Оксазинон и способ получения таксола - RU2033994C1
Код документа: RU2033994C1
Описание
Изобретение относится к новому оксазинону и способу получения таксола, включающему использование такого оксази- нона.
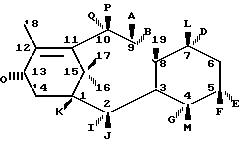
Таксановое семейство
терпенов, членом которого является таксол, привлек значительный интерес как в биологической, так и в химической отраслях техники. Таксол является многообещающим агентом для хемотерапии рака с широким
спектром антилейкемической и опухолеингибирующей активности и имеет следующую структуру
C


Благодаря своей активности таксол в настоящее время проходит клинические исследования как во Франции, так и в США. Поставка таксола для этих исследований обеспечивается в настоящее время путем использования коры нескольких видов тиса. Таксол обнаружен в коре медленно растущих вечнозеленых деревьев лишь в незначительных количествах. Вследствие этого химики в последние годы пытаются найти надежный синтетический способ получения таксолов.
Один из способов синтеза, который был предложен,
направлен на синтез тетрациклического ядра из товарных химических веществ. Известен синтез собрата таксола таксузина [1]
Однако общий синтез таксола будет многостадийным, утомительным и
дорогостоящим процессом.
Альтернативный подход к получению таксола [2] включает использование собрата таксола, 10-диацетил-баккатина III, который имеет следующую формулу:

10-Дезацетилбаккатин III является более легко доступным, чем таксол, поскольку он может быть получен из листьев Taxus baccata. Согласно способу Гриин, 10-дезацетилбаккатин III превращается в таксол с помощью присоединения С10 ацетильной группы и присоединения С13-амидосложноэфирной боковой цепи с помощью сложной этерификации С-13 спирта β-амидокарбоновой кислотой. Хотя данный подход требует относительно мало стадий, синтез β-амидокарбоновой кислоты является многостадийным процессом, который протекает с низким выходом, а реакция сочетания или конденсации является утомительной и протекает с низким выходом. Однако данная реакция является ключевой стадией, которая необходима в каждом рассматри- ваемом синтезе таксола или биологически активного производного таксола, поскольку для противоопухолевой активности требуется присутствие β-амидосложноэфирной боковой цепи в положении С13[3]
Основная трудность в синтезе таксола и других потенциальных противоопухолевых агентов заключается в недостатке или отсутствии легко доступных звеньев или молекул, которые можно было бы легко присоединить к С13 кислороду для получения β-амидосложноэфирной боковой цепи. Разработка таких веществ и способа их присоединения с достижением высокого выхода облегчило бы синтез таксола и родственных противоопухолевых агентов, имеющих модифицированный набор ядерных заместителей или модифицированной С13 боковой цепи. Данная потребность было восполнена с помощью открытия нового легкодоступного исходного продукта для боковой цепи и эффективного способа его присоединения к С13 кислороду.
Предлагаемое изобретение направлено на
предшественник боковой цепи оксазином формулы
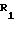

R2 и R5 независимо выбраны из водорода, алкила, алкенила, алкинила, арила, гетероарила и ОR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащищающая группа;
R3 и R6 независимо выбраны из водорода, алкила, алкенила, алкинила, арила и гетероарила.
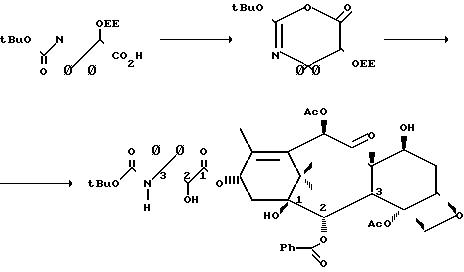
Изобретение направлено также на процесс получения таксольного промежуточного продукта, включающий контактирование спирта с оксазином формулы I в присутствии достаточного количества активирующего агента с тем, чтобы вызвать взаимо- действие оксазинов со спиртом с образованием β-амидосложного эфира, который может использоваться в качестве проме- жуточного продукта в синтезе таксола.
Оксазин (1) и его
производные имеют следующую структуру
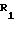

R2 и R5 независимо выбираются из водорода, алкила, алкенила, алкинила, арила, гетероарила и OR8, где R8 алкил, алкенил, алкинил, арил, гетероарил или гидроксилзащитная группа;
R3 и R6 независимо выбираются из водорода, алкила, алкенила, алкинила, арила и гетероарила.
Предпочтительно оксазинон (1) имеет структуру

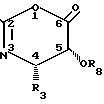

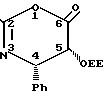
Согласно правилам EUPAC оксазиноном (2) является 2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-он.
В соответствии с настоящим изобретением представляется способ получения таксольных промежуточных продуктов, природного
таксола и не встречающихся в природе таксолов, имеющих следующую структурную формулу



Оксазиноны (1) превращаются в β-амидосложные эфиры в присутствии спирта и активирующего агента, предпочтительно третичного амина, такого как триэтиламин, диизопропилэтиламин, пиридин, N-метил-имидазол и 4-диметиламинопиридин(ДМАР). Например, оксазиноны (1) взаимодействуют с соединениями, имеющими таксановое тетрациклическое ядро и С13 гидроксильную группу в присутствии 4-диметиламинопиридина, давая вещества, имеющие β-амидосложноэфирную группу у С13.
Наиболее предпочтительно спиртом является 7-0-триэтилсилилбаккатин III, который может получить
по способу [2] или другими способами. Дезацетилбаккатин III превращается в 7-0-триэтилсилилбаккатин III в соответствии со следующей схемой реакции

В условиях, которые тщательно оптимизированы, 10-дезацетилбаккатин III подвергается взаимодействию с 20 экв (С2Н5) SiCl при 23оС в атмосфере аргона в течение 20 ч в присутствии 50 мл пиридина/ммоль 10-дезацетилбаккатина III, давая 7-триэтилсилил-10-дезацетилбаккатин III (3 а) в виде реакционного продукта с выходом 84-86% после очистки. Продукт реакции затем ацетилируется 5 экв. СН3СОСl и 25 мл пиридина/ммоль 3 а при 0оС в атмосфере аргона в течение 48 ч, давая с выходом 86% 7-0-триэтилсилилбаккатин III (3 b).
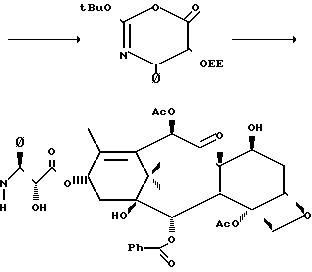
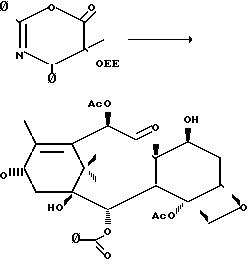
Как показано на следующей схеме реакции, 7-0-триэтилсилилбаккатин III (3 b) может подвергаться реакции с оксазиноном при комнатной температуре,
давая таксольный промежуточный продукт, в котором С-7- и С-2-гидроксильные группы защищены триэтилсилильной и этоксиэтильной защитными группами соответственно. Эти группы затем гидролизуются в мягких
условиях так, чтобы не затронуть сложноэфирную связь или заместители таксола. Синтез таксола из оксазинона (2) осуществляется следующим образом:

Хотя указанная схема направлена на синтез природного продукта таксола, процесс может использоваться с модификациями в оксазиноне или тетрациклическом спирте, который может быть получен из природных или неприродных источников для производства других синтетических таксолов.
Оксазинон (1) может превращаться в β -амидосложный эфир в присутствии активирующего агента и спирта, иного чем 7-0-триэтилсилилбаккатин III, образуя промежуточный продукт таксола. Синтез таксола может затем протекать с использованием таксольного промежуточного продукта со соответствующей схеме реакции.
Оксазиноновыми алкильными группами (одним или с различными заместителями) предпочтительно являются низший алкил, содержащий 1-6 атомов углерода в главной цепи и до 15 атомов углерода. Они могут быть прямыми или разветвленными и включают метил, этил, пропил, изопропил, бутил, изобутил, третбутил, арил, гексил и др.
Оксазиноновыми алкенильными группами (одни или с различными заместителями) предпочтительно являются низший алкенил, содержащий 2-6 атомов углерода в главной цепи и до 15 атомов углерода. Они могут быть с прямой или разветвленной цепью и включают этенил, пропенил, изопропенил, бутенил, изобутенил, арил, гексанил и др.
Оксазиноновыми алкинильными группами (одни или с различными заместителями), предпочтительно являются низший алкинил, содержащий 2 6 углеродных атомов в главной цепи и до 15 углеродных атомов. Они могут быть с прямой или разветвленной цепью и включают этинил, пропинил, бутинил, изобутинил, арил, гексинил и др.
Примеры алканоилокси оксазинона включают ацетат, пропионат, бутират, валерат, изобутирал и др. Более предпочтительным алканоилокси является ацетат.
Указанные оксазиноновые арильные фрагменты, один или в сочетании с различными заместителями, содержат 6-15 атомов углерода и включают фенил, α-нафтил или β-нафтил и пр. Заместители включают алканокси, гидрокси, галоген, алкил, арил, алкенил, ацил, ацилокси, нитро, амино, амидо и др. Фенил является более предпочтительным арилом.
Как отмечалось выше, R2 и R5 оксазинона (1) могут быть ОR8, где R8 алкил, ацил, кеталь, этоксиэтил (ЕЕ),
2,
2,2-трихлорэтоксиметил или другая гидроксилзащищающая группа, такая как ацетали и простые эфиры, т. е. метоксиметил (МОМ), бензилоксиметил, сложные эфиры, такие как ацетаты, карбонаты, такие как
метилкарбонаты, и др. Известно множество разнообразных защитных групп для гидроксильной группы в органическом синтезе [4] Выбранная гидроксилзащищающая группа должна легко удаляться в условиях,
которые являются достаточно мягкими, чтобы не затрагивать сложноэфирную связь или другие заместители таксольного промежуточного продукта. Однако R8предпочтительно представляет собой
этоксиэтил или 2,2,2-трихлорэтоксиметил, наиболее предпочтителен этоксиэтил.
Предпочтительные значения оксазиноновых заместитетелей R1-R3, R5,-R8 следующие: R1=OR7, R1=Ar, R1=n -MeOPh, R1 алкил, R1 алкенил, R1 алкинил, R1= H; R2=ОR8; R3=Rh, R3=Ar, R3 n -MeOPh, R3 алкил, R3= алкенил, R3 алкинил, R3= H; R5=H; R6=H; R7 алкил, R7 алкенил, R7=алкинил, R7=арил, R7-гетероарил; R8= ЕЕ, R8=алкил, R8=OCOR, R8 MOM, R8 =Cl3CCH2OCH2.
Поскольку оксазинон имеет несколько асимметричных атомов углерода, соединения настоящего изобретения, имеющие асимметричные атомы углерода, могут существовать в диастереомерных, рацемической или оптической активной формах. Более конкретно данное изобретение включает энантиомеры, диастереомеры, рацемические смеси и иные смеси соединений.
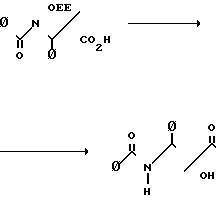
Оксазиноны (1) могут быть получены из легко доступных материалов в соответствии со следующей схемой реакции
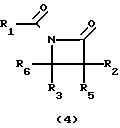




Карбоновую кислоту (5) может получить в соответствии с известным [2] способом. Бета-лактамы (4) могут быть получены из легко доступных материалов, как показано на следующей схеме реакции, где R1 и R3 фенил;
R5 и R6 водород;
R2-OR8, причем R8 этоксиэтил:
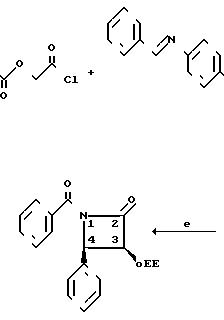


Исходные материалы являются легко доступными. Альфа-акрилоксиацетилхлорид получают из гликолевой кислоты в присутствии третичного амина, она циклоконденсируется с аминами, полученными из альдегидов и n-метоксианилина, давая 2,1-n-метоксифенил-3-ацилокси-4-арилазети-дин-2 оны.
N-Метоксифенильная группа может легко удаляться при окислении цериевым нитратом аммония, ацилокси группа может гидролизоваться в стандартных увловиях, аналогичных условиям, известным в технике для получения 3-гидрокси-4-арилазетидин-2-онов.
3-Гидроксильная группа может защищаться большим разнообразием стандартных защитных групп, таких как 1-этоксиэтильная группа. Предпочтительно, рацемический 3-гидрокси-4-арилазетидин-2-он расщепляется на чистые энантиомеры, перед защитой с помощью перекристаллизации соответствующих 2-метокси-2-(трифторметил)-фенилуксусных сложных эфиров и при получении таксола используется только правовращающий энантиомер. В любом случае 3-(1-этоксиэтокси)-4-фенил-азетидин-2-он может превращаться в β-лактам с помощью обработки основанием, предпочтительно н-бутиллитием, и ароилхлоридом при 78оС или ниже.
П р и м е р 1. Приготовление цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- он-2. Цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-он. К раствору 962 мг (4,56 ммоль) имина, полученного из бензальдегида, и n-метокси анилина и 0,85 мл (6,07 ммоль) триэтиламина в 15 мл СН2 Cl2 при 20оС добавлялся по каплям раствор 413 мг (3,04 ммоль α-ацетоксиацетилхлорида в 15 мл в метиленхлориде. Реакционную смесь оставляли нагреваться до 25оС в течение 18 ч. Затем ее разбавляли 100 мл метиленхлорида и раствор экстрагировали 30 мл 10%-ного водного раствора НСl. Органический слой промывали 30 мл воды и 30 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали для получения твердой массы. Твердое вещество обрабатывали 50 мл гексана и смесь фильтровали. Оставшееся твердое вещество перекристаллизовывали из смеси этилацетата (гексан, давая 645 мг (68%) цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 163оС.
Цис-3-ацетокси-4-фенилазетидин-2-он. К раствору 20,2 г цис-1-n-метоксифенил-3-ацетокси-4-фенилазетидин-2-она в 700 мл ацетонитрила при -10оС медленно добавляли раствор цериевого нитрата аммония в 450 мл воды в течение часа. Смесь перемешивали в течение 30 мин при 10оС и разбавляли 500 мл эфира. Водный слой экстрагировали двумя (100 мл) порциями воды, двумя (100 мл) порциями насыщенного водного раствора бисульфата натрия, двумя (100 мл) порциями насыщенного водного раствора бикарбоната натрия и концентрировали, введя 18,5 г твердого вещества. Перекристаллизация твердого вещества из смеси ацетон/гексан давала 12,3 г (92%) цис-3-ацетокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 152-154оС.
Цис-3-гидрокси-4-фенилазетидин-2-он.
К смеси 200 мл тетрагидрофурана и 280 мл раствора 1 М водной гидроокиси калия при 0оС добавляли к раствору 4,59 г (22,4 ммоль) цис-3-ацетокси-4-фенилазетидин-2-она в 265 мл тетрагидрофурана с помощью капельной воронки в течение 40 мин. Раствор перемешивали при 0оС в течение часа и добавляли 100 мл воды и 100 мл насыщенного бикарбоната натрия. Смесь экстрагировали 4 порциями по 200 мл этилацетата; объединенные органические слои сушили над сульфатом натрия и концентрировали, давая 3,54 г (97%) рацемического цис-3-гидрокси-4-фенилазетидин-2-она в виде белых кристаллов, т.пл. 147-149оС. Это вещество разделялось на его анантиомеры путем перекристаллизации его 2-метокси-2-(трифторметил) фенилуксусного эфира из смеси гексан/ацетон с последующим гидролизом (α)25 Нg 177.
Цис-3-(1-этоксиэтокси)-4-фенилазети-дин-2-он. К раствору 3,41 г (20,9 ммоль) цис-3-гидрокси-4-фенилазетидин-2-она в 15 мл тетрагидрофурана при 0оС добавляли 5 мл этилвинилового эфира и 20 мг (0,2 ммоль) метансульфоновой кислоты. Смесь перемешивали при 0оС в течение 20 мин, разбавляли 20 мл насыщенного водного бикарбоната натрия и экстрагировали тремя (40 мл) порциями этилацетата. Объединенные этилацетатные слои сушили над сульфатом натрия и концентрировали, давая 4,87 г (99%) цис-3-(1-этоксиэтокси)-5-фенилазетидин-2-она в виде бесцветного масла.
Цис-1-бензоил-3-(1-этоксиэтокси)-4-фе- нилазетидин-2-он. К раствору 2,35 г (10 ммоль) цис-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в 40 мл ТГФ при 78оС добавляли 6,1 мл (10,07 ммоль) 1,65 М раствора н-бетиллития в гексане. Смесь перемешивали в течение 10 мин при 78оС и добавляли раствор 1,42 г (10,1 ммоль) бензоилхлорида в 10 мл ТГФ. После этого смесь перемешивали при 78оС в течение 1 ч, разбавляли 70 мл насыщенного водного бикарбоната натрия и экстрагировали тремя порциями по 50 мл этилацетата. Объединенные этилацетатные экстракты сушили над сульфатом натрия и концентрировали, давая 3,45 г масла. Хроматография масла на силикагеле с элюированием смесью этилацетата и гексана получали 3,22 г (95%) цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в виде бесцветного масла.
2R, 3S1= N-бензоил-n-(1-этоксиэтил)-3 фенилизосерин. К раствору 460 мг (1,36 ммоль) цис-1-бензоил-3-(1-этоксиэтокси)-4-фенилазетидин-2-она в 20 мл ТГФ при 0оС добавляли 13,5 мл 1 М водного раствора (13,5 ммоль) гидроокиси калия. Смесь перемешивали при 0оС в течение 10 мин и ТГФ выпаривался. Смесь распределяли между 12 мл 1 н. водного раствора НСl и 30 мл хлороформа. Водный слой экстрагировали двумя дополнительными порциями по 30 мл хлороформа. Объединенные хлороформные экстракты сушили над сульфатом натрия и концентрировали, давая 416 мг (86%) 2R, 3S1 =N-бензоил-0-(1-этоксиэтил)-3-фенили- зосерина формулы (5), где R1 и R3 фенил; R2 этоксиэтил.
Цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1, 3-оксазин-6-он-2. К раствору 416 мг (1,16 ммоль) 2R, 3S1=N-бензоил-0-(1-этоксиэтил)-3-фенилизомерина в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Затем добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировался 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 265 мг (65%) цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигид-ро-1,3-оксазин-6-он-2 в виде бесцветного масла, (α)25 Hg 22 (СНСl3, с.1.55).
П р и м е р 2. Получение таксола.
В небольшой реакционный сосуд добавляли 77 мг (0,218 ммоль) (-)-цис-2,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 2,40 мг (0, 057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-го водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля и элюировали этилацетатом. Мгновенная хроматография на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 46 мг (77%) 21-0(1-этоксиэтил)-7-0-триэтилсилилтаксола в виде смеси приблизительно 2:1 диастереомеров и 9,3 мг (23% ) 7-0-триэтилсилилбаккатина III. Выход в расчете на потребленный 7-0-триэтилсилилбаккатин III был количественный.
Образец в 5 мг 21-(1-этоксиэтил)-7-0-триэтилсилилтаксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Затем раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) таксола, который был идентичен аутентичному образцу во всех отношениях.
П р и м е р 3. Получение N-дибензол-N-третбутоксикарбонил-таксола.

2-Третбутокси-4-фенил-5-(1-этоксиэток- си)-4,5-дигидро-1, 3-оксазин-6-он. К раствору 409 мг (1,16 ммоль) N-третбутоксикарбонил-С-(1-этоксиэтил)-3-фенилизосерина (3) в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Далее добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 235 мг (70%) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
N-дебензоил-N-третбутоксикарбонил-таксол. В небольшой реакционный сосуд добавляли 73 мг (0,218 ммоль) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,2-оксазин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ного водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля и элюировали этилацетатом. Мгновенной хроматографией на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана получали 44 мг (73%) N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтокси)-7-0-триэтилсилил- таксола в виде приблизительно 1:1 смеси диастереомеров и 9,3 мг (23%) 7-0-триэтилсилилбаккатина III.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-21(1-этоксиэтокси)-7-0-три-этилсилилтаксол а растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонилтаксола.
П р и м е р 4. Получение
N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-3-фе- нил-таксола
2-Третбутокси-4, 5-дифенил-5-(1-этокси- этокси)-4,5-дигидро-1,3-оксазин-6-он. К раствору 497 мг (1,16 ммоль) N-третбутоксикарбонил-0-(1-этоксиэтил)-3,3-дифенилизосери- на (3) в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Затем добавляли раствор 134 кг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата и данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 243 мг (59%) 2-третбутокси-4,4, -дифенил-5-(1-этоксиэтокси)-4,4-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
N-Дебензоил-N-третбутоксикарбонил-3-фенил-таксол. В небольшой реакционный сосуд добавляли 90 мг (0,218 ммоль) 2-третбутокси-4,4-дифенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через средство из силикагеля и элюировали этилацетатом. Мгновенной хроматографией на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана получали 44 мг (66%) N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-3-фенил-7-0-триэтил-силил -такв виде приблизительно 3:1 смеси диастереомеров.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-2-(1-этоксиэтил)-31-фенил- 7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ного водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на силикагеле с элюированием смесью этилацетата и гексана, давая 4,0 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонил-3-фенилтак-сола.
П р и м е р 5. Получение 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5 дигидро-1,3-оксазин-6-она.
К раствору 480 мг (1,16 ммоль) N-бензоил -С-(1-этоксиэтил)-2-метил-3-фенилизосерина в 20 мл ТГФ добавляли 261 мг (2,33 ммоль) твердого третбутилата калия и смесь перемешивали при 25оС в течение 30 мин. Добавляли раствор 134 мг (1,16 ммоль) метансульфонилхлорида в 3,2 мл ТГФ и смесь перемешивали при 25оС в течение 1,5 ч. Смесь разбавляли 80 мл гексана и этилацетата, данный раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия и 10 мл солевого раствора. Органическую фазу сушили над сульфатом натрия и концентрировали, давая 270 мг (76%0 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5-дигидро-1,3-оксазин-6-она в виде бесцветного масла.
П р и м е р 6. 3-метил-таксол.
В небольшой реакционный сосуд добавляли 77 мг (0,218 ммоль) 2,4-дифенил-5-(1-этоксиэтокси)-5-метил-4,5-дигидро-1,3-окса- зин-6-она, 40 мг (0,057 ммоль) 7-0-триэтилсилилбаккатина III 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (ДМАР) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через средство из силикагеля и элюировали этилацетататом. Мгновенная хроматография на силикагеле с элюированием смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана дает 32 мг (53%) 2-(1-этокси-этил)-3'-метил-7-0-триэтилсилил-таксола в виде приблизительно 1:1 смеси диастереомеров.
Образец 2'-(1-этоксиэтил)-3'-метил-7-0-триэтилсилил-таксола (5 мг) растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонке из силикагеля с элюированием смесью этилацетата и гексана, давая 3,9 кг (приблизительно 90%) 3'-метил-таксола.
Дополнительные примеры.
N-дебензоил-N-(1-нафтоил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-(1-нафтил)-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90% ) N-дебензоил-N-(1-нафтоил)-2'-(1-этоксиэто- кси)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-(1-нафтоил)-2'-(1-этоксиэтокси)-7-0-триэтилсилил- таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) N-дебензоил-N-(1-нафтоил)-таксола.
N-дебензоил-N-(2-нафтоил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-(2-нафтил)-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 64 мг (93% ) N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэто- кси)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэтокси)-7-0-триэтилсилил- таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,5 мг (приблизительно 90%) N-дебензоил-N-(2-нафтоил)-таксола.
N-дебензоил-N-пивалоил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-третбутил-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 55 мг (85% ) N-дебензоил-N-пивалоил-2'-(1-этоксиэток-си)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-пивалоил-2'-(1-этоксиэтокси)-7-0-триэтилсилил-та- ксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) N-дебензоил-N-пивалоил-таксола.
N-дебензоил-N-пентаноил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-n-бутил-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле при элюировании смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (89%) N-дебензоил-N-пентаноил-2'-(1-этоксиэток- си)-7-0-триэтилсилил-таксола.
Образец в 5 мг N-дебензоил-N-пентаноил-2'-(1-этоксиэтокси)-7-0-триэтилсилил-та- ксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) N-дебензоил-N-пентаноил-таксола.
3-Десфенил-3'-(1-нафтил)-таксол. В небольшой реакционный сосуд добавляли 125 мг (0,320 ммоль) 2-фенил-4-(1-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-(1-наф- тил)-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-(1-нафтил)-7-0-триэтилсилил-так- сола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) 3'-десфенил-3'-(1-нафтил)-таксола.
3'-десфенил-3'-(2-нафтил)-таксол. В небольшой реакционной сосуд добавляли 125 мг (0,320 ммоль) 2-фенил-4-(2-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 62 мг (90%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-(2-наф- тил)-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-(2-нафтил)-7-0-триэтилсилил-так- сола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,9 мг (приблизительно 95%) 3'-десфенил-3'-(2-нафтил)-таксола.
3'-Десфенил-3'-t-бутил-таксол. В небольшой реакционный сосуд добавляли 102 мг (0,320 ммоль) 2-фенил-4-третбутил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6- она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография из силикагеля, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (89%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-t-бутил-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-t-бутил-7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 92%) 3'-десфенил-3'-t-бутил-таксола.
3'-Десфенил-3'-р-метоксифенил-таксол. В небольшой реакционный сосуд добавляют 118 мг (0,320 ммоль) 2-фенил-4-р-метокси-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мг (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 58 мг (87%) 2'-(1-этоксиэтокси)-3'-десфенил-3'-р-метоксифенил-7-0-триэтилсилил-таксола.
Образец в 5 мг 2'-(1-этоксиэтокси)-3'-десфенил-3'-р-метоксифенил-7-0-триэтилси-лил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,6 мг (приблизительно 90%) 3'-десфенил-3'-р-метоксифенил-таксола.
N-Дебензоил-N-(2-нафтоил)-3'-десфе-нил-3'-(1-нафтил)таксол. В небольшой реакционный сосуд добавляли 140 мгл (0,320 ммоль) 2-(2-нафтил)-4-(1-нафтил)-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 45 мг (0,064 ммоль) 7-0-триэтилсилилбаккатина III, 7,8 мгл (0,064 ммоль) 4-диметиламинопиридина (DMAP) и 0,032 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагировали 20 мл 10%-ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 51 мг (70%) N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэто- кси)-3'-десфенил-3'-(1-нафтил)-7-0-триэтил- силил-таксола.
Образец в 5 мг N-дебензоил-N-(2-нафтоил)-2'-(1-этоксиэтокси)-3'-десфенил-3'-(1- нафтил)-7-0-триэтилсилил-таксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора HCl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,3 мг (приблизительно 80%) N-дебензоил-N-(2-нафтоил)-3'-десфенил-3'-(1-нафтил)-таксола.
N-дебензоил-N-третбутоксикарбонил-10-деацетил-таксол (таксотер). В небольшой реакционный сосуд добавляли 73 мг (0,218 ммоль) 2-третбутокси-4-фенил-5-(1-этоксиэтокси)-4,5-дигидро-1,3-оксазин-6-она, 44 мг (0,057 ммоль) 7,10-бис-0-триэтилсилилбаккатина III, 6,9 мг (0,057 ммоль) 4-диметиламинопиридина (DMAP) и 0,029 мл пиридина. Смесь перемешивали при 25оС в течение 12 ч и разбавляли 100 мл этилацетата. Этилацетатный раствор экстрагируют 20 мл 10% -ого водного раствора сульфата меди, сушили над сульфатом натрия и концентрировали. Остаток фильтровали через фильтровальное средство из силикагеля, элюированного этилацетатом. Мгновенная хроматография на силикагеле, элюированном смесью этилацетата и гексана, с последующей перекристаллизацией из смеси этилацетата и гексана давала 45 мг (68%) N-дебензоил-N-третбутоксикарбонил-2'-(1-этоксиэтокси)-3'-7,10-бис-0-триэтилс илилв виде приблизительно 1:1 смеси диастериомеров.
Образец в 5 мг N-дебензоил-N-третбутоксикарбонил-2'-(1-этоксиэтокси)-3'-7,10-бис-0-триэтилс илилтаксола растворяли в 2 мл этанола и добавляли 0,5 мл 0,5%-ого водного раствора НСl. Смесь перемешивали при 0оС в течение 30 ч и разбавляли 50 мл этилацетата. Раствор экстрагировали 20 мл насыщенного водного раствора бикарбоната натрия, сушили над сульфатом натрия и концентрировали. Остаток очищали с помощью хроматографии на колонне на силикагеле, элюированном смесью этилацетата и гексана, давая 3,8 мг (приблизительно 90%) N-дебензоил-N-третбутоксикарбонил-10-деацетил-таксола.
Реферат
Использование: в производстве таксола - агента для хемотерапии рака с широким спектром антилейкемической и опухолеингибирующей активностей. Сущность изобретения: продукт - оксазинон ф-лы 1, где R1 - фенил, 1- или 2-нафтил, C1-C6 -алкил, третбутоксигруппа; R2 - фенил, 1- или 2-нафтил, C1-C6 -алкил или параметоксифенил; R3 - гидроксилзащищающая группа, такая как этоксиэтил, а также таксол. Реагент 1: оксазинон ф-лы 1. Реагент 2: 10-7-0-триэтилсилилбаккатин. Условия процесса: в присутствии активирующего агента, например третичного амина, и переводом полученного бета-амидосложного эфира действием кислоты в целевой продукт. Способ позволяет упростить процесс получения таксола за счет использования нового легкодоступного исходного - оксазинона со структурной ф-лой 1:
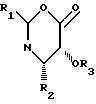
Формула

где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или T-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 -C6-алкил или p-метоксифенил;
R3 гидроксилзащищающая группа, такая как этоксиэтил.
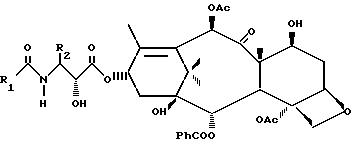
где R1 фенил, α- -или β -нафтил, C1 - C6-алкил или t-бутоксигруппа;
R2 фенил, a- -или β -нафтил, C1 C6-алкил или p-метоксифенил,
отличающийся тем, что оксазинон общей формулы

где R1, R2 и R3 имеют указанные значения, подвергают взаимодействию со спиртом формулы

в присутствии активирующего агента и полученный β -амидосложный эфир действием кислоты переводят в целевой продукт.
14.11.89 при R1 и R2 фенил, a или b -нафтил, C1 C6-алкил, R3 -гидроксизащищающая группа, такая как этоксиэтил, R2 p-метоксифенил;
30.10.90 при R1 фенил, a или b -нафтил, C1 - C6-алкил или t-бутоксигруппа, R2 фенил, a- или β -нафтил, C1 C6-алкил или p-метоксифенил и R3 - гидроксизащищающая группа, такая как этоксиэтил.
Комментарии