготовлено амидированием N-метилдиацетилколхицеина следующей формулы
сахарной карбоксильной кислоты, представленной следующей формулой:
R-COOH
где R как определено выше
или его реакционноспособным производным упомянутых соединений.
Амидирование N-метилдеацетилколхицеинамида формулы (II) сахарной карбоксильной кислотой формулы (III) или ее активным производным может быть осуществлено путем использования известной в химии пептидов реакции амидирования.
Например, соединение изобретения может быть приготовлено взаимодействием N-метилдеацетилколхицеинамида формулы (II) с галогенгидридом сахарной карбоновой кислоты формулы (III) в присутствии основания. Указанная выше реакция может быть проведена в большинстве случаев при температуре приблизительно между 0oС и 30oС, предпочтительно примерно от 0oС до комнатной температуре. Используемое количество галоидангидрида строго не лимитировано, но удобно использовать его в области обычно от 1 до 1,5 молей, особенно от 1 до 1,2 молей на моль N-метилдеацетилколхицеинамида. Далее, в качестве оснований здесь могут быть использованы, к примеру, третичные амины, такие, как триэтиламин и пиридин, карбонаты щелочных металлов (бикарбонаты), такие, как карбонат натрия, бикарбонат натрия, карбонат калия и бикарбонат калия и т.д. Их используемое количество может быть в основном в области от 1 до 1,5 молей, предпочтительно от 1 до 1,2 молей на моль N-метилдеацетилколхицеинамида.
Приведенная выше реакция может обычно проводиться в инертном растворителе, и среди используемых растворителей здесь могут быть, к примеру, упомянуты галогенированные углеводороды, такие, как метиленхлорид, хлороформ, четыреххлористый углерод, дихлорэтилен и трихлорэтилен; алифатические эфиры, такие, как этиловый эфир и метилцеллозольв, ароматические угледороды, такие, как бензол и толуол; и т.д.
Кроме того, соединение изобретения может быть приготовлено либо прямым взаимодействием N-метилдеацетилколхицеинамида с сахарной карбоксильной кислотой приведенной выше формулы (III) в присутствии конденсирующего агента, такого, как DCC (дициклогексилкарбодимид) или реакцией N-метилдеацетилколхицеинамида с эфиром (например, метиловый эфир, этиловый эфир, бутиловый эфир или тому подобные) сахарной карбоновой кислоты приведенной выше формулы (III).
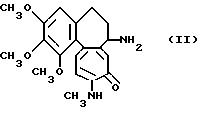
В таких описанных реакциях N-метилдеацетилколхицеинамид приведенной выше формулы (II), используемый как исходное сырье, является новым соединением, не описанным ранее в литературе, и может, к примеру, быть приготовлен взаимодействием деацетилколхицина по [J. Am. Chem. Soc.


Соединение приведенной выше формулы (I) изобретения может, кроме того, быть приготовлено взаимодействием деацетилколхицеинового производного по приведенному далее ссылочному примеру 2, описанного выше в ЕР-А-493,064 и представленного следующей формулой

где R как определено выше, с метиламином для превращения метокси группы в метиламино группу.
Реакция метиламинирования соединения формулы (IV) может быть осуществлена в соответствии с описанным ранее способом [по Patrick J. Davis: Antimicrobial Agents and Chemotherapy, Mar. 1981, стр. 465-469; J. L. Hartwell et al. J. Am. Chem. Soc.
Хотя используемое количество метиламина строго не лимитировано, но удобно использовать его в области обычно от 2 до 30 молей, преимущественно от 10 до 20 молей на моль соединения формулы (IV). Реакция обычно проводится в водной среде в закрытом сосуде.
Соединения изобретения, полученные описанными здесь способами, могут быть разделены и очищены обычными известными методами, например экстракцией, хроматографией, кристаллизацией или их комбинацией, и тому подобными.
Соединение изобретения, где в остатках сахаров, обозначенных R, присутствуют защитные группы для гидроксильных групп, в некотором случае может быть подвергнуто реакции по снятию защитных групп, например гидролизу с целью удаления защитной группы.
Кроме того, полученное таким образом производное N-метилдеацетилколхицеинамида формулы (I) может при необходимости быть переведено в такую соль, как упомянуто выше, в соответствии с известной реакцией по образованию соли, например, обработкой его соответствующей кислотой.
Производное N-метилдеацетилколхицеинамида приведенной выше формулы (I), полученное в изобретении, содержит асимметричный атом углерода в боковой цепи и может существовать в виде D-формы, L-формы или DL-формы.
Как следует из проведенного в пробирке испытания на раковых клетках, производные N-метилдеацетилколхицеинамида приведенной выше формулы (I), полученные в изобретении, обладают отличным канцеростатическим действием.
Пример 1. Испытания: тест в пробирке на ингибирование роста раковых клеток
Мышиные
лейкемические клетки р388/S и
адриамицин-устойчивые мышиные лейкемические клетки Р388/ADR, подкультурно трансплантированные в абдоминальную полость мышей, были взяты оттуда вместе с образцами
соответственно и после промывки
суспендированы в среде PPMI 1640 (каждая содержащая 10% зародышей бычьей сыворотки и 10 мкМ 2-меркаптоэтанола) до 2х105 клеток/мл, соответственно. К порциям
каждой из этих клеточных
суспензий добавлено по 0,5 об. каждого испытуемого раствора, соответственно (растворы соединения, описанного далее в примере 1, в диметилсульфоксиде), и смеси раздельно
помещены на пластину для
выращивания культур с 24 углублениями, и культивировали в течение 2 дней в сосуде для роста культур в 5% двуокиси углерода. 0,5% раствор Трипана Синего добавлен к каждому из
мясных бульонов клеточных
культур в том же количестве и путем подсчета под микроскопом числа клеток, не окрасившихся как живые клетки, была найдена 50% концентрация ингибирования пролиферации (1С50) для соединения
примера 1. Результаты эксперимента, проведенного трижды, представлены в таблице.
Пример 2.
Испытание на острую токсичность
Порции
испытуемых растворов
(растворы, полученные растворением соединения, описанного далее в примере 1, в 0,2 N соляной кислоте и затем разбавлением физиологическим раствором соли до заданных концентраций)
введены
внутрибрюшинным способом группам CDF самцов мышей, каждая группа состояла из 8 мышей, соответственно, и наблюдали в течение 10 дней вес тел, симптомы, сроки гибели и т.д. применяемые
количества
снижали и при общем соотношении 1,3 была установлена максимальная (количество) доза в 62 мг/кг, значение LD50 было рассчитано с помощью метода Litchfield Wilcoxon'a и в
результате LD50 соединения формулы (1) было 41,0 мг/кг (от 35,8 до 46,9 мг/кг).
Наблюдали симптомы острой токсичности, выражающиеся в ухудшении состояния шерсти на первый и последующие дни после введения и возникновение поноса или запора в случаях групп высокой дозы. Кроме того, среди выживших случаев в группе с введением в 47 мг/кг наблюдались случаи проявления при ходьбе отека правой ноги и паралич одной стороны тела. Далее, что касается изменения веса, вес тела в каждом случае имел тенденцию к понижению от 2 до 4 дней после введения, но после этого постепенно увеличивался.
Как очевидно из приведенных выше результатов испытания, соединения изобретения оказывают сильное ингибирующее воздействие на раковые клетки и только сравнительно низко токсичны и могут быть предложены как противораковые агенты.
Когда соединение изобретения используется как противораковое, соединение может быть введено орально или парентерально (например, внутривенной инъекцией, внутримышечной инъекцией, подкожной инъекцией или тому подобными). Его эффективная доза может варьироваться в широких пределах в зависимости от симптома, степени заболевания, веса и возраста пациента, которому соединение предназначено для введения, мнения врача и т.д. но, например, в случае инъекции может обычно быть приблизительно от 1 до 5 мг/кг/день и соединение может быть введено 1 раз в день или несколькими порциями за день.
Когда соединение изобретения используется как лекарство, эффективное количество соединения может быть сформировано вместе с фармацевтически приемлемыми носителями или разбавителем (например, эксципиенты, растворители, другие добавки и т.д.) в единичных дозах, пригодных для приема, например, дозированные формы, такие, как таблетки, порошки, гранулы, капсулы, кишечные агенты, таблетки, сиропы, эликсиры, жидкости, суспензии и эмульсии.
В качестве переносчиков или разбавителей, пригодных к употреблению по приведенной выше формулировке, могут, к примеру, быть упомянуты носители, такие, как крахмал, лактоза, сахароза, маннитол и карбоксиметилцеллюлоза; смазки, такие, как стеарат магния, лаурилсульфат натрия и тальк; связующие вещества, такие, как декстрин, микрокристаллическая целлюлоза, поливинилпирролидон, арабская камедь, кукурузный крахмал и желатин; дезинтегранты, такие, как картофельный крахмал и карбоксиметилцеллюлоза; разжижители, такие, как дистиллированная вода для инъекций, физиологическая соль, водные растворы глюкозы, растительные масла для инъекций, пропиленгликоль и полиэтиленгликоль и т.д. и далее, если это необходимо, сюда могут быть включены вкусовая добавка, краситель, тонизирующее вещество, стабилизатор, антисептик, вещество, делающее введение соединения изобретения безболезненным и т.д.
Кроме того, другие фармакологически активные вещества могут быть при необходимости включены в лекарство по изобретению.
Ссылочный
пример 1. Приготовление N-метилдеацетилколхицеинамида
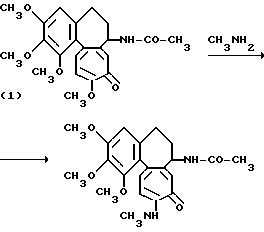
Смесь раствора колхицина (5,00 г, 12,5 ммолей) в водном 40% растворе метиламина (10 мл, 130 ммолей) и этилового спирта (10 мл) подвергалась взаимодействию при 120oС в течение 20 часов при перемешивании и в запаянной трубке. Растворитель удаляют перегонкой, добавлено 10 мл воды для осаждения и смесь экстрагируют хлороформом.
Слой хлороформа отделен, промыт водой и высушен. Растворитель выпарен и остаток разделен и очищен с помощью силикагелевой колоночной хроматографии [хлороформ: метанол (20:1)] Тем самым получено 4,2 г N-метилдеацетилколхицеинамида в виде желтых кристаллов (выход 84%).

Смесь раствора N-метилдеацетилколхицеинамида (3,00 г, 7, 5 ммолей), воды (50 мл) и концентрированной серной кислоты (15 мл) перемешивали при 100oС в течение 5 часов для осуществления гидролиза (реакция деацетилирования). Реакционный раствор подщелочили безводным карбонатом натрия при охлаждения льдом и проэкстрагировали хлороформом.
Слой хлороформа отделили, промыли водой и высушили. Растворитель выпарили досуха. Остаток разделили и очистили с помощью силикагелевой колоночной хроматографии на силикагеле [хлороформ: метанол (20:1)] Тем самым получено 2,20 г N-метилдеацетилколхицеинамида в виде желтых кристаллов.
Ссылочный
пример 2 Приготовление N-(0,0-изопропилиденглицероил)-деацетилколхицеина (IVa) [другое название: ацетонидамид деацетилколхицин-глицериновой кислоты]

Ацетонид глицерата калия (3,60 г, 20 ммолей) суспендирован в 30 мл сухого эфира, и эфирный (5 мл) раствор 2,4 г (20 ммолей) тионилхлорида добавлен к суспензии по каплям. После этого смесь нагревали до кипения с обратным холодильником в течение 3 часов. Затем смесь охлаждена до комнатной температуры, осадок отфильтрован методом отсасывания, и фильтрат сконцентрирован при пониженном давлении. Добавлен сухой метиленхлорид и растворения осадка.
Тем временем 2,96 (8,3 ммолей) деацетилколхицина и 2,02 г (20 ммолей) триэтиламина были растворены в 30 мл метиленхлорида. Смесь охлаждена до 0oС и добавлен по каплям указанный выше раствор хлорангидрида глицериновой кислоты в метиленхлориде. После перемешивания при 0oС в течение 3 часов раствор метиленхлорида промыт водным раствором бикарбоната натрия. Слой метиленхлорида высушен над сульфатом магния и затем сконцентрирован при пониженном давлении. Остаток разделен с помощью колоночной хроматогратографии на силикагеле и 1,11 г приведенного в заглавии соединения (IVa-V: L-изомер) получено из бензол-ацетон (5:1) элюата). Выход 28% т.пл. 251-253oС (с разложением).
Кроме того, 0,58 г второго указанного в заглавии соединения (IVa-2: D-изомер) получено из бензол:
ацетон (5:2) элюата. Выход 14%
Пример 1. Приготовление N'-(0,0-изопропилиденглицероил)-N-метилдиацетилколхицеинамида (1а)

40% раствор N-(0,0-изопропилиденглицероил) деацетилколхицина (IVa) (2,74 г, 5,6 ммолей) и 10 мл безводного метиламина поместили в запаянную трубку и перемешивали при нагревании от 70oС до 80oС в течение 20 часов. После завершения реакции реакционная смесь сконцентрирована упариванием при пониженном давлении и растворена в хлороформе. Слой хлороформа промыт насыщенным раствором соли и высушен над сульфатом магния. Хлороформ отогнан при пониженном давлении и остаток подвергнут разделению и очистке с помощью колоночной хроматографии на силикагеле. В результате 1,93 г описанного соединения 1а было получено из бензол: ацетон 4:1 элюата. Выход: 70%
ИК: 1660 см-1 (С=0)
ЯМР: δ1,40, 1, 44 (3Н, S), 1,49, 1,69 (3Н, S), 1,85-1,95 (2Н, m) 2,17-2,54 (3Н, m), 3,07 (3Н, d), 3,61 (3Н, S), 3,89 (3Н, S), 3,94 (3Н, S), 3,99-4,06 (1H, m), 4,18-4,29 (1Н, m) 4,41-4,51 (1Н, m), 4,63-4,72 (1Н, m), 6,54 (1Н, S) 7,05-7,44 (3Н, m)
Пример 2 Приготовление N'-(0,0-изопропилиденглицероил)-N-метилдеацетилколхицеинамида (1а)

Ацетонид глицерата натрия (3,60 г, 20 ммолей) суспендирован в сухом эфире (30 мл), и эфирный (5 мл) раствор 2,4 г (20 ммолей) тионилхлорида добавлен по каплям. После этого смесь нагревали до кипения с обратным холодильником в течение 3 часов. Смесь охлаждена до комнатной температуры, образовавшийся осадок отфильтрован методом отсасывания и фильтрат сконцентрирован при пониженном давлении. Для его растворения добавлен сухой хлороформ.
Тем временем 2,96 г (8,3 ммолей) N-метилдеацетилколхицеинамида и 2,02 г (20 ммолей) триэтиламина растворены в 30 мл хлороформа.
Смешанный раствор охлажден до 0oС и указанный выше раствор хлорангидрида ацетонида глицериновой кислоты добавлен по каплям. Полученный
хлороформный раствор перемешивали при 0oС в
течение 3 часов и затем промывали водным раствором бикарбоната натрия. Хлороформный слой высушен над безводным сульфатом магния и
сконцентрирован при пониженном давлении. Остаток подвергнут разделению
и очистке с помощью колоночной хроматографии на силикагеле. В результате получено 1,81 г целевого соединения (1а) из бензол:
ацетон (4:1) элюата. Выход 45%
Пример 3. Подготовка для
инъекции
2,0 г N'-(0,0-изопропилиденглицероил-N-метилдиацетилколхицеинамида растворено в 1 л дистиллированной воды для
инъекций при нормальной температуре, раствор был изотонирован
добавлением хлористого натрия и смесь помещена в ампулы, которые потом запаяли, 1 мл такой инъекции содержит 2 мг эффективного
ингредиента.
Пример 4.

Тионилхлорид (1,19 г 10 ммолей) прибавляют к раствору 1,81 г (5 ммолей) тетраацетата глюкуроновой кислоты в хлороформе и нагревают при рефлюксе 4 часа.
После охлаждения смеси до комнатной температуры растворитель и избыток тионилхлорида удаляют при пониженном давлении. Остаток хлорангидрида растворяют в 10 мл метиленхлорида и подвергают взаимодействию с N-метилдеацетилколхицеинамидом по методике примера 2, таким образом получая соединение (Ib).
Пример 5.
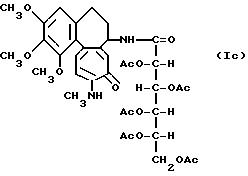
Тионилхлорид (1,50 г, 13 ммолей) добавляют к 30 мл раствора 2,57 г (6,3 ммолей) пентаацетата глюконовой кислоты и нагревают при рефлюксе в течение 3 часов. После охлаждения смеси до комнатной температуре растворитель и избыток тионилхлорида удаляют при пониженной температуре. Остаток хлорангидрида растворяют в метиленхлориде и подвергают взаимодействию с N-метилдеацетилколхицеинамидом, следуя методике примера 2 с получением соединения (Iс).
Реферат
Использование: в качестве противоопухолевого агента. Сущность
изобретения: производные N-метилдеацетилколхицеинамида формулы I:


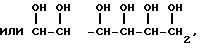
Формула

где R обозначает


гидроксильные группы, в которых могут быть защищены ацильной группой, ацетальной или кетальной группой.

амидируют хлорангидридом формулы
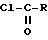
где R имеет указанные значения.

подвергают взаимодействию с метиламином.
Комментарии