Производные n-ацил-2-арилциклоалкиламина, стимулирующие синтез мелатонина, фармацевтическая композиция, способ мелатонинстимулирующего связывания - RU2162076C2
Код документа: RU2162076C2
Чертежи













Описание
Предметом настоящего изобретения являются новые производные N-ацил-2-арилциклопропилметиламина, обладающие лекарственными и биологически активными свойствами, способы их получения, содержащие их фармацевтические составы и способы их применения. Эти соединения стимулируют синтез мелатонина, что делает их полезными для лечения некоторых заболеваний.
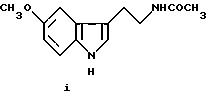
Мелатонин (i; N-ацетил-5-метокситриптамин) является гормоном, который синтезируется и выделяется, главным образом, шишковидным телом. У млекопитающих уровни мелатонина подчиняются циклическому, циркадному ритму, причем самые высокие уровни наблюдаются в темный период циркадного цикла. Мелатонин участвует в трансдукции фотопериодической информации и, по-видимому, модулирует различные функции нервной и эндокринной систем у позвоночных, в том числе половую активность, вес тела и обмен веществ у фотопериодических млекопитающих, управление циркадными ритмами и изменение физиологии сетчатки глаза.
Результаты последних исследований показали, что мелатонин осуществляет биологическое воздействие с помощью определенных рецепторов. Использование биологически активного, меченого радиоактивным изотопом агониста [i125]-2-иодмелатонина позволило идентифицировать высокоаффинные рецепторы мелатонина в центральной нервной системе животных разных видов. В литературе сообщалось о клонировании потомства одного такого высокоаффинного рецептора мелатонина из меланоцитов лягушки. В результате проведения авторадиографических исследований было установлено, что рецепторы мелатонина расположены в нескольких определенных структурах мозга млекопитающих.
Хотя существуют значительные различия в распределении рецепторов даже у близких видов, как правило, наибольшая плотность центров связывания наблюдается в несвязанных ядрах гипоталамуса. У людей характерное связывание [i125] -2-иодмелатонина в гипоталамусе полностью происходит в надхиазматических ядрах, что позволяет с большой степенью уверенности предположить, что рецепторы мелатонина расположены на участке биологических часов человека.
Было установлено, что экзогенное введение мелатонина позволяет синхронизировать циркадные ритмы у крыс (Cassone и др., J. Biol. Rhymes, 1, 219-229, 1986). Мелатонин вводят людям с целью лечения нарушений суточного ритма организма, вызываемых десинхронизацией циркадных ритмов (Arendt и др., Br. Med. J., 292, 1170, 1986). Кроме того, применение однократной дозы мелатонина в качестве снотворного рассматривалось Вуртманом в международной заявке на патент WO 94/07487.
Центры связывания мелатонина были обнаружены в разных тканях тела человека, например в сетчатке глаза, надхиазматических ядрах, селезенке и т.д. Из этого следует, что мелатонин выполняет несколько физиологических функций и не отличается высокой избирательностью. В случае применения мелатонина велика вероятность возникновения побочных явлений. Агонисты мелатонина должны оказывать более избирательное действие, чем мелатонин, и вызывать значительно меньше побочных явлений. Приемлемые агонисты мелатонина способны устранить недостатки, присущие мелатонину, в результате чего их действие становится более предсказуемым и, возможно, более продолжительным.
Агонисты мелатонина особенно полезны для лечения хронобиологических нарушений. Кроме того, их можно использовать для дальнейшего изучения воздействия рецепторов мелатонина и лечения вызываемых ими заболеваний, таких как депрессия, синдром смещения времени наибольшей работоспособности, нарушения сна, глаукома, половые расстройства, рак, нарушения иммунной и нейроэндокринной систем.
Помимо
простых
индоловых производных самого мелатонина были созданы различные амидные структуры, которые использовались в качестве лигандов мелатонина. Эти амидные структуры можно представить общей
формулой

в которой Z представляет собой арильную или гетероарильную систему, присоединяемую цепью их двух углеродных атомов к амидной группе.
Ниже приводятся несколько типичных примеров.
Йоус и др. в заявке на европейский патент EPA 527687A рассматривают в
качестве лигандов
мелатонина этиламины, имеющие циклические заместители 1,

в которой Ar' помимо прочего представляет собой замещенный или незамещенный бензо[b] тиофен-3-ил, бензимидазол-1-ил, бензо[b]фуран-3-ил, 1,2-бензизокcазол-3-ил 1,2-бензизотиазол-3-ил или индазол-3-ил;
R1 помимо прочего представляет собой алкильную или циклоалкильную группу; и
R2 является водородом или низшим алкилом.
Ланглоис и др. в заявке на патент
Австралии AU-A
48729/93 в качестве лигандов, стимулирующих синтез мелатонина, описывают арилалкил(тио)амиды формулы 2
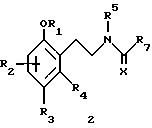
в которой R1 - водород или низший алкил;
R2 - водород, галоген или низший алкил;
R3 и R4 являются одинаковыми или разными группами, представляя среди прочего водород, галоген или низший алкил;
R5 - водород или низший алкил;
X - сера или кислород; и
R7 представляет собой среди прочего низший алкил или алкилен.
Однако в этих противопоставленных материалах не говорится о новых производных арилциклопропилметиламина, стимулирующих синтез мелатонина по настоящему изобретению.
Ранее было представлено несколько соединений, содержащих структурные элементы, характерные для соединений по настоящему изобретению, хотя ни в одном из противопоставленных материалов не указывалось на стимулирование этими соединениями синтеза мелатонина.
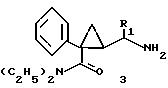
Матсуда и др. в международной заявке на патент WO 95/22521 описывают 1-фенил-2-(1-аминоалкил)-N, N-диэтилциклопропанкарбоксамиды формулы 3 в качестве антагонистов рецепторов N-метил-D-аспартата (NMDA), у которых R1 помимо прочего представляет собой C1-C5линейную насыщенную алифатическую группу, C1-C5линейную ненасыщенную алифатическую группу, разветвленную алифатическую группу или фенильную группу, которая может быть замещена одним-тремя заместителями, выбираемыми независимо друг от друга из группы, включающей галоген, C1-C4алкил, нитро, амино, гидрокси и C1-C4 алкокси.
Производные 1,2-диарилциклопропана формулы 4 рассматриваются в заявке на патент NE 6701256 в качестве веществ, стимулирующих центральную нервную систему,
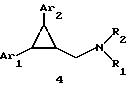
Ar1 и Ar2 представляют собой независимо или необязательно замещенный фенил;
R1 помимо прочего представляет собой водород, низший алкил или ацил;
R2 помимо прочего является алкилом, циклоалкилом или аралкилом.
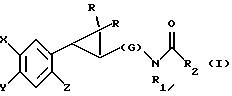
Настоящее изобретение относится к соединениям формулы I или II, которые стимулируют синтез
мелатонина и, таким
образом, могут использоваться для лечения вызываемых им заболеваний:
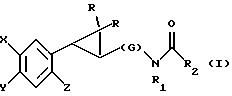
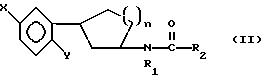
в которых X - галоген, водород, C1-4алкил или OR5, где R5 представляет собой водород, C1-4перфторалкил, C1-4фторалкил, C1-4пердейтероалкил, C1-20алкил, C4-20алкциклоалкил, C2-20карбонитрилалкил, C3-22 карбоалкоксиалкил, C3-20алкенил, C3-20алкинил, C9-20аралкил, C9-20аралкенил, C9-20алкинил, C2-20гидроксиалкил, C8-20 арилоксиалкил, C7-20пиридилалкил или C6-20пиррилалкил;
Y - водород или галоген;
z - водород, галоген, циано, арил, C7-20аралкил, C8-20 арилкинил или C2-20алкамидо;
R в обоих случаях представляет собой водород, галоген или C1-4алкил;
G - двухвалентный метилен, этилен или C1-4 алкметилен;
R1 представляет собой водород, C1-4алкил или бензил; и
R2 - C1-6алкил, C2-6алкенил, C3-6 циклоалкил, C2-4алкоксиалкил, C1-4трифторметилалкил, C2-8алкилтиоалкил или NR3R4, где
R3 и R4 независимо друг от друга выбирают из водорода и C1-4алкила, но R3 и R4 не могут быть одновременно водородом.
Предметом настоящего изобретения являются соединения формулы I или II, способы их получения и применения и составы для лечения определенных заболеваний.
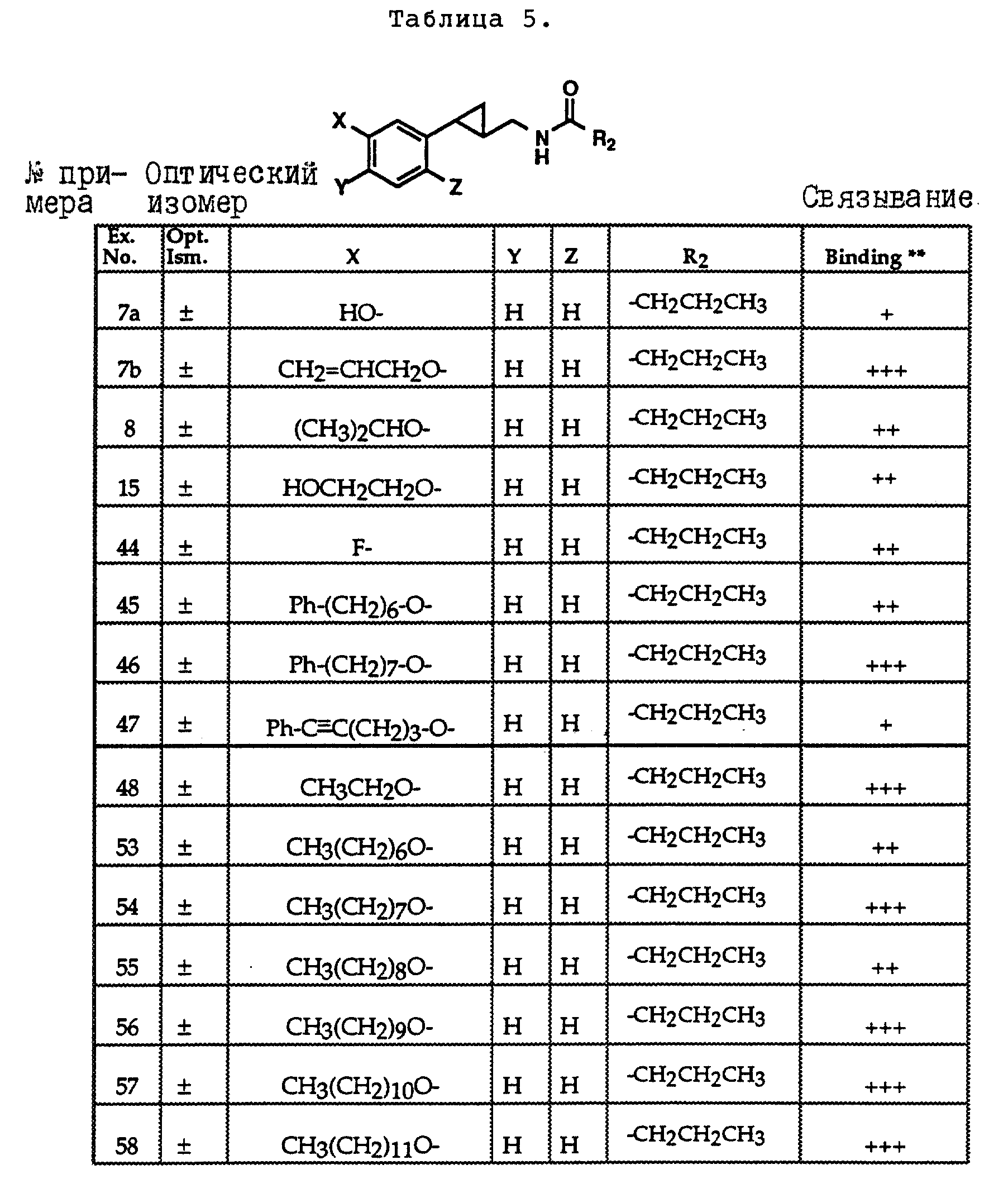
Соединениями формулы I или II являются:
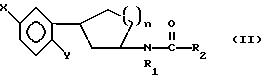
в которых X - галоген, водород, C1-4алкил или OR5, где R5 представляет собой водород, C1-4перфторалкил, C1-4фторалкил, C1-4пердейтероалкил, C1-20алкил, C4-20алкциклоалкил, C2-20карбонитрилалкил, C3-22карбоалкоксиалкил, C3-20алкенил, C3-20алкинил, C9-20аралкил, C9-20аралкенил, C9-20аралкинил, C2-20гидроксиалкил, C8-20арилоксиалкил, C7-20пиридилалкил или C6-20пиррилалкил;
X - водород или галоген;
Z - водород, галоген, циано, арил, C7-20аралкил, C8-20арилкинил или C2-20алкамидо;
P в обоих случаях представляет собой водород, галоген или C1-4 алкил;
G - двухвалентный метилен, этилен или C1-4алкметилен;
R1 - водород, C1-4алкил или бензил; и
R2 - C1-6алкил, C2-6алкенил, C3-6циклоалкил, C2-4алкоксиалкил, C1-4трифторметилалкил, C2-8алкилтиоалкил или NR3R4, где R3 и R4 независимо друг от друга выбирают из водорода и C1-4алкила, но R3 и R4 не могут одновременно быть водородом.
Следует отметить, что используемый здесь термин "галоген" означает фтор, хлор, бром и иод; термин "алкил" относится к насыщенным углеводородным радикалам с прямолинейной или разветвленной цепью; "фторалкил" означает монофторзамещенные насыщенные углеводородные радикалы с прямолинейной и разветвленной цепью; "алкенил" означает углеводородный радикал с прямолинейной и разветвленной цепью, содержащий углерод-углеродную двойную связь; "циклоалкил" означает насыщенные циклические углеводородные радикалы; "алкокси" означает алкильный радикал, присоединенный к молекуле с помощью атома кислорода; "алкилтиоалкил" означает алкильный радикал, присоединенный к другому с помощью атома серы; "арилоксиалкил" означает алкильный радикал, присоединенный к необязательно замещенной фенильной группе с помощью атома кислорода; "алкциклоалкил" означает алкильный радикал, присоединенный к насыщенному циклическому углеводородному радикалу; "карбонитрилалкил" означает алкильный радикал, присоединенный к нитрильному радикалу с помощью атома углерода; "карбоалкоксиалкил" означает алкильный радикал, присоединенный непосредственно к карбоалкоксильному радикалу; "гидроксиалкил" означает алкильный радикал, присоединенный к атому углерода с помощью атома кислорода; "пиридилалкил" означает пиридильный радикал, присоединенный непосредственно к конечному атому углерода алкильного радикала; "пиррилалкил" означает пиррильный радикал, присоединенный непосредственно к конечному атому углерода алкильного радикала; "трифторметилалкил" означает трифторзамещенную метильную группу, присоединенную непосредственно к алкильному радикалу; "перфторалкил" означает насыщенные фторуглеродные радикалы с прямолинейной и разветвленной цепью; "пердейтероалкил" означает насыщенные дейтероуглеродные радикалы с прямолинейной и разветвленной цепью; "циано" означает радикал, содержащий углерод-азотную тройную связь; "алкинил" означает углеродные радикалы с прямолинейной и разветвленной цепью, содержащие углерод-углеродную тройную связь; "аралкил", "аралкенил" и "аралкинил" (или соответственно "арилалкил", "арилалкенил" и "арилалкинил") означают радикалы, в которых необязательно замещенная фенильная группа присоединена к конечному атому углерода соответственно алкильного, алкенильного или алкинильного радикала; "алкамидо" (или "алкиламидо") относится к NC(O)-алкильным группам, содержащим указанное число атомов углерода; "алкметилен" (или "алкилметилен") означает алкильный радикал, присоединенный непосредственно к метеновому радикалу. "Бензил" или "Bn" означает фенилметильную группу, -CH2-фенил. Фенильные группы, если они имеются, содержат заместители, выбираемые из группы, включающей водород, галоген, трифторметил и C1-4 алкокси.
Числа, указанные в виде подстрочных индексов после символа "C", означают число атомов углерода в группе.
На основании биологических тестов предпочтительными были признаны следующие соединения формулы I. Все эти соединения отличаются сродством связывания в отношении рецептора мелатонина человека при значениях IC50, равных 600 нм или меньше.
Предпочтительными соединениями формулы I являются такие соединения, в которых R1 является водородом, R2 - C1-4алкилом, X и Z независимо друг от друга представляют водород или галоген, и X является OR5, где R5 - C1-20алкил, C3-20алкенил, C3-20алкинил и C9-20аралкил, аралкенил или аралкинил. Желательно, чтобы в радикале R5 отсутствовали атомы O, N и S. Особенно предпочтительны соединения, в которых R2 является C1-4алкилом.
Ниже
представлены
предпочтительные соединения по настоящему изобретению:
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]-2- метоксиацетамид,
(+)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]пропанамид,
(транс)-N-[[2-(4-хлор-3-метоксифенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]-2- метилпропанамид,
(транс)-N-[[2-[3-(6-фенилгексил)окси]фенил]циклопропил]метил] бутанамид,
(транс)-N-[[2-(2-бром-5-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]ацетамид,
(-)-(транс)-N-[[2-(2,4-дибром-5-метоксифенил)циклопропил]
метил] бутанамид,
(транс)-N-[[2-(2-иод-5-метоксифенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(2-иод-5-метоксифенил)циклопропил]метил] бутанамид,
(транс)-N-[[2-[3-(гептилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(2-пропенилокси)фенил]циклопропил]метил] бутанамид,
(транс)-N-[[2-[3-[(7-фенилгептил)окси] фенил]
циклопропил]метил] бутанамид,
(транс)-N-[[2-(3-[(этоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-октилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(нонилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-децилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-ундецилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-додецилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N- [[2-[(2-фенилэтил)окси]фенил]циклопропил]метил] бутанамид,
(транс)-N-[[2-[3-[3-(3-метоксифенил)пропокси] фенил] циклопропил] метил] бутанамид,
(-)-(транс)-N-[[2-(5-метокси-2-(фенилэтинил)фенил)циклопроп-1-ил] метил] бутанамид,
(транс)-N-[[3-(3-метоксифенил)-2,2-дифтор-1-циклопроп-1-ил] метил] бутанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопроп-1-ил]метил]циклопропан карбоксамид,
(-)-(транс)-N-[[2-[3-[3-(3-метоксифенил)пропокси] фенил] циклопроп-1-ил] метил]бутанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопроп-1-ил] метил] - N'-метилмочевина,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопроп-1-ил]метил] пропанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопроп-1- ил]метил]ацетамид,
(-)-(транс)-3,3,3-трифтор-N-[[2-(3-метоксифенил) циклопроп-1-ил]-метил] пропанамид,
(транс)-N-[[2-(3-[(3,7,
11-триметилдодека-2,6,10-триен-1- ил)-окси]фенил] циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[(4-фенилбут-1-ил)окси]фенил]циклопроп-1-ил] метил]бутанамид,
(транс)-N-[[2-[3-[(5-фенилпент-1-ил)окси] фенил] циклопроп-1-ил] метил)бутанамид,
(транс)-N-[2-[(3-тридейтерометоксифенил)циклопроп-1-ил] метил]бутанамид,
(транс)-N-[[2-[3-[(3-циклогексилпроп-1-ил)окси]фенил]циклопроп- 1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[(3-циклопентилпроп-1-ил)окси]фенил]циклопроп- 1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[2-[3-(трифторметил)фенил]этокси]фенил] циклопpoп-1-ил] метил]бутaнaмид,
(транс)-N-[[2-[3-[[2-(3-метоксифенил)циклопроп-1- ил] метокси] фенил] циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(4-хлор-3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]-2- метилтиоацетамид,
(транс)-N-[[2-[3-[3-(3-метоксифенил)пропокси] фенил] циклопропил] метил] бутанамид,
(-)-(транс)-N-[[2-(4-иoд-3-мeтoкcифeнил)циклoпpoп-1- ил] метил]-бутанамид,
(-)-(транс)-N-[[2-(5-метокси-2 (фенилэтил)циклопроп-1-ил]метил] бутанамид,
(-)-(транс)-N-[[2-(4-метокси-[1,1'-бифенил] -2-ил- циклопроп-1-ил]метил] бутанамид,
(-)-(транс)-N-[[2-(4-метокси-4'-(трифторметил)[1,1'- бифенил]-2-ил]циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-бромфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-бромфенил)циклопроп-1-ил]метил]ацетамид,
(транс)-N-[[2-(3-бромфенил)циклoпpoп-1-ил]метил]пpопанамид,
(-)-(транс)-N-[[2-[2-иод-5-[3-(3-метоксифенил)
пpoпoкcи]фенил]циклопроп-1-ил]метил]бутaнaмид,
(транс)-N-[[2-[3-[(3-фенилпpоп-1-ил)окси] фенил] циклопроп-1-ил]метил] бутанамид,
(транс)-N-[[2-[3[(3-феноксипроп-1-ил)окси] фенил]
циклопроп-1-ил]метил] бутанамид,
(транс)-N-[[2-[3-[(3-димeтилoктa-2,6-диен-1-ил) окси] фенил]циклoпpoп-1-ил]метил]бутанамид,
(транс)-N-[[2-[3-(5-метилгексилокси)фенил]
циклопроп- 1-ил]метил]бутанамид,
(транс)-N-[[2-[3-(4-метил-3-пентен-1-ил- окси)фенил] циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[(3-циклогексилбут-1-ил)окcи]
фенил]циклопроп- 1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[2-[2-(трифторметил)фенил]этокси]фенил] циклопроп-1-ил] метил]бутанамид,
(транс)-N-[[2-[3-[2-(3-фторфенил)
этoкcи]фенил]циклопроп-1-ил]-метил]бутанамид,
(транс)-N-[[2-[3-[3-(4-метоксифенил)пропокси] фенил] циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[2-(2-фторфенил)
этокcи]фенил]циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-[3-[2-(2-метоксифенил) этокси]фенил]циклопроп-1-ил]метил] бутанамид,
(-)-(транс)-N-[[2-(3-фторфенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(3-фторфенил)циклопроп-1-ил]метил] 2-метил-пропанамид,
(-)-(транс)-N-[[2-(2-бром-5-фторфенил)циклопроп-1- ил]метил]бутанамид,
(-)-(транс)-N-[[2-(4-бром-3-фторфенил)циклопроп-1-ил] метил]бутанамид,
(-)-(транс)-N-[[2-(5-фтор-2-иодфенил)циклопроп-1-ил] метил]бутанамид,
(-)-(транс)-N-[[2-(3-фтор-4-иодфенил) циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[3-(3-метокcифенил)-2,
2-дифтоp-1-циклопроп-1- ил]метил]бутанамид,
(транс)-N-[[3-(3-метоксифенил)-2,2-дифтор-1-циклопроп- 1-ил] метил]-2-метилпропанамид,
(транс)-N-[[2-(3-бромфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-бромфенил)циклoпpoп-1-ил]метил]ацетамид,
(транс)-N-[[2-(3-метилфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-бромфенил)циклопроп-1-ил]метил]пропанамид,
(транс)-N-[[2-(3-метилфенил)циклопроп-1-ил]метил]- 2-метилпропанамид,
(транс)-N-[[2-(3-метилфенил)циклопроп-1-ил]метил]ацетамид,
(транс)-N-[[2-(3-хлорфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-хлорфенил)циклопроп-1-ил]метил]пропанамид,
(транс)-N-[[2-(3-хлорфенил)циклопроп-1-ил]метил]ацетиамид,
(транс)-N-[[2-(3-хлорфенил)циклопроп-1-ил]метил] циклопропанкарбоксамид,
(транс)-N-[[2-(2,
5-дифторфенил)циклопроп-1-ил]метил]ацетамид,
(транс)-N-[[2-(2,5-дифторфенил)циклопроп-1-ил]метил]пропанамид,
(транс)-N-[[2-(2,5-дифторфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(2,5-дифторфенил)циклопроп-1-ил] метил] циклопропанкарбоксамид,
(транс)-N-[[2-[3-(пентафторэтил)фенил]циклопроп-1-ил]метил] бутанамид,
(-)-(транс)-N-[(2-фенилциклопроп-1-ил)метил]бутанамид,
(транс)-N-[(2-фенилциклопроп-1-ил)метил]ацетамид,
(транс)-N-[(2-фенилциклопроп-1-ил)метил]бутанамид,
(транс)-N-[2-[(3-этилфенил)циклопроп-1-ил]метил]ацетамид,
(транс)-N-[2-[(3-этилфенил)циклопроп-1-ил]метил]пропанамид,
(транс)-N-[2-[(3-этилфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[2-[(3-этилфенил)циклопроп-1-ил]метил] циклопропанкарбоксамид,
(транс)-N-[[2-[3-(трифторметил)фенил]циклопроп-1-ил]метил] бутанамид,
(транс)-N-[[2-(3-трифторметил)фенил] циклопроп-1-ил] метил]-2- метилпропанамид,
(-)-(транс)-N-[[2-(2-бром-5-фторфенил)циклопроп-1-ил]метил] бутанамид,
(транс)-N-[[2-(3-метилфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[2-[(3-этилфeнил)циклопроп-1-ил]метил]бутанамид,
(цис)-N-[3-метоксифенил)циклогексил]ацетамид,
(цис)-N-[3-метоксифенил)циклогексил]-2-метил-пропанамид,
(цис)-N-[3-метоксифенил)циклогексил]бутанамид,
(цис)-N-этил-N'-[3-метоксифенил)циклогексил]мочевина,
N-[3-метоксифенил)циклопентил]бутанамид.
Получение соединений формулы I.
Соединения формулы I можно получить в соответствии со следующими схемами реакций (способы получения 1-8). Группы R, R1, R2, R3, R4, X, Y, Z, G и R5 имеют вышеуказанные значения.
Способ получения 1 (см. схему 1).
В полученных соединениях формулы I R1 и R - водород, G - метилен, а другие заместители имеют вышеуказанные значения.
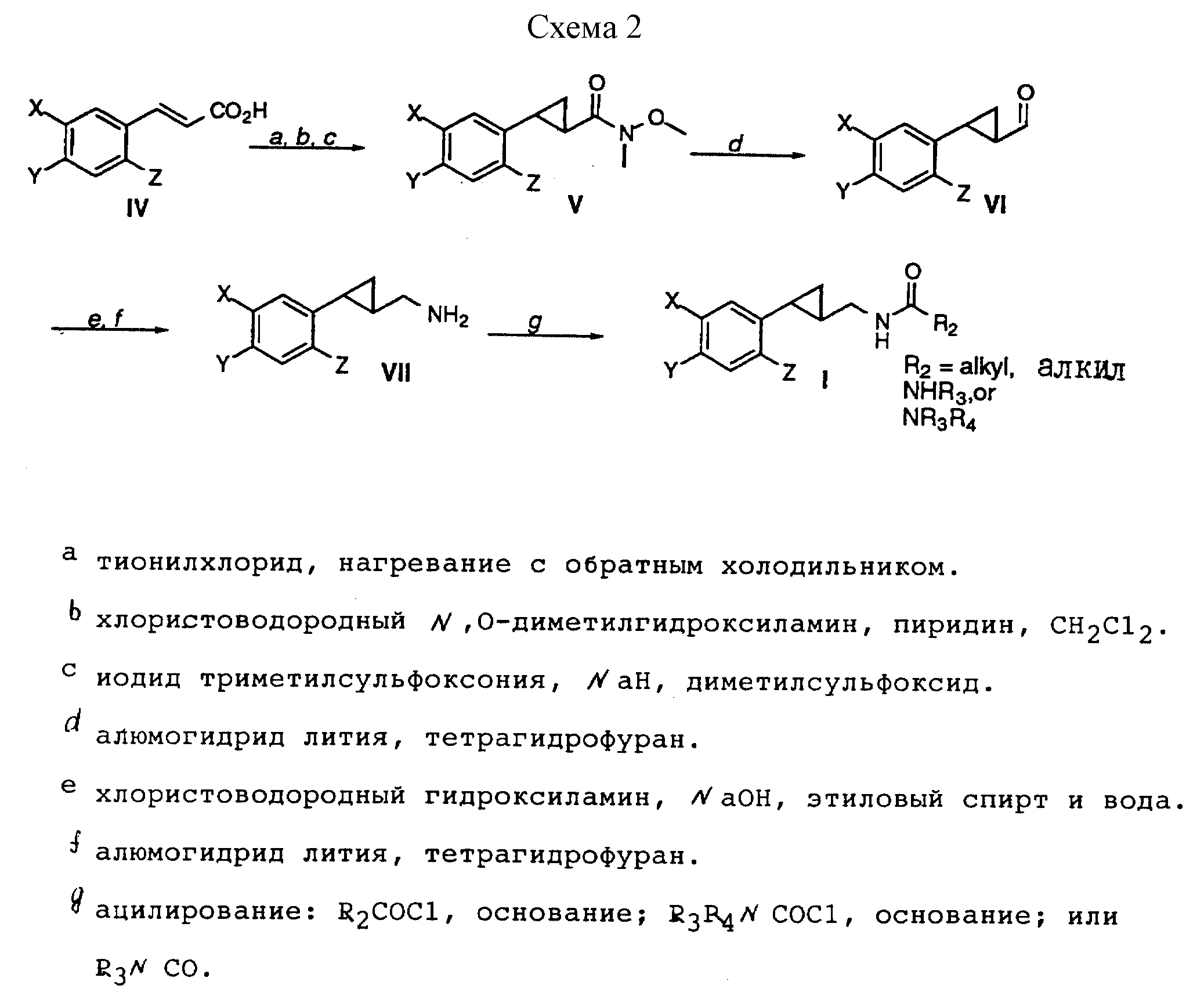
Способ получения 2 (см. схему 2).
В соединении формулы I, полученном в соответствии со способом получения 2, группа R2 - алкил, NHR3 или NR3R4 (вышеуказанные значения), G - метилен; R1 и R - водород.
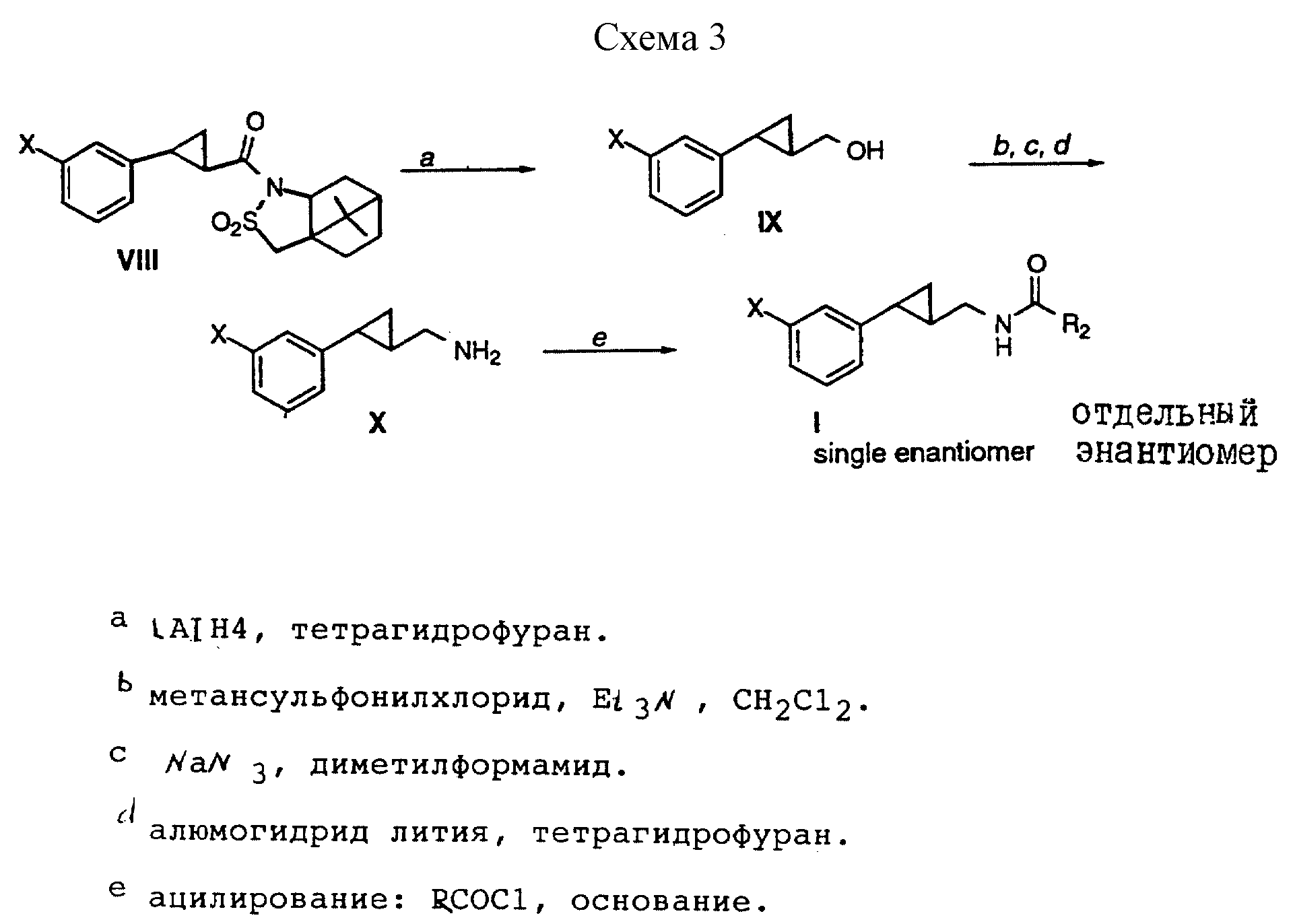
Способ получения 3 (см. схему 3).
В соединениях формулы I, полученных в соответствии со способом получения 3, R1, R, Y и Z - водород, а G - метилен.
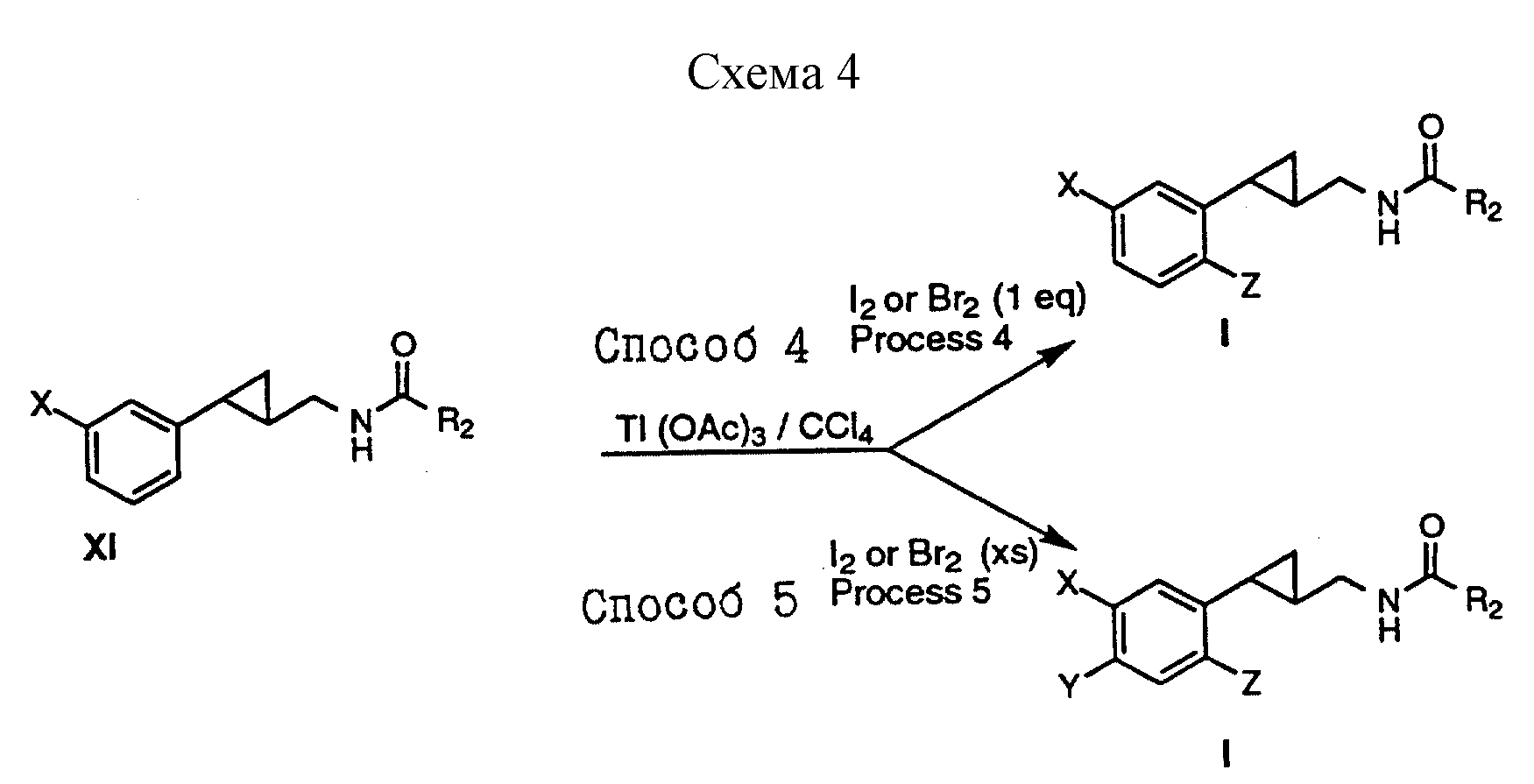
Способы получения 4, 5 (см. схему 4).
В соединениях формулы I, полученных в соответствии со способом получения 4, G - метилен; Y, R1 и R - водород, а Z - Br или I. В соединениях формулы I, полученных в соответствии со способом получения 5, Y и Z - Br или I, G - метилен, а R1 и R - водород.
Способ получения 6 (см. схему 5).
В соединениях формулы I, полученных в соответствии со способом получения 6, R1 - C1-4алкил или бензил, G - метилен, R-Н, и R2 , X, Y, Z имеют вышеуказанные значения.
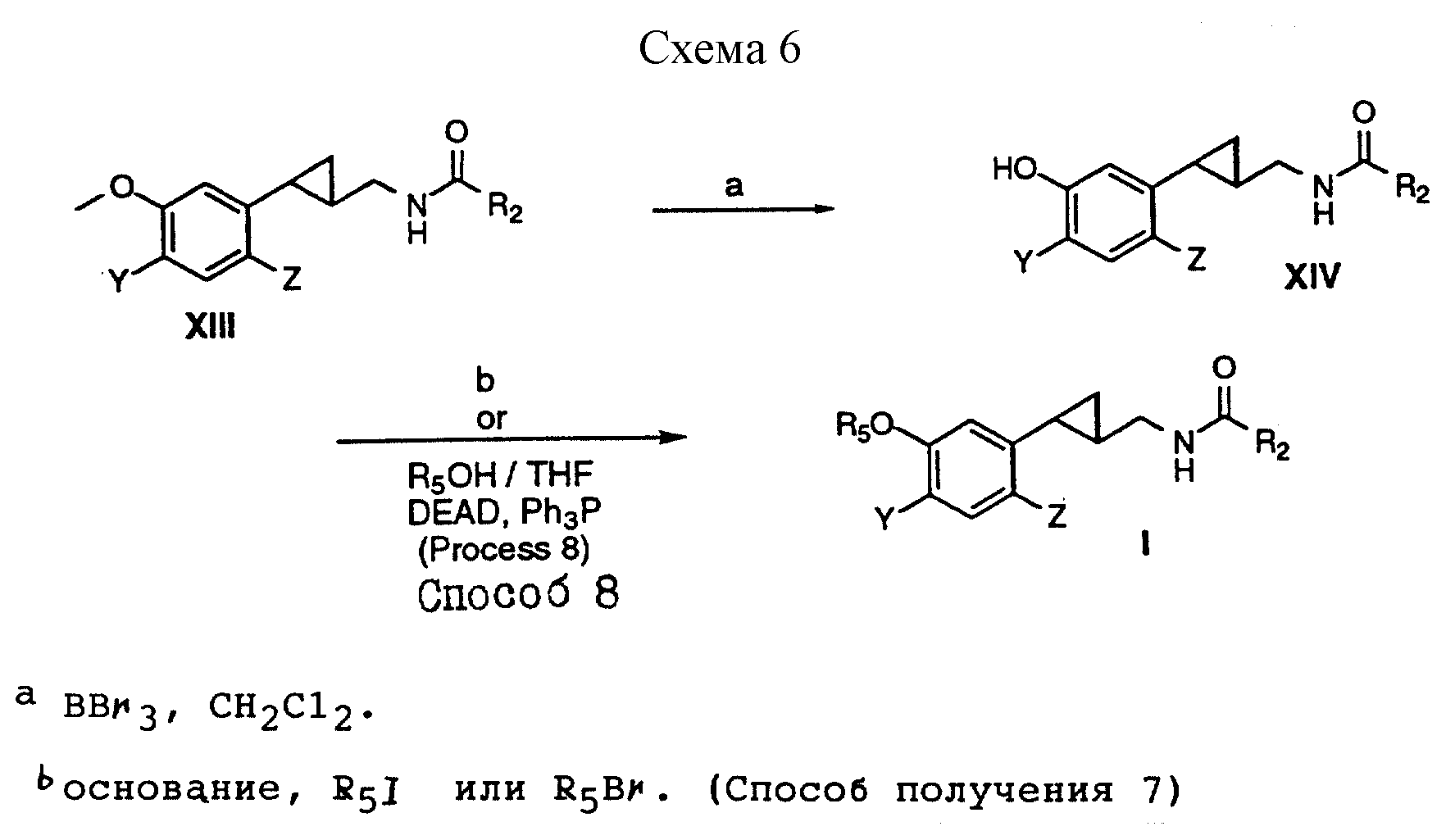
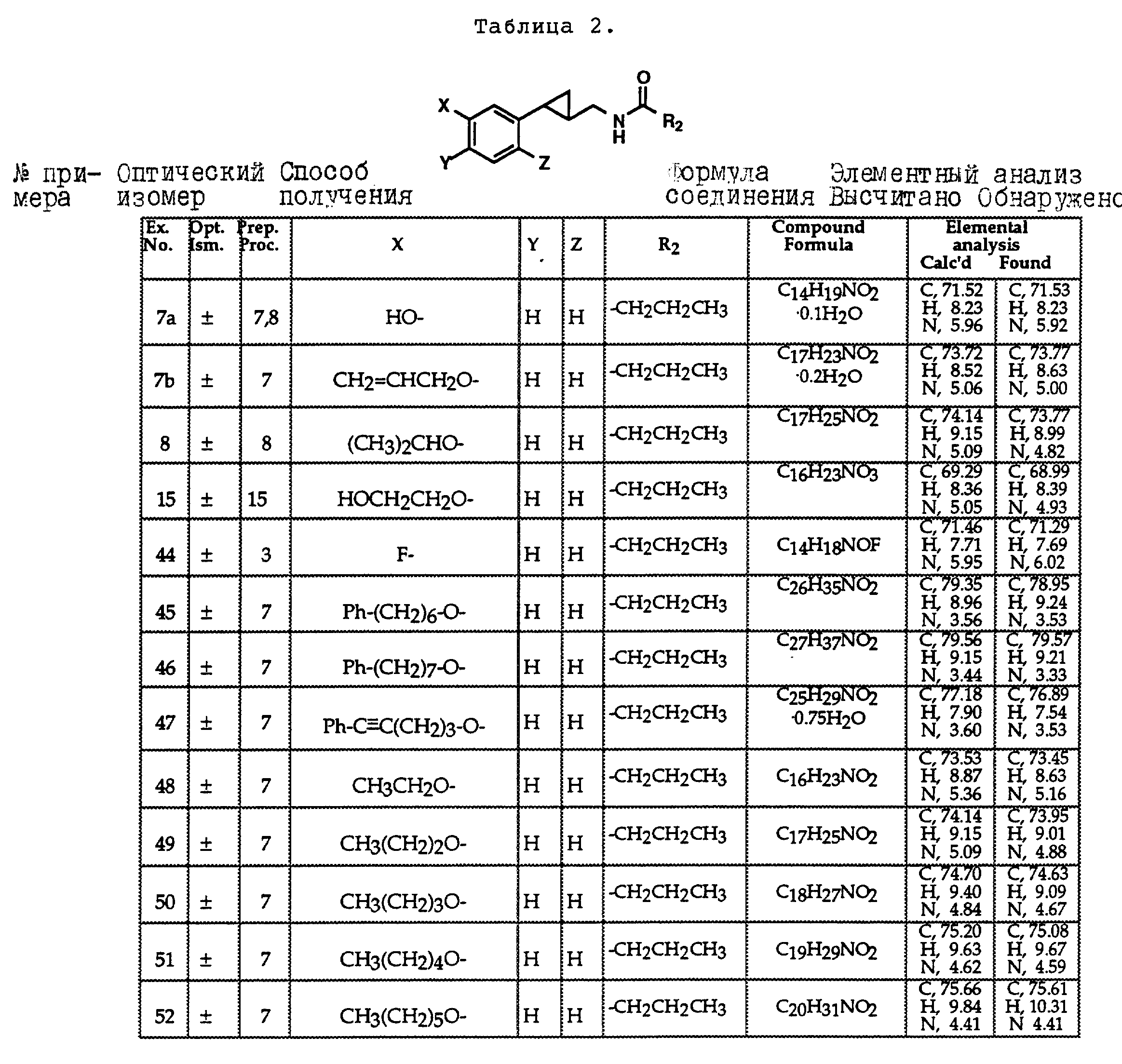
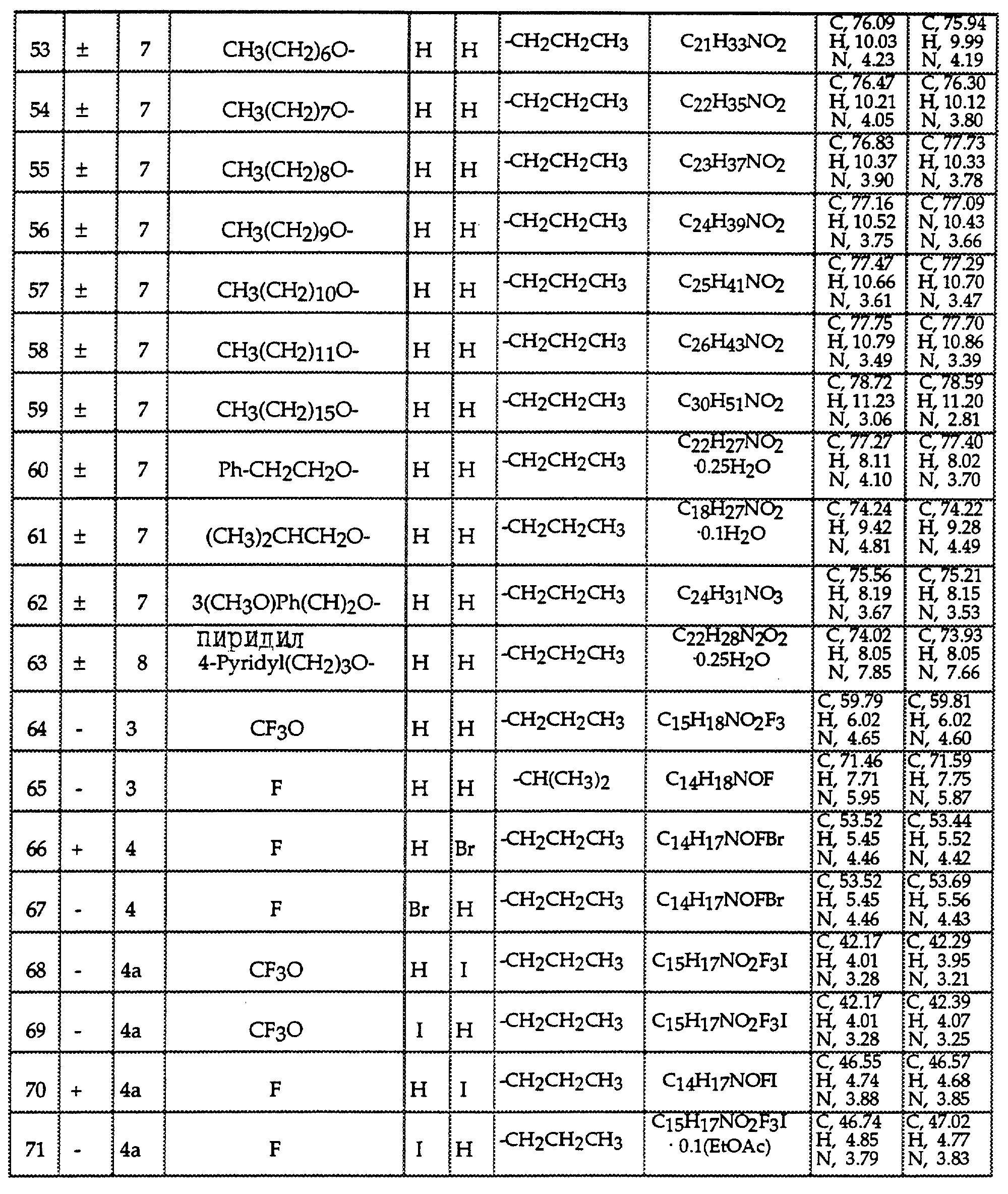
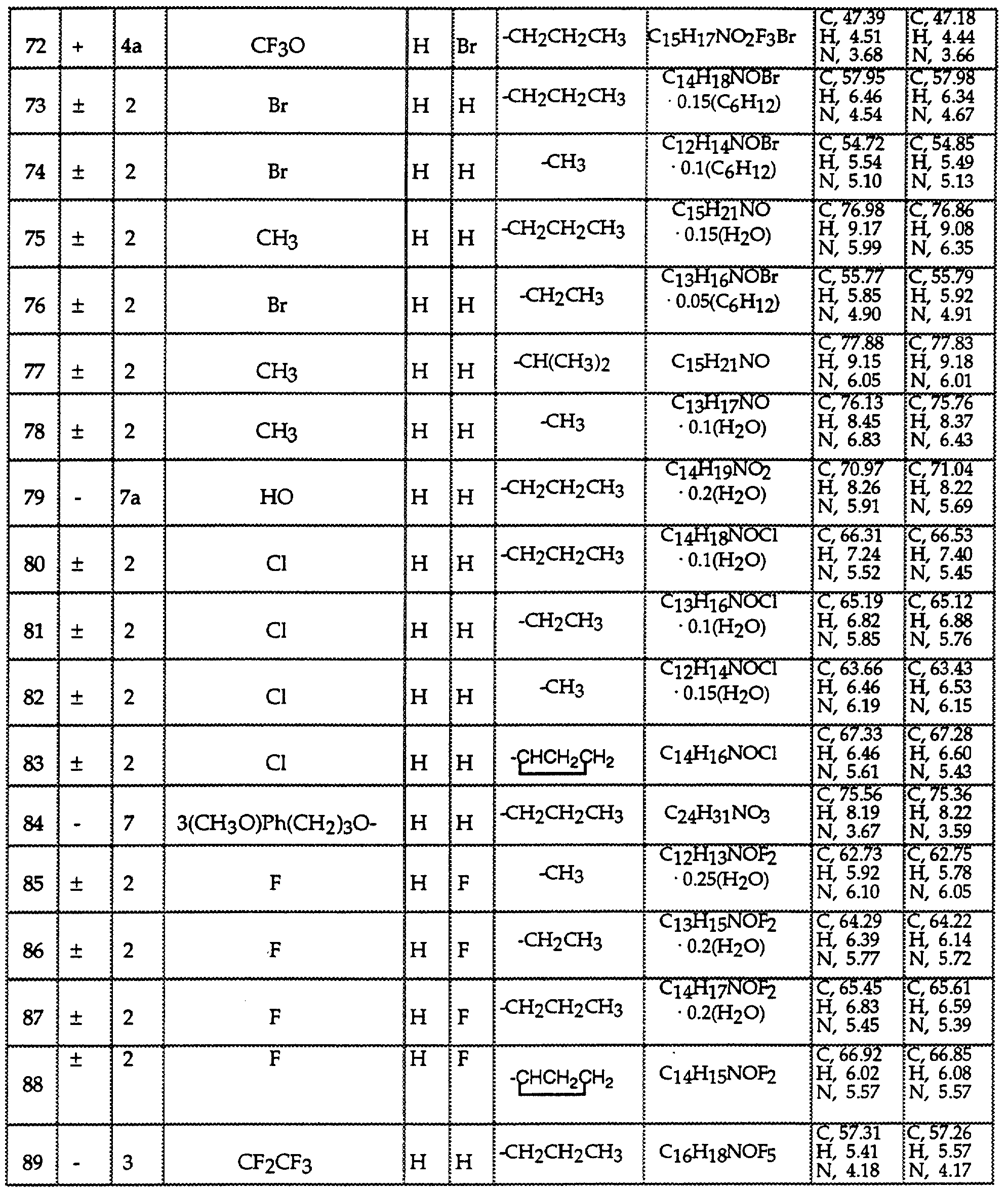
Способы получения 7,8 (см. схему 6).
В соединениях формулы I, полученных в соответствии со способами получения 7 и 8, G - метилен, X - OR5, R1 и R - водород, а Y, Z, R2 и R5 имеют вышеуказанные значения.
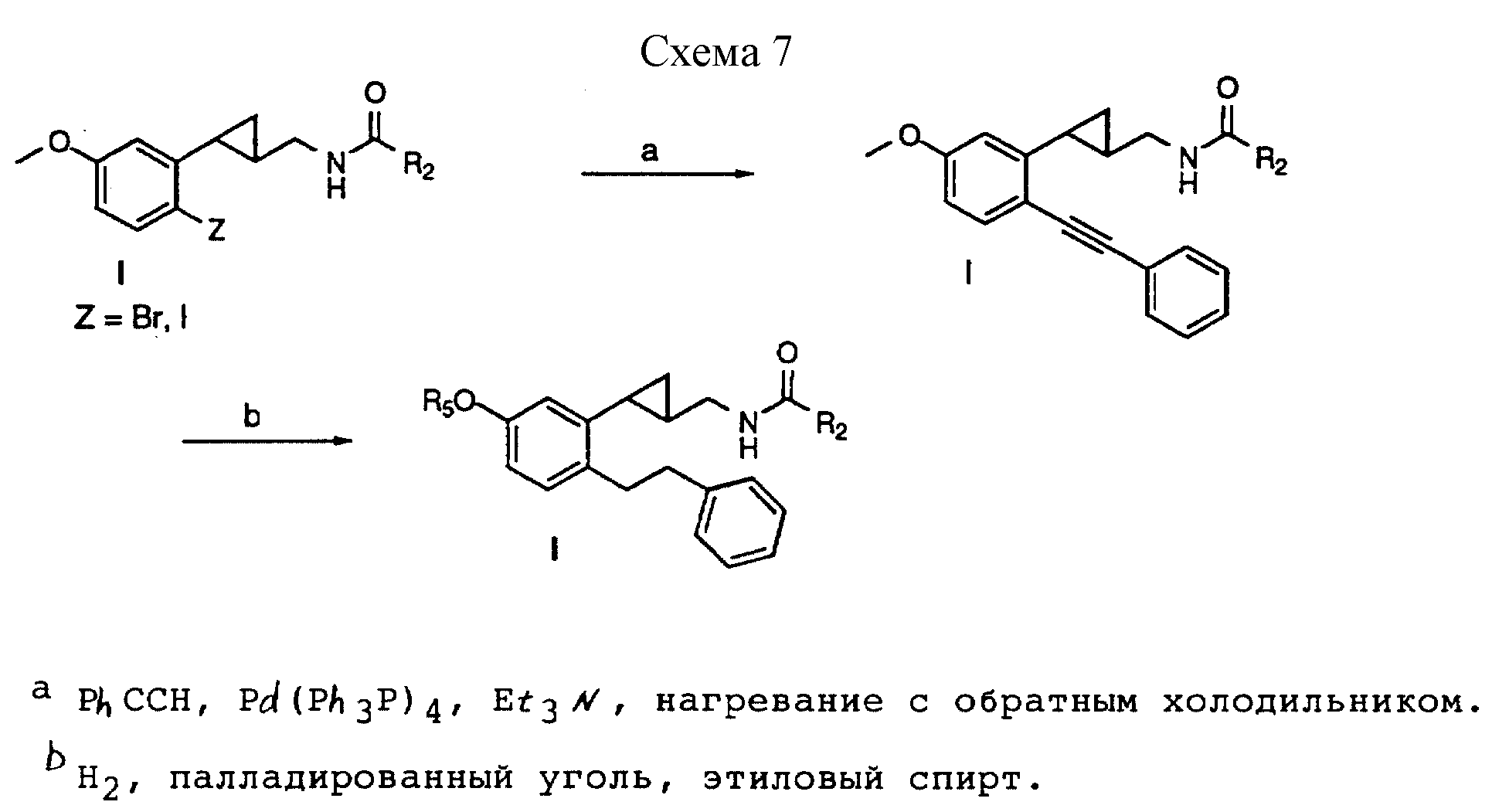
Способ получения 9 (см. схему 7).
В соединениях формулы I, полученных в соответствии со способом получения 9, X - OCH3, G - метилен, Z - фенилалкил или гидроциннамил, a R1, R и Y - водород.
Способы получения 10, 11 (см. схему 8).
В соединениях формулы I, полученных в соответствии со способом получения 10, X - OCH3, G - метилен, Z - циано, a R1, R и Y - водород. В соединениях формулы I, полученных в соответствии со способом получения 11, X - OCH3, G - метилен, Z - фенил, a R1, R и Y - водород.
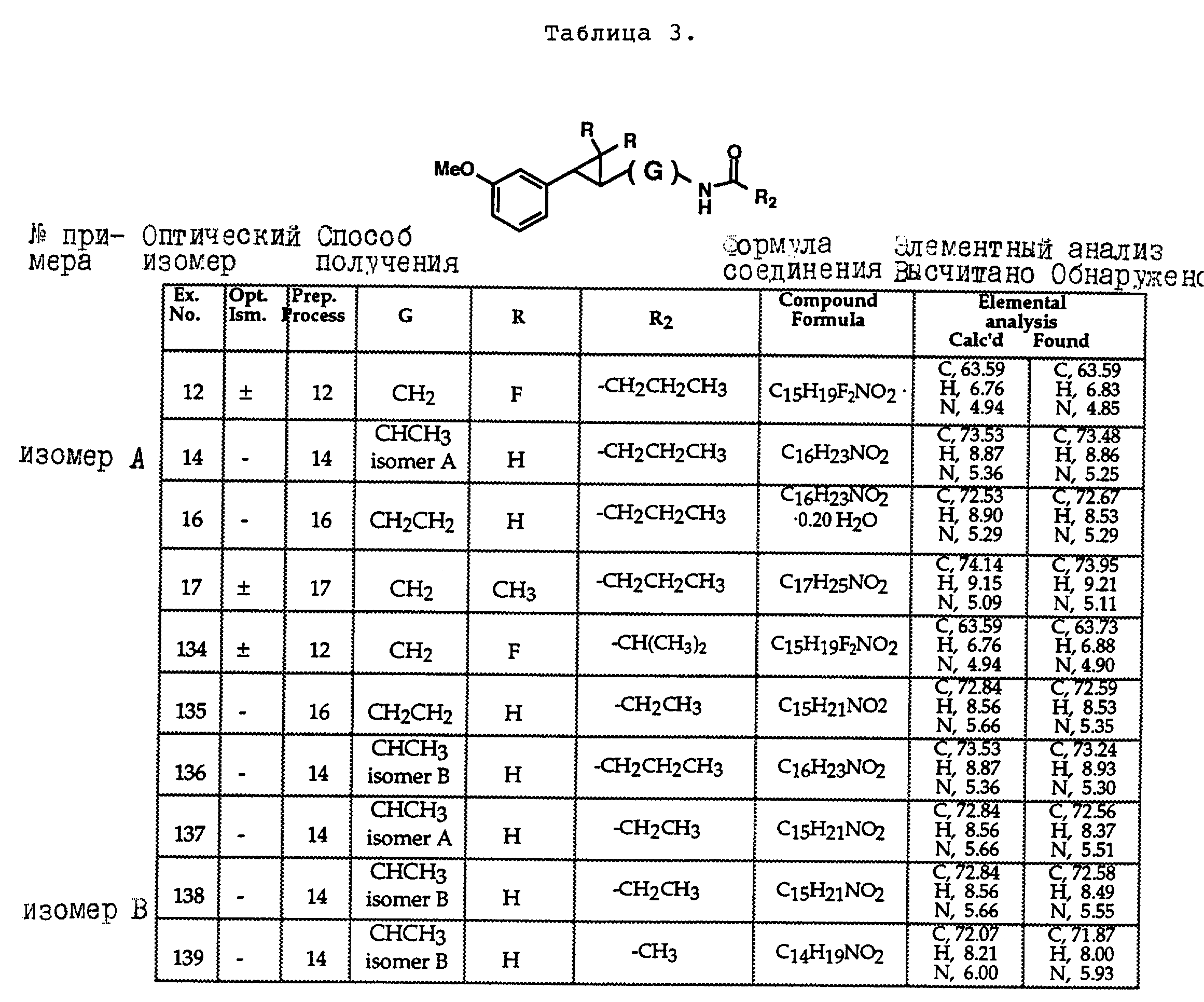
Способ получения 12 (см. схему 9).
В соединениях формулы I, полученных в соответствии со способом получения 12, X - OCH3, R - фтор, G - метилен, a Z, R1 и Y - водород.
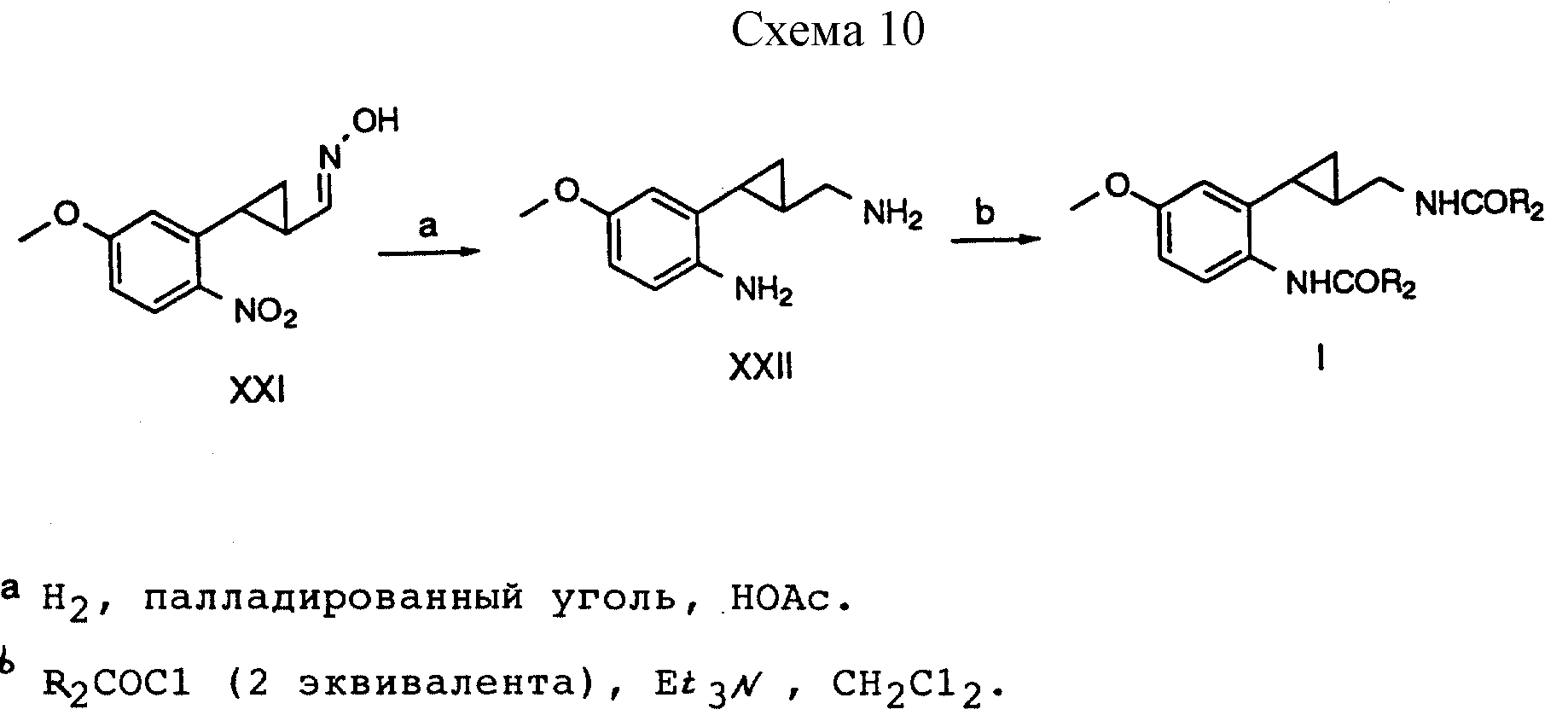
Способ получения 13 (см. схему 10).
В соединениях формулы I, полученных в соответствии со способом получения 13, X - OCH3, Z - алкамидо, G - метилен, a R, R1 и Y водород.
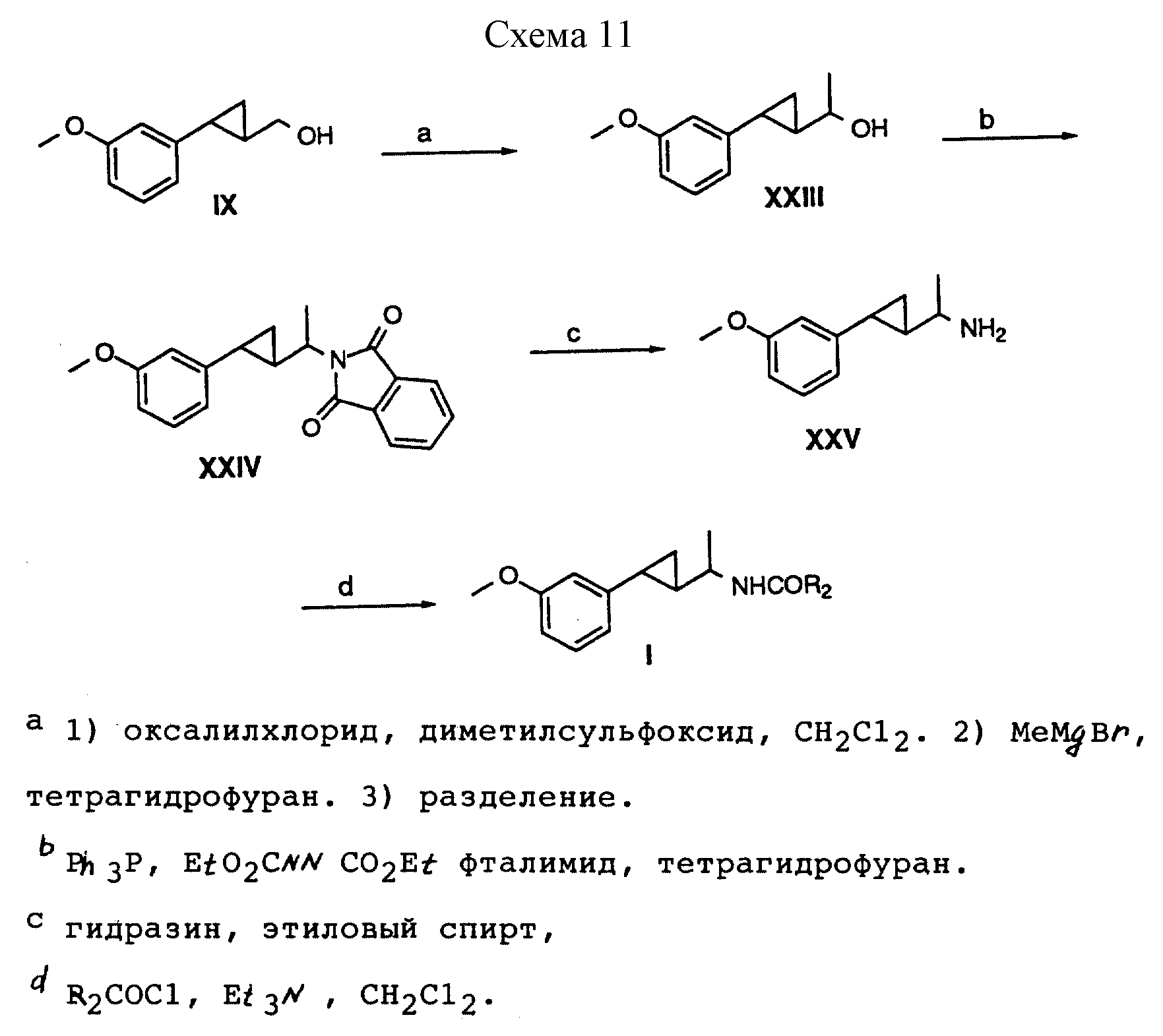
Способ получения 14 (см. схему 11).
В соединениях формулы I, полученных в соответствии со способом получения 14, X - OCH3, G - алкметилен, a R, R1, Z и Y - водород.
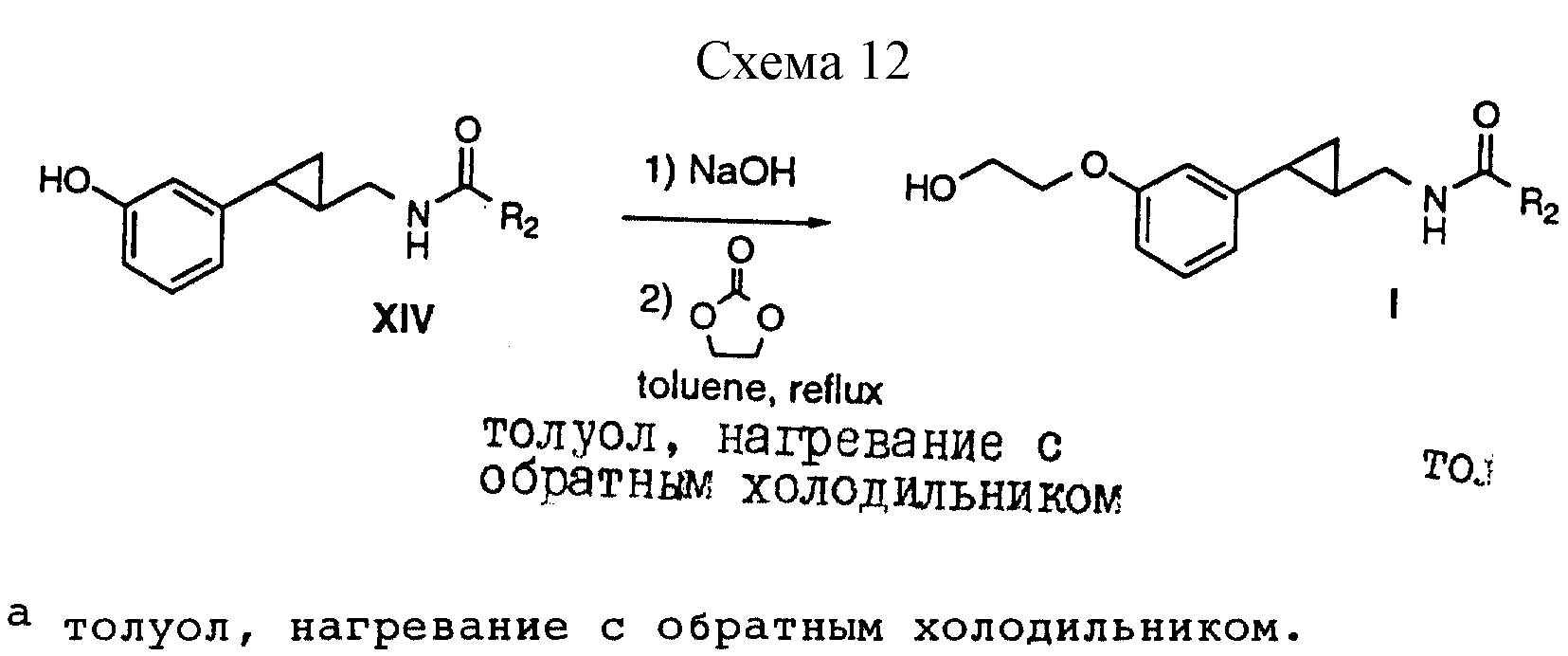
Способ получения 15 (см. схему 12).
В соединениях формулы I, полученных в соответствии со способом получения 15, X - 2-гидроксиэтил, G - метилен, a R, R1, Z и Y - водород.
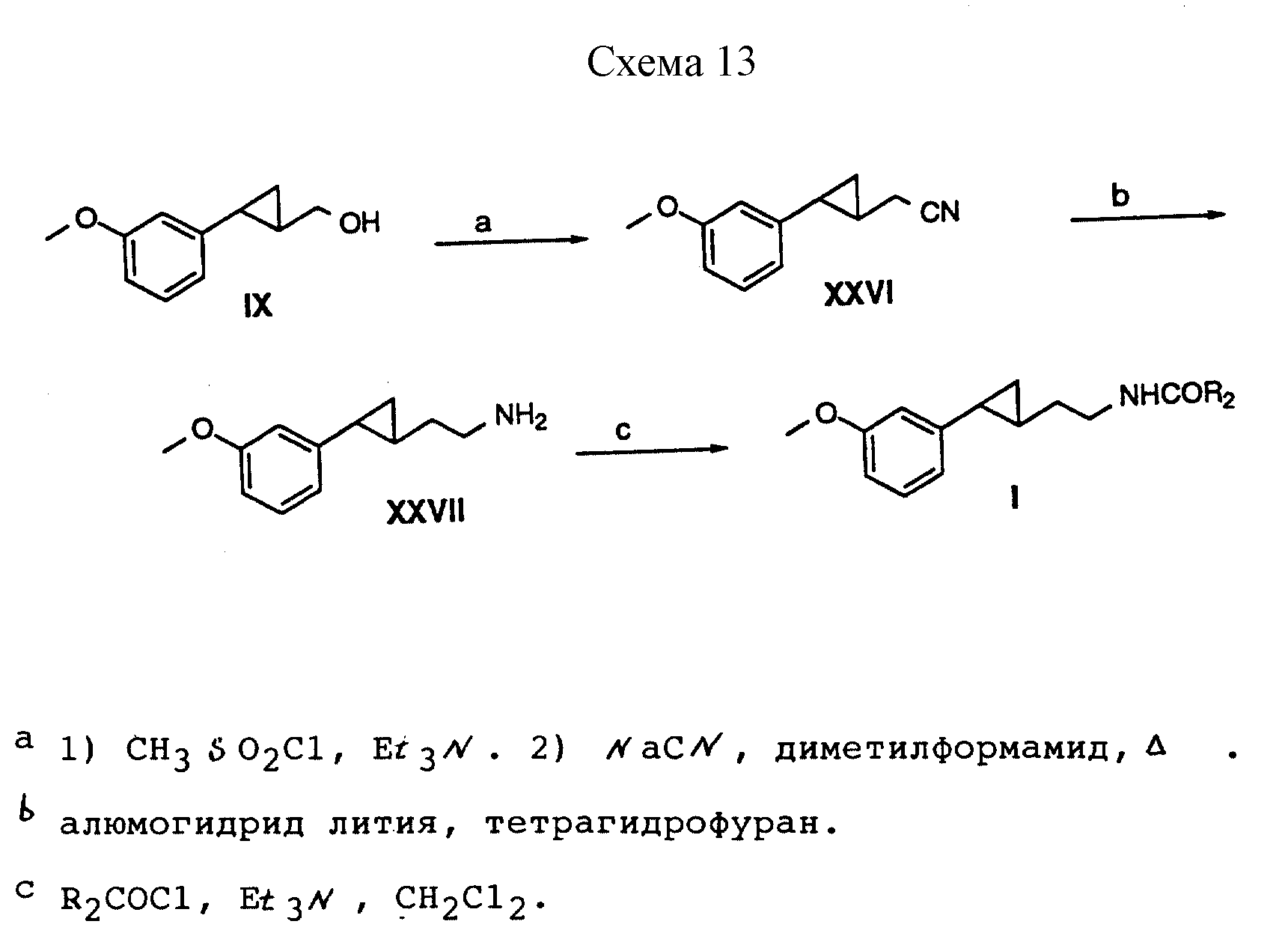
Способ получения 16 (см. схему 13).
В соединениях формулы I, полученных в соответствии со способом получения 16, X - OCH3, G - этилен, a R, R1, Z и Y - водород.
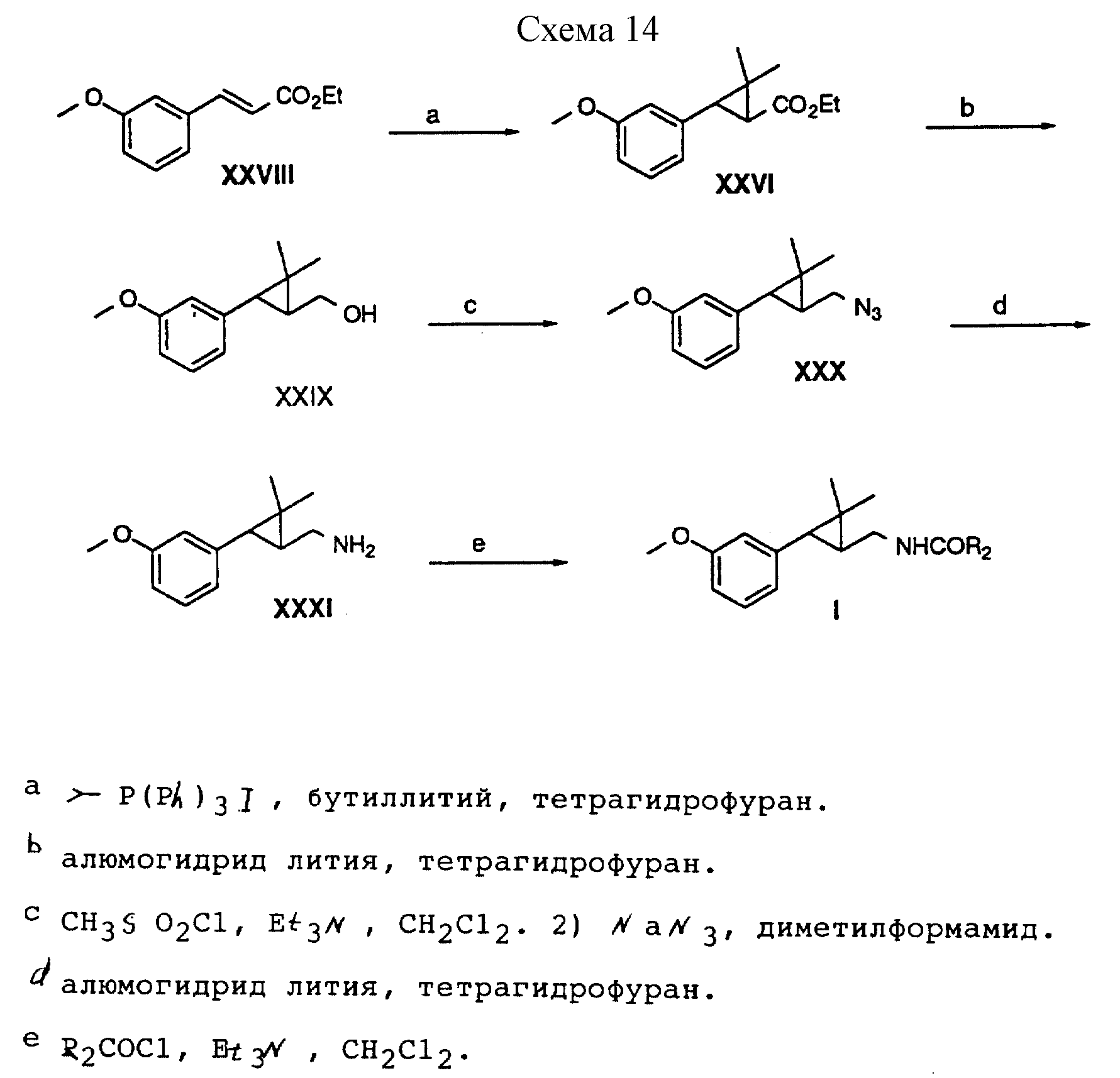
Способ получения 17 (см. схему 14).
В соединениях формулы 1, полученных в соответствии со способом получения 17, X - OCH3, G - метилен, R - метил, а R1, Z, и Y - водород.
Получение соединений формулы II.
Соединения формулы II получали по методу, описанному в способе получения 18. Металлорганическое соединение (1), например реактив Гриньяра или литиевый реагент, конденсировали с соответствующим циклоалкеноном (2) в присутствии соли меди (1), такой как CuBr, CuI, CuBr·Me2S или подобной соли, с образованием 3-арил-циклоалканона (3). Альтернативно соединение 1 может быть конденсировано с 3-алкокси-циклоалканоном (4), в результате чего после ацетализации, каталитической гидрогенизации и удаления защитной группы образуется соединение 3. Приемлемыми катализаторами для этого являются палладированный уголь и подобные катализаторы. Циклоалканон, представляющий собой соединение 3, можно превратить в соединения формулы I с помощью стандартных способов, таких как конденсация с гидроксиамином, с последующей каталитической гидрогенизацией и ацилированием. Приемлемыми катализаторами для этого являются скелетный никелевый катализатор гидрирования, палладированный уголь и подобные катализаторы. Приемлемыми ацилирующими реагентами являются галогениды карбоновой кислоты, ангидриды, ацилимидазолы, алкилизоцианаты и карбоновые кислоты в присутствии конденсирующих агентов, таких как карбонилимидазол, карбодиимиды и другие.
Способ получения 18 (см. схему 15).
Если специально не указано, все заместители в соединениях формулы I, полученных в соответствии со способами получения 1-17, могут иметь любые значения, приведенные выше для формулы I.
Получение соединений формулы I включает следующие стадии, описываемые ниже для каждого из способов 1-17.
Способ получения 1 (реакции a-d).
(i) В результате обработки выпускаемых промышленностью альдегидов формулы II гидридом натрия и цианометилфосфонатом с последующим циклопропанированием с использованием метилида диметилоксосульфония были получены промежуточные циклопропаннитрилы формулы III.
(ii) В результате каталитического восстановления нитрилов формулы III водородом в присутствии оксида платины с последующим ацилированием полученного амина соответствующим ацилхлоридом были получены соединения формулы I, в которой R1 является водородом.
Способ получения 2 (реакции a-g).
(i) В результате обработки выпускаемых промышленностью коричных кислот формулы IV тионилхлоридом, нагреваемым с обратным холодильником, с последующим ацилированием хлористоводородным N,O-диметилгидроксиламином в присутствии пиридина были получены ненасыщенные N-метил, N-метоксиамиды, которые циклопропанировали с использованием метилида диметилоксофосфония, в результате чего были получены промежуточные соединения формулы V.
(ii) В результате восстановления амидов формулы V с помощью алюмогидрида лития были получены альдегиды формулы VI.
(iii) Альдегиды формулы VI превращали в оксимы с помощью хлористоводородного гидроксиламина и гидроксида натрия и восстанавливали гидридом литийаммония, в результате чего были получены амины формулы VII.
(iv) Соединения формулы VII ацилировали с помощью соответствующих ацилирующих агентов, таких как ацилхлориды, карбамоилхлориды или изоцианаты, с образованием соединений формулы I, в которой R1 - водород, а R2 - алкил, NR3R4 или NHR3, как указывалось выше.
Способ получения 3 (реакции a-e).
(i) В результате восстановления известных гомохиральных сультамов формулы VIII алюмогидридом лития были получены спирты формулы IX в виде отдельных энантиомеров.
(ii) В результате превращения спиртов формулы IX в соответствующие сложные эфиры метансульфоновой кислоты с помощью метансульфонилхлорида и триэтиламина с последующим образованием азида при использовании азида натрия в диметилформамиде и восстановления алюмогидридом лития были получены энантиочистые амины формулы X.
(iii) Амины формулы X ацилировали соответствующими ацилхлоридами с образованием соединений формулы I, в которой R1 - водород, Y и Z - водород.
Способ получения 4.
Амиды формулы XI, полученные в соответствии со способами получения I, II или III, обрабатывали ацетатом таллия (III) и 1 эквивалентом брома или иода с образованием соединений формулы I, в которой Y - водород, Z - бром или иод, а R1 - водород.
Способ получения 5.
Амиды формулы XI, полученные в соответствии со способами получения I, II или III, обрабатывали ацетатом таллия (III) и 2 или более эквивалентами брома или иода с образованием соединений формулы I, в которой Y и Z - бром или иод, а R1 - водород.
Способ получения 6.
Амиды формулы XII, полученные в соответствии со способами получения I, II или III, обрабатывали гидридом натрия и соответствующим алкил- или бензилгалогенидом с образованием соединений формулы I, в которой R1 является C1-4алкилом или бензилом.
Способ получения 7.
(i) Амиды формулы XIII, полученные в соответствии со способами получения I, II или III, обрабатывали трибромидом бора с образованием соединений формулы XIV.
(ii) Амиды
формулы XIV обрабатывали основанием, а полученные алкоксиды алкилировали соответствующими алкилиодидами или алкилбромидами с образованием соединений формулы I, в
которой X - OR5 и
R1 - водород.
Способ получения 8.
(i) Амиды формулы XIII, полученные в соответствии со способами получения I, II или III, обрабатывали трибромидом бора с образованием соединений формулы XIV.
(ii) Амиды формулы XIV добавляли к полученному раствору пригодного спирта, трифенилфосфина и диэтилазодикарбоксилата с образованием соединений формулы I, в которой X - OR5 и R1 - водород.
Способ получения 9.
(i) Амиды формулы I, в которой Z является Br или I, полученные в соответствии со способом получения IV, соединяли с фенилацетиленом под слоем тетракис(трифенилфосфин) палладия (0) с образованием соединений формулы I, в которой Z является фенилацетиленом.
(ii) Амиды формулы I, полученные выше, гидрировали над катализатором в виде палладированного угля с образованием соединений формулы I, в которой Z является гидроциннамилом.
Способ получения 10.
(i) Амиды формулы I, в которой Z является иодом, полученные в соответствии со способом получения IV, нагревали с обратным холодильником в пиридине в присутствии цианида меди (I) с образованием соединений формулы I, в которой Z является циано.
Способ получения 11.
(i) Амиды формулы I, в которой Z является иодом, полученные в соответствии со способом получения IV, подвергали взаимодействию с осуществлением реакции сочетания по методу Мицонобу в присутствии водного раствора гидроксида бария, фенилборной кислоты, тетракис(трифенилфосфин) палладия (0) и диметоксиэтана с образованием соединений формулы I, в которой Z является фенилом.
Способ получения 12 (реакции a-h).
(i) В результате восстановления натриевым боргидридом 3-метоксикоричной кислоты (XV) с последующей обработкой иодом и хлористоводородной кислотой был получен аллиловый спирт формулы XVI.
(ii) Соединения формулы XVI ацилировали уксусным ангидридом в пиридине с образованием аллилацетата формулы XVII.
(iii) Аллилацетат формулы XVII циклопропанировали путем обработки хлордифторацетатом натрия в диглиме, нагреваемом с обратным холодильником, в результате чего был получен дифторциклопропан формулы XVIII.
(iv) Дифторциклопропан формулы XVIII обрабатывали гидроксидом калия в метаноле с образованием соответствующего спирта формулы XIX.
(v) Спирт формулы XIX превращали в мезилат путем обработки метансульфонилхлоридом в дихлорметане в присутствии триэтиламина. Мезилат вываривали вместе с диметилформамидом и обрабатывали азидом натрия с образованием соответствующего азида. Этот азид восстанавливали с помощью алюмогидрида лития с образованием амина формулы XX.
(vi) Амин формулы XX превращали в амиды формулы I, в которой R является фтором, путем ацилирования ацилхлоридом в присутствии триэтиламина.
Способ получения 13 (реакции a, b).
(i) Оксим формулы XXI, полученный в соответствии со способом получения II, превращали в диамин формулы XXII путем каталитической гидрогенизации на палладированном угле в уксусной кислоте.
(ii) Диамин формулы XXII диацилировали путем обработки двумя эквивалентами ацилхлорида в присутствии триэтиламина с образованием амидов формулы I, в которой Z является алкамидо.
Способ получения 14 (реакции a-d).
(i) Спирт формулы IX, полученный в соответствии со способом получения III, окисляли по методу Сверна, в результате чего был получен карбоксальдегид, который обрабатывали бромидом метилмагния с образованием смеси диастереометов. Эпимеры отделяли посредством хроматографии, что позволило получить чистые спирты формулы XXIII.
(ii) Спирт формулы XXIII превращали в фталимид формулы XXIV путем обработки фталимидом, диэтилазодикарбоксилатом и трифенилфосфином.
(iii) Фталимид формулы XXIV обрабатывали гидразином в этаноле с образованием амина формулы XXV.
(iv) В результате ацилирования амина формулы XXV ацилхлоридом в присутствии триэтиламина были получены соединения формулы I, в которой 6 является алкметиленом.
Способ получения 15.
(i) Фенол формулы XIV, полученный в соответствии со способом получения VII, превращали в соль натрия путем обработки гидроксидом натрия и нагревания с обратным холодильником вместе с этиленкарбонатом в толуоле, что позволило получить амиды формулы I, в которой R5 является 2-гидроксиэтилом.
Способ получения 16 (реакции a-c).
(i) Спирт формулы IX, полученный в соответствии со способом получения III, обрабатывали метансульфонилхлоридом в присутствии триэтиламина, в результате чего был получен мезилат, который затем превращали в нитрил формулы XXVI путем обработки цианидом натрия в диметилформамиде.
(ii) Нитрил формулы XXVI восстанавливали алюмогидридом лития в тетрагидрофуране с образованием амина формулы XXVI.
(iii) Амин формулы XXVII ацилировали хлорангидридом в присутствии триэтиламина с образованием амидов формулы I, в которой G является этиленом.
Способ получения 17 (реакции a-e).
(i) Выпускаемый промышленностью сложный этиловый эфир 3-метоксикоричной кислоты циклопропанировали с помощью илида, полученного из иодида изопропилтрифенилфосфония и бутиллития с образованием циклопропана формулы XXVI.
(ii) Циклопропан формулы XXVI превращали в спирт формулы XXIX путем восстановления алюмогидридом лития в тетрагидрофуране.
(iii) Спирт формулы XXIX обрабатывали метансульфонилхлоридом и триэтиламином в дихлорметане с образованием мезилата, который затем превращали в азид формулы XXX путем обработки азидом натрия в диметилформамиде.
(iv) Азид формулы XXX восстанавливали алюмогидридом лития в тетрагидрофуране с образованием амина формулы XXXI.
(v) Амин формулы XXXI ацилировали ацилгалогенидом и триэтиламином с образованием амидов формулы I, в которой R является метилом.
Реагенты, растворители и условия реакции, используемые при выполнении вышеописанных подготовительных стадий, хорошо известны специалистам в области органического синтеза. Все стадии представляют собой обычные органические реакции, многократно описанные в научной литературе.
Эти подготовительные методы могут быть изменены с целью получения других соединений, входящих в объем настоящего изобретения, но отдельно не рассматриваемых.
Кроме того, соединения формулы I включают все фармацевтически приемлемые сольваты, причем предпочтительными сольватами являются гидраты. В объем настоящего изобретения входят также геометрические и оптические изомеры, например смеси энантиомеров, отдельные энантиомеры и диастереомеры, которые образуются в результате структурной асимметрии определенных соединений данного ряда. Предпочтение, как правило, отдается транс-циклопропановым стереоизомерам. Разделение или стереоспецифический синтез отдельных изомеров производится с помощью различных методов, которые хорошо известны специалистам в этой области.
Это изобретение далее включает меченные радиоактивным изотопом соединения, рассмотренные выше, в частности соединения, меченные радиоактивным иодом, например I1232, I123, I125 или I131; такие меченные радиоактивным изотопом соединения используются в качестве определенных высокоаффинных рецепторов и, таким образом, могут применяться в анализах связывания рецепторов, авторадиографических исследованиях и в других in vitro и in vivo биологических тестах, которые выполняются для фармакологического изучения новых средств, стимулирующих синтез мелатонина.
В приведенном ниже разделе "Описание предпочтительных вариантов осуществления настоящего изобретения" дается более подробное описание синтеза соединений формулы I и промежуточных соединений формул II-XXXI.
Соединения по настоящему изобретению характеризуются сродством с рецепторами эндогенного гормона шишковидного тела, мелатонином, как было установлено в результате анализа связывания рецепторов, и являются его агонистами, о чем свидетельствуют результаты функционального анализа. Ниже описываются биологические тесты.
Как указывалось выше, мелатонин имеет непосредственное отношение к регулированию разных биологических ритмов и оказывает биологическое действие в результате взаимодействия с определенными рецепторами. Известно, что введение агонистов мелатонина является весьма полезным при лечении связанных с ним различных заболеваний. Такими заболеваниями являются депрессия, нарушение суточного ритма организма, синдром смещения времени наибольшей работоспособности, нарушения сна, глаукома, некоторые половые расстройства, рак, нарушения иммунной и нейроэндокринной систем.
Систематическое введение соединений формулы I может производиться по схеме лекарственного лечения самим мелатонином. Дозировка и схема лекарственного лечения определяются лечащим врачом с учетом таких факторов, как возраст, пол и физическое состояние больного, способ введения и характер заболевания. Можно использовать пероральный, чрескожный, подкожный, внутривенный, внутримышечный, ректальный, транс-буккальный, назальный и глазной способы введения.
Одно или несколько соединений по настоящему изобретению смешивали с фармацевтически приемлемыми количествами одного или нескольких известных фармацевтических наполнителей, в результате чего получали композицию, предназначенную для введения желаемым способом. Как правило, такие составы содержат один или несколько носителей или разбавителей. Приемлемыми носителями являются твердые, полутвердые и жидкие вещества, которые способны смешиваться или совместимы с активным агентом (агентами).
Подходящими носителями являются лактоза, декстроза, сахароза, сорбит, маннит, крахмалы, аравийская камедь, фосфат кальция, альгиниты, трагант, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, целлюлоза, вода, сироп, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния, минеральное масло и другие. Возможно применение смесей.
Другими полезными наполнителями являются смазки, увлажнители, гелеобразующие вещества, эмульгаторы, антикоагулянты, красители, ароматизаторы, высушивающие вещества и другие. Возможно применение смесей.
Как правило, композиции, включающие соединения по настоящему изобретению, содержат 0,10-10% активного вещества и 99,90-90% или другое приемлемое количество наполнителя.
Дозировка определяется лечащим врачом с учетом состояния больного. Однако суточные дозы от 0,1 мг до 100 мг оказываются полезными для лечения нарушений сна, циркадного ритма и других заболеваний.
Способы лечения с использованием соединений по настоящему изобретению включают стадию (стадии) введения одной или нескольких доз указанного соединения больному, предпочтительно млекопитающему, например человеку, нуждающемуся в таком лечении.
Соединения по настоящему изобретению, способы их получения и биологическое действие станут понятнее после рассмотрения следующих примеров, которые приводятся для иллюстрации и никоим образом не ограничивают объем настоящего изобретения.
В следующих примерах температуры выражены в градусах шкалы Цельсия (oC), температуры плавления не скорректированы. Спектральные характеристики протонного ядерного магнитного резонанса (ЯМР) относятся к химическим сдвигам (δ) выраженным в величинах м.д., по сравнению с тетраметилсиланом, используемым в качестве эталона. Относительная площадь, указанная для сигналов ЯМР при различных химических сдвигах, соответствует числу атомов водорода определенного типа в молекуле.
Мультиплетность сигналов представлена как широкий синглет, синглет, триплет или мультиплет. Спектры ЯМР получены при использовании растворов соединений в дейтеро-диметилсульфоксиде (DMSO6) или дейтеро-хлороформе (CDCl3). Описания инфракрасных спектров включают только числа абсорбционных волн (см-1), позволяющие идентифицировать функциональные группы; определения инфракрасного спектра производились с использованием в качестве разбавителя бромистого калия (KBr). Результаты элементных анализов даны в весовых процентах.
Ниже приводятся примеры, подробно описывающие получение типичных соединений формулы I и синтезированных промежуточных соединений. Специалистам в этой области хорошо известно, что в результате замены используемых веществ и способов можно получить другие соединения, описываемые в настоящем изобретении. Приведенное выше описание изобретения и нижеследующие примеры позволяют полностью осуществить настоящее изобретение.
Примеры.
В примерах 1-139 описывается получение и отличительные особенности соединений формулы I. В примерах 140-144 представлена аналогичная информация для соединений формулы II.
Пример 1 (способ получения 1).
Получение (транс)-N-[[2-(2-фтор-5-метоксифенил)-циклопропил] метил]-2-метилпропанамида.
Стадия i.
(a) (транс)-3-(2-фтор-5-метоксифенил)-2-пропеннитрил.
К суспензии NaH (6,90 г, 60% дисперсия в минеральном масле, 173 ммоля) в тетрагидрофуране (500 мл), находившейся при
0oC,
по каплям добавляли диэтилцианометилфосфонат (30,10 г, 170 ммолей). Вслед за этим по каплям добавляли раствор 2-фтор-5-метоксибензальдегида (24,5 г, 159 ммолей) в тетрагидрофуране (50
мл). Полученную
суспензию оставляли для нагревания до комнатной температуры. Через 18 часов добавляли воду (200 мл) и экстрагировали полученный раствор с помощью EtOAc. Органические фракции соединяли,
промывали
рассолом, сушили над MgSO4 и концентрировали с образованием белого воскообразного твердого вещества. Это вещество очищали путем перегонки по методу Кюгельрора, что позволило
получить
указанное в заголовке соединение, 24,3 г (86%), температура плавления 56-57oC;
спектр ЯМР1H (300 МГц, CDCl3) δ : 3.79 (с,3H), 5.98 (д, J =
16.1 Гц,
1H), 6.85-6.90 (м,1H), 6.91-6.93 (м,1H), 6.99 (т,J= 6.5 Гц,1H), 7.43 (д, J = 16.1 Гц,1H);
масс-спектр (изобутан-DCl) m/e 177.
Результаты анализа, высчитанные для
C10H8NOF:
C 67.79, H 4.55, N 7.91;
обнаружено: C 67.41, H 4.44, N 7.86.
(b) (транс)-2-(2-фтор-5-метоксифенил)циклопропанкарбонитрил.
К суспензии NaH (1,73 г, 71 ммоля) в диметилсульфоксиде (40 мл) небольшими порциями добавляли твердый йодистый триметилсульфоксоний (15,9 г, 72 ммоля). После прекращения вспенивания (40
минут) по
каплям добавляли раствор (транс)-3-(2-фтор-5-метоксифенил)-2-пропеннитрила (4,26 г, 24 ммоля) в диметилсульфоксиде (10 мл), поддерживая температуру в интервале 35 -40oC. Эту смесь
перемешивали в течение 18 часов при комнатной температуре, после чего по каплям добавляли насыщенный раствор NH4Cl (100 мл) и экстрагировали этилацетатом. Органические фракции собирали,
промывали рассолом, сушили (K2CO3) и концентрировали с образованием красного масла, которое очищали посредством хроматографии на колонке из силикагеля (смесь CH2
Cl2 и гексана в соотношении 60: 40), в результате чего было получено указанное в заголовке соединение в виде светлого масла (46%);
спектр ЯМР1H (300 МГц, CDCl3)
δ : 0.82-0.88 (м,2H), 1.65-1.75 (м,1H), 2.55-2.62 (м,1H), 3.75 (с,3H), 6.50-6.55 (м,1H), 6.60- 6.70 (м,1H), 6.92 (т, J=6.5 Гц,1H).
Стадия ii.
(c) (транс)-2-(2-фтор-5-метоксифенил)циклопропанметанамин.
Суспензию (транс)-2-(2-фтор-5-метоксифенил)циклопропанкарбонитрила (5,0 г, 26 ммолей), PtO2 (200 мг) и CHCl3 (10 мл) в этиловом спирте (65 мл) гидрировали в аппарате Парра в течение 3 часов при давлении 3,85 ат. Катализатор фильтровали через пробку из целита, а растворители удаляли. Полученную
хлористоводородную соль распределяли между CH2Cl2 и 10% K2CO3. Органический слой отделяли, сушили над K2CO3 и концентрировали с
образованием указанного в заголовке соединения в виде свободного амина, 3,8 г (74%);
спектр ЯМР1H (250 МГц, CDCl3) δ : 0.79-0.82 (м,1H), 0.90-0.95 (м,1H), 1.49
(широкий синглет,2H), 1.70-1.76 (м,1H), 1.82-1.86 (м,1H), 2.60-2.74 (м, 2H), 3.75 (с,3H), 6.35-6.42 (м,1H), 6.50-6.62(м,1H), 6.85-6.90 (м,1H).
(d) (транс)-N-[[2-(2-фтор-5-метоксифенил)циклопропил] - метил] -2-метилпpопанамид.
К перемешиваемому с помощью магнитной мешалки раствору (транс)-2-(2-фтор-5-метоксифенил)
циклопропанметанамида (600 мг, 3,1 ммоля), Et3N (909 мг, 9,0 ммолей) в сухом дихлорметане (15 мл), находившемуся при 0oC, по каплям добавляли изобутирилхлорид (352 мг, 3,3
ммоля). Полученную суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 18 часов. Растворители удаляли, а остаток растворяли в этилацетате (100 мл) и последовательно
промывали водой, 5% лимонной кислотой, 5% K2CO3, рассолом и сушили над K2CO3. После концентрирования посредством ротационного выпаривания было получено
неочищенное масло, которое затем очищали путем перегонки по методу Кюгельрора. Перекристаллизация полученного твердого вещества из смеси простого этилового эфира и гексана (1:1) позволила получить
380
мг (47%) указанного в заголовке соединения; температура плавления 93-94oC;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.82-0.88 (м,1H), 0.97-1.03 (м,1H),
1.13
(д, J= 8.1 Гц, 6H), 1.14-1.24 (м,1H), 1.85-1.91 (м,1H), 2.25-2.30 (м, 1H), 2.93-3.02 (м,1H), 3.52 (двойной триплет, J=7.8, 13.8 Гц,1H), 3.72 (c, 3H), 5.74 (широкий синглет,1H), 6.42-6.44 (м,1H),
6.59-6.64 (м,1H), 6.91 (т, J=6.5 Гц,1H);
инфракрасный спектр (пленка NaCl) 3298, 2962, 1642, 1546, 1502, 1428 см-1;
масс-спектр (изобутан-DCl) m/e 253.
Результаты анализа, высчитанные для C15H20NO2F:
C 67.90, H 7.60, N 5.28;
обнаружено: C 68.18, H 7.77, N 5.24.
Пример 2 (способ получения 2).
Получение (транс)-N-[[2-(3-метоксифенил)циклопропил]метил] бутанамида.
Стадия i.
(a) (b) (транс)-N-метокси-N-метил-3-(3-метоксифенил)- 2-пропенамид.
Раствор 3-метоксикоричной кислоты (75,0 г, 0,42 моля) в тионилхлориде (250 мл) нагревали с обратным холодильником в
течение
1 часа. Большую часть тионилхлорида отгоняли, полученный остаток обрабатывали дихлорметаном, а оставшийся тионилхлорид удаляли путем совместной перегонки с образованием хлорангидрида
3-метоксикроичной
кислоты, который имел достаточно высокую чистоту для использования без дальнейшей очистки. К раствору хлорангидрида 3-метоксикоричной кислоты (0,42 моля) в сухом дихлорметане (500
мл), находившемуся
при 0oC, добавляли пиридин (186 мл, 2,3 моля) и хлористоводородный N, O- диметилгидроксиламин (45,17 г, 0,46 моля). Полученный раствор перемешивали в течение 18 часов
при комнатной
температуре, разбавляли дихлорметаном (200 мл) и последовательно промывали 1 н. раствором HCl, насыщенным раствором бикарбоната натрия и рассолом. Затем его сушили над MgSO4
и
концентрировали в условиях вакуума с образованием указанного в заголовке соединения в виде темного масла (88,43 г, 95%);
спектр ЯМР1H (300 МГц, CDCl3) δ :
3.25
(с,3H), 3.68 (с,3H), 3.84 (с, 3H), 6.82-6.86 (м, 1H), 7.00 (д, J=16.1 Гц,1H), 7.10 (широкий синглет,1H), 7.15-7.20 (м,1H), 7.32 (т, J =6.6 Гц,1H), 7.72 (д, J =16.0 Гц,1H).
(c) (транс)-N-метокси-N-метил-2-(3-метоксифенил)- циклопропанкарбоксиамид.
К суспензии NaH (16,27 г, 0,68 моля) в диметилформамиде (375 мл) небольшими порциями добавляли твердый йодистый
триметилсульфоксоний (149,16 г, 0,68 моля). После прекращения вспенивания (40 минут) по каплям добавляли раствор (транс)-N- метокси-N-метил-3-(3-метоксифенил)-2-пропенамида (50 г, 0,23 моля) в
диметилсульфоксиде (50 мл), поддерживая температуру в интервале 35-40oC. Эту смесь перемешивали в течение 18 часов при комнатной температуре, после чего по каплям добавляли насыщенный
раствор NH4Cl (400 мл) и экстрагировали этилацетатом. Органические вещества собирали, промывали рассолом, сушили (K2CO3) и концентрировали с образованием красного
масла, которое затем очищали путем перегонки по методу Кюгельрора (130oC, 0,5 мм Hg), в результате чего было получено указанное в заголовке соединение в виде белого воска (52,47 г, 100%);
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.82-0.88 (м,1H), 1.20-1.30 (м,1H), 1.59-1.62 (м, 1H), 2.40-2.45 (м,1H), 3.22 (с,3H), 3.68 (c,3H), 3.79 (c,3H), 6.61-6.72 (м,
3H), 7.18 (т, J=6.0 Гц,1H).
Стадия ii.
(d) (транс)-2-(3-метоксифенил)циклопропанкарбоксальдегид.
К быстро перемешиваемой суспензии алюмогидрида
лития
(7,74 г, 204 ммоля) в тетрагидрофуране (800 мл), находившейся при температуре -45oC, по каплям добавляли раствор (транс)-N-метокси-N-метил-2-(3-метоксифенил) циклопропанкарбоксамида
(40 г,
171 ммоля) в тетрагидрофуране (100 мл), поддерживая температуру ниже -40oC. После окончания добавления охлаждающую ванну удаляли, а реакционную смесь оставляли для нагревания до
5oC, а потом сразу же вновь охлаждали до -45oC. По каплям осторожно добавляли раствор бисульфата калия (40 г, 300 ммолей) в воде (120 мл), поддерживая температуру ниже -30o
C. После окончания добавления охлаждающую ванну удаляли, а суспензию перемешивали при комнатной температуре в течение 30 минут. Полученную смесь фильтровали через целит и фильтровальную
лепешку
промывали простым эфиром. Соединенные фильтраты промывали холодным 1 н. раствором HCl, 5% K2CO3, рассолом и сушили над MgSO4. После концентрирования
посредством
ротационного выпаривания было получено указанное а заголовке соединение в виде светлого масла (29,9 г, 99%);
спектр ЯМР1H (300 МГц, CDCl3) δ :
1.49-1.58 (м,1H),
1.65-1.73 (м,1H), 2.10-2.19 (м,1H), 2.58-2.67 (м,1H), 3.80 (с,3H), 6.66-6.78 (м,3H), 7.21 (т, J=6.6 Гц,1H), 9.32 (д, J=2.0 Гц,1H).
Стадия iii.
(e)(f) (транс)-2-(3-метоксифенил)циклопропанметанамин.
Раствор (транс)-2-(3-метоксифенил)циклопропанкарбоксальдегида (31,0 г, 176 ммолей), хлористоводородного гидроксиламина (38,57 г,
555
молей), этанола (200 мл), воды (120 мл) и 10 н. раствора NaOH (55 мл, 555 ммолей) нагревали с обратным холодильником (паровая баня) в течение 18 часов. Этот раствор охлаждали до комнатной
температуры,
разбавляли водой (1 л), последовательно промывали 1 н. раствором HCl, водой, рассолом и сушили над K2CO3. После концентрирования путем ротационного выпаривания было
получено 30,
98 г (92%) оксима, который сразу же использовали на следующей стадии. Оксим (30,98 г, 162 ммоля) вываривали с тетрагидрофураном (50 мл) и по каплям добавляли к суспензии алюмогидрида
лития (9,2 г, 242
ммоля) в тетрагидрофуране (300 мл), которая находилась при температуре -45oC, поддерживая температуру ниже -40oC. Реакционную смесь оставляли для нагревания до
комнатной
температуры, перемешивали в течение 4 часов и вновь охлаждали до -45oC. Затем по каплям осторожно добавляли раствор бисульфата калия (55 г, 404 ммоля) в воде (200 мл).
Охлаждающую ванну
удаляли, а суспензию перемешивали при комнатной температуре в течение 30 минут. Полученную пасту фильтровали через целит, а фильтровальную лепешку промывали простым этиловым эфиром.
Соединенные
фильтраты экстрагировали 12 н. раствором HCl, кислые вытяжки подщелачивали (50% NaOH) и экстрагировали дихлорметаном. Органические вещества соединяли, промывали рассолом, сушили (K2CO3) и концентрировали в условиях вакуума с образованием указанного в заголовке соединения в виде светлого масла (17,62 г, 56%, две стадии);
спектр ЯМР1H (300
МГц, CDCl3) δ : 0.80-0.95 (м,2H), 1.31-1.35 (м,1H), 1.70-1.79 (м,1H), 2.28 (широкий синглет,2H), 2.70- 2.74 (м,2H), 3.79 (с,3H), 6.60-6.71 (м,3H), 7.17 (т, J=6.4 Гц,1H).
Стадия iv.
(g) (транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид.
К перемешиваемому с помощью магнитной мешалки раствору
(транс)-[2-(3-метоксифенил)циклопропанметанамина (1,02 г, 5,8 ммоля) и Et3N (1,60 мл, 11,5 ммоля) в сухом дихлорметане (20 мл), находившемуся при температуре 0oC, по каплям
добавляли бутирилхлорид (0,69 г, 6,4 ммоля). Полученную суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 4 часов. Растворители удаляли. Остаток вываривали с EtOAc
(100 мл) и последовательно промывали водой, 5% лимонной кислотой, 5% K2CO3, рассолом и сушили над K2CO3. После концентрирования путем ротационного
выпаривания получали неочищенный продукт, который затем очищали посредством испарительной хроматографии (силикагель, смесь 40% EtOAc и гексана) с образованием 1,01 г (71%) указанного в заголовке
соединения;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.85-0.95 (м,5H), 1.22-1.31 (м,1H), 1.62 (к, J= 7.1 Гц,2H), 1.73-1.77 (м,1H), 2.13 (т, J 7.7 Гц,2H), 3.15-3.36 (м,
2H), 3.76
(с,3H), 5.59 (с,1H), 6.55-6.69 (м,3H), 7.15 (т, J=6.5 Гц,1H);
инфракрасный спектр (пленка NaCl) 3302, 2862, 1732, 1644 см-1;
масс-спектр (изобутан-DCl) m/e
247.
Результаты анализа, высчитанные для C15H21NO2:
C 72.84, H 8.56, N 5.66;
обнаружено: C 72.71, H 8.50, N 5.62.
Пример 3 (способ получения 3).
3a. Получение (+)-(транс)-N-[[2-(3-метоксифенил)циклопропил] метил]бутанамида.
Стадия i.
(a) (+)-(транс)-2-(3-метоксифенил)циклопропанметанол.
К суспензии алюмогидрида лития (0,65 г, 17,1 ммоля) в тетрагидрофуране (45 мл), находившейся при температуре -45oC, по
каплям добавляли раствор (2'S)-N-(1S,2S)-2-(3-метоксифенил)циклопропанкарбонил борнан-10',2'-сультама (3,36 г, 8,6 ммоля), полученный по методу Валлгарда Дж., Аппельберга У., Шереха И, и Хакселя У.
(J.Chem.Soc. Penkin Trans. 1, стр. 461-470, 1994 г.), в тетрагидрофуране (20 мл). Охлаждающую ванну удаляли, а реакционную смесь оставляли для нагревания до комнатной температуры, после чего ее сразу
же вновь охлаждали до -45oC. Осторожно добавляли раствор бикарбоната калия (3,9 г, 29 ммолей) в воде (15 мл), повысив температуру до -5oC. Полученную пасту перемешивали при
комнатной температуре в течение 1 часа, фильтровали через целит, а фильтровальную лепешку промывали простым этиловым эфиром. Соединенные фильтраты промывали холодным (0oC) 1 н. раствором
HCl, 5% K2CO3, рассолом, сушили (K2CO3) и концентрировали с образованием неочищенного воска. После растирания в порошок вместе с гексаном, фильтрования
осажденного хирального вспомогательного вещества и последующего концентрирования фильтрата было получено указанное в заголовке соединение в виде светлого масла (1,33 г, 87%);
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.90-1.00 (м,2H), 1.21- 1.25 (м,1H), 1.52-1.59 (широкий синглет, 1H), 1.78-1.85 (м,1H), 3.60 (двойной дублет, J= 5.0,> 1 Гц,2H), 3.80 (с,3H),
6.60-6.72 (м, 3H), 7.20 (м, J=7.5 Гц,1H);
[α]0 55,5o (с=1,
CH2Cl2).
Стадия ii.
(b) (c) (+)-(транс)-1-(азидометил)-2-(3-метоксифенил)- циклопропан.
К раствору
(+)-(транс)-2-(3-метоксифенил)циклопропанметанола (1,33 г, 7,5 ммоля) и триэтиламина (1,57 мл, 11,3 ммоля) в дихлорметане (25 мл), находившемуся при температуре 0oC, по каплям добавляли
метансульфонилхлорид (950 мг, 8,3 ммоля). После окончания добавления ледяную баню удаляли и продолжали перемешивание в течение 30 минут. Полученный раствор обрабатывали дихлорметаном (100 мл),
промывали водой, насыщенным раствором NaHCO3 и сушили над K2CO3. После концентрирования путем ротационного выпаривания было получено 1,78 г неочищенного масла.
Мезилат
растворяли в диметилформамиде (25 мл), обрабатывали азидом натрия (980 мг, 15 ммолей) и продолжали перемешивать при комнатной температуре в течение 18 часов. Полученную смесь концентрировали,
остаток
растворяли в простом этиловом эфире, промывали водой, рассолом, а растворители удаляли посредством ротационного выпаривания, что позволило получить 1,11 г (73%, две стадии) светлого масла;
ЯМР1H (300 МГц, CDCl3) δ : 0.85-0.92 (м,2H), 1.41-1.45 (м,1H), 1.80-1.83 (м,1H), 3.25-3.38 (м,1H), 3.48-3.58 (м,1H), 3.83 (с, 3H), 6.70-6.78 (м, 3H), 7.21 (м,J=7.8
Гц,
1H).
(d) (+)-(транс)-2-(3-метоксифенил)циклопропанметанамин.
К перемешиваемой суспензии алюмогидрида лития (455 мг, 12 ммолей) в тетрагидрофуране (20 мл),
находившейся при температуре -30oC, добавляли раствор (+)-(транс)-1-(азидометил)-2-(3-метоксифенил) циклопропана (1,11 г, 5,5 ммоля) в тетрагидрофуране (10 мл), поддерживая температуру
ниже
-30oC. После окончания добавления суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 4 часов. Реакционную смесь охлаждали до -45oC и по
каплям
осторожно добавляли раствор KHSO4 (2,6 г) в воде (20 мл). Полученную суспензию перемешивали при комнатной температуре в течение 30 минут, фильтровали через целит, а фильтровальную
лепешку
хорошо промывали простым этиловым эфиром. Фильтраты собирали, экстрагировали 1 н. раствором HCl, а кислые слои подщелачивали (30% NaOH). Основный раствор экстрагировали CH2Cl2,
суши ли (K2CO3) и концентрировали с образованием 400 мг (41%) указанного в заголовке соединения в виде светлого масла;
спектр ЯМР1H (300 МГц,
CDCl3
) δ : 0.86-0.95 (м,2H), 1.24-1.31 (м,1H), 1.72-1.79 (м, 1H), 2.10-2.40 (широкий синглет,2H), 2.70- 2.74 (м,2H), 3.79 (с,3H), 6.60-6.72 (м,3H), 7.15 (м, J=7,8 Гц, 1H).
Стадия iii.
(e) (+)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид.
К перемешиваемому с помощью магнитной мешалки раствору
(+)- (транс)-2-(3-метоксифенил)циклопропанметанамина (400 мг, 2,3 ммоля) и Et3N (1,1 мл, 7,9 ммоля) в сухом дихлорметане (15 мл), находившемуся при температуре 0oC, по каплям
добавляли бутирилхлорид (269 мг, 2,5 ммоля). Полученную суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 18 часов. Растворители удаляли, остаток вываривали вместе
с
EtOAc (100 мл) и последовательно промывали водой, 5% лимонной кислотой, 5% K2CO3г рассолом и сушили над K2CO3. После концентрирования путем ротационного
выпаривания был получен неочищенный продукт, который затем очищали посредством испарительной хроматографии (силикагель, смесь 1% метилового спирта и CH2Cl2), что позволило
получить 310 мг (56%) указанного в заголовке соединения в виде светлого масла;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.86-0.95 (м,5H), 1.22- 1.31 (м,1H), 1.54-1.83
(м, 3H), 2.08 (т, J=7.7 Гц,2H), 3.16-3.36 (м, 2H), 3.76 (с,3H), 5.59 (широкий синглет,1H), 6.55-6.76 (м,3H), 7.17 (т, J=7.8 Гц,1H);
инфракрасный спектр (пленка NaCl) 3296, 2962, 1644, 1604,
1550, 1494 см-1;
масс-спектр (изобутан-DCl) m/e 247;
[α]0 +57,2o (c=1, CH2Cl2).
Результаты анализа, высчитанные для C15H21NO2 ·0,15H2O
C
72.05, H 9.59, N 5.60;
обнаружено: C 71.97, H 8.59, N 5.60.
3. (-)-(транс-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид.
Указанное в заголовке
соединение
синтезировали представленным выше способом из (2'R)-N-(1R, 2R)-2-(3- метоксифенил)-циклопропанкарбонил]борнан-10', 2'-сультама, полученного по методу Валлгарда Дж., Аппельберга У., Шереха
И. и Хакселя
У. (J. Chem. Soc. Perkin Trans.I стр. 461-470, 1994 г.);
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.86-0.95 (м,5H), 1.22-1.31 (м,1H), 1.54-1.83 (м,3H),
2.08 (к, J=7.7
Гц,2H), 3.16-3.36 (м,2H), 3.76 (c,3H), 5.59 (широкий синглет,1H), 6.55-6.76 (м,3H), 7.17 (м, 1 =7.8 Гц,1H);
инфракрасный спектр (пленка NaCl) 3294, 2962, 1644, 1604, 1559,
1495 см-1
;
масс-спектр (изобутан-DCl) m/e 247.
Пример 4 (способ получения 4).
4a. Получение (транс)-N-[[2-(2-иод-5-метоксифенил)циклопропил] метил] бутанамида.
К перемешиваемому раствору (транс)-N-[[2-(3-метоксифенил) циклопропил] метил] бутанамида (120 мг, 0,49 ммоля) и TI(OAc)3 (522 мг, 1,5 ммоля) в CCl4 (10 мл) добавляли
раствор I2 (139 мг, 0,55 ммоля) в CCl4 (20 мл). Полученную суспензию нагревали с обратным холодильником в течение 18 часов. Затем ее охлаждали до
комнатной температуры,
фильтровали через спекшуюся стекломассу с порами среднего размера, а фильтрат промывали насыщенным раствором тиосульфата натрия, рассолом и сушили (K2CO3
). После концентрирования
путем ротационного выпаривания было получено 150 мг оранжевого масла, которое очищали посредством хроматографии (силикагель, смесь 40% EtOAc и гексана), что позволило
получить 108 мг (60%) указанного
в заголовке соединения в виде белого твердого вещества, температура плавления 84 -86oC;
спектр ЯМР1H (300 МГц, CDCl3)
δ : 0.87-1.01 (м,5H), 1.07-1.23
(м,1H), 1.59-1.71 (м, 2H), 1.84-1.90 (м,1H), 2.16 (т, J=7.8 Гц, 2H), 3.16-3.25 (м, 1H), 3.46-3.53 (м,1H), 3.73 (с,3H), 5.83 (широкий синглет,1H), 6.45-6.50 (м,
2H), 7.66 (д, J=8.5 Гц,1H);
инфракрасный спектр (пленка NaCl) 3304, 2962, 1636, 1555, 1444, 1416 см-1;
масс-спектр (изобутан-DCl) m/e 373.
Результаты
анализа, высчитанные для C15
H20NO2I:
C 48.27, H 5.40, N 3.75;
обнаружено: C 48.24, H 5.41, N 3.53.
4b. Получение (транс)-N-[[2-(2-иод-5-фторфенил)-циклопропил] метил]бутанамида.
К перемешиваемому раствору (транс)-N-[[2-(3-фторфенил)- циклопропил]метил] бутанамида (1,0 г, 4,3 ммоля), TI(OAc)3 (2,5 г, 6,5 ммоля) и тетрафторамина (15
мл) в ацетонитриле (15 мл) добавляли раствор NaI (705 мг, 4,7 ммоля) в воде (2 мл). Полученную суспензию нагревали при температуре 55oC в
течение 12 часов. Затем ее охлаждали до комнатной
температуры, фильтровали через спекшуюся стекломассу с порами среднего размера, а фильтрат промывали насыщенным раствором тиосульфата натрия,
рассолом и сушили (K2CO3). После
концентрирования путем ротационного выпаривания было получено 1,26 г оранжевого масла, которое очищали посредством хроматографии (силикагель,
смесь 35% EtOAc и гексана), что позволило получить 371 мг
(25%) указанного в заголовке соединения в виде белого твердого вещества: температура плавления 66-68oC;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.91- 0.99
(м,5H), 1.00-1.15 (м,1H), 1.70 (к, J=7.5 Гц,2H), 1.89-1.92 (м,1H), 2.17 (т, J= 7.2 Гц,2H), 3.28-3.32 (м, 1H), 3.43-3.50 (м, 1H), 5.67
(широкий синглет,1H), 6.59-6.67 (м,2H), 7.71-7.76 (м,1H);
инфракрасный спектр (пленка NaCl) 3417, 3294, 2962, 1638, 1554, 1448, 1412 см-1;
масс-спектр (ESI) m/e 361.
Результаты анализа, высчитанные для C14H17NOFI:
C 46.55, H 4.74, N 3.88;
обнаружено: C 46.57, H 4.68, N 3.85.
Пример 5 (способ получения 5).
Получение (транс)-N-[(2-(2,
4-дибром-5-метоксифенил)циклопропил] метил] бутанамида
К перемешиваемому раствору N-[[2-(3-метоксифенил)циклопропил] метил]
бутанамида (190 мг, 0,77 ммоля) и TI(OAc)3 (881 мг, 2,3
ммоля) в CCl4 (20 мл), находившемуся при температуре 0oC, по каплям добавляли раствор Br2 (238 мг,
1,5 ммоля) в CCl4 (10 мл). После окончания добавления эту
суспензию фильтровали через спекшуюся стекломассу с порами среднего размера, а фильтрат промывали насыщенным раствором тиосульфата
натрия, рассолом, сушили (K2CO3) и
концентрировали с образованием светлого масла. После очистки посредством хроматографии (силикагель, смесь 35% EtOAc и гексана) было получено
185 мг (62%) указанного в заголовке соединения в виде
белого твердого вещества: температура плавления 114-115oC;
спектр ЯМР1H (300 МГц, CDCl3) δ :
0.90-1.02 (м,5H), 1.04-1.16 (м,1H), 1.60-1.69 (м,2H),
1.89-1.93 (м,1H), 2.16 (т, J=7.8 Гц,2H), 3.04-3.16 (м,1H), 3.49-3.54 (м,1H), 3.82 (с,3H), 5.77 (широкий синглет,1H), 6.44 (с,1H), 7.66 (с,1H);
инфракрасный спектр (пленка NaCl) 3290, 2960,
1632, 1550, 1474, 1442 см-1;
масс-спектр: m/e 405.
Результаты анализа, высчитанные для C15H19NO2Br2:
C 44.47,
H 4.73, N 3.46;
обнаружено: C 44.59, H 4.65, N 3.16.
Пример 6 (способ получения 6).
Получение (транс)-N-[[2-(3-метоксифенил)циклопропил] метил] -N-(фенилметил)бутанамида.
Перемешиваемый с помощью магнитной мешалки раствор (транс)-N-[[2-(3-метокcифенил)циклопропил]
метил]бутанамида (1,0 г, 4,0 ммоля) и NaH (105 мг, 4,4 ммоля)
в диметилформамиде (10 мл) обрабатывали бензилбромидом (750 мг, 4,4 ммоля). Полученную суспензию перемешивали в течение 18 часов при
комнатной температуре, разбавляли простым этиловым эфиром (100 мл)
и быстро охлаждали водой (100 мл). Слои разделяли, после чего органическую фазу промывали водой, рассолом, сушили (K2
CO3) и концентрировали с образованием неочищенного масла,
которое очищали посредством испарительной хроматографии (силикагель, смесь 45% EtOAc и гексана) с образованием 580 мг (43%)
указанного в заголовке соединения в виде светлого масла;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.78-0.98 (м,4H), 1.17-1.33 (м,1H), 1.60- 1.79 (м, 4H), 2.27-2.44 (м,
2H), 3.17-3.33 (м,1H), 3.51-3.61 (м,1H), 3.77 (широкий синглет, 3H), 4.46-4.82
(м,2H), 6.50-6.72 (м,3H), 7.10-7.34 (м,6H);
инфракрасный спектр (пленка NaCl) 2962, 1644, 1604, 1454, 1210
см-1;
масс-спектр (изобутан-DCl) m/e 337.
Результаты анализа, высчитанные для C22H27NO2 ·0,1H2O:
C
77.88, H 8.08, N 4.13;
обнаружено: C 77.87, H 8.12, N 3.89.
Пример 7 (способ получения 7).
Получение (транс)-N-[[2-[3-(2-пропенилокси)фенил]циклопропил] метил]бутанамида.
Стадия i.
(a) (транс)-N-[[2-(3-гидроксифенил)циклопропил]метил]- бутанамид.
К перемешиваемому раствору
(транс)-N-[[2-(3-метоксифенил) циклопропил] метил] бутанамида (3,50 г, 14,2 ммоля) в
дихлорметане (50 мл), находившемуся при температуре -78oC, по каплям добавляли BBr4 (28,4 мл,
1 н. раствор в CH2Cl2, 28,4 ммоля). После окончания добавления
охлаждающую ванну удаляли и продолжали перемешивать в течение 18 часов. Полученный раствор выливали на смесь льда
с водой (100 мл) и экстрагировали 2,5 н. раствором NaOH. Основные вытяжки собирали,
промывали дихлорметаном и подкисляли (концентрированная HCl). Кислый раствор экстрагировали дихлорметаном,
органические вещества собирали, промывали рассолом, сушили (MgSO4) и
концентрировали с образованием 2,49 г (75%) указанного в заголовке соединения:
спектр ЯМР1H (300
МГц, DMSO-d6) δ : 0.72-0.84 (м,5H), 1.09-1.15 (м,1H), 1.42-1.54
(м, 2H), 1.63-1.69 (м,1H), 2.02 (т, J= 5,0 Гц,2H), 2.99-3.11 (м, 2H), 6.39-6.50 (м,3H), 6.98 (т, J=7.8 Гц,1H), 7.91
(с,1H), 9.19 (широкий синглет,1H);
инфракрасный спектр (пленка NaCl) 3300,
2964, 1642, 1585, 1542, 1466 см-1;
масс-спектр (изобутан-DCl) m/e 233.
Результаты анализа, высчитанные для C14H19NO2 ·0,
1H2O:
C 71.52, H 8.23, N 5.96;
обнаружено: C 71.53, H 8.32, N 5.92.
Стадия ii.
(b) (транс)-N-[[(2-пропен-1-илоксифенил)циклопропил]- метил]бутанамид.
К быстро перемешиваемому раствору (транс)-N-[[2-(3-гидpoкcифенил)
циклoпpoпил] метил] бутанамида (0,8 г, 3,4 ммоля)
и KOH (210 мг, 3,74 ммоля) в этаноле (15 мл) добавляли аллилиодид (622 мг, 3,7 ммоля). После перемешивания в течение 18 часов суспензию разбавляли
простым этиловым эфиром (100 мл) и промывали водой, 2
н. раствором NaOH, рассолом, сушили (K2CO3) и концентрировали с образованием неочищенного остатка. Полученное вещество
очищали посредством хроматографии (силикагель, смесь 40%
EtOAc и гексана), что позволило получить 320 мг (34%) указанного в заголовке соединения;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.70-0.94 (м,5H), 1.12-1.32 (м,1H),
1.58-1.84 (м, 3H), 2.20 (т: J=6.0 Гц,2H), 3.15-3.36 (м,2H), 4.48 (д, J=5.6 Гц, 2H), 5.24 (д, J=10.0 Гц,1H), 5.40 (д, J=17.2 Гц,1H), 5.56
(широкий синглет,1H), 5.96-6.07 (м,1H), 6.57-6.70 (м,3H), 7.13
(т, J=7.2 Гц,1H);
инфракрасный спектр (пленка NaCl) 3296, 2962, 1644, 1607, 1548, 1494 см-1;
масс-спектр
m/e 273.
Результаты анализа, высчитанные для
C17H23NO2 ·0,20(H2O):
C 73.72, H 8.52, N 5.06;
обнаружено: C 73.77,
H 8.63, N 5.00.
Пример 8 (способ получения 8).
Получение (транс)-N-[[2-[3-(метилэтокси)фенил]циклопропил] метил]бутанамида.
К быстро перемешиваемому
раствору (транс)-N-[[2-(3- гидроксифенил)циклопропил] метил]
бутанамида (390 мг, 1,7 ммоля), трифенилфосфина (498 мг, 1,9 ммоля) и изопропанола (150 мг, 2,5 ммоля) в тетрагидрофуране (10 мл),
находившемуся при температуре 0oC, в виде одной порции
добавляли диэтилазодикарбоксилат (331 мг, 1,9 ммоля). После перемешивания в течение 2 часов эту суспензию разбавляли простым этиловым
эфиром (100 мл), промывали водой, 2 н. раствором NaOH и рассолом,
сушили (K2CO3) и концентрировали с образованием неочищенного воска. Полученное вещество очищали посредством
хроматографии (силикагель, смесь 35% EtOAc и гексана), что позволило
получить 144 мг (31%) указанного в заголовке соединения;
спектр ЯМР1H (300 МГц, CDCl3) δ :
0.85-0.94 (м,5H), 1.22-1.24 (м,1H), 1.29 (д, J= 6.7 Гц,6H), 1.58-1.68
(м,2H), 1.70-1.77 (м,1H), 2.14 (т, J=6.0 Гц, 2H), 3.14-3.36 (м, 2H), 4.44-4.54 (м,1H), 5.81 (широкий синглет,1H), 6.53-6.67 (м,
3H), 7.13 (т, J=7.2 Гц,1H);
инфракрасный спектр (пленка NaCl)
3294, 2974, 1644, 1610, 1550, 1492 см-1;
масс-спектр m/e 275.
Результаты анализа,
высчитанные для C17H25NO2:
C 74.14, H
9.15, N 5.09;
обнаружено: C 73.77, H 8.99, N 4.82.
Пример 9 (способ получения 9).
9a. Получение (-)-(транс)-N-[[2-(5-метокси-2-фенилэтинил)фенил] циклопропил]метил]бутанамида.
(a) (-)-(транс)-N-[[2-(5-метокси-2-фенилэтинил)фенилциклопропил] метил] бутанамид.
Перемешиваемую суспензию
(транс)-N-[[2-(2-иод-5-метоксифенил) циклопропил]метил]бутанамида (400 мг, 1,1 ммоля), фенилацетилена (133 мг, 1,3 ммоля) и Pd(Ph3
P)4 (63 мг, 0,05 ммоля) в триэтиламине (10 мл)
нагревали с обратным холодильником в течение 18 часов, охлаждали до комнатной температуры и разбавляли простым этиловым эфиром (100 мл),
промывали водой, рассолом, сушили (K2CO3)
и концентрировали в условиях вакуума с образованием 400 мг красного масла. После очистки посредством хроматографии (силикагель, смесь
30% EtOAc и гексана) было получено 220 мг белого твердого вещества
(58%): температура плавления 107-108oC;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.78 (т,
J=7.5 Гц,3H), 0.94-0.98 (м, 1H), 1.14-1.16 (м,2H), 1.40- 1.56 (м,
2H), 1.78-1.93 (м, 2H), 2.25-2.31 (м, 1H), 2.80-2.99 (м,1H), 3.74-3.83 (м,1H), 3.79 (с,3H), 5.84 (широкий синглет, 1H), 6.41 (с,1H),
6.71 (д,J=4 Гц, 1H), 7.16-7.61 (м,6H);
инфракрасный спектр
(пленка NaCl) 3310, 2960, 2214, 1638, 1544, 1502, 1428 см-1;
масс-спектр (ESl) m/e 347.
Результаты анализа, высчитанные для C23H25NO2:
C 79.51, H 7.25, N 4.03;
обнаружено: C 79.28, H 7.21, N 3.78.
9b. Получение
(-)-(транс)-N-[[2-(5-метокси-2- фенилэтил)фенил]циклопропил]метил]бутанамидa
(b) (-)-(транс)-N-[[2-(5-метокси-2-фенилэтил]фенил]- циклопропил]метил] бутанамид.
Суспензию
(-)-(транс)-N-[[2-(5-метокси-2-фенилэтинил)фенил] циклопропил] метил] бутанамида
(100 мг, 0,28 ммоля), 20% палладированного угля (50 мг) в этаноле (25 мл) встряхивали под слоем водорода (аппарат
Парра, 3,5 ат) в течение 18 часов. Полученную суспензию фильтровали через целит и
концентрировали в условиях вакуума с образованием светлого масла (100 мг, 100%):
спектр ЯМР1H
(300 МГц, CDCl3) δ : 0.90-0.94 (м,5H), 1.25-1.30 (м,1H), 1.60-1.66
(м, 1H), 1.71-1.79 (м, 2H), 2.05-2.15 (м,2H), 2.88-2.98 (м,4H), 3.20-3.35 (м, 2H), 3.77 (с, 3H), 5.57 (широкий
синглет,1H), 6.47 (с,1H), 6.66-6.68 (м,1H), 7.05 (д,J=4 Гц,1H), 7.17-7.32 (м,5H);
инфракрасный спектр (пленка NaCl) 3294, 2960, 1644, 1548, 1498, 1454 см-1;
масс-спектр
(ESI) m/e 351.
Результаты анализа высчитанные для C23H29NO2 ·0,25(H2O):
C 77.60, H 8.41, N 3.58;
обнаружено: C
77.48, H 8.41, N 3.58.
Пример 10 (способ получения 10).
Получение (-)-(транс)-N-[[2-(2-циано-5-метоксифенил)циклопропил] метил] бутанамида.
В 100 мл
колбу с круглым основанием, оснащенную обратным холодильником и хлоркальциевой трубкой,
вводили (транс) - N-[[2-(2-иод-5-метоксифенил)циклопропил] метил]бутанамид (200 мг, 0,6 ммоля), порошкообразный
цианид меди (I) (106 мг, 1,2 ммоля) и безводный пиридин (2 мл). Эту смесь затем
нагревали при температуре 185oC в течение 18 часов. Реакционную смесь быстро охлаждали путем добавления 50%
водного раствора аммиака (30 мл), после чего экстрагировали толуолом (3х50 мл).
Органические вещества соединяли, промывали водой, 1 н. раствором HCl, рассолом и сушили над MgSO4. После
фильтрования и концентрирования в условиях вакуума было получено 310 мг неочищенного
масла, которое очищали посредством хроматографии (силикагель, смесь 45% EtOAc и гексана) с образованием светлого
масла (120 мг, 74%):
спектр ЯМР1H (300 МГц, CDCl3)
δ : 0.93 (т, J=6.0 Гц,3H), 0.96- 0.99 (м, 1H), 1.10-1.16 (м,1H), 1.20-1.28 (м,1H), 1.64-1.71 (м,2H),
2.03-2.10 (м,1H), 2.20 (т, J=7.8 Гц,2H), 2.72-2.79 (м,1H), 3.82 (с,3H), 3.85-3.92 (м,1H),
6.27 (широкий синглет, 1H), 6.56 (с,1H), 6.73 (д, J=8.4 Гц,1H), 7.53 (д, J=8.7 Гц,1H);
инфракрасный
спектр (пленка NaCl) 3300, 2964, 2218, 1646, 1606, 1564 см-1;
масс-спектр (ESI) m/e 272.
Результаты анализа, высчитанные для C16H20N2O2·0,25(H2O):
C 69.41, H 7.46, N 10.12,
обнаружено: C 69.41, H 7.46, N 10.00.
Пример 11 (способ получения 11).
Получение (-)-(транс)-N-[[2-(2-фенил-5-метоксифенил)циклопропил] метил] бутанамида.
К перемешиваемой суспензии (транс)-N-[[2-(2-иод-5-метоксифенил) циклопропил} метил} бутанамида
(210 мг, 0,56 ммоля), гидроксида бария (265 мг, 0,84 ммоля) и воды (1 мл) в
диметиловом эфире (6 мл) в виде одной порции добавляли смесь Pd(Ph3P)4 (12 мг, 0,01 ммоля) и
фенилборной кислоты (75 мг, 0,62 ммоля). Полученную смесь нагревали с обратным
холодильником в течение 18 часов, разбавляли толуолом, промывали водой, рассолом, сушили над K2CO3
и концентрировали в условиях вакуума с образованием красного воска. После
очистки посредством хроматографии (силикагель, смесь 35% EtOAc и гексана) было получено 145 мг (85%) светлого масла;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.69- 0.79
(М,1H), 0.89 (т, J= 7.2 Гц, 3H), 0.93-0.98 (м,1H), 1.01-1.08 (м,1H), 1.51-1.64 (м,2H), 1.79-1.86 (м, 1H), 2.01 (т: J=7.2 Гц,
2H), 2.78-2.86 (м,1H), 3.21- 3.30 (м,1H), 3.79 (с, 3H), 5.04 (широкий
синглет,1H), 6.50 (с,1H), 6.74 (д, J=8.4 Гц,1H), 7.12 (д, J=8.5 Гц,1H), 7.30-7.43 (м,5H);
инфракрасный спектр (пленка
NaCl) 3296, 2962, 1644, 1610, 1550, 1482 см-1;
масс-спектр (ESI) m/e 323.
Результаты анализа, высчитанные для C21H25NO2 ·0,
5(H2O):
C 75.87, H 7.88, N 4.21;
обнаружено: C 75.95, H 7.86, N 4.15.
Пример 12 (способ получения 12).
Получение (±)-(транс)-[2, 2-дифтор-3-(3-метоксифенил) циклопропилметил] бутанамида.
Стадия i.
(a) (±)-(транс)-3-(3-метоксифенил)-проп-2-ен-1-ол.
Раствор
3-метокси-коричной кислоты (50,24 г, 281 ммоль) в 400 мл безводного
тетрагидрофурана добавляли к перемешиваемой с помощью магнитной мешалки суспензии борогидрида натрия (12,80 г, 338 ммолей) в 500 мл
безводного тетрагидрофурана, находившейся при комнатной температуре.
Суспензию перемешивали до тех пор, пока не прекращалось выделение газа, охлаждали до 0oC и в течение 1 часа добавляли
раствор иода (35,77 г, 141 ммоля) в безводном тетрагидрофуране (500
мл). Эту суспензию перемешивали в течение 3 часов и по каплям добавляли 4 н. раствор хлористоводородной кислоты (100 мл).
Полученную суспензию экстрагировали простым диэтиловым эфиром (3х500 мл) и
соединенные органические порции промывали 3 н. раствором NaOH (3х500 мл), рассолом (500 мл), сушили (MgSO4),
фильтровали и концентрировали в условиях вакуума. После перегонки по методу
Кюгельрора (0,5 мм) получили указанное в заголовке соединение в виде светлой жидкости (23,12 г, 50%);
спектр ЯМР1H (300 МГц, CDCl3) δ : 3.82 (с,3H), 4.34 (д,J=6
Гц, 2H), 6.37 (двойной триплет, J=15.6 Гц,1H), 6.60 (д,J=15 Гц,1H), 6.80 (двойной дублет, J= 8,1=1 Гц,1H), 6.93 (т,J=1 Гц,
1H), 7.00 (д,J=8 Гц,1H), 7.24 (т, J=8 Гц,1H).
Стадия iii.
(b) (3-(3-метоксифенил)аллиловый эфир (±)-(транс)-уксусной кислоты.
В перемешиваемый
с помощью магнитной мешалки раствор (±
)- (транс)-3-(3-метоксифенил)-проп-2-ен-1-ола (22,35 г, 136 ммолей) в безводном пиридине (115 мл), находившийся при температуре 0oC, по каплям
добавляли уксусный ангидрид (16,0 мл, 170
ммолей). После окончания добавления ледяную баню удаляли, раствор оставляли для нагревания до комнатной температуры и перемешивали в течение 24 часов.
Полученный раствор концентрировали с условиях
вакуума, а остаток вываривали вместе с простым диэтиловым эфиром (300 мл), промывали 1 н. раствором HCl (2х300 мл), 5% NaHCO3 (200 мл), водой
(200 мл), рассолом (200 мл), сушили над
MgSO4, фильтровали и концентрировали в условиях вакуума с образованием светлой жидкости. После испарительной хроматографии (элюирование смесью 5%
простого диэтилового эфира и гексана) и
перегонки полученной жидкости по методу Кюгельрора (0,5 мм) был получен требуемый продукт в виде светлой жидкости (16,95 г, 60%);
спектр ЯМР1H (300 МГц, CDCl3) δ
: 2.11 (с,3H), 3.81 (с,3H), 4.73 (д,J=6 Гц, 2H), 6.28 (двойной триплет, J=15.6 Гц,1H), 6.63 (д, J=15 Гц,1H), 6.82 (двойной дублет, J=8, 1 Гц,1H), 6.92 (т,
J=1 Гц, 1H), 6.99 (д, J=8 Гц,1H), 7.24 (т,
J=8 Гц,1H).
Стадия iii.
(c) 2,2-дифтор-3-(3-метоксифенил)циклопропилметиловый эфир (±)-(транс)-уксусной кислоты.
Раствор (3-(3-метоксифенил)
аллилового эфира (±)-(транс)- уксусной кислоты (1,63 г, 7.90 ммоля) в безводном диглиме нагревали с обратным холодильником, а затем в
течение 1 часа по каплям добавляли раствор
хлордифторацетата натрия (9,02 г, 59,2 ммоля) в безводном диглиме (25 мл). Полученную суспензию охлаждали до комнатной температуры и выливали на 200 мл
льда. Эту суспензию экстрагировали простым
диэтиловым эфиром (4х200 мл), а соединенные органические экстракты промывали водой (3х400 мл), рассолом (400 мл), сушили (MgSO4), фильтровали и
концентрировали в условиях вакуума. После
перегонки по методу Кюгельрора (0,5 мм) было получено 1,59 г (79%) продукта в виде светлого масла:
спектр ЯМР1H (300 МГц, CDCl3) δ : 2.10 (с,3H), 2.24 (дважды
двойной триплет, J= 15, 7.5, 7 Гц,1H), 2.63 (двойной дублет, J=15, 7.5 Гц,1H), 3.80 (с, 3H), 4.19-4.25 (м,1H), 4.31-4.37 (м,1H), 6.74-6.83 (м,3H), 7.25
(т, J=8 Гц,1H).
Стадия iv.
(d) (±)-(транс)-(2,2-дифтор-3-(3-метоксифенил)циклопропил] метанол.
Перемешиваемый с помощью магнитной мешалки
раствор гидроксида калия (2,39 г, 42,6 ммоля) в
смеси метанола и тетра-гидрофурана с соотношением 3:1 (88 мл) обрабатывали 2,2-дифтор-3- (3-метоксифенил) циклопропилметиловым эфиром (±
)-(транс)-уксусной кислоты (4,16 г, 16,2 ммоля). Этот
раствор перемешивали в течение 2 часов и концентрировали в условиях вакуума. Остаток вываривали вместе с простым диэтиловым эфиром (200 мл) и
водой (200 мл), слои разделяли, водный слой экстрагировали
простым диэтиловым эфиром (2х200 мл), органические экстракты промывали насыщенным раствором NaHCO3 (300 мл), водой (300 мл),
рассолом (300 мл), сушили (MgSO4), фильтровали и и
концентрировали в условиях вакуума с образованием 3,42 г (99%) светлого масла, которое использовали без дальнейшей очистки:
спектр ЯМР1H (300 МГц, CDCl3) δ :
1.60 (широкий синглет,1H), 2.15-2.36 (м, 1H), 2.60 (дважды двойной дублет, J=15, 7.5, 1.5 Гц, 1H), 3.81 (с,3H), 3.83-3.98 (м, 2H), 6.77
(т, J=1 Гц,1H), 6.81 (д,J= 8 Гц, 1H), 6.82 (двойной дублет, J=8,
1 Гц,1H), 7.26 (т, J=8 Гц,1H).
Стадия v.
(e) (f) (a) (±)-(транс)-[2, 2-дифтор-3-(3-метоксифенил) циклопропил]метанамин.
К перемешиваемому с
помощью магнитной мешалки раствору спирта (3,27 г, 15,3 ммоля), триэтиламина (3,14 г, 31,0 ммоль) и CH2Cl2 (50 мл), находившемуся при 0oC под слоем азота, в течение 15
минут по каплям добавляли метансульфонилхлорид (2,8 г, 24,4 ммоля). Раствор перемешивали в течение 30
минут, разбавляли CH2Cl2 (200 мл) и последовательно промывали водой (200 мл)
и насыщенным раствором NaHCO3 (200 мл), сушили (K2CO3), фильтровали
и концентрировали в условиях вакуума при температуре 5oC. Остаток вываривали с безводным
диметилформамидом (50 мл), обрабатывали азидом натрия (1,96 г, 30,1 ммоля) и перемешивали в течение
14 часов. Полученный раствор выливали в воду (500 мл), экстрагировали простым диэтиловым эфиром
(4х250 мл), соединенные экстракты промывали водой (4х500 мл), рассолом (2х300 мл), сушили (MgSO4), фильтровали и концентрировали в условиях вакуума (осторожно !) с образованием светлого
масла. Это масло вываривали с безводным простым диэтиловым эфиром (40 мл) и по каплям добавляли к
перемешиваемой с помощью магнитной мешалки суспензии алюмогидрида лития (1,16 г, 30,6 ммоля) в
безводном простом диэтиловом эфире (60 мл), находившейся при температуре -30oC. Суспензию
оставляли для нагревания в течение 3 часов, вновь охлаждали до -30oC и по каплям
добавляли раствор KHSO4 (2,6 г, 19 ммолей) в воде (20 мл). Полученную суспензию оставляли для
нагревания до комнатной температуры, перемешивали в течение 1 часа и фильтровали через целит с
последующим элюированием простым диэтиловым эфиром (400 мл). Слои разделяли и органический слой
экстрагировали 1 н. раствором HCl (3х100 мл), соединенные водные экстракты подщелачивали 50% NaOH и
экстрагировали CH2Cl2 (3х150 мл). Органическую часть промывали рассолом (200
мл), сушили
(K2CO3), фильтровали и концентрировали в условиях вакуума с
образованием 1,23 г (38%) светлого масла;
спектр ЯМР1H (300 МГц, CDCl3)
δ : 1.42 (широкий синглет,2H), 1.97-2.08 (м, 1H), 2.44- 2.52 (м,1H), 2.91-3.10 (м,2H),
3.80 (с,3H), 6.76 (т, J=1 Гц, 1H), 6.81 (д, J=8 Гц,1H), 6.81 (двойной дублет, J=8, 1 Гц,1H), 7.23 (т, J=8
Гц,1H).
Стадия v.
(h) (±)-(транс)-[2, 2-дифтор-3-(3-метоксифенил)циклопропилметил] бутанамид.
К перемешиваемому с помощью магнитной мешалки
раствору (±)- (транс)-[2,2-дифтор-3-(3-метоксифенил)циклопропил]
метанамина (672 мг, 3,16 ммоля) и триэтиламина (1,5 мл) в безводном CH2Cl2 (20 мл) по каплям добавляли
раствор бутирилхлорида (370 мг, 3,47 ммоля) в безводном CH2
Cl2 (3 мл). Раствор перемешивали в течение 48 часов и концентрировали в условиях вакуума. Остаток вываривали с CH2Cl2 (100 мл) и промывали 1 н. раствором HCl (100 мл),
1 н. раствором NaOH (100 мл), рассолом (100 мл), сушили (K2CO3), фильтровали и концентрировали в условиях
вакуума. После испарительной хроматографии (элюирование смесью гексанов
и этилацетата с соотношением 2:1) был получен требуемый продукт в виде светлого масла, который после выстаивания затвердевал и
превращался в воскообразное вещество:
спектр ЯМР1H
(300 МГц, CDCl3) δ : 0.92 (т, J=7.4 Гц,3H), 1.64 (секстет, J= 7.4 Гц,2H), 2.09-2.21 (м,1H), 2.16 (т,J=7.4 Гц,
2H), 2.51 (двойной дублет, J= 13.8, 7.4 Гц, 1H), 3.19-3.22 (м,1H), 3.76
(с,3H), 3.77-3.85 (м,1H),5.90 (широкий синглет, 1H), 6.70 (с,1H), 6.75 (д, J=8 Гц,1H), 6.77 (двойной дублет,J=8, 1 Гц,1H), 7.20
(т, J=8 Гц,1H);
инфракрасный спектр (пленка NaCl) 3302, 2960,
1644, 1552, 1288, 1266, 1250, 1160 см-1;
масс-спектр (изобутан-DCl) m/e 282 (M-H-).
Результаты анализа, высчитанные для C15H19
NO2F2:
C 63.59, H 6.76, N 4.94;
обнаружено: C 63.59, H 6.83, N 4.85.
Пример 13 (способ получения 13).
Получение (транс)-N-[[2-(2-бутириламино-5-метоксифенил) циклопропил] метил]бутанамидa.
Стадия i.
(a) (транс)-N-[[2-(2-амино-5-метоксифенил)циклопропан] метило]амин.
В сосуд Парра выливали суспензию (транс)-2-(2-нитро-5- метоксифенил)циклопропанкарбоксальдегидоксима (1,0 г, 4,2 ммоля),
10% палладированного угля (100 мг) и HOAc (25 мл) и в течение 18
часов встряхивали ее в атмосфере водорода (3,5 ат). Полученное вещество фильтровали через целит, вываривали с водой (300 мл) и
подщелачивали 10 н. раствором NaOH. Этот продукт затем экстрагировали с
помощью CH2Cl2, экстракты соединяли, сушили над K2CO3 и концентрировали в условиях
вакуума с образованием 800 мг (100%) красного масла:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.78-0.85 (м,1H), 1.34-1.39 (м,1H), 1,59-1.69 (м, 1H), 1.77-1.83 (м,1H),
1.88 (широкий синглет, 2H), 3.10-3.38 (м, 2H), 3.66 (с,3H), 6.39 (д,J=6.0
Гц,1H), 6.50-6.59 (м,1H), 6.61 (широкий синглет, 2H), 6.78-6.97 (м,1H).
Стадия ii.
(b) (транс)-N-[[2-(2-бутириламино-5-метоксифенил)циклопропил] метил]бутанамид.
К перемешиваемому с помощью магнитной мешалки раствору (транс)-N-[[2-(2-амино-5-метоксифенил)циклопропан]
метил]амина (400 мг, 2,2 ммоля) и Et3N (0,86 мл, 6,6 ммоля) в сухом
CH2Cl2 (15 мл), находившемуся при температуре 0oC, по каплям добавляли бутирилхлорид
(256 мг, 2,4 ммоля). Полученную суспензию оставляли для нагревания до комнатной
температуры и перемешивали в течение 18 часов. После концентрирования путем ротационного выпаривания был получен
продукт, который затем очищали посредством испарительной хроматографии (силикагель,
смесь 30% EtOAc и гексана), что позволило получить 250 мг (44%) светлого масла;
спектр ЯМР1H
(300 МГц, CDCl3) δ : 0.75-1.04 (м,3H), 1.50-1.76 (м,5H), 2.10 (т, J=
8.1 Гц,2H), 2.45 (т, J=8.0 Гц,2H), 2.68- 1.78 (м,1H), 3.69 (c, 3H), 3.72-3.78 (м, 2H), 6.27 (д, J=2.0 Гц,1H),
6.36 (широкий синглет,1H), 6.63 (двойной дублет, J= 7.8, 2.7 Гц, 1H), 7.50 (д, J=8.7 Гц,
1H), 7.89 (широкий синглет,1H);
инфракрасный спектр (пленка NaCl) 3290, 2961, 1647, 1534, 1425 см-1;
масс-спектр (ESI) m/e 332.
Результаты анализа,
высчитанные для C19H28N2O3·0,4H2O:
C 67.19, H
8.54, N 8.24;
обнаружено: C 67.58, H 8.19, N 7.77.
Пример 14 (способ получения 14).
Получение (-)-(транс)-N-[1-2-(3-метоксифенил)циклoпpoпил]этил] бутaнaмидa (изомер A).
Стадия i.
(a) (-)-(транс)-1-[2-(3-метоксифенил)циклопропил] этанол (изомер A) и (-)-(транс)-1-[2-(3-метоксифенил)циклопропил]этанол (изомер B).
Диметилсульфоксид (3,98 мл, 56 ммолей) по каплям
добавляли к раствору оксалилхлорида (2 М раствор в CH2Cl2, 19,5 мл, 39 ммолей) в метиленхлориде (20 мл),
находившемуся при температуре -78oC. Вслед за этим по каплям
добавляли раствор (-)-(транс)-[2-(3-метоксифенил)циклопропил]метанола (5,0 г, 28 ммолей) в дихлорметане (10 мл). Этот раствор
перемешивали в течение 30 минут, после чего медленно добавляли триэтиламин
(16,2 мл, 112 ммоля) и полученную суспензию оставляли для нагревания до комнатной температуры. Эту суспензию перемешивали в
течение 30 минут, разбавляли CH2Cl2 (100 мл),
промывали водой, сушили над K2CO3 и концентрировали в условиях вакуума с образованием 5,8 г альдегида,
который использовали без дальнейшей очистки. Альдегид вываривали с
тетрагидрофураном (10 мл) и по каплям добавляли к раствору бромида метилмагния (3 М раствор в простом этиловом эфире, 23,3 мл, 70
ммолей) в тетрагидрофуране (25 мл), находившемуся при температуре
0oC. Этот раствор перемешивали при 0oC в течение 2 часов, после чего добавляли 2 н. раствор HCl (20 мл) в
EtOAc. Полученную суспензию промывали водой, рассолом, сушили (K2CO3) и концентрировали в условиях вакуума с образованием 5,5 г неочищенного масла. После очистки посредством
хроматографии на силикагеле (смесь 25% EtOAc и гексана) были получены два
диастереомера, (изомер А) 1,66 г, первая полоса, и (изомер В) 1,64 г, вторая полоса:
(изомер A) Rf= 0,45
(смесь 25% EtOAc с гексанами);
спектр ЯМР1H (300 МГц,
CDCl3) δ : 0.84-0.94 (м,2H), 1.19-1.27 (м,1H), 1.30 (д, J= 6,3 Гц, 3H), 1.83-2.02 (м,1H), 3.32-3.38 (м,1H),
3.77 (с,3H), 6.60-6.73 (м,3H), 7.12 (т,J=7.8 Гц,1H);
[α
]0 -49,0o (c=1, CH2Cl2).
(изомер В)
Rf = 0,40 (смесь 25% EtOAc с гексанами);
спектр ЯМР1H (300 МГц, CDCl3) δ :
0.93-1.00 (м,2H), 1,21-1.29 (м,1H), 1.32 (д, J= 6.3 Гц, 3H), 1.75-1.78 (м,
1H), 3.33-3.38 (м,1H), 3.77 (с,3H), 6.59-6.69 (м,3H), 7.15 (т,J=7.8 Гц,1H);
[α]0 -55,3o (с=1, CH2Cl2).
Стадия ii.
(b) (-)-(транс)-2-[1-[2-(3-метоксифенил)циклопропил] этил] изоиндол-1,3-дион (изомер A).
Диэтилазодикарбоксилат (1,79 г, 10,3 ммоля) по каплям добавляли к перемешиваемому раствору
(-)-(транс)-1-[2-(3- метоксифенил)циклопропил]этанола (изомер A) (1,66 г, 8,6 ммоля),
трифенилфосфина (2,69 г, 10,3 ммоля) и фталимида (1,52 г, 10,3 ммоля) в тетрагидрофуране (75 мл), находившемуся
при 0oC. Реакционную смесь оставляли для нагревания до комнатной температуры
и перемешивали в течение 18 часов. Растворитель удаляли в условиях вакуума, полученный остаток вываривали с
EtOAc (150 мл) и последовательно промывали 1 н. раствором HCl, 1 н. раствором NaOH и
рассолом. После сушки над K2CO3, фильтрования и концентрирования в условиях вакуума было
получено 2,6 г (94%) продукта в виде воскообразного белого твердого вещества:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.80-0.92 (м,2H), 1.48-1.60 (м,4H), 1.81-1.88 (м,
1H), 1.91-2.00 (м,1H), 3.76 (с,3H), 6.58-6.70 (м,3H), 7.15 (т, J=7,8 Гц,1H), 7.24-7.80
(м,4H).
Стадия iii.
(c) (-)-(транс)-1-[2-(3-метоксифенил)циклопропил]этиламин (изомер A).
Раствор (-)-(транс)-2-[1-[2-(3-метоксифенил) циклопропил]
этил] изоиндол-1,3-диона (изомер A) (2,6 г, 8,1 ммоля) и гидразина (827 г, 25,6 ммоля) в этаноле (75 мл)
перемешивали при комнатной температуре в течение 18 часов. Полученную белую пасту разбавляли
этанолом (100 мл), обрабатывали 10 н. раствором HCl (10 мл) и перемешивали в течение 1 часа. Этот раствор
обрабатывали EtOAc (200 мл) и экстрагировали 1 н. раствором HCl. Кислые вытяжки соединяли,
промывали простым этиловым эфиром, подщелачивали (10 н. раствор NaOH) и экстрагировали CH2Cl2. Органические вещества сушили над K2CO3 и концентрировали в
условиях вакуума с образованием 1,1 г неочищенного масла. После очистки посредством хроматографии на
силикагеле (смесь 10% метилового спирта и CH2Cl2) было получено светлое масло
(600 мл, 40%):
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.85-0.98
(м,2H), 1.10- 1.20 (м,1H), 1.25 (д, J=6.0 Гц,3H), 1.70-1.79 (м,1H), 2.30 (широкий синглет,2H),
2.48-2.52 (м,1H), 3.75 (с,3H), 6.58-6.75 (м,3H), 7.15 (т,J=7.0 Гц,1H).
Стадия iv.
(d) (-)-(транс)-N-[1-[2-(3-метоксифенил)циклопропил] этил] бутанамин (изомер A).
К перемешиваемому с помощью магнитной мешалки раствору
(-)-(транс)-1-[2-(3-метоксифенил)циклопропил]этиламина (изомер A) (200 мг, 1,1 ммоля) и E3 (0,46 мл, 3,3 ммоля) в сухом
дихлорметане (15 мл), находившемуся при 0oC, по каплям
добавляли бутирилхлорид (127 мг, 1,2 ммоля). Полученную суспензию оставляли для нагревания до комнатной температуры и перемешивали в
течение 4 часов. Растворитель удаляли в условиях вакуума, остаток
вываривали с EtOAc (100 мл), последовательно промывали водой, 5% лимонной кислотой, 5% K2CO3, рассолом и сушили
над K2CO3. После фильтрования и
концентрирования в условиях вакуума был получен неочищенный продукт, который очищали посредством испарительной хроматографии (силикагель, смесь
40% EtOAc и гексана), что позволило получить 100 мг
(39%) указанного в заголовке соединения: Rf = 0,75 (смесь 10% метилового спирта и CDCl3);
спектр ЯМР1H
(300 МГц, CDCl3) δ : 0.85-0.97 (м,
4H), 1.04-1.17 (м,2H), 1.24 (д, J=6.6 Гц,3H), 1.61-1.73 (м,2H), 1.77-1.83 (м,1H), 2.19 (т,J=7.6 Гц, 2H), 3.54-3.66 (м,1H), 3.77 (с,3H), 5.59 (с,1H),
6.57-6.71 (м,3H), 7.15 (т, J=7.6 Гц,1H);
инфракрасный спектр (пленка NaCl) 3291, 2963, 1640, 1543, 1455 см-1;
масс-спектр (ESI) m/e 261.
Результаты
анализа, высчитанные для C16H23
NO2:
C 73.53, H 8.87, N 5.36;
обнаружено: C 73.48, H 8.86, N 5.25.
Пример 15 (способ получения 15).
Получение
(транс)-N-[[2-(3-(метилокси)фенил]циклопропи]метил] бутанамида
Раствор (транс)-N-[[2-(3-гидpoкcифeнил)циклопропил] метил] - бутанамида (342 мг, 1,47 ммоля) и
гидроксида натрия (58 мг, 1,45
ммоля) в безводном метаноле (10 мл) концентрировали в условиях вакуума, а остаток измельчали в порошок под слоем безводного толуола (3 мл). Эту суспензию обрабатывали
раствором этиленкарбоната (259
мг, 2,94 ммоля) в безводном толуоле (5 мл). Полученную смесь нагревали с обратным холодильником в течение 12 часов, охлаждали до комнатной температуры и обрабатывали 3
н. раствором NaOH (100 мл),
льдом (50 г) и простым диэтиловым эфиром (100 мл). Водный слой отделяли и экстрагировали свежим простым диэтиловым эфиром (2х100 мл). Соединенные органические экстракты
промывали водой (200 мл),
рассолом (200 мл), сушили (K2CO3), фильтровали и концентрировали в условиях вакуума. После испарительной хроматографии полученного масла (смесь гексана
и этилацетата с
соотношением 1:2) было получено 178 мг (44%) указанного в заголовке соединения в виде светлого масла;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.84-0.92
(м,2H), 0.91
(т,J=7.4 Гц, 2H), 1.21-1.32 (м,1H), 1.63 (секстет, J=7.4 Гц,2H), 1.72-1.78 (м,1H), 2.13 (т, J=7.4 Гц, 2H), 2.30 (широкий синглет, 1H), 3.16-3.34 (м,2H), 3.91 (т, J= 4.5 Гц,2H), 4.03 (т,
J=4.5 Гц, 2H),
5.75 (широкий синглет,1H), 6.58 (т, J=1.5 Гц, 1H), 6.63 (д, J=8.0 Гц,1H), 6.68 (дважды двойной дублет, 0=8.0, 1.5, 1.5 Гц, 1H), 7.13 (т,J=8.0 Гц,1H);
инфракрасный спектр
(пленка NaCl) 3300,
3076, 2962, 1644, 1610, 1582, 1552 см-1;
масс-спектр m/e 276 (M-H-).
Результаты анализа, высчитанные для C16H23
NO3:
C 69.29, H 8.36, N 5.05;
обнаружено: C 68.99, H 8.39, N 4.93.
Пример 16 (способ получения 16).
Получение (-)-(транс)-N-[[2-(3-метоксифенил)цикпопропил] этил] бутанамида.
Стадия i.
(a) (-)-(транс)-1-(цианометил)-2-(3-метоксифенил)циклопропан.
К перемешиваемому с помощью магнитной мешалки раствору (-)-(транс)-2-(3-метоксифенил)циклопропанметанола (1,60 г, 8,8 ммоля) и триэтиламина (1,9 мл, 13,2 ммоля) в дихлорметане (150 мл), находившемуся при 0oC, по каплям добавляли метансульфонилхлорид (1,21 г, 10,6 ммоля). После окончания добавления ледяную баню удаляли и продолжали перемешивать в течение 1 часа. Этот раствор обрабатывали дихлорметаном (150 мл), промывали насыщенным раствором NaHCO3 и сушили K2CO3. После концентрирования в условиях вакуума было получено 1,78 г неочищенного масла, которое использовали без дальнейшей очистки.
Мезилат растворяли в диметилформамиде (100 мл), обрабатывали цианидом натрия (862 мг, 17,6 ммоля) и перемешивали на паровой бане в
течение
4 часов. Этот раствор концентрировали в условиях вакуума, остаток растворяли в простом этиловом эфире (100 мл), промывали водой, рассолом и удаляли растворитель в условиях вакуума с
образованием 1,41
г (85%) светлого масла;
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.90-1.09 (м,2H), 1.23-1.30 (м,1H), 1.81-1.92 (м, 1H), 2.52-2.60 (м,2H)., 3.78 (с,
3H), 6.58- 6.72 (м,
3H), 7.19 (т, J=8.0 Гц,1H).
(b) (-)-(транс)-2-(3-метоксифенил)циклопропанэтиламин.
К перемешиваемой суспензии алюмогидрида лития (650 мг, 17
ммолей) в
тетрагидрофуране (50 мл), находившейся при температуре -45oC, добавляли раствор (-)-(транс)-1-(цианометил)-2-(3- метоксифенил)циклопропана (1,41 г, 7,5 ммоля) в тетрагидрофуране
(25 мл),
поддерживая температуру ниже -30oC. После окончания добавления суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 4 часов. Затем ее вновь
охлаждали до
-45oC и по каплям осторожно добавляли 1 н. раствор HCl (50 мл). Полученную суспензию перемешивали при комнатной температуре в течение 30 минут, фильтровали через целит и
тщательно промывали
фильтровальную лепешку простым этиловым эфиром. Фильтраты соединяли, экстрагировали 1 н. раствором HCl, а кислые слои подщелачивали (30% NaOH). Основный раствор экстрагировали с
помощью CH2
Cl2, сушили (K2CO3) и концентрировали в условиях вакуума с образованием 500 мг (39%) светлого масла:
спектр ЯМР1H (300 МГц,
CDCl3)
δ : 0.75-0.82 (м,1H), 0.88-0.96 (м,1H), 1.00-1.11 (м, 1H), 1.45-1.70 (м,3H), 2.62 (широкий синглет,2H), 2.88 (т, J= 7.5 Гц,2H), 3.77 (с,3H), 6.52-6.70 (м,3H), 7.18 (т, J=8.0
Гц,1H).
(c) (-)-(транс)-N-[[2-(3-метоксифенил)циклопропил]этил] бутанамид.
К перемешиваемому с помощью магнитной мешалки раствору
(-)- (транс)-2-(3-метоксифенил)циклопропанэтиламина (400 мг, 2,2 ммоля) и Et3N (0,86 мл, 6,6 ммоля) в сухом дихлорэтане (15 мл), находившемуся при 0oC, по каплям добавляли
бутирилхлорид (256 мг, 24 ммоля). Полученную суспензию оставляли для нагревания до комнатной температуры и перемешивали в течение 18 часов. После концентрирования в условиях вакуума был получен
неочищенный продукт, который затем очищали посредством испарительной хроматографии (силакагель, смесь 30% EtOAc и гексана), что позволило получить 250 мг (44%) светлого масла:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.70-0.81 (м,1H), 0.88-1.01 (м,5H), 1.54- 1.68 (м, 5H), 2.12 (т,J=7.2 Гц,2H), 3.29-3.36 (м, 2H), 3.77 (с,3H), 5.75 (широкий синглет,1H), 6.54-6.71
(м,
3H), 7.14 (т,J=7.8 Гц,1H);
инфракрасный спектр (пленка NaCl) 3295, 2962, 1644, 1604, 1551, 1494 см-1;
масс-спектр (ESI) m/e 261;
[α]0 -66,3o (с=1, CH2Cl2).
Результаты анализа,
высчитанные для
C16H26NO2·0,20H2O:
C 72.53, H 6.90, N 5.29;
обнаружено: C 72.67, H 9.03, N 5.29.
Пример 17 (способ получения 17).
Получение (транс)-N-[[3-(3-метоксифенил)-2,2-диметил-1- циклопропил]метил]бутанамида.
Стадия i.
(a) Этиловый эфир (транс)-3-(3-метоксифенил)-2, 2- диметилциклопропанкарбоновой кислоты.
К перемешиваемому раствору иодида изопропилтрифенилфосфония (50 г, 116 ммолей) в тетрагидрофуране (250 мл),
находившемуся при 0oC,
по каплям добавляли бутиллитий (2,3 М раствор в гексане, 42,2 мл, 115,6 ммоля), поддерживая температуру ниже 5oC. Этот раствор перемешивали в течение 1
часа, по каплям вливали раствор
этилового эфира коричной кислоты (18,33 г, 89 ммолей) в тетрагидрофуране (50 мл), оставляли для нагревания до комнатной температуры и перемешивали в течение 4 часов.
Полученную суспензию быстро
охлаждали насыщенным раствором NH4Cl (200 мл) и экстрагировали этилацетатом. Органические экстракты соединяли, промывали рассолом, сушили (K2CO3) и концентрировали с
образованием светло-коричневого твердого вещества. Это твердое вещество очищали путем перегонки по методу Кюгельрора (110oC, 0,5 мм) с образованием 13,0 г
(59%) светлого масла:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.95 (с,3H), 1.12 (т, J=6.5 Гц, 3H), 1.36 (с,3H), 1.95 (д, J=6.0 Гц,1H), 2.62 (д, =6.0 Гц,1H), 3.80 (с,
3H), 4.12 (к,J=7.0Гц,2H),
6.65-6.68 (м,2H), 7.18-7.26 (м,2H).
Стадия ii.
(b) (транс)-3-(3-метоксифенил)-2,2-диметил-циклопропил - метанол.
К
суспензии алюмогидрида лития (4,21
г, 111 ммолей) в тетрагидрофуране (200 мл), находившейся при температуре -45oC, по каплям добавляли раствор этилового эфира (транс)-3-(3- метоксифенил)
-2,2-диметил-циклопропанкарбоновой
кислоты (12,5 г, 50,4 ммоля) в тетрагидрофуране (20 мл). Охлаждающую ванну удаляли и реакционную смесь оставляли для нагревания до комнатной температуры, после чего
сразу же вновь охлаждали до -45oC. Осторожно добавляли раствор KHSO4 (24 г, 177 ммолей) в воде (50 мл), повышая температуру до -5oC. Полученную пасту перемешивали при
комнатной температуре в течение 1
часа, фильтровали через целит, а фильтровальную лепешку промывали простым этиловым эфиром. Соединенные фильтраты промывали холодным (0oC) 1 н. раствором
HCl (3 раза), 5% K2CO3 (1 раз), рассолом (1 раз), сушили (K2CO3) и концентрировали с образованием 10,1 г (97%) светлого масла:
спектр ЯМР1H (300 МГц, CDCl3)
δ : 0.85 (с,3H), 1.25 (с,3H), 1.78 (д, J= 6.0 Гц,1H), 3.62-3.72 (м,3H), 3.79 (с,3H), 3.82 (двойной дублет, J=7, 10 Гц, 1H), 6.70-6.78 (м,2H), 7.20 (т,
J=7.5 Гц, 1H), 7.35 (м,1H).
Стадия iii.
(c) (транс)-(3-(3-метоксифенил)-2,2-диметил-циклопропил) -метил) азид.
К раствору (транс)-(3-(3-метоксифенил)-2,2-диметил- циклопропил)метанола (10,0 г, 48,5 ммоля) и триэтиламина (10,1 мл, 72,8 ммоля) в дихлорметане (100 мл), находившемуся при 0oC, по каплям добавляли метансульфонилхлорид (6,11 г, 53,4 ммоля). После окончания добавления ледяную баню удаляли и продолжали перемешивать в течение 30 минут. Реакционную смесь разбавляли дихлорметаном (100 мл), промывали водой, насыщенным раствором NaHCO3 и сушили над K2CO3. После концентрирования в условиях вакуума было получено 13,09 г окрашенного масла, пригодного для использования на следующей стадии.
Вышеуказанный мезилат
растворяли в диметилформамиде (100 мл), обрабатывали азидом натрия (3,78 г, 58,2 ммоля) и перемешивали при комнатной температуре в течение
18 часов. Эту смесь концентрировали, а остаток растворяли в
простом этиловом эфире, промывали водой (1 раз), рассолом (1 раз) и удаляли растворители в условиях вакуума, что позволило получить 10,1 г
светлого масла, пригодного для использования без дальнейшей
очистки:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.85 (с,3H), 1.30 (с,3H), 1.39-1.42 (м, 1H), 1.80 (д,
J=6.0 Гц,1H), 3.30 (двойной дублет, J=8,10 Гц,1H), 3.51 (двойной
дублет, J=6.0, 8.3 Гц, 1H), 3.80 (с,3H), 6.68-6.79 (м,3H), 7.20 (т, J=7.5 Гц,1H).
Стадия iv.
(d) (транс)-(3-(3-метоксифенил)-2,2-диметил-циклопропил) -метил)амин.
Перемешиваемую суспензию алюмогидрида лития (4,80 г, 126,6 ммоля) в тетрагидрофуране (100 мл) при температуре -30oC обрабатывали раствором (транс)-(3-(3-метоксифенил)-2,
2- диметилциклопропил метил азида (6,48 г, 30,14 ммоля) в тетрагидрофуране (10 мл), поддерживая температуру ниже -30oC. После
окончания добавления суспензию оставляли для нагревания до
комнатной температуры и перемешивали в течение 4 часов. Эту суспензию охлаждали до -45oC и по каплям (осторожно!) добавляли
раствор KHSO4 (16 г) в воде (20 мл). Полученную
суспензию перемешивали при комнатной температуре в течение 30 минут, фильтровали через целит, а фильтровальную лепешку тщательно промывали
простым этиловым эфиром. Фильтраты соединяли, экстрагировали
1 н. раствором HCl (2 раза) и кислые слои подщелачивали (30% NaOH). Основный раствор экстрагировали CH2Cl2 (3 раза),
сушили (K2CO3) и концентрировали с
образованием 2,80 г (47%) светлого масла:
спектр ЯМР1H (300 МГц, CDCl3) δ : 0.82 (с,3H), 1.22 (с,3H),
1.60-1.68 (м, 4H), 2.75-2.92 (м,2H), 3.78 (с,3H), 6.68-6.75
(м,3H), 7.15 (т,J=7.5Гц, 1H).
Стадия v.
(e) (транс)-N-[[3-(3-метоксифенил)-2, 2-диметил-1- циклопропил]метил]бутанамид.
(3-(3-метоксифенил)-2,
2-диметилциклопропил)метил)амин (620 мг, 3,0 ммоля) ацилировали бутирилхлоридом (0,34 мл, 3,3 ммоля), как описывалось
в примере 1, что позволило получить 550 мг (67%) светлого масла:
спектр
ЯМР1H (300 МГц, CDCl3) δ : 0.80 (с,3H), 0.90 (т,J=7.4 Гц, 3H), 1.23 (с, 3H), 1.23-1.30
(м,1H), 1.58-1.71 (м,3H), 2.14 (т, J= 7.3 Гц,2H), 3.39 (т,0=5.4 Гц,2H), 3.76 (с,3H),
5.50 (широкий синглет,1H), 6.65-6.71 (м, 3H), 7.15 (т, J=7.8 Гц,1H);
инфракрасный спектр (пленка NaCl)
3294, 2964, 1642, 1550, 1490, 1456, 1274 см-1;
масс-спектр m/e 275,
39.
Результаты анализа, высчитанные для C17H25NO2:
C
74.14, H 9.15, N 5.09;
обнаружено: C 73,95, H 9.21, N 5.11.
В таблице 1 приведены химические данные для соединений формулы I, в которой X - OCH3, R - водород, G - метилен, а другие заместители имеют указанные в таблице значения.
В таблице 2 приведены химические данные для соединений формулы I, в которой R1 и R - водород, G - метилен, а R2, X, Y и Z имеют значения, указанные в таблице.
В таблице 3 приведены химические данные для соединений формулы I, в которой X - OCH3, Y и Z - водород и R1 - водород. Заместители R, G и R2 имеют значения, указанные в таблице.
Получение соединений формулы II из 2-циклоалкенона.
Пример 140: Смесь цис- и транс-N-[3-(3-метоксифенил)циклопентил] бутанамида.
Раствор 2-циклопентанона (8,21 г, 0,1 моля) в тетрагидрофуране (50 мл) по каплям добавляли к реактиву Гриньяра, полученному из 3-броманизола (18,7 г, 0,1 моля) и металлического магния (2,5 г) в тетрагидрофуране (200 мл). Раствор перемешивали в течение 18 часов, после чего охлаждали путем медленного добавления 3 н. раствора HCl (75 мл). Эту смесь перемешивали в течение 1 часа и разбавляли простым этиловым эфиром (200 мл). Органический слой отделяли, дважды экстрагировали водой и дважды рассолом. Органический раствор концентрировали до образования янтарного масла, которое хроматографировали на силикагеле с использованием в качестве элюента смеси CH2Cl2 и гексанов, что позволило получить требуемый циклопентанон (2,0 г, 10,5%, инфракрасный спектр: 1734 см-1).
Раствор вышеуказанного циклопентанона (1,9 г, 10 ммолей), хлористоводородного гидроксиламина (2,8 г, 40 ммолей) и гидроксида натрия (4 мл 10 н. раствора, 40 ммолей) в этаноле (150 мл) нагревали с обратным холодильником в течение 18 часов. Эту смесь охлаждали, а затем гидрировали на скелетном никелевом катализаторе под давлением 4,2 ат в течение 4 часов. Смесь фильтровали, а фильтрат концентрировали в условиях вакуума. Остаток растворяли в ацетонитриле (100 мл) и HCl (2 мл 12 н. раствора), после чего раствор концентрировали в условиях вакуума. Остаток растирали в порошок с ацетонитрилом, в результате чего было получено липкое твердое вещество, которое перекристаллизовывали из этанола с образованием хлористоводородного амина в виде белого порошка (0, 5 г, 26,2%).
Раствор промежуточного вещества, представляющего собой соль амина (0.5 г, 2,62 ммоля), в пиридине (10 мл) охлаждали в ледяной бане, добавляя при этом бутирилхлорид (0,363 мл, 3,5 ммоля). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, а затем концентрировали. Остаток растворяли в метиленхлориде (50 мл) и дважды экстрагировали 1 н. раствором HCl (30 мл). Органический слой концентрировали и неочищенный продукт хроматографировали на силикагеле с использованием в качестве элюента смеси метиленхлорида и этилацетата, что позволило получить смесь цис- и транс-N-[3- (3-метоксифенил)циклопентил]бутанамида в виде светлого масла (0,3 г, 43,6%).
Высчитано для C16H23NO2: C 74.53, H 6.67, N 5.36.
Обнаружено: C 73.18, H 8.79, N 5.36.
Масс-спектр (изобутан-DCl): 262 (M+Н).
Инфракрасный спектр (пленка): 3288, 2960, 1638, 1548, 1264, 698 см-1.
Получение соединений формулы II из 3-алкокси-циклоалкенона.
Пример 141: (Цис)-N-[3-(3-метоксифенил)циклогексил]бутанамид.
Раствор 3-(2-пропилокси)-1-циклогексенона (28,0 г, 0,182 моля) в тетрагидрофуране (100 мл) по каплям добавляли к реактиву Гриньяра, полученному из 3-броманизола (37,4 г, 0,2 моля) и металлического магния (6, 08 г, 0,25 моля), в тетрагидрофуране (400 мл). Этот раствор перемешивали в течение 3 часов, а затем охлаждали путем медленного добавления 3 н. раствора HCl (150 мл). Эту смесь перемешивали в течение 10 часов и разбавляли простым этиловым эфиром (400 мл). Органический слой отделяли, дважды экстрагировали водой (100 мл) и дважды рассолом. Органический раствор концентрировали до образования янтарного масла, которое перегоняли по методу Кюгельрора с образованием промежуточного энона в виде бледно-янтарного масла (35,1 г, 95,5%).
Спектр ПМР (CDCl3, 300 МГц)
δ : 2.17 (м,2H), 2.50 (т,2H), 2.77 (т,2H), 3.85 (с, 3H),
6.41 (с,1H), 6.97 (двойной дублет,1H), 7.06 (т,1H), 7.15 (д, 1H), 7.30 (т,1H);
инфракрасный спектр (пленка): 1663 см-1.
Смесь вышеуказанного янтарного масла (35, 1 г, 0,174 моля), этиленгликоля (15 г, 0,242 моля) и пара-толуолсульфокислоты (0,5 г) в толуоле (200 мл) нагревали с обратным холодильником под конденсационным горшком Дина-Старка в течение 18 часов. Добавляли еще одну порцию пара-толуолсульфокислоты (0,5 г) и продолжали нагревание в течение 24 часов. Полученный раствор охлаждали и дважды экстрагировали насыщенным раствором карбоната натрия (100 мл), дважды водой и дважды рассолом. Органический слой концентрировали до образования коричневого масла, которое перегоняли по методу Кюгельрора с образованием промежуточного кеталя в виде светлого масла, которое отверждали.
Спектр ПМР (CDCl3), 300 МГц) δ : 1.85 (т,2H), 2.4- 2.5 (м,2H), 2.66 (широкий синглет, 2H), 3.84 (c,3H), 4.04 (c,4H), 6.17 (м,1H), 6.80 (двойной дублет,1H), 8.95 (т,1H), 7.00 (д,1H), 7.25 (т,1H).
Раствор промежуточного масла в этаноле (200 мл) гидрировали на 10% палладированном угле (1 г) под давлением 4.2 ат в течение 18 часов. Эту смесь фильтровали, а фильтрат перемешивали с 3 н. раствором HCl (75 мл) в течение 4 часов. Раствор концентрировали и трижды экстрагировали этилацетатом (200 мл). Органические экстракты сушили вместе с рассолом и концентрировали. Маслянистый остаток перегоняли по методу Кюгельрора с образованием промежуточного кетона в виде светлого масла (29,97 г, 95%).
Спектр ПМР (CDCl3), 300 МГц) δ : 1.73-1.95 (м,2H), 2.05-2.2 (м,2H), 2.33-2.63 (м, 4H), 2.95-3.07 (м,1H), 3.87 (с,3H), 6.70-6.85 (наложение,3H), 7.20-7.30 (м,1H).
Инфракрасный спектр (пленка): 1711 см-1.
Раствор вышеуказанного кетона (10,86 г, 53,24 ммоля) в этаноле (150 мл) добавляли к смеси сульфата гидроксиламина (12,31 г, 0,15 ммоля) и 10 н. раствора гидроксида натрия (15 мл), после чего нагревали с обратным холодильником в течение 6 часов. Эту смесь охлаждали и гидрировали на скелетном никелевом катализаторе (2 г) под давлением 4,2 ат в течение 16 часов. Полученную смесь фильтровали, а фильтрат концентрировали до образования липкого остатка. Этот остаток распределяли между этилацетатом и водой. Органический слой отделяли, дважды экстрагировали водой (75 мл) и трижды 3 н. раствором HCl (в общей сложности 100 мл). Кислые вытяжки подщелачивали 50% гидроксидом натрия и трижды экстрагировали этилацетатом (100 мл). Органические экстракты сушили над рассолом и концентрировали. Остаток растворяли в ацетонитриле (50 мл) и по каплям добавляли 12 н. раствор HCl, что позволило получить промежуточный хлористоводородный амин в виде белого осадка, который фильтровали и сушили на воздухе (2,54 г, 19,8%).
Раствор промежуточной соли амина (0,53 г, 2,59 ммоля) в пиридине (10 мл) охлаждали в ледяной бане, добавляя с помощью шприца бутирилхлорид (0,33 мл, 3,2 ммоля). Реакционную смесь перемешивали в течение 2 часов при комнатной температуре, а потом концентрировали. Остаток растворяли в метиленхлориде (30 мл) и дважды экстрагировали 1 н. раствором HCl (30 мл). Органический слой концентрировали и неочищенный продукт хроматографировали на силикагеле с использованием в качестве элюента смеси метиленхлорида и этилацетата, что позволило получить (±)-цис-N- [3-(3-метоксифенил)циклогексил]бутирамид в виде светлого масла (0,45 г, 62,7%).
Высчитано для C17H25NO2·0,2H2O: C 73.18, H 9.18, N 5.02.
Обнаружено: C 73.27, H 9.17, N 5.01.
Масс-спектр (изобутан-DCl): 276 (M+H).
Инфракрасный спектр (пленка): 3286, 2932, 1640, 1548, 1268, 698 см-1.
Спектр CMP (CDCl3) δ : 13.68, 19.25, 25.05, 32.93, 38.93, 41.10, 43.12, 48.43, 55.12, 111.18, 112.70, 119.11, 129.31, 147.87, 159.60, 171.98 частей на миллион.
Пример 142: (± )-цис-N-[3-(3-метоксифенил)-циклогексил] 2-метил-пропанамид.
Это соединение было получено в соответствии со способом, описанным в примере 2, и выделено в виде белого порошка (0,49 г, 68,3%, температура плавления 89 -92oC).
Высчитано для C17H25NO2·0,09H2O: C 73.71, H 9.16, N 5.06.
Обнаружено: C 73.78, H 9.16, N 5.01.
Масс-спектр (изобутан-DCl): 276 (M+H).
Инфракрасный спектр (пленка): 3290, 2930, 1642, 1544, 1236, 698 см-1 .
Спектр CMP (CDCl3) δ : 19.60, 19.67, 25.08, 32.91, 32.96, 35.78, 41.17, 43.16, 48.31, 55.15, 111.23, 112.73, 119.14, 129.33, 147.91, 159.66, 175.97 частей на миллион.
Пример 143: (±)-цис-N-[3-(3-метоксифенил)- циклогексил]ацетамид.
Это соединение было получено в соответствии со способом, описанным в примере 2, и выделено в виде светлого масла (0,25 г, 38,8%).
Высчитано для C15H21NO2·0,1H2O: C 72.29, H 8.59, N 5.60.
Обнаружено: C 72.31, H 8.58, N 5.62.
Масс-спектр (изобутан-DCl): 248 (M+H).
Инфракрасный спектр (пленка): 3284, 2932, 1652, 1554, 1270, 698 см-1.
Спектр CMP (CDCl3) δ : 23.56, 25.07, 32.93, 33.01, 40.95, 43.12, 48.71, 55.15, 111.23, 112.73, 119.13, 129.34, 147.84, 159.65, 169.08 частей на миллион.
Пример 144: (Цис)-N-этил-N'-[3-(3-метоксифенил)-циклогекси] мочевина.
Соединение было получено в соответствии со способом, описанным в примере 2, и было выделено в виде светлого масла (0,36 г, 49,2%).
Высчитано для C16H24N2O2· 0,35H2O: C 67.98, H 8.81, N 9.91.
Обнаружено: C 67.92, H 9.01, N 9.99.
Масс-спектр (изобутан-DCl): 277 (M+H).
Инфракрасный спектр (пленка): 2930, 1632, 1570, 1268, 1156, 1048 см-1.
Спектр CMP (CDCl3) δ : 15.39, 25.15, 33.07, 33.58, 35.35, 41.61, 43.25, 49.67, 55.12, 111.13, 112.13, 119.13, 129.30, 147.93, 157.67, 159.58 частей на миллион.
Пример 145 (измерение мелатонинстимулирующего связывания).
Мелатонинстимулирующее связывание, вызываемое соединениями формулы 1, определяли с помощью модифицированного метода Репперта С.М. и др. (Neuron, т. 13, стр. 1177-1185, 1994). Активными считались соединения со значениями IC50, равными 600 нм или меньше. Ниже описываются реактивы, мембраны и другие параметры, используемые при выполнении этого анализа.
Реагенты.
(a) 50 ммолей трис-буфера, содержащего 12,5 ммоля MgCl2 и 2 ммоля этилендиаминтетрауксусной кислоты (pH 7,4 при температуре 37oC).
(b) Промывочный буфер: 20 ммолей трис-основания, содержащего 2 ммоля MgCl2 (pH 7,4 при комнатной температуре).
(c) c-Хлормелатонин (конечная концентрация, равная 10-5 молей).
(d) 2-(I125)-Иодмелатонин (конечная концентрация, равная 100 пм).
(Источник NEN).
Приготовление мембраны.
кДНК человека (MLIA человека) в пкДНКИ вводили в COS-1 клетки по методу введения диэтиламиноэтил-декстрина. Через три дня после удаления сред планшеты промывали забуференным физиологическим раствором; клетки удаляли с помощью сбалансированного солевого раствора Хэнкса и осаждали центрифугированием. Надосадочную жидкость удаляли, а капли осадка замораживали. Для получения мембранных гомогенатов капли осадка оттаивали и вновь суспендировали в триметилолэтановом буфере, трис-основании, MgCl2, этилендиаминтетрауксусной кислоте (pH 7,4 при температуре 37oC) и добавляли апротинин, лейпептин и фенилметилсульфонилфторид. Клетки гомогенизировали и центрифугировали, полученный осадок вновь суспендировали в триметилолэтане и замораживали. Во время анализа небольшую аликвоту оттаивали на льду и вновь суспендировали в триметилолэтановом буфере.
Инкубация.
При температуре 37oC в течение 1 часа. Реакцию прекращали фильтрованием.
В таблице 4 представлены показатели мелатонинстимулирующего действия соединений формулы I, в которой X - OCH3, R - водород, G - метилен, а другие заместители имеют значения, указанные в таблице.
В таблице 5 представлены показатели мелатонинстимулирующего действия соединений формулы I, в которой R1 и R - водород, G - метилен, a R2, X, Y и Z имеют значения, указанные в таблице.
В таблице 6 представлены показатели мелатонинстимулирующего действия соединений формулы I, в которой X - OCH3, Y и Z - водород и R1 - водород. Заместители R, G и R2 имеют значения, указанные в таблице.
В объем настоящего изобретения входят все обоснованные варианты, которые могут быть осуществлены специалистом в этой области.
Реферат
Описываются новые производные N-ацил-2-арилциклоалкиламина формулы I или II, стимулирующие синтез мелатонина, в которой X - галоген, водород, C1-4 -алкил или OR5, где R5 представляет собой водород, C1-4-перфторалкил, C1-4-фторалкил, C1-4-пердейтероалкил, C1-20-алкил, C4-20-алкциклоалкил, C2-20-карбонитрилоалкил, C3-22-карбоалкоксиалкил, C3-20-алкенил, C3-20-алкинил, C9-20-фенилалкил, C9-20 -фенилалкенил, C9-20-фенилалкинил, C2-20-гидроксиалкил, C8-20-фенилоксиалкил, C7-20-пиридилалкил или C6-20-пиррилалкил; Y - водород или галоген; Z - водород, галоген, циано, фенил, C7-20-фенилалкил, C8-20-фенилалкинил или C2-20-алкамидо; R в обоих случаях является водородом, галогеном или C1-4 -алкилом; n равно 1 или 2; G - двухвалентный метилен, этилен или C1-4-алкметиленовый фрагмент; R1 - водород; C1-4-алкил или бензил; R2 - C1-6 -алкил, C2-6-алкенил, C3-6-циклоалкил, C2-4-алкоксиалкил, C1-4-трифторметилалкил, C2-8-алкилтиоалкил или NR3R4, где R3 и R4 независимо друг от друга выбирают из водорода и C1-4-алкила, но R3 и R4 не могут одновременно быть водородом. Новые производные N-ацил-2-арилциклопропилметиламина обладают лекарственными и биологически активными свойствами. Эти соединения стимулируют синтез мелатонина, что делает их полезными для лечения некоторых заболеваний. Описывается также способ их получения. 3 с. и 16 з.п.ф-лы, 6 табл.

или

Формула

в которой Х - галоген, водород, С1-4-алкил или OR5 где R5 представляет собой водород, С1-4-перфторалкил, С1-4-фторалкил, С1-4-пердейтероалкил, С1-20-алкил, С4-20-алкциклоалкил, С2-20-карбонитрилоалкил, С3-22-карбоалкоксиалкил, С3-20 -алкенил, С3-20-алкинил, С9-20-фенилалкил, С9-20-фенилалкенил, С9-20-фенилалкинил, С2-20-гидроксиалкил, С8-20-фенилоксиалкил, С7-20-пиридилалкил или С6-20-пиррилалкил;
Y - водород или галоген;
Z - водород, галоген, циано, фенил, С7-20-фенилалкил, С8-20-фенилалкинил или С2-20-алкамидо;
R в обоих случаях является водородом, галогеном или С1-4-алкилом;
n равно 1 или 2;
G - двухвалентный метилен, этилен или С1-4 -алкметиленовый фрагмент;
R1 - водород, С1-4-алкил или бензил;
R2 - С1-6-алкил, C2-6-алкенил, С3-6 -циклоалкил, С2-4-алкоксиалкил, С1-4-трифторметилалкил, С2-8-алкилтиоалкил или N R3 R4, где R3 и R4 независимо друг от друга выбирают из водорода и С1-4-алкила, но R3 и R4 не могут одновременно быть водородом.
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]пропанамид,
(+)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]ацетамид,
(-)-(транс)-N-[[2-(3-метоксифенил)циклопропил]метил]-2-метилпропанамид.
(транс)-N-[[2-(4-хлор-3-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(4-хлор-5-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(2,4-дибром-5-метоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(2-иод-5-метоксифенил)циклопропил]метил]бутанамид,
(-)-(транс)-N-[[2-(2-иод-3-метоксифенил)циклопропил]метил]бутанамид.
(транс)-N-[[2-(3-[(этоксифенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-(октилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-(децилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-ундецилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[(3-додецилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(гептилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(нонилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(2-пропенилокси)фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-(6-фенилгексил)окси]фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-[(7-фенилгептил)окси]фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-[(2-фенилэтил)окси]фенил]циклопропил]метил]бутанамид,
(транс)-N-[[2-[3-[(3-(3-метоксифенил)пропокси] фенил] циклопропил]метил] бутанамид.
(транс)-N-[[2-(3-хлорфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-бромфенил)циклопроп-1-ил]метил]бутанамид,
(-)-(транс)-N-[[2-(3-фторфенил)циклопропил]метил]бутанамид,
(транс)-N-[[2-(2,5-дифторфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(2-бром-5-фторфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-метилфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-этилфенил)циклопроп-1-ил]метил]бутанамид,
(транс)-N-[[2-(3-трифторметокси)фенил]циклопроп-1-ил]метил]бутанамид.
(цис)-N-[3-(3-метоксифенил)циклогексил]ацетамид,
(цис)-N-[3-(3-метоксифенил)циклогексил]-2-метил-пропанамид,
(цис)-N-[3-(3-метоксифенил)циклогексил]бутанамид,
(цис)-N-этил-N'-[3-(3-метоксифенил)циклогексил], мочевину,
N-[3-(3-метоксифенил)циклопентил]бутанамид.
Комментарии