Ингибиторы гистондезацетилазы - RU2453536C2
Код документа: RU2453536C2
Описание
Область изобретения
Описаны соединения формулы (I), их производные, аналоги, таутомерные формы, стереоизомеры, геометрические изомеры, полиморфы, гидраты, сольваты, промежуточные соединения, фармацевтически приемлемые соли, фармацевтические композиции, метаболиты и пролекарства.

В описании раскрыт способ получения вышеуказанных новых стильбеноподобных соединений формулы (I), их производных, аналогов, таутомерных форм, стереоизомеров, полиморфов, гидратов, сольватов, фармацевтически приемлемых солей, фармацевтических композиций, метаболитов и пролекарств.
Раскрытые в описании соединения являются ингибиторами гистондезацетилазы (HDAC), а также останавливают клеточный рост неопластических клеток, ингибируя таким образом пролиферацию. Они могут применяться в качестве терапевтических агентов при заболеваниях, в которые вовлечен клеточный рост, таких как злокачественные опухоли, аутоиммунные заболевания, кожные заболевания, инфекции и т.д.
Предпосылки изобретения
Регуляция транскрипции является главным событием в клеточной дифференциации, пролиферации и апоптозе. Активация транскрипции набора генов определяет назначение клетки, и по этой причине транскрипция жестко регулируется разнообразием факторов. Одним из ее регуляторных механизмов, вовлеченных в процесс, является альтерация третичной структуры ДНК, которая влияет на факторы транскрипции в их целевых участках ДНК. Нуклеосомная целостность регулируется состоянием ацетилирования ядерного гистона, результатом является разрешение транскрипции.
Считают, что виды регуляции фактора транскрипции включаются изменениями в структуре хроматина. Изменения сродства белков гистона к свернутой ДНК в нуклеосомах изменяет структуру хроматина. Полагают, что гипоацилированные гистоны имеют большее сродство к ДНК и образуют тесно связанный комплекс ДНК-гистон, и делают ДНК недоступной для регуляции транскрипции. Состояние ацилирования гистона управляется равновесием активностей гистонацетилтрансферазы (НАТ) и гистондезацетилазы (HDAC).
Первое выделение гистондезацетилазы из неочищенных клеточных экстрактов было описано в 1964 году, но молекулярная характеризация изоформ фермента была осуществлена только недавно. Ингибиторами гистондезацетилазы (HDAC) являются цинкгидролазы, ответственные за дезацетилирование остатков N-ацетил-лизина в гистоне и негистоновых белковых субстратов. HDAC человека классифицируют по двум различным классам: HDAC и сиртуины. HDAC разделяют на два подкласса на основе их родства с гистондезацетилазами дрожжей: RPD 3 (класс I включает HDAC 1, 2, 3, 8 и 11) и Hda 1 (класс II включает HDAC 4, 6, 7, 9 и 10). Все указанные HDAC имеют сильно преобразованный цинк-зависимый каталитический домен. Растет число доказательств того, что состояние ацетилирования белков, и таким образом - семейства HDAC, играет решающую роль в модуляции ряда биологических процессов, включая транскрипцию и клеточный цикл.
Недавно было обнаружено, что ингибиторы HDAC останавливают клеточный рост и апоптоз у некоторых типов раковых клеток, включая рак прямой кишки, Т-клеточную лимфому и эритролейкемические клетки (M. Paris, et al., J. Med. Chem., 2008, 51, 1505-1529).
Было установлено, что ингибитор HDAC MG3290 является мощным, селективным к грибкам потенциатором некоторых азольных противогрибковых средств на видах Aspergillus и Candida, включая C. glabrata, и также было найдено, что он потенцирует азол-резистентный мутант C. glabrata (WO 2008/021944 и US 2008/0139673).
Принимая, что апоптоз является решающим фактором прогрессирования рака, ингибиторы HDAC являются многообещающими реагентами для раковой терапии в качестве эффективных индукторов апоптоза.
Недавно субероиланилидгидроксамовая кислота (SAHA) была выпущена как противораковый агент для лечения кожной Т-клеточной лимфомы (CTCL), и она известна как ингибитор HDAC. Было идентифицировано несколько структурных классов ингибиторов HDAC, обзор дан в Marks, P.A. et al., J. Natl. Cancer Inst., 2000, 92, 1210-1215. Более конкретно, в патентных публикациях WO 98/55449 и US 5369108 сообщается о алканоилгидроксаматах с HDAC-ингибирующей активностью. Другими соединениями, которые способны ингибировать активность HDAC, являются трихостатин A (TSA), PXD101, тропоксин (TPX), бутират натрия (NaB), вальпроат натрия (VPA), пептиды, содержащие циклическую гидроксамовую кислоту (CHAP), депсипептиды FK-228, MGCD0103 и MS-275 могут подавлять такие гены, что приводит к антипролиферативным эффектам in vitro и противораковым эффектам in vivo.
1) В WO 2001038322 раскрыты соединения и способы для ингибирования ферментной активности гистондезацетилазы, которые имеют следующие формулы I и II.

где Cy представляет циклоалкил, арил, гетероарил или гетероциклил, любой из которых может быть необязательно замещен; L2 представляет C1-C6 насыщенный алкилен или C1-C6 алкенилен, где алкилен или алкенилен могут быть необязательно замещенными; Ar представляет арилен, где указанный арилен может быть необязательно дополнительно замещен. Y2 представляет химическую связь или насыщенный алкилен с нормальной или разветвленной цепью, который может быть необязательно замещен; Z выбран из группы, состоящей из анилинила, пиридила, тиадиазолила и -O-M, M является H. L3 выбран из группы, состоящей из C1-C6 алкилена или C1-C6 алкенилена, где алкилен или алкенилен могут быть необязательно замещенными; Y3 представляет C2-С3 алкенилен или C2-С3 алкинилен;
2) В US 6624197 B1 раскрыт класс дифенилэтиленов формулы A
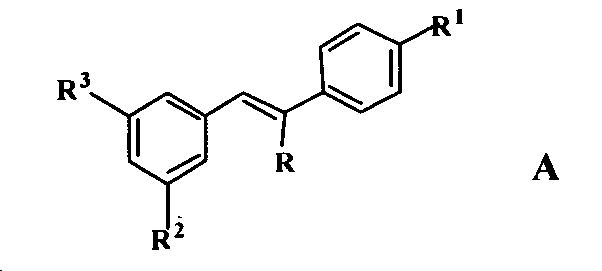
где R представляет водород или -CO2Z, Z представляет водород или катион; и R1, R2 и R3, каждый независимо, представляют H, -OH или -OR4, где R4 представляет нормальный или разветвленный алкил с 1-12 атомами углерода; при условии, что когда R представляет водород, а R2=R3=-OMe, тогда R1 не является -OH. Конфигурация при двойной связи может быть E/Z. Также предложен класс стиренов формулы B;
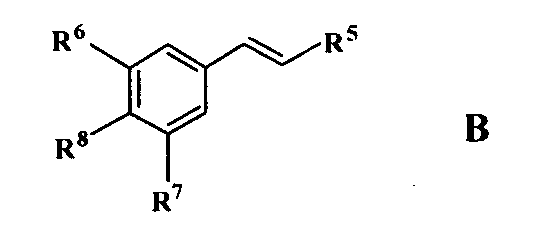
где R5 представляет водород или метил; R6 и R7 независимо представляют водород или OMe; R8 представляет водород или гидрокси. Конфигурация при двойной связи может быть E/Z. Предложены фармацевтические композиции соединений формулы A или B для лечения диабета, включающие терапевтически эффективное количество соединений в физиологически приемлемом носителе. Также предложен способ лечения диабета, включающий стадию орального введения субъекту, страдающему диабетическим состоянием, терапевтически эффективного количества соединения формулы A или B.
3) В US 20050038125 описан способ лечения и/или профилактики расстройств с повышенными уровнями PGE2 (таких как артрит, фибромиалгия и боль) и/или LTB4 (таких как астма, аллергия, артрит, фибромиалгия и воспаление), включающий введение млекопитающему эффективного количества птеростильбенового компонента (PS компонента), фармацевтически приемлемой соли PS компонента или предшественника PS компонента, где PS компонент имеет формулу C.
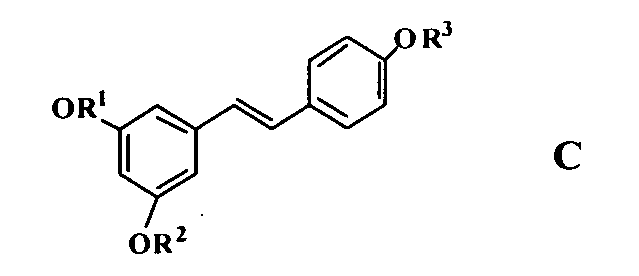
в которой R1, R2 и R3 независимо выбраны из водорода, C1-50 гидрокарбила, C1-50 замещенного гидрокарбила, C1-50 гетерогидрокарбила, C1-50 замещенного гетерогидрокарбила; и где, по меньшей мере, один из R1 и R2 не является водородом.
4) В US 2004/0077726 раскрыты некоторые активные соединения карбаминовой кислоты, которые ингибируют активность HDAC и имеют следующую формулу D,
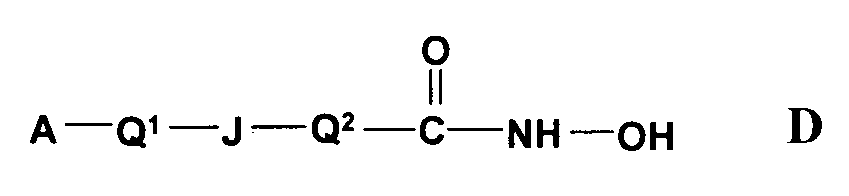
в которой A представляет арильную группу; Q1 представляет ковалентную связь или арильную лидерную группу; J представляет сульфонамидный линкер, выбранный из: -S(=O)2NR1- и -NR1S(=O)2-; R1 представляет заместитель в сульфонамиде; и Q2 представляет кислотную лидерную группу; при условии, что если J представляет -S(=O)2NR1-, то Q1 представляет арильную лидерную группу; и их фармацевтически приемлемые соли, сольваты, амиды, сложные эфиры, простые эфиры, химически защищенные формы и пролекарства. Описаны фармацевтические композиции, включающие такие соединения, и их применение для ингибирования пролиферативных состояний. Также описаны соединения формулы E, в которой Q1 представляет ковалентную связь, J представляет -NR1SO2-, Q2 представляет фенилен-мета-транс-этилен. RB представляет фтор, хлор, метил, этил, изопропил, трет-бутил, трифторметил, гидрокси, метокси, этокси, изопропокси, метилтио, амино, диметиламино, диэтиламино, морфолино, ацетамидо, нитро и фенил, m является целым числом от 0 до 4.

5) В WO 2008/054154 раскрыто нафталинилоксипропенильное производное в качестве ингибиторов HDAC формулы 1a-1d, в котором R1 представляет замещенные или незамещенные алкильные группы с одним или более заместителями.

Краткое описание
Новые замещенные ингибиторы HDAC формулы (I)
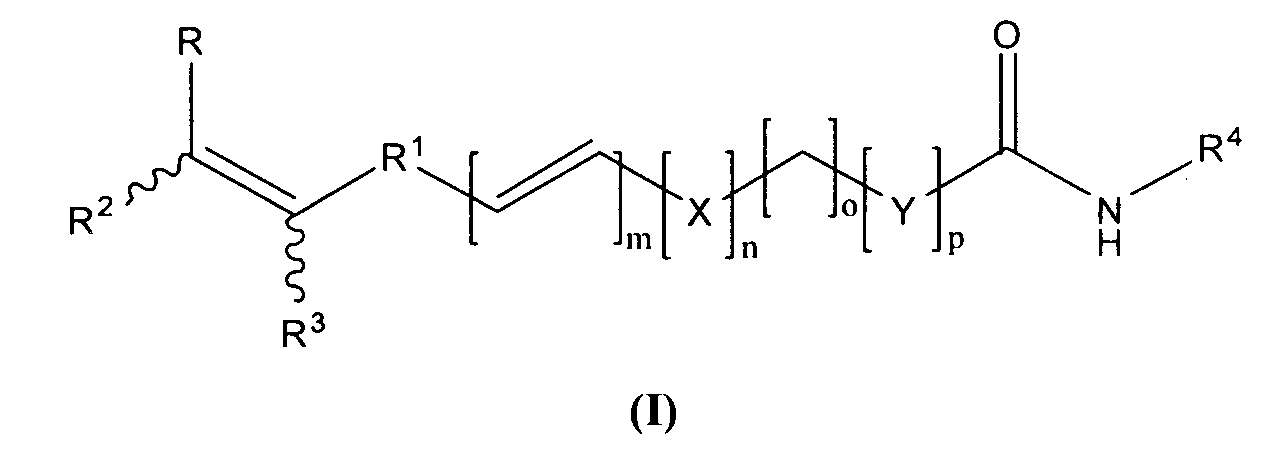
их производные, аналоги, таутомерные формы, стереоизомеры, полиморфы, сольваты, промежуточные соединения, фармацевтически приемлемые соли, фармацевтические композиции, метаболиты и пролекарства,
в которых конфигурация при двойной связи может быть E/Z;
R представляет замещенные или незамещенные группы, выбранные из арила, циклоалкила, гетероарила, арилалкила, арилалкенила, арилалкинила, гетероциклила, гетероарилалкила, гетероарилалкенила и гетероарилалкинила;
R1 представляет замещенные или незамещенные группы, выбранные из арильных и гетероарильных групп;
R2 и R3 независимо представляют водород, алкил, -COOR5, -CONR5R6, -CH2NR5R6, -CH2CH2NR5R6, -CH2CH2OR5, -CH2OR5, -CH2OCONR5R6 и -CH2NR5COR6; в которых один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом;
R5 и R6 независимо представляют водород, замещенные или незамещенные группы, выбранные из алкила, циклоалкила, гетероциклила, арила, арилалкила, арилалкенила, арилалкинила, гетероарила, гетероарилалкила, гетероарилалкенила и гетероарилалкинила, или R5 или R6 могут быть объединены с образованием 3-8-членного кольца, имеющего 0-2 гетероатома, таких как N, O или S;
R4 представляет OR7, арил, орто-замещенный анилин, аминоарил и аминогетероарил, которые могут быть далее замещены; где R7 представляет водород, -COR8, замещенные или незамещенные группы, выбранные из алкила, циклоалкила, арила, гетероарила и гетероциклила; где R8 представляет замещенные или незамещенные группы, выбранные из алкила, арила, гетероарила и гетероциклила; X представляет -O-, -NR7-, -CONR7-, -NR7SO2-, -SO2NR7-, -SO2O-, О-SO2-, -CH2NR7-, -NR7CONR7- и -NR7CO-;
Y представляет арил, арилалкенил и гетероарил;
m является целым числом от 0-3; n является целым числом от 0-1; o является целым числом от 0-7; и p является целым числом от 0-1,
при условии, что если n, o и p=0, тогда m=0-1; и
при условии, что если n=1, o=3-7 и p=0, тогда m=0-1; и
при условии, что если n, o и p=1, тогда m=0-1.
Краткое описание фигур
Фиг. 1 показывает эффективность соединения 105 на модели ксенографта НСТ-116.
Подробное описание
Новые соединения формулы (I)

их производные, аналоги, таутомерные формы, стереоизомеры, полиморфы, сольваты, промежуточные соединения, фармацевтически приемлемые соли, фармацевтические композиции, метаболиты и пролекарства,
в которых конфигурация при двойной связи может быть E/Z;
R представляет замещенные или незамещенные группы, выбранные из арила, циклоалкила, гетероарила, арилалкила, арилалкенила, арилалкинила, гетероциклила, гетероарилалкила, гетероарилалкенила и гетероарилалкинила;
R1 представляет замещенные или незамещенные группы, выбранные из арила и гетероарила;
R2 и R3 независимо представляют водород, алкил, -COOR5, -CONR5R6, -CH2NR5R6, -CH2CH2NR5R6, -CH2CH2OR5, -CH2OR5, -CH2OCONR5R6 и -CH2NR5COR6; в которых один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом;
R5 и R6 независимо представляют водород, замещенные или незамещенные группы, выбранные из алкила, циклоалкила, гетероциклила, арила, арилалкила, арилалкенила, арилалкинила. гетероарила, гетероарилалкила, гетероарилалкенила и гетероарилалкинила; или R5 и R6 могут быть объединены с образованием насыщенного или ненасыщенного 3-8-членного кольца, имеющего 0-2 гетероатома, таких как N, O или S;
R4 представляет OR7, арил, орто-замещенный анилин, аминоарил и аминогетероарил, которые могут быть далее замещены; где R7 представляет водород, -COR8, необязательно замещенный алкил, циклоалкил, арил, гетероарил и гетероциклил; где R8 представляет необязательно замещенный алкил, арил, гетероарил и гетероциклил;
X представляет -O-, -NR7-, -CONR7-, -NR7SO2-, -SO2NR7-, -SO2O-, О-SO2-, -CH2NR7-, -NR7CONR7- и -NR7CO-;
Y представляет арил, арилалкенил и гетероарил;
m является целым числом от 0-3; n является целым числом от 0-1; o является целым числом от 0-7; и p является целым числом от 0-1,
когда группы R, R1, R5, R6, R7 и R8 являются замещенными, заместители, которых может быть один или более, выбраны из галогенов, таких как фтор, хлор, бром, йод; гидрокси; нитро; циано; оксо (=O); тиоксо (=S); азидо; нитрозо; амино; гидразино; формила; алкила; алкокси; арила; галогеналкильной группы, такой как трифторметил, трибромметил и трихлорметил; галогеналкокси, включая -OCH2Cl; арилалкокси, включая бензилокси и фенилэтокси; циклоалкила; -O-циклоалкила; арила; алкокси; гетероциклила: гетероарила; алкиламино; -O-CH2-циклоалкила; -COORa; -C(O)Rb; -C(S)Ra: -C(O)NRaRb; -NRaC(O)NRbRc; -N(Ra)SORb; -N(Ra)SO2Rb; -NRaC(O)ORb; -NRaRb; -NRaC(O)Rb-; NRaC(S)Rb-; -SONRaRb-; -SO2NRaRb-; -ORa; -ORaC(O)ORb-; -OC(O)NRaRb; OC(O)Ra; -OC(O)NRaRb-; -RaNRbRc; -RaORb-; -SRa; -SORa и -SO2Ra; Ra, Rb и Rc каждый независимо представляет атом водорода; замещенные или незамещенные группы, выбранные из алкила; арила; арилалкила; циклоалкила; гетероциклила; гетероарила и гетероарилалкила;
заместители, в свою очередь, далее замещены галогенами, такими как фтор, хлор, бром и йод; гидрокси; нитро; циклоалкилом; циано; азидо; нитрозо, амино, гидразино, формилом; алкилом; галогеналкильной группой, такой как трифторметил и трибромэтил;
при условии, что если n, o и p=0, тогда m=0-1; и
при условии, что если n=1, o=3-7 и p=0, тогда m=0-1; и
при условии, что если n, o и p=1, тогда m=0-1.
Термин «алкил» относится к алифатическим углеводородным группам с нормальной или разветвленной цепью, имеющим указанное число атомов углерода, которые присоединены к остатку молекулы единственным атомом. Примеры таких алкильных групп включают, но без ограничения, метил, этил, н-пропил, изопропил, бутил, изобутил, трет-бутил, пентил, гексил, гептил и октил.
Термин «арил» относится к ароматическим радикалам, имеющим от 6 до 14 атомов углерода, таким как фенил, нафтил, бифенил, инданил, замещенным или незамещенным ариленовым группам, таким как фенилен, бифенилен, нафтилен, антраценилен, фенантрилен и инданилен.
Термин «арилалкил» относится к арильной группе, непосредственно связанной с алкильной группой, примеры таких арилалкильных групп включают, но без ограничения, бензил и фенэтил.
Термин «гетероциклил» относится к стабильному 3-15-членному кольцевому радикалу, который состоит из атомов углерода и от одного до пяти гетероатомов, выбранных из азота, фосфора, кислорода и серы. Для целей данного изобретения гетероциклический кольцевой радикал может быть моноциклической, бициклической или трициклической системами колец, а атомы азота, фосфора, углорода, кислорода или серы в гетероциклическом кольцевом радикале могут быть необязательно окислены до различных состояний окисления. В дополнение, атом азота может быть необязательно кватернизован; и кольцевой радикал может быть частично или полностью насыщенным. Примеры таких гетероциклических кольцевых радикалов включают, но без ограничения, азетидинил, акридинил, бензодиоксолил, бензодиоксанил, бензофуранил, карбазолил, циннолинил, диоксоланил, индолизинил, нафтиридинил, пергидроазепинил, феназинил, фенотиазинил, феноксазинил, фталазинил, пиридил, птеридинил, пуринил, хиназолинил, хиноксалинил, хинолинил, изохинолинил, тетразолил, имидазолил, тетрагидроизохинолил, пиперидинил, пиперазинил, гомопиперазинил, 2-оксоазепинил, азепинил, пирролил, 4-пиперидонил, пирролидинил, пиразинил, пиримидинил, пиридазинил, оксазолил, оксазолинил, триазолил, инданил, изоксазолил, изоксазолидинил, тиазолил, тиазолинил, тиазолидинил, изотиазолил, хинуклидинил, изотиазолидинил, индолил, изоиндолил, индолинил, изоиндолинил, октагидроиндолил, октагидроизоиндолил, хинолил, изохинолил, декагидроизохинолил, бензимидизолил, тиадиазолил, бензопиранил, бензотиазолил, бензооксазолил, тиенил, морфолинил, тиоморфолинил, сульфоксид тиоморфолинила, фурил, тетрагидрофурил, тетрагидропиранил, хроманил и изохроманил. Гетероциклический кольцевой радикал может быть присоединен к основной структуре по любому гетероатому или атому углерода, что приводит к образованию устойчивой структуры.
Термин «гетероарил» относится к ароматическому гетероциклическому кольцевому радикалу, определенному выше. Гетероароматический кольцевой радикал может быть присоединен к основной структуре по любому гетероатому или атому углерода, что приводит к образованию устойчивой структуры.
Термин «гетероарилалкил» относится к гетероарильному кольцевому радикалу, определенному выше, непосредственно связанному с алкильной группой. Гетероарилалкильный радикал может быть присоединен к основной структуре по любому атому углерода алкильной группы.
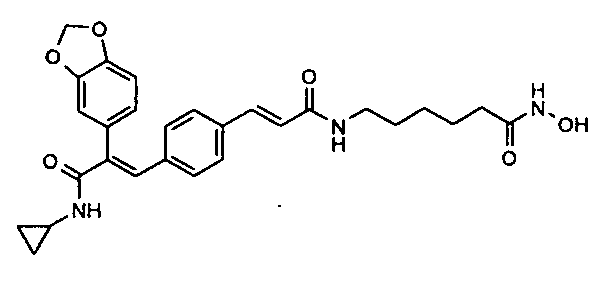
Термин «циклоалкил» относится к неароматической моно- или полициклической кольцевой системе из примерно от 3 до 12 атомов углерода. Примеры циклоалкильных групп включают, но без ограничения, циклопропил, циклобутил, циклопентил, циклогексил, циклооктанил, а примеры полициклических колец включают пергидронафтильную, адамантильную и норборнильную группы, мостиковые циклические группы или спиробициклические группы, например, спиро[4.4]-нон-2-ил.
Термин «алкенил» относится к алифатической углеводородной группе, содержащей двойную связь углерод-углерод, которая может иметь нормальную или разветвленную цепь из от примерно 2 до 10 атомов углерода, и примеры алкенильных групп включают, но без ограничения, этенил, 1-пропенил, 2-пропенил, изо-пропенил, 2-метил-1-пропенил, 1-бутенил и 2-бутенил.
Термин «арилалкенил» относится к ароматическому кольцевому радикалу, непосредственно связанному с алкенильной группой. Арильный радикал может быть присоединен к основной структуре по любому атому углерода алкенильной группы. Примеры таких арилалкенильных групп включают, но без ограничения, фенилэтенил и фенилпропенил.
Термин «гетероарилалкенил» относится к гетероарильному кольцевому радикалу, непосредственно связанному с алкенильной группой. Гетероарильный радикал может быть присоединен к основной структуре по любому атому углерода алкенильной группы. Примеры таких арилалкенильных групп включают, но без ограничения, тиенилпропенил, индолилпропенил, пиридинилэтенил и индолилпропенил.
Термин «алкокси» относится к алкильной группе, присоединенной через атом кислорода к остатку молекулы. Представительные примеры таких групп включают, но без ограничения, -OCH3 и -OC2H5.
Термин «арилокси» относится к арильной группе, присоединенной через атом кислорода к остатку молекулы. Представительные примеры таких групп включают, но без ограничения, -O-фенил и -O-бифенил.
Термин «алкиламино» относится к алкильной группе, определенной выше, присоединенной через аминогруппу к остатку молекулы. Представительные примеры таких групп включают, но без ограничения, -NHCH3 и -N(CH3)2.
Термин «алкинил» относится к гидрокарбильной группе с нормальной или разветвленной цепью, имеющей, по меньшей мере, одну тройную связь углерод-углерод и содержащей атомы углерода в интервале 2-12 атомов. Представительные примеры таких групп включают, но без ограничения, этинил, пропинил и бутинил.
Термин «арилалкинил» относится к ароматическому кольцевому радикалу, непосредственно связанному с алкинильной группой. Арильный радикал может быть присоединен к основной структуре по любому атому углерода алкинильной группы.
Термин «гетероарилалкинил» относится к гетероарильному радикалу, непосредственно связанному с алкинильной группой. Гетероарильный радикал может быть присоединен к основной структуре по любому атому углерода алкинильной группы.
Далее, соединением формулы (I) могут быть его производные, аналоги, таутомерные формы, стереоизомеры, геометрические изомеры, полиморфы, сольваты, промежуточные соединения, фармацевтически приемлемые соли, фармацевтические композиции, метаболиты и пролекарства.
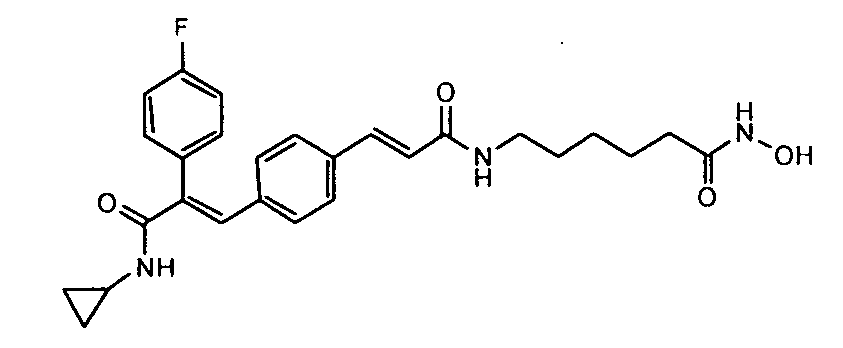
Фармацевтически приемлемые сольваты могут быть гидратами или включать другие кристаллизационные растворители, такие как спирты.
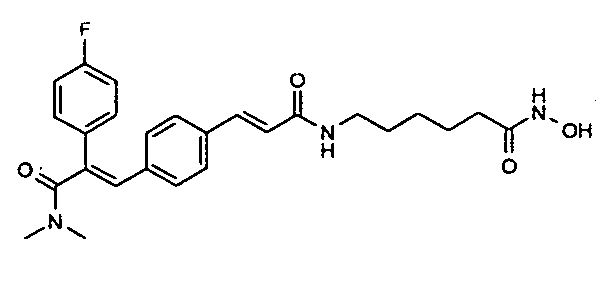
Соединения, раскрытые в описании, могут быть в форме Е или Z геометрических изомеров, и в некоторых случаях также могут присутствовать их смеси. В случаях, когда в формуле I присутствуют две или более двойных связи, это может приводить к более чем двум геометрическим изомерам, и в таких случаях изобретение охватывает все изомеры.
Понятно, что соединения, включенные в семейство соединений формулы (I), являются изомерными формами, включая таутомеры и стереоизомеры (диастереомеры, энантиомеры и геометрические изомеры в виде конфигурационных изомеров E или Z или смеси изомеров E или Z). Также понятно, что некоторые изомерные формы, такие как диастереомеры, энантиомеры и геометрические изомеры, могут быть разделены специалистами в данной области физическими и/или химическими способами.
Соединения, раскрытые в описании, могут существовать в виде единственных стереоизомеров, рацематов или смесей энатиомеров и/или диастереомеров. Все такие стереоизомеры, рацематы и их смеси рассматриваются как входящие в объем описанного объекта изобретения.
Фраза «фармацевтически приемлемый» относится к соединениям или композициям, которые являются физиологически переносимыми и обычно не дают аллергической или подобной нежелательной реакции, включая, но без ограничения, расстройство желудка или головокружение, при ведении млекопитающему.
Фармацевтически приемлемые соли включают соли, производные неорганических оснований, таких как Li, Na, K, Ca, Mg, Fe, Cu, Zn и Mn; соли органических оснований, таких как N,N'- диацетилэтилендиамин, глюкамин, триэтиламин, холин, дициклогексиламин, бензиламин, триалкиламин и тиамин, гуанидин, диэтаноламин, α-фенилэтиламин, пиперидин, морфолин, пиридин, гидроксиэтилпирролидин, гидроксиэтилпиперидин и тому подобные, соли аммония или замещенного аммония, соли алюминия. Соли также включают соли аминокислот, таких как глицин, аланин, цистеин, цистин, лизин, аргинин, фенилаланин, гуанидин и т.д. Если выгодно, то соли могут включать аддитивные соли кислот, которыми являются сульфаты, нитраты, фосфаты, перхлораты, бораты, гидрогалогениды, ацетаты, тартраты, малеаты, цитраты, сукцинаты, пальмоаты, метансульфонаты, тозилаты, бензоаты, салицилаты, гидроксинафтоаты, бензолсульфонаты, аскорбаты, глицерофосфаты и кетоглутараты.
В описании раскрыты пролекарства соединений формулы (I), которые при введении претерпевают химическое превращение вследствие метаболических процессов перед тем, как стать активными веществами. В общем, такие пролекарства могут быть функциональными производными соединений по изобретению, которые способны легко превращаться in vivo в соединение по изобретению.
«Пролекарство» обозначает соединение, которое способно превращаться in vivo метаболическими средствами (то есть, путем гидролиза, восстановления или окисления) в соединение формулы (I). Например, сложноэфирное пролекарство соединения формулы (I), содержащего гидроксильную группу, может быть превращено гидролизом in vivo в родительскую молекулу.
Раскрытые активные соединения также могут быть приготовлены в любой твердой или жидкой форме, например, соединение может быть в кристаллической форме, в аморфной форме и может иметь любой размер частиц. Далее, частицы соединения могут быть микронизированы или нанонизированы, или могут быть агломерированы, приготовлены в виде гранул, порошков, масел, масляных суспензий или в любом другом виде твердой или жидкой физической формы.
Также в описании раскрыты фармацевтические композиции, содержащие одно или более соединений общей формулы (I), определенной выше, их производных, аналогов, таутомерных форм, стереоизомеров, полиморфов, гидратов, метаболитов, пролекарств, фармацевтически приемлемых солей, фармацевтически приемлемых сольватов в комбинации с обычными применяемыми в фармации носителями, разбавителями и тому подобным, полезные для лечения и/или профилактики пролиферативных расстройств.
Фармацевтическая композиция может находиться в обычно используемой форме, такой как таблетки, капсулы, порошки, сиропы, растворы, суспензии и тому подобное, может содержать ароматизаторы, подсластители и т.д. в подходящих твердых или жидких носителях или разбавителях или в подходящих стерильных средах для получения инъекционных растворов или суспензий. Композиции могут быть получены способами, известными в данной области. Подходящие фармацевтически приемлемые носители включают твердые наполнители или разбавители и стерильные водные или органические растворы. Активное соединение будет присутствовать в таких фармацевтических композициях в количестве, достаточном для обеспечения желаемой дозировки в диапазоне, раскрытом выше. Подходящие пути введения включают системное введение, такое как оральное, или парентеральное, такое как чрескожное, внутримышечное, внутривенное и подкожное введение. Так, для орального введения соединения могут быть объединены с подходящим твердым или жидким носителем или разбавителем с получением капсул, таблеток, порошков, сиропов, растворов, суспензий и тому подобного. Если желательно, фармацевтические композиции могут содержать дополнительные компоненты, такие как ароматизаторы, подсластители, эксципиенты и тому подобное. Для парентерального введения соединения могут быть объединены со стерильными водными или органическими средами с образованием растворов или суспензий для инъекций. Например, могут применяться растворы в кунжутном масле или масле земляного ореха, водном пропиленгликоле и тому подобном, а также водные растворы водорастворимых фармацевтически приемлемых аддитивных солей кислот, солей соединений со щелочными или щелочноземельными металлами. Растворы для инъекций, полученные таким образом, могут быть затем введены внутривенно, внутрибрюшинно, подкожно или внутримышечно.
Соединения формулы (I) могут также быть введены в виде фармацевтической композиции в фармацевтически приемлемом носителе, предпочтительно составленных для орального введения.
Соединения, раскрытые в описании, могут также проявлять полиморфизм. Данное изобретение далее включает различные полиморфы соединений. Термин «полиморф» относится к конкретному кристаллическому состоянию вещества, имеющему определенные физические свойства, такие как дифракция рентгеновских лучей, ИК-спектры, температура плавления и тому подобное.
Данное изобретение, в дополнение к перечисленным выше соединениям, охватывает применение гомологов и аналогов таких соединений. В данном контексте гомологами являются молекулы, имеющие существенное структурное сходство с описанными выше соединениями, а аналогами являются молекулы, имеющие существенное биологическое сходство безотносительно к структурному сходству.
Термин «ингибитор гистондезацетилазы» применяют для идентификации соединения, которое способно взаимодействовать с гистондезацетилазой и ингибировать ее активность, более конкретно - ферментную активность. Ингибирование ферментной активности гистондезацетилазы означает уменьшение способности гистондезацетилазы к удалению ацетильной группы из гистона. Предпочтительно такое ингибирование является специфическим, то есть ингибитор гистондезацетилазы уменьшает способность гистондезацетилазы к удалению ацетильной группы из гистона при концентрации, которая ниже, чем концентрация ингибитора, требуемая для достижения другого, не связанного с этим, биологического эффекта.
Термин «гистондезацетилаза» и «HDAC» относится к одному из семейств ферментов, которые удаляют ацетильные группы у остатков ε-аминогрупп лизина на N-конце гистона. Если в контексте не указано иного, то термин «гистон» подразумевает любой гистонный белок, включая Н1, Н2А, Н2В, Н3, Н4 и Н5, любых видов. HDAC белки человека или генные продукты включают, но без ограничения, HDAC-1, HDAC-2, HDAC-3, HDAC-4, HDAC-5, HDAC-6, HDAC-7, HDAC-8, HDAC-9 и HDAC-10. Гистондезацетилаза также может быть произведена из протозойного или грибкового источника.
Изобретение также обеспечивает способ лечения рака у пациента, включающий введение терапевтически эффективного количества соединения формулы (I).
Настоящее изобретение обеспечивает способ лечения расстройства, вызванного, связанного или сопровождающегося нарушениями клеточной пролиферации и/или ангиогенеза, включающий введение терапевтически эффективного количества соединения формулы (I).
Расстройство является пролиферативным расстройством или выбрано из группы, состоящей из рака, воспалительного/иммунного расстройства, фибротических заболеваний (например, фиброза печени), диабета, аутоиммунного заболевания, хронического и острого нейродегенеративного заболевания, болезни Хантингтона и инфекционного заболевания, но не ограничивающейся перечисленными.
Соединения, раскрытые в описании, применяют для лечения или профилактики рака. Рак может включать солидные опухоли или гематологические злокачественные состояния.
Настоящее изобретение предоставляет способ лечения расстройства, заболевания или состояния, которое можно лечить ингибированием ферментов HDAC, включающий введение терапевтически эффективного количества соединения формулы (I).
Изобретение предоставляет способ лечения рака у пациента, включающий введение терапевтически эффективного количества соединения формулы (I). Рак может быть гематологическим злокачественным состоянием, и эта форма гематологического злокачественного состояния выбрана из группы, состоящей из В-клеточной лимфомы, Т-клеточной лимфомы и лейкемии. В случае солидных опухолей опухоли выбраны из группы, состоящей из рака груди, рака легкого, рака яичника, рака предстательной железы, рака головы, рака шеи, рака носа, рака желудка, рака прямой кишки, рака поджелудочной железы и рака мозга.
Термин «терапевтически эффективное количество» или «эффективное количество» обозначает количество, достаточное для благотворного действия или достижения желаемого результата. Эффективное количество может быть введено за одно или более введений. Эффективное количество обычно досточно для облегчения, улучшения, стабилизации, обращения, замедления или отсрочки прогрессирования болезненного состояния.
В другом аспекте соединение может быть введено комбинированной терапией в комбинации соединения формулы (I) с одним или более отдельных агентов, не ограниченных такими мишенями, как HDAC, метилтрансфераза ДНК, киназы белков теплового шока (например, HSP90) и другими металлопротеазами матрикса.
«Комбинированная терапия» включает введение рассматриваемых соединений в комбинации с другими биологически активными ингредиентами (такими как, но без ограничения, различные антинеопластические агенты) и с нелекарственными видами лечения (такими как, но без ограничения, хирургия или лучевая терапия). Соединения, раскрытые в описании, могут применяться в комбинации с другими фармацевтически активными соединениями, предпочтительно - с усиливающими эффект соединений изобретения. Соединения могут быть введены одновременно или последовательно с лечением другими лекарствами.
В другом аспекте рассматриваемые соединения могут быть объединены с антинеопластическими агентами (например, малыми молекулами, моноклональными антителами, антисмысловыми РНК и слитными белками), которые ингибируют одну или более биологических мишеней. Такая комбинация может усилить терапевтическую эффективность по сравнению с эффективностью, достигаемой только одним из агентов, и может предотвратить или отсрочить появление резистентных вариантов.
В другом аспекте рассматриваемые соединения могут быть объединены с противогрибковыми агентами (например, азолами), которые ингибируют одну или более биологических мишеней. Такая комбинация может усилить терапевтическую эффективность по сравнению с эффективностью, достигаемой только одним из агентов, и может предотвратить или отсрочить появление резистентных вариантов.
Соединения изобретения вводят в комбинации с химиотерапевтическими агентами. Химиотерапевтические агенты составляют обширную область терапевтического лечения в онкологии. Эти агенты вводят на различных стадиях заболевания с целью сокращения опухолей, разрушения оставшихся раковых клеток после хирургии, индуцирования ремиссии, поддержания ремиссии и/или ослабления симптомов, относящихся к раку или его лечению.
Подразумевается, что термин «субъект», применяемый в описании, включает всех млекопитающих и, в частности, человека, нуждающихся в лечении. Терапевтически эффективное количество будет изменяться в зависимости от субъекта и подвергаемого лечению болезненного состояния, массы и возраста субъекта, тяжести болезненного состояния, конкретного выбранного соединения формулы (I), режима дозирования, которого следует придерживаться, графика введения, способа введения и тому подобного, все вышеназванное может быть легко определено средним специалистом в данной области.
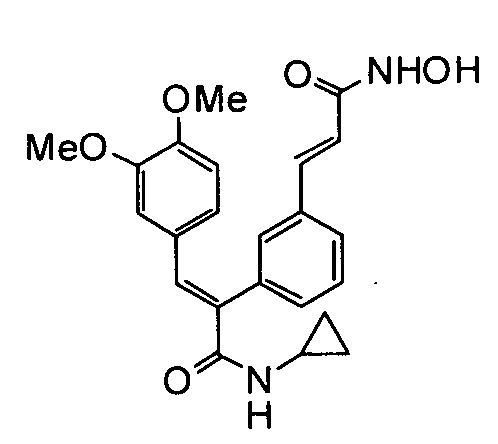
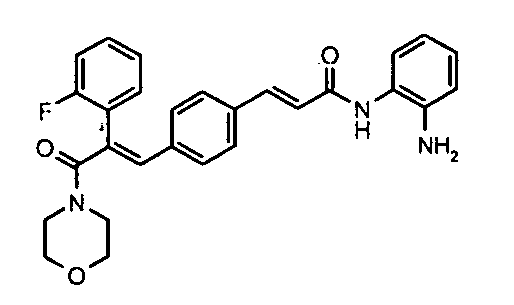
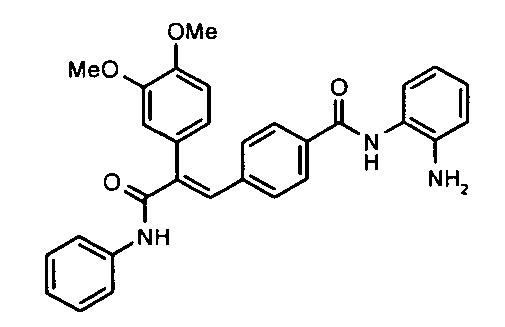
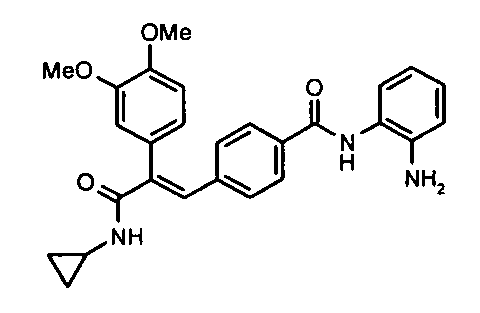
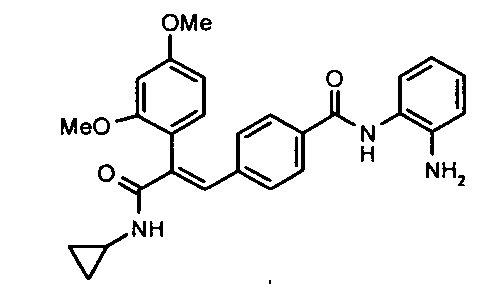
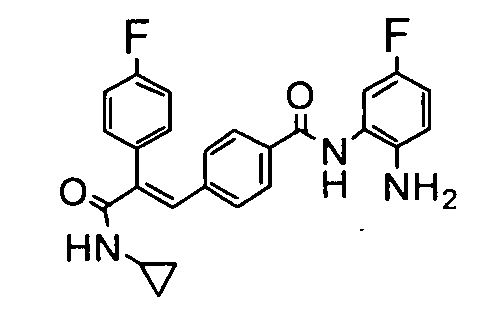
Представительные соединения включают:
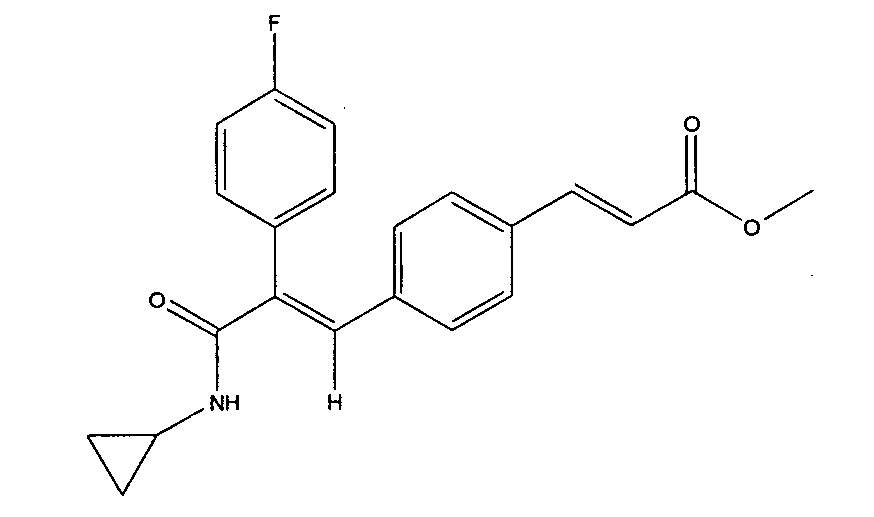
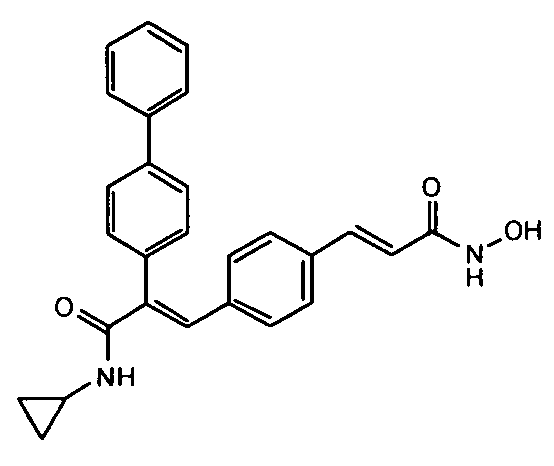
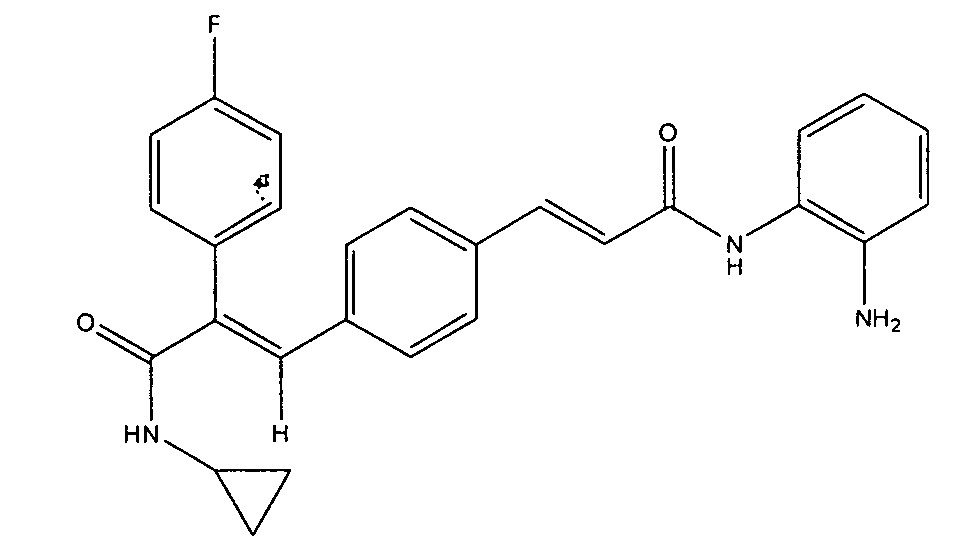
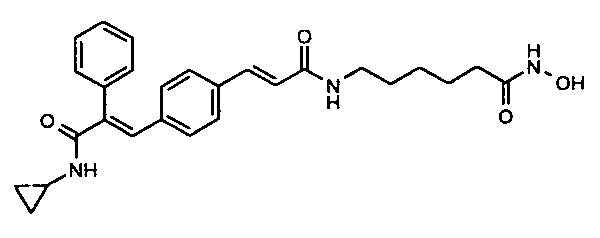
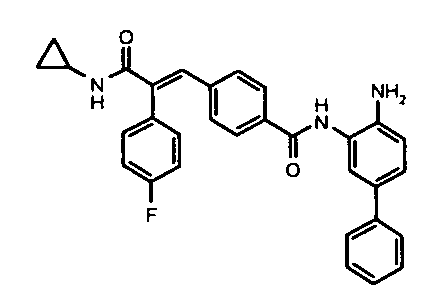
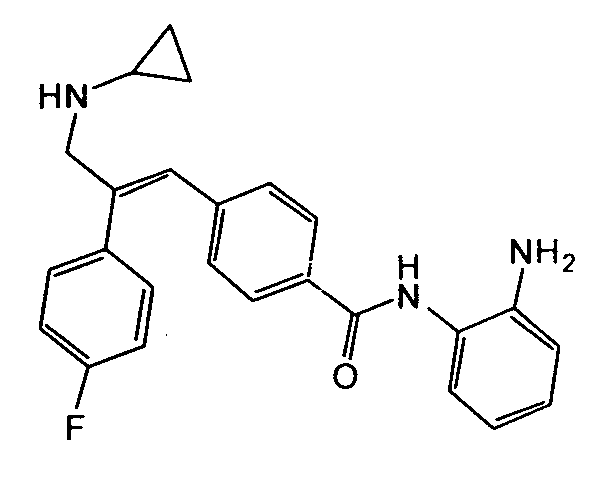
1. N-Циклопропил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
2. N-Метил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
3. N,N-Диметил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
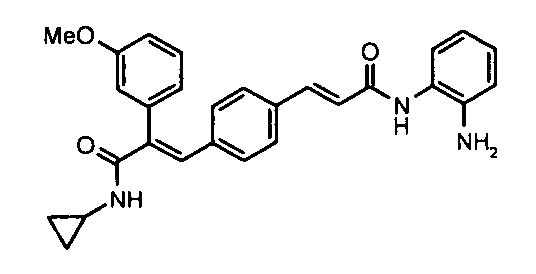
4. 2-Фенил-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
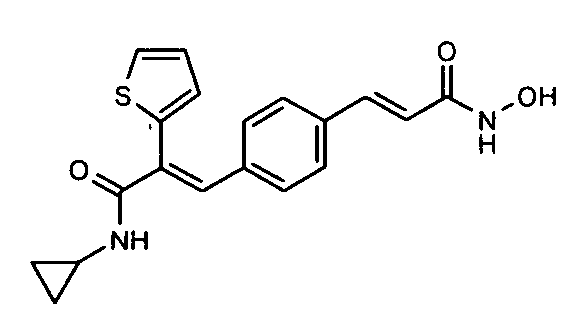
5. N-Циклопропил-2-(тиофен-2-ил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
6. N-Циклопропил-2-фенил-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
7. N-Циклопропил-2-(4-трифторметилфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
8. N-Циклопропил-2-(пиридин-3-ил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
9. N-Циклопропил-2-(4-метоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
10. N-Циклопропил-2-(2-хлорфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
11. N-Циклопропил-2-(2-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
12. N-Циклопропил-2-(3-хлорфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
13. N-Циклопропил-2-[бензодиоксол-5-ил]-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
14. N-Циклопропил-2-(4-метилфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
15. N-Морфолино-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
16. N-Морфолино-2-(2-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
17. N-Морфолино-2-(3-метоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
18. N-Тиоморфолино-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
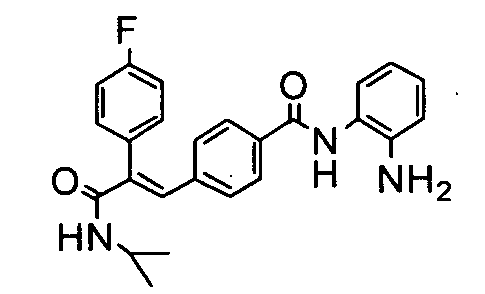
19. N-Циклооктил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
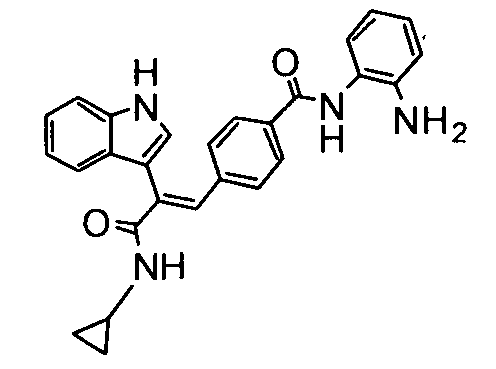
20. N-Циклопропил-2-(3-метоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
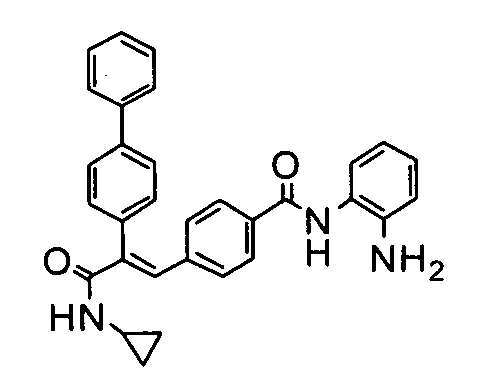
21. N-Циклопропил-2-(3-фторфенил)-3-(4-((lE)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
22. N-Изопропил-2-(3-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
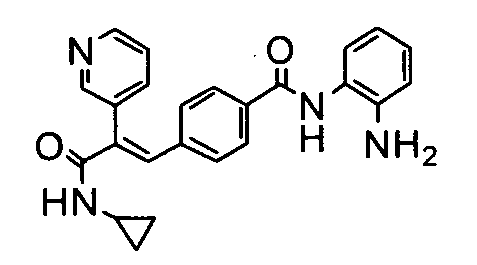
23. N-Изопропил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
24. N-Изопропил-2-(3,4-дифторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
25. N-Циклопропил-2-(3-фтор-4-метоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
26. N-Изопропил-2-(3-фтор-4-метоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
27. N-Циклопропил-2-(3,4-дифторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
28. 2-(4-Фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
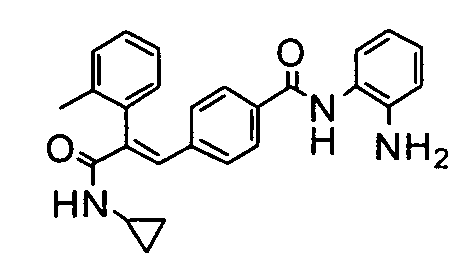
29. 2-(4-Фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламид;
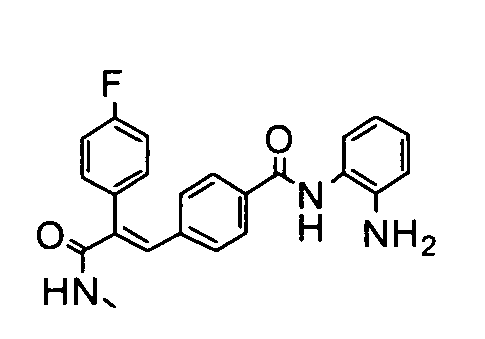
30. N-Пирролидино-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
31. N-Циклопропил-2-(4-Циклопропилметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
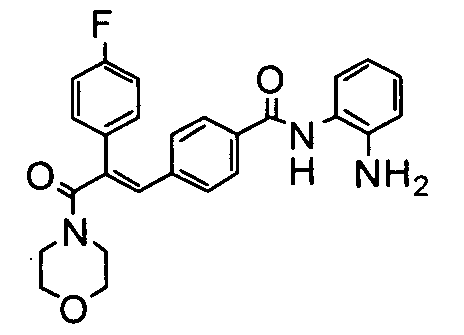
32. N-Циклопропил-2-(4-бензилоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
33. N-Циклопропил-2-(4-циклопентилоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
34. N-(4-Фторбензил)-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
35. N-Циклопропил-2-(2,4-диметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
36. N-Циклопропил-2-(3,4-диметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
37. N-Циклопропил-2-(индол-3-ил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
38. N-Циклопропил-2-(тиофен-3-ил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
39. N-Циклопропил-3-(4-фторфенил)-2-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
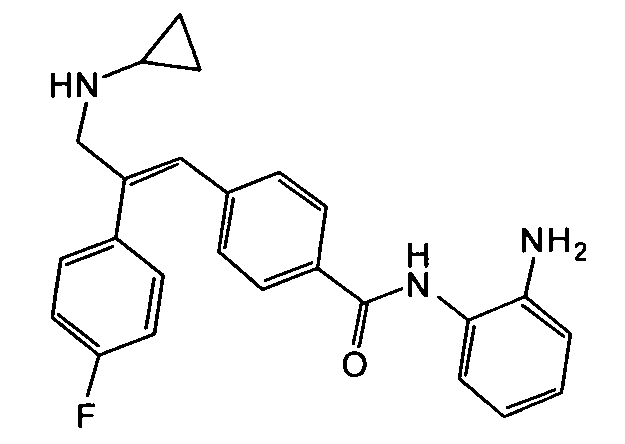
40. N-Циклопропил-3-(4-фторфенил)-2-(3-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
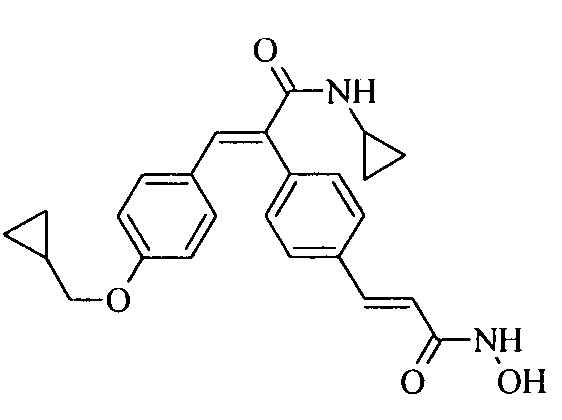
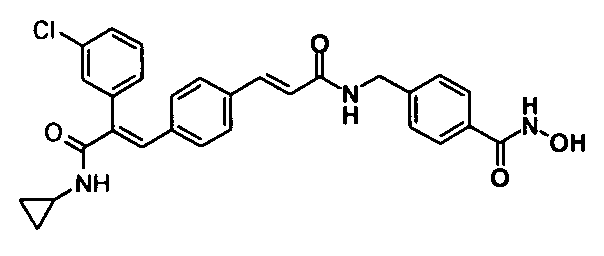
41. N-Циклопропил-2-(3-циклопропилметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
42. 2-(3-Циклопропилметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламид;
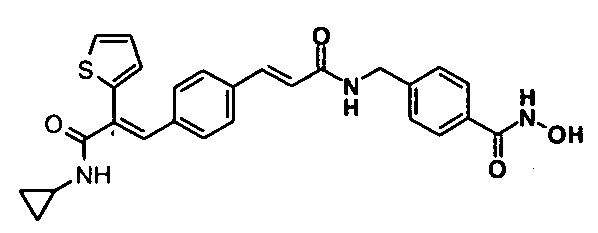
43. N-Циклопропил-2-(3-циклопентилоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
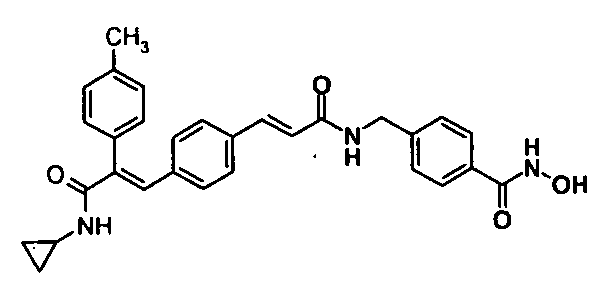
44. 2-(3-Циклопентилоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламид;
45. N-Циклопропил-2-(бифенил-4-ил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
46. 2-(4-Циклопропилметоксифенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламид;
47. N-Циклопропил-3-(3,4-диметоксифенил)-2-(3-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
48. N-Циклопропил-3-(4-метоксифенил)-2-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
49. N-Циклопропил-3-(4-циклопропилметоксифенил)-2-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
50. N-Циклопропил-3-(4-циклопентилоксифенил)-2-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
51. N-Циклопропил-2-(4-фторфенил)-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)бут-2-енамид;
52. 2-[4-(Диметиламино)фенил]-3-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-циклопропилакриламид;
53. N-Циклопропил-3-(4-фторфенил)-2-(4-(3-(гидроксиамино)-3-оксопропил)фенил)акриламид;
54. N-Циклопропил-2-(4-фторфенил)-3-(3-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламид;
55. 3-(4-((1E)-3-(Циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)фенил)-N-гидроксиакриламид;
56. 3-(4-((1E)-3-(Циклопропиламино)-2-фенилпроп-1-ен-1-ил)фенил)-N-гидроксиакриламид;
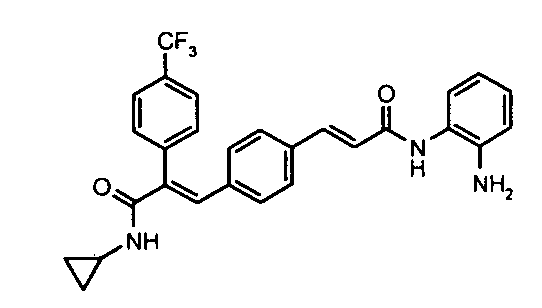
57. 3-(4-((1E)-2-(3-Циклопентилоксифенил)-3-(циклопропиламино)проп-1-ен-1-ил)фенил)-N-гидроксиакриламид;
58. 3-(4-((1E)-2-(3-Хлорфенил)-3-(циклопропиламино)проп-1-ен-1-ил)фенил)-N-гидроксиакриламид;
59. N-Циклопропил-3-(4-(3-(2-аминофениламино)-3-оксопроп-1-ен-1-илфенил)-2-(4-фторфенил)акриламид;
60. 3-(4-((1E)-3-(2-Аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-фторфенил)-N,N-диметилакриламид;
61. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-(трифторметил)фенил)акриламид;
62. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(пиридин-3-ил)акриламид;
63. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-хлорфенил)акриламид;
64. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-[бензодиоксол-5-ил]акриламид;
65. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-фторфенил)акриламид;
66. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3-хлорфенил)акриламид;
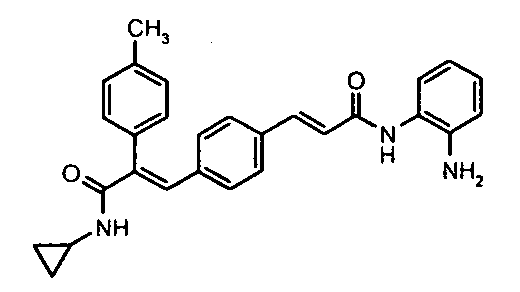
67. (E)-N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-метилфенил)акриламид;
68. N-Морфолино-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-фторфенил)акриламид;
69. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3-метоксифенил)акриламид;
70. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-фенилакриламид;
71. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(тиофен-2-ил)акриламид;
72. N-Морфолино-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-фторфенил)акриламид;
73. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3,4-дифторфенил)акриламид;
74. N-Циклопропил-3-(4-((1E)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3,4-диметоксифенил)акриламид;
75. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
76. 6-((1E)-3-(4-(3-(N,N-Диметиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
77. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
78. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
79. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
80. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
81. 6-((1E)-3-(4-(3-Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
82. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-енил)фенил)акриламидо)-N-гидроксигексанамид;
83. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-енил)фенил)акриламидо)-N-гидроксигексанамид;
84. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
85. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
86. 6-((1E)-3-(4-(3-Морфолино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)акриламидо)-N-гидроксигексанамид;
87. 6-((1E)-3-(4-(3-(Морфолино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
88. 6-((1E)-3-(4-(3-(Циклопропиламино)-2-(3-фтор-4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамид;
89. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
90. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
91. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
92. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
93. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
94. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
95. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
96. 4-(((lE)-3-(4-(3-(Морфолино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
97. 4-(((1E)-3-(4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
98. 4-(((1E)-3-(4-(3-(Морфолино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамид;
99. 4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид;
100. 4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид;
101. 4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид;
102. 4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид;
103. 4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамид;
104. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензамид;
105. (E)-N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
106. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-трифторметилфенил)-3-оксопроп-1-ен-1-ил)бензамид;
107. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)бензамид;
108. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
109. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
110. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
111. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(1,3-бензодиоксол-5-ил)-3-оксопроп-1-ен-1-ил)бензамид;
112. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)бензамид;
113. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-ен-1-ил)бензамид;
114. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)бензамид;
115. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3,4-дифторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
116. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
117. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-хлор-4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
118. N-(2-Аминофенил)-4-(3-(фениламино)-2-(3,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
119. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
120. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
121. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-нафтил)-3-оксопроп-1-ен-1-ил)бензамид;
122. N-(2-Аминофенил)-4-(3-фениламино-2-(2,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамид;
123. N-(2-Амино-4-фторфенил)-4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
124. N-(2-Аминофенил)-4-(3-(циклопропиламино)-3-оксо-2-(1H-индол-3-ил)проп-1-ен-1-ил)бензамид;
125. N-(2-Аминофенил)-4-(3-(циклопропиламино)-3-оксо-2-бифенил-4-ил-проп-1-ен-1-ил)бензамид;
126. 4-(2-(4-Фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)-N-(2-гидроксифенил)бензамид;
127. N-(2-Аминофенил)-4-[3-(циклопропиламино)-3-оксо-2-пиридин-3-ил-проп-1-ен-1-ил]бензамид;
128. N-(2-Аминофенил)-4-(2-(4-гидроксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
129. N-(2-Аминофенил)-4-(2-(2,6-дифторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
130. N-(2-Аминофенил)-4-(2-(2,5-дифторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
131. N-(2-Аминофенил)-4-(2-(4-фторфенил)-3-(изопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
132. N-(2-N-(4-Аминобифенил-3-ил)-4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)бензамид;
133. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-метилфенил)-3-оксопроп-1-ен-1-ил)бензамид;
134. N-(2-Аминофенил)-4-(3-(метиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
135. (Z)-N-(2-Аминофенил)-4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
136. N-(2-Аминофенил)-4-[2-(4-фторфенил)-3-морфолин-4-ил-3-оксопроп-1-ен-1-ил]бензамид;
137. N-(2-Аминофенил)-4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
138. N-(2-Аминофенил)-3-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
139. N-(2-Аминофенил)-4-(3-(фениламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
140. 4-[3-Амино-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил]-N-(2-аминофенил)бензамид;
141. N-(2-Аминофенил)-4-(2-(4-циклопентилоксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
142. N-(2-Аминофенил)-4-(2-(4-циклопропилметоксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамид;
143. N-(2-Аминофенил)-4-(3-(бензиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамид;
144. N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)бензамид;
145. 4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамид;
146. 4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамид;
147. 4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)-N-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамид;
148. 4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
149. 4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
150. 4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
151. 4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
152. 4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
153. 4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
154. 4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
155. 4-(3-(Циклопропиламино)-2-(2-хлор-4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
156. 4-(3-(Циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
157. 4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
158. 4-(3-(Циклопропиламино)-2-(4-трифторметилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
159. 4-(3-(Циклопропиламино)-2-(3,4-дифторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамид;
160. 4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамид;
161. 4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(6-гидроксиамино)-6-оксогексил)бензамид;
162. 4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамид;
163. 4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамид;
164. 4-(3-(Циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамид;
165. N-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)-N-гидроксиоктандиамид и
166. N-(2-Аминофенил)-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензамид.
Также предложен способ получения соединений формулы (I), в которой все группы определены ранее, показанный на следующей схеме-1.
Указанный способ получения соединений формулы (I), в которой R2=СООН, включает стадии:
A) Конденсации соединения формулы 1a с соединением формулы 1b в уксусном ангидриде в присутствии органического основания с получением соединения формулы 1c, в которой R3=H или незамещенный алкил, R, R1, X, Y, m, n, o и p определены ранее;

B) 1) Реакции соединения формулы 1c с агентом, активирующим кислоту, таким как гидрохлорид 1-этил-3-(3-диметиламинопропил)-карбодиимида (EDCI), 1-гидроксибензотриазол (HOBt) и тому подобным и с амином NHR5R6 с получением соединения формулы 2a, в которой R, R1, X, Y, m, n, o и p определены ранее.
2) Реакции соединения формулы 1c с подходящим агентом, активирующим карбоновую кислоту, и с основанием с получением ангидрида in situ, который, по восстановлении подходящим восстанавливающим агентом, дает соединение формулы 2b, в которой R, R1, R3, X, Y, m, n, o и p определены ранее.
3) Окисления 2b подходящим окисляющим агентом, дающего соответствующий альдегид, который, по восстановительном аминировании HNR5R6, дает соединение формулы 2c, в которой R, R1, X, Y, m, n, o и p определены ранее.
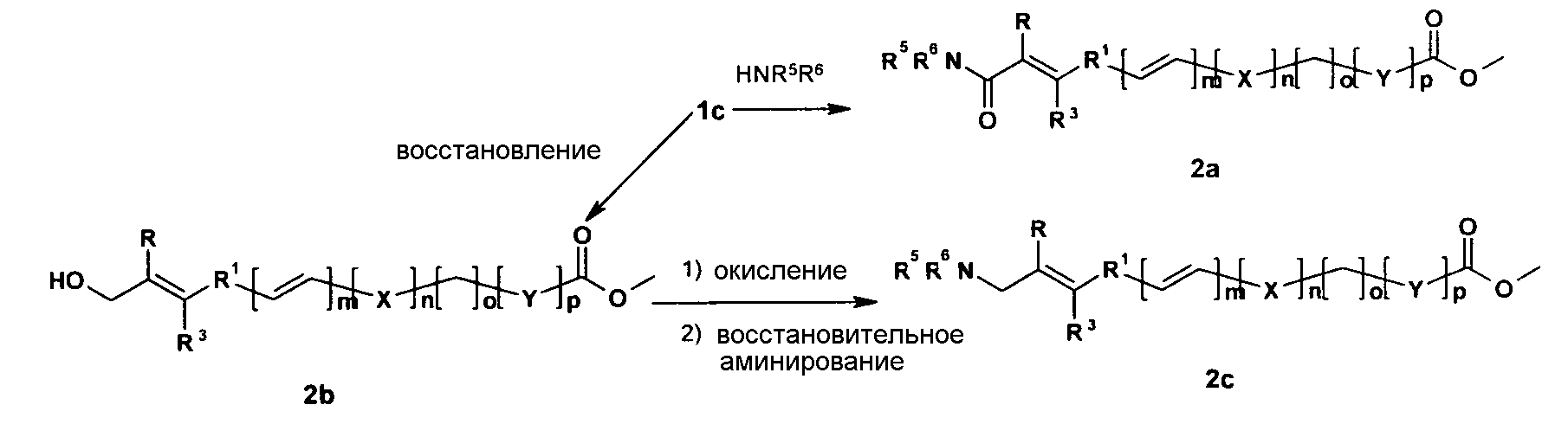
C) Гидролиза соединений формул 2a или 2b или 2c основанием с получением соответствующей кислоты. Сочетание кислоты с активирующими агентами, такими как EDCI, HOBt и тому подобными, в присутствии соответствующего амина R4NH2 с получением соединения общей формулы (I), или реакции соединения формул 2a или 2b или 2c с R4NH2 в присутствии основания с получением соединения общей формулы (I), в которой R, R1, R2, R3, R4, R5, R6, X, Y, m, n, o и p определены ранее.

В описании также предложен способ получения соединения формулы (I) из соединения формулы (II), в которой, когда один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом, R4, R3, R2, R1, R, X, Y, m, n, o и p определены ранее.

Тому же способу следуют при синтезе соединения формулы (I), в которой R2=H или незамещенный алкил, а R3=COOH, соответствующим образом выбирая кислоты и карбонильные соединения, применяемые на стадиях A-C вышеуказанной схемы синтеза.
Все вышеуказанные альтернативные реакции можно проводить при температуре от 0°C до комнатной температуры, а продолжительность реакции может изменяться от 2 до 24 часов.
Получены фармацевтически приемлемые соли соединений формулы (I). Аддитивные соли кислот получают обработкой кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, азотная кислота, серная кислота, фосфорная кислота, п-толуолсульфоновая кислота, метансульфоновая кислота, уксусная кислота, лимонная кислота, малеиновая кислота, салициловая кислота, гидроксинафтойная кислота, аскорбиновая кислота, пальмитиновая кислота, янтарная кислота, бензойная кислота, бензолсульфоновая кислота, винная кислота и тому подобными, в растворителях, таких как этилацетат, эфир, спирты, ацетон, тетрагидрофуран (THF), диоксан и т.д. Также можно применять смесь растворителей.
Примеры, приведенные ниже, представлены исключительно в иллюстративных целях, а потому их не следует рассматривать как ограничивающие объем изобретения.
Пример 1:
Синтез N-циклопропил-2-(4-фторфенил)-3-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-yl)фенил)акриламида.

Стадия-I
Получение метил-(E)-3-(4-формилфенил)акрилата

Суспензию (E)-3-(4-формилфенил)акриловой кислоты (2 г, 10,5 ммоль) в метаноле (30 мл) охлаждают до 5°C, и затем при перемешивании добавляют концентрированную H2SO4 (3 мл) и нагревают при 60°C в течение 2 часов. Растворитель удаляют выпариванием, и полученное соединение 15 минут перемешивают с водой (100 мл). Выпавшее твердое вещество белого цвета отфильтровывают, промывают водой (300 мл) и сушат с получением чистого продукта (1,9 г, выход 86%).
Стадия-II
Получение 2-(4-фторфенил)-3-(4-((E)-3-метокси-3-оксопроп-1-ен-1-ил)фенил)акриловой кислоты

Смесь 4-фторфенилуксусной кислоты (2,5 г, 13,2 ммоль) и метил-(E)-3-(4-формилфенил)акрилата (2,03 г, 13,2 ммоль) при перемешивании растворяют в уксусном ангидрида (8 мл). К этой смеси добавляют диизопропилэтиламин (DIPEA) (3,4 мл, 19,7 ммоль) и 2 часа перемешивают при 30°C. По завершении реакции (что наблюдают на ТСХ, применяя 100% этилацетат в качестве элюента), реакционную смесь выливают в воду и устанавливают pH равным 1, применяя разбавленную HCl (1:1). Водный слой экстрагируют этилацетатом (2×150 мл). Объединенный слой этилацетата промывают водой до нейтральной реакции промывных вод и осушают над безводным Na2SO4. Слой этилацетата выпаривают досуха с получением липкого соединения и затем растирают с холодным дихлорметаном (DCM) с получением твердого вещества белого цвета. Полученное твердое вещество отфильтровывают и сушат в вакууме, получая указанное в заголовке соединение (2 г, выход 47%).
Стадия-III
Получение метил-3-(E)(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акрилата
Смесь 2-(4-фторфенил)-3-(4-((E)-3-метокси-3-оксопроп-1-ен-1-ил)фенил)акриловой кислоты (0,23 г, 0,71 ммоль) и циклопропиламина (0,03 г, 0,60 ммоль), EDCl (0,27 г, 1,4 ммоль), HOBt (0,10 г, 0,71 ммоль) при перемешивании растворяют в N,N-диметилформамиде (DMF) (6 мл). При постоянном перемешивании в реакционную смесь по каплям добавляют триэтиламин (TEA) (0,75 мл, 36 ммоль) и перемешивают ее при 30°C в течение 2 часов. После этого реакционную смесь разбавляют этилацетатом и последовательно промывают водой (3×50 мл) и насыщенным раствором соли (3×50 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая чистое соединение (0,25 г, выход 96%).
Стадия-IV
Получение N-циклопропил-2-(4-фторфенил)-3-(4-((E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида

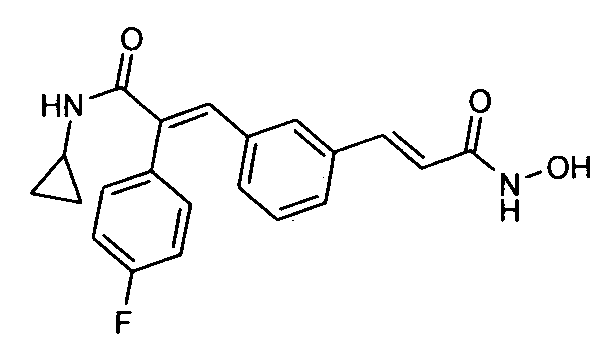
Гидрохлорид гидроксиламина (0,86 г, 12,3 ммоль) в метаноле (3 мл) смешивают с KOH (0,69 г, 12,3 ммоль) в метаноле (3 мл) при 0°C и обрабатывают ультразвуком в течение 2 минут, полученный осадок белого цвета отфильтровывают. Фильтрат добавляют к метил-3-(E)(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акрилату (0,25 г, 0,68 ммоль) в DCM (1,5 мл) и смесь 30 минут перемешивают при комнатной температуре. Реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (2×150 мл). Слой этилацетата осушают над безводным Na2SO4 и концентрируют, получая липкое соединение, которое растирают с DCM (15 мл). Полученное твердое вещество бледно-коричневого цвета отфильтровывают и промывают DCM (3×5 мл), получая указанное в заголовке соединение (0,07 г, выход 28%).1H ЯМР (DMSO-d6) δ (м.д.): 0,49-0,53 (2H, дд, -CH2), 0,61-0,66 (2H, м, -CH2), 2,72-2,77 (1H, м, -CH), 6,38-6,42 (1H, д, =CH), 7,00-7,02 (2H, д, Ar-H), 7,16-7,27 (5H, м, Ar-H и =CH), 7,33-7,43 (3H, м, Ar-H и =CH), 7,81-7,82 (1H, д, -NH), 9,04 (1H, с, -OH), 10,73 (1H, с, -NH). MC m/z: 367,1 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 1.



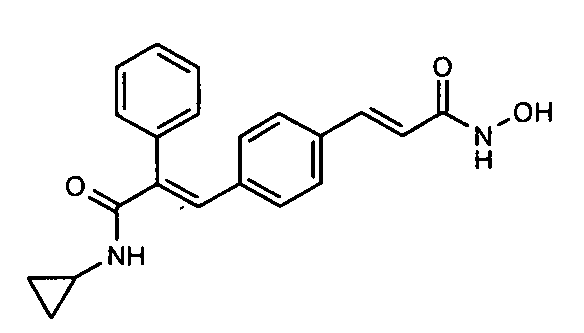
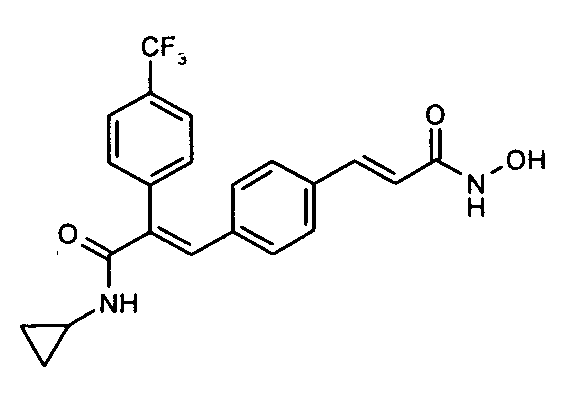




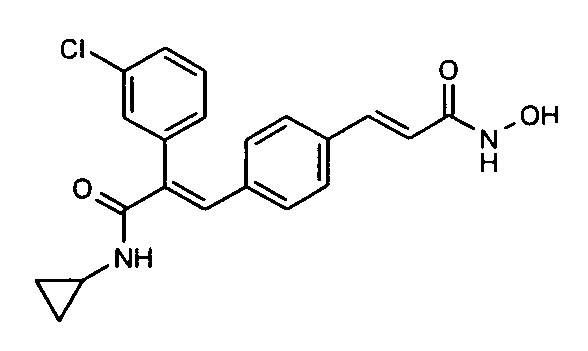
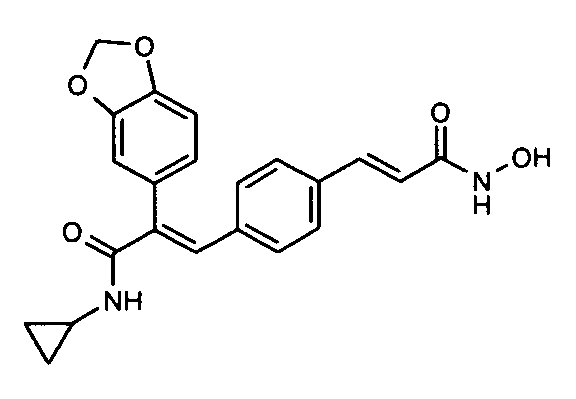





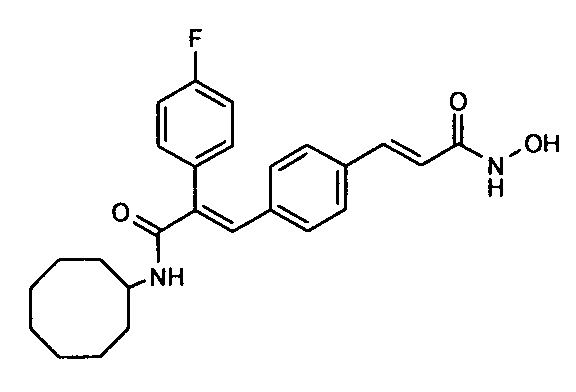
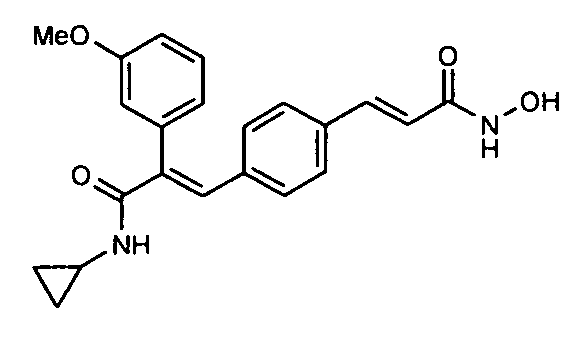



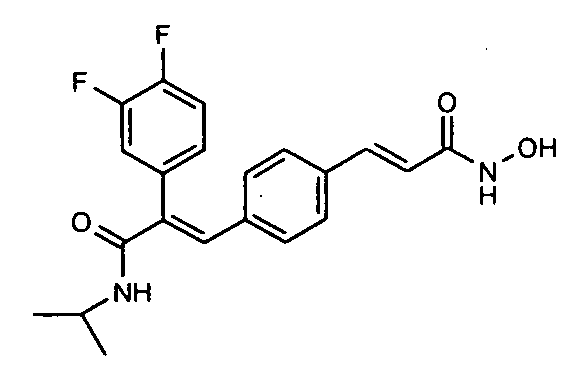

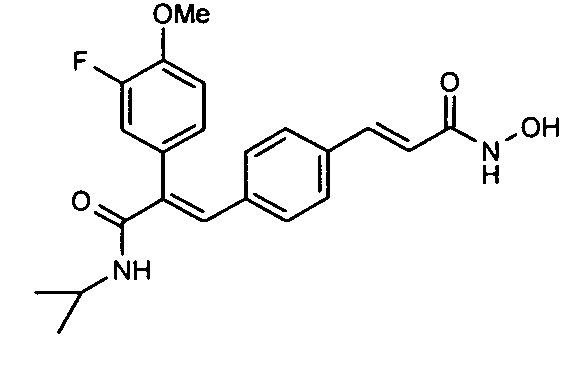





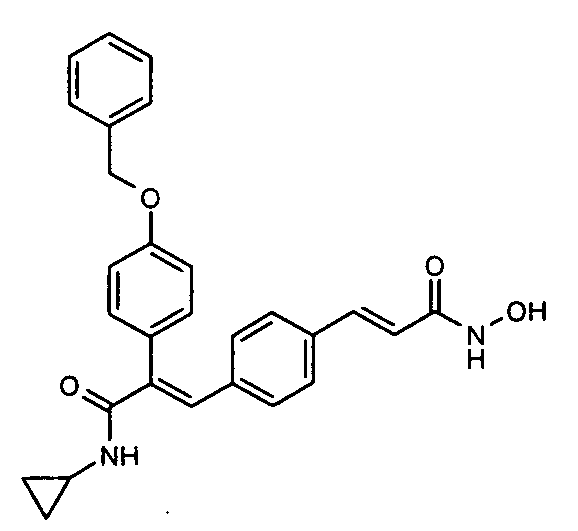
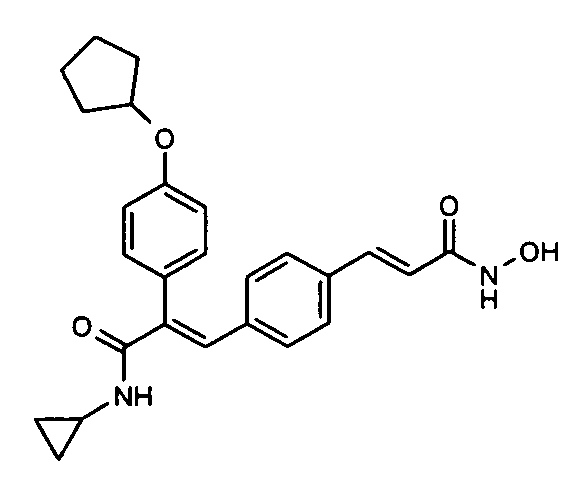
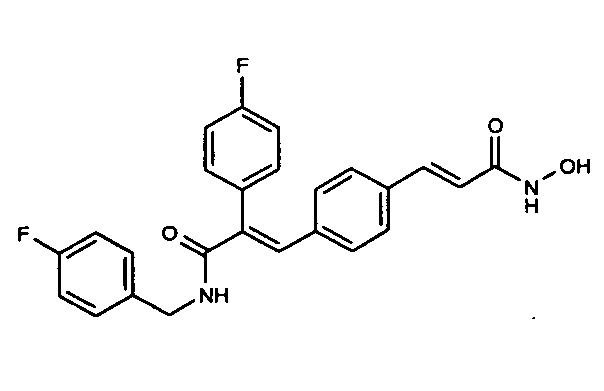
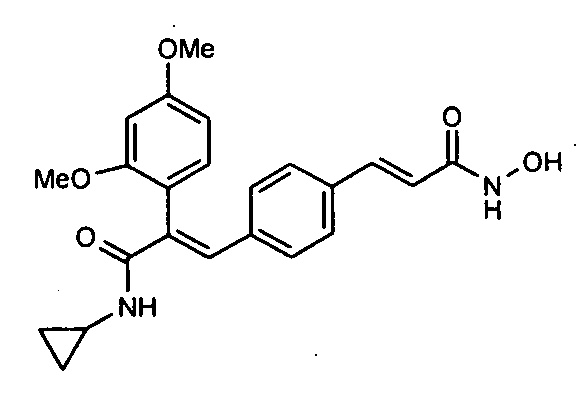
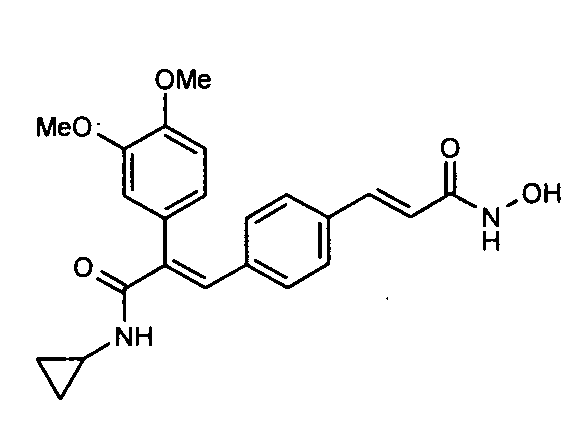

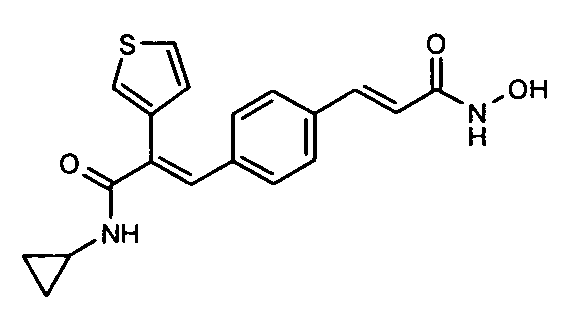


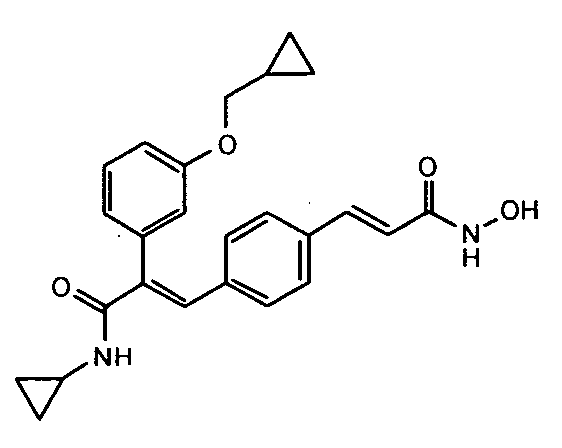


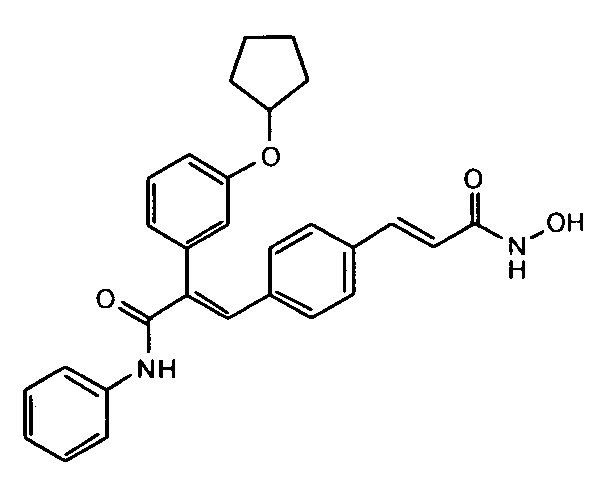






Пример 55:
Синтез (1E)-3-(4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)фенил)-N-гидроксиакриламида
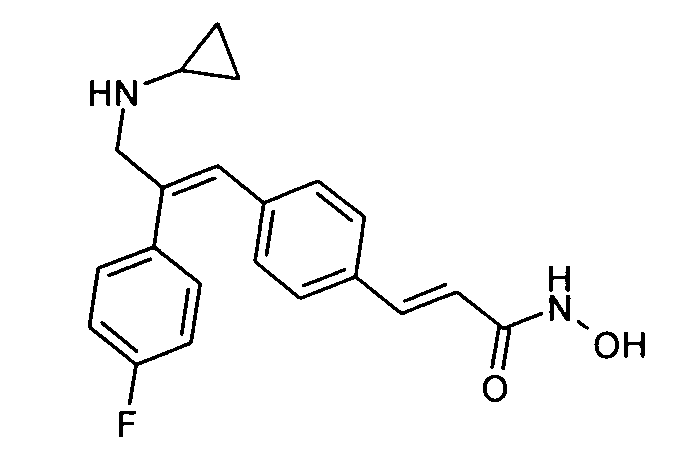
Стадия-I
Получение метил-3-(1E)(4-(2-(4-фторфенил)-3-гидроксипроп-1-ен-1-ил)фенил)акрилата
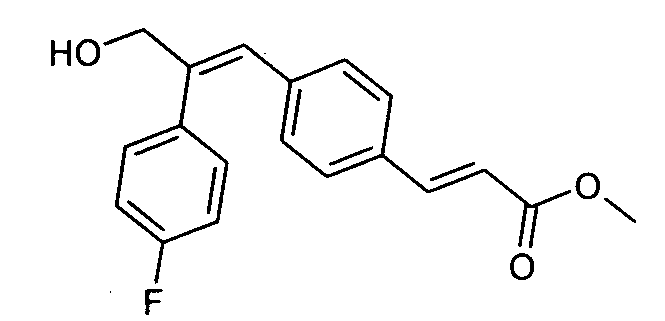
К суспензии 2-(4-фторфенил)-3-(4-(3-(E)-метокси-3-оксопроп-1-ен-1-ил)фенил)акриловой кислоты (2 г, 6,1 ммоль, получена по методике, описанной в Примере 1, стадия-II) в THF (10 мл) при постоянном перемешивании добавляют триэтиламин (0,85 мл, 6,67 ммоль) при 5°C. В течение 30 минут при 5°С в этот раствор по каплям добавляют метилхлорформиат (0,53 мл. 6,67 ммоль) и перемешивают 30 минут при той же температуре. К этой реакционной смеси при перемешивании в один прием добавляют боргидрид натрия (0,9 г, 24,5 ммоль) и метанол (5 мл) и реакционную смесь перемешивают 2 часа при 30°C. По завершении реакции реакционную смесь разбавляют этилацетатом (300 мл) и последовательно промывают водой (2×100 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение, которое очищают колоночной хроматографией, применяя 12% этилацетат/гексаны в качестве элюента, получая чистое соединение в виде твердого вещества белого цвета (1,5 г, выход 79%).
Стадия-II
Получение метил-3-(1E)(4-(2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акрилата

К суспензии хлорхромата пиридиния (PCC, 0,75 г, 3,5 ммоль) в дихлорметане (20 мл) при постоянном перемешивании по каплям добавляют раствор метил-3-(E)(4-(2-(4-фторфенил)-3- гидроксипроп-1-ен-1-ил)фенил)акрилата (0,9 г, 2,9 ммоль) в дихлорметане (5 мл) и реакционную смесь перемешивают 1 час при комнатной температуре. Реакционную массу разбавляют диэтиловым эфиром (200 мл) и фильтруют через слой Celite, фильтрат последовательно промывают насыщенным водным раствором NaHCO3 (3×100 мл) и водой (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая чистое соединение, указанное в заголовке, в виде твердого вещества белого цвета (0,5 г, выход 56%).
Стадия-III
Получение метил-3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)фенил)акрилата

Смесь метил-3-(1Е)(4-(2-(4-фторфенил)-3-гидроксипроп-1-ен-1-ил)фенил)акрилата (0,44 г, 1,4 ммоль) и циклопропиламина (0,14 г, 2,4 ммоль) перемешивают с MeOH (40 мл) 3 часа. К реакционной смеси добавляют боргидрид натрия (0,09 г, 2,3 ммоль) и перемешивают 30 минут. После этого реакционную смесь разбавляют этилацетатом (300 мл) и последовательно промывают водой (2×100 мл) и насыщенным раствором соли (1×100 мл). Органический слой сушат над безводным Na2SO4 и концентрируют, получая чистое соединение, указанное в заголовке, в виде липкого соединения бледно-желтого цвета (0,4 г, выход 80%).
Стадия-IV
Получение (1Е)-3-(4-((3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)фенил)-N-гидроксиакриламида

Гидрохлорид гидроксиламина (5 г, 71,8 ммоль) в метаноле (20 мл) смешивают с KOH (4 г, 71,8 ммоль) в метаноле (18 мл) при 0°C и 2 минуты обрабатывают ультразвуком и фильтруют. Фильтрат добавляют к метил-3-(1E)(4-(3-(циклопропиламино)-2-(4-фторфенил)-проп-1-ен-1-ил)фенил)акрилату (1,4 г, 4 ммоль) в дихлорметане (5 мл) и добавляют KOH (0,67 г, 12 ммоль). Реакционную смесь перемешивают 30 минут при комнатной температуре. Реакционную смесь концентрируют, получая липкое соединение желтого цвета, которое растворяют в воде (200 мл) и устанавливают pH равным 8, применяя разбавленную уксусную кислоту, экстрагируют этилацетатом (2×150 мл). Слой этилацетата осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение, которое очищают флэш-хроматографией, применяя 0,4% DCM:MeOH в качестве элюента, получая чистое соединение, указанное в заголовке, в виде твердого вещества желтого цвета (0,56 г, выход 40%).1H ЯМР (DMSO-d6) δ (м.д.): 0,74-0,76 (2H, д, -CH2), 0,85 (2H, с, -CH2), 2,73 (1H, с, -CH), 4,13 (2H, с, -CH2), 6,38-6,42 (1H, д, =CH), 6,96-6,98 (3H, д, Ar-H), 7,23-7,28 (2H, т, Ar-H и =CH), 7,34-7,40 (5H, м, Ar-H и =CH), 9,00 (2H, д, -NH и -OH), 10,79 (1H, с, -NH). MC m/z: 352,9 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 55.


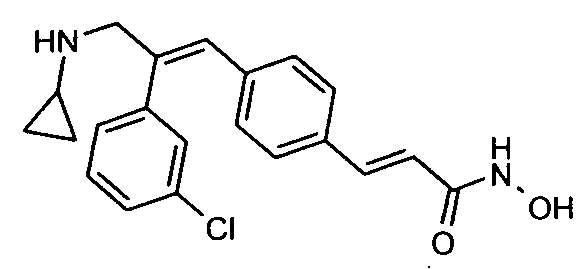
Пример 59:
Синтез N-циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-илфенил)-2-(4-фторфенил)-акриламида

Стадия-I
Получение 3-(1Е)-(4-(3-циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1- ил)фенил)акриловой кислоты
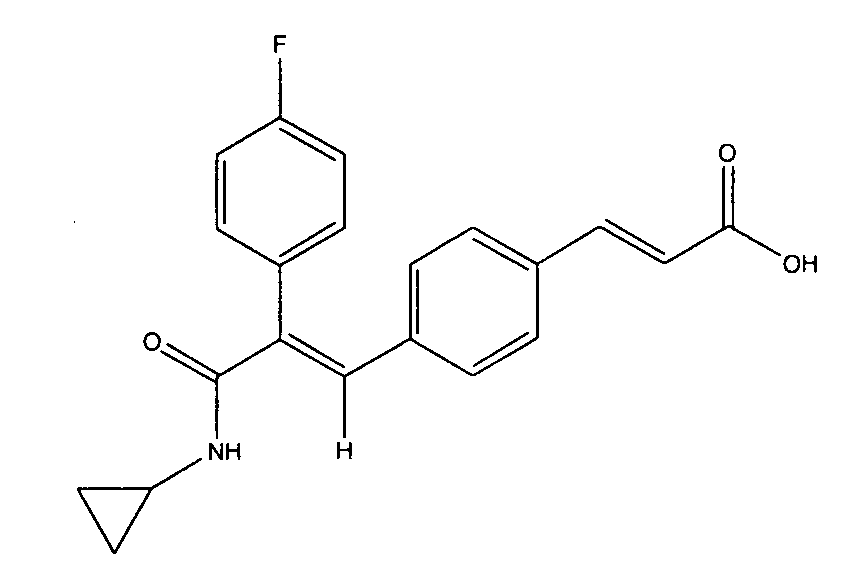
К раствору метил-3-(1Е)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акрилата (1 г, 2,7 ммоль) в метаноле (20 мл) добавляют раствор NaOH (0,44 г, 4,4 ммоль) в воде (1 мл). Реакционную смесь перемешивают 2 часа при 70°C. после этого растворитель полностью удаляют выпариванием, разбавляют водой (50 мл) и экстрагируют этилацетатом (2×50 мл). Водный слой подкисляют до pH 2 разбавленной водной HCl (1:1) и оставляют стоять при 4°C на 30 минут, выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая ожидаемый продукт в виде твердого вещества белого цвета (0,67 г, выход 70%).
Стадия-II
Получение N-циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-илфенил)-2-(4-фторфенил)-акриламида
К суспензии 3-(1Е)-(4-(3-циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)акриловой кислоты (0,21 г, 0,6 ммоль) в DMF (5 мл) добавляют EDCI (0,23 г, 1,2 ммоль), HOBt (0,08 г, 0,6 ммоль), o-фенилендиамин (0,06 г, 0,54 ммоль), после чего добавляют триэтиламин (0,25 мл, 1,8 ммоль). Реакционную смесь перемешивают 1 час, после чего смесь добавляют в холодную воду (20 мл). Водный слой экстрагируют этилацетатом (1×150 мл), промывают водой (2×50 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение. Неочищенное, окрашенное в желтый цвет соединение растирают с этилацетатом (20 мл), получая указанное в заголовке соединение в виде твердого вещества желтого цвета (0,06 г, выход 24%).1H ЯМР (DMSO-d6) δ (м.д.): 0,52 (2H, м, -CH2), 0,64-0,65 (2H, д, -CH2), 2,75 (1H, м, -CH), 4,93 (2H, с, -NH2), 6,57 (1H, т, =CH), 6,75 (1H, д, Ar-H), 6,82-6,86 (1H, д, =CH), 6,91 (1H, т, Ar-H), 7,03-7,05 (2H, д, Ar-H), 7,19-7,22 (4H, м, Ar-H), 7,29 (1H, с, =CH), 7,31-7,33 (1H, д, Ar-H), 7,43-7,47 (3H, т, Ar-H), 7,97-7,98 (1H, д, -NH), 9,35 (1H, с, -NH). MC m/z: 442,2 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 59.











Пример 75:
Синтез 6-(3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида

Стадия-I
Получение метил-6-(3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)гексаноата

К суспензии 3-(1E)-(4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)фенил)акриловой кислоты (получена в соответствии с методикой, описанной в Примере 59, стадия-I) (0,35 г, 1 ммоль) в DMF (15 мл) добавляют EDCI (0,83 г, 2 ммоль), HOBt (0,13 г, 1 ммоль), метил-6-аминокапронат (0,16 г, 0,9 ммоль), после чего добавляют триэтиламин (0,4 мл, 3 ммоль). Реакционную смесь перемешивают 2 часа, после чего смесь добавляют в холодную воду (50 мл). Водный слой экстрагируют этилацетатом (1×150 мл), промывают водой (2×50 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение. Неочищенное соединение, окрашенное в желтый цвет, промывают смесью этилацетат/гексан (0,5/9,5; 2×20 мл), получая указанное в заголовке соединение в виде твердого вещества желтого цвета (0,25 г, выход 52%).
Стадия-II
Получение 6-(3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида
Гидрохлорид гидроксиламина (0,63 г, 9 ммоль) в метаноле (3 мл) смешивают с KOH (0,51 г, 12,3 ммоль) в метаноле (3 мл) при 0°C и 2 минуты обрабатывают ультразвуком, полученный осадок белого цвета отфильтровывают. Фильтрат добавляют в метил-6-(3-(E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)гексаноату (0,24 г, 0,5 ммоль) в DCM (1,5 мл) и смесь перемешивают при комнатной температуре 30 минут. Реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (1×200 мл). Слой этилацетата промывают водой (100 мл), осушают над безводным Na2SO4 и концентрируют, получая липкое соединение, которое затем растирают с DCM (15 мл), получая твердое вещество. Полученное твердое вещество отфильтровывают и промывают DCM (5 мл), получая указанное в заголовке соединение (0,075 г, выход 32%).1H ЯМР (DMSO-d6) δ (м.д.): 0,51 (2H, м, -CH2), 0,62-0,64 (2H, т, -CH2), 1,24 (2H, м, -CH2), 1,40-1,49 (4H, м, -CH2), 1,91-1,93 (2H, д, -CH2), 2,74-2,76 (1H, м, -CH), 3,12-3,13 (2H, д, -CH2), 6,53-6,57 (1H, д, =CH), 6,99-7,01 (2H, д, Ar-H), 7,18-7,22 (4H, м, Ar-H), 7,27 (1H, с, =CH), 7,28-7,32 (1H, д, =CH), 7,37-7,39 (2H, д, Ar-H), 7,82-7,83 (1H, д, -NH), 8,06 (1H, с, -NH), 8,67 (1H, с, -OH), 10,34 (1H, с, -NH). MC m/z: 480,3 (M++1).
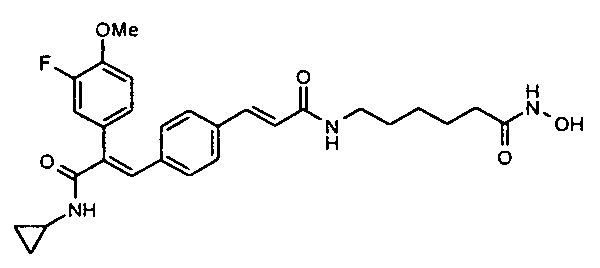
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 75.









Пример 89:
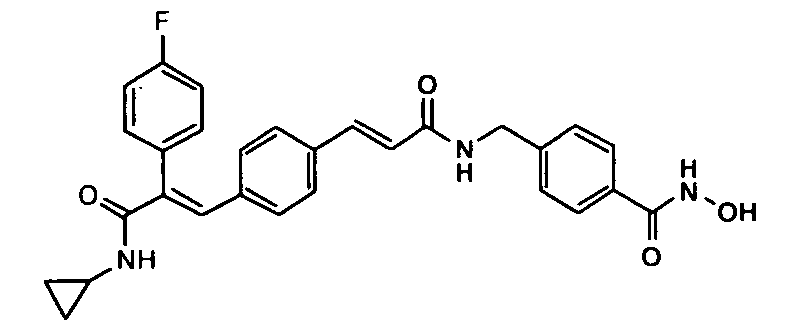
Синтез 4-((3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида
Стадия-I
Получение метил-4-((3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)бензоата

К суспензии (1E)-3-(4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)фенил)акриловой кислоты (получена в соответствии с методикой, описанной в Примере 59, стадия-II, 0,35 г, 1 ммоль) в DMF (15 мл) добавляют EDCI (0,83 г, 2 ммоль), HOBt (0,13 г, 1 ммоль) и гидрохлоридную соль метил-4-аминометилбензоата (0,18 г, 0,9 ммоль), затем добавляют триэтиламин (0,4 мл, 3 ммоль). Реакционную смесь перемешивают при комнатной температуре 1,5 часа, после чего смесь добавляют в холодную воду (50 мл). Водный слой экстрагируют этилацетатом (1× 150 мл) и промывают водой (2×50 мл), 10% разбавленной HCl (50 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение (0,225 г, выход 53,5%).
Стадия-II
Получение 4-((3-(1E)-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида
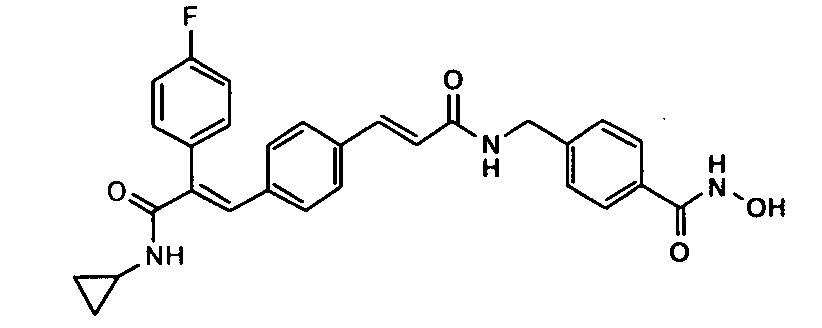
Гидрохлорид гидроксиламина (0,55 г, 8 ммоль) в метаноле (2 мл) смешивают с KOH (0,45 г, 8 ммоль) в метаноле (2 мл) при 0°C и 2 минуты обрабатывают ультразвуком, полученный белый осадок отфильтровывают. Фильтрат добавляют в метил-4-((3-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)акриламидо)метил)бензоат (0,22 г, 0,44 ммоль) в DCM (1,5 мл) и смесь перемешивают при комнатной температуре 1,5 часа. Реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (1×200 мл). Слой этилацетата промывают водой (100 мл), высушивают над безводным Na2SO4 и концентрируют, получая неочищенное соединение, с последующим растиранием с DCM (15 мл). Полученное твердое вещество отфильтровывают и промывают DCM (5 мл), получая указанное в заголовке соединение (0,044 г, выход 20%).1H ЯМР (DMSO-d6) δ (м.д.): 0,51 (2H, м, -CH2), 0,62-0,65 (2H, м, -CH2), 2,73-2,75 (1H, м, -CH), 4,37-4,38 (2H, д, -CH2), 6,62-6,66 (1H, д, =CH), 7,00-7,02 (2H, д, Ar-H), 7,15-7,19 (3H, м, Ar-H), 7,21-7,27 (3H, д, Ar-H и =CH), 7,35 (2H, д, Ar-H), 7,38-7,40 (2H, м, Ar-H и NH), 7,69-7,71 (2H, д, Ar-H), 7,83-7,84 (1H, д, =CH), 8,63 (1H, т, NH). MC m/z: 500,1 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 89.






Пример 99:
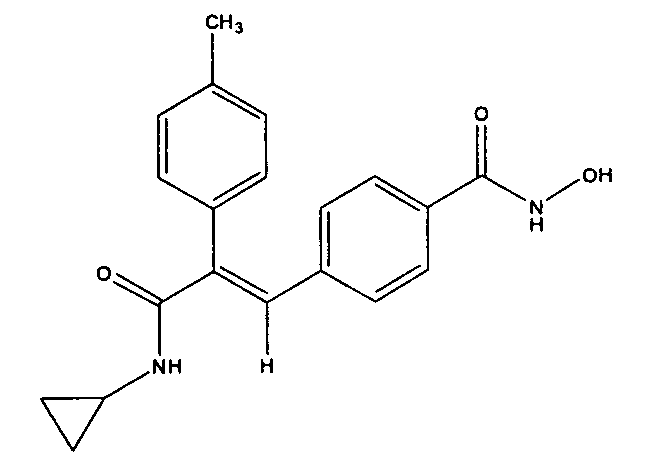
Синтез 4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида
Стадия-I
Получение 3-(4-(метоксикарбонил)фенил)-2-(4-метилфенил)акриловой кислоты

Смесь 4-метилфенилуксусной кислоты (3 г, 20 ммоль) и метил-4-формилбензоата (3,3 г, 20 ммоль) при перемешивании растворяют в Ac2O (8 мл). К этой смеси добавляют диизопропилэтиламин (DIPEA) (5,2 мл, 30 ммоль) и 6 часов перемешивают при 30°C. По завершении реакции, контролируемой с помощью ТСХ, применяя 100% этилацетат в качестве элюента, реакционную смесь выливают в воду и устанавливают pH равным 3 с помощью разбавленной HCl (1:1). Водный слой экстрагируют этилацетатом (2×150 мл). Объединенный слой этилацетата промывают водой до нейтральной реакции промывных вод и осушают над безводным Na2SO4. Слой этилацетата выпаривают досуха, получая липкое соединение, которое растирают с холодным дихлорметаном (DCM) с получением твердого вещества бледно-желтого цвета. Его отфильтровывают и сушат в вакууме, получая указанное в заголовке соединение (3,88 г, выход 66%).
Стадия-II
Получение метил-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензоата

Смесь 3-(4-(метоксикарбонил)фенил)-2-(4-метилфенил)акриловой кислоты (0,23 г, 0,71 ммоль), циклопропиламина (3,88 г, 13 ммоль), EDCl (5 г, 26 ммоль), HOBt (1,8 г, 13 ммоль) при перемешивании растворяют в N,N-диметилформамиде (DMF) (6 мл). К указанной реакционной смеси при постоянном перемешивании по каплям добавляют триэтиламин (TEA) (5,5 мл, 39 ммоль). Реакционную смесь перемешивают 4 часа при 30°C. После этого реакционную смесь выливают в ледяную воду (150 мл), при стоянии при комнатной температуре в течение 1 часа образуется осадок белого цвета, который отфильтровывают и промывают гексаном (100 мл), сушат в вакууме, получая чистое соединение (2,9 г, выход 66%).
Стадия-III
Получение 4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензойной кислоты

К раствору метил-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензоата (4 г, 12 ммоль) в метаноле (10 мл) добавляют раствор NaOH (1,4 г, 36 ммоль) в воде (1 мл). Реакционную смесь два часа кипятят с обратным холодильником при 70°C. Растворитель удаляют выпариванием, выливают в ледяную воду. Водный слой подкисляют до pH 3 лимонной кислотой и оставляют стоять при 4°C на 30 минут, выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая твердое вещество бледно-желтого цвета (3,2 г, выход 83%).
Стадия-IV
Получение 4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида

К суспензии 4-(3-(циклопропиламино)-2-(п-толил)-3-оксопроп-1-ен-1-ил)бензойной кислоты (0,1 г, 0,3 ммоль) в DMF (3 мл) добавляют гексафторфосфат бензотриазол-1-илокси-трис(диметиламино)фосфония (BOP-реагент, 0,23 г, 0,55 ммоль), HOBt (0,04 г, 0,3 ммоль), гидрохлорид гидроксиламина (0,03 г, 0,35 ммоль), затем добавляют DIPEA (0,16 мл, 0,9 ммоль). Реакционную смесь перемешивают 1 час, после чего смесь добавляют в холодную воду (100 мл) и выдерживают 1 час при 0°C, образуется твердое вещество белого цвета. Полученное твердое вещество отфильтровывают и промывают водой (50 мл), сушат в вакууме, получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,080 г, выход 77%).1H ЯМР (DMSO-d6) δ (м.д.): 0,46-0,51 (2H, м, -CH2), 0,58-0,60 (2H, м, -CH2), 2,29 (3H, с, CH3), 2,69-2,70 (1H, м, -CH), 6,81 (4H, м, Ar-H), 6,97-7,03 (3H, м, Ar-H), 7,36-7,38 (1+1H, д, Ar-H и =CH), 7,70 (1H, д, NH), 8,99 (1H, с, OH), 11,10 (1H, с, NH). MC m/z: 337,1 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 99.

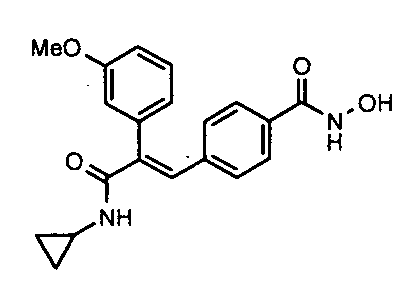
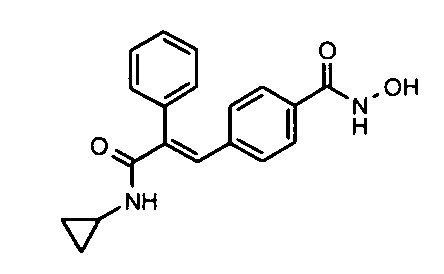

Пример 104:
Синтез N-(2-аминофенил)-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензамида
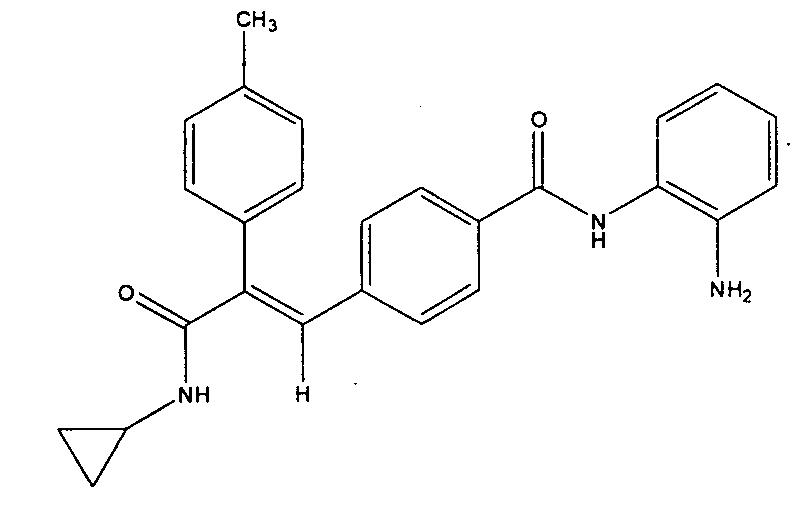
Стадия-I
Получение N-(2-аминофенил)-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензамида

К суспензии 4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензойной кислоты (0,2 г, 0,6 ммоль, получена в соответствии с методикой, описанной в Примере 99, стадии I-III) в DMF (3 мл) добавляют EDCI (0,23 г, 1,1 ммоль), HOBt (0,08 г, 5 ммоль), o-фенилендиамин (0,08 г, 0,7 ммоль), затем добавляют TEA (0,23 мл, 15 ммоль). Реакционную смесь перемешивают 4 часа, после чего смесь добавляют в холодную воду (100 мл) и выдерживают 1 час при 0°C, получая твердое вещество бледно-желтого цвета. Твердое вещество отфильтровывают и промывают водой (50 мл), сушат в вакууме, получая указанное в заголовке соединение (0,110 г, выход 45%).1H ЯМР (DMSO-d6) δ (м.д.): 0,51 (2H, м, -CH2), 0,63-0,65 (2Н, м, -CH2), 2,33 (3H, с, -CH3), 2,67-2,75 (1H, м, -CH), 4,87 (2H, с, -NH2), 6,55-6,58 (1H, м, Ar-H), 6,74-6,76 (1H, м, Ar-H), 6,93-6,97 (1H, м, Ar-H), 7,04 (2H, д, Ar-H), 7,11 (3H, м, Ar-H), 7,19 (2H, м, Ar-H), 7,25 (1H, с, =CH), 7,76-7,78 (3H, м, Ar-H и -NH), 9,57 (1H, с, -NH). MC m/z: 412,2 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 104.

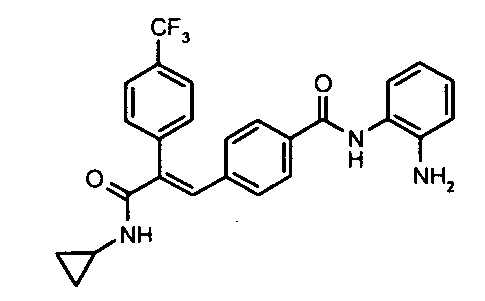

























Пример 144:
Синтез N-(2-аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-енил)бензамида
Стадия-I
Получение метил-3-(4-(2-(4-фторфенил)-3-гидроксипроп-1-ен-1-ил)фенил)бензоата

К суспензии 2-(4-фторфенил)-3-(4-(метоксикарбонил)фенил)акриловой кислоты (3 г, 10 ммоль) (получена в соответствии с методикой, описанной в Примере 99, стадия-I) в THF (30 мл) при постоянном перемешивании добавляют триэтиламин (1,5 мл, 12 ммоль) при 5°C. К этому раствору по каплям добавляют метилхлорформиат (0,86 мл, 12 ммоль) при 5°C и перемешивают 30 минут при той же температуре. К этой реакционной смеси в один прием добавляют борогидрид натрия (1,5 г, 40 ммоль) и по каплям - метанол (20 мл), и реакционную смесь перемешивают 1 час при 30°C. По завершении реакции реакционную смесь разбавляют этилацетатом (200 мл) и промывают водой (100 мл) и насыщенным раствором соли (100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение. Неочищенный продукт очищают колоночной хроматографией, применяя 10% этилацетат/гексан в качестве элюента, получают чистое соединение в виде твердого вещества белого цвета (1,4 г, выход 51%).
Стадия-II
Получение метил-4-(2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензоата

Хлорхромат пиридиния (PCC) (1,12 г, 5,2 ммоль) растворяют в дихлорметане (20 мл). При постоянном перемешивании по каплям добавляют раствор метил-3-(4-(2-(4-фторфенил)-3-гидроксипроп-1-ен-1-ил)фенил)бензоата (1,14 г, 4 ммоль) в дихлорметане (4 мл) и реакционную смесь перемешивают 1 час при комнатной температуре. Реакционную смесь разбавляют диэтиловым эфиром (100 мл) и фильтруют через Celite; фильтрат промывают насыщенным водным раствором NaHCO3 (2×100 мл) и водой (100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая чистое указанное в заголовке соединение в виде твердого вещества белого цвета (0,58 г, выход 53%).
Стадия-III
Получение метил-4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)бензоата

Смесь метил-4-(2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензоата (0,568 г, 2 ммоль) и циклопропиламина (0,17 г, 3 ммоль) перемешивают с MeOH (50 мл) 3 часа. В реакционную смесь добавляют NaBH4 (0,114 г, 3 ммоль) и ее перемешивают 30 минут. После этого реакционную смесь разбавляют этилацетатом (300 мл) и промывают водой (3×50 мл) и насыщенным раствором соли (100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая чистое указанное в заголовке соединение в виде липкого соединения бледно-желтого цвета (0,51 г, выход 76%).
Стадия-IV
Получение 4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)бензойной кислоты

К раствору метил-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензоата (0,5 г, 1,5 ммоль) в метаноле (10 мл) добавляют раствор NaOH (0,088 г, 3,8 ммоль) в воде (0,5 мл). Реакционную смесь кипятят с обратным холодильником 1 час при 70°C. Растворитель удаляют выпариванием, и остаток выливают в ледяную воду. Водный слой подкисляют до pH 3 лимонной кислотой, выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая твердое вещество бледно-желтого цвета (0,4 г, выход 77%).
Стадия-V
Получение N-(2-аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-енил)бензамида
К раствору 4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)бензойной кислоты (0,4 г, 1,3 ммоль) в DMF (5 мл) добавляют EDCI (0,49 г, 2,6 ммоль), HOBt (0,175 г, 1,3 ммоль) и TEA (0,54 мл, 3,9 ммоль), после чего добавляют o-фенилендиамин (0,280 г, 2,6 ммоль). Реакционную смесь перемешивают 2 часа при комнатной температуре, после чего остаток выливают в воду и экстрагируют этилацетатом (300 мл) и промывают водой (3×50 мл) и насыщенным раствором соли (100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение. Полученное соединение очищают флэш-хроматографией, применяя 15% этилацетат/гексан в качестве элюента, чистую фракцию выпаривают, получая указанное в заголовке соединение (0,040 г, выход 9%).1H ЯМР (DMSO-d6) δ (м.д.): 0,24-0,25 (2H, м, -CH2), 0,37-0,39 (2H, м, -CH2), 2,13-2,14 (1H, м, -CH), 3,57 (2H, с, -CH2), 4,86 (2H, с, -NH2), 6,55-6,59 (1H, т, Ar-H), 6,70 (1H, с, =CH), 6,74-6,76 (1H, д, Ar-H), 6,93-6,95 (1H, т, Ar-H), 7,02-7,04 (2H, м, Ar-H), 7,10-7,12 (1H, д, Ar-H), 7,16-7,26 (4H, м, Ar-H), 7,74-7,76 (2H, д, Ar-H), 9,52 (1H, с, -NH). MC m/z: 402,2 (M++1).
Пример 145:
Синтез 4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)-N-(4-((1E)-3-(гидроксиламино)-3-оксопроп-1-ен-1-ил)бензил)бензамида

Стадия-I
Получение метил-3(4-((4-((1E)-3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамидо)метил)фенил)акрилата

К суспензии 4-((1E)-3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензойной кислоты (0,3 г, 0,9 ммоль, получена в соответствии с методикой, описанной в Примере 99, стадия-III) в DMF (5 мл) добавляют EDCI (0,35 г, 1,8 ммоль), HOBt (0,12 г, 0,9 ммоль), метил-4-аминометилциннамат (0,237 г, 1,1 ммоль), после чего добавляют триэтиламин (0,4 мл, 3 ммоль). Реакционную смесь перемешивают 8 часов, после чего смесь выливают в холодную воду (100 мл), полученный осадок белого цвета отфильтровывают, промывают водой (1×150 мл), сушат в вакууме, получая указанное в заголовке соединение в виде твердого вещества желтого цвета (0,4 г, выход 87%).
Стадия-II
Получение 4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)-N-(4-((1E)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамида
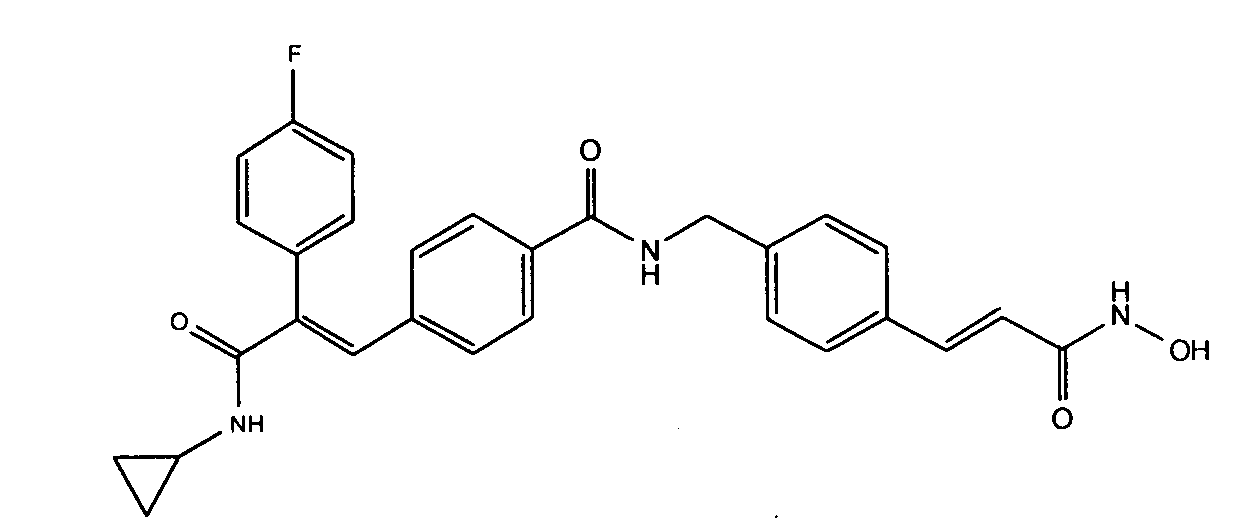
Гидрохлорид гидроксиламина (0,5 г, 7,2 ммоль) в метаноле (2 мл) смешивают с KOH (0,4 г, 7,2 ммоль) в метаноле (2 мл) при 0°C и 2 минуты обрабатывают ультразвуком, полученный осадок белого цвета отфильтровывают. Фильтрат добавляют в метил-3-(4-((4-(1E)-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)бензамидо)метил)фенил)акрилат (0,20 г, 0,4 ммоль) в DCM (1,5 мл) и смесь перемешивают 30 минут при комнатной температуре. Реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (3×100 мл). Слои этилацетата промывают водой (100 мл), осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение, которое растирают с DCM (15 мл), получая твердое вещество, которое отфильтровывают и промывают DCM (5 мл), получая указанное в заголовке соединение (0,050 г, выход 25%).1H ЯМР (DMSO-d6) δ (м.д.): 0,51-0,52 (2H, м, -CH2), 0,63-0,65 (2H, м, -CH2), 2,74-2,75 (1H, м, -CH), 4,43-4,44 (2H, д, -CH2), 6,39-6,43 (1H, д, =CH), 7,05-7,07 (2H, д, Ar-H), 7,16-7,22 (4H, м, Ar-H), 7,29-7,32 (3H, м, Ar-H и =CH), 7,40-7,44 (1H, д, =CH), 7,49-7,51 (2H, д, Ar-H), 7,68-7,70 (2H, д, Ar-H), 7,89-7,90 (1H, д, -NH), 9,00 (2Н, м, NH и OH), 10,73 (1H, с, NH). MC m/z: 498,1 (M+-1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 145.


Пример 148:
Синтез 4-3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида
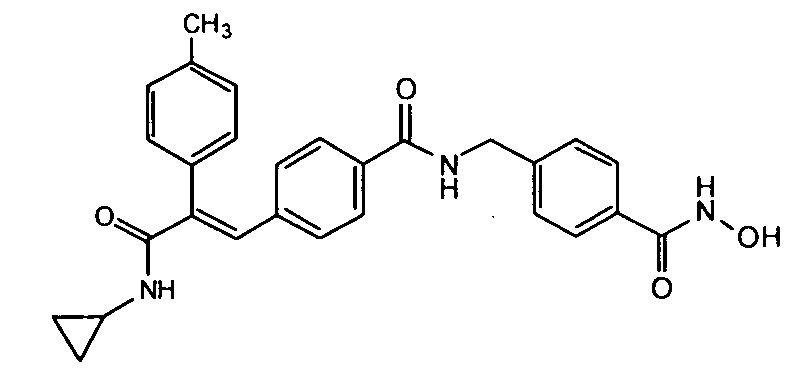
Стадия-I
Получении 4-((4-3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензамидо)метил)бензойной кислоты

К раствору метилового эфира 3-(4-((2-(4-метилфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)фенил)бензойной кислоты (0,26 г, 0,5 ммоль, получена по аналогии с методикой, описанной в Примере 145, стадия-II с подходящими реагирующими веществами) в метаноле (10 мл) добавляют раствор NaOH (0,06 г, 1,6 ммоль) в воде (0,5 мл). Реакционную смесь перемешивают 3 часа при 70°C. Растворитель полностью удаляют выпариванием, разбавляют водой (50 мл) и экстрагируют этилацетатом (2×50 мл). Водный слой подкисляют до pH 3 разбавленной водной HCl (1:1) и оставляют стоять на 30 минут при 4°C, выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая твердое вещество желтого цвета (0,22 г, выход 88%).
Стадия-II
Получение 4-3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида
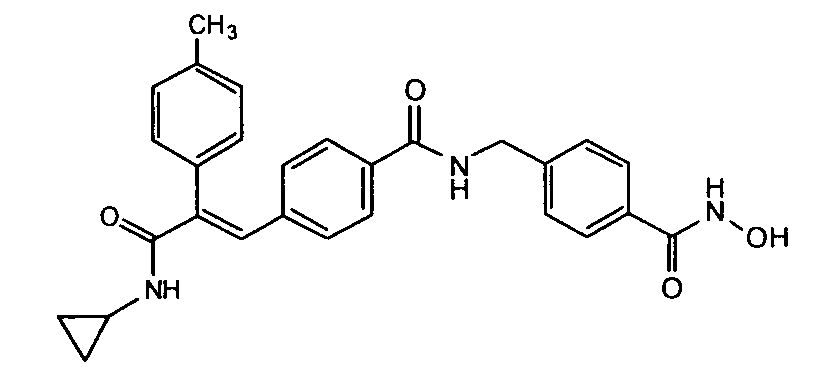
К суспензии 4-3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-енил)бензойной кислоты (0,22 г, 0,5 ммоль) в DMF (5 мл) добавляют EDCI (0,4 г, 0,9 ммоль), HOBt (0,06 г, 0,5 ммоль), гидрохлорид гидроксиламина (0,05 г, 0,7 ммоль), после чего добавляют триэтиламин (0,25 мл, 1,4 ммоль). Реакционную смесь перемешивают 1 час, после чего смесь добавляют в холодную воду (20 мл). При стоянии при комнатной температуре в течение 10 минут образуется осадок, который отфильтровывают, промывают (20 мл) водой и сушат в вакууме, получая указанное в заголовке соединение (0,14 г, выход 61%).1H ЯМР (DMSO-d6) δ (м.д.): 0,51 (2H, м, -CH2), 0,62-0,64 (2H, м, -CH2), 2,32 (3H, с, -CH3), 2,74-2,75 (1H, м, -CH), 4,45 (2H, д, -CH2), 7,02 (2H, с, Ar-H), 7,06 (2H, д, Ar-H), 7,18 (2H, д, Ar-H), 7,22 (1H, с, =CH), 7,33 (2H, д, Ar-H), 7,67-7,77 (4H, м, Ar-H), 7,78 (1H, д, NH), 9,02 (2H, м, NH и OH), 11,17 (1H, с, NH). MC m/z: 470,4 (M++1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 148.




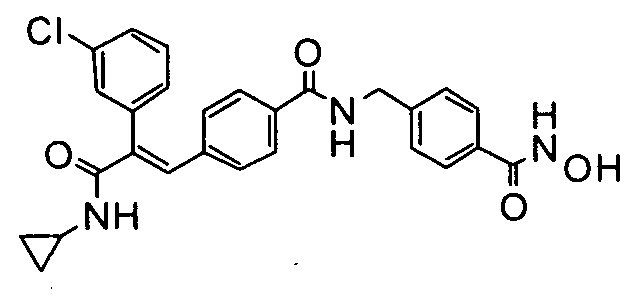

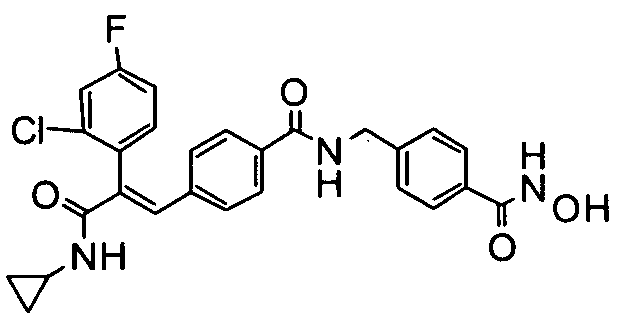




Пример 160:
Синтез 4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида

Стадия-I
Получение метил-6-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамидо)гексаноата
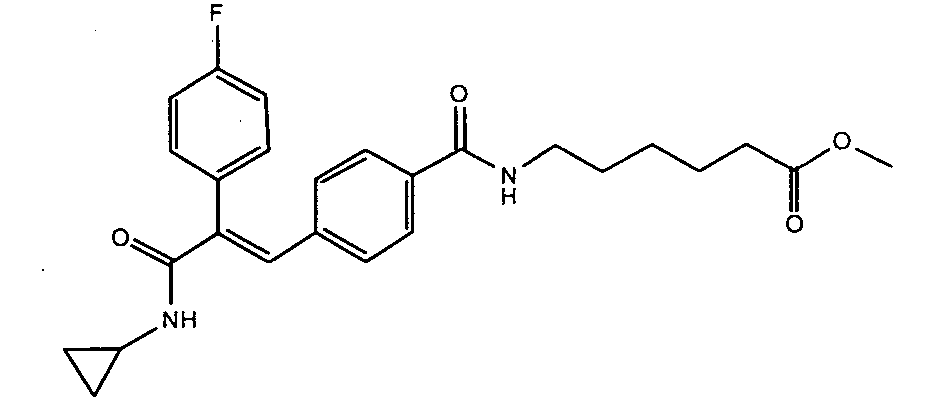
К суспензии 3-(4-2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензойной кислоты (0,4 г, 1,2 ммоль, получена в соответствии с методикой, описанной в Примере 99, стадия-III) в DMF (5 мл) добавляют EDCI (0,47 г, 2,4 ммоль), HOBt (0,17 г, 1,2 ммоль), метил-6-аминокапронат (0,27 г, 1,4 ммоль), после чего добавляют триэтиламин (0,5 мл, 3,6 ммоль). Реакционную смесь перемешивают 8 часов, после чего смесь добавляют в холодную воду (50 мл). Водный слой экстрагируют этилацетатом (1×150 мл), промывают водой (2×50 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение, которое промывают гексаном (2×20 мл), получая указанное в заголовке соединение в виде твердого вещества белого цвета (0,5 г, выход 89%).
Стадия-II
Получение 4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида

Гидрохлорид гидроксиламина (0,55 г, 8 ммоль) в метаноле (3 мл) смешивают с KOH (0,45 г, 8 ммоль) в метаноле (3 мл) при 0°C и 2 минуты обрабатывают ультразвуком, полученный осадок белого цвета отфильтровывают. Фильтрат добавляют в метил-6-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)бензамидо)гексаноат (0,2 г, 0,4 ммоль) в DCM (1,5 мл) и смесь 30 минут перемешивают при комнатной температуре. Реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (3×100 мл). Слой этилацетата промывают водой (100 мл), осушают над безводным Na2SO4 и концентрируют, получая указанное в заголовке соединение (0,040 г, выход 20%).1H ЯМР (DMSO-d6) δ (м.д.): 0,52 (2H, м, -CH2), 0,63-0,64 (2H, м, -CH2), 1,23- 1,25 (2H, м, -CH2), 1,44-1,50 (4H, м, -CH2), 1,90-1,94 (2H, м, -CH2), 2,74-2,75 (1H, м, -CH), 3,17-3,19 (2H, м, -CH2), 7,04 (2H, д, Ar-H), 7,16-7,22 (4H, м, Ar-H), 7,28 (1H, с, =CH), 7,63 (2H, д, Ar-H), 7,89 (1H, д, NH), 8,36 (1H, т, NH), 8,66 (1H, с, -OH), 10,32 (1H, с, NH). MC m/z: 452,2 (M+-1).
Следующие соединения были получены в соответствии с методикой, приведенной в Примере 160.
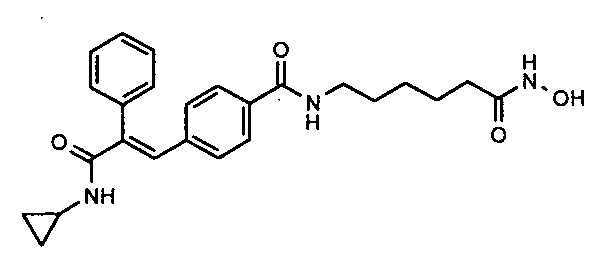

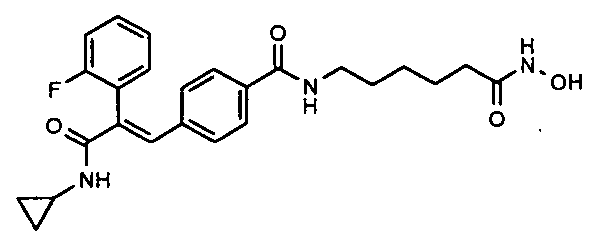
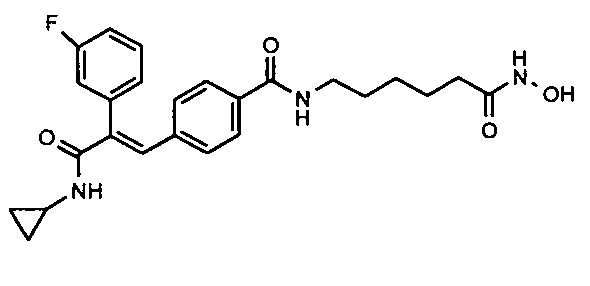
Пример 165:
Синтез N-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)-N'-гидроксиоктандиамина

Стадия-I
Получение 2-(4-аминофенил)-N-циклопропил-3-(4-фторфенил)акриламида

К раствору трет-бутил-4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенилкарбамата (0,8 г, 2 ммоль, получен по аналогии с методикой, описанной в Примере 1, стадии II-III, применяя соответствующие реагирующие вещества) в дихлорметане (10 мл) добавляют трифторуксусную кислоту (0,44 мл, 6 ммоль). Реакционную смесь перемешивают 2 часа, после чего выпаривают DCM, и ее разбавляют этилацетатом (100 мл) и промывают 10% раствором бикарбоната натрия (3×50 мл), водой (3×50 мл). Органический слой осушают над безводным Na2SO4 и выпаривают, получая неочищенное соединение, указанное в заголовке (0,53 г, выход 90%).
Стадия-II
Получение этил-8-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)-8-оксооктаноата

К суспензии 2-(4-аминофенил)-N-циклопропил-3-(4-фторфенил)акриламида (0,53 г, 1,7 ммоль) в DMF (6 мл) добавляют BOP-реагент (1,5 г, 3,4 ммоль), HOBt (0,2 г, 1,7 ммоль), этилсуберат (0,34 г, 1,7 ммоль), после чего добавляют триэтиламин (0,7 мл, 5,1 ммоль). Реакционную смесь перемешивают при комнатной температуре 2,5 часа. По завершении реакции смесь добавляют в холодную воду (50 мл). Водный слой экстрагируют этилацетатом (1×150 мл), промывают водой (2×50 мл) и насыщенным раствором соли (1×100 мл). Органический слой осушают над безводным Na2SO4 и концентрируют, получая неочищенное соединение (0,3 г, выход 35%).
Стадия-III
Получение N-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)-N'-гидроксиоктандиамида
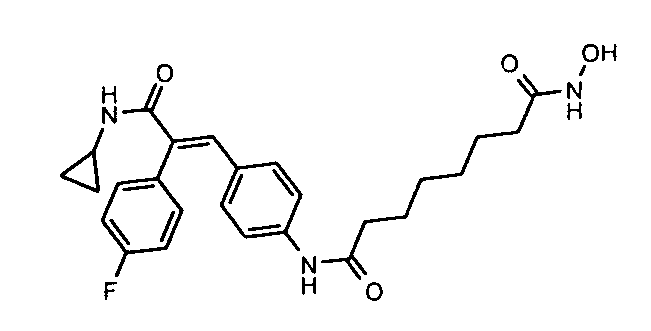
Гидрохлорид гидроксиламина (0,78 г, 11,2 ммоль) в метаноле (2 мл) смешивают с KOH (0,63 г, 11,2 ммоль) в метаноле (2 мл) при 0°C и 2 минуты обрабатывают ультразвуком, полученный осадок белого цвета отфильтровывают. Фильтрат добавляют в этил-8-(4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)-8-оксооктаноат (0,3 г, 0,62 ммоль) в метаноле (1,5 мл) и смесь перемешивают 1,5 часа при комнатной температуре. После этого реакционную смесь разбавляют водой (200 мл) и экстрагируют этилацетатом (1×200 мл). Слой этилацетата промывают водой (100 мл), осушают над безводным Na2SO4 и концентрируют с получением неочищенного соединения, которое очищают флэш-хроматографией, применяя 1,2% MeOH/DCM в качестве элюента, выпаривают чистую фракцию, получая указанное в заголовке соединение (0,056 г, выход 20%).1H ЯМР (DMSO-d6) δ (м.д.): 0,47-0,51 (2H, м, -CH2), 0,60-0,64 (2H, м, -CH2), 1,25-1,29 (4H, м, -CH2), 1,48-1,53 (2H, м, -CH2), 1,54-1,59 (2H, м, -CH2), 1,92-1,96 (2H, т, -CH2), 2,29-2,32 (2H, т, -CH2), 2,73-2,74 (1H, м, -CH), 7,04-7,06 (6H, м, Ar-H), 7,21 (1H, с, =CH), 7,58-7,60 (3H, м, Ar-H и NH), 8,66 (1H, с, -NH), 9,97 (1H, с, -OH), 10,34 (1H, с, -NH). MC m/z: 468,3 (M++1).
Пример 166:
Синтез N-(2-аминофенил)-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензамида

Стадия-I
Получение метил-4-((4-(3-(циклопропил)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензоата

К раствору 2-(4-аминофенил)-N-циклопропил-3-(4-фторфенил)акриламида (0,34 г, 1,15 ммоль, получен в соответствии с методикой, описанной в Примере 165, стадия-I) в дихлорметане (25 мл) при перемешивании при 37°С добавляют метил-4-формилбензоат (0,185 г, 1,15 ммоль). После перемешивания в течение 5 минут к реакционной смеси добавляют триацетоксиборогидрид натрия (0,39 г, 1,85 ммоль), а затем добавляют уксусную кислоту (0,3 мл). Реакционную смесь перемешивают 8 часов при комнатной температуре. После этого реакционную смесь обрабатывают системой этилацетат:вода (1:1, 100 мл) и экстрагируют этилацетатом (3×50 мл). Органический слой промывают насыщенным раствором соли (100 мл) и высушивают над безводным сульфатом натрия и выпаривают, получая указанное в заголовке соединение (0,42 г, выход 80%)
Стадия-II
Получение 4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензойной кислоты

К раствору метил-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензоата (0,42 г, 0,9 ммоль) в метаноле (10 мл) добавляют раствор NaOH (0,075 г, 18 ммоль) в воде (0,5 мл). Реакционную смесь кипятят с обратным холодильником 1 час при 70°C. Растворитель удаляют выпариванием, и остаток выливают в ледяную воду. Водный слой подкисляют лимонной кислотой до pH 3 и оставляют стоять при 4°C на 30 минут, выпавшее твердое вещество отфильтровывают и сушат в вакууме, получая твердое вещество бледно-желтого цвета (0,22 г, выход 51%).
Стадия-III
Получение N-(2-аминофенил)-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензамида

К суспензии 4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензойной кислоты (0,215 г, 0,5 ммоль) в DMF (3 мл) добавляют CDCl (0,23 г, 1,1 ммоль), HOBt (0,08 г, 0,55 ммоль), o-фенилендиамин (0,086 г, 0,7 ммоль), после чего добавляют TEA (0,2 мл, 1,5 ммоль). Реакционную смесь перемешивают 4 часа, после чего смесь добавляют в холодную воду (100 мл) и выдерживают 1 час при 0°C. Образовавшееся твердое вещество бледно-желтого цвета отфильтровывают и промывают этилацетатом (15 мл), сушат в вакууме, получая указанное в заголовке соединение (0,020 г, выход 8%).1H ЯМР (DMSO-d6) δ (м.д.): 0,46-0,47 (2H, м, -CH2), 0,60-0,62 (2H, м, -CH2), 2,70-2,71 (1H, м, -CH), 4,36-4,37 (2H, д, -CH2), 4,88 (2H, с, -NH2), 6,55-6,61 (4H, м, Ar-H и -NH), 6,76-6,82 (3H, м, Ar-H), 6,93-6,94 (1H, т, Ar-H), 6,98-7,04 (3H, м, Ar-H), 7,06-7,17 (3Н, м, Ar-H), 7,48-7,50 (3H, М, Ar-H и =CH), 7,93-7,95 (2H, д, Ar-H и -NH), 9,61 (1H, с, -NH). MC m/z: 521,1 (M++1).
Противораковые экспериментальные методы
Противораковый скрининг:
Экспериментальные лекарства подвергли скринингу против раковой активности на трех клеточных линиях, используя пять концентриций для каждого соединения. Клеточные линии НСТ 116 (прямая кишка), NClH460 (легкое) и U251 (глиома) хранили в DMEM, содержащем 10% плодной сыворотки коров. 96-Луночные микротитровые планшеты инокулировали клетками в виде 100 мкл суспензии (5×104 клеток/мл) в течение 24 часов при 37°С, 5% СО2, 95% воздуха и 100% относительной влажности. Отдельные планшеты с этими клеточными линиями также инокулировали для определения жизнеспособности клеток перед добавлением соединений (Т0).
Добавление экспериментальных лекарств:
После 24-часовой инкубации исследуемые соединения добавляли в 96-луночные планшеты. Каждый планшет содержит одну из вышеназванных клеточных линий, и следующие образцы были приготовлены трижды: пять различных разбавлений (0,01, 0,1, 1, 10 и 100 мкМ) четырех исследуемых соединений, подходящие разбавления цитотоксичного стандарта и ростовой среды (необработанные) лунки. Соединения растворяли в ДМСО для приготовления 20 мМ исходного раствора в день добавления лекарства, и готовили последовательные разбавления в полной ростовой среде при 2× крепости, так, чтобы 100 мкл, добавляемых в лунки, давали конечные концентрации (0,01, 0,1, 1, 10 и 100 мкМ) в лунке. В этих исследованиях в качестве стандартного лекарства применяли SAHA.
Изменение в конечной точке:
Для измерений Т0, через 24 часа после засева клеток, к планшету Т0 добавляли 20 мкл раствора 3-(4,5-диметил-2-тиазолил)-2,5-дифенил-2H-тетразолия (MTT) и инкубировали 3 часа при 37°С в инкубаторе с СО2. Планшет, содержащий клетки и исследуемые соединения, обрабатывали подобным образом через 48 часов после инкубации. Через 3 часа после добавления МТТ содержимое лунки осторожно отсасывали, после чего добаляли 150 мкл ДМСО на лунку. Планшеты встряхивали, чтобы гарантировать растворение кристаллов формазана в ДМСО, и считывали поглощение при 570 нм (А570).
Расчет GI50, TGI и LC50:
Процент роста (PG) вычисляли по отношению к контролю и лункам нулевого измерения (Т0) следующим образом:
PG=(A570test-A570T0)/(A570control-A570T0)×100
(если A570test>A570T0)
PG=(A570test-A570T0)/(A570T0)×100
(если A570test
Значения PG представляли на графике против концентрации лекарства, чтобы вычислить следующее: GI50 является концентрацией, требуемой для снижения PG на 50% против контроля; TGI является концентрацией, требуемой для снижения PG на 100% против контроля, и LC50 является концентрацией, требуемой для снижения PG на 50% против Т0. (Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. (J. Immunol. Methods. 1983, 65 (1-2), 55-63); Anne Monks et al. "Feasibility of high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines" (JNCI, Vol.83, No.11, 1991)).
Результаты ингибирования роста синтезированными соединениями приведены в Таблице 1.
Скрининг активности HDAC:
Исследование ингибирования гистондезацетилазы (HDAC) с применением субстрата Boc-Lys (Ac)-AMC: ингибирование HDAC вовлечено в модуляцию транскрипции и индуцирование апоптоза или дифференциации раковых клеток. Флуориметрическое исследование предоставляет быстрый метод, основанный на флуоресценции, который исключает радиоактивность, экстракции или хроматографию, применяемые в традиционных методах. Метод основан на двух стадиях. Сначала флуориметрический субстрат HDAC, который включает ацетилированную боковую цепь лизина, инкубируют с образцом, имеющим активность HDAC (экстракт мышиной печени). Дезацетилирование субстрата делает субстрат чувствительным на следующей стадии; обработка трипсиновым стоп-раствором дает флуорофор, который может быть легко определен с применением флуоресцентного считывающего устройства для планшетов.
Исследование выполняли в черном 96-луночном микропланшете, а общий объем для исследования составлял 100 мкл. Фермент мышиной печени (10 мг/мл) разбавляли 1:6 буфером HDAC. Готовили ферментный коктейль из 10 мкл разбавленного фермента и 30 мл буфера HDAC. В каждую лунку добавляли 40 мкл ферментного коктейля, а затем добавляли 10 мкл исследуемого соединения (1 мкМ и 10 мкМ) или буфера (контроль). Планшет прединкубировали 5 минут при 37°С. Реакцию HDAC начинали добавлением 50 мкл HDAC субстрата Boc-Lys (Ac)-AMC (Bachem AG, Switzerland). Планшет инкубировали 30 минут при 37°С. Реакцию останавливали добавлением 100 мкл трипсинового стоп-раствора и инкубировали 15-30 минут при 37°С. Измерением флуоресценции при длине волны возбуждения 360 нм и длине волны испускания 460 нм отслеживали выделение АМС. Чистый буфер и чистый субстрат служили холостыми опытами. Для выбранных соединений исследованиями в широком диапазоне концентраций: 0,001, 0,01, 0,1, 1 и 10 мкМ, была определена IC50 (концентрация, ингибирующая 50% HDAC) (Dennis Wegener et al., Anal. Biochem., 321, 2003, 202-208).
Результаты ингибирования HDAC при 1 и 10 мкМ и значения IC50 приведены в Таблице 1.
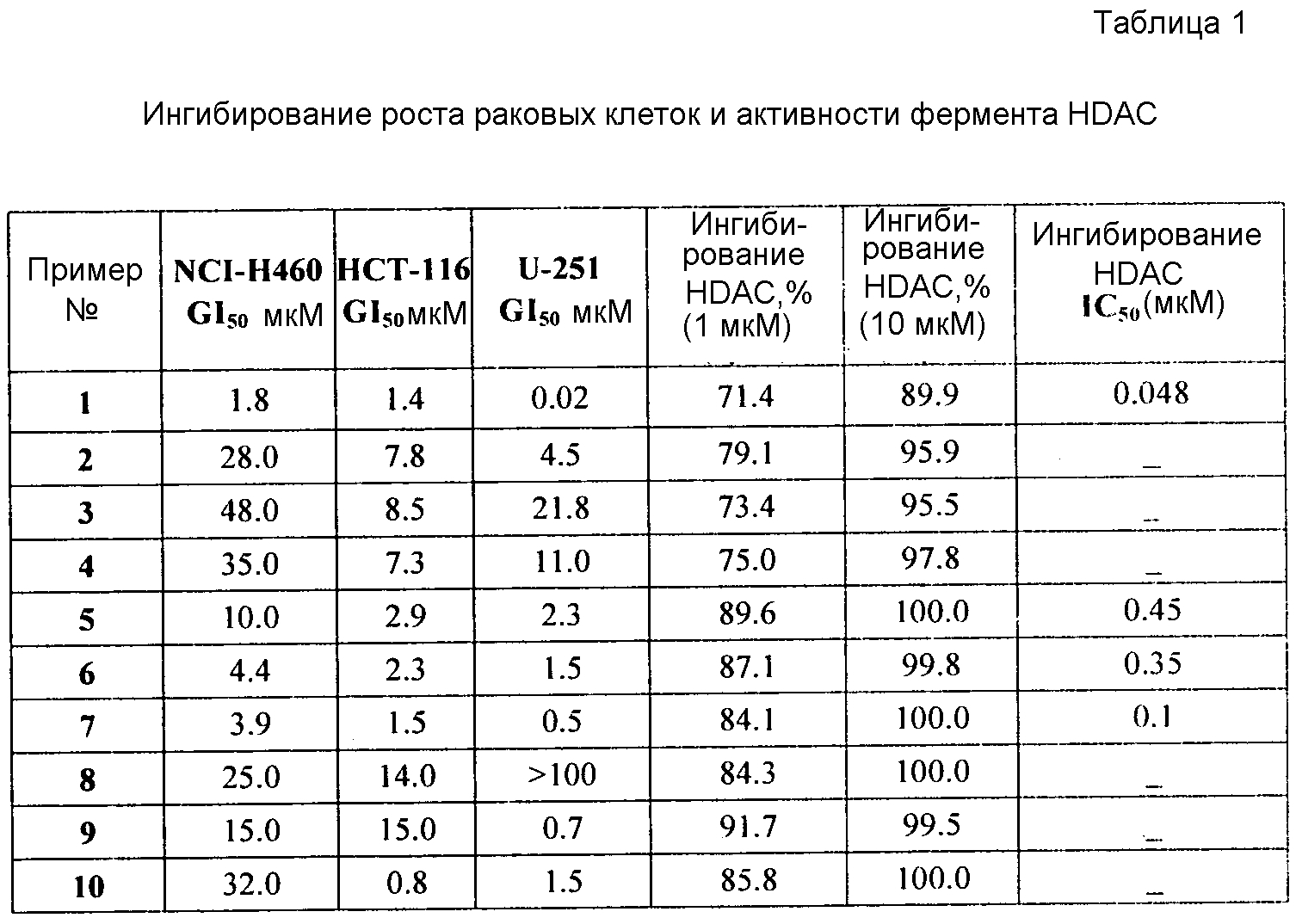
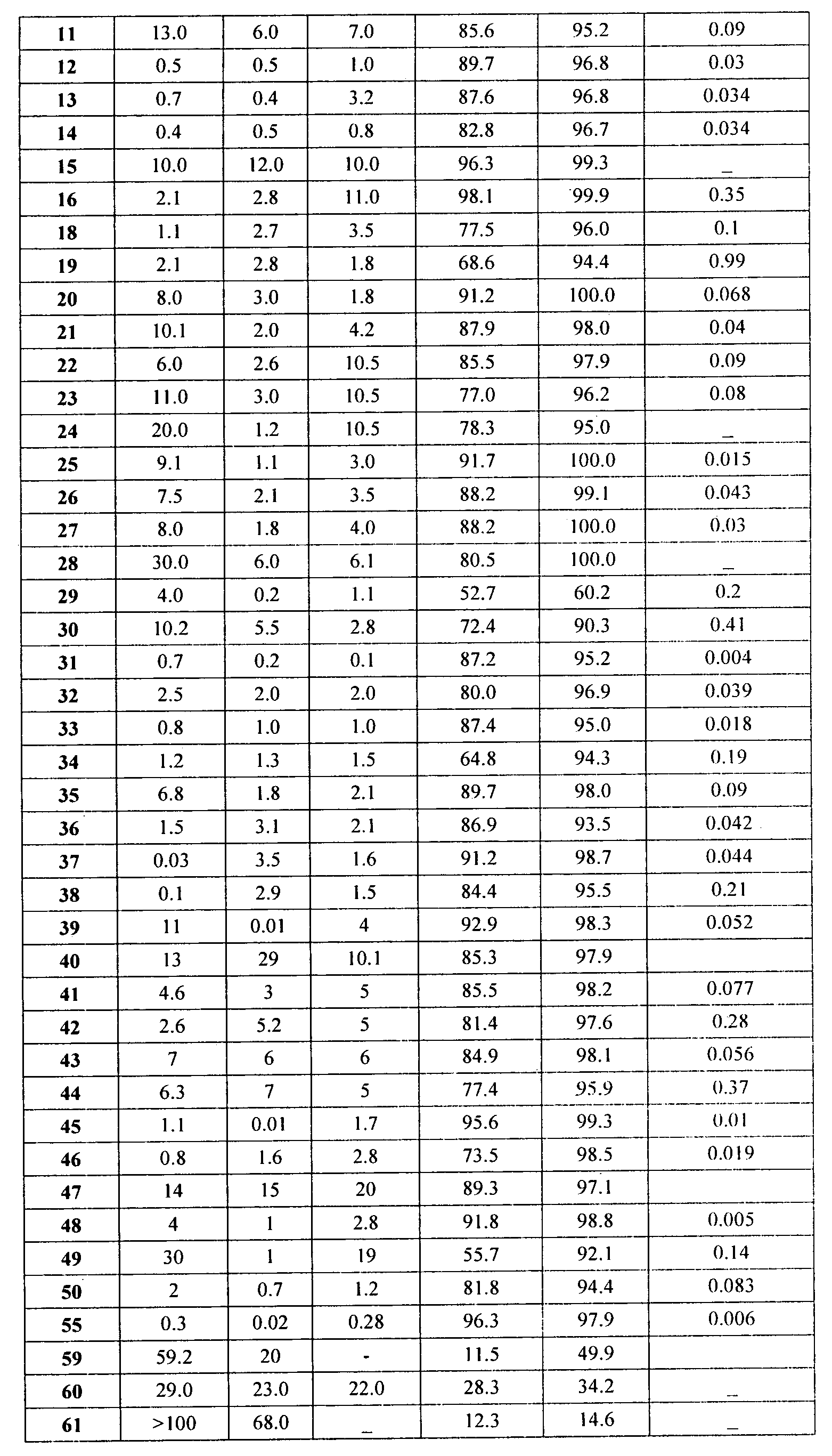


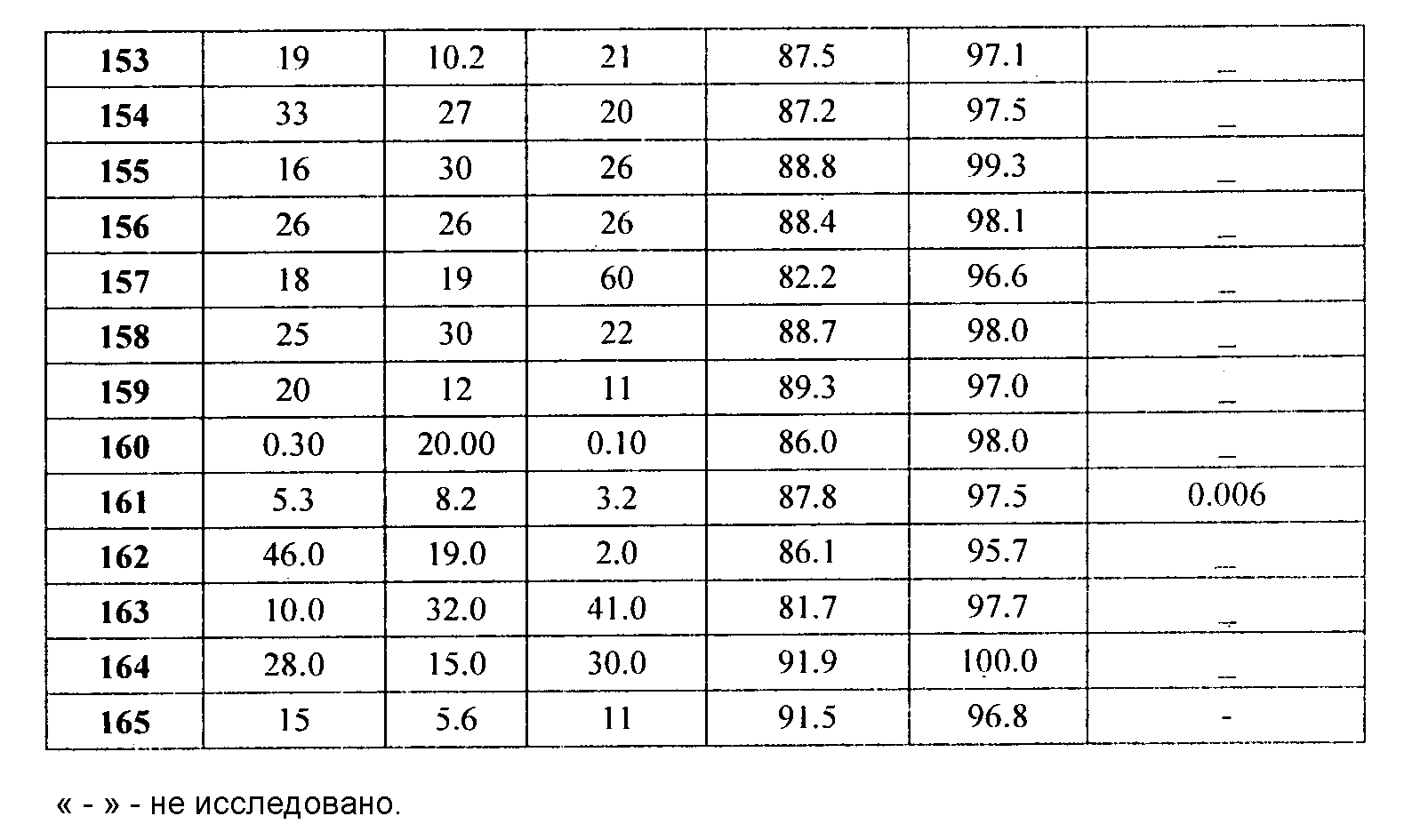
Селективность к изоформам HDAC:
Поскольку известно, что соединения типа бензамидов имеют потенциал специфичности к HDAC класса 1, активные соединения были протестированы на ингибиторную активность в отношении HDAC1. Исследование проводили, как описано ранее, с применением рекомбинантного фенмента HDAC1 (BIOMOL. USA), следуя инструкциям производителя. Для определения значений IC50 соединения исследовали при пяти различных концентрациях (0,001, 0,01, 0,1, 1 и 10 мкМ). Результаты, приведенные в Таблице-2, показывают, что эти соединения ингибируют фермент HDAC1 при наномолярных концентрациях, что существенно ниже по сравнению с активностью HDAC на планшетах с ферментом мышиной печени, что указывает на специфическую активность к изоформам HDAC.
Определение ацетилирования гистона (Н3), ацетилирования тубулина и индукции р21:
Уровни ацетилированного гистона (Н3), ацетилированного тубулина и р21 определяли в клеточном лизате сэндвичным методом ELISA (Cell Signaling Technology, USA, номер в каталоге: 7232, 7204 и 7167, соответственно), следуя инструкциям производителя. Вкратце, клетки рака прямой кишки (НСТ116, 10000/лунка) 4 часа инкубировали с исследуемым соединением (1 и 10 мкМ) или средой (контроль) при 37°С в инкубаторе с СО2. Для индукции р21 инкубирование длилось 18 часов. Посли инкубирования в буфере для лизиса клеток обработкой ультразвуком на льду готовили клеточные лизаты. Лизаты собирали после центрифугирования и подвергали процедуре исследования ELISA. 100 мкл каждого разбавленного клеточного лизата в буфере для разбавления (1:1) добавляли в микролунки, покрытые подходящим захватывающим антителом, и инкубировали в течение ночи при 4°С. После промывки добавляли 100 мкл антитела для определения на 1 час при 37°С. После второй промывки добавляли 100 мкл HRP-связанного вторичного антитела на 30 минут при 37°С. Наконец, после необходимой промывки добавляли 100 мкл субстрата TMB на 10 минут при 37°С, после чего добавляли 100 мкл стоп-раствора. Поглощение каждой лунки измеряли с помощью спектрофотометра при 450 нм (А450). Результаты выразили как кратность роста (A450test/А450контроль) по сравнению с контролем, и они представлены в Таблице 3. В этих исследованиях были протестированы выбранные соединения, и было найдено, что они вызывают ацетилирование гистона и тубулина и индуцируют экспрессию р21 в раковых клетках прямой кишки в несколько раз выше по сравнению с необработанным контролем. Таким образом, эти соединения продемонстрировали хорошую активность к HDAC в клетках в дополнение к активности в выделенных ферментных препаратах.
Метаболическая стабильность в микросомах печени in vitro:
Метаболическую стабильность определяют как процент родительского соединения, теряемый со временем в присутствии микросом печени, S9 печени, или гепатоцитов, в зависимости от цели исследования. На основании представлений о метаболической стабильности недавно открытых соединений эти соединения могут быть ранжированы для дальнейших исследований, а потенциал лекарства-кандидата, не продвинутого в разработку вследствие фармакокинетических причин, может быть понижен.
Готовят фосфатный буфер (рН 7,4) и исходные растворы исследуемых соединений (обычно в ДМСО или воде). Инкубируют реакционную смесь, включающую криоконсервированные микросомы печени мыши или человека (1 мг/мл), исследуемое соединение (50 мкМ) и NADPH в течение различных периодов времени, например 10, 15, 30 и 60 минут, или единственного периода времени, например 60 минут. Реакцию начинают добавлением NADPH и останавливают сразу или через 60 минут скринингового исследования, или через 5, 15, 30 или 60 минут для более точной оценки клиренса добавлением охлажденного льдом ацетонитрила, после чего готовят образцы. Определение потери родительского соединения (по сравнению с нулевой временной точкой контроля и/или NADPH-контролем) проводят методами ВЭЖХ или ЖХ-МС. Метаболизм выражают как процент исследуемого соединения, метаболизировавшегося спустя определенное время. Применяемым качественным критерием метаболической способности микросом является маркерная реакция и маркерный субстрат (например, тестостерон) (Rodrigues, A.D., Use of in vitro human metabolism studies in drug development. An industrial perspective. Biochem. Pharm., 48(12):2147-2156, 1994). Метаболическую стабильность выражают как % метаболизма соединения через 30 минут инкубирования в присутствии активных микросом. Соединение, имеющее метаболизм менее 30%, определяют как высокостабильное. Соединение, имеющее метаболизм от 30% до 60%, определяют как умеренно стабильное, а соединения, показавшие % метаболизма выше 60%, определяют как малостабильные. Было установлено, что некоторые соединения являются высоко и умеренно стабильными.
Противораковая активность in vivo:
Были проведены исследования с использованием самок бестимусных мышей SCID (тяжелый сочетанный иммунодефицит, Severe Combined Immune Deficient) в возрасте 6-8 недель. Мышей содержали в индивидуально вентилируемых клетках (IVC) при постоянных температуре (22±3°С) и влажности (50±20%). Мыши имели свободный доступ к корму и воде. Опухоли получали из ATCC, USA и поддерживали in vivo подкожным введением (s.c.) фрагментов опухоли (прибл. 30 мг) здоровым мышам в соответствии со стандартными опубликованными методиками. Все протоколы исследований на животных утверждены Institutional Animal Ethics Committee, ORLL, Chennai. Каждая экспериментальная группа включала 6-8 мышей, несущих s.c. опухоли. Опухоли имплантировали пункцией в срединную область с помощью Trocar, и рост опухоли отслеживали, измеряя диаметры опухоли штангенциркулем Vernier. Объем опухоли (TV) рассчитывали по следующей формуле:
TV (мм3)=L x W2×0,5,
где L и W являются наибольшим и наименьшим диаметрами опухоли, соответственно. Обработку соединениями начинали, когда опухоль становилась пальпируемой (150-200 мм3).
Исследуемое соединение вводили оральным скармливанием в объеме 5-10 мл/кг. Лекарства вводили один раз в день ежедневно в течение 21 дня. Контрольным мышам вводили носитель в эквивалентном объеме. Размер опухоли измеряли дважды в неделю, а массу тела записывали ежедневно перед введением.
Эффективность исследуемого соединения (Т) оценивали, вычисляя несколько параметров, основанных на объеме опухоли (TV) по отношению к необработанному контролю (С). Обычными оцениваемыми параметрами были T/C % [TVиссл./TVконтроль×100] и ингибирование объема опухоли (TVI=1-T/C%). Другими параметрами были относительный объем опухоли, процентное изменение объема опухоли, задержка опухоли и логарифм гибели клеток.
Токсические эффекты лечения оценивали по потере массы тела (в %). Летальную токсичность определяли как любой случай смерти в обработанных группах, имевший место перед любым случаем смерти в контрольной группе. Мышей осматривали ежедневно на предмет смертности и клинических признаков токсичности.
Результаты изучения ксенографта:
Соединение 105 показало хорошую противораковую активность in vivo на модели ксенографта НСТ116 (прямая кишка). Обработка соединением 105 (100 мг/кг, перорально, ежедневно в течение 21 дня) привела к максимальному ингибированию объема опухоли (TVI) 39,4% по сравнению с контролем, получавшим носитель, в течение курса исследования (Фиг. 1). Более того, обработка соединением не привела к значимой потере массы тела или смертности, связанной с обработкой, по сравнению с контролем.
Реферат
Изобретение относится к соединениям формулы (I), обладающие ингибиторной активностью к ферменту гистондезацетилазе (HDAC), их стереоизомерам, гидратам, сольватам и фармацевтически приемлемым солям, соединениям формулы (II) соединениям выбранным из списка, способу получения соединения формулы (I), фармацевтической композиции, способу ингибирования и способам лечения с использованием соединений формулы (I). В формуле (I) и (II) ! ! ! R представляет замещенные или незамещенные группы, выбранные из (С6-С10)арила, (С3-С12)циклоалкила, гетероарила, (С6-С10)арил, (С1-С6)алкила и гетероциклила; где гетероциклил здесь и далее отвечает 5-10-членному кольцевому радикалу, который состоит из атомов углерода и от одного до пяти гетероатомов, выбранных из азота кислорода и серы, а гетероарил здесь и далее отвечает ароматическому гетероциклилу, и каждый арил, циклоалкил, гетероарил, арилалкил и гетероциклил может быть замещен одним или более заместителями, выбранными из галогенов, включающих фтор, хлор, бром, йод, (С1-С6)алкил, (С1-С6)алкокси, (С6-С10)арил, галоген(С1-С6)алкил, (С6-С10)арил(С1-С6)алкокси, -O-(С3-С12)циклоалкил, -O-СН2-(С3-С12)циклоалкил, гидроксила, NRaRb и ORa, где Ra и Rb независимо представляют собой (С1-С6)алкил и арил; R1 представляет собой (С6-С10)арил; R2 и R3 независимо представляют водород, (С1-С6)алкил, -COOR5, -CONR5R6, -CH2NR5R6, -СН2CH2NR5R6, -СН2СН2ОН или -СН2OН; при условии, что один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом; R5 и R6 независимо представляют водород, (С1-С6)алкил, (С3-С12)циклоалкил, (С6-С10)арил, (С6-С10)арил(С1-С6)алкил, гетероарил или гетероарил(С1-С6)алкил, которые могут быть незамещенными или замеще
Формула

его таутомерные формы, стереоизомеры, полиморфы, гидраты, сольваты и фармацевтически приемлемые соли,
в которой конфигурация при двойной связи может быть E/Z;
R представляет замещенные или незамещенные группы, выбранные из (С6-С10)арила, (С3-С12)циклоалкила, гетероарила, (С6-С10)арил(С1-С6)алкила и гетероциклила;
где гетероциклил здесь и далее отвечает 5-10-членному кольцевому радикалу, который состоит из атомов углерода и от одного до пяти гетероатомов, выбранных из азота кислорода и серы, а гетероарил здесь и далее отвечает ароматическому гетероциклилу, и каждый арил, циклоалкил, гетероарил, арилалкил и гетероциклил может быть замещен одним или более заместителями, выбранными из галогенов, включающих фтор, хлор, бром, йод, (С1-С6)алкил, (С1-С6)алкокси, (С6-С10)арил, галоген(С1-С6)алкил, (С6-С10)арил(С1-С6)алкокси, -O-(С3-С12)циклоалкил, -O-СН2-(С3-С12)циклоалкил, гидроксила, NRaRb и ORa, где Ra и Rb независимо представляют собой (С1-С6)алкил и арил;
R1 представляет собой (С6-С10)арил;
R2 и R3 независимо представляют водород, (С1-С6)алкил,
-COOR5, -CONR5R6, -CH2NR5R6, -CH2CH2NR5R6, -CH2CH2OH или
-CH2OH; при условии, что один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом;
R5 и R6 независимо представляют водород, (С1-С6)алкил, (С3-С12)-циклоалкил, (С6-С10)арил, (С6-С10)арил(С1-С6)алкил, гетероарил или гетероарил(С1-С6)алкил, которые могут быть незамещенными или замещенными; или R5 и R6 могут быть объединены с образованием насыщенного или ненасыщенного 3-8-членного кольца, имеющего 0-2 гетероатома, включая N, О или S;
причем гетероарил здесь и далее отвечает 5-10-членному кольцевому радикалу, который состоит из атомов углерода и от одного до пяти гетероатомов, выбранных из азота, кислорода и серы, и каждый алкил, циклоалкил, арил, арилалкил, гетероарил или гетероарилалкил могут быть замещены одним или более заместителями, выбранными из галогена, содержащего хлор, фтор, бром или йод, (С1-С6)алкокси и NRaRb;
R4 представляет ОН, (С6-С10)арил, орто-замещенный анилин или амино (С6-С10)арил, которые могут быть необязательно замещены одной или более группами, выбранными из галогенов, содержащих фтор, хлор, бром, йод, гидроксила, аминогруппы или (С6-С10)арила;
Х представляет собой -NR7-, -CONR7- или -NR7CO-;
R7 представляет собой водород или (C1-С6) алкил;
Y представляет (С6-С10)арил или (С6-С10)арил(С2-С6)алкенил;
m является целым числом от 0-1; n является целым числом от 0-1; о является целым числом от 0-7; и p является целым числом от 0-1.
R представляет замещенные или незамещенные группы, выбранные из (С6-С10)арильной группы, включающей фенил, нафтил и инданил; (С3-С12)циклоалкильной группы, включающей циклопропил, циклобутил, циклопентил, циклогексил и циклооктанил; гетероарильной группы, включающей пиридинил, пиридазинил, пиримидил, триазинил, пирролил, индолил, пиразолил, имидазолил, пиразинил, пиримидинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил и хинолинил; (С6-С10)арил(С1-С6)алкильной группы, включающей бензил и фенилэтил;
гетероциклильной группы, включающей бензодиоксолил, бензодиоксанил и бензофуранил;
R1 представляет (С6-С10)арильную группу, включающую фенил, нафтил и инданил;
R2 и R3 независимо представляют водород, (С1-С6)алкильную группу, включая метил, этил, н-пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил; -COOR5, -CONR5R6, CH2NR5R6, -CH2CH2NR5R6, -СН2СН2ОН или -СН2ОН; при условии, что один из R2 или R3 является водородом или незамещенным алкилом, другой не является ни водородом, ни незамещенным алкилом;
R5 и R6 независимо представляют водород, замещенные или незамещенные группы, выбранные из (С1-С6)алкильной группы, включая метил, этил, н-пропил, изопропил, бутил, изобутил, трет-бутил, пентил и гексил; (С3-С12)циклоалкильной группы, включая циклопропил, циклобутил, циклопентил, циклогексил и циклооктил; (С6-C10)арильной группы, включая фенил, нафтил и инданил; (С6-С10)арил(С1-С6)алкильной группы, включая бензил и фенилэтил; гетероарильной группы, включая пиридинил, пиридазинил, пиримидил, триазинил, пирролил, пиразолил, имидазолил, пиразинил, пиримидинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, индолил и хинолинил; гетероарилалкильной группы, включая тиенилпропил, пиридинилэтил и индолилпропил; или R5 и R6могут быть объединены с образованием насыщенного или ненасыщенного 3-8-членного кольца, имеющего 1-2 гетероатома, включая N, О или S;
R4 представляет ОН, (С6-С10)арил, включая фенил, нафтил и инданил; орто-замещенный анилин или амино(С6-С10)арил, которые необязательно замещены;
X представляет собой -NR7-, -CONR7- или -NR7CO-;
R7 представляет собой водород или (C1-С6)алкил;
Y представляет (С6-С10) арильную группу, включая фенил, нафтил и инданил; или (С6-С10)арил(С2-С6)алкенильную группу, включая фенилэтенил и фенилпропенил.
N-Циклопропил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Метил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N, N-Диметил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-Фенил-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(тиофен-2-ил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-фенил-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-трифторметилфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(пиридин-3-ил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-метоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(2-хлорфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(2-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3-хлорфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-[бензодиоксол-5-ил]-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-метилфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Морфолино-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Морфолино-2-(2-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Mopфoлинo-2-(3-метоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Тиоморфолино-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклооктил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3-метоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Изопропил-2-(3-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Изопропил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Изопропил-2-(3,4-дифторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3-фтор-4-метоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Изопропил-2-(3-фтор-4-метоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3,4-дифторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-(4-Фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-(4-Фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламида;
N-Пирролидино-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклoпpoпил-2-(4-Циклoпpoпилмeтoкcифeнил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-бензилоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-циклопентилоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-(4-Фторбензил)-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(2,4-диметоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3,4-диметоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(индол-3-ил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(тиофен-3-ил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-3-(4-фторфенил)-2-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-3-(4-фторфенил)-2-(3-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(3-циклопропилметоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-(3-Циклопропилметоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламида;
N-Циклопропил-2-(3-циклопентилоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-(3-Циклопентилоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламида;
N-Циклопропил-2-(бифенил-4-ил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
2-(4-Циклопропилметоксифенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-фенилакриламида;
N-Циклопропил-3-(3,4-диметоксифенил)-2-(3-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-3-(4-метоксифенил)-2-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-3-(4-циклопропилметоксифенил)-2-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-3-(4-циклопентилоксифенил)-2-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
N-Циклопропил-2-(4-фторфенил)-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)бут-2-енамида;
2-[4-(Диметиламино)фенил]-3-(4-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)-N-циклопропилакриламида;
N-Циклопропил-3-(4-фторфенил)-2-(4-(3-(гидроксиамино)-3-оксопропил)фенил)акриламида;
N-Циклопропил-2-(4-фторфенил)-3-(3-((1Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)фенил)акриламида;
3-(4-((1Е)-3-(Циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)фенил)-N-гидроксиакриламида;
3-(4-((1Е)-3-(Циклопропиламино)-2-фенилпроп-1-ен-1-ил)фенил)-N-гидроксиакриламида;
3-(4-((1Е)-2-(3-Циклопентилоксифенил)-3-(Циклопропиламино)проп-1-ен-1-ил)фенил)-N-гидроксиакриламида;
3-(4-((1Е)-2-(3-Хлорфенил)-3-(Циклопропиламино)проп-1-ен-1-ил)фенил)-N-гидроксиакриламида;
N-Циклопропил-3-(4-(3-(2-аминофениламино)-3-оксопроп-1-ен-1-илфенил)-2-(4-фторфенил)акриламида;
3-(4-((1Е)-3-(2-Аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-фторфенил)-N,N-диметилакриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-(трифторметил)фенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(пиридин-3-ил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-хлорфенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-[бензодиоксол-5-ил]акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-фторфенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3-хлорфенил)акриламида;
(Е)-N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-метилфенил)акриламида;
N-Морфолино-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(4-фторфенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3-метоксифенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-фенилакриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(тиофен-2-ил)акриламида;
N-Морфолино-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(2-фторфенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3,4-дифторфенил)акриламида;
N-Циклопропил-3-(4-((1Е)-3-(2-аминофениламино)-3-оксопроп-1-ен-1-ил)фенил)-2-(3,4-диметоксифенил)акриламида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(N,N-Диметиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-енил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-Морфолино)-2-(4-фторфенил)-3-оксопроп-1-енил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Морфолино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
6-((1Е)-3-(4-(3-(Циклопропиламино)-2-(3-фтор-4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)-N-гидроксигексанамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Морфолино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(((1Е)-3-(4-(3-(Морфолино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)акриламидо)метил)-N-гидроксибензамида;
4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида;
4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида;
4-(3-(Циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида;
4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида;
4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-гидроксибензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)бензамида;
(Е)-N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-трифторметилфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-метоксифенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(1,3-бензодиоксол-5-ил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(тиофен-2-ил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3,4-дифторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-метоксифенил)3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-хлор-4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(фениламино)-2-(3,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(3,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2,4-диметоксифенил)-3-оксопроп-1 -ен-1 -ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-нафтил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-фениламино-2-(2,4-диметоксифенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Амино-4-фторфенил)-4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-3-оксо-2-(1Н-индол-3-ил)проп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-3-оксо-2-бифенил-4-ил-проп-1-ен-1-ил)бензамида;
4-(2-(4-Фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)-N-(2-гидроксифенил)бензамида;
N-(2-Аминофенил)-4-[3-(циклопропиламино)-3-оксо-2-пиридин-3-ил-проп-1-ен-1-ил]бензамида;
N-(2-Аминофенил)-4-(2-(4-гидроксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(2-(2,6-дифторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(2-(2,5-дифторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(2-(4-фторфенил)-3-(изопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-(4-Аминобифенил-3-ил)-4-(3-(циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-енил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(2-метилфенил)3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(метиламино)-2-(4-фторфенил)-3 -оксопроп-1-ен-1-ил)бензамида;
(Z)-N-(2-Аминофенил)-4-(2-(4-фторфенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-[2-(4-фторфенил)-3-морфолин-4-ил-3-оксопроп-1-ен-1-ил]бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-3-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(фениламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
4-[3-Амино-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил]-N-(2-аминофенил)бензамида;
N-(2-Аминофенил)-4-(2-(4-циклопентилоксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(2-(4-циклопропилметоксифенил)-3-(циклопропиламино)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(бензиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)бензамида;
N-(2-Аминофенил)-4-(3-(циклопропиламино)-2-(4-фторфенил)проп-1-ен-1-ил)бензамида;
4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-((Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(4-((Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)-N-(4-((Е)-3-(гидроксиамино)-3-оксопроп-1-ен-1-ил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(2-хлорфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(3-хлорфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(4-метоксифенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(2-хлор-4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-[бензодиоксол-5-ил]-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(4-трифторметилфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(3,4-дифторфенил)-3-оксопроп-1-ен-1-ил)-N-(4-(гидроксикарбамоил)бензил)бензамида;
4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида;
4-(3-(Циклопропиламино)-2-фенил-3-оксопроп-1-ен-1-ил)-N-(6-гидроксиамино)-6-оксогексил)бензамида;
4-(3-(Циклопропиламино)-2-(4-метилфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида;
4-(3-(Циклопропиламино)-2-(2-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида;
4-(3-(Циклопропиламино)-2-(3-фторфенил)-3-оксопроп-1-ен-1-ил)-N-(6-(гидроксиамино)-6-оксогексил)бензамида;
N-(4-(3-(Циклопропиламино)-2-(4-фторфенил)-3-оксопроп-1-ен-1-ил)фенил)-N-гидроксиоктандиамида и
N-(2-Аминофенил)-4-((4-(3-(циклопропиламино)-1-(4-фторфенил)-3-оксопроп-1-ен-2-ил)фениламино)метил)бензамида.
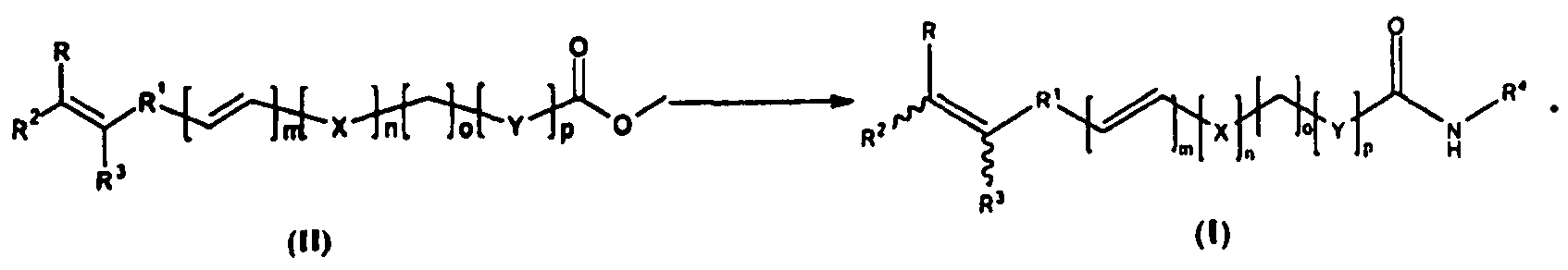

Документы, цитированные в отчёте о поиске
Гидроксаматные производные, применимые в качествеингибиторов дезацетилазы
Комментарии